 Wenfeng Wang1†
Wenfeng Wang1† Hua Yan
Hua Yan- 1Department of Cardiology, Wuhan Asia General Hospital Affiliated to Wuhan University of Science and Technology, Wuhan, Hubei, China
- 2Department of Cardiology, Ezhou Central Hospital, Ezhou, Hubei, China
Diabetic kidney disease (DKD), a major microvascular complication of diabetes, is closely associated with functional imbalances in ion channels regulating sodium (Na+), calcium (Ca2+), potassium (K+), and chloride (Cl–). This review systematically examines the roles of ion channels in glomerular filtration barrier dysfunction, tubular reabsorption, and fibrotic processes in DKD, with emphasis on the pathological relevance of sodium-glucose cotransporter 2 (SGLT2), epithelial sodium channels (ENaC), transient receptor potential (TRP) channels, chloride channels, aquaporins (AQPs), and PIEZO channels. We further evaluate the clinical efficacy and challenges of ion channel-targeted therapies, including SGLT2 inhibitors and mineralocorticoid receptor antagonists. Emerging strategies integrating ion channel omics, machine learning, engineered biomaterials, and exosome-based delivery systems are proposed to shift DKD treatment paradigms from disease progression delay to pathological reversal. Interdisciplinary collaboration is critical to achieving personalized precision medicine, offering novel perspectives for DKD diagnosis and management.
1 Introduction
Diabetic kidney disease (DKD), one of the most severe microvascular complications of diabetes, has become the leading cause of end-stage renal disease (ESRD) worldwide. Epidemiological data indicate that approximately 40% of diabetic patients develop chronic kidney disease (CKD), with type 2 diabetes posing a higher risk. Between 1990 and 2017, diabetes-related CKD cases surged by 74%, from 1.4 million to 2.4 million (1–3). The proportion of ESRD cases attributed to diabetes increased from 19% in 2000 to 29.7% in 2015, with further increases anticipated (3). The Western Pacific region (e.g., China, Japan) and the United States exhibit the highest incidence of diabetes-related ESRD, with rates 2–3 times greater than those in Europe (4). China alone has over 110 million diabetic patients, of whom 32.5% with type 2 diabetes develop CKD, translating to over 30 million DKD cases. However, awareness and screening rates remain alarmingly low at 26% and < 55%, respectively (4–7). DKD now represents the primary etiology of CKD-related hospitalizations in China and dominates new hemodialysis cases. These trends underscore the immense global healthcare and economic burden of CKD.
The core pathological features of DKD include glomerular hyperfiltration, podocyte injury, tubulointerstitial fibrosis, and chronic inflammatory microenvironments. Disease progression is driven not only by metabolic dysregulation but also by complex molecular network imbalances (8–12). While conventional therapies targeting glycemic control, blood pressure management, and renin-angiotensin-aldosterone system (RAAS) inhibition remain foundational, approximately 30% of DKD patients experience relentless renal function decline despite intensive treatment, highlighting the urgent need for novel therapeutic targets (7–9, 13).
This review specifically addresses this therapeutic gap by examining the pivotal role of ion channel dysfunction in DKD pathogenesis—a mechanism insufficiently targeted by current treatments. Recent studies implicate ion channel dysfunction as a pivotal driver of DKD pathogenesis. The kidney, a hub for ion transport, relies on precise regulation of Na+, K+, and Ca2+ channels. For instance, SGLT2 overexpression in proximal tubules exacerbates Na+ and glucose reabsorption, inducing glomerular hypertension and oxidative stress, thereby accelerating podocyte detachment and basement membrane thickening (14). Conversely, aberrant ENaC activation in distal nephrons promotes Na+ retention, hypertension, and tubulointerstitial fibrosis via a “metabolic-hemodynamic” vicious cycle (15–17). Additionally, pathological Ca2+ influx mediated by TRP channels (e.g., TRPC6, TRPV4) triggers podocyte cytoskeletal remodeling, mitochondrial dysfunction, and apoptotic signaling, directly contributing to proteinuria (17–19).
Mounting evidence demonstrates that SGLT2 inhibitors confer renal protection through three distinct yet complementary mechanisms: First, by suppressing excessive glucose and sodium reabsorption in proximal tubules, these agents restore tubuloglomerular feedback and ameliorate glomerular hyperfiltration (20). Second, they modulate tissue oxygenation homeostasis while attenuating inflammatory and fibrotic pathways (21). Third, they promote blood pressure reduction (mean decrease 3.3 mmHg) and vascular resistance improvement through enhanced urinary sodium and glucose excretion (20). Clinical trials confirm that SGLT2 inhibitors reduce the risk of composite renal endpoints by 30% (including end-stage renal disease and serum creatinine doubling), accompanied by modest reductions in HbA1c (0.25%) and body weight (0.8 kg) (20). Notably, these nephroprotective effects appear mediated through common mechanisms in both diabetic and non-diabetic CKD populations, while providing additional cardiovascular benefits including amelioration of atherosclerosis and mitigation of uremic toxin-induced cardiac damage (22, 23).
Representative SGLT2 inhibitors (e.g., dapagliflozin) exert their effects by reducing intraglomerular pressure and restoring tubuloglomerular feedback, thereby optimizing renal energy metabolism. The DAPA-CKD trial demonstrated that dapagliflozin could delay ESRD progression by 6.6 years (24–26). Similarly, the selective mineralocorticoid receptor antagonist finerenone, by inhibiting ENaC overactivation, achieves a 32% reduction in urinary albumin-to-creatinine ratio (UACR) and a 23% decrease in kidney-related events, marking a therapeutic breakthrough in DKD management (27). Emerging therapeutic strategies, including TRPC5 inhibitors, have shown promise in reversing podocyte injury in preclinical models (28, 29), highlighting the substantial clinical potential of ion channel-targeted therapies. However, challenges persist due to the complexity and tissue specificity of ion channel regulation. For example, SGLT2 inhibitors may induce euglycemic ketoacidosis, while ENaC antagonists require careful monitoring of hyperkalemia risk (30). Future research must integrate single-cell omics and structural biology (e.g., cryo-electron microscopy) to elucidate dynamic ion channel expression profiles across renal cell populations and develop subtype-specific modulators. Recent structural resolution of the Cav2.1 calcium channel subunit (31) provides a molecular blueprint for selective drug design, a strategy applicable to DKD-related channels.
Ion channel dysregulation exhibits intricate associations with inflammatory and fibrotic pathways in DKD. A comprehensive elucidation of these molecular networks will enable targeted therapeutic interventions to achieve disease remission. Our purpose is thus threefold: (1) Elucidate molecular linkages between ion channel dysregulation and DKD progression; (2) Evaluate clinical challenges in translating channel-targeted therapies; (3) Propose precision strategies (e.g., single-cell omics, structural pharmacology) to develop subtype-specific modulators that mitigate off-target risks while maximizing renoprotection. By integrating these insights, we aim to advance ion channel therapeutics from disease management toward personalized, pathology-reversing regimens for DKD.
2 Literature search strategies
We conducted a systematic narrative literature review utilizing PubMed, Web of Science, Google Scholar, and EMBASE databases (2022–2025) with the following search terms: “ion channels,” “diabetic kidney disease,” “SGLT2 inhibitors,” “TRP channels,” and “precision therapy.” The review incorporated preclinical studies, clinical trials, and relevant review articles. Furthermore, we critically examine future research directions and clinical prospects regarding ion channel modulation in DKD, while providing expert perspectives. This review synthesizes current evidence concerning ion channel mechanisms, therapeutic targets, and innovative multidisciplinary approaches (including multi-omics integration, machine learning applications, engineered nanocarrier systems, and structural pharmacology) to transform DKD treatment paradigms from disease progression delay to potential pathological reversal.
3 Pathogenesis and current research in DKD
The pathogenesis of DKD involves multifactorial interactions among energy metabolic dysregulation, oxidative stress, inflammatory responses, fibrosis, genetic susceptibility, renin angiotensin system (RAS) intestinal flora and metabolites forming a complex “metabolic-inflammatory-fibrotic” cascade network. Current mechanistic research has shifted from a singular metabolic perspective to multidimensional network regulation, integrating interdisciplinary technologies to dissect the “gene-environment-Gut Microbiota-metabolism” interplay, which is critical for elucidating the underlying mechanisms of DKD (Figure 1).
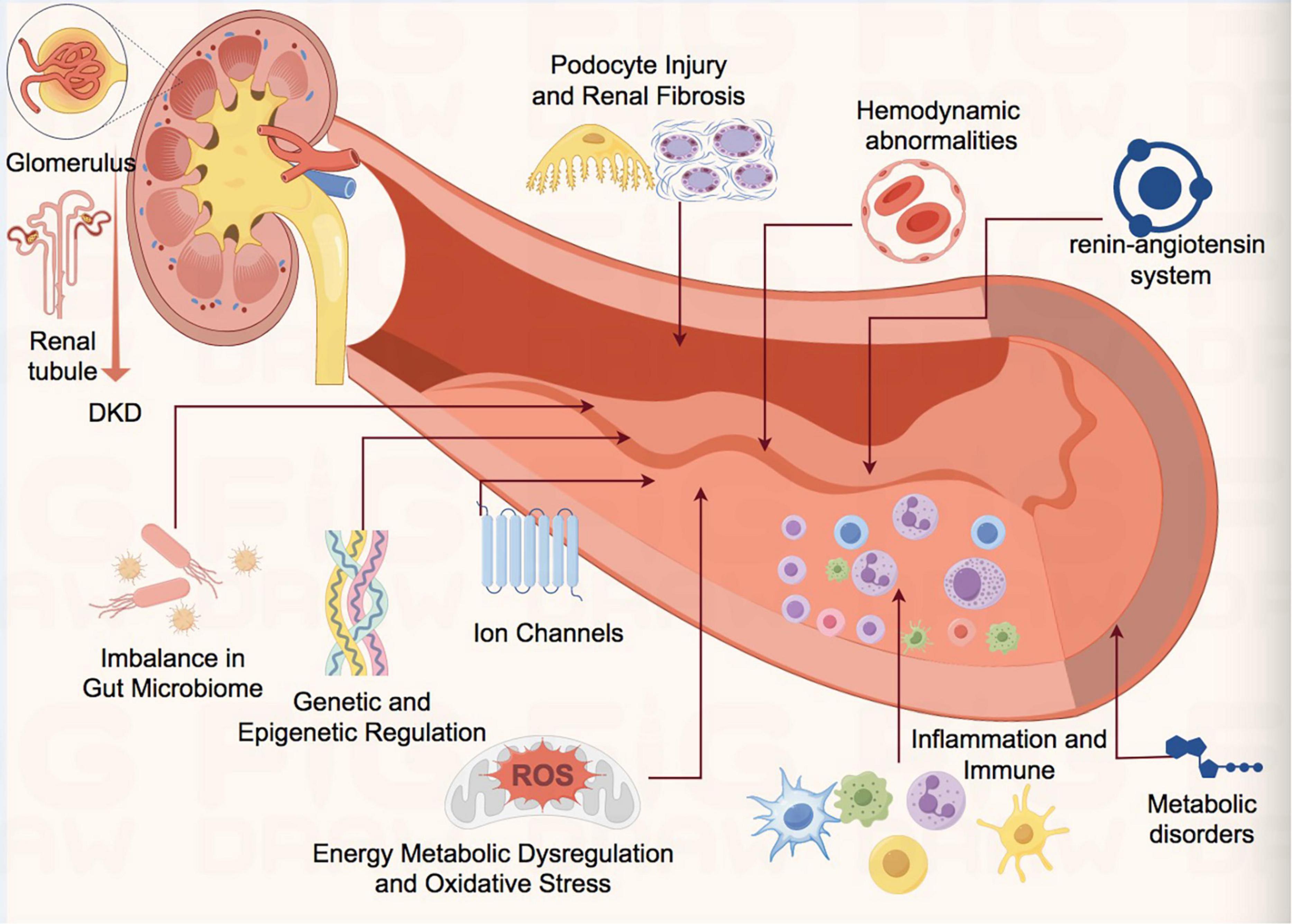
Figure 1. Schematic diagram of pathophysiological mechanism of DKD. DKD, diabetic kidney disease; ROS, reactive oxygen species.
3.1 Energy metabolic dysregulation and oxidative stress
Hyperglycemia activates the polyol pathway, protein kinase C (PKC) signaling, and accumulation of advanced glycation end products (AGEs), inducing renal oxidative stress and energy metabolism abnormalities (32–37). AGE binding to its receptor (RAGE) activates NADPH oxidase, leading to reactive oxygen species (ROS) overproduction, mitochondrial dysfunction, and DNA damage, thereby promoting podocyte apoptosis and glomerular basement membrane thickening (35–38). Additionally, hyperglycemia suppresses ATP-sensitive potassium (KATP) channels, exacerbating membrane depolarization and calcium overload, further damaging tubular epithelial cells (39).
3.2 Inflammation and immune dysregulation
Chronic inflammation is a key driver of DKD progression. Hyperglycemia activates NF-κB and TGF-β1 pathways, promoting macrophage infiltration and release of pro-inflammatory cytokines (e.g., TNF-α, IL-6, IL-1β), thereby fostering an inflammatory microenvironment (39–41). Overexpression of monocyte chemoattractant protein-1 (MCP-1) and fibronectin (FN) accelerates glomerulosclerosis, while Th17/Treg cell imbalance impairs anti-inflammatory responses, perpetuating a vicious cycle (42–44).
3.3 Podocyte injury and renal fibrosis
Podocytes, critical components of the glomerular filtration barrier, undergo injury that underlies proteinuria and renal functional loss (45, 46). Hyperglycemia induces aberrant expression of long non-coding RNAs (e.g., lncRNA evf-2) in podocytes, which bind heterogeneous nuclear ribonucleoprotein U (hnRNPU) to regulate cell cycle re-entry and inflammatory responses, exacerbating podocyte detachment (47). Pathological calcium influx mediated by transient receptor potential (TRP) channels (e.g., TRPC6, TRPV4) triggers podocyte cytoskeletal remodeling and apoptosis, further disrupting the filtration barrier (48–50). TGF-β1 signaling activation drives epithelial-mesenchymal transition (EMT), promoting tubulointerstitial fibrosis and extracellular matrix (ECM) deposition (51).
