 Selma Menabit
Selma Menabit Paris Lavin
Paris Lavin Tatiana Begun
Tatiana Begun Adrian Teacă
Adrian Teacă Mihaela Mureşan
Mihaela Mureşan Cristina Purcarea
Cristina Purcarea- 1Department of Biology-Ecology, National Institute for Research and Development on Marine Geology and Geoecology-GeoEcoMar, Bucharest,, Romania
- 2Department of Microbiology, Institute of Biology Bucharest of the Romanian Academy, Bucharest, Romania
- 3Facultad de Ciencias del Mar y Recursos Biológicos, Departamento de Biotecnología, Universidad de Antofagasta, Antofagasta, Chile
- 4Centro de Investigacion en Inmunologia y Biotecnologia Biomedica de Antofagasta (CIIBBA), Universidad de Antofagasta, Antofagasta, Chile
Bacteria colonizing bivalves play a critical role in host health by supporting digestion, nutrient cycling, and immune defense. While the microbiomes of marine bivalves have been studied globally, their diversity and functional roles across specific organs remain underexplored. This study investigates the structural and predicted functional diversity of bacterial communities associated with different organs (siphon, gills, and stomach) of the marine bivalve Mya arenaria Linnaeus, 1758, along with the surrounding sediments from the Romanian Black Sea coast, using 16S rRNA gene sequencing with Illumina technology. Bacterial communities within the bivalve differed markedly from those in the sediments and varied across organs. Sediment samples exhibited greater taxonomic diversity (19 phyla) than bivalve organs (14–15 phyla). Verrucomicrobiota dominated the siphon and gills, Spirochaetota were most abundant in the stomach, and Desulfobacterota predominated in sediments. Nitrate-reducing bacteria, particularly those from the genus Persicirhabdus, were prevalent in all organs and may contribute to host resilience under hypoxic conditions. The presence of Sulfurimonas in the stomach suggests a possible nutritional association, while halotolerant Woeseia species identified in sediments likely play a role in environmental nutrient cycling. Predictive functional profiling indicated potential bacterial involvement in various metabolic processes, including carbohydrate, amino acid, and energy metabolism. Additionally, pathways related to xenobiotic degradation and antibiotic biosynthesis were inferred across all sample types, indicating a potential capacity for broader ecological and possibly biotechnological roles. However, these functions were inferred from 16S rRNA data and require further validation through metagenomic or transcriptomic approaches. To our knowledge, this is the first detailed analysis of microbiome variability across different organs of M. arenaria, offering new insights into host–microbe interactions in this species.
1 Introduction
Microorganisms are widespread and essential components in marine ecosystems, establishing diverse and often intimate associations with a wide range of animals (Fraune and Bosch, 2010; Krishnaveni et al., 2025). Marine benthic invertebrates, like all multicellular organisms, engage in complex interactions with a diverse array of microbes, including bacteria (Knowlton and Rohwer, 2003; Li et al., 2023a, b), and these associations can take the form of mutualism, commensalism, or parasitism. Bacteia colonizing marine invertebrates play crucial roles in maintaining host health, supplying essential nutrients, and offering protection against pathogens. Moreover, they contribute significantly to the overall functioning and adaptive responses of hosts to environmental change (Bourne et al., 2016; Pita et al., 2018). Hosts may possess species-specific bacterial communities distinct from their surrounding environment, which can be acquired vertically, from conspecifics, or from the environment (Koskella et al., 2017). The degree of specificity of these communities varies among host species, with some exhibiting only a few highly specific symbionts, whereas others share bacteria with the surrounding environment (Lemieux-Labonté et al., 2016; Lemay et al., 2018; Roth‐Schulze et al., 2018).
Bivalves harbor a wide variety of microorganisms, including protozoa, fungi, viruses, and bacteria (Rey-Campos et al., 2022), with bacteria being the most extensively studied group (Masanja et al., 2023). The microbiome of bivalves plays a critical role in the host health and disease, with the bacterial component involved in processes such as digestion, nutrient cycling, and immune defense (Pierce and Ward, 2018; Timmins-Schiffman et al., 2021). Certain bivalve-colonizing bacteria have been found to produce antimicrobial compounds that help protect the host from pathogens (Destoumieux-Garzón et al., 2020; Balbi et al., 2021). In addition, some species belonging to the families Phyllobacteriaceae and Hyphomicrobiaceae (Pseudomonadota) and Flavobacteriaceae (Bacteroidota) are associated with enhanced disease resistance in bivalves (Dubé et al., 2019). These microorganisms produce enzymes that break down complex carbohydrates and contribute to the immune response of their host (Moruf et al., 2020).
The structure of microbial communities colonizing marine invertebrates, including bivalves, is highly variable, with different body regions harboring distinct microbial communities (Masanja et al., 2023). For instance, the bacterial composition of Eriocheir sinensis (Chinese crab) was dominated by Tenericutes and Pseudomonadota in the gut, whereas Actinobacteria, Pseudomonadota, and Bacteroidetes predominated in the gill samples (Zhang et al., 2016). Similarly, Mytilus galloprovincialis possesses a microbiota with tissue-specific variations: where the digestive gland is characterized by Ruminococcaceae and Lachnospiraceae, the stomach is dominated by Flavobacteriaceae, and the foot by Spirochaetaceae representatives (Musella et al., 2020).
The Boreal-Atlantic bivalve Mya arenaria Linnaeus, 1758, is native to the North Atlantic, off the coasts of America and Canada (Strasser, 1998). Invasive in Europe, it has been reported in the Barents Sea (Galkin, 1998), White (Russanova, 1963), Baltic (Kube, 1996), North (Strasser, 1998), Wadden (Smidt, 1951), Mediterranean seas (Stora et al., 1995), and along the European coasts of the Atlantic Ocean (Conde et al., 2011). In the Black Sea, it was first recorded in 1966 near Odessa (Beshevli and Kolyagin, 1967; Gomoiu and Porumb, 1969), later identified near Berezan Island (Zambriborshch et al., 1968), and within a relatively short time became a common species in the northwestern part of the basin. Considering the age of the identified specimens, as well as the time required for larval development and reaching sexual maturity, Zambriborshch et al. (1968) suggested that the species entered the Black Sea in the early 50s. In the Romanian Black Sea region, this species was first reported in the late 1960s, with a limited number of specimens identified near Zaton and Tomis Harbor (Constanţa) (Gomoiu and Porumb, 1969), and was later observed on the Romanian continental shelf at depths of up to 35 m (Skolka and Gomoiu, 2004).
This bivalve thrives in the organic-rich muds of the Black Sea, particularly in sedimentary habitats influenced by inputs from the Danube River, which carry high bacterial loads, including potential pathogens (Ene et al., 2025). Although not currently consumed in Romania, M. arenaria is recognized for its edible potential and is successfully harvested and farmed in other regions of the world (Wheaton et al., 2008). In the Black Sea, it serves as prey for several commercially important benthic fish species, potentially raising public health concerns. Despite exposure to bacterial pathogens, the species appears to remain unaffected, likely due to immune adaptations that enable tolerance to challenging environmental conditions such as hypoxia, rapid fluctuations and high organic pollution. Some studies have shown that the soft-shell clam hosts a distinct microbiome, suggesting a possible symbiotic relationship between the clam and its associated microorganisms (Cabelli and Heffernan, 1970; Liu et al., 2020).
While extensive research has been conducted on bacterial communities colonizing benthic organisms in marine environments (Gilbertson et al., 2012; Fuirst et al., 2021; Li et al., 2023b; Liu et al., 2023), including bivalves (Musella et al., 2020; Akter et al., 2023), only a few studies have addressed the microbiome of these invertebrates from the Black Sea, notably in the bivalves Mytilaster lineatus (Onishchenko and Kiprianova, 2006) and Donax trunculus (Ibryamova et al., 2022), as well as the tube-dwelling polychaete Melinna palmata (Menabit et al., 2024).
Microbial ecology studies carried out in the Black Sea ecosystem have predominantly focused on bacterial communities from seawater (Jørgensen et al., 1991; Sorokin et al., 1995; Glaubitz et al., 2010; Bryukhanov et al., 2015; Ruginescu et al., 2022) and sediments (Schulz et al., 1999; Thamdrup et al., 2000; Leloup et al., 2007; Schäfer et al., 2007; Coolen and Shtereva, 2009; Schippers et al., 2012), with limited comparison between their diversity and that of invertebrate-associated communities (Onishchenko and Kiprianova, 2006; Menabit et al., 2024).
In this context, the present study aims to assess the structural and functional bacterial diversity associated with the marine bivalve M. arenaria, focusing on organ-specific (siphon, gills, and stomach) microbiomes and their distinctions from surrounding sediment communities using a high-throughput Illumina 16S rRNA gene sequencing approach. The study was based on the hypothesis that M. arenaria harbors a microbiome that facilitates its adaptation to stressful estuarine environments and organic pollution, including bacterial contaminants. Rather than using classical microbiological methods, we applied molecular techniques that enabled the identification of bacterial genes potentially involved in diverse metabolic functions, including antibiotic resistance. This investigation represents the first characterization of microbiome variability across different organs of the bivalve M. arenaria.
2 Materials and methods
2.1 Study area and sample collection and preparation
The north-western Black Sea covers 127,000 km2, accounting for 94% of the shelf’s total area, and approximately 1.2% of the total Black Sea water volume. This region receives significant freshwater input from the Danube, Dniester, Dnieper, and Southern Bug rivers with the Danube being the largest and most important water and sediment supplier, strongly influencing sedimentation in the north-western part of the basin (Panin and Jipa, 2002).
Sediment samples were collected during a one-time field campaign in July 2020 from two stations along the Romanian coast of the Black Sea (Figure 1), located 36.3 km apart, at water depths of 19.0 m (P8-20) (44°47,920’N; 29°37,850’E) and 27.0 m (CT-02) (44°17,420’N; 28°72,310’E), respectively (Figure 1). Station P8–20 is located within the area influenced by Danube freshwater (Figure 1).
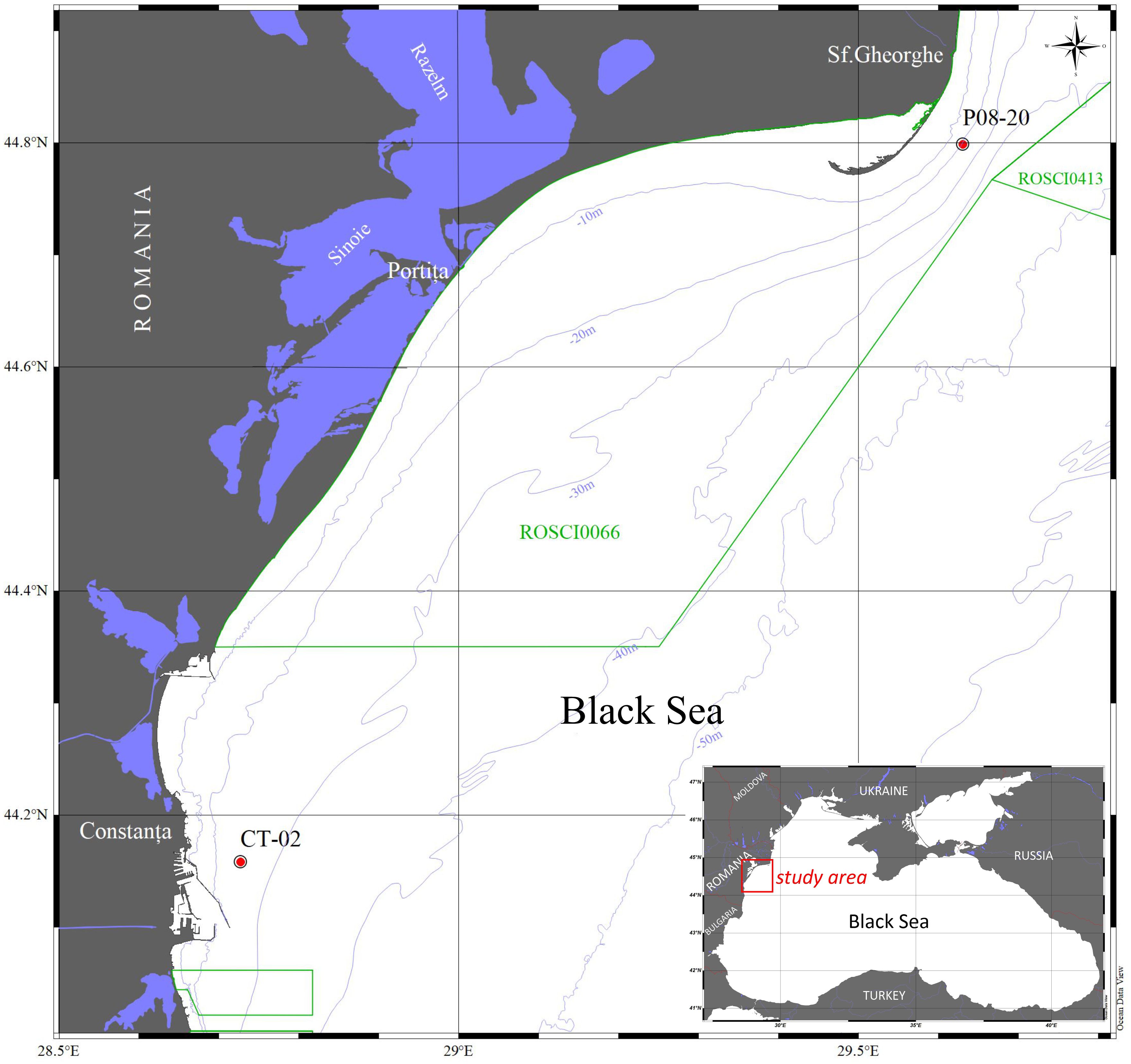
Figure 1. Map of the Romanian sector of the Black Sea showing the study area and sampling stations CT-02 and P08-20.
Sediments were collected using a Van Veen grab sampler with a surface area of 0.135 m2 (Todorova and Konsulova, 2005). The sedimentary material containing M. arenaria specimens from both sites consisted of mud, which derived from alluvial deposits composed of silt and clay particles. To evaluate the microbial communities, two sediment subsamples and two specimens of M. arenaria were collected from each station. The low number of samples was due to limitations related to sampling constraints. Typically, only one or two samples fall within habitats characteristic of this species, with no more than a few specimens captured per sample. After removing the sediment fraction that had been in contact with the sampling equipment to minimize contamination, the remaining sediment was transferred into sterile containers and stored at −20°C until further processing.
For bivalve DNA extraction, each selected specimen was washed with sterile water and preserved in 200 μl of Tris-EDTA buffer (pH 8.0) at −20°C (Ross et al., 1990).
