 Maxime Gauberti
Maxime Gauberti Fanny Potzeha1
Fanny Potzeha1 Denis Vivien
Denis Vivien Sara Martinez de Lizarrondo
Sara Martinez de Lizarrondo- 1Normandie Univ, UNICAEN, Institut National de la Santé et de la Recherche Médicale UMR-S U1237, “Physiopathology and Imaging of Neurological Disorders” PhIND, Caen, France
- 2Department of Diagnostic Imaging and Interventional Radiology, Centre Hospitalier Universitaire Caen Côte de Nacre, Caen, France
- 3Department of Clinical Research, Centre Hospitalier Universitaire Caen, Caen, France
Ischemic stroke is one of the leading causes of death and disability worldwide. Current medical management in the acute phase is based on the activation of the fibrinolytic cascade by intravenous injection of a plasminogen activator (such as tissue-type plasminogen activator, tPA) that promotes restauration of the cerebral blood flow and improves stroke outcome. Unfortunately, the use of tPA is associated with deleterious effects such as hemorrhagic transformation, symptomatic brain edema, and angioedema, which limit the efficacy of this therapeutic strategy. Preclinical and clinical evidence suggests that intravenous thrombolysis generates large amounts of bradykinin, a peptide with potent pro-inflammatory, and pro-edematous effects. This tPA-triggered generation of bradykinin could participate in the deleterious effects of thrombolysis and is a potential target to improve neurological outcome in tPA-treated patients. The present review aims at summarizing current evidence linking thrombolysis, bradykinin generation, and neurovascular damage.
Introduction
Stroke represents the second leading cause of mortality in the world, the third leading cause of reduced disability-adjusted life years and is a major drain on public health-care funding (1). Most strokes are ischemic in nature (80%) and occur when an artery that supplies blood to the brain is blocked by a blood clot (2, 3). The aim of the initial medical management is to limit the extension of brain damage by recanalizing the occluded artery (4). To date, thrombolysis (injection of tissue-type plasminogen activator (tPA), sometimes combined with endovascular thrombectomy), is the only pharmacological treatment for acute ischemic stroke (5). It acts by activating an inactive zymogen, plasminogen, into its active form, plasmin, which is itself able to degrade fibrin, the main constituent of blood clots (6).
In clinical trials, intravenous tPA injection has been shown to improve neurological outcome when injected up to 4.5 h after the appearance of symptoms (7). Yet, tPA administration is not devoid of side effects (6, 8). For instance, when tPA is administered after 6 h, meta-analysis of clinical trials showed that it worsens the neurological outcome (9). Moreover, even when injected in the right therapeutic window, current evidence supports that tPA increases by up to 60% the mortality of ischemic stroke patients in the first 7 days (8). This increased mortality is partly explained by an increased rate of hemorrhagic transformation. It has also been demonstrated that stroke patients treated with tPA present more severe brain edema, with malignant edema being 2.7 fold more frequent after tPA administration in patients with occlusion of the internal carotid artery (10). These data suggest that besides its beneficial effects on the cerebral blood flow, tPA administration also exerts deleterious effects on the neurovascular unit (11). Therefore, blockade of these deleterious effects may significantly improve the clinical efficacy of tPA mediated thrombolysis and increase its therapeutic window. However, to date, the mechanisms driving the deleterious effects of tPA remain poorly understood and probably include both plasmin-dependent (12) and plasmin-independent processes (11).
There is now compelling evidence supporting that tPA-induced bradykinin generation could be one of the drivers of the deleterious effects of tPA (13–15). Bradykinin is a potent pro-inflammatory and pro-edematous peptide, that is generated by proteolytic cleavage from its precursor high-molecular weight kininogen (HMWK) (16, 17). HMWK cleavage is classically triggered by the activation of the contact system, which is also composed of factor XII (FXII), factor XI (FXI), and plasma prekallikrein (PPK). The contact system is started by FXII binding to negatively charged surfaces such as platelet polyphosphate and mast cell heparin (18). Notably, there is a significant generation of bradykinin during thrombolysis in stroke patients (14, 19). Given the multiple roles for bradykinin in the neurovascular unit described so far including increase in BBB permeability, inflammatory cytokines upregulation, release of glutamate by astrocytes and microglial activation (all potential pathogenic players in ischemic stroke) (17), it is tempting to speculate that the generation of bradykinin triggered by tPA administration participates in its deleterious effects.
The aim of this present review is to summarize current evidence linking tPA administration, bradykinin generation, and neurovascular damage in stroke patients. In the first part of this review, we will present clinical evidence supporting the existence of deleterious effects of tPA on the neurovascular unit. Then, we will describe the molecular pathways linking tPA administration to bradykinin generation. Finally, we will recapitulate the most recent preclinical evidence on the effects of bradykinin in the ischemic brain.
The Effects of tPA on the Neurovascular Unit: Clinical Evidence
Intravenous thrombolysis is now an established stroke treatment for patients presenting within 4.5 h after stroke onset (20). Indeed, large, multicenter, randomized, and controlled clinical trials demonstrated that intravenous tPA administration improves stroke outcome in this subset of patients (7). Although tPA is beneficial for most patients, there is a growing body of evidence that tPA may also harm, in particular when tPA is administered more than 6 h after symptom onset. It is therefore tempting to speculate that the outcome after tPA administration in stroke patients depends on a balance between its beneficial and deleterious effects. Exhaustive literature reviews have been published on the preclinical effects of tPA on the neurovascular unit (6, 11, 21). Here, we will focus only on clinical evidence and extract the data that are relevant to the understanding of the pathophysiology of stroke in the context of tPA administration.
Beneficial Effects
Regarding the beneficial effects of tPA, they appear mainly dependent on its ability to promote arterial recanalization. For instance, in patients presenting occlusion of the termination of the internal carotid artery, in which the rate of recanalization is estimated to be <5% (22), the ICARO study (case-control study) demonstrated that there is no beneficial effect of tPA administration vs. placebo (10). Additionally, patients who recanalize after tPA administration have a far better outcome than patients without recanalization as demonstrated by a large number of studies (23). However, tPA seems also to benefit patients without visible arterial occlusion at presentation (24). Although this can be explained by the presence of distal microemboli not seen on angiography, we cannot exclude the existence of beneficial effects of tPA that are independent of its ability to promote arterial recanalization (25). These putative fibrinolysis-independent effects are relevant for the design of adjuvant treatments for tPA, since these treatments should aim at mitigating the deleterious effects of tPA without jeopardizing its beneficial effects.