3.4 Genetic and epigenetic regulation
Genetic polymorphisms (e.g., ACE insertion/deletion polymorphism) and epigenetic modifications (e.g., DNA methylation, histone acetylation) significantly influence DKD susceptibility (52–58). Genetic predispositions conferred by polymorphisms in ELMO1, AGTR1, and P2 × 7 also play critical roles in DKD pathogenesis (53–55). Furthermore, lncRNAs (e.g., H19, MALAT1) modulate chromatin remodeling to influence inflammation-related gene expression, while miRNAs (e.g., miR-21, miR-29) target fibrotic pathway genes, contributing to renal fibrosis (56–62). These findings underscore the pivotal role of genetic factors in DKD development.
3.5 Multiple regulatory effects of renin angiotensin system (RAS)
The renin-angiotensin(Ang)-aldosterone system (RAAS) plays a critical role in the pathogenesis of both acute and chronic kidney diseases.
Recent studies have demonstrated that abnormal activation of the renin-angiotensin system (RAS) not only promotes glomerular injury through the classical Ang II/AT1R axis but also involves novel mechanisms including RhoA/ROCK signaling pathway-mediated cytoskeletal remodeling (63–65). RAS activation upregulates the RhoA/ROCK signaling pathway, resulting in increased endothelial permeability and cytoskeletal reorganization. Similar mechanisms may be triggered by mechanical stress (such as glomerular hypertension) in DKD. Inhibition of RhoA/ROCK can reduce podocyte detachment and mesangial matrix deposition, indicating this pathway serves as a crucial downstream effector of RAS (63). Additionally, emerging research suggests that the histone deacetylase SIRT6 can attenuate RAS-induced profibrotic and oxidative stress responses by inhibiting the Wnt/β-catenin signaling pathway, thereby ameliorating podocyte injury and glomerulosclerosis (66). Ellagic acid has been shown to significantly reduce TNF-α levels in cisplatin-induced nephrotoxicity models while suppressing the ERK1/2-NF-κB inflammatory pathway through SIRT6 activation (67). Additionally, non-classical RAS components contribute to renal injury. The imbalance of the Ang-(1–7)/Mas axis exacerbates oxidative stress, while aldosterone promotes fibrosis through upregulation of ENaC and TGF-β1. Novel non-steroidal mineralocorticoid receptor antagonists (e.g., Finerenone) can partially reverse these pathological processes (68).
3.6 Mechanisms of interaction between gut microbiota dysbiosis and the gut-kidney axis
The gut microbiota, as the largest microbial ecosystem in the human body, plays a pivotal role in host metabolic processes. Dysbiosis of the gut microbiota is closely associated with metabolic disorders such as diabetes. Patients with diabetic nephropathy (DN) exhibit significant differences in both the compositional distribution of gut microbiota and the levels of gut-derived metabolites compared to healthy individuals (69). Microbial metabolites, including short-chain fatty acids (SCFAs), bile acids, hydrogen sulfide, and uremic toxins, function as chemical messengers (70). These metabolites influence renal function in DN by modulating immune responses, inflammatory cascades, and oxidative stress pathways.
The latest research has revealed the key role of intestinal flora imbalance in DKD and potential intervention strategies: SGLT2 inhibitors (such as dapagliflozin) can delay the progress of DKD by regulating bile acid metabolism spectrum, enhancing antioxidant capacity, and dynamically improving the structure of intestinal flora (71). Additionally, probiotic intervention (such as lactic acid bacteria) can produce indole derivatives through tryptophan metabolism, inhibit the activation of aryl hydrocarbon receptor (AHR) pathway, reduce glomerular basement membrane damage and podocyte apoptosis, and provide a new treatment approach for membranous nephropathy complicated with DKD (72). Clinical studies further found that the characteristics of intestinal flora in patients with DKD (especially in ESRD stage) were significantly correlated with deterioration of renal function (such as decreased eGFR, accumulation of uremic toxins) and psychological distress (73). These findings provide a theoretical foundation for multi-dimensional interventions targeting the gut-kidney axis, including microbiota remodeling and metabolite regulation.
3.7 Interorgan regulatory networks in metabolic disorders
Metabolic disorders such as diabetes are characterized by nutrient metabolism dysregulation. Recent studies highlight the critical role of amino acids in metabolic homeostasis. Branched-chain amino acids (BCAAs)—leucine, isoleucine, and valine—are essential amino acids with potent metabolic regulatory effects. Elevated plasma BCAA levels in DKD patients, coupled with downregulation of catabolic genes (e.g., BCKDK), exacerbate mitochondrial dysfunction and oxidative stress via AMPK pathway suppression (74, 75).
Furthermore, the latest research reveals that intestinal flora dysbiosis in DKD patients leads to abnormal metabolite profiles, manifested by reduced short-chain fatty acids (such as acetic acid and propionic acid) and accumulated uremic toxins (such as TMAO and indoxyl sulfate). These changes aggravate renal inflammation and fibrosis by activating the AHR pathway and pro-inflammatory factors (IL-6 and TNF-α) (72, 75). SGLT2 inhibitors (such as dapagliflozin) can enhance antioxidant capacity and delay DKD progression by regulating bile acid metabolic profiles and maintaining bacterial population balance. Simultaneously, probiotics (such as lactic acid bacteria) inhibit the AHR pathway through tryptophan-derived indole metabolites, thereby reducing kidney injury (66, 71, 72). Traditional Chinese medicine (such as Tangshen formula) promotes SCFA production and reduces uremic toxins by remodeling microbial composition (including Firmicutes/Bacteroidetes ratio regulation), with mechanisms involving multi-target modulation of the gut-kidney axis (72). Multigroup analyses further confirm that Blautia abundance and bile acid dysmetabolism in hypertensive DKD models drive disease progression via the microbial-intestinal-metabolic axis, suggesting that targeted microbial metabolic microenvironment interventions (such as FMT and metabolite supplementation) represent novel therapeutic directions for DKD (73). Future studies should employ multi-omics technologies to analyze flora-metabolite interaction networks for optimizing precision intervention strategies.
4 Role of ion channels in DKD pathogenesis and targeted intervention strategies: from mechanistic insights to precision therapy
Ion channels, as core regulators of renal electrolyte balance and signal transduction, are critically implicated in the pathological progression of DKD (76–81). Dysfunction of ion channels in DKD exhibits spatiotemporal specificity and network-dependent regulation, intricately intertwined with metabolic disturbances, oxidative stress, inflammatory fibrosis, and podocyte injury. Hyperglycemia-induced activation of the polyol pathway, protein kinase C (PKC), and advanced glycation end product (AGE) signaling initiates a vicious cycle of ion channel dysfunction and oxidative stress. This section systematically examines the physiological roles and dysregulation of sodium (Na+), calcium (Ca2+), potassium (K+), and chloride (Cl–) channels across nephron segments in DKD, integrating recent preclinical and clinical advances. The following table shows the latest studies on ion channel-targeted therapies in DKD models and their corresponding mechanisms in DKD (Table 1).
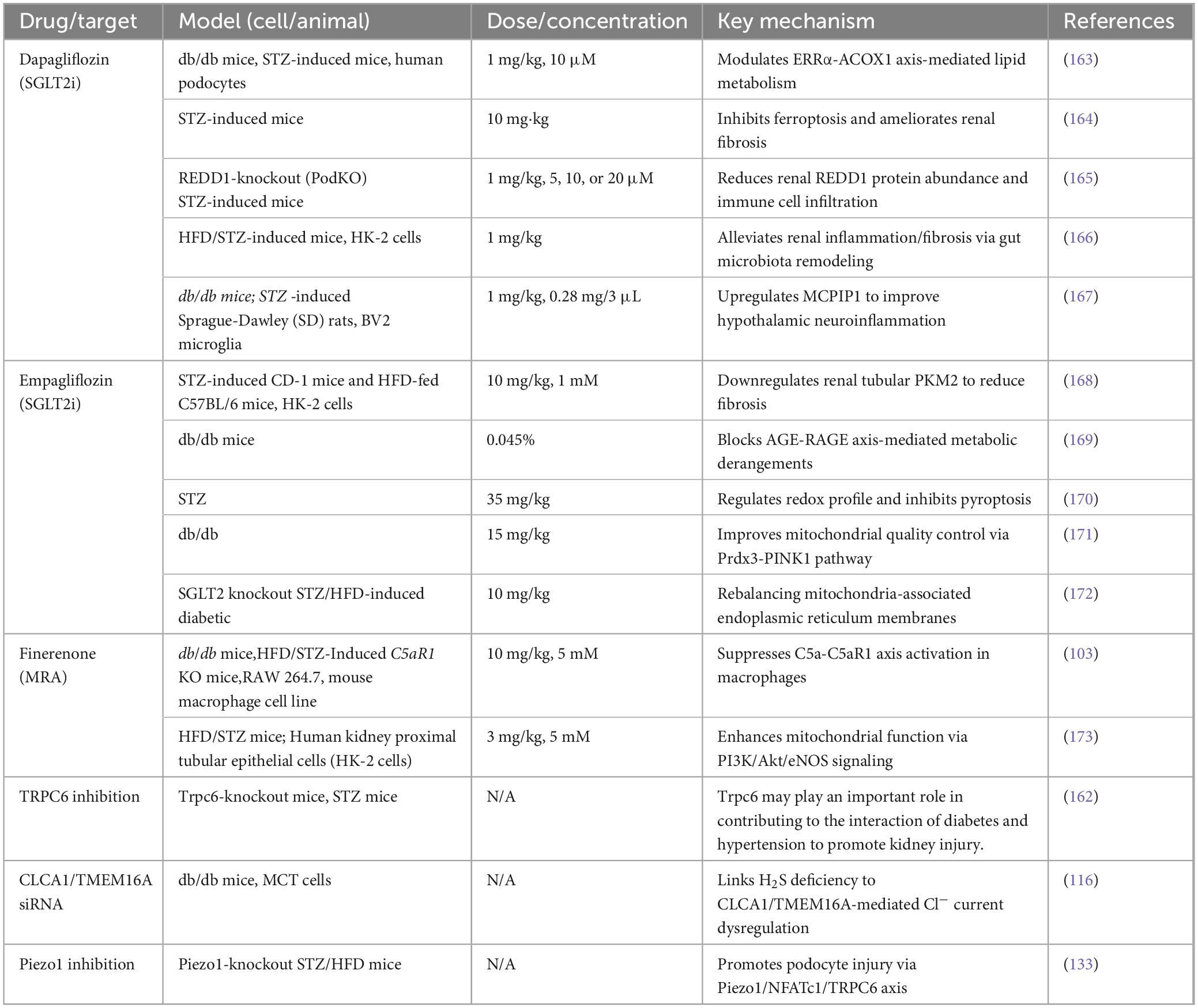
Table 1. Ion channel-targeted therapies in DKD models.
4.1 Sodium channels
Sodium channels are pivotal molecular components for maintaining systemic sodium homeostasis and blood pressure regulation, distributed across distinct nephron segments to execute specialized functions. This section focuses on sodium channels critically involved in DKD pathophysiology.
4.1.1 Sodium-glucose cotransporter 2 (SGLT2)
Sodium-glucose cotransporters (SGLTs) in renal proximal tubules, particularly SGLT2, play a central role in glucose homeostasis by reabsorbing 90% of filtered glucose. SGLT2, localized to the apical membrane of proximal tubule S1/S2 segments, actively transports one glucose molecule with two sodium ions, serving as the rate-limiting step for glomerular glucose reabsorption. In DKD, hyperglycemia upregulates SGLT2 expression via hypoxia-inducible factor-1α (HIF-1α)- and reactive oxygen species (ROS)-dependent transcriptional regulation, exacerbating sodium-glucose reabsorption and glomerular hyperfiltration. This “tubuloglomerular feedback imbalance” not only elevates intraglomerular capillary pressure but also induces podocyte hypertrophy and basement membrane thickening through mTORC1 activation (82–84).
SGLT2 inhibitors (e.g., dapagliflozin) exhibit renoprotective effects beyond glucose lowering (85–87). The DAPA-CKD trial, the first renal outcome study of SGLT2 inhibitors in CKD patients (with or without type 2 diabetes), demonstrated that dapagliflozin significantly reduced: Primary composite endpoint risk (eGFR decline ≥ 50%, progression to ESRD, renal/cardiovascular death) by 39%. Renal-specific composite endpoint risk (eGFR decline ≥ 50%, progression to ESRD, renal death) by 44%. Cardiovascular composite endpoint risk (cardiovascular death or heart failure hospitalization) by 29%. All-cause mortality risk by 31% (87–90). Mechanisms include: (1) hemodynamic improvement: Inhibiting proximal tubule sodium reabsorption, activating macula densa pressure sensors, restoring tubuloglomerular feedback, and reducing intraglomerular pressure (10–15 mmHg); (2) metabolic reprogramming: Activating AMPK/SIRT1 to enhance fatty acid oxidation and reduce lipotoxicity; (3) anti-inflammatory effects: Suppressing NLRP3 inflammasome activation and lowering IL-1β/IL-18 levels (86, 87).
However, SGLT2 inhibitors may rarely induce euglycemic ketoacidosis (incidence: 0.1–0.3%), attributed to increased proximal tubule ketone reabsorption and pancreatic α-cell glucagon dysregulation (91, 92). Dual SGLT1/2 inhibitors (e.g., sotagliflozin), which inhibit intestinal SGLT1 to reduce glucose absorption, may mitigate this risk but require further validation (93–95).
