Abstract
With a high frequency and a poor prognosis, combined pre-and post-capillary pulmonary hypertension (Cpc-PH) is a significant subtype of pulmonary hypertension linked to left-sided heart disease (PH-LHD). The complicated pathophysiology of Cpc-PH is primarily characterized by elevated pulmonary venous pressure leading to an increase in retrocapillary pressure, which is followed by elevated pulmonary artery pressure and a marked rise in pulmonary vascular resistance (PVR). There is currently no well-defined treatment plan for Cpc-PH, and there are numerous obstacles to overcome. In patients with Cpc-PH, the effectiveness of targeted medications for pulmonary hypertension is limited and debatable. Recent research has revealed that the prevalence and progression of Cpc-PH may be influenced by genetic factors, metabolic syndrome, oxidative stress, and fibrosis. To help doctors better manage and treat patients with Cpc-PH, this review provides a detailed description of the disease’s epidemiology, pathogenesis, diagnostic techniques, current treatment status, and potential therapeutic targets.
Introduction
The 2022 European guidelines for the diagnosis and treatment of pulmonary hypertension (PH), the 2022 ESC/ERS Guidelines for the Diagnosis and Treatment of Pulmonary Arterial Hypertension, define PH as the mean pulmonary artery pressure (mPAP) measured by right heart catheterization (RHC) at sea level and at rest ≥20 mmHg, and pulmonary artery wedge pressure (PAWP) > 15 mmHg (1). Compared with the 2015 guidelines, the threshold for mPAP was lowered from 25 mmHg to 20 mmHg, and the threshold for PVR was lowered from 3 WU to 2 WU, which is more conducive to the early detection and diagnosis of patients with PH. Pulmonary arterial hypertension associated with left-sided heart disease (PH-LHD) is the most common form, accounting for approximately 68.5% of PH cases (2). PH-LHD is divided into two subtypes: Isolated postcapillary PH (Ipc-PH): mPAP>20 mmHg, PAWP>15 mmHg, PVR ≤ 2 WU; Combined pre-and post-capillary PH (Cpc-PH): mPAP>20 mmHg, PAWP>15 mmHg, PVR > 2 WU (3). Combined pre-and postcapillary pulmonary hypertension (Cpc-PH) is a specific phenotype of left-sided heart disease-associated pulmonary hypertension (PH-LHD, WHO category 2) characterized by the coexistence of retrocapillary hypertension (increased pulmonary venous pressure) and pulmonary vascular remodeling (precapillary hypertension) due to left-sided heart disease.
In hemodynamics, Cpc-PH is usually associated with left heart disease or lung disease, which leads to elevated pulmonary venous pressure and subsequently affects pulmonary arterial pressure. In the early stage, the elevation of PAP is mainly due to the passive transmission of left atrial pressure. At this stage, PVR usually remains within the normal range and has not yet caused significant changes in pulmonary vascular structure. As the left atrial pressure continues to rise, changes begin to occur in the pulmonary vascular structure: veins become more permeable, with collagen deposition, leading to thickening of the vessel walls and narrowing of the lumen; capillaries and small arteries, under long-term high-pressure stimulation, develop intimal fibrosis and medial hypertrophy of the pulmonary arteries, ultimately resulting in an increase in PVR. When PVR exceeds 2 WU, Cpc-PH is established. The probability of hospitalization for heart failure or all-cause death increased by 2% for every 1 mmHg increase in mPAP, and by 7% for every 1 WU increase in PVR (4). According to the new definition, the ideal PVR threshold is nearly 2 WU, or 2.2 WU. Combined capillary pulmonary hypertension is difficult to diagnose and treat; a customized treatment strategy must be created, taking into account the effects of lung and left heart disease (Figure 1). This holds significant importance in comprehending the pathophysiological process of pulmonary hypertension and enhancing patient outcomes.
Figure 1

WHO classification, etiological classification, and hemodynamic classification of pulmonary arterial hypertension.
Epidemiology
About 1% of people worldwide have pulmonary hypertension, and among those 65 and older, the frequency rises to 10%. In developed countries (eg, Europe and the United States), the annual incidence of Cpc-PH is about 5–10 cases per 100,000 people, accounting for 5–10% of all new cases of pulmonary hypertension. The age of onset is more common in the elderly (> 65 years) (1–5), which is associated with a high incidence of left-sided heart disease, and the proportion of women is slightly higher. Hypertension (>70%), diabetes, and obesity (BMI > 30%) are major risk factors in patients with underlying medical conditions, and the incidence of Cpc-PH is higher in patients with valvular disease (eg, mitral regurgitation) and atrial fibrillation. Mortality is significantly higher in patients with Cpc-PH than in PH-LHD alone (2-year survival rate is about 50–60%, 5-year survival rate is about 40–50%, vs. 70–80%), and the risk of death is increased by 20–30% for every 1 WU increase in PVR (3–6). In studies of various types of pulmonary hypertension, mildly elevated mPAP (21–24 mmHg) was significantly associated with increased mortality (3–7). Several studies of patients with systemic sclerosis have found that patients with 24 mmHg ≥ mPAP ≥ 21 mmHg not only have higher mortality but are more likely to progress to pulmonary hypertension (mPAP ≥ 25 mmHg) (3). A total of 5–12% patients with left-sided heart disease (PH-LHD) developed Cpc-PH. According to EU-US and US cohort studies, 1.5–3% of patients with heart failure with reduced ejection fraction (HFrEF) also developed Cpc-PH; and between 2 and 5% of patients with Heart Failure with Preserved Ejection Fraction (HFpEF) developed Cpc-PH (due to pulmonary vascular disease and common metabolic syndrome). Pulmonary hypertension is linked to a twofold increase in mortality in people with both HFrEF and HFpEF. The 3-and 5-year death rates for PH-LHD were 42.3 and 52.6%, respectively, in a cohort study of 4,621 patients with the condition (8). However, all of these PH-LHD clinical cohort studies are from Western nations. From Ipc-PH to Cpc-PH, the annual conversion rate is about 3–6% (9). In certain groups of people with systemic sclerosis, left-sided heart disease and PAH may coexist, increasing the risk of Cpc-PH. The elderly, diabetic patients (30–40%) and patients with chronic kidney disease (GFR < 60 mL/min/1.73 m2) are more likely to have mixed pH (10).
China currently lacks representative data on PH-LHD, particularly in the form of long-term follow-up reports. Therefore, multi-center registration studies and the creation of a PH-LHD/Cpc-PH database based on RHC as the standard diagnosis in China are urgently needed.
Pathophysiological mechanisms
Cpc-PH involves complex pathophysiological mechanisms. The increase of PAWP reflects left atrial dysfunction. This increase in pressure will be transmitted to the pulmonary circulation, leading to the increase of PAP and PVR, and the decrease of pulmonary artery compliance (PAC). It will eventually lead to endothelial dysfunction, pulmonary vascular remodeling and further increase the afterload of the right ventricle.
Pulmonary vascular remodeling and fibrosis mechanisms
Pulmonary vascular remodeling is the core pathological feature of Cpc-PH, characterized by intimal proliferation, medial hypertrophy, and adventitial fibrosis of the pulmonary arterioles. In the early stages, elevated left ventricular filling pressure leads to impaired pulmonary venous return, increased pulmonary vascular pressure, but no significant structural changes, termed IPC-PH (passive PH) (11). As the disease progresses, structural and functional changes in the pulmonary arteries occur, with thickening and hardening of the vascular walls, narrowing of the lumen, increased resistance, and persistent elevation of pulmonary vascular resistance leading to right heart failure, entering the Cpc-PH phase (12). Patients with Cpc-PH have a higher incidence of right ventricular dysfunction and pulmonary artery remodeling, with significant features of distal pulmonary artery hypertrophic remodeling, fibrosis, and luminal occlusion (13, 14).
Macrophages interact with pulmonary vascular endothelial cells to drive pulmonary vascular remodeling, with an increase in CD68+ cells (M2 macrophage subtype) in the adventitial layer, and adventitial fibroblasts may be the primary mediators of their activation and polarization. Fatty acid-binding protein 5 (FABP5) exacerbates pulmonary artery fibrosis by activating the Wnt/β-catenin signaling pathway, significantly upregulated in Cpc-PH mouse models, and closely related to pulmonary artery fibrosis. In vitro experiments have shown that inhibiting FABP5 (via siRNA or antagonists) can attenuate TGF-β1-induced fibrotic responses. Specific alveolar capillary endothelial cells (Plvap+) can transiently activate mesenchymal gene aSMA, affecting extracellular matrix remodeling, vascular integrity, and cell-to-cell interactions (15, 16).
SBFI-26 is a selective FABP5 inhibitor that can inhibit fibrosis by modulating the Wnt/β-catenin signaling pathway, significantly suppressing pulmonary artery fibrosis and remodeling in animal models, reducing pulmonary artery wall thickness and collagen deposition. Circular RNA circALMS1 is downregulated in patients with pulmonary arterial hypertension, and its overexpression can inhibit the proliferation and migration of pulmonary microvascular endothelial cells by suppressing the miR-17-3p/YTHDF2 pathway, improving right heart function (17, 18).
Neurohumoral mechanisms
These include sympathetic overactivation, activation of the renin-angiotensin-aldosterone system (RAAS), inflammatory cell infiltration, and oxidative stress responses. Angiotensin II (Ang II) activates the RhoA/Rho kinase signaling pathway, inhibits myosin light chain phosphatase (MLCP) and endothelial nitric oxide synthase (eNOS) activity, leading to vasoconstriction and endothelial dysfunction (19). The ACE2-Ang (1–7)-Mas axis promotes the release of nitric oxide (NO) and prostaglandins, exerting vasodilatory, anti-proliferative, and anti-inflammatory effects, with enhanced activity improving pulmonary hemodynamics (20). Recombinant human soluble ACE2 (rhACE2) can improve pulmonary artery pressure and reduce oxidative stress (21, 22).
Left atrial dysfunction
HFpEF, HFrEF, and VHD can lead to elevated left atrial pressure (LAP), increased volume, and subsequent left atrial enlargement, impaired contractility, and interstitial fibrosis as part of the remodeling process (23). Left atrial remodeling weakens its barrier function, passively transmitting pressure to the pulmonary vasculature, causing elevated pulmonary venous pressure and pulmonary congestion (24). Sudden increases in LAP can lead to “alveolar-capillary stress failure,” disrupting the alveolar-capillary barrier and causing pulmonary edema (25). Under the influence of endothelial dysfunction, neurohumoral factors, and inflammatory cell infiltration, persistent changes in LAP lead to structural abnormalities in the pulmonary vasculature and increased pulmonary vascular resistance (26–28).
Pulmonary vascular endothelial dysfunction
Endothelial cell injury leads to vasoconstriction, inflammatory responses, and fibrosis. Vasoreactive substances such as nitric oxide (NO), prostacyclin (PGI2), and endothelin-1 (ET-1) collectively regulate pulmonary vasodilation and constriction (29). NO is synthesized and released by eNOS, dilating blood vessels through the NO-sGC-cGMP-PKG pathway, inhibiting smooth muscle cell proliferation, and slowing pulmonary vascular remodeling (30). Elevated aldosterone levels can induce oxidative stress, impair ET-B receptor signaling, reduce NO synthesis and bioavailability, and simultaneously, excessive ET-1 production can inhibit eNOS expression, decreasing NO secretion (31, 32).
Inflammation and chemotaxis mechanisms
CXCL8 (IL-8) and its receptors CXCR1 and CXCR2 are upregulated in pulmonary arterial hypertension, with CXCL8 promoting the recruitment and activation of inflammatory cells through binding to CXCR1/2, exacerbating pulmonary artery inflammation and remodeling. The interaction between CXCL10 and its receptor CXCR3 plays a significant role in the pathogenesis of PH, with CXCL10 inducing chemotaxis of T cells and natural killer cells, participating in pulmonary artery inflammatory responses. The signaling pathways of CXCL12 and its receptors CXCR4 and ACKR3 also play important roles in PH, with the CXCL12-CXCR4 axis not only regulating the migration of inflammatory cells but also promoting the proliferation of PASMCs through the PI3K/Akt signaling pathway (33–36).
Genetic factors and molecular signaling pathway mechanisms
BMPR-II is a member of the TGF-β superfamily, with heterozygous mutations present in 80% of familial PH patients and 20% of sporadic PH patients, making it a key pathogenic gene for primary and hereditary PH (37–39). It influences cell proliferation, differentiation, tissue repair, inflammation, and angiogenesis through Smad1/5/8 and non-Smad signaling pathways. BMP9 gene mutation rates are as high as 6.7% in East Asian populations, with mutations reducing circulating BMP9 levels, weakening vascular anti-apoptotic and anti-damage capabilities, and increasing the risk of idiopathic PH by over 22 times (40–42). The PPARγ-p53 pathway regulates cell proliferation, apoptosis, and inflammatory responses, affecting pulmonary vascular remodeling (43–47). Other gene mutations (such as SMAD1, SMAD4, SMAD9, CAV1, and KCNK3) have also been identified in BMPR-II mutation-negative families (48–50). CLIC4 is highly expressed under stress conditions, promoting BMPR-II internalization and degradation, inhibiting the BMP signaling pathway, and inducing a phenotypic shift in endothelial cells toward anti-apoptotic and proliferative states (51–57).
With the in-depth study of the pathogenesis of Cpc-PH, more and more evidence suggests that mutations in multiple functional genes (such as BMPRII, ALK1, CAV1, TBX4, KCNK3, SMAD8, EIF2AK4, etc.) play an important role in the pathogenesis of the disease. At present, researchers generally believe that the occurrence of PH is similar to tumors, with a “secondary strike” mechanism, where genetic and environmental factors jointly promote the occurrence and development of the disease.