2.2 Physicochemical parameters
In situ measurements of temperature, salinity, dissolved oxygen (DO) concentration, and pH in the bottom water level above the seabed were conducted using an EXO2 multi-parameter probe (YSI Incorporated, Yellow Springs, USA).
2.3 DNA extraction, 16S rRNA gene sequencing and sequence analysis
Genomic DNA was extracted from two M. arenaria individuals and two sediment samples from each sampling site. Mya specimens were rinsed with sterile water, dissected under aseptic conditions. DNA was isolated from the siphon, gills, and stomach of each individual using the DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany), following an optimized protocol that included an initial cell disruption step (Iancu et al., 2015). Tissue samples were resuspended in Tris-EDTA buffer (pH 8) and homogenized at 20°C for 12 min in a SpeedMill PLUS homogenizer at 50 Hz (Analytik Jena, Jena, Germany), with 5 ZR BashingBead 0.2 mm lysis matrix (Zymo Research, Irvine, CA, USA), and subsequently processed according to the manufacturer’s protocol. For sediment samples, DNA was isolated using the DNeasy PowerSoil Pro Kit (Qiagen, Hilden, Germany).The V3–V4 hypervariable regions of the 16S rRNA gene were amplified using the primer pair 341F/805R (Takahashi et al., 2014) and sequenced on an Illumina MiSeq platform with 300 bp paired-end reads (Macrogen, Seoul, South Korea).
The resulting DNA sequences were processed using the DADA2 package (v1.8) implemented in R (v4.0.2) (Callahan et al., 2016). After removing the forward and reverse primer sequences with cutadapt (v4.2.2) (Martin, 2011), the reads were trimmed and filtered (Lu et al., 2023). Amplicon sequence variants (ASVs) were inferred from the de-replicated sequences, and chimeras were removed using the consensus assignment method (Lu et al., 2023). Taxonomic identification of ASVs was performed using the SILVA v138 16S rRNA database (silva.nr. v138). Analyses were conducted using MicrobiomeAnalyst 2.0 (Lu et al., 2023). Standard data filtering procedures were applied to improve the quality of downstream analyses. A low-count filter was implemented using a minimum threshold of four reads, retaining only those features present in at least 20% of the samples (prevalence filter). Additionally, a low-variance filter was applied to remove features showing minimal variation across experimental conditions, based on the interquartile range (IQR). These filters aimed to eliminate low-abundance and noninformative features, which are often the result of sequencing noise or background contamination.
The 16S rRNA gene sequences of bacteria colonizing M. arenaria and associated sediments were deposited in the NCBI Sequence Read Archive (SRA) under the BioProject accession number PRJNA922888.
Pattern search analysis based on Spearman correlation (Legendre and Legendre, 2012) was performed to assess whether specific taxa from particular organs and surrounding sediments were consistently associated, thereby enabling the identification of statistically significant relationships between data matrices.
The estimation of potential microbial functions was conducted by assessing gene abundances according to KEGG metabolism, using the TAx4Fun2 program (Lu et al., 2023).
2.4 Statistical analyses
Alpha and beta diversity was calculated using the pyloseq package (McMurdie and Holmes, 2013). Alpha diversity of ASVs was assessed using the Chao1, Shannon, Evenness, and Fisher indices. Bray–Curtis dissimilarity was used to assess beta diversity by comparing diversity between samples and microbial communities. The ordination-based method, Principal Coordinate Analysis (PCoA), was used to visualize the matrix in a 2-D plot, where each point represents the entire microbiome of a single sample. The statistical significance (p <0.05) of the clustering patterns in ordination plots was evaluated using permutational ANOVA (PERMANOVA) and analysis of group similarities (ANOSIM). Statistical analyses were performed using MicrobiomeAnalyst 2.0 (Lu et al., 2023). To compare alpha diversity indices (Chao1, Shannon, and Fisher) among groups (organs and sediments), the non-parametric Kruskal–Wallis test was applied, as it does not require assumptions of normality or homogeneity of variances. When significant differences were found (p <0.05), post hoc multiple comparisons were performed using Dunn’s test with Bonferroni correction to control for Type I error. All analyses were conducted in R (v4.5.1) using the FSA and PMCMRplus packages (Mangiafico, 2015; Pohlert, 2014).
LDA Effect Size (LEfSe) was based on the non-parametric Kruskal–Wallis test, which identifies significant differences in terms of taxa abundance across the analyzed sample types and ranks taxa according to the Linear Discriminant Analysis (LDA) (Segata et al., 2011). The Student’s t-test was employed to statistically compare the mean gene abundance across samples (Mishra et al., 2019).
3 Results
3.1 Environmental characteristics of Mya arenaria habitat
In situ measurements of water parameters—temperature, salinity, dissolved oxygen (DO), and pH—at the two sampling stations (P8–20 and CT-02) revealed minimal variation in temperature and salinity, ranging from 11.01°C to 11.96°C and 17.92 PSU to 18.26 PSU, respectively (Table 1). A slightly alkaline pH was recorded at both sites, with an average of 8.09 ± 0.15 across the two stations (Table 1). Meanwhile, a lower DO value (4.2 mg L−1) was recorded at the shallower P8–20 station, located under the Danube Delta influence area as compared to CT-02 (6.1 mg L−1) (Table 1). These data suggest an overall similar physicochemical environment of the bivalve habitat across both sampling sites, regardless of water depth.
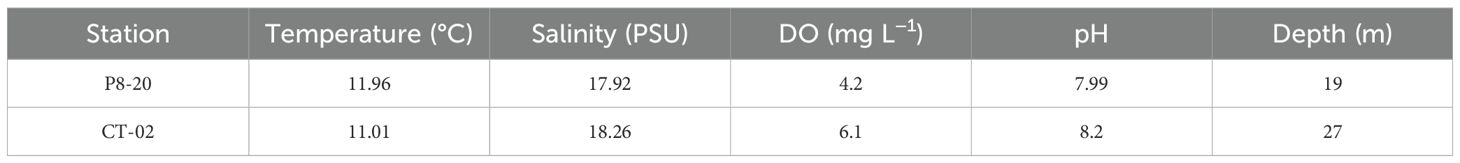
Table 1. Physicochemical parameters of seawater from the sampling sites.
3.2 Bacterial diversity colonizing the organs of M. arenaria and sediments
To assess the microbial diversity and composition of M. arenaria organs and the surrounding environment, 14 DNA samples obtained from the siphon (4), gills (4), stomach (2), and sediments (4) were analyzed, based on sequencing quality of their corresponding 16S rRNA gene amplicons (Table 2).
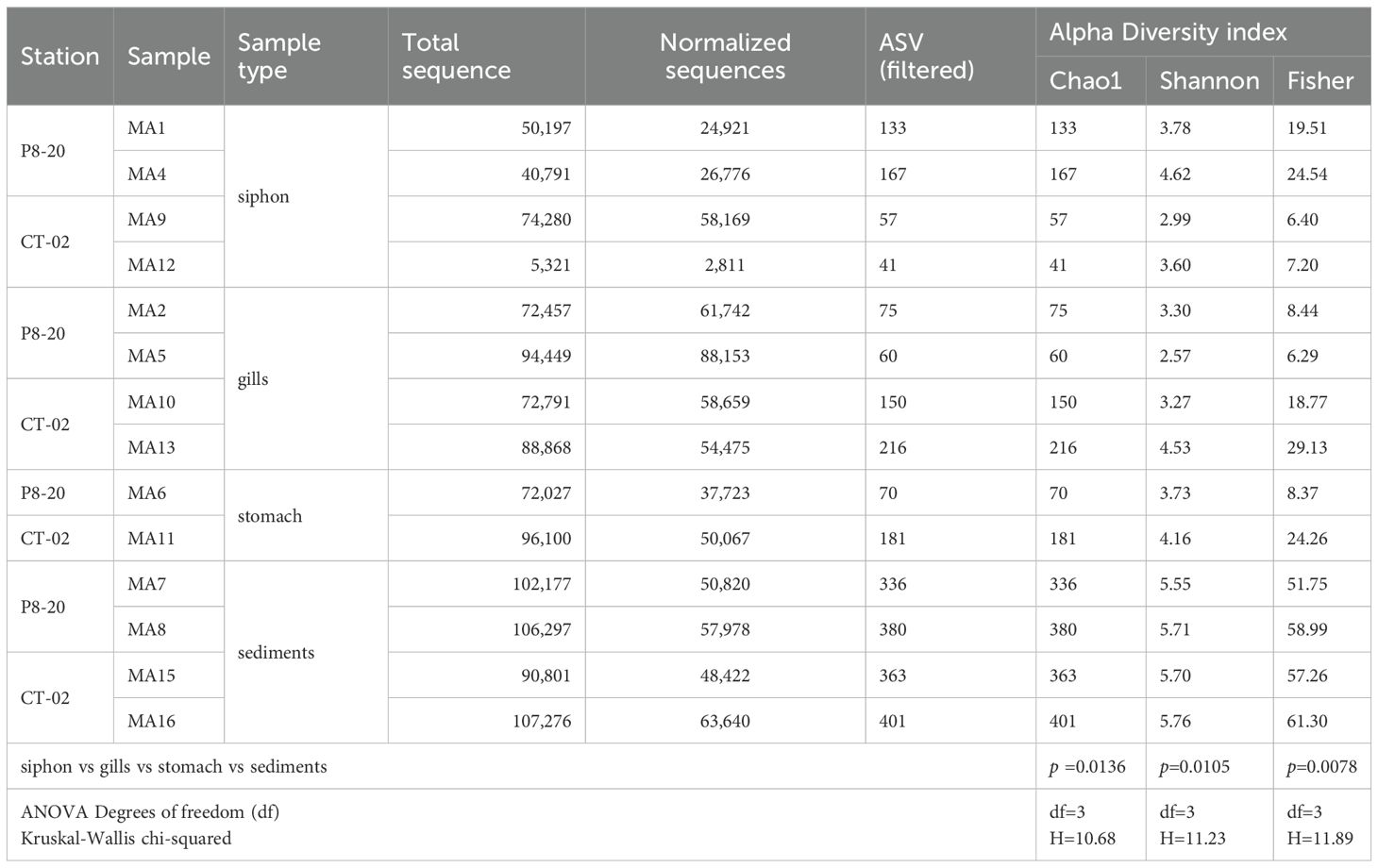
Table 2. Number of ASVs and alpha diversity indices of bacterial communities from M. arenaria organs and associated sediments, with statistical comparisons (Kruskal–Wallis test, p-values).
Illumina sequencing of the 16S rRNA gene revealed a total of 1,073,832 DNA sequences and 684,356 normalized sequences, corresponding to 2,630 unique bacterial amplicon sequence variants (ASVs), ranging from 401 (sample MA16) to 41 (sample MA12), with an average of 187.9 ASVs.
Rarefaction curves for all analyzed samples from the P8–20 and CT-02 stations indicated a fully characterized bacterial community, capturing a representative segment of the microbial diversity within bivalve organs and sediments (Supplementary Figure S1).
Alpha diversity analysis based on the Shannon index revealed significant differences between the tissue samples and sediments (p=0.0105; [ANOVA] df-value=3) (Figure 2A, Table 2). Sediment-associated bacterial communities from both locations exhibited high microbial diversity, with Shannon values ranging from 5.55 to 5.76, and a mean of 5.68 ± 0.09 (Table 2). In host tissues, the Shannon diversity index for bacterial communities ranged from 2.99 to 4.62 in the siphon (mean: 3.75 ± 0.67) and from 2.57 to 4.53 in the gills (mean: 3.41 ± 0.81), while the stomach-associated bacterial communities ranged from 3.73 to 4.53, with a mean of 3.94 ± 0.3 (Figure 2A, Table 2). Although the highest mean was observed in the stomach, followed by the siphon and then the gills, the differences were not statistically significant (Figure 2A, Table 2). The corresponding Chao1, Shannon, and Fisher diversity metrics reflected a similar trend (Chao1 p=0.0136; [ANOVA] df-value=3; Shannon p=0.0105; [ANOVA] df-value=3; Fisher p=0.0078; [ANOVA] df-value=3), indicating lower diversity in tissue-colonizing bacteria compared to surrounding sediments (Table 2; Supplementary Tables S1, S2). In contrast, no statistically significant differences were observed between these communities across depths ([t-test] statistic=−0.375, p=0.714) (Figure 2B).
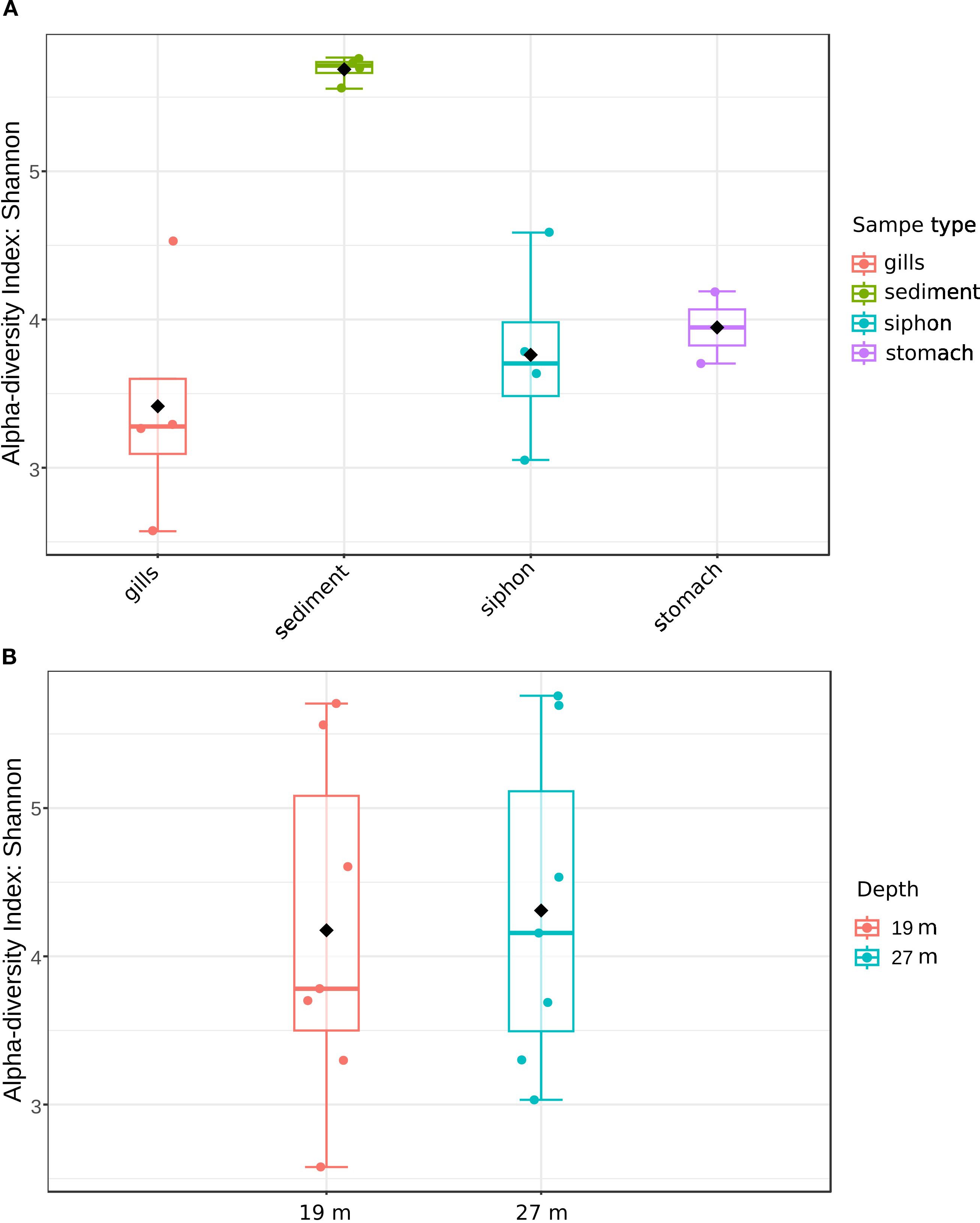
Figure 2. Alpha diversity of bacterial communities from M. arenaria organs and sediments based on Shannon index. (A) sample type (siphon, gills, stomach, sediments); (B) depth (m) of the sampling site.