Symptomatic Intracranial Hemorrhage
The main deleterious effects of tPA highlighted by clinical studies are symptomatic intracranial hemorrhage (sICH), symptomatic brain edema and angioedema. sICH is probably the most feared complication of tPA administration with a mortality rate approaching 50% (26). The risk of sICH after tPA administration ranges from 2 to 7% according to the definition used. Most of them occur within 24 h after tPA administration with a median time ranging from 5 to 10 h. sICH are related to the dose of tPA administered, as recently demonstrated in a randomized study comparing the standard dose of tPA (0.9 mg/kg) to a lower dose (0.6 mg/kg) that showed sICH rates of 2.1% vs. 1.0% respectively (27). In contrast, time from symptom onset to tPA administration is not significantly associated with sICH risk according to a large meta-analysis (28), although there is a trend for a higher risk when tPA administration is delayed (7, 9). Interestingly, an increased risk of sICH has also been demonstrated after administration of other plasminogen activators such as streptokinase or urokinase (29, 30). Since these plasminogen activators are structurally unrelated, this suggests that the pro-hemorrhagic effect of tPA is dependent on plasminogen activation. Reperfusion and subsequent reperfusion-injury do not seem to explain alone the pro-hemorrhagic effect of tPA since hemorrhage can occur in areas remote from the infarcted tissue (31) and in the absence of reperfusion (32). Given that human imaging studies demonstrated that breakdown of the blood brain barrier (BBB) precedes sICH (33), these data altogether suggest that tPA administration leads to BBB breakdown and subsequent sICH by a plasminogen dependent mechanism. As a concomitant phenomenon, tPA administration induces a coagulopathy characterized by fibrinogen depletion and prolongation of prothrombin and partial thromboplastin times which may also increase the risk of sICH (34, 35).
Symptomatic Brain Edema
Symptomatic brain edema is a less known complication of tPA treatment, although its effect on mortality is significant (36). In the IST-3 trial for instance, 3% of tPA-treated patients (47/1515) died from symptomatic swelling of the ischemic lesion. Symptomatic brain edema is usually defined as a severe brain swelling with mass effect on imaging. The independent effects of tPA on brain edema are difficult to measure since the reperfusion achieved using tPA is associated with a reduced ischemic lesion size and therefore, a reduced brain swelling. In this regard, the case-control ICARO study that involved stroke patients with occlusion of the internal carotid artery is interesting, since the recanalization rate is expected to be <5% in this subset of patients, allowing to observe the effects of tPA almost independently of arterial recanalization (10). In this study involving 253 patients per group, the rate of fatal symptomatic brain edema (referred as malignant edema) was 8.3% in tPA-treated patients vs. 3.1% in controls (p = 0.013). As a comparison, the rate of fatal sICH was only 2.8% in tPA-treated patients, suggesting that the contribution of symptomatic brain edema to mortality is larger than sICH in this patient subset. In a cohort of 943 tPA-treated patients, Strbian et al. studied the parameters associated with the development of brain edema (36). They found imaging evidence of brain edema in 28% of tPA-treated patients and severe forms in 10%. The factors associated with edema development were higher baseline National Institutes of Health Stroke Scale, presence of hyperdense cerebral artery sign or early infarct signs, and longer treatment delays. Altogether, these data suggest that tPA administration promotes brain edema, especially in patients showing early sign of brain infarction on imaging and experiencing longer onset-to-tPA delays. Whether this pro-edema effect is dependent on plasminogen activation cannot be easily retrieved from clinical data.
Angioedema
Angioedema is a rare complication of tPA administration, occurring in ~2% of patients with ischemic stroke (37). It manifests usually as a hemi-orolingual edema during or shortly after tPA administration that can be life threatening. Angioedema is associated with the use of angiotensin-converting enzyme (ACE) inhibitors, which is found in 45% of patients with angioedema (19). This finding unraveled bradykinin as the causative factor for tPA-induced angioedema. Indeed, ACE is not only able to process angiotensin into angiotensin-II, it is also responsible for the degradation of bradykinin. Thus, the plasma half-life of bradykinin is extended in patients taking ACE inhibitors, thereby increasing the plasmatic concentration in case of bradykinin generation, that can more easily reach a level triggering angioedema (38). Although the occurrence of angioedema does not significantly influence neurological outcome, patients with angioedema tend to have more sICH and more malignant brain edema according to the largest series published so far (37). Overall, these clinical data support the following mechanism: tPA administration triggers bradykinin generation, which is itself able to promote angioedema, especially in patients taking ACE inhibitors. In line with the tendency for a higher incidence of sICH and malignant brain edema in patients presenting angioedema, it is tempting to speculate that the effects of tPA-induced bradykinin generation are not restricted to the occurrence of angioedema but also influence the evolution of the ischemic lesion in the brain.
Molecular Mechanisms of Bradykinin Generation After tPA Administration
The first convincing evidence of bradykinin generation after tPA administration was published by Agostoni et al. They showed that intravenous administration of tPA or streptokinase in patients with acute myocardial infarction was associated with a cleavage of HMWK, the precursor of bradykinin (13). Interestingly, more HMWK was cleaved after streptokinase than after tPA administration, pointing toward plasmin as a key player in this mechanism, since it has been demonstrated that streptokinase generates more plasmin than tPA after injection (39). Later, the same results were obtained in ischemic stroke patients treated with tPA (14). To be able to interfere with the generation of bradykinin after tPA administration, it is therefore relevant to understand first how tPA generates plasmin and then, how plasmin generates bradykinin (40).
tPA-Induced Plasmin Generation
tPA cleaves plasminogen at its Arg561—Val562 peptide bond, into the active fibrinolytic enzyme, plasmin (41). Although tPA can activate plasminogen into plasmin in the plasma, its efficiency is 100 times higher in the presence of fibrin as a cofactor (42). This role for fibrin as a cofactor for plasminogen activation explains the relative fibrin specificity of tPA compared to streptokinase. This provides a mechanistic explanation for the above mentioned higher systemic generation of plasmin when streptokinase is used as a fibrinolytic agent. Moreover, this implies that, at the level of the thrombus which contains large amounts of fibrin, there is a massive generation of plasmin and therefore, a potentially intense generation of bradykinin. Accordingly, this locally generated bradykinin could act on the ischemic brain downstream of the occlusion site.
