 Hongwei Li
Hongwei Li Yuan Chen1†
Yuan Chen1† Zhongrong Xia
Zhongrong Xia- 1School of Life Science, Huizhou University, Huizhou, China
- 2Guangdong Huidong Sea Turtle National Nature Reserve Bureau, Huizhou, China
- 3Key Laboratory of Marine Biological Genetic Resources, Third Institute of Oceanography, Ministry of Natural Resources, Xiamen, China
Introduction: The critically endangered hawksbill turtle (Eretmochelys imbricata) is essential for healthy coral reef ecosystems, and its gut microbiota plays a vital role in host nutrition and overall health. A comprehensive understanding of this microbial diversity is crucial for effective conservation, yet current knowledge remains limited. This study aimed to characterize the bacterial taxa present in the fecal samples of hawksbill turtles using culture-dependent methods.
Methods: Fecal samples from hawksbill turtles were cultured under both aerobic and anaerobic conditions using two distinct bacterial media. A total of 161 bacterial strains were isolated and identified using standard microbiological techniques and 16S rRNA gene sequencing.
Results: The isolated strains were classified into three phyla: Pseudomonadota (formerly Proteobacteria), Actinomycetota, and Bacillota (formerly Firmicutes). The most frequently identified species were Psychrobacter celer, Shewanella algae, Sulfitobacter pontiacus, Vibrio mytili, Enterococcus hirae, and Psychrobacter maritimus. Ten isolates were identified as potentially representing six novel bacterial species.
Discussion: This study expands our understanding of the microbial diversity associated with hawksbill turtles. It demonstrates the utility of fast and simple culture-dependent approaches for characterizing the microbiota of endangered species, providing a foundation for future research into the roles of these bacteria in turtle health and reef ecosystem function.
1 Introduction
The hawksbill sea turtle (Eretmochelys imbricata) is classified as Critically Endangered on the International Union for Conservation of Nature Red List of Threatened Species due to intense commercial exploitation for its shell and meat, as well as the degradation of its nesting and marine habitats (IUCN, 2021). Although generally categorized as an omnivore, the hawksbill primarily feeds on sponges within coral reef ecosystems (Meylan, 1988). Unlike green sea turtles, which possess a cecum housing a rich microflora (Bjorndal, 1985), hawksbills lack this anatomical feature (Bjorndal, 1985). Emerging research highlights the mutualistic relationship between hosts and their resident microbiota, wherein hosts derive nutrients from microbial activity, and diet influences the gut microbiota composition and abundance (Flint et al., 2012; Valdes et al., 2018). For instance, juvenile green turtles fast acquired polysaccharide fermenting gut microbiota after settlement into coastal habitats (Campos et al., 2018). An investigation showed that the bacterial microbiota of sea turtles is heavily influenced by geography (Scheelings et al., 2020). Additionally, a study indicated that despite the differences in origin, size and conditions of the animals, Firmicutes, Bacteroidetes, and Proteobacteria were dominant in the gut microbiome of all stranded sea turtles (Arizza et al., 2019). Similarly, gut microbiota enable hosts to ferment non-digestible substrates, fostering the growth of specialized microbial communities (Valdes et al., 2018). Lower bacterial diversity has been consistently observed in human diseases compared to healthy controls (Manichanh et al., 2006; Wang et al., 2008; Turnbaugh et al., 2009; Schippa et al., 2010; de Goffau et al., 2013; Lambeth et al., 2015; Scher et al., 2015), suggesting that gut microbiota diversity may serve as an indicator of host health (Sommer et al., 2017). These findings emphasize the critical roles of gut microbial communities in host nutrition and health. Consequently, a deeper understanding of gut microbiota diversity is vital for future conservation efforts targeting hawksbill turtles.
Although metagenomic approaches enable comprehensive sequencing of the entire metagenome, providing species-level assignments and genomic assemblies (Ye et al., 2019), microbial cultivation remains indispensable for elucidating microbial diversity and understanding their functional roles in hosts (Capri et al., 2023; McNally et al., 2021; Grigorescu et al., 2018). Notably, culture-dependent techniques can detect microorganisms present in low concentrations that may escape detection through metagenomic methods (Chen et al., 2022a).
In our previous study, high-throughput sequencing revealed that Bacteroidetes, Firmicutes, and Fusobacteria were the dominant gut microbial phyla in hawksbill sea turtles, reflecting the characteristics of both herbivorous and carnivorous sea turtles (Chen et al., 2022b). Additionally, enrichment analysis identified a high abundance of genes related to glycosyltransferases and auxiliary activities in the gut microbiota of hawksbill turtles (Lagier et al., 2012). However, these findings remain insufficient for a comprehensive understanding of gut microbiota diversity. Laboratory-based cultivation of gut bacteria offers a promising avenue for elucidating their roles in hawksbill turtles.
In this study, we used culture-dependent methods to characterize the bacterial taxa isolated from hawksbill fecal samples, with the aim of providing a more detailed understanding of gut microbiota diversity in this critically endangered species.
2 Materials and methods
2.1 Animals and sampling
This study was approved by the Committee on Biomedical Ethics of Huizhou University. We collected 14 fecal samples from hawksbill turtles (Eretmochelys imbricata) residing in the Sea Turtles National Nature Reserve, Huidong, China (22°33′15′′N; 114°52′33′′E). A total of 14 hawksbills inhabit the reserve. The healthy hawksbills were hosted in tanks, and were fed mainly small sea fish with a small amount of vegetables. The ages of the first and second batches of hawksbill turtles were 7, 12 years, respectively. Each batch included five females and two males. Fecal samples were collected from each turtle near their feeding time (stools produced after feeding), which made sampling difficult. Therefore, we decided to collect fecal samples in two batches. Seven samples were collected on August 4, 2022 (first batch), and the remaining seven were collected on August 9, 2022 (second batch). All sampling tools were sterilized at high temperature (121°C) and high pressure (105 kPa) for 30 min before being oven-dried. To minimize contamination from the surrounding water, the fecal pellet cores were accessed using sterilized forceps and sampled with sterile swabs. Buffers were not used during collection. After sampling, fecal samples were immediately placed in cryogenic vials, flash-frozen in liquid nitrogen, transported to the laboratory, and stored at −80°C until further processing.
2.2 Culture-dependent identification of bacteria in hawksbill feces
The culture-dependent isolation of bacterial samples from hawksbill feces was conducted following the method described by Griffith et al. (2018), with minor modifications. Strict sterile procedures were followed throughout the process. Both anaerobic and aerobic conditions were used to obtain a greater variety of microorganisms. Under aerobic conditions: 1) Two grams of each fecal sample were homogenized in 20 mL of sterile seawater using a sterilized motorized pellet pestle until the samples were thoroughly broken down into small particles. 2) A 100-µL aliquot of the homogenized solution was serially diluted up to a 10-9 dilution in sterile seawater. Aliquots from dilutions of 10-7, 10-8, and 10-9 were plated onto two types of agar-based solid media: 2216E supplemented with 1 g/L sodium acetate (M1) and 2216E supplemented with 25 g/L jellyfish extract (M2) (2216E; Table 1), and then incubated at 28°C. 1) and 2) were performed on a laminar flow bench to avoid contamination. Anaerobic conditions: 1) were the same as aerobic conditions; and 2) aliquots from dilutions of 10-7, 10-8, and 10-9 were plated onto M1 agar-based solid media, placed in an anaerobic bag, and incubated at 28°C. Here, 1) and 2) were performed on Baker Ruskinn Consept 400.
