Abstract
The complexity and diversity of the immune response in patients with asthma, chronic obstructive pulmonary disease (COPD), and asthma-COPD overlap present significant challenges for disease management. Relying on a limited number of biomarkers and clinical data is insufficient to fully reveal the immunopathogenesis of these diseases. However, in vitro technologies such as cell analysis, cytokine investigation, and nucleic acid sequencing have provided new insights into the underlying mechanisms of these diseases, leading to the discovery of several biomarkers—including cell degranulation, cell function, secreted cytokines, and single nucleotide polymorphisms—that have potential clinical implications. This paper reviews the immunopathogenesis in asthma, chronic obstructive pulmonary disease, and asthma-COPD overlap and examines the applications of recent in vitro models to detect candidate biomarkers that could enhance diagnostic precision, predict severity, monitor treatments, and develop new treatment strategies. A deeper understanding of the immune response in these diseases, along with the integration of in vitro models into clinical practice, could greatly improve the management of these respiratory diseases, making approaches more personalized and efficient.
Introduction
Asthma and chronic obstructive pulmonary disease (COPD) are complex diseases associated with high morbidity and mortality rates worldwide. According to the Global Initiative for Asthma (GINA) 2023 (1), they serve as umbrella terms for heterogeneous characteristics that overlap in some older patients, resulting in the previously identified asthma-COPD overlap (ACO) syndrome. In 2019, the prevalence of ACO ranged from 1.4% to 2.6% in the general population; among individuals with asthma and COPD, it varied from 19.5% to 33.6% and from 19.3% to 39.9%, respectively (2). The coexistence of these two diseases leads to a specific phenotype influenced by various individual factors, including medical conditions, immune responses, and genetic variation, which contribute to worse clinical outcomes and complicate identification and treatment compared to asthma or COPD alone (3).
The majority of ACO cases often present airway obstruction and respiratory symptoms due to inflammation from excessive immune responses, which are linked to worse outcomes (4, 5). Environmental exposures to microbes, allergens, and air pollution—including cigarette smoke, electronic cigarettes, and particulate matter with a diameter of 2.5 μm or smaller (PM2.5)—induce cooperation between innate and adaptive immune responses, leading to persistent airway inflammation and remodeling by recruiting immune cells and releasing cytokines that differ in inflammatory characteristics (6, 7) (Table 1, Figure 1).
Table 1
| Immune response | Disease characteristics | Dysfunction's effect on immunopathogenesis | References | |||
|---|---|---|---|---|---|---|
| Asthma | COPD | ACO | ||||
| Innate immune cells | ||||||
| Eosinophil | ++++ | + | ++++ | •Correlate with exacerbation frequency •Airway hyperresponsive •Increase type-2 cytokine production •Tissue damage •Mucus hypersecretion •Airway remodeling | (10, 15, 163–166) | |
| Neutrophil | ++ | ++++ | ++++ | •Tissue damage and remodeling •Associate with emphysema •Cytotoxic activity | (46, 164, 165) | |
| Mast cell | +++ | +++ | ++ | •Correlation with the allergy phenotype •Increase airway hyperresponsiveness and type-2 cytokines •Induce airway epithelial barrier disruption •Contribute to fibrosis and airway remodeling via fibroblast crosstalk and epithelial migration | (9, 167–171) | |
| ILC1 | ++ | +++ | N/A | •Increase neutrophil activation via Th1 cytokines | (59, 67, 68, 172, 173) | |
| ILC2 | ++++ | + | N/A | •Increase type 2 inflammation via Th2 cytokines | (68, 172, 173) | |
| ILC3 | ++ | ++ | N/A | •Increase neutrophil activation via Th17 cytokines | (59, 67, 73, 172) | |
| Macrophage | +++ | ++ | ++++ | •Tissue damage •Fibroblast activation leading to airway remodeling •Th1 and Th2 polarization | (9, 174) | |
| Dendritic cell | cDC1 | ++ | +++ | N/A | •Initiate T-cell activation via MHC molecule and co-stimulating cytokines | (175–179) |
| cDC2 | + | ++ | N/A | |||
| moDC | N/A | N/A | N/A | |||
| pDC | + | ++ | N/A | |||
| Adaptive immunity | ||||||
| Th1 | ++ | +++ | N/A | •Drive non-type 2 inflammation phenotype and cell recruitment via pro-inflammatory cytokines and chemokines | (81, 83, 85, 180, 181) | |
| Th2 | +++ | + | N/A | •Drive type-2 inflammation phenotype and cell recruitment via pro-inflammatory cytokines and chemokines | (81, 83, 85, 172, 180) | |
| Th17 | ++ | +++ | N/A | •Drive non-type 2 inflammation phenotype and cell recruitment via pro-inflammatory cytokines and chemokines | (81, 83, 180–183) | |
| Treg | ++ | + | N/A | •Reduce immune suppressive function, leading to immune overactivation | (83, 180, 182, 183) | |
| Tc | + | ++++ | ++ | •Increase cytotoxic activity, causing tissue damage and emphysema | (9, 183–185) | |
| B-cell | ++ | + | N/A | •Increase IgE secretion associated with airway hyperresponsiveness •Autoantibody production | (185, 186) | |
| Cytokines | ||||||
| IL-4 | +++ | + | ++++ | •Th2 polarization •Airway hyper-responsiveness •Tissue remodeling •IgE and IgG1 switching •Mast cell proliferation | (163, 187) | |
| IL-5 | +++ | + | ++ | •Eosinophils proliferation, maturation, survival, and activation | (10, 87, 187, 188) | |
| IL-13 | ++++ | + | ++ | •Th2 polarization •Airway hyper-responsiveness •Tissue remodeling •Goblet cell hyperplasia •ASM hypertrophy and hyperplasia | (87) | |
| IL-8 (CXCL-8) | +++ | ++++ | ++++ | •Neutrophil recruitment | (87, 163) | |
| IL-17 | + | ++++ | ++ | •Fibrocyte proliferation •Neutrophil recruitment and activation •ILC3 polarization | (87, 189) | |
| IL-6 | ++++ | ++ | +++ | •Neutrophil activation | (10, 87, 188) | |
| TNF-α | ++++ | ++ | +++ | •Neutrophil activation •Macrophage activation | (87, 163, 189) | |
| IFN-γ | + | +++ | ++ | •Neutrophil activation | (187) | |
| TGF-β | ++ | ++++ | +++ | •Airway remodeling •Treg polarization •M2c differentiation | (10, 187) | |
| Immune mediators | ||||||
| Periostin | ++++ | ++ | +++ | •Mucus hypersecretion •goblet cell hyperplasia •eosinophil recruitment •Airway remodeling | (15, 87) | |
| YKL-40 | ++ | ++++ | +++ | •Airway remodeling •Airway inflammation | (10, 15, 166) | |
| Nitric oxide | ++++ | ++ | ++ | •Type 2 inflammation •Vasodilation •Bronchodilator | (87, 190) | |
Role and characteristics of inflammatory players in asthma, COPD, and ACO.
N/A, not applicable; + to ++++, from suggested to the proven role compared between asthma, COPD, and ACO.
Figure 1

Immunopathogenesis of airway inflammation in ACO. Figure created with www.BioRender.com. Environmental exposures initiate ACO immunopathogenesis by increasing immune cell infiltration and cytokine secretion, which leads to airway inflammation. In the early stages, various cytokine chain reactions activate effector cells, resulting in tissue damage, bronchoconstriction, and mucus hypersecretion. The inflammatory signals at the damaged sites attract and activate fibroblasts, which move more slowly than immune cells. The prolonged period of chronic inflammation and repeated cycles of tissue injury and abnormal repair processes induce airway remodeling by increasing the extracellular matrix (ECM), airway smooth muscle (ASM) hyperplasia/hypertrophy, and goblet cell hyperplasia, ultimately causing permanent obstruction. DC, Dendritic cell; TCR, T-cell receptor; TSLP, Thymic stromal lymphopoietin; EET, Extracellular matrix; GABA, Gamma-aminobutyric acid; CGRP, Calcitonin gene-related peptide; Th1, T-helper cell type 1; Th2, T-helper cell type 2; Th17, T-helper cell type 17; Treg, regulatory T-cell; Tc, Cytotoxic T-cell; ILC1, Innate lymphoid cell type 1; ILC2, Innate lymphoid cell type 2; ILC3, Innate lymphoid cell type 3; Nϕ, Neutrophil; MC, Mast cell; M1, Alveolar macrophage type 1; M2, Alveolar macrophage type 2 ECM, extracellular matrix; ASM, airway smooth muscle.
This article focuses on novel data regarding immunopathogenesis, laboratory examination, and biomarkers in ACO patients based on in vitro models. Although techniques such as flow cytometry, enzyme-linked immunosorbent assay (ELISA), cytology, histology, and molecular methods are well established in clinical research for understanding ACO diversity, they also shed light on their potential for future clinical applications (8–10). A better understanding of ACO immunopathogenesis through these models aims to establish more effective stratification criteria and identify accurate biomarkers, ultimately improving diagnosis, prediction, prognosis, monitoring, and the identification of novel treatment targets.
The innate immune response in asthma, COPD, and ACO
Airway epithelial cells (AECs)
AECs respond to environmental exposures by releasing alarmins, including thymic stromal lymphopoietin (TSLP), IL-25, and IL-33, which activate type 2 innate lymphoid cells (ILC2s), eosinophils, and mast cells (MCs), driving inflammation and immune cell recruitment (11). In asthma-chronic obstructive pulmonary disease overlap, AECs increase the secretion of other Damage-Associated Molecular Patterns (DAMPs), such as high mobility group box 1 protein (HMGB1), 70-kilodalton heat shock proteins (HSP70), cathelicidin LL-37 (LL-37), and calcium-binding protein A8 (A8), leading to the recruitment of effector cells and the initiation of inflammation (12). Additionally, epithelial cells and other cell types, such as basal, club, ciliated, goblet, ionocytes, endothelial, and fibroblast cells, are induced by IL-4, IL-13, and TGF-β to secrete periostin. The overexpression of periostin results in mucus hypersecretion, goblet cell hyperplasia, and increased matrix metalloproteinase (MMP)-9 secretion, contributing to eosinophil inflammation and permanent airway obstruction (13). IL-4 and IL-13 also trigger AECs to produce eotaxin, which attracts fibroblasts and eosinophils to the inflamed site (14). Moreover, activated AECs, neutrophils, and macrophages can secrete YKL-40, correlating with airway inflammation and remodeling (15).
Cilia on AECs are vital in respiratory disease. Motile cilia play a critical role in lung homeostasis by beating the mucus layer, which traps inhaled pathogens, toxins, and particles out of the airway. Abnormalities in ciliary function, quantity, and structure impair mucus clearance, increasing susceptibility to infections and promoting chronic inflammation (16, 17). Primary cilia act as cellular antennas, influencing the differentiation, migration, and cell fate control of AECs, airway smooth muscle (ASM) cells, and fibroblasts (18). Interestingly, although the quantity of primary cilia does not differ in normal epithelium, it significantly increases in COPD, specifically in the remodeling area (18). The increased antennas may contribute to dysregulated tissue repair, leading to abnormal differentiation and proliferation, ultimately resulting in airway thickening.
In the alveoli, alveolar type 1 cells (AT1) facilitate gas exchange, while alveolar type 2 cells (AT2) repair tissue damage through self-renewal, differentiation into AT1, secretion of TGF-β, and production of surfactant. Cigarette smoke and PM2.5 have been shown to induce apoptosis in AECs and disrupt the differentiation of AT2 into AT1, leading to pulmonary emphysema in COPD murine models (19).
Dendritic cells (DCs)
DCs identify ligands through pattern recognition receptors (PRRs), which trigger them to co-activate naïve helper T cells (Th0) via antigen presentation and cytokine secretion. The cytokines produced by DCs depend on the types of antigens and the subpopulations of DCs, creating the microenvironment around the site of inflammation (20). cDCs can be classified into conventional DCs type 1 (cDC1), conventional DCs type 2 (cDC2), plasmacytoid DCs (pDCs), and monocyte-derived DCs (moDCs) (21). cDC1s and moDCs are the primary sources of IL-12, IFN-α, and IFN-β, which activate T helper type 1 (Th1) polarization. Additionally, they cross-present antigens to CD8+ cytotoxic T cells (Tc), leading to tissue damage in the lungs (20, 21). In contrast, studies in asthmatic mice have shown that cDC2s and moDCs play significant roles in T helper type 2 (Th2) polarization (22). cDC2s also contribute to promoting T helper type 17 (Th17) polarization in lung asthmatic mouse models (23). pDCs produce type-I IFN, TNF-α, and IL-6, inducing Th1, Th17, and regulatory T cell (Treg) polarization (24).
Dysfunction of DCs contributes to the progression of asthma, COPD, and ACO. An imbalance in DC subpopulations correlates with COPD severity, as indicated by the ratio of OX40L to programmed death-ligand 1 (PD-L1) (25). Moreover, reduced DC activation leads to a decrease in Th2 and Th17 signaling, as demonstrated by a reduction in mucus production and cellular recruitment (26).
