Abstract
The Conus subgenus Tesseliconus, whose members are believed to be primarily worm-hunters, phylogenetically clusters closely with piscivorous groups relative to most other vermivorous subgenera. A previous study even documented the Tesseliconus species C. tessulatus to opportunistically prey on fish. Here, we identified and analyzed putative conopeptide sequences from the venom gland transcriptome of C. tessulatus and its sister species C. eburneus. From the set of assembled sequences with predicted complete coding sequences, we identified 260 C. ebureneus and 339 C. tessulatus transcripts for which assignment to a conopeptide gene superfamily and/or cysteine framework was possible. In addition, we identified over 50 transcripts per species that are highly similar to previously reported disulfide-poor conopeptides. Agglomerative clustering (75% similarity threshold) of the predicted signal sequences revealed the presence of 18 possibly novel gene superfamilies, alongside 10 known gene superfamily clusters. Inter- and intra-species variations in conopeptide diversity and expression were also observed, hinting to a number of potential but not necessarily exclusive scenarios. In particular, we hypothesize that the Tesseliconus species investigated in this study might be targeting a more diverse prey type than previously thought, and that individuals even of the same species may exhibit subtle differences in prey preference that allows them to better coexist within a given environment.
1 Introduction
The marine gastropod genus Conus is a highly diverse group of predatory snails, with over 800 species registered to date (WoRMS Editorial Board, 2024). These marine snails are carnivorous in nature, feeding mainly on worms, fish, and other mollusks. Among the three major prey types, worms appear to be the ancestral cone snail diet, with the piscivores and molluscivores evolving multiple times from different vermivorous lineages that were accompanied by various behavioral and physiological changes (Olivera et al., 2015).
The subgenus Tesseliconus is particularly interesting because members of this clade, believed to be primarily vermivorous, were phylogenetically closer to fish-hunting groups relative to most other worm hunters (Kraus et al., 2011; Olivera et al., 2015). A study on Conus tessulatus revealed that although this Tesseliconus species preferred to target worms, mainly nereid polychaetes, they also appear to consider fish as a secondary prey (Aman et al., 2015). The same study also reported the presence of δ-conotoxins obtained from C. tessulatus and Conus eburneus that exhibit striking sequence similarities with their counterparts from piscivorous cone snail species, but are relatively distant from those in mollucivores – providing insights into the possible origins of piscivory in cone snails (Aman et al., 2015).
A recent study investigating χ-conotoxins as an evolutionary innovation of mollusk-hunting cone snails placed the subgenus Tesseliconus as the phylogenetically closest group to the monophyletic molluscivorous clade (Espino et al., 2024). This observation therefore puts the Tesseliconus clade as a sister lineage to molluscivores and one of the piscivorous clusters, making its conopeptide composition even more interesting. The Tesseliconus species C. tessulatus is also arguably the most successful Conus species in terms of geographic range, found throughout the Indo-Pacific region, suggesting that this species, and other Tesseliconus members for that matter, may have the potential to prey on a wider array of organisms.
Nonetheless, most of the molecular studies involving Tesseliconus species were done at the protein or proteome level (Aman et al., 2015; Itang et al., 2020; Yang et al., 2017), probably with the exception of a study involving C. eburneus in which conotoxins were identified from cDNA library constructs (Liu et al., 2012). However, more recent studies on Conus venom duct transcriptomes using high-throughput sequencing technologies, such as RNASeq, were able uncover much greater conopeptide diversity (Barghi et al., 2015b; Gao et al., 2018; Li et al., 2017; Peng et al., 2016; Yao et al., 2019).
Thus, in this study, total RNA extracts from the venom ducts of two Tesseliconus species, C. eburneus and C. tessulatus, were subjected to high-throughput sequencing and subsequent transcriptomics analysis. The predicted conopeptide transcripts were investigated in terms of interspecific diversity, intraspecific variations in expression, as well as similarities with previously characterized peptides. Possibly novel conopeptide gene superfamilies and mature peptide configurations were also explored and reported.
2 Materials and methods
2.1 Sample collection, total RNA extraction and sequencing
Three adult specimens of C. eburneus (Ce1, Ce2, and Ce3) and C. tessulatus (Ct1, Ct2, and Ct3) were collected by the contracted local fishermen in Caw-oy Lapu-Lapu City, Cebu, Philippines. The specimens were initially identified by morphological examination of the shell. Prior to dissection, live snails were acclimatized for 24 hours in an aerated (improvised) aquarium filled with sea water. The snails were dissected directly on ice, and venom ducts were carefully obtained and stored in 1 ml of RNAlater® (Invitrogen) at 4°C prior to long term storage at -80°C. The total RNA was isolated using the Trizol extraction method. Briefly, thawed venom duct was homogenized using 2.0 mm of ZR bashing beads (ZYMO research) in a bead beater (Precellys, Berlin Technologies) with 1 ml TRIzol reagent (Sigma-Aldrich TRI reagent®). The total RNA was extracted from the homogenate using chloroform and precipitated using isopropanol. The pellet was washed twice with 75% EtOH and air dried. The pelleted RNA was resuspended in 50 ul of RNAase-free water, and clean-up was done using the Qiagen purification kit following the manufacturers protocol. The quality and quantity of total RNA was evaluated using the Agilent TapeStation and Qubit, respectively (Supplementary Figure S5). Individual library was constructed in each sample using the TruSeq Stranded mRNA Library prep kit and the sequencing was performed in Illumina NextSeq550 using Mid Output v2.5 with 300 cycles. All library preparations and sequencing were done in the sequencing facility of PCARI-SGCL, University of the Philippines.
2.2 Quality control of raw sequence reads
Six sets of paired-end sequence reads from three individuals of C. eburneus and C. tessulatus were pre-processed mainly using the suggested best practices for de novo transcriptome assembly with Trinity by the Harvard FAS Informatics group (https://informatics.fas.harvard.edu/best-practices-for-). Briefly, the initial and final quality assessment of raw and filtered sequence data were done using Fastqc (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Erroneous base calls were then corrected based on k-mer composition using Rcorrector (Song and Florea, 2015), and reads that were deemed unfixable were subsequently removed using the FilterUncorrectabledPEfastq.py script obtained from the Transcriptome Assembly Tools repository of the Harvard Informatics group (https://github.com/harvardinformatics/TranscriptomeAssemblyTools). Adapter and quality trimming of the error corrected reads was implemented afterwards using the tool Trim Galore (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/). The remaining reads were further mapped against the SILVA database of rRNA sequences (Quast et al., 2013) using Bowtie2 (Langmead and Salzberg, 2012), and only reads that did not map to any of the rRNA sequences were retained. For each of the individual samples, >90% of the reads passed the filtering step (Supplementary Table S7).
2.3 De novo assembly of venom duct transcripts and abundance estimation
Quality filtered paired-end read pairs from three individuals of each species were co-assembled using three different tools: Trinity (Haas et al., 2013) implemented with k-mers 25 and 31, Trans-ABySS (Robertson et al., 2010), and rnaSPAdes (Bushmanova et al., 2019) – the latter two using default parameters. The resulting assemblies from each of the tools and parameters were then combined and clustered at 100% sequence similarity threshold using CD-HIT (Fu et al., 2012).
Transcripts from the resulting combined assemblies (one each for C. eburneus and C. tessulatus) were further assessed and filtered using TransRate (Smith-Unna et al., 2016), and only those included in the set of good contigs were used in the subsequent analyses. From the set of good contigs, transcript expression levels for each of the individual samples were estimated using Salmon (Patro et al., 2017).
2.4 Conopeptide identification and precursor sequence analysis
Open reading frames (ORFs) were detected from each of the assembled transcripts using Transdecoder (https://github.com/TransDecoder/TransDecoder), which also outputs the corresponding amino acid sequences for the predicted coding regions. Note that only the best coding region per transcript was retained, either by being the longest predicted ORF for that particular transcript or by having significant sequence similarities with known conopeptides as determined by running the blastp algorithm of the Basic Local Alignment and Search Tool (BLAST) (Altschul et al., 1990) against a custom curated database of conopeptide sequences collected from the following data sources: GenBank (National Center for Biotechnology Information, USA), UniProt (Swiss Institute of Bioinformatics, Switzerland), and ConoServer (Kaas et al., 2012).