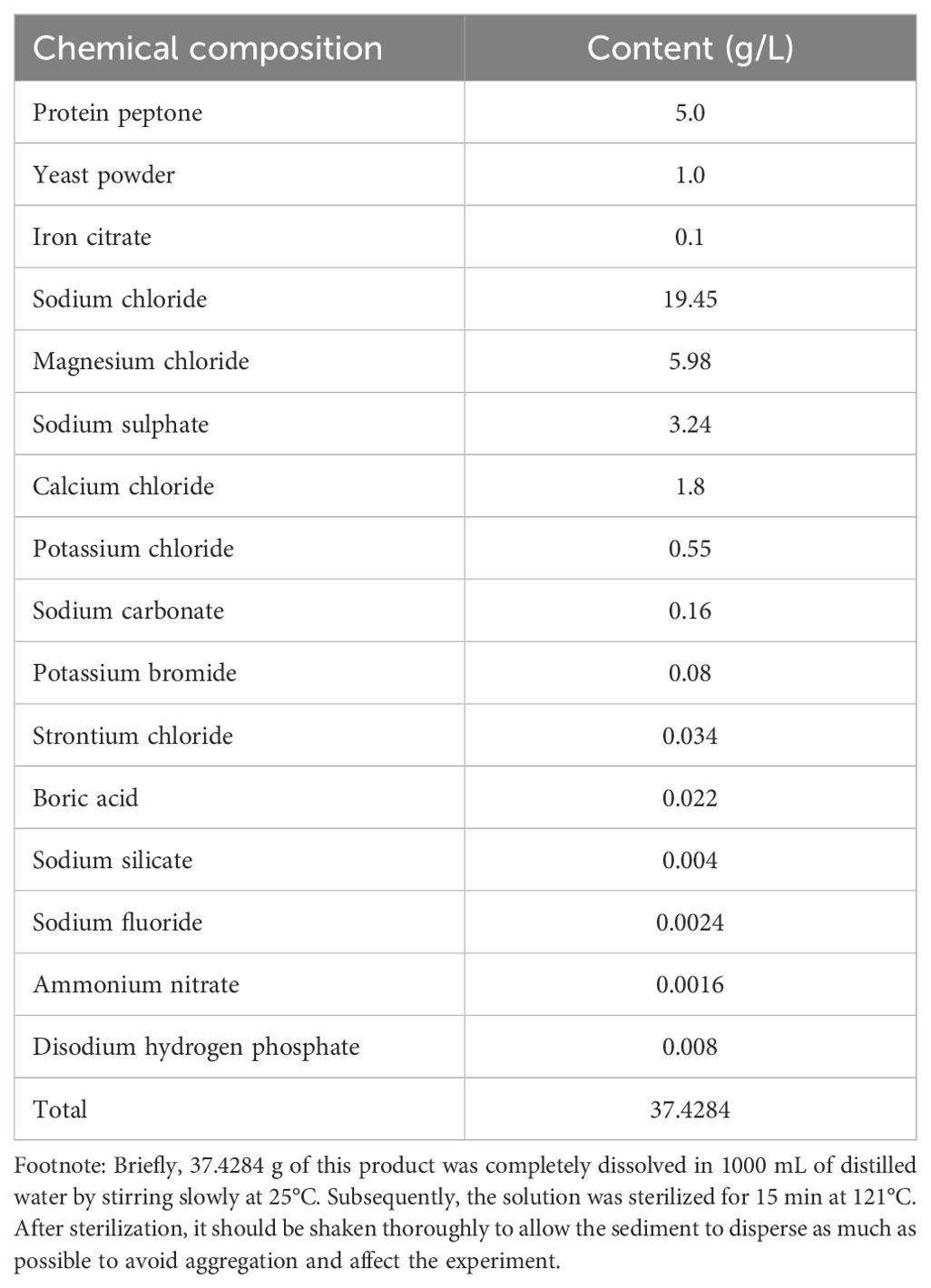
Table 1. Composition of the 2216E medium used to cultivate marine bacteria.
The plates were monitored daily for bacterial colony growth. Multiple diluted plates of the same sample were used for colony comparison, and different strains were selected. A purified single colony was obtained by repeated streaking on a plate. A certain amount of a single colony was then added to TE buffer from the purified plate for subsequent DNA extraction.
2.3 16S rDNA-based taxonomic characterization of bacterial clones
DNA was extracted from bacterial cells using the Silicone Bacterial Genomic DNA Extraction Kit (Shanghai SBS Genetechnology Co., Ltd.). The extracted DNA was amplified by polymerase chain reaction (PCR) using the primers described by Liu and Shao (2005): 27F (5′-AGAGTTTGATCCTGGCTCAG-3′) and 1492R (5′-ACGGCTACCTTGTTACGACT-3′). Each 50 µL PCR reaction contained 1 µL DNA template (> 20 ng), 0.2 µL Taq polymerase, 5 µL 10× reaction buffer, 4 µL 2.5 mM dNTPs, 1 µL each of 10 µM forward and reverse primers, 4 µL of 25 mM MgCl2, and 20.7 µL dH2O. The thermal cycling conditions were as follows: initial denaturation at 94°C for 1 min; 30 cycles of denaturation at 94°C for 1 min, annealing at 55°C for 1 min, and extension at 72°C for 1.5 min; final elongation at 72°C for 10 min; and hold at 10°C. The PCR products were sequenced using the Illumina NovaSeq 6000 system (Novogene, Beijing, China). The EzTaxon database contains sequences of type strains of prokaryotic species with validly published names. Type strains serve as the nomenclatural types for bacterial and archaeal species in taxonomy. They are used as reference points for identifying and classifying new isolates. Therefore, the 16S rRNA gene sequence data were compared and analyzed using the EzTaxon bacterial type species database (http://www.ezbiocloud.net/eztaxon/ify) to construct a phylogenetic tree. A comparison was also made at the NCBI database (http://blast.ncbi.nl.nih.gov/Blast.cgi) to check whether there were other type species that had not been included in EzTaxon. Following Chun et al. (2018), species identification was conducted using a combination of 16S rRNA sequence similarity and DNA–DNA hybridization (DDH) similarity. First, a 16S-based search (Yoon et al., 2017) identified candidate species for comparison to the strain of interest. Only species with ≥ 98.7% 16S similarity were selected (Kim et al., 2014). DDH techniques are the gold standard for the genomic similarity analyses of pairwise strain sets for classification. Subsequently, DDH similarity was assessed, with ≥ 70% similarity used as the threshold for species boundaries (Meier-Kolthoff et al., 2014). If DDH similarity was < 70%, the isolate was considered a potential new species, even if the 16S similarity was ≥ 98.7%. New species candidates were further analyzed through complete genome sequencing.
2.4 Complete genome sequencing of potential new species
Genomic DNA was extracted from bacterial strains identified as potential new species using the OMEGA Bacterial Genomic DNA Extraction Kit. DNA was quantified using a TBS-380 fluorometer (Turner BioSystems Inc., Sunnyvale, CA). High-quality DNA samples (OD260/280 = 1.8–2.0, > 6 µg) were used to construct DNA libraries. Genomic DNA (1 µg) was fragmented into 300–500 bp segments using a Covaris M220 ultrasonicator. The fragmented DNA was end-repaired, adenylated at the 3′ ends, and ligated with indexed adapters using the TruSeq™ Nano DNA Sample Prep Kit. The resulting DNA library was enriched by PCR amplification for eight cycles. Target bands were recovered using 5.2% Certified Low Range Ultra Agarose gel electrophoresis and quantified using the TBS-380 fluorometer (Picogreen). The enriched libraries were proportionally pooled for sequencing. Bridge PCR amplification was performed on a cBot solid-phase carrier to generate clusters, and the libraries were sequenced using the Cyclone SEQ platform (www.biozeron.com).
2.5 Data analysis and genome assembly
The original image data were converted into sequence data using Base Calling, with results stored in the FASTQ file format. The raw data were filtered using the Trimmomatic tool (ILLUMINACLIP:adapters.fa:2:30:10 SLIDINGWINDOW:4:15 MINLEN:75) (http://www.usadellab.org/cms/index.php?page=trimmomatic) to improve the accuracy of the subsequent assembly. The filtering process involved several steps: 1) removing adapter sequences from the reads and trimming non-AGCT bases at the 5′ end; 2) trimming the ends of reads with low sequencing quality (Q-value < Q20); 3) eliminating reads containing more than 10% ambiguous bases (N); and 4) discarding small fragments < 75 bp after adapter removal and quality trimming. The resulting clean reads were assembled de novo using ABySS v2.0.2 (http://www.bcgsc.ca/platform/bioinfo/software/abyss) with optimized K-mer parameters to generate the best possible assembly sequences. Further assembly refinement included filling gaps and correcting base errors using GapCloser v1.12 (https://sourceforge.net/projects/soapdenovo2/files/GapCloser/). Clean reads were aligned with the assembled genome sequences to calculate the GC content and coverage depth. The reliability and normality of the assembly results were assessed by determining the overall GC content distribution and coverage depth.
2.6 Gene annotation and phylogenetic tree construction
The predicted protein sequences of genes were aligned against the NR, KEGG, eggNOG, STRING, and GO databases using BLAST+2.7.1 with an alignment threshold of E-value ≤ 1e-5. This process provided a comprehensive annotation of the predicted genes. OrthoMCL v2.0.3 was used to align the amino acid (or nucleotide) sequences of all the analyzed species to identify homologous genes. Similarity clustering was performed with the parameters BLASTP E-value ≤ 1e-5 and MCL_INFLATION=1.5, yielding a list of homologous gene clusters. Phylogenetic analysis was based on GTDB-Tk and FastTree (Chaumeil et al., 2019). Using GTDB-Tk v2.4.0 (database release R220), we extracted and aligned sequences of 120 bacterial marker genes from the target genomes. A maximum-likelihood tree was constructed using FastTree 2.1.11 with default parameters. Owing to resolution limitations in the figure, the bootstrap support values are not clearly visible; however, the corresponding Newick-format tree file is provided as Supplementary Data.
3 Results
3.1 Bacterial taxa from hawksbill feces cultured on M1 medium under aerobic conditions
Bacterial isolates from the first batch of samples cultured on M1 medium under aerobic conditions were subjected to 16S rRNA gene sequencing. Sequence alignment identified 77 bacterial isolates, which were assigned to 39 distinct species. At the phylum level, 46 isolates were classified as Pseudomonadota, 10 as Bacteroidota, 13 as Actinomycetota, and eight as Bacillota. The isolates were predominantly classified at the genus level as Psychrobacter, Tenacibaculum, Glutamicibacter, Shewanella, Vibrio, and Pseudoalteromonas. The most frequently isolated species included Psychrobacter celer, Shewanella algae, Tenacibaculum discolor, Tenacibaculum mesophilum, Pseudoalteromonas tetraodonis, Vibrio neocaledonicus, Alcaligenes faecalis subsp. phenolicus, and Rheinheimera aquimaris, with Psychrobacter celer being the most common isolate (Figure 1).
