Abstract
Alzheimer’s disease (AD) is the most frequent cause of dementia worldwide, and it is estimated that the number of patients will increase to 131 million by 2050. Most of the current methods of dealing with AD are designed to alleviate the symptoms, and there is no effective way of stopping the progression of the disease. Personalized immunotherapy has the potential to be highly effective and cut down on side effects because it can be targeted accurately and intervened early. Considering the genetic factors, many studies are increasingly looking at taking the immune status into account. This article further discusses the genetic and immune characteristics of AD, the methods of integrating multiple histological data, the identification of biomarkers, the stratification of patients, the precise treatment plans, and the application and future trends of immunotherapy, giving new directions for the future treatment of AD. In this mini-review, the authors address the critical role that genetic background and immune status play in shaping therapeutic strategies for AD, noting that there is a unique immune response in carriers of the APOEε4 allele compared to non-carriers, and that this difference may affect the course of the disease as well as the efficacy of immunotherapy. The aim of this review is to give an overview of the current understanding of the influence of genetic and immune factors on each other in AD, focusing on the impact of the APOEε4 allele on the immune response and its implications for immunotherapy.
1 Introduction
Alzheimer’s disease (AD) is the most common form of dementia, affecting approximately 47 million people worldwide, with projections indicating an increase to 131 million cases by 2050 (1–5). The growing prevalence of AD places a significant burden on healthcare systems and social support services, while also imposing substantial economic pressures (6). Core pathologic features of AD include the deposition of Aβ plaques in the brain and the hyperphosphorylation of tau protein in neurofibrillary tangles (7). Aβ is produced through abnormal cleavage by β- and γ-secretases, forming Aβ40 and Aβ42 monomers, which subsequently aggregate into amyloid plaques (8). These plaques, at high concentrations, trigger microglial infiltration and activation. However, microglia activation also plays a role in exacerbating neuronal injury through excessive immune responses and inflammation (9–12).
Current pharmacological treatments of AD are represented essentially by acetylcholinesterase inhibitors and the glutamate antagonist memantine (13). They inhibit acetylcholinesterase to reduce the breakdown rate of acetylcholine and therefore enhance central cholinergic neurotransmission (14). They may even exert an additional beneficial effect, such as delaying cognitive decline or improving functional activities in everyday life in the first year of treatment. However, they cannot impede the progress of the disease, and the cognitive functions continue to deteriorate rapidly after drug withdrawal (15–17). Besides, great variability concerning their efficacy is exhibited among different patients, and the risk of side effects associated with prolonged administration limits its long-term clinical application (18–20).
So far, immunotherapy aimed at controlling immune function has achieved striking successes in treating tumors, including but not limited to immune checkpoint inhibitors and CAR-T cell therapy (21–24). In AD, immunotherapy aims to stimulate the immune system to target and clear Aβ deposits, offering promise as a disease-modifying therapy (25). Personalized immunotherapy, in particular, holds significant potential for targeted precision, early intervention, and minimizing side effects (26–29). Early detection, facilitated by biomarker-based diagnostics, allows for timely interventions that may prevent or delay neuronal damage (30). Accordingly, tailored treatment plans in accordance with patient profiles, by adjusting dosages and treatment schedules, can ensure optimal immune response and reduction in side effects. This will definitely improve patient compliance and tolerance (31). By elucidation of the pathophysiological mechanism of AD, personalized immunotherapy will be incorporated into the benefits and innovations of genomics, proteomics, and other interdisciplinary disciplines that enable more appropriate and effective disease management (32). This review specifically examines the role of genetic and immune characteristics in AD, focusing on how the APOEε4 allele affects the immune response and disease progression, and we discuss how this knowledge can lead to the development of personalized immunotherapies that take into account individual genetic and immune differences, which can improve efficacy and cut down on adverse effects.
2 Genetic background and immune system in AD
APOE4 encoded by APOE ε4 is very different from APOE3 in lipid metabolism, resulting in disturbances of cholesterol transport and distribution in the brain, thereby affecting the stability and function of neuronal membranes (33). APOE4 is easier to bind with Aβ, promoting the aggregation and deposition of Aβ to form amyloid plaques, which is one of the typical pathologic features of AD. The interaction of APOE4 with mitochondria triggers mitochondrial dysfunction, which is manifested as increased mitochondrial calcium levels and increased reactive oxygen species generation, further aggravating energy metabolism disorders and oxidative stress and impairing neuronal survival (34). From the perspective of the immune response, compared with other APOE isoforms, APOE4 is more susceptible to proteolytic hydrolysis under stress conditions, and the generated products promote neurofibrillary tangle formation, affect the function of immune cells including microglia, and decrease the efficiency of Aβ clearance, thereby contributing to AD. APOE4 is able to translocate into the nucleus, where it drives the expression of genes involved in senescence, Aβ production, inflammation, and apoptosis, further exacerbating the pathologic changes in AD (35–39).
Familial Alzheimer’s Disease (FAD) is primarily caused by mutations in the APP, PSEN1, and PSEN2 genes, which encode proteins critical to Aβ generation and clearance (40, 41). Mutations in the APP gene result in the abnormal accumulation of Aβ following the cleavage of amyloid precursor protein by β- and γ-secretases at the cell membrane, thus promoting the formation of amyloid plaques (42, 43). The PSEN1 and PSEN2 genes encode the key components of the γ-secretase complex, and mutations in these genes lead to abnormal γ-secretase activity, increasing Aβ generation and deposition. Notably, mutations in the PSEN1 gene are the most common pathogenic factor in FAD (Figure 1) (44–47). While the occurrence of PSEN2 gene mutations is seldom observed to be causing AD, it functions much like PSEN1; that is, Aβ tends to accrue abnormally.
FIGURE 1

The APOEε4 allele disrupts lipid metabolism, promotes Aβ aggregation, impairs mitochondrial function, alters immune responses, and regulates gene expression. These changes further lead to the formation of amyloid plaques, oxidative stress, impairment in energy metabolism, chronic inflammation, and neuronal damage. Mutations within the APP, PSEN1, and PSEN2 genes promote abnormal Aβ production and γ-secretase activities, thus acting to further promote the amyloid plaque formation and neurodegeneration (by Figdraw).
Genes regulate the intensity of immune responses by influencing the development and differentiation of immune cells, as well as by modulating cellular signaling pathways (48–50). Studies suggest that the TREM2 gene, expressed in microglial cells, plays a crucial role in regulating their phagocytic function and inflammatory response. By contrast, PSEN1 mutations modulate γ-secretase activity and hence Aβ generation and immune cell activation (51–54). Some of them induce an inappropriate expression of immune markers and lead or maintain inflammatory responses. For example, APOE ε4 allele is associated with higher levels of inflammation markers such as IL-6 and TNF-α whereas mutations in TREM2 impact the inflammatory response in microglial cells (55–58).
AD involves a dual function of microglia and astrocytes in the pathologic process. These glial cells promote tissue repair and maintain neurological homeostasis through clearing of Aβ plaques and neurofibrillary tangles, secretion of anti-inflammatory cytokines such as IL-10 and TGF-β (59–61). On the contrary, over-activated microglia and astrocytes lead to the chronic inflammatory response that results from releasing a huge amount of pro-inflammatory cytokines such as TNF-α and IL-1β (62–64). These pro-inflammatory factors, while promoting further neuroinflammation, causing neuronal damage and death, promote Aβ production and Tau proteins over-phosphorylation via activating several signaling pathways such as NF-κB, thereby setting up a vicious circle (65, 66).
3 Integration of genetic and immune profiles
3.1 Integration methods of multi-omics data
In this respect, GWAS in genomics found genetic variants that are associated with AD and definitely confirmed that APOE ε4 is a major genetic risk of the disease. Besides regulating Aβ metabolism, these variants also enhance neuroinflammation by modulating the activity of immune cells (67–70). While transcriptomics offers information on dynamic gene expression changes, single-cell RNA sequencing has indeed enabled cell-type-specific investigation into AD-dependent shifts in gene expression at previously unparalleled resolution (71, 72). Indeed, microglia from AD brains have been found to express characteristic transcriptional phenotypes. It was noted that AD patients had a particular microglial transcriptional state in the brain that was strongly associated with inflammatory responses and lipid processing. Genomic and transcriptomic information is further complemented by proteomics, which examines protein expression and function, representing the end product of gene expression and their roles in diseased conditions (73, 74). Proteome-wide association studies integrate GWAS results with proteomic data to identify genes that influence AD risk by altering protein abundance. Bioinformatics tools and algorithms are thus very important for the integration of multi-omics data, enabling researchers to uncover more holistic insights into disease mechanisms (75). Weighted gene co-expression network analysis is one of the most popular methods used to identify gene modules associated with AD and their key hub genes. By constructing gene co-expression networks, researchers can pinpoint modules closely linked to disease progression and further explore their biological significance through functional annotation (76). Moreover, machine learning and artificial intelligence technologies have become crucial in the integration and analysis of multi-omics data (77). Approaches like deep learning can automatically identify patterns and trends from complex multi-omics datasets, enabling the discovery of novel biomarkers and the prediction of disease outcomes (Figure 2) (78).