Gene therapy and molecular signaling pathway mechanisms
Experiments based on p53 nanoparticle delivery and AAV vector-mediated delivery of normal BMPR2 genes have shown promising results. Lipid nanoparticles (LNPs) delivering CRISPR-Cas9 systems can repair gene mutations in PASMCs (58–60). Canagliflozin alleviates PH symptoms by activating PPARγ and inhibiting its S225 phosphorylation, showing good protective effects in animal models. Maintaining pulmonary vascular homeostasis by deubiquitination to activate the ALK2-Smad1/5/9-PPARγ axis, with upregulation of BRCC3, can alleviate PH symptoms (61–64) (Figure 2).
Figure 2
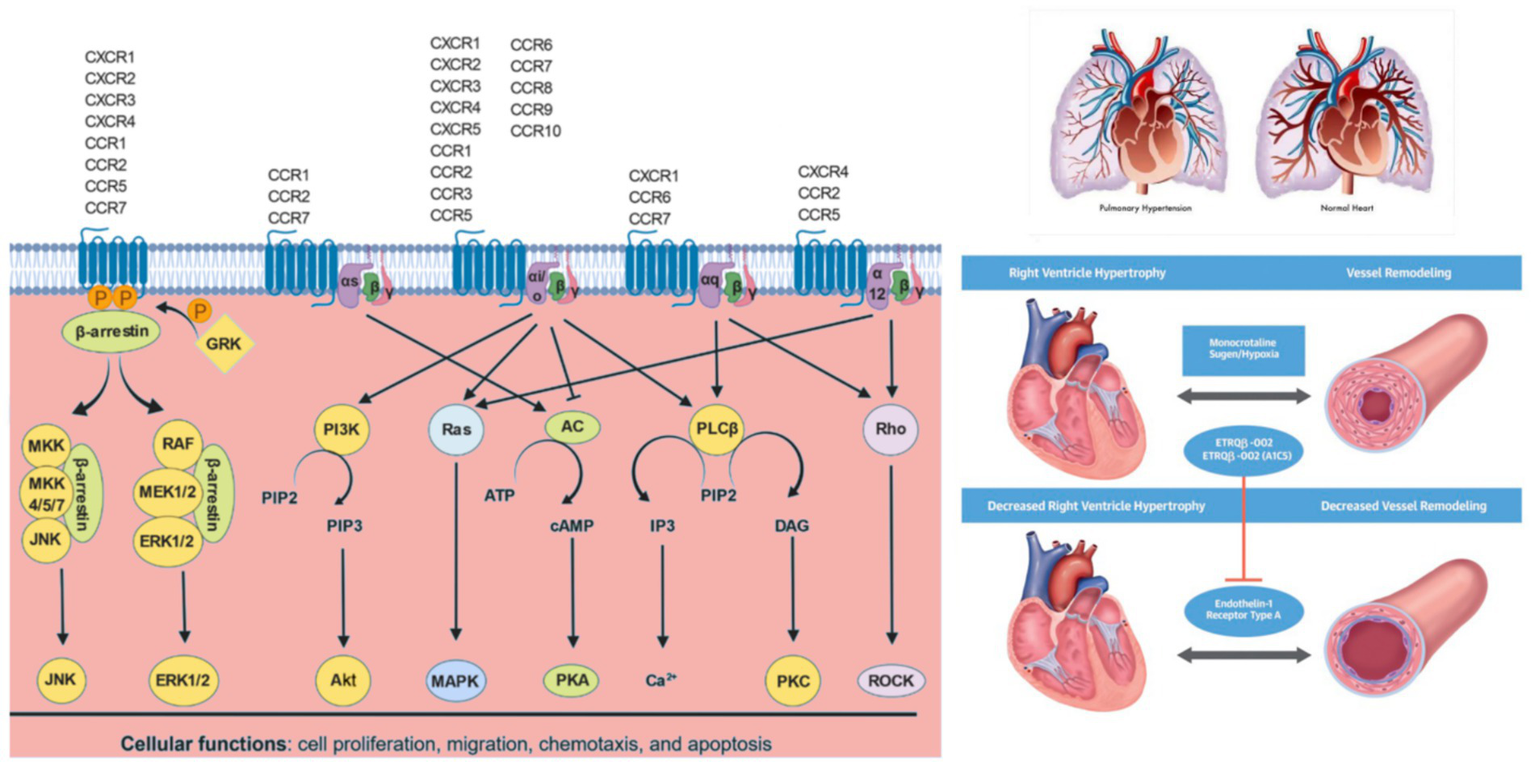
Chemokine signaling pathways: Chemokines bind to their cognate chemokine receptors expressed on different cell types. Upon receptor activation, the G protein dissociates into Gα and Gβγ subunits. Depending on the specific Gα subunit(s) to which the chemokine receptor is coupled, various downstream signaling pathways are regulated. Gαs protein activates adenylate cyclase (AC) to produce cAMP, which activates cAMP-dependent PKA. Gαi/o inactivates AC limiting cAMP levels and PKA activity. Gαi/o proteins activate PI3Ks, which converts phosphatidylinositol 4,5-bisphosphate (PIP2) to phosphatidylinositol (3,4,5)-trisphosphate (PIP3), which in turn activates PKB (Akt). Gαi/o and Gα12 proteins activate Ras, which further activates various MAPKs. Gαi/o and Gαq proteins activate PLCβ, which catalyzes PIP2 to IP3 and DAG. IP3 further regulates intracellular free calcium (Ca2+) levels, while DAG activates PKC. Gαq and Gα12 proteins also activates Rho family of GTPases (Rho), which further activates Rho-associated PK (ROCK). GPCR kinases (GRK) phosphorylate GPCRs, which enable β-arrestins to bind and internalize GPCRs, which can result in receptor recycling, if receptor phosphorylation is reversed by protein phosphatase 2 (PP2A) or degradation in lysosomes. β-arrestin bound to GPCRs can also activate MAPK pathways such as JNK and ERKs 1 and 2 (ERK1/2).
Diagnostic methods
Diagnosis of Cpc-PH is based on a combination of hemodynamic, radiographic, and clinical findings. Right Heart Catheterization (RHC) remains the gold standard for distinguishing PH-LHD/Cpc-PH from other forms of PH, with direct measurement of mPAP, PAWP, PVR, and DPG were directly measured to determine the hemodynamic classification. Diastolic Pressure Gradient (DPG) = Pulmonary Diastolic Blood Pressure-PAWP, DPG ≥ 7 mmHg indicates mixed pulmonary hypertension; Transpulmonary Pressure Gradient (TPG) = mPAP-PAWP, TPG ≥ 12 mmHg may indicate a mixed form. A growing body of data suggests that the ratio of stroke volume to pulmonary artery pulse pressure, a surrogate measure of pulmonary artery compliance (PAC), is associated with poor outcomes in patients with heart failure (65), suggesting that PACs should be given greater attention in the evaluation of early pulmonary vascular disease (66) (Figure 3).
Figure 3
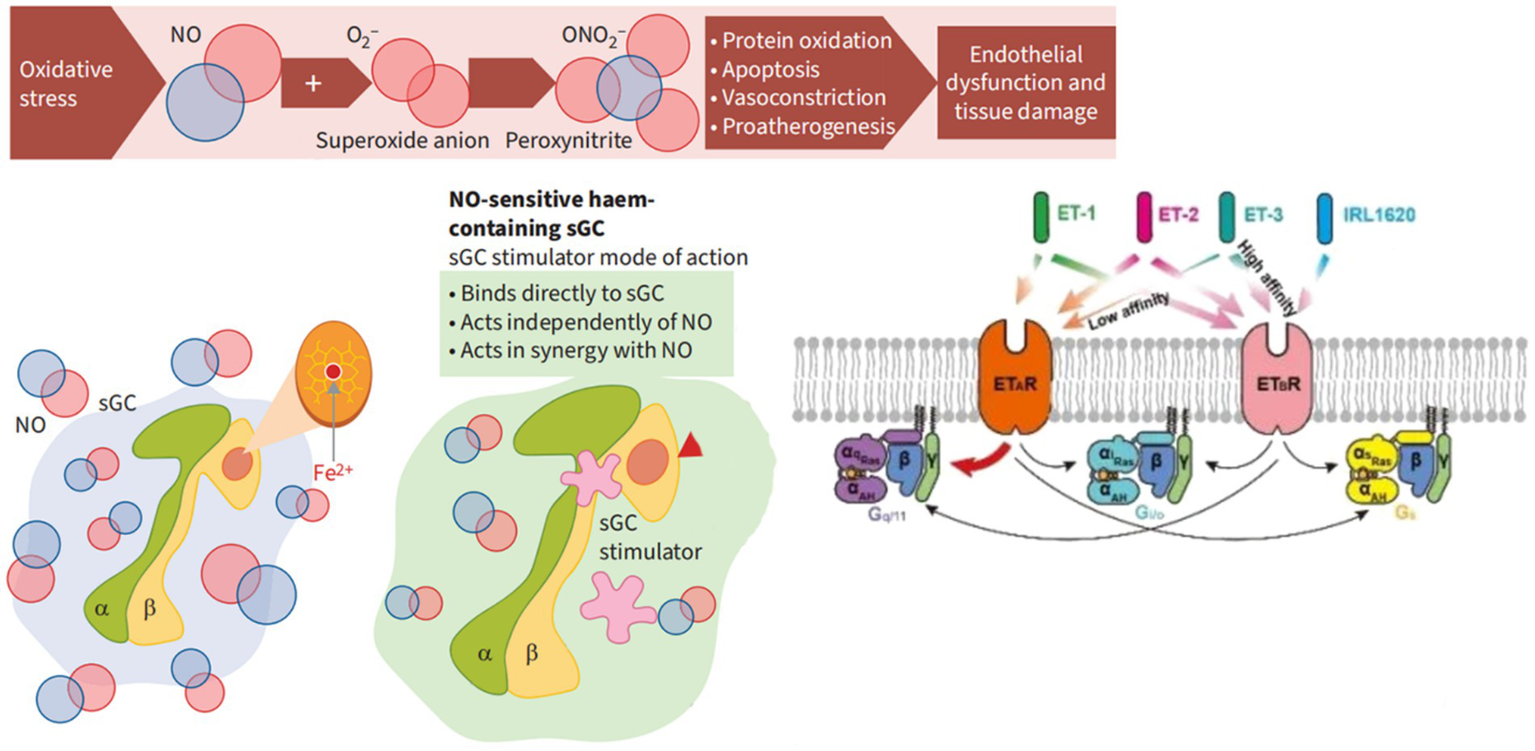
The mechanism of action of soluble guanylate cyclase (sGC) stimulants and activators. α, SGC α subunit; β value, sGC β subunit; cGMP, Cyclic guanosine monophosphate; Fe2+, Iron ions oxidize in a + 2 state; NO, Nitric oxide; O2-, Superoxide anion; ONO2-, Peroxynitrite.
Although right heart catheterization is the gold standard for diagnosing PH, it is invasive. Future research will focus on the development of more precise noninvasive diagnostic techniques, such as the use of advanced imaging techniques (eg, electrocardiogram, magnetic resonance imaging, computed tomography) and biomarker testing, to more accurately assess the hemodynamic status and pulmonary vascular architecture of patients (67). Transthoracic echocardiography (TTE) is the most important screening item used to measure the severity of PH. TTE identifies right ventricular dilation, the presence of valvular regurgitation, and the presence of valvular regurgitation Left ventricular systolic or diastolic dysfunction (68). Ventilation/perfusion scans are also recommended when heart failure (HF) presents with hypercoagulability. Ventilation-perfusion scans remain essential in the clinical workflow, as chronic thromboembolic pulmonary hypertension is unlikely if perfusion is normal (69). Chestx-ray may show cardiomegaly and dilated pulmonary arteries, as well as lung parenchyma or chest wall abnormalities. Chest CT angiography, although considered less sensitive than ventilation-perfusion scanning, may reveal signs of chronic thromboembolic disease, such as filling defects or wedge-shaped or irregular linear opacities due to previous thrombosis. Used to determine the presence of pulmonary artery stenosis or occlusive lesions (70). Serum amino-terminal natriuretic peptide (BNP) levels can be included in risk stratification because they correlate strongly with the severity of pulmonary hypertension and can be used to predict survival (71). The 6MWT reflects the overall exercise tolerance and daily activity ability, and is suitable for assessing functional status and rehabilitation effect. BNP quantifies cardiac stress and the degree of injury, and is suitable for assessing pathophysiological severity and treatment efficacy. The combination of the two can improve the comprehensiveness of the evaluation of patients with Cpc-PH. In patients with PH, elevated BNP reflects right ventricular pressure overload and progression of right heart failure (72, 73).
CardioMEMS is an implantable pressure monitoring system that is implanted into the pulmonary artery via a catheter to monitor PAP, CO, mPAP, PAWP, and others in real time to guide the treatment of heart failure and PH (74). The doctor can remotely monitor the patient’s condition and promptly modify the treatment plan thanks to the data being wirelessly transferred to an external device. This is essential for the evaluation of the condition. In one study, patients treated with CardioMEMS experienced a significant reduction in hospitalization and a significant improvement in quality of life. The CHAMPION study is a multicenter, randomized, controlled trial evaluating CardioMEMS in patients with heart failure (75). The results showed that patients using CardioMEMS had a significant reduction in hospitalization and a significant improvement in quality of life within 6 months. The PULSAR study further explored the use of CardioMEMS in patients with pulmonary hypertension. CardioMEMS was found to significantly improve 6-min walking distance (6MWD) and NT-proBNP levels (76). In a long-term follow-up study, patients treated with CardioMEMS demonstrated sustained improvement in pulmonary artery pressure and cardiac function over 2 years of follow-up, with no device-related serious adverse events (77).