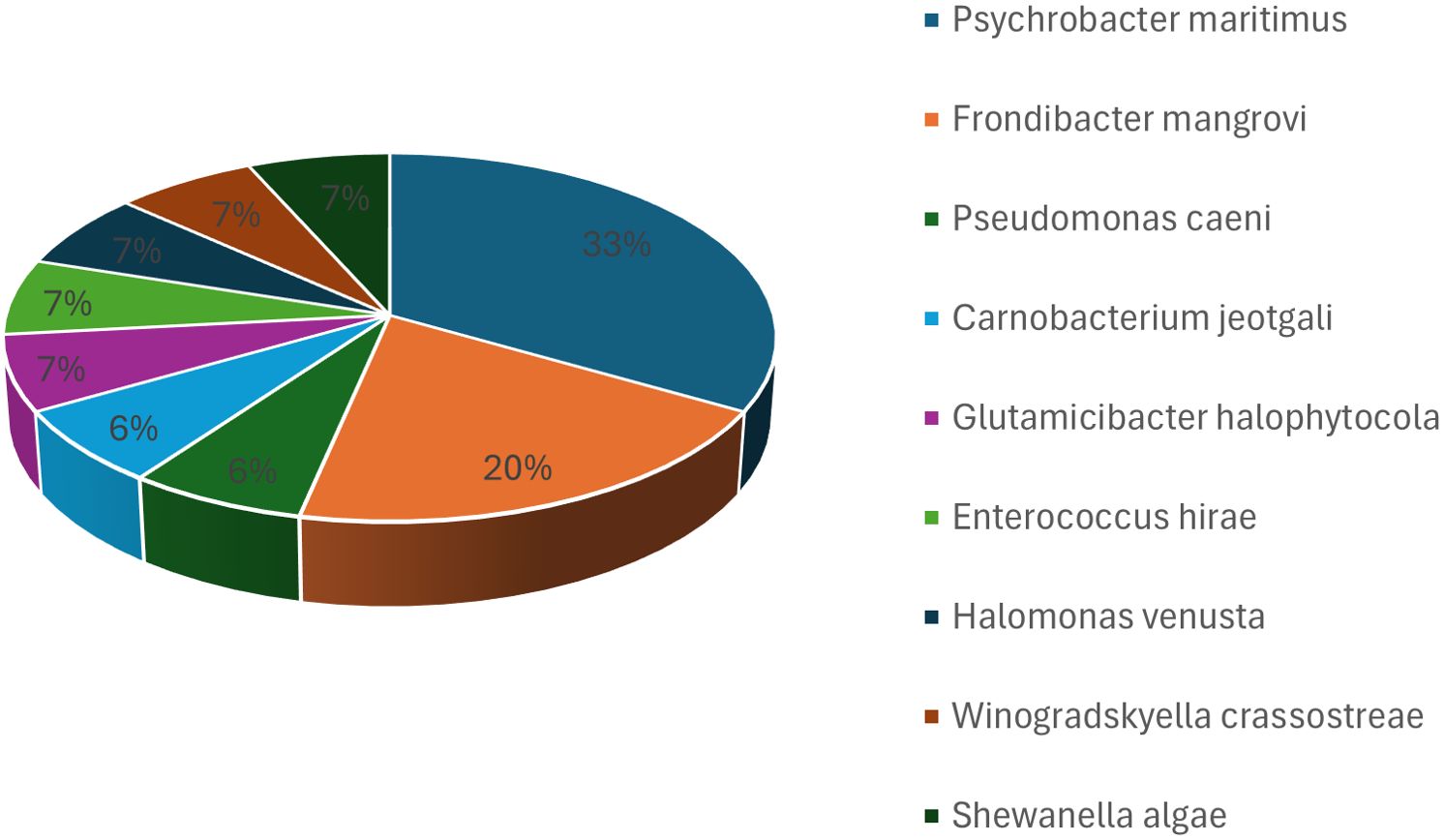
Figure 1. Bacterial taxa cultured from hawksbill feces in the first batch using M1 medium under aerobic conditions. The top identified species were Psychrobacter celer, Shewanella algae, Tenacibaculum discolor, Tenacibaculum mesophilum, Pseudoalteromonas tetraodonis, Vibrio neocaledonicus, Alcaligenes faecalis subsp. phenolicus, and Rheinheimera aquimaris, with Psychrobacter celer being the most frequently isolated species.
Among the 77 isolates, six displayed ≥ 98.7% 16S rRNA gene sequence similarity with known species, including Glutamicibacter arilaitensis (98.79%), Mesonia sediminis (98.25%), Rhodococcus phenolicus (98.92%), and Alcanivorax borkumensis (98.94%). However, DDH analysis revealed a similarity of < 70% for these six isolates, suggesting that they may represent novel species. These isolates were subsequently selected for complete genomic sequencing. The 16S rRNA genes of bacterial isolates from the second batch of samples were amplified and sequenced under aerobic conditions. Sequence alignment identified 31 isolates, which were assigned to 17 distinct species. At the phylum level, 23 isolates belonged to Pseudomonadota, five to Bacillota, two to Bacteroidota, and one to Campylobacterota. The isolates were primarily assigned to Sulfitobacter, Psychrobacter, Sporosarcina, and Pseudoalteromonas at the genus level. The most frequently isolated species included Sulfitobacter pontiacus, Sporosarcina aquimarina, Pseudoalteromonas carrageenovora, Psychrobacter piscatorii, and Psychrobacter cibarius, with Sulfitobacter pontiacus being the most common isolate (Figure 2).
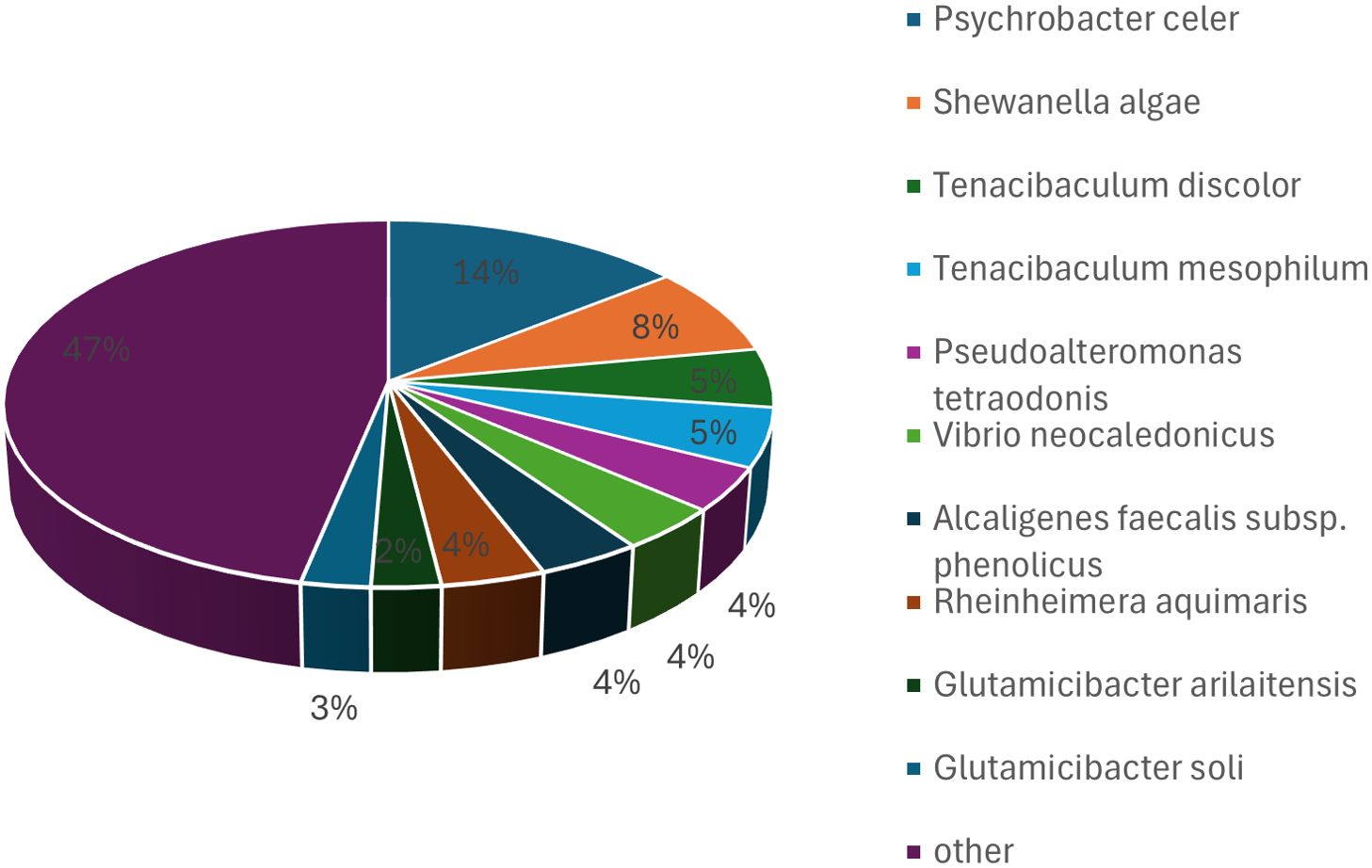
Figure 2. Bacterial taxa cultured from hawksbill feces in the second batch using M1 medium under aerobic conditions. The predominant species identified were Sulfitobacter pontiacus, Sporosarcina aquimarina, Pseudoalteromonas carrageenovora, Psychrobacter piscatorii, and Psychrobacter cibarius, with Sulfitobacter pontiacus being the most common isolate.
