 Ankita Dua1
Ankita Dua1 Rashmi Kumari2
Rashmi Kumari2 Mona Singh3
Mona Singh3 Roushan Kumar4
Roushan Kumar4 Sunila Pradeep3,5Akinyemi I. Ojesina3,5,6*Roshan Kumar3,4,5*
Sunila Pradeep3,5Akinyemi I. Ojesina3,5,6*Roshan Kumar3,4,5*- 1Department of Zoology, Shivaji College, University of Delhi, New Delhi, India
- 2Department of Zoology, College of Commerce, Arts & Science, Patliputra University, Patna, Bihar, India
- 3Department of Obstetrics and Gynecology, Division of Gynecologic Oncology, Medical College of Wisconsin, Milwaukee, WI, United States
- 4Post-Graduate Department of Zoology, Magadh University, Bodh Gaya, Bihar, India
- 5Medical College of Wisconsin Cancer Center, Milwaukee, WI, United States
- 6Department of Microbiology and Immunology, Medical College of Wisconsin, Milwaukee, WI, United States
The global prevalence of Metabolic Dysfunction-Associated Steatotic Liver Disease (MASLD) has reached alarming levels, affecting nearly one-third of the world's population. This review analyzes current evidence on the intricate relationships between MASLD, insulin resistance, and type 2 diabetes mellitus (T2DM), with particular emphasis on gut microbiome interactions. As MASLD progresses from simple steatosis to Metabolic Dysfunction-Associated Steatohepatitis (MASH), it can lead to severe complications including fibrosis, cirrhosis, and hepatocellular carcinoma. The pathogenesis of MASLD is multifactorial, involving hepatic lipid accumulation, oxidative stress, inflammation, and dysregulation of the gut-liver axis. Insulin resistance is a central driver of disease progression, closely linked to obesity and metabolic syndrome. Recent research highlights how gut microbiome dysbiosis exacerbates MASLD through mechanisms such as increased intestinal permeability, systemic inflammation, and altered metabolic signaling. Identification of microbial signatures offers promise for novel diagnostic and therapeutic strategies. By integrating metabolic, inflammatory, and microbial perspectives, this review provides a comprehensive overview of MASLD pathogenesis and its association with obesity, insulin resistance, and T2DM.
Introduction
MASLD has emerged as the most common liver disease worldwide and has become a major health burden in both developed and emerging countries (1, 2). Its global prevalence is high, affecting roughly 30% of the population, and has shown an alarming 50.4% relative increase between 1990 and 2019 (3). MASLD is a disorder characterized by hepatic steatosis [fat deposition in > 5% hepatocytes (4)]; when no other cause for secondary fat accumulation like excess alcohol consumption can be identified. It can be diagnosed in a patient meeting one out of five cardiovascular risk factors (5). It ranges from benign non-inflammatory condition (NAFL) to severe MASLD which includes portal and lobular inflammation (6). Without intervention, MASLD may progress to fibrosis, cirrhosis, or even hepatocellular carcinoma (HCC) (7, 8). MASLD is the hepatic manifestation of metabolic syndrome (metS), driven by genetic variants like PNPLA3 rs738409 G and strongly associated with metabolic comorbidities, including obesity, T2DM, hyperlipidaemia and hypertension (8–10). MASLD is now recognized as a multifactorial disease, and recent literature proposes this renaming to better reflect its metabolic origins and remove alcohol-related exclusions (11).
The beginning of the concept of non-alcoholic fatty liver disease (NAFLD) was reported in the year 1980 by Ludwig et al. as a condition that can progress to cirrhosis without consumption of significant alcohol (12). The patients had diabetes mellitus and were obese. There have been significant changes in this nomenclature over the years giving the strong association of NAFLD with various metabolic factors. A new term for the condition “Metabolic (dysfunction) associated fatty liver disease (MAFLD)” was coined by Eslam et al. in 2020, that correlated with hepatic steatosis that could be diagnosed with at least two metabolic risk abnormalities, obesity, blood biomarkers or in the presence of T2DM (13). An important factor in this new nomenclature was to avoid any reference to alcohol in the MAFLD acronym. This has been further supported by the Asian Pacific Association for the Study of the Liver (APASL), multiple national societies including the Malaysian Society of Gastroenterology and Hepatology and a wide range of global stakeholders (11, 14, 15).
Several years later, the term MASLD was proposed, and its diagnosis can be done based on the patient meeting one of five cardiovascular risk factors, unlike MAFLD, which underlines a requirement that the patients meet two of seven parameters of metabolic dysfunction (5). MetALD is a term coined for patients with MASLD along with consumption of alcohol (140–350 g/week and 210–420 g/week for females and males, respectively).
The severe form of MASLD is MASH, a replacement of the term non-alcoholic steatohepatitis (NASH), characterized by the presence of lobular inflammation and ballooning of hepatocytes and is associated with a greater risk of fibrosis progression (Figure 1). A multi-society Delphi consensus statement on a new fatty liver disease nomenclature was published in 2023, thereby introducing the term metabolic dysfunction-associated steatotic liver disease (MASLD) and effectively letting go of the term NAFLD and MAFLD (16). The nomenclature was based on diagnostic criteria that was non-stigmatizing and aims to improve patient awareness.
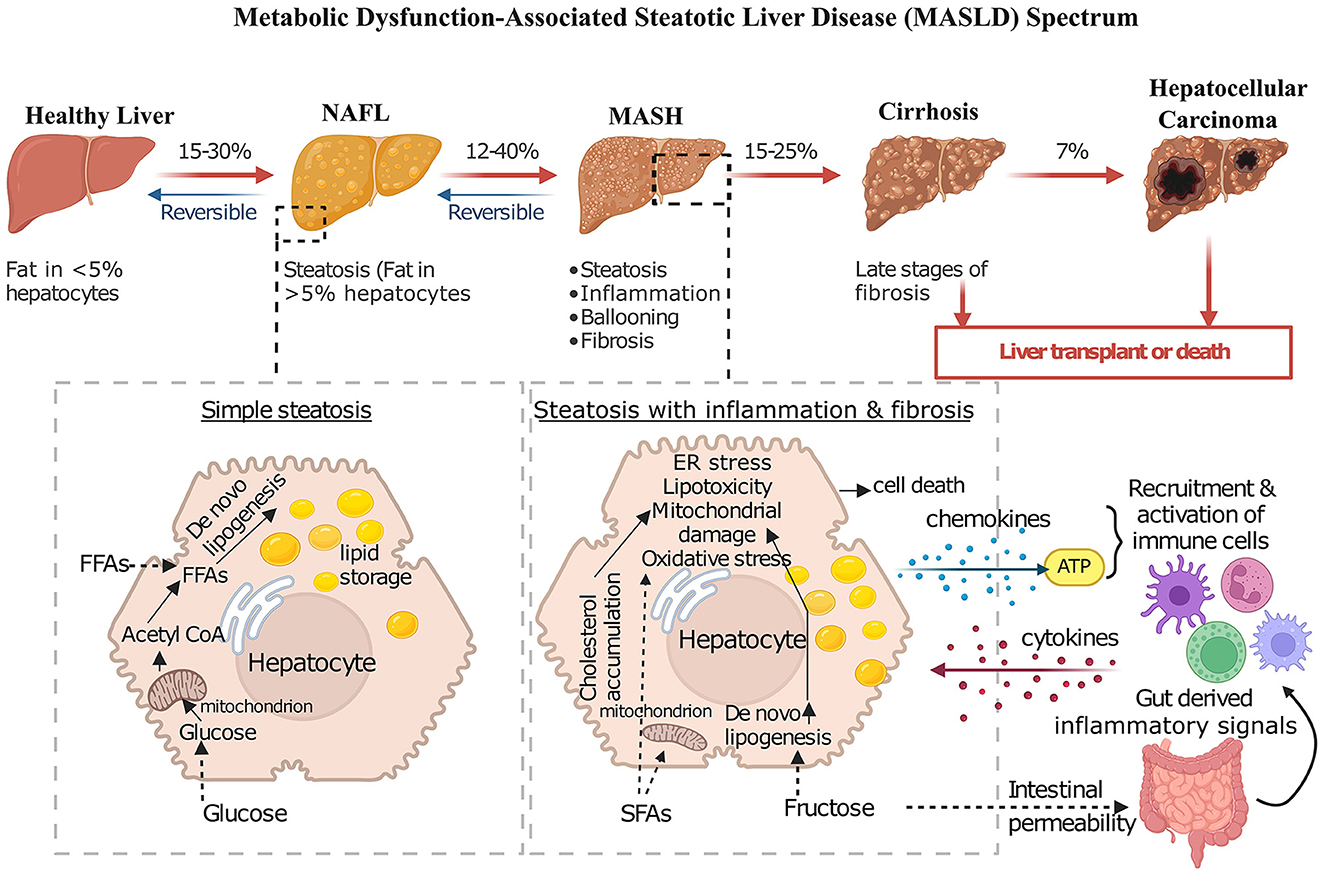
Figure 1. Progressive stages of MASLD and its complications. This diagram shows the progressive nature of MASLD, encompassing a spectrum of liver conditions ranging from simple steatosis to advanced stages such as MASH, cirrhosis, and hepatocellular carcinoma. In healthy individuals, fat content within hepatocytes typically remains low. In NAFL, fat accumulation increases, but the condition is often reversible with lifestyle modifications. MASH, however, involves not only fat accumulation but also inflammation, increasing degeneration of hepatocytes, and the formation of fibrosis. Cirrhosis, the most advanced stage, is characterized by extensive scarring that disrupts liver function, leading to serious complications like liver failure and increasing the risk of developing hepatocellular carcinoma. The diagram also depicts key pathological mechanisms contributing to MASH progression, including lipid accumulation due to increased uptake of free fatty acids and de novo lipogenesis (DNL), mitochondrial dysfunction, ER stress, oxidative stress, and inflammation mediated by immune cell activation and cytokine release.
Dysbiosis of the gut microbiota has been consistently linked to both obesity and T2DM—two metabolic disorders closely associated with MASLD. By examining the gut-liver axis as the central integrative pathway, this review explores how microbial dysbiosis mechanistically contributes to MASLD initiation and progression, and how hepatic metabolic dysfunction reciprocally alters gut microbiome composition. Our goal is to integrate essential factors such as obesity, insulin resistance, the gut-liver axis, immune system changes, and the role of the microbiome, providing a comprehensive overview of this increasingly common and complex metabolic disorder.
Pathophysiology of MASLD/MASH
MASLD is characterized by excessive fat accumulation in the liver, with a spectrum ranging from simple steatosis to MASH and potentially cirrhosis (Figure 1).