The multidimensional beta diversity analysis of bacterial communities, based on sample type, revealed significant differences in microbial composition. The PERMANOVA and ANOSIM results supported this finding: PERMANOVA indicated that approximately 47% of the variation (R²=0.468) was explained by differences between tissues (gills, siphon, stomach) and sediments, underscoring the key role of habitat in structuring these communities. Additionally, ANOSIM (R=0.564) confirmed clear segregation between groups, with greater homogeneity within each sample type (e.g., sediment samples) than between sample types. The accompanying PCoA plot, in which axes 1 and 2 collectively explain approximately 62% of total variability, shows distinct clustering between tissue and sediment samples. The sediment samples appeared more homogeneous, indicating lower internal variability compared to mollusk tissues (Figure 3).
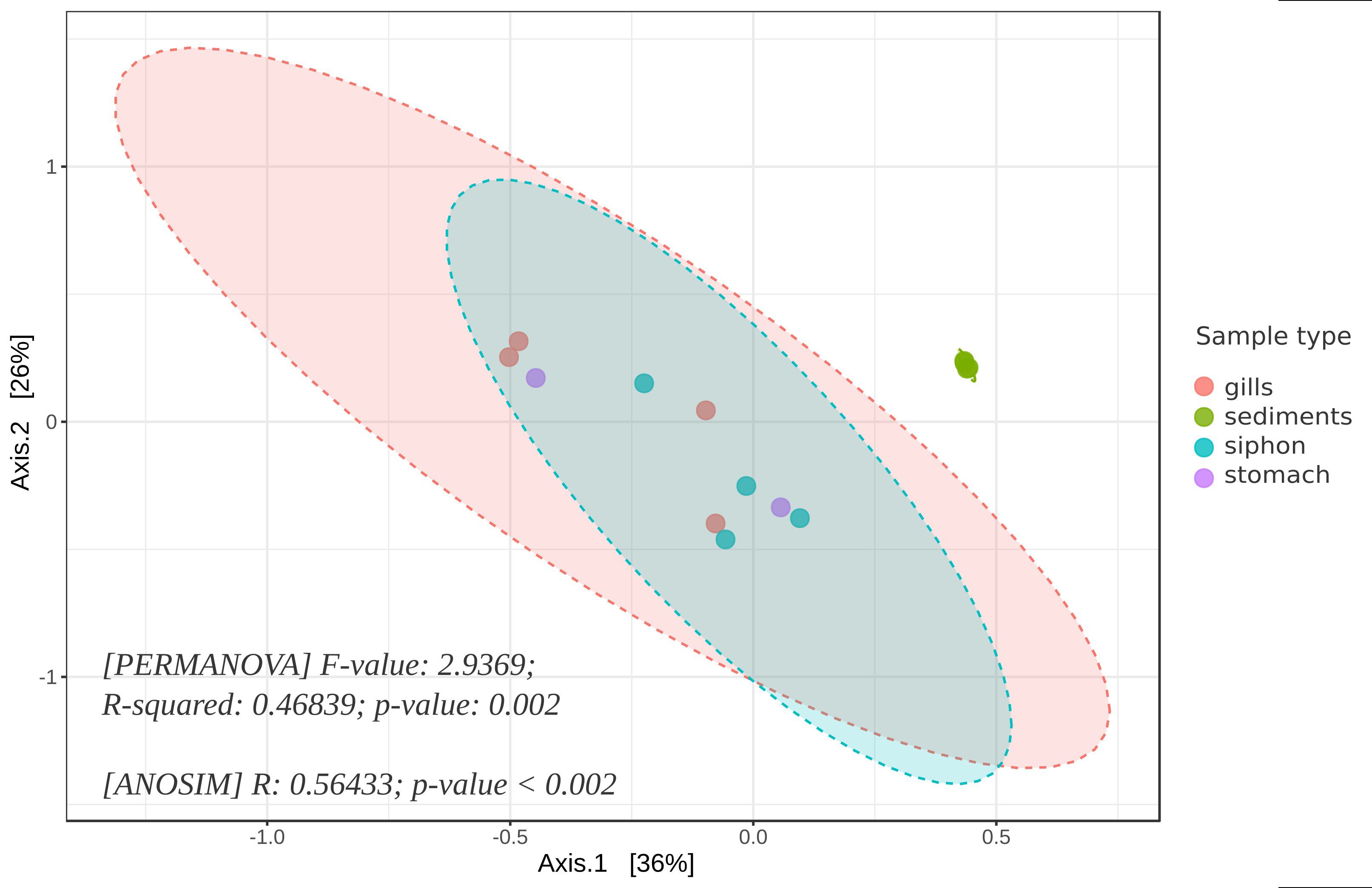
Figure 3. PCoA analysis of the beta diversity of bacterial communities from M. arenaria organs (siphon, gills, stomach) and adjacent sediments, visualized at ASV level; ellipses represent 95% confidence intervals for each sample group, and statistical significance was confirmed by PERMANOVA (F=2.94, R²=0.47, p=0.002) and ANOSIM (R=0.56, p <0.002).
3.3 Taxonomic profile of bacteria associated with M. arenaria (siphon, gills, stomach) and sediments
The bacterial taxonomic assignment of M. arenaria organs revealed a diverse community encompassing 16 phyla, 23 classes, 34 orders, 41 families, and 43 genera in the siphon. The gill microbiome comprised 15 phyla, 21 classes, 33 orders, 39 families, and 35 genera, whereas the stomach-colonizing bacteria were assigned to 14 phyla, 22 classes, 30 orders, 29 families, and 31 genera. Meanwhile, the sediment microbiota consisted of 20 phyla, 38 classes, 50 orders, 48 families, and 54 genera (Supplementary Table 1).
At the phylum level, siphon-associated communities were dominated by Verrucomicrobiota (28.2%), Spirochaetota (26.5%), Pseudomonadota I (13%), and Desulfobacterota (8.7%) (Figure 4A). The gill-associated bacteria were dominated by Verrucomicrobiota (48.4%), followed by representatives of Spirochaetota (26%), Pseudomonadota (7.9%), and Campylobacterota (7.1%). In contrast, the stomach was dominated by Spirochaetota (32%), along with Verrucomicrobiota (23.5%), Planctomycetota (12.9%), and Pseudomonadota (9.6%) (Figure 4A). Meanwhile, sediment communities showed comparable proportions of Desulfobacterota (27.9%) and Proteobacteria (27%), along with Bacteroidota (9.3%) and Chloroflexi (7.4%) (Figure 4A).
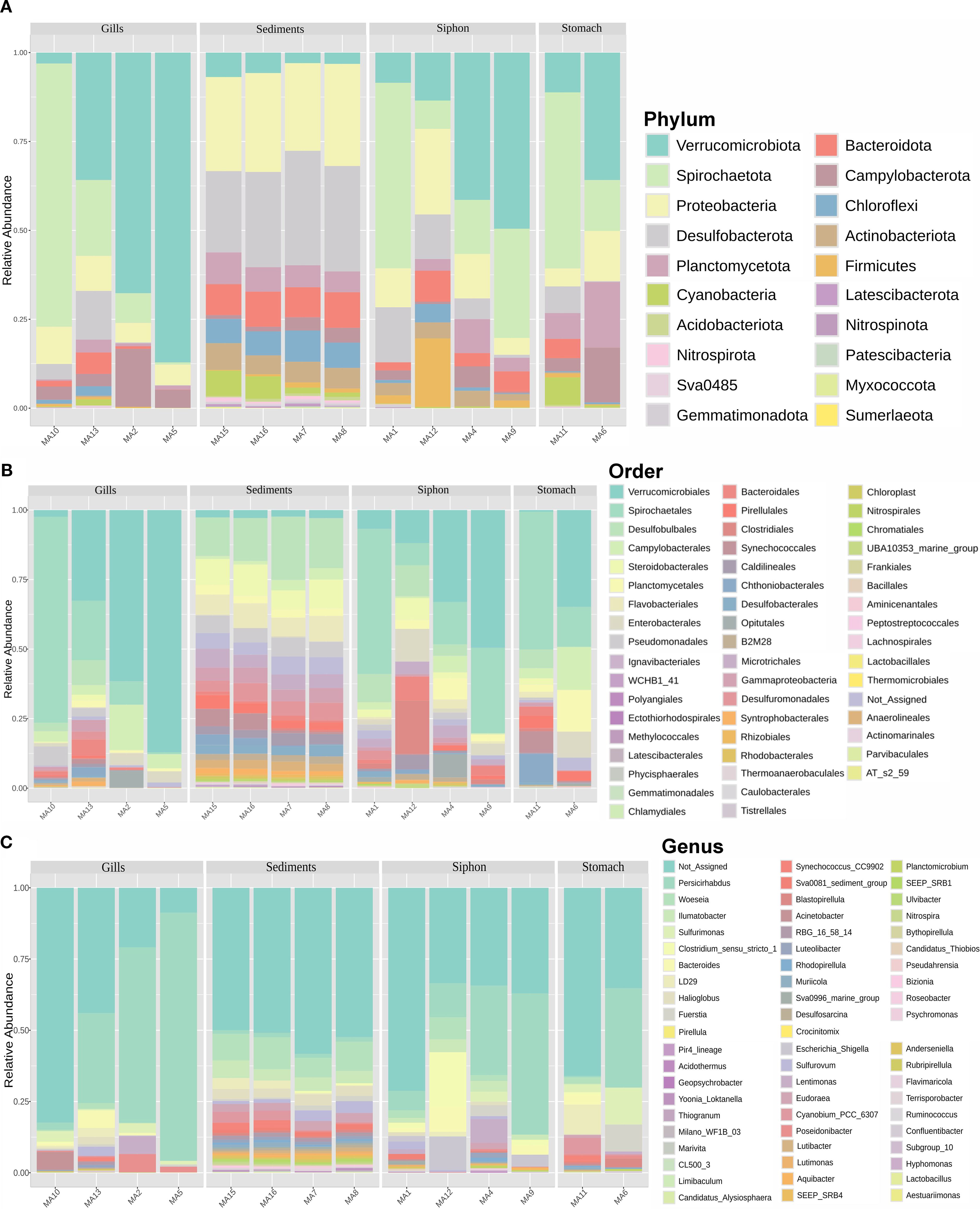
Figure 4. Relative abundance profile of bacterial taxa associated with M. arenaria organs (siphon, gills, stomach) and surrounding sediments. (A) phyla; (B) order; (C) genera.
The composition of bacterial classes (Supplementary Figure S2) differed substantially among tissues and environmental samples. While Verrucomicrobiae (28.2%) and Spirochaetia (26.5%) were the dominant taxa in the siphon, and the gills were primarily composed of Verrucomicrobiae (48.4%) and Spirochaetia (26.1%), the stomach hosted a bacterial community dominated by Spirochaetia (31.9%) and Verrucomicrobiae (23.4%) along with a notable contributions from Planctomycetes (12.8%) and Campylobacteria (9.5%). In contrast, the sediment microbiome was predominantly composed of Gammaproteobacteria (25.4%) and Desulfobulbia (17.5%), with notable contributions from Bacteroidia (9.2%) and Anaerolineae (6.9%) (Supplementary Figure S2).
At the order level (Figure 4B), bacteria colonizing the siphon were dominated by Spirochaetales (26.2%), followed by Verrucomicrobiales (24.7%), Desulfobulbales (6.2%), Clostridiales (5.6%), Enterobacterales (4.7%), and Planctomycetales (3.3%). The gills and stomach samples were mainly composed of Verrucomicrobiales, accounting for an average of 45.9%, and 17.8%, respectively. Campylobacterales were also highly represented in these two organs, with average relative abundances of 7.2%, and 9.5%, respectively. Moreover, notable abundances of Spirochaetales (27.4%) were observed in the gills, with lower contributions from Enterobacterales (2.4%) and Bacteroidales (1.9%). Meanwhile, Planctomycetales (8.8%), Chthoniobacterales (5.2%), Enterobacterales (4.7%), and Synechococcales (3.8%) were better represented in the stomach (Figure 4B). For the sediment samples, Desulfobulbales dominated (16.9%), followed by representatives of Steroidobacterales (9.6%), Flavobacteriales (8.0%), and Pseudomonadales (5.2%) (Figure 4B).
The taxa belonging to the families Spirochaetaceae and Rubritaleaceae families were identified at high abundances in the three analyzed organs (Supplementary Figure S3). Thus, in the siphon samples, they accounted for an average of 26.2% and 24.7%, respectively; in the gills, 45.9% and 27.4%, and in the stomach, 32.5% and 17.7%. Desulfobulbaceae (4.2%) and Enterobacteriaceae (4.3%) were better represented in the siphon, Arcobacteraceae (4.2%) in gills, while Sulfurimonadaceae (7.8%) and Chthoniobacteraceae (5.2%) exhibited higher relative abundances in the stomach. Meanwhile, bacterial species of Desulfobulbaceae (12.1%) dominated the sediment samples, followed by Woseiaceae (9.6% average) and Flavobacteriaceae (8.0%) (Supplementary Figure S3).