Alveolar macrophages (AMs)
AMs capture and eliminate foreign antigens while also repairing damaged tissue. Upon activation, they polarize into two subtypes: classically activated macrophages (M1) and alternatively activated macrophages (M2) (27). M1 is triggered by lipopolysaccharide (LPS) and cytokines such as TNF-α, IFN-γ, and IL-12. This subclass can eliminate bacteria, damaged cells, and tumors, and it plays a role in Th1 activation through the secretion of IL-1β, IL-6, IL-12, IL-18, IL-23, TNF-α, inducible nitric oxide synthase (iNOS), reactive oxygen species (ROS), CXCL-1, CXCL-2, CXCL-3, CXCL-9, and CXCL-10 (27–30). Conversely, M2 is notable for its role in tissue repair and maintaining immune homeostasis through the secretion of IL-10, TGF-β, and vascular endothelial growth factor (VEGF) (29, 30). M2 can be divided into four main subgroups: M2a, M2b, M2c, and M2d. M2a polarization, influenced by IL-4 and IL-13, functions in tissue repair by secreting TGF-β, insulin-like growth factor, and fibronectin. M2b is induced by immune complexes, toll-like receptor (TLR) ligands, or IL-1R ligands and regulates the immune response through IL-10. M2c requires IL-10 and TGF-β to differentiate, producing the anti-inflammatory cytokine IL-10 and the tissue remodeling cytokine TGF-β. M2d, induced by IL-6, TLR ligands, and the A2 adenosine receptor (A2R), produces IL-10, TGF-β, monocyte colony-stimulating factor (M-CSF), and VEGF (31–33).
At a steady state of lung tissue, the major population of AMs exhibits a hybrid of M1 and M2 surface markers, allowing them to polarize swiftly (34). However, dysregulation in M1/M2 polarization is implicated in asthma, COPD, and ACO. Studies of small airway tissue and bronchoalveolar lavage (BAL) samples from smokers and COPD populations indicate that M1 predominates in the airway wall, while M2 phenotypes and their associated cytokines are elevated in the luminal space compared to normal controls. This suggests that the overexpression of M2 switching in the lumen produces excessive tissue repair following M1-mediated tissue damage, contributing to lung fibrosis and permanent obstruction (35).
Eosinophils
Eosinophils respond to IL-3, IL-5, and granulocyte-macrophage colony-stimulating factor (GM-CSF), all of which are crucial for their proliferation, maturation, survival, and activation. They are also activated by TSLP and IL-33. This activation leads to inflammation, tissue injury, and airway remodeling through their adhesion, degranulation, cytokine secretion, and chemotaxis involving CCL-1, CCL-5, CCL-7, and CCL-8 (36–39). Eosinophil granules contain proteins, including major basic protein (MBP), eosinophil cation protein (ECP), eosinophil peroxidase (EPO), and eosinophil-derived neurotoxin (EDN), which affect the immunopathogenesis of ACO through their cytotoxic functions and induce inflammation (38, 40, 41). Moreover, recent reports indicate that cadherin L, derived from eosinophils, also promotes emphysema in COPD with eosinophilia (42).
Eosinophil-derived mediators, such as TGF-β1, cysteinyl leukotrienes, IL-4, and IL-13, drive the proliferation of ASM and fibroblasts, leading to long-term structural changes (38). Additionally, eosinophils interact with the neuroimmune system by forming eosinophil extracellular traps (EETs), which activate pulmonary neuroendocrine cells (PNCEs) to secrete calcitonin gene-related peptide (CGRP) and γ-aminobutyric acid (GABA), promoting cell infiltration and cytokine release, thereby increasing inflammation and mucus production (4). These non-selective mechanisms of eosinophil granule toxicity cause tissue damage and hyperresponsiveness, resulting in an increased exacerbation rate of ACO (40, 41, 43).
Neutrophils
Neutrophils are stimulated by extracellular matrix (ECM) proteins, cytokines, and microorganisms at inflammatory sites. Optimal activation requires a two-step process involving priming and activating stimuli. Several factors, such as TNF-α, GM-CSF, IFN-γ, IL-6, IL-17, CXCL-1, and CXCL-8, have been identified as priming agents that facilitate full activation alongside DAMPs and pathogen-associated molecular patterns (PAMPs) (44, 45). Activated neutrophils release serine proteases, including neutrophil elastase (NE), cathepsin G (CG), proteinase 3 (PR3), reactive oxygen species (ROS), myeloperoxidase (MPO), and neutrophil extracellular traps (NETs). These proteases, along with ROS formation and NETosis, degrade elastin, collagen, fibronectin, and proteoglycans, contributing to epithelial cell destruction and structural changes (46). Moreover, neutrophils can initiate tissue repair through heat shock signaling, controlling the movement of the matrix from healthy areas into injured tissue (47).
Recent research shows that ECM proteins influence neutrophil functions, migration, ROS production, MPO secretion, and NET formation. Type III collagen in lung tissue reduces neutrophil migration while enhancing ROS production, leading to tissue damage and prolonged neutrophil presence in the lung tissue, contributing to long-term inflammation and airway remodeling. This may relate to the increased production of type I and type III collagen in the airway during the early stages of COPD (48). ECM also determines the velocity and direction of neutrophil migration, guiding them to the injury site and causing them to remain in the tissue (49). These neutrophil functions may explain the severity of ACO patients due to increased neutrophil attraction and activation at the inflamed site.
Mast cells (MCs)
MC activation occurs due to various stimuli, including IL-3, IL-9, IL-33, IgE, and DAMPs (50). Once activated, they produce histamine, tryptase, chymase, leukotrienes C4, and prostaglandins D2, IL-4, IL-5, IL-13, VEGF-A, VEGF-C, and CXCL-1. These molecules are associated with inflammation and airway remodeling (50–52). Interestingly, MC-released mediators can crosstalk with neurons, inducing them to release the neuropeptide substance P, thereby triggering MC degranulation (51).
MCs have two main subpopulations defined by their content granules of tryptase and chymase: MCT (tryptase-positive) and MCTC (both tryptase and chymase-positive). In asthma and COPD, MCTC populations are increased, affecting bronchial epithelial cells by altering their migration, velocity, proliferation, and morphology, thereby disrupting the epithelial barrier through the production of tryptase and chymase (53). The granules of MCs play a significant role in airway hyperresponsiveness, inflammation, and airway thickening. Histamine induces ASM contraction, while leukotrienes and prostaglandins mediate bronchoconstriction and remodeling (54, 55).
Innate lymphoid cells (ILCs)
ILCs are innate immune cells that differentiate into five subsets: ILC1, ILC2, ILC3, NK cells, and lymphoid tissue inducer (LTi) cells. They express cytokines similar to Th1, Th2, and Th17 but are distinguished from T cells by their lack of specific antigen receptors, especially T cell receptors (TCRs) (56). Although the mechanisms of ILCs in ACO are still being explored, their cytokines can induce several cells to function, such as neutrophils, eosinophils, MCs, AMs, T cells, and B cells, leading to airway inflammation and remodeling in asthma and COPD. ILC1 and NK cells share the expression of IFN-γ and TNF-α via T-bet driven by IL-12 (52, 57). The cytokines released by ILC1s, such as IFN-γ and TNF-α, increase neutrophilic inflammation (58, 59). NK cells contribute to tissue damage by releasing perforin and granzyme A and B, which correlates with the risk of COPD exacerbation (60). ILC2s release IL-4, IL-5, IL-9, and IL-13 through GATA-3 activation when exposed to TSLP, IL-25, and IL-33 (56, 61). ILC2 and type 2 cytokines are increased in the peripheral blood of COPD patients (62). The increased eosinophil count correlates with ILC2 levels in asthmatic patients (63). Moreover, ILC2 also plays a role in epithelial barrier destruction, inducing inflammation in murine asthma lung tissue (64). ILC3 and LTi secrete IL-17 and IL-22 through ROR-γt activation when stimulated with IL-2 and IL-23, contributing to neutrophilic inflammation and recruitment (52, 65, 66). A positive correlation has been observed between the ILC3 population, neutrophils, and AM1 in severe asthma (67).
These ILC subtypes are not permanent and can switch to other subtypes (68). Research has found that ILCs increasingly transition into the ILC1 subclass, with the elevated ILC1 population associated with smoking status and disease severity in COPD (59). Interestingly, ILC2s can polarize into the ILC1 subtype in response to infections or exposure to toxic agents in COPD (68). This explains why individuals with the same phenotype exhibit different characteristics.
In asthma, the elevated number of ILC2s and the overexpression of type 2 cytokines correlate with greater disease severity and progression (63, 68–71). In contrast, the levels of ILC1s and ILC3s cytokines are elevated in ACO and COPD patients (72). Moreover, an increased ILC3 population and neutrophil inflammation are observed in the neutrophil asthma and COPD groups (66, 73). These changes are affected by the overexpression of specific cytokines, which can excessively drive ILC plasticity into subgroups under different conditions.
Roles of adaptive immunity in asthma, COPD, and ACO
T-cells
Helper T-cells (Th) can polarize into subsets such as Th1, Th2, Th17, and Treg. Th1 cells, activated by IL-12 through STAT-4, express IFN-γ, TNF-α, and IL-2, inducing neutrophil inflammation (74). Th2 cells express IL-4, IL-5, IL-9, and IL-13 through the activation of GATA-3 upon exposure to IL-4. Th2 cytokines play an important role in disease pathophysiology by regulating eosinophil proliferation, maturation, survival, and activation through IL-5. IL-4 and IL-13 induce airway hyper-responsiveness (AHR), attracting eosinophils and neutrophils, resulting in bronchial contraction, increased bronchial ECM, and the secretion of TGF-β1 from fibrocytes (45, 75, 76). Th17 differentiation, driven by IL-6, IL-21, IL-23, and TGF-β, results in the secretion of IL-17A, IL-21, and IL-22 through STAT-3 activation. This process induces fibrocytes to release TNF-α, CXCL-1, and CXCL-8, contributing to neutrophil inflammation (45, 77). Treg cells, activated by IL-2, IL-10, and TGF-β, prevent excessive tissue damage by releasing important cytokines, such as IL-10, IL-35, and TGF-β, to inhibit T-cell polarization and promote Treg differentiation (77, 78). Reduced IL-10 levels are found in patients with poor lung function (79). Tc cells produce IFN-γ, TNF-α, perforin, and granzyme B after receiving antigens on MHC class I and co-stimulation with IL-12, IL-15, and IL-18, inducing epithelial cell apoptosis and contributing to emphysema (80).
In respiratory diseases, T-cell imbalance affects the pathogenesis of asthma and COPD (81–85). COPD patients exhibit elevated Th1 and IFN-γ levels, which are associated with decreased lung function (85). Another study involving asthma patients shows that a high level of Th17 correlates with COPD and asthma severity by promoting neutrophil infiltration, thus contributing to airway remodeling (83, 86). Moreover, COPD patients with high Th1 and low Th17 experience increased severity and frequency of exacerbations compared to those with high levels. These patients have inadequate Th1 and Th17 responses to combat infections (82). This may explain why some patients with a Th1 phenotype have frequent exacerbations due to infections. In addition, asthmatic patients demonstrate an imbalance with an elevated Th2/Th1 ratio, highlighting the Th2 role in eosinophil inflammation and airway remodeling (84, 87, 88).
Moreover, the absence of regulatory mechanisms for Treg maintains inflammation in the long run. COPD patients with rapid declines in lung function exhibit low levels of Treg (89). The decrease in the function of Treg leads to dysregulation in the immune response, highlighting the need for future studies on Treg function and personalized approaches for asthma, COPD, and ACO (87, 90).
B-cells
B cells produce antibodies against antigens, activated via surface antigen receptors and signals from Th cells and CD40. Once activated, B cells proliferate and retain high-affinity B-cell receptors as memory B cells residing in tissue. In lung tissue, resident B cells are stimulated by local antigens and release immunoglobulins such as IgE, which are sensitized on mast cells for degranulation (91). Moreover, B cells may contribute to airway inflammation through IgA secretion after receiving signals from epithelial cells, including IL-6, B-cell activating factor (BAFF), and a proliferation-inducing ligand (APRIL) (92, 93). An increase in IgA+ memory B cells correlates with decreased lung function in severe asthma (94). Despite their role in disease progression, recent data highlight their regulatory function in inflammation and airway remodeling processes, suggesting potential therapeutic uses in managing diseases (95, 96).
Novel tools for assessing immunopathogenesis in asthma, COPD, and ACO
Current guidelines from GINA and the Global Initiative for Chronic Obstructive Lung Disease (GOLD) primarily rely on clinical data for managing asthma, COPD, and ACO, including spirometry and a few biomarkers such as blood and sputum eosinophils, IgE, and the fractional exhaled nitric oxide test (FeNO). These limited details prove insufficient for effective disease management and highlight the immune diversity present in asthma, COPD, and ACO (97). Recent in vitro tools provide comprehensive information that enhances disease management through a better understanding of immunopathophysiology, enabling clinicians to implement more precise and personalized treatment strategies based on potential biomarkers for diagnosis, monitoring, prognosis, and treatment.
Epithelial function study
Air–liquid interface (ALI) cell culture
The ALI model mimics the lung environment in vitro, enabling cells to differentiate and form a functional epithelial layer. This model allows researchers to directly investigate the effects of drugs and substances on epithelial cells without dilution from culture media. Moreover, the ALI model provides insights into the hydration status of the mucus layer under confocal microscopy (98).
High-speed digital video microscopy
High-speed digital video microscopy analyzes the functions of motile cilia by recording video and playing it back in slow motion to detect abnormalities in cilia beating. This technique allows us to quantify cilia beat frequency (CBF) and evaluate the abnormality of the cilia beat pattern as a percentage of dyskinetic cilia, referred to as the dyskinetic index (DI). Additionally, the population of immotile cilia is measured and reported as a percentage, known as the immotile index (16).
Transmitted electron microscopy (TEM)
TEM can investigate abnormalities in the structure of cilia and epithelial cells, including defects in ciliary axonemes such as microtubule and dynein arm defects. It can assess disruptions in epithelial integrity by evaluating cellular extrusion and cytoplasmic blebbing and visualize damage to the mitochondria, which serve as the energy source for cilia to function normally (16).