FIGURE 2
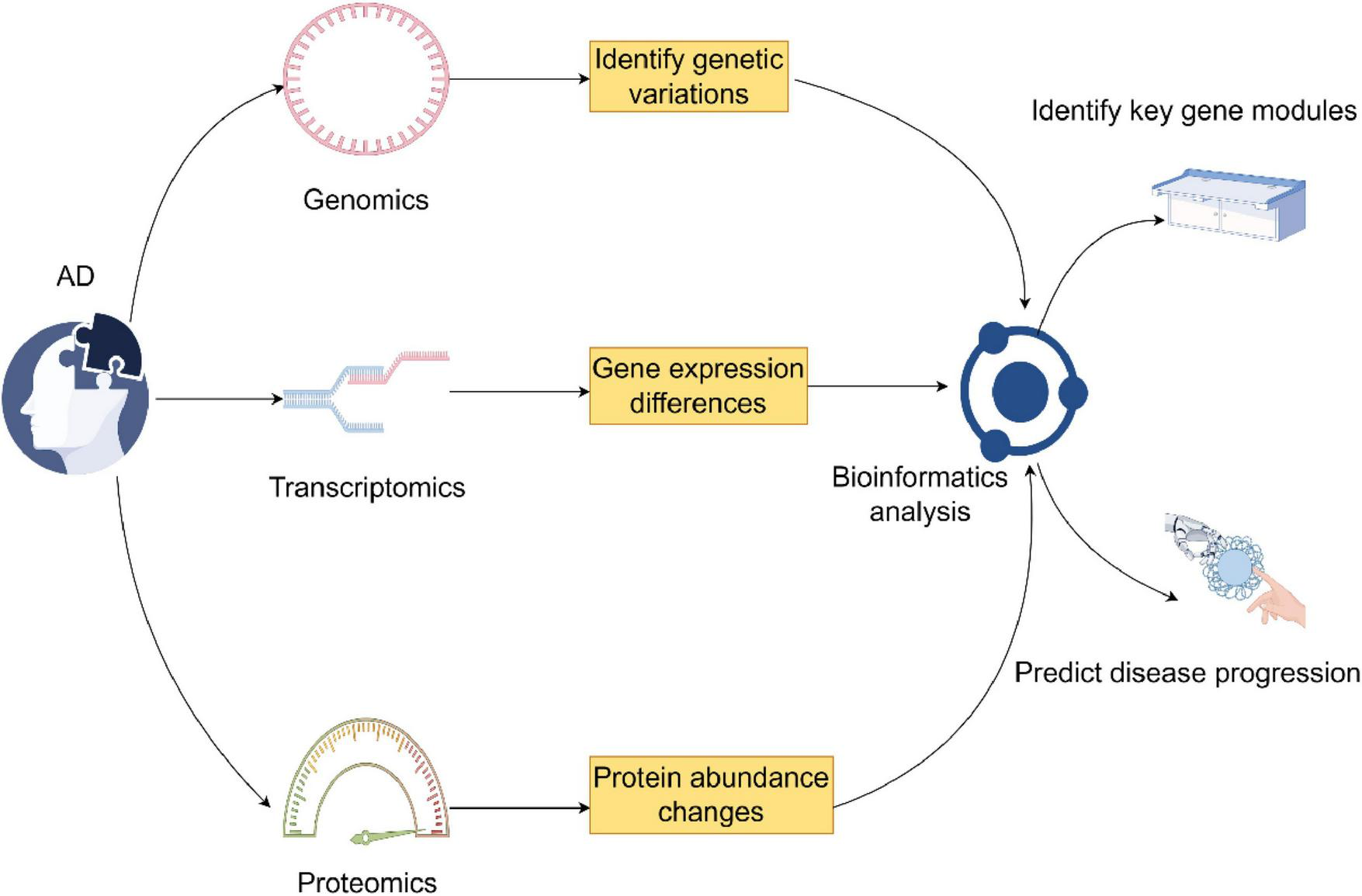
This figure combines genomics, transcriptomics, and proteomics in the investigation of Alzheimer’s disease. It identifies genetic variations through genomics, transcriptomics through differences in gene expression, and changes in protein abundance through proteomics. Bioinformatics analysis links them in the identification of the key gene modules and prediction of the disease outcome (by Figdraw).
3.2 Limitations of current omics technologies and barriers to implementation in clinics
Even with the significant development of multi-omics technologies, there are still some limitations and obstacles to their implementation in the clinical setting, one of which is the high cost and complexity of multi-omics technologies, which may affect their large-scale application in normal clinical practice (79, 80). Single-cell RNA sequencing, for example, contains a huge amount of information but requires specialized instrumentation and expertise, making it difficult to perform in small clinical laboratories. Furthermore, the sheer volume of data generated by these technologies requires sophisticated bioinformatics and computing resources, which are not readily available in all clinical settings (81). Another shortcoming is the lack of a standardized process for data collection, processing, and analysis, which can lead to different or even conflicting results from different studies or laboratories (82). Moreover, because of differences in data types, formats, and quality, fusion of multi-omics data from different sources may be challenging, requiring the creation of robust and flexible bioinformatics programs to ensure accurate and reliable data fusion (83). Translating the new insights from genomics into clinically feasible insights remains a major problem. Biological systems are complex and AD is highly variable, and it is difficult to identify clear and actionable biomarkers that can be used to make diagnoses, predict disease progression, and determine therapeutic response (84). The most common biomarkers are those that can be used for diagnostic purposes and that can be used in a clinical setting. Overcoming these limitations will require researchers, clinicians, and bioinformaticians to work together to develop more accessible, standardized, and clinically relevant genomic techniques and data analysis tools (85).
3.3 Identification and validation of biomarkers
High-throughput genomics technologies have made it possible to identify genes associated with AD and their expression pattern (86). Machine learning algorithms enhance the efficiency and accuracy of the analysis of gene data. By using machine learning algorithms like support vector machines and random forests, predictive models could be developed which could differentiate AD patients from healthy controls based on their sEV profiles (87–90). The immune system is a very important player in the pathology of AD. Inflammation markers, like sTREM2 and YKL-40, have been reported to be increased in AD patients and serve as biomarkers of neuroinflammation (91). Moreover, immune cell activation, especially microglia, is highly related to AD development, and proteins involved in immune signaling pathways can also be used as potential biomarkers. Immunomics analysis combined with machine learning methods could more effectively screen and validate immune-related biomarkers (92, 93). Further, it has been shown that plasma levels of p-tau181 provide high diagnostic accuracy for the early stages of AD (94, 95). Further sensitivity and specificity can be obtained by using an integrated diagnostic model that combines multiple biomarkers with clinical data. Such a model will help not only in the early identification of AD patients but also form a basis for developing personalized treatment plans, ultimately improving the prognosis and quality of life of patients (96, 97).
3.4 Patient stratification and precision therapy
APOE ε4 allele is the most important genetic risk factor for AD, and the patients carrying this allele have a higher incidence and faster development of the disease (98). The identification of such genetic alterations allows the division of patients according to different genetic subtypes and provides a rationale for personalized treatment approaches (99, 100). Inflammatory responses have been identified as one of the early features of AD and are closely related to disease progression. According to the immune cell types and levels of inflammatory factors, patients can be divided into different immune subtypes (101). High levels of pro-inflammatory factors, including IL-1β and TNF-α, indicate an active inflammatory process. Higher levels of anti-inflammatory factors, such as IL-10, reflect the patient’s immune regulation status (102). In patients with high-risk genetic mutations, lifestyle intervention, cognitive training, and other approaches may delay disease onset and progression. Personalized treatment plans should take both the patient’s genetic and immune profiles into account, often employing multi-target drug combination therapies (103). Given the long and complex course of AD, patient conditions change over time (104), necessitating dynamic adjustments to treatment plans based on ongoing assessments. Regular monitoring of genetic and immune biomarkers can provide critical insights into treatment efficacy and disease progression (105). Personalized treatment means the precise adjustment of medication doses and methods according to personal genetic background and medication metabolism capability, therefore (106).
3.5 Structural variability of Aβ and tau aggregates and its implications for personalized immunotherapy
Recent studies have shown that Aβ plaques and tau protein aggregates in the brains of AD patients have distinct individual and subtype-specific structural differences, a phenomenon known as “strain.” This conformational diversity alters the pathogenicity and spread of aggregates in the brain, and this structural change can make biomarker detection extremely difficult, as conventional diagnostic tests may not detect all forms of aggregates (107). From an immunotherapeutic perspective, antibodies targeting a specific conformation may not be effective in all patients, even to the extent that they do not work at all in those with different aggregation types (108). Therefore, future personalized immunotherapy strategies will have to take into account the biophysical properties of pathological aggregates, perhaps in combination with specific antibodies, aggregate sequencing technologies, and aggregate resolving imaging to achieve a higher degree of accuracy, which will also further subdivide patient stratification, thus driving the development of more effective and personalized therapies (109).