Amino acid sequences of transcripts with conopeptide matches were further analyzed using the ConoPrec tool in ConoServer (http://www.conoserver.org/?page=conoprec) in order to determine the precursor peptide structure, particularly the signal and mature peptide sequences. The same tool also assigns gene superfamily and cysteine framework classifications to each of the putative conopeptide sequences.
2.5 Inter- and intra-specific conopeptide expression and diversity analysis
Putative conopeptides with known gene superfamily or cysteine framework classifications were gathered together with their corresponding expression estimates. Scatter plots of cumulative expression levels per gene superfamily or cysteine framework plotted against the number of unique conopeptide sequences for each of the given classifications were generated using custom scripts implemented in R (R Core Team, 2021). Venn diagrams of shared conopeptides within and between species were generated also using R.
2.6 Identification of possibly novel conopeptide superfamilies
In order to identify possibly novel gene superfamilies, signal sequences for each of the putative conopeptides as predicted by ConoPrec were clustered via agglomerative clustering at 75% similarity threshold with minimum distance single linkage using USEARCH (Edgar, 2010). The resulting cluster tree was then viewed and manually inspected using FigTree (http://tree.bio.ed.ac.uk/software/figtree/). Cluster of signal sequences with unknown gene superfamily having at least four member sequences were then classified as putative novel superfamilies. For each putative novel superfamily cluster, member signal sequences were aligned using the L-INS-I algorithm in MAFFT (Katoh et al., 2002). The resulting alignments were then used to generate sequence profiles using WebLogo (Crooks et al., 2004).
2.7 Detection of putative disulfide-poor conopeptide
Putative conopeptide sequences without known cysteine framework classifications were analyzed for sequence similarity with previously identified disulfide-poor conopeptide sequences in the ConoServer database using the blastp algorithm in BLAST. In this case, conopeptides are deemed disulfide-poor if they belong to any of the following classes: conopressin, contryphan, conoCAP, conoGAY, conantokin, contulakin, conorfamide, conophan, conomap, conomarphin, conolysin, cono-NPY, and hormone-like including insulin (Lebbe and Tytgat, 2016). Alignment of putative conantokin precursor sequences classified under the B1 conopeptide superfamily was also done and viewed using Aliview (Larsson, 2014).
2.8 Tesseliconus diet investigation
Predicted mature peptide sequences were used as BLAST queries against a database of mature conopeptide sequences obtained from Conus species with known diet preference based on ConoServer information. Sequences from C. eburneus and C. tessulatus were excluded from the custom database in order to get the nearest conopeptide match from a different species.
Sequences of delta conopeptides, believed to be involved in the transition from vermivorous to piscivorous diet, were also clustered using USEARCH (75% similarity threshold, minimum distance single linkage). The delta conopeptides from this study, as well as those from the UniProt database, with precursor sequence lengths of at least 40 a.a. were included in the analysis. Agglomerative clustering was done for both full precursor and predicted mature sequences, and the resulting cluster trees were viewed using FigTree.
3 Results
3.1 Conopeptide prediction from Tesseliconus venom duct transcriptome
From the non-redundant merged assemblies, a total of 201,656 good quality contigs were obtained from the transcriptome of C. eburneus and 282,501 from C. tessulatus. Among these assembled transcripts, 82,170 were identified to contain candidate coding regions from C. eburneus and 119,373 from C. tessulatus; of which, only 33,414 and 51,981 were predicted to have complete open reading frames, respectively. Nevertheless, based on the statistics reported by the transcript abundance estimation tool Salmon, an overall mapping rate of around 97% was observed for the quasi-mapping of sequence reads from all individual samples against the assemblies corresponding to their respective species. The pertinent assembly metrics are shown in Table 1.
Table 1
| METRIC | C. eburneus | C. tessulatus |
|---|---|---|
| Merged Assembly | ||
| Total number of transcripts | 201,656 | 282,501 |
| Total number of bases (bp) | 121,742,504 | 185,835,200 |
| GC content (%) | 41.68 | 41.54 |
| N50 (bp) | 1,133 | 1,182 |
| Average Salmon mapping rate (%) | 97.73 | 97.59 |
| Coding Region Prediction | ||
| With putative coding region | 82,170 | 119,373 |
| With predicted complete coding sequence | 33,414 | 51,981 |
| With partial 3′; region | 7,631 | 11,280 |
| With partial 5′; region | 26,141 | 37,401 |
| With partial 3′; and 5′; regions (internal) | 14,984 | 18,711 |
| Conopeptide sequence similarity search | ||
| Transcripts with conopeptide sequence match | 1,517 | 1,640 |
| Non-redundant peptide sequences | 954 | 1,063 |
| With predicted complete coding sequence | 424 | 537 |
| With known superfamily or framework | 260 | 339 |
De novo assembly and conopeptide identification metrics from C. eburneus and C. tessulatus venom duct transcriptome.
Sequence similarity search against a custom curated database, composed of conopeptide sequences from NCBI GenBank, UniProt and ConoServer, identified significant matches with 1,517 C. eburneus transcripts and 1,640 C. tessulatus transcripts, which corresponds to 954 and 1,063 unique peptide sequences, respectively. Among these, 424 sequences from C. eburneus were tagged to have complete coding regions, with 260 of these sequences predicted to carry a known conopeptide superfamily or cysteine framework. For C. tessulatus, 537 were tagged to have complete coding regions, with known superfamilies or cysteine frameworks identified in a subset of 339 sequences.
Note that a preliminary set of proteomic data from C. eburneus and C. tessulatus (unpublished) yielding a set of 26 and 20 peptide fragments, respectively, were all mapped to 22 unique transcript sequences from this study (8 from C. eburneus and 14 from C. tessulatus). However, because only a small fraction of the conopeptide sequences can be observed in the proteome due to the sheer throughput of transcriptomic analysis, we would like to caution that some of the transcripts reported in this study might not be translated into functional venom peptides. Similar to most other transcriptomic studies, the presence of assembly artifacts cannot be fully discounted as well. Nevertheless, we only included high confidence precursor conopeptide predictions in subsequent downstream analyses by filtering for those with significant sequence similarities with known conopeptides, as well as those with either a known gene superfamily or cysteine framework.
3.2 Interspecific diversity of conopeptides with known superfamily or cysteine framework
Among those with predicted complete coding sequences, the set of putative conopeptide transcripts with known superfamilies or cysteine frameworks were observed to be mostly different between C. eburneus and C. tessulatus. Based on sequence similarities of the predicted mature peptide regions, only 23 sequences overlap between the two sister species at 70% identity threshold, 12 of which have matching regions that are 100% identical (Supplementary Figure S1). The comparison of estimated expression levels, predicted cysteine frameworks and gene superfamilies, as well as the actual peptide sequences of these 12 highly identical conopeptides shared by both Tesseliconus species are shown in Table 2. Six of these putative conopeptides were predicted to have unknown, possibly novel, gene superfamily classifications.
