 Luca Vitanza1
Luca Vitanza1 Anna Maria Coccia1Alessia Peluso1David Brandtner2
Anna Maria Coccia1Alessia Peluso1David Brandtner2 Anna Muratore1
Anna Muratore1 Fulvio Ferrara1
Fulvio Ferrara1 Luca Lucentini1Andrea Piccioli3
Luca Lucentini1Andrea Piccioli3 Giuseppina La Rosa1
Giuseppina La Rosa1 Rossella Briancesco1* and The sea care team
Rossella Briancesco1* and The sea care team- 1National Center for Water Safety (CeNSiA), Istituto Superiore di Sanità, Rome, Italy
- 2Department of Infectious Disease, Istituto Superiore di Sanità, Rome, Italy
- 3Office of the Director General, Istituto Superiore di Sanità, Rome, Italy
Introduction: This study provides a comprehensive analysis of the microbial communities in marine environments across different geographical regions, including the Mediterranean Sea (Northwest and Southwest), the Atlantic Ocean, the Arctic Ocean, and the Indian Ocean. The goal of this research is to examine the diversity and composition of microbial life in these ecosystems and investigate the impact of environmental factors on microbial communities.
Materials and methods: High-throughput NGS sequencing techniques of the V3-V4 variable region of the 16S rRNA gene was employed to identify bacterial composition of the epipelagic communities in 45 water samples, using the MiSeq rRNA amplicon sequencing protocol (Illumina). Shannon index was used to described population diversity in samples.
Results: We identified significant differences in the bacterial composition of these ecosystems, highlighting the dominance of Proteobacteria, Cyanobacteria, and specific genera such as SAR11, Alteromonas, and Synechococcus. Variations in microbial communities were strongly influenced by environmental factors such as temperature and salinity. Notably, the Mediterranean exhibited the highest microbial diversity, while the Atlantic displayed thelowest.
Discussion: Our results reveal the complex interplay between microbial life and environmental conditions, emphasizing the need for long-term monitoring of these communities. These baseline data are essential for understanding the impacts of climate change and anthropogenic pollution on marine ecosystems. By improving our understanding of microbial biodiversity and its connection to ecological and human health, this study contributes to the broader goal of planetary health, offering a foundation for future efforts to mitigate the effects of environmental alterations.
1 Introduction
Marine microbes live in a dynamic equilibrium, interacting in numerous ways with each other and with the surrounding environment, through complex trophic networks, ruled by cross-feeding and ecological successions. Numerous factors influence the structure of marine bacterial communities such as inorganic nutrient concentration, phosphorus, nitrogen, seasonal variations, depth, oxygen levels, salinity, algal composition, and particulate organic carbon. Environmental variables drive genetic and functional diversity across locations, selecting for the most suitable metabolic capabilities under specific conditions (Morales and Holben, 2011). In marine habitats, mutualism and competition are obligate strategies shaping the structure of the bacterial community at any given time, following seasonal succession in prokaryotes and picoeukaryotes species abundance (Giordano et al., 2024; Korlević et al., 2022). Through the interaction network called Microbial loop, microorganisms contribute to global biogeochemical cycles, the recycling of organic debris, and the maintenance of balance and productivity within oceanic food chains (Cherabier and Ferrière, 2022). Moreover, in natural ecosystems, microorganisms play a critical role in the biodegradation of pollutants, while the high diversity within microbial communities enhances their resilience to the spread of antimicrobial resistance, both of which are vital for maintaining planetary homeostasis (Klümper et al., 2024).
Microbes are key players in horizontal gene transfer, which enables entire bacterial communities to rapidly adapt to environmental fluctuations, acting as a superorganism that responds to real-time changes. Furthermore, secondary metabolites, produced in a diverse range of molecules, serve crucial ecological roles by providing selective advantages and facilitating chemical communication between species and their environment (Giordano et al., 2015).
The functioning of ecosystems has been dramatically altered in last decades by human activities and pollutants (Hajji and Lucas, 2024). In the marine environment, xenobiotic molecules of human origin have a profound impact on the stability of microbial communities, which should be considered a fundamental standard for maintaining water quality, preserving ecosystem health, and advancing sustainable development (Nimnoi and Pongsilp, 2020). Anthropogenic pollution, including excess nutrients, microplastics, heavy metals, and chemicals, significantly impacts marine microbial communities, especially in coastal areas, where increased exposure leads to disruptions in community structure, diversity, and ecosystem functions. Iron fertilization of the seas is widely used, not without consequences for algal blooms (Buesseler et al., 2008). In such a vast and interconnected habitat, marine trade contributes to the spread of bacterial strains from allochthonous sites, implementing the core microbiota of the ocean and thus attenuating biogeographic specificity. Changes in the physiology or abundance of one organism alter ecosystems, affecting other taxa and reshaping communities (Hennon et al., 2018). Artificial substrates (e.g. microplastics) floating in the oceans or sinking for degradation are a major environmental concern, both through the diffusion of chemical pollutants and through microbial adhesion to suspended particles in the water column (Harbeitner et al., 2024), with a demonstrated ability to spread antimicrobial resistance genes and strains to new areas via horizontal gene transfer (Bowley et al., 2021; Stevenson et al., 2024; Yang et al., 2019).
Of particular concern are the effects of bacterial population shifting on aspects directly or indirectly related to human health. Climate alteration is enhancing infection patterns for certain diseases previously occurring mainly in warm regions: pathogenic bacteria are thriving in their familiar habitats and adapting to new areas as their ranges expand. Some species of the genus Vibrio are among the most potentially dangerous human pathogens (Baker-Austin et al., 2016; Semenza et al., 2017; Vezzulli, 2023).
Research efforts have primarily focused on coastal regions within specific geographical areas, leaving much of the open ocean underexplored in terms of its microbial communities and their interactions with environmental biotic and abiotic factors. Moreover, our understanding of the whole network of relationships between bacterial groups cannot be sufficiently supported by in vitro studies, since most marine bacteria remain unculturable under canonical laboratory conditions (Stulberg et al., 2016) although interesting perspectives have been proposed by some authors (Berdy et al., 2017; Rodrigues and de Carvalho, 2022).
Sequencing technology and computational methods for analysing large DNA sequence datasets have significantly enhanced our understanding of marine microbial physiology and interactions (Moran, 2015). High-throughput next-generation sequencing (NGS) technology targeting the taxonomically informative 16S rRNA gene is the standard method for exploring the entire bacterial community in marine environments. While it provides detailed insights into the composition and diversity of microbial communities, its resolution is generally limited to broader taxonomic levels, such as genus. It is estimated that only a small fraction of the microbial species in the ocean have been catalogued, with the true extent of diversity likely far exceeding what is represented in current databases, believed to encompass less than 5% of oceanic microbial communities (Sunagawa et al., 2015).
Building on previous global marine projects (e.g. TARA Oceans, 2009-2013) (PANGAEA, 2017), this research aims to enhance our understanding of oceanic microbial communities by investigating their diversity and key physico-chemical drivers, and looking forward to how changes in these factors might affect the structure and dynamics of marine microbial communities, including any shifts in their composition.
This research is part of the Sea Care project, a three-year investigation (2022-2025), that is a collaborative initiative between the Italian Military Navy and the National Institute of Health - Istituto Superiore di Sanità (ISS). The aim of this global project is to study the health risks associated with emerging contaminants in the marine environment and climate change, from a ‘planetary health’ perspective, covering the world’s oceans.
The data presented in this study, obtained from the first-year project, are crucial for understanding the ocean’s microbiome. This knowledge provides a valuable baseline for understanding ocean microbiome composition and variability across regions and may contribute to future efforts in long-term ecological monitoring, ocean health assessments, and the development of informed strategies for sustainable marine governance, in line with the objectives of the UN2030 Agenda.
2 Materials and methods
2.1 Water sampling and filtration
Between May 2022 and January 2023, during four sampling campaigns, a total of 45 marine water samples (Supplementary Table S1; Figure 1) were collected aboard 4 military naval vessels: Amerigo Vespucci, Caio Duilio, Alliance, and Paolo Thaon di Revel. The routes crossed the Mediterranean Sea, the Indian Ocean (including Persian Gulf, the Gulf of Oman, the Gulf of Aden and the Red Sea), the Atlantic Ocean and the Arctic Ocean. Sampling procedures were adapted to the operational conditions of each vessel and to the prevailing weather and sea state. Whenever possible, the ship was brought to a complete stop and seawater was collected manually from the starboard side near the bow, after temporarily shutting off all nearby discharges to minimize contamination risk. Alternatively, when sea conditions permitted, a small boat (launch) was deployed to collect samples at a short distance from the main vessel. These precautions were taken to avoid any contamination from the ship’s structure or engine. At each site, 3–5 L of surface seawater were collected from approximately 30 cm depth using a dedicated bucket that was thoroughly sanitized after each use. To focus on the free-living microbial fraction and minimize membrane clogging, a two-step filtration procedure was performed on board using a vacuum filtration system. Seawater was first pre-filtered through a 5 µm mesh filter (mixed cellulose esters, Millipore) to remove larger particles. The resulting filtrate was then passed through a 0.22 µm sterile membrane filter (mixed cellulose esters, 47 mm diameter, Millipore) to retain microbial cells. Each 0.22 µm filter was placed in a sterile 2 mL tube and stored at −20 °C until DNA extraction.
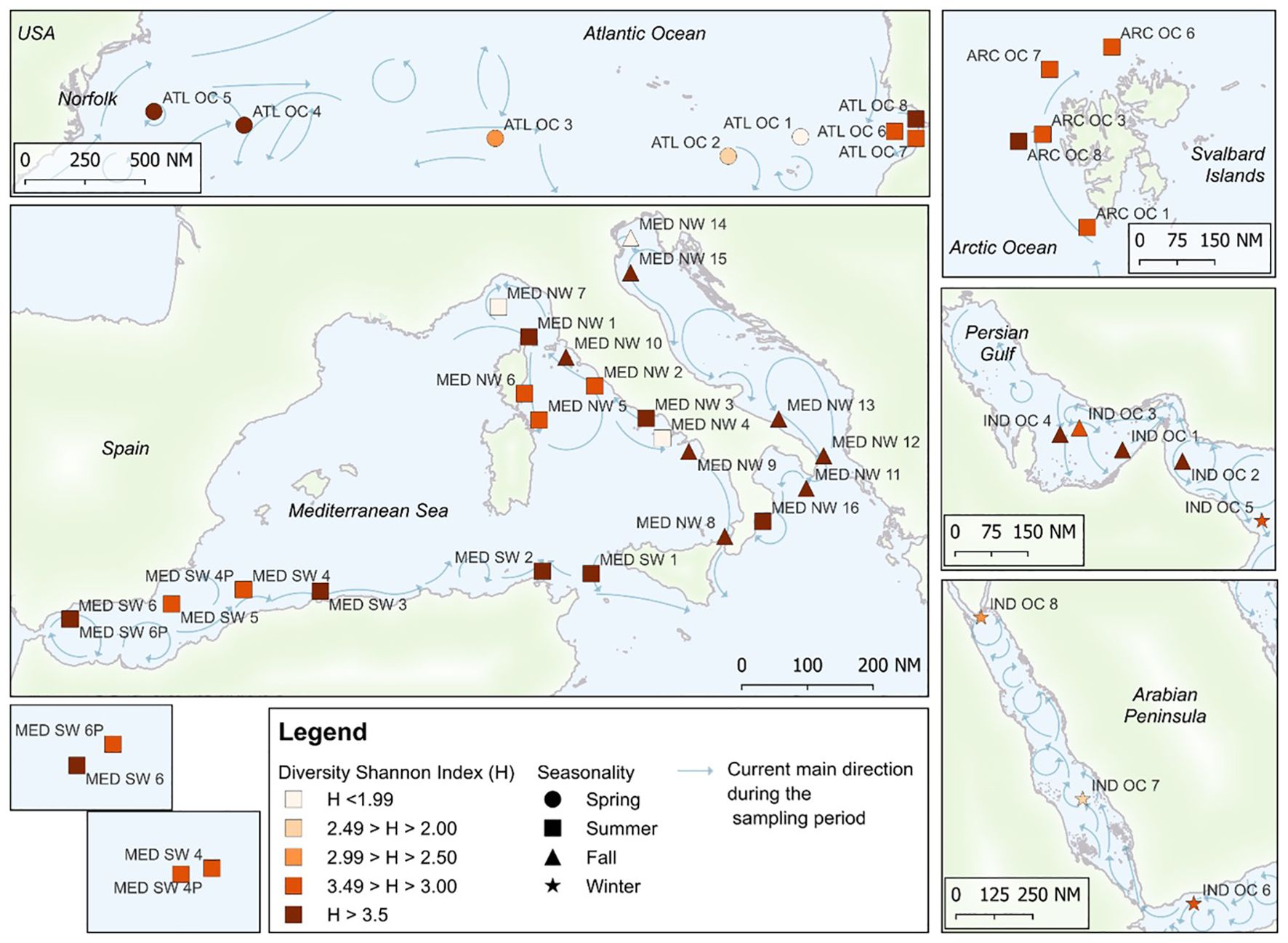
Figure 1. Sampling points from the first-year campaign of the Sea Care project are provided, along with the Shannon Diversity Index (H), the season of sampling, and the direction of marine currents at the time of sampling. The main currents direction is represented as monthly average during the sampling period and is elaborated starting from data by Copernicus Marine Service (CMEMS https://marine.copernicus.eu/, database DOI: 10.48670/moi-00016).