4 Current status and prospects of immunotherapy in AD
4.1 Types and mechanisms of immunotherapeutic approaches
Before elaborating on the types of immunotherapy for AD, it is important to clarify the fundamental differences between active and passive immunotherapy. Active immunotherapy relies on triggering the patient’s own immune system to recognize and fight against specific antigens, and this type of therapy often involves the use of vaccines or other medications that can trigger an immune response. Passive immunotherapy, on the other hand, refers to the injection of pre-prepared antibodies or other immune components directly into the patient, without the need for a response from the patient’s own immune system (110). These antibodies bind to and neutralize pathogenic proteins to reduce their harmful effects, and both approaches are aimed at improving the progression of AD, but they differ greatly in their principles of operation and possible side effects (111). The most important difference between the two approaches is that they have been shown to be effective in improving the progression of AD. Passive immunotherapy accelerates the process of pathologic protein removal through injecting certain monoclonal antibodies directly into a patient’s body. Aducanumab, Lecanemab, and Donanemab were permitted by the U.S. FDA for treating early AD (112). These antibodies delay cognitive decline by binding Aβ and promoting the removal of Aβ. Some adverse reactions, including brain edema and microhemorrhages, have been developed in clinical trials. Therefore, structural optimization and delivering strategy development to optimize safety and efficacy will probably be a more critical direction of the antibody-related therapy in future research (113).
Active immunotherapy, in contrast, elicits an immune response from the body by the administration of antigens or vaccines, which then clears the pathological proteins. Early research involving Aβ vaccines seemed promising, as vaccination in animal models led to a decrease in Aβ deposition and improved cognitive function. However, early clinical trials in humans were halted because some patients developed meningoencephalitis. With improved vaccine design and adjuvant selection, incidences of such side effects have been relatively rare in recent years (114). Several studies have demonstrated that vaccination against Aβ deposition, using antigens composed of non-Aβ peptides folded into a conformation similar to that of Aβ oligomers, effectively reduces Aβ deposition and improves cognitive function. Meanwhile, active immunotherapies do face some other major problems in clinical practices, including individual variation within immune responses, and the major call of inducing long-term immunity for many diseases (115, 116). Tau pathology is one of the key drivers of neurodegenerative, cognitive decline in AD, and an increasing number of studies are demonstrating the importance of tau pathology. Several monoclonal antibodies (semorinemab, gosuranemab, zagotenemab) have entered clinical trials that attempt to target extracellular tau proteins, but with mixed results (117). The results of this study have been reported in a number of studies. It is important to note that tau vaccines and antisense oligonucleotides (e.g., BIIB080) have the potential to reduce pathologic tau burden and slow cognitive decline in early trials, and other investigational treatments such as synaptic modulators, neuroprotectants, metabolic modulators, and anti-inflammatory drugs are changing the landscape of AD treatment (118). The current study of tau vaccines and antisense oligonucleotides has shown that they are effective in reducing pathologic tau burden.
Cell therapies belong to a new direction in the field of AD treatment with great potential, involving the transplantation of certain cells with the aim of strengthening the immune response or repairing damaged neural tissues, the two main types of cell therapies undergoing research are listed below: CAR-T cell therapy involves engineering the patient’s T cells so that these T cells exhibit chimeric antigen receptors that specifically recognize and destroy cells bearing disease-associated antigens (Figure 3). In the setting of AD, CAR-T cells can be programmed to specifically target Aβ or tau proteins, and CAR-T therapies have had significant success in cancer treatment, although their use in AD is still in the early stages of research. Clinical studies have shown that CAR-T cells shrink Aβ plaques and optimize cognitive function in animal models (119) (Table 1). iPSCs can differentiate into a variety of cell types containing neurons and glial cells, which can be used as cellular replacement therapies, and iPSC-derived cells give the possibility of replacing damaged neurons and supporting neurologic repair. Numerous studies have shown that iPSC-derived neurons can be incorporated into the brain and improve cognitive function in animal models of AD to judge the safety and efficacy of iPSC-derived cell therapies in AD patients (120, 121).
FIGURE 3

The mechanisms of three immunotherapies for AD. Passive Immunotherapy involves the direct administration of monoclonal antibodies to bind with Aβ for clearance, which may cause side effects such as edema and microhemorrhage. Active Immunotherapy stimulates the immune system through vaccination to produce antibodies that clear Aβ deposits, requiring optimized antigen design to reduce side effects. Cell Therapy uses stem or immune cell transplantation to repair damaged neural tissue or clear pathological proteins, although its clinical application is still in early stages. All three therapies target Aβ to slow the pathological progression of AD.
TABLE 1
| Therapy type | Mechanism description | Clinical application and challenges | References |
| Passive immunotherapy | Injection of monoclonal antibodiesto clear pathological proteins. | Approved for early AD treatment, optimization needed for safety. | (112) |
| Active immunotherapy (Vaccine) | Injection of antigens or vaccines to stimulate immune response. | Improved to reduce side effects, individual differences and immune memory maintenance are still challenges. | (114) |
| Cell therapy | Transplantation of immune cells or stem cells to repair neural tissue. | In early stages, further validation of safety and efficacy is required. | (122) |
Therapeutic strategies for AD.
4.2 Review and analysis of clinical trials
Clinical Evaluation of Aducanumab: Primarily Two Phase III Studies-EMERGE and ENGAGE Both of these studies were designed as a randomized, double-blind, placebo-controlled, multicenter trials to evaluate the efficacy and safety of Aducanumab in early AD patients. The EMERGE and ENGAGE trials were designed to enroll thousands of patients with mild cognitive impairment and mild AD, respectively, for 78-week-long studies using variable dosing of Aducanumab for treatment, including high dosing at 10 mg/kg (123). In the EMERGE study, the high-dose Aducanumab group demonstrated a 30% reduction in the Clinical Dementia Rating-Sum of Boxes (CDR-SB) score at 78 weeks, a clinically significant symptom benefit. However, the ENGAGE trial failed to achieve the difference in the CDR-SB score statistically, comparing Aducanumab treatment and the placebo group. Potential causes may come from various aspects such as patients with baseline characteristics, different disease stages, and a progressive condition or even dosages during the treatment (124). The results of the EMERGE and ENGAGE trials have been the subject of much debate, particularly with regard to the U.S. Food and Drug Administration’s (FDA) granting of accelerated approval for Aducanumab, a pathway that permits approval of a drug on the basis of surrogate endpoints that may be indicative of clinical benefit, and which on the one hand is widely lauded for granting access to potential therapeutic treatments. On the other hand, it has been criticized for perhaps curtailing standards for drug efficacy and safety (125). Differences in the results of the EMERGE and ENGAGE trials have led to an enduring debate about the reliability of the accelerated approval process, as well as the need for more stringent post-marketing surveillance of Aducanumab’s long-term safety and efficacy (126). The results of the EMERGE and ENGAGE trials were not the same as those of the ENGAGE trial, which led to an enduring debate about the reliability of the accelerated approval process.
The Phase III clinical trial, Clarity AD, of lecanemab, in a double-blind randomized placebo-controlled fashion in approximately 2,000 patients with early-stage AD, was conducted. This single trial was designed to assess the effects of Lecanemab on cognitive function and Aβ deposition, with a fixed-dose, 18-month duration of treatment (127). The results of the Clarity AD trial revealed that Lecanemab was able to slow the rate of cognitive and functional decline by 27%, while significantly reducing Aβ levels in patients. Furthermore, lecanemab showed statistically significant benefit for three alternate cognition and function measures. Clinical benefits for Lecanemab in slowing the course of AD would therefore seem possible from these results (128).
The success of Lecanemab may be due to precision targeting and improved clinical trial design, where Lecanemab selectively neutralizes soluble and toxic Aβ aggregates, cuts down on non-specific binding, and reduces side effects, as well as a rigorous patient selection and stratification strategy that ensures credible trial results, factors that contributed to the success of Lecanemab in clinical trials (129). In contrast, the inconsistent results of the Aducanumab trial have generated much debate, and have demonstrated the importance of personalized treatment planning and rigorous trial design to prevent inconsistent efficacy due to patient heterogeneity, and that future clinical trials should incorporate genetic and immune profiling to better predict patient response and improve treatment outcomes (130).
4.3 Strategies and prospects of personalized immunotherapy
These advances in genomics have, in one way, helped to identify specific genetic variants linked with AD and have thus allowed the design of targeted approaches for treatment. In the case of genetic mutations in Aβ and tau proteins, specific small molecules or monoclonal antibodies can be developed. Moreover, new gene-editing technologies such as CRISPR/Cas9 offer new possibilities in the field of personalized gene therapy by allowing the direct repair or replacement of pathogenic genes. This approach not only increases the specificity of treatment but also reduces the destruction of normal cells, giving the patients more precise therapeutic options (131–134). Development of immune-modulating drugs is another important part of personalized immunotherapy. Very recently, immune checkpoint inhibitors including PD-1/PD-L1 and CTLA-4 inhibitors have shown tremendous achievements in several diseases. By modulating the immune response against Aβ and tau proteins, these drugs improve pathological protein clearance (135). Relevant studies have demonstrated that personalized nanovaccines, designed using tumor cell membrane vesicles from a patient, induce immune responses against patient-specific antigens. Such an approach enhances the immunogenicity and specificity of the vaccine, hence offering more effective options for the treatment of patients (136–138). Case studies related to integrated personalized strategies have pointed out that collaborative efforts and data sharing between disciplines hold immense potential to drive the development of personalized immunotherapies further. These have potential in the elucidation of AD with integrated genomic, proteomic data, and clinical information for personalized patient treatment options (139, 140). Establishment of a data-sharing platform: Clinical trial data, biomarkers, response to treatments-all this information can be shared on the data-sharing platform. This will help hasten the further development and validation of new therapies (141–143) (Figure 4). Further acceleration can be achieved by developing and validating new therapies through integration of patient genomic, proteomic data, and clinical information.