A comparison of isolates from the first and second batches revealed a dominance of isolates from the phyla Pseudomonadota, Actinomycetota, and Bacillota in both batches. Differences were primarily observed at the genus and species levels, reflecting variations in microbial composition based on sampling time. Overall, these results demonstrated an unexpectedly high diversity of bacterial taxa identified using the culture-dependent approach.
3.2 Bacterial taxa cultured from hawksbill feces on M1 under anaerobic conditions
A total of 38 bacterial strains, representing 18 species, were isolated from the first batch of samples under anaerobic cultivation conditions. At the phylum level, 24 isolates belonged to Pseudomonadota, 11 to Bacillota, and three to Bacteroidota. The isolates were primarily assigned to Shewanella, Vibrio, Enterococcus, and Frondibacter at the genus level. Among these, the most frequently identified species were Shewanella algae, Vibrio mytili, Enterococcus hirae, Frondibacter mangrovi, and Paraclostridium benzoelyticum, with Shewanella algae being the most prevalent isolate (Figure 3). Seven species were found under both anaerobic and aerobic conditions on M1 media: Psychrobacter celer, Shewanella algae, Alcaligenes faecalis subsp. phenolicus, Enterococcus hirae, Vibrio plantisponsor, Psychrobacter alimentarius, and Vibrio mytili. A comparison of the microorganisms isolated under anaerobic and aerobic conditions revealed both similarities and differences. In both conditions, most isolates belonged to the phylum Pseudomonadota. However, notable differences were observed at the genus and species levels, highlighting the impact of cultivation conditions on the diversity of isolates.
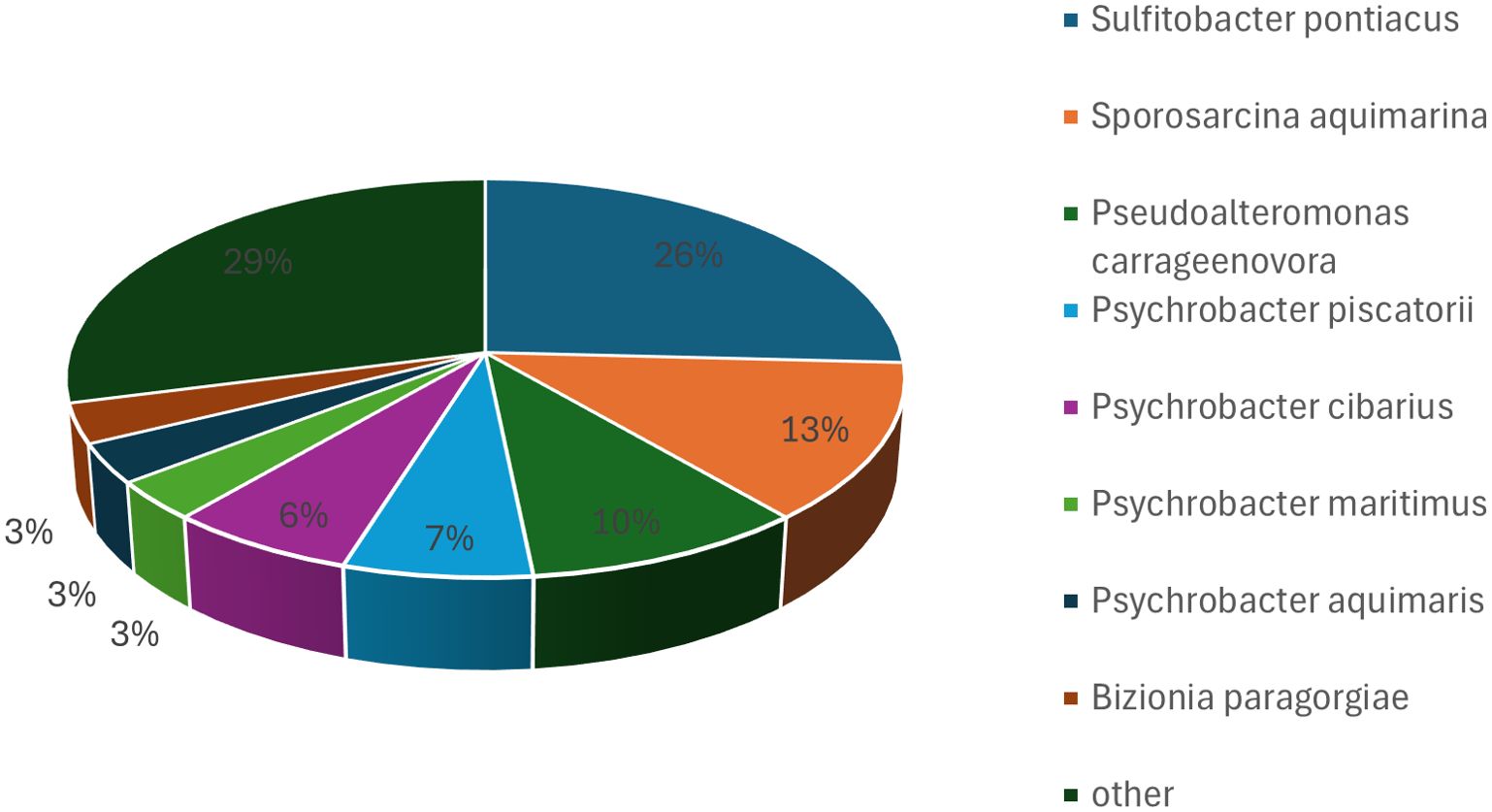
Figure 3. Bacterial taxa cultured from hawksbill feces using M1 medium under anaerobic conditions. The most frequently identified species were Shewanella algae, Vibrio mytili, Enterococcus hirae, Frondibacter mangrovi, and Paraclostridium benzoelyticum, with Shewanella algae being the most abundant isolate.
3.3 Bacterial taxa cultured from hawksbill feces on M2 under aerobic conditions
The 16S rRNA genes of bacterial isolates grown on aerobic M2 medium were amplified and sequenced. Sequence alignment revealed that 15 bacterial strains were isolated, representing 9 distinct species. At the phylum level, eight isolates belonged to Pseudomonadota, four to Bacteroidota, two to Bacillota, and one to Actinomycetota. At the genus level, the isolates were primarily assigned to Psychrobacter and Frondibacter. Among the 15 isolates, the most frequently identified species was Psychrobacter maritimus, which accounted for five isolates (33.33%). The second most common species was Frondibacter mangrovi (Figure 4). Notably, four isolates showed 16S rRNA gene similarity to Frondibacter mangrovi (94.63%) and Glutamicibacter halophytocola (98.99%), but DDH similarity was < 70%, indicating that these isolates represent potentially novel species. These isolates were selected for further analysis, including complete genomic sequencing. A comparison of bacterial isolates based on the medium composition (M1 versus M2) revealed both similarities and differences. At the phylum level, Pseudomonadota was the most commonly identified group in isolates from both media. However, the addition of jellyfish extract to M2 resulted in a significant reduction in the number of isolates compared to M1 under aerobic conditions. Differences were also observed at the genus and species levels.