4.1.2 Epithelial sodium channel (ENaC)
The epithelial sodium channel (ENaC), also known as the amiloride-sensitive sodium channel (ASSC), is a Na+-permeable ion channel located in the apical membranes of epithelial cells in the kidney, lung, colon, and other tissues. ENaC-mediated Na+ transport is critical for regulating salt and water absorption across epithelial surfaces (1, 2, 8–10, 15). Composed of α, β, and γ subunits, ENaC in the distal nephron (distal convoluted tubule and collecting duct) mediates aldosterone-dependent sodium reabsorption (15–17). Hyperglycemia alters ENaC gating, disrupting renal electrolyte homeostasis, exacerbating hypertension, and accelerating DKD progression (15–17, 96). In DKD, mineralocorticoid receptor (MR) overactivation phosphorylates ENaC via serum/glucocorticoid-regulated kinase 1 (SGK1), increasing membrane expression and open probability, leading to sodium retention and elevated blood pressure (97). Additionally, ROS suppress Nedd4-2, a ubiquitin ligase responsible for ENaC γ-subunit degradation, further enhancing channel activity (15–17, 96–98).
Mineralocorticoid Receptor Antagonists (MRAs) targeting the ENaC-MR axis offer precision therapy for DKD. Traditional steroidal MRAs (e.g., spironolactone, eplerenone) showed limited long-term renal benefits and safety concerns, prompting the development of non-steroidal MRAs. Finerenone, a selective non-steroidal MRA, significantly reduces: Major endpoint incidence (ESRD, sustained eGFR decline, or death). Secondary endpoints (cardiovascular death, non-fatal myocardial infarction, non-fatal stroke, or heart failure hospitalization); with a lower incidence of hyperkalemia compared to steroidal MRAs (99–102). Renoprotective mechanisms include: (1) anti-fibrotic effects: Downregulating TGF-β1/Smad3, profibrotic growth factors (CCN2, CTGF); (2) endothelial protection: Restoring NO bioavailability and reducing endothelin-1 levels. Finerenone also alleviates macrophage overactivation via regulation of the complement C5a-C5aR1 axis (G protein subunit alpha i2) and ameliorates high glucose-induced podocyte epithelial-mesenchymal transition through Krüppel-like factor 5 (KLF5) modulation (103–105).
4.2 Calcium channels
The pathogenesis of DKD involves a complex interplay between calcium signaling imbalance and energy metabolism dysregulation, synergistically driving renal injury progression. Podocytes, as critical components of the glomerular filtration barrier, develop ion channel abnormalities—such as disrupted mitochondrial-endoplasmic reticulum calcium cycling and cytoskeletal dynamics—that underlie proteinuria. Hyperglycemia-induced intracellular calcium overload activates calcium-dependent proteases (e.g., calpain) and TRPC6 channels, leading to mesangial extracellular matrix deposition and podocyte cytoskeletal collapse, which exacerbate proteinuria and glomerulosclerosis (1, 2, 8–13, 19). Concurrently, endoplasmic reticulum calcium dyshomeostasis and mitochondrial calcium overload trigger oxidative stress and inflammatory responses, promoting apoptosis of renal tubular epithelial cells (1, 2, 8, 11, 12, 19).
At the metabolic level, mitochondrial dysfunction (e.g., excessive ROS production, impaired fatty acid oxidation) and AMPK/mTOR pathway imbalance induce cellular energy crises. Enhanced glycolysis exacerbates microenvironment acidosis and metabolic memory effects, further accelerating fibrosis (1, 2, 10, 13, 19). Notably, calcium signaling and energy metabolism form an intersecting regulatory network via mitochondria-associated ER membranes (MAMs) and calcium-dependent metabolic enzymes (e.g., pyruvate dehydrogenase, PDH), where oxidative stress and NF-κB-driven inflammation create a self-perpetuating cycle that amplifies renal damage. Therapeutic strategies targeting voltage-gated calcium channels (VGCCs), TRP channels, AMPK activation, mTOR inhibition, or mitochondrial antioxidants show potential clinical value. Future research should explore multi-target interventions to disrupt this pathological loop, advancing precision therapy for DKD.
4.2.1 Transient receptor potential (TRP) channels
The TRP channel family (e.g., TRPC5, TRPC6, TRPV4) plays a central role in podocyte injury in DKD (18, 29, 50, 80, 106). Hyperglycemia activates angiotensin II (Ang II)/AT1R signaling, upregulating TRPC6 expression and inducing pathological calcium influx (intracellular Ca+ concentration > 500 nM), which drives the following pathological effects: Podocyte cytoskeletal disintegration: Calcium-dependent protease calpain cleaves synaptopodin and nephrin, disrupting slit diaphragm integrity; Mitochondrial damage: Calcium overload opens mitochondrial permeability transition pores (mPTP), reducing ATP synthesis by 50–70%. Apoptotic signaling: Calcium/calmodulin -dependent kinase II (CaMKII) phosphorylates FOXO1, suppressing anti-apoptotic genes (e.g., Bcl-2) (28, 106, 107). Salemkour et al. demonstrated that dual inhibition of TRPC6 and calpain restores glomerular autophagy flux, reducing renal injury with high clinical translational potential (50). Additionally, the TRPC5 inhibitor GFB-887 is under investigation for safety and efficacy in focal segmental glomerulosclerosis, refractory minimal change disease, and DKD (28).
4.2.2 Voltage-gated calcium channels (VGCCs)
VGCCs mediate calcium influx during membrane depolarization and are essential for neurotransmission, muscle contraction, membrane excitability, synaptic plasticity, and gene expression (108). In DKD, hyperglycemia aberrantly activates L-type VGCCs (e.g., Cav1.2) in mesangial cells, increasing calcium current density (2–3 fold) and driving fibrosis via: (1) inflammatory cytokine release: Calcium signaling activates nuclear factor of activated T cells (NFAT), promoting TGF-β1, connective tissue growth factor (CTGF), and IL-6 synthesis; (2) extracellular matrix (ECM) deposition: Upregulating collagen IV and fibronectin expression; (3) cellular proliferation: Activating ERK1/2 pathways to induce pathological mesangial cell hyperplasia (108). Peng et al. revealed that low-dose nifedipine can enhance EPCs’ angiogenic potential and implied that chronic treatment with low-dose nifedipine may be a safe and economic manner to prevent ischemic diseases in diabetes (109). Preclinical and clinical studies suggest that L-type calcium channel blockers (e.g., manidipine, nifedipine) mitigate renal dysfunction, highlighting the need for kidney-specific VGCC modulators (110).
4.3 Potassium ion channels
Potassium ion channels play a pivotal role in electrolyte balance and urine concentration in the kidneys, predominantly localized in renal tubular epithelial cells (e.g., principal cells of the distal convoluted tubule and collecting duct). By regulating potassium secretion and reabsorption (e.g., ROMK channel-mediated potassium secretion), these channels maintain serum potassium homeostasis. Under pathological conditions, dysfunctional potassium channels (due to genetic mutations, pharmacological inhibition, or chronic kidney disease) may lead to hyperkalemia (impaired excretion) or hypokalemia (excessive loss), triggering systemic physiological abnormalities.
4.3.1 Renal outer medullary potassium channel (ROMK)
ROMK, located on the apical membrane of the thick ascending limb (TAL) of the loop of Henle, mediates potassium recycling to sustain NKCC2 (Na+-K+-2Cl– cotransporter) activity, which is critical for the urine-concentrating mechanism (1, 2, 8–10). Hyperglycemia suppresses ATP-sensitive potassium channel (KATP) opening via the AGEs-RAGE pathway, inducing depolarization of renal tubular epithelial cell membranes, mitochondrial electron transport chain uncoupling, and a 3–5-fold increase in ROS production. ROS further activates the NLRP3 inflammasome, promoting IL-1β and IL-18 release and establishing an “oxidative stress-inflammation” cascade. This cascade activates NF-κB and TGF-β1 signaling pathways, driving macrophage infiltration and collagen deposition (111). Single-cell RNA sequencing has revealed significant downregulation of fatty acid oxidation-related genes and upregulation of injury markers (e.g., Havcr1, Vcam1) and pro-inflammatory factors (e.g., SPP1) in proximal tubular cells of DKD patients, suggesting that ion channel dysfunction exacerbates renal microenvironment deterioration via metabolic reprogramming and dysregulated intercellular communication (112, 113). In DKD, hyperglycemia phosphorylates ROMK at the S44 residue (2-fold increase in phosphorylation) via the PKC pathway, leading to channel internalization and degradation. Consequences include: (1) impaired potassium secretion: Elevated serum potassium (> 5.0 mmol/L) activates ENaC, aggravating sodium retention. (2) TAL dysfunction: Reduced NKCC2 activity diminishes urinary concentrating capacity, worsening polyuria (96). ROMK gene (KCNJ1) mutations cause Bartter syndrome type II (hypokalemic alkalosis), whose pathological phenotype mirrors ROMK inhibition in DKD as a “mirror phenomenon.” The lack of selective ROMK inhibitors has hindered therapeutic exploration. Single-cell electrophysiology combined with CRISPR screening has identified aberrant crosstalk between ROMK and Kir4.1/5.1 channels in DKD renal tubular cells, suggesting the need for multi-target potassium channel modulators to restore electrolyte homeostasis (114).
4.4 Chloride ion channels
Chloride channels are widely distributed in glomerular and tubular epithelial cells, regulating cell volume, transmembrane potential, ion transport, and signaling (79, 114). Emerging evidence highlights the critical role of chloride channels (ClCs) in DKD pathogenesis via ion dysregulation, inflammation, oxidative stress, and fibrotic signaling. In diabetes, hyperglycemia-induced mitochondrial dysfunction and endoplasmic reticulum (ER) stress impair ClC activity (e.g., downregulated CLC-5 in proximal tubules), disrupting lysosomal acidification and protein degradation, thereby exacerbating cellular injury (115). Dysfunctional ClCs, including calcium-activated TMEM16A (ANO1) and volume-regulated VRAC (LRRC8), trigger NLRP3 inflammasome activation, excessive ROS production, and TGF-β/Smad-driven epithelial-mesenchymal transition (EMT), promoting glomerulosclerosis and interstitial fibrosis (116). Notably, TMEM16A upregulation in diabetic models correlates with elevated pro-inflammatory cytokines and extracellular matrix deposition, while its inhibition ameliorates proteinuria and renal damage (116). Impaired Cl– transport via CLC-5 disrupts autophagic flux, accelerating podocyte apoptosis. These channels also modulate hemodynamic stress by influencing renal vascular tone and sodium retention (117). Additionally, the cystic fibrosis transmembrane conductance regulator (CFTR), a cAMP-activated ATP-gated Cl– channel, has recently been linked to DKD pathology. Studies suggest CFTR upregulation suppresses the Wnt/β-catenin pathway, improving tubular lesions during DKD (115, 117). Although challenges remain in subtype selectivity and long-term safety, targeting specific ClCs (e.g., TMEM16A inhibitors or CLC-K antagonists) shows therapeutic promise in preclinical studies. Spatiotemporal regulation of ClCs may unveil novel strategies to mitigate DKD progression (118).
4.5 Aquaporins
Aquaporins (AQPs), a family of membrane proteins regulating transmembrane water and solute transport, are integral to diverse physiological processes (119, 120). Recent evidence underscores their role in DKD pathogenesis, particularly in water homeostasis, metabolic dysregulation, and cellular injury (119–121). In diabetes, hyperglycemia-induced AQP1 downregulation in proximal tubules disrupts water reabsorption, exacerbating tubular dysfunction and proteinuria. Impaired AQP7 expression in adipocytes contributes to dyslipidemia and lipotoxicity, while defective glycerol transport via AQP7 aggravates insulin resistance and ectopic lipid deposition, linking metabolic imbalance to DKD progression (122). Preclinical studies demonstrate that berberine-mediated PI3K/Akt activation restores AQP1, improving renal function in diabetic models (123). Single-cell sequencing by Tsai et al. identified AQP4 as a key mediator of early DKD signaling and intercellular crosstalk (124). Despite progress, the molecular mechanisms of AQP-mediated renal protection remain incompletely defined, necessitating further research into tissue-specific regulatory networks and translational strategies targeting AQPs to alleviate DKD.
4.6 Mechanosensitive ion channels
DKD is characterized by glomerulosclerosis, mesangial expansion, and proteinuria, with podocyte injury playing a central role. Podocytes endure significant mechanical stress (e.g., circumferential wall stress, filtration slit shear stress, and Bowman’s capsule fluid shear stress) due to their anatomical structure (125). Piezo proteins (Piezo1/2), key mammalian mechanotransducers, convert mechanical stimuli into intracellular chemical signals, regulating diverse physiological processes and disease progression (126). Given their mechanosensory properties and DKD pathology, Piezo1 likely acts as a critical mediator linking mechanical stress (e.g., glomerular hypertension, hemodynamic shear forces) and metabolic disturbances to downstream inflammatory, oxidative, and fibrotic pathways. Analogous to fibrosis in liver, lung, and intestine, glomerular hyperfiltration-induced mechanical stress (e.g., shear forces, intraglomerular hypertension) may activate Piezo1 in endothelial cells and podocytes, triggering sustained Ca2+ influx. This activates the ROS-NLRP3 inflammasome axis, promotes IL-1β release, and synergizes with TGF-β/Smad signaling to drive EMT and extracellular matrix deposition, accelerating renal fibrosis (126–130). Additionally, metabolic disturbances (e.g., lipotoxicity) may enhance Piezo sensitivity via altered membrane fluidity, amplifying oxidative-inflammatory cascades, podocyte detachment, and filtration barrier disruption. Notably, aberrant Piezo1 activation in immune cells (e.g., platelets, neutrophils) exacerbates microthrombosis and NETosis-related inflammation, forming a “metabolic-mechanical-immune” network (131, 132). Li et al. reported Piezo1 upregulation in DKD, where the Piezo1/NFATc1/TRPC6 axis promotes podocyte injury. Podocyte-specific Piezo1 knockout attenuates DKD progression (133). Current studies show that selective inhibitors (e.g., GsMTx4) or genetic targeting of Piezo1 ameliorates proteinuria, podocyte injury, and fibrosis in diabetic and hypertensive models. However, tissue-specific strategies (e.g., avoiding systemic bleeding risks) and dynamic effects across disease stages require further exploration (133, 134). Integrating mechanobiology and metabolomics may elucidate spatiotemporal regulation of Piezo1 in renal cells, offering novel targets for DKD intervention.