In a study of 208 patients with chronic heart failure, BNP was significantly inversely associated with 6MWD (r = −0.61), and the combined predictive efficacy of the two measures was superior to that of a single measure (78). In patients with PH, 6MWD < 300 m and BNP > 300 pg./mL indicate very high risk and require intensive therapy. Studies have shown that for every 50 m reduction in 6MWD, the risk of death in patients with PH is significantly increased; A decline of >50 m in 6 MWD over 24 weeks is associated with at least a four-fold increase in mortality (79). 6MWT is widely used to evaluate the efficacy of targeted drugs (e.g., Sotatercept), surgery, or exercise rehabilitation. As an example, a mean increase of 40.8 m in 6 MWD followed by pharmacotherapy was accompanied by simultaneous improvement in pulmonary vascular resistance and BNP levels (80). An improvement of 30–50 m in 6MWD is considered the threshold for clinical benefit in patients with PH and is referred to as the minimum clinically important difference (MCID) (81). The peak oxygen uptake cannot be directly measured by 6MWT, and the results are easily affected by factors such as age, gender, and corridor length, so it needs to be comprehensively judged in combination with other indicators. BNP levels were shown to increase significantly with NYHA cardiac function class (Grade I: 186 ± 22 pg./mL; Grade IV: 266 ± 165 picograms/mL) (82). ROC Curve analysis showed that BNP predicted adverse events in patients with chronic heart failure with an area under the curve of 0.914 (sensitivity 0.778, specificity 0.977) (83, 84).
Current medical and surgical treatments
Based on existing evidence, PH-LHD targeted therapy does not have significant effectsin Cpc-PH patients. Conventional treatment modalities include strengthening the control of underlying diseases, targeted drug therapy, and surgical intervention. Its complexity requires precise hemodynamic assessment and risk stratification, while being alert to the potential risks of targeted drugs. It is recommended to use first-line drugs, including beta blockers, RAAS inhibitors, soluble guanylate cyclase stimulants, and sodium glucose cotransporter 2 inhibitors, in a reasonable combination to improve patient symptoms, delay disease progression, and reduce mortality. For Cpc-PH patients with metabolic syndrome and other diseases, they should also be managed. Although there is limited evidence for the targeted drug application of Cpc-PH, targeted drugs should still be considered in specific situations. The trial outcomes of several medication classes that may be utilized to treat Cpc-PH are reviewed in the sections that follow (Tables 1–3). For Ipc-PH, targeted drug therapy may be harmful because its pulmonary vascular remodeling has not yet occurred, and the increase in pulmonary artery pressure is passive. For Cpc-PH, although targeted drugs may be beneficial for some patients, there is still a lack of clear screening criteria. Future research should focus on screening PH-LHD patients who are sensitive to targeted drugs, especially Cpc-PH patients with high pulmonary artery pressure even after improvement in left heart function (85).
Table 1
| The trial | Drug | Duration | Primary and secondary endpoints | Treatment effects and adverse events |
|---|---|---|---|---|
| Endothelin receptor antagonists | ||||
| MELODY-1 | Macitentan | 12 weeks | PVR, PAWP, Average right atrial pressure | No significant improvement in PVR, PAWP or Average right atrial pressure, fluid retention, NYHA functional grading worsening. |
| HEAT | Darusentan | 3 weeks | CI, PVR, mPAP, PAWP, Average right atrial pressure | No significant reduction in PVR, mPAP, PAWP or right atrial pressure. The incidence of adverse events in the high-dose group is higher. |
| EARTH | Darusentan | 24 weeks | Cardiac remodeling, clinical status, echocardiography parameters, etc. | No improvement in cardiac remodeling or clinical status, and no obvious benefit was seen in patients with echocardiography suggesting PH. |
| PDE 5 inhibitor | ||||
| RELAX | Sildenafil | 24 weeks | 6MWD, PVR, NT-proBNP, WHO-FC | 6MWD significantly increased by 40.8 meters, PVR significantly decreased, NT-proBNP level decreased significantly, and WHO functional grading improved. |
| SOVIAC | Sildenafil | 12 weeks | mPAP, mPCWP, CO, peak VO₂ | Sildenafil leads to worse clinical outcomes such as death and heart failure hospitalization in patients with persistent PH after valve surgery. |
| Macitentan/Tadalafil combination study | Macitentan/Tadalafil | 16 weeks | PVR | The reduction of PVR in the M/T FDC group was significantly higher than that in the monotherapy group. Adverse events leading to discontinuation, serious AEs, and special concerns (anemia, hypotension, and edema) are more common. |
Endothelin receptor antagonists and PDE 5 inhibitor.
Table 2
| The trial | Type | Study object | Number | Time | Target | Drugs | Endpoints | Conclusion |
|---|---|---|---|---|---|---|---|---|
| PATENT-1 | Multi-center double blind random placebo | PH | 341 | 12 weeks | NO-sGC-cGMP | Riociguat | 6MWD, PVR, NT-proBNP, WHO-FC, TTP, QOL | Improve the exercise capacity and functional indexes of patients with PH, improve the clinically relevant primary and secondary endpoints, delay the occurrence of clinical deterioration, and significantly reduce NT-proBNP. |
| PATENT-2 | Double-blind Random placebo | PH | 396 | 24 months | NO-sGC-cGMP | Riociguat | 6MWD, AE, TTP, NT-proBNP, WHO-FC | It is a well-tolerated and effective treatment with sustained improvement in 6MWD and WHO-FC. |
| CHEST-1 | Multi-center open label double blind placebo | CTEPH | 243 | 16 weeks | NO-sGC-cGMP | Riociguat | 6MWD, PVR, NT-proBNP, WHO-FC, TTCW, Borg, EQ-5D, LPH | It is effective in improving exercise capacity and cardiac function in patients with inoperable or postoperative recurrent/persistent CTEPH. |
| CHEST-2 | Multi-center opening label double-blind placebo | CTEPH | 237 | 24 months | NO-sGC-cGMP | Riociguat | 6MWD, LPH, PVR, EQ-5D, NT-proBNP, WHO-FC, TTCW, Borg, | It can sustainably improve exercise capacity and cardiac function in patients with persistent/recurrent CTEPH after inoperable or PEA, and is clinically and well tolerated. |
| VICTORIA | Multi-center random double-blind parallel placebo phase III | NYHA II-IV, EF < 45%, BNP ≥ 300 pg./ml, NT-proBNP ≥ 1,000 pg./mL | 5,050 | 10.8 months | sGC-cGMP | Vericiguat | CV Death, First HF, Hospitalization, All-cause Death | Reduce the composite endpoint risk of cardiovascular death or HF hospitalization in patients with HFrEF by reducing recent heart failure exacerbation events. |
| VITALITY-HFpEF | Multi-center double-blind placebo phase II.b | HFpEF, NYHA II-III, EF ≥ 45% | 789 | 24 weeks | sGC-cGMP | Vericiguat | KCCQ-PLS, 6MWD | There was no improvement in KCCQ-PLS in patients with HFpEF. |
| DILATE-1 | Double blind random placebo control parallel groups stage II.a | LVEF>50%, mPAP ≥ 25 mmHg, PAWP > 15 mmHg | 36 | 30 days | NO-sGC-cGMP | Riociguat | mPAP, SV, SBP, PVR | Riociguat was well tolerated, with 2 mg of riociguat significantly increasing SV and cardiac index CI and decreasing systolic blood pressure, SVR, and RVED area without altering HR, TPG, or PVR |
| LEPHT | Phase II.b, multi-center double blind random placebo | HFpEF, LVEF ≤ 40%, mPAP ≥ 25 mm Hg | 201 | 16 weeks | NO-sGC-cGMP | Riociguat | mPAP, CI, SV, PVR | Riociguat was well tolerated in patients with PH-1 HFrEF and did not significantly improve mPAP, but significantly improved cardiac index, stroke volume index, and pulmonary vascular resistance in the highest dose group |
| SOCRATES-PRESERVED | Forward-looking, random double blind placebo phase II.b | HFpEF, LVEF > 45% | 477 | 12 weeks | sGC-cGMP | Vericiguat | NT-proBNP, LAV, KCCQ | Failed to change NT-proBNP, LAV. The KCCQ score improved significantly. |
| SOCRATES-REDUCED | Multi-center double-blind random placebo control | HFrEF, LVEF≤40% | 2,707 | 12 weeks | sGC-cGMP | Vericiguat | NT-proBNP, Echocardiographic Parameters, All-cause mortality, Cardiac death, Heart failure hospitalization | Vericiguat was well tolerated but did not significantly reduce NT-proBNP levels and did not improve echocardiographic parameters or primary clinical endpoints |
Soluble guanylate cyclase stimulator.
CTEPH, Chronic thromboembolic pulmonary hypertension; PH, Pulmonary hypertension; PVR, pulmonary vascular resistance; NT-proBNP, N-terminal brain sodium peptide precursor; EF, Left ventricular ejection fraction; LAV, Left atrial volume; NYHA, New York heart function classification; WHO-FC, WHO heart function classification; QOL, Quality of life; KCCQ-PLS, Kansas City Cardiomyopathy questionnaire activity restricted score; EQ-5D, Quality of life score; LPH, Pulmonary hypertension quality of life; AE, Adverse events; TTP, Time to clinical deterioration; TTCW, Clinical deterioration events; Borg, Changes in dyspnea score; CV Death, cardiovascular death; CI, cardiac index; CO, cardiac output; 6MWD, 6-min walking distance; VO2, peak oxygen uptake; LHD, left heart disease; NT-proBNP, N-terminal pro-B-type natriuretic peptide; LVEF, left ventricular ejection fraction; HF, heart failure; Cpc-PH, Combined post-and pre-capillary pulmonary hypertension; HFpEF, heart failure with preserved ejection fraction; LAV, left atrial volume; LVESV, left ventricular end systolic volume; mPAP, mean pulmonary artery pressure; NYHA, New York Heart Association; PH, pulmonary hypertension; SBP, systolic blood pressure; PAWP, pulmonary artery wedge pressure; SPAP, systolic pulmonary artery pressure.
Table 3
| The trial | Drug | Number | Duration | Endpoints | Conclusion |
|---|---|---|---|---|---|
| FREEDOM-C | Treprostinil | 350 | 16 weeks | WHO functional class, Borg dyspnea score, dyspnea fatigue index score | Oral Treprostinil for 16 weeks could not significantly improve the activity tolerance, but could significantly change the dyspnea fatigue index and Borg dyspnea classification. |
| TRIUMPH-I | Treprostinil | 235 | 12 weeks | 6MWD, Borg dyspnea score, WHO functional class | Inhaled Treprostinil combined with bosentan or sildenafil treatment significantly improved the 6-min walking distance (6MWD) at 12 weeks. |
| FIRST | Epoprostenol | 471 | 1 year | Survival, clinical events, congestive heart failure symptoms, 6MWD, quality-of-life measures | CI increased significantly (from 1.81 to 2.61 L/min/m2), while PAWP and PVR decreased significantly. However, in patients with heart failure, the survival rate of the Epoprostenol treatment group decreased. |
| STEP | Iloprost | 34 | 16 weeks | NYHA Functional Class, 6MWD | The combined use of bosentan and iloprost significantly improved the exercise tolerance (6MWD increase) and delayed the disease progression. |
| GRIPHON | Selexipag | 1,156 | 63 weeks | Survival, clinical events, 6MWD, PVR | The incidence of pulmonary hypertension related events (such as hospitalization, disease progression, death) was significantly reduced, and 6MWD increased by 12.62 meters on average. |
PGI2 Drugs.
Endothelin receptor antagonists
So far, four types of ERAs have been clinically tested, namely bosentan, anlisentan, Masitentan, and Sitaxentan. Among them, Sitaxentan has been withdrawn from the market worldwide due to fatal liver injury (75). Endothelin-1 (ET-1) participates in vascular constriction and cell proliferation by activating endothelin receptors (ET-A and ET-B) (86). Cochrane studies have shown that using endothelin receptor antagonists for 3–6 months significantly improves patients’ exercise capacity, symptoms, and cardiorespiratory hemodynamic indicators (87). However, it is currently unclear whether these drugs can significantly reduce the mortality rate of patients (88). In PH-LHD, increased expression of endothelin-β1 is associated with disease progression, making it a potential therapeutic target (89). However, ERA has limited efficacy in PH-LHD and is associated with a high risk of adverse reactions (90). Common adverse events include headaches, anemia, and edema, which are usually dose-dependent (91). Bosentan may cause liver dysfunction, but it can usually be restored after reduction or discontinuation of the medication (92). New ERAs, such as Masitentan, have reduced the incidence of these adverse reactions by optimizing their pharmacokinetic properties (93). In the study of patients with advanced HFrEF, bosentan failed to improve NYHA functional grading, systolic pulmonary artery pressure (PAP), tricuspid regurgitation velocity, etc., causing fluid retention and peripheral edema, and increasing the hospitalization rate of heart failure (94). Macitentan did not significantly improve PVR, PAWP, or mRAP after 12 weeks of treatment for Cpc-PH patients, according to the MELODY-1 trial; however, the difference was not statistically significant (95). Although daluxostane therapy did not significantly lower PAWP, mPAP, PVR, or RAP, it did improve cardiac index in HF patients after 3 weeks in the HEAT study. In the high-dose group, adverse events were more common. The EARTH trial also showed that, even in individuals with echocardiographic evidence of PH, Darusentan treatment for 24 weeks did not improve clinical status or cardiac remodeling in patients with chronic HF. In clinical trials of PH-LHD, ERA has generally demonstrated a high incidence of side events; several trials were prematurely stopped because of safety concerns. Therefore, currently ERA is not recommended for the treatment of PH-LHD/Cpc-PH.
