Abstract
The Northern Gulf of Alaska (NGA) is a highly productive and diverse marine ecosystem. Differences in nutrient supply and physical circulation between nearshore and offshore waters in the NGA result in a mosaic of water masses with distinct biogeochemical signatures. We hypothesized that microbial communities in these regions not only differ in composition but also in the ecological interaction networks they support. We used amplicon sequencing of the 16S (V4) and 18S (V9) rRNA genes to characterize the microbial community differences between nearshore, continental shelf, and offshore regions in the NGA in summers 2018-2021. We observed significantly different community assemblages by region (MRPP, p = 0.001), with higher relative abundances and cell counts of heterotrophic bacteria and Synechococcus nearshore, elevated Alphaproteobacteria and SAR11 clades offshore, and greater dinoflagellates and Spirotrich ciliates on the shelf. Co-occurrence networks of operational taxonomic units (OTUs) of prokaryotes and eukaryotes were constructed for each region using statistically significant correlations (Spearman rank >0.8, Bonferroni corrected p< 0.05). Overall, the offshore network had higher centralization (0.331) and density (0.112), indicating higher connectivity and therefore more potential interactions compared to the shelf (0.191, 0.069) and nearshore (0.165, 0.041) networks. The nearshore network was characterized by higher proportions of potentially parasitic taxa such as Cryothecomonas aestivalis, Syndiniales Dino Group I, and MAST-1C and parasitoid bacteria Bdellovibrio and like organisms, suggesting that nearshore conditions may increase parasitoid/predator success through increased contact rates. Significant correlations between cryptophyte Plagioselmis prolonga and ciliate Oligotrichida were identified in all three regions, supporting previous findings that kleptoplasty is likely an important strategy across the NGA. Eukaryotic taxa that had the highest degree centrality across all three regions; P. prolonga and Phaeocystis are known to be mixotrophs, suggesting a role for bacterivory in forging a high number of interactions between protists and bacteria. This study represents the first region-specific co-occurrence network analysis across nearshore to offshore gradients in the NGA. By identifying highly connected taxa and potential trophic strategies, our findings provide new insight into how microbial interactions shape community structure and resilience in a dynamic subarctic ecosystem.
1 Introduction
Oceans are experiencing rapid changes in temperature, stratification, and nutrient input because of climate change (Behrenfeld et al., 2006; Boyd et al., 2008; Hutchins and Fu, 2017). High-latitude regions, particularly the Arctic, are warming at a rate of approximately two to three times faster than the global average (Serreze et al., 2009; Screen and Simmonds, 2010; Cohen et al., 2014). Marine microbes make up more than 90% of ocean biomass and play critical roles in marine food webs, primary and secondary production, and the flow of energy and nutrients, making them significant drivers of biogeochemical cycles and environmental processes (Abirami et al., 2021). Variation in sea surface temperature has been shown to affect microbial biodiversity (Hutchins and Fu, 2017; Abirami et al., 2021; Cohen, 2022) and has the potential to alter important microbial interactions and distributions (Cram et al., 2015; Fuhrman et al., 2015; Lima-Mendez et al., 2015; Worden et al., 2015).
The ocean’s large microbial populations and diversity likely support a complex network of interactions, from mutualism to antagonism, that shape marine ecosystems (Amin et al., 2012; Faust and Raes, 2012; Braga et al., 2016; Arandia-Gorostidi et al., 2022). Parasitoid bacteria like Bdellovibrio and like organisms (BALOs) can influence community structure by preying on small Gram-negative bacteria and helping regulate their populations (Sockett, 2009; Negus et al., 2017; Cohen et al., 2021; Mookherjee and Jurkevitch, 2022). Kleptoplasty, where microbes steal plastids from prey to temporarily acquire photosynthesis, is another important interaction, seen in marine ecosystems involving Teleaulax amphioxeia, Mesodinium rubrum, Dinophysis, and ciliates like Strombidium (Stoecker and Silver, 1990; McManus et al., 2012; Rial et al., 2012; Rusterholz et al., 2017; Cruz and Cartaxana, 2022). At the same time, mutualistic interactions are essential for maintaining microbial community structure and ecosystem services. For example, Prochlorococcus relies on helper bacteria like Alteromonas to remove reactive oxygen species (Morris et al., 2008, 2011; Hennon et al., 2018). These relationships are critical for ecosystem function, and shifts in ocean chemistry due to climate change could potentially destabilize these interactions (Hennon et al., 2018).
Although microbial interactions are important in shaping marine ecosystems, they are difficult to identify (Berry and Widder, 2014; Cagua et al., 2019; MatChado et al., 2021; Wang et al., 2021). Many marine microbes remain uncultured, and most of what is known about the diversity and distribution of these microbial communities comes from molecular techniques such as amplicon sequencing of the 16S and 18S rRNA genes (Hofer, 2018; Bodor et al., 2020). Network-based analytical approaches are useful for identifying potential microbial interactions based on co-occurrence or co-abundances of microbial taxa (Fuhrman, 2009; Needham et al., 2013; Berry and Widder, 2014; Cui et al., 2019; Li et al., 2021; MatChado et al., 2021; Costas-Selas et al., 2024). Using a co-occurrence network-based approach, significant correlations between microbial taxa are interpreted as potential interactions between those pairs (González et al., 2010; Berry and Widder, 2014; Cagua et al., 2019). Additionally, node-specific and global network measurements can provide statistical measurements about differences in putative interaction networks that help to uncover differences between co-occurrence networks’ structure and function (Faust and Raes, 2012; Berry and Widder, 2014; Cagua et al., 2019). These methods can also identify microbial taxa with high numbers of potential interactions within the system, termed keystone microbes, whose removal could disproportionately affect network stability (González et al., 2010; Cagua et al., 2019; MatChado et al., 2021).
In this study, we examined how microbial communities, co-occurrence networks and keystone microbes differ by region in the Northern Gulf of Alaska (NGA) Longterm Ecological Research (LTER) site. The NGA LTER represents an important subarctic region in the North Pacific Ocean that supports culturally and economically important fisheries (Holen, 2014; Szymkowiak, 2020). However, there are few published datasets examining microbial community diversity and dynamics, particularly for prokaryotes. Variations in water masses and high seasonality in primary production result in a mosaic of nutrient regions across the NGA. Nearshore communities are influenced by the Alaska Coastal Current (ACC), which is driven by along-shore winds and characterized by a low-salinity core from freshwater inputs (Strom et al., 2006; Stabeno et al., 2016). These nearshore waters are high in micronutrients such as iron, but are often limited by nitrate (Strom et al., 2006; Aguilar-Islas et al., 2016; Stabeno et al., 2016). The Alaska Current – Alaskan Stream is a cyclonic boundary current along the shelf break/slope that together with mesoscale eddies often found moving westward in this region (Ladd et al., 2007) promote mixing of offshore iron-deplete, but nitrate-replete waters with shelf waters, which in summer tend to be replete in iron relative to nitrate, creating patches that can support higher primary production (Aguilar-Islas et al., 2016; Stabeno et al., 2016; Coyle et al., 2019). Natural gradients of iron and macronutrients in the NGA create contrasting nutrient-limited conditions for microbial communities and their interactions. Based on these nutrient differences, we hypothesized that nearshore regions, which tend to be rich in micronutrients, would have lower network connectivity and a greater presence of antagonistic interactions. In environments where key nutrients are more readily available, microbes may not need to rely as heavily on cooperative interactions to meet their metabolic needs (Morris et al., 2012; D’Souza et al., 2018). In contrast, offshore regions are often macronutrient-replete but iron-limited, and we expected microbial communities in these waters to form more tightly connected networks. Under limiting conditions, microbes may be more dependent on mutualistic or metabolically complementary interactions to access essential resources such as iron, which is often exchanged through cross-feeding or shared uptake strategies (Amin et al., 2012; Zelezniak et al., 2015; Bertrand et al., 2015).
2 Methods
2.1 Sample site
Water samples for DNA and flow cytometry analysis were collected within the NGA LTER site at various depths, including surface, ten meters, and at the depth of the deep chlorophyll maximum (DCM) (Cohen, 2022). Three transects: the Seward line, Middleton Island line, and Kodiak Island line were sampled to characterize the spatial variation of water properties throughout the NGA (Figure 1). Sampling of the Seward and Middleton Island lines was conducted in summers 2018–2021 while the Kodiak Island line was sampled summers 2018–2019 and 2021 (Supplementary Table 1). In this study, summer refers to sampling conducted between June 25 and July 19 of each year, consistent with the seasonal cruise schedule of the NGA LTER (Strom et al., 2006; Aguilar-Islas et al., 2016). The 2018 cruise was conducted onboard R/V Woldstad and the other cruises were onboard R/V Sikuliaq. Regions were a priori selected based on distance from shore and their proximity to currents and bathymetric features (Figure 1). Nearshore stations were within 30 nautical miles or 55.6 km from shore in a region influenced by the ACC. This region is characterized by low-salinity, iron- and copper-rich waters driven by freshwater discharge and terrestrial inputs from systems like the Copper River plume (Stabeno et al., 2016; Ortega et al., 2025; Reister et al., 2024). Shelf stations greater than 30 nautical miles from shore and over the continental shelf as defined here as areas shallower than 300 meters depth. Offshore stations were defined as those over the continental slope (> 300 m bottom depth) and are influenced by the Alaska Current - Alaskan Stream and mesoscale eddies, which facilitate offshore-shelf exchange and contribute to nutrient patchiness (Okkonen et al., 2001; Strom et al., 2010; Coyle et al., 2019). These spatial divisions are consistent with hydrographic and nutrient patterns described in previous studies of the NGA, including variations in salinity structure, current systems, and nutrient distributions (Stabeno et al., 2016; Aguilar-Islas et al., 2016; Strom et al., 2006; Reister et al., 2024; Ortega et al., 2025). The map of sample locations was produced using Ocean Data View (Schlitzer, 2022).
Figure 1
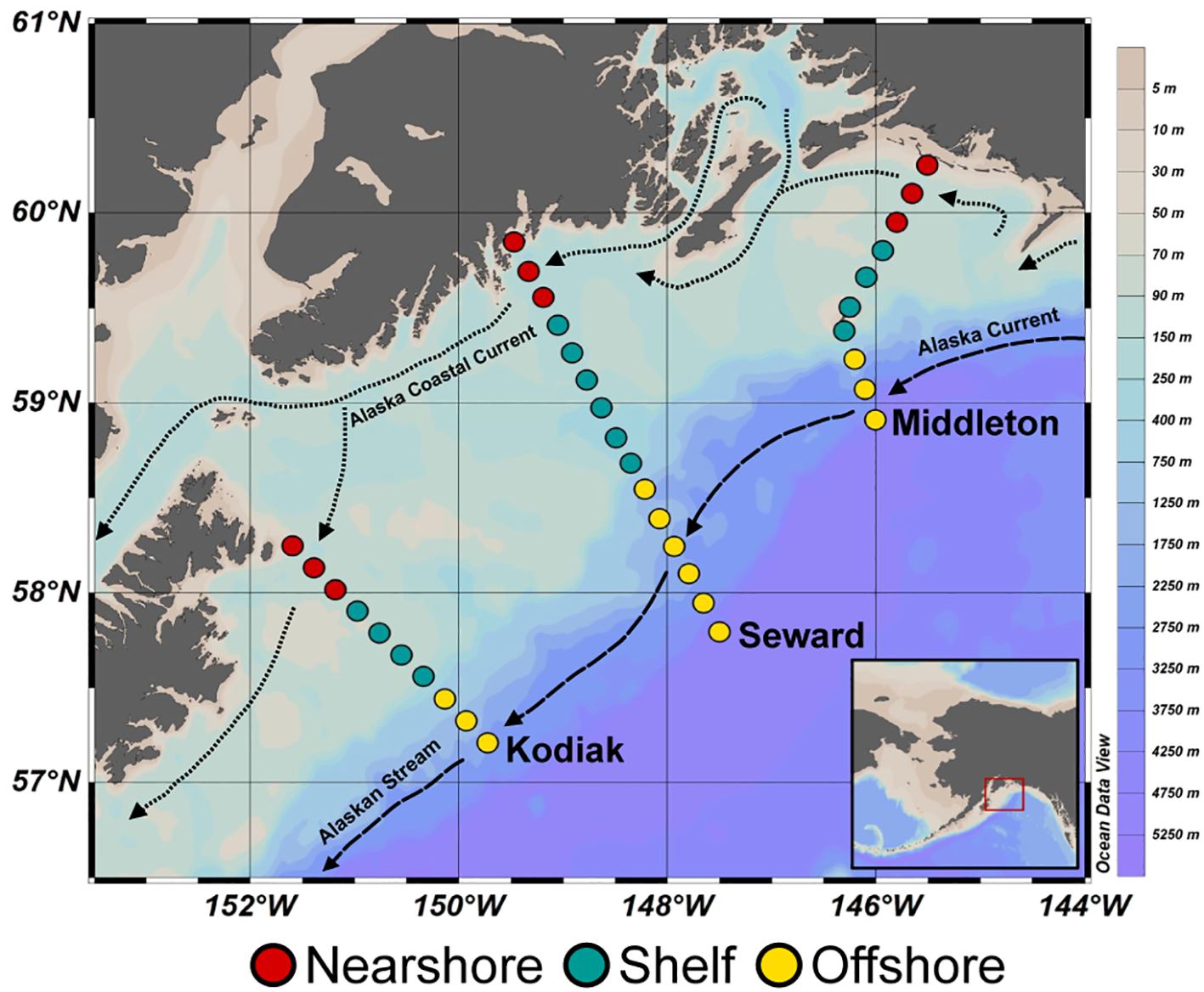
Stations for the Northern Gulf of Alaska (NGA). Sample categories based on location are differentiated by color; nearshore (red), shelf (teal), and offshore (yellow). Approximate locations of currents are indicated with dashed lines with directions indicated by arrows. Background color represents ocean bathymetry.
2.2 Flow cytometry
Whole seawater was collected for flow cytometry in cryovials and preserved with a final concentration of 0.5% glutaraldehyde. Samples were fixed in the dark for ten minutes, flash-frozen in liquid nitrogen, and stored at -80°C until analysis. EasyCheck Beads were used to calibrate the flow cytometer, following a 20x dilution and running three replicates (Cytek, Fremont, USA). For DNA analysis, samples were stained with SYBR Green (100x dilution), incubated in the dark for ten minutes, and analyzed alongside unstained controls using a Guava 5ST easyCyte flow cytometer equipped with a 488-nm laser and detectors for side scatter, forward scatter, red, green, and yellow fluorescence (Luminex, Austin, USA). Following known optical properties, heterotrophic bacteria, and Synechococcus were manually gated using Guava InCyte (Luminex, Austin, USA).