Plasmin-Induced Bradykinin Generation
The mechanism by which plasmin generates bradykinin is debated. The first described mechanism is based on the fact that plasmin is able to directly cleave HMWK and generates bradykinin in vitro (14, 19). This has been demonstrated using electrophoresis of HMWK after incubation with tPA, plasminogen, tPA plus plasminogen and plasmin. Whereas no cleavage occurred in the presence of tPA or plasminogen when added alone, a significant cleavage of HMWK occurred in the presence of plasmin or tPA plus plasminogen. This cleavage was accompanied with bradykinin release as assessed by quantification of immunoreactive bradykinin. Although this direct mechanism is apparent in vitro, whether it occurs in vivo and participates in bradykinin generation after intravenous injection of tPA remains unknown.
An alternative, indirect mechanism of bradykinin generation after plasmin formation has been described by Simão et al. They first observed that the incubation of human plasma with tPA leads to the formation of active plasma kallikrein (PKal), the enzyme of the contact phase responsible for “physiologic” HMWK cleavage (43). Interestingly, the increase in PKal activity after addition of tPA was lost in Factor XII (FXII) and plasminogen deficient plasma (15). A set of experiments allowed to dissect the complete cascade: tPA activates plasminogen into plasmin, plasmin activates FXII into FXIIa and finally, FXIIa activates plasma prekallikrein into PKal. In line with these in vitro findings, intravenous injection of tPA in mice leads to an increase in PKal activity. As expected, this increased PKal activity leads to a cleavage of HMWK. This indirect mechanism of bradykinin formation is probably the dominant mechanism after tPA administration, since tPA does not induce HMWK cleavage in FXII or plasma prekallikrein deficient mice, even if there is plasmin generation in these animals.
Besides activation of the contact phase and subsequent bradykinin generation, there is also a growing body of evidence suggesting that tPA administration induces a widespread hemovascular dysfunction also involving activations of the coagulation cascade and the complement system (Figure 1) (44–46). All these biochemical cascades can participate in the deleterious effects of tPA on the neurovascular unit in the acute stroke settings.
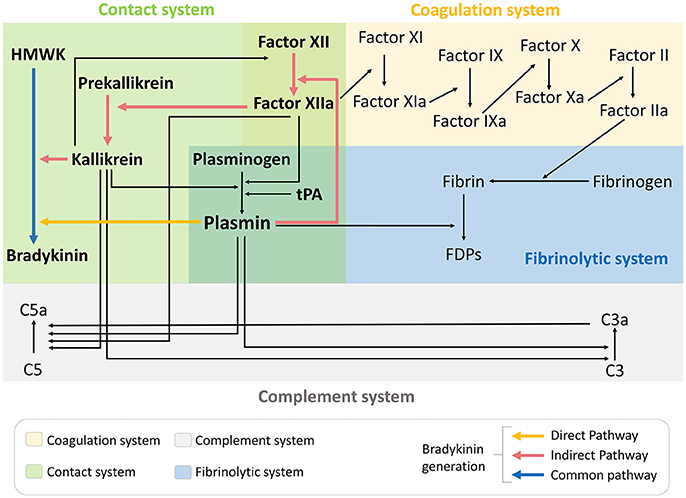
Figure 1. tPA administration drives hemovascular dysfunction. Thrombolytic therapy in the acute phase of ischemic stroke promotes activation not only of the fibrinolytic pathway but also coagulation, complement and contact phase systems. FDPs, fibrin degradation products; HMWK, high molecular weight kininogen; tPA, tissue-type plasminogen activator.
Bradykinin Generation as a Unifying Mechanism for the Effects of tPA on the Neurovascular Unit
Bradykinin is a nonapeptide (Arg1-Pro2-Pro3-Gly4-Phe5-Ser6-Pro7-Phe8-Arg9) released from HMWK after its cleavage. Bradykinin acts through two receptors: kinin 1 receptor (B1R) and kinin 2 receptor (B2R). It requires further processing to Des-Arg9-Bradykinin by peptidases (such as carboxypeptidase N) to act through B1R. B2R is almost ubiquitously expressed in the brain (including in the cerebral cortex) whereas B1R is inducible and overexpressed in pathological conditions. B1R and B2R have been described on all the main cellular cell types of the brain including neurons, astrocytes, microglia, oligodendrocytes, and endothelial cells (47). Interestingly, both receptors are upregulated in the ischemic brain (17). Thus, it has been demonstrated that B1R and B2R are significantly overexpressed at the protein level as early as 4 h post-stroke onset in mice (48). It is therefore tempting to speculate that the effects of tPA-induced bradykinin generation on the neurovascular unit are time-dependent with a larger effect size when tPA administration is delayed. B2R activation has also been shown to induce tPA release from endothelial cells, potentially further amplifying bradykinin generation (49).