2.2 Determination of seawater environmental parameters
Seawater temperatures, pH, salinity and chlorophyll were measured on site at each sampling location using a handheld multi-parameter water quality probe (IDRONAUT, Ocean Seven 316 Plus) equipped with dedicated sensors for each parameter.
2.3 DNA extraction, sequencing, bioinformatics, and statistical analysis
Total DNA from the prokaryotic biomass retained on the membrane filters was extracted in the laboratory using the DNeasy PowerWater Kit (Qiagen) according to the manufacturer’s instruction. DNA purity and concentration were determined using a Qubit 4.0 Fluorometer with the Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific), following the manufacturer’s instructions.
The V3–V4 region was amplified using the primer pair 341F (5′-CCTACGGGNGGCWGCAG-3′) and 785R (5′-GACTACHVGGGTATCTAATCC-3′), originally designed by Klindworth et al. (2013), which target a ~460 bp fragment of the 16S rRNA gene. The V3–V4 variable region was sequenced following the MiSeq rRNA Amplicon Sequencing protocol (Illumina, San Diego, CA) with some modifications. The detailed methodology for DNA extraction, PCR amplification, and sequencing, including reagent concentrations, cycling conditions, and sequencing protocol, is provided in the Supplementary Materials.
Sequencing was performed on an Illumina MiSeq platform. Bioinformatics analyses were performed in Quantitative Insights into Microbial Ecology v2022.2 (QIIME2) software suite and R packages, the DADA2 algorithm, and an in-house trained classifier for taxonomy assignment. The Silva database, release 138.1 was used to identify the bacterial flora in each sample and to compare them between groups.
The relative abundance rate of the bacteria was determined at both the phylum and genus level, focusing on taxa that accounted for at least 1% of the total microbial community in each water sample.
The alpha diversity index was calculated for each library, and the analysis refers to the variety and complexity of species in a community. Shannon index was used to describe population diversity in samples (Sarmiento et al., 2019).
Statistically significant differences in alpha diversity (Shannon) in pairwise comparisons between bacterial communities from different geographical areas were estimated using the Kruskal-Wallis pairwise test (QIIME2). The correlation between physico-chemical parameters and bacterial genera abundance was determined using Pearson’s correlation. Due to the compositional nature of relative abundance data, Pearson correlation analysis required appropriate data transformation to avoid spurious results. Following Gloor et al. (2017), relative abundance data were transformed using centered log-ratio (CLR) transformation implemented in the “compositions” package (version v.4.3.1).
3 Results
3.1 Sequence read summaries
Details on the sequence read summaries, including the total number of filtered reads, geographic group distributions, and amplicon sequence variant (ASV) clustering, are provided in the Supplementary Materials (Supplementary Table S2). Sequences were submitted to the NCBI Sequence Read Archive (SRA) (link to be provided at proof stage, currently under review).
3.2 Marine microbiome
A total of 31 distinct phyla were identified. The most prevalent phyla, along with their occurrence percentages across the 45 sampling sites, were: Proteobacteria, Actinobacteriota, and Bacteroidota (100%), Cyanobacteria (95.5%), Verrucomicrobiota (93.3%), Marinimicrobia (82.2%), Campylobacterota (75.5%), and Firmicutes (15.5%). These phyla appeared in varying proportions across the samples. Proteobacteria were the most abundant at all sites, with relative frequencies ranging from 31.50% to 94.30%, followed by Bacteroidota (0.20%–48.11%), Cyanobacteria (0.19%-46.20%), Actinobacteriota (1.11%-18.59%), Firmicutes (0.09%–14.30%), Verrucomicrobiota (0.07%-3.84%), Campylobacterota (0.07%-2.80%), and Marinimicrobia (0.08 -1.77%).
Approximately 400 bacterial genera were identified within the 45 microbial communities examined. The heat map in Supplementary Figure S4 represents the relative abundance of bacterial genera identified in seawater samples collected from different geographical regions, as determined by 16S rRNA gene sequencing. Only genera with a relative abundance greater than 1% were included in this heat map to simplify visualization, highlight the most significant taxa, and minimize the influence of low-abundance taxa. A core group of 15 genera is shared by the Atlantic, Mediterranean and Indian Oceans, while the Arctic Ocean has a different composition, with a smaller number of genera.
The north-western (NW) Mediterranean shows a greater diversification of genus composition, as seen in the central part of the map. SAR11 clade Ia was the most widespread genus identified in this study, with higher relative abundances in Mediterranean and Atlantic water samples. Even at lower abundance levels, SAR11 clade II remained widely distributed. A high abundance of Alteromonas and Pseudoalteromonas was observed in some samples from the NW Mediterranean Sea and from the Atlantic Ocean, respectively. These two genera were predominant in number of ASVs (Alteromonas 28253 ASVs in the sample MED NW7; Pseudoalteromonas 19432 ASVs in ATL OC1). Except for the Arctic Ocean, where Synechococcus_CC9902 was detected sporadically and in trace amounts (below 1% relative abundance), the genus was present in all other geographic areas considered, with the highest relative abundance in Mediterranean Sea samples. Two other genera frequently encountered are AEGEAN-169_marine_group and SAR86.
The microbial community structures across different geographical areas are presented in Figures 2–6. For each geographical area, the detected bacterial phyla and genera are presented in bar-coded form, with the results described individually. For clarity and consistency, taxonomic composition plots at the genus level (Figures 2-6, lower panels) display only those ASVs that could be classified at the genus level. ASVs assigned only at the family level or higher were excluded from these visualizations.
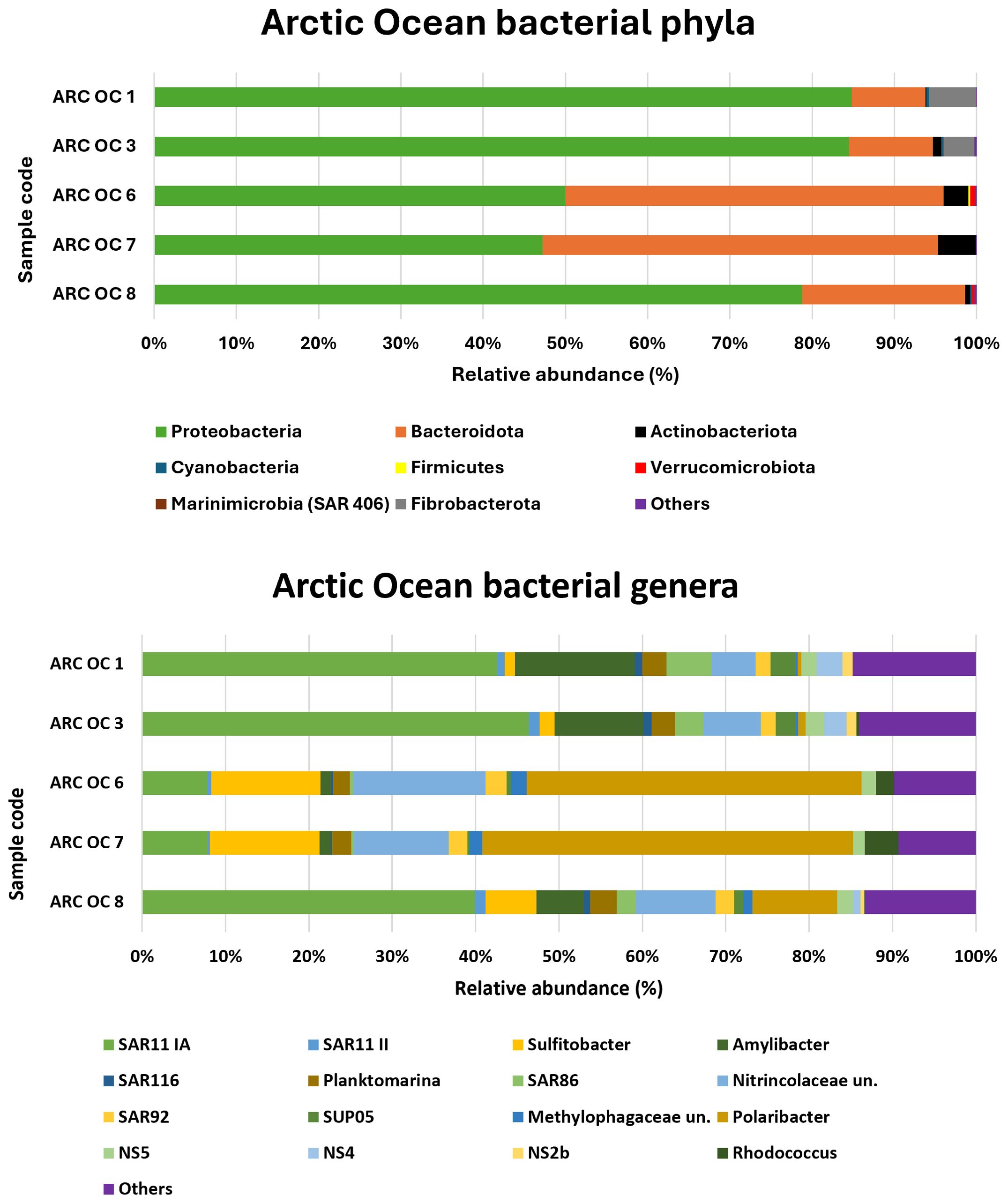
Figure 2. Relative abundance of bacterial phyla and genera in the Arctic Ocean.
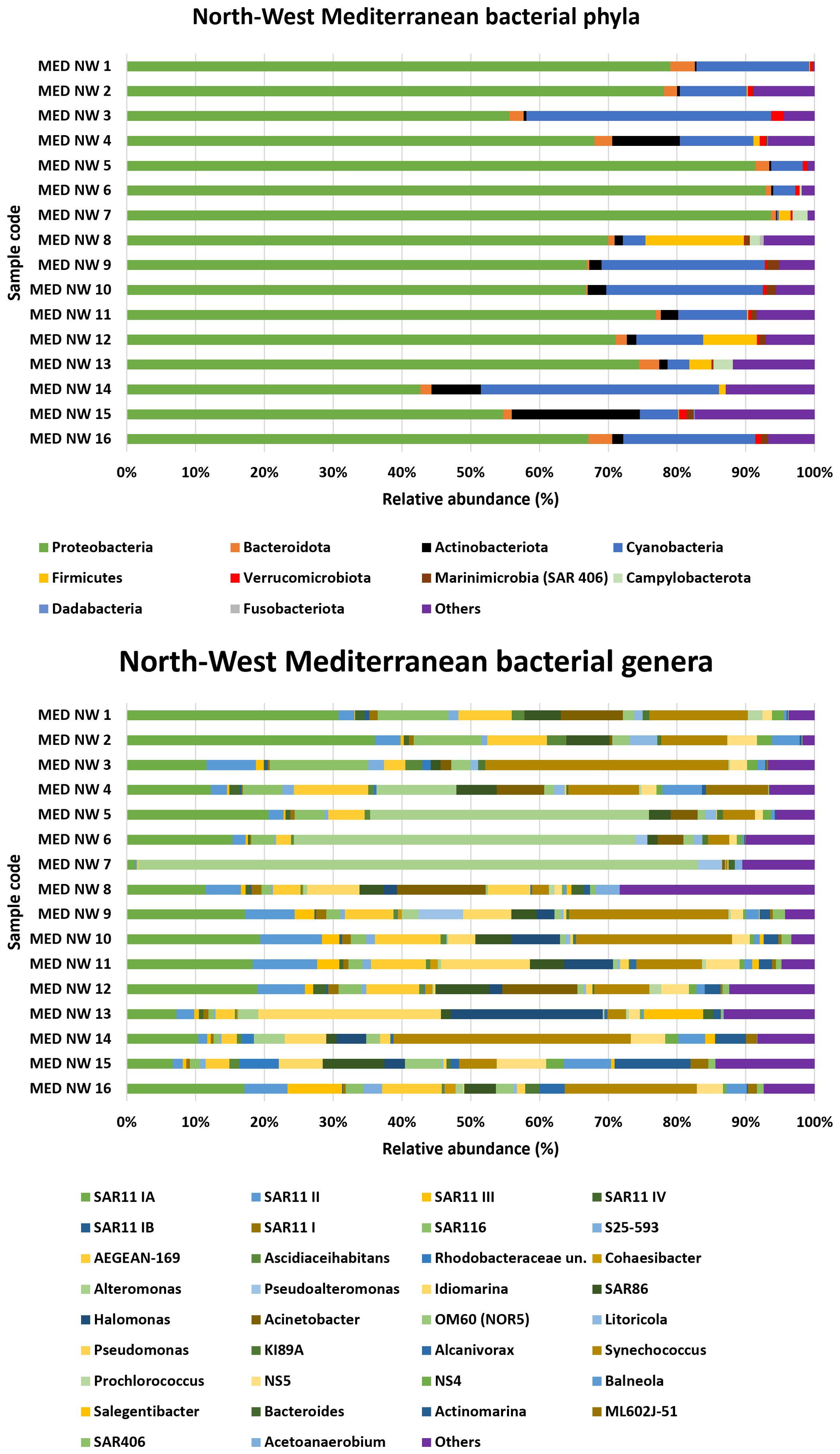
Figure 3. Relative abundance of bacterial phyla and genera in the Northwest Mediterranean Sea.