4.7 Voltage-dependent anion channels (VDAC)
VDACs (voltage-dependent anion-selective channels), also termed mitochondrial porins, are the most abundant proteins in the outer mitochondrial membrane (OMM). They mediate ion (e.g., Ca2+) and metabolite (e.g., ATP, tRNA, DNA) exchange between mitochondria and the cytosol, ensuring mitochondrial complex function and energy production (135). In DKD, upregulated expression of the ER-resident protein reticulon-1A (RTN1A) exacerbates tubular epithelial cell (TEC) injury via ER stress. TEC-specific RTN1A overexpression worsens DKD phenotypes, including tubular damage, interstitial fibrosis, and renal dysfunction. Mechanistically, RTN1A disrupts ER-mitochondria contacts by interacting with mitochondrial hexokinase-1 (HK-1) and VDAC1, dissociating their binding. HK-1/VDAC1 dissociation activates apoptosis and inflammasome pathways, driving TEC loss (136).
4.8 Toward precision ion channel therapeutics
The aforementioned ion channels play central roles in DKD pathogenesis by modulating glomerular function, podocyte homeostasis, and mitochondrial metabolism. Non-selective cation channels (e.g., TRPV4, TRPC5) drive endothelial injury and podocyte apoptosis via calcium overload, oxidative stress, and inflammation, while mitochondrial channel dysfunction exacerbates metabolic imbalance (76–81). Emerging precision strategies include: (1) Small-molecule inhibitors: Targeting channel activity (e.g., TRPV4 antagonists) or upstream regulators (e.g., finerenone for mineralocorticoid receptors) to improve proteinuria and renal function. (2) Multi-target therapies: Synergistically regulating metabolic and ionic homeostasis (e.g., SGLT2 inhibitors combined with TRPC6 antagonists). (3) Biomarker-guided approaches: Leveraging urinary exosomal miRNAs (e.g., miR-29c) or DNA methylation signatures for early diagnosis and treatment stratification.
Advanced delivery systems: Utilizing kidney-targeted nanoparticles or engineered exosomes to enhance therapeutic specificity. Despite progress, key challenges remain: (1) deciphering crosstalk between ion channel networks and metabolic/inflammatory pathways. (2) Validating clinical efficacy of novel targets (e.g., Piezo1, VDAC1) in diverse patient cohorts. (3) Optimizing personalized regimens through multi-omics integration (genomics, proteomics, metabolomics) (137–139).
By bridging mechanistic insights with scalable clinical tools, ion channel-targeted therapies are transitioning from disease delay to pathomechanistic reversal, offering a transformative framework for precision nephrology.
5 Current challenges and future directions
Despite significant progress in ion channel-targeted therapies for DKD, clinical translation faces multiple challenges. Current strategies have evolved from single-receptor blockade to multidimensional precision interventions. Future efforts should focus on: (1) Subtype-selective drug design: Leveraging cryo-electron microscopy (cryo-EM) and AI-based predictions to resolve dynamic channel conformations. (2) Spatiotemporal-specific modulation: Utilizing nanotechnology for localized, high-concentration drug delivery to renal tissues. (3) Multi-target synergistic interventions: Combining SGLT2 inhibitors, MRAs, and TRP antagonists to overcome compensatory signaling activation. These strategies aim to shift DKD treatment paradigms from “disease delay” to “pathological reversal.” This section systematically analyzes current limitations—spanning technical bottlenecks, safety optimization, and therapeutic innovation—and proposes a breakthrough tripartite strategy integrating precision medicine, multi-omics technologies, and optimized drug safety.
5.1 Integration of precision medicine and multi-omics
The convergence of precision medicine and multi-omics technologies is advancing DKD intervention toward cell-specific and dynamic regulation. Single-cell sequencing and spatial transcriptomics reveal upregulated SGLT2 and ENaC expression in proximal tubular epithelial cells and abnormal TRPC5/TRPC6 ratios in podocytes of DKD patients, highlighting the need for subtype/cell-specific inhibitors (140–143). Metabolomics identifies branched-chain amino acid (BCAA) accumulation as a driver of mitochondrial dysfunction via KATP channel inhibition (74). Machine learning algorithms are increasingly applied to integrate multi-omics data, systematically capturing complex interactions and establishing robust data linkages. AI-driven multi-omics platforms can decode “ion channel-metabolite-epigenetic” networks, enhancing patient stratification and therapeutic efficacy (144). To mitigate systemic side effects (e.g., ketoacidosis with SGLT2 inhibitors, renal limitations of non-steroidal MRAs), novel delivery systems—such as kidney-targeted nanocarriers (e.g., PEGylated liposomes achieving 10-fold higher renal drug concentration) and exosomes—enable precise delivery, minimizing off-target effects (145–147). Furthermore, multi-target therapies (e.g., SGLT2 inhibitors combined with finerenone) demonstrate synergistic benefits by dual inhibition of sodium reabsorption and TGF-β1/Smad3 signaling, reducing UACR. However, cross-scale models (e.g., PhysiCell) are required to quantify synergy and toxicity risks (148, 149). Future studies must integrate multi-omics data with dynamic pharmacodynamic models to transition DKD treatment from “single-target intervention” to “spatiotemporal precision modulation.”
5.2 Cutting-edge technological innovations
Cutting-edge research is driving systemic innovation from molecular mechanisms to clinical paradigms, primarily manifested in the following aspects:
5.2.1 Structure-guided drug design
Cryo-electron microscopy (cryo-EM)-resolved TRPC5-calmodulin complexes, combined with AI-driven molecular dynamics simulations (e.g., AlphaFold 3), empower structural pharmacology to develop conformation-selective inhibitors. For instance, non-competitive antagonists targeting TRPC5 allosteric sites are being designed to circumvent cardiovascular side effects associated with traditional pore-blocking agents (150, 151).
5.2.2 Dynamic channel network analysis
Integration of single-cell multi-omics with optogenetic techniques (e.g., ChR2 optogenetic systems) enables real-time dissection of ion channel dynamics. Light-activated modulation allows precise activation or inhibition of specific channels, achieving spatiotemporal control over channel behavior and revealing their regulatory patterns (152).
5.2.3 Advanced drug screening platforms
Kidney organoid co-culture systems coupled with microelectrode arrays replicate DKD pathological microenvironments and monitor ionic current fluctuations in real time, significantly enhancing drug screening efficiency. Concurrently, machine learning models (e.g., AlphaFold 3) accelerate virtual drug discovery by predicting dynamic ion channel conformations (151, 153).
5.3 Toward precision spatiotemporal modulation
The interdisciplinary integration of these technologies propels DKD therapeutics from “static target inhibition” to “precision spatiotemporal modulation.” Ion channel network regulation offers a novel strategy to disrupt the “metabolic-inflammatory-fibrotic” cycle in DKD. While targeted therapies (e.g., SGLT2 inhibitors, TRP antagonists) show clinical promise, challenges persist in subtype selectivity, delivery efficiency, and patient heterogeneity. Future integration of cryo-EM structural insights, single-cell multi-omics, and AI predictive analytics will unravel channel regulatory networks, enabling patient-tailored regimens. Through global collaborations (e.g., the DKD Channelome Initiative), standardized omics pipelines, validated biomarkers, and ethical AI guidelines can democratize access to precision nephrology, ultimately transitioning DKD care from delayed intervention to pathological reversal.
6 Discussion and perspectives
6.1 Core pathogenic role of ion channel dysregulation
The pathological progression of DKD is fundamentally driven by a vicious cycle of ion channel network dysfunction and “metabolic-inflammatory-fibrotic” cascades. This review systematically demonstrates that ion channel dysregulation—via calcium overload, oxidative stress, and inflammatory microenvironments—serves as a core driver of podocyte injury, glomerulosclerosis, and interstitial fibrosis (1, 2, 9–14). Clinically validated therapies targeting SGLT2, ENaC, and TRP channels (e.g., dapagliflozin, finerenone, GSK2193874) have marked a paradigm shift in DKD management from “symptom control” to “mechanism-based intervention” (82–92, 99–102).
6.2 Critical bottlenecks for pathological reversal
However, achieving pathological reversal requires overcoming the following bottlenecks and advancing interdisciplinary innovation.
6.2.1 Molecular mechanism elucidation
Transitioning from static targets to dynamic networks, the spatiotemporal specificity and regulatory dynamics of ion channels are central to DKD complexity. Cryo-EM and single-cell multi-omics technologies can unravel “channel-metabolism” crosstalk as novel therapeutic targets.
6.2.2 Therapeutic strategy innovation
Establishment of a three-tiered precision intervention framework. (1) Early-stage intervention (high-risk screening): AI models integrating ion channel gene polymorphisms and metabolic biomarkers to predict critical targets and identify high-risk individuals. (2) Mid-stage reversal (targeting core pathology): Precision strategies for renal-targeted drug accumulation. (3) Late-stage synergy (multi-pathway blockade): Combinatorial therapies to enhance treatment efficacy.
6.2.3 Technological challenges and interdisciplinary breakthroughs
(1) Drug delivery limitations: Current nanocarriers (e.g., liposomes, gold nanoparticles) exhibit ∼40% renal targeting efficiency, with unresolved long-term biocompatibility issues (e.g., hepatosplenic accumulation toxicity) (145–147). In contrast, exosomes—with superior biocompatibility, target specificity, and extended half-life—are emerging as promising delivery vehicles. Engineered exosomes modified with renal tubular-specific markers significantly enhance targeting efficiency (146, 147). (2) Dynamic monitoring: Traditional electrophysiology fails to track ion channel activity in live tissues. Novel techniques, such as optogenetics (e.g., ChR2-mediated KATP channel activation) combined with two-photon microscopy, enable real-time monitoring of podocyte calcium signaling and filtration barrier permeability (152).
6.3 Future directions: bridging discovery to clinical impact
Precision drug design, powered by cryo-EM structural insights and AI-driven molecular dynamics (AlphaFold 3), will yield conformation-selective inhibitors (150–153). Global consortia like the DKD Channelome Initiative aim to standardize omics pipelines, validate target engagement biomarkers, and establish ethical AI guidelines for vulnerable populations.
Future research should prioritize precision medicine approaches to bridge ion channel discoveries and clinical applications. First, artificial intelligence-driven multi-omics integration—incorporating urinary exosomal RNA profiles, single-cell epigenetic data, and wearable device metrics—will enable prediction of individual channelopathies (e.g., concurrent TRPC6/Piezo1 dysregulation) and optimization of SGLT2 inhibitor therapeutic responses. Simultaneously, biodegradable nanocarriers (e.g., megalin-targeted PLGA nanoparticles) and customized exosomes (e.g., CD133-engineered vesicles for glomerular targeting) could improve renal drug delivery efficiency while reducing off-target effects. Second, international collaborations such as the DKD Channelome Initiative should establish standardized biomarker protocols (e.g., TRPV4 autoantibody detection) for diverse populations, resolving genetic variability while ensuring target validation through blockchain-protected data exchange. Advanced structural biology tools (cryo-EM and AlphaFold 3) will elucidate glucose-altered ion channel architectures to develop highly selective (> 100-fold) allosteric drugs, complemented by patient-derived organoid-microfluidic systems for customized drug testing. Ultimately, innovative trial designs integrating computational patient avatars and adaptive Bayesian methods could halve DKD progression rates by 2,030 via coordinated modulation of metabolic, inflammatory, and fibrotic pathways.
6.4 Multitarget regulation by traditional chinese medicine (TCM) in DKD management
Traditional Chinese Medicine (TCM) and integrated TCM-Western medicine strategies exhibit substantial therapeutic potential in the diagnosis and management of DKD. As a central manifestation of diabetic microvascular complications, DKD necessitates combined interventions targeting metabolic regulation and renal protection. TCM modulates multiple pathological pathways in DKD: Alpiniae Oxyphyllae Fructus ameliorates inflammation and oxidative stress by activating podocyte autophagy, regulating non-coding RNAs, and restoring gut microbiota balance (154, 155). Poria cocos and its active components attenuate renal fibrosis through TGF-β1/Smad and NF-κB signaling pathway inhibition (156). Jingui Shenqi Pills, when combined with Western medications, reduce urinary albumin-to-creatinine ratio (ACR), modulate gut microbiota composition (e.g., increasing Prevotella abundance), and suppress inflammatory cytokines such as IL-2 (157). TCM also corrects epigenetic dysregulation (e.g., suppressing DNMT1-mediated DNA methylation), potentially reversing “metabolic memory” to delay DKD progression (158, 159).
In contrast, SGLT2 inhibitors (e.g., dapagliflozin) primarily inhibit proximal tubular sodium-glucose reabsorption, ameliorate glomerular hyperfiltration, and exert metabolic (e.g., glycemic and blood pressure control) and anti-inflammatory or anti-fibrotic effects. Clinical trials confirm their efficacy in slowing end-stage renal disease progression, independent of glucose-lowering mechanisms. While both TCM and SGLT2 inhibitors demonstrate renoprotective properties, TCM’s multi-dimensional regulation (e.g., epigenetics, microbiome) may address broader pathological networks, whereas SGLT2 inhibitors predominantly improve hemodynamic abnormalities.
Integrated therapies further enhance DKD management: Early-stage DKD patients benefit from combined angiotensin system inhibitors (ACEI/ARB), SGLT2 inhibitors (e.g., dapagliflozin), and TCM formulas (e.g., Tang Shen Fang), which synergistically reduce proteinuria, improve metabolic profiles, and restore gut microbiota homeostasis (160). High-risk patients with nephrotic-range proteinuria achieve superior renal outcomes (12-month proteinuria reduction: 7,289.25 mg vs. 4,512.79 mg) and fewer glycemic fluctuations with low-dose multi-target immunosuppressive regimens (glucocorticoids + tacrolimus + mycophenolate mofetil) compared to cyclophosphamide-based protocols (161).