Phosphodiesterase (PDE 5 inhibitor)
PDE5 inhibitors can improve PH and right ventricular function by inhibiting PDE5 enzymes, leading to vasodilation. A Cochrane review suggests that PDE5 inhibitor treatment can increase patients’ average 6-min walking distance by 48 meters, while improving their functional level and reducing the risk of hospitalization associated with PAH. The AMBITION trial showed that the initial combination of Ambrisentan (ERA) and Tadalafil (PDE5 inhibitor) significantly reduced the risk of clinical failure. In HFrEF patients, multiple early small-scale studies have shown that sildenafil significantly reduces pulmonary artery systolic pressure (PASP), mPAP, PVR, 6MWT, and improves right ventricular function (96, 97). Sildenafil did not increase exercise capacity, left ventricular mass, or clinical composite endpoints (death, cardiac/renal hospitalization, increased heart failure symptoms, etc.) as compared to placebo treatment in a large randomized controlled trial (n = 216). The sildenafil group saw a modest increase in vascular adverse events, including headache, flushing, and hypotension, however these were not statistically significant (98, 99). The efficacy of PDE5 inhibitors in HFpEF patients is not yet clear, especially in patients without right ventricular dysfunction. Right ventricular dysfunction may be an important predictor of the benefit of PDE5 inhibitor therapy (98). HFpEF patients with concomitant right ventricular failure may benefit from PDE5 inhibitor therapy, such as reducing right atrial pressure and improving right heart function. HFpEF patients without right ventricular dysfunction did not show significant benefits (100). Multiple meta-analyses have summarized the efficacy of PDE5 inhibitors (including sildenafil) in PH-LHD. In HFrEF patients, PDE5 inhibitors significantly improved mPAP PVR, LVEF, physical ability and quality of life (101, 102). PDE5 inhibitors, such as sildenafil, have shown certain clinical benefits in HFrEF patients, particularly in improving hemodynamics and exercise capacity (103). In the SOVIAC study, sildenafil even led to worse clinical outcomes such as death and hospitalization for heart failure in patients with persistent PH after valve surgery (104). The TRITON study evaluated the efficacy of triple combination therapy (Masitentan, Tadalafil, and Selexipag) versus dual combination therapy (Masitentan and Tadalafil) in newly diagnosed, untreated PH patients, and the results showed no significant difference in the primary endpoint of PVR between the two. A cohort analysis of the Spanish PH registry investigated the predictive factors of PDE5 inhibitor treatment response. The results showed that male gender, diagnosis of portal pulmonary hypertension (PoPH), or HIV-PAH were independent predictors of favorable response to PDE5 inhibitors (105). And carbon monoxide dispersibility (DLco) ≤ 40% of the predicted value is associated with adverse reactions. For patients who have received PDE5 inhibitors but have not achieved the desired clinical efficacy, conversion to soluble guanylate cyclase (sGC) stimulants may be considered.
Soluble guanyl cyclase stimulators
Soluble guanyl cyclase stimulators promote vasodilation by enhancing the production of cyclic guanosine monophosphate (cGMP), complementing the PDE5 inhibitor (which works by reducing cGMP degradation) mechanism, and have received extensive attention in the field of heart failure (HF) and pulmonary hypertension (PH) in recent years. Vericiguat was well tolerated in patients with HFrEF in the SOCRATES-REDUCED study, but did not significantly reduce NT-proBNP levels or improve echocardiographic parameters (106–109). In the SOCRATES-PRESERVED study, vericiguat did not significantly reduce NT-proBNP levels, but it was well tolerated and associated with improved quality of life (110). In the VICTORIA study, vericiguat significantly reduced the composite endpoint of cardiovascular death or hospitalization for heart failure in patients with symptomatic worsening HFrEF (111). In the LEPHT trial, riociguat was well tolerated in patients with HFrEF, significantly improving cardiac index, stroke volume index, and PVR in the high-dose group (112). In the DILATE-1 trial, Riociguat improved stroke volume and systolic blood pressure, but had no significant effect on mPAP and PVR (113). The LEPHT study evaluated the safety and efficacy of riociguat in patients with HFrEF and pulmonary hypertension (PH) (114). The study showed that after 16 weeks of treatment with riociguat (2 mg, TID), patients had an increase in cardiac index and a significant decrease in pulmonary vascular resistance (PVR) and systemic vascular resistance (SVR) compared with the placebo group. The DILATE-1 study was in patients with HFpEF and PH (LVEF > 50%, mPAP ≥ 25 mmHg, PAWP > 15 mmHg). The results showed that after 6 h of treatment, the levociguat 2 mg group showed improvements in stroke volume and right ventricular end-diastolic area, although there was no significant decrease in mean pulmonary artery pressure (mPAP) (115). The COMPERA 2.0 study evaluated the safety and efficacy of riociguat in patients with pulmonary hypertension and cardiometabolic comorbidities. The PASSION study is planned to evaluate the effects of riociguat on exercise tolerance, cardiac function indexes, and WHO functional classification in patients with heart failure and pulmonary hypertension. Riociguat has shown potential benefits in patients with HF-related Pulmonary Hypertension in some studies, such as improved PVR, SVR, 6MWD, suggesting that it may have a positive effect on exercise tolerance (116). In a study of 61 patients with pulmonary hypertension (PH) who had an inadequate response to PDE-5 inhibitors, switching to riociguat resulted in an increase in 6 MWD at 24 weeks, an improvement in WHO grade, and a decrease in NT-proBNP levels (117, 118). In patients with heart failure, vericiguat has shown significant clinical benefit in patients with HFrEF, particularly in reducing the risk of cardiovascular events, but has limited effect in patients with HFpEF (119–121). Higher doses of riociguat improve PVR and systemic vascular resistance in patients with Cpc-PH, but have limited efficacy in patients with Ipc-PH (122, 123). The study, which included data from PATENT-1, PATENT-2, PATENT PLUS and REPLACE, showed that the incidence and severity of adverse events were similar to those in the placebo group in the riociguat group (124).
PGI2 drugs
Prostacyclin is mainly produced by endothelial cells and has vasodilation, antithrombotic and antiproliferative effects, which can improve hemodynamics and cardiac function. Prostacyclin drugs include prostacyclin analogs (such as Epoprostenol, Treprostinil, Iloprost, Beraprost) and prostacyclin IP receptor agonists (such as Selexipag). Eprostol works by reducing PVR and improving right ventricular function, but long-term use may activate harmful neurohormonal systems (such as the renin-angiotensin system), leading to worsening of heart failure. In the FIRST trial, although epprostol significantly improved cardiac index, pulmonary wedge pressure (PAWP) and systemic vascular resistance (SVR), the trial was terminated early due to the decline in patient survival. Treprolinil is a novel prostacyclin analog that may improve metabolism and cardiac function by activating the AMPK pathway in skeletal muscle and right ventricle. In animal models, Trepronnier improved metabolic syndrome and reduced pulmonary arterial pressure, showing potential to prevent the development of heart failure (HFpEF) with ejaculation fractions. Unlike heart failure (HFrEF), which has reduced ejection fraction, HFpEF is often accompanied by metabolic syndrome and diabetes, and the beneficial effects of prostacyclin analogs on metabolism and pulmonary blood vessels make it possible that it is more suitable for the treatment of PH-HFpEF. In patients with Cpc-PH, targeted pulmonary hypertension therapy (such as phosphodiesterase 5 inhibitors and endothelin receptor antagonists) fails to improve symptoms, but instead increases morbidity and death Rate. A meta-analysis showed that although PH-targeted therapy may improve exercise capacity in patients with left heart disease (LHD), the risk of adverse events is higher. A meta-analysis showed that although pH targeted therapy can improve the exercise ability of patients with left heart disease (LHD), the risk of adverse events is higher, but the efficacy of human pH HFPEF patients still needs further clinical trials to verify, and the treatment strategy of skeletal muscle and right ventricular AMPK pathway may be a potential direction for the prevention of PH-LHD (125–131).
Ralinepag, as a novel prostacyclin receptor (IP receptor) agonist, exhibits significant vasodilation effects, which can effectively reduce PVR while inhibiting vascular smooth muscle cell proliferation and platelet aggregation. Its drug has a half-life of up to 24 h, which gives it a potential advantage in the treatment of PH. In Phase II clinical trials, Ralinepag has shown good efficacy, and common adverse reactions include headache, nausea and diarrhea. At present, the Phase III clinical trial of Ralinepag is being promoted, and the preliminary long-term data of the ADVANCE EXTENSION study have been released at relevant academic conferences (132). At present, prostacyclin drugs have limited their widespread use in clinical practice due to their poor stability and many side effects. Therefore, the development of novel prostacyclin drugs with higher receptor selectivity, fewer side effects, more stable, easier to preserve and more easily accepted by patients has become the research direction (133).
Sotatercep
Sotatercept is a fusion protein of the type IIA activin receptor (ActRIIA). Results from the PULSAR study show that Sotatercept significantly outperforms placebo in reducing PVR and improving 6MWD, as well as in secondary endpoints such as NT-proBNP levels, WHO functional classification, and clinical worsening events. Results from the STELLAR study indicate that after 24 weeks of treatment, patients in the Sotatercept group saw an increase of 40.8 meters in 6MWD compared to a mere 1.0 - meter increase in the placebo group. Other secondary endpoints, including reductions in NT-proBNP levels, improvements in WHO functional classification, and decreases in PVR, were also significantly better in the Sotatercept group. With a median follow - up of 32.7 weeks, Sotatercept reduced the risk of clinical worsening or death by 84%. Additionally, Sotatercept improved right - heart function by lowering mPAP and reducing the workload on the right side of the heart. In terms of safety, adverse events in the Sotatercept group mainly included capillary dilation, epistaxis, gingival bleeding, and thrombocytopenia, but most of these events were mild. In the post-hoc analysis of the STELLAR study, Sotatercept was found to reduce the size of the right side of the heart and improve right - ventricular function and hemodynamic parameters after 24 weeks of treatment. Ongoing trials such as SOTERIA, HYPERION, ZENITH, CADENCE, MK - 7962-020, and MOONBEAM will further explore the long - term safety and effectiveness of Sotatercept in diverse patient populations, providing more comprehensive clinical evidence for the future treatment of pulmonary arterial hypertension. Compared with traditional small - molecule compounds, Sotatercept has higher efficacy and fewer adverse effects. By rebalancing the BMPR2 signaling pathway and inhibiting the Smad2/3 signaling pathway, Sotatercept reduces the proliferation of pulmonary vascular smooth muscle cells and collagen deposition, thereby reversing pulmonary vascular remodeling. This goal is not achievable by most currently available targeted drugs. Moreover, Sotatercept’s dosing schedule of once every 3 weeks and its subcutaneous injection route are more convenient than those of traditional medications (Table 4). With its unique mechanism of action and significant clinical benefits, Sotatercept offers a new treatment option for Cpc-PH patients and has the potential to transform the current treatment landscape (134–138).
Table 4
| Trial name | Clinical Trials.gov ID | Type | Trial population | Primary endpoint | Secondary endpoints | Trial results | Number | Duration |
|---|---|---|---|---|---|---|---|---|
| PULSAR | NCT04811092 | Randomized, double-blind, placebo-controlled, Phase III | Newly diagnosed intermediate- and high-risk PAH patients | PVR | 6MWD, NT-proBNP levels, WHO FC, Clinical worsening events | Sotatercept significantly reduced PVR, increased 6MWD, improved NT-proBNP and WHO functional classification | 106 | 24 weeks |
| STELLAR | NCT04896008 | Randomized, double-blind, placebo-controlled, Phase III | WHO functional class III or IV high-risk PAH patients | Change in 6MWD at 24 weeks | Multicomponent improvement, PVR, NT-proBNP, WHO functional classification, clinical worsening events | Sotatercept significantly increased 6MWD, reduced PVR, improved NT-proBNP and WHO functional classification | 323 | 24 weeks |
| HYPERION | NCT04811092 | Randomized, double-blind, placebo-controlled, Phase III | Newly diagnosed intermediate- and high-risk PAH patients | Time to clinical worsening | 6MWD, NT-proBNP levels, WHO functional classification | Terminated early due to significant interim results | 662 | Estimated completion in January 2030 |
| ZENITH | NCT04896008 | Randomized, double-blind, placebo-controlled, Phase III | WHO FC III or IV high-risk PAH patients | Time to clinical worsening | Overall survival, transplant-free survival | Interim analysis showed significant reduction in clinical worsening events, terminated early | 166 | Estimated completion in November 2025 |
| SOTERIA | NCT04796337 | Open-label, long-term extension study | Adult PAH patients who completed a sotatercept parent study | Number of adverse events and time to treatment discontinuation | Long-term safety, tolerability, and efficacy | Ongoing, expected to enroll 700 participants | 700 | Long-term (ongoing) |
| CADENCE | NCT04945460 | Randomized, double-blind, placebo-controlled, Phase II | PH patients with heart failure and preserved ejection fraction | Change in PVR | Change in 6MWD | Ongoing, expected to enroll 150 participants | 150 | Estimated completion in February 2027 |
| MK-7962-020 | NCT05818137 | Non-randomized, open-label, Phase III | Japanese PAH patients | Efficacy and safety | – | Ongoing, expected to enroll 35 participants | 35 | Estimated completion in August 2025 |
| MOONBEAM | NCT05587712 | Open-label, Phase II | PAH children aged 1–18 years | Safety, tolerability, pharmacokinetics, and pharmacodynamics | – | Ongoing, expected to enroll 42 children | 42 | Estimated completion in 2028 |
Sotatercept.
Ongoing clinical trials
Many patients with cardiovascular diseases often have metabolic syndrome (MS), such as obesity, dyslipidemia, insulin resistance, and diabetes (Table 5). MS can induce systemic inflammatory responses, and inflammatory factor infiltration and imbalance in immunomodulation are key pathogenic drivers of vascular remodeling. Therefore, immunotherapy may become a new therapeutic strategy for PH-LHD/Cpc-PH (139–142) (Table 6).