2.3 Gene sequencing and bioinformatic analysis
Approximately 1–5 liters of seawater was filtered through a 0.2 µm pore Sterivex filter (Fisher Scientific, Hampton, USA) and frozen at -80°C until extraction. DNA was extracted from Sterivex filters using DNeasy PowerWater Sterivex kit (Qiagen, Valencia, CA, USA, Cat. No. 14600-50-NF) following the manufacturer’s protocol. Microbial community composition was analyzed by amplifying the 16S rRNA and 18S rRNA gene sequences using TaggiMatrix primer multiplexing (Glenn et al., 2019). Prokaryotic communities were amplified using the 16S (V4 region) 515F and 806R primers (Parada et al., 2016). Eukaryotic communities were amplified using the 18S (V9 region) 1389F and 1510R primers (Amaral-Zettler et al., 2009). PCR was performed using the HiFi HotStart PCR Kit (Kapa Biosystems, Wilmington, MA, USA) on a Veriti 96-Well Thermal Cycler (Applied Biosystems, Waltham, MA, USA). The thermocycling profile consisted of an initial denaturation at 98°C for 1 minute, followed by 25 cycles of 98°C for 20 seconds, 55°C for 20 seconds, and 72°C for 30 seconds, with a final extension at 72°C for 5 minutes. Amplicons were cleaned using a HighPrep PCR Clean-up System (MagBio, Gaithersburg, MD, USA), and a second PCR was performed to add Illumina adapters and dual indices. Samples were sequenced using the Illumina MiSeq Platform (Illumina, San Diego, USA). A total of 232 prokaryotic and 198 eukaryotic samples were sequenced (PRJNA887083). The difference in sample numbers between prokaryotic and eukaryotic datasets reflects sample loss due to sequencing failure or low read counts that did not meet quality thresholds after rarefaction.
Raw sequences from Illumina were demultiplexed using Mr. Demuxy and processed using QIIME2 (version 2020.8) (Cock et al., 2009; Bolyen et al., 2019). Sequences were quality filtered (Phred score of 20) and denoised using DADA2 (Callahan et al., 2016). Operational taxonomic units (OTUs) were de novo clustered to 97% nucleotide identity and assigned taxonomy with the feature-classifier (Bokulich et al., 2018) using the Silva version 138 and PR2 reference databases (Guillou et al., 2012; Quast et al., 2013). OTUs were used instead of ASVs because preliminary analyses showed that minor nucleotide differences (particularly among eukaryotic sequences) led to over-splitting of biologically similar taxa, which were then excluded from downstream analysis. This seemed to disproportionately reduce eukaryotic representation in the networks and biased results toward prokaryotes. Clustering at 97% identity allowed for more balanced representation of microbial domains while maintaining ecological relevance for network construction. Non-target 16S and 18S sequences, such as those identified as metazoan, chloroplast, and mitochondrial sequences were removed before further analysis. Samples were rarefied to a depth of 5,000 reads for the 16S rRNA gene and 2,500 reads for the 18S rRNA gene. These rarefaction depths are consistent with other marine studies using amplicon community analysis for eukaryotes, which can yield fewer sequences per sample due to differences in primer specificity and template abundance (Logares et al., 2014; Massana et al., 2015; Needham and Fuhrman, 2016). Rarefaction curves confirmed that these thresholds captured the majority of observed diversity (Supplementary Figure 3).
For spatial comparisons of microbial community composition and co-occurrence network construction, samples were grouped by region (nearshore, shelf, and offshore) (Supplementary Table 1). Although individual samples were collected and sequenced at distinct depths, including surface, ten meters, and a deep chlorophyll maximum (DCM), samples from each depth were grouped by region during network construction to evaluate broader spatial patterns. This approach maintained depth-level resolution during sequencing while enabling regional-scale ecological analysis.
2.4 Co-occurrence network analysis
Microbial taxon-taxon co-occurrence networks were constructed using relative read abundance data derived from rarefied OTU tables. Relative abundance was used to normalize differences in sequencing depth and emphasize proportional relationships among taxa across samples. While relative abundance helped normalize sequencing depth, we recognize that rRNA gene copy number varies widely, especially among eukaryotes. We did not apply copy number correction because our goal was to compare co-occurrence patterns not absolute abundance and robust correction methods for mixed environmental datasets are still limited. This approach is consistent with other cross-domain network studies (Lima-Mendez et al., 2015; Faust et al., 2015; Tipton et al., 2018). Networks were constructed for samples with significant Spearman rank correlations (ρ > 0.8), with p-values adjusted for multiple testing using the Bonferroni correction method (adjusted p< 0.05) in R using the Hmisc package (Harrell and Dupont, 2019). Networks were visualized in Cytoscape (Shannon et al., 2003) and subnetworks and a merged network were constructed in Cytoscape using MetScape, a Cytoscape plugin (Basu et al., 2017). Nodes represent all OTUs with significant correlations, which are represented by edges. Network and node-based measurements were calculated using NetworkAnalyzer, a built-in Cytoscape plugin (Cytoscape version 3.8.2; Assenov et al., 2008). Degree centrality (connectivity) was calculated as the number of edges linked to each node (Diestel, 2005). Potential keystone microbes were identified as the top ten OTUs with the highest degree centrality values in each network. Degree centrality reflects the number of significant co-occurrence relationships a taxon has and is commonly used as a proxy for ecological influence in microbial networks (Berry and Widder, 2014; Banerjee et al., 2018; Herren and McMahon, 2017). While additional centrality metrics such as closeness were calculated, degree centrality was prioritized due to its interpretability, simplicity, and established use in identifying highly connected taxa that likely structure microbial communities. Closeness centrality of each node was calculated as the reciprocal of the average shortest path length (Newman, 2003), where the average shortest path length is the mean length of the shortest path between the node and all other nodes. Co-occurrence fragmentation (f) was calculated for each network as:
where f is the fragmentation index, CL is the number of disconnected subgraphs (components), and N is the total number of nodes in the network. This metric describes network stability, with values closer to 0 indicating a more cohesive network, and values closer to 1 indicating higher fragmentation and reduced connectivity (Berry and Widder, 2014; Yang et al., 2024; Pilosof et al., 2014). A full set of network statistic definitions can be found in Supplementary Table 2.
2.5 Statistical analysis
Non-metric multidimensional scaling ordination (NMDS) and significance of the multiple regression using environmental fit (envfit) with Bonferroni correction were conducted to test significant differences in microbial communities using the R package vegan (version 2.6-4, R version 4.2.1) (Dixon, 2003). Multiple response permutation procedure (MRPP) was conducted to test significant differences between microbial community composition using vegan (version 2.4-2) (Dixon, 2003). Differences in network measurements between sampling regions were compared using the non-parametric Wilcoxon rank sum test with the ggpubr package (Kassambara, 2020).
3 Results
3.1 Microbial community structure
Summer spatial variability in microbial communities and their interactions across the Northern Gulf of Alaska was investigated using seawater samples collected from three regions: nearshore, shelf, and offshore (Figure 1). DNA was extracted and amplified for the 16S and 18S rRNA genes for taxonomic identification of microbial communities. After rarefying samples to 5,000 reads for 16S and 2,500 reads for 18S, 175 prokaryotic and 145 eukaryotic samples remained for downstream analysis. These depths were selected based on rarefaction curves (Supplementary Figure 3) and are consistent with marine microbial diversity studies (Logares et al., 2014; Massana et al., 2015; Needham and Fuhrman, 2016). Summary statistics on sequencing output, OTU counts, and taxonomic composition are provided in Supplementary Table 3. Barplots summarizing the relative abundance of major prokaryotic and eukaryotic taxa across regions are presented in Supplementary Figure S1. Both prokaryotic (16S) and eukaryotic (18S) communities differed significantly by region (Figure 2, Bonferroni adjusted p< 0.05). The robustness of regional differences in community composition were further tested with multiple response permutation procedure (MRPP) and communities were found to differ significantly by region in both prokaryotes (p = 0.001) and eukaryotes (p = 0.001). Additionally, shifts in prokaryotic and eukaryotic communities were significantly correlated with environmental variables: temperature, salinity, nitrate, and silicic acid concentrations (Figure 2, Bonferroni adjusted, p< 0.05). MRPP analysis also showed significant differences by depth and year in both datasets (p ≤ 0.008). The strongest depth-related shifts occurred between surface and DCM samples for both prokaryotes and eukaryotes. Interannual differences were also detected, with particularly distinct clustering among eukaryotic communities across years. These patterns align with previous reports of temporal variability in the NGA (Cohen, 2022). Taxonomic shifts across depth and year were primarily driven by changes in the relative abundance of Alphaproteobacteria, Gammaproteobacteria, Syndiniales, and diatoms, while groups like Bdellovibrionota and Group I Syndiniales remained relatively stable across environmental gradients.
Figure 2
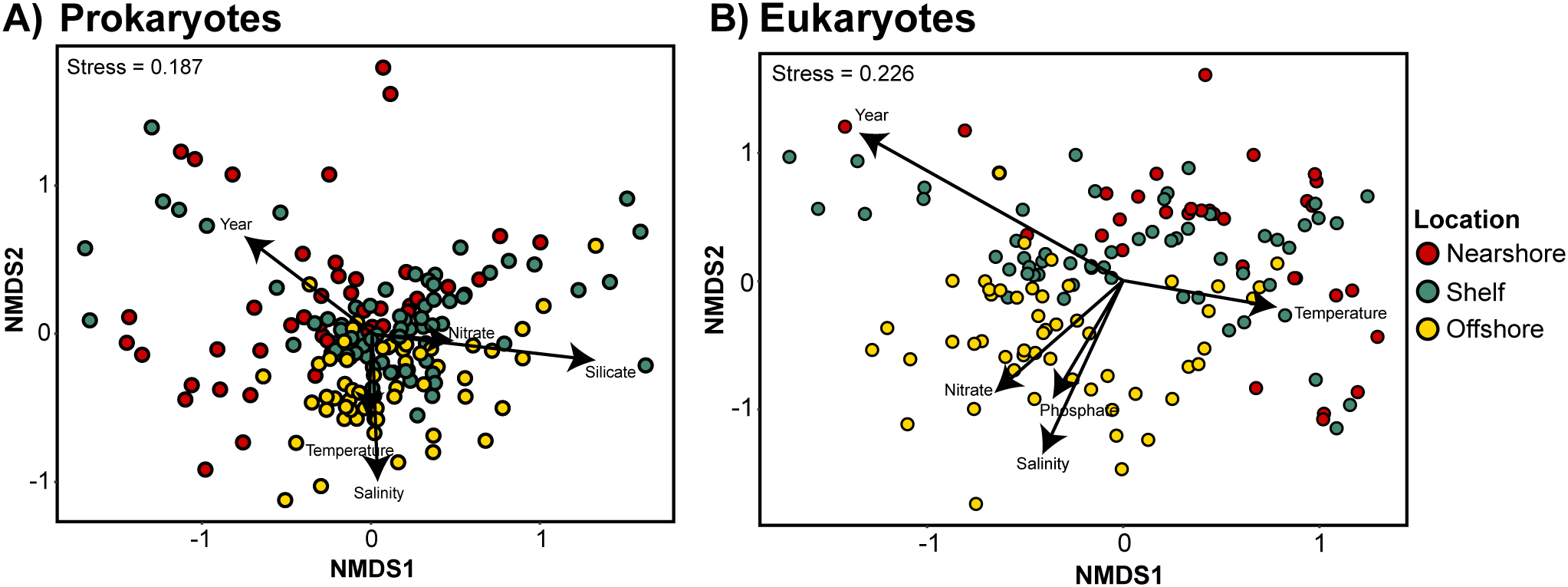
Nonmetric multidimensional scaling (NMDS) ordination of variation in (A) prokaryotic and (B) eukaryotic water community structure. Color represents sample locations: Nearshore (red), Shelf (teal), and Offshore (yellow). Vectors represent significant correlations (p < 0.05) with environmental variables.
Shifts in the prokaryotic community were characterized by shifts in Alphaproteobacteria and Gammaproteobacteria. Alphaproteobacteria were the most abundant offshore (19.58%) and least abundant nearshore (14.45%) (Supplementary Figure S1A). Pelagibacter ubique (SAR11), a prominent Alphaproteobacteria, varied by region, with clades I and II highest nearshore, clade IV highest offshore, and lowest levels on the shelf (Supplementary Figure S1C). Gammaproteobacteria were most abundant nearshore (21.91%) and less abundant offshore and shelf. SAR86 within Gammaproteobacteria had the highest abundance offshore. Bdellovibrionota, also known as Bdellovibrio and like organisms (BALOs), showed relatively consistent abundance across nearshore and offshore regions (2.53%).
Shifts in summertime eukaryotic communities were characterized by changes in dinoflagellates, diatoms, and prymnesiophytes. Dinoflagellates (phylum Dinoflagellata) were most abundant on the shelf (42.59%), with slightly lower relative abundance offshore (41.73%) and nearshore (37.07%) (Supplementary Figure S1B). Within this group, the genus Dinophysis, a mixotrophic dinoflagellate, showed highest relative abundance offshore and lowest nearshore. The class Syndiniales, composed of parasitic dinoflagellates, was the most abundant across all regions. Group I Syndiniales were more common nearshore, while Groups II, III, and unclassified Syndiniales were more abundant offshore (Supplementary Figure S1D). Gyrista, a recently defined phylum that includes diatoms and other stramenopiles, was most abundant offshore (24.57%), with lower relative abundance nearshore (13.59%) and on the shelf (12.30%). Within this group, pennate diatoms (class Bacillariophyceae) were more abundant nearshore (5.79%) and were primarily composed of the genus Pseudo-nitzschia (Supplementary Figure S1E).
3.2 Differences in network statistics by region
Differences in microbial interactions by region were investigated by constructing microbial co-occurrence networks for each region using OTUs as nodes connected by statistically significant Spearman rank correlations (edges) (Figure 3; Table 1). The nearshore network contained the greatest number of nodes (n = 352), followed closely by the shelf network (n = 350), while the offshore network had the fewest (n = 226). In contrast, the shelf network had the highest number of edges (n = 1682), with the most positive correlations (n = 1552). The nearshore network had slightly fewer total edges (n = 1288), but still a high number of positive correlations (n = 1139). The offshore network had the fewest edges overall (n = 1127) and the highest proportion of negative correlations (n = 242). Eukaryotes dominated all three networks, representing the largest proportion of nodes in each region (Figure 3).