Enzyme-linked immunosorbent assay (ELISA)
ELISA is a technique used to monitor inflammation in respiratory diseases by quantifying substances such as proteins and cytokines. For example, sputum periostin indicates fixed airflow obstruction and correlates with elevated levels of sputum IL-13 and eosinophils (13, 99). ELISA can also detect soluble cytokine receptors, such as sIL-2R levels, which are linked to disease severity in the COPD group (100). This method also measures the levels of EETs and NETs (4). Additionally, the ELISA method can detect specific IgE (sIgE), which is essential for allergy evaluation (101).
Flow cytometry
Flow cytometry is a technology used to analyze and classify cells based on their expressions, such as DNA/RNA content, CD markers, transcription factors, cytokines, and cell receptors. This provides more information about cell state, cell subtype, cell function, and cytokine production (4, 9).
Flow cytometry effectively quantifies the total number of specific cell types by highlighting their distinguishing features (34) (Figures 2A, B). It also reveals the percentages of surface receptors present on immune cells (Figures 2C, D). Markers such as IL-4R, IL-5R, CXCR1, and IL-17R are essential in severe obstructive lung disease, serving as targets for biological therapy that can potentially influence patients' dose response (102).
Figure 2

Flow cytometry data demonstrating clinical utility in asthma, COPD, and ACO. (A) Identification of eosinophil and neutrophil populations based on surface protein expression. (B) Classification of T-cell subsets based on both surface and intracellular protein expression. (C) Analysis of eosinophil receptor expression; higher fluorescent intensity indicates the percentage of positive cells (D). In the analysis of neutrophil receptor expression, increased fluorescence intensity signifies a higher percentage of receptor-positive cells. (E) Cytokine analysis using bead-based assays; beads A and B are distinguished by size. Each detector bead has a unique fluorescence intensity (Bead-APC), and cytokine concentrations are quantified based on the fluorescence intensity of the secondary antibody. Eos, Eosinophils; Neu, Neutrophils.
Furthermore, flow cytometry detects biomarkers using multiplex bead-based indirect immunofluorescence assays, determining biomarker levels based on fluorescent intensity on cytokine-specific beads (Figure 2E) (103). It also tracks cell proliferation, such as in the lymphocyte transformation test (LTT), which uses carboxyfluorescein diacetate succinimidyl ester (CFSE) dye to monitor lymphocyte generation. Flow cytometry also provides cell function analysis, such as the MCs activation test (MAT), which can improve the diagnosis of IgE-mediated allergic phenotypes and analyze EETs and NETs formation in specific cell populations (4, 104).
Immune cell function tests
Adhesion assay
An adhesion assay evaluates how effectively stimulated cells, such as eosinophils and neutrophils, attach to the airway structure using their surface receptors. The results are reported as the percentage of adherent cells detected through their cell functions or total cell viability from all added cells. The increase in eosinophil and neutrophil adhesion correlates with higher eosinophil and neutrophil counts and infiltration into inflamed lung structures in obstructive lung diseases, which leads to airway remodeling in persistent inflammation (38).
Degranulation assay
Currently, several methods for investigating degranulation activity are available, such as flow cytometry and ELISA. Flow cytometry techniques detect molecule production during degranulation, noting surface markers (e.g., CD63, CD107a, and CD203c) and intracellular changes (e.g., histamine and calcium concentration) (105). Additionally, the ELISA method can be used to directly detect degranulation-specific proteins, such as ECP, MPO, and tryptase (105, 106).
Chemotaxis assay
A chemotaxis assay analyzes cell direction, migration, and velocity in response to a chemoattractant. The dysfunction of cell migration leads to severe chronic inflammation and increases airway remodeling in obstructive lung disease (107). An outstanding feature in recent years is microfluidic-based migration assays, which can simulate the 3D microenvironments of human lung tissue. Unlike the 2D system that only enables cell adhesion and migration on surfaces, the 3D design mimics lung tissue conditions by allowing cells to interact with an ECM-like matrix. Cell migration is recorded through live-cell videos and images to measure migration percentage, total distance, direction, speed, and velocity (49).
Molecular analysis
Molecular technologies, such as nucleic acid sequencing and real-time polymerase chain reaction (RT-PCR), shed light on how single nucleotide polymorphisms (SNPs) play a role in immune system dysregulation in various diseases. SNPs are variations in a single nucleotide within human DNA that can lead to differences in individual phenotypes. Numerous studies have demonstrated the significance of SNPs in assessing disease risk and severity, as well as in informing treatments for asthma, COPD, and ACO, which are critical for future clinical practice (108). For example, gene variations associated with the IL-4/IL-13 pathway are linked to asthma susceptibility. Chinese patients with four SNPs, including IL4 (rs2243250C>T), IL13 (rs1800925C>T), IL4R (rs1805010G>A), and STAT6 (rs3224011T>C), show an association with a high-risk genotype for asthma vulnerability by elevating the expression levels of IL4, IL13, and STAT6 (108). Variations in TNF (rs1800629G>A) are associated with an increased risk of COPD in Asian populations (109). The haplotype of VEGFA (rs833068G>A, rs833070T>C, rs3024994C>T, rs3024997G>A, and rs3025000C>T), designated as GCCAT, significantly increases COPD risk in the Mongolian population, with an odds ratio (OR) of 3.409 (110). Furthermore, research on type-2 phenotype asthma and COPD among the Japanese population indicates that IL4R (rs8832A > G), associated with IL-4Rα levels and frequent exacerbations, may predict the effectiveness of IL-4Rα antagonist drugs (111).
Clinical implications of asthma, COPD, and ACO immunopathogenesis findings
To date, the clinical information obtained from routine medical practice remains insufficient for effectively managing and improving the clinical outcomes of asthma, COPD, and ACO patients. However, the majority of clinical treatment recommendations and practice guidelines depend on the phenotypes rather than the endotypes of asthma, COPD, and ACO (99).
Laboratory investigations, including biomarkers that represent the immunopathogenesis of asthma, COPD, and ACO, are sophisticated tools used to reveal the underlying individual characteristics in clinical practice. These phenotypic approaches and biomarkers, such as eosinophil count, sIgE, and FeNO, are widely integrated into routine clinical practice (112). The purpose of these biomarkers is to diagnose and classify obstructive airway diseases, such as type-2 inflammatory patterns that include allergic and eosinophilic phenotypes among type-2 high asthmatic patients (113). Moreover, they help predict the outcomes or prognosis of the diseases. Elevated levels of type-2 biomarkers, despite standard care in severe asthma, are associated with poor clinical outcomes (114). More importantly, biomarkers are also utilized to monitor the magnitude of underlying systemic and local disease activity (115). Ultimately, their clinical applicability lies in predicting treatment response with disease-specific treatment modalities (116, 117). The immune response is a critical determinant influencing disease outcomes. Given the current insights into immunopathogenesis, there is a growing recognition of the vital role that in vitro models play in enhancing clinical practice. Therefore, in vitro tools are emerging to fill the gap in our understanding of these complexities and clarify clinical management by identifying and targeting individual characteristics (118) (Table 2, Figure 3).
Table 2
| Clinical application | Biomarkers/Targets | Phenotypes | Role/potential use | |
|---|---|---|---|---|
| Diagnosis | IL-8 (CXCL-8) | Non-type 2 | Show diagnostic performance in the ACO group with the area under the curve (AUC) of 0.68 (164). | |
| NAGL | Non-type 2 | Identifies ACO from asthma with an AUC of 0.75 (166). | ||
| TNF | rs1800629G>A | Non-type 2 | Higher risk genotype for COPD susceptibility in Asian populations. The A allele is associated with a 2.45-fold increased risk of COPD compared to the G allele in a meta-analysis (109, 191). | |
| Periostin | Type 2 | Surrogate marker for type-2 high phenotypes (15, 99). Show diagnostic performance in asthma with an AUC of 0.87 (192). | ||
| IL13 | rs1800925C>T | Type 2 | Higher risk genotype for asthma susceptibility from higher expression levels of IL4, IL13, and STAT6 (108). | |
| IL4R | rs1805010G>A | |||
| IL4 | rs2243250C>T | |||
| STAT6 | rs3224011T>C | |||
| YKL-40 | Type 2 | Identifies ACO from COPD with an AUC of 0.71 (166). | ||
| VEGFA | rs833068G>A | Type 2/Non-type 2 | The haplotype of GCCAT significantly increases COPD risk with an odds ratio (OR) of 3.409 in the Mongolian population (110). | |
| VEGFA | rs833070T>C | |||
| VEGFA | rs3024994C>T | |||
| VEGFA | rs3024997G>A | |||
| VEGFA | rs3025000C>T | |||
| VEGF-A | Type 2/Non-type 2 | Show diagnostic performance in the ACO group with an AUC of 0.65 (164). | ||
| Prognosis | ILC1 and ILC3 | Non-type 2 | Prognosis severity linked to cigarette smoke (59, 67). | |
| TGF-β:IL-35 | Non-type 2 | Prognosis marker for fibrosis development (78). | ||
| IL4R | rs8832A>G | Type 2 | Correlated with frequent exacerbations (111). | |
| Eosinophil adhesion | Type 2 | Marker for exacerbation and airway remodeling (38). | ||
| Periostin | Type 2 | Marker for airway remodeling prediction (15). | ||
| IL-10 | Type 2/Non-type 2 | Potential predictor for severity of obstructive lung disease (79). | ||
| Monitoring | NET | Non-type 2 | Potential biomarker for monitoring ICS treatment in obstructive lung disease. Patients who have regular ICS show lower levels than those who do not have regular ICS (5). | |
| Periostin | Type 2 | Useful for monitoring anti-IL-13 and anti-IgE treatments (15, 99). | ||
| Therapeutic | IL4R | rs8832A>G | Type 2 | Predict the efficiency of IL-4Rα antagonist drugs (111). |
| HSF | Non-type 2 | Target for decreasing ECM excess from matrix transfer during lung tissue injury (47). | ||
| IL-35 | Non-type 2 | Reduces fibrosis expression by preventing TGF-β binding and Th17 differentiation (78). | ||
| Neutrophil integrins | Non-type 2 | Novel therapy for non-type-2 inflammation phenotype (49). | ||
| Periostin | Type 2 | Target for reducing mucus secretion and eosinophilic inflammation (15). | ||
| Siglec-8 | Type 2 | Target marker for enhancing apoptosis of eosinophil via ADCC activity and inhibiting mast cell functions (193). | ||
| Eosinophil adhesion | Type 2 | Target for reducing survivability and pro-proliferative effects on the lung structure (38). | ||
| IL-3 | Type 2 | Alternative target for controlling eosinophil activation in anti-IL5 non-responders (39). | ||
| IL-33 | Type 2 | Target for decreasing eosinophil functions and allergic processes (37). | ||
| Mast cells | Type 2 | Mast-cell-depleting monoclonal antibodies decrease disease exacerbation (51). | ||
| PD-1 | Type 2 | Target for decreasing ILC2 activation and functions (61). | ||
| IL-9 | Type 2/Non-type 2 | Target for decreasing in Th2 and Th17 cytokine expression, eosinophil infiltration, goblet cell hyperplasia, and the proliferation of ILC2, Th2, Th17, and mast cells (76). | ||
Utility of example biomarkers in asthma, COPD, and ACO.
Figure 3

In vitro models bridging immunopathogenesis insights to clinical practice. Figure created with www.BioRender.com. In vitro models translate complex immune pathophysiology into clinical practice for asthma, COPD, and ACO. These models improve diagnostic accuracy, endotype identification, therapeutic target discovery, treatment-response prediction, intensive monitoring, and precise prognostic outcomes.
For asthma, eosinophils and type-2 inflammatory cytokines play a vital role in disease pathogenesis. Persistently high type-2 biomarkers are associated with worse asthma outcomes and predict the response to treatment with type-2 biologics such as omalizumab, mepolizumab, benralizumab, and dupilumab (119–121). Conventional systemic biomarkers, including blood eosinophilia, serum periostin, and serum sIgE, have been proven to be predictive markers for the response to omalizumab in landmark studies (122–124). The level of blood eosinophilia has been shown to respond to IL-5 receptor antagonists, such as benralizumab, in a dose-dependent fashion. A significant reduction in asthma exacerbations was noted with increased blood eosinophilia (125, 126).
Exhaled biomarkers such as FeNO are becoming increasingly promising as markers for investigating the upregulation of the IL-4/IL-13 pathway in airway epithelial cells (115, 127). An increase in FeNO, along with higher blood eosinophilia, serves predictive roles in response to IL-4 receptor antagonists or dupilumab for reducing exacerbations (128–130). These findings emphasize the predictive significance of biomarkers related to the immunopathogenesis of the type-2 asthma phenotype.
In contrast to type-2 high asthma, characterized by eosinophilic and allergic inflammation, type-2 low asthma includes not only neutrophilic asthma but also mixed granulocytic and pauci-granulocytic asthma (131, 132). Currently, there is no consensus or clear definition of type-2 low asthma in clinical practice (133). As a result, no neutrophilic biomarkers are available for clinical use (134). Clinical trials have shown that neutrophilic-targeted biologic agents for severe asthma, such as anti-IL-17 and anti-TNF-α antagonists, are ineffective (135, 136). Despite specific antagonists targeting various asthma phenotypes based on inflammatory profiles, an unmet need persists (131). To date, the anti-TSLP monoclonal antibody tezepelumab has shown clinical efficacy in both type-2 high and type-2 low asthma by modulating the upstream cytokine pathways involved in asthma pathogenesis (137–140). However, critical factors for clinical application include cost-effectiveness, availability, accessibility of these treatments, and patient selection (141, 142).