Clear differences were observed in the abundance of bacterial genera across the bivalve organs and associated sediments (Figure 4C). In the siphon, the community was dominated by unassigned sequences (44.1%), followed by the genus Persicirhabdus (24.9%), with minor contributions from Clostridium_sensu_stricto_1 (5.7%) and Escherichia/Shigella (4.5%). The gills showed a high abundance of Persicirhabdus (45.7%) and unassigned sequences (39.0%), while Sulfurimonas (2.2%) and Poseidonibacter (2.1%) emerged as the next most relevant genera. In the stomach, unassigned sequences were predominant (50.6%), followed by Persicirhabdus (17.7%) and Sulfurimonas (7.8%), along with LD29 (5.2%), a poorly characterized genus. Finally, in sediments, unassigned sequences dominated (52.9%), followed by Woeseia (9.4%), Ilumatobacter (4.8%), and Halioglobus (3.5%), genera associated with marine environments and sulfur cycling (Figure 4C).
Heatmap analysis of the distribution of bacterial phyla in clam organs and sediments highlighted similarities and differences among bacterial communities according to the sample type and water depth (Figure 5). The analysis revealed that bacteria colonizing the organs of M. arenaria formed a distinct cluster compared to those associated with sediments (Figure 5). Moreover, no association pattern among these communities with respect to water depth was observed (Figure 5).
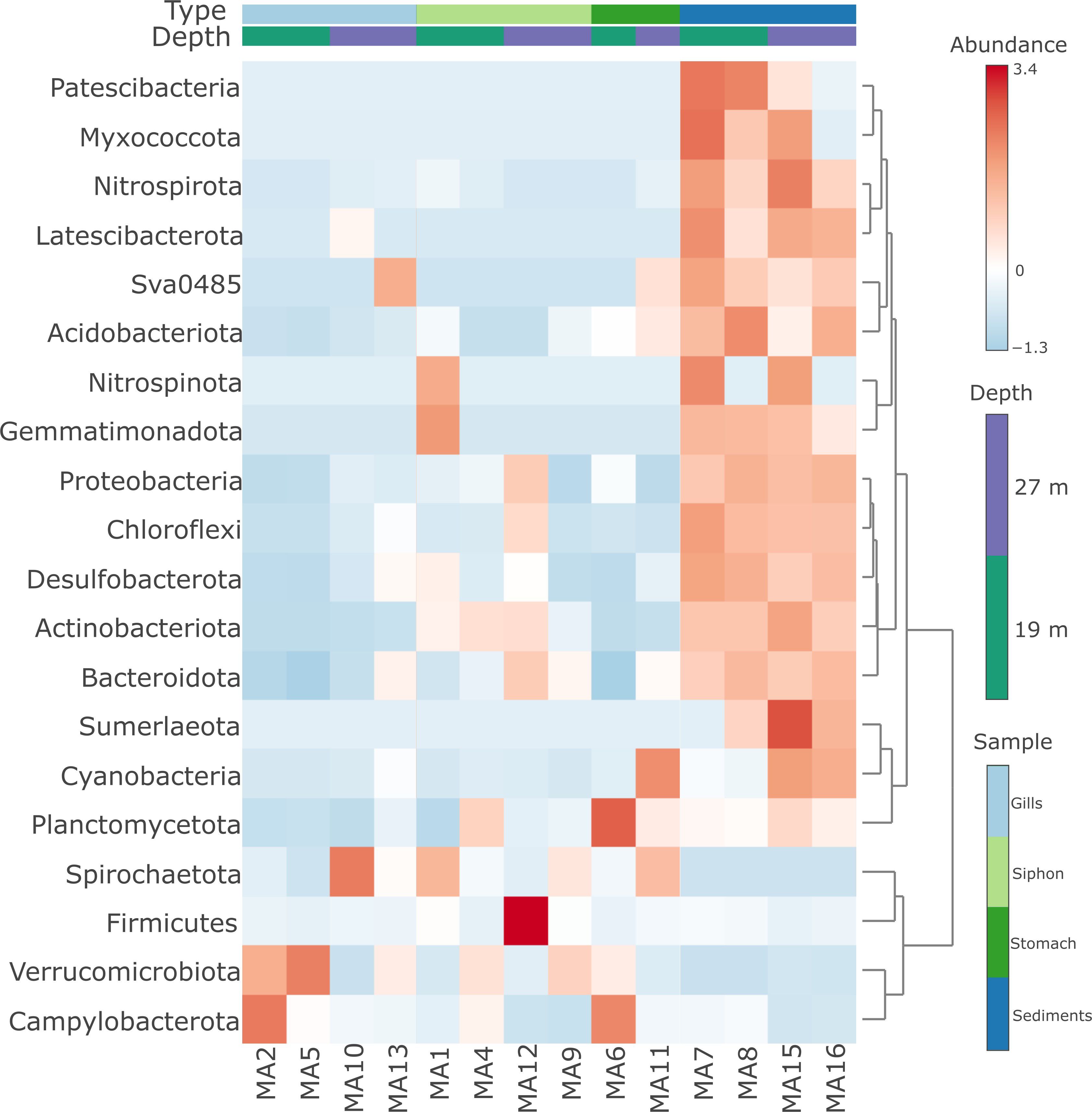
Figure 5. Heatmap distribution of bacterial phyla from M. arenaria organs (siphon, gills, stomach) and sediments, in relation to water depth.
Pattern search analysis of bacteria phyla from clam organs and sediments (Figure 6) revealed a positive correlation between Verrucomicrobiota and gills, as well as between sediment samples and Latescibacterota, Sva0485, Chloroflexi, Myxococcota, Nitrospirota, Patescibacteria, and Desulfobacterota, which were either absent or present at very low abundances in the analyzed organs. Campylobacterota was also positively correlated with the stomach samples (Figure 6). Conversely, a negative correlation was observed between phyla Planctomycetota and Firmicutes and the gill samples, as well as between Cyanobacteria and the siphon samples (Figure 6).
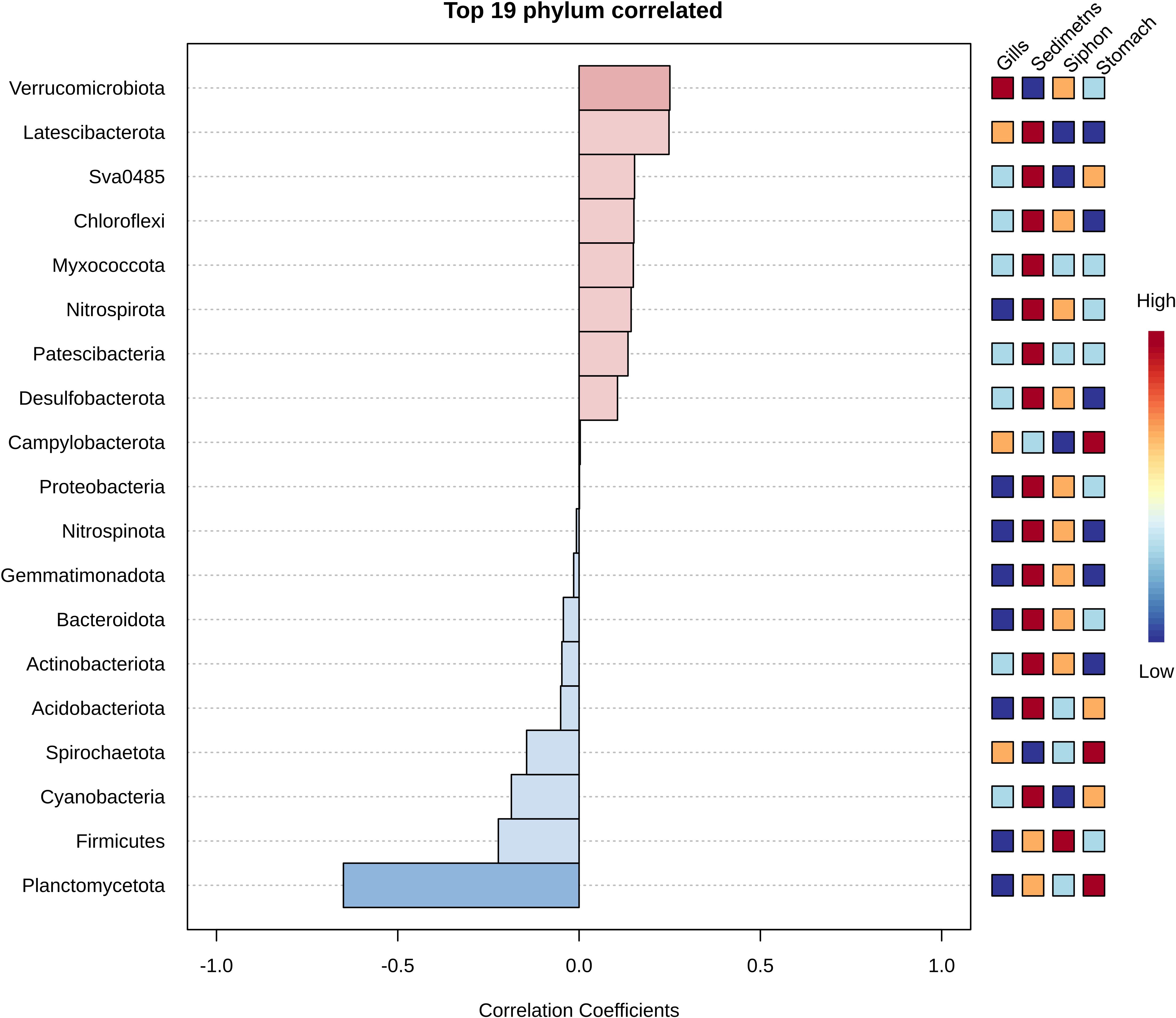
Figure 6. Phylum-level correlation analysis between bacterial taxa and sample types in M. arenaria, showing the strength and direction of associations for the top 19 most correlated phyla; positive correlations (red) indicate enrichment in specific organs or sediments; negative correlations (blue) indicate depletion, as determined by Spearman’s rank correlation.
The LDA Effect Size (LefSe) analysis of microbial taxa with significant differential abundance among the sample types (Supplementary Figure S4) identified Spirochaetota, Planctomycetota, and Cyanobacteria as significant biomarkers differentiating the analyzed sample types (LDA score >3), highlighting their dominance in the stomach. Moreover, the prevalence of other phyla including Desulfobacterota, Pseudomonadota, Bacteroidota, Chloroflexi, and Actinobacteriota (LDA score >3), was observed in the sediment samples (Supplementary Figure S4).
3.4 Predicted functional role of the microbiome of M. arenaria from different organs and associated sediments
The 16S rRNA gene sequencing data was used to estimate the functional profile of the corresponding bacteria based on sequence similarities to organisms with fully sequenced and annotated genomes, using Tax4Fun with SILVA annotated features for functional profiling.
The predicted functional profile analysis showed a comparable distribution of the relative abundance of various genes based on Kyoto Encyclopedia of Genes and Genomes (KEGG) metabolic pathways (Figure 7). In this respect, genes related to carbohydrate, amino acid, and energy metabolism were prevalent across all samples, followed by those involved in the metabolism of cofactors, vitamins, and nucleotides, which showed comparable values between organs and sediments (Figure 7). Although not among the dominant pathways, several associated with xenobiotic biodegradation and antibiotic biosynthesis were identified (Figure 7). Thus, the analysis revealed a slight increase in the expression of genes associated with most of the identified degradation pathways within organs (e.g., degradation of toluene, ethylbenzene, xylene, chlorocyclohexane, and chlorobenzene), suggesting differential metabolic activity between the two types of matrices analyzed (Figure 7). With regard to antibiotic synthesis, no major differences in gene abundances were observed between organs and sediments, with the dominant functions being related to streptomycin and monobactam biosynthesis, which were slightly more prevalent in sediment-colonizing bacteria. Meanwhile, the analyzed organs showed high abundances of genes involved in penicillin and cephalosporin biosynthesis (Figure 7).
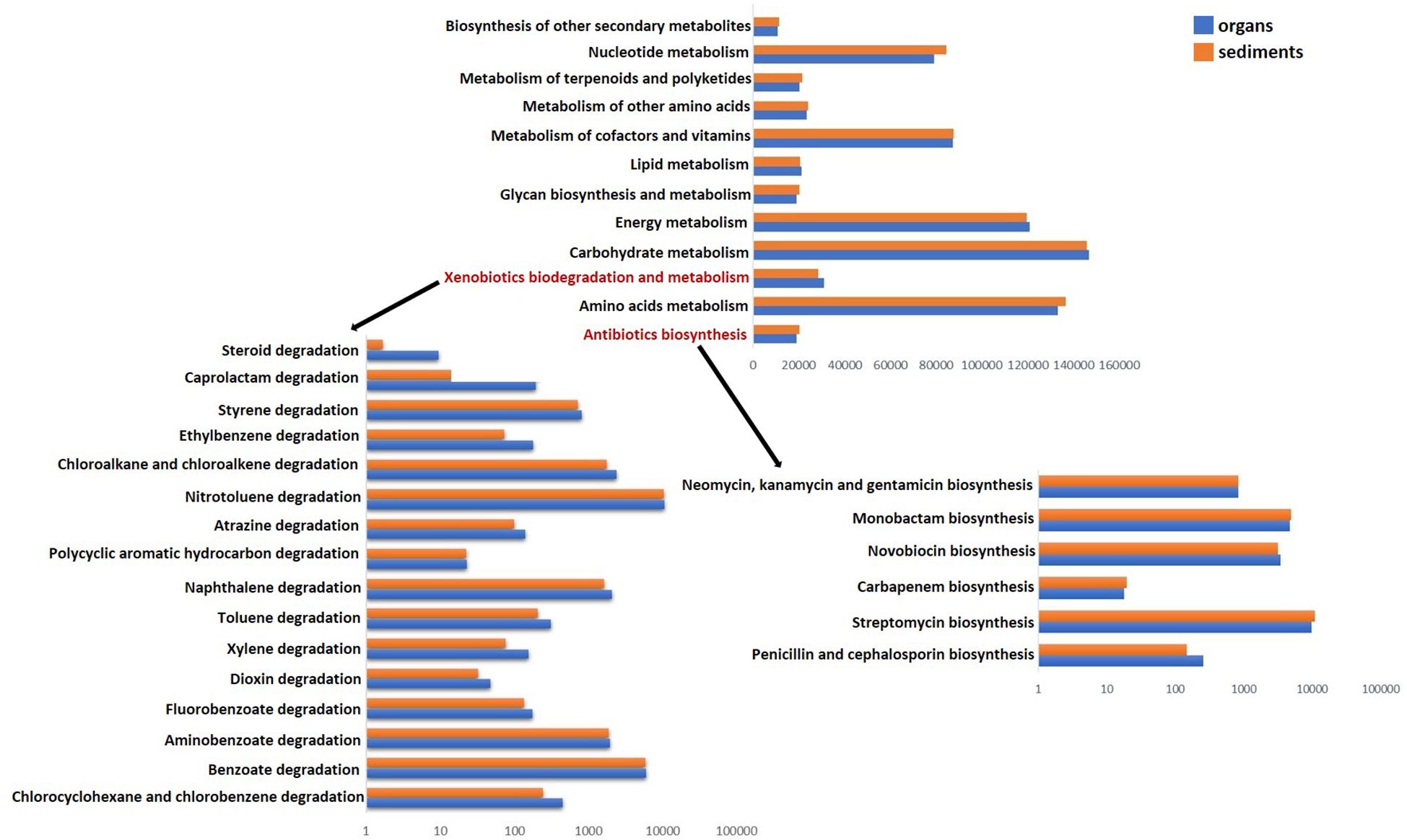
Figure 7. Predicted functional profile of bacterial communities colonizing the organs (blue) and sediments (red) based on KEGG metabolic pathways. Detailed gene profile xenobiotic biodegradation and metabolism (left) and antibiotic synthesis (right). Number of predicted functional genes corresponds to ASVs identified in the analyzed matrices. Functional prediction was made by linking ASV taxonomy to KEGG gene orthologs, reflecting the potential metabolic capabilities of the microorganism.