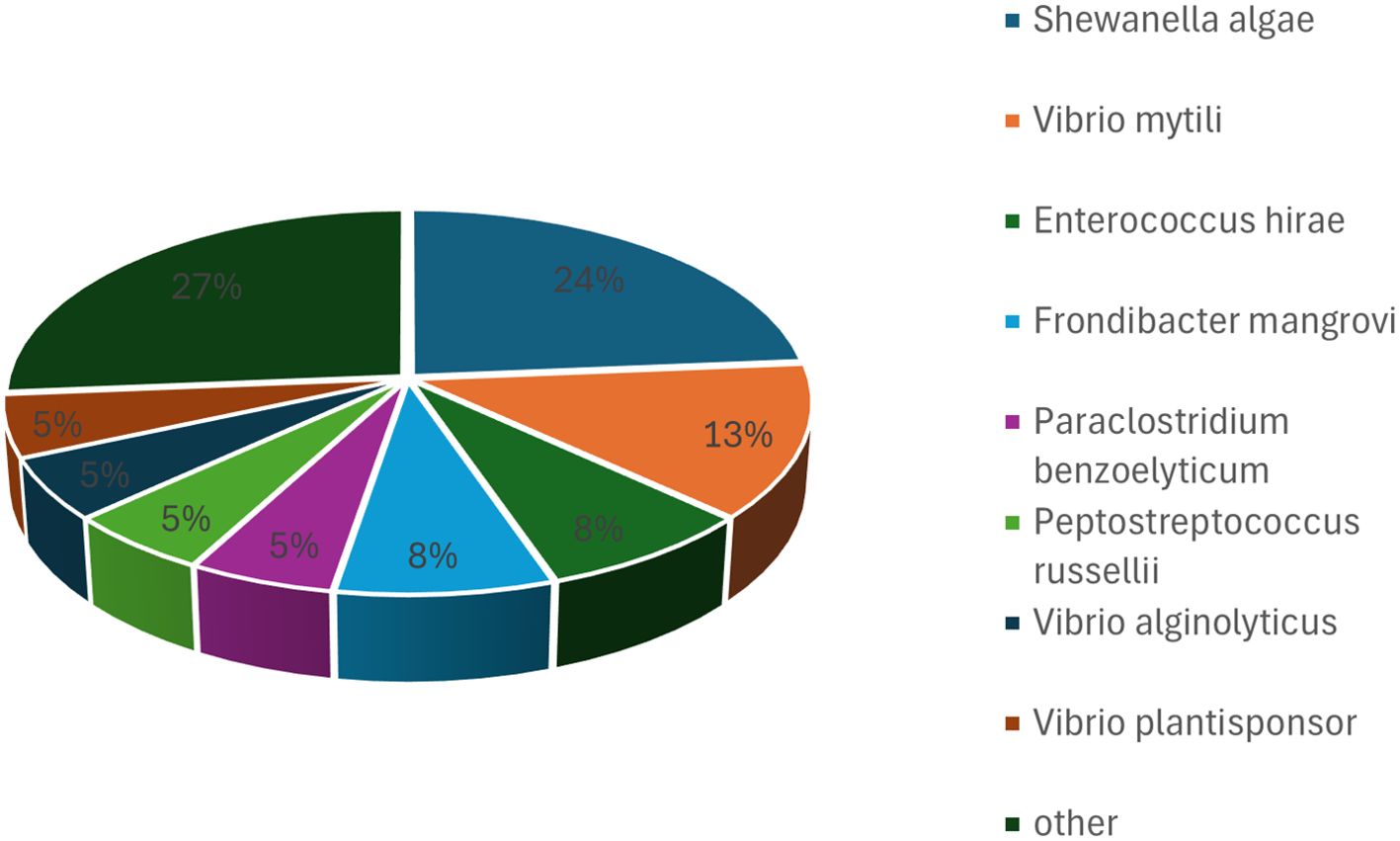
Figure 4. Bacterial taxa cultured from hawksbill feces using M2 medium under aerobic conditions. Among the 15 isolates, the dominant species was Psychrobacter maritimus, accounting for 5 isolates (33.33%), followed by Frondibacter mangrovi.
3.4 Phylogenetic analysis and gene enrichment for potential new species
Phylogenetic analysis revealed that six potential new species isolated from hawksbill feces were grouped into five distinct clusters (Figure 5). Among them, four species (designated as DM7, DM50, DM51, and DM55) were isolated from hawksbill feces on M1 medium under aerobic conditions, whereas two species (DS3 and DS5) were isolated from hawksbill feces on M2 medium under aerobic conditions. Interestingly, isolates from different media did not exhibit a distinct phylogenetic distribution. For instance, both DM7 and DS3 belonged to the genus Glutamicibacter. DM50, DM51, and DM55 were classified under the genera Mesonia, Rhodococcus, and Alcanivorax, respectively. Notably, DS5 was assigned to the genus Enterococcus_B, which differed from its initial classification based on 16S similarity. Further functional analyses were conducted using the whole genomic sequences of these potential new species, with gene functions annotated through KEGG pathway analysis. Genes from DS3 and DS5 showed partial similarity, both being enriched in carbohydrate metabolism pathways, although they also exhibited functional differences (Supplementary Figures 1, 2). Similarly, genes from DM7, DM50, DM51, and DM55 shared some common features, particularly their enrichment in amino acid metabolism pathways, while also displaying unique functional characteristics (Supplementary Figures 3-6). Notably, the gene functions of DS3 and DM7 exhibited a high degree of similarity, with both being enriched in pathways related to amino acid metabolism, carbohydrate metabolism, membrane transport, nucleotide metabolism, and translation. This observation suggests that species within the same genus may share highly similar functional profiles.
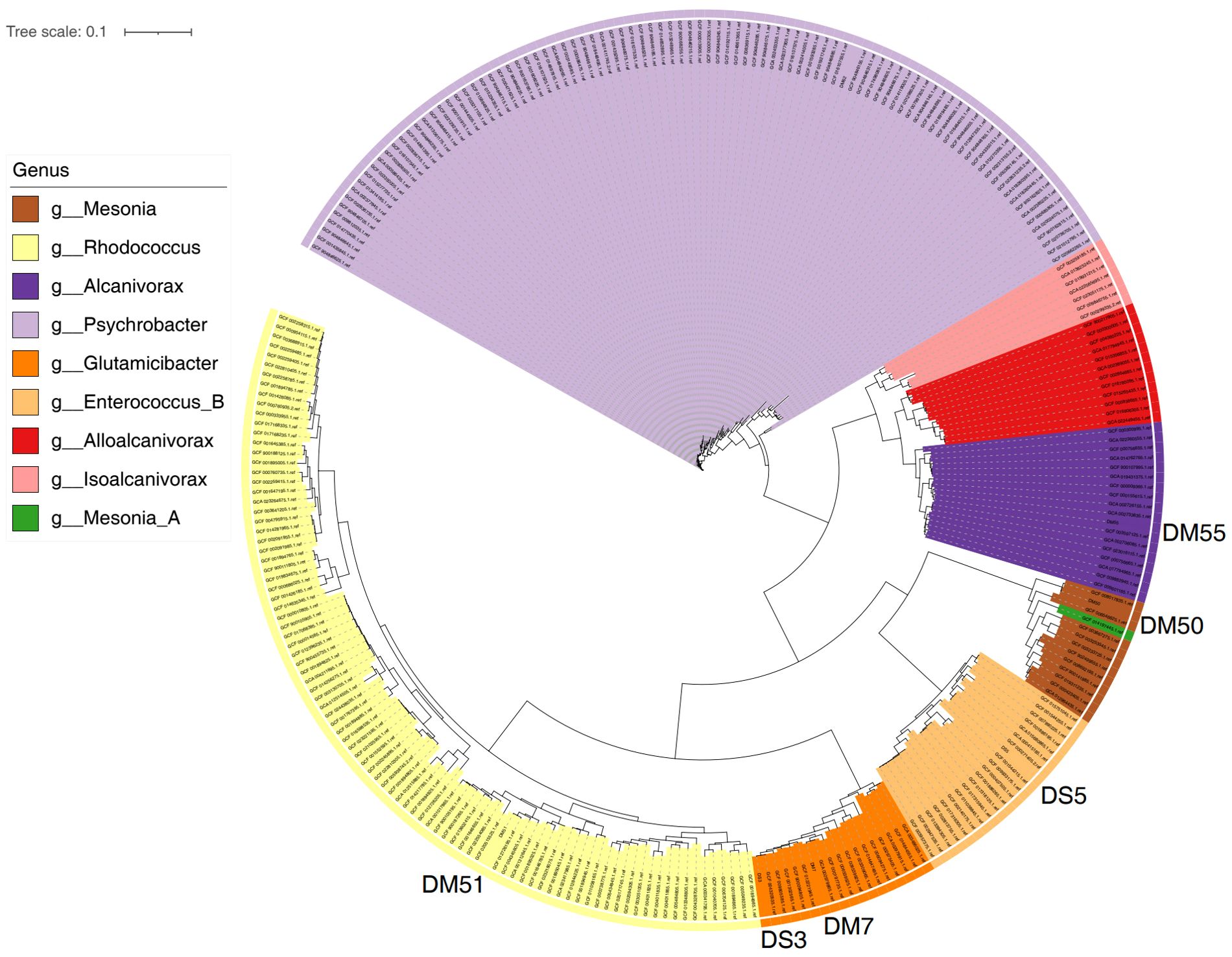
Figure 5. Phylogenetic analysis of novel bacterial species isolated from hawksbill feces. Four new species (DM7, DM50, DM51, and DM55) were identified from samples cultured on M1 medium under aerobic conditions, and two new species (DS3 and DS5) were isolated from samples cultured on M2 medium under aerobic conditions.