Table 2
| Transcript ID | Ave. Expression (TPM) | Cysteine Framework | Gene Superfamily | Precursor Sequence |
|---|---|---|---|---|
| Ce-0603 | 3.5 | I | A | MGMRMMFTVYLLVVLATTVVSFTSDRAPDGRNAAATDGRNAAAKAFGLITPTVRDGCCSNPACMLNNPNQCG |
| Ct-0680 | 7.88 | I | A | MGMRMMFTVFLLVVLATTVDSFTSDRAPDGRNAAATDGRNAAAKAFGLITPTVRDGCCSNPACMLNNPNQCG |
| Ce-0358 | 1986.9 | III | M | MMSKLGVLLTICLLLFPLTAVPLDGDQPADQPAERKQNEQHPLFDQKRGCCRWPCPSRCGMARCCSS |
| Ct-0773 | 442.24 | III | M | MMSKLGVLLTICLLLFPLTAVPLDGDQPADQPAERTQNEQHPLFDQKRGCCRWPCPSRCGMARCCSS |
| Ce-0236 | 98.46 | XV | ? (1) | MSTLGMVLLLLLLLLPLGNSDGDGDRQAMDRDRTASEARSAPRLRLRRHAVHGRSANKRCSTKICGDDCCSSSACECEVHGGTSNEVGCSCPVMILL |
| Ct-0926 | 128.49 | XV | ? (1) | MSTLGMVLLLLLLLLPLGNSDGDGDRQAMDRDRIASDERSAPRLHLRRHVAHGRLANKRCSTKICGDDCCSSSACECEVHGGTSNEVGCSCP |
| Ce-0400 | 1024.92 | VI/VII | O2 | MEKLTILLLVAAVLMSTLFLAQGVGEKTQKAKIDLFKARKLSENKQTRGECVGWSAYCGPWNNPPCCDWYVCEGVYCALDWD |
| Ct-1001 | 3230.97 | VI/VII | O2 | MEKLTILLLVAAVLMSTLFLAQGVGEKTQKAKIDLFKARKLSENKQTRGECVGWSAYCGPWNNPPCCDWYVCEGVYCALDWD |
| Ce-0321 | 69.46 | XIV | ? (3) | MNFSVMFIVALVLTLSMTDGFIRPAENGGRTFRQHSPDAKDLQTHQIKTRDLCPHCPNGCHVDRTCI |
| Ct-0942 | 129.94 | XIV | ? (3) | MNFSVVFIVALVLTLSMTDGFIRPAENGGRAFGQHGPDAKDLQTRQIKTRDLCPHCPNGCHVDRTCIE |
| Ce-0382 | 47.96 | XVI | T | MLCLPVFIILLLLASPAAPNPLERRIQSDLIRAALEDADMKSEKGILSIMGKLGKVVNIGGIASSILCSVCTSCCSTE |
| Ct-0305 | 44.17 | XVI | T | MLCLPVFIILLLLASPAAPNPLERRIQSDLIRAALEDADMKSEKGILSIMGKLGKVVNIGGIASSILCSVCTSCCSTE |
| Ce-0477 | 845.57 | XIV | ? (3) | MKLSVMFIVFLMLTMPVIDAGHSRRAANGGEAGVLAGDRAANLMALLQERQCPPSCQSCSNC |
| Ct-0675 | 158.73 | XIV | ? (3) | MKLSVMFIVFLMLTMPVIDAGHSRRAANEGEAGMLADDRAANLMALLQERQCPPSCQSCSNC |
| Ce-0559 | 5.76 | V | ? (13) | MATNLWMTLSMLVMVVMATAVSDSTPVHETKARSAPWEVRSLARQPVSCCLLVLLIEWCCPG |
| Ct-0043 | 6.77 | V | ? (13) | MATNLWMTLSMLVMVIIATAVTDSTPVHETKARSAPWEVRSLARQPVSCCLLVLLIEWCCPG |
| Ce-0245 | 8.17 | V | ? (T) | MLRLPIFLILLLSLSSAAGFPAESELQRDLALQSPKDFGMRTDHLLLKRVGDDCCVDGHIGTCCKK |
| Ct-0607 | 10.18 | V | ? (T) | MLCLPIFLILLLSLSSAAGFSVESELQRDLALQSPKDFGMRTDHLLLKKVGDDCCVDGHIGTCCKK |
| Ce-0881 | 5.83 | VI/VII | O1 | MKLTCMMIVAVMFLTAWTFVTADDSINGLEDRGIWGEPLSKARDEMNPEASKLNKRCIPNSELCDIPTQCCSGICLVVCMP |
| Ct-0126 | 10.18 | VI/VII | ? (O1) | MKLTGMMIVAVLFLTAWTFITADDSINGLENRGIWGEPLSKARDKMNPEASKLNKRCIPNSELCDIPTQCCSGICLVVCMP |
| Ce-0750 | 1167.53 | III | M | MLKMGVVLFTFLVLFPLATLQLDADQPVERYAENKQDLNPDERMKFILHALGQRRCCISPACNDTCYCCQDR |
| Ct-0003 | 244.19 | III | M | MLKMGVLLFTFLVLFPLATLQLDADQPVERYAENKQGLNPDERMKFILHALGQRRCCISPACNDTCYCCQDR |
| Ce-0154 | 248.22 | XXVII | ? (6) | MRSHLLLTVMLLLTLFTGGDAGPRRANRLEKHFVNRDCQSGCVGCHNPAGCCCGNQVCVNNNHCEPSSLWF |
| Ct-0295 | 195.17 | XXVII | ? (6) | MRYHLMLTVILLLTLFTGGDAGPRRANRLEKHFVNRDCQSGCVGCHNPAGCCCGNQVCVNNNHCEPSSLWF |
Conopeptides shared by both C. eburneus (Ce) and C. tessulatus (Ct) with 100% sequence identity at the matching mature peptide regions.
Expression estimates are average TPM values from three individuals per species. The predicted sequence structures, as well as cysteine framework and superfamily classifications, were obtained using the ConoPrec precursor analysis tool in ConoServer (http://conoserver.org/?page=conoprec). Unknown superfamily classifications were denoted with the ‘?’ symbol, followed by a classification in parenthesis if the signal sequence clustered with a known or proposed novel (numeric notation) gene superfamily based on agglomerative clustering (75% similarity cutoff). The signal (underlined, regular black font) and mature (underlined, bold orange font) sequences are also annotated.
Using the number of unique precursor conopeptide sequences as diversity estimate, it appears that conopeptides with cysteine framework VI/VII had the highest diversity in both C. eburneus and C. tessulatus individuals sampled in this study (Figures 1A, B). However, in terms of cumulative average expression, framework XIV was observed to be more abundantly expressed than VI/VII in both species. The majority of conopeptides predicted to have the XIV cysteine framework have unknown superfamily classifications – some of which have signal sequences forming exclusive clusters of possibly novel gene superfamilies (Clusters 3, 7 and 15), as described further in the latter sections of this paper, whereas those with superfamily classifications all fall under the A superfamily. Notably, the most highly expressed cysteine framework in C. eburneus, framework III, had substantially lower expression and diversity in C. tessulatus.
Figure 1
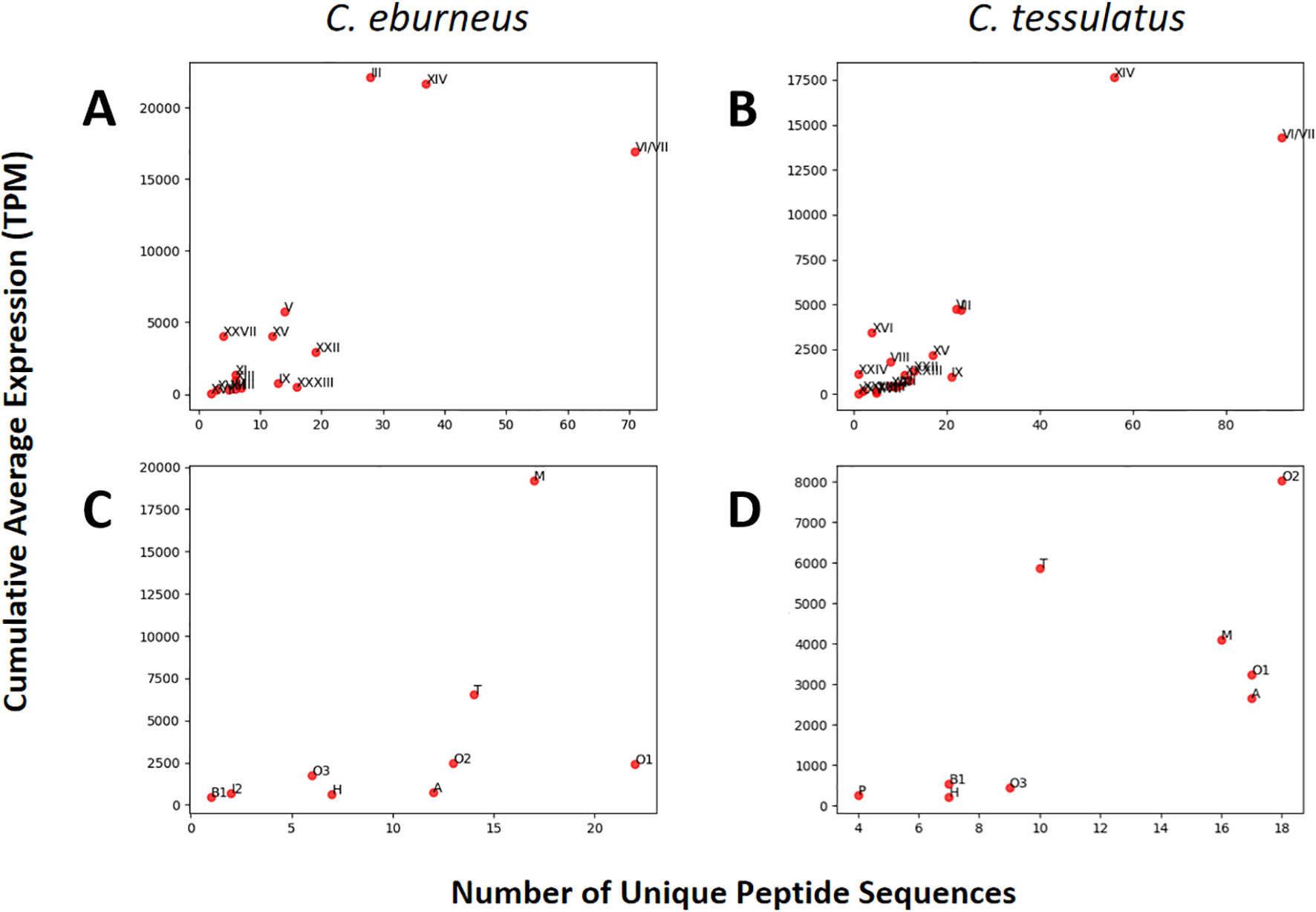
Cumulative average expression (three individuals per species) plotted against the number of unique peptide sequences (diversity estimate) for each of the known cysteine framework (A, B) and superfamily (C, D) classifications. Cysteine framework VI/VII appears to be the most diverse but not the most highly expressed, with framework XIV registering higher expression levels in both species. For C. eburneus, in particular, framework III is the most abundantly expressed. In terms of gene superfamilies, M and O2 are the most expressed for C. ebureneus and C. tessulatus, respectively. Superfamily O1 is highly diverse in both species, but O2 also has the highest number of unique peptide sequences in C. tessulatus.