Contradictory Findings Regarding Bradykinin Impact in Stroke
The implication of the kallikrein-kinin system in neurological disorders has been recently reviewed (17). Here, we will focus mostly on the effects of bradykinin relevant to thrombolysis and stroke. The effects of endogenous bradykinin on the ischemic brain are numerous and the results of experimental studies are often contradictory (47). For instance, genetic deletion or pharmacological blockade of B2R has been shown to protect from stroke in a number of studies, mainly performed using the transient mechanical vascular occlusion model of stroke (filament) (50–52). In contrast, some studies reported the absence or even deleterious effects of B2R inhibition using similar experimental models (48, 53). In addition, intravenous injection or overexpression of tissue kallikrein (able to generate kallidin, a bradykinin analog, after cleavage of low-molecular kininogen) has beneficial effects in stroke, which are dependent on B2R activation (54, 55). Regarding B1R, the data are more homogenous since most studies described deleterious effects on ischemic lesion size, brain edema and post-stroke inflammation (48, 56–58). It is however difficult to isolate the contribution of bradykinin on each of these pathophysiological processes, since reduction of lesion size itself is associated with reduced brain edema and inflammation. In this context and in the absence of experimental studies investigating the effects of bradykinin receptor blockade after tPA administration, we can only make hypotheses regarding the impact of bradykinin in the deleterious effects of tPA (Figure 2), as summarized thereafter.
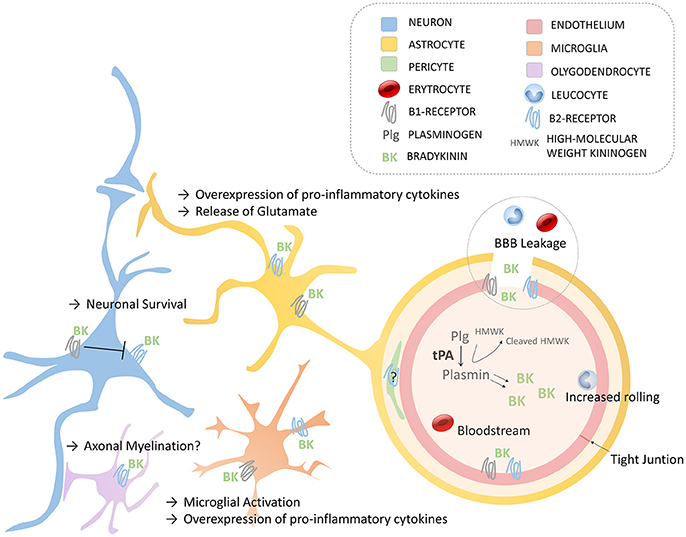
Figure 2. Schematic of the main roles of bradykinin in the Neurovascular Unit via B1- and B2- Receptors. Endothelium (with tight junctions), astrocytes, neurons, oligodendrocytes, microglia, and pericytes compose the neurovascular unit. BBB, blood-brain barrier; BK, bradykinin; HMWK, High molecular Weight Kininogen; Plg, plasminogen; tPA, tissue-type plasminogen activator.
Bradykinin Enhances BBB Permeability
The most documented effect of bradykinin on the neurovascular unit is an increase in the permeability of the BBB. From a mechanistical point of view, bradykinin binding on either B1R or B2R promotes intracellular calcium release, leading to downregulation of claudin-5 (one of the main mediators of BBB integrity) and subsequent BBB leakage (17). Accordingly, B2R agonists (and to a lesser extent, B1R agonists) have been used as a mean to transiently increase BBB permeability for maximizing brain bioavailability of chemotherapeutic drugs (59, 60). In the context of stroke, this bradykinin-induced BBB leakage is considered deleterious since it allows macromolecules to reach the brain parenchyma, thereby promoting brain edema, and weakens the BBB, thereby increasing the risk of hemorrhagic transformation (61). In line with these hypotheses, in mice with thrombotic occlusion of the middle cerebral artery, tPA administration increases hemorrhage transformation, infarct volume and edema (15). Of note, these deleterious effects of tPA are prevented by a PKal inhibitor, pointing toward contact phase activation (and possibly bradykinin generation) as the main pathophysiological player. This might explain why tPA-treated patients with angioedema, who supposedly generate more bradykinin, tend to have more sICH and more malignant brain edema (37). When tPA administration is delayed, the increased expression of bradykinin receptors by the ischemic brain might also explain the higher rate of symptomatic brain edema and the trend for a higher risk of hemorrhagic transformation (9, 36).
Pro-inflammatory Effects of Bradykinin
Beside increasing BBB permeability, bradykinin displays pro-inflammatory effects in the cerebral circulation (17). For instance, direct intra-carotid injection of bradykinin in gerbils induces an increased rolling of leukocytes and adhesion of platelets on the brain endothelium, an effect dependent on B2R (62). The mechanisms by which bradykinin mediates post-stroke inflammation is classically described as involving the activation of B2R (on endothelial cells, astrocytes, neurons, microglia), the release of arachidonic acid and the activation of cyclo-oxygenase (COX) enzymes (63–65). Bradykinin is also a potent stimulator of the expression of other inflammatory mediators such as cytokines and acts as a leucocyte chemoattractant (66). More recently, inhibition of B1R (but not B2R) has been shown to reduce post-ischemic inflammation in a transient middle cerebral artery occlusion model, in terms of proinflammatory cytokines expression and extent of brain invasion by immune cells (48), but these results can be biased by the dramatic reduction of ischemic lesion size in case of B1R inhibition. Overall, it is reasonable to assume that bradykinin has pro-inflammatory effects through both B1R and B2R activations (17). Bradykinin may also activate mast cells, that play a significant role in tPA-induced brain edema and hemorrhagic transformation (67, 68). All these bradykinin-mediated mechanisms could, at the very least, participate to the pro-inflammatory effects of tPA, as observed in several experimental studies (69). However, brain inflammation after stroke is a dual-edged sword, with inflammatory cascades stimulating deleterious but also potentially beneficial pathways, including post-injury repair processes (70). Thus, the net effect of additional bradykinin generation triggered by tPA-administration in the context of acute ischemic stroke is difficult to predict and could explain the Janus-faced nature of tPA in this context (6).