Although the beta diversity analysis (Figure 3) revealed significant differences between clam tissues and sediment microbiota, their predicted functions based on 16S rRNA gene sequences suggested only limited variation between the two matrices. In this regard, further investigations using metagenomic and metatranscriptomic analyses are required to gain deeper insight into the functional variability of these communities (Aguiar-Pulido et al., 2016), which may provide a more detailed understanding of their metabolic profiles.
4 Discussion
This study aimed to characterize the bacterial communities of the bivalve M. arenaria collected from the Romanian part of the Black Sea, and is the first to examine the microbiome of different organs (siphon, gills, and stomach) of this marine invertebrate, as well as the associated sediments.
Previous research on M. arenaria-associated bacteria has primarily focused on microbial contamination, including studies on the kinetics of coliform bacteria uptake and removal by this bivalve (Cabelli and Heffernan, 1970), and, more recently, reports of significant contamination with pathogens such as Arcobacter, Cryptosporidium parvum, Giardia, and Salmonella in clams collected from the north shore of the St. Lawrence River (Quebec, Canada) (Lévesque et al., 2006). The present study applied culture-independent Illumina sequencing of the 16S rRNA gene to characterize the taxonomic and functional diversity of bacterial communities colonizing the siphon, gills, and stomach of M. arenaria, as well as associated sediments from two locations in the Black Sea. This represents a more comprehensive and targeted molecular characterization compared to a previous NGS study of M. arenaria microbiome, which only characterized the bacterial diversity of the whole organisms collected from two locations on the east coast of Canada (Liu et al., 2020). Moreover, this study investigated the hypothesis that M. arenaria hosts a microbiome that may help it cope with stressful estuarine conditions and organic pollution, including bacterial contaminants. This molecular approach also enabled the identification of bacterial genes potentially involved in diverse metabolic functions, including antibiotic resistance, providing insight into functional traits that allow the clam to cope with environmental stressors.
Our data showed that the bivalve microbiota was well differentiated from that of its physical habitat, and exhibited compositional variations depending on the organ, with greater taxonomic diversity (19 phyla) in the sediments as compared to the bivalve organs (14–15 phyla), independent of water depth. The phylum Verrucomicrobiota dominated the siphon and gill samples, Spirochaetota was the most abundant in the stomach, and the sediment communities were mainly composed of Desulfobacterota.
No significant differences in bacterial diversity were observed between samples from different depths, likely reflecting similar environmental conditions across sites. All samples were collected from circalittoral sandy mud habitats, which are characterized by benthic communities dominated by detritivores and suspension feeders such as oligochaetes, polychaetes, mollusks, and bacterivorous nematodes (Nenciu et al., 2023; Mureşan et al., 2019; Teacă et al., 2019, 2020). These results align with other studies showing that microbial communities in similar habitats are comparable, even across different geographical regions (Lauber et al., 2009).
Recent investigations in the Black Sea revealed the dominance of Actinobacteriota and Chloroflexi in sediments from areas with physico-chemical water characteristics and substrate composition comparable to those observed in our research (Menabit et al., 2024). While these bacteria were also detected, the most abundant taxa colonizing the sediments in this study were assigned to Pseudomonadota and Desulfobacterota. Shifts in microbial composition might be attributed to the distinct habitat types (Teacă et al., 2020), as such changes in the structure of the zoobenthic community could influence microbial populations. While prior research (Reveillaud et al., 2014) has demonstrated that microbial communities often reflect host and environmental differences across large spatial scales, our study found relatively minor differences in bacterial composition between the two sampling sites, suggesting limited spatial variation in this context.
Bivalves, like many other organisms, host microbial communities that are specific to various organs and tissues. Previous data have reported the digestive system as containing the highest concentrations of bacteria, particularly the stomach, gastric juice, crystalline style and digestive diverticula (Kueh and Chan, 1985). The results obtained in this survey indicated a specific microbiota in this bivalve, distinct from its physical habitat, with compositional variations depending on the organ. Thus, representatives of the order Spirochaetales showed the highest abundances in siphon and gill samples, with the Verrucomicrobiales group being better represented in gills. Taxa belonging to Spirochaetales have also been identified in the digestive tract of invertebrates such as arthropods (Berlanga et al., 2007) and mollusks (Romero and Espejo, 2001), as well as in bivalve gills (Duperron et al., 2007). A recent study in New Zealand showed these bacteria to be abundant in the siphon and digestive glands of the bivalve Paphies australis (Biessy et al., 2020), consistent with our data, which revealed a high representation of Spirochaetales taxa in the siphon samples. Bacteria classified in the order Verrucomicrobiales have been identified in various environments such as the water column (Yoon et al., 2007), sediments (Yoon et al., 2008), in association with marine invertebrates, such as the polychaete Periserrula leucophryna (Yang et al., 2007) and different organs (gills, stomach, foot) of the bivalve M. galloprovincialis (Musella et al., 2020). The results also indicated a statistical correlation between the phylum Planctomycetales and stomach samples, consistent with data on the stomach microbiome of C. gigas, which showed a high abundance of taxa belonging to this group (Offret et al., 2020).
The present data revealed distinct, organ-specific distributions of bacterial taxa within M. arenaria. Members of the order Bacteroidales were predominantly associated with the siphon, while bacteria from Campylobacterales and Synechococcales exhibited higher relative abundances in the stomach samples. The enrichment of Bacteroidales is consistent with the high representation of this phylum in other marine invertebrates, including the crab Callinectes sapidus (Givens et al., 2013), the gastropod Haliotis diversicolor (Zhao et al., 2018), and the bivalve M. galloprovincialis (Musella et al., 2020). Similarly, Campylobacterales that prevailed in the stomach of M. arenaria have been reported in the digestive tracts of various marine organisms, such as the sea urchin Lytechinus variegatus (Hakim et al., 2015) and the ascidian Ciona intestinalis (Dishaw et al., 2014), suggesting a possible functional role in digestion. The presence of Synechococcales in stomach tissues may be linked to the dietary habits of M. arenaria, as these cyanobacteria are known to constitute a significant nutritional component for other marine taxa, including sipunculid worms (Li et al., 2023a; Liu et al., 2023) and bivalves (Qiao et al., 2022).
Despite variations in composition, our research showed that the Verrucomicrobiota and Spirochaetota phyla exhibited high abundances in all analyzed organs. In addition, a notable proportion of sequences belonging to Planctomycetota was recovered from the stomach samples. A study conducted at two locations on the East Coast of Canada demonstrated that soft-shell clams from Nova Scotia had a higher abundances of Pseudomonadota and Acidobacteria, but a lower abundance of Actinobacteria than those from Quebec. Furthermore, samples from Nova Scotia exhibited greater microbial diversity than those from Quebec (Liu et al., 2020). These findings suggest that environmental factors may shape the microbial communities of this bivalve, influencing both composition and diversity.
The genus Persicirhabdus encompasses nitrate-reducing bacteria (Yoon et al., 2008), which, under certain conditions, such as low oxygen levels, can reduce nitrates to nitric oxide (Strahl and Abele, 2020). Studies have shown that the clam Arctica islandica may harbor nitrate-reducing bacteria, and that nitric oxide could play a crucial role in reducing the metabolic activity of A. islandica during self-burial and shell closure. Under these conditions, nitric oxide contributes to the initiation of metabolic depression, which may help conserve energy during hypoxia or anoxia and protect against oxidative stress (Strahl and Abele, 2020). Accordingly, the high abundance of these bacteria in the analyzed organs of M. arenaria could support the survival and adaptation of this bivalve in the low-oxygen conditions of the investigated area.
The genus Sulfurimonas is characterized as sulfur-oxidizing bacteria, capable of growing chemoautotrophically with reduced sulfur compounds, such as sulfide, thiosulfate and elemental sulfur (Sievert et al., 2008), and has been isolated from various environments, including coastal marine sediments (Hoor, 1975; Cai et al., 2014; Wang et al., 2020), polychaete nests (Inagaki et al., 2003; Takai et al., 2006), and deep-sea hydrothermal vents (Hu et al., 2021). While sulfur-oxidizing bacteria are typically found in the gills of marine bivalves (Dando et al., 1985; Caro et al., 2007), recent investigations have revealed a persistent population of these microbes in the digestive tract of adult hosts lucinid clam Loripes orbiculatus, which contribute to the host’s nutrition by fixing carbon from sulfide oxidation (Alcaraz et al., 2024). These findings are consistent with our investigations, where we detected higher abundances of Sulfurimonas colonizing the stomach samples, further suggesting a role of these bacteria in digestion or other processes that contribute to the host’s nutrition.
Bacteroides, identified in all analyzed organs of M. arenaria, was previously documented in the digestive tract of the mussel M. edulis and the oyster C. virginica, and plays an important role in digestion, including the breakdown of complex carbohydrates and the production of essential amino acids (Pierce and Ward, 2019).
Poseidonibacter is considered a synonym or subgroup within the genus Arcobacter (On et al., 2021), members of which are known pathogens linked to human and animal diseases, previously detected in various water bodies, including wastewater, seawater, and freshwater systems, often associated with fecal contamination (Ghaju Shrestha et al., 2022), as well as colonizing soft-shell clams collected from the north shore of the St. Lawrence River, and other bivalves such as M. galloprovincialis (Ottaviani et al., 2016). In our study, we observed high abundances of these bacteria in the gills of M. arenaria specimens collected from the station located in the Danube influence area, which was not surprising given that the river is permanently subject to anthropogenic pressures such as industrial activities, intensive farming, and agricultural runoff (Gasparotti, 2014). Moreover, the Escherichia/Shigella genera were identified in all analyzed organs. Escherichia includes both commensal and pathogenic species (Braz et al., 2020), whereas Shigella is highly infectious and responsible for significant global morbidity and mortality (Schroeder and Hilbi, 2008). Bivalves, as filter-feeding organisms, concentrate bacteria from the environment and serve as suitable bioindicators of water contamination. Consequently, the presence of these genera in bivalve tissues may pose serious public health risks through seafood consumption and highlight underlying threats to ecosystem health, including microbial pollution and reduced water quality.
Unlike the microorganisms associated with M. arenaria, those colonizing the sediments were mainly represented by taxa belonging to the orders Desulfobulbales, Steroidobacterales, and Flavobacteriales orders. The high abundance of the Desulfobulbales group, mainly composed of sulfur-reducing bacteria thriving in anoxic environments (Ward et al., 2021), could be correlated with the low dissolved oxygen concentrations characterizing the Romanian part of the Black Sea. These particularities could also explain the presence of taxa belonging to Steroidobacterales in sediments, which are known for their ability to oxidize sulfur (Mußmann et al., 2017). Previous studies have demonstrated that Chloroflexi species account for 25.5% to 41.3% of total 16S rRNA sequences in marine sediments globally (Hoshino et al., 2020). Some representatives of this phylum are also known for decomposing organic matter (Landry et al., 2017). In this study, the prevalence of this group in sediment samples could be associated with the marine habitat, characterized by high organic matter content, from which the samples were collected (Bucşe et al., 2020; Teacă et al., 2020). Moreover, the genus Woeseia was consistently detected across all sediment samples. This group comprises halotolerant bacteria capable of sulfur oxidation and denitrification (Du et al., 2016; Mußmann et al., 2017) and has previously been isolated from both coastal and deep-sea surface sediments (Mußmann et al., 2017; Hoffmann et al., 2020).
A preliminary assessment of the functional profile of the microbiome of the bivalve M. arenaria and adjacent sediments was performed by determining the functional diversity of the bacterial communities based on16S rRNA gene sequencing data, using the Tax4Fun platform (Langille et al., 2013). While this tool cannot replace the assessment of gene functionality through metagenomic analysis, studies have shown that it can provide insight into the functions of microorganisms in diverse habitats such as biofilms (Koo et al., 2017), the water column (Bowman and Ducklow, 2015), and sediments (Su et al., 2018). However, the effectiveness of this method depends on the genomic information available in databases, which in some cases does not reflect the functional diversity of the microorganisms in the investigated ecosystem (Wemheuer et al., 2020).