In terms of gene superfamily classification, O1 had the highest diversity in C. eburneus and second highest in C. tessulatus (Figures 1C, D). For C. tessulatus, the highest diversity and expression was observed for the O2 superfamily, whereas the most abundantly expressed superfamily in C. eburneus was M. Other notable differences include those of the A and T superfamilies whose expression levels were substantially higher in C. tessulatus than in C. eburneus, as well as the P superfamily whose representatives were detected exclusively in C. tessulatus.
3.3 Intraspecific variations in expression of conopeptides with known superfamily or cysteine framework
Among the 260 predicted conopeptides with known cysteine framework or gene superfamily in C. eburneus, we found that 102 (39.23%) were expressed in all three individuals of the said species included in this study. An additional 91 (35.00%) were shared by two individuals, and 67 (25.77%) were uniquely expressed (Figure 2A). A relatively similar pattern was observed for C. tessulatus, with 142 (41.89%) of the 339 predicted conopeptides with known superfamily or framework found to be expressed in all three individuals, 110 (32.45%) shared by two samples, and 87 (25.66%) were unique to a particular individual (Figure 2B).
Figure 2
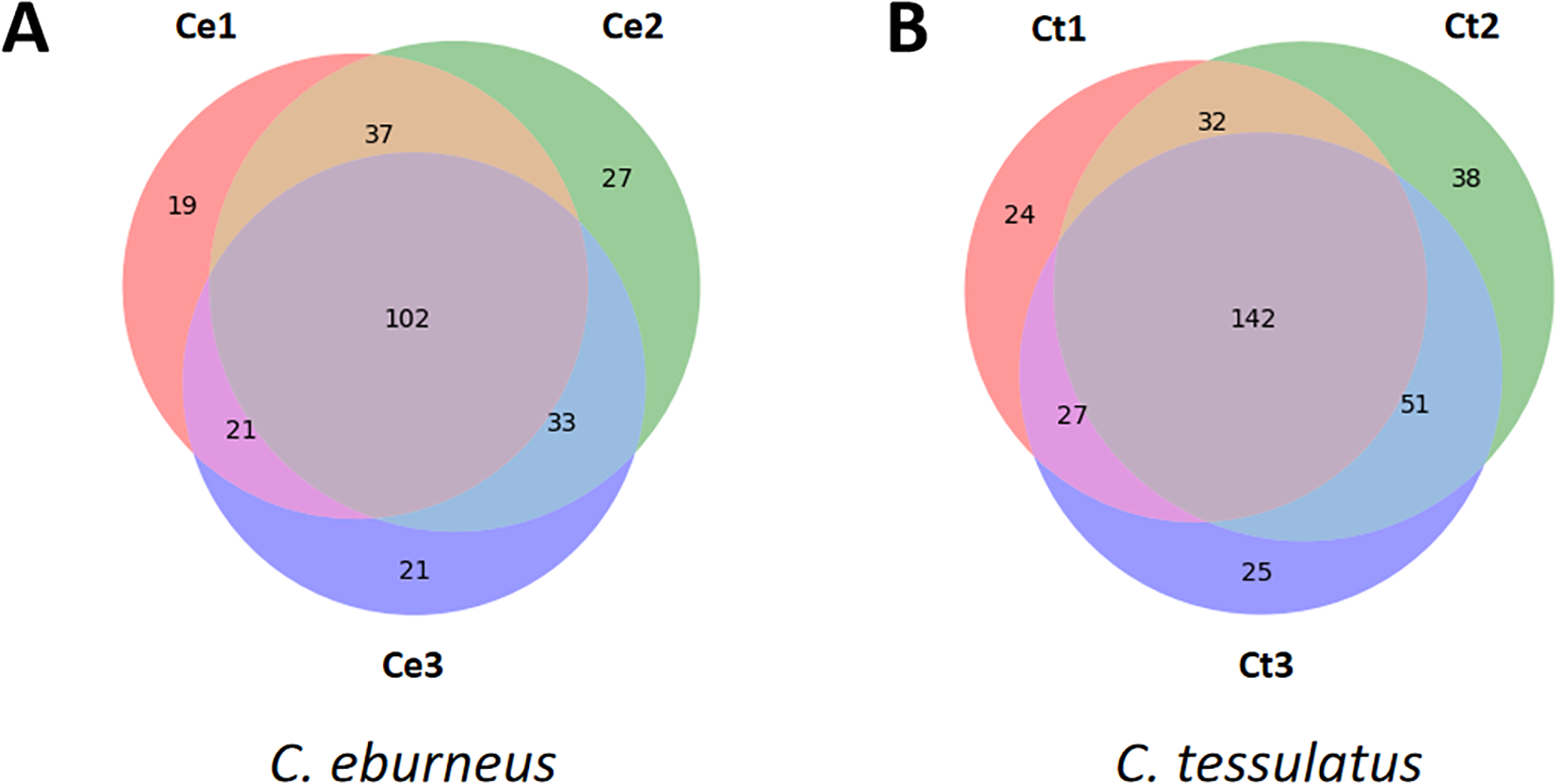
Venn diagram showing commonly expressed conopeptides, with known cysteine framework or superfamily classifications, in three different individuals of (A) C. eburneus and (B) C. tessulatus. Although the majority of these predicted conopeptides were expressed (≥ 1 TPM) in at least two samples of the same species, about 25% of the conopeptides in each of the species appear to be unique to an individual sample.
Although most of the conopeptides detected were shared by individuals of the same species, their relative expression levels appear to differ. Supplementary Figures S2, S3 show the cumulative expression of conopeptides per cysteine framework or gene superfamily for both C. eburneus and C. tessulatus, respectively. For C. eburneus, individual 3 (Ce3) registered a relatively different pattern of cumulative expression, particularly for peptides with cysteine frameworks VI/VII (higher expression) and XIV (lower expression), compared to those observed in Ce1 and Ce2. Similarly, C. tessulatus individual Ct2 was observed to have higher VI/VII and lower XIV cumulative expression relative to Ct1 and Ct3. Other notable differences in expression levels across individuals of the same species include the following: O2 and T superfamilies and V cysteine framework in C. eburneus, as well as the T and M superfamilies and the III, V and XVI cysteine frameworks in C. tessulatus.