Figure 3
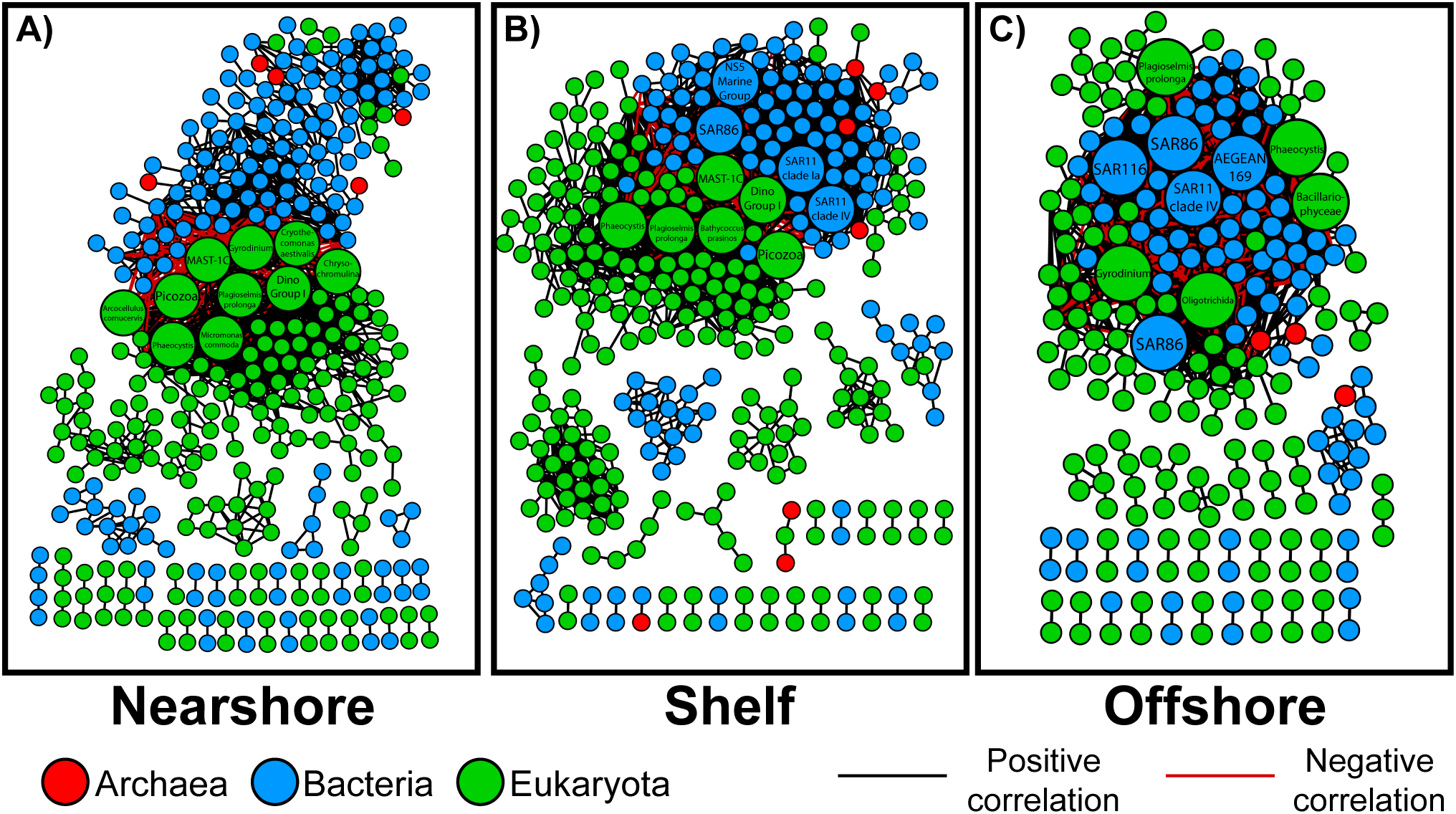
Co-occurrence networks of operational taxonomic units (OTUs) (nodes) with statistically significant Spearman rank correlations (> 0.8, Bonferroni corrected p< 0.05) including positive (black) and negative (red) edges. Nodes are color-coded by taxonomy: Archaea (red), Bacteria (blue), and Eukaryota (green). Node size reflects degree centrality, with larger nodes having more connections. Panel labels (A-C) correspond to the Nearshore, Shelf, and Offshore networks.
Table 1
| Network statistic | Nearshore | Shelf | Offshore |
|---|---|---|---|
| Nodes | 352 | 350 | 226 |
| Edges | 1288 | 1682 | 1127 |
| Positive Edges | 1139 | 1552 | 885 |
| Negative Edges | 149 | 130 | 242 |
| Average Neighbors | 9.883 | 13.852 | 15.29 |
| Diameter | 24 | 12 | 9 |
| Radius | 12 | 6 | 5 |
| Path Length | 6.474 | 3.652 | 2.887 |
| Clustering Coefficient | 0.478 | 0.481 | 0.481 |
| Density | 0.041 | 0.069 | 0.112 |
| Heterogeneity | 1.064 | 0.926 | 1.021 |
| Centralization | 0.165 | 0.191 | 0.331 |
| Fragmentation | 0.63 | 0.59 | 0.6 |
| Components | 39 | 31 | 33 |
Co-occurrence network statistics for nearshore, shelf, and offshore networks.
Degree centrality was not significantly different between nearshore and offshore networks (Figure 4A). However, the shelf network had a significantly higher degree centrality than the other two networks. Closeness centrality was significantly different between all three networks (Figure 4B). The offshore network had significantly higher closeness centrality compared to nearshore and shelf. The average shortest path length of nodes was significantly higher in nearshore compared to both shelf and offshore (Figure 4C). Additionally, offshore and shelf had slightly lower fragmentation values (0.60, 0.59) compared to nearshore (0.63), although no statistical test was performed since fragmentation is a single summary metric per network (Table 1).
Figure 4
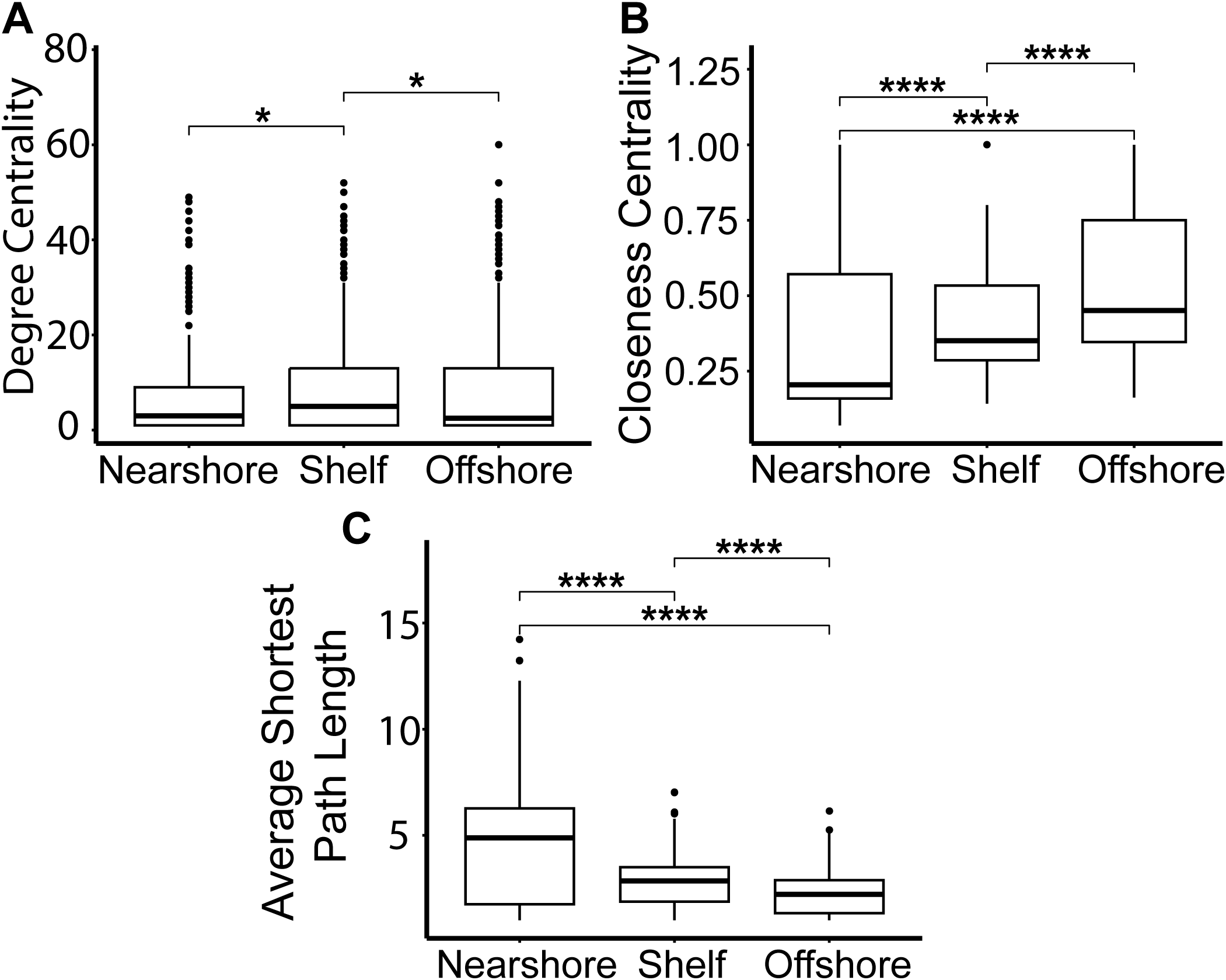
Whole network summary statistics from co-occurrence networks; nearshore, shelf, and offshore. Degree (A), Closeness (B), and Average Shortest Path Length (C) measurements. The number of asterisks represent p-values of Wilcoxon rank sum test [*(p ≤ 0.05), ****(p ≤ 0.0001)].
3.3 Top microbes in network connectivity
Key microbes are those likely to play critical roles within a given network, such that their removal could disrupt the network by severing interactions among microbes. To identify these key microbes within each interaction network, we focused on the top ten microbes with the highest degree centrality, or the most connected nodes in each network (Table 2). This cutoff was chosen as a consistent way to highlight the most highly connected and potentially influential taxa across networks, following previous microbial network studies (Berry and Widder, 2014; Banerjee et al., 2018). These microbes are likely to play critical roles within the networks, such that their removal could disrupt the network by severing the interactions between many microbes. Nearshore and shelf networks were the most similar, having five microbes in common. While nearshore and offshore only had three microbes in common. Only shelf and offshore networks had bacteria in the top ten highest degree centrality. Two of these bacteria were in common, both being within the Proteobacteria phyla including the Gammaproteobacteria SAR86 and the Alphaproteobacteria SAR11 clade IV. Two OTUs were in the top ten in all three networks. Both are within the eukaryote domain: with closest hits to the cryptophyte P. prolonga and the haptophyte Phaeocystis. These shared taxa are known to be ecologically important in marine systems and may contribute to different processes depending on local environmental conditions. For example, SAR11 and SAR86 are ubiquitous oligotrophic heterotrophic bacteria that may fulfill similar roles in utilizing dissolved organic matter (DOM), while cryptophytes and Phaeocystis are high-latitude primary producers associated with complex trophic interactions and bloom dynamics (Giovannoni, 2017; Schoemann et al., 2005).
Table 2
| Region | Organism | Degree | Closeness |
|---|---|---|---|
| Nearshore | Eukaryota; Cryothecomonas aestivalis | 49 | 0.237 |
| Nearshore | Eukaryota; Dino-Group-I | 48 | 0.237 |
| Nearshore | Eukaryota; MAST-1C | 46 | 0.242 |
| Nearshore | Eukaryota; Chrysochromulina sp. | 44 | 0.235 |
| Nearshore | Eukaryota; Picozoa sp. | 42 | 0.235 |
| Nearshore | Eukaryota; Arcocellulus cornucervis | 42 | 0.234 |
| Nearshore | Eukaryota; Plagioselmis prolonga | 42 | 0.234 |
| Nearshore | Eukaryota; Gyrodinium | 40 | 0.239 |
| Nearshore | Eukaryota; Phaeocystis | 39 | 0.232 |
| Nearshore | Eukaryota; Micromonas commoda | 34 | 0.237 |
| Shelf | Eukaryota; Dino-Group-I | 52 | 0.433 |
| Shelf | Eukaryota; Phaeocystis | 50 | 0.413 |
| Shelf | Eukaryota; Plagioselmis prolonga | 47 | 0.417 |
| Shelf | Bacteria; SAR11 Clade IV | 45 | 0.397 |
| Shelf | Eukaryota; MAST-1C | 45 | 0.406 |
| Shelf | Eukaryota; Picozoa sp. | 44 | 0.401 |
| Shelf | Eukaryota; Bathycoccus prasinos | 43 | 0.398 |
| Shelf | Bacteria; SAR11 Clade Ia | 42 | 0.402 |
| Shelf | Bacteria; SAR86 | 42 | 0.397 |
| Shelf | Bacteria; NS5 marine group | 40 | 0.392 |
| Offshore | Eukaryota; Gyrodinium | 60 | 0.559 |
| Offshore | Bacteria; AEGEAN-169 | 52 | 0.5 |
| Offshore | Eukaryota; Bacillariophyceae | 52 | 0.515 |
| Offshore | Eukaryota; Phaeocystis | 48 | 0.523 |
| Offshore | Eukaryota; Plagioselmis prolonga | 47 | 0.521 |
| Offshore | Bacteria; SAR116 | 46 | 0.491 |
| Offshore | Bacteria; SAR11 Clade IV | 45 | 0.489 |
| Offshore | Eukaryota; Oligotrichida | 45 | 0.498 |
| Offshore | Bacteria; SAR86 | 44 | 0.482 |
| Offshore | Bacteria; SAR86 | 43 | 0.486 |
Potential keystone microbes with the highest degree centrality from each network; Nearshore, Shelf, and Offshore.
Degree centrality was used to identify the top 10 most connected nodes in each network. Closeness centrality is included for comparison, as it reflects a different aspect of network structure.
Microbes with the top ten highest degree centrality in the nearshore network were all eukaryotes (Table 2; Figure 3). Three microbes were only found in the top ten most connected taxa in the nearshore network: the haptophyte Chrysochromulina sp., diatom Arcocellulus cornucervis, and the green algae Micromonas commode. The shelf network, in contrast, had four bacteria in the top ten highest degree centrality OTUs. Two of the four bacteria were Alphaproteobacteria, SAR11 clade IV and SAR11 clade Ia, while the third was a Gammaproteobacteria, SAR86, and the fourth, NS5 marine group differed as a Flavobacteria within the Bacteridota phylum. The offshore network differed the most, having the least common top ten most connected microbes to both nearshore and shelf networks. Bacteria were highest in the offshore network making up half of the ten most connected. All five bacteria were within the Proteobacteria phyla: three Alphaproteobacteria (AEGEAN-169, SAR116, SAR11 clade IV) and two Gammaproteobacteria (SAR86). An unidentified diatom within the Bacillariophyceae class and a ciliate within the Oligotrichida order were also within the top ten most connected microbes in the offshore network.
4 Discussion
In the ocean, microbial interactions shape the structure and function of microbial communities, making them critical to marine ecosystem processes (Braga et al., 2016; Arandia-Gorostidi et al., 2022). However, many marine microbes remain uncultivated, and advancing our understanding of their interactions is challenging, particularly because some taxa require co-culturing to survive and grow in laboratory conditions (Lok, 2015; Hofer, 2018; Wang et al., 2021). Network-based analytical approaches such as co-occurrence or co-abundance are a useful alternative, since these can infer potential associations between microbes (Berry and Widder, 2014; Cagua et al., 2019; Yang et al., 2022) and assess ecosystem stability (Widder et al., 2014; Banerjee et al., 2018; Wang et al., 2023; Yang et al., 2024). To uncover potential interactions between microbes in the NGA, we constructed co-occurrence networks of archaea, bacteria, and eukaryotes (Figure 3) and explored network topological features, which allowed us to determine which microbes played an outsized role in the interaction network by being most highly connected (Table 2).