The data obtained in this study on the functional diversity of the bacterial communities associated with the bivalve M. arenaria and surrounding sediments are consistent with previous investigations emphasizing the role of marine bacteria in carbohydrate metabolism (Gavriilidou et al., 2020). Thus, studies have shown that bacteria associated with some bivalves produce enzymes that break down complex carbohydrates, and contribute to the immune response of these organisms (Dubé et al., 2019). In addition, representatives of the orders Verrucomicrobiales and Bacteroidales orders, mainly identified in M. arenaria organs, are involved in the hydrolysis of various polysaccharides (Martinez-Garcia et al., 2012). Likewise, Dunkai et al. (2023) demonstrated that bacterial strains isolated from the digestive system of the bivalve Crenomytilus grayanus can degrade various nutrient substrates (sugars, amino acids, and polysaccharides) as well as xenobiotic substances (petroleum hydrocarbons, bisphenol A, and atrazine). Therefore, the presence of genes involved in xenobiotic metabolism in the microbiota of M. arenaria could play an important role in maintaining the health and adaptability of this species in diverse environments, including polluted areas. Several studies have highlighted the presence and potential of microbial communities colonizing marine sediments to degrade aromatic hydrocarbons, including toluene, ethylbenzene, xylene, chlorocyclohexane, and chlorobenzene (Moore et al., 2020; Suarez-Moo et al., 2020; Nikolova et al., 2021). Similar degradation pathways which have also been identified in the current study, highlighting the potential of these bacteria as valuable resources for bioremediation and the pharmaceutical industry. Moreover, research undertaken by Šyvokienė and Butrimavičienė (2013) demonstrated that bacteria associated with the digestive systems of swollen river mussels (Unio tumidus) and zebra mussels (Dreissena polymorpha) from the Curonian Lagoon (Winters et al., 2011) are involved in the biodegradation of petroleum hydrocarbons. Mollusks, including bivalves, are emerging as a promising source of novel marine bioactive compounds, with certain species containing substances that could have diverse applications across industries (Sousa and Hinzmann, 2020). The current investigation demonstrates that bacteria associated with both the organs of M. arenaria and the surrounding sediments harbor genes involved in antibiotic synthesis. This finding aligns with other research, such as studies from the Red Sea, where the detection of biosynthetic gene clusters polyketide synthase (PKS) and non-ribosomal peptide synthase (NRPS) in marine invertebrate-associated bacteria indicates their potential to produce bioactive compounds, including antibiotics (El Samak et al., 2018).
5 Conclusion
This study revealed significant differences in bacterial community diversity between the organs of the bivalve M. arenaria and the adjacent sediments, with greater diversity in the sediments. The bivalve harbors a distinct microbiota that varies by organ and differs clearly from that of its physical habitat. Notably, the phylum Verrucomicrobiota predominated in the siphon and gills, while Spirochaetota were more abundant in the stomach. In contrast, Desulfobacterota dominated the sediment-associated microbial communities. Persicirhabdus bacteria were consistently abundant across all examined organs and may contribute to the clam’s metabolic depression and adaptation to low-oxygen conditions. Additionally, sulfur-oxidizing Sulfurimonas species in the stomach likely contribute to the host’s nutrition. Sediments, in contrast, were primarily colonized by Woeseia species, which are key players in the degradation of organic matter. Functional diversity analyses suggested that bacteria inhabiting both bivalve tissues and adjacent sediments participate in diverse metabolic processes, with a particular emphasis on carbohydrate, amino acid, and energy metabolism. Several pathways were associated with xenobiotic degradation and antibiotic biosynthesis, highlighting the biotechnological potential of these microbial communities for applications such as bioremediation and pharmaceutical development.
This survey, providing the first organ-specific characterization of the microbiome associated with M. arenaria revealed clear structural differences among the siphon, gills, and stomach, as well as a distinct separation from the surrounding sediment microbial communities. The observed compositional divergence highlighted the presence of a specialized and compartmentalized microbiome within the bivalve, likely shaped by organ-specific functions and external environmental exposures. Notably, the internal microbial communities do not mirror the environmental microbiota, suggesting a non-random, potentially symbiotic assemblage with functional relevance to host biology. These results offer a foundation for future investigations into the ecological and physiological roles of the bivalve microbiome, particularly in the context of environmental stress and host adaptation. Further studies employing metagenomic and metatranscriptomic approaches will be essential to elucidate the mechanistic interactions between M. arenaria, its microbiota, and the surrounding habitat. Nevertheless, the small number of specimens analyzed could potentially bias interpretations of the structural and functional diversity within the bivalve microbiome.
Data availability statement
All data supporting the findings of this study are provided within the manuscript. The 16S rRNA gene sequences from this study were deposited in the NCBI Sequence Read Archive (SRA) under BioProject accession number PRJNA922888 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA922888/).
Ethics statement
The manuscript presents research on animals that do not require ethical approval for their study.
Author contributions
SM: Investigation, Methodology, Validation, Writing – original draft, Writing – review & editing. PL: Writing – review & editing, Writing – original draft, Software, Methodology. TB: Writing – review & editing, Resources. AT: Writing – review & editing, Software, Resources. MM: Writing – review & editing, Resources. CP: Writing – review & editing, Writing – original draft, Validation, Supervision, Formal Analysis, Conceptualization.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. Financial support was provided by the National Core Programme PN 23 30 02–02 of the Romanian Ministry of Education and Research, Romanian Academy project RO1567−IBB05/2024, and HORIZON EUROPE—MARBEFES/101060937—MARine Biodiversity and Ecosystem Functioning leading to Ecosystem Services.
Acknowledgments
The authors thank Lavinia Iancu for technical support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2025.1659674/full#supplementary-material
References
Aguiar-Pulido V., Huang W., Suarez-Ulloa V., Cickovski T., Mathee K., and Narasimhan G. (2016). Metagenomics, metatranscriptomics, and metabolomics approaches for microbiome analysis. Evol. Bioinform. Online 12, 5–16. doi: 10.4137/EBO.S36436, PMID: 27199545
Akter S., Wos-Oxley M. L., Catalano S. R., Hassan M. M., Li X., Qin J. G., et al. (2023). Host species and environment shape the gut microbiota of cohabiting marine bivalves. Microb. Ecol. 86, 1755–1772. doi: 10.1007/s00248-023-02192-z, PMID: 36811710
Alcaraz C. M., Séneca J., Kunert M., Pree C., Sudo M., and Petersen J. M. (2024). Sulfur-oxidizing symbionts colonize the digestive tract of their lucinid hosts. ISME J. 18, wrae200. doi: 10.1093/ismejo/wrae200, PMID: 39388223
Balbi T., Auguste M., Ciacci C., and Canesi L. (2021). Immunological responses of marine bivalves to contaminant exposure: contribution of the-omics approach. Front. Immunol. 12. doi: 10.3389/fimmu.2021.618726, PMID: 33679759
Berlanga M., Paster B. J., and Guerrero R. (2007). Coevolution of symbiotic spirochete diversity in lower termites. Int. Microbiol. 10, 133–139., PMID: 17661292
Beshevli L. E. and Kolyagin V. A. (1967). A find of mollusk mya arenaria L. (Bivalvia) in northwestern blackSea. Vestn. Zool 3, 82–84.
Biessy L., Pearman J. K., Smith K. F., Hawes I., and Wood S. A. (2020). Seasonal and spatial variations in bacterial communities from tetrodotoxin-bearing and non-tetrodotoxin-bearing clams. Front. Microbiol. 5. doi: 10.3389/fmicb.2020.01860, PMID: 32849450
Bourne D. G., Morrow K. M., and Webster N. S. (2016). Insights into the coral microbiome: underpinning the health and resilience of reef ecosystems. Annu. Rev. Microbiol. 70, 317–340. doi: 10.1146/annurev-micro-102215-095440, PMID: 27482741
Bowman J. S. and Ducklow H. W. (2015). Microbial communities can be described by metabolic structure: a general framework and application to a seasonally variable, depth-stratified microbial community from the coastal West Antarctic peninsula. PloS One 10, e0135868. doi: 10.1371/journal.pone.0135868, PMID: 26285202
Braz V. S., Melchior K., and Moreira C. G. (2020). Escherichia coli as a multifaceted pathogenic and versatile bacterium. Front. Cell. Infect. Microbiol. 10. doi: 10.3389/fcimb.2020.548492, PMID: 33409157
Bryukhanov A. L., Korneeva V. A., and Pimenov N. V. (2015). Detection of anaerobic sulfate reducing bacteria in oxygen containing upper water layers of the Black and Baltic seas. Moscow Univ. Biol. Sci. Bull. 70, 184–188. doi: 10.3103/S0096392515040057
Bucşe A., Vasiliu D., Balan S., Parvulescu O. C., and Dobre T. (2020). Heavy metal spatial distribution and pollution assessment in the surface sediments of the north – western black sea shelf. Rev. Chim. 71, 155–170. doi: 10.37358/Rev.Chim.1949
Cabelli V. J. and Heffernan W. P. (1970). Elimination of bacteria by the soft-shell clam, mya arenaria. J. Fisheries Res. Board Canada 27, 1579–1587. doi: 10.1139/f70-179
Cai L., Shao M.-F., and Zhang T. (2014). Non- contiguous finished genome sequence and description of Sulfurimonas hongkongensis sp. nov., a strictly anaerobic denitrifying, hydrogen- and sulfur-oxidizing chemolithoautotroph isolated from marine sediment. Stand Genomic Sci. 9, 1302–1310. doi: 10.4056/sigs.4948668, PMID: 25197498
Callahan B. J., McMurdie P. J., Rosen M. J., Han A. W., Johnson A. J. A., and Holmes S. P. (2016). DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869, PMID: 27214047
Caro A., Gros O., Got P., De Wit R., and Troussellier M. (2007). Characterization of the population of the sulfur-oxidizing symbiont of Codakia orbicularis (Bivalvia, Lucinidae) by single-cell analyses. Appl. Environ. Microbiol. 73, 2101–2109. doi: 10.1128/AEM.01683-06, PMID: 17259363
Conde A., Aira M., Novais J., and Dominguez J. (2011). On an early record of the alien clam Mya arenaria in the Iberian Peninsula and its likely confusion with Scrobicularia plana (Bivalvia). Vie Milieu 61, 1–7.
Coolen M. J. L. and Shtereva G. (2009). Vertical distribution of metabolicaly active eukaryotes in the water column and sediments of the Black Sea. FEMS Microbiol. Ecol. 70, 525–539. doi: 10.1111/j.1574-6941.2009.00756.x, PMID: 19732144
Dando P. R., Southward A. J., Southward E. C., Terwilliger N. B., and Terwilliger R. C. (1985). Sulphur-oxidising bacteria and haemoglobin in gills of the bivalve mollusc Myrtea spinifera. Mar. Ecol. Prog. Ser. 23, 85–98. doi: 10.3354/meps023085
Destoumieux-Garzón D., Canesi L., Oyanedel D., Travers M. A., Charrière G. M., Pruzzo C., et al. (2020). Vibrio–bivalve interactions in health and disease. Environ. Microbiol. 22, 4323–4341. doi: 10.1111/1462-2920.15055, PMID: 32363732
Dishaw L. J., Flores-Torres J., Lax S., Gemayel K., Leigh B., Melillo D., et al. (2014). The gut of geographically disparate Ciona intestinalis harbors a core microbiota. PloS One 9, e93386. doi: 10.1371/journal.pone.0093386, PMID: 24695540
Du Z. J., Wang Z. J., Zhao J. X., and Chen G. J. (2016). Woeseia oceani gen. Nov., sp. Nov., a chemoheterotrophic member of the order Chromatiales, and proposal of Woeseiaceae fam. Nov. Int. J. Syst. Evol. Microbiol. 66, 107–112. doi: 10.1099/ijsem.0.000683, PMID: 26474827
Dubé C. E., Ky C. L., and Planes S. (2019). Microbiome of the black-lipped pearl oyster Pinctada margaritifera, a multi-tissue description with functional profiling. Front. Microbiol. 10. doi: 10.3389/fmicb.2019.01548, PMID: 31333634
Dunkai T. I., Bogatyrenko E. A., and Kim A. V. (2023). Biodiversity and metabolic properties of bacterial communities from the digestive system of the bivalve crenomytilus grayanus. Microbiology 92, 552–563. doi: 10.1134/S0026261723600982
Duperron S., Fiala-Médioni A., Caprais J.-C., Olu K., and Sibuet M. (2007). Evidence for chemoautotrophic symbiosis in a Mediterranean cold seep clam (Bivalvia: Lucinidae): comparative sequence analysis of bacterial 16S rRNA, APS reductase and RubisCO genes. FEMS Microbiol. Ecol. 59, 64–70. doi: 10.1111/j.1574-6941.2006.00194.x, PMID: 17233745
El Samak M., Solyman S. M., and Hanora A. (2018). Antimicrobial activity of bacteria isolated from Red Sea marine invertebrates. Biotechnol. Rep. 19, e00275. doi: 10.1016/j.btre.2018.e00275, PMID: 30197871
Ene A., Teodorof L., Chiţescu C. L., Burada A., Despina C., Bahrim G. E., et al. (2025). Surface water contaminants (Metals, nutrients, pharmaceutics, endocrine disruptors, bacteria) in the danube river and black sea basins, SE Romania. Appl. Sci. 15, 5009. doi: 10.3390/app15095009
Fraune S. and Bosch T. C. (2010). Why bacteria matter in animal development and evolution. Bioessays 32, 571–580. doi: 10.1002/bies.200900192, PMID: 20544735
Fuirst M., Ward C. S., Schwaner C., Diana Z., Schultz T., and Rittschof D. (2021). Compositional and functional microbiome variation between tubes of an intertidal polychaete and surrounding marine sediments. Front. Mar. Sci. 8. doi: 10.3389/fmars.2021.656506
Galkin Y. (1998). Long-term changes in the distribution of molluscs in the Barents Sea related to the climate. Ber. Polarforsch. 287, 100–143.
Gasparotti C. (2014). The main factors of water pollution in Danube River basin. EuroEconomica. Danubius Univ. Galati. 1, 91–106.
Gavriilidou A., Gutleben J., Versluis D., Forgiarini F., van Passel M. W. J., Inghan C. J., et al. (2020). Comparative genomic analysis of Flavobacteriaceae: insights into carbohydrate metabolism, gliding motility and secondary metabolite biosynthesis. BMC Genomics 21, 569. doi: 10.1186/s12864-020-06971-7, PMID: 32819293
Ghaju Shrestha R., Tanaka Y., and Haramoto E. (2022). A review on the prevalence of arcobacter in aquatic environments. Water 14, 1266. doi: 10.3390/w14081266
Gilbertson W. W., Gilbertson Solan M., and Prosser J. I. (2012). Differential effects of microorganism–invertebrate interactions on benthic nitrogen cycling. FEMS Microbiol. Ecol. 82, 11–22. doi: 10.1111/j.1574-6941.2012.01400.x, PMID: 22533682
Givens C. E., Burnett K. G., Burnett L. E., and Hollibaugh J. T. (2013). Microbial communities of the carapace, gut, and hemolymph of the Atlantic blue crab, Callinectes sapidus. Mar. Biol. 160, 2841–2851. doi: 10.1007/s00227-013-2275-8
Glaubitz S., Labrenz M., Jost G., and Jurgens K. (2010). Diversity of active chemolithoautotrophic prokaryotes in the sulfidic zone of a Black Sea pelagic redox cline as determined by rRNA-based stable isotope probing. FEMS Microbiol. Ecol. 74, 32–41. doi: 10.1111/j.1574-6941.2010.00944.x, PMID: 20649907
Gomoiu M.-T. and Porumb I. (1969). Mya arenaria L. @ a bivalve recently penetrated into the Black Sea. Rev. Roum. Biol. Serie Zool. Ed. Acad. Bucuresti. 14, 199–202.