The development of MASLD occurs in a coordinated fashion and was proposed earlier as a two-hit hypothesis (17). The first hit is the steatosis through de novo lipogenesis (DNL) in the liver which increases the insulin resistance (17, 18). The second hit means the progression from MASLD to MASH, representing a critical escalation in liver disease severity involving additional cellular and molecular stresses like endoplasmic reticulum (ER) stress, mitochondrial damage, oxidative stress [involving the production of reactive oxygen species (ROS)] (19, 20). Accumulation of saturated fatty acids (SFAs) increases DNL resulting from increased fructose uptake, or cholesterol accumulation in the ER leading to cellular stress (Figure 1) (17, 21). Increased fructose levels are also one of the key contributors to the progression of MASLD to MASH by increasing gut permeability, which initiates a cascade of inflammatory responses by releasing cytokines and promoting microbiota dysbiosis (22). This dysregulation is compounded by heightened activation of hepatic toll-like receptors (TLRs), changes in bile acid metabolism, and local alcohol production by gut microbes, all of which can exacerbate inflammation and tissue damage (23). Elevated blood ethanol levels in MASH patients further indicate the presence of alcohol-producing bacteria, which potentially elevate the production of ROS, adding another layer of hepatic inflammation and stress (24).
However, at present it is referred as the “multi-hit hypothesis” as the development and progression of the disease arises from a combination of factors that are interconnected and contribute to the advancement of the disease.
Lipogenesis is fuelled by the uptake of glucose and free fatty acids (FFAs) and their incorporation into lipid-synthesis pathways. In most cases, steatosis is an early event in MASLD, but it does not necessarily transition to MASH. Lipogenesis in the liver and lipolysis of the adipose tissue result in elevated circulating FFA and their metabolites which can induce inflammatory responses. This further activates TLR4 signaling that activates the NF-kB pathway; these are crucial to progression to MASH (5). This leads to inflammation, fibrosis and hepatocarcinogenesis (25). Beyond lipid accumulation, insulin resistance is important in MASLD as its presence leads to increased FFA in blood and lipid accumulation. Interleukins and cytokines are released by adipocytes and ROS are generated due to oxidation of excess FFA (26, 27). Oxidative stress in such conditions is a step forward during development of fibrosis in MASH. Another factor that contributes to progression of MASLD is autophagic dysfunction in cellular degradative organelles. Damaged mitochondria during MALSD, unable to undergo autophagy also contribute to build-up of ROS and lowering the defense system of the liver (28). Ultimately, dysbiosis of the gut microbiota plays a key role in driving hepatic fibrogenesis. Excess lipid accumulation compromises the gut barrier, and hence microbial toxins translocate into the bloodstream. The cumulative effect of these pathogenic processes promotes hepatic inflammation and triggers apoptosis of hepatocytes. These cellular damages lead to release of pro-inflammatory mediators such as ATP, extracellular vesicles, and chemokines, subsequently reinforcing the inflammatory process and leading to the development of fibrosis. Further, the prolonged inflammation may progress to cirrhosis and ultimately HCC.
The interplay between MASLD, insulin resistance, and metabolic syndrome
MASLD is strongly associated with metabolic syndrome, which includes obesity, T2DM, dyslipidemia, and hypertension (Figure 2) (29–32). As the global prevalence of obesity and T2DM rises, so does the prevalence of MASLD, emphasizing its connection to metabolic dysfunction (33–35). Given its nature, the disease's progression is marked by insulin resistance (IR), particularly in the liver. Impaired ability of insulin to suppress endogenous glucose synthesis, 45–50% reductions in glucose disposal and a measure of whole-body insulin sensitivity are the indicative of hepatic insulin resistance (30, 36).
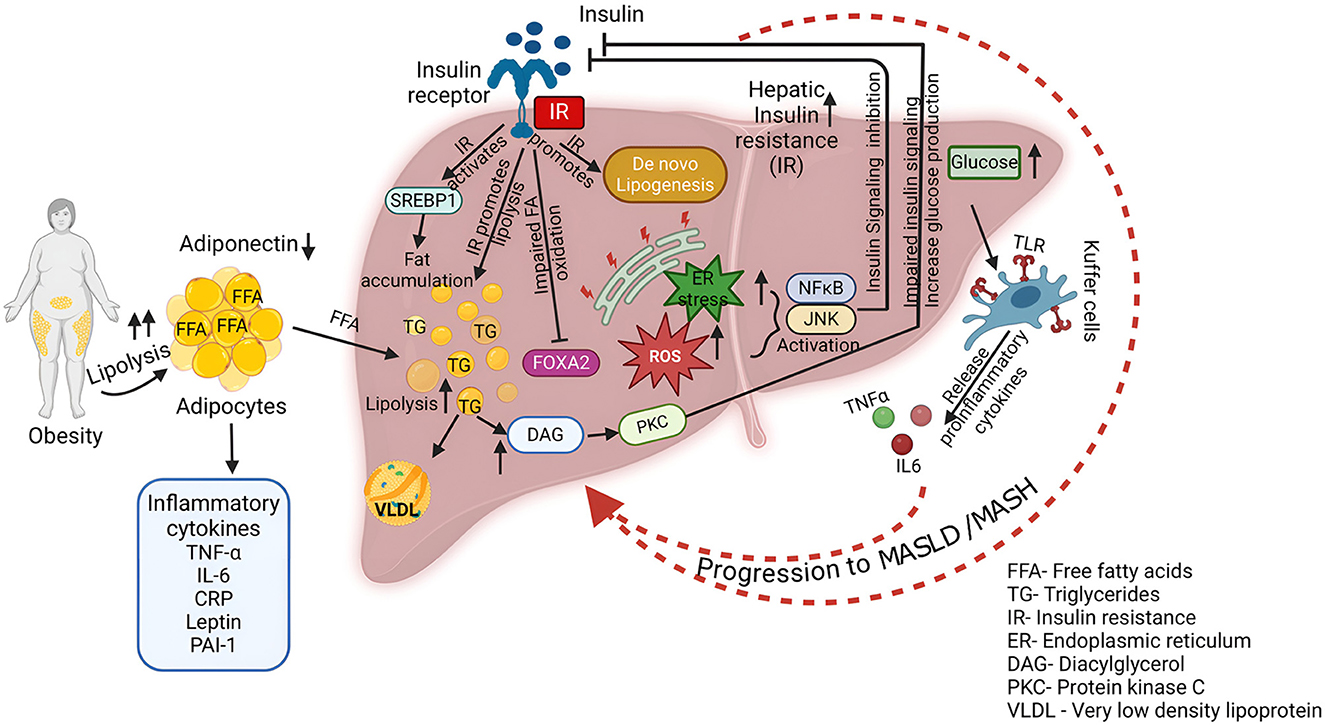
Figure 2. Association of MASLD with metabolic syndrome. This diagram shows the complex interplay of factors contributing to the development and progression of MASLD/MASH. Key processes include increased lipolysis in obese individuals, leading to elevated FFA levels. Insulin resistance impairs hepatic insulin signaling, promoting DNL, reducing fatty acid oxidation, and increasing glucose production. Excessive FFA influx, coupled with impaired fatty acid oxidation and increased DNL, results in triglyceride accumulation within the liver. This lipid accumulation triggers ER stress, leading to the activation of inflammatory pathways. Activated Kupffer cells release pro-inflammatory cytokines such as TNF-α and IL-6, exacerbating inflammation and tissue damage. Furthermore, increased fatty acid oxidation generates ROS, contributing to oxidative stress and hepatocellular damage.
Obesity is widely recognized as a key factor in promoting systemic inflammation, which is closely linked to the development of IR (37). Specifically, abdominal visceral fat plays a significant role in both peripheral and hepatic IR in individuals with T2DM, while excessive subcutaneous fat in men has also been associated with IR in the liver and peripheral tissues (38). Adiponectin, an adipokine secreted by adipocytes, shows an inverse relationship with the amount of fat in the abdomen and liver, which is closely tied to both hepatic and peripheral IR (39, 40). Obesity activates various proinflammatory pathways, which includes elevated level of cytokines such as tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), C-reactive protein (CRP), plasminogen activator inhibitor-1 (PAI-1), and leptin (Figure 2) (37, 41). These molecules are the main causative factors in the pathophysiology of IR. Conditions of obesity or excessive nutrient intake induce ER stress, production of ROS, and accumulation of ceramides, which activate the NF-κB and JNK pathways, leading to insulin signaling inhibition (42–45). Inhibition of JNK1 and IKK-β (which activates NF-κB) in mouse models has shown improvements in IR both locally in the liver and systemically, highlighting the role of inflammation in exacerbating IR (46, 47). This mutual reinforcement of inflammation and IR creates a vicious cycle that worsens both conditions (48).
IR is considered a central mechanism in the development and progression of MASLD to MASH and cirrhosis, impairing the liver's ability to manage fat (49–51). The liver's supply of fatty acids, a primary source of triglyceride (TG) synthesis, comes from dietary fat, lipolysis in adipocytes, and DNL (Figure 2) (49). Both high-fat and high-carbohydrate diets promote fat accumulation in the liver, with FFAs from adipocytes playing a critical role (52). Normally, insulin suppresses lipolysis, but in the state of IR, this suppression is impaired, leading to excessive FFAs that accumulate in the liver (53, 54). Additionally, hyperinsulinemia, a hallmark of IR, further exacerbates liver fat accumulation by promoting DNL through the activation of sterol regulatory element-binding protein 1c (SREBP-1c) (55). In addition to increasing liver fat synthesis, hyperinsulinemia elevates triglyceride production and promotes very-low-density lipoprotein (VLDL) synthesis. However, insulin resistance impairs fatty acid oxidation, a process regulated by forkhead box protein A2 (FOXA2) (55). In normal conditions, FOXA2 promotes lipid metabolism, but it remains inactive in hyperinsulinemic states, leading to the accumulation of fat in the liver. As part of the liver's adaptation to excessive FFAs, mitochondrial respiration rates increase, yet excessive fatty acid oxidation results in oxidative stress and hepatocellular damage, contributing to the progression to MASH (56). While IR is a major cause of fatty liver, some evidence suggests that fatty liver itself can exacerbate IR (57, 58). The influx of FFAs into the liver leads to an abnormal increase in long-chain fatty acyl-CoA and diacylglycerol (DAG), which in turn activates protein kinase C-δ (PKC-δ) (59). This activation disrupts insulin signaling and promotes glucose production in the liver. FFAs also activate the IKK-β and JNK pathways, further exacerbating IR via PKC-θ activation (60). These inflammatory pathways, activated by fatty acid influx, play a central role in the vicious cycle of liver fat accumulation and IR. In MASLD, a paradoxical relationship exists in which increased DNL coexists with inappropriately elevated gluconeogenesis despite hyperinsulinemia. This has led to the concept of pathway-specific hepatic insulin resistance, where the insulin activation pathway involving protein kinase B/forkhead box protein O1 is inhibited, while the SREBP-1c pathway remains activated (61). Activation of carbohydrate response element-binding protein (ChREBP) also induces an increase in precursors of DNL and an increase in enzymes that further aggravate hepatic steatosis, especially under exposure to lipogenic substrates (62).