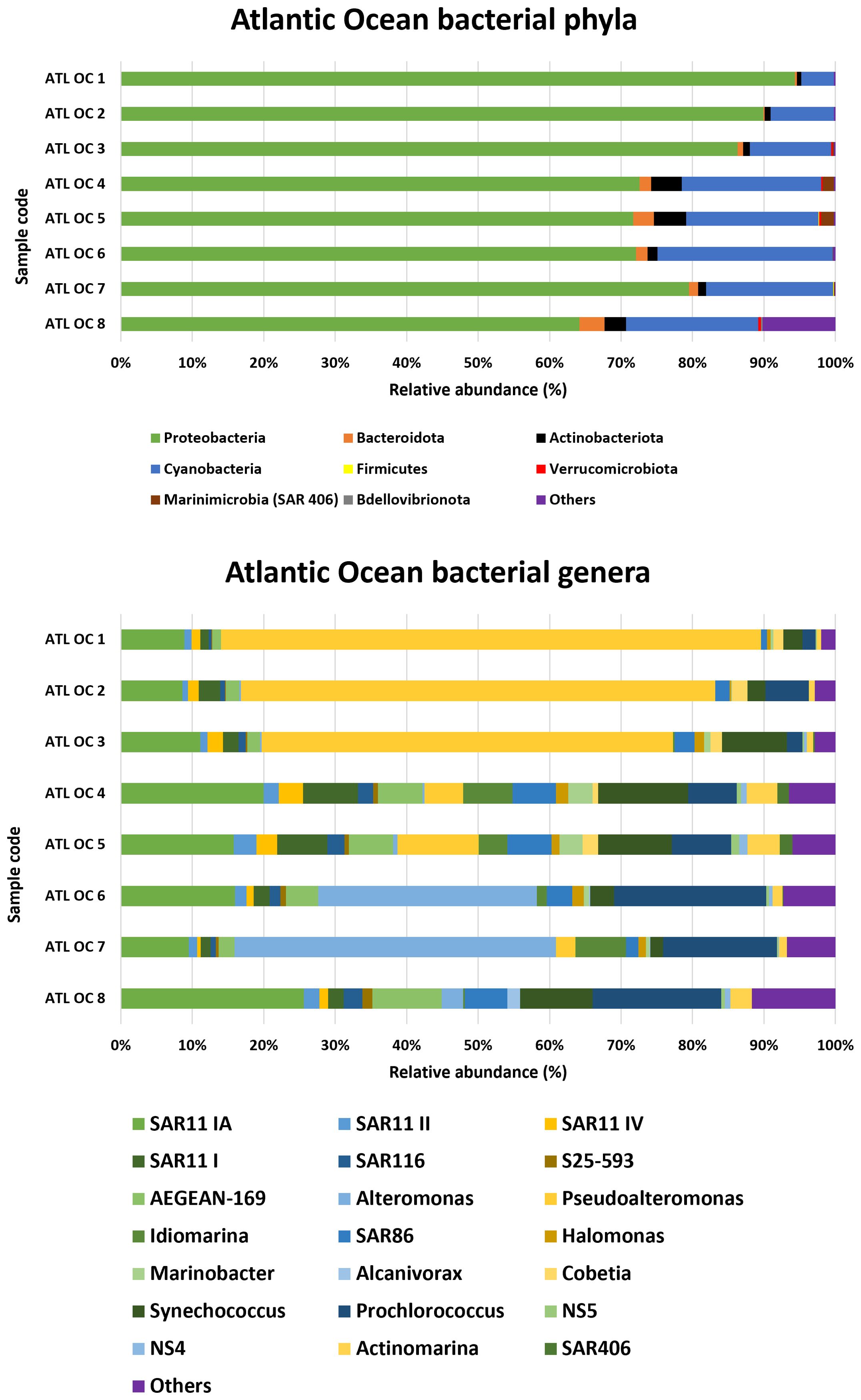
Figure 4. Relative abundance of bacterial phyla and genera in the Southwest Mediterranean Sea.
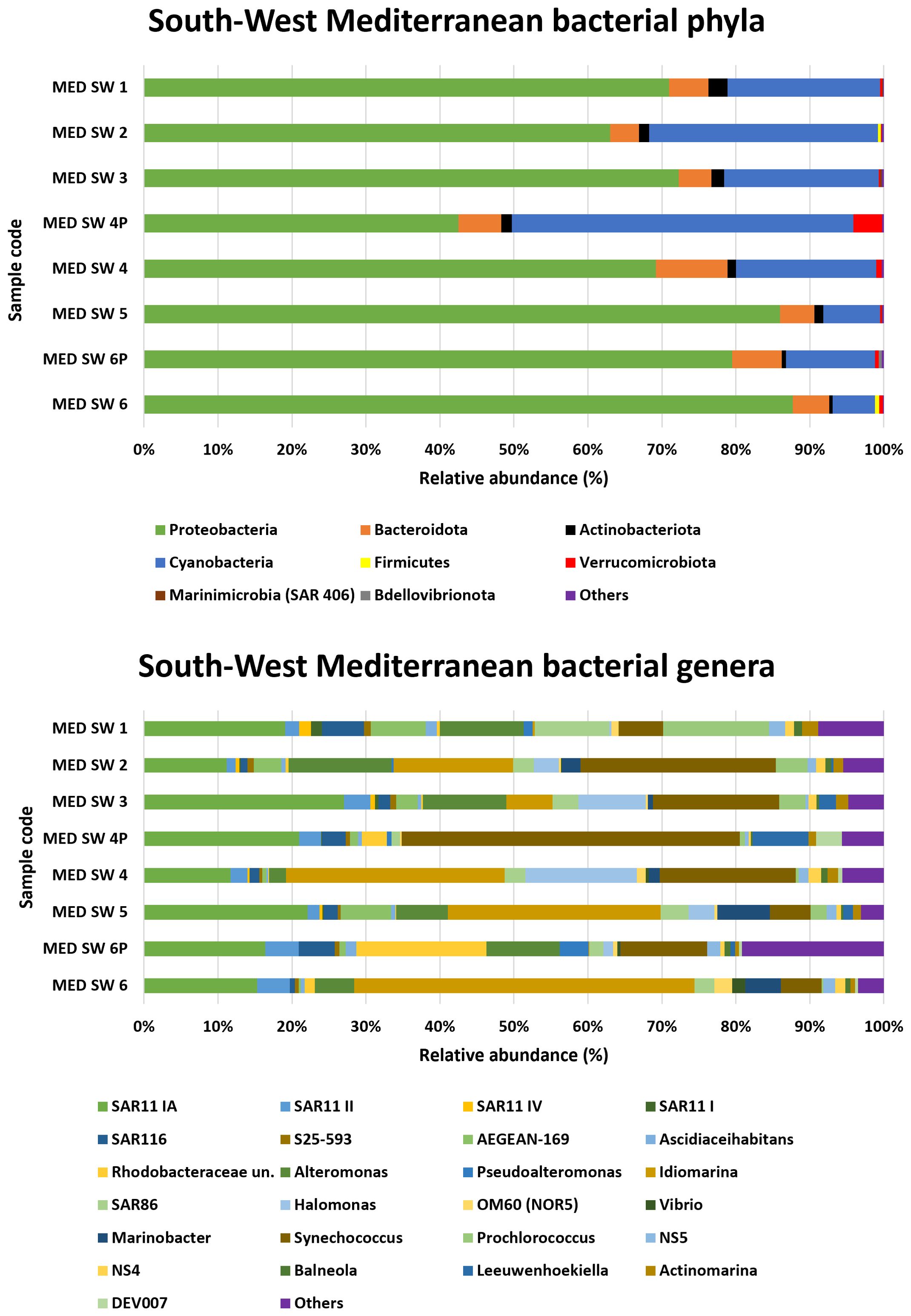
Figure 5. Relative abundance of bacterial phyla and genera in the Atlantic Ocean.
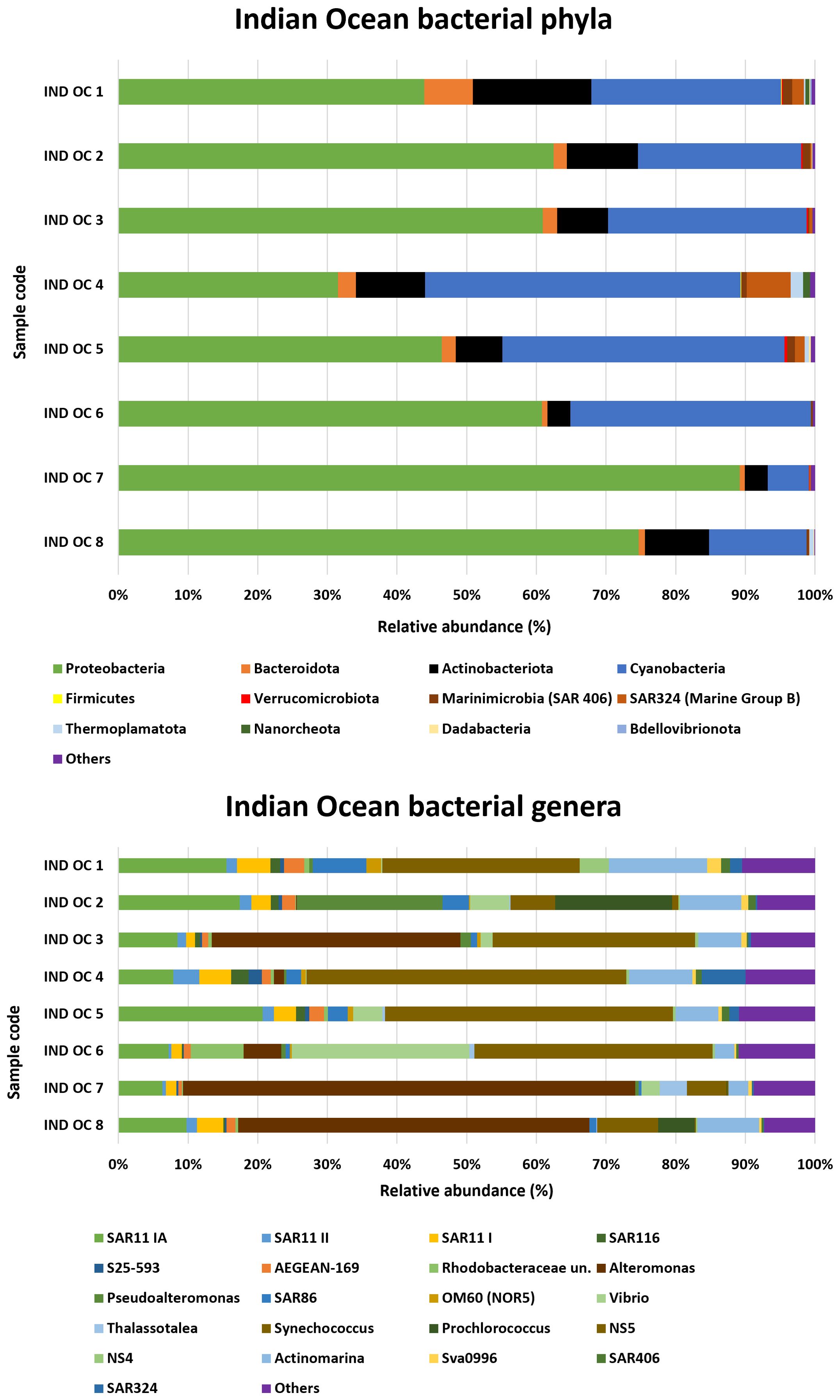
Figure 6. Relative abundance of bacterial phyla and genera in the Indian Ocean.