FIGURE 4
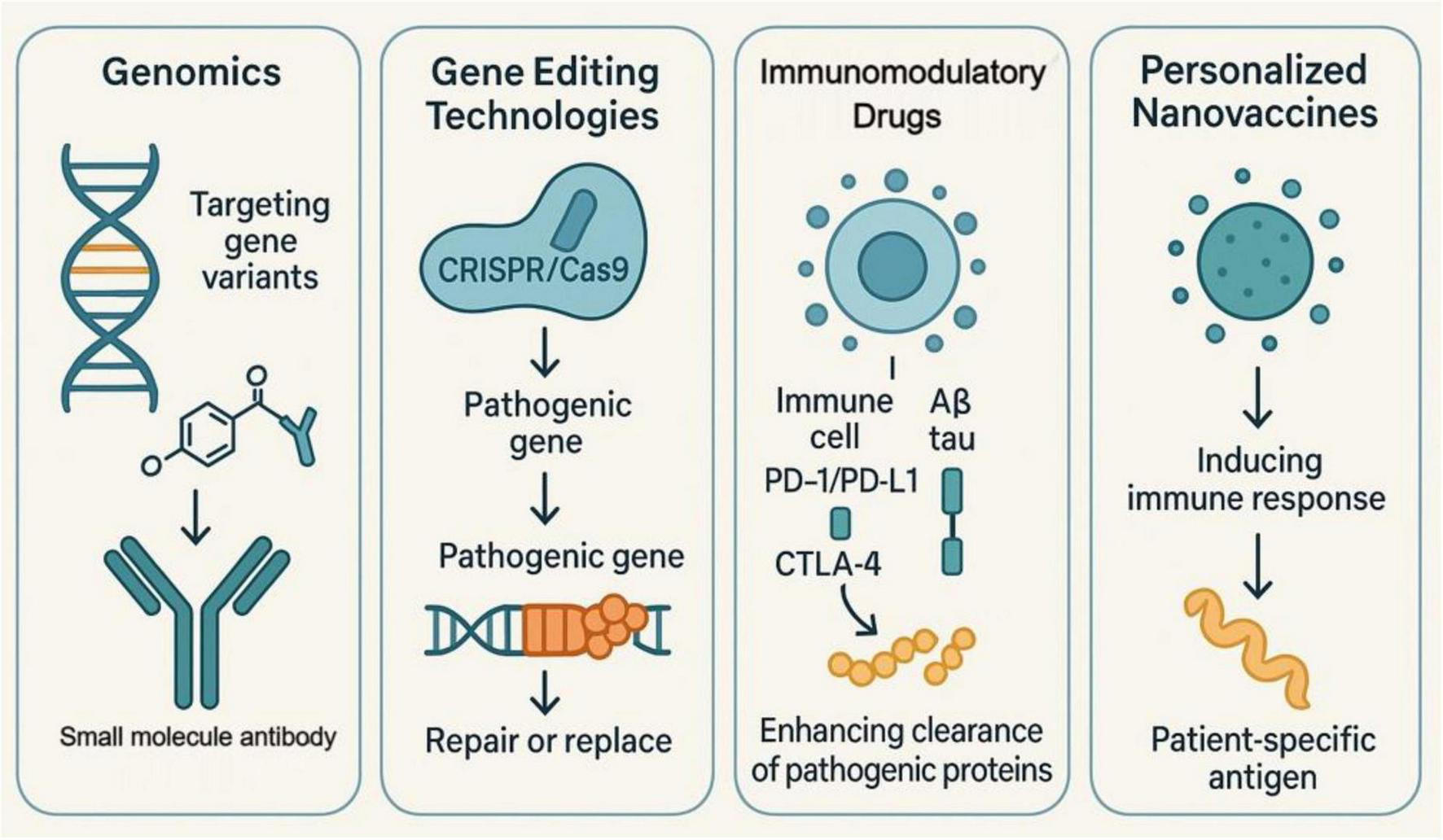
Four mechanisms of personalized immunotherapy in AD treatment. Genomics involves identifying gene variants associated with AD to design small molecule antibodies targeting Aβ and tau proteins. Gene editing technologies such as CRISPR/Cas9 enhance precision by repairing or replacing pathogenic genes, thereby reducing side effects. Immunomodulatory drugs, including PD-1/PD-L1 and CTLA-4 inhibitors, modulate the immune system’s response to pathological proteins, enhancing their clearance. Personalized nanovaccines induce an immune response using patient-specific antigens, improving the vaccine’s immunogenicity and specificity.
5 Conclusion and perspectives
In recent years, individualized immunotherapy has slowly become a key part of AD treatment. Individualized protocols based on each patient’s unique immune status and genetic profile can improve treatment efficiency and reduce side effects. Genetic situation and immune status play a very crucial role in the integration of AD salvage, and patients who carry the APOEε4 allele have a different immune response than non-carriers. Patient’s immune status also alters the efficacy of immune salvage. To better understand the mechanism of individualized strategies for AD immune salvage and to improve the salvage plan, there is an urgent need to explore in depth the effects of different genotypes and immune statuses on immune salvage response, and to combine the latest bioinformatics analysis techniques and AI algorithms, which are useful to facilitate the creation of accurate stratification and salvage prognostic models. Clinical trial design should be more individualized, and multicenter, large-scale clinical studies can be used to re-validate whether individualized immunotherapy is safe and effective. Regarding the future direction of research, we propose to carry out some specific actions, such as increasing the integration of multi-omics in clinical trials, forming biomarker clusters, adopting artificial intelligence and machine learning technologies, focusing on patient categorization and personalized trial design, implementing longitudinal observation and adopting adaptive trial design, and creating a platform for data sharing. It is also necessary to strengthen the search for basic research on genes and immune mechanisms, so as to give stronger theoretical support to the development of immunotherapy, and to pay attention to the study of the genetic backgrounds and immune characteristics of different races, so as to ensure the universality and usability of therapeutic strategies. In drug research and development, attention should be paid to the design and improvement of multi-target drugs to achieve all-round therapeutic results. In clinical practice, a long-term follow-up mechanism should be created to judge the long-term safety and efficacy of immunotherapy, thereby improving the quality of life of Alzheimer’s disease patients. For future research directions, we will pay special attention to the integration of multi-omics technology in clinical trials. Researchers should consider comprehensive multi-omics analysis as a standard procedure in clinical trials, including genomics, transcriptomics, proteomics and metabolomics, etc., so as to fully grasp the mechanism of disease and reflect the differences in the responses of individuals to treatments. Moreover, the integration of multi-omics data is useful for identifying new biomarkers and predicting disease outcomes more accurately, and the creation of multi-omics databases and data sharing platforms will enhance collaboration among researchers and accelerate the translation of research results into the clinic.
Statements
Author contributions
CH: Writing – original draft, Conceptualization, Data curation. YS: Data curation, Writing – review & editing. MZ: Writing – review & editing, Data curation. XZ: Conceptualization, Funding acquisition, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by the Shenzhen Municipal Science and Technology Innovation Commission (No. JCYJ20210324122210028) and the National Natural Science Foundation of China: (No. 82174509).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1.
Global Nutrition Target Collaborators. Global, regional, and national progress towards the 2030 global nutrition targets and forecasts to 2050: A systematic analysis for the global burden of disease study 2021. Lancet. (2025) 404:2543–83. 10.1016/S0140-673601821-X
2.
Synnott P Majda T Lin P Ollendorf D Zhu Y Kowal S . Modeling the population equity of alzheimer disease treatments in the US.JAMA Netw Open. (2024) 7:e2442353. 10.1001/jamanetworkopen.2024.42353
3.
Safiri S Ghaffari Jolfayi A Fazlollahi A Morsali S Sarkesh A Daei Sorkhabi A et al Alzheimer’s disease: A comprehensive review of epidemiology, risk factors, symptoms diagnosis, management, caregiving, advanced treatments and associated challenges. Front Med (Lausanne). (2024) 11:1474043. 10.3389/fmed.2024.1474043
4.
Lennon M Lipnicki D Lam B Crawford J Schutte A Peters R et al Blood pressure, antihypertensive use, and late-life Alzheimer and non-Alzheimer dementia risk: An individual participant data meta-analysis. Neurology. (2024) 103:e209715. 10.1212/WNL.0000000000209715
5.
Landeiro F Harris C Groves D O’Neill S Jandu K Tacconi E et al The economic burden of cancer, coronary heart disease, dementia, and stroke in england in 2018, with projection to 2050: An evaluation of two cohort studies. Lancet Healthy Longev. (2024) 5:e514–23. 10.1016/S2666-756800108-9
6.
GBD 2021 Us Burden of Disease Collaborators. The burden of diseases, injuries, and risk factors by state in the USA, 1990-2021: A systematic analysis for the global burden of disease study 2021. Lancet. (2024) 404:2314–40. 10.1016/S0140-673601446-6
7.