As MASLD progresses, hepatic inflammation, particularly involving M1 macrophages (Kupffer cells), becomes a key factor in disease progression (63). These macrophages, when activated by TLR ligands and interferon-γ, release proinflammatory cytokines such as TNF-α and IL-6, which contribute to the progression of MASLD and systemic IR by modulating other immune cells (64) (Table 1).
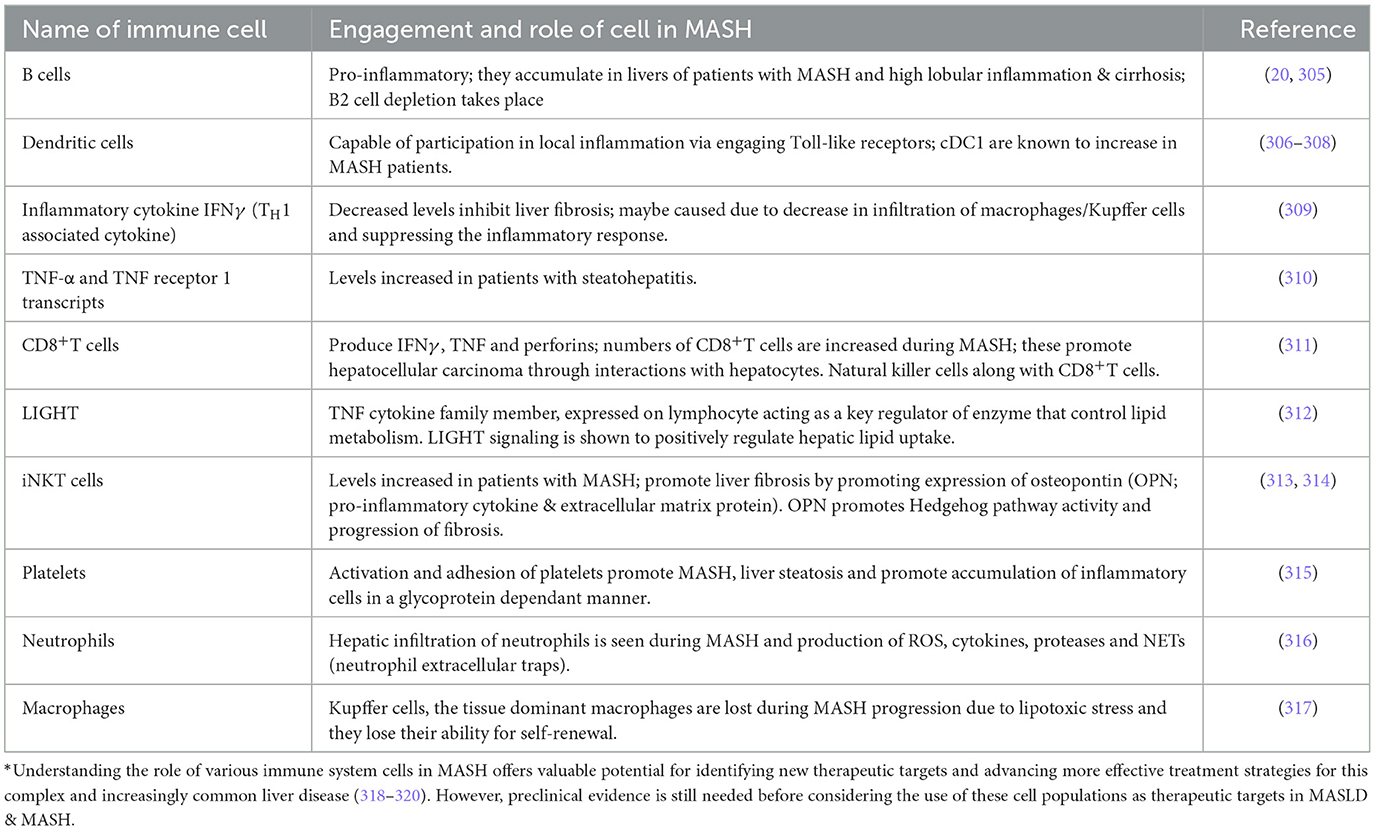
Table 1. Engagement of different cells of the immune system in severity and progression of MASH.*
Gut microbiome and MASLD: a key connection
Building on the understanding of MASLD and its progression to MASH, the role of the gut microbiome has emerged as a significant factor in disease pathogenesis. Reviewing microbial signatures with respect to various stages of progression of liver disease would be very useful for predicting biomarkers as well for designing therapeutic approaches (Table 2). Gut microbiota, the collective genome of gut-residing microbes, is increasingly recognized as an environmental factor that influences metabolic health by impacting energy balance, inflammation, and IR (65–70). It has been demonstrated in both mice and human studies that obesity and T2DM are associated with changes in gut microbiota, though it is not clear whether these are the cause or effect of the underlying metabolic changes (71). Specifically, obese people and mice have fewer Bacteroidetes and more Firmicutes compared with their lean counterparts. More importantly, the relative proportion of these two major bacterial divisions is positively correlated with body weight, i.e., a higher proportion of Firmicutes and a lower proportion of Bacteroidetes. This altered ratio is associated with increased energy harvest from food, potentially exacerbating obesity (71). High-fat diets (HFD) in mice have also been shown to reduce beneficial bacteria such as Bifidobacteria, which improve mucosal barrier function, reducing gut permeability and inflammation (72). Such microbiota shifts promote inflammation and metabolic dysfunction, creating a cycle that worsens insulin resistance and metabolic syndrome (71, 73).
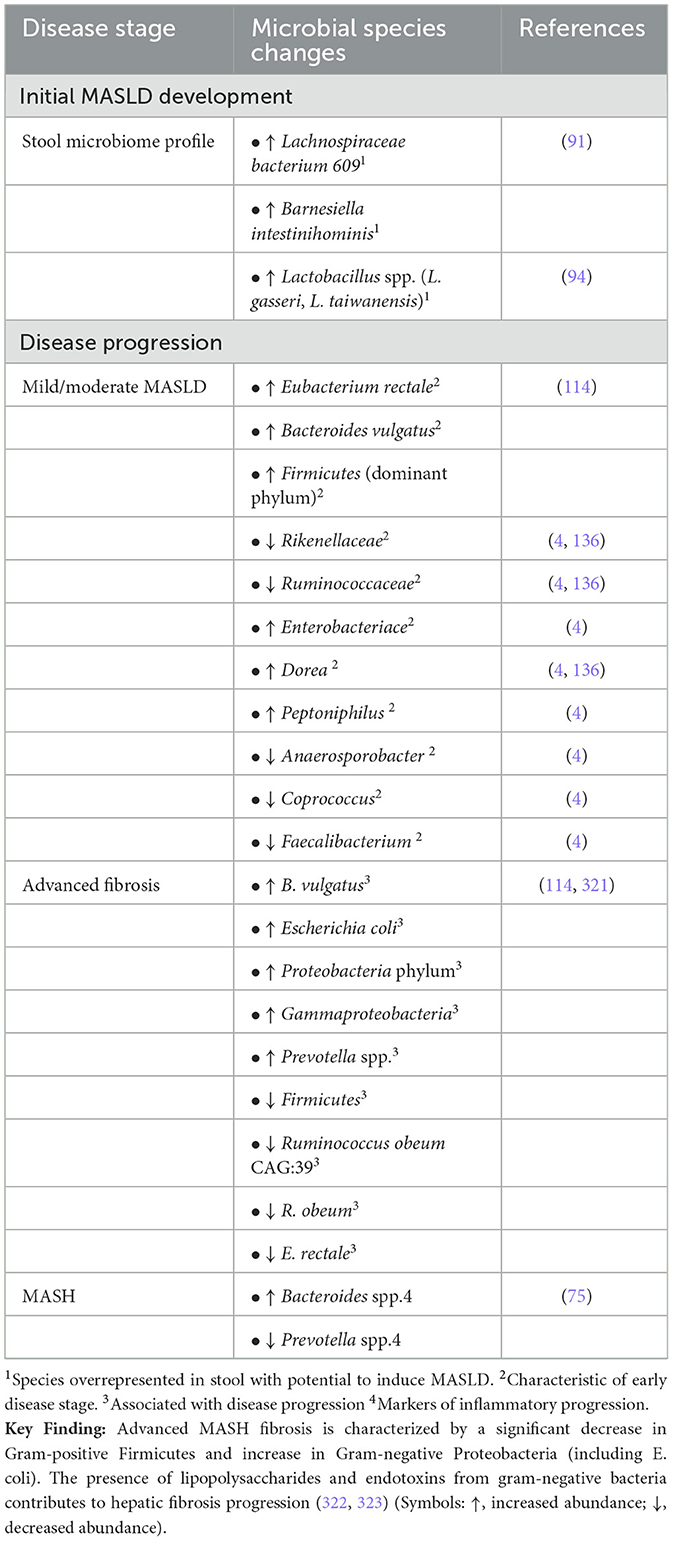
Table 2. Microbial species distribution across different stages of MASLD and MASH.
The genome of the microbiome bears information to coding of several enzymes that are absent in the human host, and they cooperate with contribution to individualized traits in the host (74). Obesity and improper eating habits have been significant co-occurrences with MASLD; however, presence/absence of many bacterial species have altered the microbiome in such conditions. Studies have shown increased abundance of bacteria from the Proteobacteria phylum, especially within the Enterobacteriaceae family and the Escherichia genus in individuals with MASLD and MASH compared to their healthy counterparts (24). An increased abundance of Bacteroides genus was reported in MASH (75). Pathogenesis of MASH has been influenced by increased fecal content of deoxycholic acid, raffinose, choline, D-pinitol and stachyose in patients.
A 16S rRNA cohort study of biopsy proven Asian population was done to explore microbial markers for assessment of severity of fibrosis between obese and non-obese subjects (76). An increase in levels of total bile acid, especially primary bile acids and ursodeoxycholic acid (UDCA), and propionate levels was seen in stool samples of subjects with worsening fibrosis. A dominance of bacterial population of Veillonellaceae was found in non-obese individuals with MASLD. Six genera of Gram- negative bacteria (Megasphaera, Veillonella, Dialister, Allisonella, Anaeroglobus, and Negativicoccus) are a part of the family Veillonellaceae and these are known to produce propionate-and can utilize lactate as a substrate. It has been proposed that accumulation of these populations lead to more propionate production that is absorbed into the liver and hence progression to MASLD. On the contrary, Ruminococcaceae members were found to decrease in numbers in fibrosis in MASLD non-obese patients and are responsible for maintaining homeostasis of the gut microbial environment (Table 2). R. faecis exerted a protective effect on liver damage. It was concluded that assessment of gut microbes and stool metabolites could be used for diagnosis of fibrosis in non-obese subjects with MASLD.