Bradykinin and Neuronal Cell Death
Bradykinin can also directly influence neuronal cell fate in the ischemic brain, as recently reviewed (71). In particular, B2R protects against neuronal cell death via multiple molecular mechanisms including anti-apoptotic, anti-oxidative, anti-inflammatory, anti-autophagic, and anti-excitotoxic effects (excitotoxicity refers to the damage triggered by excessive stimulation of neurons by neurotransmitters). Bradykinin displays also a B2R dependent vasodilatory effect on brain microvessels that could improve cerebral perfusion and indirectly mitigate neuronal cell death (63). Thus, bradykinin-mediated B2R activation may participate in the beneficial effects of tPA administration in patients without visible arterial occlusion (24). In vitro experiments suggest that there is a crosstalk between B1R and B2R during pro-excitotoxic challenges in neurons (72). It has been reported that bradykinin triggers a neuroprotective cascade via B2R activation in hippocampal neurons conferring protection against excitotoxicity. Notably, co-activation of B1R blocks this protective effect. Since carboxypeptidases allowing bradykinin processing and subsequent B1R activation are highly expressed in the brain (47), this suggests that the beneficial effects of B2R activation in stroke might be blocked by concomitant B1R activation after bradykinin processing. This suggests that B1R or carboxypeptidase blockade could promote neuroprotection, especially in the context of thrombolysis for acute ischemic stroke. Since B1R expression is induced by brain ischemia, this may also explain why the beneficial effects of tPA administration are larger when thrombolysis is performed in the first hours following stroke onset (9).
Conclusion and Therapeutic Perspectives
Even if there are probably several molecular effectors of the deleterious effects of tPA on the neurovascular unit in acute ischemic stroke, current pathophysiological knowledge suggests that bradykinin could be one of the most important players. In particular, the pro-inflammatory and pro-edematous effects of bradykinin could explain the increased incidence of sICH and symptomatic edema in tPA-treated patients. If true, angioedema would be only the tip of the iceberg of bradykinin effects in tPA-treated stroke patients. In line with this hypothesis, inhibition of bradykinin receptors or bradykinin processing peptidases could improve the efficacy of tPA and stroke outcome. The challenge resides in the adequate choice of the inhibitors in order to block the deleterious effects without jeopardizing the beneficial effects of bradykinin. The repercussion of such strategy could be large not only in acute ischemic stroke but also in other diseases where pharmacological thrombolysis is indicated including intraventricular brain hemorrhage, acute myocardial infarction, pulmonary embolism, and limb ischemia. This should also stimulate the development of new thrombolytic strategies that act independently of plasmin generation, such as a disintegrin and metalloprotease with thrombospondin type I repeats-13 (ADAMTS-13) or N-Acetylcysteine (73, 74).
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by the Institut National de la Santé Et de la Recherche Médicale (INSERM).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Feigin VL, Norrving B, Mensah GA. Global burden of stroke. Circ Res. (2017) 120:439–48. doi: 10.1161/CIRCRESAHA.116.308413
2. Sudlow CL, Warlow CP. Comparable studies of the incidence of stroke and its pathological types: results from an international collaboration. International stroke incidence collaboration. Stroke (1997) 28:491–9. doi: 10.1161/01.STR.28.3.491
3. Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, et al. Executive summary: heart disease and stroke statistics−2016 update: a report from the american heart association. Circulation (2016) 133:447–54. doi: 10.1161/CIR.0000000000000366
4. Moussaddy A, Demchuk AM, Hill MD. Thrombolytic therapies for ischemic stroke: triumphs and future challenges. Neuropharmacology (2018) 134(Pt B):272–9. doi: 10.1016/j.neuropharm.2017.11.010
5. Broderick JP, Palesch YY, Demchuk AM, Yeatts SD, Khatri P, Hill MD, et al. Endovascular therapy after intravenous t-PA versus t-PA alone for stroke. N Engl J Med. (2013) 368:893–903. doi: 10.1056/NEJMoa1214300
6. Vivien D, Gauberti M, Montagne A, Defer G, Touze E. Impact of tissue plasminogen activator on the neurovascular unit: from clinical data to experimental evidence. J Cereb Blood Flow Metab. (2011) 31:2119–34. doi: 10.1038/jcbfm.2011.127
7. Wardlaw JM, Murray V, Berge E, Del Zoppo G, Sandercock P, Lindley RL, et al. Recombinant tissue plasminogen activator for acute ischaemic stroke: an updated systematic review and meta-analysis. Lancet (2012) 379:2364–72. doi: 10.1016/S0140-6736(12)60738-7
8. Sandercock P, Wardlaw JM, Lindley RI, Dennis M, Cohen G, Murray G, et al. The benefits and harms of intravenous thrombolysis with recombinant tissue plasminogen activator within 6 h of acute ischaemic stroke (the third international stroke trial [IST-3]): a randomised controlled trial. Lancet (2012) 379:2352–63. doi: 10.1016/S0140-6736(12)60768-5
9. Lees KR, Bluhmki E, Von Kummer R, Brott TG, Toni D, Grotta JC, et al. Time to treatment with intravenous alteplase and outcome in stroke: an updated pooled analysis of ECASS, ATLANTIS, NINDS, and EPITHET trials. Lancet (2010) 375:1695–703. doi: 10.1016/S0140-6736(10)60491-6
10. Paciaroni M, Balucani C, Agnelli G, Caso V, Silvestrelli G, Grotta JC, et al. Systemic thrombolysis in patients with acute ischemic stroke and Internal Carotid ARtery occlusion: the ICARO study. Stroke (2012) 43:125–30. doi: 10.1161/STROKEAHA.111.630624