COPD is characterized by corticosteroid insensitivity (143). Both neutrophilic and eosinophilic inflammation underlies the disease pathogenesis (144). The increased blood eosinophilia in COPD is associated with a higher frequency of exacerbations. Therefore, COPD patients with eosinophilia and frequent exacerbations, despite maximizing bronchodilator therapy, are indicators for ICS-containing regimens according to treatment recommendations (145). Nevertheless, the predictive roles of blood eosinophilia for ICS in patients with COPD are widely accepted (146, 147). The role of type-2 inflammation, particularly in COPD, concerning elevated blood eosinophilia has been debated. Recent clinical trials have demonstrated clinical benefits in treating type-2 inflammation with biologics, including IL-4/IL-13, IL-5, and IL-5R modulation. Despite these findings, the clinical trial results for mepolizumab and benralizumab have shown inconsistent outcomes regarding the reduction of exacerbations in COPD patients with blood eosinophilia; however, dupilumab can reduce exacerbations and improve lung function (148–150). Additionally, anti-inflammatory treatments for COPD, including low-dose azithromycin, have been shown to reduce COPD exacerbations (151, 152). However, the precise immunomodulating mechanisms underlying these effects have not been fully elucidated.
Finally, ACO, the distinct clinical entities of pure asthma and COPD have been proposed for two decades (153). The immunopathogenesis of coexisting asthma and COPD has not been fully clarified (154). Since the definitive diagnosis of ACO depends on the presence of both clinical features of asthma and COPD in patients with airway diseases, it remains non-standardized. Consequently, clinical biomarkers for ACO have been insufficient (155–158). More importantly, the primary treatment consists of ICS-containing regimens, which rely on managing asthma and COPD with eosinophilic inflammation as a cornerstone. To date, there is no specific targeted treatment for ACO, and there is a lack of promising biomarkers to predict clinical responsiveness to ACO treatments (159, 160).
The novel molecular and cellular biomarkers that are promising for determining the nature of the disease represent unmet needs and are urgently required for clinical application. The majority of biomarkers used in clinical practice depend on ease of use and convenience in clinical contexts. However, the mechanistic correlation with the pathogenesis of diseases, the correlation with disease severity, and the predictive capability regarding clinical outcomes are lacking (112). More importantly, the majority of clinical trials for targeted therapies for asthma and COPD rely on conventional biomarkers, such as blood eosinophils, serum total IgE, and FeNO (161). Therefore, the off-target approach significantly impacts the clinical efficacy and effectiveness of these treatments. The lack of molecular and target specificity for immunopathogenesis mechanism underlying biologic agents, which modulate both type-2 high and type-2 low phenotypes, remains an unmet clinical need (154).
The present article review focuses on the immunopathogenesis of asthma, COPD, and ACO, emphasizing the role of circulating cytokine assays, cytokine receptor expression on the surface of inflammatory cells, and functional responses in laboratories. Determining whether these factors become targets of type-2 and non-type-2 biological treatment requires further clinical studies. Both the modulation of circulating cytokines and cytokine receptor expression can be measured and applied in future preclinical and clinical studies. Their roles in assessing the efficacy and effectiveness of these targeted treatments for asthma, COPD, and ACO show promise as novel biomarkers (154, 162).
Despite the use of conventional biomarkers in the clinical field to investigate their roles in airway diseases, they do not encompass all patient characteristics (162) (Figure 4). Developing ideal and novel biomarkers is pivotal to improving the outcomes of airway disease management in the future. The goal of these biomarkers is to comprehensively capture individual traits, particularly concerning the immunopathogenesis of obstructive diseases (154). Furthermore, these novel biomarkers may serve as surrogates to guide the development of more treatable targets.
Figure 4
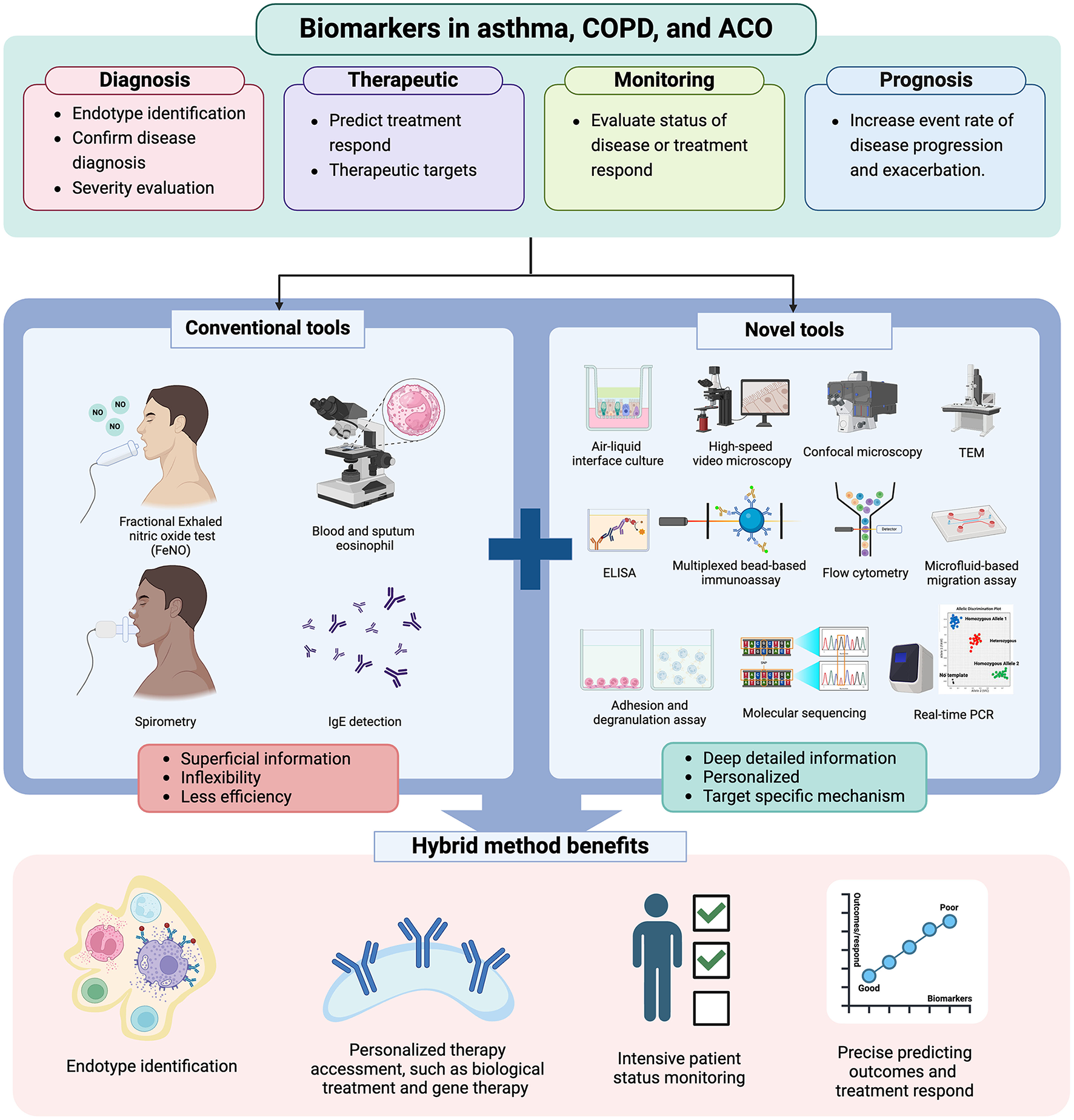
The role of novel tools in future clinical practice. Figure created with www.BioRender.com. Only conventional tools, such as FeNO, blood and sputum eosinophil counts, spirometry, and IgE detection, are insufficient to provide a comprehensive understanding of the patient's actual profile. Therefore, combining novel and conventional tools provides more detailed patient profiles, enabling clinicians to enhance diagnostic accuracy, select the most effective treatments, monitor patient status closely, and predict adverse outcomes or treatment responses. TEM, transmitted electron microscopy; real-time PCR, real-time polymerase chain reaction.
Consequently, the integration of biomarkers into the real-world management of airway diseases, including ACO, informs approaches to patient care and ultimately enhances clinical outcomes. Novel biomarkers should be easily accessible, cost-effective, and minimally invasive, offering more detailed information about an individual's traits for accurate diagnosis, forecasting treatment outcomes, adjusting doses, prognosticating further outcomes, and tailoring personalized treatment (154, 162) (Figures 3, 4).
Conclusion
In conclusion, using in vitro tools highlights that understanding the underlying immunopathogenesis of disease can improve management, paving the way for personalized patient care based on immune characteristics. Further research is essential to better understand the complexity of ACO and identify potential markers for its management in clinical practice.
Statements
Author contributions
KS: Writing – original draft. TK: Writing – original draft. JV: Writing – review & editing. PD: Writing – review & editing. PL: Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was financially supported by the “PHC SIAM” program (Project number: 49515ZF), implemented by the French Ministry for Europe and Foreign Affairs, the French Ministry of Higher Education and Research and the Thai Ministry of Higher Education, Science, Research and Innovation. Additionally, this work was partially supported by the “Scholarships for Ph.D. Students” (Project number: PHD/0002/2565) from Mahidol University, Thailand. The article processing charge was funded by the Research Administration Unit, Faculty of Medicine Ramathibodi Hospital, Mahidol University, Thailand.
Acknowledgments
We greatly thank all the staff of the Clinical Immunology Laboratory, Department of Pathology, Ramathibodi Hospital, Mahidol University, Thailand; Inserm UMR-S 1250 unit; the University Hospital of Reims (CHU de Reims); and the University of Reims Champagne-Ardenne, France, for providing valuable laboratory data and knowledge.
Conflict of interest
KS declares having received research funds from Mahidol University for Ph.D. students (Scholarship for Ph.D. students). JV reports receiving speaker and consultancy fees from AstraZeneca, HpVac, L'Oréal, Novartis, and Thermo Fisher Scientific over the past 5 years, in addition to reimbursement for travel expenses related to an international meeting outside the submitted work. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
A2R, A2 adenosine receptor; ACO, asthma-COPD overlap; AECs, airway epithelial cells; AHR, airway hyperresponsiveness; ALI, air–liquid interface; AMs, alveolar macrophages; APRIL, a proliferation-inducing ligand; ASM, airway smooth muscle; AT1, alveolar type 1 cells; AT2, alveolar type 2 cells; BAFF, B-cell activating factor; BAL, bronchoalveolar lavage; CBF, ciliary beat frequency; cDC1, conventional dendritic cells type 1; cDC2, conventional dendritic cells type 2; CG, cathepsin G; CGRP, calcitonin gene-related peptide; COPD, chronic obstructive pulmonary disease; CFSE, carboxyfluorescein diacetate succinimidyl ester; CXCL, C-X-C motif; DAMPs, damage-associated molecular patterns; DCs, dendritic cells; DI, dyskinetic index; ECM, extracellular matrix; ECP, eosinophil cation protein; EDN, eosinophil-derived neurotoxin; EETs, eosinophil extracellular traps; ELISA, enzyme-linked immunosorbent assay; EPO, eosinophil peroxidase; FeNO, fractional exhaled nitric oxide; GABA, γ-aminobutyric acid; GATA-3, GATA-binding protein 3; GINA, global initiative for asthma; GM-CSF, granulocyte-macrophage colony-stimulating factor; GOLD, global initiative for chronic obstructive lung disease; HMGB1, high mobility group box 1 protein; HSP70, 70-kilodalton heat shock proteins; ICS, inhaled corticosteroid; IFN-α, interferon α; IL, interleukin; ILC1, type 1 innate lymphoid cell; ILC2, type 2 innate lymphoid cell; ILC3, type 3 innate lymphoid cell; iNOS, inducible nitric oxide synthase; LL-37, cathelicidin LL-37; LPS, lipopolysaccharide; LTi, lymphoid tissue inducer; LTT, lymphocyte transformation test; M-CSF, monocyte colony-stimulating factor; M1 macrophages, classically activated macrophages; M2 macrophages, alternatively activated macrophages; MAT, mast cell activation test; MBP, major basic protein; MCs, mast cells; MCT, mast cells tryptase-positive; MCTC, mast cells tryptase- and chymase-positive; MMP-9, matrix metalloprotease 9; moDCs, monocyte-derived dendritic cells; MPO, myeloperoxidase; NE, neutrophil elastase; NETs, neutrophil extracellular traps; NK cells, natural killer cells; OX40L, tumor necrosis factor ligand superfamily member 4; PAMPs, pathogen-associated molecular patterns; PD-L1, programmed death-ligand 1; pDCs, plasmacytoid dendritic cells; PM2.5, particulate matter particles with a diameter of 2.5 μm or smaller; PNCEs, pulmonary neuroendocrine cells; PR3, proteinase 3; PRRs, pattern recognition receptors; ROR-γt, retinoic-acid–receptor-related orphan nuclear receptor gamma t; ROS, reactive oxygen species; RT-PCR, real-time polymerase chain reaction; S100A8, S100 calcium-binding protein A8; sIgE, specific IgE; SNPs, single nucleotide polymorphisms; STAT-3, signal transducer and activator of transcription 3; STAT-4, signal transducer and activator of transcription 4; STAT-6, signal transducer and activator of transcription 6; Tc, cytotoxic T-cells; TCRs, T cell receptors; TEM, transmission electron microscopy; TGF-β, transforming growth factor beta β; Th, helper T-cells; Th0, naïve helper T cell; Th1, T helper type 1; Th17, T helper type 17; Th2, T helper type 2; TLRs, toll-like receptors; Tregs, regulatory T cells; TSLP, thymic stromal lymphopoietin; VEGF, vascular endothelial growth factor; YKL-40, chitinase-3-like protein 1.