The impact of diet on gut microbiota composition further influences MASLD progression, as evidenced by studies showing that Bacteroides thrive on high-fat animal-based diets, while Prevotella is more prevalent with plant-based polysaccharide diets (77, 78). Contents of branched chain fatty acids due to Bacteroides population produced by fermentation of amino acids are correlated with insulin resistance that further gives impetus to development of MASH. A study conducted with stool samples of mice with MASLD-MCC revealed that oral administration of a species of Bifidobacterium pseudolongum was successful in preventing hepatocellular carcinogenesis (10). Ramos et al. performed a detailed analysis of the gut microbiome of patients with biopsy proven MASLD and their study concluded an enrichment of Parabacteroides distasonis and Alistipes putredenis species in MASLD patients. They also found that Prevotella copri, was a dominant species for MASLD disease progression also linked to higher intestinal permeability (79). Iljazovic et al. (80) demonstrated that Prevotella populations, previously associated with colitis in animal studies, can worsen intestinal inflammation and potentially lead to systemic autoimmune responses condition (24). This may occur by reducing IL-18 production, which further intensifies gut inflammation. Increased population of Prevotella has been linked to mucosal sites with inflammation and increase in T-helper type 17 cells mediating this process (81). These bacteria have also been implicated in activation of TLR-2, that lead to production of Th17- polarizing cytokines by antigen presenting cells such as IL-23 and IL-1. They mediate spreading of inflammatory mediators and bacterial products. Additionally, elevated blood ethanol levels in MASH patients indicate the presence of alcohol-producing bacteria, which contributes to the production of ROS, further increasing hepatic inflammation and oxidative stress.
In patients with MASH, gut microbiota changes extend beyond metabolic influences to play a direct role in liver pathology. The composition of gut microbiota can affect the liver due to the portal circulation of venous blood from the gut to the liver. Changes in gut microbes and their derived products can have effects on systemic and hepatic immunity, inflammation, and liver architecture and function (82–88). It is now recognized that MASLD is the hepatic expression of metabolic syndrome and given the association of gut microbiota with obesity and insulin resistance, several studies have sought to investigate the role of gut microbiota in MASLD (89–91). The most abundant species traced during advanced fibrosis are E.coli and Bacteroides vulgatus (4, 79). B. vulgatus is known to increase with obesity, increment in BMI, Hb1Ac and insulin resistance (92). The bacterial populations in advanced stages of cirrhosis undergo a major transformation, with an increase in pathogenic bacteria and decrease in beneficial bacteria. A reduction in Faecalibacterium prausnitzii, an anti-inflammatory species has been reported in subjects with cirrhosis (92, 93). Its decrease is also seen in other conditions such as obesity, T2DM and bowel diseases (4).
Several animal studies have analyzed the changes in gut microbiota in response to high fat feeding (73, 91, 94). Mice deficient in the hormone leptin are called ob/ob and the ones deficient in leptin receptor are called db/db that mimic conditions of obese and diabetic strains respectively. In the ob/ob mouse, the changes in microbiota occurred rapidly following the administration of a high fat diet resulting in an increase in proportional weight in Firmicutes and a decrease in Bacteroidetes (71, 85) and an increase in intestinal permeability. A decrease in Bacteroidetes was also seen in the TLR4 knockout mouse whilst there were no differences in wild type mice. More recent studies have found that high fat diets in mice lead to increased presence of gram-negative bacteria, malonaldehyde (MDA) modified end products, and reduced defensin expression. An MDA rich environment may cause induction of bacterial cytolysins and affect antimicrobial defense mechanisms in the gut. Several studies have examined diet-induced changes in gut microbiota in mice strains fed a high fat or high sucrose diet and, in each case, a significant proportional increase in Firmicutes and decrease in Bacteroidetes was seen (95–99). High fat or high sucrose feeding also led to increased Dorea and Eubacterium rectale and decreased Bifidobacterium (100–103). To date, the only human dietary intervention study found that weight loss by energy restriction with a Mediterranean or low-fat diet increased Firmicutes and reduced Bacteroidetes: Eubacterium rectale in obese individuals (101, 104–106). This is important as it shows that specific changes in human gut microbiota can be linked to dietary patterns and may be relevant to the etiology of MASLD (90).
Gut barrier dysfunction and endotoxemia
Intestine is one of the most crucial internal barriers and its disturbance leads to an immune response as bacterial products pass through the gut, and evidence shows that the immune systems of patients with MASLD are primed toward a pro-inflammatory state. This explains why the severity of MASLD is often associated with the presence of an inflammatory state, and the immune response is closely linked with the mechanism of liver damage and inflammation. Intestinal permeability refers to ability of the extracellular barrier to allow any exchange between tissues and intestinal lumen. The gut barrier limits passage of potentially pathogenic molecules and microorganisms to the systemic circulation (107). Passage of bacteria and their products from the gut lumen to the bloodstream and liver and spleen is known as intestinal bacterial translocation. Livers of Healthy individuals have an exposure of small bacterial products such as lipopolysaccharides (LPS), a dominant molecule on surface of Gram-negative bacteria. Increased levels of LPS are found in patients of inflammatory diseases. Hepatic inflammation is resultant of a complex interaction of Kupffer cells, neutrophils, hepatocytes and sinusoidal cells. Metabolic dysfunction in the liver takes place because of interaction of hepatocytes and Kupffer cells with pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) and initiating a series of inflammatory events.
Increased blood endotoxin levels have been detected in patients with MASLD on comparison with healthy liver controls which also suggests that these could be used an indicative biomarker for progression of liver disease (108). The LPS present on the outer cell membrane of Gram-negative bacteria in the intestine constitute these endotoxins that can induce inflammatory activities. Dysbiosis coupled with disturbance of the intestinal barrier leads to release of endotoxins from the lumen of the gut into the circulation and it enters the liver via the portal vein (Figure 3). Increase in levels of endotoxins is also coupled with a simultaneous increase in C- reactive protein (CRP) that is considered as a marker for systemic inflammation (Figure 3). Excessive growth of aerobic and anaerobic gram-negative bacteria is known to cause a condition called small intestinal bacterial overgrowth (SIBO) (109). This contribution of SIBO can be due to increased oxidative stress, insulin resistance, increased ethanol production and by modulating choline metabolism by promoting the excessive conversion of choline into trimethylamine (TMA) by gut bacteria, leading to potential choline deficiency and contributing to the development of MASLD. Increase in gut permeability due to disruption of the tight cell junctions in SIBO affected individuals would lead to entry of bacteria and their products—endotoxins. This in turn induces expression of nuclear kappa B expression that activates TLR-4 and proinflammatory cytokines such as TNF-α and IL-6 and IL-8 (Figure 3) (110, 111).
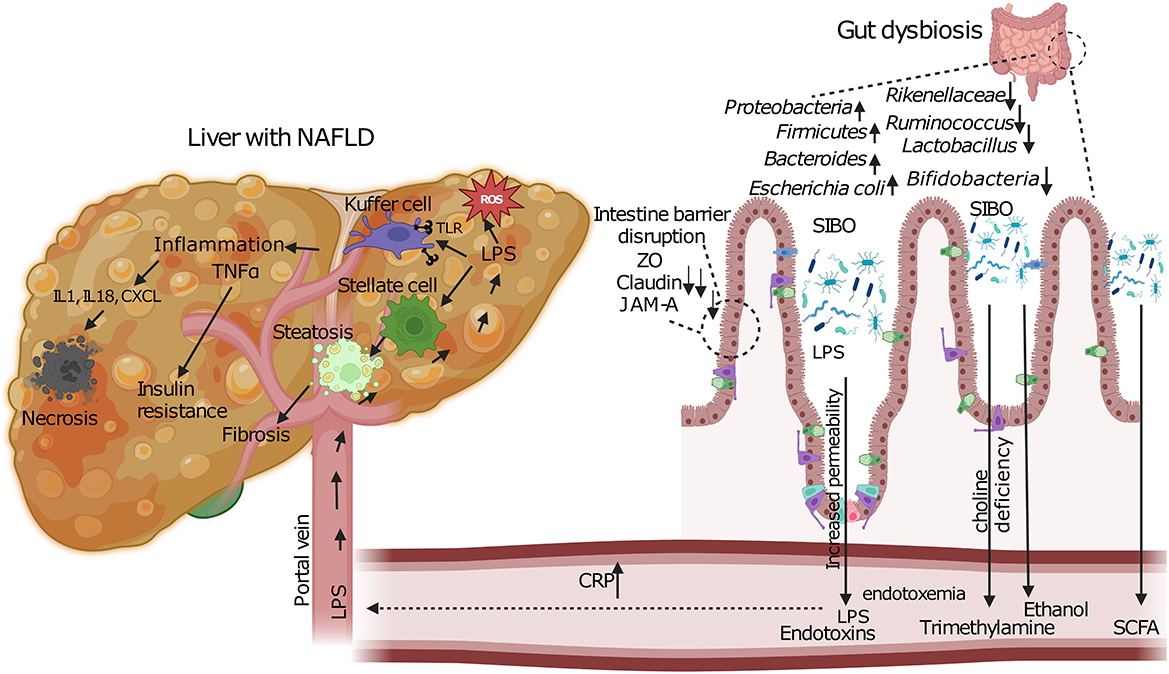
Figure 3. Pathogenic mechanisms linking gut dysbiosis to MASLD progression. Intestinal dysbiosis and SIBO lead to barrier dysfunction through disruption of tight junction proteins (ZO, Claudin, JAM-A). This results in increased intestinal permeability and translocation of bacterial endotoxins (LPS) via the portal vein. In the liver, LPS activates Kupffer and stellate cells, triggering inflammatory cascades (TNFα, IL1, IL18, CXCL) and oxidative stress (ROS). Concurrent metabolic alterations include increased ethanol production, altered SCFA metabolism, and choline deficiency due to enhanced bacterial conversion to trimethylamine. These pathways collectively promote hepatic steatosis, inflammation, and fibrosis characteristic of MASLD.