Mechanistic studies reveal that Astragaloside IV alleviates podocyte oxidative injury via JNK/ERK1/2 pathway suppression, triptolide inhibits complement C5b-9 deposition through p38MAPK modulation, and Panax notoginseng oral liquid activates Nrf2/HO-1 to enhance antioxidant defenses, collectively preserving renal function (159). These advancements underscore the promise of targeting “metabolic memory” and multi-pathway interventions for DKD, warranting further validation of TCM’s mechanistic precision and long-term safety in multicenter trials.
7 Conclusion
In conclusion, ion channel network modulation offers a novel therapeutic paradigm for disrupting the “metabolic-inflammatory-fibrotic” vicious cycle in DKD. While targeted therapies (e.g., SGLT2 inhibitors and TRP channel antagonists) demonstrate clinical efficacy, persistent challenges remain regarding subtype selectivity, drug delivery efficiency, and patient heterogeneity. The convergence of cryo-electron microscopy (cryo-EM), single-cell multi-omics profiling, and AI-based predictive modeling will elucidate comprehensive channel regulatory networks, while parallel exploration of traditional medicine compounds may enable truly personalized therapeutics (Figure 2). Through multidisciplinary collaboration and innovative pharmacologic development, we can potentially transform DKD management—from merely delaying disease progression to achieving pathological reversal of ion imbalance-induced renal injury.
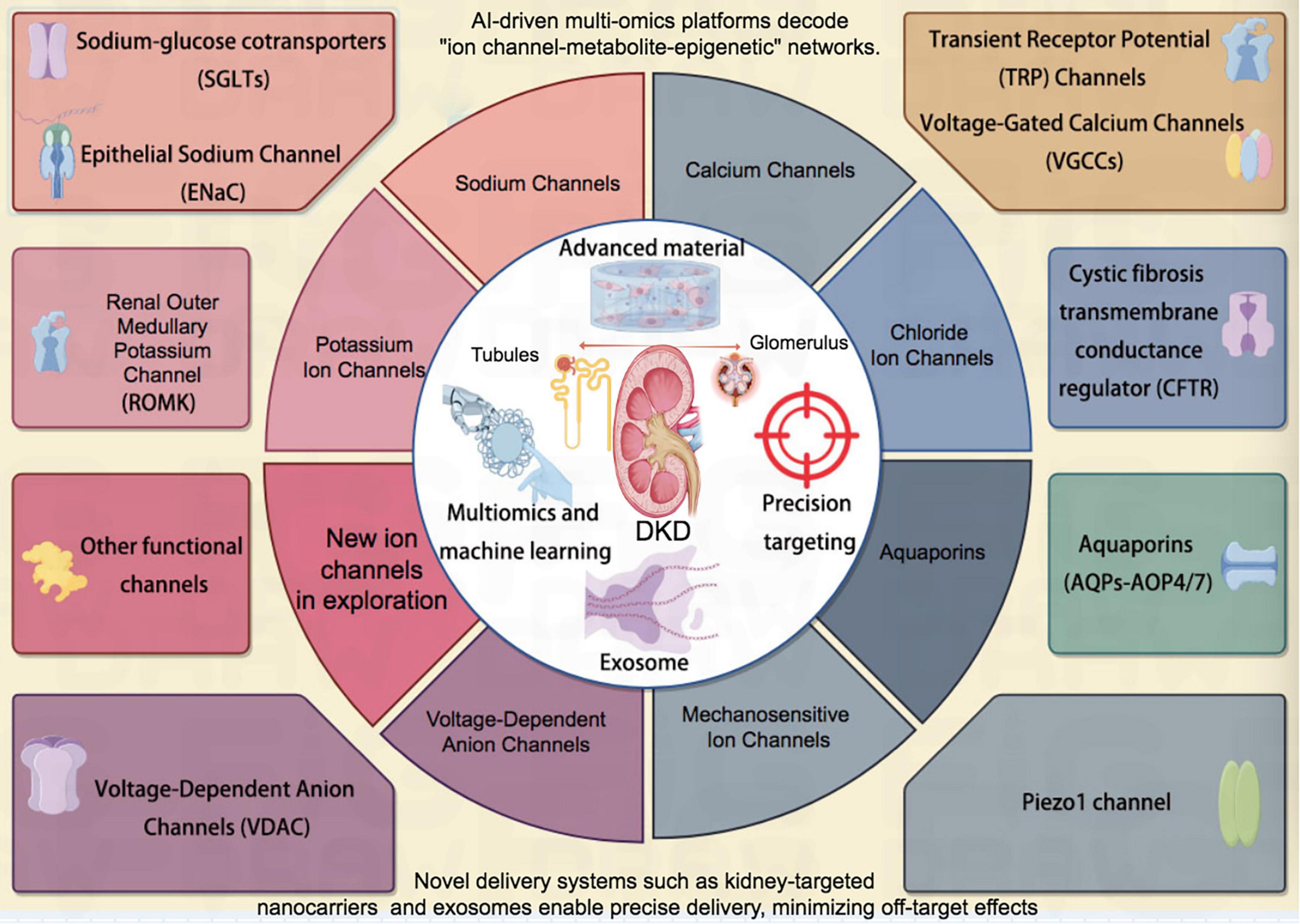
Figure 2. Schematic diagram of a series of ion channels related to DKD. DKD, diabetic kidney disease.
Author contributions
WW: Conceptualization, Methodology, Writing – original draft, Data curation, Investigation. BK: Conceptualization, Methodology, Writing – original draft, Formal Analysis. CW: Methodology, Writing – original draft, Investigation. XX: Investigation, Methodology, Writing – original draft, Data curation. XF: Data curation, Writing – original draft, Conceptualization, Formal Analysis, Writing – review & editing. HY: Conceptualization, Writing – original draft, Writing – review & editing, Methodology, Project administration.
Funding
The authors declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
Some cartoon components were from www.figdraw.com for model drawing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Gupta S, Dominguez M, Golestaneh L. Diabetic kidney disease: An update. Med Clin North Am. (2023) 107:689–705. doi: 10.1016/j.mcna.2023.03.004
2. Zhao M, Cao Y, Ma L. New insights in the treatment of DKD: Recent advances and future prospects. BMC Nephrol. (2025) 26:72. doi: 10.1186/s12882-025-03953-3
3. Alobaid T, Karalliedde J, O’Connell M, Gnudi L, Sheehan K, Lim K, et al. The prevalence and progression of microvascular complications and the interaction with ethnicity and socioeconomic status in people with type 2 diabetes: A systematic review and meta-analysis. J Diabetes Res. (2025) 2025:3307594. doi: 10.1155/jdr/3307594
4. Jia W, Yu R, Wang L, Zhu D, Guo L, Weng J, et al. China National Diabetic Chronic Complications (DiaChronic) Study Group. Prevalence of chronic kidney disease among Chinese adults with diabetes: A nationwide population-based cross-sectional study. Lancet Reg Health West Pac. (2025) 55:101463. doi: 10.1016/j.lanwpc.2024.101463
5. Chaudhry K, Karalliedde J. Chronic kidney disease in type 2 diabetes: The size of the problem, addressing residual renal risk and what we have learned from the credence trial. Diabetes Obes Metab. (2024) 26(Suppl5):25–34. doi: 10.1111/dom.15765
6. Selby N, Taal M. What every clinician needs to know about chronic kidney disease: Detection, classification and epidemiology. Diabetes Obes Metab. (2024) 26(Suppl 6):3–12. doi: 10.1111/dom.15683
7. Chowdhury T, Mukuba D, Casabar M, Byrne C, Yaqoob M. Management of diabetes in people with advanced chronic kidney disease. Diabet Med. (2025) 42:e15402. doi: 10.1111/dme.15402
8. Efiong E, Bazireh H, Fuchs M, Amadi P, Effa E, Sharma S, et al. Crosstalk of hyperglycaemia and cellular mechanisms in the pathogenesis of diabetic kidney disease. Int J Mol Sci. (2024) 25:10882. doi: 10.3390/ijms252010882
9. Tuttle K, Agarwal R, Alpers C, Bakris G, Brosius F, Kolkhof P, et al. Molecular mechanisms and therapeutic targets for diabetic kidney disease. Kidney Int. (2022) 102:248–60. doi: 10.1016/j.kint.2022.05.012
10. Watanabe K, Sato E, Mishima E, Miyazaki M, Tanaka T. What’s new in the molecular mechanisms of diabetic kidney disease: Recent advances. Int J Mol Sci. (2022) 24:570. doi: 10.3390/ijms24010570
11. Chen J, Liu Q, He J, Li Y. Immune responses in diabetic nephropathy: Pathogenic mechanisms and therapeutic target. Front Immunol. (2022) 13:958790. doi: 10.3389/fimmu.2022.958790
12. Dong R, Xu Y. Glomerular cell cross talk in diabetic kidney diseases. J Diabetes. (2022) 14:514–23. doi: 10.1111/1753-0407.13304
13. Boima V, Agyekum A, Ganatra K, Agyekum F, Kwakyi E, Inusah J, et al. Advances in kidney disease: Pathogenesis and therapeutic targets. Front Med. (2025) 12:1526090. doi: 10.3389/fmed.2025.1526090
14. Gaggini M, Sabatino L, Suman A, Chatzianagnostou K, Vassalle C. Insights into the Roles of GLP-1, DPP-4, and SGLT2 at the crossroads of cardiovascular, renal, and metabolic pathophysiology. Cells. (2025) 6:387. doi: 10.3390/cells14050387
15. Kirabo A, Masenga S, Kleyman T. Epithelial Na+ channels, immune cells, and salt. Annu Rev Physiol. (2025) 87:381–95. doi: 10.1146/annurev-physiol-022724-105050
16. Aufy M, Hussein A, Stojanovic T, Studenik C, Kotob M. Proteolytic activation of the epithelial sodium channel (ENaC): Its mechanisms and implications. Int J Mol Sci. (2023) 24:17563. doi: 10.3390/ijms242417563
17. Chen Y, Yu X, Yan Z, Zhang S, Zhang J, Guo W. Role of epithelial sodium channel-related inflammation in human diseases. Front Immunol. (2023) 14:1178410. doi: 10.3389/fimmu.2023.1178410
18. Khan S, Khan S, Suleman M, Khan M, Alsuhaibani A, Refat M, et al. The multifunctional TRPC6 protein: Significance in the field of cardiovascular studies. Curr Probl Cardiol. (2024) 49(1 Pt B):102112. doi: 10.1016/j.cpcardiol.2023.102112
19. Yan P, Ke B, Fang X. Ion channels as a therapeutic target for renal fibrosis. Front Physiol. (2022) 13:1019028. doi: 10.3389/fphys.2022.1019028
20. Liu D, Chen X, He W, Lu M, Li Q, Zhang S, et al. Update on the pathogenesis, diagnosis, and treatment of diabetic tubulopathy. Integr Med Nephrol Androl. (2024) 11:e23–9. doi: 10.1097/IMNA-D-23-00029
21. Dai Z, Chen J, Zou R, Liang X, Tang J, Yao C. Role and mechanisms of SGLT-2 inhibitors in the treatment of diabetic kidney disease. Front Immunol. (2023) 14:1213473. doi: 10.3389/fimmu.2023.1213473
22. Lentini P, Zanoli L, Ronco C, Benedetti C, Previti A, Laudadio G, et al. The vascular disease of diabetic kidney disease. Cardiorenal Med. (2023) 13:202–10. doi: 10.1159/000527274
23. Han J, Luo L, Wang Y, Miyagishi M, Kasim V, Wu S. SGLT2 inhibitor empagliflozin promotes revascularization in diabetic mouse hindlimb ischemia by inhibiting ferroptosis. Acta Pharmacol Sin. (2023) 44:1161–74. doi: 10.1038/s41401-022-01031-0
24. Rodriguez-Valadez J, Tahsin M, Masharani U, Park M, Hunink M, Yeboah J, et al. Potential mediators for treatment effects of novel diabetes medications on cardiovascular and renal outcomes: A meta-regression analysis. J Am Heart Assoc. (2024) 13:e032463. doi: 10.1161/JAHA.123.032463
25. McEwan P, Gabb P, Davis J, Garcia Sanchez J, Sjöström C, Barone S, et al. The long-term effects of dapagliflozin in chronic kidney disease: A time-to-event analysis. Nephrol Dial Transplant. (2024) 39:2040–7. doi: 10.1093/ndt/gfae106
26. Wheeler D, Jongs N, Stefansson B, Chertow G, Greene T, Hou F, et al. Safety and efficacy of dapagliflozin in patients with focal segmental glomerulosclerosis: A prespecified analysis of the dapagliflozin and prevention of adverse outcomes in chronic kidney disease (DAPA-CKD) trial. Nephrol Dial Transplant. (2022) 37:1647–56. doi: 10.1093/ndt/gfab335
27. Young T, Toussaint N, Di Tanna G, Arnott C, Hockham C, Kang A, et al. Risk factors for fracture in patients with coexisting chronic kidney disease and type 2 diabetes: An observational analysis from the CREDENCE trial. J Diabetes Res. (2022) 2022:9998891. doi: 10.1155/2022/9998891
28. Xu Y, Ren Y, Zhang J, Niu B, Liu M, Xu T, et al. Discovery of pyridazinone derivatives bearing tetrahydroimidazo[1,2-a]pyrazine scaffold as potent inhibitors of transient receptor potential canonical 5 to ameliorate hypertension-induced renal injury in rats. Eur J Med Chem. (2024) 275:116565. doi: 10.1016/j.ejmech.2024.116565
29. Kumaş-Kulualp M, Gepdiremen A, Karataş E, Eşrefoğlu M. Clemizole hydrochloride, a potent TRPC5 calcium channel inhibitor, prevents cisplatin-induced nephrotoxicity in Spraque-Dawley rats. J Biochem Mol Toxicol. (2023) 37:e23372. doi: 10.1002/jbt.23372
30. Inose R, Muraki Y. Sodium-glucose cotransporter-2 inhibitor treatment has differential effects on the incidence of various malignancies: Evidence from a spontaneous adverse reaction database. Int J Clin Pharmacol Ther. (2025) 63:98–104. doi: 10.5414/CP204645
31. Ho W, Hsiao C, Wu P, Chen J, Tu Y, Wu V. Comparison of different medical treatments for primary hyperaldosteronism: A systematic review and network meta-analysis. Ther Adv Chronic Dis. (2024) 15:20406223241239775. doi: 10.1177/20406223241239775
32. Li L, Zou J, Zhou T, Liu X, Tan D, Xiang Q. mTOR-mediated nutrient sensing and oxidative stress pathways regulate autophagy: A key mechanism for traditional Chinese medicine to improve diabetic kidney disease. Front Pharmacol. (2025) 16:1578400. doi: 10.3389/fphar.2025.1578400
33. Ma X, Ma J, Leng T, Yuan Z, Hu T, Liu Q. Advances in oxidative stress in pathogenesis of diabetic kidney disease and efficacy of TCM intervention. Ren Fail. (2023) 45:2146512. doi: 10.1080/0886022X.2022.2146512
34. Feng A, Yin R, Xu R, Zhang B, Yang L. An update on renal tubular injury as related to glycolipid metabolism in diabetic kidney disease. Front Pharmacol. (2025) 16:1559026. doi: 10.3389/fphar.2025.1559026
35. Takasu M, Kishi S, Nagasu H, Kidokoro K, Brooks C, Kashihara N. The role of mitochondria in diabetic kidney disease and potential therapeutic targets. Kidney Int Rep. (2024) 10:328–42. doi: 10.1016/j.ekir.2024.10.035
36. Narongkiatikhun P, Choi Y, Hampson H, Gotzamanis J, Zhang G, van Raalte D, et al. Unraveling diabetic kidney disease: The roles of mitochondrial dysfunction and immunometabolism. Kidney Int Rep. (2024) 9:3386–402. doi: 10.1016/j.ekir.2024.09.019
37. Liu P, Zhu W, Wang Y, Ma G, Zhao H, Li P. Chinese herbal medicine and its active compounds in attenuating renal injury via regulating autophagy in diabetic kidney disease. Front Endocrinol. (2023) 14:1142805. doi: 10.3389/fendo.2023.1142805