References
1.
Global Initiative for Asthma . Global Strategy for Asthma Management and Prevention (2023). Available online at: www.ginasthma.org (accessed September 29, 2023).
2.
Hosseini M Almasi-Hashiani A Sepidarkish M Maroufizadeh S . Global prevalence of asthma-COPD overlap (ACO) in the general population: a systematic review and meta-analysis. Respir Res. (2019) 20:229. 10.1186/s12931-019-1198-4
3.
Hardin M Cho M McDonald ML Beaty T Ramsdell J Bhatt S et al . The clinical and genetic features of COPD-asthma overlap syndrome. Eur Respir J. (2014) 44:341–50. 10.1183/09031936.00216013
4.
Lu Y Huang Y Li J Huang J Zhang L Feng J et al . Eosinophil extracellular traps drive asthma progression through neuro-immune signals. Nat Cell Biol. (2021) 23:1060–72. 10.1038/s41556-021-00762-2
5.
Gal Z Gezsi A Pallinger E Visnovitz T Nagy A Kiss A et al . Plasma neutrophil extracellular trap level is modified by disease severity and inhaled corticosteroids in chronic inflammatory lung diseases. Sci Rep. (2020) 10:4320. 10.1038/s41598-020-61253-2
6.
Yang W Yang X Jiang L Song H Huang G Duan K et al . Combined biological effects and lung proteomics analysis in mice reveal different toxic impacts of electronic cigarette aerosol and combustible cigarette smoke on the respiratory system. Arch Toxicol. (2022) 96:3331–47. 10.1007/s00204-022-03378-z
7.
Paplinska-Goryca M Misiukiewicz-Stepien P Proboszcz M Nejman-Gryz P Gorska K Zajusz-Zubek E et al . Interactions of nasal epithelium with macrophages and dendritic cells variously alter urban PM-induced inflammation in healthy, asthma and COPD. Sci Rep. (2021) 11:13259. 10.1038/s41598-021-92626-w
8.
Colombo SAP Brown SL Hepworth MR Hankinson J Granato F Kitchen SJ et al . Comparative phenotype of circulating versus tissue immune cells in human lung and blood compartments during health and disease. Discov Immunol. (2023) 2:kyad009. 10.1093/discim/kyad009
9.
Dey S Lu W Haug G Chia C Larby J Weber HC et al . Airway inflammatory changes in the lungs of patients with asthma-COPD overlap (ACO): a bronchoscopy endobronchial biopsy study. Respir Res. (2023) 24:221. 10.1186/s12931-023-02527-x
10.
Ghosh N Choudhury P Kaushik SR Arya R Nanda R Bhattacharyya P et al . Metabolomic fingerprinting and systemic inflammatory profiling of asthma COPD overlap (ACO). Respir Res. (2020) 21:126. 10.1186/s12931-020-01390-4
11.
Kabata H Flamar AL Mahlakoiv T Moriyama S Rodewald HR Ziegler SF et al . Targeted deletion of the TSLP receptor reveals cellular mechanisms that promote type 2 airway inflammation. Mucosal Immunol. (2020) 13:626–36. 10.1038/s41385-020-0266-x
12.
Huang X Tan X Liang Y Hou C Qu D Li M et al . Differential DAMP release was observed in the sputum of COPD, asthma and asthma-COPD overlap (ACO) patients. Sci Rep. (2019) 9:19241. 10.1038/s41598-019-55502-2
13.
Burgess JK Jonker MR Berg M Ten Hacken NTH Meyer KB van den Berge M et al . Periostin: contributor to abnormal airway epithelial function in asthma?Eur Respir J. (2021) 57:2001286. 10.1183/13993003.01286-2020
14.
Ito Y Al Mubarak R Roberts N Correll K Janssen W Finigan J et al . IL-13 induces periostin and eotaxin expression in human primary alveolar epithelial cells: comparison with paired airway epithelial cells. PLoS One. (2018) 13:e0196256. 10.1371/journal.pone.0196256
15.
Shirai T Hirai K Gon Y Maruoka S Mizumura K Hikichi M et al . Combined assessment of serum periostin and YKL-40 may identify asthma-COPD overlap. J Allergy Clin Immunol Pract. (2019) 7:134–45 e1. 10.1016/j.jaip.2018.06.015
16.
Thomas B Koh MS O'Callaghan C Allen JC Rutman A Hirst RA et al . Dysfunctional Bronchial Cilia Are a Feature of Chronic Obstructive Pulmonary Disease (COPD). COPD. (2021) 18:657–63. 10.1080/15412555.2021.1963695
17.
Gohy S Carlier FM Fregimilicka C Detry B Lecocq M Ladjemi MZ et al . Altered generation of ciliated cells in chronic obstructive pulmonary disease. Sci Rep. (2019) 9:17963. 10.1038/s41598-019-54292-x
18.
Perotin JM Coraux C Lagonotte E Birembaut P Delepine G Polette M et al . Alteration of primary cilia in COPD. Eur Respir J. (2018) 52:1800122. 10.1183/13993003.00122-2018
19.
Yu H Lin Y Zhong Y Guo X Lin Y Yang S et al . Impaired AT2 to AT1 cell transition in PM2. 5-induced mouse model of chronic obstructive pulmonary disease. Respir Res. (2022) 23:70. 10.1186/s12931-022-01996-w
20.
Zhang S Chopin M Nutt SL . Type 1 conventional dendritic cells: ontogeny, function, and emerging roles in cancer immunotherapy. Trends Immunol. (2021) 42:1113–27. 10.1016/j.it.2021.10.004
21.
Rosa FF Pires CF Kurochkin I Halitzki E Zahan T Arh N et al . Single-cell transcriptional profiling informs efficient reprogramming of human somatic cells to cross-presenting dendritic cells. Sci Immunol. (2022) 7:eabg5539. 10.1126/sciimmunol.abg5539
22.
Plantinga M Guilliams M Vanheerswynghels M Deswarte K Branco-Madeira F Toussaint W et al . Conventional and monocyte-derived CD11b(+) dendritic cells initiate and maintain T helper 2 cell-mediated immunity to house dust mite allergen. Immunity. (2013) 38:322–35. 10.1016/j.immuni.2012.10.016
23.
Izumi G Nakano H Nakano K Whitehead GS Grimm SA Fessler MB et al . CD11b(+) lung dendritic cells at different stages of maturation induce Th17 or Th2 differentiation. Nat Commun. (2021) 12:5029. 10.1038/s41467-021-25307-x
24.
Ogata M Ito T Shimamoto K Nakanishi T Satsutani N Miyamoto R et al . Plasmacytoid dendritic cells have a cytokine-producing capacity to enhance ICOS ligand-mediated IL-10 production during T-cell priming. Int Immunol. (2013) 25:171–82. 10.1093/intimm/dxs103
25.
Stoll P Ulrich M Bratke K Garbe K Virchow JC Lommatzsch M . Imbalance of dendritic cell co-stimulation in COPD. Respir Res. (2015) 16:19. 10.1186/s12931-015-0174-x
26.
Nobs SP Pohlmeier L Li F Kayhan M Becher B Kopf M . GM-CSF instigates a dendritic cell-T-cell inflammatory circuit that drives chronic asthma development. J Allergy Clin Immunol. (2021) 147:2118–33 e3. 10.1016/j.jaci.2020.12.638
27.
Robbe P Draijer C Borg TR Luinge M Timens W Wouters IM et al . Distinct macrophage phenotypes in allergic and nonallergic lung inflammation. Am J Physiol Lung Cell Mol Physiol. (2015) 308:L358–67. 10.1152/ajplung.00341.2014
28.
Wu X Wang Z Shi J Yu X Li C Liu J et al . Macrophage polarization toward M1 phenotype through NF-kappaB signaling in patients with Behcet's disease. Arthritis Res Ther. (2022) 24:249. 10.1186/s13075-022-02938-z
29.
Kohler JB Cervilha DAB Riani Moreira A Santana FR Farias TM Alonso Vale MIC et al . Microenvironmental stimuli induce different macrophage polarizations in experimental models of emphysema. Biol Open. (2019) 8:bio040808. 10.1242/bio.040808
30.
Kohno K Koya-Miyata S Harashima A Tsukuda T Katakami M Ariyasu T et al . Inflammatory M1-like macrophages polarized by NK-4 undergo enhanced phenotypic switching to an anti-inflammatory M2-like phenotype upon co-culture with apoptotic cells. J Inflamm. (2021) 18:2. 10.1186/s12950-020-00267-z
31.
Wang Q Ni H Lan L Wei X Xiang R Wang Y . Fra-1 protooncogene regulates IL-6 expression in macrophages and promotes the generation of M2d macrophages. Cell Res. (2010) 20:701–12. 10.1038/cr.2010.52
32.
Arora S Dev K Agarwal B Das P Syed MA . Macrophages: Their role, activation and polarization in pulmonary diseases. Immunobiology. (2018) 223:383–96. 10.1016/j.imbio.2017.11.001
33.
Wang LX Zhang SX Wu HJ Rong XL Guo J . M2b macrophage polarization and its roles in diseases. J Leukoc Biol. (2019) 106:345–58. 10.1002/JLB.3RU1018-378RR
34.
Mitsi E . Kamng'ona R, Rylance J, Solorzano C, Jesus Reine J, Mwandumba HC, et al. Human alveolar macrophages predominately express combined classical M1 and M2 surface markers in steady state. Respir Res. (2018) 19:66. 10.1186/s12931-018-0777-0
35.
Eapen MS Hansbro PM McAlinden K Kim RY Ward C Hackett TL et al . Abnormal M1/M2 macrophage phenotype profiles in the small airway wall and lumen in smokers and chronic obstructive pulmonary disease (COPD). Sci Rep. (2017) 7:13392. 10.1038/s41598-017-13888-x
36.
Uchida AM Ro G Qiang L Peterson KA Round J Dougan M et al . Human differentiated eosinophils release IL-13 in response to IL-33 stimulation. Front Immunol. (2022) 13:946643. 10.3389/fimmu.2022.946643
37.
Angulo EL McKernan EM Fichtinger PS Mathur SK . Comparison of IL-33 and IL-5 family mediated activation of human eosinophils. PLoS ONE. (2019) 14:e0217807. 10.1371/journal.pone.0217807
38.
Januskevicius A Janulaityte I Kalinauskaite-Zukauske V Gosens R Malakauskas K . The enhanced adhesion of eosinophils is associated with their prolonged viability and pro-proliferative effect in asthma. J Clin Med. (2019) 8:1274. 10.3390/jcm8091274
39.
Nelson RK Brickner H Panwar B Ramirez-Suastegui C . Herrera-de la Mata S, Liu N, et al. Human Eosinophils Express a Distinct Gene Expression Program in Response to IL-3 Compared with Common beta-Chain Cytokines IL-5 and GM-CSF. J Immunol. (2019) 203:329–37. 10.4049/jimmunol.1801668
40.
Ben-Zimra M Bachelet I Seaf M Gleich GJ Levi-Schaffer F . Eosinophil major basic protein activates human cord blood mast cells primed with fibroblast membranes by integrin-beta1. Allergy. (2013) 68:1259–68. 10.1111/all.12232
41.
Wang Z DiDonato JA Buffa J Comhair SA Aronica MA Dweik RA et al . Eosinophil peroxidase catalyzed protein carbamylation participates in asthma. J Biol Chem. (2016) 291:22118–35. 10.1074/jbc.M116.750034
42.
Xu X Yu T Dong L Glauben R Wu S Huang R et al . Eosinophils promote pulmonary matrix destruction and emphysema via Cathepsin L. Signal Transduct Target Ther. (2023) 8:390. 10.1038/s41392-023-01698-9
43.
Soragni A Yousefi S Stoeckle C Soriaga AB Sawaya MR Kozlowski E et al . Toxicity of eosinophil MBP is repressed by intracellular crystallization and promoted by extracellular aggregation. Mol Cell. (2015) 57:1011–21. 10.1016/j.molcel.2015.01.026
44.
Mol S Hafkamp FMJ Varela L Simkhada N Taanman-Kueter EW Tas SW et al . Efficient neutrophil activation requires two simultaneous activating stimuli. Int J Mol Sci. (2021) 22:10106. 10.3390/ijms221810106
45.
Bellini A Marini MA Bianchetti L Barczyk M Schmidt M Mattoli S . Interleukin (IL)-4, IL-13, and IL-17A differentially affect the profibrotic and proinflammatory functions of fibrocytes from asthmatic patients. Mucosal Immunol. (2012) 5:140–9. 10.1038/mi.2011.60
46.
Varricchi G Modestino L Poto R Cristinziano L Gentile L Postiglione L et al . Neutrophil extracellular traps and neutrophil-derived mediators as possible biomarkers in bronchial asthma. Clin Exp Med. (2022) 22:285–300. 10.1007/s10238-021-00750-8
47.
Fischer A Wannemacher J Christ S Koopmans T Kadri S Zhao J et al . Neutrophils direct preexisting matrix to initiate repair in damaged tissues. Nat Immunol. (2022) 23:518–31. 10.1038/s41590-022-01166-6
48.
Harju T Kinnula VL Paakko P Salmenkivi K Risteli J Kaarteenaho R . Variability in the precursor proteins of collagen I and III in different stages of COPD. Respir Res. (2010) 11:165. 10.1186/1465-9921-11-165
49.
Kraus RF Gruber MA Kieninger M . The influence of extracellular tissue on neutrophil function and its possible linkage to inflammatory diseases. Immun Inflamm Dis. (2021) 9:1237–51. 10.1002/iid3.472
50.