Impact of dysbiosis in MASLD
Gut dysbiosis disrupts the bile acid metabolism pathway, which in turn causes dysfunction of the gut-liver axis (112). Evidence showing an increase in potential harmful bacteria (e.g. Escherichia coli, and Bacteroides) and a decrease in beneficial bacteria (e.g. Bifidobacteria and Lactobacillus), strengthened the fact that dysbiosis is associated with MASLD (113–116). Studies have shown MASLD alterations like hepatic triglyceride elevated levels, upregulation of genes related to lipid uptake and lipogenesis in germ free mice upon fecal matter transplant (FMT) from hepatic steatosis suffering obese mice (91). In another study, it was found that inflammasome-mediated gut dysbiosis can cause hepatic steatosis in wild-type mice when cohoused with MASH-affected mice (117). The metabolites produced by gut microbiome are essential component that can modulate the pathophysiology of MASLD and MASH. One of the most common metabolites produced by gut bacteria in response to dietary fiber breakdown is SCFA which plays a crucial role in maintaining metabolic, nervous, and immune system (118). By influencing host epigenetics, activating G protein-coupled receptors, and preventing pathogenic microbial infections, SCFAs function as vital mediators between the gut microbiota and the host, acting as energy substrates for intestinal epithelial cells and preserving homeostasis in host immune and energy metabolism (119). Acetic acid, propionic acid, and butyric acid are the most common SCFA accounting for 90–95% of the colon's total SCFA content (118). The acetate boosts liver fat oxidation by facilitating changes in mitochondria and activating AMP-activated protein kinase (120), while propionate may promote the release of leptin, which helps to suppress the formation of new lipids (121). Butyrate is primarily utilized by colon cells as their main energy source and displays anti-inflammatory properties (122). Butyric acid has also the ability to hinder the activation of ChREBP and SREBP-1, then, suppress the process of lipogenesis (123). Researchers have found in high-fat-fed mice, commensal microbe that produces acetate can suppress MASLD progression by modulating free fatty acid receptor 2 (FFAR2) signaling in the liver (124). Several other studies have demonstrated that butyrate can regulate gut microbiota, hepatic Glucagon-like peptide-1 (GLP-1) receptor expression, TLR4 pathways and intestinal tight junctions thus attenuating the development of MASLD (125–128). Decreased production of butyrate results in increased intestinal inflammation, increased gut permeability, endotoxemia and systemic inflammation (129). In addition to SCFA, bile acid and ethanol are other metabolites that plays a vital role in MASLD. Preclinical trials have shown that microbiota derived endogenous ethanol can accelerate liver steatosis and inflammation (24, 130). Additionally, there are evidence showing increased level of blood ethanol in MASLD patients (131). Gut microbiota is also involved in Bile acid metabolism. They have the capacity to convert primary bile acid into secondary bile acid. However, due to decreased abundance of related bacteria this ability is compromised in case of MASLD (132). By targeting genes linked to fatty acid synthesis and oxidative stress, a lower amount of deconjugated bile acid can further reduce taurine production and cause hepatic steatosis and inflammation (133). In addition, the receptor for bile acids, farnesoid X receptor (FXR) is found to be downregulated in MASLD (132). The decrease in the level of intestinal FXR is correlated with decrease in the secretion of an enterokine that regulate synthesis of hepatic bile acid fibroblast growth factor 15/19 (FGF15/19), which in turn can reduce liver steatosis (134). Amino acids and choline are some other gut microbial metabolites that are reported to modulate MASLD (129).
Diet plays a pivotal role in shaping the composition and function of the gut microbiome. A Western-style diet, rich in fats and refined carbohydrates, has been shown to induce dysbiosis and compromise gut barrier integrity. Interestingly, it has been found by investigators that obese infant mice with a western diet have excess weight gain and accelerate the progression of MASLD (135). It is hypothesized that onset of MASLD is triggered by high fat diet which induces an increase in FFAs and LPS which are derived from the gut anaerobic bacteria (114, 136). Endotoxemia occurs when there are elevated levels of LPS in the blood and is commonly observed in states of obesity, insulin resistance and in MASLD patients. Increased endotoxemia occurs due to higher levels of gut derived LPS and translocation of bacteria, particularly due to a high fat diet. In response to LPS, Kupffer cells, which are resident liver macrophages, and hepatic stellate cells are activated and release pro-inflammatory cytokines, this further elevates insulin resistance and promotes hepatic inflammation and fibrosis, thus having a central role in MASLD progression (137–139). LPS also induces fat accumulation in the liver and ROS production. Kupffer cells along with liver sinusoidal endothelial cells (LSECs), hepatic stellate cells (HSCs), and local immune cells, specifically unconventional T cells, natural killer (NK) cells, and hepatic dendritic cells make up the nonparenchymal liver cells, which are significant chemokine sources and responders. PAMPs or DAMPs are released when hepatocytes are damaged, whether by infection or other causes. These signals attach to TLR4 and other TLRs on Kupffer cells and trigger the release of proinflammatory cytokines, such as TNF, chemokines, and reactive oxygen and nitrogen species (140). TNF released by Kupffer cells is thought to play a key role in exacerbating liver damage, primarily by causing hepatocyte death but also by degrading the hepatic microcirculation by causing endothelial cells to swell and become activated, which leads to sinusoidal platelet aggregation and makes it easier for peripheral immune cells to enter. Interleukin (IL)-1β and CXC chemokines, including CXCL1, CXCL2, and CXCL8 (IL-8), are secreted by activated Kupffer cells. Key chemokines CXCL1, CXCL2, and CXCL8 draw neutrophils primarily through the chemokine receptors CXCR1 and CXCR2, which release proteases and ROS, causing hepatocyte necrosis (141).
Therapeutic approaches
The existing diagnosis of MASLD relies on clinical evaluations as well as biopsy results, with liver biopsy being the only diagnostic method that can accurately determine its severity (142). Nonetheless, this invasive technique carries the risk of serious and potentially fatal complications. As a result, effectively forecasting and promptly taking measures to avert the onset of MASLD continues to be a challenge. Several studies which focus on blood biochemical markers, gut microbiota, and fecal SCFAs, uncover a close association between gut microbiota and the progression of MASLD, thereby improving its clinical diagnosis (142–144). While we emphasizes on improving clinical diagnosis using microbial signatures, we acknowledge that future validation studies should also include head-to-head comparisons with established non-invasive tests such as FIB-4 and elastography to determine the relative diagnostic performance and clinical utility of microbiome-based approaches.
As discussed, gut–liver axis is an important bridge between gut and liver. The dysbiosis/malfunction of the gut–liver axis plays one of the most important roles in the onset and progression of MASLD by altering the intestinal permeability, increasing the level of portal toxic metabolites, promoting hepatic inflammation. Thus, microbiota based pharmacological modulation of gut–liver axis is an emerging and promising therapeutic method for MASLD treatment (145–148). There are numerous pathways by which microbiota can affect liver health, and several approaches that have been proposed to target these in order to improve liver health (148–153). There are several drug candidates that are in later stage clinical trials which includes PPAR agonists, anti-fibrotic therapies, anti-inflammatory agents, antioxidants, and treatments targeting the gut-liver axis (154). The gut microbiota can be therapeutically modulated through several approaches, including antibiotic therapy, probiotic supplementation, prebiotic administration, synbiotic interventions (155, 156) and FMT (157, 158). Obesity and T2DM are linked to substantial compositional and functional alterations of the gut microbiota. Therefore, modulation of the gut microbiota represents an attractive approach for the management of diabetes in the context of MASLD (68, 91, 159, 160).
Modulation of gut microbiota
Gut microbiota may be altered using prebiotics, probiotics or their combination known as synbiotic. According to FAO/WHO, Probiotics are live non-pathogenic microorganisms, which when administered in adequate amounts, confer a health benefit on the host (161, 162). The most widely used bacterial populations are of Lactobacillus, Bifidobacterium and Streptococcus that are capable of suppressing growth of Gram-negative pathogens (163, 164). These beneficial bacteria can reduce lipid deposition, endotoxemia, oxidative stress, and inflammation by regulating the expression levels of TNF-α, NF-κB, and collagen (162). Improving the gut barrier is the primary way that probiotics protect against MASLD in the gut–liver axis. Lactobacillus rhamnosus, L. acidophilus, L. plantarum, and Streptococcus thermophilus are a few probiotics that have shown the ability to activate tight junction proteins to improve the intestinal permeability (165). A recent randomized controlled trial has shown that probiotics stabilizes the mucosal immune function that in turn protects the MASLD patients from increased intestinal permeability (166). A variety of probiotics, particularly well-known like Lactobacillus, Bifidobacterium, and Streptococci, have been studied clinically in relation to the prevention and treatment of MASLD. Wong et al. treated MASH patients for 6 months with a variety of probiotics and discovered that the subjects receiving probiotics had a considerably lower liver fat level than the placebo group (167). According to clinical data, probiotics can help MASLD patients' liver histology and liver injury indices like alanine aminotransferase (ALT) and aspartate aminotransferase (AST) (168). But according to a different clinical study conducted during the same time, giving multiple-strain probiotics to MASLD patients only improved liver steatosis and not liver enzymes (169). Probiotics have consistently been shown to reduce liver enzyme levels (e.g., ALT, AST) in MASLD patients, but improvements in liver histology like inflammation, fibrosis, or steatosis have not been demonstrated in biopsy-confirmed settings. Several meta-analyses confirm this pattern (162, 169–177). A 2024 network meta-analysis of 37 randomized controlled trials (RCTs, n=1,921) found that probiotics significantly lowered ALT and AST, and improved liver stiffness and steatosis based on elastography and controlled attenuation parameter (CAP) scores but did not show histological resolution (178). Similarly, a 2019 meta-analysis reported significant reductions in liver enzymes and steatosis by ultrasound, but histological effects were unassessed (162, 174, 179). Most trials rely on biochemical or imaging measures (e.g., ultrasound or FibroScan), and rarely include sequential liver biopsies to confirm tissue-level changes. Probiotics effectively lower liver enzymes in MASLD without proven histological improvements, likely due to non-tailored approaches. Personalized microbiome modulation using patient-specific microbial profiling, strain selection, prebiotic support, and mechanistic biomarkers offers a promising path to bridge this efficacy gap. Ongoing research in precision microbiome interventions (pharmacomicrobiomics, host-microbe profiling) will be critical for developing such customized therapeutic strategies (180).
Prebiotics are non-digestible food ingredients that have beneficial effects on the host by selectively stimulating the growth and/or activity of one or a limited number of bacteria in the colon, and thus improving host health (181, 182). Prebiotics are capable of increasing activity of good bacteria and resisting growth of detrimental species. Fructooligosaccharides (FOS), inulin, transgalactooligosaccharides (TOS), and lactulose are examples of common prebiotics. Prebiotics are a safe and efficient way to control the gut microbiota since they can boost the growth and activity of probiotics (183). Prebiotics can prevent the growth of harmful bacteria like Salmonella enteritidis, Klebsiella pneumoniae, and Escherichia coli while simultaneously activating the advantageous bacteria (184). This characteristic can enhance the gut barrier, support gut microbial homeostasis, and ultimately slow the advancement of MASLD. Through fermentation, prebiotics can also protect against MASLD by producing SCFAs, which have been shown to protect against MASLD and the gut-liver axis (183). Larch wood arabinogalactan (LA-AG), a novel complex soluble dietary fiber was discovered by Sun et al., as a potential prebiotic. By promoting the fermentation of organic acids, LA-AG was able to reduce the activity of harmful bacteria and enhance intestinal health (185). Therefore, by controlling the gut-liver axis, LA-AG may be effective in preventing MASLD.