Knecht L Dalsbol K Simonsen A Pilchner F Ross J Winge K et al Autoantibody profiles in alzheimer s, parkinson s, and dementia with Lewy bodies: Altered igg affinity and Igg/Igm/Iga Responses to alpha-synuclein, amyloid-beta, and tau in disease-specific pathological patterns. J Neuroinflamm. (2024) 21:317. 10.1186/s12974-024-03293-3
8.
Mroke P Goit R Rizwan M Tariq S Rizwan A Umer M et al Implications of the Gut microbiome in Alzheimer’s disease: A narrative review. Cureus. (2024) 16:e73681. 10.7759/cureus.73681
9.
Zhang Z Bai J Zhang S Wang R Zhu S Li T et al The relationship between alzheimer’s disease and pyroptosis and the intervention progress of traditional chinese medicine. Int J Gen Med. (2024) 17:4723–32. 10.2147/IJGM.S478479
10.
Wang W Gong Z Wang Y Zhao Y Lu Y Sun R et al Mutant Notch3ecd triggers defects in mitochondrial function and mitophagy in cadasil cell models. J Alzheimers Dis. (2024) 100:1299–314. 10.3233/JAD-240273
11.
Wu S Chen N Wang C . Frontiers and hotspots evolution in anti-inflammatory studies for Alzheimer’s disease.Behav Brain Res. (2024) 472:115178. 10.1016/j.bbr.2024.115178
12.
Liu P Liu X Ren M Liu X Shi X Li M et al Neuronal cathepsin s increases neuroinflammation and causes cognitive decline Via Cx3cl1-Cx3cr1 axis and Jak2-Stat3 pathway in aging and Alzheimer’s disease. Aging Cell. (2024) 24:e14393. 10.1111/acel.14393
13.
Gutierre R Rocha P Graciani A Coppi A Arida R . Tau, amyloid, iron, oligodendrocytes ferroptosis, and inflammaging in the hippocampal formation of aged rats submitted to an aerobic exercise program.Brain Res. (2024) 1850:149419. 10.1016/j.brainres.2024.149419
14.
Kang J Khatib L Heston M Dilmore A Labus J Deming Y et al Gut microbiome compositional and functional features associate with Alzheimer’s disease pathology. medRxiv [Preprint]. (2024). 10.1101/2024.09.04.24313004
15.
Alfaro-Ruiz R Martin-Belmonte A Aguado C Moreno-Martinez A Fukazawa Y Lujan R . Selective disruption of synaptic NMDA receptors of the hippocampal trisynaptic circuit in abeta pathology.Biol Res. (2024) 57:56. 10.1186/s40659-024-00537-7
16.
Ali J Choe K Park J Park H Kang H Park T et al The interplay of protein aggregation, genetics, and oxidative stress in Alzheimer’s disease: Role for natural antioxidants and immunotherapeutics. Antioxidants (Basel). (2024) 13:862. 10.3390/antiox13070862
17.
Wang L Li W Wu W Liu Q You M Liu X et al Effects of electroacupuncture on microglia phenotype and epigenetic modulation of C/Ebpbeta in Samp8 mice. Brain Res. (2024) 1849:149339. 10.1016/j.brainres.2024.149339
18.
Haessler A Candlish M Hefendehl J Jung N Windbergs M . Mapping cellular stress and lipid dysregulation in Alzheimer-related progressive neurodegeneration using label-free raman microscopy.Commun Biol. (2024) 7:1514. 10.1038/s42003-024-07182-6
19.
Chilton P Ghare S Charpentier B Myers S Rao A Petrosino J et al Age-associated temporal decline in butyrate-producing bacteria plays a key pathogenic role in the onset and progression of neuropathology and memory deficits in 3xtg-Ad mice. Gut Microbes. (2024) 16:2389319. 10.1080/19490976.2024.2389319
20.
Qian X Ding G Chen S Liu X Zhang M Tang H . Blood cathepsins on the risk of Alzheimer’s disease and related pathological biomarkers: Results from observational Cohort and Mendelian randomization study.J Prev Alzheimers Dis. (2024) 11:1834–42. 10.14283/jpad.2024.107
21.
Vida H Sahar M Nikdouz A Arezoo H . Chemokines in neurodegenerative diseases.Immunol Cell Biol. (2024) 103:275–92. 10.1111/imcb.12843
22.
Luo X Luo B Fei L Zhang Q Liang X Chen Y et al Ms4a superfamily molecules in tumors, Alzheimer’s and autoimmune diseases. Front Immunol. (2024) 15:1481494. 10.3389/fimmu.2024.1481494
23.
Hammers D Eloyan A Thangarajah M Taurone A Beckett L Gao S et al Differences in baseline cognitive performance between participants with early-onset and late-onset Alzheimer’s disease: Comparison of leads and Adni. Alzheimers Dement. (2024) 21:e14218. 10.1002/alz.14218
24.
Alqabandi J David R Abdel-Motal U ElAbd R Youcef-Toumi K . An innovative cellular medicine approach via the utilization of novel nanotechnology-based biomechatronic platforms as a label-free biomarker for early melanoma diagnosis.Sci Rep. (2024) 14:30107. 10.1038/s41598-024-79154-z
25.
Hrybouski S Das S Xie L Brown C Flamporis M Lane J et al Bold amplitude correlates of preclinical Alzheimer’s disease. medRxiv [Preprint]. (2024). 10.1101/2024.10.27.24316243
26.
Heneka M Morgan D Jessen F . Passive anti-amyloid beta immunotherapy in Alzheimer’s disease-opportunities and challenges.Lancet. (2024) 404:2198–208. 10.1016/S0140-673601883-X
27.
Zou Y Wang C Li H Zhong M Lin J Hu Y et al Epileptic seizures induced by pentylenetetrazole kindling accelerate Alzheimer-like neuropathology in 5xfad mice. Front Pharmacol. (2024) 15:1500105. 10.3389/fphar.2024.1500105
28.
Chen Y Qi Y Hu Y Qiu X Qiu T Li S et al Integrated cerebellar radiomic-network model for predicting mild cognitive impairment in Alzheimer’s disease. Alzheimers Dement. (2024) 21:e14361. 10.1002/alz.14361
29.
Aisen P Bateman R Crowther D Cummings J Dwyer J Iwatsubo T et al The case for regulatory approval of amyloid-lowering immunotherapies in Alzheimer’s disease based on clearcut biomarker evidence. Alzheimers Dement. (2024) 21:e14342. 10.1002/alz.14342
30.
Nosheny R Miller M Conti C Flenniken D Ashford M Diaz A et al The Adni administrative core: Ensuring Adni’s success and informing future ad clinical trials. Alzheimers Dement. (2024) 20:9004–13. 10.1002/alz.14311
31.
Menendez-Gonzalez M . Therapeutic challenges derived from the interaction among apolipoprotein E, cholesterol, and amyloid in Alzheimer’s disease.Int J Mol Sci. (2024) 25:2029. 10.3390/Ijms252212029
32.
Carracedo S Launay A Dechelle-Marquet P Faivre E Blum D Delarasse C et al Purinergic-associated immune responses in neurodegenerative diseases. Prog Neurobiol. (2024) 243:102693. 10.1016/j.pneurobio.2024.102693
33.
Qu L Xu S Lan Z Fang S Xu Y Zhu X . Apolipoprotein E in Alzheimer’s disease: Focus on synaptic function and therapeutic strategy.Mol Neurobiol. (2024) 62:3040–52. 10.1007/s12035-024-04449-1
34.
Zhang L Xia Y Gui Y . Neuronal Apoe4 in Alzheimer’s disease and potential therapeutic targets.Front Aging Neurosci. (2023) 15:1199434. 10.3389/fnagi.2023.1199434
35.
Sun Y Wang Z Huang H . Roles of Apoe4 on the pathogenesis in Alzheimer’s disease and the potential therapeutic approaches.Cell Mol Neurobiol. (2023) 43:3115–36. 10.1007/s10571-023-01365-1
36.
de Oliveira F . The problem of multiple adjustments in the assessment of minimal clinically important differences.Alzheimers Dement (NY). (2025) 11:e70032. 10.1002/trc2.70032
37.
Kannan L Lelo de Larrea-Mancera ES Maniglia M Vodyanyk MM Gallun FJ Jaeggi SM et al Multidimensional Relationships between sensory perception and cognitive aging. Front Aging Neurosci. (2024) 16:1484494. 10.3389/fnagi.2024.1484494
38.
Sabouri S Rostamirad M Dempski R . Unlocking the Brain’s zinc code: Implications for cognitive function and disease.Front Biophys. (2024) 2:1406868. 10.3389/frbis.2024.1406868
39.
Xing X Liu H Zhang M Li Y . Mapping the current trends and hotspots of extracellular vesicles in Alzheimer’s disease: A bibliometric analysis.Front Aging Neurosci. (2024) 16:1485750. 10.3389/fnagi.2024.1485750
40.
Valdes P Caldwell A Liu Q Fitzgerald M Ramachandran S Karch C et al Integrative multiomics reveals common endotypes across psen1, psen2, and app mutations in familial Alzheimer’s disease. Alzheimers Res Ther. (2025) 17:5. 10.1186/s13195-024-01659-6
41.