11. Docagne F, Parcq J, Lijnen R, Ali C, Vivien D. Understanding the functions of endogenous and exogenous tissue-type plasminogen activator during stroke. Stroke (2015) 46:314–20. doi: 10.1161/STROKEAHA.114.006698
12. Niego B, Medcalf RL. Plasmin-dependent modulation of the blood-brain barrier: a major consideration during tPA-induced thrombolysis? J Cereb Blood Flow Metab. (2014) 34:1283–96. doi: 10.1038/jcbfm.2014.99
13. Agostoni A, Gardinali M, Frangi D, Cafaro C, Conciato L, Sponzilli C, et al. Activation of complement and kinin systems after thrombolytic therapy in patients with acute myocardial infarction. A comparison between streptokinase and recombinant tissue-type plasminogen activator. Circulation (1994) 90:2666–70. doi: 10.1161/01.CIR.90.6.2666
14. Marcos-Contreras OA, Martinez De Lizarrondo S, Bardou I, Orset C, Pruvost M, Anfray A, et al. Hyperfibrinolysis increases blood-brain barrier permeability by a plasmin- and bradykinin-dependent mechanism. Blood (2016) 128:2423–34. doi: 10.1182/blood-2016-03-705384
15. Simao F, Ustunkaya T, Clermont AC, Feener EP. Plasma kallikrein mediates brain hemorrhage and edema caused by tissue plasminogen activator therapy in mice after Stroke (2017) 129:2280–90. doi: 10.1182/blood-2016-09-740670
16. Costa-Neto CM, Dillenburg-Pilla P, Heinrich TA, Parreiras-E-Silva LT, Pereira MG, Reis RI, et al. Participation of kallikrein-kinin system in different pathologies. Int Immunopharmacol. (2008) 8:135–42. doi: 10.1016/j.intimp.2007.08.003
17. Nokkari A, Abou-El-Hassan H, Mechref Y, Mondello S, Kindy MS, Jaffa AA, et al. Implication of the kallikrein-kinin system in neurological disorders: quest for potential biomarkers and mechanisms. Prog Neurobiol (2018) 165-7:26–50. doi: 10.1016/j.pneurobio.2018.01.003
18. Weidmann H, Heikaus L, Long AT, Naudin C, Schluter H, Renne T. The plasma contact system, a protease cascade at the nexus of inflammation, coagulation and immunity. Biochim Biophys Acta (2017) 1864:2118–27. doi: 10.1016/j.bbamcr.2017.07.009
19. Molinaro G, Gervais N, Adam A. Biochemical basis of angioedema associated with recombinant tissue plasminogen activator treatment: an in vitro experimental approach. Stroke (2002) 33:1712–6. doi: 10.1161/01.STR.0000017284.77838.87
20. Powers WJ, Rabinstein AA, Ackerson T, Adeoye OM, Bambakidis NC, Becker K, et al. Guidelines for the early management of patients with acute ischemic stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke (2018) 49:e46–110. doi: 10.1161/STR.0000000000000158
21. Fredriksson L, Lawrence DA, Medcalf RL. tPA modulation of the blood-brain barrier: a unifying explanation for the pleiotropic effects of tPA in the CNS. Semin Thromb Hemost. (2017) 43:154–68. doi: 10.1055/s-0036-1586229
22. Bhatia R, Hill MD, Shobha N, Menon B, Bal S, Kochar P, et al. Low rates of acute recanalization with intravenous recombinant tissue plasminogen activator in ischemic stroke: real-world experience and a call for action. Stroke (2010) 41:2254–8. doi: 10.1161/STROKEAHA.110.592535
23. Zangerle A, Kiechl S, Spiegel M, Furtner M, Knoflach M, Werner P, et al. Recanalization after thrombolysis in stroke patients: predictors and prognostic implications. Neurology (2007) 68:39–44. doi: 10.1212/01.wnl.0000250341.38014.d2
24. Lahoti S, Gokhale S, Caplan L, Michel P, Samson Y, Rosso C, et al. Thrombolysis in ischemic stroke without arterial occlusion at presentation. Stroke (2014) 45:2722–7. doi: 10.1161/STROKEAHA.114.005757
25. Correa F, Gauberti M, Parcq J, Macrez R, Hommet Y, Obiang P, et al. Tissue plasminogen activator prevents white matter damage following stroke. J Exp Med. (2011) 208:1229–42. doi: 10.1084/jem.20101880
26. Yaghi S, Willey JZ, Cucchiara B, Goldstein JN, Gonzales NR, Khatri P, et al. Treatment and outcome of hemorrhagic transformation after intravenous alteplase in acute ischemic stroke: a scientific statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke (2017) 48:e343–61. doi: 10.1161/STR.0000000000000152
27. Anderson CS, Robinson T, Lindley RI, Arima H, Lavados PM, Lee TH, et al. Low-Dose versus standard-dose intravenous alteplase in acute ischemic stroke. N Engl J Med. (2016) 374:2313–23. doi: 10.1056/NEJMoa1515510
28. Messe SR, Pervez MA, Smith EE, Siddique KA, Hellkamp AS, Saver JL, et al. Lipid profile, lipid-lowering medications, and intracerebral hemorrhage after tPA in get with the guidelines-stroke. Stroke (2013) 44:1354–9. doi: 10.1161/STROKEAHA.111.671966
29. Jaillard A, Cornu C, Durieux A, Moulin T, Boutitie F, Lees KR, et al. Hemorrhagic transformation in acute ischemic stroke. The MAST-E study. MAST-E Group. Stroke (1999) 30:1326–32. doi: 10.1161/01.STR.30.7.1326
30. Kidwell CS, Saver JL, Carneado J, Sayre J, Starkman S, Duckwiler G, et al. Predictors of hemorrhagic transformation in patients receiving intra-arterial thrombolysis. Stroke (2002) 33:717–24. doi: 10.1161/hs0302.104110
31. Strbian D, Sairanen T, Meretoja A, Pitkaniemi J, Putaala J, Salonen O, et al. Patient outcomes from symptomatic intracerebral hemorrhage after stroke thrombolysis. Neurology (2011) 77:341–8. doi: 10.1212/WNL.0b013e3182267b8c
32. Ogata J, Yutani C, Imakita M, Ishibashi-Ueda H, Saku Y, Minematsu K, et al. Hemorrhagic infarct of the brain without a reopening of the occluded arteries in cardioembolic stroke. Stroke (1989) 20:876–83. doi: 10.1161/01.STR.20.7.876
33. Kim EY, Na DG, Kim SS, Lee KH, Ryoo JW, Kim HK. Prediction of hemorrhagic transformation in acute ischemic stroke: role of diffusion-weighted imaging and early parenchymal enhancement. AJNR Am J Neuroradiol. (2005) 26:1050–5.