Cristinziano L Poto R Criscuolo G Ferrara AL Galdiero MR Modestino L et al . IL-33 and superantigenic activation of human lung mast cells induce the release of angiogenic and lymphangiogenic factors. Cells. (2021) 10:145. 10.3390/cells10010145
51.
Plum T Wang X Rettel M Krijgsveld J Feyerabend TB Rodewald HR . Human mast cell proteome reveals unique lineage, putative functions, and structural basis for cell ablation. Immunity. (2020) 52:404–16 e5. 10.1016/j.immuni.2020.01.012
52.
Xie M Zhang M Dai M Yue S Li Z Qiu J et al . IL-18/IL-18R signaling is dispensable for ILC development but constrains the growth of ILCP/ILCs. Front Immunol. (2022) 13:923424. 10.3389/fimmu.2022.923424
53.
Berlin F Mogren S Tutzauer J Andersson CK . Mast Cell Proteases tryptase and chymase induce migratory and morphological alterations in bronchial epithelial cells. Int J Mol Sci. (2021) 22:5250. 10.3390/ijms22105250
54.
Soma T Uchida Y Nakagome K Hoshi R Nagata M . Eicosanoids seasonally impact pulmonary function in asthmatic patients with Japanese cedar pollinosis. Allergol Int. (2020) 69:594–600. 10.1016/j.alit.2020.04.014
55.
Chhabra J Li YZ Alkhouri H Blake AE Ge Q Armour CL et al . Histamine and tryptase modulate asthmatic airway smooth muscle GM-CSF and RANTES release. Eur Respir J. (2007) 29:861–70. 10.1183/09031936.00106306
56.
Mazzurana L Czarnewski P Jonsson V Wigge L Ringner M Williams TC et al . Tissue-specific transcriptional imprinting and heterogeneity in human innate lymphoid cells revealed by full-length single-cell RNA-sequencing. Cell Res. (2021) 31:554–68. 10.1038/s41422-020-00445-x
57.
Klose CSN Flach M Mohle L Rogell L Hoyler T Ebert K et al . Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell. (2014) 157:340–56. 10.1016/j.cell.2014.03.030
58.
Jonckheere AC Seys SF Steelant B Decaesteker T Dekoster K Cremer J et al . Innate lymphoid cells are required to induce airway hyperreactivity in a murine neutrophilic asthma model. Front Immunol. (2022) 13:849155. 10.3389/fimmu.2022.849155
59.
Blomme EE Provoost S De Smet EG De Grove KC Van Eeckhoutte HP De Volder J et al . Quantification and role of innate lymphoid cell subsets in chronic obstructive pulmonary disease. Clin Transl Immunology. (2021) 10:e1287. 10.1002/cti2.1287
60.
Osterburg AR Lach L Panos RJ Borchers MT . Unique natural killer cell subpopulations are associated with exacerbation risk in chronic obstructive pulmonary disease. Sci Rep. (2020) 10:1238. 10.1038/s41598-020-58326-7
61.
Helou DG Shafiei-Jahani P Lo R Howard E Hurrell BP Galle-Treger L et al . PD-1 pathway regulates ILC2 metabolism and PD-1 agonist treatment ameliorates airway hyperreactivity. Nat Commun. (2020) 11:3998. 10.1038/s41467-020-17813-1
62.
Jiang M Tao S Zhang S Wang J Zhang F Li F et al . Type 2 innate lymphoid cells participate in IL-33-stimulated Th2-associated immune response in chronic obstructive pulmonary disease. Exp Ther Med. (2019) 18:3109–16. 10.3892/etm.2019.7924
63.
Yu QN Tan WP Fan XL Guo YB Qin ZL Li CL et al . Increased group 2 innate lymphoid cells are correlated with eosinophilic granulocytes in patients with allergic airway inflammation. Int Arch Allergy Immunol. (2018) 176:124–32. 10.1159/000488050
64.
Sugita K Steer CA Martinez-Gonzalez I Altunbulakli C Morita H Castro-Giner F et al . Type 2 innate lymphoid cells disrupt bronchial epithelial barrier integrity by targeting tight junctions through IL-13 in asthmatic patients. J Allergy Clin Immunol. (2018) 141:300–10 e11. 10.1016/j.jaci.2017.02.038
65.
Starkey MR Plank MW Casolari P Papi A Pavlidis S Guo Y et al . IL-22 and its receptors are increased in human and experimental COPD and contribute to pathogenesis. Eur Respir J. (2019) 54:1800174. 10.1183/13993003.00174-2018
66.
He LX Yang L Liu T Li YN Huang TX Zhang LL et al . Group 3 innate lymphoid cells secret neutrophil chemoattractants and are insensitive to glucocorticoid via aberrant GR phosphorylation. Respir Res. (2023) 24:90. 10.1186/s12931-023-02395-5
67.
Ham J Kim J Sohn KH Park IW Choi BW Chung DH et al . Cigarette smoke aggravates asthma by inducing memory-like type 3 innate lymphoid cells. Nat Commun. (2022) 13:3852. 10.1038/s41467-022-31491-1
68.
Silver JS Kearley J Copenhaver AM Sanden C Mori M Yu L et al . Inflammatory triggers associated with exacerbations of COPD orchestrate plasticity of group 2 innate lymphoid cells in the lungs. Nat Immunol. (2016) 17:626–35. 10.1038/ni.3443
69.
Smith SG Chen R Kjarsgaard M Huang C Oliveria JP O'Byrne PM et al . Increased numbers of activated group 2 innate lymphoid cells in the airways of patients with severe asthma and persistent airway eosinophilia. J Allergy Clin Immunol. (2016) 137:75–86 e8. 10.1016/j.jaci.2015.05.037
70.
Christianson CA Goplen NP Zafar I Irvin C Good JT Jr ., Rollins DR, et al. Persistence of asthma requires multiple feedback circuits involving type 2 innate lymphoid cells and IL-33. J Allergy Clin Immunol. (2015) 136:59–68 e14. 10.1016/j.jaci.2014.11.037
71.
Bartemes KR Kephart GM Fox SJ Kita H . Enhanced innate type 2 immune response in peripheral blood from patients with asthma. J Allergy Clin Immunol. (2014) 134:671–8 e4. 10.1016/j.jaci.2014.06.024
72.
Svetlana S Larisa P Marina B Nailja K Timur L Igor K et al . Serum IL-17 and IL-18 levels in asthma-COPD overlap syndrome patients. Eur Respir J. (2015) 46:PA4886. 10.1183/13993003.congress-2015.PA4886
73.
De Grove KC Provoost S Verhamme FM Bracke KR Joos GF Maes T et al . Characterization and Quantification of Innate Lymphoid Cell Subsets in Human Lung. PLoS ONE. (2016) 11:e0145961. 10.1371/journal.pone.0145961
74.
Powell MD Read KA Sreekumar BK Jones DM Oestreich KJ . IL-12 signaling drives the differentiation and function of a T(H)1-derived T(FH1)-like cell population. Sci Rep. (2019) 9:13991. 10.1038/s41598-019-50614-1
75.
Manson ML Safholm J James A Johnsson AK Bergman P Al-Ameri M et al . IL-13 and IL-4, but not IL-5 nor IL-17A, induce hyperresponsiveness in isolated human small airways. J Allergy Clin Immunol. (2020) 145:808–17 e2. 10.1016/j.jaci.2019.10.037
76.
Li Y Lan F Yang Y Xu Y Chen Y Qin X et al . The absence of IL-9 reduces allergic airway inflammation by reducing ILC2, Th2 and mast cells in murine model of asthma. BMC Pulm Med. (2022) 22:180. 10.1186/s12890-022-01976-2
77.
Harbour SN DiToro DF Witte SJ Zindl CL Gao M Schoeb TR et al . T(H)17 cells require ongoing classic IL-6 receptor signaling to retain transcriptional and functional identity. Sci Immunol. (2020) 5:eaaw2262. 10.1126/sciimmunol.aaw2262
78.
Osuna-Gomez R Barril S Mulet M Zamora Atenza C Millan-Billi P Pardessus A et al . The immunoregulatory role of IL-35 in patients with interstitial lung disease. Immunology. (2023) 168:610–21. 10.1111/imm.13596
79.
LeVan TD Romberger DJ Siahpush M Grimm BL Ramos AK Johansson PL et al . Relationship of systemic IL-10 levels with proinflammatory cytokine responsiveness and lung function in agriculture workers. Respir Res. (2018) 19:166. 10.1186/s12931-018-0875-z
80.
Freeman CM Han MK Martinez FJ Murray S Liu LX Chensue SW et al . Cytotoxic potential of lung CD8(+) T cells increases with chronic obstructive pulmonary disease severity and with in vitro stimulation by IL-18 or IL-15. J Immunol. (2010) 184:6504–13. 10.4049/jimmunol.1000006
81.
Zhao Y Yang J Gao YD Guo W . Th17 immunity in patients with allergic asthma. Int Arch Allergy Immunol. (2010) 151:297–307. 10.1159/000250438
82.
Yu Y Zhao L Xie Y Xu Y Jiao W Wu J et al . Th1/Th17 cytokine profiles are associated with disease severity and exacerbation frequency in COPD patients. Int J Chron Obstruct Pulmon Dis. (2020) 15:1287–99. 10.2147/COPD.S252097
83.
Xiong XF Zhu M Wu HX Fan LL Cheng DY . Immunophenotype in acute exacerbation of chronic obstructive pulmonary disease: a cross-sectional study. Respir Res. (2022) 23:137. 10.1186/s12931-022-02058-x
84.
Woodruff PG Modrek B Choy DF Jia G Abbas AR Ellwanger A et al . T-helper type 2-driven inflammation defines major subphenotypes of asthma. Am J Respir Crit Care Med. (2009) 180:388–95. 10.1164/rccm.200903-0392OC
85.
Chen G Mu Q Meng ZJ . Cigarette smoking contributes to Th1/Th2 cell dysfunction via the cytokine milieu in chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. (2023) 18:2027–38. 10.2147/COPD.S426215
86.
Zhou T Huang X Zhou Y Ma J Zhou M Liu Y et al . Associations between Th17-related inflammatory cytokines and asthma in adults: a case-control study. Sci Rep. (2017) 7:15502. 10.1038/s41598-017-15570-8
87.
de Llano LP Cosio BG Iglesias A de Las Cuevas N Soler-Cataluna JJ Izquierdo JL et al . Mixed Th2 and non-Th2 inflammatory pattern in the asthma-COPD overlap: a network approach. Int J Chron Obstruct Pulmon Dis. (2018) 13:591–601. 10.2147/COPD.S153694
88.
Vieira Braga FA Kar G Berg M Carpaij OA Polanski K Simon LM et al . A cellular census of human lungs identifies novel cell states in health and in asthma. Nat Med. (2019) 25:1153–63. 10.1038/s41591-019-0468-5
89.
Eriksson Strom J Pourazar J Linder R Blomberg A Lindberg A Bucht A et al . Airway regulatory T cells are decreased in COPD with a rapid decline in lung function. Respir Res. (2020) 21:330. 10.1186/s12931-020-01593-9
90.
Bai Y Zhou Q Fang Q Song L Chen K . Inflammatory cytokines and T-lymphocyte subsets in serum and sputum in patients with bronchial asthma and chronic obstructive pulmonary disease. Med Sci Monit. (2019) 25:2206–10. 10.12659/MSM.913703
91.
Allie SR Bradley JE Mudunuru U Schultz MD Graf BA Lund FE et al . The establishment of resident memory B cells in the lung requires local antigen encounter. Nat Immunol. (2019) 20:97–108. 10.1038/s41590-018-0260-6
92.
Ladjemi MZ Lecocq M Weynand B Bowen H Gould HJ Van Snick J et al . Increased IgA production by B-cells in COPD via lung epithelial interleukin-6 and TACI pathways. Eur Respir J. (2015) 45:980–93. 10.1183/09031936.00063914
93.
Ladjemi MZ Martin C Lecocq M Detry B Nana FA Moulin C et al . Increased IgA expression in lung lymphoid follicles in severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2019) 199:592–602. 10.1164/rccm.201802-0352OC
94.
Habener A Grychtol R Gaedcke S DeLuca D Dittrich AM Happle C et al . IgA(+) memory B-cells are significantly increased in patients with asthma and small airway dysfunction. Eur Respir J. (2022) 60:2102130. 10.1183/13993003.02130-2021
95.
Ghosh S Hoselton SA Asbach SV Steffan BN Wanjara SB Dorsam GP et al . B lymphocytes regulate airway granulocytic inflammation and cytokine production in a murine model of fungal allergic asthma. Cell Mol Immunol. (2015) 12:202–12. 10.1038/cmi.2014.103
96.
Habener A Happle C Grychtol R Skuljec J Busse M Daluge K et al . Regulatory B cells control airway hyperreactivity and lung remodeling in a murine asthma model. J Allergy Clin Immunol. (2021) 147:2281–94 e7. 10.1016/j.jaci.2020.09.041
97.
Global Initiative for Asthma and Global Initiative for Chronic Obstructive Pulmonary Disease . Diagnosis and Initial Treatment of Asthma, COPD, and Asthma-COPD Overlap 2017. Available online at: www.goldcopd.org (accessed February 2, 2024).
98.
Astrand AB Hemmerling M Root J Wingren C Pesic J Johansson E et al . Linking increased airway hydration, ciliary beating, and mucociliary clearance through ENaC inhibition. Am J Physiol Lung Cell Mol Physiol. (2015) 308:L22–32. 10.1152/ajplung.00163.2014
99.