3.2.1 Arctic ocean
The relative abundances of bacterial phyla and genera detected in the Arctic Ocean are shown in Figure 2. The phyla Proteobacteria and Bacteroidota accounted for more than 90% of the relative abundance. Fibrobacterota and Actinobacteriota were also present with relative abundances of approximately 6% in the samples that were richer in these two minority phyla (ARC OC 1 and ARC OC 7, respectively). Cyanobacteria were only present in traces in some of these samples. At the genus level, the predominance of the SAR11 clade Ia was characteristic of samples ARC OC 1, ARC OC 3, and ARC OC 8. In the northern most samples ARC OC 6 and ARC OC 7, the genus Polaribacter, belonging to the phylum Bacteroidota, was predominant with a relative abundance of more than 40%. The highest relative abundance for the genus Sulfitobacter was found in the sample ARC OC 7 (13.2%) and ARC OC 6 (13.1%), while the lowest was found in ARC OC 1 (1.2%). The Nitrincolaceae were consistently present in the analysed samples, with percentages ranging from ~5% to ~16%, with the highest percentages observed in the ARC OC 6 and 7 samples. The genus Rhodococcus, belonging to the family of Nocardiaceae, detected in samples ARC OC 3-6-7 and 8, had the highest relative abundance in sample ARC OC 7 (4%).
3.2.2 Northwest Mediterranean sea
At the phylum level, the NW Mediterranean Sea was dominated by Proteobacteria, which accounted for more than 90% of the total population in some samples (MED NW 5, 6, and 7) (Figure 3). The second most abundant phylum was Cyanobacteria, with the highest percentages found in samples MED NW 3 (35.6%) and MED NW 14 (34.6%).
The dominant group across all samples was the SAR11 clade Ia, with more modest yet notable contributions from other clades such as II, III, IV, and Ib. Notably, clade II is the second most abundant after Ia, reaching 9.4% in sample MED NW 11.
Among the Alphaproteobacteria, the SAR116 group stood out for its relevance, with a maximum percentage of 14.3% in sample MED NW 13. Among the Gammaproteobacteria, Alteromonas was one of the most common genera with a relative abundance value reaching 81.2%, alongside the SAR86 group (9%). The genus Idiomarina was also significant: although absent in samples MED NW 1 through MED NW 7, it was present in all the other samples with varying percentages, reaching 26.6% in sample MED NW 13.
Regarding bacteria of the family Flavobacteriaceae, the genus Salegentibacter was found, with a presence varying from 0.1% (MED NW 9) to 8.6% (MED NW 16). The genus Vibrio was identified only in a few samples, with a presence of 0.4% in samples MED NW 6 and MED NW 7, and 0.2% in sample MED NW 9.
Finally, the genus Acinetobacter was present in high percentages in four samples: MED NW 8 (12.9%), MED NW 12 (10.9%), MED NW 1 (9%), and MED NW 4 (6.9%).
3.2.3 Southwest Mediterranean sea
The analysis of bacterial phyla in the southwestern Mediterranean is shown in Figure 4. Proteobacteria were the dominant phylum in all samples, with relative abundances ranging from 42.5% in the MED SW 4P sample to 88% in MED SW 6.
Cyanobacteria represented a substantial proportion of the bacterial community in certain samples, peaking at 46.2% in MED SW 4P and reaching its lowest level of 5.7% in MED SW 6.
Sample MED SW 4P exhibited the lowest contribution of Proteobacteria, the highest abundance of Cyanobacteria, and the maximum detection of Verrucomicrobiota (3.9%).
Bacteroidota was consistently present across all samples, with percentages ranging from 3.9% in MED SW 2 to 9.7% in MED SW 4. SAR11 clade Ia was the most prominent genus, with its peak in sample MED SW 3 (27.1%) and the lowest abundance in MED SW 4 (11.7%). SAR11 clade II contributed to the microbial community, with a maximum of 4.5% in MED SW 6P. SAR116 showed moderate representation, reaching 5.7% in MED SW 1. Among the Gammaproteobacteria, the genus Alteromonas was remarkable, especially abundant in MED SW 2 (13.9%) but absent in MED SW 4P. Another noteworthy genus was Idiomarina, which dominated certain samples such as MED SW 6 (46%) and MED SW 4 (29.6%). Synechococcus and Prochlorococcus were the most abundant genera. Synechococcus was particularly prominent in sample MED SW 4P (45.8%). Prochlorococcus, although less abundant, showed its highest representation in MED SW 1 (14.4%). Finally, Vibrio was detected in 50% of the samples.
3.2.4 Atlantic ocean
In the Atlantic Ocean samples (Figure 5), Proteobacteria was the most abundant phylum, with the highest proportion observed in ATL OC 1 (94.3%) and the lowest in ATL OC 8 (64.2%). Cyanobacteria was the second most prevalent phylum, with a maximum relative abundance recorded in ATL OC 6 (24.5%) and a minimum in ATL OC 1 (4.6%). The Marinimicrobia (SAR 406) phylum was present in small but notable proportions, particularly in ATL OC 4 and ATL OC 5, with values of 1.6% and 1.8% respectively. At the genus level, Synechococcus was found consistently among the most abundant cyanobacterial genera across Atlantic samples, although its relative abundance varied markedly among stations, peaking at ATL OC 4 (12.6%) and ATL OC 5 (10.3%), and showing lower levels at ATL OC 7 (1.8%). Pseudoalteromonas reached a high relative abundance in ATL OC 1 (75.6%), where it represented a dominant component of the community. Alteromonas was also well represented across multiple sites, particularly in ATL OC 7 (45%) and ATL OC 6 (30.6%). Additional genera, such as SAR11 clade Ia, Prochlorococcus and AEGEAN-169 marine group were recurrently detected and contributed to the overall diversity.
3.2.5 Indian ocean
Two phyla from the Archaea domain, Thermoplasmatota and Nanoarchaeota, were detected with percentages never exceeding 2% (Figure 6). The bacterial phyla Proteobacteria, Bacteroidota and Actinobacteriota were well represented in the Indian Ocean samples, with peak abundances of 89.2% (IND OC 7), 7% (IND OC 1) and 17% (IND OC 1), respectively, while Nanoarchaeota and Thermoplasmatota were detected with percentages never exceeding 2%.
Proteobacteria abundance ranged from a high of 89.2% in IND OC 7 to a low of 31.5% in IND OC 4. Analysis of bacterial genera in the Indian Ocean samples revealed the occurrence of SAR11 clade I, SAR 11 II, and SAR 11 IV (the latter less than 1%) across all samples.
Synechococcus was the most abundant and widespread genus, detected in all the samples examined. The genus Alteromonas was predominant in most samples, surpassing Pseudoalteromonas in all cases except for IND OC 2, where Pseudoalteromonas peaked at 20.9%. The genus SAR86 was widespread in this area, although its abundance varied: it reached a maximum in IND OC 1 (7.7%), but declined in IND OC 7, where it was almost absent (0.4%). Prochlorococcus was found in 3 samples reaching a relative abundance of 16.8% in sample IND OC 2. IND OC 6 had the highest proportion of the genus Vibrio among all Indian Ocean samples with 25.5%, followed by IND OC2, IND OC 5, IND OC 6 and IND OC3 with 5.6%, 4.2%, 2.6% and 1.7% respectively.
3.3 Alpha diversity
Figure 7 shows the Shannon alpha diversity index of microbial communities across the different geographic regions, highlighting variations in community richness and evenness. Statistically significant differences among oceanic regions are reported in Supplementary Table S3 (Kruskal–Wallis, p < 0.05).
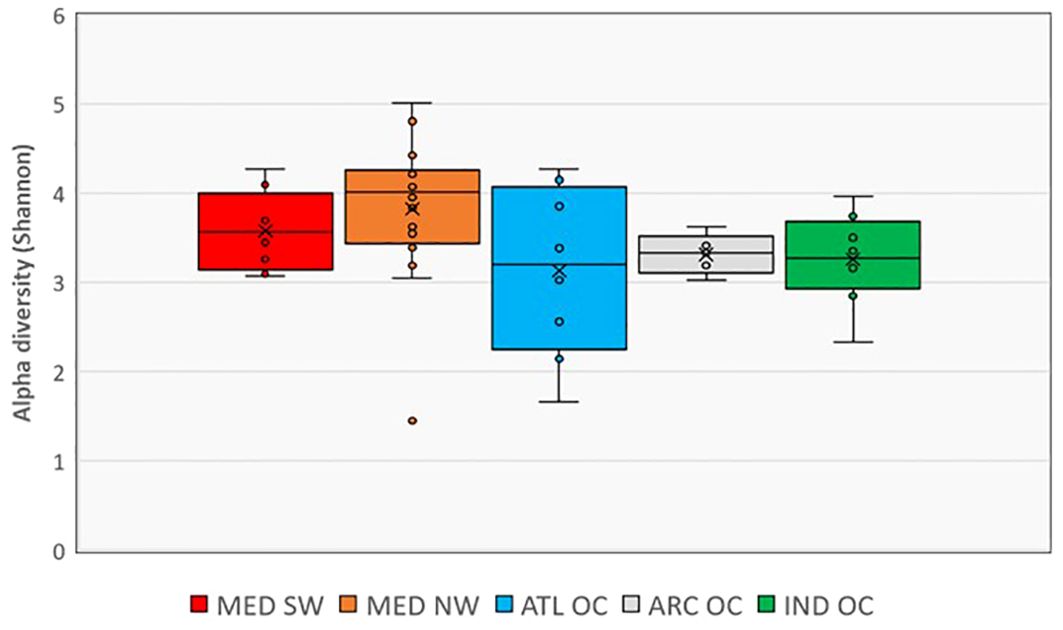
Figure 7. Alpha-diversity Shannon index of microbial communities from different geographic areas.
The Mediterranean Sea exhibited the highest alpha diversity among all sampled areas. Both subregions—northwestern (MED NW) and southwestern (MED SW)—had significantly higher Shannon values than the Arctic Ocean and the Indian Ocean, with a particularly marked difference between MED NW and the Indian Ocean (p < 0.01), and a significant difference between MED SW and the Indian Ocean (p < 0.05). Among the two Mediterranean subregions, MED NW showed slightly higher median diversity than MED SW.
The Atlantic Ocean showed the broadest variability in Shannon values across sites, as reflected by the large interquartile range. While some samples had high diversity, the median value was comparable to MED SW but lower than MED NW.
The Arctic Ocean displayed the lowest alpha diversity, with minimal variation between samples. The Indian Ocean, which includes semi-enclosed basins like the Persian Gulf, Gulf of Oman, Gulf of Aden, and Red Sea, also showed significantly lower alpha diversity compared to both Mediterranean subregions.
The spatial arrangement of sampling sites, overlaid with Shannon diversity classes, is shown in Figure 1, offering insight into the geographic structuring of microbial alpha diversity.