Table 5
| Drug name | Drug type | Mechanism of action | Development stage |
|---|---|---|---|
| TX45 | Long-acting Fc-Relaxin fusion protein | Improves pulmonary hemodynamics and left ventricular function | Phase Ib: Single intravenous infusion significantly reduced PCWP by 17.9% and PVR by 30%. Phase II APEX Trial: Ongoing, with topline data expected in 2026. |
| Seralutinib | Tyrosine kinase inhibitor | Inhibits relevant signaling pathways to improve pulmonary arterial hypertension | TORREY trial (phase II trial): PVR decreased by 14% (p = 0.0310), and in patients with more severe symptoms, PVR decreased by 21% (p = 0.0427) PROSERA trial (Phase III trial) is currently underway |
| Imatinib | Tyrosine kinase inhibitor | Non-vasodilator, mechanism not fully elucidated | Inhalation formulation undergoing Phase II and III clinical trials. |
| Macitentan | Endothelin receptor antagonist | Blocks endothelin signaling pathways, reducing pulmonary arterial pressure | Approved, better efficacy when used in combination with Tadalafil. |
| MK-5475 | Soluble guanylate cyclase activator | Activates guanylate cyclase, increases cGMP levels, and relaxes vascular smooth muscle | INSIGNIA-PAH Phase II clinical trial results published in September 2024. |
| Inhaled Treprostinil | Prostacyclin analog | Vasodilation, improvement of pulmonary arterial hypertension | Post-hoc analysis and extension studies of the INCREASE trial show improved survival in patients with IPF and pulmonary arterial hypertension. |
| MRE-269 | Selective IP receptor agonist | Main metabolite of Selexipag, activates IP receptors to increase intracellular cAMP levels, leading to pulmonary vasodilation | Under investigation. |
| T26A | Prostaglandin transporter (PGT) inhibitor | Reduces intracellular concentration of PGE2, targeting pulmonary vasodilation | Under investigation. |
| MN-08 | Nitrate ester derivatives of memantine | Antagonism of N-methyl-D-aspartate (NMDA) receptors and release of NO | Approved for clinical trials by China’s NMPA in January 2025, Phase II clinical trial to commence soon. |
| KER-012 | ACVR2B ligand trapper | Blocks signaling of TGF-β superfamily members, reversing pulmonary vascular remodeling | Ongoing Phase II clinical trial. |
| Recombinant MPB9 | Recombinant protein | Targets BMP9, modulates bone morphogenetic protein | In development stage. |
| Letrozole, Tamoxifen, DHEA | Sex hormones | Regulates sex hormone levels, potentially beneficial for PH | Under investigation. |
| Metformin, Trimetazidine | Metabolic targeting drugs | Modulates metabolic processes, potentially beneficial for PH | Under investigation. |
| Canagliflozin | SGLT-2 inhibitor | Inhibits SGLT-2 receptor, reduces right ventricular systolic pressure, improves PH | Under investigation. |
| Empagliflozin | SGLT-2 inhibitor | Inhibits SGLT-2 receptor | EMBRACE-HF study showed a reduction in pulmonary artery diastolic pressure by 1.7 mmHg compared to placebo at Week 12. |
| Celastramycin | Benzoylpyrrole compound | Reduces secretion of inflammatory factors and reactive oxygen species, targets anti-proliferative effects on PH-PASMC | Not yet in clinical trials, but animal experiments have shown potential for improving symptoms of pulmonary arterial hypertension. |
Ongoing clinical trials.
Table 6
| Author | Title | Result | Date | Potential therapeutic target |
|---|---|---|---|---|
| Hou et al. (143) | Targeting Fibroblast Activation Protein for Molecular Imaging of Fibrotic Remodeling in Pulmonary Arterial Hypertension | 18F-FAPI PET/CT imaging is feasible for visualizing the remodeling of the PA and the RV in PAH. Although it offers promise for assessing disease-related changes, its role in evaluating disease severity and monitoring therapeutic efficacy requires further investigation. | 2025 | fibroblast activation protein inhibitor (FAPI) |
| Zhang et al. (144) | Endogenous hydrogen sulfide persulfidates endothelin type A receptor to inhibit pulmonary arterial smooth muscle cell proliferation | Endogenous H2S persulfidated ETAR at Cys69 to inhibit the binding of ET-1 to ETAR, subsequently suppressed PASMC proliferation, and antagonized pulmonary vascular structural remodeling. |
2025 | ETAR over sulfurization modification |
| Li et al. (63) | Genetic recording of transient endothelial activation in distinct alveolar capillary cells during pulmonary fibrosis | Transient EndoMT activation of specific endothelial cells (Plvap+) during pulmonary arterial hypertension and pulmonary fibrosis may affect extracellular matrix remodeling and vascular integrity. | 2024 | EndoMT |
| Shen et al. (145) | BRCC3 Regulation of ALK2 in vascular smooth muscle cells implication in pulmonary hypertension | BRCC3 regulates the BMP signaling pathway and maintains pulmonary vascular homeostasis by deubiquitinating K472 and K475 sites of ALK2.BRCC3 deficiency exacerbates pulmonary arterial hypertension, and PPARγ agonists can partially alleviate its effects. | 2024 | BRCC3-ALK2-Smad1/5/9-PPARy |
| Harvey et al. (146) | Lysosomal dysfunction and inflammatory sterol metabolism in pulmonary arterial hypertension | NCOA7 regulates lysosome function and cholesterol metabolism, inhibits endothelial cell immune activation, and alleviates pulmonary arterial hypertension. Small molecule compound 958ami can activate NCOA7 and improve the pathological phenotype of PAH model. | 2025 | NCOA7 |
Studies uncovering potential therapeutic targets.
Potential new therapeutic targets
Stem cell therapy, as an emerging therapeutic approach, has attracted much attention for its ability to self-renew, proliferate and differentiate into a variety of specific cells. Stem cells commonly used to treat pulmonary hypertension (PH) include endothelial progenitor cells (EPCs) and mesenchymal stem cells (MSCs). Among them, EPCs are oligona stem cells that tend to differentiate into endothelial cells (EC), but may promote the occurrence and development of PH in a pathological state. For example, EPCs in hypoxic newborn calves showed stronger migration and ductal capacity. To correct the defects of EPCs, cell infusion becomes a direct approach. However, in PH progression, EPCs may reactivate their hematopoietic tendencies by abnormally expressing hematopoietic transcription factors, resulting in excessive infiltration of immune cells. Liang et al. successfully blocked the EHT process and reversed the decline in EPCs levels using the endothelial-hematopoietic transition (EHT) inhibitor Runx1 in a single-dose monoclonal toxin (MCT). In addition, erythropoietin (EPO) has also been shown to have vascular protection effects, which restores the number of circulating EPCs and reverses vascular remodeling by promoting the expression of heme oxygenase-1 (HO-1). Mesenchymal stem cells (MSCs) are pluripotent stem cells that tend to differentiate into mesenchymal cells such as osteoblasts, adipocytes, chondrocytes and myocytes. MSCs show significant potential in the treatment of PH, especially in rat models induced by chronic hypoxia and single-dose monoclonal toxin (SuHx), where MSCs are able to reverse collagen deposition and reduce peripulmonary vasculo-hemophilia factors (vWF) and α-smooth muscle actin (α-SMA). In addition, MSCs also improve PH symptoms by regulating intestinal flora. Studies have shown that both SuHx and MCT can destroy the homeostasis of the intestinal microbiota in mice, while MSCs treatment can restore the anti-inflammatory bacterial level and improve the immune-regulating functional bacterial population (147–151).
Progress in interventional treatment
An emerging interventional therapy technique called pulmonary artery denervation (PADN), which is based on catheter ablation, reduces pulmonary artery resistance, improves right heart function, and delays pulmonary artery disease by inhibiting sympathetic nerve activity by ablation of nerve-intensive areas in or near the pulmonary artery. High pressure (PH) progress. Clinical research and animal experimentation have both produced positive outcomes. According to recent research, PADN can reverse the downregulation of β-adrenergic receptors and the overexpression of α-adrenergic receptors in rat lung tissues in the preclinical model of pulmonary hypertension-left heart disease (PH-LHD). PADN significantly decreased mean pulmonary arterial pressure (mPAP) and 6-min walking distance in a phase II trial of mixed etiology PH, including PH-LHD. In addition, multi-center trials for Cpc-PH also showed that PADN can reduce systolic and diastolic pulmonary artery pressure, increase cardiac output and 6-min walking distance, while significantly reducing clinical deterioration and hospitalization. Rate. A clinical study in 98 patients with Cpc-PH further confirmed that PADN significantly reduces pulmonary vascular resistance (PVR) and improves exercise tolerance. Although PADN shows good prospects in different types of PH, further studies are needed to evaluate its long-term safety and clinical outcomes (152–155).
In the management of heart failure, ARB is one of the standard treatment drugs, which can indirectly have a positive effect on Cpc-PH by reducing cardiac load and improving cardiac function. In the PADN-5 study, the efficacy of PADN combined with standard drug therapy for heart failure (including ARB) was evaluated in Cpc-PH patients, with significant effects in reducing the incidence of clinical deterioration, improving hemodynamic indicators (such as PVR, PAWP), and enhancing patient exercise endurance. Although there have been studies exploring the potential role of ARB in the treatment of heart failure and related pulmonary arterial hypertension (PH), there is currently no clear clinical research directly confirming the effectiveness and safety of ARB combined with targeted drugs in the treatment of post capillary pulmonary hypertension (Cpc-PH).
In the management of Cpc-PH patients, it is crucial to expand the range of management strategies. A comprehensive assessment should be conducted for each patient, including medical history, physical examination, imaging studies, and hemodynamic monitoring. Existing risk stratification models should be used to rigorously assess the risk level of each patient. A variety of treatment options, such as pharmacological therapy and surgical interventions, should be tailored to the individual patient’s condition. Surgical interventions include pulmonary endarterectomy (PEA), atrial septostomy, pulmonary balloon angioplasty (PBAV), atrial septal defect closure, ventricular septal defect closure, patent ductus arteriosus closure, and lung transplantation. For patients with more complex conditions, emerging treatment technologies, such as PADN or stem cell and other biological therapies, can be explored. A multidisciplinary team should be organized to jointly participate in the diagnosis and treatment process of the patient. By strengthening follow-up, the patient’s condition changes can be monitored in a timely manner, and the treatment plan can be adjusted accordingly. Through this comprehensive management strategy, we can better address the complexity of Cpc-PH and provide more comprehensive treatment support for patients.
Future
The most prevalent kind of PH is Cpc-PH, and unfavorable prognoses rise in tandem with the patient population’s increased risk of morbidity and mortality. Although there are clear similarities to PH in many areas, the current guidelines do not advocate targets for PH because there are currently no clinical trials that can demonstrate the safety and efficacy of these targeted medications in patients with Cpc-PH. In order to identify novel therapeutic targets, create targeted treatment plans for Cpc-PH, and implement evidence-based strategies to keep HF patients from developing PH, we must thereby expand our understanding of the pathophysiology, etiology, and genetic components of the condition. In order to make sure that treatment approaches can do more than just support physiological processes, we will conduct patient-centered clinical trials as new treatments are developed. These trials should assess the beneficial effects of treatment on patient survival, readmission, and quality of life. Significant clinical improvement can also result from recovery.
Statements
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author contributions
ML: Methodology, Writing – original draft. BW: Conceptualization, Project administration, Writing – review & editing. CR: Supervision, Writing – review & editing. YW: Resources, Software, Writing – review & editing. WZ: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1.
Humbert M Kovacs G Hoeper MM Badagliacca R Berger RMF Brida M et al . ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J. (2022) 43:3618–731. doi: 10.1093/eurheartj/ehac237
2.
Thomas CA Anderson RJ Condon DF de Jesus Perez VA . Diagnosis and Management of Pulmonary Hypertension in the modern era: insights from the 6th world symposium. Pulmonary Ther. (2020) 6:9–22. doi: 10.1007/s41030-019-00105-5
3.
Fauvel C Damy T Berthelot E Bauer F Eicher JC de Groote P et al . Post-capillary pulmonary hypertension in heart failure: impact of current definition in the PH-HF multicentre study. Eur Heart J. (2024) 45:3274–88. doi: 10.1093/eurheartj/ehae467
4.
Maron BA Bortman G De Marco T Huston JH Lang IM Rosenkranz SH et al . Pulmonary hypertension associated with left heart disease. Eur Respir J. (2024) 64:2401344. doi: 10.1183/13993003.01344-2024
5.
Hoeper MM Humbert M Souza R Idrees M Kawut SM Sliwa-Hahnle K et al . A global view of pulmonary hypertension. Lancet Respir Med. (2016) 4:306–22. doi: 10.1016/S2213-2600(15)00543-3
6.
Titz A Mayer L Appenzeller P Müller J Schneider SR Tamm M et al . Long-term outcome of patients with combined post-and pre-capillary pulmonary hypertension. Eur Heart J Open. (2023) 3:oead069. doi: 10.1093/ehjopen/oead069
7.
Leary PJ Lindstrom M Johnson CO Emmons-Bell S Rich S Corris PA et al . Global, regional, and national burden of pulmonary arterial hypertension, 1990-2021: a systematic analysis for the global burden of disease study 2021. Lancet Respir Med. (2025) 13:69–79. doi: 10.1016/s2213-2600(24)00295-9
8.
Anderson JJ Lau EM . Pulmonary hypertension definition, classification, and epidemiology in Asia. JACC Asia. (2022) 2:538–46. doi: 10.1016/j.jacasi.2022.04.008
9.
Karia N Howard L Johnson M Kiely DG Lordan J McCabe C et al . Predictors of outcomes in mild pulmonary hypertension according to 2022 ESC/ERS guidelines: the EVIDENCE-PAH UK study. Eur Heart J. (2023) 44:4678–91. doi: 10.1093/eurheartj/ehad532
10.
Babu G Annis JS Garry JD Freiberg MS Hemnes AR Brittain EL . Clinical features do not identify risk of progression from isolated postcapillary pulmonary hypertension to combined pre-and postcapillary pulmonary hypertension. Pulmonar. Circ. (2023) 13:e12249. doi: 10.1002/pul2.12249
11.
Guignabert C Aman J Bonnet S Dorfmüller P Olschewski AJ Pullamsetti S et al . Pathology and pathobiology of pulmonary hypertension: current insights and future directions. Eur Respir J. (2024) 64:2401095. doi: 10.1183/13993003.01095-2024
12.
Tobal R Potjewijd J van Empel VPM Ysermans R Schurgers LJ Reutelingsperger CP et al . Vascular remodeling in pulmonary arterial hypertension: the potential involvement of innate and adaptive immunity. Front Med. (2021) 8:806899. doi: 10.3389/fmed.2021.806899
13.
Ruopp NF Cockrill BA . Diagnosis and treatment of pulmonary arterial hypertension: a review. JAMA. (2022) 327:1379–91. doi: 10.1001/jama.2022.4402
14.