3.4 Detection of possibly novel conopeptide gene superfamilies
Agglomerative clustering (75% similarity threshold) of predicted conopeptide signal sequences from both C. eburneus and C. tessulatus revealed 10 known superfamily clusters and 18 possibly novel gene superfamilies with at least four representative Tesseliconus conopeptide sequences (Figure 3). The O1 and M superfamilies form the largest sequence clusters, while O2, T, A and O3 also have relatively large representations. On the other hand, clusters 1, 3, 4, and 6 contain the highest number of protein precursors among the novel clusters. Table 3 lists the designated major gene superfamily clusters (both known and novel), the cysteine frameworks associated with these superfamilies, and the number of precursor peptides comprising these clusters. Sequences comprising the potentially novel gene superfamily clusters are also indicated in Supplementary Tables 1, 2, designated as cluster_01 to cluster_18 indicating their tentative numeric superfamily classifications.
Figure 3

Agglomerative clustering of signal sequences predicted from the set of putative Tesseliconus conopeptides with a known superfamily or cysteine framework. known gene superfamily clusters are labeled accordingly. Numbered clusters (red) indicate possibly novel gene superfamilies (with ≥4 member precursors), with the sequence profiles of the eight largest clusters also shown. Note that the profile for Cluster 09 has been manually truncated at the N-terminal end based on alignment data.
Table 3
| Gene superfamily | Cysteine framework | Putative protein precursors* | ||
|---|---|---|---|---|
| C. eburneus | C. tessulatus | TOTAL | ||
| Known superfamilies | ||||
| O1 | VI/VII | 32 (10) | 37 (20) | 69 (30) |
| M | III, VIII, IX | 29 (12) | 26 (10) | 55 (22) |
| O2 | VI/VII, XV | 22 (9) | 24 (6) | 46 (15) |
| T | V, XVI | 16 (2) | 19 (9) | 35 (11) |
| A | I, XIV, XXII | 12 (0) | 20 (3) | 32 (3) |
| O3 | VI/VII, XIV | 10 (4) | 10 (1) | 20 (5) |
| H | VI/VII | 7 (0) | 7 (0) | 14 (0) |
| I2 | XI | 5 (3) | 3 (3) | 8 (6) |
| B1 | None | 1 (0) | 7 (0) | 8 (0) |
| P | IX | 0 (0) | 7 (3) | 7 (3) |
| Possibly novel superfamilies (≥4 precursor sequences)** | ||||
| 1 | VI/VII, XV, XVII, XXII | 12 | 23 | 35 |
| 2 | XXXIII | 3 | 2 | 5 |
| 3 | XIV | 12 | 14 | 26 |
| 4 | XIII, XXII, XXXIII | 11 | 10 | 21 |
| 5 | VI/VII | 4 | 4 | 8 |
| 6 | VI/VII, IX, XII, XXVII | 16 | 22 | 38 |
| 7 | XIV | 2 | 8 | 10 |
| 8 | IX | 3 | 4 | 7 |
| 9 | XXXIII | 3 | 7 | 10 |
| 10 | XI | 0 | 4 | 4 |
| 11 | VIII | 2 | 2 | 4 |
| 12 | VIII | 2 | 2 | 4 |
| 13 | V | 4 | 7 | 11 |
| 14 | VI/VII | 1 | 3 | 4 |
| 15 | XIV | 4 | 7 | 11 |
| 16 | VIII | 2 | 3 | 5 |
| 17 | XII | 2 | 3 | 5 |
| 18 | XXII | 2 | 7 | 9 |
List of predicted known and possibly novel Tesseliconus gene superfamilies, their associated cysteine framework/s and the number of precursor peptides observed.
*Values inside a parenthesis indicate the number of signal sequences with unknown superfamily classifications (based on ConoPrec analysis) but clustered with other sequences assigned to a known gene superfamily. **Signal sequence profiles for the eight largest novel clusters (counts in boldface, underlined) are shown in Figure 3.
Relatively larger superfamily clusters with at least 20 precursor peptides were mainly associated with multiple (three to four) cysteine frameworks, except for the O1 superfamily that has been associated exclusively with framework VI/VII. We do note, however, that some of the O1 sequences have unknown framework classifications. Altogether, we found a total of 16 cysteine frameworks associated with the designated major gene superfamilies (known and novel). Notably, all eight sequences classified under the B1 superfamily have no cysteine framework classifications.
3.5 Detection of disulfide-poor conopeptides
A total of eight (one from C. eburneus and seven from C. tessulatus) predicted conopeptides classified as belonging to the B1 gene superfamily. Upon further analysis, the predicted mature peptide sequences from these transcripts were found to be highly similar with previously reported conantokin precursor peptide sequences (Supplementary Table S3). Interestingly, one conantokin precursor in the database isolated from C. eburneus was not observed in the transcripts obtained from samples of the same species in this study, but was the top hit for three C. tessulatus B1 precursor peptides with over 90% precursor sequence similarity. Furthermore, alignment of the Tesseliconus B1 precursor sequences showed that although the signal and pro regions of the peptides are relatively conserved, the mature regions appear to be much more divergent (Supplementary Figure S4).
Apart from the B1 superfamily conopeptides, we were also able to predict putative conopeptide precursors without known gene superfamily and cysteine framework classifications that have high amino acid sequence similarities with previously reported disulfide-poor conopeptides in the ConoServer database. In particular, we were able to identify a total of 54 putative disulfide-poor conopeptide precursors in C. eburneus and 56 in C. tessulatus. Table 4 shows a summary of the additional disulfide-poor conopeptide classes predicted from the two Tesseliconus species, while the individual sequence and ConoServer match details for all these putative conopeptides from C. eburneus and C. tessulatus are listed in Supplementary Tables S4, S5, respectively.
Table 4
| Disulfide-poor conopeptide class | C. eburneus | C. tessulatus | ||
|---|---|---|---|---|
| # of Precursors | %Identity range | # of Precursors | %Identity range | |
| Conantokin | 25 | 48.08% - 100.00% | 21 | 55.03% - 100.00% |
| Conomarphin | 21 | 52.00% - 100.00% | 20 | 46.67% - 98.00% |
| Conopressin | 5 | 73.00% - 77.00% | 3 | 75.00% - 78.00% |
| Contryphan | 3 | 87.50% - 100.00% | 11 | 72.73%S - 100.00% |
| Insulin | 0 | N/A | 1 | 55.26% |
Putative disulfide-poor conopeptide precursors predicted from C. eburneus and C. tessulatus.
The range of percent sequence identities observed from each of the predicted mature peptide sequences and their top match in the ConoServer database are also shown.
3.6 Tesseliconus diet investigation
For both C. eburneus and C. tessulatus, most (>50%) of the mature peptide were found to have high sequence similarities with those obtained from species with known vermivorous diets, while the remaining peptides have top matches that are almost evenly split between those obtained from Conus species with piscivorous and molluscivorous diets (Table 5). In terms of expression, conopeptides that are highly similar to those found in other vermivorous species registered the highest cumulative average expression in both Tesseliconus species (Supplementary Table S6). However, we found that the cumulative average expression of piscivorous-like conopeptides is substantially higher in C. tessulatus than in C. eburneus, whereas the converse is true for molluscivorous-like conopeptides.
Table 5
| Diet of top conoServer match | C. eburneus | C. tessulatus |
|---|---|---|
| Vermivorous | 466 (137) | 550 (179) |
| Piscivorous | 206 (65) | 250 (88) |
| Molluscivorous | 250 (62) | 233 (63) |
Known primary diets of the Conus species wherein the top conopeptide matches were sourced.
BLAST analysis was performed using a database of mature peptide sequences from ConoServer, with the previously reported conopeptides from C. eburneus and C. tessulatus excluded, as well as those without known diet information. Primary counts are for all predicted unique mature conopeptide sequences (including those with truncated precursors), whereas values inside the parentheses are counts for mature peptides predicted to have precursors with complete coding sequences.