4.1 Regional nutrient gradients structure microbial community composition in the NGA
Microbial community composition in both prokaryotes and eukaryotes differed by region in the NGA (Figure 2). Previous research in the tropical Pacific Ocean and Sargasso Sea has observed similar distinct nearshore, shelf, and offshore prokaryotic communities (Wang et al., 2019; Tucker et al., 2021) driven by cross-shelf gradients in salinity and temperature. In the NGA, shifts in prokaryotic and eukaryotic communities were significantly correlated with shifts in salinity and temperature (Figure 2). Prokaryotic communities exhibited interannual variability, consistent with previous observations of community shifts during the 2019 marine heatwave, which favored small celled phytoplankton (Cohen, 2022; Strom, 2023). Other studies in the NGA have reported regional differences in eukaryotic plankton communities, including protists such as dinoflagellates and microzooplankton like ciliates, often linked to cross-shelf salinity gradients and freshwater input from rivers (Coyle and Pinchuk, 2005; Beamer et al., 2016; Strom et al., 2024). We observed a similar trend in our amplicon data, with both prokaryotic and eukaryotic communities clustering by region (Figure 2). Nitrate and phosphate concentrations were highest in offshore and shelf regions and lowest nearshore, indicating stronger macronutrient limitation in surface nearshore waters (Supplementary Table 1). This pattern is consistent with prior observations (Strom et al., 2006; Aguilar-Islas et al., 2016; Stabeno et al., 2016; Coyle et al., 2019) and supports the idea that regional niche partitioning of microbial communities is influenced by water mass characteristics.
The ACC has a low-salinity core and transports micronutrients from terrestrial runoff, including inputs from the Copper River plume, to nearshore waters (Ortega et al., 2025). Although rich in micronutrients like iron and copper, the ACC is often limited by macronutrients such as nitrate during summer (Strom et al., 2006; Aguilar-Islas et al., 2016; Coyle et al., 2019). Past studies have observed chain-forming diatoms dominating these nearshore waters, particularly during summer stratification (Strom et al., 2006). Our amplicon data supported this pattern, with high relative read abundance of Bacillariophyceae diatoms, including Pseudo-nitzschia spp. and Arcocellulus spp., in nearshore samples (Supplementary Figure S1E). Pseudo-nitzschia is a chain-forming pennate diatom frequently associated with elevated nutrient concentrations and high productivity (Zhang et al., 2021; Moreno et al., 2022), traits characteristic of the ACC (Strom et al., 2006; Stabeno et al., 2016). Some Pseudo-nitzschia species are also known to produce domoic acid, though toxin levels in the NGA are generally low. In contrast, offshore waters showed high relative abundance of Actinocyclus, a large centric diatom. While often associated with nutrient-rich coastal regions, Actinocyclus can also appear in offshore waters when mesoscale eddies or cross-shelf exchange events introduce nutrient pulses to otherwise iron-limited surface waters (Stabeno et al., 2004; Crawford et al., 2007; Ladd et al., 2007). This regional distinction in diatom assemblages likely reflects niche partitioning shaped by nutrient availability along cross-shelf gradients and water masses.
4.2 Co-occurrence network suggests the importance of kleptoplasty and parasitoids in NGA
To examine microbial interactions shared across environmental gradients, we constructed a merged network of significant correlations and nodes found in all three regions. Co-occurrence networks infer potential interactions based on patterns of co-presence or mutual exclusion, but these patterns can result from various ecological processes, including direct interactions, shared environmental preferences, or indirect associations (Fuhrman, 2009; Weiss et al., 2016). Positive correlations may suggest mutualism, shared environmental preferences, or metabolic exchange, but do not necessarily reflect direct interactions (Hibbing et al., 2010; Ghoul and Mitri, 2016). However, correlation alone does not imply causation, and method limitations such as the lack of temporal resolution and potential indirect effects must be considered when interpreting these networks (Needham et al., 2013; Kodera et al., 2022). Despite these constraints, co-occurrence networks are useful hypothesis-generating tools that can identify recurring ecological patterns and guide future experimental investigations.
The merged network revealed significant correlations and nodes shared across all three regions. Although there were significant differences between nearshore and offshore networks and different potential keystone microbes, some significant correlations were found in all three networks (Figure 5). The cryptophyte P. prolonga was a potential keystone microbe in all three networks (Figure 3; Table 2). P. prolonga is a common marine cryptophyte that is an important plastid donor in kleptoplastic interactions with ciliates and dinoflagellates (Altenburger et al., 2020; Cruz and Cartaxana, 2022). In our networks, P. prolonga was consistently associated with ciliates and dinoflagellates, including a grouping found in all three regions involving the ciliate order Oligotrichida and the dinoflagellate Gyrodinium (Figures 5, 6). Kleptopasty allows the host to obtain plastids from ingested photosynthetic prey (Tsuchiya et al., 2020; Cruz and Cartaxana, 2022). Marine oligotrich ciliates have been found to retain prey plastids which remain functional for days to weeks giving them the temporary ability to perform photosynthesis in addition to phagotrophy, making them non-constitutive mixotrophs (Stoecker et al., 1988; Jonsson, 1989; McManus et al., 2012). Ciliates within Oligotrichida have been identified in high latitude environments with the ability to perform mixotrophy (Stoecker and Lavrentyev, 2018; O’Hara, 2023; Strom et al., 2024), suggesting this strategy may be especially advantageous in cold, seasonally variable systems like the NGA. Mixotrophy enhances survival under low-prey or low-light conditions and supports vertical and horizontal transfer of organic matter (Stoecker et al., 2017). It can also lengthen food chains and improve trophic transfer efficiency by bridging microbial and metazoan consumers (Stoecker et al., 2017). Recent work highlights the ecological significance of mixotrophs in the NGA, where environmental gradients in light and nutrients may select for flexible nutritional modes (Strom et al., 2024). The consistent correlations between P. prolonga and Oligotrichida nodes suggest kleptoplasty is a prevalent nutritional strategy in the NGA, contributing to primary production and enhancing trophic connectivity in this high-latitude ecosystem.
Figure 5
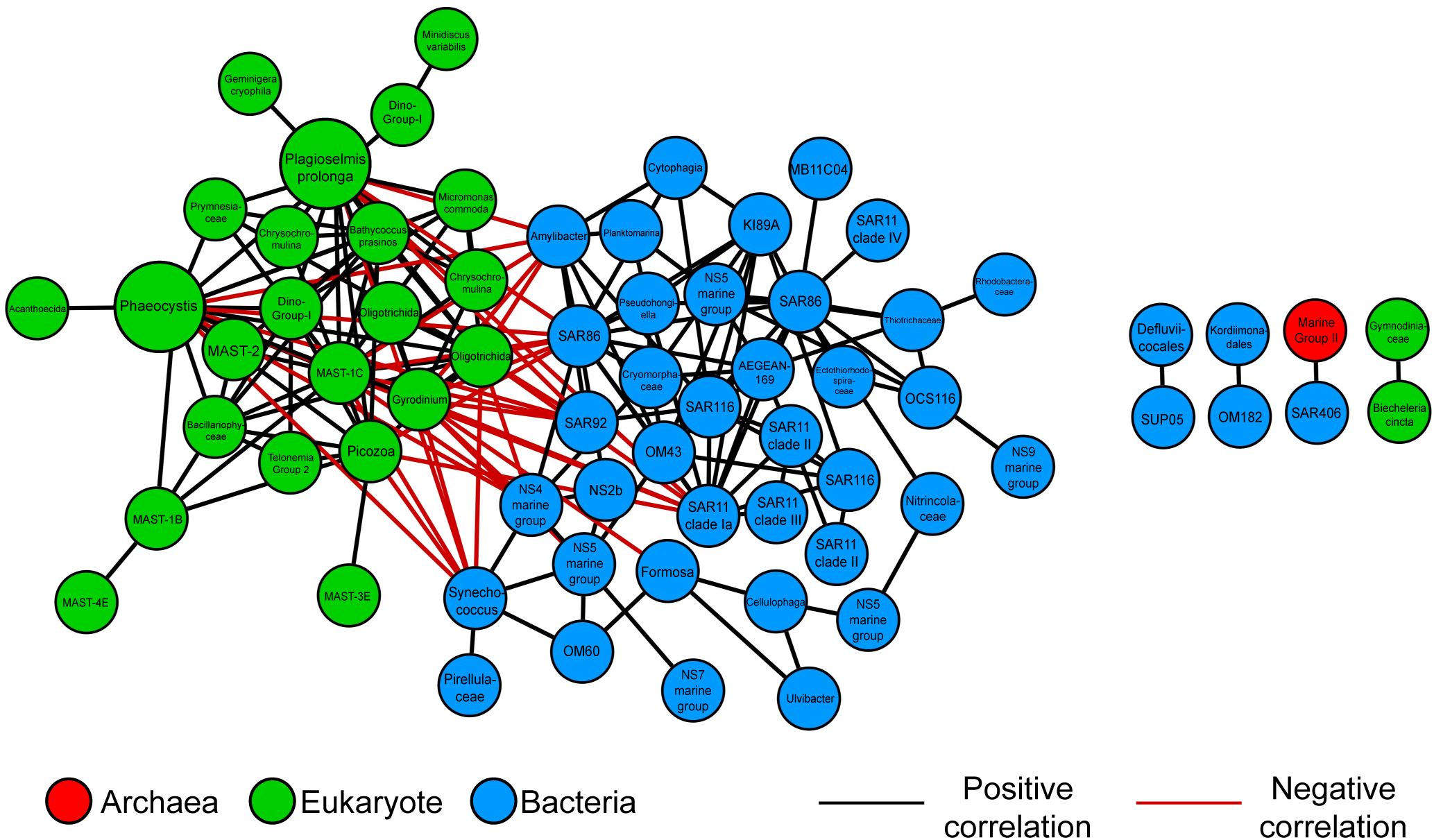
Merged network of significant correlations and nodes found in all three networks (Nearshore, Shelf, and Offshore). Black edges represent positive significant correlations, while red represents negative correlations. Nodes are color-coded by taxonomy: Archaea (red), Bacteria (blue), and Eukaryota (green). Node size reflects degree centrality, with larger nodes having more co-occurrence relationships across regions. Larger nodes are potential keystone microbes from global networks.
Figure 6
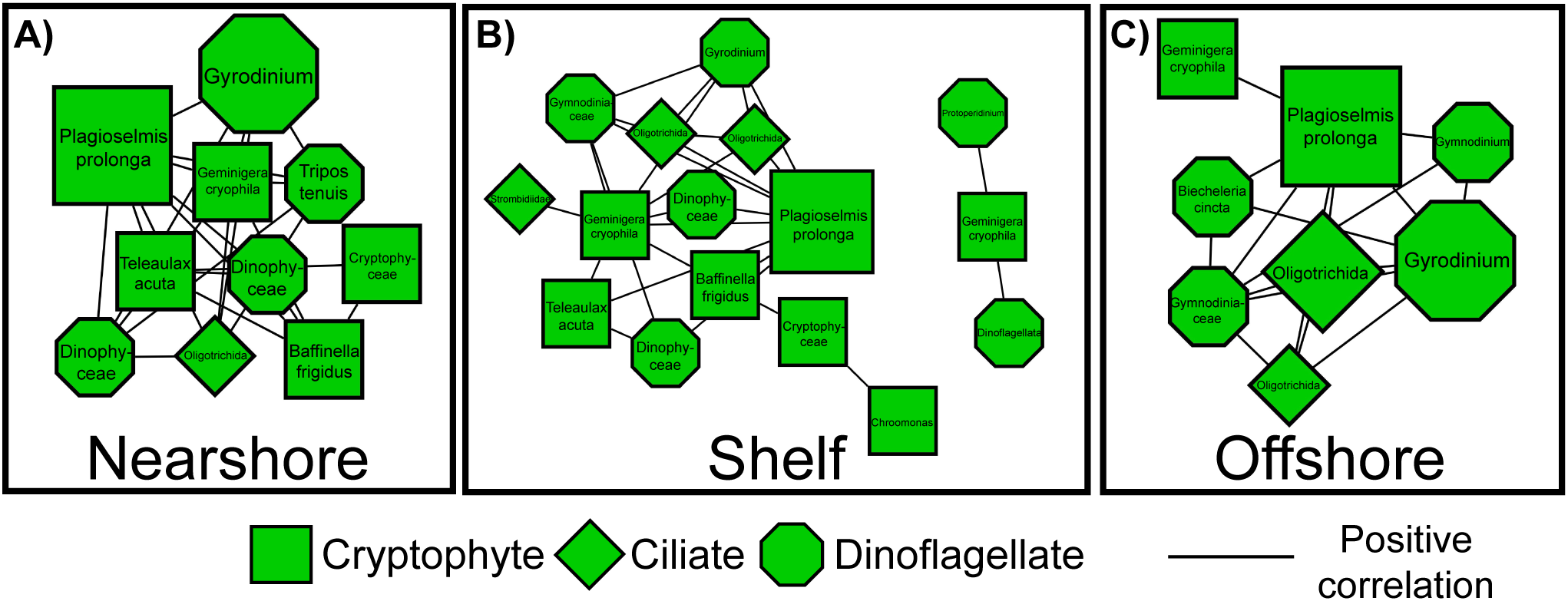
Subnetworks of cryptophyte species (squares) and significantly correlated ciliate (diamonds) and dinoflagellate (octagons) nodes for (A) Nearshore, (B) Shelf, and (C) Offshore regions. Node size reflects degree centrality, with larger nodes having more co-occurrence relationships across regions.
Within the nearshore network microbes associated with parasitism and predation were more abundant and connected (Figure 3). This includes potential keystone microbes and predatory BALOs (Figure 7; Table 2). BALOs are parasitoid bacteria that feed upon and reproduce within gram-negative bacteria using bdelloplasts until the prey cell lyses releasing new BALO cells (Sockett, 2009; Negus et al., 2017; Laloux, 2019). BALOs have two life phases a motile attack phase where they can swim toward prey and a proliferative phase after attachment to prey (Sockett, 2009; Laloux, 2019). Subnetworks showed BALOs (including OM27) significantly correlated with flavobacteria such as NS7 marine group, Cryomorphaceae, and others in all three regions (Figure 7). These flavobacteria are often particle-associated marine microbes (Mitulla et al., 2016; Milici et al., 2017). Since BALOs contain two different life phases, they may move toward particles where there is a higher abundance of potential prey (Lambert et al., 2006, 2011; Iida et al., 2009). Flow cytometry data showed higher heterotrophic bacteria and Synechococcus nearshore cell counts compared to offshore (Supplementary Figure S2). Prey density has been linked to predator density as well as predator-prey interactions (Holling, 1959; Dick et al., 2014; Cuthbert et al., 2021). A minimum prey density is therefore required to support a predator population (Holling, 1959; Dick et al., 2014; Cuthbert et al., 2021). Higher cell counts could increase cell contact rates and therefore give parasitic and predatory microbes higher success in the nearshore network (Hu et al., 2013).