In a clinical trial Bomhof et al. (186) showed, using oligofructose as an example, that giving patients with MASH a supplement of the prebiotic can improve their liver steatosis and non-alcoholic fatty liver activity score (NAS) (186). Furthermore, according to a meta-analysis prebiotic treatment can enhance anthropometric and biochemical parameters such as body mass index (BMI), ALT, AST, fasting insulin, and insulin resistance in individuals with MASLD (187).
Apart from probiotic and prebiotics, their combination called synbiotic can be used that is capable of boosting metabolism of heathy bacteria and modulate the gut microbiome (161). Several studies have shown the protective effect of synbiotics on liver and cardio related disorders (171, 179, 188–190). More studies to define the health benefits of pre and probiotics in the context of MASLD and T2DM are required. If these therapies are shown to be beneficial, an important issue will be the best strains of probiotics or types of prebiotics to use, and the optimal duration of therapy. This microbe directed therapy for MASLD, and diabetes could also involve the use of antibiotics with selective action in the intestine, although this is unlikely to be an attractive strategy for patients or doctors. The idea that antibiotics can reduce the effects of microbiota and their metabolites on host metabolism via the gut–liver axis is the foundation for their use in the treatment of MASLD. Preclinical trials have shown that by inhibiting gut bacteria, antibiotics can control the amount of portal secondary bile acid, reducing liver fibrosis and inflammation and preventing the progression of MASLD (191). Another study demonstrated that neomycin and polymyxin B can significantly lower hepatic lipid build-up by decreasing the translocation of endotoxin in a MASLD mouse model (192). In a Phase II clinical trial, the powerful next-generation macrolide antibiotic Solithromycin was shown to lower the ALT and NAS of MASH patients (193). There are several studies in animal models demonstrating that broad-spectrum antibiotics can prevent and reverse MASLD, although the side effects of long-term antibiotic use are considerable (194–196). Antibiotics should be used with caution as they may eradicate certain bacterial species linked to good health and result in the emergence of some antibiotic-resistant bacteria (197).
An exciting potential future alternative is the use of FMT from a healthy lean donor to a patient with MASLD and obesity or diabetes (174, 198, 199). FMT is an effective therapeutic option for liver and metabolic diseases associated with intestinal microbiota dysbiosis (85, 198, 200). There have been several studies demonstrating the therapeutic effects of FMT on ulcerative colitis, T2DM and patients, which were associated with improved insulin resistance, restored healthy microbiota, and normalized blood lipid levels (201–205). Several investigations have demonstrated that FMT is an effective bacteriotherapy for MASLD as well. Zhou et al. discovered in an early preclinical investigation that FMT might reduce High fat diet (HFD)-induced MASH by enhancing the gut barrier, raising SCFA levels, and controlling gut microbiota (206). Another study in 2021 by Zhang et al., demonstrated that germ-free (GF) mice receiving FMT had less hepatic lipid accumulation and inflammation than normal chow-fed animals in contrast to mice fed high-fat/high-cholesterol (HFHC) and receiving FMT (207). Recent human trials have also shown that FMT can lower intestinal permeability and hepatic steatosis in MASLD patients, which is in line with animal investigations (208). However, some side effects, like bacteremia and perforations, have still been documented in FMT (209). Therefore, additional clinical trials must be carried out to increase the effectiveness and lower the negative effects of FMT treatment in MASLD/MASH.
Microbiota-based therapies, such as probiotics and FMT, are being explored as adjunct treatments for MASLD, primarily due to the gut-liver axis's role in disease progression. However, current evidence reveals several critical limitations.
For probiotics, therapeutic effects vary widely depending on the strain, dose, and treatment duration. While some studies report reductions in liver enzymes and steatosis, results are inconsistent, and few trials assess histological endpoints (174, 178, 210). Moreover, the adult gut microbiome exhibits strong ecological resilience, often reverting to its original state after probiotic intervention (174), limiting long-term efficacy. There's also no consensus on optimal strains or treatment regimens (211).
FMT shows some promise in early MASLD trials (212, 213), but results are mixed. Engraftment of donor microbes is unpredictable and often influenced by host factors, such as baseline microbiota composition and diet (214). Safety concerns have also been raised—cases of extended spectrum beta lactamase (ESBL)-producing E. coli infection following FMT prompted FDA safety alerts (215), underscoring the need for rigorous donor screening.
Furthermore, both therapies lack standardized protocols regarding delivery method, donor/strain selection, and outcome measurement. Most trials are short-term and fail to evaluate long-term outcomes like fibrosis reversal. Given that MASLD is a complex, multifactorial disease, targeting the microbiome alone may be insufficient without concurrent lifestyle or metabolic interventions (4, 216).
In conclusion, while microbiota-based therapies hold promise as adjunctive treatments for MASLD, they are currently limited by inconsistent efficacy, methodological heterogeneity, safety concerns, and incomplete understanding of long-term outcomes. Well-designed RCT's with standardized protocols, mechanistic endpoints, and extended follow-up periods are urgently needed to clarify their role. Until such data are available, these therapies should be considered experimental and used with caution in the clinical setting.
Other promising agents as adjunctive therapy
Using FXR agonists
FXR agonists are a class of drugs that have been reported to decrease hepatic steatosis and improving insulin sensitivity and hence a promise in treatment of various gastrointestinal diseases (217). FXRs are nuclear receptors present in liver, kidney, intestine, pancreas and adipose tissue and are actively involved in bile acid, lipid and glucose metabolism and inflammation. The FXR agonists bind to the receptors and activate them and regulate target genes involved in the biological pathways. These agonists have been a part of successful clinical trials which showed that they assist in improvement in liver inflammation as well as insulin sensitivity. These receptors are activated endogenously by bile acids and are regulators of bile acid production, conjugation, and transport.
Several FXR agonists that have been assessed in clinical trials and their effects are:
• Obeticholic Acid (OCA): A derivative/synthetic variant of bile acid, it improves insulin sensitivity, liver inflammation, hepatocellular ballooning and reduces fibrosis (218). When bound to the FXR receptors, lipophilic bile acids decrease gluconeogenesis and triglycerides in the liver, promote insulin sensitivity. This also increases the expression of hepatic scavenger receptors (SRB1), a liver protein crucial for cholesterol homeostasis (219). It is responsible for reverse cholesterol transport by increasing the clearance of HDL by liver cells (220). Several studies have also reported a side effect of its use: pruritus as well limiting its use (218).
• Cilofexor: Non-steroidal molecule that reduces steatosis, downstages hepatic fibrosis (221).
• Tropifexor: Novel and highly potent agonist of FXR and is being used in stage 2 human clinical trials in patients (222).
• Vonafexor: A non-steroidal FXR agonist that has an action of reduction of liver fat content, fibrosis biomarkers, body weight and improving kidney function (223).
Using SGLT inhibitors and incretin-based approaches
Incretin-based therapies and sodium-glucose cotransporter 2 (SGLT-2) inhibitors are now being worked on as novel classes of glucose-lowering drugs used in the management of T2DM and are proving to be playing a simultaneous role in improving liver health (224) (Table 3).
• SGLT-2 inhibitors are antihyperglycemic agents that target SGLT-2 proteins expressed in the proximal convoluted tubules of the kidneys, where they normally mediate the reabsorption of glucose from the urine; by inhibiting this process, these agents promote increased urinary glucose excretion and help lower blood glucose levels (225).
• These drugs exert their effect by preventing the reabsorption of filtered glucose from the tubular lumen.
• Their beneficial effects range from weight loss, regulation of stress in the endoplasmic reticulum, oxidative stress, low-grade inflammation, apoptosis and autophagy (226).
• Incretins are hormones derived from the intestinal mucosa that play a key role in regulation of blood sugar levels as they stimulate secretion of insulin from pancreas post glucose intake (227).
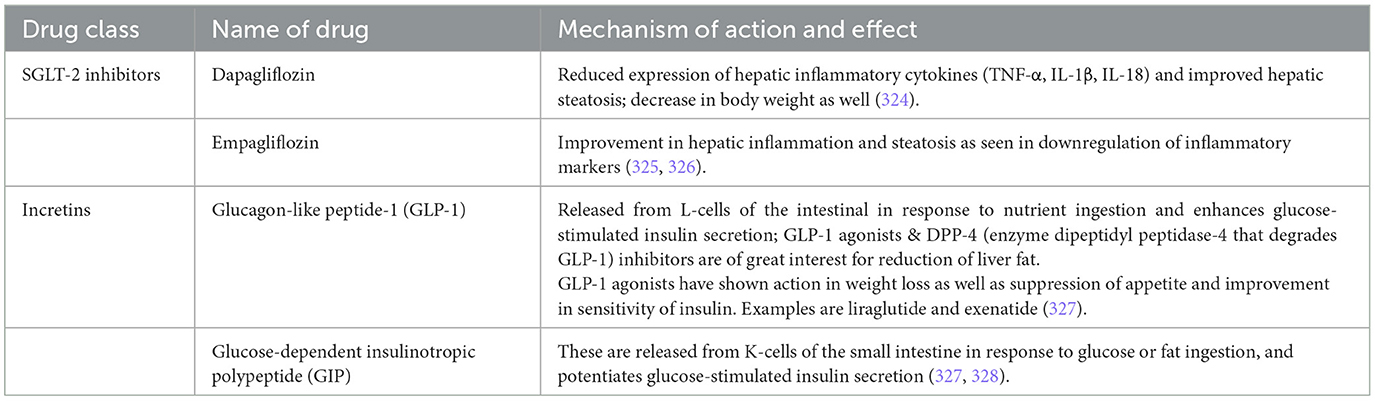
Table 3. Antidiabetic drugs with potential hepatic benefits.
Role of AMP-activated protein kinase (AMPK)
AMPK activators are now gathering attention as alternatives to conventional treatments (228). These are involved in modulating energy metabolism under conditions of increased AMP:ATP ratio during energy deprivation. It further inhibits DNL gene expression by suppressing the actions of the enzyme acetyl-CoA carboxylase 1 pathway and promotes lipolysis through activation of the carnitine palmitoyl transferase 1 pathway in the liver.
Various agents that function as AMPK activators can be classified broadly into two categories:
A. Direct activators: Examples such as: A-769662 (229), Salicylate [both implied for improving liver function and reduction of hepatic fat].
B. Agents that mimics AMPK's downstream activity: Such as metformin (230) and aramchol (231) [reduction of hepatic fat content and inhibition of fatty acid synthesis in liver respectively].