38. Ma Y, Wang X, Lin S, King L, Liu L. The potential role of advanced glycation end products in the development of kidney disease. Nutrients. (2025) 17:758. doi: 10.3390/nu17050758
39. Merrins M, Kibbey R. Glucose regulation of β-Cell KATP channels: It is time for a new model! Diabetes. (2024) 73:856–63. doi: 10.2337/dbi23-0032
40. Hauwanga W, Abdalhamed T, Ezike L, Chukwulebe I, Ko Oo A, Wilfred A, et al. The pathophysiology and vascular complications of diabetes in chronic kidney disease: A comprehensive review. Cureus. (2024) 16:e76498. doi: 10.7759/cureus.76498
41. Chu C, Huang Y, Cao L, Ji S, Zhu B, Shen Q. Role of macrophages in peritoneal dialysis-associated peritoneal fibrosis. Ren Fail. (2025) 47:2474203. doi: 10.1080/0886022X.2025.2474203
42. Dousdampanis P, Aggeletopoulou I, Mouzaki A. The role of M1/M2 macrophage polarization in the pathogenesis of obesity-related kidney disease and related pathologies. Front Immunol. (2025) 15:1534823. doi: 10.3389/fimmu.2024.1534823
43. Xu C, Ha X, Yang S, Tian X, Jiang H. Advances in understanding and treating diabetic kidney disease: Focus on tubulointerstitial inflammation mechanisms. Front Endocrinol. (2023) 14:1232790. doi: 10.3389/fendo.2023.1232790
44. Njeim R, Merscher S, Fornoni A. Mechanisms and implications of podocyte autophagy in chronic kidney disease. Am J Physiol Renal Physiol. (2024) 326:F877–93. doi: 10.1152/ajprenal.00415.2023
45. Jiang H, Shen Z, Zhuang J, Lu C, Qu Y, Xu C, et al. Understanding the podocyte immune responses in proteinuric kidney diseases: From pathogenesis to therapy. Front Immunol. (2024) 14:1335936. doi: 10.3389/fimmu.2023.1335936
46. Nakamichi R, Hishikawa A, Chikuma S, Yoshimura A, Sasaki T, Hashiguchi A, et al. DNA-damaged podocyte-CD8 T cell crosstalk exacerbates kidney injury by altering DNA methylation. Cell Rep. (2023) 42:112302. doi: 10.1016/j.celrep.2023.112302
47. Zhang C, Zhao H, Yan Y, Li Y, Lei M, Liu Y. LncRNA evf-2 exacerbates podocyte injury in diabetic nephropathy by inducing cell cycle re-entry and inflammation through distinct mechanisms triggered by hnRNPU. Adv Sci. (2024) 11:e2406532. doi: 10.1002/advs.202406532
48. Márquez-Nogueras K, Vuchkovska V, Kuo I. Calcium signaling in polycystic kidney disease- cell death and survival. Cell Calcium. (2023) 112:102733. doi: 10.1016/j.ceca.2023.102733
49. Staruschenko A, Alexander R, Caplan M, Ilatovskaya D. Calcium signalling and transport in the kidney. Nat Rev Nephrol. (2024) 20:541–55. doi: 10.1038/s41581-024-00835-z
50. Salemkour Y, Yildiz D, Dionet L, Hart D, Verheijden KA, Saito R, et al. Podocyte injury in diabetic kidney disease in mouse models involves TRPC6-mediated calpain activation impairing autophagy. J Am Soc Nephrol. (2023) 34:1823–42. doi: 10.1681/ASN.0000000000000212
51. Kim J, Lee S, Jang H, Jung S, Jung M, Yun J, et al. Transcriptional intermediary factor 1γ-induced irisin in skeletal muscle attenuates renal fibrosis in diabetic nephropathy. J Cachexia Sarcopenia Muscle. (2025) 16:e13810. doi: 10.1002/jcsm.13810
52. Martínez-Nava Y, Ogaz-Escarpita M, Reza-López S, Leal-Berumen I. Diabetic kidney disease and polymorphisms of the ELMO1 and AGTR1 genes: Systematic review. Nefrologia. (2025) 45:194–213. doi: 10.1016/j.nefroe.2025.02.005
53. Mohammedi K, Marre M, Alhenc-Gelas F. Genetic predisposition to nephropathy and associated cardiovascular disease in people with type 1 diabetes: Role of the angiotensinI-converting enzyme (ACE), and beyond; A narrative review. Cardiovasc Diabetol. (2024) 23:453. doi: 10.1186/s12933-024-02544-0
54. Ali A, Pham C, Morahan G, Ekinci E. Genetic risk scores identify people at high risk of developing diabetic kidney disease: A systematic review. J Clin Endocrinol Metab. (2024) 109:1189–97. doi: 10.1210/clinem/dgad704
55. Song J, Ni J, Yin X. The genetic side of diabetic kidney disease: A review. Int Urol Nephrol. (2023) 55:335–43. doi: 10.1007/s11255-022-03319-w
56. Zheng M, Yang Z, Shi L, Zhao L, Liu K, Tang N. The role of lncRNAs in AKI and CKD: Molecular mechanisms, biomarkers, and potential therapeutic targets. Genes Dis. (2024) 12:101509. doi: 10.1016/j.gendis.2024.101509
57. Puri B, Majumder S, Gaikwad A. LncRNA MALAT1 as a potential diagnostic and therapeutic target in kidney diseases. Pathol Res Pract. (2025) 266:155783. doi: 10.1016/j.prp.2024.155783
58. Tan R, Jia J, Li T, Wang L, Kantawong F. A systematic review of epigenetic interplay in kidney diseases: Crosstalk between long noncoding RNAs and methylation, acetylation of chromatin and histone. Biomed Pharmacother. (2024) 176:116922. doi: 10.1016/j.biopha.2024.116922
59. Ramanathan K, Fekadie M, Padmanabhan G, Gulilat H. Long noncoding RNA: An emerging diagnostic and therapeutic target in kidney diseases. Cell Biochem Funct. (2024) 42:e3901. doi: 10.1002/cbf.3901
60. Liu Z, Fu Y, Yan M, Zhang S, Cai J, Chen G, et al. microRNAs in kidney diseases: Regulation, therapeutics, and biomarker potential. Pharmacol Ther. (2024) 262:108709. doi: 10.1016/j.pharmthera.2024.108709
61. Bakinowska E, Olejnik-Wojciechowska J, Kiełbowski K, Skoryk A, Pawlik A. Pathogenesis of sarcopenia in chronic kidney disease-the role of inflammation, metabolic dysregulation, gut dysbiosis, and microRNA. Int J Mol Sci. (2024) 25:8474. doi: 10.3390/ijms25158474
62. Rajabi S, Saberi S, Najafipour H, Askaripour M, Rajizadeh M, Shahraki S, et al. Interaction of estradiol and renin-angiotensin system with microRNAs-21 and -29 in renal fibrosis: Focus on TGF-β/smad signaling pathway. Mol Biol Rep. (2024) 51:137. doi: 10.1007/s11033-023-09127-4
63. Saleh M, Shaaban A, Talaat I, Elmougy A, Adra S, Ahmad F. RhoA/ROCK inhibition attenuates endothelin-1-induced glomerulopathy in the rats. Life Sci. (2023) 323:121687. doi: 10.1016/j.lfs.2023.121687
64. Cai A, Meng Y, Zhou H, Cai H, Shao X, Wang Q, et al. Podocyte pathogenic bone morphogenetic protein-2 pathway and immune cell behaviors in primary membranous nephropathy. Adv Sci. (2024) 11:e2404151. doi: 10.1002/advs.202404151
65. Chen Y, Huang C, Duan Z, Chen Y, Xu C. LncRNA NEAT1 accelerates renal fibrosis progression via targeting miR-31 and modulating RhoA/ROCK signal pathway. Am J Physiol Cell Physiol. (2023) 324:C292–306. doi: 10.1152/ajpcell.00382.2021
66. Miao H, Wang Y, Su W, Zou L, Zhuang S, Yu X, et al. Sirtuin 6 protects against podocyte injury by blocking the renin-angiotensin system by inhibiting the Wnt1/β-catenin pathway. Acta Pharmacol Sin. (2024) 45:137–49. doi: 10.1038/s41401-023-01148-w
67. Peng K, Zeng C, Gao Y, Liu B, Li L, Xu K, et al. Overexpressed SIRT6 ameliorates doxorubicin-induced cardiotoxicity and potentiates the therapeutic efficacy through metabolic remodeling. Acta Pharm Sin B. (2023) 13:2680–700. doi: 10.1016/j.apsb.2023.03.019
68. Sanz-Gómez M, Manzano-Lista F, Vega-Martín E, González-Moreno D, Alcalá M, Gil-Ortega M, et al. Finerenone protects against progression of kidney and cardiovascular damage in a model of type 1 diabetes through modulation of proinflammatory and osteogenic factors. Biomed Pharmacother. (2023) 168:115661. doi: 10.1016/j.biopha.2023.115661
69. Hu Y, Ni X, Chen Q, Qu Y, Chen K, Zhu G, et al. Predicting diabetic kidney disease with serum metabolomics and gut microbiota. Sci Rep. (2025) 15:12179. doi: 10.1038/s41598-025-91281-9
70. Liu P, Jin M, Hu P, Sun W, Tang Y, Wu J, et al. Gut microbiota and bile acids: Metabolic interactions and impacts on diabetic kidney disease. Curr Res Microb Sci. (2024) 7:100315. doi: 10.1016/j.crmicr.2024.100315
71. Wu J, Chen Y, Yang H, Gu L, Ni Z, Mou S, et al. Sodium glucose co-transporter 2 (SGLT2) inhibition via dapagliflozin improves diabetic kidney disease (DKD) over time associatied with increasing effect on the gut microbiota in db/db mice. Front Endocrinol. (2023) 14:1026040. doi: 10.3389/fendo.2023.1026040
72. Zhao H, Zhao T, Li P. Gut microbiota-derived metabolites: A new perspective of traditional Chinese medicine against diabetic kidney disease. Integr Med Nephrol Androl. (2024) 11:e23–4. doi: 10.1097/IMNA-D-23-00024
73. Lu J, Gong X, Zhang C, Yang T, Pei D. A multi-omics approach to investigate characteristics of gut microbiota and metabolites in hypertension and diabetic nephropathy SPF rat models. Front Microbiol. (2024) 15:1356176. doi: 10.3389/fmicb.2024.1356176
74. Deng X, Tang C, Fang T, Li T, Li X, Liu Y. Disruption of branched-chain amino acid homeostasis promotes the progression of DKD via enhancing inflammation and fibrosis-associated epithelial-mesenchymal transition. Metabolism. (2025) 162:156037. doi: 10.1016/j.metabol.2024.156037
75. Xu J, Wang N, Yang L, Zhong J, Chen M. Intestinal flora and bile acid interactions impact the progression of diabetic kidney disease. Front Endocrinol. (2024) 15:1441415. doi: 10.3389/fendo.2024.1441415
76. Shen Y, Cheng L, Zhang D. TRPV1: A novel target for the therapy of diabetes and diabetic complications. Eur J Pharmacol. (2024) 984:177021. doi: 10.1016/j.ejphar.2024.177021
77. Ma R, Tao Y, Wade M, Mallet R. Non-voltage-gated Ca2+ channel signaling in glomerular cells in kidney health and disease. Am J Physiol Renal Physiol. (2024) 327:F249–64. doi: 10.1152/ajprenal.00130.2024
78. Tu Y, Shu H, Sun L, Liao Q, Feng L, Ren M, et al. The physiopathologic roles of calcium signaling in podocytes. Front Biosci. (2023) 28:240. doi: 10.31083/j.fbl2810240
79. Liu J, Li X, Xu N, Han H, Li X. Role of ion channels in the mechanism of proteinuria (Review). Exp Ther Med. (2022) 25:27. doi: 10.3892/etm.2022.11726
80. Dryer S, Kim E. The Effects of TRPC6 knockout in animal models of kidney disease. Biomolecules. (2022) 12:1710. doi: 10.3390/biom12111710
81. Staruschenko A, Ma R, Palygin O, Dryer S. Ion channels and channelopathies in glomeruli. Physiol Rev. (2023) 103:787–854. doi: 10.1152/physrev.00013.2022
82. Smeijer J, Kohan D, Dhaun N, Noronha I, Liew A, Heerspink H. Endothelin receptor antagonists in chronic kidney disease. Nat Rev Nephrol. (2025) 21:175–88. doi: 10.1038/s41581-024-00908-z
83. Hung C, Lu L. New Insights into the Role of SGLT-2 inhibitors in the prevention of dementia. Neurol Int. (2024) 16:1717–30. doi: 10.3390/neurolint16060124
84. Viggiano D, Joshi R, Borriello G, Cacciola G, Gonnella A, Gigliotti A, et al. SGLT2 Inhibitors: The first endothelial-protector for diabetic nephropathy. J Clin Med. (2025) 14:1241. doi: 10.3390/jcm14041241
85. Speedtsberg E, Tepel M. Narrative review investigating the nephroprotective mechanisms of sodium glucose cotransporter type 2 inhibitors in diabetic and nondiabetic patients with chronic kidney disease. Front Endocrinol. (2023) 14:1281107. doi: 10.3389/fendo.2023.1281107