Figure 7
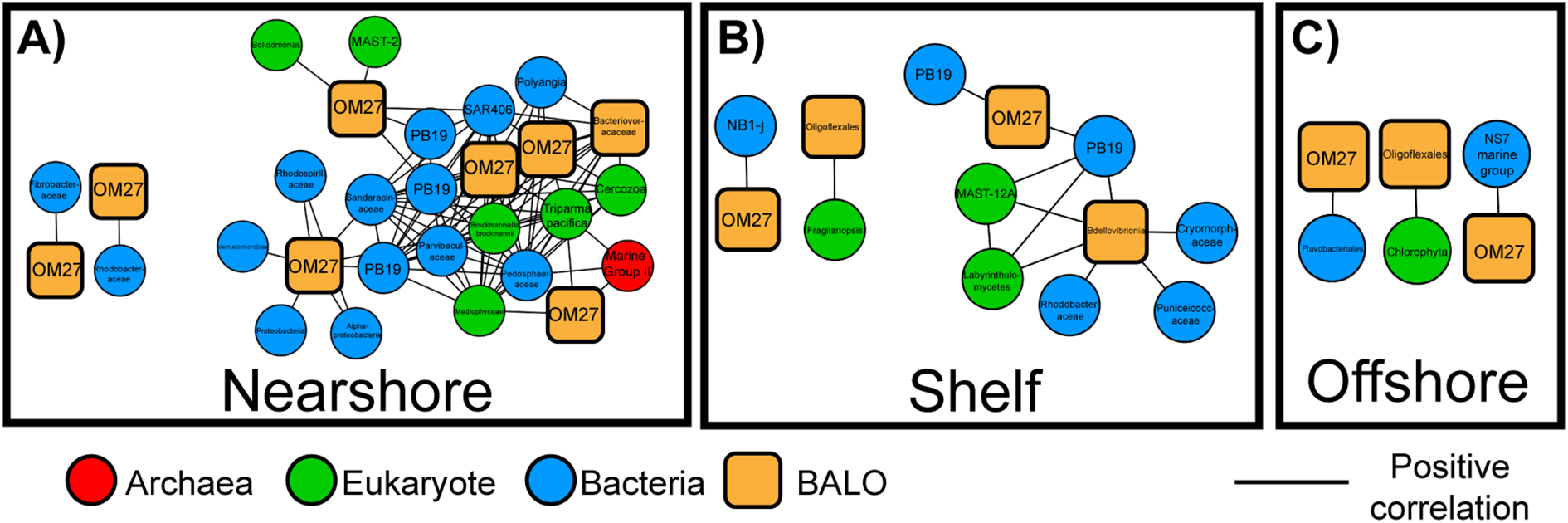
Subnetwork of predatory Bdellovibrio and like organisms (BALOs) (orange squares) and significantly correlated nodes, including potential bacterial prey (blue circles) and associations with eukaryotes (green circles) for (A) Nearshore, (B) Shelf, and (C) Offshore regions.
Interestingly, eukaryotic parasitoids, such as Cryothecomonas aesticalis, a nanoflagellate parasitizing diatoms (Drebes et al., 1996; Catlett et al., 2023), were the most connected microbes in the nearshore network (Table 2), highlighting how complex microbial interactions like parasitism can dominate under dynamic, high-biomass conditions even when surface nutrients are seasonally depleted (Worden et al., 2015). Other highly connected microbes in the nearshore, such as Syndiniales Dino-Group I and MAST-1C, are also known or suspected parasites (Guillou et al., 2008; Massana et al., 2009; Lin et al., 2012; Cleary and Durbin, 2016; Clarke et al., 2019). These eukaryotic parasites were notably absent as the most connected microbes in the offshore network, where microbial parasites were not considered keystone species. The nearshore environment typically has higher micronutrient availability; however, in summer, surface macronutrient depletion may limit primary production, allowing excess micronutrients to persist. These conditions could still support abundant microbial communities particularly in subsurface layers or particle-rich zones that serve as hosts for parasites (Mitulla et al., 2016; Milici et al., 2017). Additionally, freshwater inputs nearshore often create a shallow pycnocline, which may concentrate potential hosts in the surface layer and enhance contact rates with parasites (Lucas et al., 1999). Greater environmental variability in salinity and temperature nearshore may also increase host susceptibility to infection, as physiological stress can compromise host defenses (Harvell et al., 2002). In addition, in macronutrient-depleted surface waters, some microbes may occupy deeper layers to access nutrients, where reduced light imposes further metabolic stress that could increase vulnerability to parasitism (Strom et al., 2006; Worden et al., 2015; Stoecker et al., 2017). In contrast, offshore waters are typically colder and iron-limited, with high nitrate but low chlorophyll concentrations characteristic of high-nutrient, low-chlorophyll (HNLC) systems (Aguilar-Islas et al., 2016; Coyle et al., 2019). Host density in these waters may be limited by iron availability, reducing opportunities for parasitic lifestyles (Boyd, 2007; Strom et al., 2024). However, episodic mixing events such as eddies or intrusions from the Alaska Current and Alaskan Stream may intermittently deliver iron to surface waters, potentially creating localized conditions favorable for parasitism (Ladd et al., 2007; Coyle et al., 2019). These ecological factors may help explain the stronger prominence of parasitic eukaryotes in the nearshore compared to offshore ecosystems.
4.3 What do network characteristics suggest about resilience and stability of the NGA?
Network analysis of microbial co-occurrences provides valuable insights into community structure, resilience, and stability by identifying keystone taxa, those microbes most crucial to maintaining ecological functions. Using measures like degree centrality, which reflects a taxon’s connectivity within the network, we can determine which species, if removed, would be most likely to disrupt community dynamics (Berry and Widder, 2014; Banerjee et al., 2018). The loss of these keystone microbes can lead to significant shifts in microbial community structure and function, as their absence can disproportionately affect ecological processes (Berry and Widder, 2014; Cagua et al., 2019). Network fragmentation, which occurs when key correlations between taxa are lost, further exacerbates this disruption, leading to communities that are less resilient to disturbances (Widder et al., 2014; Wang et al., 2023; Yang et al., 2024). Communities exhibiting higher functional redundancy and lower fragmentation are more likely to maintain stability and recover from perturbations, as they have a greater capacity to compensate for the loss of keystone taxa (Wang et al., 2023). Understanding these key interactions and network properties is essential when evaluating the stability and adaptability of microbial communities, especially in varying environmental conditions.
Potential keystone microbes were identified as the top ten taxa in each network with the highest degree centrality values (Table 2; Figure 3). This cutoff follows previous microbial network studies and was selected to enable consistent cross-network comparison, though we acknowledge that degree centrality alone may not capture all aspects of keystone behavior. Our results suggest that potential keystone microbes shifted from predominantly eukaryotic taxa nearshore to an equal mix of bacteria and eukaryotes offshore (Table 2; Figure 3). Offshore keystone microbes included many oligotrophic bacteria such as SAR11, SAR86, and SAR116 (Brown et al., 2012; Giovannoni, 2017; Hoarfrost et al., 2020; Roda-Garcia et al., 2021). While some of these taxa are also highly abundant, which may contribute to their high centrality, their consistent appearance as central nodes across samples suggests they may also play important ecological roles. Despite being abundant and widespread, many bacteria with oligotrophic lifestyles are adapted to low-nutrient conditions and remain poorly understood due to their uncultivated status (Faust and Raes, 2012; Zelezniak et al., 2015; Yan et al., 2023). These traits may confer an advantage in the iron-limited HNLC offshore waters of the NGA. Studies have shown that interactions among such streamlined taxa, including cross-feeding and metabolite exchange, can structure microbial communities and enhance ecosystem function, highlighting the importance of further investigating their ecological roles (Braakman et al., 2017).
The negative correlations between bacteria and eukaryotic keystone microbes found in our datasets (Figure 3) are possibly driven by bacterivory, with eukaryotic microbes phagocytizing bacterial prey (Sakka et al., 2000; Christaki et al., 2002; Strom, 2002; Sarmento et al., 2013; Arandia-Gorostidi et al., 2022; Follett et al., 2022). For example, the cryptophyte Plagioselmis prolonga, a keystone microbe in the nearshore network, had exclusively negative correlations with bacteria (Figure 5). Many cryptophytes are mixotrophs, supplementing autotrophy with phagotrophy of bacteria (O’Hara, 2023; Šimek et al., 2023), suggesting that P. prolonga’s role as a keystone microbe may be linked to its dual trophic strategy. Similarly, Phaeocystis, another keystone microbe present across all regions, had a high number of negative correlations with bacteria, possibly due to bacterial degradation of bloom remains, such as carbon released from cell lysis (Brussaard et al., 1995, 1996) or due to the ability of Phaeocystis to phagocytize bacteria (Koppelle et al., 2022). By linking microbial primary production and heterotrophic consumption, mixotrophs like P. prolonga and Phaeocystis may connect different parts of the microbial network by occupying different trophic modes. This metabolic flexibility likely contributes to their high centrality and supports their potential roles as keystone microbes in our dataset (Li et al., 2024). Similar patterns have been observed in other marine systems, where mixotrophy is associated with ecological versatility and central roles in microbial networks (Flynn et al., 2013; Mitra et al., 2014; Ward and Follows, 2016).
Network-wide stability metrics indicated that the offshore network exhibited nodes with significantly higher closeness centrality (Figure 4B) compared to the networks in the other two regions. Higher closeness centrality suggests that microbes in the offshore network are more interconnected, meaning disturbances may propagate more quickly, but the overall network is more resistant to fragmentation (Assenov et al., 2008; Berry and Widder, 2014; Cagua et al., 2019; MatChado et al., 2021). While we cannot directly infer specific interaction types from correlation-based networks, higher connectivity in other ecological systems has been linked to community stability and robustness (Jeong et al., 2000; Poisot et al., 2011; Hannigan et al., 2018; Qian and Akçay, 2020). Some studies have also found that highly connected networks are more common in systems where cooperative or mutually beneficial interactions prevail (Thébault and Fontaine, 2010; Montesinos-Navarro et al., 2017; Costas-Selas et al., 2024). Further investigation is needed to confirm whether similar dynamics are occurring in the NGA. In contrast, lower connectivity in the nearshore and shelf networks could indicate reduced stability, potentially driven by a greater prevalence of antagonistic interactions such as parasitism or competition. Although these interactions still contribute to network structure, they may result in more specialized or short-lived associations that fragment networks rather than broadly linking them (Weitz and Dushoff, 2008; Coyte et al., 2015). Antagonistic dynamics also tend to be more specific and variable than mutualistic ones, which could make strong correlations less consistent over time. This fits with ecological theory showing that mutualistic networks are often more cohesive and nested, while antagonistic ones can be more modular and fragmented (Thébault and Fontaine, 2010; Allesina and Tang, 2012). Alternatively, the lower connectivity we observed may reflect greater environmental variability in nearshore and shelf waters compared to offshore. These regions experience stronger fluctuations in temperature, salinity, and freshwater input due to river discharge, stratification, and glacial melt (Beamer et al., 2016; Ortega et al., 2025; Reister et al., 2024; Strom et al., 2024). Prior studies have linked that kind of variability to reduced stability and coherence in microbial networks (Shade et al., 2012; Tylianakis and Morris, 2017). Taken together, these patterns suggest that while the offshore network may be more resistant to perturbation due to its higher connectivity, the lower connectivity observed in nearshore and shelf communities could make them more vulnerable to environmental change or better able to reorganize in response to it through faster turnover or more flexible associations (Shade et al., 2012).
Defining a stable ecosystem, especially a marine one, is difficult since the system is inherently dynamic. Researchers have used fragmentation measurements of microbial co-occurrence networks as an indicator of ecological network sensitivity, with higher fragmentation suggesting greater instability (Widder et al., 2014; Banerjee et al., 2018; Yang et al., 2024). All three co-occurrence networks had fragmentation values greater than 0.5, suggesting microbial communities in all three regions of the NGA are more fragmented and potentially less stable than those observed in other marine ecosystems with lower fragmentation, such as freshwater lakes, the East China Sea, and seagrass meadows (Widder et al., 2014; Wang et al., 2023; Yang et al., 2024), though differences in taxonomic focus and how the networks were built make direct comparisons harder to interpret. Among the three regions, the nearshore network had the highest fragmentation value and the lowest connectivity (Table 1), which could indicate greater vulnerability to disturbance. This region also experiences the highest environmental variability, such fragmentation may also reflect greater flexibility in microbial associations rather than instability per se. This region also contained a greater proportion of parasitic and predatory taxa, such as parasitic eukaryotes and BALOs, which have been shown to influence microbial community structure through top-down interactions and host-parasite dynamics (Thingstad and Lignell, 1997; Chow et al., 2013; Lima-Mendez et al., 2015; Worden et al., 2015). The presence of these taxa suggests strong top-down control, which may contribute to increased fragmentation. According to the kill-the-winner hypothesis, strain-specific predators like viruses can promote microbial diversity by targeting the most competitive taxa, preventing dominance and enabling coexistence (Thingstad, 2000; Winter et al., 2010; Thingstad et al., 2014). If parasitism disproportionately affects highly connected or central taxa, it may destabilize the network by weakening key interactions (Weitz and Dushoff, 2008; Chow et al., 2013; Lima-Mendez et al., 2015). Alternatively, top down pressure could allow less dominant taxa to emerge as new keystone species, potentially reshaping the structure and function of the microbial community. The high fragmentation in the nearshore network could reflect this where top-down control both supports diversity and increases the risk of instability.
Differences in network characteristics between nearshore and offshore regions may be driven by two key factors. First, microbial community composition and keystone taxa vary significantly between regions (Figure 2; Table 2), influencing interaction types and network structure. Antagonistic interactions, such as parasitism or predation, may be more prevalent in the nearshore, where exploitative relations could dominate, whereas offshore microbial communities may experience a more complex mix of interactions. These interactions might not necessarily be mutualistic but could involve processes like micronutrient sharing or cross-feeding that may foster greater network cohesion, which we interpret here as one form of structural stability (Amin et al., 2012; Bertrand et al., 2015; Cooper and Smith, 2015; Arandia-Gorostidi et al., 2022). Second, geochemical differences between regions, driven by the mixing of water masses in the Northern Gulf of Alaska, impact nutrient availability and primary productivity (Strom et al., 2010; Aguilar-Islas et al., 2016; Coyle et al., 2019). Offshore microbial communities may be more stable, potentially due to selective pressures favoring micronutrient exchange processes such as cross-feeding or siderophore exchange, which could reinforce network stability (Amin et al., 2012; Morris et al., 2012; Zelezniak et al., 2015; D’Souza et al., 2018). At the same time, antagonistic interactions can also contribute to ecological stability by regulating population dynamics, suppressing dominance, and promoting turnover (Shade et al., 2012; Coyte et al., 2015; Tylianakis and Morris, 2017). However, the relative roles of different interaction types in promoting network stability remain debated in the ecological literature, and the link between interaction type and stability is likely context-dependent. In contrast, the nearshore environment, characterized by more variable micronutrient and freshwater inputs, may foster a greater prevalence of exploitative interactions, since environmental variability and resource pulses have been shown to push microbial networks toward more antagonistic or short-lived associations (Hawkes and Keitt, 2015; Mickalide and Kuehn, 2019). The differences suggest that moderate variability in nutrient availability and a mosaic environmental structure could promote microbial diversity and stability, but this remains speculative and contrasts with the observed fragmentation in the nearshore network. Ultimately, these findings raise questions about the potential for ecosystem transitions in the NGA. In 2019, a shift in microbial community composition was observed, including a notable increase in chlorophytes and oligotrophic bacteria (Cohen, 2022). The challenge moving forward is to identify early warning signs that might precede tipping points in ecosystem functioning. Identifying these signs could help predict future transitions and offer valuable insights for understanding the resilience of the system (Chisholm and Filotas, 2009; Scheffer et al., 2012).