AMPK is a critical regulator of cellular energy metabolism and oxidative stress defense. It has been identified as a central protein capable of mitigating cytotoxicity, suppressing inflammation, and preventing fibrosis. Hence, it has been positioned as a promising therapeutic target for addressing the primary drivers of MASLD.
Targeting liver health
Currently, most of the effective therapeutic methods are targeting liver health. MASLD can't only be thought of as a precursor to T2DM, as liver damage has been shown to exacerbate diabetes by causing insulin resistance and beta cell failure. There are several molecular pathways that are thought to be involved in the process, and these are potential targets for therapeutic intervention. First, hepatocyte lipid overload and the presence of fat metabolites have been shown to activate serine kinase cascades that cause insulin resistance (44, 62, 232–235). So, blockade of these kinases may prevent the progression from MASLD to T2DM. There is an ongoing trial to assess the effectiveness of pioglitazone in treating advanced liver disease due to its insulin sensitizing effects, while lifestyle modification is a rational and safe therapy for MASLD with T2DM (236, 237). However, diabetes is typically characterized by compromised antioxidant capacity and increased oxidative stress. In patients with concurrent MASLD and diabetes, this oxidative imbalance exacerbates hepatic inflammation and fibrogenesis, worsening disease prognosis and dramatically increasing the risk of hepatocellular carcinoma and other liver diseases, while also elevating the risk of microvascular and macrovascular diabetic complications (238–244). High-dose vitamin E therapy has been shown to improve all aspects of liver histology in adults with MASLD (236, 245). It has also been shown to prevent the onset of T2DM in adults with metabolic syndrome and/or diabetes. High-dose statin therapy has been suggested as a treatment of MASLD. Triglyceride-lowering effects and improvement of aminotransferase levels were seen in early studies. However, recent evidence suggests that there is a risk of further liver damage (246, 247). These drug therapies are probably inappropriate for mild liver disease in T2DM and will have to be balanced with the risks and benefits. Ultimately, drug therapy for the liver in T2DM must be tailored to the individual.
Lifestyle interventions and diabetes management
Lifestyle variables like excessive consumption of foods high in calories and a decrease in physical activity and exercise, are closely linked to the development of MASLD. Despite many negative effects as stated earlier, currently there are no pharmacological treatments for MASLD. Therefore, healthy lifestyle is the most important management of MASLD which involves diet, exercise and weight loss (248). Healthy lifestyle for both adults and children include eating a diet high in fruits, nuts, seeds, whole grains, fish, poultry along with regular physical exercise and avoiding excessive intake of red meat, ultra-processed foods, sugar-sweetened beverages, and meals fried at high temperatures (248). Several randomized controlled trials have shown lifestyle interventions reduce body weight, improve hepatic triglyceride content and improve MASLD activity score in patients suffering from MASLD (249–254). Furthermore, most studies show that changes in lifestyle are associated with improvements in cardiovascular disease (CVD) risk variables, including insulin resistance and blood cholesterol levels. Several clinical practice guidelines promote weight loss through calorie restriction as the best evidence-based strategy to improve MASLD across the disease spectrum (250, 252, 255). Comprehensive lifestyle modification should include dietary change to lower calorie intake, lifestyle and behavioral training, and increase physical activity. Avoiding smoking should also be part of the changes, as it has been linked to MASLD, fibrosis progression and HCC (256). Although several hypo energetic diets can reduce liver fat and promote weight reduction, the Mediterranean diet (MD) offers additional cardiometabolic benefits related to CVD risk reduction, which is the leading cause of mortality in most people with MASLD (257). However, their real-world feasibility across diverse, resource-constrained populations hinges on overcoming significant implementation barriers.
A mixed-methods trial in Northern England involving 19 MASLD patients demonstrated that after 12 weeks of MD counseling using meal plans and recipes, adherence increased from moderate to high, yielding modest weight loss (~2.4 kg) and HDL improvements. However, significant obstacles emerged, including an obesogenic environment, everyday stress, demand for convenience foods, and limited understanding of MASLD's health implications factors that adversely affected commitment to dietary changes (258–260). Similarly, a qualitative study in Australia with multicultural participants revealed that while the MD was perceived as more enjoyable and sustainable than a low-fat diet, barriers included access to culturally appropriate foods and sustaining changes post-interventions (260). A Tunisian study found patients could adhere to MD principles when fresh ingredients were affordable, and recipes were culturally tailored. Tunisian NAFLD patients had low MD adherence due to financial constraints and dietary adaptation challenges (261).
Systematic reviews highlight recurrent themes affecting MD adoption beyond Mediterranean regions: economic constraints, such as higher costs of fresh produce and olive oil; limited availability in local markets; cultural mismatches, and low nutrition literacy hindering behavior change. In regions with low socioeconomic status, MD adherence is strongly associated with greater food costs and younger age, while access to affordable, healthy food options especially in food deserts poses practical limitations (259, 262, 263).
Incorporating physical activity and exercise with dietary changes should be emphasized in the treatment of MASLD. Numerous randomized controlled trials have shown that exercise alone lowers liver fat in people with MASLD (264), whereas inadequate physical activity is linked to an increased risk for MASLD progression (265). Additionally, several recent studies have shown that a higher level of physical activity is linked to a lower risk of cirrhosis, liver fibrosis, and all-cause mortality (266–268). Exercise regardless of weight loss has hepatic and cardiometabolic benefits and it should be regularly advised and customized to patients' physical capabilities and preferences (269). Individuals with sedentary lifestyle and no physical activity should set achievable goals minimum of at least 150 min per week (30 min per day on 5 days per week) of moderate activity that includes anything that will raise the heart best and break a sweat still allowing to talk (269). According to current recommendations, a mix of resistance (also known as “strength,” like weight-lifting) and aerobic (often known as “cardio,” like brisk walking, cycling, and swimming) exercise should be employed (265, 269). Dietary and lifestyle changes must be adopted for life to prevent the progression of MASLD and its common comorbidities—namely, CVD and type 2 diabetes.
Treatment approaches for MASLD in diabetic patients primarily focus on improving insulin control and reducing liver fat accumulation. Achieving better glycemic control, as indicated by lower HbA1c levels, has been associated with reductions in ALT levels, which serve as a marker of liver inflammation (270, 271). For T2DM, effective management of hyperglycemia and improved insulin sensitivity are critical for lowering rates of DNL and reducing hepatic fat. These changes are difficult to achieve in practice and there are no current treatments that are highly effective. One promising therapeutic avenue involves enhancing the activity of peroxisome proliferator-activated receptor gamma (PPARγ). Thiazolidinediones (TZDs), which target this receptor, have shown potential in improving insulin sensitivity and reducing ALT levels in MASH patients (272, 273). However, due to their cardiovascular side effects, TZDs are not recommended for MASLD treatment (274). Additionally, anabolic hormone therapy to correct deficiencies or counteract the muscle wasting associated with MASLD/MASH represents a compelling area for future research.
Low glycemic index diets rich in monounsaturated fatty acids are associated with improvement in hepatic steatosis and ALT levels, and in a recent study, ad libitum low carbohydrate diets resulted in greater weight loss and improvement in insulin resistance compared to energy-restricted high carbohydrate diets (275–277). Weight loss has been associated with improvements in liver enzymes and histology in a number of different patient populations, and the associated metabolic improvements are likely to be mediated via reduction in adipose tissue inflammation and secretion of pro-inflammatory adipokines (278–281).
Animal studies have shown that a combination of exercise and dietary modification results in an alteration of the intestinal microbiota, an increase in the production of intestinal mucins, and a reduction of endotoxin and inflammatory cytokine production, which in turn prevents the development of steatosis (74, 143, 282–285). In human studies, increased physical activity has been associated with a reduced prevalence of hepatic steatosis, and a recent randomized control trial has shown that 12 months of moderate-intensity exercise in patients with T2DM resulted in reduced hepatic triglyceride content (286–289). Lifestyle interventions such as physical activity, dietary modification, and weight loss are the first line of therapy for the management of T2DM and are the most effective interventions for prevention of diabetes in high-risk populations (279, 290–292). Evidence is now emerging to suggest that these interventions may be effective in the prevention and management of MASLD and, in doing so, may influence the intestinal microbiota (286, 293, 294).
Altogether, these studies suggest that the beneficial effects of lifestyle interventions on both diabetes and MASLD could be mediated via modulation of the gut microbiota, and as our understanding of the mechanisms involved increases, it may be possible to make targeted therapeutic recommendations (154).
Can conclusions drawn from animal studies sufficiently support translation to human pathophysiology?
While animal models especially mouse such as diet induced, deficiency induced, toxin induced, genetically induced or as a mixture of these modalities have provided foundational insights into the mechanisms of MASLD and MASH, the translation of these findings to human pathophysiology remains limited due to significant biological and metabolic disparities between species (295, 296).
The ob/ob and db/db models, for instance, exhibit key features of human metabolic syndrome and develop hepatic steatosis on a standard diet. When exposed to a secondary insult (e.g., methionine- and choline-deficient [MCD] diet), these models can progress to MASH like phenotypes. However, their translational relevance is restricted by the fact that congenital leptin deficiency (as seen in ob/ob mice) or leptin resistance (db/db mice) is extremely rare in humans (297–299). This limits their capacity to represent the etiology of human obesity, insulin resistance, and hepatic steatosis.
Moreover, the MCD diet model, while useful in inducing liver injury and inflammation, leads to metabolic alterations such as weight loss and decreased insulin levels that contrast starkly with human MASH, which is typically associated with obesity and insulin resistance. This explains the fact that many animal models either replicate histopathological features or metabolic features but rarely both (296).
Significantly, human clinical data have highlighted the limitations of these models. A study utilizing a human liver chimeric mouse model revealed striking differences in molecular responses between murine and human hepatocytes when exposed to a Western diet, indicating species-specific liver functions and responses (300). Another study revealed there was partial overlap in liver transcriptome profiles between mice and humans, the gene expression patterns in mouse models remained distinctly different from those in humans, indicating that the pathophysiology in mice does not fully replicate human MASLD (301). In another study Vacca et al., did a retrospective study and assessed mouse models using a human proximity score derived from metabolic phenotypes, liver histopathology and transcriptomic similarity to human liver data (302). They concluded from the study that Western style diets especially those with added cholesterol and longer feeding are the closest match across metabolic, histological, and molecular layers and Choline-deficient or genetically driven models may help elucidate specific fibrosis mechanisms but do not recapitulate the full human disease spectrum (302).
Thus, animal models remain invaluable for understanding discrete mechanisms or stages of MASLD/MASH. However, no single mouse model accurately recapitulates the integrated metabolic, histological, and molecular complexity of human disease. While existing models continue to guide mechanistic research and therapeutic testing, further development of models that more closely align with human pathophysiology is crucial to improve translational validity (295, 303, 304).