He D Zhang X Chen S Dai C Wu Q Zhou Y et al . Dynamic changes of metabolic syndrome Alter the risks of cardiovascular diseases and all-cause mortality: evidence from a prospective cohort study. Front. Cardiovascular Med. (2021) 8:706999. doi: 10.3389/fcvm.2021.706999
15.
Baldwin JG Silveira CRF Gattinoni L . Restoring the supply lines: removing roadblocks to fatty acid uptake enhances T cell-driven cancer fight. Signal Transduct Target Ther. (2025) 10:53. doi: 10.1038/s41392-025-02151-9
16.
Qian J Zhao Z Ma L Liu W Song Y . USP14 targets FABP5-mediated ferroptosis to promote proliferation and cisplatin resistance of HNSCC. Clin Transl Oncol Off Pub Federation Spanish Oncol Soc Nat Cancer Inst Mexico. (2025) 10:1–16. doi: 10.1007/s12094-025-03857-6
17.
Zhang J Li Y Chen Y Zhang J Jia Z He M et al . O(8)G site-specifically modified tRF-1-AspGTC: a novel therapeutic target and biomarker for pulmonary hypertension. Circ Res. (2024) 135:76–92. doi: 10.1161/CIRCRESAHA.124.324421
18.
Gao P Lu Y Tang K Wang W Wang T Zhu Y et al . Ficolin-1 ameliorates pulmonary fibrosis via directly binding to TGF-β1. J Transl Med. (2024) 22:1051. doi: 10.1186/s12967-024-05894-1
19.
Chang JX Sun N Wu BX . Application value of assessing the pulmonary vascular resistance in the evaluation of patients with pulmonary arterial hypertension. Zhonghua Xin Xue Guan Bing Za Zhi. (2021) 49:96–101. doi: 10.3760/cma.j.cn112148-20200520-00416
20.
Frid MG Brunetti JA Burke DL Carpenter TC Davie NJ Reeves JT et al . Hypoxia-induced pulmonary vascular remodeling requires recruitment of circulating mesenchymal precursors of a monocyte/macrophage lineage. Am J Pathol. (2006) 168:659–69. doi: 10.2353/ajpath.2006.050599
21.
Mascolo A Scavone C Rafaniello C De Angelis A Urbanek K di Mauro G et al . The role of renin-angiotensin-aldosterone system in the heart and lung: focus on COVID-19. Front Pharmacol. (2021) 12:667254. doi: 10.3389/fphar.2021.667254
22.
Pei Y Ren D Yin Y Shi J Ai Q Hao W et al . Endothelial FUNDC1 deficiency drives pulmonary hypertension. Circ Res. (2025) 136:e1–e19. doi: 10.1161/CIRCRESAHA.124.325156
23.
Gau YC Yeh TJ Hsu CM Hsiao SY Hsiao HH . Pathogenesis and treatment of myeloma-related bone disease. Int J Mol Sci. (2022) 23:112. doi: 10.3390/ijms23063112
24.
Steinhauser ML Maron BA . Viewing pulmonary arterial hypertension pathogenesis and opportunities for disease-modifying therapy through the Lens of biomass. JACC Basic Trans Sci. (2024) 9:1252–63. doi: 10.1016/j.jacbts.2024.04.009
25.
Papamichalis M Xanthopoulos A Papamichalis P Skoularigis J Triposkiadis F . Adult congenital heart disease with pulmonary arterial hypertension: mechanisms and management. Heart Fail Rev. (2020) 25:773–94. doi: 10.1007/s10741-019-09847-5
26.
Yung LM Nikolic I Paskin-Flerlage SD Pearsall RS Kumar R Yu PB . A selective transforming growth factor-β ligand trap attenuates pulmonary hypertension. Am J Respir Crit Care Med. (2016) 194:1140–51. doi: 10.1164/rccm.201510-1955OC PMID:
27.
Zhang RY Liu J Sun Y Wang W Wang C . metabolic reprogramming in pulmonary hypertension. Chin J Tuberculosis Resp Dis. (2022) 45:313–7. doi: 10.3760/cma.j.cn112147-20210820-00578
28.
Fajardo RJ Manoharan RK Pearsall RS Davies MV Marvell T Monnell TE et al . Treatment with a soluble receptor for activin improves bone mass and structure in the axial and appendicular skeleton of female cynomolgus macaques (Macaca fascicularis). Bone. (2010) 46:64–71. doi: 10.1016/j.bone.2009.09.018
29.
Yong HE Murthi P Wong MH Kalionis B Cartwright JE Brennecke SP et al . Effects of normal and high circulating concentrations of activin a on vascular endothelial cell functions and vasoactive factor production. Pregnancy Hypertension. (2015) 5:346–53. doi: 10.1016/j.preghy.2015.09.006
30.
Bousseau S Sobrano Fais R Gu S Frump A Lahm T . Pathophysiology and new advances in pulmonary hypertension. BMJ Med. (2023) 2:e000137. doi: 10.1136/bmjmed-2022-000137
31.
McMurtry IF Abe K Ota H Fagan KA Oka M . Rho kinase-mediated vasoconstriction in pulmonary hypertension. Adv Exp Med Biol. (2010) 661:299–308. doi: 10.1007/978-1-60761-500-2_19
32.
van Duin RWB Stam K Cai Z Uitterdijk A Garcia-Alvarez A Ibanez B et al . Transition from post-capillary pulmonary hypertension to combined pre-and post-capillary pulmonary hypertension in swine: a key role for endothelin. J Physiol. (2019) 597:1157–73. doi: 10.1113/JP275987
33.
Szulcek R Sanchez-Duffhues G Rol N Pan X Tsonaka R Dickhoff C et al . Exacerbated inflammatory signaling underlies aberrant response to BMP9 in pulmonary arterial hypertension lung endothelial cells. Angiogenesis. (2020) 23:699–714. doi: 10.1007/s10456-020-09741-x
34.
Le TT Karmouty-Quintana H Melicoff E Le TT Weng T Chen NY et al . Blockade of IL-6 trans signaling attenuates pulmonary fibrosis. J Immunol. (1950) 193:3755–68. doi: 10.4049/jimmunol.1302470
35.
Hiraide T Tsuda N Momoi M Shinya Y Sano M Fukuda K et al . CXCL12/CXCR4 pathway as a novel therapeutic target for RNF213-associated pulmonary arterial hypertension. Sci Rep. (2024) 14:26604. doi: 10.1038/s41598-024-77388-5
36.
Weng Y Siciliano SJ Waldburger KE Sirotina-Meisher A Staruch MJ Daugherty BL et al . Binding and functional properties of recombinant and endogenous CXCR3 chemokine receptors. J Biol Chem. (1998) 273:18288–91. doi: 10.1074/jbc.273.29.18288 PMID:
37.
Li Y Wei X Xiao R Chen Y Xiong T Fang ZM et al . SMYD2-methylated PPARγ facilitates hypoxia-induced pulmonary hypertension by activating Mitophagy. Circ Res. (2024) 135:93–109. doi: 10.1161/CIRCRESAHA.124.323698
38.
Vivanco I Sawyers CL . The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev Cancer. (2002) 2:489–501. doi: 10.1038/nrc839
39.
Sciarretta S Volpe M Sadoshima J . Mammalian target of rapamycin signaling in cardiac physiology and disease. Circ Res. (2014) 114:549–64. doi: 10.1161/CIRCRESAHA.114.302022
40.
Babicheva A Makino A Yuan JX . mTOR signaling in pulmonary vascular disease: pathogenic role and therapeutic target. Int J Mol Sci. (2021) 22:2144. doi: 10.3390/ijms22042144
41.
Rashid J Alobaida A Al-Hilal TA Hammouda S McMurtry IF Nozik-Grayck E et al . Repurposing rosiglitazone, a PPAR-γ agonist and oral antidiabetic, as an inhaled formulation, for the treatment of PAH. J Control Release. (2018) 280:113–23. doi: 10.1016/j.jconrel.2018.04.049
42.
Zhou H Li D Zhu P Hu S Hu N Ma S et al . Melatonin suppresses platelet activation and function against cardiac ischemia/reperfusion injury via PPARγ/FUNDC1/mitophagy pathways. J Pineal Res. (2017) 63:12438. doi: 10.1111/jpi.12438
43.
Chaikuad A Thangaratnarajah C von Delft F Bullock AN . Structural consequences of BMPR2 kinase domain mutations causing pulmonary arterial hypertension. Sci Rep. (2019) 9:18351. doi: 10.1038/s41598-019-54830-7
44.
Lotsios NS Keskinidou C Dimopoulou I Kotanidou A Langleben D Orfanos SE et al . Effects of modulating BMP9, BMPR2, and AQP1 on BMP signaling in human pulmonary microvascular endothelial cells. Int J Mol Sci. (2024) 25:8043. doi: 10.3390/ijms25158043
45.
Yamaji-Kegan K Takimoto E Zhang A Weiner NC Meuchel LW Berger AE et al . Hypoxia-induced mitogenic factor (FIZZ1/RELMα) induces endothelial cell apoptosis and subsequent interleukin-4-dependent pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. (2014) 306:L1090–103. doi: 10.1152/ajplung.00279.2013
46.
Hara H Takeda N Morita H Hatano M Amiya E Maki H et al . Three novel BMPR2 mutations associated with advanced pulmonary arterial hypertension. Hum Genome variation. (2017) 4:17010. doi: 10.1038/hgv.2017.10
47.
Wang J Zhang C Liu C Wang W Zhang N Hadadi C et al . Functional mutations in 5'UTR of the BMPR2 gene identified in Chinese families with pulmonary arterial hypertension. Pulmonary Circulation. (2016) 6:103–8. doi: 10.1086/685078
48.
Okur Z Schlauri N Bitsikas V Panopoulou M Ortiz R Schwaiger M et al . Control of neuronal excitation-inhibition balance by BMP-SMAD1 signalling. Nature. (2024) 629:402–9. doi: 10.1038/s41586-024-07317-z
49.
Dehorter N Marichal N Marín O Berninger B . Tuning neural circuits by turning the interneuron knob. Curr Opin Neurobiol. (2017) 42:144–51. doi: 10.1016/j.conb.2016.12.009
50.
Xiao L Michalski N Kronander E Gjoni E Genoud C Knott G et al . BMP signaling specifies the development of a large and fast CNS synapse. Nat Neurosci. (2013) 16:856–64. doi: 10.1038/nn.3414
51.
Tian W Jiang X Sung YK Shuffle E Wu TH Kao PN et al . Phenotypically silent bone morphogenetic protein receptor 2 mutations predispose rats to inflammation-induced pulmonary arterial hypertension by enhancing the risk for Neointimal transformation. Circulation. (2019) 140:1409–25. doi: 10.1161/CIRCULATIONAHA.119.040629
52.
Hautefort A Mendes-Ferreira P Sabourin J Manaud G Bertero T Rucker-Martin C et al . Bmpr2 mutant rats develop pulmonary and cardiac characteristics of pulmonary arterial hypertension. Circulation. (2019) 139:932–48. doi: 10.1161/CIRCULATIONAHA.118.033744
53.
Sawada H Saito T Nickel NP Alastalo TP Glotzbach JP Chan R et al . Reduced BMPR2 expression induces GM-CSF translation and macrophage recruitment in humans and mice to exacerbate pulmonary hypertension. J Exp Med. (2014) 211:263–80. doi: 10.1084/jem.20111741
54.
Theobald V Benjamin N Seyfarth HJ Halank M Schneider MA Richtmann S et al . Reduction of BMPR2 mRNA expression in peripheral blood of pulmonary arterial hypertension patients: a marker for disease severity?Genes. (2022) 13:759. doi: 10.3390/genes13050759
55.
Maddaloni C Ronci S De Rose DU Bersani I Campi F Di Nardo M et al . Neonatal persistent pulmonary hypertension related to a novel TBX4 mutation: case report and review of the literature. Ital J Pediatr. (2024) 50:41. doi: 10.1186/s13052-024-01575-3
56.
Haarman MG Kerstjens-Frederikse WS Berger RMF . TBX4 variants and pulmonary diseases: getting out of the 'Box'. Curr Opin Pulm Med. (2020) 26:277–84. doi: 10.1097/MCP.0000000000000678
57.
Ma L Chung WK . The role of genetics in pulmonary arterial hypertension. J Pathol. (2017) 241:273–80. doi: 10.1002/path.4833
58.
Sydykov A Mamazhakypov A Maripov A Kosanovic D Weissmann N Ghofrani HA et al . Pulmonary hypertension in acute and chronic high altitude maladaptation disorders. Int J Environ Res Public Health. (2021) 18:1692. doi: 10.3390/ijerph18041692
59.
Sherratt SCR Libby P Dawoud H Bhatt DL Mason RP . Eicosapentaenoic acid improves endothelial nitric oxide bioavailability via changes in protein expression during inflammation. J Am Heart Assoc. (2024) 13:e034076. doi: 10.1161/JAHA.123.034076
60.
Zhang Z Huang W Huang D Xu Z Xie Q Tan X et al . Repurposing of phosphodiesterase-5 inhibitor sildenafil as a therapeutic agent to prevent gastric cancer growth through suppressing c-MYC stability for IL-6 transcription. Commun Biol. (2025) 8:85. doi: 10.1038/s42003-025-07519-9
61.
Hartupee J Mann DL . Neurohormonal activation in heart failure with reduced ejection fraction. Nat Rev Cardiol. (2017) 14:30–8. doi: 10.1038/nrcardio.2016.163
62.
Patel KP Zhang K . Neurohumoral activation in heart failure: role of paraventricular nucleus. Clin Exp Pharmacol Physiol. (1996) 23:722–6. doi: 10.1111/j.1440-1681.1996.tb01765.x
63.
Li H Zhang S Huang X Zhang Z Liu K Wang QD et al . Genetic recording of transient endothelial activation in distinct alveolar capillary cells during pulmonary fibrosis. Cell Discovery. (2024) 10:119. doi: 10.1038/s41421-024-00745-1
64.