4 Discussion
The animal gut microbiota, primarily composed of bacteria, plays a crucial role in influencing the host’s fitness, phenotype, and health by performing various functions traditionally attributed to the host itself (Rath and Dorrestein, 2012; Falony et al., 2016). However, the sheer abundance and diversity of bacteria in the gut pose significant challenges for researchers attempting to identify their taxa. Although metagenomic methods enable the sequencing of entire metagenomes, offering species-level assignments and complete genomic assemblies (Chen et al., 2022a), culture-dependent approaches seem redundant. Nonetheless, culture-dependent techniques remain invaluable for detecting microorganisms present in low concentrations that may be undetected by metagenomics. In this study, we observed significant differences in bacterial taxa identified via culture-dependent techniques compared to those identified using metagenomic methods. Our previous metagenomic studies revealed that the gut bacteria of hawksbills primarily belong to the phyla Bacteroidetes, Firmicutes, and Fusobacteria (Chen et al., 2022b). In contrast, the majority of bacterial isolates obtained through culture-dependent methods belonged to the phyla Pseudomonadota (formerly Proteobacteria), Actinomycetota, and Bacillota (formerly Firmicutes). Pseudomonadota is among the predominant phyla in natural environments, including marine ecosystems (Higgins et al., 2018; Ferro et al., 2019; Scaccia et al., 2021), and on the carapace of hawksbill turtles (Loghmannia et al., 2023), yet remains relatively underexplored compared to other groups (Zhang et al., 2018; Radaic and Kapila, 2021). Similarly, Actinomycetota, which are widely distributed across diverse ecosystems such as soil, freshwater, and marine environments (Stach and Bull, 2005; Mohammadipanah and Wink, 2015), are recognized as a valuable source of antibiotics (Aguilar et al., 2024). Bacillota, another major phylum, is strongly associated with marine organisms and is ubiquitous in terrestrial and aquatic environments (Viju et al., 2021). At the genus level, metagenomic analyses from our previous studies identified Cetobacterium, Bacteroides, and Paludibacter as the most abundant genera in hawksbill gut microbiota (Chen et al., 2022b). In contrast, Psychrobacter emerged as the dominant genus in our culture-dependent results, followed by Glutamicibacter and Vibrio. These discrepancies between the two approaches underscore the complementary nature of metagenomic and culture-dependent techniques.
In this study, more isolates were obtained from M1 medium under aerobic conditions than under anaerobic conditions. Under aerobic conditions, the strains isolated from M1 medium predominantly belonged to three species: Psychrobacter celer, Sulfitobacter pontiacus, and Shewanella algae. Psychrobacter celer is of particular interest because of its psychrophilic or psychrotolerant characteristics, which make it a promising candidate for industrial applications (Rodrigues et al., 2009). Therefore, hawksbill fecal samples could serve as a valuable resource for isolating Psychrobacter celer. Another species, Sulfitobacter pontiacus, previously isolated from the Black Sea, is well known for producing a highly active, soluble AMP-independent sulfite oxidase. This enzyme has significant potential for use in biosensor systems to detect sulfite in food and beverages (Muffler and Ulber, 2008). Thus, the isolation of Sulfitobacter pontiacus from hawksbill fecal samples represents a practical approach for accessing this biotechnologically valuable species. Under anaerobic conditions, Shewanella algae was the most frequently isolated species. Shewanella spp. are known for their ability to thrive in remarkably diverse environmental conditions (Ivanova et al., 2001; Yang et al., 2007; Caro-Quintero et al., 2011; Naghoni et al., 2017; Cha et al., 2020). However, it is important to note that Shewanella algae is increasingly recognized as a conditionally pathogenic bacterium, capable of causing infections in humans and aquatic animals (Han et al., 2017; Lemaire et al., 2020). This highlights the need for caution when studying its presence in hawksbill turtles. Vibrio mytili was another notable species isolated from the M1 medium under aerobic conditions. Various studies have reported Vibrio spp. infections in fish worldwide (Destoumieux-Garzón et al., 2020). Additionally, Vibrio mytili has demonstrated high resistance to erythromycin in groupers (Amalina et al., 2019). Further investigation is necessary to assess the potential impact of this bacterium on the health of hawksbill turtles.
The top-hit species isolated from strains cultured on M2 medium included Psychrobacter maritimus, along with other notable species such as Pseudomonas caeni, Carnobacterium jeotgali, Enterococcus hirae, Halomonas venusta, Winogradskyella crassostreae, and Shewanella algae. The addition of jellyfish to the medium appeared to significantly reduce the number of isolates, potentially due to the effects of their venom (Premmaneesakul and Sithisarankul, 2019). Psychrobacter maritimus, originally isolated from coastal sea-ice and sediment samples (Romanenko et al., 2004), is a psychrophilic Bacillus. This genus has been proposed as a potential probiotic to enhance feed utilization, digestive enzyme activity, and innate immunity in groupers (Yang et al., 2011). Future research should explore the effects of Psychrobacter maritimus on hawksbill turtle health. Notably, Pseudomonas caeni is a carbapenem- and tigecycline-resistant bacterium that was previously isolated from chicken feces (Lu et al., 2023). Carnobacterium jeotgali, initially identified in Korean traditional fermented food (Kim et al., 2009), has also been detected in the rumen microbiota (Islam et al., 2021). Enterococcus hirae, the first reported anticancer probiotic (Viaud et al., 2013), has more recently emerged as one of the most common opportunistic pathogenic bacteria in poultry (Avberšek et al., 2021). Halomonas venusta is a moderately halophilic marine bacterium (Martinez-Abarca et al., 2021), whereas Winogradskyella species are widely associated with marine organisms (Schellenberg et al., 2017; Franco et al., 2018). Winogradskyella has also been linked to amebic-induced fish gill diseases (Embar-Gopinath et al., 2005, 2006). However, the specific role of Winogradskyella crassostreae in hawksbill turtles remains unclear. Further investigations are needed to determine its potential effects on the health of hawksbill turtles.
Further, based on 16S rRNA gene sequencing, we found a notable difference in species diversity between the first and second batches of bacterial isolates cultured on M1 medium under aerobic conditions. The ages of the first and second batches of hawksbill turtles were different, which may lead to differences in the diversity of isolated species. Indeed, Filek et al. (2024) reported that cloacal bacterial diversity and structure changes with age in loggerhead sea turtles.
Similarity analysis based on 16S rRNA gene sequencing and DDH indicated that ten isolated strains were likely novel species. Subsequently, these strains were selected for complete genome sequencing. Phylogenetic analysis revealed that the ten isolates belonged to five distinct genera. Gene enrichment analysis further suggested that species within the same genus exhibited highly similar functional profiles. In summary, culture-dependent techniques are essential and a valuable complement to metagenomic approaches. Finally, our study had some limitations. First, our samples did not allow us to identify the gut bacteria of wild hawksbill turtles (Eretmochelys imbricata) because all samples were collected from the hawksbill turtles (Eretmochelys imbricata) in a nature reserve. Further studies are needed to comparatively analyze the fecal bacteria of captive and wild hawksbill turtles (Eretmochelys imbricata) to identify and differentiate between the original and introduced bacteria.
5 Conclusion
In this study, we employed culture-dependent techniques to detect bacterial taxa that may be overlooked using metagenomic methods. A total of 161 bacterial strains were successfully isolated using two types of media under aerobic and anaerobic conditions. The most frequently identified species included Psychrobacter celer, Shewanella algae, Sulfitobacter pontiacus, Vibrio mytili, Enterococcus hirae, and Psychrobacter maritimus. In addition, six new species were identified based on 16S rRNA gene sequence similarity and DDH analysis. These findings suggest that culture-dependent methods may be a valuable complement to metagenomic approaches for the detection and identification of bacterial isolates.
Data availability statement
The original sequence data for the six new species are available at the SRA under the accession number PRJNA1262675 (https://www.ncbi.nlm.nih.gov/sra/PRJNA1262675).
Ethics statement
The animal studies were approved by the Committee on Biomedical Ethics of Huizhou University. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent was obtained from the owners for the participation of their animals in this study.
Author contributions
HL: Supervision, Methodology, Conceptualization, Validation, Investigation, Writing – review & editing, Formal Analysis, Writing – original draft, Project administration, Funding acquisition. YC: Investigation, Funding acquisition, Writing – review & editing, Conceptualization, Formal Analysis, Data curation, Methodology. ZX: Writing – review & editing, Investigation, Conceptualization, Resources, Visualization. YL: Investigation, Writing – review & editing, Resources, Validation, Formal Analysis, Conceptualization, Methodology. RX: Visualization, Resources, Investigation, Conceptualization, Writing – review & editing, Software, Methodology, Formal Analysis.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This study was supported by the Educational Commission of Guangdong Province, China (Grant No. 2020ZDZX1050) and the Science and Technology Planning Project of Huizhou City, China (Grant No. 2020SC0302019).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2025.1671894/full#supplementary-material
Supplementary Figure 1 | KEGG enrichment analysis of genes from DS3.
Supplementary Figure 2 | KEGG enrichment analysis of genes from DS5.
Supplementary Figure 3 | KEGG enrichment analysis of genes from DM7.
Supplementary Figure 4 | KEGG enrichment analysis of genes from DM50.
Supplementary Figure 5 | KEGG enrichment analysis of genes from DM51.
Supplementary Figure 6 | KEGG enrichment analysis of genes from DM55.