3.4 Environmental parameters
Detailed results for temperature, salinity, and chlorophyll in the seawater samples are provided in the Supplementary Materials. Supplementary Figure S1 illustrates seawater temperatures across the sampling sites, ranging from nearly 0°C in the Arctic Ocean samples to approximately 30°C in the Indian Ocean. Supplementary Figure S2 shows salinity values (PSU) across the sampling sites, ranging from approximately 30 PSU in Arctic Ocean samples to values approaching 40 PSU in tropical waters and in a few samples from the northwestern Mediterranean Sea. In the southwestern Mediterranean, salinity values were lower, reaching approximately 37.5 PSU. Supplementary Figure S3 illustrates chlorophyll concentrations (µg/L) across the sampling sites. The values show a high degree of variability, with peaks observed in certain samples from the North-Western Mediterranean and the Atlantic Ocean, reaching up to 17.8 µg/L, while most other sites exhibit lower concentrations.
3.5 Correlation of bacterial relative abundance with environmental parameters
The analysis of correlations between the relative abundance of microbial genera and environmental variables revealed distinct patterns associated with salinity, chlorophyll, and temperature, as shown in Figure 8. Regarding salinity, many genera exhibited negative correlations, indicating higher abundance in lower salinity waters. Notably, Marine Group II, Candidatus Actinomarina, Thalassotalea, and Synechococcus CC9902 showed pronounced negative associations with salinity. Conversely, some taxa such as the SAR116 clade and KI89A clade were more abundant in environments characterized by higher salinity.
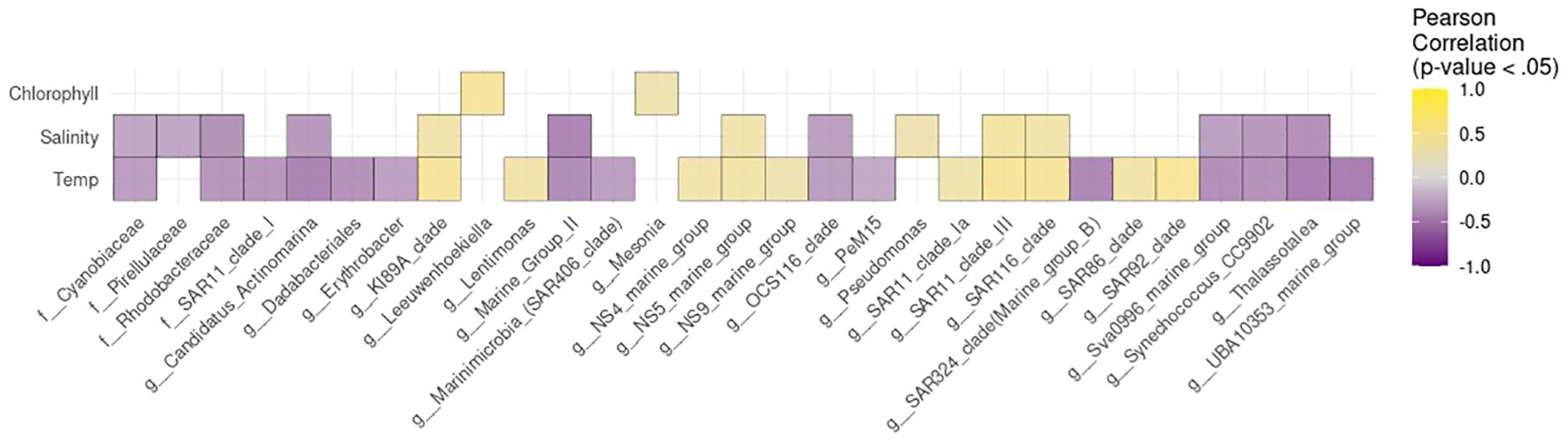
Figure 8. Pearson correlation heatmap between the relative abundance of bacterial genera/clades and environmental variables (temperature, salinity, and chlorophyll) at the sampling sites. Total relative abundance data were used, without applying any thresholds. Taxa are reported at the genus level according to SILVA classification. Only in cases where the genus was annotated as uncultured, the corresponding higher taxonomic rank is shown. The prefixes “g” and “f” indicate taxonomic assignments at the genus and family levels, respectively.
In relation to chlorophyll, only Leeuwenhoekiella and Mesonia were weekly positively correlated.
Correlations with temperature were more variable across taxa. Groups like the SAR86 clade, SAR116 clade, and SAR92 clade showed positive correlations with temperature, while others such as SAR11 clade I, and UBA10353 marine group correlated negatively, indicating differing temperature preferences. Although the main correlation analysis was conducted on the entire dataset (n = 45) to ensure adequate statistical power, additional correlation heatmaps stratified by geographic region are presented in Supplementary Figure S4, allowing a more detailed inspection of area-specific patterns.
4 Discussion
This study provides a global overview of the diversity and composition of epipelagic bacterial communities across multiple oceanic regions. The selection of the sequenced 16S rRNA region was driven by its widespread use in marine microbiome studies and its proven compatibility with the Illumina MiSeq platform, enabling direct comparison with a large number of publicly available datasets (Klindworth et al., 2013; Caporaso et al., 2012; Sunagawa et al., 2015; Choi et al., 2021; Parada et al., 2016).
The analysis of bacterial communities across the 45 sampling sites revealed a high microbial diversity, identifying 31 distinct phyla and approximately 400 genera. The dominance of Proteobacteria, Actinobacteriota, and Bacteroidota, which were present in all samples, underscores their ecological significance in marine environments.
The wide range of relative abundances of Proteobacteria reflects their adaptability to diverse environmental conditions and their central role in nutrient cycling and organic matter degradation (Sunagawa et al., 2015). Actinobacteriota, a group of microorganisms widely distributed in the marine environment, has developed different metabolic strategies with greater potential for carbohydrate and recalcitrant compounds degradation. These bacteria can rapidly sense and respond to external environmental cues needed to survive environmental changes. In this context, a specific transcriptional regulator involved in iron-sulphur cluster-bound proteins has been described (Roda-Garcia et al., 2023). This group has also been studied for years as a producer of various bioactive substances with biotechnological and therapeutic potential (Manivasagan et al., 2014; Sheikh et al., 2018).
Bacteroidota, detected across all samples, contribute to the degradation of complex organic matter, reinforcing their importance in nutrient recycling and ecosystem functioning (Fernández-Gómez et al., 2013).
The high abundance of Cyanobacteria at sites with favorable light and temperature conditions highlights their importance as primary producers (Scanlan et al., 2009). The most abundant genera were Synechococcus (detected in 43 out of 45 sites) and Prochlorococcus (26/45), the most important photosynthetic organisms in seawaters. In particular, Synechococcus thrives in well-lit, nutrient-poor conditions, making it a key primary producer (Scanlan et al., 2009). Prochlorococcus, while characterized by low relative abundance in the analysed samples, prefers stable, nutrient-limited environments, contributing significantly to carbon fixation (Flombaum et al., 2013; Kent et al., 2019; Mella-Flores et al., 2012).
SAR11 clade Ia emerged as the most widespread and abundant genus, but other SAR11 clades were frequently detected in this order of abundance: clade II, clade IV, clade III and clade Ib. This Alphaproteobacteria genus was defined using 16S rRNA gene and internal transcribed spacer (ITS) analyses, which allowed members to be grouped into several clades with distinct spatiotemporal abundance patterns (Haro-Moreno et al., 2020; Tucker et al., 2021). As the most abundant group of planktonic bacteria globally, SAR11 thrives in nutrient-poor environments due to its small cell size and streamlined genome (Morris et al., 2002). Its ability to oxidize organic compounds to CO2 and facilitate nutrient fluxes underscores its pivotal role in the global carbon cycle (Giovannoni, 2017).
Among Alphaproteobacteria, another common genus found everywhere was the SAR116 group; it is notable for its involvement in carbon and sulfur cycles, linking microbial activity to broader biogeochemical processes (Roda-Garcia et al., 2021).
Alteromonas and Pseudoalteromonas, the two most frequent Gammaproteobacteria found, stood out for their high abundances in the northwestern Mediterranean Sea and Atlantic Ocean, respectively. These genera are known for their algicidal properties, regulating algal blooms and preventing eutrophication. Their ability to produce bioactive compounds, such as enzymes and toxins, not only helps to limit the growth of other microbial species but also positions them as important contributors to nutrient remineralization and biodegradation. Their biotechnological potential, including applications in bioremediation and antibiotic production, underscores their ecological and practical importance (Cho, 2012; He W. et al., 2023; Offret et al., 2016).
Two other Proteobacteria genera that were consistently detected were AEGEAN-169_marine_group and SAR86, suggesting significant contributions to marine ecosystem functions, although their specific ecological roles remain less explored (Dupont et al., 2012; Getz et al., 2023).
To simplify, the results obtained are discussed for each of the areas, highlighting particular findings.
4.1 Arctic ocean
The bacterial community structure in the Arctic Ocean reflects the adaptations necessary to survive in one of the most extreme marine environments.
Key taxa such as SAR11, Polaribacter, Sulfitobacter, SAR 92, and Amylibacter drive essential biogeochemical processes, including carbon and sulfur cycling and nutrient fluxes, underscoring their significance in polar ecosystems.
Polaribacter, identified as a major degrader of polysaccharides (Avcı et al., 2020; Deming and Eric Collins, 2017), is particularly abundant in northern samples, emphasizing its role in processing organic matter derived from phytoplankton blooms. Similarly, Sulfitobacter, a key player in the sulfur cycle, interacts with marine microalgae to degrade algal-derived organic sulfur compounds (Zeng et al., 2020). SAR 92 bacteria are potential degraders of organosulfur molecules and sources of climate-active gases in the marine environment, especially in polar regions (He X.-Y. et al., 2023).
The consistent detection of Nitrincolaceae and Amylibacter highlights their contributions to nitrogen cycling and organic matter degradation, respectively. The exclusive presence of Amylibacter in Arctic samples, with relative abundances ranging from 1.3% (ARC OC 6-7) to over 14% (ARC OC 1), suggests its adaptation in cold environments (Wietz et al., 2021).
The detection of Fibrobacter, the only one genus comprised in the phylum Fibrobacterota traditionally associated with lignocellulose degradation in herbivore guts (Ransom-Jones et al., 2012), suggests a broader ecological role in Arctic waters, contributing to organic matter breakdown.
Rhodococcus, belonging to Actinobacteriota, previously identified in polar regions, underscores the unique metabolic versatility of Arctic microbial communities (Ferrera-Rodríguez et al., 2013). These bacteria are involved in the removal of hydrocarbons from Arctic seawater and coastal soils, and some authors suggest their potential application in bioaugmentation strategies for the remediation of oil-contaminated polar environments (Semenova et al., 2022).
As expected, the low abundance of Cyanobacteria, reflects their limited success in polar waters due to environmental constraints such as low temperatures, reduced light availability, and grazing pressure (Vincent, 2000; Zakhia et al., 2008). These factors inhibit their growth, making them less competitive compared to other bacterial taxa.
4.2 Northwestern Mediterranean
Among all the samples analysed, this area exhibits the highest presence of the Gram-positive phylum Firmicutes, especially in MED NW 8 (14.3%) and MED NW 12 (7.8%).
Although at low abundance, Campylobacterota phylum was detected only in this geographic area in 25% of the analysed samples. This group play an important role in the cycling of nitrogen and sulphur in the cold seeps of the deep ocean (Sun et al., 2023).
The genus Idiomarina, found only in this area and in the Atlantic Ocean, is associated with the breakdown of amino acids, hydrocarbons and the nitrogen cycle; its detection further highlights the role of these bacteria in maintaining ecosystem function (Hou et al., 2004; Rizzo et al., 2022).
Among the Flavobacteriaceae, Salegentibacter (McCammon and Bowman, 2000) has only been found in these sea waters, at sites not far from the coast. The occasional presence of Vibrio (3/16), known for its role in nutrient cycling and potential pathogenicity (Zhang et al., 2018), further adds to the functional diversity.
The high relative abundance of Acinetobacter in certain samples highlights its importance in bioremediation, particularly in degrading hydrocarbons and organic pollutants (Denaro et al., 2021). Its role in pollution mitigation underlines its ecological and practical relevance in particularly anthropized Mediterranean coastal waters.
4.3 Southwestern Mediterranean sea
Cyanobacteria, although less prevalent overall, showed significant variability, with high abundances in certain samples such as MED SW 4P. The unique microbial composition of MED SW 4P, with a lower contribution of Proteobacteria, a peak in Cyanobacteria and the highest detection of Verrucomicrobiota, suggests localised factors such as nutrient hotspots or water stratification. The role of Verrucomicrobiota in polysaccharide degradation further emphasizes the ecological relevance of this outlier (Martinez-Garcia et al., 2012; Sichert et al., 2020).