4.4 Conclusions
This study found microbial communities, potential keystone microbes, and their correlations differ across the shelf/slope of the NGA. Potential keystone microbes that were the most connected in the micronutrient-rich nearshore network were all eukaryotes. However, as distance increased to offshore waters, there was a shift in keystone microbes from eukaryotes to smaller bacteria often found in oligotrophic waters. The offshore network exhibited higher connectivity and less fragmentation compared to the nearshore, suggesting it may be more stable either due to greater microbial interconnectivity or as a result of the less variable environmental conditions offshore. This stability may be further promoted by more cross-domain interactions and a more balanced representation between bacteria and eukaryotes as keystone microbes in the offshore network. While the nearshore network seemed to have a greater prevalence of possible antagonistic interactions, the offshore network likely involves a more complex mix of interactions, with processes like micronutrient sharing or cross-feeding potentially playing a role in fostering network cohesion. The difference in potential interactions suggests a shift from more exploitative relationships nearshore to a more cooperative system offshore where low micronutrient concentrations can be an environmental stressor. Additionally, this study identified two potential key microbial players found in all regions, the mixotrophic nanoflagellates Plagioselmis prolonga and Phaeocystis sp., suggesting these microbes strategies are well suited to environmental gradients across the shelf/slope domains, and may contribute disproportionately to microbial interaction network stability. Overall, different network characteristics and potential interactions were apparent by region, but further research is needed to determine how microbial community composition and environmental variability influence ecosystem stability. These findings contribute to the understanding of microbial ecology during summer conditions in the NGA, and could have important implications for understanding the resilience or vulnerability of the NGA in the face of climate change.
Statements
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA887083.
Author contributions
MB: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Visualization, Writing – original draft, Writing – review & editing. JC: Investigation, Methodology, Resources, Writing – review & editing. BB: Conceptualization, Funding acquisition, Methodology, Resources, Supervision, Validation, Writing – review & editing. GH: Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This research was supported by the National Science Foundation through the following awards: Biological Oceanography (OCE-1937715), the Northern Gulf of Alaska Long Term Ecological Research program (OCE-1656070 and OCE-2322806), and the Office of Polar Programs (OPP-1937595).
Acknowledgments
We thank the captains and crew of the R/V Sikuliaq and R/V Woldstad, Dr. Suzanne Strom and the members of the Strom lab, Dr. R. Eric Collins, Dr. Ana Aguilar-Islas and the UAF Water Nutrient Analytical Facility, and Dr. Russell Hopcroft lead PI of the NGA-LTER project.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2025.1646100/full#supplementary-material
References
1
Abirami B. Radhakrishnan M. Kumaran S. Wilson A. (2021). Impacts of global warming on marine microbial communities. Sci. Total Environ.791, 147905. doi: 10.1016/j.scitotenv.2021.147905
2
Aguilar-Islas A. M. Séguret M. J. M. Rember R. Buck K. N. Proctor P. Mordy C. W. et al . (2016). Temporal variability of reactive iron over the Gulf of Alaska shelf. Deep Sea Res. Part II: Topical Stud. Oceanography132, 90–106. doi: 10.1016/j.dsr2.2015.05.004
3
Allesina S. Tang S. (2012). Stability criteria for complex ecosystems. Nature483, 205–208. doi: 10.1038/nature10832
4
Altenburger A. Blossom H. E. Garcia-Cuetos L. Jakobsen H. H. Carstensen J. Lundholm N. et al . (2020). Dimorphism in cryptophytes—The case of Teleaulax amphioxeia/Plagioselmis prolonga and its ecological implications. Sci. Adv.6, eabb1611. doi: 10.1126/sciadv.abb1611
5
Amaral-Zettler L. A. McCliment E. A. Ducklow H. W. Huse S. M. (2009). A method for studying protistan diversity using massively parallel sequencing of V9 hypervariable regions of small-subunit ribosomal RNA genes. PloS One4, e6372. doi: 10.1371/journal.pone.0006372
6
Amin S. A. Parker M. S. Armbrust E. V. (2012). Interactions between diatoms and bacteria. Microbiol. Mol. Biol. Rev.76, 667–684. doi: 10.1128/MMBR.00007-12
7
Arandia-Gorostidi N. Krabberød A. K. Logares R. Deutschmann I. M. Scharek R. Morán X. A. G. et al . (2022). Novel interactions between phytoplankton and bacteria shape microbial seasonal dynamics in coastal ocean waters. Front. Mar. Sci.9. doi: 10.3389/fmars.2022.901201
8
Assenov Y. Ramírez F. Schelhorn S.-E. Lengauer T. Albrecht M. (2008). Computing topological parameters of biological networks. Bioinformatics24, 282–284. doi: 10.1093/bioinformatics/btm554
9
Banerjee S. Schlaeppi K. van der Heijden M. G. A. (2018). Keystone taxa as drivers of microbiome structure and functioning. Nat. Rev. Microbiol.16, 567–576. doi: 10.1038/s41579-018-0024-1
10
Basu S. Duren W. Evans C. R. Burant C. F. Michailidis G. Karnovsky A. (2017). Sparse network modeling and MetScape-based visualization methods for the analysis of large-scale metabolomics data. Bioinformatics33, 1545–1553. doi: 10.1093/bioinformatics/btx012
11
Beamer J. P. Hill D. F. Arendt A. Liston G. E. (2016). High-resolution modeling of coastal freshwater discharge and glacier mass balance in the Gulf of Alaska watershed. Water Resour. Res.52, 3888–3909. doi: 10.1002/2015WR018457
12
Behrenfeld M. J. O’Malley R. T. Siegel D. A. McClain C. R. Sarmiento J. L. Feldman G. C. et al . (2006). Climate-driven trends in contemporary ocean productivity. Nature444, 752–755. doi: 10.1038/nature05317
13
Berry D. Widder S. (2014). Deciphering microbial interactions and detecting keystone species with co-occurrence networks. Front. Microbiol.5. doi: 10.3389/fmicb.2014.00219
14
Bertrand E. M. McCrow J. P. Moustafa A. Zheng H. McQuaid J. B. Delmont T. O. et al . (2015). Phytoplankton–bacterial interactions mediate micronutrient colimitation at the coastal Antarctic sea ice edge. Proc. Natl. Acad. Sci.112, E6–E6. doi: 10.1073/pnas.1501615112
15
Bodor A. Bounedjoum N. Vincze G. E. Erdeiné Kis Á. Laczi K. Bende G. et al . (2020). Challenges of unculturable bacteria: environmental perspectives. Rev. Environ. Sci. Bio/Technology19, 1–22. doi: 10.1007/s11157-020-09522-4
16
Bokulich N. A. Kaehler B. D. Rideout J. R. Dillon M. Bolyen E. Knight R. et al . (2018). Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome6, 90. doi: 10.1186/s40168-018-0470-z
17
Bolyen E. Rideout J. R. Dillon M. R. Bokulich N. A. Abnet C. C. Al-Ghalith G. A. et al . (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol.37, 852–857. doi: 10.1038/s41587-019-0209-9
18
Boyd P. W. (2007). Biogeochemistry: iron findings. Nature446, 989–991. doi: 10.1038/446989a
19
Boyd P. W. Doney S. C. Strzepek R. Dusenberry J. Lindsay K. Fung I. (2008). Climate-mediated changes to mixed-layer properties in the Southern Ocean: assessing the phytoplankton response. Biogeosciences5, 847–864. doi: 10.5194/bg-5-847-2008
20
Braakman R. Follows M. J. Chisholm S. W. (2017). Metabolic evolution and the self-organization of ecosystems. Proc. Natl. Acad. Sci.114, E3091–E3100. doi: 10.1073/pnas.1619573114
21
Braga R. M. Dourado M. N. Araújo W. L. (2016). Microbial interactions: ecology in a molecular perspective. Braz. J. Microbiol.47, 86–98. doi: 10.1016/j.bjm.2016.10.005
22
Brown M. V. Lauro F. M. DeMaere M. Z. Muir L. Wilkins D. Thomas T. et al . (2012). Global biogeography of SAR11 marine bacteria. Mol. Syst. Biol.8, 595. doi: 10.1038/msb.2012.28
23
Brussaard C. Gast G. J. Duyl F. Riegman R. (1996). Impact of phytoplankton bloom magnitude on a pelagic microbial food web. Mar. Ecol. Prog. Ser.144, 211–221. doi: 10.3354/meps144211
24
Brussaard C. Riegman R. Noordeloos A. Cadee G. Witte H. Kop A. J. et al . (1995). Effects of grazing, sedimentation and phytoplankton cell lysis on the structure of a coastal pelagic food web. Mar. Ecol. Prog. Ser.123, 259–271. doi: 10.3354/meps123259
25
Cagua E. F. Wootton K. L. Stouffer D. B. (2019). Keystoneness, centrality, and the structural controllability of ecological networks. J. Ecol.107, 1779–1790. doi: 10.1111/1365-2745.13147
26
Callahan B. J. McMurdie P. J. Rosen M. J. Han A. W. Johnson A. J. A. Holmes S. P. (2016). DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods13, 581–583. doi: 10.1038/nmeth.3869
27
Catlett D. Peacock E. Crockford E. Futrelle J. Batchelder S. Stevens B. et al . (2023). Temperature dependence of parasitoid infection and abundance of a diatom revealed by automated imaging and classification. Proc. Natl. Acad. Sci. United States America120, e2303356120. doi: 10.1073/pnas.2303356120
28
Chisholm R. A. Filotas E. (2009). Critical slowing down as an indicator of transitions in two-species models. J. Theor. Biol.257, 142–149. doi: 10.1016/j.jtbi.2008.11.008
29
Chow C.-E. T. Sachdeva R. Cram J. A. Steele J. A. Needham D. M. Patel A. et al . (2013). Temporal variability and coherence of euphotic zone bacterial communities over a decade in the Southern California Bight. ISME J.7, 2259–2273. doi: 10.1038/ismej.2013.122
30
Christaki U. Courties C. Karayanni H. Giannakourou A. Maravelias C. Kormas K. A. et al . (2002). Dynamic characteristics of Prochlorococcus and Synechococcus consumption by bacterivorous nanoflagellates. Microbial Ecol.43, 341–352. doi: 10.1007/s00248-002-2002-3
31
Clarke L. J. Bestley S. Bissett A. Deagle B. E. (2019). A globally distributed Syndiniales parasite dominates the Southern Ocean micro-eukaryote community near the sea-ice edge. ISME J.13, 734–737. doi: 10.1038/s41396-018-0306-7
32
Cleary A. C. Durbin E. G. (2016). Unexpected prevalence of parasite 18S rDNA sequences in winter among Antarctic marine protists. J. Plankton Res.38, 401–417. doi: 10.1093/plankt/fbw005
33
Cock P. J. A. Antao T. Chang J. T. Chapman B. A. Cox C. J. Dalke A. et al . (2009). Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics25, 1422–1423. doi: 10.1093/bioinformatics/btp163
34
Cohen J. (2022). Shifts in microbial community composition during the 2019 Pacific marine heatwave in the northern Gulf of Alaska (Fairbanks, AK: University of Alaska Fairbanks). Available online at: http://hdl.handle.net/11122/13115. (Accessed June 11, 2024).
35
Cohen J. Screen J. A. Furtado J. C. Barlow M. Whittleston D. Coumou D. et al . (2014). Recent Arctic amplification and extreme mid-latitude weather. Nat. Geosci.7, 627–637. doi: 10.1038/ngeo2234
36
Cohen Y. Pasternak Z. Müller S. Hübschmann T. Schattenberg F. Sivakala K. K. et al . (2021). Community and single cell analyses reveal complex predatory interactions between bacteria in high diversity systems. Nat. Commun.12, 5481. doi: 10.1038/s41467-021-25824-9
37
Cooper M. B. Smith A. G. (2015). Exploring mutualistic interactions between microalgae and bacteria in the omics age. Curr. Opin. Plant Biol.26, 147–153. doi: 10.1016/j.pbi.2015.07.003
38
Costas-Selas C. Martínez-García S. Delgadillo-Nuño E. Justel-Díez M. Fuentes-Lema A. Fernández E. et al . (2024). Linking the impact of bacteria on phytoplankton growth with microbial community composition and co-occurrence patterns. Mar. Environ. Res.193, 106262. doi: 10.1016/j.marenvres.2023.106262
39
Coyle K. O. Hermann A. J. Hopcroft R. R. (2019). Modeled spatial-temporal distribution of productivity, chlorophyll, iron and nitrate on the northern Gulf of Alaska shelf relative to field observations. Deep Sea Res. Part II: Topical Stud. Oceanography165, 163–191. doi: 10.1016/j.dsr2.2019.05.006
40
Coyle K. O. Pinchuk A. I. (2005). Seasonal cross-shelf distribution of major zooplankton taxa on the northern Gulf of Alaska shelf relative to water mass properties, species depth preferences and vertical migration behavior. Deep Sea Res. Part II: Topical Stud. Oceanography52, 217–245. doi: 10.1016/j.dsr2.2004.09.025
41
Coyte K. Z. Schluter J. Foster K. R. (2015). The ecology of the microbiome: networks, competition, and stability. Science350, 663–666. doi: 10.1126/science.aad2602
42
Cram J. A. Chow C.-E. T. Sachdeva R. Needham D. M. Parada A. E. Steele J. A. et al . (2015). Seasonal and interannual variability of the marine bacterioplankton community throughout the water column over ten years. ISME J.9, 563–580. doi: 10.1038/ismej.2014.153
43
Crawford W. Brickley P. Thomas A. (2007). Mesoscale eddies dominate surface phytoplankton in northern Gulf of Alaska. Prog. Oceanography75, 287–303. doi: 10.1016/j.pocean.2007.08.016
44
Cruz S. Cartaxana P. (2022). Kleptoplasty: Getting away with stolen chloroplasts. PloS Biol.20, e3001857. doi: 10.1371/journal.pbio.3001857
45
Cui Y. Chun S.-J. Baek S. H. Lee M. Kim Y. Lee H.-G. et al . (2019). The water depth-dependent co-occurrence patterns of marine bacteria in shallow and dynamic Southern Coast, Korea. Sci. Rep.9, 9176. doi: 10.1038/s41598-019-45512-5
46
Cuthbert R. N. Dalu T. Wasserman R. J. Sentis A. Weyl O. L. F. Froneman P. W. et al . (2021). Prey and predator density-dependent interactions under different water volumes. Ecol. Evol.11, 6504–6512. doi: 10.1002/ece3.7503
47
D’Souza G. Shitut S. Preussger D. Yousif G. WasChina S. Kost C. (2018). Ecology and evolution of metabolic cross-feeding interactions in bacteria. Natural Product Rep.35, 455–488. doi: 10.1039/C8NP00009C
48
Dick J. T. A. Alexander M. E. Jeschke J. M. Ricciardi A. MacIsaac H. J. Robinson T. B. et al . (2014). Advancing impact prediction and hypothesis testing in invasion ecology using a comparative functional response approach. Biol. Invasions16, 735–753. doi: 10.1007/s10530-013-0550-8
49
Diestel R. (2005). Graph Theory (New York: Springer).