References
Aguilar C., Alwali A., Mair M., Rodriguez-Orduña L., Contreras-Peruyero H., Modi R., et al. (2024). Actinomycetota bioprospecting from ore-forming environments. Microb. Genom. 10, 1253. doi: 10.1099/mgen.0.001253
Amalina N. Z., Santha S., Zulperi D., Amal M. N. A., Yusof M. T., Zamri-Saad M., et al. (2019). Prevalence, antimicrobial susceptibility and plasmid profiling of Vibrio spp. isolated from cultured groupers in Peninsular Malaysia. BMC Microbiol. 19, 251. doi: 10.1186/s12866-019-1624-2
Arizza V., Vecchioni L., Caracappa S., Sciurba G., Berlinghieri F., Gentile A., et al. (2019). New insights into the gut microbiome in loggerhead sea turtles Caretta caretta stranded on the Mediterranean coast. PloS One 14 (8), e0220329. doi: 10.1371/journal.pone.0220329
Avberšek J., Mićunović J., Šemrov N., and Ocepek M. (2021). Surveillance of the source of poultry infections with Enterococcus hirae and Enterococcus cecorum in Slovenia and E. hirae antibiotic resistance patterns.w. New Microbiol. 44, 210–216.
Bjorndal K. A. (1985). Nutritional ecology of sea turtles. Copeia 1985, 736–751. doi: 10.2307/1444767
Campos P., Guivernau M., Prenafeta-Boldú F. X., and Cardona L. (2018). Fast acquisition of a polysaccharide fermenting gut microbiome by juvenile green turtles Chelonia mydas after settlement in coastal habitats. Microbiome 6, 69. doi: 10.1186/s40168-018-0454-z
Capri F. C., Prazzi E., Casamento G., Gambino D., Cassata G., and Alduina R. (2023). Correlation between microbial community and hatching failure in loggerhead sea turtle Caretta caretta. Microb. Ecol. 86, 1923–1933. doi: 10.1007/s00248-023-02197-8
Caro-Quintero A., Deng J., Auchtung J., Brettar I., Höfle M. G., Klappenbach J., et al. (2011). Unprecedented levels of horizontal gene transfer among spatially co-occurring Shewanella bacteria from the Baltic Sea. ISME J. 5, 131–140. doi: 10.1038/ismej.2010.93
Cha Q. Q., Ren X. B., Sun Y. Y., He X. Y., Su H. N., Chen X. L., et al. (2020). Shewanella polaris sp. nov., a psychrotolerant bacterium isolated from Arctic brown algae. Int. J. Syst. Evol. Microbiol. 70, 2096–2102. doi: 10.1099/ijsem.0.004022
Chaumeil P. A., Mussig A. J., Hugenholtz P., and Parks D. H. (2019). GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 36, 1925–1927. doi: 10.1093/bioinformatics/btz848
Chen Y., Xia Z., and Li H. (2022a). Comparative analysis of the fecal bacterial communities of hawksbill sea turtles (Eretmochelys imbricata) and green sea turtles (Chelonia mydas). FEMS Microbiol. Lett. 369, fnac073. doi: 10.1093/femsle/fnac073
Chen Y., Xia Z., and Li H. (2022b). Metagenomic comparison of gut communities between hawksbills (Eretmochelys imbricata) and green sea turtles (Chelonia mydas). Arch. Microbiol. 204, 450. doi: 10.1007/s00203-022-03073-8
Chun J., Oren A., Ventosa A., Christensen H., Arahal D. R., da Costa M. S., et al. (2018). Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int. J. Syst. Evol. Microbiol. 68, 461–466. doi: 10.1099/ijsem.0.002516
de Goffau M. C., Luopajärvi K., Knip M., Ilonen J., Ruohtula T., Härkönen T., et al. (2013). Fecal microbiota composition differs between children with β-cell autoimmunity and those without. Diabetes 62, 1238–1244. doi: 10.2337/db12-0526
Destoumieux-Garzón D., Canesi L., Oyanedel D., Travers M. A., Charrière G. M., Pruzzo C., et al. (2020). Vibrio-bivalve interactions in health and disease. Environ. Microbiol. 22, 4323–4341. doi: 10.1111/1462-2920.15055
Embar-Gopinath S., Butler R., and Nowak B. (2005). Influence of salmonid gill bacteria on development and severity of amoebic gill disease. Dis. Aquat. Organ. 67, 55–60. doi: 10.3354/dao067055
Embar-Gopinath S., Crosbie P., and Nowak B. F. (2006). Concentration effects of Winogradskyella sp. on the incidence and severity of amoebic gill disease. Dis. Aquat. Organ. 73, 43–47. doi: 10.3354/dao073043
Falony G., Joossens M., Vieira-Silva S., Wang J., Darzi Y., Faust K., et al. (2016). Population level analysis of gut microbiome variation. Science 352, 560–564. doi: 10.1126/science.aad3503
Ferro P., Vaz-Moreira I., and Manaia C. M. (2019). Betaproteobacteria are predominant in drinking water: are there reasons for concern? Crit. Rev. Microbiol. 45, 649–667. doi: 10.1080/1040841X.2019.1680602
Filek K., Vuković B. B., Žižek M., Kanjer L., Trotta A., Di Bello A., et al. (2024). Loggerhead sea turtles as hosts of diverse bacterial and fungal communities. Microb. Ecol. 87, 79. doi: 10.1007/s00248-024-02388-x
Flint H. J., Scott K. P., Louis P., and Duncan S. H. (2012). The role of the gut microbiota in nutrition and health. Nat. Rev. Gastroenterol. Hepatol. 9, 577–589. doi: 10.1038/nrgastro.2012.156
Franco A., Busse H. J., Schubert P., Wilke T., Kämpfer P., and Glaeser S. P. (2018). Winogradskyella pocilloporae sp. nov., isolated from healthy tissue of the coral Pocillopora damicornis. Int. J. Syst. Evol. Microbiol. 68, 1689–1696. doi: 10.1099/ijsem.0.002731
Griffith B. C., Weiss B. L., Aksoy E., Mireji P. O., Auma J. E., Wamwiri F. N., et al. (2018). Analysis of the gut-specific microbiome from field-captured tsetse flies, and its potential relevance to host trypanosome vector competence. BMC Microbiol. 18, 146. doi: 10.1186/s12866-018-1284-7
Grigorescu A. S., Renoz F., Sabri A., Foray V., Hance T., and Thonart P. (2018). Accessing the hidden microbial diversity of aphids: an illustration of how culture-dependent methods can be used to decipher the insect microbiota. Microb. Ecol. 75, 1035–1048. doi: 10.1007/s00248-017-1092-x
Han Z., Sun J., Lv A., Sung Y., Shi H., Hu X., et al. (2017). Isolation, identification and characterization of Shewanella algae from reared tongue sole, Cynoglossus semilaevis Günther. Aquaculture 468, 356–362. doi: 10.1016/j.aquaculture.2016.10.038
Higgins D., Pal C., Sulaiman I. M., Jia C., Zerwekh T., Dowd S. E., et al. (2018). Application of high-throughput pyrosequencing in the analysis of microbiota of food commodities procured from small and large retail outlets in a U.S. metropolitan area – a pilot study. Food Res. Int. 105, 29–40. doi: 10.1016/j.foodres.2017.10.057
Islam M., Kim S. H., Son A. R., Ramos S. C., Jeong C. D., Yu Z., et al. (2021). Seasonal influence on rumen microbiota, rumen fermentation, and enteric methane emissions of Holstein and Jersey steers under the same total mixed ration. Anim. (Basel) 11, 1184. doi: 10.3390/ani11041184
IUCN (2021). The IUCN red list of threatened species. Available online at: https://www.iucnredlist.org/.