In both Tesseliconus species, the most highly expressed vermivorous-like conopeptides closely match conomarphin precursor sequences obtained from Conus betulinus. This is consistent with a previous proteomic study that identified a number of conomarphin peptides from C. eburneus exhibiting high degrees of similarity with those from C. betulinus (Itang et al., 2020). Some of the highly expressed molluscivorous-like conopeptides had significant sequence similarities with those obtained from Conus victoriae and Conus marmoreus, particularly peptides classified under the M gene superfamily with cysteine framework III. On the other hand, piscivorous-like conopeptides with relatively high average expression levels appeared to have significant similarities with those found in Conus magus (A superfamily), Conus catus (O1 and O2 superfamilies), Conus ermineus (cysteine framework XXII) and Conus californicus (divergent superfamily).
Agglomerative clustering of delta conopeptide sequences, believed to be one of the conopeptide families that enabled the shift from vermivorous to piscivorous behavior (Aman et al., 2015), showed that mature sequences from piscivores and vermivores indeed clustered more closely than their molluscivorous homologs (Supplementary Figure S5A). Interestingly, at the precursor peptide level, the molluscivorous and piscivorous clusters appear to be more closely related, with the vermivorous cluster forming an outgroup (Supplementary Figure S5B). However, it must be noted that there are only five representative molluscivorous delta sequences included in this analysis, three of which were observed from Conus textile.
4 Discussion
In this study, we analyzed whole transcriptome data sets from two Tesseliconus species: C. eburneus and C. tessulatus. We found that the diversity of putative conopeptides are highly different between the two Tesseliconus species, sharing only 23 peptides with at least 75% amino acid sequence similarity at the mature region. This observation is quite surprising considering that C. eburneus and C. tessulatus are arguably two of the most closely related Conus species whose transcriptome profiles have been compared to date, based on a previously published comprehensive phylogeny of Conus species (Puillandre et al., 2014). A previous study on two Splinoconus species, Conus tribblei and Conus lenavati, found 67 orthologous conopeptides shared by the sister species, 21 of which have identical mature regions (Barghi et al., 2015a). Another study on closely related species under the Turriconus subgenus, Conus andremenezi and Conus praecellens, identified 68 orthologous conopeptides at 95% similarity threshold, but only 9 of which shared 100% identity (Li et al., 2017). Comparison of conopeptides obtained from the venom duct transcriptomes of Conus flavidus and Conus frigidus (cryptic species under the Virgiconus clade) similarly identified 68 shared transcripts (Himaya et al., 2022). Lastly, two cryptic species under the Virroconus subgenus, Conus judaeus and Conus ebraeus, were observed to share 129 conopeptide transcripts (Pardos-Blas et al., 2022), the highest number of shared conopeptides reported as of this writing. Note that all of the aforementioned Conus subgenera are comprised of species believed to be primarily worm hunters.
Considering the relatively high number of putative conopeptides identified in the current study (about double the number of conopeptides reported for Splinoconus, Turriconus and Virgiconus) but with less overlaps between the sister species, the aforementioned observations hint at two general possibilities: (i) the individual Tesseliconus samples might have been exposed to highly variable environments, which can affect the abundance/scarcity of prey as well as the presence of competitors and (ii) the prey preference of C. eburneus and C. tessulatus are possibly more diverse than previously observed. In addition, the lower number of similar peptides, notwithstanding the higher number of identified peptides and the arguably closer affinity of C. eburneus and C. tessulatus compared to the other previously analyzed sister species, may also indicate a faster rate of divergence and hence adaptation in Tesseliconus, consistent with exposure to a more variable environment.
Tesseliconus species are mostly found along the intertidal and subtidal zones that constantly experience drastic environmental changes to which they must quickly adapt. For instance, tidal fluctuations may restrict or promote the movement of certain organisms that may result in sudden changes in the predator-prey dynamics of a given environment. We therefore hypothesize that the greater conopeptide repertoire observed in Tesseliconus species enable them to more readily adapt to such drastic changes. However, Virgiconus and Virroconus species that are also primarily found in intertidal zones registered a substantially higher proportion of shared conopeptides. The similarities observed for these subgenera can possibly be explained by the fact that the subjects compared are cryptic species, suggesting that their likeness may not only be morphological but also physiological. On the other hand, Splinoconus and Turriconus species are deep water inhabitants and are therefore less likely to frequently experience drastic environmental variabilities.
In Conus, dietary breadth has been positively associated with the observed conopeptide diversity (Phuong et al., 2016). Thus, the high number of unique conopeptides obtained from this study indicates that the two Tesseliconus species are not exclusively vermivorous but can also target other prey types. This is consistent with a previous observation that although C. tessulatus is primarily a worm hunter, it appears to opportunistically prey on fish as well (Aman et al., 2015). In addition, even though a number of previous studies have classified both C. tessulatus and C. eburneus as vermivorous (Duda et al., 2001; Puillandre et al., 2014), a relatively recent study tagged C. eburneus as a molluscivorous species (Li et al., 2020).
Nonetheless, we found that more than half of the conopeptides identified in this study have mature regions that are more similar to those obtained from other vermivorous species (excluding C. eburneus and C. tessulatus), whereas the remaining were almost evenly distributed between those that are more similar with conopeptides obtained from piscivorous and molluscivorous species. The identification of mainly vermivorous-like conopeptides in the samples may provide additional evidence supporting the fact that Tesseliconus species are primarily worm hunters, but could have secondary feeding behaviors. The concurrent expression of piscivorous-like and molluscivorous-like conopeptides, albeit at lower diversities and cumulative expression, may however signify two possible scenarios. The first is that there may be a scarcity of the preferred prey in the environment where the samples were collected and that they are opportunistically targeting fish and other mollusks as prey. The second is that the subjects are using these peptides as deterrent to fish and other molluscan competitors present in their surroundings (Olivera et al., 2014). However, we note a couple of crucial limitations in this type of analysis: sequence similarity alone does not provide sufficient evidence for a conopeptide’s physiological role, and current databases may lack comprehensive representation of conopeptide diversity. Nevertheless, considering the unique phylogenetic position of the subgenus Tessiliconus as a sister clade to both molluscivores and piscivores, we believe that the relevance of the reported observations are warranted.
Akin to what has been reported in Splinoconus, Turriconus, Virgiconus and Virroconus, the expression of conopeptides in Tesseliconus species, notwithstanding their diversity, are mainly dominated by only a few gene superfamilies in terms of expression levels (M, O2, T, O1, A in this study). However, unlike in Splinoconus and Turriconus wherein the P superfamily has been abundantly observed (Li et al., 2017), conopeptide precursors classified under the said superfamily were only identified in C. tessulatus at very low diversity and expression, and were not detected in C. eburneus. Similarly, the P superfamily has been observed at low levels in Virroconus and Virgiconus, suggesting that their abundant expression might be linked to the deep water habitat of the Splinoconus and Turriconus species.
Furthermore, C. tessulatus have a relatively high cumulative expression of the A superfamily precursor peptides, which were previously observed to be absent in Splinoconus and Turriconus but have very high expression levels in the fish-hunting species Conus geographus (Li et al., 2017). The A superfamily precursors were also observed in C. eburneus, but with a much lower cumulative expression. These observations further allude to the secondary fish-hunting behavior of C. tessulatus and possibly of C. eburneus and other Tesseliconus species in general. We also note that the A superfamily conopeptides were observed in Virroconus and with relatively high diversity in Virgiconus.
The relatively high expression of conomarphin precursors in both Tesseliconus species suggests that the samples may have also been attacking other gastropods because these conopeptides were previously shown to cause paralysis in mollusks (Mendoza et al., 2019). As to whether this illustrates a secondary molluscivorous behavior or a defense mechanism remains to be determined. Interestingly, conomarphins were not reported in the transcriptomes of previously studied Splinoconus, Turriconus, Virroconus and Virgiconus sister species.
Agglomerative clustering of delta-like conopeptide sequences revealed interesting patterns that may shed light in the evolution of this pharmacological family, which is believed to have facilitated the shift to piscivorous behavior in certain Conus species. In particular, the clustering of full precursor sequences further supports the ancestral vermivorous behavior of cone snails. However, the evolution of the mature delta peptide sequences to facilitate piscivorous behavior appeared to have required less changes than what was necessary for a molluscivorous adaptation. This is somewhat consistent with a previous suggestion that delta conopeptides were already being used by an ancestral lineage of vermivorous cone snails as deterrent against fish competitors (Aman et al., 2015; Olivera et al., 2014).