Bacteroidota, although less dominant than Proteobacteria and Cyanobacteria, were present in all samples from this area and showed a higher relative abundance when compared to samples from the northwestern Mediterranean.
Firmicutes were found less frequently in samples from the south-western Mediterranean than in the north-western area.
Idiomarina, already described above, had the highest measured abundance values in these samples.
Halomonas, a common genus in the Mediterranean and the Atlantic, was found in 75% of the sampled sites in this area. Members of the Halomonas genus are widespread in the biosphere, colonizing common to extreme environments. These bacteria display broad physiological plasticity and metabolic versatility and have developed specific adaptations that allow them to maintain or grow under extreme physical, chemical, and energetic conditions (Kim et al., 2013).
The observed pattern of Proteobacteria dominance and variable Cyanobacteria contribution is consistent with the findings of Quéméneur et al. (2020), who analysed prokaryotic communities in Tunisian coastal waters in the southwestern Mediterranean region. They identified Alphaproteobacteria (particularly SAR11), Gammaproteobacteria (including Alteromonas) and Cyanobacteria as major components of the microbial assemblage. They also found that spatial variability in community composition was associated with environmental gradients, such as salinity and nutrient availability. These observed community patterns may also be influenced by hydrodynamic processes and the inflow of Atlantic waters through the Strait of Gibraltar, which have been shown to affect microbial diversity and stratification in the Alboran Sea and across Mediterranean basins (Sebastián et al., 2021).
4.4 Atlantic ocean
The bacterial communities in the Atlantic Ocean exhibit distinct spatial patterns driven by environmental gradients and proximity to coastal or open-ocean regions.
The presence of Marinimicrobia (SAR406) in samples closer to the American continent underscores its role in the degradation of complex organic matter and nutrient cycling in coastal or deeper waters (Wright et al., 2012). Its local enrichment may indicate distinct biogeochemical processes influenced by terrestrial runoff or proximity to the continental shelf.
In the open ocean samples (ATL OC 1-3), Pseudoalteromonas emerges as the dominant genus with relative abundances above 50%, but its presence decreases significantly as one moves closer to coastal areas. Conversely, Alteromonas displays a contrasting pattern: this genus becomes increasingly abundant in samples taken closer to the European coast (ATL OC 6–8). The dominance of Pseudoalteromonas in open ocean samples is consistent with its ecological role in nutrient cycling and organic matter remineralisation, while the abundance of Alteromonas in samples near the European coast (ATL OC 6-8) reflects its adaptation to the nutrient-rich conditions often found in coastal regions (Offret et al., 2016).
Synechococcus is notably prevalent in samples from the open ocean (except for ATL OC 2), whereas Prochlorococcus exhibits a higher relative abundance in samples collected near the European coast. While Prochlorococcus is typically associated with oligotrophic open-ocean environments (Flombaum et al., 2013), local environmental conditions—such as stratification or the presence of specific ecotypes—may explain its coastal enrichment in this region.
Finally, the genus Cobetia, which is involved in the marine sulphur cycle (Geng et al., 2024), was detected only in this area.
4.5 Indian ocean
Microbial communities in the Indian Ocean exhibit remarkable diversity, with the highest number of phyla detected among all sampled regions. These include two archaeal phyla, Thermoplasmatota and Nanoarchaeota, albeit in low proportions, which may in part be attributable to methodological limitations previously discussed in this study. Within the Thermoplasmatota phylum, the genus Marine Group II was identified, while the genus Woesearchaeales was associated with the Nanoarchaeota phylum. Members of the order Nanoarchaeota are obligate symbionts, meaning that they require attachment to another archaeal host to grow. The detection of genera such as Marine Group II and Woesearchaeales highlights their potential role in methanogenesis and adaptability to diverse marine habitats, including the photic zone (Amils, 2011; St. John and Reysenbach, 2019).
Notably, SAR324 (Marine Group B), previously classified as Deltaproteobacteria, shows significant enrichment in sample IND OC 4. The metabolic versatility of this group, including genes for RuBisCO-mediated carbon fixation, highlights its ecological importance in carbon cycling in diverse marine habitats (Boeuf et al., 2021; Quero et al., 2020). Interestingly, this sample has the lowest representation of Proteobacteria.
Vibrio was found in 87% of the samples analysed, with the highest proportion in IND OC 6, highlighting its ecological flexibility and potential role in biogeochemical cycles (Zhang et al., 2018).
4.6 Alpha diversity
The differences in alpha diversity observed across oceanic regions reflect the influence of environmental gradients and ecological conditions that limit microbial community composition and richness. The low diversity in the Arctic Ocean is consistent with the harsh polar environment—characterized by low temperatures, limited nutrient availability, and strong seasonal light variation—which restricts microbial growth and ecosystem complexity.
In contrast, the high alpha diversity observed in the Mediterranean Sea may be explained by its dynamic hydrology, semi-enclosed structure, and biogeochemical variability (Sebastián et al., 2021). These features create a wide range of ecological niches that support diverse microbial communities.
The Atlantic and Pacific Oceans showed high spatial variability in alpha diversity, likely driven by large-scale oceanographic features such as currents, depth profiles, and upwelling zones. This variability highlights how local environmental drivers can modulate microbial diversity, sometimes overriding broader latitudinal patterns.
Reduced diversity in the Indian Ocean, particularly in semi-enclosed areas like the Persian Gulf and Red Sea, may result from strong physicochemical stressors and anthropogenic impacts such as high salinity, pollution, and eutrophication (Sheppard et al., 2010; Ismail and Al Shehhi, 2022; Qian et al., 2010). These factors likely favour a few stress-tolerant taxa, leading to lower overall evenness.
Together, these observations underline the importance of both regional conditions and large-scale gradients in shaping marine microbial communities and reinforce the value of geographically extensive surveys to understand global patterns of microbial diversity.
4.7 Correlation analysis
The observed correlations highlight temperature as a key structuring factor of bacterial communities, with clear distinctions between taxa associated with warm oligotrophic environments (the SAR86 clade, SAR116 clade, and SAR92) and those favoring colder conditions as SAR11 clade I, and UBA10353). Salinity-driven patterns were more variable, reflecting ecological differences between clades, including Marine Group II, often reported in high-salinity surface layers. The weak correlations with chlorophyll (Leeuwenhoekiella and Mesonia) suggest that primary production alone does not explain microbial community structure across broad oceanographic gradients, reinforcing the importance of including multiple physicochemical and biological parameters when interpreting bacterioplankton biogeography. Indeed, beyond the measured parameters, other factors may also influence the observed microbial distributions, including nutrient availability (e.g., phosphate, nitrate, silicate), the presence of dissolved organic matter, hydrodynamic processes (such as upwelling or stratification), interactions with viruses and grazers, and anthropogenic inputs, especially in semi-enclosed basins like the Mediterranean. These unmeasured or temporally variable drivers likely contribute to shaping bacterial communities in ways not fully captured by the environmental parameters assessed here.
5 Limitation of the study
While this study provides valuable insights into marine microbial diversity, several limitations should be acknowledged. First, the use of the V3–V4 hypervariable region of the 16S rRNA gene, while widely adopted in marine microbial ecology, has limitations, particularly in its reduced ability to detect archaeal taxa. This underrepresentation has been well documented in comparative studies (Parada et al., 2016; Wear et al., 2018), and stems in part from the primer pair used, which was originally optimized for broad bacterial coverage. However, the widespread use of this primer set also offers the advantage of compatibility with a large number of existing datasets, facilitating comparative analyses and meta-analytical integration across studies and regions. Looking ahead, to overcome current limitations and enhance taxonomic resolution—particularly for Archaea and less abundant clades—we plan to incorporate full-length 16S rRNA gene sequencing using PacBio HiFi technology in future campaigns of the Sea Care project.
Another limitation is the single-time point sampling design, which may not capture the full temporal variability of microbial communities. This limitation arises from the necessity to follow naval routes during the Sea Care project campaigns.
Additionally, the focus on only a few environmental parameters (salinity, temperature, chlorophyll) may not fully account for other key factors, such as nutrients, trace elements or pollutants, that could influence microbial diversity. However, in future campaigns, we plan to expand the range of environmental parameters and anthropogenic contaminants monitored, to better understand their role in shaping microbial diversity.
6 Conclusions
This study provides a comprehensive overview of marine microbial diversity across multiple oceanic regions, emphasizing the importance of global-scale monitoring. The innovative use of several naval vessels, each covering different routes across the oceans, allowed for extensive sampling across diverse marine ecosystems. Significant regional differences were observed, with the Mediterranean exhibiting the highest genus-level diversity and the Arctic the lowest. Microbial communities, primarily dominated by Proteobacteria and Cyanobacteria, are central to biogeochemical cycles and serve as sensitive indicators of environmental changes. These findings underline the critical role of microbial diversity in maintaining ecosystem balance, directly linking marine health to planetary health. The study highlights the need for continuous monitoring to detect shifts in microbial communities driven by climate change and human activities. Future research will expand on these results by incorporating additional environmental parameters and emerging contaminants, using advanced sequencing technologies to deepen our understanding of marine ecosystems and their response to global environmental challenges, ultimately contributing to sustainable strategies for preserving planetary health.
Data availability statement
The sequencing data have been deposited in the NCBI Sequence Read Archive (SRA) under the BioProject accession number PRJNA1294399 (https://www.ncbi.nlm.nih.gov/sra/PRJNA1294399).
Author contributions
LV: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. AC: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Visualization, Writing – original draft, Writing – review & editing. APe: Data curation, Formal analysis, Methodology, Writing – original draft, Writing – review & editing. DB: Data curation, Formal analysis, Software, Writing – original draft, Writing – review & editing. AM: Data curation, Formal analysis, Methodology, Software, Writing – original draft, Writing – review & editing. FF: Investigation, Project administration, Resources, Supervision, Validation, Writing – original draft, Writing – review & editing. LL: Funding acquisition, Project administration, Resources, Supervision, Visualization, Writing – original draft, Writing – review & editing. APi: Funding acquisition, Project administration, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. GR: Conceptualization, Data curation, Investigation, Methodology, Validation, Visualization, Writing – original draft, Writing – review & editing. RB: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
Group members of The sea care team:
Laura BARONE - Department of Chemistry, University of Rome “Sapienza”
Sara BOGIALLI – Department of Chemical Sciences, University of Padua, Padua, Italy
Lucia BONADONNA formerly affiliated with the National Centre of Water Safety, ISS, Rome, Italy
Giusy BONANNO Ferraro - National Centre of Water Safety, ISS, Rome, Italy
Giuseppe BORTONE –Environmental Protection Agency of Emilia-Romagna, Bologna, Italy
Eleonora BRANCALEONE - Department of Environment and Health, ISS, Rome, Italy
Roberto CAMMARATA – Central Directorate of Human and Economic Resources, ISS, Rome, Italy
Mario CERRONI – National Centre of Water Safety, ISS, Rome, Italy
Fortunato D’ANCONA – Department of Infectious Disease, ISS, Rome, Italy
Stefania DE ANGELIS - National Centre of Water Safety, ISS, Rome, Italy
Simonetta DELLA LIBERA - National Centre of Water Safety, ISS, Rome, Italy
Roberta DI GIOIA - National Centre of Water Safety, ISS, Rome, Italy
Antonio DONDOLINI POLI – Italian Navy’s Health Inspectorate, Rome, Italy
Antonella FILIPPI – National Centre of Water Safety, ISS, Rome, Italy
Giuseppina GULLIFA – Department of Chemistry, University of Rome “Sapienza”
Marcello IACONELLI - National Centre of Water Safety, ISS, Rome, Italy
Pamela MANCINI - National Centre of Water Safety, ISS, Rome, Italy
Camilla MARCHIAFAVA – National Centre of Water Safety, ISS, Rome, Italy
Daniela MATTEI – National Centre of Water Safety, ISS, Rome, Italy
Giorgia MATTEI – National Centre of Water Safety, ISS, Rome, Italy
Cristina MAZZIOTTI – Oceanographic Facility “Daphne”, Environmental Protection Agency of Emilia-Romagna, Bologna, Italy
Susanna MURTAS – National Centre of Water Safety, ISS, Rome, Italy
Federica NIGRO DI GREGORIO – National Centre of Water Safety, ISS, Rome, Italy
Elena PAPA – Department of Chemistry, University of Rome “Sapienza”, Rome, Italy
Flavia RICCARDO – Department of Infectious Disease, ISS, Rome, Italy
Roberta RISOLUTI – Department of Chemistry University of Rome “Sapienza”, Rome, Italy
Clara SETTE – National Centre of Water Safety, ISS, Rome, Italy.