Hakim J. A., Koo H., Dennis L. N., Kumar R., Ptacek T., Morrow C. D., et al. (2015). An abundance of Epsilonproteobacteria revealed in the gut microbiome of the laboratory cultured sea urchin, Lytechinus variegatus. Front. Microbiol. 13. doi: 10.3389/fmicb.2015.01047, PMID: 26528245
Hoffmann K., Bienhold C., Buttigieg P. L., Knittel K., Laso-Pérez R., Rapp J. Z., et al. (2020). Diversity and metabolism of Woeseiales bacteria, global members of marine sediment communities. ISME J. 14, 1042–1056. doi: 10.1038/s41396-020-0588-4, PMID: 31988474
Hoor T. T. A. (1975). A new type of thiosulphate oxidizing, nitrate reducing microorganism: Thiomicrospira denitrificans sp. nov. Net J. Sea Res. 9, 344–350. doi: 10.1016/0077-7579(75)90008-3
Hoshino T., Doi H., Uramoto G. I., Wörmer L., Adhikari R. R., Xiao N., et al. (2020). Global diversity of microbial communities in marine sediment. Proc. Natl. Acad. Sci. U S A. 117, 27587–27597. doi: 10.1073/pnas.1919139117, PMID: 33077589
Hu Q., Wang S., Lai Q., Shao Z., and Jiang L. (2021). Sulfurimonas indica sp. nov., a hydrogen- and sulfur-oxidizing chemolithoautotroph isolated from a hydrothermal sulfide chimney in the Northwest Indian Ocean. Int. J. Syst. Evol. Microbiol. 71, 4575. doi: 10.1099/ijsem.0.004575, PMID: 33263512
Iancu L., Carter D. O., Junkins E. N., and Purcarea C. (2015). Using bacterial and necrophagous insect dynamics for postmortem interval estimation during cold season: Novel case study in Romania. Forensic Sci. Int. 254, 106–117. doi: 10.1016/j.forsciint.2015.07.024, PMID: 26217916
Ibryamova S., Pavlova B., Stanachkova E., Salim S., Lyatif A., Dimitrov D., et al. (2022). “Seasonal variations of the microflora of wedge clam Donax trunculus (Linnaeus 1758) from the region of Arkutino (Bulgarian Black Sea aquatory),” in Current trends of ecology, vol. 17 . Eds. Chankova S., Peneva V., Metcheva R., Beltcheva M., Vassilev K., Radeva G., and Danova K. (Sofia, Bulgaria: BioRisk), 253–262. doi: 10.3897/biorisk.17.77097
Inagaki F., Takai K., Kobayashi H., Nealson K. H., and Horikoshi K. (2003). Sulfurimonas autotrophica gen. nov., sp. nov., a novel sulfur-oxidizing epsilon- proteobacterium isolated from hydrothermal sediments in the Mid-Okinawa Trough. Int. J. Syst. Evol. Microbiol. 53, 1801–1805. doi: 10.1099/ijs.0.02682-0, PMID: 14657107
Jørgensen B. B., Fossing H., Wirsen C. O., and Jannasch H. W. (1991). Sulfide oxidation in the anoxic Black Sea chemocline. International Commission for the Protection of the Danube River. Vienna International Centre D0412. Deep-Sea Res. 38, 1083–S1103. doi: 10.1016/S0198-0149(10)80025-1
Knowlton N. and Rohwer F. (2003). Multispecies microbial mutualisms on coral reefs: the host as a habitat. Am. Nat. 162, 51–62. doi: 10.1086/378684, PMID: 14583857
Koo H., Hakim J. A., Morrow C. D., Eipers P. G., Davila A., Andersen D. T., et al. (2017). Comparison of two bioinformatics tools used to characterize the microbial diversity and predictive functional attributes of microbial mats from Lake Obersee, Antarctica. J. Microbiol. Methods 140, 15–22. doi: 10.1016/j.mimet.2017.06.017, PMID: 28655556
Koskella B., Hall L. J., and Metcalf C. J. E. (2017). The microbiome beyond the horizon of ecological and evolutionary theory. Nat. Ecol. Evol. 1, 1606–1615. doi: 10.1038/s41559-017-0340-2, PMID: 29038487
Krishnaveni M., Venkatesh S., Asha S., Rathika R., Sobia P. M., Iyyadurai M., et al. (2025). “20 - Marine invertebrate-microbe interactions: An insight into immunogenomics through metagenomic approach,” in Metagenomics, 2nd ed. (Cambridge, MA: Academic Press), 461–491. doi: 10.1016/B978-0-323-91631-8.00004-4
Kube J. (1996). Spatial and temporal variations in the population structure of the soft-shell clam Mya arenaria in the Pomeranian Bay (southern Baltic Sea). J. Sea Res. 35, 335–344. doi: 10.1016/S1385-1101(96)90760-1
Kueh C. S. and Chan K. Y. (1985). Bacteria in bivalve shellfish with special reference to the oyster. J. Appl. Bacteriol. 59, 41–47. doi: 10.1111/j.1365-2672.1985.tb01773.x, PMID: 4030530
Landry Z., Swan B. K., Herndl G. J., Stepanauskas R., and Giovannoni S. J. (2017). SAR202 genomes from the dark ocean predict pathways for the oxidation ofrecalcitrant dissolved organic matter. mBio 8, e00413–e00417. doi: 10.1128/mBio.00413-17, PMID: 28420738
Langille M. G. I., Zaneveld J., Caporaso J. G., McDonald D., Knights D., Reyes J. A., et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotech. 31, 814–821. doi: 10.1038/nbt.2676, PMID: 23975157
Lauber C., Hamady M., Knight R., and Fierer N. (2009). Pyrosequencing based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl. Environ. Microbiol. 75, 5111–5120. doi: 10.1128/AEM.00335-09, PMID: 19502440
Legendre P. and Legendre L. (2012). Numerical Ecology, Developments in Environmental Modelling. 3rd Edition (Amsterdam: Elsevier), 419.
Leloup J., Loy A., Knab N. J., Borowski C., Wagner M., and Jørgensen B. B. (2007). Diversity and abundance of sulfate-reducing microorganisms in the sulfate and methane zones of a marine sediment, Black Sea. Environ. Microbiol. 9, 131–142. doi: 10.1111/j.1462-2920.2006.01122.x, PMID: 17227418
Lemay M. A., Martone P. T., Keeling P. J., Burt J. M., Krumhansl K. A., Sanders R. D., et al. (2018). Sympatric kelp species share a large portion of their surface bacterial communities. Environ. Microbiol. 20, 658–670. doi: 10.1111/1462-2920.13993, PMID: 29124859
Lemieux-Labonté V., Tromas N., Shapiro B. J., and Lapointe F. J. (2016). Environment and host species shape the skin microbiome of captive neotropical bats. PeerJ 4, e2430. doi: 10.7717/peerj.2430, PMID: 27688962
Lévesque B., Gagnon F., Valentin A., Cartier J.-F., Chevalier P., Cardinal P., et al. (2006). Étude de la contamination microbienne des myes (Mya arenaria) de la rive nord de l’estuaire maritime du fleuve Saint-Laurent (Québec, Canada). Can. J. Microbiol. 52, 984–991. doi: 10.1139/w06-061, PMID: 17110967
Li J., Chen S., Wu P., Zhu C., Hu R., Li T., et al. (2023a). Insights into the relationship between intestinal microbiota of the aquaculture worm Sipunculus nudus and surrounding sediments. Fishes 8, 32. doi: 10.3390/fishes8010032
Li J., Zhang Y., Sun J., Thompson F., and Zhang Y. (2023b). Interaction between marine invertebrates and symbiotic microbes in a changing environment: Community structure and ecological functions. Front. Mar. Sci. 9. doi: 10.3389/fmars.2022.1128906
Liu C., Liu C., Gao F., Wang A., Wang H., Yang Y., et al. (2023). Composition of Particulate Matter and Bacterial Community in Gut Contents and Surrounding Sediments of Three Sipunculan Species (Siphonosoma australe, Phascolosoma arcuatum, and Sipunculus nudus). Int. J. Mol. Sci. 24, 6001. doi: 10.3390/ijms24066001, PMID: 36983074
Liu X., Teixeira J. S., Ner S., Ma K. V., Petronella N., Banerjee S., et al. (2020). Exploring the potential of the microbiome as a marker of the geographic origin of fresh seafood. Front. Microbiol. 11. doi: 10.3389/fmicb.2020.00696, PMID: 32362885
Lu Y., Zhou G., Ewald J., Pang Z., Shiri T., and Xia J. (2023). MicrobiomeAnalyst 2.0: comprehensive statistical, functional and integrative analysis of microbiome data. Nucleic Acids Res. 51, 310–318. doi: 10.1093/nar/gkad407, PMID: 37166960
Mangiafico S. S. (2015). An R companion for the handbook of biological statistics (Rutgers Cooperative Extension, New Brunswick, NJ, USA). Available online at: http://rcompanion.org (Accessed August 06, 2025).
Martin M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 10–12. doi: 10.14806/ej.17.1.200
Martinez-Garcia M., Brazel D. M., Swan B. K., Arnosti C., Chain P. S. G., Reitenga K. G., et al. (2012). Capturing single cell genomes of active polysaccharide degraders: an unexpected contribution of verrucomicrobia. PloS One 7, e35314. doi: 10.1371/journal.pone.0035314, PMID: 22536372
Masanja F., Yang K., Xu Y., He G., Liu X., Xu X., et al. (2023). Bivalves and microbes: a mini-review of their relationship and potential implications for human health in a rapidly warming ocean. Front. Mar. Sci. 10. doi: 10.3389/fmars.2023.1182438
McMurdie P. J. and Holmes S. (2013). Phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PloS One 8, e61217. doi: 10.1371/journal.pone.0061217, PMID: 23630581
Menabit S., Lavin P., Mureşan M., Begun T., Teacă A., and Purcarea C. (2024). First screening of bacteria assemblages associated with the marine polychaete Melinna palmata Grube 1870 and adjacent sediments. Front. Mar. Sci. 10. doi: 10.3389/fmars.2023.1279849
Mishra P., Singh U., Pandey C. M., Mishra P., and Pandey G. (2019). Application of student’s t-test, analysis of variance, and covariance. Ann. Card Anaesth. 22, 407–411. doi: 10.4103/aca.ACA_94_19, PMID: 31621677
Moore B. M., Chadhain S.M.Ní., Miller J. L., Jones S. H., and Launen L. A. (2020). Metagenome sequences from tidal marsh and marine sediment from the great bay estuary of new hampshire. Microbiol. Resour. Announc. 9, e00038–e00020. doi: 10.1128/MRA.00038-20, PMID: 32139581
Moruf R. O., Okunade G. F., and Elegbeleye O. W. (2020). Bivalve mariculture in two–way interaction with phytoplankton: a review of feeding mechanism and nutrient recycling. Bull. Univ. Agric. Sci. Vet. Med. Cluj-Napoca. Anim. Sci. Biotechnol. 77, 1. doi: 10.15835/buasvmcn-asb:2020.0010
Mußmann M., Pjevac P., Krüger K., and Dyksma S. (2017). Genomic repertoire of the Woeseiaceae/JTB255, cosmopolitan and abundant core members of microbial communities in marine sediments. ISME J. 11, 1276–1281. doi: 10.1038/ismej.2016.185, PMID: 28060363
Mureşan M., Teacă A., Popa A., and Begun T. (2019). Free-living marine nematodes community structural changes within a post-dredging site at the Romanian shelf. J. Environ. Prot. Ecol. 20, 753–760.
Musella M., Wathsala R., Tavella T., Rampelli S., Barone M., Palladino G., et al. (2020). Tissue-scale microbiota of the Mediterranean mussel (Mytilus galloprovincialis) and its relationship with the environment. Sci. Total Environ. 717, 137209. doi: 10.1016/j.scitotenv.2020.137209, PMID: 32084687
Nenciu M., Niţă V., Teacă A., Popa A., and Begun T. (2023). An assessment of potential beam trawling impact on north-western black sea benthic habitats aiming at a sustainable fisheries management. Water 15, 2241. doi: 10.3390/w15122241
Nikolova C.N., Ijaz U.Z., Magill C., Kleindienst S., Joye S.B., and Gutierrez T.. (2021). Response and oil degradation activities of a northeast Atlantic bacterial community to biogenic and synthetic surfactants. Microbiome 9, 191. doi: 10.1186/s40168-021-01143-5, PMID: 34548108
Offret C., Paulino S., Gauthier O., Château K., Bidault A., Corporeau C., et al. (2020). The marine intertidal zone shapes oyster and clam digestive bacterial microbiota. FEMS Microbiol. Ecol. 96, fiaa078. doi: 10.1093/femsec/fiaa078, PMID: 32353873
On S. L. W., Miller W. G., Biggs P. J., Cornelius A. J., and Vandamme P. (2021). Aliarcobacter, Halarcobacter, Malaciobacter, Pseudarcobacter and Poseidonibacter are later synonyms of Arcobacter: Transfer of Poseidonibacter parvus, Poseidonibacter antarcticus, ‘Halarcobacter arenosus’, and ‘Aliarcobacter vitoriensis’ to Arcobacter as Arcobacter parvus comb. nov., Arcobacter antarcticus comb. nov., Arcobacter arenosus comb. nov. and Arcobacter vitoriensis comb. nov. Int. J. Syst. Evol. Microbiol. 71, 005133. doi: 10.1099/ijsem.0.005133, PMID: 34825881
Onishchenko O. M. and Kiprianova E. A. (2006). Shewanella genus bacteria isolated from the Black Sea water and molluscs. Mikrobiol. Z. 68, 12–21., PMID: 16786624
Ottaviani D., Mosca F., Chierichetti S., Tiscar P. G., and Leoni F. (2016). Genetic diversity of Arcobacter isolated from bivalves of Adriatic and their interactions with Mytilus galloprovincialis hemocytes. Microbiologyopen 6, e00400. doi: 10.1002/mbo3.400, PMID: 27650799
Panin N. and Jipa D. (2002). Danube river Sediment Input and its Interaction with the North-western Black Sea. Estuar. Coast. Shelf Sci. 54, 551–562. doi: 10.1006/ecss.2000.0664
Pierce M. L. and Ward J. E. (2018). Microbial ecology of the bivalvia, with an emphasis on the family ostreidae. J. Shellfish Res. 37, 793–806. doi: 10.2983/035.037.0410
Pierce M. L. and Ward J. E. (2019). Gut microbiomes of the eastern oyster (Crassostrea virginica) and the blue mussel (Mytilus edulis): temporal variation and the influence of marine aggregate-associated microbial communities. Msphere 4, e00730–e00719. doi: 10.1128/mSphere.00730-19, PMID: 31826972
Pita L., Rix L., Slaby B. M., Franke A., and Hentschel U. (2018). The sponge holobiont in a changing ocean: from microbes to ecosystems. Microbiome 6, 46. doi: 10.1186/s40168-018-0428-1, PMID: 29523192
Pohlert T. (2014). The pairwise multiple comparison of mean ranks package (PMCMR) (R package). Available online at: https://cran.r-project.org/web/packages/PMCMRplus (Accessed August 06, 2025).