50
Dixon P. (2003). VEGAN, a package of R functions for community ecology. J. Vegetation Sci.14, 927–930. doi: 10.1111/j.1654-1103.2003.tb02228.x
51
Drebes G. Kühn S. F. Gmelch A. Schnepf E. (1996). Cryothecomonas aestivalis sp. nov., a colourless nanoflagellate feeding on the marine centric diatom Guinardia delicatula (Cleve) Hasle. Helgoländer Meeresuntersuchungen50, 497–515. doi: 10.1007/BF02367163
52
Faust K. Raes J. (2012). Microbial interactions: from networks to models. Nat. Rev. Microbiol.10, 538–550. doi: 10.1038/nrmicro2832
53
Faust K. Lima-Mendez G. Lerat J. S. Sathirapongsasuti J. F. Knight R. Huttenhower C. et al . (2015). Cross-biome comparison of microbial association networks. Front. Microbiol. 6, 1200. doi: 10.3389/fmicb.2015.01200
54
Flynn K. J. Mitra A. Anestis K. Anschütz A. A. Calbet A. Ferreira G. D. et al . (2013). Mixotrophic protists and a new paradigm for marine ecology: where does plankton research go now? J. Plankton Res.35, 97–104. doi: 10.1093/plankt/fbs062
55
Follett C. L. Dutkiewicz S. Ribalet F. Zakem E. Caron D. Armbrust E. V. et al . (2022). Trophic interactions with heterotrophic bacteria limit the range of Prochlorococcus. Proc. Natl. Acad. Sci. United States America119, e2110993118. doi: 10.1073/pnas.2110993118
56
Fuhrman J. A. (2009). Microbial community structure and its functional implications. Nature459, 193–199. doi: 10.1038/nature08058
57
Fuhrman J. A. Cram J. A. Needham D. M. (2015). Marine microbial community dynamics and their ecological interpretation. Nat. Rev. Microbiol.13, 133–146. doi: 10.1038/nrmicro3417
58
Ghoul M. Mitri S. (2016). The ecology and evolution of microbial competition. Trends Microbiol.24, 833–845. doi: 10.1016/j.tim.2016.06.011
59
Giovannoni S. J. (2017). SAR11 bacteria: The most abundant plankton in the oceans. Annu. Rev. Mar. Sci.9, 231–255. doi: 10.1146/annurev-marine-010814-015934
60
Glenn T. C. Pierson T. W. Bayona-Vásquez N. J. Kieran T. J. Hoffberg S. L. Thomas J. C. IV et al . (2019). Adapterama II: universal amplicon sequencing on Illumina platforms (TaggiMatrix). PeerJ7, e7786. doi: 10.7717/peerj.7786
61
González A. M. M. Dalsgaard B. Olesen J. M. (2010). Centrality measures and the importance of generalist species in pollination networks. Ecol. Complexity7, 36–43. doi: 10.1016/j.ecocom.2009.03.008
62
Guillou L. Bachar D. Audic S. Bass D. Berney C. Bittner L. et al . (2012). The Protist Ribosomal Reference database (PR2): a catalog of unicellular eukaryote Small Sub-Unit rRNA sequences with curated taxonomy. Nucleic Acids Res.41, D597–D604. doi: 10.1093/nar/gks1160
63
Guillou L. Viprey M. Chambouvet A. Welsh R. M. Kirkham A. R. Massana R. et al . (2008). Widespread occurrence and genetic diversity of marine parasitoids belonging to Syndiniales (Alveolata). Environ. Microbiol.10, 3349–3365. doi: 10.1111/j.1462-2920.2008.01731.x
64
Hannigan G. D. Duhaime M. B. Koutra D. Schloss P. D. (2018). Biogeography and environmental conditions shape bacteriophage–bacteria networks across the human microbiome. PloS Comput. Biol.14, e1006099. doi: 10.1371/journal.pcbi.1006099
65
Harrell F. Jr. Dupont C. (2019). Hmisc: Harrell Miscellaneous. R package version. Available online at: https://CRAN.R-project.org/package=Hmisc. (Accessed May 10, 2023).
66
Harvell C. D. Mitchell C. E. Ward J. R. Altizer S. Dobson A. P. Ostfeld R. S. et al . (2002). Climate warming and disease risks for terrestrial and marine biota. Science296, 2158–2162. doi: 10.1126/science.1063699
67
Hawkes C. V. Keitt T. H. (2015). Resilience vs. historical contingency in microbial responses to environmental change. Ecol. Lett.18, 612–622. doi: 10.1111/ele.12451
68
Hennon G. M. Morris J. J. Haley S. T. Zinser E. R. Durrant A. R. Entwistle E. et al . (2018). The impact of elevated CO2 on Prochlorococcus and microbial interactions with “helper” bacterium Alteromonas. ISME J.12, 520–531. doi: 10.1038/ismej.2017.189
69
Herren C. M. McMahon K. D. (2017). Keystone taxa predict compositional change in microbial communities. Environ. Microbiol.19, 2206–2217. doi: 10.1111/1462-2920.13647
70
Hibbing M. E. Fuqua C. Parsek M. R. Peterson S. B. (2010). Bacterial competition: surviving and thriving in the microbial jungle. Nat. Rev. Microbiol.8, 15–25. doi: 10.1038/nrmicro2259
71
Hoarfrost A. Nayfach S. Ladau J. Yooseph S. Arnosti C. Dupont C. L. et al . (2020). Global ecotypes in the ubiquitous marine clade SAR86. ISME J.14, 178–188. doi: 10.1038/s41396-019-0516-7
72
Hofer U. (2018). The majority is uncultured. Nat. Rev. Microbiol.16, 716–717. doi: 10.1038/s41579-018-0097-x
73
Holen D. (2014). Fishing for community and culture: the value of fisheries in rural Alaska. Polar Rec.50, 403–413. doi: 10.1017/S0032247414000205
74
Holling C. S. (1959). Some characteristics of simple types of predation and parasitism. Can. Entomologist91, 385–398. doi: 10.4039/Ent91385-7
75
Hu H. Nigmatulina K. Eckhoff P. (2013). The scaling of contact rates with population density for the infectious disease models. Math. Biosci.244, 125–134. doi: 10.1016/j.mbs.2013.04.013
76
Hutchins D. A. Fu F. (2017). Microorganisms and ocean global change. Nat. Microbiol.2, 17058. doi: 10.1038/nmicrobiol.2017.58
77
Iida Y. Hobley L. Lambert C. Fenton A. K. Sockett R. E. Aizawa S.-I. (2009). Roles of multiple flagellins in flagellar formation and flagellar growth post bdelloplast lysis in Bdellovibrio bacteriovorus. J. Mol. Biol.394, 1011–1021. doi: 10.1016/j.jmb.2009.10.003
78
Jeong H. Tombor B. Albert R. Oltvai Z. N. Barabási A.-L. (2000). The large-scale organization of metabolic networks. Nature407, 651–654. doi: 10.1038/35036627
79
Jonsson P. (1989). Photosynthetic assimilation of inorganic carbon in marine oligotrich ciliates (Ciliophora, OligotriChina). Mar. Microbial Food Webs2, 55–68.
80
Kassambara A. (2020). ggplot2 Based Publication Ready Plots. Available online at: https://rpkgs.datanovia.com/ggpubr/ (Accessed June 3, 2021).
81
Kodera S. M. Das P. Gilbert J. A. Lutz H. L. (2022). Conceptual strategies for characterizing interactions in microbial communities. iScience25, 103775. doi: 10.1016/j.isci.2022.103775
82
Koppelle S. López-Escardó D. Brussaard C. P. D. Huisman J. Philippart C. J. M. Massana R. et al . (2022). Mixotrophy in the bloom-forming genus Phaeocystis and other haptophytes. Harmful Algae117, 102292. doi: 10.1016/j.hal.2022.102292
83
Ladd C. Mordy C. Kachel N. Stabeno P. (2007). Northern Gulf of Alaska eddies and associated anomalies. Deep-Sea Res. Part I: Oceanographic Res. Papers54, 487–509. doi: 10.1016/j.dsr.2007.01.006
84
Laloux G. (2019). Shedding light on the cell biology of the predatory bacterium Bdellovibrio bacteriovorus. Front. Microbiol.10. doi: 10.3389/fmicb.2019.03136
85
Lambert C. Evans K. J. Till R. Hobley L. Capeness M. Rendulic S. et al . (2006). Characterizing the flagellar filament and the role of motility in bacterial prey-penetration by Bdellovibrio bacteriovorus. Mol. Microbiol.60, 274–286. doi: 10.1111/j.1365-2958.2006.05081.x
86
Lambert C. Fenton A. K. Hobley L. Sockett R. E. (2011). Predatory Bdellovibrio bacteria use gliding motility to scout for prey on surfaces. J. Bacteriology193, 3139–3141. doi: 10.1128/JB.00224-11
87
Li Y. Kan J. Liu F. Lian K. Liang Y. Shao H. et al . (2024). Depth shapes microbiome assembly and network stability in the Mariana Trench. Microbiol. Spectr.12, e02110–e02123. doi: 10.1128/spectrum.02110-23
88
Li L. Pujari L. Wu C. Huang D. Wei Y. Guo C. et al . (2021). Assembly processes and co-occurrence patterns of abundant and rare bacterial community in the Eastern Indian Ocean. Front. Microbiol.12. doi: 10.3389/fmicb.2021.616956
89
Lima-Mendez G. Faust K. Henry N. Decelle J. Colin S. Carcillo F. et al . (2015). Determinants of community structure in the global plankton interactome. Science348, 1262073. doi: 10.1126/science.1262073
90
Lin Y.-C. Campbell T. Chung C.-C. Gong G.-C. Chiang K.-P. Worden A. Z. (2012). Distribution patterns and phylogeny of marine stramenopiles in the North Pacific Ocean. Appl. Environ. Microbiol.78, 3387–3399. doi: 10.1128/AEM.06952-11
91
Logares R. Audic S. Bass D. Bittner L. Boutte C. Christen R. et al . (2014). Patterns of rare and abundant marine microbial eukaryotes. Curr. Biol.24, 813–821. doi: 10.1016/j.cub.2014.02.050
92
Lok C. (2015). Mining the microbial dark matter. Nature522, 270–273. doi: 10.1038/522270a
93
Lucas L. Koseff J. Cloern J. Monismith S. Thompson J. (1999). Processes governing phytoplankton blooms in estuaries. I: The local production-loss balance. Mar. Ecol. Prog. Ser.187, 1–15. doi: 10.3354/meps187001
94
Massana R. Gobet A. Audic S. Bass D. Bittner L. Boutte C. et al . (2015). Marine protist diversity in European coastal waters and sediments as revealed by high-throughput sequencing. Environ. Microbiol.17, 4035–4049. doi: 10.1111/1462-2920.12955
95
Massana R. Unrein F. Rodríguez-Martínez R. Forn I. Lefort T. Pinhassi J. et al . (2009). Grazing rates and functional diversity of uncultured heterotrophic flagellates. ISME J.3, 588–596. doi: 10.1038/ismej.2008.130
96
MatChado M. S. Lauber M. Reitmeier S. Kacprowski T. Baumbach J. Haller D. et al . (2021). Network analysis methods for studying microbial communities: A mini review. Comput. Struct. Biotechnol. J.19, 2687–2698. doi: 10.1016/j.csbj.2021.05.001
97
McManus G. B. Schoener D. Haberlandt K. (2012). Chloroplast symbiosis in a marine ciliate: ecophysiology and the risks and rewards of hosting foreign organelles. Front. Microbiol.3. doi: 10.3389/fmicb.2012.00321
98
Mickalide H. Kuehn S. (2019). Higher-order interaction inhibits bacterial invasion of a phototroph–consortium community. Nat. Ecol. Evol.3, 381–389. doi: 10.1038/s41559-018-0793-0
99
Milici M. Vital M. Tomasch J. Badewien T. H. Giebel H.-A. Plumeier I. et al . (2017). Diversity and community composition of particle-associated and free-living bacteria in mesopelagic and bathypelagic Southern Ocean water masses: Evidence of dispersal limitation in the Bransfield Strait. Limnology Oceanography62, 1080–1095. doi: 10.1002/lno.10487
100
Mitra A. Flynn K. J. Tillmann U. Raven J. A. Caron D. A. Stoecker D. K. et al . (2014). The role of mixotrophic protists in the biological carbon pump. Biogeosciences11, 995–1005. doi: 10.5194/bg-11-995-2014
101
Mitulla M. Dinasquet J. Guillemette R. Simon M. Azam F. Wietz M. (2016). Response of bacterial communities from California coastal waters to alginate particles and an alginolytic Alteromonas macleodii strain. Environ. Microbiol.18, 4369–4377. doi: 10.1111/1462-2920.13314
102
Montesinos-Navarro A. Hiraldo F. Tella J. L. Blanco G. (2017). Network structure embracing mutualism–antagonism continuums increases community robustness. Nat. Ecol. Evol.1, 1661–1669. doi: 10.1038/s41559-017-0320-6
103
Mookherjee A. Jurkevitch E. (2022). Interactions between Bdellovibrio and like organisms and bacteria in biofilms: beyond predator–prey dynamics. Environ. Microbiol.24, 998–1011. doi: 10.1111/1462-2920.15844
104
Moreno A. R. Anderson C. Kudela R. M. Sutula M. Edwards C. Bianchi D. (2022). Development, calibration, and evaluation of a model of Pseudo-nitzschia and domoic acid production for regional ocean modeling studies. Harmful Algae118, 102296. doi: 10.1016/j.hal.2022.102296
105
Morris J. J. Johnson Z. I. Szul M. J. Keller M. Zinser E. R. (2011). Dependence of the cyanobacterium Prochlorococcus on hydrogen peroxide scavenging microbes for growth at the ocean’s surface. PloS One6, e16805. doi: 10.1371/journal.pone.0016805
106
Morris J. J. Kirkegaard R. Szul M. J. Johnson Z. I. Zinser E. R. (2008). Facilitation of robust growth of Prochlorococcus colonies and dilute liquid cultures by “helper” heterotrophic bacteria. Appl. Environ. Microbiol.74, 4530–4534. doi: 10.1128/AEM.02479-07
107
Morris J. J. Lenski R. E. Zinser E. R. (2012). The Black Queen Hypothesis: Evolution of dependencies through adaptive gene loss. mBio3, e00036–e00012. doi: 10.1128/mBio.00036-12
108
Needham D. M. Chow C.-E. T. Cram J. A. Sachdeva R. Parada A. Fuhrman J. A. (2013). Short-term observations of marine bacterial and viral communities: patterns, connections and resilience. ISME J.7, 1274–1285. doi: 10.1038/ismej.2013.19
109
Needham D. M. Fuhrman J. A. (2016). Pronounced daily succession of phytoplankton, archaea and bacteria following a spring bloom. Nat. Microbiol.1, 16005. doi: 10.1038/nmicrobiol.2016.5
110
Negus D. Moore C. Baker M. Raghunathan D. Tyson J. Sockett R. E. (2017). Predator versus pathogen: how does predatory bdellovibrio bacteriovorus interface with the challenges of killing gram-negative pathogens in a host setting? Annu. Rev. Microbiol.71, 441–457. doi: 10.1146/annurev-micro-090816-093618
111
Newman M. E. J. (2003). A measure of betweenness centrality based on random walks. Soc. Networks27, 39–54. doi: 10.1016/j.socnet.2004.11.009
112
O’Hara R. B. (2023). EcolUtils: Utilities for community ecology analysis. R package version 0.1. Available online at: https://cran.r-project.org/package=EcolUtils (Accessed February 7, 2024).