Intra-specific variations in conopeptide expression levels may be due to the relatively different external pressures being experienced by each of the samples at the time of collection. Alternatively, these differences in expression may also be accounted for by the possibility that even individuals of the same species may have subtle differences in prey preference to facilitate coexistence. This is an extension of the notion that Conus species tend to specialize particularly in environments with high species diversity (Kumar et al., 2015). However, we cannot fully discount the influence of systemic differences in the experimental handling of the samples leading to the observed intra-specific expression level variations, although steps were actively taken to minimize this possibility.
The identification of the 18 possibly novel gene superfamilies in this study together with more than 50 (per species) disulfide-poor conopeptides, in addition to precursors with known gene superfamilies and cysteine frameworks, is indicative of the fact that the true diversity of conopeptides is still currently grossly under-estimated. Nevertheless, more recent studies (Barghi et al., 2015b; Gao et al., 2018; Li et al., 2017; Yao et al., 2019) employing high-throughput sequencing technologies similar to this work have proved to be very useful in uncovering this hidden conopeptide diversity.
5 Conclusions
In this study, we identified and analyzed the diversity and variations in expression of conopeptides obtained from the venom duct of two Tesseliconus species, Conus eburneus and Conus tessulatus. Apart from conopeptides with known gene superfamilies and cysteine frameworks, the identification of various possibly novel gene superfamilies and disulfide-poor conopeptides suggest that the true diversity of these peptide group is still grossly underestimated. This also alludes to the utility of high-throughput RNA sequencing approaches to uncover the hidden diversity of conopeptides. The observed inter- and intra-species variations in expression levels of different conopeptide groups suggest a number of possible scenarios that are not necessarily exclusive. These variations could be due to the difference in environmental pressures being experienced by each of the sampled individuals. The high diversity of conopeptides found in this study might also suggest that C. eburneus and C. tessulatus have a more diverse target pool, which includes different species of worm, fish and other mollusks. Part of the venom cocktail may have also been allocated to peptides that enable the samples to deter competitors. The observed variations may also hint to subtle differences in prey preference even in individuals of the same species in order to better coexist – an extension to a previously held notion that Conus species tend to specialize in highly diverse environments.
Statements
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
Ethical approval was not required for the study involving animals in accordance with the local legislation and institutional requirements because the collection of samples used in this study has already been approved by the Department of Agriculture - Bureau of Fisheries and Aquatic Resources (DA-BFAR), Republic of the Philippines, under gratuitous permit no. 0111–16. All experimental procedures involving animal handling were done according to standard laboratory practices.
Author contributions
FT: Visualization, Formal analysis, Resources, Conceptualization, Writing – review & editing, Methodology, Investigation, Writing – original draft. DM: Writing – review & editing, Methodology, Investigation, Resources. AL: Resources, Funding acquisition, Supervision, Writing – review & editing, Conceptualization.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study received partial financial support from the Department of Science and Technology - Philippine Council for Health Research and Development through a research grant to AL (under the Discovery and Development of Health Products research program).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2025.1616692/full#supplementary-material
Supplementary Table 1Predicted conopeptide sequences from C. eburneus with either a known gene superfamily or cysteine framework. The predicted sequence structures, as well as cysteine framework and gene superfamily classifications, were obtained using the ConoPrec precursor analysis tool in ConoServer (http://conoserver.org/?page=conoprec). Sequences comprising potentially novel gene superfamily clusters were labeled cluster_01 to cluster_18, denoting their designated cluster numbers. The blastp hits against a custom database of conopeptide sequences collected from ConoServer, Genbank, and Swissprot are also shown.
Supplementary Table 2Predicted conopeptide sequences from C. tessulatus with either a known gene superfamily or cysteine framework. The predicted sequence structures, as well as cysteine framework and gene superfamily classifications, were obtained using the ConoPrec precursor analysis tool in ConoServer (http://conoserver.org/?page=conoprec). Sequences comprising potentially novel gene superfamily clusters were labeled cluster_01 to cluster_18, denoting their designated cluster numbers. The blastp hits against a custom database of conopeptide sequences collected from ConoServer, Genbank, and Swissprot are also shown.
Supplementary Table 3List of conopeptides from the B1 gene superfamily without cysteine framework classifications. All of the said putative conopeptides were found to have high sequence similarities with previously reported conantokin precursor peptide sequences.
Supplementary Table 4Putative disulfide-poor conopeptides predicted from C. eburneus. The amino acid sequence (signal, pre, mature, post), predicted cysteine framework and gene superfamily classifications, closest ConoServer match of the mature peptide and their percent identity, as well as the disulfide-poor conopeptide class are also shown.
Supplementary Table 5Putative disulfide-poor conopeptides predicted from C. tessulatus. The amino acid sequence (signal, pre, mature, post), predicted cysteine framework and gene superfamily classifications, closest ConoServer match of the mature peptide and their percent identity, as well as the disulfide-poor conopeptide class are also shown.
Supplementary Table 6Cumulative average expression of predicted conopeptides classified based on the diet of the Conus species wherein the top mature region match was obtained. Primary values are for all unique mature conopeptide sequences (including those with truncated precursors), whereas values inside the parentheses are expression estimates for mature peptides predicted to have precursors with complete coding sequences.
Supplementary Table 7Quality filtering of raw sequence data. The quality control procedure includes read error correction, adapter and quality trimming, and removal of rRNA-mapped sequence reads.
References
1
Altschul S. F. Gish W. Miller W. Myers E. W. Lipman D. J. (1990). Basic local alignment search tool. J. Mol. Biol.215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
2
Aman J. W. Imperial J. S. Ueberheide B. Zhang M. M. Aguilar M. Taylor D. et al . (2015). Insights into the origins of fish hunting in venomous cone snails from studies of Conus tessulatus. Proc. Natl. Acad. Sci. United States America112, 5087–5092. doi: 10.1073/pnas.1424435112
3
Barghi N. Concepcion G. P. Olivera B. M. Lluisma A. O. (2015a). Comparison of the venom peptides and their expression in closely related conus species: Insights into adaptive post-speciation evolution of conus exogenomes. Genome Biol. Evol.7, 1797–1814. doi: 10.1093/gbe/evv109
4
Barghi N. Concepcion G. P. Olivera B. M. Lluisma A. O. (2015b). High conopeptide diversity in Conus tribblei revealed through analysis of venom duct transcriptome using high-throughput sequencing platforms. Mar Biotechnol. 17(1), 81–98. doi: 10.1007/s10126-014-9595-7
5