34. Matosevic B, Knoflach M, Werner P, Pechlaner R, Zangerle A, Ruecker M, et al. Fibrinogen degradation coagulopathy and bleeding complications after stroke thrombolysis. Neurology (2013) 80:1216–24. doi: 10.1212/WNL.0b013e3182897015
35. Vandelli L, Marietta M, Gambini M, Cavazzuti M, Trenti T, Cenci MA, et al. Fibrinogen decrease after intravenous thrombolysis in ischemic stroke patients is a risk factor for intracerebral hemorrhage. J Stroke Cerebrovasc Dis. (2015) 24:394–400. doi: 10.1016/j.jstrokecerebrovasdis.2014.09.005
36. Strbian D, Meretoja A, Putaala J, Kaste M, Tatlisumak T. Cerebral edema in acute ischemic stroke patients treated with intravenous thrombolysis. Int J Stroke (2013) 8:529–34. doi: 10.1111/j.1747-4949.2012.00781.x
37. Myslimi F, Caparros F, Dequatre-Ponchelle N, Moulin S, Gautier S, Girardie P, et al. Orolingual angioedema during or after thrombolysis for cerebral ischemia. Stroke (2016) 47:1825–30. doi: 10.1161/STROKEAHA.116.013334
38. Hofman Z, De Maat S, Hack CE, Maas C. Bradykinin: inflammatory product of the coagulation system. Clin Rev Allergy Immunol. (2016) 51:152–61. doi: 10.1007/s12016-016-8540-0
39. Nagai N, Vanlinthout I, Collen D. Comparative effects of tissue plasminogen activator, streptokinase, and staphylokinase on cerebral ischemic infarction and pulmonary clot lysis in hamster models. Circulation (1999) 100:2541–6. doi: 10.1161/01.CIR.100.25.2541
40. De Maat S, Bjorkqvist J, Suffritti C, Wiesenekker CP, Nagtegaal W, Koekman A, et al. Plasmin is a natural trigger for bradykinin production in patients with hereditary angioedema with factor XII mutations. J Allergy Clin Immunol. (2016) 138:1414–23.e1419. doi: 10.1016/j.jaci.2016.02.021
41. Parry MA, Zhang XC, Bode I. Molecular mechanisms of plasminogen activation: bacterial cofactors provide clues. Trends Biochem Sci. (2000) 25:53–9. doi: 10.1016/S0968-0004(99)01521-2
42. Nesheim M, Walker J, Wang W, Boffa M, Horrevoets A, Bajzar L. Modulation of fibrin cofactor activity in plasminogen activation. Ann NY Acad Sci. (2001) 936:247–60. doi: 10.1111/j.1749-6632.2001.tb03513.x
43. Schmaier AH. Assembly, activation, and physiologic influence of the plasma kallikrein/kinin system. Int Immunopharmacol. (2008) 8:161–5. doi: 10.1016/j.intimp.2007.08.022
44. Cap AP. Plasmin: a driver of hemovascular dysfunction. Blood (2016) 128:2375–6. doi: 10.1182/blood-2016-09-735720
45. Zhao XJ, Larkin TM, Lauver MA, Ahmad S, Ducruet AF. Tissue plasminogen activator mediates deleterious complement cascade activation in stroke. PLoS ONE (2017) 12:e0180822. doi: 10.1371/journal.pone.0180822
46. Irmscher S, Doring N, Halder LD, Jo EAH, Kopka I, Dunker C, et al. Kallikrein cleaves C3 and activates complement. J Innate Immun. (2018) 10:94–105. doi: 10.1159/000484257
47. Negraes PD, Trujillo CA, Pillat MM, Teng YD, Ulrich H. Roles of kinins in the nervous system. Cell Transplant. (2015) 24:613–23. doi: 10.3727/096368915X687778
48. Austinat M, Braeuninger S, Pesquero JB, Brede M, Bader M, Stoll G, et al. Blockade of bradykinin receptor B1 but not bradykinin receptor B2 provides protection from cerebral infarction and brain edema. Stroke (2009) 40:285–93. doi: 10.1161/STROKEAHA.108.526673
49. Joseph K, Tholanikunnel BG, Kaplan AP. Cytokine and estrogen stimulation of endothelial cells augments activation of the prekallikrein-high molecular weight kininogen complex: implications for hereditary angioedema. J Allergy Clin Immunol. (2017) 140:170–6. doi: 10.1016/j.jaci.2016.09.032
50. Zausinger S, Lumenta DB, Pruneau D, Schmid-Elsaesser R, Plesnila N, Baethmann A. Effects of LF 16-0687 Ms, a bradykinin B(2) receptor antagonist, on brain edema formation and tissue damage in a rat model of temporary focal cerebral ischemia. Brain Res. (2002) 950:268–78. doi: 10.1016/S0006-8993(02)03053-6
51. Groger M, Lebesgue D, Pruneau D, Relton J, Kim SW, Nussberger J, et al. Release of bradykinin and expression of kinin B2 receptors in the brain: role for cell death and brain edema formation after focal cerebral ischemia in mice. J Cereb Blood Flow Metab. (2005) 25:978–89. doi: 10.1038/sj.jcbfm.9600096
52. Su J, Cui M, Tang Y, Zhou H, Liu L, Dong Q. Blockade of bradykinin B2 receptor more effectively reduces postischemic blood-brain barrier disruption and cytokines release than B1 receptor inhibition. Biochem Biophys Res Commun. (2009) 388:205–11. doi: 10.1016/j.bbrc.2009.07.135
53. Xia CF, Smith RS Jr, Shen B, Yang ZR, Borlongan CV, Chao L, et al. Postischemic brain injury is exacerbated in mice lacking the kinin B2 receptor. Hypertension (2006) 47:752–61. doi: 10.1161/01.HYP.0000214867.35632.0e
54. Tang Y, Shao Y, Su J, Zhou H, Liu L, Ren H, et al. The protein therapy of kallikrein in cerebral ischemic reperfusion injury. Curr Med Chem. (2009) 16:4502–10. doi: 10.2174/092986709789760689
55. Liu L, Liu H, Yang F, Chen G, Zhou H, Tang M, et al. Tissue kallikrein protects cortical neurons against hypoxia/reoxygenation injury via the ERK1/2 pathway. Biochem Biophys Res Commun. (2011) 407:283–7. doi: 10.1016/j.bbrc.2011.02.112