113
Okkonen S. Jacobs G. Metzger E. Hurlburt H. Shriver J. (2001). Mesoscale variability in the boundary currents of the Alaska Gyre. Continental Shelf Res.21, 1219–1236. doi: 10.1016/S0278-4343(00)00085-6
114
Ortega E. L. S. Reister I. Danielson S. L. Aguilar-Islas A. M. (2025). Surface macro- and micro-nutrients within the Copper River plume region respond to along-shore winds. Mar. Chem.270, 104508. doi: 10.1016/j.marchem.2025.104508
115
Parada A. E. Needham D. M. Fuhrman J. A. (2016). Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol.18, 1403–1414. doi: 10.1111/1462-2920.13023
116
Pilosof S. Porter M. A. Pascual M. Kéfi S. (2014). The multilayer nature of ecological networks. Proc. Natl. Acad. Sci.111, 14430–14435. doi: 10.1073/pnas.1411723111
117
Poisot T. Lepennetier G. Martinez E. Ramsayer J. Hochberg M. E. (2011). Resource availability affects the structure of a natural bacteria–bacteriophage community. Biol. Lett.7, 201–204. doi: 10.1098/rsbl.2010.0774
118
Qian J. J. Akçay E. (2020). The balance of interaction types determines the assembly and stability of ecological communities. Nat. Ecol. Evol.4, 356–365. doi: 10.1038/s41559-020-1121-x
119
Quast C. Pruesse E. Yilmaz P. Gerken J. Schweer T. Yarza P. et al . (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res.41, D590–D596. doi: 10.1093/nar/gks1219
120
Reister I. Danielson S. L. Aguilar-Islas A. M. (2024). Perspectives on Northern Gulf of Alaska salinity field structure, freshwater pathways, and controlling mechanisms. Prog. Oceanography229, 103373. doi: 10.1016/j.pocean.2023.103373
121
Rial P. Garrido J. L. Jaén D. Rodríguez F. (2012). Pigment composition in three Dinophysis species (Dinophyceae) and the associated cultures of Mesodinium rubrum and Teleaulax amphioxeia. J. Plankton Res.35, 433–437. doi: 10.1093/plankt/fbs099
122
Roda-Garcia J. J. Haro-Moreno J. M. Huschet L. A. Rodriguez-Valera F. López-Pérez M. (2021). Phylogenomics of SAR116 clade reveals two subclades with different evolutionary trajectories and an important role in the ocean sulfur cycle. mSystems6, e00944–e00921. doi: 10.1128/mSystems.00944-21
123
Rusterholz P. M. Hansen P. J. Daugbjerg N. (2017). Evolutionary transition towards permanent chloroplasts? – Division of kleptochloroplasts in starved cells of two species of Dinophysis (Dinophyceae). PloS One12, e0177512. doi: 10.1371/journal.pone.0177512
124
Sakka A. Legendre L. Gosselin M. Delesalle B. (2000). Structure of the oligotrophic planktonic food web under low grazing of heterotrophic bacteria: Takapoto Atoll, French Polynesia. Mar. Ecol. Prog. Ser.197, 1–17. doi: 10.3354/meps197001
125
Sarmento H. Romera-Castillo C. Lindh M. Pinhassi J. Sala M. M. Gasol J. M. et al . (2013). Phytoplankton species-specific release of dissolved free amino acids and their selective consumption by bacteria. Limnology Oceanography58, 1123–1135. doi: 10.4319/lo.2013.58.3.1123
126
Scheffer M. Carpenter S. R. Lenton T. M. Bascompte J. Brock W. Dakos V. et al . (2012). Anticipating critical transitions. Science338, 344–348. doi: 10.1126/science.1225244
127
Schlitzer R. (2022). Ocean Data View. Available online at: https://odv.awi.de/ (Accessed July 19, 2024).
128
Schoemann V. Becquevort S. Stefels J. Rousseau V. Lancelot C. (2005). Phaeocystis blooms in the global ocean and their controlling mechanisms: a review. J. Sea Res.53, 43–66. doi: 10.1016/j.seares.2004.01.008
129
Screen J. A. Simmonds I. (2010). The central role of diminishing sea ice in recent Arctic temperature amplificationNat.464, 1334–1337. doi: 10.5194/tc-3-11-20092009
130
Serreze M. C. Barret A. P. Stroeve J. C. Kindig D. N. Holland M. M . (2009). The emergence of surface-based Arctic amplification. Cryosphere3, 11–19. doi: 10.5194/tc-3-11-2009
131
Shade A. Peter H. Allison S. D. Baho D. L. Berga M. Bürgmann H. et al . (2012). Fundamentals of microbial community resistance and resilience. Front. Microbiol.3. doi: 10.3389/fmicb.2012.00417
132
Shannon P. Markiel A. Ozier O. Baliga N. S. Wang J. T. Ramage D. et al . (2003). Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res.13, 2498–2504. doi: 10.1101/gr.1239303
133
Šimek K. Mukherjee I. Szöke-Nagy T. Haber M. Salcher M. M. Ghai R. (2023). Cryptic and ubiquitous aplastidic cryptophytes are key freshwater flagellated bacterivores. ISME J.17, 84–94. doi: 10.1038/s41396-022-01326-4
134
Sockett R. E. (2009). Predatory lifestyle of Bdellovibrio bacteriovorus. Annu. Rev. Microbiol.63, 523–539. doi: 10.1146/annurev.micro.091208.073346
135
Stabeno P. J. Bell S. Cheng W. Danielson S. Kachel N. B. Mordy C. W. (2016). Long-term observations of Alaska Coastal Current in the northern Gulf of Alaska. Deep Sea Res. Part II: Topical Stud. Oceanography132, 24–40. doi: 10.1016/j.dsr2.2015.12.016
136
Stabeno P. J. Bond N. A. Hermann A. J. Kachel N. B. Mordy C. W. Overland J. E. (2004). Meteorology and oceanography of the northern gulf of Alaska. Continental Shelf Res.24, 859–897. doi: 10.1016/j.csr.2004.02.007
137
Stoecker D. K. Hansen P. J. Caron D. A. Mitra A. (2017). Mixotrophy in the marine plankton. Annu. Rev. Mar. Sci.9, 311–335. doi: 10.1146/annurev-marine-010816-060617
138
Stoecker D. K. Lavrentyev P. J. (2018). Mixotrophic plankton in the polar seas: A Pan-Arctic review. Front. Mar. Sci.5. doi: 10.3389/fmars.2018.00292
139
Stoecker D. K. Silver M. W. (1990). Replacement and aging of chloroplasts in Strombidium capitatum (Ciliophora: Oligotrichida). Mar. Biol.107, 491–502. doi: 10.1007/BF01313434
140
Stoecker D. K. Silver M. W. Michaels A. E. Davis L. H. (1988). Obligate mixotrophy in Laboea strobila, a ciliate which retains chloroplasts. Mar. Biol.99, 415–423. doi: 10.1007/BF02112135
141
Strom S. (2002). Novel interactions between phytoplankton and microzooplankton: their influence on the coupling between growth and grazing rates in the sea. Hydrobiologia480, 41–54. doi: 10.1023/A:1021224832646
142
Strom S. (2023). Recent marine heatwaves affect marine ecosystems from plankton to seabirds in the northern Gulf of Alaska. Oceanography36, 9. doi: 10.5670/oceanog.2023.s1.9
143
Strom S. Bright K. Fredrickson K. (2024). Widespread ciliate and dinoflagellate mixotrophy may contribute to ecosystem resilience in a subarctic sea: the northern Gulf of Alaska. Aquat. Microbial Ecol.90, 1–21. doi: 10.3354/ame02005
144
Strom S. L. Macri E. L. Fredrickson K. A. (2010). Light limitation of summer primary production in the coastal Gulf of Alaska: physiological and environmental causes. Mar. Ecol. Prog. Ser.402, 45–57. doi: 10.3354/meps08456
145
Strom S. Olson M. Macri E. Mord C. (2006). Cross-shelf gradients in phytoplankton community structure, nutrient utilization, and growth rate in the coastal Gulf of Alaska. Mar. Ecol. Prog. Ser.328, 75–92. doi: 10.3354/meps328075
146
Szymkowiak M. (2020). Adaptations and well-being: Gulf of Alaska fishing families in a changing landscape. Ocean Coast. Manage.197, 105321. doi: 10.1016/j.ocecoaman.2020.105321
147
Thébault E. Fontaine C. (2010). Stability of ecological communities and the architecture of mutualistic and trophic networks. Science329, 853–856. doi: 10.1126/science.1188321
148
Thingstad T. F. (2000). Elements of a theory for the mechanisms controlling abundance, diversity, and biogeochemical role of lytic bacterial viruses in aquatic systems. Limnology Oceanography45, 1320–1330. doi: 10.4319/lo.2000.45.6.1320
149
Thingstad T. F. Lignell R. (1997). Theoretical models for the control of bacterial growth rate, abundance, diversity and carbon demand. Aquat. Microbial Ecol.13, 19–27. doi: 10.3354/ame013019
150
Thingstad T. F. Våge S. Storesund J. E. Sandaa R.-A. Giske J. (2014). A theoretical analysis of how strain-specific viruses can control microbial species diversity. Proc. Natl. Acad. Sci.111, 7813–7818. doi: 10.1073/pnas.1400909111
151
Tipton L. Müller C. L. Kurtz Z. D. Huang L. Kleerup E. Morris A. et al . (2018). Fungi stabilize connectivity in the lung and skin microbial ecosystems. Microbiome6, 12. doi: 10.1186/s40168-018-0519-9
152
Tsuchiya M. Miyawaki S. Oguri K. Toyofuku T. Tame A. Uematsu K. et al . (2020). Acquisition, maintenance, and ecological roles of kleptoplasts in Planoglabratella opercularis (Foraminifera, Rhizaria). Front. Mar. Sci.7. doi: 10.3389/fmars.2020.00585
153
Tucker S. J. Freel K. C. Monaghan E. A. Sullivan C. E. S. Ramfelt O. Rii Y. M. et al . (2021). Spatial and temporal dynamics of SAR11 marine bacteria across a nearshore to offshore transect in the tropical Pacific Ocean. PeerJ9, e12274. doi: 10.7717/peerj.12274
154
Tylianakis J. M. Morris R. J. (2017). Ecological networks across environmental gradients. Annu. Rev. Ecology Evolution Systematics48, 25–48. doi: 10.1146/annurev-ecolsys-110316-022821
155
Wang Z. Juarez D. L. Pan J.-F. Blinebry S. K. Gronniger J. Clark J. S. et al . (2019). Microbial communities across nearshore to offshore coastal transects are primarily shaped by distance and temperature. Environ. Microbiol.21, 3862–3872. doi: 10.1111/1462-2920.14734
156
Wang F. Li M. Huang L. Zhang X.-H. (2021). Cultivation of uncultured marine microorganisms. Mar. Life Sci. Technol.3, 117–120. doi: 10.1007/s42995-021-00093-z
157
Wang P. Li S.-P. Yang X. Si X. Li W.-J. Shu W. et al . (2023). Spatial scaling of soil microbial co-occurrence networks in a fragmented landscape. mLife2, 209–215. doi: 10.1002/mlf2.12073
158
Ward B. A. Follows M. J. (2016). Marine mixotrophy increases trophic transfer efficiency, mean organism size, and vertical carbon flux. Proc. Natl. Acad. Sci. United States America113, 2958–2963. doi: 10.1038/ismej.2015.235
159
Weiss S. Van Treuren W. Lozupone C. Faust K. Friedman J. Deng Y. et al . (2016). Correlation detection strategies in microbial data sets vary widely in sensitivity and precision. ISME J. 10, 1669–1681. doi: 10.1038/ismej.2015.235
160
Weitz J. S. Dushoff J. (2008). Alternative stable states in host–phage dynamics. Theor. Ecol.1, 13–19. doi: 10.1007/s12080-007-0001-1
161
Widder S. Besemer K. Singer G. A. Ceola S. Bertuzzo E. Quince C. et al . (2014). Fluvial network organization imprints on microbial co-occurrence networks. Proc. Natl. Acad. Sci. United States America111, 12799–12804. doi: 10.1073/pnas.1411723111
162
Winter C. Bouvier T. Weinbauer M. G. Thingstad T. F. (2010). Trade-offs between competition and defense specialists among unicellular planktonic organisms: the “killing the winner” hypothesis revisited. Microbiol. Mol. Biol. Rev.74, 42–57. doi: 10.1128/MMBR.00034-09
163
Worden A. Z. Follows M. J. Giovannoni S. J. Wilken S. Zimmerman A. E. Keeling P. J. (2015). Environmental science. Rethinking the marine carbon cycle: factoring in the multifarious lifestyles of microbes. Science347, 1257594. doi: 10.1126/science.1257594
164
Yan C. Owen J. S. Seo E.-Y. Jung D. He S. (2023). Microbial interaction is among the key factors for isolation of previous uncultured microbes. J. Microbiol.61, 655–662. doi: 10.1007/s12275-023-00063-3
165
Yang Y. Shi Y. Fang J. Chu H. Adams J. M. (2022). Soil microbial network complexity varies with pH as a continuum, not a threshold, across the north China plain. Front. Microbiol.13. doi: 10.3389/fmicb.2022.895687
166
Yang M. Zhao L. Yu X. Shu W. Cao F. Liu Q. et al . (2024). Microbial community structure and co-occurrence network stability in seawater and microplastic biofilms under prometryn pollution in marine ecosystems. Mar. pollut. Bull.199, 115960. doi: 10.1016/j.marpolbul.2023.115960
167
Zelezniak A. Andrejev S. Ponomarova O. Mende D. R. Bork P. Patil K. R. (2015). Metabolic dependencies drive species co-occurrence in diverse microbial communities. Proc. Natl. Acad. Sci. United States America112, 6449–6454. doi: 10.1073/pnas.1421834112
168
Zhang S. Zheng T. Lundholm N. Huang X. Jiang X. Li A. et al . (2021). Chemical and morphological defenses of Pseudo-nitzschia multiseries in response to zooplankton grazing. Harmful Algae104, 102033. doi: 10.1016/j.hal.2021.102033
Summary
Keywords
co-occurrence network, keystone microbes, trophic interactions, kleptoplasty, parasitoid, subarctic, metabarcoding
Citation
Brauner M, Cohen J, Briggs BR and Hennon GMM (2025) Identifying potential keystone microbes from co-occurrence networks in the Gulf of Alaska. Front. Mar. Sci. 12:1646100. doi: 10.3389/fmars.2025.1646100
Received
12 June 2025
Accepted
04 August 2025
Published
10 September 2025
Volume
12 - 2025
Edited by
Jin Zhou, Tsinghua University, China
Reviewed by
Xiaolei Wang, Ocean University of China, China
Sangwook Lee, Hong Kong University of Science and Technology, Hong Kong SAR, China
Updates
Copyright
© 2025 Brauner, Cohen, Briggs and Hennon.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gwenn M. M. Hennon, gmhennon@alaska.edu
†ORCID: Megan Brauner, orcid.org/0009-0000-1366-2917; Jacob Cohen, orcid.org/0000-0003-3768-4840; Brandon R. Briggs, orcid.org/0000-0002-3808-8269; Gwenn M. M. Hennon, orcid.org/0000-0002-5232-3843
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.