Xiao X Liu H Liu X Zhang W Zhang S Jiao B . App, Psen1, and Psen2 variants in Alzheimer’s disease: Systematic re-evaluation according to ACMG guidelines.Front Aging Neurosci. (2021) 13:695808. 10.3389/fnagi.2021.695808
42.
Mullan M Crawford F Buchanan J . technical feasibility of genetic testing for Alzheimer’s disease.Alzheimer Dis Assoc Disord. (1994) 8:102–15. 10.1097/00002093-199408020-00007
43.
Hutton M Hardy J . The presenilins and Alzheimer’s disease.Hum Mol Genet. (1997) 6:1639–46. 10.1093/hmg/6.10.1639
44.
Gao L Zhang Y Sterling K Song W . Brain-derived neurotrophic factor in Alzheimer’s disease and its pharmaceutical potential.Transl Neurodegener. (2022) 11:4. 10.1186/s40035-022-00279-0
45.
Weninger S Irizarry M Fleisher A Leon T Maruff P Miller D et al Alzheimer’s disease drug development in an evolving therapeutic landscape. Alzheimers Dement (NY). (2024) 10:e70015. 10.1002/trc2.70015
46.
Dhakal U Briceno E Sharma U Bogati U Sharma A Shrestha L et al Health Care systems and policies for older adults in nepal: New challenges for a low-middle income country. Discov Public Health. (2024) 21:256. 10.1186/s12982-024-00382-9
47.
Ng T Udeh-Momoh C Lim M Gleerup H Leifert W Ajalo C et al Guidelines for the standardization of pre-analytical variables for salivary biomarker studies in Alzheimer’s disease research: An updated review and consensus of the salivary biomarkers for dementia research working group. Alzheimers Dement. (2024) 21:e14420. 10.1002/alz.14420
48.
Zgorzynska E . Trem2 in Alzheimer’s disease: Structure, function, therapeutic prospects, and activation challenges.Mol Cell Neurosci. (2024) 128:103917. 10.1016/j.mcn.2024.103917
49.
Wijesinghe P Li H Ai Z Campbell M Chen S Xi J et al Apolipoprotein E dysfunction in Alzheimer’s disease: A study on miRNA regulation, glial markers, and amyloid pathology. Front Aging Neurosci. (2024) 16:1495615. 10.3389/fnagi.2024.1495615
50.
Wang S Xi J Zhang M Wang J . Identification of potential therapeutic targets for Alzheimer’s disease from the proteomes of plasma and cerebrospinal fluid in a multicenter Mendelian randomization study.Int J Biol Macromol. (2025) 294:139394. 10.1016/j.ijbiomac.2024.139394
51.
Roveta F Bonino L Piella E Rainero I Rubino E . Neuroinflammatory biomarkers in Alzheimer’s disease: From pathophysiology to clinical implications.Int J Mol Sci. (2024) 25:11941. 10.3390/ijms252211941
52.
Zhao Y Guo Q Tian J Liu W Wang X . Trem2 bridges microglia and extracellular microenvironment: Mechanistic landscape and therapeutical prospects on Alzheimer’s disease.Ageing Res Rev. (2025) 103:102596. 10.1016/j.arr.2024.102596
53.
Yu Z Liu J Liu Z Liu X Tuo J Li J et al Roles of blood monocytes carrying Trem2(R47h) mutation in pathogenesis of Alzheimer’s Disease and its therapeutic potential in App/Ps1 MICE. Alzheimers Dement. (2024) 21:e14402. 10.1002/alz.14402
54.
Beydoun M Beydoun H Li Z Hu Y Noren Hooten N Ding J et al Alzheimer’s disease polygenic risk, the plasma proteome, and dementia incidence among UK older adults. Geroscience. (2024) 47:2507–23. 10.1007/s11357-024-01413-8
55.
Bagyinszky E An S . Haploinsufficiency and Alzheimer’s disease: The possible pathogenic and protective genetic factors.Int J Mol Sci. (2024) 25:1959. 10.3390/ijms252211959
56.
Baligacs N Albertini G Borrie S Serneels L Pridans C Balusu S et al Homeostatic microglia initially seed and activated microglia later reshape amyloid plaques in alzheimer’s disease. Nat Commun. (2024) 15:10634. 10.1038/s41467-024-54779-w
57.
Wang Y Huang L Guo H Liu M Zhang Y Zhang Z et al Higher Csf Strem2 attenuates Apoe Epsilon4-related risk for amyloid pathology in cognitively intact adults: The cable study. J Neurochem. (2025) 169:e16273. 10.1111/jnc.16273
58.
Yao M Miller G Vardarajan B Baccarelli A Guo Z Liu Z . Deciphering proteins in Alzheimer’s disease: A new Mendelian randomization method integrated with Alphafold3 for 3d structure prediction.Cell Genom. (2024) 4:100700. 10.1016/j.xgen.2024.100700
59.
Guenoun D Blaise N Sellam A Roupret-Serzec J Jacquens A Steenwinckel J et al Microglial depletion, a new tool in neuroinflammatory disorders: Comparison of pharmacological inhibitors of the Csf-1r. Glia. (2024) 73:686–700. 10.1002/glia.24664
60.
Weinstock M . Therapeutic agents for Alzheimer’s disease: A critical appraisal.Front Aging Neurosci. (2024) 16:1484615. 10.3389/fnagi.2024.1484615
61.
Song X Wang C Ding Q Li P Sun S Wei W et al Modulation of Beta secretase and neuroinflammation by biomimetic nanodelivery system for Alzheimer’s disease therapy. J Control Release. (2024) 378:735–49. 10.1016/j.jconrel.2024.12.060
62.
Gonul C Kiser C Yaka E Oz D Hunerli D Yerlikaya D et al Microglia-like cells from patient monocytes demonstrate increased phagocytic activity in probable Alzheimer’s disease. Mol Cell Neurosci. (2024) 132:103990. 10.1016/j.mcn.2024.103990
63.
Meng L Gu T Yu P Zhang Z Wei Z . The role of microglia in neuroinflammation associated with cardiopulmonary bypass.Front Cell Neurosci. (2024) 18:1496520. 10.3389/fncel.2024.1496520
64.
Flury A Aljayousi L Park H Khakpour M Mechler J Aziz S et al A neurodegenerative cellular stress response linked to dark microglia and toxic lipid secretion. Neuron. (2024) 113:554–571.e14. 10.1016/j.neuron.2024.11.018
65.
Haessler A Gier S Jung N Windbergs M . The Abeta42: Abeta40 ratio modulates aggregation in beta-amyloid oligomers and drives metabolic changes and cellular dysfunction.Front Cell Neurosci. (2024) 18:1516093. 10.3389/fncel.2024.1516093
66.
Vasilopoulou F Piers T Wei J Hardy J Pocock J . Amelioration of signaling deficits underlying metabolic shortfall in Trem2(R47h) human Ipsc-derived microglia.FEBS J. (2024) 292:1743–62. 10.1111/febs.17353
67.
Bello-Corral L Seco-Calvo J Molina Fresno A Gonzalez A Llorente A Fernandez-Lazaro D et al Prevalence of Apoe alleles in a spanish population of patients with a clinical diagnosis of Alzheimer’s disease: An observational case-control study. Medicina (Kaunas). (2024) 60:1941. 10.3390/medicina60121941
68.
Grau-Jurado P Mostafaei S Xu H Mo M Petek B Kalar I et al Medications and cognitive decline in Alzheimer’s disease: Cohort cluster analysis of 15,428 patients. J Alzheimers Dis. (2025) 103:931–40. 10.1177/13872877241307870
69.
Visscher P Gyngell C Yengo L Savulescu J . Heritable polygenic editing: The next frontier in genomic medicine?Nature. (2025) 637:637–45. 10.1038/s41586-024-08300-4
70.
Graham A Bellou E Harwood J Yaman U Celikag M Magusali N et al Human longevity and Alzheimer’s disease variants act via microglia and oligodendrocyte gene networks. Brain. (2025) 148:969–84. 10.1093/brain/awae339
71.
Wang Q Zhu H Deng L Xu S Xie W Li M et al Spatial transcriptomics: Biotechnologies, computational tools, and neuroscience applications. Small Methods. (2025) 6:e2401107. 10.1002/smtd.202401107
72.
Reid A Jayadev S Prater K . Microglial responses to Alzheimer’s disease pathology: Insights from “omics” studies.Glia. (2025) 73:519–38. 10.1002/glia.24666
73.
Shwab E Man Z Gingerich D Gamache J Garrett M Serrano G et al Comparative mapping of single-cell transcriptomic landscapes in neurodegenerative diseases. bioRxiv [Preprint]. (2024). 10.1101/2024.12.13.628436
74.
He Y Lu W Zhou X Mu J Shen W . Unraveling Alzheimer’s disease: Insights from single-cell sequencing and spatial transcriptomic.Front Neurol. (2024) 15:1515981. 10.3389/fneur.2024.1515981
75.
Azargoonjahromi A Alzheimer’s Disease Neuroimaging Initiative. Serotonin enhances neurogenesis biomarkers, hippocampal volumes, and cognitive functions in Alzheimer’s disease. Mol Brain. (2024) 17:93. 10.1186/s13041-024-01169-4
76.