Gillich A Zhang F Farmer CG Travaglini KJ Tan SY Gu M et al . Capillary cell-type specialization in the alveolus. Nature. (2020) 586:785–9. doi: 10.1038/s41586-020-2822-7
65.
Ear J Huang H Wilson T Tehrani Z Lindgren A Sung V et al . RAP-011 improves erythropoiesis in zebrafish model of diamond-Blackfan anemia through antagonizing lefty1. Blood. (2015) 126:880–90. doi: 10.1182/blood-2015-01-622522
66.
De Rosa G Andolfo I Marra R Manna F Rosato BE Iolascon A et al . RAP-011 rescues the disease phenotype in a cellular model of congenital Dyserythropoietic Anemia type II by inhibiting the SMAD2-3 pathway. Int J Mol Sci. (2020) 21:577. doi: 10.3390/ijms21155577
67.
Latson LA Prieto LR . Congenital and acquired pulmonary vein stenosis. Circulation. (2007) 115:103–8. doi: 10.1161/CIRCULATIONAHA.106.646166
68.
Rosenkranz S Gibbs JS Wachter R De Marco T Vonk-Noordegraaf A Vachiéry JL . Left ventricular heart failure and pulmonary hypertension. Eur Heart J. (2016) 37:942–54. doi: 10.1093/eurheartj/ehv512
69.
Brittain EL Thenappan T Huston JH Agrawal V Lai YC Dixon D et al . Elucidating the clinical implications and pathophysiology of pulmonary hypertension in heart failure with preserved ejection fraction: a call to action: a science advisory from the American Heart Association. Circulation. (2022) 146:e73–88. doi: 10.1161/CIR.0000000000001079
70.
Alushi B Beckhoff F Leistner D Franz M Reinthaler M Stähli BE et al . Pulmonary hypertension in patients with severe aortic stenosis: prognostic impact after Transcatheter aortic valve replacement: pulmonary hypertension in patients undergoing TAVR. J Am Coll Cardiol Img. (2019) 12:591–601. doi: 10.1016/j.jcmg.2018.02.015
71.
Maron BA Kovacs G Vaidya A Bhatt DL Nishimura RA Mak S et al . Cardiopulmonary hemodynamics in pulmonary hypertension and heart failure: JACC review topic of the week. J Am Coll Cardiol. (2020) 76:2671–81. doi: 10.1016/j.jacc.2020.10.007
72.
Rosenkranz S Howard LS Gomberg-Maitland M Hoeper MM . Systemic consequences of pulmonary hypertension and right-sided heart failure. Circulation. (2020) 141:678–93. doi: 10.1161/CIRCULATIONAHA.116.022362
73.
Usman MS Van Spall HGC Greene SJ Pandey A McGuire DK Ali ZA et al . The need for increased pragmatism in cardiovascular clinical trials. Nat Rev Cardiol. (2022) 19:737–50. doi: 10.1038/s41569-022-00705-w
74.
Abraham WT Adamson PB Bourge RC Aaron MF Costanzo MR Stevenson LW et al . Wireless pulmonary artery haemodynamic monitoring in chronic heart failure: a randomised controlled trial. Lancet. (2011) 377:658–66. doi: 10.1016/S0140-6736(11)60101-3
75.
Jones TK McElhinney DB Vincent JA Hellenbrand WE Cheatham JP Berman DP et al . Long-term outcomes after Melody Transcatheter pulmonary valve replacement in the US investigational device exemption trial. Circ Cardiovasc Interv. (2022) 15:e010852. doi: 10.1161/CIRCINTERVENTIONS.121.010852
76.
Simonneau G Montani D Celermajer DS Denton CP Gatzoulis MA Krowka M et al . Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. (2019) 53:1801913. doi: 10.1183/13993003.01913-2018
77.
Kovacs G Berghold A Scheidl S Olschewski H . Pulmonary arterial pressure during rest and exercise in healthy subjects: a systematic review. Eur Respir J. (2009) 34:888–94. doi: 10.1183/09031936.00145608
78.
Maron BA Hess E Maddox TM Opotowsky AR Tedford RJ Lahm T et al . Association of Borderline Pulmonary Hypertension with Mortality and Hospitalization in a large patient cohort: insights from the veterans affairs clinical assessment, reporting, and tracking program. Circulation. (2016) 133:1240–8. doi: 10.1161/CIRCULATIONAHA.115.020207
79.
Douschan P Kovacs G Avian A Foris V Gruber F Olschewski A et al . Mild elevation of pulmonary arterial pressure as a predictor of mortality. Am J Respir Crit Care Med. (2018) 197:509–16. doi: 10.1164/rccm.201706-1215OC
80.
Maron BA Kleiner DE Arons E Wertheim BM Sharma NS Haley KJ et al . Evidence of advanced pulmonary vascular remodeling in obstructive hypertrophic cardiomyopathy with pulmonary hypertension. Chest. (2023) 163:678–86. doi: 10.1016/j.chest.2022.09.040
81.
Huston JH Maron BA French J Huang S Thayer T Farber-Eger EH et al . Association of Mild Echocardiographic Pulmonary Hypertension with Mortality and Right Ventricular Function. JAMA Cardiol. (2019) 4:1112–21. doi: 10.1001/jamacardio.2019.3345
82.
Strange G Stewart S Celermajer DS Prior D Scalia GM Marwick TH et al . Threshold of pulmonary hypertension associated with increased mortality. J Am Coll Cardiol. (2019) 73:2660–72. doi: 10.1016/j.jacc.2019.03.482
83.
Fayyaz AU Edwards WD Maleszewski JJ Konik EA DuBrock HM Borlaug BA et al . Global pulmonary vascular remodeling in pulmonary hypertension associated with heart failure and preserved or reduced ejection fraction. Circulation. (2018) 137:1796–810. doi: 10.1161/CIRCULATIONAHA.117.031608
84.
Maron BA Brittain EL Hess E Waldo SW Barón AE Huang S et al . Pulmonary vascular resistance and clinical outcomes in patients with pulmonary hypertension: a retrospective cohort study. Lancet Respir Med. (2020) 8:873–84. doi: 10.1016/S2213-2600(20)30317-9
85.
Xanthouli P Jordan S Milde N Marra A Blank N Egenlauf B et al . Haemodynamic phenotypes and survival in patients with systemic sclerosis: the impact of the new definition of pulmonary arterial hypertension. Ann Rheum Dis. (2020) 79:370–8. doi: 10.1136/annrheumdis-2019-216476
86.
Stevenson DJ Avery AJ Coupland C Hobbs FDR Kendrick D Moore MV et al . Recruitment to a large scale randomised controlled clinical trial in primary care: the Helicobacter eradication aspirin trial (HEAT). Trials. (2022) 23:140. doi: 10.1186/s13063-022-06054-w
87.
Vachiery J-L Delcroix M Al-Hiti H Efficace M Hutyra M Lack G et al . Melody-1: a pilot study of MACITENTAN in pulmonary hypertension due to left ventricular dysfunction. J Am Coll Cardiol. (2017) 69:1880. doi: 10.1016/S0735-1097(17)35269-5
88.
Young PJ Saxena MK Bellomo R Freebairn RC Hammond NE van Haren FM et al . The HEAT trial: a protocol for a multicentre randomised placebo-controlled trial of IV paracetamol in ICU patients with fever and infection. Critical Care Resuscitation J Austr Acad Critic Care Med. (2012) 14:290–6. PMID:
89.
Dumbleton JS Avery AJ Coupland C Hobbs FD Kendrick D Moore MV et al . The Helicobacter eradication aspirin trial (HEAT): a large simple randomised controlled trial using novel methodology in primary care. EBioMedicine. (2015) 2:1200–4. doi: 10.1016/j.ebiom.2015.07.012
90.
Kreutzer J Armstrong AK Rome JJ Zellers TM Balzer DT Zampi JD et al . Comparison of the investigational device exemption and post-approval trials of the Melody transcatheter pulmonary valve. Catheter Cardiovasc Interv. (2021) 98:E262–e274. doi: 10.1002/ccd.29657
91.
Packer M McMurray JJV Krum H Kiowski W Massie BM Caspi A et al . Long-term effect of endothelin receptor antagonism with Bosentan on the morbidity and mortality of patients with severe chronic heart failure: primary results of the ENABLE trials. JACC Heart Failure. (2017) 5:317–26. doi: 10.1016/j.jchf.2017.02.021
92.
Kaluski E Cotter G Leitman M Milo-Cotter O Krakover R Kobrin I et al . Clinical and hemodynamic effects of Bosentan dose optimization in symptomatic heart failure patients with severe systolic dysfunction, associated with secondary pulmonary hypertension – a multi-center randomized study. Cardiology. (2008) 109:273–80. doi: 10.1159/000107791
93.
Vachiéry JL Delcroix M Al-Hiti H Efficace M Hutyra M Lack G et al . Macitentan in pulmonary hypertension due to left ventricular dysfunction. Eur Respir J. (2018) 51:1701886. doi: 10.1183/13993003.01886-2017
94.
Lüscher TF Enseleit F Pacher R Mitrovic V Schulze MR Willenbrock R et al . Hemodynamic and neurohumoral effects of selective endothelin a (ET(a)) receptor blockade in chronic heart failure: the heart failure ET(a) receptor blockade trial (HEAT). Circulation. (2002) 106:2666–72. doi: 10.1161/01.cir.0000038497.80095.e1 PMID:
95.
Masarone D Valente F Verrengia M Contaldi C di Palma V Falco L et al . Pharmacologic treatment of pulmonary hypertension due to heart failure with preserved ejection fraction: are there more arrows on our Bow?J Clin Med. (2024) 13:6867. doi: 10.3390/jcm13226867
96.
Rao SD Adusumalli S Mazurek JA . Corrigendum to: pulmonary hypertension in heart failure patients. Card Fail Rev. (2020) 6:e30. doi: 10.15420/cfr.2020.1.1
97.
Bauersachs J Olsson KM . Targeting pulmonary hypertension in patients with heart failure and preserved ejection fraction: rather static than DYNAMIC development?Eur Heart J. (2022) 43:3414–6. doi: 10.1093/eurheartj/ehac387
98.
Karedath J Dar H Ganipineni VDP Gorle SA Gaddipati S Bseiso A et al . Effect of Phosphodiesterase-5 (PDE-5) inhibitors on clinical outcomes in patients with pulmonary hypertension: a Meta-analysis of randomized control trials. Cureus. (2023) 15:e33363. doi: 10.7759/cureus.33363
99.
Wang RS Huang S Waldo SW Hess E Gokhale M Johnson SW et al . Elevated pulmonary arterial compliance is associated with survival in pulmonary hypertension: results from a novel network medicine analysis. Am J Respir Crit Care Med. (2023) 208:312–21. doi: 10.1164/rccm.202211-2097OC
100.
Vikholm P Schiller P Hellgren L . A modified Glenn shunt reduces venous congestion during acute right ventricular failure due to pulmonary banding: a randomized experimental study. Interact Cardiovasc Thorac Surg. (2014) 18:418–25. doi: 10.1093/icvts/ivt547
101.
Mauri L D'Agostino RB Sr . Challenges in the design and interpretation of noninferiority trials. N Engl J Med. (2017) 377:1357–67. doi: 10.1056/NEJMra1510063
102.
Pallmann P Bedding AW Choodari-Oskooei B Dimairo M Flight L Hampson LV et al . Adaptive designs in clinical trials: why use them, and how to run and report them. BMC Med. (2018) 16:29. doi: 10.1186/s12916-018-1017-7
103.
Chen Z Zhao K Xiao C He Z Liu S Wu X et al . Phosphodiesterase inhibitor for heart failure with preserved ejection fraction: a systematic review and meta-analysis. SPJ Off Pub. (2022) 30:1079–87. doi: 10.1016/j.jsps.2022.05.012
104.
Elliott J Menakuru N Martin KJ Rahaghi FN Rischard FP Vanderpool RR . iCPET calculator: a web-based application to standardize the calculation of alpha Distensibility in patients with pulmonary arterial hypertension. J Am Heart Assoc. (2023) 12:e029667. doi: 10.1161/JAHA.123.029667
105.
Sanchez Palacios GM Schmidt C Wichman T . Targeted therapy with phosphodiesterase 5 inhibitors in patients with pulmonary hypertension due to heart failure and elevated pulmonary vascular resistance: a systematic review. Pulmonary Circ. (2020) 10:2045894020948780. doi: 10.1177/2045894020948780
106.
Zhan Y Li L Zhou J Ma Y Guan X Wang S et al . Efficacy of vericiguat in patients with chronic heart failure and reduced ejection fraction: a prospective observational study. BMC Cardiovasc Disord. (2025) 25:83. doi: 10.1186/s12872-025-04477-2
107.
Stasch JP Hobbs AJ . NO-independent, haem-dependent soluble guanylate cyclase stimulators. Handb Exp Pharmacol. (2009) 11:277–308. doi: 10.1007/978-3-540-68964-5_13
108.
Das BB . Novel therapies for right ventricular failure. Curr Cardiol Rep. (2025) 27:26. doi: 10.1007/s11886-024-02157-9
109.
Rasheed A Aslam S Sadiq HZ Ali S Syed R Panjiyar BK . New and emerging therapeutic drugs for the treatment of pulmonary arterial hypertension: a systematic review. Cureus. (2024) 16:e68117. doi: 10.7759/cureus.68117
110.
Kuroda K Akagi S Nakamura K Sarashina T Ejiri K Ito H . Successful transition from Phosphodiesterase-5 inhibitors to Riociguat without a washout period in patients with pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension: a pilot cohort study. Heart Lung Circ. (2020) 29:331–6. doi: 10.1016/j.hlc.2019.01.013
111.
Armstrong PW Pieske B Anstrom KJ Ezekowitz J Hernandez AF Butler J et al . Vericiguat in patients with heart failure and reduced ejection fraction. N Engl J Med. (2020) 382:1883–93. doi: 10.1056/NEJMoa1915928
112.