Ono J Takai M Kamei A Ohta S Nair P Izuhara K et al . A novel assay for improved detection of sputum periostin in patients with asthma. PLoS ONE. (2023) 18:e0281356. 10.1371/journal.pone.0281356
100.
Zhang Y Ren L Sun J Han F Guo X . Increased serum soluble interleukin-2 receptor associated with severity of acute exacerbation of chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. (2021) 16:2561–73. 10.2147/COPD.S321904
101.
Toyota H Sugimoto N Kobayashi K Suzuki Y Takeshita Y Ito A et al . Comprehensive analysis of allergen-specific IgE in COPD: mite-specific IgE specifically related to the diagnosis of asthma-COPD overlap. Allergy Asthma Clin Immunol. (2021) 17:13. 10.1186/s13223-021-00514-9
102.
Wilson TM Maric I Shukla J Brown M Santos C Simakova O et al . IL-5 receptor alpha levels in patients with marked eosinophilia or mastocytosis. J Allergy Clin Immunol. (2011) 128:1086–92 e1-3. 10.1016/j.jaci.2011.05.032
103.
Lehmann JS Rughwani P Kolenovic M Ji S Sun B . Chapter Nine - LEGENDplex™: Bead-assisted multiplex cytokine profiling by flow cytometry. In:GalluzziLRudqvistN-P, editors. Methods Enzymol, vol. 629. Cambridge: Academic Press (2019). p. 151–76.
104.
Bahri R Custovic A Korosec P Tsoumani M Barron M Wu J et al . Mast cell activation test in the diagnosis of allergic disease and anaphylaxis. J Allergy Clin Immunol. (2018) 142:485–96 e16. 10.1016/j.jaci.2018.01.043
105.
Elst J van der Poorten MM Van Gasse AL De Puysseleyr L Hagendorens MM Faber MA et al . Mast cell activation tests by flow cytometry: a new diagnostic asset?Clin Exp Allergy. (2021) 51:1482–500. 10.1111/cea.13984
106.
Talal S Mona K Karem A Yaniv L Reut HM Ariel S et al . Neutrophil degranulation and severely impaired extracellular trap formation at the basis of susceptibility to infections of hemodialysis patients. BMC Med. (2022) 20:364. 10.1186/s12916-022-02564-1
107.
Galarza S Kim H Atay N Peyton SR Munson JM . 2D or 3D? How cell motility measurements are conserved across dimensions in vitro and translate in vivo. Bioeng Transl Med. (2020) 5:e10148. 10.1002/btm2.10148
108.
Li J Lin LH Wang J Peng X Dai HR Xiao H et al . Interleukin-4 and interleukin-13 pathway genetics affect disease susceptibility, serum immunoglobulin E levels, and gene expression in asthma. Ann Allergy Asthma Immunol. (2014) 113:173–9 e1. 10.1016/j.anai.2014.05.004
109.
Zhang L Gu H Gu Y Zeng X . Association between TNF-alpha−308 G/A polymorphism and COPD susceptibility: a meta-analysis update. Int J Chron Obstruct Pulmon Dis. (2016) 11:1367–79. 10.2147/COPD.S105394
110.
Gao X Wang X Jiao N Chen J Sun D . Association of VEGFA polymorphisms with chronic obstructive pulmonary disease in Chinese Han and Mongolian populations. Exp Physiol. (2021) 106:1839–48. 10.1113/EP089523
111.
Hyodo K Masuko H Oshima H Shigemasa R Kitazawa H Kanazawa J et al . Common exacerbation-prone phenotypes across asthma and chronic obstructive pulmonary disease (COPD). PLoS ONE. (2022) 17:e0264397. 10.1371/journal.pone.0264397
112.
Sharma S Khurana S Federman AD Wisnivesky J Holguin F . Asthma-chronic obstructive pulmonary disease overlap. Immunol Allergy Clin North Am. (2020) 40:565–73. 10.1016/j.iac.2020.07.002
113.
Habib N Pasha MA Tang DD . Current understanding of asthma pathogenesis and biomarkers. Cells. (2022) 11:2764. 10.3390/cells11172764
114.
Hirano T Matsunaga K . Measurement of blood eosinophils in asthma and chronic obstructive pulmonary disease. Intern Med. (2023) 62:21–5. 10.2169/internalmedicine.9339-22
115.
Escamilla-Gil JM Fernandez-Nieto M Acevedo N . Understanding the cellular sources of the fractional exhaled nitric oxide (FeNO) and its role as a biomarker of type 2 inflammation in asthma. Biomed Res Int. (2022) 2022:5753524. 10.1155/2022/5753524
116.
Benson VS Hartl S Barnes N Galwey N Van Dyke MK Kwon N . Blood eosinophil counts in the general population and airways disease: a comprehensive review and meta-analysis. Eur Respir J. (2022) 59:2004590. 10.1183/13993003.04590-2020
117.
Rupani H Kent BD . Using fractional exhaled nitric oxide measurement in clinical asthma management. Chest. (2022) 161:906–17. 10.1016/j.chest.2021.10.015
118.
Svenningsen S Nair P . Asthma endotypes and an overview of targeted therapy for asthma. Front Med. (2017) 4:158. 10.3389/fmed.2017.00158
119.
Nagase H Suzukawa M Oishi K Matsunaga K . Biologics for severe asthma: the real-world evidence, effectiveness of switching, and prediction factors for the efficacy. Allergol Int. (2023) 72:11–23. 10.1016/j.alit.2022.11.008
120.
Porsbjerg CM Townend J Bergeron C Christoff GC Katsoulotos GP Larenas-Linnemann D et al . Association between pre-biologic T2-biomarker combinations and response to biologics in patients with severe asthma. Front Immunol. (2024) 15:1361891. 10.3389/fimmu.2024.1361891
121.
Agache I Beltran J Akdis C Akdis M Canelo-Aybar C Canonica GW et al . Efficacy and safety of treatment with biologicals (benralizumab, dupilumab, mepolizumab, omalizumab and reslizumab) for severe eosinophilic asthma. A systematic review for the EAACI Guidelines - recommendations on the use of biologicals in severe asthma. Allergy. (2020) 75:1023–42. 10.1111/all.14221
122.
Hanania NA Wenzel S Rosen K Hsieh HJ Mosesova S Choy DF et al . Exploring the effects of omalizumab in allergic asthma: an analysis of biomarkers in the EXTRA study. Am J Respir Crit Care Med. (2013) 187:804–11. 10.1164/rccm.201208-1414OC
123.
Humbert M Beasley R Ayres J Slavin R Hebert J Bousquet J et al . Benefits of omalizumab as add-on therapy in patients with severe persistent asthma who are inadequately controlled despite best available therapy (GINA 2002 step 4 treatment): INNOVATE. Allergy. (2005) 60:309–16. 10.1111/j.1398-9995.2004.00772.x
124.
Casale TB Luskin AT Busse W Zeiger RS Trzaskoma B Yang M et al . Omalizumab effectiveness by biomarker status in patients with asthma: evidence from PROSPERO, a prospective real-world study. J Allergy Clin Immunol Pract. (2019) 7:156–64 e1. 10.1016/j.jaip.2018.04.043
125.
Kroes JA de Jong K Hashimoto S Zielhuis SW van Roon EN Sont JK et al . Clinical response to benralizumab can be predicted by combining clinical outcomes at 3 months with baseline characteristics. ERJ Open Res. (2023) 9:00559-2022. 10.1183/23120541.00559-2022
126.
FitzGerald JM Bleecker ER Menzies-Gow A Zangrilli JG Hirsch I Metcalfe P et al . Predictors of enhanced response with benralizumab for patients with severe asthma: pooled analysis of the SIROCCO and CALIMA studies. Lancet Respir Med. (2018) 6:51–64. 10.1016/S2213-2600(17)30344-2
127.
Maniscalco M Fuschillo S Mormile I Detoraki A Sarnelli G Paulis A et al . Exhaled nitric oxide as biomarker of type 2 diseases. Cells. (2023) 12:2518. 10.3390/cells12212518
128.
Pavord ID Deniz Y Corren J Casale TB FitzGerald JM Izuhara K et al . Baseline FeNO independently predicts the dupilumab response in patients with moderate-to-severe asthma. J Allergy Clin Immunol Pract. (2023) 11:1213–20 e2. 10.1016/j.jaip.2022.11.043
129.
Busse WW Wenzel SE Casale TB FitzGerald JM Rice MS Daizadeh N et al . Baseline FeNO as a prognostic biomarker for subsequent severe asthma exacerbations in patients with uncontrolled, moderate-to-severe asthma receiving placebo in the LIBERTY ASTHMA QUEST study: a post-hoc analysis. Lancet Respir Med. (2021) 9:1165–73. 10.1016/S2213-2600(21)00124-7
130.
Tohda Y Nakamura Y Fujisawa T Ebisawa M Arima K Miyata M et al . Dupilumab efficacy and safety in Japanese patients with uncontrolled, moderate-to-severe asthma in the phase 3 LIBERTY ASTHMA QUEST study. Allergol Int. (2020) 69:578–87. 10.1016/j.alit.2020.04.002
131.
Shilovskiy IP Nikolskii AA Kurbacheva OM Khaitov MR . Modern view of neutrophilic asthma molecular mechanisms and therapy. Biochemistry. (2020) 85:854–68. 10.1134/S0006297920080027
132.
Chung KF . Type-2-low severe asthma endotypes for new treatments: the new asthma frontier. Curr Opin Allergy Clin Immunol. (2023) 23:199–204. 10.1097/ACI.0000000000000899
133.
Hinks TSC Levine SJ Brusselle GG . Treatment options in type-2 low asthma. Eur Respir J. (2021) 57:2000528. 10.1177/00145246211054274
134.
Fitzpatrick AM Chipps BE Holguin F Woodruff PG . T2-“Low” asthma: overview and management strategies. J Allergy Clin Immunol Pract. (2020) 8:452–63. 10.1016/j.jaip.2019.11.006
135.
Busse WW Holgate S Kerwin E Chon Y Feng J Lin J et al . Randomized, double-blind, placebo-controlled study of brodalumab, a human anti-IL-17 receptor monoclonal antibody, in moderate to severe asthma. Am J Respir Crit Care Med. (2013) 188:1294–302. 10.1164/rccm.201212-2318OC
136.
Wenzel SE Barnes PJ Bleecker ER Bousquet J Busse W Dahlen SE et al . A randomized, double-blind, placebo-controlled study of tumor necrosis factor-alpha blockade in severe persistent asthma. Am J Respir Crit Care Med. (2009) 179:549–58. 10.1164/rccm.200809-1512OC
137.
O'Byrne PM Panettieri RA Taube C Brindicci C Fleming M Altman Altman P Development of an inhaled anti-TSLP therapy for asthma . Pulm Pharmacol Ther. (2023) 78:102184. 10.1016/j.pupt.2022.102184
138.
Menzies-Gow A Corren J Bourdin A Chupp G Israel E Wechsler ME et al . Tezepelumab in adults and adolescents with severe, uncontrolled asthma. N Engl J Med. (2021) 384:1800–9. 10.1056/NEJMoa2034975
139.
Corren J Menzies-Gow A Chupp G Israel E Korn S Cook B et al . Efficacy of tezepelumab in severe, uncontrolled asthma: pooled analysis of the PATHWAY and NAVIGATOR clinical trials. Am J Respir Crit Care Med. (2023) 208:13–24. 10.1164/rccm.202210-2005OC
140.
Menzies-Gow A Wechsler ME Brightling CE Korn S Corren J Israel E et al . Long-term safety and efficacy of tezepelumab in people with severe, uncontrolled asthma (DESTINATION): a randomised, placebo-controlled extension study. Lancet Respir Med. (2023) 11:425–38. 10.1016/S2213-2600(22)00492-1
141.
McGregor MC Krings JG Nair P Castro M . Role of biologics in asthma. Am J Respir Crit Care Med. (2019) 199:433–45. 10.1164/rccm.201810-1944CI
142.
Krings JG McGregor MC Bacharier LB Castro M . Biologics for severe asthma: treatment-specific effects are important in choosing a specific agent. J Allergy Clin Immunol Pract. (2019) 7:1379–92. 10.1016/j.jaip.2019.03.008
143.
Lewis BW Ford ML Rogers LK Britt RD Jr . Oxidative stress promotes corticosteroid insensitivity in asthma and COPD. Antioxidants. (2021) 10:1335. 10.3390/antiox10091335
144.
David B Bafadhel M Koenderman L De Soyza A . Eosinophilic inflammation in COPD: from an inflammatory marker to a treatable trait. Thorax. (2021) 76:188–95. 10.1136/thoraxjnl-2020-215167
145.
Papaporfyriou A Bakakos P Hillas G Papaioannou AI Loukides S . Blood eosinophils in COPD: friend or foe?Expert Rev Respir Med. (2022) 16:35–41. 10.1080/17476348.2021.2011219
146.
Cheng SL . Blood eosinophils and inhaled corticosteroids in patients with COPD: systematic review and meta-analysis. Int J Chron Obstruct Pulmon Dis. (2018) 13:2775–84. 10.2147/COPD.S175017
147.
Harries TH Rowland V Corrigan CJ Marshall IJ McDonnell L Prasad V et al . Blood eosinophil count, a marker of inhaled corticosteroid effectiveness in preventing COPD exacerbations in post-hoc RCT and observational studies: systematic review and meta-analysis. Respir Res. (2020) 21:3. 10.1186/s12931-019-1268-7
148.
Pavord ID Chanez P Criner GJ Kerstjens HAM Korn S Lugogo N et al . Mepolizumab for eosinophilic chronic obstructive pulmonary disease. N Engl J Med. (2017) 377:1613–29. 10.1056/NEJMoa1708208
149.