Ivanova E. P., Sawabe T., Gorshkova N. M., Svetashev V. I., Mikhailov V. V., Nicolau D. V., et al. (2001). Shewanella japonica sp. nov. Int. J. Syst. Evol. Microbiol. 51, 1027–1033. doi: 10.1099/00207713-51-3-1027
Kim M., Oh H. S., Park S. C., and Chun J. (2014). Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int. J. Syst. Evol. Microbiol. 64, 346–351. doi: 10.1099/ijs.0.059774-0
Kim M. S., Roh S. W., Nam Y. D., Yoon J. H., and Bae J. W. (2009). Carnobacterium jeotgali sp. nov., isolated from a Korean traditional fermented food. Int. J. Syst. Evol. Microbiol. 59, 3168–3171. doi: 10.1099/ijs.0.010116-0
Lagier J. C., Armougom F., Million M., Hugon P., Pagnier I., Robert C., et al. (2012). Microbial culturomics: paradigm shift in the human gut microbiome study. Clin. Microbiol. Infect. 18, 1185–1193. doi: 10.1111/1469-0691.12023
Lambeth S. M., Carson T., Lowe J., Ramaraj T., Leff J. W., Luo L., et al. (2015). Composition, diversity and abundance of gut microbiome in prediabetes and type 2 diabetes. J. Diabetes Obes. 2, 1–7. doi: 10.15436/2376-0949.15.031
Lemaire O. N., Méjean V., and Iobbi-Nivol C. (2020). The Shewanella genus: ubiquitous organisms sustaining and preserving aquatic ecosystems. FEMS Microbiol. Rev. 44, 155–170. doi: 10.1093/femsre/fuz031
Liu C. and Shao Z. (2005). Alcanivorax dieselolei sp. nov., a novel alkane-degrading bacterium isolated from sea water and deep-sea sediment. Int. J. Syst. Evol. Microbiol. 55, 1181–1186. doi: 10.1099/ijs.0.63443-0
Loghmannia J., Nasrolahi A., and Dobretsov S. (2023). Epibiotic bacteria on the carapace of hawksbill and green sea turtles. Biofouling 39, 385–398. doi: 10.1080/08927014.2023.2220275
Lu X., Zhang L., Peng K., Wang Q., Liu R., Wang Z., et al. (2023). Characterisation of a novel tigecycline resistance Gene tet (X22) and its coexistence with blaNDM-1 in a Pseudomonas caeni isolate. Int. J. Antimicrob. Agents 62, 106961. doi: 10.1016/j.ijantimicag.2023.106961
Manichanh C., Rigottier-Gois L., Bonnaud E., Gloux K., Pelletier E., Frangeul L., et al. (2006). Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut 55, 205–211. doi: 10.1136/gut.2005.073817
Martinez-Abarca F., Hernández-Soto L. M., Ramírez-Saad H. C., and Aguirre-Garrido J. F. (2021). Complete genome sequence of Halomonas venusta type strain DSM 4743, a moderately halophilic marine bacterium. Microbiol. Resour. Announc. 10, e00144–e00121. doi: 10.1128/MRA.00144-21
McNally K. L., Bowen J. L., Brisson J. O., Kennedy A., and Innis C. J. (2021). Evaluation of the respiratory microbiome and the use of tracheal lavage as a diagnostic tool in Kemp’s ridley sea turtles (Lepidochelys kempii). Anim. (Basel) 11, 2927. doi: 10.3390/ani11102927
Meier-Kolthoff J. P., Klenk H. P., and Göker M. (2014). Taxonomic use of DNA G+C content and DNA-DNA hybridization in the genomic age. Int. J. Syst. Evol. Microbiol. 64, 352–356. doi: 10.1099/ijs.0.056994-0
Meylan A. (1988). Spongivory in hawksbill turtles: a diet of glass. Science 239, 393–395. doi: 10.1126/science.239.4838.393
Mohammadipanah F. and Wink J. (2015). Actinobacteria from arid and desert habitats: Diversity and biological activity. Front. Microbiol. 6. doi: 10.3389/fmicb.2015.01541
Muffler K. and Ulber R. (2008). Fed-batch cultivation of the marine bacterium Sulfitobacter pontiacus using immobilized substrate and purification of sulfite oxidase by application of membrane adsorber technology. Biotechnol. Bioeng. 99, 870–875. doi: 10.1002/bit.21631
Naghoni A., Emtiazi G., Amoozegar M. A., Cretoiu M. S., Stal L. J., Etemadifar Z., et al. (2017). Microbial diversity in the hypersaline Lake Meyghan, Iran. Sci. Rep. 7, 11522. doi: 10.1038/s41598-017-11585-3
Premmaneesakul H. and Sithisarankul P. (2019). Toxic jellyfish in Thailand. Int. Marit. Health 70, 22–26. doi: 10.5603/IMH.2019.0004
Radaic A. and Kapila Y. L. (2021). The oralome and its dysbiosis: new insights into oral microbiome-host interactions. Comput. Struct. Biotechnol. J. 19, 1335–1360. doi: 10.1016/j.csbj.2021.02.010
Rath C. M. and Dorrestein P. C. (2012). The bacterial chemical repertoire mediates metabolic exchange within gut microbiomes. Curr. Opin. Microbiol. 15, 147–154. doi: 10.1016/j.mib.2011.12.009
Rodrigues D. F., da C Jesus E. C., Ayala-del-Río H. L., Pellizari V. H., Gilichinsky D., Sepulveda-Torres L., et al. (2009). Biogeography of two cold-adapted genera: Psychrobacter and Exiguobacterium. Int. Soc Microb. Ecol. J. 3, 658–665. doi: 10.1038/ismej.2009.25
Romanenko L. A., Lysenko A. M., Rohde M., Mikhailov V. V., and Stackebrandt E. (2004). Psychrobacter maritimus sp. nov. and Psychrobacter arenosus sp. nov., isolated from coastal sea ice and sediments of the Sea of Japan. Int. J. Syst. Evol. Microbiol. 54, 1741–1745. doi: 10.1099/ijs.0.63096-0
Scaccia N., Vaz-Moreira I., and Manaia C. M. (2021). The risk of transmitting antibiotic resistance through endophytic bacteria. Trends Plant Sci. 26, 1213–1226. doi: 10.1016/j.tplants.2021.09.001
Scheelings T., Moore R., Van T., Klaassen M., and Reina R. (2020). The gut bacterial microbiota of sea turtles differs between geographically distinct populations. Endangered Species Res. 42, 95–108. doi: 10.3354/esr01042
Schellenberg J., Busse H. J., Hardt M., Schubert P., Wilke T., Kämpfer P., et al. (2017). Winogradskyella haliclonae sp. Nov., isolated from a marine sponge of the genus Haliclona. Int. J. Syst. Evol. Microbiol. 67, 4902–4910. doi: 10.1099/ijsem.0.002192
Scher J. U., Ubeda C., Artacho A., Attur M., Isaac S., Reddy S. M., et al. (2015). Decreased bacterial diversity characterizes the altered gut microbiota in patients with psoriatic arthritis, resembling dysbiosis in inflammatory bowel disease. Arthritis Rheumatol. 67, 128–139. doi: 10.1002/art.38892
Schippa S., Iebba V., Barbato M., Di Nardo G., Totino V., Checchi M. P., et al. (2010). A distinctive ‘microbial signature’ in celiac pediatric patients. BMC Microbiol. 10, 175. doi: 10.1186/1471-2180-10-175
Sommer F., Anderson J. M., Bharti R., Raes J., and Rosenstiel P. (2017). The resilience of the intestinal microbiota influences health and disease. Nat. Rev. Microbiol. 15, 630–638. doi: 10.1038/nrmicro.2017.58
Stach J. E. M. and Bull A. T. (2005). Estimating and comparing the diversity of marine Actinobacteria. Antonie Leeuwenhoek. 87, 3–9. doi: 10.1007/s10482-004-6524-1
Turnbaugh P. J., Hamady M., Yatsunenko T., Cantarel B. L., Duncan A., Ley R. E., et al. (2009). A core gut microbiome in obese and lean twins. Nature 457, 480–484. doi: 10.1038/nature07540
Valdes A. M., Walter J., Segal E., and Spector T. D. (2018). Role of the gut microbiota in nutrition and health. BMJ 361, k2179. doi: 10.1136/bmj.k2179
Viaud S., Saccheri F., Mignot G., Yamazaki T., Daillère R., Hannani D., et al. (2013). The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science 342, 971–976. doi: 10.1126/science.1240537
Viju N., Punitha S. M. J., and Satheesh S. (2021). An analysis of biosynthesis gene clusters and bioactivity of marine bacterial symbionts. Curr. Microbiol. 78, 2522–2533. doi: 10.1007/s00284-021-02535-4
Wang M., Karlsson C., Olsson C., Adlerberth I., Wold A. E., Strachan D. P., et al. (2008). Reduced diversity in the early fecal microbiota of infants with atopic eczema. J. Allergy Clin. Immunol. 121, 129–134. doi: 10.1016/j.jaci.2007.09.011
Yang S. H., Lee J. H., Ryu J. S., Kato C., and Kim S. J. (2007). Shewanella donghaensis sp. nov., a psychrophilic, piezosensitive bacterium producing high levels of polyunsaturated fatty acid, isolated from deep-sea sediments. Int. J. Syst. Evol. Microbiol. 57, 208–212. doi: 10.1099/ijs.0.64469-0
Yang H.-L., Sun Y.-Z., Ma R.-L., Zhang C.-X., and Lin W.-Y. (2011). Effect of dietary administration of Psychrobacter sp. on the growth, feed utilization, digestive enzymes and immune responses of grouper Epinephelus coioides. Aquacult. Nutr. 17, e733–e740.
Ye S. H., Siddle K. J., Park D. J., and Sabeti P. C. (2019). Benchmarking metagenomics tools for 568 taxonomic classification. Cell 178, 779–794. doi: 10.1016/j.cell.2019.07.010
Yoon S. H., Ha S. M., Kwon S., Lim J., Kim Y., Seo H., et al. (2017). Introducing EzBioCloud: a taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 67, 1613–1617. doi: 10.1099/ijsem.0.001755
Keywords: culture-dependent methods, 16S rRNA gene similarity, DDH similarity, bacterial isolates, gut microbiota
Citation: Li H, Chen Y, Xia Z, Lin Y and Xu R (2025) Analysis of the gut-specific microbiome from critically endangered hawksbill sea turtles (Eretmochelys imbricata) using a culture-dependent approach. Front. Mar. Sci. 12:1671894. doi: 10.3389/fmars.2025.1671894
Received: 23 July 2025; Accepted: 15 September 2025;
Published: 03 October 2025.
Edited by:
Vikash Kumar, Central Inland Fisheries Research Institute (ICAR), IndiaReviewed by:
Javad Loghmannia, Shahid Beheshti University, IranBiswajit Mandal, Central Inland Fisheries Research Institute (ICAR), India
Copyright © 2025 Li, Chen, Xia, Lin and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongwei Li, bGh3Y2F1QDE2My5jb20=
†These authors share first authorship