56. Raslan F, Schwarz T, Meuth SG, Austinat M, Bader M, Renne T, et al. Inhibition of bradykinin receptor B1 protects mice from focal brain injury by reducing blood-brain barrier leakage and inflammation. J Cereb Blood Flow Metab. (2010) 30:1477–86. doi: 10.1038/jcbfm.2010.28
57. Sang H, Liu L, Wang L, Qiu Z, Li M, Yu L, et al. Opposite roles of bradykinin B1 and B2 receptors during cerebral ischaemia-reperfusion injury in experimental diabetic rats. Eur J Neurosci. (2016) 43:53–65. doi: 10.1111/ejn.13133
58. Sang H, Qiu Z, Cai J, Lan W, Yu L, Zhang H, et al. Early increased bradykinin 1 receptor contributes to hemorrhagic transformation after ischemic stroke in type 1 diabetic rats. Trans Stroke Res. (2017) 8:597–611. doi: 10.1007/s12975-017-0552-4
59. Borlongan CV, Emerich DF. Facilitation of drug entry into the CNS via transient permeation of blood brain barrier: laboratory and preliminary clinical evidence from bradykinin receptor agonist, Cereport. Brain Res Bull. (2003) 60:297–306. doi: 10.1016/S0361-9230(03)00043-1
60. Cote J, Savard M, Neugebauer W, Fortin D, Lepage M, Gobeil F. Dual kinin B1 and B2 receptor activation provides enhanced blood-brain barrier permeability and anticancer drug delivery into brain tumors. Cancer Biol Ther. (2013) 14:806–11. doi: 10.4161/cbt.25327
61. Simao F, Feener EP. The effects of the contact activation system on hemorrhage. Front Med. (2017) 4:121. doi: 10.3389/fmed.2017.00121
62. Waldner MJ, Baethmann A, Uhl E, Lehmberg J. Bradykinin-induced leukocyte- and platelet-endothelium interactions in the cerebral microcirculation. Brain Res. (2012) 1448:163–9. doi: 10.1016/j.brainres.2012.02.010
63. Sobey CG, Heistad DD, Faraci FM. Mechanisms of bradykinin-induced cerebral vasodilatation in rats. Evidence that reactive oxygen species activate K+ channels. Stroke (1997) 28:2290–4. discussion: 2295. doi: 10.1161/01.STR.28.11.2290
64. Sarker MH, Hu DE, Fraser PA. Acute effects of bradykinin on cerebral microvascular permeability in the anaesthetized rat. J Physiol. (2000) 528 (Pt 1):177–87. doi: 10.1111/j.1469-7793.2000.00177.x
65. Brian JE Jr, Faraci FM, Moore SA. COX-2-dependent delayed dilatation of cerebral arterioles in response to bradykinin. Am J Physiol Heart Circ Physiol. (2001) 280:H2023–9. doi: 10.1152/ajpheart.2001.280.5.H2023
66. Sobey CG. Bradykinin B2 receptor antagonism: a new direction for acute stroke therapy? Br J Pharmacol. (2003) 139:1369–71. doi: 10.1038/sj.bjp.0705415
67. Strbian D, Karjalainen-Lindsberg ML, Kovanen PT, Tatlisumak T, Lindsberg PJ. Mast cell stabilization reduces hemorrhage formation and mortality after administration of thrombolytics in experimental ischemic stroke. Circulation (2007) 116:411–8. doi: 10.1161/CIRCULATIONAHA.106.655423
68. Oschatz C, Maas C, Lecher B, Jansen T, Bjorkqvist J, Tradler T, et al. Mast cells increase vascular permeability by heparin-initiated bradykinin formation in vivo. Immunity (2011) 34:258–68. doi: 10.1016/j.immuni.2011.02.008
69. Mehra A, Ali C, Parcq J, Vivien D, Docagne F. The plasminogen activation system in neuroinflammation. Biochim Biophys Acta (2016) 1862:395–402. doi: 10.1016/j.bbadis.2015.10.011
70. Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. (2011) 17:796–808. doi: 10.1038/nm.2399
71. Ji B, Cheng B, Pan Y, Wang C, Chen J, Bai B. Neuroprotection of bradykinin/bradykinin B2 receptor system in cerebral ischemia. Biomed Pharmacother. (2017) 94:1057–63. doi: 10.1016/j.biopha.2017.08.042
72. Martins AH, Alves JM, Perez D, Carrasco M, Torres-Rivera W, Eterovic VA, et al. Kinin-B2 receptor mediated neuroprotection after NMDA excitotoxicity is reversed in the presence of kinin-B1 receptor agonists. PLoS ONE (2012) 7:e30755. doi: 10.1371/journal.pone.0030755
73. Denorme F, Langhauser F, Desender L, Vandenbulcke A, Rottensteiner H, Plaimauer B, et al. ADAMTS13-mediated thrombolysis of t-PA-resistant occlusions in ischemic stroke in mice. Blood (2016) 127:2337–45. doi: 10.1182/blood-2015-08-662650
Keywords: contact phase, fibrinolysis, angioedema, inflammation, brain edema, blood-brain barrier, factor XII, kininogen
Citation: Gauberti M, Potzeha F, Vivien D and Martinez de Lizarrondo S (2018) Impact of Bradykinin Generation During Thrombolysis in Ischemic Stroke. Front. Med. 5:195. doi: 10.3389/fmed.2018.00195
Received: 19 March 2018; Accepted: 14 June 2018;
Published: 03 July 2018.
Edited by:
Joost Meijers, University of Amsterdam, NetherlandsReviewed by:
Coen Maas, University Medical Center Utrecht, NetherlandsHeiko Herwald, Lund University, Sweden
Jonas Emsley, University of Nottingham, United Kingdom
Robert Lindsay Medcalf, Monash University, Australia
Copyright © 2018 Gauberti, Potzeha, Vivien and Martinez de Lizarrondo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sara Martinez de Lizarrondo, c21hcnRpbmV6QGN5Y2Vyb24uZnI=