Carolina VENERI - National Centre of Water Safety, ISS, Rome, Italy
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This study was funded by the “Sea Care Project (2022–2025): Health, Environment and Climate Research in the Vision of Planetary Health,” supported by the Italian Navy in collaboration with the National Institute of Health (ISS).
Acknowledgments
We gratefully acknowledge all members of the sampling team for their valuable contribution to the success of the study. The sampling team included researchers and technicians of the Istituto Superiore di Sanità (ISS), Rome, researchers from the Regional Environmental Protection Agency of Emilia-Romagna, PhD students in Analytical Chemistry from the Sapienza University of Rome, and Professors of the Chemical Faculty of the University of Padua. All participants underwent specific training to ensure a consistent methodological approach throughout activities. Very special thanks go to the Italian Navy and its Chief of Staff, Vice Admiral Enrico Credendino, for their role in the Memorandum of Understanding between the Italian Navy and the ISS. Special recognition is due to Rear Admiral Cristiano Nervi (Head of the Naval Support and Experimental Centre of the Italian Navy and of the Operational Structure of the National Pole of the Underwater Dimension) for promoting and supporting the project’s approval. We also thank Vice Admiral Roberto Dattola (EP&OHS General Office of Navy General Staff) for ensuring maximum support for the project’s activities and success, and Rear Admiral (LH) Massimiliano Lauretti (Head of the Third Division “Plans, Operations and Maritime Strategy) for providing the decisive stimulus for implementing the project during the Amerigo Vespucci’s World Tour Campaign. Our gratitude goes to the commanders of the naval vessels that hosted the Sea Care project teams: Capt. Jacopo Rollo (Commanding Officer of the Destroyer Caio Duilio), Capt. Massimiliano Siragusa, Capt. Luigi Romagnoli, Capt. Giuseppe Lai (Commanding Officers of the Training Ship Amerigo Vespucci), Cdr. Maurizio Demarte (Commanding Officer of the Oceanographic Vessel Alliance, Italian Navy Hydrographic Institute), Cdr. Emanuele Morea, Cdr Alessandro Serrani (Commanding Officers of the Offshore Patrol Vessel Paolo Thaon di Revel). The team’s expertise and professionalism were crucial to the successful completion of all planned activities, especially considering that military vessels are not typically equipped for research purposes. We would also like to express our gratitude to Cdr. Giuseppe Aceto (Head of the Environmental Protection Office, Manager of Operation of the Sea Care Project) for his invaluable support in coordinating and facilitating relations within the Naval General Staff and High Commands, as well as with all the naval vessels in which the scientific teams were embedded. His problem-solving skills were greatly appreciated. We would like to thank all the military personnel, both on board and on shore, for their support, passion, and professionalism in ensuring the success of this project. Finally, we thank Michele Sonnessa from the Bioinformatics service of Biofab for the support provided.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2025.1625011/full#supplementary-material
References
Amils R. (2011). “Nanoarchaeota,” in Encyclopedia of Astrobiology (Springer Berlin Heidelberg, Berlin, Heidelberg), 1106–1106. doi: 10.1007/978-3-642-11274-4_1040
Avcı B., Krüger K., Fuchs B. M., Teeling H., and Amann R. I. (2020). Polysaccharide niche partitioning of distinct Polaribacter clades during North Sea spring algal blooms. ISME J. 14, 1369–1383. doi: 10.1038/s41396-020-0601-y
Baker-Austin C., Trinanes J. A., Taylor N. G. H., Hartnell R., Siitonen A., and Martinez-Urtaza J. (2016). Erratum: Corrigendum: Emerging Vibrio risk at high latitudes in response to ocean warming. Nat. Clim. Change 6, 802–802. doi: 10.1038/nclimate3024
Berdy B., Spoering A. L., Ling L. L., and Epstein S. S. (2017). In situ cultivation of previously uncultivable microorganisms using the ichip. Nat. Protoc. 12, 2232–2242. doi: 10.1038/nprot.2017.074
Boeuf D., Eppley J. M., Mende D. R., Malmstrom R. R., Woyke T., and DeLong E. F. (2021). Metapangenomics reveals depth-dependent shifts in metabolic potential for the ubiquitous marine bacterial SAR324 lineage. Microbiome 9, 172. doi: 10.1186/s40168-021-01119-5
Bowley J., Baker-Austin C., Porter A., Hartnell R., and Lewis C. (2021). Oceanic hitchhikers - assessing pathogen risks from marine microplastic. Trends Microbiol. 29, 107–116. doi: 10.1016/j.tim.2020.06.011
Buesseler K. O., Doney S. C., Karl D. M., Boyd P. W., Caldeira K., Chai F., et al. (2008). Environment: Ocean iron fertilization - Moving forward in a sea of uncertainty. Science 319, 162–164. doi: 10.1126/science.1154305
Caporaso JG., Lauber CL., Walters WA., Berg-Lyons D., Lozupone CA., Turnbaugh PJ., et al. (2012). Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 6, 1621–1624. doi: 10.1038/ismej.2012.8
Cherabier P. and Ferrière R. (2022). Eco-evolutionary responses of the microbial loop to surface ocean warming and consequences for primary production. ISME J. 16, 1130–1139. doi: 10.1038/s41396-021-01166-8
Cho J. Y. (2012). Algicidal activity of marine Alteromonas sp. KNS-16 and isolation of active compounds. Biosci. Biotechnol. Biochem. 76, 1452–1458. doi: 10.1271/bbb.120102
Choi A. R., Lee S. A., and Cho J. C. (2021). Latitudinal gradients in bacterioplankton community structure of the surface ocean across the South Pacific Ocean. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.678943
Deming J. W. and Eric Collins R. (2017). “Sea ice as a habitat for Bacteria, Archaea and viruses,” in Sea Ice. Ed. Thomas D. N. (Wiley, Hoboken), 326–351. doi: 10.1002/9781118778371.ch13
Denaro R., Di Pippo F., Crisafi F., and Rossetti S. (2021). “Biodegradation of hydrocarbons in marine environment,” in Water Pollution and Remediation: Organic Pollutants. Environmental Chemistry for a Sustainable World, vol. 54 . Eds. Inamuddin, Ahamed M. I., and Lichtfouse E. (Springer, Cham). doi: 10.1007/978-3-030-52395-4_7
Dupont C. L., Rusch D. B., Yooseph S., Lombardo M.-J., Alexander Richter R., Valas R., et al. (2012). Genomic insights to SAR86, an abundant and uncultivated marine bacterial lineage. ISME J. 6, 1186–1199. doi: 10.1038/ismej.2011.189
Fernández-Gómez B., Richter M., Schüler M., Pinhassi J., Acinas S. G., González J. M., et al. (2013). Ecology of marine Bacteroidetes: a comparative genomics approach. ISME J. 7, 1026–1037. doi: 10.1038/ismej.2012.169
Ferrera-Rodríguez O., Greer C. W., Juck D., Consaul L. L., Martínez-Romero E., and Whyte L. G. (2013). Hydrocarbon-degrading potential of microbial communities from Arctic plants. J. Appl. Microbiol. 114, 71–83. doi: 10.1111/jam.12020
Flombaum P., Gallegos J. L., Gordillo R. A., Rincón J., Zabala L. L., Jiao N., et al. (2013). Present and future global distributions of the marine Cyanobacteria Prochlorococcus and Synechococcus. PNAS 110, 9824–9829. doi: 10.1073/pnas.1307701110
Geng X.-M., Cai S.-N., Zhu H.-X., Tang Z.-G., Li C.-Y., Fu H.-H., et al. (2024). Genomic analysis of Cobetia sp. D5 reveals its role in marine sulfur cycling. Mar. Genomics 75, 101108. doi: 10.1016/j.margen.2024.101108
Getz E. W., Lanclos V. C., Kojima C. Y., Cheng C., Henson M. W., Schön M. E., et al. (2023). The AEGEAN-169 clade of bacterioplankton is synonymous with SAR11 subclade V (HIMB59) and metabolically distinct. mSystems 8. doi: 10.1128/msystems.00179-23
Giordano D., Chiellini C., Mengoni A., and Bacci G. (2015). Phylogenetic structure and community composition of the culturable bacterial fraction of the benthic mat of Lake Untersee, East Antarctica. Front. Microbiol. 6, 1254. doi: 10.3389/fmicb.2015.01254
Giordano N., Gaudin M., Trottier C., Delage E., Nef C., Bowler C., et al. (2024). Genome-scale community modelling reveals conserved metabolic cross-feedings in epipelagic bacterioplankton communities. Nat. Commun. 15, 2721. doi: 10.1038/s41467-024-46374-w
Giovannoni S. J. (2017). SAR11 bacteria: the most abundant plankton in the oceans. Ann. Rev. Mar. Sci. 9, 231–255. doi: 10.1146/annurev-marine-010814-015934
Gloor G. B., Macklaim J. M., Pawlowsky-Glahn V., and Egozcue J. J. (2017). Microbiome datasets are compositional: And this is not optional. Front. Microbiol. 8. doi: 10.3389/fmicb.2017.02224
Hajji A. L. and Lucas K. N. (2024). Anthropogenic stressors and the marine environment: From sources and impacts to solutions and mitigation. Mar. pollut. Bull. 205, 116557. doi: 10.1016/j.marpolbul.2024.116557
Harbeitner R. C., Wittmers F., Yung C. C. M., Eckmann C. A., Hehenberger E., Blum M., et al. (2024). Gradients of bacteria in the oceanic water column reveal finely-resolved vertical distributions. PloS One 19, e0298139. doi: 10.1371/journal.pone.0298139
Haro-Moreno J. M., Rodriguez-Valera F., Rosselli R., Martinez-Hernandez F., Roda-Garcia J. J., Gomez M. L., et al. (2020). Ecogenomics of the SAR11 clade. Environ. Microbiol. 22, 1748–1763. doi: 10.1111/1462-2920.14896
He X.-Y., Liu N.-H., Liu J.-Q., Peng M., Teng Z.-J., Gu T.-J., et al. (2023). SAR92 clade bacteria are potentially important DMSP degraders and sources of climate-active gases in marine environments. MBio 14. doi: 10.1128/mbio.01467-23
He W., Xue H.-P., Liu C., Zhang A. H., Huang J.-K., and Zhang D.-F. (2023). Biomineralization of struvite induced by indigenous marine bacteria of the genus Alteromonas. Front. Mar. Sci. 10. doi: 10.3389/fmars.2023.1085345
Hennon G. M. M., Morris J. J., Haley S. T., Zinser E. R., Durrant A. R., Entwistle E., et al. (2018). The impact of elevated CO2 on Prochlorococcus and microbial interactions with ‘helper’ bacterium Alteromonas. ISME J. 12, 520–531. doi: 10.1038/ismej.2017.189
Hou S., Saw J. H., Lee K. S., Freitas T. A., Belisle C., Kawarabayasi Y., et al. (2004). Genome sequence of the deep-sea gamma-proteobacterium Idiomarina loihiensis reveals amino acid fermentation as a source of carbon and energy. Proc. Natl. Acad. Sci. U.S.A. 101, 18036–18041. doi: 10.1073/pnas.0407638102
Ismail K. A. and Al Shehhi M. R. (2022). Upwelling and nutrient dynamics in the Arabian Gulf and Sea of Oman. PloS One 17, e0276260. doi: 10.1371/journal.pone.0276260
Kent A. G., Baer S. E., Mouginot C., Huang J. S., Larkin A. A., Lomas M. W., et al. (2019). Parallel phylogeography of Prochlorococcus and Synechococcus. ISME J. 13, 430–441. doi: 10.1038/s41396-018-0287-6
Kim K. K., Lee J.-S., and Stevens D. A. (2013). Microbiology and epidemiology of Halomonas species. Future Microbiol. 8, 1559–1573. doi: 10.2217/fmb.13.108
Klindworth A., Pruesse E., Schweer T., Peplies J., Quast C., Horn M., et al. (2013). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41, e1. doi: 10.1093/nar/gks808
Klümper U., Gionchetta G., Catão E., Bellanger X., Dielacher I., Elena A. X., et al. (2024). Environmental microbiome diversity and stability is a barrier to antimicrobial resistance gene accumulation. Commun. Biol. 7, 706. doi: 10.1038/s42003-024-06338-8
Korlević M., Markovski M., Herndl G. J., and Najdek M. (2022). Temporal variation in the prokaryotic community of a nearshore marine environment. Sci. Rep. 12, 16859. doi: 10.1038/s41598-022-20954-6
Manivasagan P., Kang K.-H., Sivakumar K., Li-Chan E. C. Y., Oh H.-M., and Kim S.-K. (2014). Marine actinobacteria: an important source of bioactive natural products. Environ. Toxicol. Pharmacol. 38, 172–188. doi: 10.1016/j.etap.2014.05.014
Martinez-Garcia M., Brazel D. M., Swan B. K., Arnosti C., Chain P. S. G., Reitenga K. G., et al. (2012). Capturing single cell genomes of active polysaccharide degraders: an unexpected contribution of verrucomicrobia. PloS One 7, e35314. doi: 10.1371/journal.pone.0035314
McCammon S. A. and Bowman J. P. (2000). Taxonomy of Antarctic Flavobacterium species: description of Flavobacterium gillisiae sp. nov., Flavobacterium tegetincola sp. nov., and Flavobacterium xanthum sp. nov., nom. rev. and reclassification of [Flavobacterium] salegens as Salegentibacter salegens gen. nov., comb. nov. Int. J. Syst. Evol. Microbiol. 50, 1055–1063. doi: 10.1099/00207713-50-3-1055
Mella-Flores D., Six C., Ratin M., Partensky F., Boutte C., Le Corguillé G., et al. (2012). Prochlorococcus and synechococcus have evolved different adaptive mechanisms to cope with light and UV stress. Front. Microbiol. 3. doi: 10.3389/fmicb.2012.00285
Morales S. E. and Holben W. E. (2011). Linking bacterial identities and ecosystem processes: can ‘omic’ analyses be more than the sum of their parts? FEMS Microbiol. Ecol. 75, 2–16. doi: 10.1111/j.1574-6941.2010.00938.x
Moran M. A. (2015). The global ocean microbiome. Science 350, 1262–1263. doi: 10.1126/science.aac8455
Morris R. M., Rappé M. S., Connon S. A., Vergin K. L., Siebold W. A., Carlson C. A., et al. (2002). SAR11 clade dominates ocean surface bacterioplankton communities. Nature 420, 806–810. doi: 10.1038/nature01240
Nimnoi P. and Pongsilp N. (2020). Marine bacterial communities in the upper gulf of Thailand assessed by Illumina next-generation sequencing platform. BMC Microbiol. 20, 19. doi: 10.1186/s12866-020-1701-6
Offret C., Desriac F., Le Chevalier P., Mounier J., Jégou C., and Fleury Y. (2016). Spotlight on antimicrobial metabolites from the marine bacteria pseudoalteromonas: chemodiversity and ecological significance. Mar. Drugs 14, 129. doi: 10.3390/md14070129
Parada A. E., Needham D. M., and Fuhrman J. A. (2016). Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 18, 1403–1414. doi: 10.1111/1462-2920.13023
Qian W., Liao B. Y., Chang A. Y., and Zhang J. (2010). Maintenance of duplicate genes and their functional redundancy by reduced expression. Trends in Genetics 26, 425–430.