Qiao L., Chang Z., Li J., and Li T. (2022). Selective feeding of three bivalve species on the phytoplankton community in a marine pond revealed by high-throughput sequencing. Sci. Rep. 12, 6163. doi: 10.1038/s41598-022-08832-7, PMID: 35418129
Reveillaud J., Maignien L., Eren A. M., Huber J. A., Apprill A., Sogin M. L., et al. (2014). Host-specificity among abundant and rare taxa in the sponge microbiome. ISME J. 6), 1198–1209. doi: 10.1038/ismej.2013.227, PMID: 24401862
Rey-Campos M., Ríos-Castro R., Gallardo-Escárate C., Novoa B., and Figueras A. (2022). Exploring the potential of metatranscriptomics to describe microbial communities and their effects in molluscs. Int. J. Mol. Sci. 23, 16029. doi: 10.3390/ijms232416029, PMID: 36555669
Romero J. and Espejo R. (2001). The prevalence of noncultivable bacteria in oysters (Tiostrea Chilensis, Philippi 1845). J. Shellfish Res. 20, 1235–1240.
Ross K. S., Haites N. E., and Kelly K. F. (1990). Repeated freezing and thawing of peripheral blood and DNA in suspension: Effects on DNA yield and integrity. J. Med. Genet. 27, 569–570. doi: 10.1136/jmg.27.9.569, PMID: 2231649
Roth-Schulze A. J., Pintado J., Zozaya-Valdés E., Cremades J., Ruiz P., Kjelleberg S., et al. (2018). Functional biogeography and host specificity of bacterial communities associated with the Marine Green Alga Ulva spp. Mol. Ecol. 27, 1952–1965. doi: 10.1111/mec.14529, PMID: 29420863
Ruginescu R., Lavin P., Iancu L., Menabit S., and Purcarea C. (2022). Bioprospecting for novel bacterial sources of hydrolytic enzymes and antimicrobials in the Romanian littoral zone of the Black Sea. Microorganisms 10, 2468. doi: 10.3390/microorganisms10122468, PMID: 36557721
Russanova M. N. (1963). “Short notes on biology of some massinvertebrate species in the vicinity of the cape Kartesh,” in Materials of integrated research of the White Sea. 2 (in Russian) (USSR Academy of Science, Moscow-Leningrad), 53–65.
Schäfer H., Ferdelman T. G., Fossing H., and Muyzer G. (2007). Microbial diversity in deep sediments of the Benguela Upwelling System. Aquat. Microb. Ecol. 50, 1–9. doi: 10.3354/ame01164
Schippers A., Kock D., Höft C., Köweker G., and Siegert M. (2012). Quantification of microbial communities in subsurface marine sediments of the Black Sea and off Namibia. Front. Microbiol. 3. doi: 10.3389/fmicb.2012.00016, PMID: 22319518
Schroeder G. N. and Hilbi H. (2008). Molecular pathogenesis of Shigella spp.: controlling host cell signaling, invasion, and death by type III secretion. Clin. Microbiol. Rev. 21, 134–156. doi: 10.1128/CMR.00032-07, PMID: 18202440
Schulz H. N., Brinkhoff T., Ferdelman T. G., Mariné M. H., Teske A., and Jørgensen B. B. (1999). Dense populations of a giant sulfur bacterium in Namibian shelf sediments. Science 284, 493–495. doi: 10.1126/science.284.5413.493, PMID: 10205058
Segata N., Izard J., Waldron L., Gevers D., Miropolsky L., Garrett W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12, R60. doi: 10.1186/gb-2011-12-6-r60, PMID: 21702898
Sievert S. M., Scott K. M., Klotz M. G., Chain P. S. G., Hauser L. J., Hemp J., et al (2008). Genome of the epsilonproteobacterial chemolithoautotroph Sulfurimonas denitrificans. Appl Environ Microbiol. 74, 1145–1156. doi: 10.1128/AEM.01844-07., PMID: 18065616
Skolka M. and Gomoiu M.-T. (2004). Specii invazive în Marea Neagră. Impactul ecologic al patrunderii de noi specii in ecosistemele acvatice (Constanța, Romania: Ovidius University Press Constanta), 180.
Smidt E. L. B. (1951). Animal Production in the Danish Wadden Sea. Medd. Komm. Dan. Fisk. Havunders. Set. Fisk. (Copenhagen: C.A. Reitzel) 11, 151.
Sorokin Y. I., Sorokin P. Y., Avdeev V. A., Sorokin D. Y., and Ilchenko S. V. (1995). Biomass, production and activity of bacteria in the Black Sea, with special reference to chemosynthesis and the sulfur cycle. Hydrobiologia 308, 61–76. doi: 10.1007/BF00037788
Sousa H. and Hinzmann M. (2020). Antibacterial components of the Bivalve’s immune system and the potential of freshwater bivalves as a source of new antibacterial compounds. Fish Shellfish Immunol. 98, 971–980. doi: 10.1016/j.fsi.2019.10.062, PMID: 31676427
Stora G., Arnoux A., and Galas M. (1995). Time and spatial dynamics of Mediterranean lagoon macrobenthos during an exceptionally pro-longed interruption of freshwater inputs. Hydrobiologia 300/301, 123–132. doi: 10.1007/BF00024454
Strahl J. and Abele D. (2020). Nitric oxide mediates metabolic functions in the bivalve Arctica islandica under hypoxia. PloS One 15, e0232360. doi: 10.1371/journal.pone.0232360, PMID: 32379772
Strasser M. (1998). Mya arenaria — an ancient invader of the North Sea coast. Helgolander Meeresunters 52, 309–324. doi: 10.1007/BF02908905
Su Z., Dai T., Tang Y., Tao Y., Huang B., Mu Q., et al. (2018). Sediment bacterial community structures and their predicted functions implied the impacts from natural processes and anthropogenic activities in coastal area. Mar. pollut. Bull. 131, 481–495. doi: 10.1016/j.marpolbul.2018.04.052, PMID: 29886974
Suarez-Moo P., Lamelas A., García-Bautista I., Barahona-Pérez L., Sandoval-Flores G., Valdes D., et al. (2020). Characterization of sediment microbial communities at two sites with low hydrocarbon pollution in the southeast Gulf of Mexico. PeerJ 8, e10339. doi: 10.7717/peerj.10339, PMID: 33354414
Šyvokienė J. and Butrimavičienė L. (2013). Aquatic organisms and petroleum hydrocarbon degrading bacteria associated with their digestive system. Environ. Research Eng. Manage. 4, 6–19. doi: 10.5755/j01.erem.66.4.5300
Takahashi S., Tomita J., Nishioka K., Hisada T., and Nishijima M. (2014). Development of a prokaryotic universal primer for simultaneous analysis of bacteria and archaea using next-generation sequencing. PloS One 9, e105592. doi: 10.1371/journal.pone.0105592, PMID: 25144201
Takai K., Suzuki M., Nakagawa S., Miyazaki M., Suzuki Y., Inagaki F., et al. (2006). Sulfurimonas paralvinellae sp. nov., a novel mesophilic, hydrogen- and sulfur-oxidizing chemolithoautotroph within the Epsilonproteobacteria isolated from a deep-sea hydrothermal vent polychaete nest, reclassification of Thiomicrospira denitrificans as Sulfurimonas denitrificans comb. nov. and emended description of the genus Sulfurimonas. Int. J. Syst. Evol. Microbiol. 56, 1725–1733. doi: 10.1099/ijs.0.64255-0, PMID: 16901999
Teacă A., Mureşan M., Begun T., Popa A., and Ion G. (2019). Marine benthic habitats within a physical disturbed site from the Romanian Coast of the Black Sea. J. Environ. Prot. Ecol. 20, 723–732.
Teacă A., Mureşan M., Menabit S., Bucse A., and Begun T. (2020). Assessment of Romanian circalittoral soft bottom benthic habitats under Danube River influence. Regional Stud. Mar. Sci. 40, 101523. doi: 10.1016/j.rsma.2020.101523
Thamdrup B., Rosselló-Mora R., and Amann R. (2000). Microbial manganese and sulfate reduction in Black Sea shelf sediments. Appl. Environ. Microbiol. 66, 2888–2897. doi: 10.1128/AEM.66.7.2888-2897.2000, PMID: 10877783
Timmins-Schiffman E., White S. J., Thompson R. E., Vadopalas B., Eudeline B., Nunn B. L., et al. (2021). Coupled microbiome analyses highlights relative functional roles of bacteria in a bivalve hatchery. Environ. Microbiome 16, 1–12. doi: 10.1186/s40793-021-00376-z, PMID: 33902744
Todorova V. and Konsulova T. (2005). Manual for quantitative sampling and sample treatment of marine soft-bottom macrozoobenthos (PO Box 152, 9000 Varna, Bulgaria: Institute of Oceanology, Bulgarian Academy of Sciences), 38. Available online at: www.blacksea-commission.org (Accessed February 12, 2025).
Wang S. S., Jiang L. J., Liu X. W., Yang S. P., and Shao Z. Z. (2020). Sulfurimonas xiamenensis sp. nov. and Sulfurimonas lithotrophica sp. nov., hydrogen- and sulfur-oxidizing chemolithoautotrophs within the Epsilonproteobacteria isolated from coastal sediments, and an emended description of the genus Sulfurimonas. Int. J. Syst. Evol. Microbiol. 70, 2657–2663. doi: 10.1099/ijsem.0.004087, PMID: 32134372
Ward L. M., Bertran E., and Johnston D. T. (2021). Expanded genomic sampling refines current understanding of the distribution and evolution of sulfur metabolisms in the desulfobulbales. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.666052, PMID: 34093483
Wemheuer F., Taylor J. A., Daniel R., Johnston E., Meinicke P., Thomas T., et al. (2020). Tax4Fun2: prediction of habitat-specific functional profiles and functional redundancy based on 16S rRNA gene sequences. Environ. Microbiome 15, 1–12. doi: 10.1186/s40793-020-00358-7, PMID: 33902725
Wheaton F. W., Schaffer G. U., Ingling A., and Douglass L. (2008). Physical properties of soft shell clams, Mya arenaria. Aquacult. Eng. 38, 181–188. doi: 10.1016/j.aquaeng.2008.03.002
Winters A. D., Marsh T. L., and Faisal M. (2011). Heterogeneity of bacterial communities within the zebra mussel (Dreissena polymorpha) in the Laurentian Great Lakes Basin. J. Great Lakes Res. 37, 318–324. doi: 10.1016/j.jglr.2011.01.010
Yang S.-J., Choo Y.-J., and Cho J.-C. (2007). Lutimonas vermicola gen. nov., sp nov., a member of the family Flavobacteriaceae isolated from the marine polychaete Periserrula leucophryna. Int. J. Systematic Evolutionary Microbiol. 57, 1679–1684. doi: 10.1099/ijs.0.65060-0, PMID: 17684236
Yoon J., Matsuo Y., Adachi K., Nozawa M., Matsuda S., Kasai H., et al. (2008). Description of Persicirhabdus sediminis gen. nov., sp. nov., Roseibacillus ishigakijimensis gen. nov., sp. nov., Roseibacillus ponti sp. nov., Roseibacillus persicicus sp. nov., Luteolibacter pohnpeiensis gen. nov., sp. nov. and Luteolibacter algae sp. nov., six marine members of the phylum ‘Verrucomicrobia’, and emended descriptions of the class Verrucomicrobiae, the order Verrucomicrobiales and the family Verrucomicrobiaceae. Int. J. Syst. Evol. Microbiol. 58, 998–1007. doi: 10.1099/ijs.0.65520-0, PMID: 18398209
Yoon J., Yasumoto-Hirose M., Matsuo Y., Nozawa M., Matsuda S., Kasai H., et al. (2007). Pelagicoccus mobilis gen. nov., sp. nov., Pelagicoccus albus sp. nov. and Pelagicoccus litoralis sp. nov., three novel members of subdivision 4 within the phylum ‘Verrucomicrobia’, isolated from seawater by in situ cultivation. Int. J. Syst. Evol. Microbiol. 57, 1377–1385. doi: 10.1099/ijs.0.64970-0, PMID: 17625161
Zambriborshch F. S., Marchenko A. S., and Telegin O. N. (1968). New occurrence and distribution of mya arenaria L. @ in northwestern black sea. Gidrobiol. Zh. 4, 48–51.
Zhang M., Sun Y., Chen L., Cai C., Qiao F., Du Z., et al. (2016). Symbiotic bacteria in gills and guts of Chinese mitten crab (Eriocheir sinensis) differ from the free-living bacteria in water. PloS One 11, e0148135. doi: 10.1371/journal.pone.0148135, PMID: 26820139
Keywords: invertebrates’ bacteria, Mya arenaria microbiome, Black Sea bivalve, Illumina sequencing, 16S rRNA gene
Citation: Menabit S, Lavin P, Begun T, Teacă A, Mureşan M and Purcarea C (2025) Organ-specific bacterial communities of the soft-shell clam Mya arenaria (Linnaeus, 1758) and adjacent sediments in the Black Sea. Front. Mar. Sci. 12:1659674. doi: 10.3389/fmars.2025.1659674
Received: 04 July 2025; Accepted: 27 August 2025;
Published: 17 September 2025.
Edited by:
Valerio Mazzella, Anton Dohrn Zoological Station Naples, ItalyReviewed by:
Aadil H. Bhat, University of California, Los Angeles, United StatesEsam Almuhaideb, King Saud University, Saudi Arabia
Copyright © 2025 Menabit, Lavin, Begun, Teacă, Mureşan and Purcarea. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cristina Purcarea, Y3Jpc3RpbmEucHVyY2FyZWFAaWJpb2wucm8=