86. Myasoedova V, Bozzi M, Valerio V, Moschetta D, Massaiu I, Rusconi V, et al. Anti-Inflammation and anti-oxidation: The key to unlocking the cardiovascular potential of SGLT2 inhibitors and GLP1 receptor agonists. Antioxidants. (2023) 13:16. doi: 10.3390/antiox13010016
87. Liang I, Chang H, Lai Y, Chan C, Sung C, Pu C, et al. Update on the efficacy and safety of sodium-glucose co-transporter 2 inhibitors in patients with chronic diseases: a systematic review and meta-analysis. Medicina. (2025) 61:202. doi: 10.3390/medicina61020202
88. Zhang Z, Heerspink H, Chertow G, Correa-Rotter R, Gasparrini A, Jongs N, et al. Ambient heat exposure and kidney function in patients with chronic kidney disease: A post-hoc analysis of the DAPA-CKD trial. Lancet Planet Health. (2024) 8:e225–e233. doi: 10.1016/S2542-5196(24)00026-3
89. Bae J. SGLT2 Inhibitors and GLP-1 receptor agonists in diabetic kidney disease: Evolving evidence and clinical application. Diabetes Metab J. (2025) 49:386–402. doi: 10.4093/dmj.2025.0220
90. Heerspink H, Kiyosue A, Wheeler D, Lin M, Wijkmark E, Carlson G, et al. Zibotentan in combination with dapagliflozin compared with dapagliflozin in patients with chronic kidney disease (ZENITH-CKD): A multicentre, randomised, active-controlled, phase 2b, clinical trial. Lancet. (2023) 402:2004–17. doi: 10.1016/S0140-6736(23)02230-4
91. Chow E, Clement S, Garg R. Euglycemic diabetic ketoacidosis in the era of SGLT-2 inhibitors. BMJ Open Diabetes Res Care. (2023) 11:e003666. doi: 10.1136/bmjdrc-2023-003666
92. Kamath S, Kumar U, Shrivastava V. Sodium-glucose cotransporter 2 inhibitor-induced euglycemic diabetic ketoacidosis: The other side of the coin! Cureus. (2024) 16:e58341. doi: 10.7759/cureus.58341
93. Xu L, Wu Y, Li J, Ding Y, Chow J, Li L, et al. Efficacy and safety of 11 sodium-glucose cotransporter-2 inhibitors at different dosages in type 2 diabetes mellitus patients inadequately controlled with metformin: A Bayesian network meta-analysis. BMJ Open. (2025) 15:e088687. doi: 10.1136/bmjopen-2024-088687
94. Wang H, Zu Q, Lu M, Chen R, Tang Z, Yang Z. Cardiovascular outcomes in patients with complex type 2 diabetes mellitus treated with the dual sglt inhibitor sotagliflozin: A meta-analysis. Diabetes Ther. (2025) 16:485–98. doi: 10.1007/s13300-025-01696-w
95. Liao J, Chen Y, Ling Z, Pürerfellner H, Martinek M, Derndorfer M, et al. Effects of sodium-glucose co-transporter inhibitors on individual clinical endpoints and quality of life. ESC Heart Fail. (2025) 12:1271–82. doi: 10.1002/ehf2.15136
96. Wu P, Li S, Shu T, Mao Z, Fu W, Yang Y, et al. Impaired distal renal potassium handling in streptozotocin-induced diabetic mice. Am J Physiol Renal Physiol. (2024) 327:F158–70. doi: 10.1152/ajprenal.00240.2023
97. Jia G, Lastra G, Bostick B, LahamKaram N, Laakkonen J, Ylä-Herttuala S, et al. The mineralocorticoid receptor in diabetic kidney disease. Am J Physiol Renal Physiol. (2024) 327:F519–31. doi: 10.1152/ajprenal.00135.2024
98. Butt J, Jhund P, Henderson A, Claggett B, Desai A, Viswanathan P, et al. Finerenone and new-onset diabetes in heart failure: A prespecified analysis of the FINEARTS-HF trial. Lancet Diabetes Endocrinol. (2025) 13:107–18. doi: 10.1016/S2213-8587(24)00309-7
99. Filippatos G, Pitt B, Agarwal R, Farmakis D, Ruilope L, Rossing P, et al. Finerenone in patients with chronic kidney disease and type 2 diabetes with and without heart failure: A prespecified subgroup analysis of the FIDELIO-DKD trial. Eur J Heart Fail. (2022) 24:996–1005. doi: 10.1002/ejhf.2469
100. Agarwal R, Tu W, Farjat A, Farag Y, Toto R, Kaul S, et al. Impact of finerenone-induced albuminuria reduction on chronic kidney disease outcomes in type 2 diabetes : A mediation analysis. Ann Intern Med. (2023) 176:1606–16. doi: 10.7326/M23-1023
101. Eissing T, Goulooze S, van den Berg P, van Noort M, Ruppert M, Snelder N, et al. Pharmacokinetics and pharmacodynamics of finerenone in patients with chronic kidney disease and type 2 diabetes: Insights based on FIGARO-DKD and FIDELIO-DKD. Diabetes Obes Metab. (2024) 26:924–36. doi: 10.1111/dom.15387
102. Mazzieri A, Timio F, Patera F, Trepiccione F, Bonomini M, Reboldi G. Aldosterone synthase inhibitors for cardiorenal protection: Ready for prime time? Kidney Blood Press Res. (2024) 49:1041–56. doi: 10.1159/000542621
103. Li Z, Sun Z, Tang S, Zhao M, Chen M, Chang D. Finerenone alleviates over-activation of complement C5a-C5aR1 axis of macrophages by regulating G protein subunit alpha i2 to improve diabetic nephropathy. Cells. (2025) 14:337. doi: 10.3390/cells14050337
104. De P, Khine M, Frankel A, Goldet G, Banerjee D, Montero R, et al. Finerenone in the management of diabetes kidney disease. BMC Nephrol. (2025) 26:63. doi: 10.1186/s12882-025-03985-9
105. Shu J, Chen D, Chen W, Zhang X, Wang S, Chong N, et al. Finerenone ameliorates high glucose-induced podocytes epithelial-mesenchymal transition through the regulation of krüppel-like factor 5 in diabetic nephropathy. Diabetes Metab Syndr Obes. (2025) 18:637–51. doi: 10.2147/DMSO.S503133
106. Yao X, Guo H, Sun M, Meng S, Zhu B, Fang J, et al. Klotho ameliorates podocyte injury through targeting TRPC6 channel in diabetic nephropathy. J Diabetes Res. (2022) 2022:1329380. doi: 10.1155/2022/1329380
107. Sun R, Han M, Liu Y, Su Y, Shi Q, Huang L, et al. Trpc6 knockout protects against renal fibrosis by restraining the CN-NFAT2 signaling pathway in T2DM mice. Mol Med Rep. (2024) 29:13. doi: 10.3892/mmr.2023.13136
108. Catterall W. Voltage gated sodium and calcium channels: Discovery, structure, function, and Pharmacology. Channels. (2023) 17:2281714. doi: 10.1080/19336950.2023.2281714
109. Peng C, Yang L, Zhang C, Jiang Y, Shang L, He J, et al. Low-dose nifedipine rescues impaired endothelial progenitor cell-mediated angiogenesis in diabetic mice. Acta Pharmacol Sin. (2023) 44:44–57. doi: 10.1038/s41401-022-00948-w
110. Hajdys J, Fularski P, Leszto K, Majchrowicz G, Stabrawa M, Młynarska E, et al. New insights into the nephroprotective potential of lercanidipine. Int J Mol Sci. (2023) 24:14048. doi: 10.3390/ijms241814048
111. Zhu D, Ni Y, Chen C, Dong Z, Wang L, Zhang W. Geniposide ameliorates diabetic nephropathy in type 2 diabetic mice by targeting AGEs-RAGE-dependent inflammatory pathway. Phytomedicine. (2024) 135:156046. doi: 10.1016/j.phymed.2024.156046
112. Wu H, Gonzalez Villalobos R, Yao X, Reilly D, Chen T, Rankin M. Mapping the single-cell transcriptomic response of murine diabetic kidney disease to therapies. Cell Metab. (2022) 34:1064–78.e6. doi: 10.1016/j.cmet.2022.05.010
113. Tan W, Chen J, Wang Y, Xiang K, Lu X, Han Q, et al. Single-cell RNA sequencing in diabetic kidney disease: A literature review. Ren Fail. (2024) 46:2387428. doi: 10.1080/0886022X.2024.2387428
114. Montalbetti N, Przepiorski A, Shi S, Sheng S, Baty C, Maggiore J, et al. Functional characterization of ion channels expressed in kidney organoids derived from human induced pluripotent stem cells. Am J Physiol Renal Physiol. (2022) 323:F479–91. doi: 10.1152/ajprenal.00365.2021
115. Zhang H, Liu H, Xiang J, Zhou X, Wang D, Chen R. Alpha lipoamide inhibits diabetic kidney fibrosis via improving mitochondrial function and regulating RXRα expression and activation. Acta Pharmacol Sin. (2023) 44:1051–65. doi: 10.1038/s41401-022-00997-1
116. Lee H, Sun Y, Das F, Ju W, Nair V, Kevil C, et al. The CLCA1/TMEM16A/Cl- current axis associates with H2S deficiency in diabetic kidney injury. JCI Insight. (2025) 10:e174848. doi: 10.1172/jci.insight.174848
117. Priante G, Ceol M, Gianesello L, Bizzotto D, Braghetta P, Calò L, et al. Emerging perspectives on the rare tubulopathy dent disease: Is glomerular damage a direct consequence of ClC-5 dysfunction? Int J Mol Sci. (2023) 24:1313. doi: 10.3390/ijms24021313
118. Andrini O, Eladari D, Picard N. ClC-K kidney chloride channels: From structure to pathology. Handb Exp Pharmacol. (2024) 283:35–58. doi: 10.1007/164-2023-635
119. Dutta A, Das M. Deciphering the role of aquaporins in metabolic diseases: A mini review. Am J Med Sci. (2022) 364:148–62. doi: 10.1016/j.amjms.2021.10.029
120. Guo X, Kong Y, Kwon T, Li C, Wang W. Autophagy and regulation of aquaporins in the kidneys. Kidney Res Clin Pract. (2023) 42:676–85. doi: 10.23876/j.krcp.22.247
121. Guo Y, Qiao R, Xie G, Yao Y, Du C, Shao Y, et al. Activation of TGR5 increases urine concentration by inducing AQP2 and AQP3 expression in renal medullary collecting ducts. Kidney Dis. (2024) 10:181–92. doi: 10.1159/000538107
122. Zhang Z, Wang Z, Kan M, Tian M, Zhang Z. Novel dual-emissive up-conversion fluorescent probe for imaging ectopic lipid accumulation in diabetes mellitus. ACS Sens. (2025) 10:1959–69. doi: 10.1021/acssensors.4c03149
123. Zhang J, Li Y, Zhang F, Zhang Y, Zhang L, Zhao Y, et al. Hirudin delays the progression of diabetic kidney disease by inhibiting glomerular endothelial cell migration and abnormal angiogenesis. Biomed Pharmacother. (2024) 179:117300. doi: 10.1016/j.biopha.2024.117300
124. Tsai Y, Kuo M, Huang J, Chang W, Wu L, Huang Y, et al. Single-cell transcriptomic profiles in the pathophysiology within the microenvironment of early diabetic kidney disease. Cell Death Dis. (2023) 14:442. doi: 10.1038/s41419-023-05947-1
125. Kanbay M, Copur S, Bakir C, Covic A, Ortiz A, Tuttle K. Glomerular hyperfiltration as a therapeutic target for CKD. Nephrol Dial Transplant. (2024) 39:1228–38. doi: 10.1093/ndt/gfae027
126. Lacroix J, Wijerathne TD. PIEZO channels as multimodal mechanotransducers. Biochem Soc Trans. (2025) 53:BST20240419. doi: 10.1042/BST20240419
127. Zhang L, Liu Q, Yang X, Su C, Ding H, Hu J, et al. Mechanosensitive ion channel PIEZO1 as a key regulator of intestinal fibrosis in Crohn’s Disease. Inflamm Bowel Dis. (2025) 7:izaf041. doi: 10.1093/ibd/izaf041
128. Luo S, Zhao X, Jiang J, Deng B, Liu S, Xu H, et al. Piezo1 specific deletion in macrophage protects the progression of liver fibrosis in mice. Theranostics. (2023) 13:5418–34. doi: 10.7150/thno.86103
129. Drobnik M, Smólski J, Grądalski Ł, Niemirka S, Młynarska E, Rysz J, et al. Mechanosensitive cation channel piezo1 is involved in renal fibrosis induction. Int J Mol Sci. (2024) 25:1718. doi: 10.3390/ijms25031718
130. Ogino S, Yoshikawa K, Nagase T, Mikami K, Nagase M. Roles of the mechanosensitive ion channel Piezo1 in the renal podocyte injury of experimental hypertensive nephropathy. Hypertens Res. (2024) 47:747–59. doi: 10.1038/s41440-023-01536-z
131. Wang Y, Liu L, Li J, You Y, Xiao S, Feng J. Involvement of Piezo 1 in inhibition of shear-induced platelet activation and arterial thrombosis by ginsenoside Rb1. Br J Pharmacol. (2025) 182:1957–74. doi: 10.1111/bph.17434