Quéméneur M., Bel Hassen M., Armougom F., Khammeri Y., Lajnef R., and Bellaaj-Zouari A. (2020). Prokaryotic diversity and distribution along physical and nutrient gradients in the Tunisian coastal waters (South Mediterranean Sea). Front. Microbiol. 11. doi: 10.3389/fmicb.2020.593540
Quero G. M., Celussi M., Relitti F., Kovačević V., Del Negro P., and Luna G. M. (2020). Inorganic and organic carbon uptake processes and their connection to microbial diversity in meso- and bathypelagic arctic waters (Eastern Fram Strait). Microb. Ecol. 79, 823–839. doi: 10.1007/s00248-019-01451-2
Ransom-Jones E., Jones D. L., McCarthy A. J., and McDonald J. E. (2012). The fibrobacteres: an important phylum of cellulose-degrading bacteria. Microb. Ecol. 63, 267–281. doi: 10.1007/s00248-011-9998-1
Rizzo C., Papale M., and Lo Giudice A. (2022). Idiomarina sp. Isolates from cold and temperate environments as biosurfactant producers. J. Mar. Sci. Eng. 10, 1135. doi: 10.3390/jmse10081135
Roda-Garcia J. J., Haro-Moreno J. M., Huschet L. A., Rodriguez-Valera F., and López-Pérez M. (2021). Phylogenomics of SAR116 clade reveals two subclades with different evolutionary trajectories and an important role in the ocean sulfur cycle. mSystems 6. doi: 10.1128/msystems.00944-21
Roda-Garcia J. J., Haro-Moreno J. M., and López-Pérez M. (2023). Evolutionary pathways for deep-sea adaptation in marine planktonic Actinobacteriota. Front. Microbiol. 14. doi: 10.3389/fmicb.2023.1159270
Rodrigues C. J. C. and de Carvalho C. C. C. R. (2022). Cultivating marine bacteria under laboratory conditions: Overcoming the “unculturable” dogma. Front. Bioeng. Biotechnol. 10. doi: 10.3389/fbioe.2022.964589
Sarmiento R. T., Mercado J., and Mercado J. A. (2019). Land use changes and their influence in the conservation of plant diversity within a small Binaba watershed. J. Biodivers. Environ. Sci. 14, 139–150. Available online at: http://www.innspub.net (Accessed May 5, 2025).
Scanlan D. J., Ostrowski M., Mazard S., Dufresne A., Garczarek L., Hess W. R., et al. (2009). Ecological genomics of marine picocyanobacteria. Microbiol. Mol. Biol. Rev. 73, 249–299. doi: 10.1128/MMBR.00035-08
Sebastián M., Ortega-Retuerta E., Gómez-Consarnau L., Zamanillo M., Álvarez M., Arístegui J., et al. (2021). Environmental gradients and physical barriers drive the basin-wide spatial structuring of Mediterranean Sea and adjacent eastern Atlantic Ocean prokaryotic communities. Limnol. Oceanogr. 66, 4077–4095. doi: 10.1002/lno.11944
Semenova E. M., Babich T. L., Sokolova D. S., Ershov A. P., Raievska Y. I., Bidzhieva S. K., et al. (2022). Microbial communities of seawater and coastal soil of Russian arctic region and their potential for bioremediation from hydrocarbon pollutants. Microorganisms 10. doi: 10.3390/microorganisms10081490
Semenza J. C., Trinanes J., Lohr W., Sudre B., Löfdahl M., Martinez-Urtaza J., et al. (2017). Environmental suitability of vibrio infections in a warming climate: an early warning system. Environ. Health Perspect. 125, 107004. doi: 10.1289/EHP2198
Sheikh M., Singh Rathore D., Gohel S. D., and Singh S. P. (2018). Marine actinobacteria associated with the invertebrate hosts: a rich source of bioactive compounds: a review. J. Cell. Tissue Res. 18, 6361–6374. doi: 10.1128/mSystems.00171-18
Sheppard C., Al-Husiani M., Al-Jamali F., Al-Yamani F., Baldwin R., Bishop J., et al. (2010). The Gulf: A young sea in decline. Mar. pollut. Bull. 60, 13–38. doi: 10.1016/j.marpolbul.2009.10.017
Sichert A., Corzett C. H., Schechter M. S., Unfried F., Markert S., Becher D., et al. (2020). Verrucomicrobia use hundreds of enzymes to digest the algal polysaccharide fucoidan. Nat. Microbiol. 5, 1026–1039. doi: 10.1038/s41564-020-0720-2
Stevenson E. M., Buckling A., Cole M., Lindeque P. K., and Murray A. K. (2024). Selection for antimicrobial resistance in the plastisphere. Sci. Total Environ. 908, 168234. doi: 10.1016/j.scitotenv.2023.168234
St. John E. and Reysenbach A.-L. (2019). Hydrothermal Vent Microbiology. In: Schmidt T. M. and Schaechter M. (eds.) Encyclopedia of Microbiology (Fourth Edition). (Oxford: Elsevier), 482–495. doi: 10.1016/B978-0-12-809633-8.20768-7
Stulberg E., Fravel D., Proctor L. M., Murray D. M., LoTempio J., Chrisey L., et al. (2016). An assessment of US microbiome research. Nat. Microbiol. 1, 15015. doi: 10.1038/nmicrobiol.2015.15
Sun Q.-L., Xu K., Cao L., Du Z., Wang M., and Sun L. (2023). Nitrogen and sulfur cycling driven by Campylobacterota in the sediment–water interface of deep-sea cold seep: A case in the South China Sea. mBio 14 (4), e0011723. doi: 10.1128/mbio.00117-23
Sunagawa S., Coelho L. P., Chaffron S., Kultima J. R., Labadie K., Salazar G., et al. (2015). Structure and function of the global ocean microbiome. Science 348. doi: 10.1126/science.1261359
Tucker S. J., Freel K. C., Monaghan E. A., Sullivan C. E. S., Ramfelt O., Rii Y. M., et al. (2021). Spatial and temporal dynamics of SAR11 marine bacteria across a nearshore to offshore transect in the tropical Pacific Ocean. PeerJ 9, e12274. doi: 10.7717/peerj.12274
Vezzulli L. (2023). Global expansion of Vibrio spp. in hot water. Environ. Microbiol. Rep. 15, 77–79. doi: 10.1111/1758-2229.13135
Vincent W. F. (2000). “Cyanobacterial dominance in the polar regions,” in The Ecology of Cyanobacteria (Kluwer Academic Publishers, Dordrecht), 321–340. doi: 10.1007/0-306-46855-7_12
Wear E. K., Wilbanks E. G., Nelson C. E., and Carlson C. A. (2018). Primer selection impacts specific population abundances but not community dynamics in a monthly time-series 16S rRNA gene amplicon analysis of coastal marine bacterioplankton. Environ. Microbiol. 20, 2709–2726. doi: 10.1111/1462-2920.14091
Wietz M., Bienhold C., Metfies K., Torres-Valdés S., von Appen W.-J., Salter I., et al. (2021). The polar night shift: seasonal dynamics and drivers of Arctic Ocean microbiomes revealed by autonomous sampling. ISME Commun. 1. doi: 10.1038/s43705-021-00074-4
Wright J. J., Konwar K. M., and Hallam S. J. (2012). Microbial ecology of expanding oxygen minimum zones. Nat. Rev. Microbiol. 10, 381–394. doi: 10.1038/nrmicro2778
Yang Y., Liu G., Song W., Ye C., Lin H., Li Z., et al. (2019). Plastics in the marine environment are reservoirs for antibiotic and metal resistance genes. Environ. Int. 123, 79–86. doi: 10.1016/j.envint.2018.11.061
Zakhia F., Jungblut A.-D., Taton A., Vincent W. F., and Wilmotte A. (2008). “Cyanobacteria in cold ecosystems,” in Psychrophiles: From Biodiversity to Biotechnology (Springer, Berlin Heidelberg), 121–135. doi: 10.1007/978-3-540-74335-4_8
Zeng Y.-X., Zhang Y.-H., Li H.-R., and Luo W. (2020). Complete genome of Sulfitobacter sp. BSw21498 isolated from seawater of Arctic Kongsfjorden. Mar. Genomics 53, 100769. doi: 10.1016/j.margen.2020.100769
Keywords: bacterial diversity, environmental factors, marine metagenomics, microbial ecology, seawater
Citation: Vitanza L, Coccia AM, Peluso A, Brandtner D, Muratore A, Ferrara F, Lucentini L, Piccioli A, La Rosa G, Briancesco R and The sea care team (2025) Global mapping and environmental drivers of epipelagic bacterial communities in the open oceans. Front. Mar. Sci. 12:1625011. doi: 10.3389/fmars.2025.1625011
Received: 08 May 2025; Accepted: 04 August 2025;
Published: 08 September 2025.
Edited by:
Alexander Eiler, University of Oslo, NorwayReviewed by:
Mauro Celussi, National Institute of Oceanography and Applied Geophysics, ItalyAmel Bellaaj Zouari, National Institute of Marine Sciences and Technologies, Tunisia
Copyright © 2025 Vitanza, Coccia, Peluso, Brandtner, Muratore, Ferrara, Lucentini, Piccioli, La Rosa, Briancesco and The sea care team. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rossella Briancesco, cm9zc2VsbGEuYnJpYW5jZXNjb0Bpc3MuaXQ=