Bushmanova E. Antipov D. Lapidus A. Prjibelski A. D. (2019). RnaSPAdes: A de novo transcriptome assembler and its application to RNA-Seq data. GigaScience8, 1–13. doi: 10.1093/gigascience/giz100
6
Crooks G. E. Hon G. Chandonia J. M. Brenner S. E. (2004). WebLogo: a sequence logo generator. Genome Res.14, 1188–1190. doi: 10.1101/gr.849004
7
Duda T. F. Kohn A. J. Palumbi S. R. (2001). Origins of diverse feeding ecologies within Conus, a genus of venomous marine gastropods. Biol. J. Linn. Soc.73, 391–409. doi: 10.1006/bijl.2001.0544
8
Edgar R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics26, 2460–2461. doi: 10.1093/bioinformatics/btq461
9
Espino S. Watkins M. Probst R. Koch T. L. Chase K. Imperial J. et al . (2024). χ-conotoxins are an evolutionary innovation of mollusk-hunting cone snails as a counter-adaptation to prey defense. Mol. Biol. Evol.41, msae226. doi: 10.1093/molbev/msae226
10
Fu L. Niu B. Zhu Z. Wu S. Li W. (2012). CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics28, 3150–3152. doi: 10.1093/bioinformatics/bts565
11
Gao B. Peng C. Zhu Y. Sun Y. Zhao T. Huang Y. et al . (2018). High throughput identification of novel conotoxins from the vermivorous oak cone snail (Conus quercinus) by transcriptome sequencing. Int. J. Mol. Sci.19(12), 3901. doi: 10.3390/ijms19123901
12
Haas B. J. Papanicolaou A. Yassour M. Grabherr M. Blood P. D. Bowden J. et al . (2013). De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc.8, 1494–1512. doi: 10.1038/nprot.2013.084
13
Himaya S. W. A. Arkhipov A. Yum W. Y. Lewis R. J. (2022). Comparative Venomics of C. flavidus and C. frigidus and Closely Related Vermivorous Cone Snails. Mar. Drugs20, 209. doi: 10.3390/md20030209
14
Itang C. E. M. M. Gaza J. T. Masacupan D. J. M. Batoctoy D. C. R. Chen Y. J. Nellas R. B. et al . (2020). Identification of conomarphin variants in the conus eburneus venom and the effect of sequence and PTM variations on conomarphin conformations. Mar. Drugs18(10), 503. doi: 10.3390/md18100503
15
Kaas Q. Yu R. Jin A. H. Dutertre S. Craik D. J. (2012). ConoServer: Updated content, knowledge, and discovery tools in the conopeptide database. Nucleic Acids Res.40, 325–330. doi: 10.1093/nar/gkr886
16
Katoh K. Misawa K. Kuma K. Miyata T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res.30, 3059–3066. doi: 10.1093/nar/gkf436
17
Kraus N. J. Corneli P. S. Watkins M. Bandyopadhyay P. K. Seger J. Olivera B. M. (2011). Against expectation: A short sequence with high signal elucidates cone snail phylogeny. Mol. Phylogenet. Evol.58, 383–389. doi: 10.1016/j.ympev.2010.11.020
18
Kumar P. S. Kumar D. S. Umamaheswari S. (2015). A perspective on toxicology of Conus venom peptides. Asian Pacific J. Trop. Med.8, 337–351. doi: 10.1016/S1995-7645(14)60342-4
19
Langmead B. Salzberg S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods9, 357–359. doi: 10.1038/nmeth.1923
20
Larsson A. (2014). AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinf. (Oxford England)30, 3276–3278. doi: 10.1093/bioinformatics/btu531
21
Lebbe E. K. M. Tytgat J. (2016). In the picture: Disulfide-poor conopeptides, a class of pharmacologically interesting compounds. J. Venomous Anim. Toxins Including Trop. Dis.22, 1–15. doi: 10.1186/s40409-016-0083-6
22
Li Q. Barghi N. Lu A. Fedosov A. E. Bandyopadhyay P. K. Lluisma A. O. et al . (2017). Divergence of the venom exogene repertoire in two sister species of Turriconus. Genome Biol. Evol.9, 2211–2225. doi: 10.1093/gbe/evx157
23
Li X. Chen W. Zhangsun D. Luo S. (2020). Diversity of conopeptides and their precursor genes of conus litteratus. Mar. Drugs18, 464. doi: 10.3390/md18090464
24
Liu Z. Li H. Liu N. Wu C. Jiang J. Yue J. et al . (2012). Diversity and evolution of conotoxins in Conus virgo, Conus eburneus, Conus imperialis and Conus marmoreus from the South China Sea. Toxicon60, 982–989. doi: 10.1016/j.toxicon.2012.06.011
25
Mendoza C. B. Masacupan D. J. M. Batoctoy D. C. R. Yu E. T. Lluisma A. O. Salvador-Reyes L. A. (2019). Conomarphins cause paralysis in mollusk: Critical and tunable structural elements for bioactivity. J. Pept. Sci.25, e3179. doi: 10.1002/psc.3179
26
Olivera B. M. Seger J. Horvath M. P. Fedosov A. E. (2015). Prey-capture strategies of fish-hunting cone snails: Behavior, neurobiology and evolution. Brain Behav. Evol.86, 58–74. doi: 10.1159/000438449
27
Olivera B. M. Showers Corneli P. Watkins M. Fedosov A. (2014). Biodiversity of cone snails and other venomous marine gastropods: Evolutionary success through neuropharmacology. Annu. Rev. Anim. Biosci.2, 487–513. doi: 10.1146/annurev-animal-022513-114124
28
Pardos-Blas J. R. Tenorio M. J. Galindo J. C. G. Zardoya R. (2022). Comparative Venomics of the Cryptic Cone Snail Species Virroconus ebraeus and Virroconus judaeus. Mar. Drugs20, 149. doi: 10.3390/md20020149
29
Patro R. Duggal G. Love M. I. Irizarry R. A. Kingsford C. (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods14, 417–419. doi: 10.1038/nmeth.4197
30
Peng C. Yao G. Gao B. M. Fan C. X. Bian C. Wang J. et al . (2016). High-throughput identification of novel conotoxins from the Chinese tubular cone snail (Conus betulinus) by multi-transcriptome sequencing. GigaScience5, 1–14. doi: 10.1186/s13742-016-0122-9
31
Phuong M. A. Mahardika G. N. Alfaro M. E. (2016). Dietary breadth is positively correlated with venom complexity in cone snails. BMC Genomics17, 1–15. doi: 10.1186/s12864-016-2755-6
32
Puillandre N. Bouchet P. Duda T. F. Kauferstein S. Kohn A. J. Olivera B. M. et al . (2014). Molecular phylogeny and evolution of the cone snails (Gastropoda, Conoidea). Mol. Phylogenet. Evol.78, 290–303. doi: 10.1016/j.ympev.2014.05.023
33
Quast C. Pruesse E. Yilmaz P. Gerken J. Schweer T. Yarza P. et al . (2013). The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res.41, 590–596. doi: 10.1093/nar/gks1219
34
R Core Team (2021). R: A Language and Environment for Statistical Computing (Vienna, Austria: R Foundation for Statistical Computing). Available online at: https://www.r-project.org/. (Accessed September 30, 2021)
35
Robertson G. Schein J. Chiu R. Corbett R. Field M. Jackman S. D. et al . (2010). De novo assembly and analysis of RNA-seq data. Nat. Methods7, 909–912. doi: 10.1038/nmeth.1517
36
Smith-Unna R. Boursnell C. Patro R. Hibberd J. Kelly S. (2016). TransRate: Reference-free quality assessment of de novo transcriptome assemblies. Genome Res.26, 1134–1144. doi: 10.1101/gr.196469.115
37
Song L. Florea L. (2015). Rcorrector: efficient and accurate error correction for Illumina RNA-seq reads. GigaSci. 4 (48). doi: 10.1186/s13742-015-0089-y
38
WoRMS Editorial Board (2024). World Register of Marine Species. Available online at: https://www.marinespecies.org.
39
Yang M. Zhao S. Min X. Shao M. Chen Y. Chen Z. et al . (2017). A novel μ-conotoxin from worm-hunting Conus tessulatus that selectively inhibit rat TTX-resistant sodium currents. Toxicon130, 11–18. doi: 10.1016/j.toxicon.2017.02.013
40
Yao G. Peng C. Zhu Y. Fan C. Jiang H. Chen J. et al . (2019). High-throughput identification and analysis of novel conotoxins from three vermivorous cone snails by transcriptome sequencing. Mar. Drugs17(3), 193. doi: 10.3390/md17030193
Summary
Keywords
Conus , Tesseliconus, C. eburneus , C. tessulatus , conopeptides, transcriptome
Citation
Tablizo FA, Masacupan DJM and Lluisma AO (2025) Analysis of venom gland transcriptomes from two Tesseliconus species, Conus eburneus and Conus tessulatus, reveals inter- and intra-specific variations in conopeptide diversity and expression as well as putative novel gene superfamilies and disulfide-poor venom components. Front. Mar. Sci. 12:1616692. doi: 10.3389/fmars.2025.1616692
Received
23 April 2025
Accepted
06 June 2025
Published
26 June 2025
Volume
12 - 2025
Edited by
Bingmiao Gao, Hainan Medical University, China
Reviewed by
Linlin Ma, Griffith University, Australia
Khaled Mohammed Geba, Menoufia University, Egypt
Yabing Zhu, Beijing Genomics Institute (BGI), China
Updates
Copyright
© 2025 Tablizo, Masacupan and Lluisma.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Arturo O. Lluisma, aolluisma@up.edu.ph
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.