Hu R Yu Q Zhou S Yin Y Hu R Lu H et al Co-expression network analysis reveals novel genes underlying Alzheimer’s disease pathogenesis. Front Aging Neurosci. (2020) 12:605961. 10.3389/fnagi.2020.605961
77.
He X Liu X Zuo F Shi H Jing J . Artificial intelligence-based multi-omics analysis fuels cancer precision medicine.Semin Cancer Biol. (2023) 88:187–200. 10.1016/j.semcancer.2022.12.009
78.
Ballard J Wang Z Li W Shen L Long Q . Deep learning-based approaches for multi-omics data integration and analysis.BioData Min. (2024) 17:38. 10.1186/s13040-024-00391-z
79.
Ashenden A Chowdhury A Anastasi L Lam K Rozek T Ranieri E et al The multi-omic approach to newborn screening: Opportunities and challenges. Int J Neonatal Screen. (2024) 10:42. 10.3390/ijns10030042
80.
Micheletti C Bonetti G Madeo G Gadler M Benedetti S Guerri G et al Omics sciences and precision medicine in glioblastoma. Clin Ter. (2023) 174:77–84. 10.7417/CT.2023.2474
81.
Tasca P van den Berg B Rabelink T Wang G Heijs B van Kooten C et al Application of spatial-omics to the classification of kidney biopsy samples in transplantation. Nat Rev Nephrol. (2024) 20:755–66. 10.1038/s41581-024-00861-x
82.
Strelnikova P Zakharova N Kononikhin A Bugrova A Indeykina M Mitkevich V et al Blood plasma proteomic markers of Alzheimer’s disease, current status and application prospects. Expert Rev Proteomics. (2025) 22:11–8. 10.1080/14789450.2025.2450804
83.
He C Simpson C Cossentino I Zhang B Tkachev S Eddins D et al Cell signaling pathways discovery from multi-modal data. bioRxiv [Preprint]. (2025). 10.1101/2025.02.06.636961
84.
Lopez-Ortiz S Caruso G Emanuele E Menendez H Penin-Grandes S Guerrera C et al Digging into the intrinsic capacity concept: Can it be applied to Alzheimer’s disease? Prog Neurobiol. (2024) 234:102574. 10.1016/j.pneurobio.2024.102574
85.
Shah J Rahman Siddiquee M Krell-Roesch J Syrjanen J Kremers W Vassilaki M et al Neuropsychiatric symptoms and commonly used biomarkers of Alzheimer’s disease: A literature review from a machine learning perspective. J Alzheimers Dis. (2023) 92:1131–46. 10.3233/JAD-221261
86.
Bolivar D Mosquera-Heredia M Vidal O Barcelo E Allegri R Morales L et al Exosomal mRNA signatures as predictive biomarkers for risk and age of onset in Alzheimer’s disease. Int J Mol Sci. (2024) 25:2293. 10.3390/ijms252212293
87.
Ibanez L Liu M Beric A Timsina J Kholfeld P Bergmann K et al Benchmarking of a multi-biomarker low-volume panel for Alzheimer’s disease and related dementia research. medRxiv [Preprint]. (2024). 10.1101/2024.06.13.24308895
88.
Mastenbroek S Sala A Vallez Garcia D Shekari M Salvado G Lorenzini L et al Continuous beta-amyloid Csf/Pet imbalance model to capture Alzheimer disease heterogeneity. Neurology. (2024) 103:e209419. 10.1212/WNL.0000000000209419
89.
Li D Xie Q Xie J Ni M Wang J Gao Y et al Cerebrospinal fluid proteomics identifies potential biomarkers for early-onset Alzheimer’s disease. J Alzheimers Dis. (2024) 100:261–77. 10.3233/JAD-240022
90.
Tosun D Hausle Z Iwaki H Thropp P Lamoureux J Lee E et al A cross-sectional study of alpha-synuclein seed amplification assay in Alzheimer’s disease neuroimaging initiative: Prevalence and associations with Alzheimer’s disease biomarkers and cognitive function. Alzheimers Dement. (2024) 20:5114–31. 10.1002/alz.13858
91.
Salvado G Horie K Barthelemy N Vogel J Pichet Binette A Chen C et al Disease staging of Alzheimer’s disease using a CSF-based biomarker model. Nat Aging. (2024) 4:694–708. 10.1038/s43587-024-00599-y
92.
Huang X Li Y Fowler C Doecke J Lim Y Drysdale C et al Leukocyte surface biomarkers implicate deficits of innate immunity in sporadic Alzheimer’s disease. Alzheimers Dement. (2023) 19:2084–94. 10.1002/alz.12813
93.
Howe M Britton K Joyce H Menard W Emrani S Kunicki Z et al Clinical application of plasma P-Tau217 to assess eligibility for amyloid-lowering immunotherapy in memory clinic patients with early Alzheimer’s disease. Alzheimers Res Ther. (2024) 16:154. 10.1186/s13195-024-01521-9
94.
Malaguarnera M Cabrera-Pastor A . Emerging role of extracellular vesicles as biomarkers in neurodegenerative diseases and their clinical and therapeutic potential in central nervous system pathologies.Int J Mol Sci. (2024) 25:68. 10.3390/ijms251810068
95.
Gotze K Vrillon A Dumurgier J Indart S Sanchez-Ortiz M Slimi H et al Plasma neurofilament light chain as prognostic marker of cognitive decline in neurodegenerative diseases, a clinical setting study. Alzheimers Res Ther. (2024) 16:231. 10.1186/s13195-024-01593-7
96.
Musso G Gabelli C Puthenparampil M Cosma C Cagnin A Gallo P et al Blood biomarkers for Alzheimer’s disease with the lumipulse automated platform: Age-effect and clinical value interpretation. Clin Chim Acta. (2025) 565:120014. 10.1016/j.cca.2024.120014
97.
Liampas I Kyriakoulopoulou P Karakoida V Kavvoura P Sgantzos M Bogdanos D et al Blood-based biomarkers in frontotemporal dementia: A narrative review. Int J Mol Sci. (2024) 25:1838. 10.3390/ijms252111838
98.
Huda T Diaz M Gozlan E Chobrutskiy A Chobrutskiy B Blanck G . Immunogenomics parameters for patient stratification in Alzheimer’s disease.J Alzheimers Dis. (2022) 88:619–29. 10.3233/JAD-220119
99.
Rathore S Higgins I Wang J Kennedy I Iaccarino L Burnham S et al Predicting regional tau accumulation with machine learning-based tau-pet and advanced radiomics. Alzheimers Dement (N Y). (2024) 10:e70005. 10.1002/trc2.70005
100.
Smits L Pijnenburg Y Koedam E van der Vlies A Reuling I Koene T et al Early onset Alzheimer’s disease is associated with a distinct neuropsychological profile. J Alzheimers Dis. (2012) 30:101–8. 10.3233/JAD-2012-111934
101.
Calvo-Rodriguez M Garcia-Rodriguez C Villalobos C Nunez L . Role of toll like receptor 4 in Alzheimer’s disease.Front Immunol. (2020) 11:1588. 10.3389/fimmu.2020.01588
102.
Zhang Q Yang G Luo Y Jiang L Chi H Tian G . Neuroinflammation in Alzheimer’s disease: Insights from peripheral immune cells.Immun Ageing. (2024) 21:38. 10.1186/s12979-024-00445-0
103.
Rochon E Sy M Phillips M Anderson E Plys E Ritchie C et al Bio-experiential technology to support persons with dementia and care partners at home (tend): Protocol for an intervention development study. JMIR Res Protoc. (2023) 12:e52799. 10.2196/52799
104.
Murfield J Moyle W O’Donovan A . Planning and designing a self-compassion intervention for family carers of people living with dementia: A person-based and co-design approach.BMC Geriatr. (2022) 22:53. 10.1186/s12877-022-02754-9
105.
Navathe A Volpp K Caldarella K Bond A Troxel A Zhu J et al Effect of financial bonus size, loss aversion, and increased social pressure on physician pay-for-performance: A randomized clinical trial and cohort study. JAMA Netw Open. (2019) 2:e187950. 10.1001/jamanetworkopen.2018.7950
106.
Hawkins J Catalano R Miller J . Risk and protective factors for alcohol and other drug problems in adolescence and early adulthood: Implications for substance abuse prevention.Psychol Bull. (1992) 112:64–105. 10.1037/0033-2909.112.1.64
107.
Hagar H Fernandez-Vega V Wang K Jordan L Shumate J Scampavia L et al Hyperphosphorylated tau-based Alzheimer’s disease drug discovery: Identification of inhibitors of tau aggregation and cytotoxicity. SLAS Discov. (2025) 33:100235. 10.1016/j.slasd.2025.100235
108.
Pichet Binette A Mammana A Wisse L Rossi M Strandberg O Smith R et al Associations between misfolded alpha-synuclein aggregates and Alzheimer’s disease pathology in vivo. Alzheimers Dement. (2024) 20:7624–34. 10.1002/alz.14225
109.
Fan X Chen H He W Zhang J . Emerging microglial biology highlights potential therapeutic targets for Alzheimer’s disease.Ageing Res Rev. (2024) 101:102471. 10.1016/j.arr.2024.102471
110.