Criner GJ Celli BR Brightling CE Agusti A Papi A Singh D et al . Benralizumab for the prevention of COPD exacerbations. N Engl J Med. (2019) 381:1023–34. 10.1056/NEJMoa1905248
150.
Bhatt SP Rabe KF Hanania NA Vogelmeier CF Bafadhel M Christenson SA et al . Dupilumab for COPD with blood eosinophil evidence of type 2 inflammation. N Engl J Med. (2024) 390:2274–83. 10.1056/NEJMoa2401304
151.
Albert RK Connett J Bailey WC Casaburi R Cooper JA Criner GJ et al . Azithromycin for prevention of exacerbations of COPD. N Engl J Med. (2011) 365:689–98. 10.1056/NEJMoa1104623
152.
Uzun S Djamin RS Kluytmans JA Mulder PG . van't Veer NE, Ermens AA, et al. Azithromycin maintenance treatment in patients with frequent exacerbations of chronic obstructive pulmonary disease (COLUMBUS): a randomised, double-blind, placebo-controlled trial. Lancet Respir Med. (2014) 2:361–8. 10.1016/S2213-2600(14)70019-0
153.
Choi JY Rhee CK Yoo KH Jung KS Lee JH Yoon HK et al . Heterogeneity of asthma-chronic obstructive pulmonary disease (COPD) overlap from a cohort of patients with severe asthma and COPD. Ther Adv Respir Dis. (2023) 17:17534666231169472. 10.1177/17534666231169472
154.
Hudler A Holguin F Sharma S . Pathophysiology of asthma-chronic obstructive pulmonary disease overlap. Immunol Allergy Clin North Am. (2022) 42:521–32. 10.1016/j.iac.2022.04.008
155.
Wang J Wang W Lin H Huan C Jiang S Lin D et al . Role of pulmonary function and FeNO detection in early screening of patients with ACO. Exp Ther Med. (2020) 20:830–7. 10.3892/etm.2020.8762
156.
Shi F Qiu C Yu J Yang Y Li B Feng M et al . Comparison of fractional exhaled nitric oxide in elderly patients with asthma-chronic obstructive pulmonary disease overlap and other airway inflammatory diseases. Iran J Allergy Asthma Immunol. (2018) 17:232–9.
157.
Li M Yang T He R Li A Dang W Liu X et al . The value of inflammatory biomarkers in differentiating asthma-COPD overlap from COPD. Int J Chron Obstruct Pulmon Dis. (2020) 15:3025–37. 10.2147/COPD.S273422
158.
Takayama Y Ohnishi H Ogasawara F Oyama K Kubota T Yokoyama A . Clinical utility of fractional exhaled nitric oxide and blood eosinophils counts in the diagnosis of asthma-COPD overlap. Int J Chron Obstruct Pulmon Dis. (2018) 13:2525–32. 10.2147/COPD.S167600
159.
Shim JS Kim H Kwon JW Park SY Kim S Kim BK et al . A comparison of treatment response to biologics in asthma-COPD overlap and pure asthma: Findings from the PRISM study. World Allergy Organ J. (2023) 16:100848. 10.1016/j.waojou.2023.100848
160.
Barnes PJ . Therapeutic approaches to asthma-chronic obstructive pulmonary disease overlap syndromes. J Allergy Clin Immunol. (2015) 136:531–45. 10.1016/j.jaci.2015.05.052
161.
Kuruvilla ME Lee FE Lee GB . Understanding asthma phenotypes, endotypes, and mechanisms of disease. Clin Rev Allergy Immunol. (2019) 56:219–33. 10.1007/s12016-018-8712-1
162.
Guida G Bagnasco D Carriero V Bertolini F Ricciardolo FLM Nicola S et al . Critical evaluation of asthma biomarkers in clinical practice. Front Med. (2022) 9:969243. 10.3389/fmed.2022.969243
163.
Huang AX Lu LW Liu WJ Huang M . Plasma Inflammatory Cytokine IL-4, IL-8, IL-10, and TNF-alpha levels correlate with pulmonary function in patients with asthma-chronic obstructive pulmonary disease (COPD) overlap syndrome. Med Sci Monit. (2016) 22:2800–8. 10.12659/MSM.896458
164.
Ding Q Sun S Zhang Y Tang P Lv C Ma H et al . Serum IL-8 and VEGFA are two promising diagnostic biomarkers of asthma-COPD overlap syndrome. Int J Chron Obstruct Pulmon Dis. (2020) 15:357–65. 10.2147/COPD.S233461
165.
Gava G Nunez A Esquinas C Sarasate M Loeb E Pirina P et al . Analysis of blood biomarkers in patients with chronic obstructive pulmonary disease (COPD) and with asthma-COPD overlap (ACO). COPD. (2020) 17:306–10. 10.1080/15412555.2020.1761314
166.
Wang J Lv H Luo Z Mou S Liu J Liu C et al . Plasma YKL-40 and NGAL are useful in distinguishing ACO from asthma and COPD. Respir Res. (2018) 19:47. 10.1186/s12931-018-0755-6
167.
Liu G Jarnicki AG Paudel KR Lu W Wadhwa R Philp AM et al . Adverse roles of mast cell chymase-1 in COPD. Eur Respir J. (2022) 60:2101431. 10.1183/13993003.01431-2021
168.
Siddhuraj P Jonsson J Alyamani M Prabhala P Magnusson M Lindstedt S et al . Dynamically upregulated mast cell CPA3 patterns in chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. Front Immunol. (2022) 13:924244. 10.3389/fimmu.2022.924244
169.
Bagher M Rosmark O Elowsson Rendin L Nybom A Wasserstrom S Muller C et al . Crosstalk between mast cells and lung fibroblasts is modified by alveolar extracellular matrix and influences epithelial migration. Int J Mol Sci. (2021) 22:506. 10.3390/ijms22020506
170.
Xisto DG Farias LL Ferreira HC Picanco MR Amitrano D Lapa ESJR et al . Lung parenchyma remodeling in a murine model of chronic allergic inflammation. Am J Respir Crit Care Med. (2005) 171:829–37. 10.1164/rccm.200408-997OC
171.
Tiotiu A Badi Y Kermani NZ Sanak M Kolmert J Wheelock CE et al . Association of differential mast cell activation with granulocytic inflammation in severe asthma. Am J Respir Crit Care Med. (2022) 205:397–411. 10.1164/rccm.202102-0355OC
172.
Shrestha Palikhe N Wu Y Konrad E Gandhi VD Rowe BH Vliagoftis H et al . Th2 cell markers in peripheral blood increase during an acute asthma exacerbation. Allergy. (2021) 76:281–90. 10.1111/all.14543
173.
Dhariwal J Cameron A Wong E Paulsen M Trujillo-Torralbo MB Del Rosario A et al . Pulmonary innate lymphoid cell responses during rhinovirus-induced asthma exacerbations in vivo: a clinical trial. Am J Respir Crit Care Med. (2021) 204:1259–73. 10.1164/rccm.202010-3754OC
174.
Gao J Zhou W Chen B Lin W Wu S Wu F . Sputum cell count: biomarkers in the differentiation of asthma, COPD and asthma-COPD overlap. Int J Chron Obstruct Pulmon Dis. (2017) 12:2703–10. 10.2147/COPD.S142466
175.
Spears M McSharry C Donnelly I Jolly L Brannigan M Thomson J et al . Peripheral blood dendritic cell subtypes are significantly elevated in subjects with asthma. Clin Exp Allergy. (2011) 41:665–72. 10.1111/j.1365-2222.2010.03692.x
176.
Vroman H Tindemans I Lukkes M van Nimwegen M de Boer GM Tramper-Stranders GA et al . Type II conventional dendritic cells of asthmatic patients with frequent exacerbations have an altered phenotype and frequency. Eur Respir J. (2020) 55:1900859. 10.1183/13993003.00859-2019
177.
Pallazola AM Rao JX Mengistu DT Morcos MS Toma MS Stolberg VR et al . Human lung cDC1 drive increased perforin-mediated NK cytotoxicity in chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol. (2021) 321:L1183–L93. 10.1152/ajplung.00322.2020
178.
Stoll P Bahker A Ulrich M Bratke K Garbe K Christian Virchow J et al . The dendritic cell high-affinity IgE receptor is overexpressed in both asthma and severe COPD. Clin Exp Allergy. (2016) 46:575–83. 10.1111/cea.12664
179.
Galgani M Fabozzi I Perna F Bruzzese D Bellofiore B Calabrese C et al . Imbalance of circulating dendritic cell subsets in chronic obstructive pulmonary disease. Clin Immunol. (2010) 137:102–10. 10.1016/j.clim.2010.06.010
180.
Shi YH Shi GC Wan HY Jiang LH Ai XY Zhu HX et al . Coexistence of Th1/Th2 and Th17/Treg imbalances in patients with allergic asthma. Chin Med J (Engl). (2011) 124:1951–6.
181.
Xu W Li R Sun Y . Increased IFN-gamma-producing Th17/Th1 cells and their association with lung function and current smoking status in patients with chronic obstructive pulmonary disease. BMC Pulm Med. (2019) 19:137. 10.1186/s12890-019-0899-2
182.
Zheng X Zhang L Chen J Gu Y Xu J Ouyang Y . Dendritic cells and Th17/Treg ratio play critical roles in pathogenic process of chronic obstructive pulmonary disease. Biomed Pharmacother. (2018) 108:1141–51. 10.1016/j.biopha.2018.09.113
183.
Wang H Ying H Wang S Gu X Weng Y Peng W et al . Imbalance of peripheral blood Th17 and Treg responses in patients with chronic obstructive pulmonary disease. Clin Respir J. (2015) 9:330–41. 10.1111/crj.12147
184.
Urboniene D Babusyte A Lotvall J Sakalauskas R Sitkauskiene B . Distribution of gammadelta and other T-lymphocyte subsets in patients with chronic obstructive pulmonary disease and asthma. Respir Med. (2013) 107:413–23. 10.1016/j.rmed.2012.11.012
185.
Kim WD Kim WS Koh Y Lee SD Lim CM Kim DS et al . Abnormal peripheral blood T-lymphocyte subsets in a subgroup of patients with COPD. Chest. (2002) 122:437–44. 10.1378/chest.122.2.437
186.
Chambers ES Nanzer AM Pfeffer PE Richards DF Martineau AR Griffiths CJ et al . Dendritic cell phenotype in severe asthma reflects clinical responsiveness to glucocorticoids. Clin Exp Allergy. (2018) 48:13–22. 10.1111/cea.13061
187.
Choudhury P Biswas S Singh G Pal A Ghosh N Ojha AK et al . Immunological profiling and development of a sensing device for detection of IL-13 in COPD and asthma. Bioelectrochemistry. (2022) 143:107971. 10.1016/j.bioelechem.2021.107971
188.
Uzan GC Borekci S Doventas YE Koldas M Gemicioglu B . The relationship between inflammatory markers and spirometric parameters in ACOS, Asthma, and COPD. J Asthma. (2020) 57:1273–9. 10.1080/02770903.2019.1652644
189.
Kubysheva N Boldina M Eliseeva T Soodaeva S Klimanov I Khaletskaya A et al . Relationship of Serum Levels of IL-17, IL-18, TNF-alpha, and Lung Function Parameters in Patients with COPD, Asthma-COPD Overlap, and Bronchial Asthma. Mediators Inflamm. (2020) 2020:4652898. 10.1155/2020/4652898
190.
Khatri SB Iaccarino JM Barochia A Soghier I Akuthota P Brady A et al . Use of fractional exhaled nitric oxide to guide the treatment of asthma: an official American Thoracic Society Clinical Practice Guideline. Am J Respir Crit Care Med. (2021) 204:e97–e109. 10.1164/rccm.202109-2093ST
191.
Shi C Zhao H . Association between tumor necrosis factor-308 G/A polymorphism and chronic obstructive pulmonary disease risk in chinese population: evidence from a meta-analysis. Clin Lab. (2019) 65. 10.7754/Clin.Lab.2019.190313
192.
Jia X Li S Xu T Ji N Huang M . Diagnostic accuracy of periostin in predicting asthma: a systematic review and Meta-analysis. J Asthma. (2021) 58:307–15. 10.1080/02770903.2019.1684518
193.
Kerr SC Gonzalez JR Schanin J Peters MC Lambrecht BN Brock EC et al . An anti-siglec-8 antibody depletes sputum eosinophils from asthmatic subjects and inhibits lung mast cells. Clin Exp Allergy. (2020) 50:904–14. 10.1111/cea.13681
Summary
Keywords
asthma, asthma-COPD overlap, chronic obstructive pulmonary disease, in vitro models, pathogenesis, biomarkers
Citation
Simmalee K, Kawamatawong T, Vitte J, Demoly P and Lumjiaktase P (2025) Exploring the pathogenesis and clinical implications of asthma, chronic obstructive pulmonary disease (COPD), and asthma-COPD overlap (ACO): a narrative review. Front. Med. 12:1514846. doi: 10.3389/fmed.2025.1514846
Received
21 October 2024
Accepted
26 March 2025
Published
17 April 2025
Volume
12 - 2025
Edited by
Ulrich Matthias Zissler, University of Applied Sciences Rosenheim, Germany
Reviewed by
Ramcés Falfán-Valencia, National Institute of Respiratory Diseases-Mexico (INER), Mexico
Serap Duru, Dişkapi Yildirim Training and Research Hospital, Türkiye
Updates
Copyright
© 2025 Simmalee, Kawamatawong, Vitte, Demoly and Lumjiaktase.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pascal Demoly pascal.demoly@inserm.frPutthapoom Lumjiaktase putthapoom@gmail.com
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.