Bonderman D Ghio S Felix SB Ghofrani HA Michelakis E Mitrovic V et al . Riociguat for patients with pulmonary hypertension caused by systolic left ventricular dysfunction: a phase IIb double-blind, randomized, placebo-controlled, dose-ranging hemodynamic study. Circulation. (2013) 128:502–11. doi: 10.1161/CIRCULATIONAHA.113.001458
113.
Bonderman D Pretsch I Steringer-Mascherbauer R Jansa P Rosenkranz S Tufaro C et al . Acute hemodynamic effects of riociguat in patients with pulmonary hypertension associated with diastolic heart failure (DILATE-1): a randomized, double-blind, placebo-controlled, single-dose study. Chest. (2014) 146:1274–85. doi: 10.1378/chest.14-0106
114.
Corrigendum for efficacy and safety of riociguat in combination therapy for patients with pulmonary arterial hypertension (PATENT studies). Pulmonary Circ. (2023) 13:e12250. doi: 10.1002/pul2.12250
115.
Galiè N Humbert M Vachiery JL Gibbs S Lang I Torbicki A et al . 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension. The joint task force for the diagnosis and treatment of pulmonary hypertension of the European society of cardiology (Esc) and the European respiratory society (ERS). Eur Respir J. (2015) 46:903–75. doi: 10.1183/13993003.51032-2015
116.
Waehre A Vistnes M Sjaastad I Nygård S Husberg C Lunde IG et al . Chemokines regulate small leucine-rich proteoglycans in the extracellular matrix of the pressure-overloaded right ventricle. J Appl Physiol. (1985) 112:1372–82. doi: 10.1152/japplphysiol.01350.2011
117.
Lajoie AC Bonnet S Provencher S . Combination therapy in pulmonary arterial hypertension: recent accomplishments and future challenges. Pulmonary Circ. (2017) 7:312–25. doi: 10.1177/2045893217710639
118.
Humbert M Sitbon O Chaouat A Bertocchi M Habib G Gressin V et al . Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation. (2010) 122:156–63. doi: 10.1161/CIRCULATIONAHA.109.911818
119.
Ostasevicius V Jurenas V Venslauskas M Kizauskiene L Zigmantaite V Stankevicius E et al . Low-frequency ultrasound for pulmonary hypertension therapy. Respir Res. (2024) 25:70. doi: 10.1186/s12931-024-02713-5
120.
Polhemus D Almodiel D Harb T Ziogos E Amat-Codina N Ranek M et al . Vericiguat prevents high glucose-mediated impaired vascular smooth muscle cGMP production and vasorelaxation. Sci Rep. (2025) 15:4939. doi: 10.1038/s41598-025-88938-w
121.
Khaybullina D Patel A Zerilli T . Riociguat (adempas): a novel agent for the treatment of pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension. P&T Peer-reviewed J Form Manage. (2014) 39:749–58. PMID:
122.
Wood KC Gladwin MT Straub AC . Sickle cell disease: at the crossroads of pulmonary hypertension and diastolic heart failure. Heart. (2020) 106:562–8. doi: 10.1136/heartjnl-2019-314810
123.
Wang L Halliday G Huot JR Satoh T Baust JJ Fisher A et al . Treatment with Treprostinil and metformin normalizes hyperglycemia and improves cardiac function in pulmonary hypertension associated with heart failure with preserved ejection fraction. Arterioscler Thromb Vasc Biol. (2020) 40:1543–58. doi: 10.1161/ATVBAHA.119.313883
124.
Khanna D Allanore Y Denton CP Kuwana M Matucci-Cerinic M Pope JE et al . Riociguat in patients with early diffuse cutaneous systemic sclerosis (RISE-SSc): randomised, double-blind, placebo-controlled multicentre trial. Ann Rheum Dis. (2020) 79:618–25. doi: 10.1136/annrheumdis-2019-216823
125.
Ghadimi K Cappiello JL Wright MC Levy JH Bryner BS DeVore AD et al . Inhaled Epoprostenol compared with nitric oxide for right ventricular support after major cardiac surgery. Circulation. (2023) 148:1316–29. doi: 10.1161/CIRCULATIONAHA.122.062464
126.
Califf RM Adams KF McKenna WJ Gheorghiade M Uretsky BF McNulty SE et al . A randomized controlled trial of epoprostenol therapy for severe congestive heart failure: the Flolan international randomized survival trial (FIRST). Am Heart J. (1997) 134:44–54. doi: 10.1016/S0002-8703(97)70105-4
127.
Lang IM Gaine SP . Recent advances in targeting the prostacyclin pathway in pulmonary arterial hypertension. Eur Resp Rev Off J Eur Resp Soc. (2015) 24:630–41. doi: 10.1183/16000617.0067-2015
128.
Feenstra J Hopkins P Trotter M O'Brien K . Effectiveness of epoprostenol in the management of pulmonary arterial hypertension: findings of an Australian retrospective chart review. Intern Med J. (2020) 50:1377–84. doi: 10.1111/imj.14668
129.
Guay CA Morin-Thibault LV Bonnet S Lacasse Y Lambert C Lega JC et al . Pulmonary hypertension-targeted therapies in heart failure: a systematic review and meta-analysis. PLoS One. (2018) 13:e0204610. doi: 10.1371/journal.pone.0204610
130.
Moghaddam N Swiston JR Tsang MYC Levy R Lee L Brunner NW . Impact of targeted pulmonary arterial hypertension therapy in patients with combined post-and precapillary pulmonary hypertension. Am Heart J. (2021) 235:74–81. doi: 10.1016/j.ahj.2021.01.003
131.
Tapson VF Torres F Kermeen F Keogh AM Allen RP Frantz RP et al . Oral treprostinil for the treatment of pulmonary arterial hypertension in patients on background endothelin receptor antagonist and/or phosphodiesterase type 5 inhibitor therapy (the FREEDOM-C study): a randomized controlled trial. Chest. (2012) 142:1383–90. doi: 10.1378/chest.11-2212
132.
Torres F Farber H Ristic A McLaughlin V Adams J Zhang J et al . Efficacy and safety of ralinepag, a novel oral IP agonist, in PAH patients on mono or dual background therapy: results from a phase 2 randomised, parallel group, placebo-controlled trial. Eur Respir J. (2019) 54:1901030. doi: 10.1183/13993003.51030-2019
133.
Ghofrani HA Gomberg-Maitland M Zhao L Grimminger F . Mechanisms and treatment of pulmonary arterial hypertension. Nat Rev Cardiol. (2025) 22:105–20. doi: 10.1038/s41569-024-01064-4
134.
Humbert M McLaughlin V Gibbs JSR Gomberg-Maitland M Hoeper MM Preston IR et al . Sotatercept for the treatment of pulmonary arterial hypertension. N Engl J Med. (2021) 384:1204–15. doi: 10.1056/NEJMoa2024277
135.
Hoeper MM Badesch DB Ghofrani HA Gibbs JSR Gomberg-Maitland M McLaughlin VV et al . Phase 3 trial of Sotatercept for treatment of pulmonary arterial hypertension. N Engl J Med. (2023) 388:1478–90. doi: 10.1056/NEJMoa2213558
136.
Souza R Badesch DB Ghofrani HA Gibbs JSR Gomberg-Maitland M McLaughlin VV et al . Effects of sotatercept on haemodynamics and right heart function: analysis of the STELLAR trial. Eur Respir J. (2023) 62:2301107. doi: 10.1183/13993003.01107-2023
137.
Kopeć G . Sotatercept as a next-generation therapy for pulmonary arterial hypertension: insights from the STELLAR trial. Cardiovasc Res. (2023) 119:e155–7. doi: 10.1093/cvr/cvad121
138.
Doggrell SA . Is sotatercept, which traps activins and growth differentiation factors, a new dawn in treating pulmonary arterial hypertension (PAH)?Expert Opin Biol Ther. (2023) 23:589–93. doi: 10.1080/14712598.2023.2221784
139.
Wang JJ Jin S Zhang H Xu Y Hu W Jiang Y et al . Molecular recognition and activation of the prostacyclin receptor by anti-pulmonary arterial hypertension drugs. Sci Adv. (2024) 10:eadk5184. doi: 10.1126/sciadv.adk5184
140.
Norlander AE Abney M Cephus JY Roe CE Irish JM Shelburne NJ et al . Prostaglandin I(2) therapy promotes regulatory T cell generation in patients with pulmonary arterial hypertension. Am J Respir Crit Care Med. (2023) 208:737–9. doi: 10.1164/rccm.202304-0716LE
141.
Kurosawa R Satoh K Kikuchi N Kikuchi H Saigusa D Al-Mamun ME et al . Identification of Celastramycin as a novel therapeutic agent for pulmonary arterial hypertension. Circ Res. (2019) 125:309–27. doi: 10.1161/CIRCRESAHA.119.315229
142.
Zolty R . Novel experimental therapies for treatment of pulmonary arterial hypertension. J Exp Pharmacol. (2021) 13:817–57. doi: 10.2147/JEP.S236743
143.
Hou P Chen H Liang S Guo W Zhao R Pan H et al . Targeting Fibroblast Activation Protein for Molecular Imaging of Fibrotic Remodeling in Pulmonary Arterial Hypertension. Journal of nuclear medicine: official publication. Society of Nuclear Medicine. (2025) 66:98–103. doi: 10.2967/jnumed.124.268376
144.
Zhang Y Tian X Chen L Zhao S Tang X Liu X et al . Huang, Endogenous hydrogen sulfide persulfidates endothelin type A receptor to inhibit pulmonary arterial smooth muscle cell proliferation. Redox Biology. (2025) 80:103493. doi: 10.1016/j.redox.2025.103493
145.
Shen H Gao Y Ge D Tan M Yin Q Wei TW et al . BRCC3 Regulation of ALK2 in Vascular Smooth Muscle Cells: Implication in Pulmonary Hypertension. Circulation. (2024) 150:132–150. doi: 10.1161/circulationaha.123.066430
146.
Harvey LD Alotaibi M Tai YY Tang Y Kim HJ Kelly X et al . Lysosomal dysfunction and inflammatory sterol metabolism in pulmonary arterial hypertension. Science. (2025) 387:eadn7277. doi: 10.1126/science.adn7277
147.
Hansmann G Chouvarine P Diekmann F Giera M Ralser M Mülleder M et al . Human umbilical cord mesenchymal stem cell-derived treatment of severe pulmonary arterial hypertension. Nat Cardiovas Res. (2022) 1:568–76. doi: 10.1038/s44161-022-00083-z
148.
Zhang Q Yaoita N Tabuchi A Liu S Chen SH Li Q et al . Endothelial heterogeneity in the response to autophagy drives small vessel muscularization in pulmonary hypertension. Circulation. (2024) 150:466–87. doi: 10.1161/CIRCULATIONAHA.124.068726
149.
Guignabert C Savale L Boucly A Thuillet R Tu L Ottaviani M et al . Serum and pulmonary expression profiles of the Activin signaling system in pulmonary arterial hypertension. Circulation. (2023) 147:1809–22. doi: 10.1161/CIRCULATIONAHA.122.061501
150.
van der Horst G van Bezooijen RL Deckers MM Hoogendam J Visser A Löwik CW et al . Differentiation of murine preosteoblastic KS483 cells depends on autocrine bone morphogenetic protein signaling during all phases of osteoblast formation. Bone. (2002) 31:661–9. doi: 10.1016/s8756-3282(02)00903-1 PMID:
151.
Van der Feen DE Kurakula K Tremblay E Boucherat O Bossers GPL Szulcek R et al . Multicenter preclinical validation of BET inhibition for the treatment of pulmonary arterial hypertension. Am J Respir Crit Care Med. (2019) 200:910–20. doi: 10.1164/rccm.201812-2275OC
152.
Zhang H Kan J Zhang J Xie D Li X Zhou W et al . 3-year outcome in patients with combined precapillary and Postcapillary pulmonary hypertension: results from PADN-5 trial. JACC Heart Failure. (2023) 11:1135–46. doi: 10.1016/j.jchf.2023.05.016
153.
Zhang H Zhang J Chen M Xie DJ Kan J Yu W et al . Pulmonary artery denervation significantly increases 6-min walk distance for patients with combined pre-and post-capillary pulmonary hypertension associated with left heart failure: the PADN-5 study. JACC Cardiovasc Interv. (2019) 12:274–84. doi: 10.1016/j.jcin.2018.09.021
154.
Chen SL Zhang FF Xu J Xie DJ Zhou L Nguyen T et al . Pulmonary artery denervation to treat pulmonary arterial hypertension: the single-center, prospective, first-in-man PADN-1 study (first-in-man pulmonary artery denervation for treatment of pulmonary artery hypertension). J Am Coll Cardiol. (2013) 62:1092–100. doi: 10.1016/j.jacc.2013.05.075
155.
Cai X Wei Z Shen Y Qian L Cai J Yang Y et al . Pulmonary artery denervation by noninvasive stereotactic radiotherapy: a pilot study in swine models of pulmonary hypertension. Nat Commun. (2025) 16:558. doi: 10.1038/s41467-025-55933-8
Summary
Keywords
combined pre-and postcapillary pulmonary hypertension (Cpc-PH), pathophysiological mechanisms, targeted therapy, current medical and surgical treatments, clinical trials, pulmonary vascular resistance (PVR)
Citation
Lu M, Wang B, Rong C, Wang Y and Zhang W (2025) Therapeutic challenges and new therapeutic targets for combined capillary pulmonary hypertension: a review. Front. Med. 12:1579112. doi: 10.3389/fmed.2025.1579112
Received
18 February 2025
Accepted
03 April 2025
Published
02 May 2025
Volume
12 - 2025
Edited by
Alessandra Cuomo, University of Naples Federico II, Italy
Reviewed by
Chidinma Ejikeme, Houston Methodist Hospital, United States
Sridevi Chigullapalli, Hospital and Research Centre, India
Updates
Copyright
© 2025 Lu, Wang, Rong, Wang and Zhang.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weihua Zhang, weihua@jlu.edu.cn
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.