Brody D Holtzman D . Active and passive immunotherapy for neurodegenerative disorders.Annu Rev Neurosci. (2008) 31:175–93. 10.1146/annurev.neuro.31.060407.125529
111.
Solomon B . Immunotherapeutic strategies for Alzheimer’s disease treatment.Sci World J. (2009) 9:909–19. 10.1100/tsw.2009.99
112.
Schreiner T Croitoru C Hodorog D Cuciureanu D . Passive anti-amyloid beta immunotherapies in Alzheimer’s disease: From mechanisms to therapeutic impact.Biomedicines. (2024) 12:1096. 10.3390/biomedicines12051096
113.
Yi L Tan E Zhou Z . Passive immunotherapy for Alzheimer’s disease: Challenges & future directions.J Transl Med. (2024) 22:430. 10.1186/s12967-024-05248-x
114.
Winblad B Graf A Riviere M Andreasen N Ryan J . Active immunotherapy options for Alzheimer’s disease.Alzheimers Res Ther. (2014) 6:7. 10.1186/alzrt237
115.
Lacosta A Pascual-Lucas M Pesini P Casabona D Perez-Grijalba V Marcos-Campos I et al Safety, tolerability and immunogenicity of an active anti-abeta vaccine (Abvac40) in patients with Alzheimer’s disease: A randomised, double-blind, placebo-controlled, phase I trial. Alzheimers Res Ther. (2018) 10:12. 10.1186/s13195-018-0340-8
116.
Lambracht-Washington D Rosenberg R . Active DNA Abeta42 vaccination as immunotherapy for Alzheimer disease.Transl Neurosci. (2012) 3:307–13. 10.2478/s13380-012-0037-6
117.
Guo Y Cai C Zhang B Tan B Tang Q Lei Z et al Targeting Usp11 regulation by a novel lithium-organic coordination compound improves neuropathologies and cognitive functions in Alzheimer transgenic mice. EMBO Mol Med. (2024) 16:2856–81. 10.1038/s44321-024-00146-7
118.
Wongsodirdjo P Caruso A Yong A Lester M Vella L Hung Y et al Messenger RNA-encoded antibody approach for targeting extracellular and intracellular tau. Brain Commun. (2024) 6:fcae100. 10.1093/braincomms/fcae100
119.
Duncan B Dunbar C Ishii K . Applying a clinical lens to animal models of car-T cell therapies.Mol Ther Methods Clin Dev. (2022) 27:17–31. 10.1016/j.omtm.2022.08.008
120.
McKinney C . Using induced pluripotent stem cells derived neurons to model brain diseases.Neural Regen Res. (2017) 12:1062–7. 10.4103/1673-5374.211180
121.
Duan Y Lyu L Zhan S . Stem cell therapy for Alzheimer’s disease: A scoping review for 2017-2022.Biomedicines. (2023) 11:120. 10.3390/biomedicines11010120
122.
Ou C Xue W Liu D Ma L Xie H Ning K . Stem cell therapy in Alzheimer’s disease: Current status and perspectives.Front Neurosci. (2024) 18:1440334. 10.3389/fnins.2024.1440334
123.
Mallinckrodt C Tian Y Aisen P Barkhof F Cohen S Dent G et al Investigating partially discordant results in phase 3 studies of aducanumab. J Prev Alzheimers Dis. (2023) 10:171–7. 10.14283/jpad.2023.6
124.
Budd Haeberlein S Aisen P Barkhof F Chalkias S Chen T Cohen S et al Two randomized phase 3 studies of aducanumab in early Alzheimer’s disease. J Prev Alzheimers Dis. (2022) 9:197–210. 10.14283/jpad.2022.30
125.
Barenholtz Levy H . Accelerated approval of aducanumab: Where do we stand now?Ann Pharmacother. (2022) 56:736–9. 10.1177/10600280211050405
126.
Rabinovici G . Controversy and progress in Alzheimer’s disease – FDA approval of aducanumab.N Engl J Med. (2021) 385:771–4. 10.1056/NEJMp2111320
127.
Tarawneh R Pankratz V . The search for clarity regarding “clinically meaningful outcomes” in Alzheimer disease clinical trials: Clarity-ad and beyond.Alzheimers Res Ther. (2024) 16:37. 10.1186/s13195-024-01412-z
128.
Honig L Sabbagh M van Dyck C Sperling R Hersch S Matta A et al Updated safety results from phase 3 lecanemab study in early Alzheimer’s disease. Alzheimers Res Ther. (2024) 16:105. 10.1186/s13195-024-01441-8
129.
Cohen S van Dyck C Gee M Doherty T Kanekiyo M Dhadda S et al Lecanemab clarity Ad: Quality-of-life results from a randomized, double-blind phase 3 trial in early Alzheimer’s disease. J Prev Alzheimers Dis. (2023) 10:771–7. 10.14283/jpad.2023.123
130.
Cummings J Gould H Zhong K . Advances in designs for Alzheimer’s disease clinical trials.Am J Neurodegener Dis. (2012) 1:205–16.
131.
Moore A Ritchie M . Is the relationship between cardiovascular disease and alzheimer’s disease genetic? A scoping review.Genes (Basel). (2024) 15:1509. 10.3390/genes15121509
132.
Pettigrew C Nazarovs J Soldan A Singh V Wang J Hohman T et al Alzheimer’s disease genetic risk and cognitive reserve in relationship to long-term cognitive trajectories among cognitively normal individuals. Alzheimers Res Ther. (2023) 15:66. 10.1186/s13195-023-01206-9
133.
Feldner A Turner A Simpson J Estus S . Skipping of Fcer1g Exon 2 is common in human brain but not associated with the Alzheimer’s disease genetic risk factor Rs2070902.J Alzheimers Dis Rep. (2023) 7:1313–22. 10.3233/ADR-230076
134.
Whitson H Potter G Feld J Plassman B Reynolds K Sloane R et al Dual-task gait and Alzheimer’s disease genetic risk in cognitively normal adults: A pilot study. J Alzheimers Dis. (2018) 64:1137–48. 10.3233/JAD-180016
135.
Arai H Suzuki H Yoshiyama T . Vanutide cridificar and the Qs-21 adjuvant in japanese subjects with mild to moderate Alzheimer’s disease: Results from two phase 2 studies.Curr Alzheimer Res. (2015) 12:242–54. 10.2174/1567205012666150302154121
136.
Morgan D Landreth G Bickford P . The promise and perils of an alzheimer disease vaccine: A video debate.J Neuroimmune Pharmacol. (2009) 4:1–3.
137.
Sterner R Takahashi P Yu Ballard A . Active vaccines for alzheimer disease treatment.J Am Med Dir Assoc. (2016) 17:862. 10.1016/j.jamda.2016.06.009
138.
Kwan P Konno H Chan K Baum L . Rationale for the development of an alzheimer’s disease vaccine.Hum Vaccin Immunother. (2020) 16:645–53. 10.1080/21645515.2019.1665453
139.
Xue D Blue E Conomos M Fohner A . The power of representation: Statistical analysis of diversity in us Alzheimer’s disease genetics data.Alzheimers Dement (NY). (2024) 10:e12462. 10.1002/trc2.12462
140.
Ray N Kumar A Zaman A Del Rosario P Mena P Manoochehri M et al Disentangling the genetic underpinnings of neuropsychiatric symptoms in Alzheimer’s disease in the Alzheimer’s disease sequencing project: Study design and methodology. Alzheimers Dement (Amst). (2024) 16:e70000. 10.1002/dad2.70000
141.
Greenfest-Allen E Valladares O Kuksa P Gangadharan P Lee W Cifello J et al Niagads Alzheimer’s Genomicsdb: A resource for exploring Alzheimer’s disease genetic and genomic knowledge. Alzheimers Dement. (2024) 20:1123–36. 10.1002/alz.13509
142.
Toga A Phatak M Pappas I Thompson S McHugh C Clement M et al The pursuit of approaches to federate data to accelerate alzheimer’s disease and related dementia research: Gaain, Dpuk, and Addi. Front Neuroinform. (2023) 17:1175689. 10.3389/fninf.2023.1175689
143.
Hao X Li X Zhang G Tao C Schulz P et al Alzheimer’s Disease Neuroimaging Initiative. An ontology-based approach for harmonization and cross-cohort query of Alzheimer’s disease data resources.BMC Med Inform Decis Mak. (2023) 23:151. 10.1186/s12911-023-02250-z
Summary
Keywords
Alzheimer’s disease, personalized immunotherapy, genetic-immune integration, biomarker identification, precision treatment
Citation
He C, Shen Y, Zhang M and Zhou X (2025) Integrating genetic and immune profiles for personalized immunotherapy in Alzheimer’s disease. Front. Med. 12:1603553. doi: 10.3389/fmed.2025.1603553
Received
04 April 2025
Accepted
12 May 2025
Published
02 June 2025
Volume
12 - 2025
Edited by
Sagar Gaikwad, University of Texas Medical Branch at Galveston, United States
Reviewed by
Sudipta Senapati, University of Texas Medical Branch at Galveston, United States
Updates
Copyright
© 2025 He, Shen, Zhang and Zhou.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoqing Zhou, 2233093015@qq.com
†These authors have contributed equally to this work
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.