132. Liu L, Li J, Wang Y, Gong P, Feng J, Xiao S, et al. Effects of Panax notoginseng saponins on alleviating low shear induced endothelial inflammation and thrombosis via Piezo1 signalling. J Ethnopharmacol. (2024) 335:118639. doi: 10.1016/j.jep.2024.118639
133. Li W, Zhang Z, Peng Z, Hu H, Cui X, Zhu Z, et al. PIEZO1-mediated calcium signaling and podocyte injury in diabetic kidney disease. J Am Soc Nephrol. (2025) [Online ahead of print]. doi: 10.1681/ASN.0000000634
134. Zhang Y, Lv L, Zhou Z, Zhang H, Li Q, Yang S, et al. Piezo1 facilitates the initiation and progression of renal fibrosis by mediating cell apoptosis and mitochondrial dysfunction. Ren Fail. (2024) 46:2415519. doi: 10.1080/0886022X.2024.2415519
135. Belosludtseva N, Dubinin M, Belosludtsev K. Pore-forming VDAC proteins of the outer mitochondrial membrane: Regulation and pathophysiological role. Biochemistry. (2024) 89:1061–78. doi: 10.1134/S0006297924060075
136. Xie Y, Jing E, Cai H, Zhong F, Xiao W, Gordon R, et al. Reticulon-1A mediates diabetic kidney disease progression through endoplasmic reticulum-mitochondrial contacts in tubular epithelial cells. Kidney Int. (2022) 102:293–306. doi: 10.1016/j.kint.2022.02.038
137. Waqar M. A comprehensive review on recent advancements in drug delivery via selenium nanoparticles. J Drug Target. (2025) 33:157–70. doi: 10.1080/1061186X.2024.2412142
138. Bhayana S, Schytz P, Bisgaard Olesen E, Soh K, Das V. Single-cell advances in investigating and understanding chronic kidney disease and diabetic kidney disease. Am J Pathol. (2025) 195:55–68. doi: 10.1016/j.ajpath.2024.07.007
139. Mazzieri A, Porcellati F, Timio F, Reboldi G. Molecular targets of novel therapeutics for diabetic kidney disease: A new era of nephroprotection. Int J Mol Sci. (2024) 25:3969. doi: 10.3390/ijms25073969
140. Zhang B, Wu Y, Wang Z, Gao S, Liu H, Lin Y, et al. Unveiling macrophage dynamics and efferocytosis-related targets in diabetic kidney disease: Insights from single-cell and bulk RNA-sequencing. Front Immunol. (2025) 16:1521554. doi: 10.3389/fimmu.2025.1521554
141. Zhou Y, Fang X, Huang L, Wu P. Transcriptome and single-cell profiling of the mechanism of diabetic kidney disease. World J Diabetes. (2025) 16:101538. doi: 10.4239/wjd.v16.i2.101538
142. Hu M, Deng Y, Bai Y, Zhang J, Shen X, Shen L, et al. Identifying key biomarkers related to immune response in the progression of diabetic kidney disease: Mendelian randomization combined with comprehensive transcriptomics and single-cell sequencing analysis. J Inflamm Res. (2025) 18:949–72. doi: 10.2147/JIR.S482047
143. Zhu Z, Luan G, Wu S, Song Y, Shen S, Wu K, et al. Single-cell atlas reveals multi-faced responses of losartan on tubular mitochondria in diabetic nephropathy. J Transl Med. (2025) 23:90. doi: 10.1186/s12967-025-06074-5
144. Wang L, Zhou W, Chen H, Jia X, Zheng P, Jiang H, et al. Barcoded screening identifies nanocarriers for protein delivery to kidney. Nat Commun. (2025) 16:899. doi: 10.1038/s41467-025-56257-3
145. Li J, Duan J, Hua C, Pan S, Li G, Feng Q, et al. Nanomedicine embraces the treatment and prevention of acute kidney injury to chronic kidney disease transition: Evidence, challenges, and opportunities. Burns Trauma. (2024) 12:tkae044. doi: 10.1093/burnst/tkae044
146. Han L, Cai X, Zhou H. Exosomal microRNAs: Potential nanotherapeutic targets for diabetic kidney disease. Nanomedicine. (2023) 18:1669–80. doi: 10.2217/nnm-2023-0023
147. Yang Y, Ren S, Xue J, Dong W, He W, Luo J, et al. DeSUMOylation of RBMX regulates exosomal sorting of cargo to promote renal tubulointerstitial fibrosis in diabetic kidney disease. J Adv Res (2024) 26, [Online ahead of print]. doi: 10.1016/j.jare.2024.09.021
148. Empa-Kidney Collaborative Group. Design, recruitment, and baseline characteristics of the EMPA-KIDNEY trial. Nephrol Dial Transplant. (2022) 37:1317–29. doi: 10.1093/ndt/gfac040
149. Asirvatham A, Asirvatham A, Mahadevan S. SGLT2 inhibitors and finerenone: A friendly duo in the treatment of diabetic kidney disease? J Assoc Physicians India. (2024) 72:e35–41. doi: 10.59556/japi.72.0759
150. Yang Y, Wei M, Chen L. Structural identification of riluzole-binding site on human TRPC5. Cell Discov. (2022) 8:67. doi: 10.1038/s41421-022-00410-5
151. Abramson J, Adler J, Dunger J, Evans R, Green T, Pritzel A, et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature. (2024) 630:493–500. doi: 10.1038/s41586-024-07487-w
152. Kashyap P, Bertelli S, Cao F, Kostritskaia Y, Blank F, Srikanth N, et al. An optogenetic method for the controlled release of single molecules. Nat Methods. (2024) 21:666–72. doi: 10.1038/s41592-024-02204-x
153. Kim K, Lee Y, Jung K, Kim Y, Jang E, Lee M. Highly stretchable 3D microelectrode array for noninvasive functional evaluation of cardiac spheroids and midbrain organoids. Adv Mater. (2025) 37:e2412953. doi: 10.1002/adma.202412953
154. Ren F, Lin J, Zhu M, Ma R, Zhang M, Chen W, et al. Polysaccharides from Alpinia oxyphylla fruit prevent hyperuricemia by inhibiting uric acid synthesis, modulating intestinal flora and reducing renal inflammation. Int J Biol Macromol. (2024) 278:134782. doi: 10.1016/j.ijbiomac.2024.134782
155. Yan Z, Zhang L, Kang Y, Liu S, Li X, Li L, et al. Integrating serum pharmacochemistry and network pharmacology to explore potential compounds and mechanisms of Alpiniae oxyphyllae fructus in the treatment of cellular senescence in diabetic kidney disease. Front Med. (2024) 11:1424644. doi: 10.3389/fmed.2024.1424644
156. Guo Z, Wu X, Zhang S, Yang J, Miao H, Zhao Y. Poria cocos: Traditional uses, triterpenoid components and their renoprotective pharmacology. Acta Pharmacol Sin. (2025) 46:836–51. doi: 10.1038/s41401-024-01404-7
157. Zhang C, Yue D, Wang D, Wu F. Effects of Bifidobacterium bifidum tetragonum tablets and Jin Gui Ren Qi Pill on intestinal flora and metabolism in patients with diabetic kidney disease. Front Pharmacol. (2024) 15:1346168. doi: 10.3389/fphar.2024.1346168
158. Ma L, Feng Z, Li P. New insights into the use of traditional chinese medicine for treating diabetic kidney disease by regulating DNA methylation. Integr Med Nephrol Androl (2024) 11:e24-00018. doi: 10.1097/IMNA-D-24-00018
159. Zhang W, Hu A, Wang J, Wang Y, Yu X. Use of chinese herbal medicine to inhibit podocyte damage as therapeutic strategy for membranous nephropathy. Integr Med Nephrol Androl. (2023) 10:e00004. doi: 10.1016/j.biopha.2019.108904
160. Bi J, Guo W, Ji P, Wang X, Xie Y. Diagnosis and treatment of membranous nephropathy in integrative medicine. Integr Med Nephrol Androl. (2024) 11:e23-00014. doi: 10.1097/IMNA-D-23-00014
161. Wang Y, Cui C, Tian X, Wang L, Ma X, Ge H, et al. The combination therapy of glucocorticoids, tacrolimus, and mycophenolate mofetil in primary membranous nephropathy coexisting with type 2 diabetes mellitus: A retrospective study. Integr Med Nephrol Androl. (2023) 10:1. doi: 10.1097/IMNA-D-22-00010
162. Wang Z, Fu Y, do Carmo J, da Silva A, Li X, Mouton A. Transient receptor potential cation channel 6 contributes to kidney injury induced by diabetes and hypertension. Am J Physiol Renal Physiol. (2022) 322:F76–88. doi: 10.1152/ajprenal.00296.2021
163. Hu H, Wang J, Peng Z, Fan Y, Yang Q, Hu J. Dapagliflozin attenuates diabetes-induced podocyte lipotoxicity via ERRα-Mediated lipid metabolism. Free Radic Biol Med. (2025) 234:178–91. doi: 10.1016/j.freeradbiomed.2025.04.028
164. Zhang Z, Li L, Dai Y, Lian Y, Song H, Dai X, et al. Dapagliflozin inhibits ferroptosis and ameliorates renal fibrosis in diabetic C57BL/6J mice. Sci Rep. (2025) 15:7117. doi: 10.1038/s41598-025-91278-4
165. Sunilkumar S, Subrahmanian S, Yerlikaya E, Toro A, Harhaj E, Kimball S, et al. REDD1 expression in podocytes facilitates renal inflammation and pyroptosis in streptozotocin-induced diabetic nephropathy. Cell Death Dis. (2025) 16:79. doi: 10.1038/s41419-025-07396-4
166. Ni Y, Du H, Ke L, Zheng L, Nan S, Ni L, et al. Gut-kidney interaction reinforces dapagliflozin-mediated alleviation in diabetic nephropathy. Am J Physiol Cell Physiol. (2025) 328:C452–66. doi: 10.1152/ajpcell.00651.2024
167. Da J, Xu Y, Tan Y, Zhang J, Yu J, Zhao J, et al. Central administration of Dapagliflozin alleviates a hypothalamic neuroinflammatory signature and changing tubular lipid metabolism in type 2 diabetic nephropathy by upregulating MCPIP1. Biomed Pharmacother. (2023) 168:115840. doi: 10.1016/j.biopha.2023.115840
168. Cai X, Cao H, Wang M, Yu P, Liang X, Liang H, et al. SGLT2 inhibitor empagliflozin ameliorates tubulointerstitial fibrosis in DKD by downregulating renal tubular PKM2. Cell Mol Life Sci. (2025) 82:159. doi: 10.1007/s00018-025-05688-8
169. Matsui T, Sotokawauchi A, Nishino Y, Koga Y, Yamagishi S. Empagliflozin ameliorates renal and metabolic derangements in obese type 2 diabetic mice by blocking advanced glycation end product-receptor axis. Mol Med. (2025) 31:88. doi: 10.1186/s10020-025-01138-0
170. Machado-Junior P, Lass A, de Bortolo J, Anizelli L, Rocha M, Proença H, et al. Sodium-glucose cotransporter-2 inhibitor improves renal injury by regulating the redox profile, inflammatory parameters, and pyroptosis in an experimental model of diabetic kidney disease. ACS Pharmacol Transl Sci. (2025) 8:1270–81. doi: 10.1021/acsptsci.4c00552
171. Guo C, Zhang T, Du L, Yu K, Zeng S, Li M, et al. Empagliflozin attenuates renal damage in diabetic nephropathy by modulating mitochondrial quality control via Prdx3-PINK1 pathway. Biochem Pharmacol. (2025) 235:116821. doi: 10.1016/j.bcp.2025.116821
172. Li X, Li Q, Jiang X, Song S, Zou W, Yang Q. Inhibition of SGLT2 protects podocytes in diabetic kidney disease by rebalancing mitochondria-associated endoplasmic reticulum membranes. Cell Commun Signal. (2024) 22:534. doi: 10.1186/s12964-024-01914-1
Keywords: ion channels, diabetic kidney disease, targeted therapy, precision medicine, sodium-glucose cotransporter 2
Citation: Wang W, Ke B, Wang C, Xiong X, Feng X and Yan H (2025) Targeting ion channel networks in diabetic kidney disease: from molecular crosstalk to precision therapeutics and clinical innovation. Front. Med. 12:1607701. doi: 10.3389/fmed.2025.1607701
Received: 08 April 2025; Accepted: 09 June 2025;
Published: 26 June 2025.
Edited by:
Ying-Yong Zhao, Northwest University, ChinaReviewed by:
Tingting Zhao, China-Japan Friendship Hospital, ChinaLing Chen, Shanghai Municipal Hospital of Traditional Chinese Medicine, China
Alessio Mazzieri, University of Perugia, Italy
Copyright © 2025 Wang, Ke, Wang, Xiong, Feng and Yan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hua Yan, MTgxODYwNTQ0NDVAMTYzLmNvbQ==; Xiuyuan Feng, Znh5MTIzNDEwQDE2My5jb20=
†These authors have contributed equally to this work