Conclusions and perspectives
The multifaceted nature of MASLD emerges through its complex interactions with IR and T2DM, with the gut microbiome serving as a central orchestrator of disease progression. Current cross-sectional studies inadequately address fundamental relationships between gut dysbiosis and hepatic dysfunction, demanding comprehensive longitudinal microbiome investigations with standardized protocols to delineate temporal disease progression patterns. Rigorous randomized controlled trials evaluating microbiome-targeted therapies, including FMT and precision probiotics, are essential for clinical translation of emerging mechanistic insights. The complex host-microbe-metabolic interactions underlying MASLD pathophysiology require sophisticated multi-omics integration strategies, employing advanced computational approaches to identify novel biomarkers and therapeutic targets. These complementary research strategies will accelerate the development of precision diagnostics and mechanism-based interventions, transforming MASLD management from reactive treatment to proactive, personalized medicine approaches. Given projected increases in global MASLD prevalence, this integrated research framework represents our most promising pathway toward effective therapeutic solutions for this increasingly prevalent metabolic liver disorder.
Author contributions
AD: Data curation, Investigation, Writing – original draft, Writing – review & editing. RasK: Data curation, Investigation, Writing – original draft, Writing – review & editing. MS: Investigation, Visualization, Writing – review & editing. RouK: Data curation, Validation, Writing – review & editing. SP: Supervision, Writing – review & editing. AO: Resources, Supervision, Writing – review & editing. RosK: Conceptualization, Investigation, Supervision, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. Roshan Kumar gratefully acknowledges financial support from the University Grants Commission Basic Scientific Research Start-Up Grant [No. F.30-527/2020(BSR)].
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Gen AI was used in the creation of this manuscript.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
ALT, Alanine Aminotransferase; AST, Aspartate Aminotransferase; BMI, Body Mass Index; CAP, Controlled Attenuation Parameter; ChREBP, Carbohydrate Response Element-Binding Protein; CRP, C-Reactive Protein; CVD, Cardiovascular Disease; DAG, Diacylglycerol; DAMPs, Damage-Associated Molecular Patterns; DNL, De Novo Lipogenesis; ER, Endoplasmic Reticulum; FFAs, Free Fatty Acids; FFAR2, Free Fatty Acid Receptor 2; FGF15/19, Fibroblast Growth Factor 15/19; FMT, Fecal Matter Transplant; FOS, Fructooligosaccharides; FOXA2, Forkhead Box Protein A2; FXR, Farnesoid X Receptor; GF, Germ-Free; GLP-1, Glucagon-Like Peptide-1; HbA1c, Hemoglobin A1c; HCC, Hepatocellular Carcinoma; HFD, High-Fat Diet; HFHC, High-Fat/High-Cholesterol; HSCs, Hepatic Stellate Cells; IECs, Intestinal Epithelial Cells; IL, Interleukin; IKK-β, Inhibitor of Nuclear Factor Kappa B Kinase Subunit Beta; IR, Insulin Resistance; JNK, c-Jun N-Terminal Kinase; LA-AG, Larch Wood Arabinogalactan; LPS, Lipopolysaccharides; LSECs, Liver Sinusoidal Endothelial Cells; MD, Mediterranean Diet; MDA, Malonaldehyde; MAFLD, Metabolic-Associated Fatty Liver Disease; MASLD, Metabolic Dysfunction-Associated Steatotic Liver Disease; MASH, Metabolic Dysfunction-Associated Steatohepatitis; metS, Metabolic Syndrome; NAFLD, Non-Alcoholic Fatty Liver Disease; NAFLD-HCC, NAFLD-Associated Hepatocellular Carcinoma; NAS, Non-Alcoholic Fatty Liver Activity Score; NASH, Non-Alcoholic Steatohepatitis; NETs, Neutrophil Extracellular Traps; NF-κB, Nuclear Factor Kappa B; NK, Natural Killer; OPN, Osteopontin; PAI-1, Plasminogen Activator Inhibitor-1; PAMPs, Pathogen-Associated Molecular Patterns; PKC, Protein Kinase C; PNPLA3, Patatin-like Phospholipase Domain Containing 3; PPARγ, Peroxisome Proliferator-Activated Receptor Gamma; RCT's, Randomized Controlled Trials; ROS, Reactive Oxygen Species; SCFAs, Short-Chain Fatty Acids; SFAs, Saturated Fatty Acids; SIBO, Small Intestinal Bacterial Overgrowth; SREBP-1c, Sterol Regulatory Element-Binding Protein 1c; T2DM, Type 2 Diabetes Mellitus; TG, Triglyceride; TLRs, Toll-Like Receptors; TMA, Trimethylamine; TNF-α, Tumor Necrosis Factor Alpha; TOS, Transgalactooligosaccharides; TZDs, Thiazolidinediones; UDCA, Ursodeoxycholic Acid; VLDL, Very-Low-Density Lipoprotein.
References
1. Lazarus JV, Mark HE, Anstee QM, Arab JP, Batterham RL, Castera L, et al. Advancing the global public health agenda for NAFLD: a consensus statement. Na Rev Gastroenterol Hepatol. (2022) 19:60–78. doi: 10.1016/S0168-8278(23)00603-7
2. Teng ML, Ng CH, Huang DQ, Chan KE, Tan DJ, Lim WH, et al. Global incidence and prevalence of nonalcoholic fatty liver disease. Clin Mol Hepatol. (2023) 29:S32–42. doi: 10.3350/cmh.2022.0365
3. Younossi ZM, Golabi P, Paik JM, Henry A, Van Dongen C, Henry L. The global epidemiology of nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH): a systematic review. Hepatology. (2023) 77:1335–47. doi: 10.1097/HEP.0000000000000004
4. Aron-Wisnewsky J, Vigliotti C, Witjes J, Le P, Holleboom AG, Verheij J, et al. Gut microbiota and human NAFLD: disentangling microbial signatures from metabolic disorders. Nat Rev Gastroenterol Hepatol. (2020) 17:279–97. doi: 10.1038/s41575-020-0269-9
5. Chen L, Tao X, Zeng M, Mi Y, Xu L. Clinical and histological features under different nomenclatures of fatty liver disease: NAFLD, MAFLD, MASLD and MetALD. J Hepatol. (2024) 80:e64–6. doi: 10.1016/j.jhep.2023.08.021
6. Streba LA, Vere CC, Rogoveanu I, Streba CT. Nonalcoholic fatty liver disease, metabolic risk factors, and hepatocellular carcinoma: an open question. World J Gastroenterol. (2015) 21:4103–10. doi: 10.3748/wjg.v21.i14.4103
7. Angulo P. Nonalcoholic fatty liver disease. N Engl J Med. (2002) 346:1221–31. doi: 10.1056/NEJMra011775
8. Xu Q, Feng M, Ren Y, Liu X, Gao H, Li Z, et al. From NAFLD to HCC: advances in noninvasive diagnosis. Biomed Pharmacother. (2023) 165:115028. doi: 10.1016/j.biopha.2023.115028
9. Nadasdi A, Gal V, Masszi T, Somogyi A, Firneisz G. PNPLA3 rs738409 risk genotype decouples TyG index from HOMA2-IR and intrahepatic lipid content. Cardiovasc Diabetol. (2023) 22:64. doi: 10.1186/s12933-023-01792-w
10. Song Q, Zhang X, Liu W, Wei H, Liang W, Zhou Y, et al. Bifidobacterium pseudolongum-generated acetate suppresses non-alcoholic fatty liver disease-associated hepatocellular carcinoma. J Hepatol. (2023) 79:1352–65. doi: 10.1016/j.jhep.2023.07.005
11. Fouad Y, Waked I, Bollipo S, Gomaa A, Ajlouni Y, Attia D. What's in a name? Renaming ‘NAFLD' to ‘MAFLD'. Liver Int. (2020) 40:1254–61. doi: 10.1111/liv.14478
12. Ludwig J, Viggiano TR, Mcgill DB, Oh BJ. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin Proc. (1980) 55:434–8. doi: 10.1016/S0025-6196(24)00530-5
13. Eslam M, Newsome PN, Sarin SK, Anstee QM, Targher G, Romero-Gomez M, et al. A new definition for metabolic dysfunction-associated fatty liver disease: an international expert consensus statement. J Hepatol. (2020) 73:202–9. doi: 10.1016/j.jhep.2020.07.045
14. Eslam M, George J. Reply to: Correspondence on “A new definition for metabolic associated fatty liver disease: an international expert consensus statement”: MAFLD: moving from a concept to practice. J Hepatol. (2020) 73:1268–9. doi: 10.1016/j.jhep.2020.06.036
15. Tilg H, Effenberger M. From NAFLD to MAFLD: when pathophysiology succeeds. Nat Rev Gastroenterol Hepatol. (2020) 17:387–8. doi: 10.1038/s41575-020-0316-6
16. Rinella ME, Lazarus JV, Ratziu V, Francque SM, Sanyal AJ, Kanwal F, et al. A multisociety Delphi consensus statement on new fatty liver disease nomenclature. J Hepatol. (2023) 79:1542–56. doi: 10.1097/HEP.0000000000000696
17. Paschos P, Paletas K. Non alcoholic fatty liver disease and metabolic syndrome. Hippokratia. (2009) 13:9–19.
18. Schreuder TC, Verwer BJ, Van Nieuwkerk CM, Mulder CJ. Nonalcoholic fatty liver disease: an overview of current insights in pathogenesis, diagnosis and treatment. World J Gastroenterol. (2008) 14:2474–86. doi: 10.3748/wjg.14.2474
19. Seki S, Kitada T, Yamada T, Sakaguchi H, Nakatani K, Wakasa K. In situ detection of lipid peroxidation and oxidative DNA damage in non-alcoholic fatty liver diseases. J Hepatol. (2002) 37:56–62. doi: 10.1016/S0168-8278(02)00073-9
20. Bruzzi S, Sutti S, Giudici G, Burlone ME, Ramavath NN, Toscani A, et al. B2-Lymphocyte responses to oxidative stress-derived antigens contribute to the evolution of nonalcoholic fatty liver disease (NAFLD). Free Radic Biol Med. (2018) 124:249–59. doi: 10.1016/j.freeradbiomed.2018.06.015
21. Han J, Kaufman RJ. The role of ER stress in lipid metabolism and lipotoxicity. J Lipid Res. (2016) 57:1329–38. doi: 10.1194/jlr.R067595
22. Begriche K, Igoudjil A, Pessayre D, Fromenty B. Mitochondrial dysfunction in NASH: causes, consequences and possible means to prevent it. Mitochondrion. (2006) 6:1–28. doi: 10.1016/j.mito.2005.10.004
23. Tsay CJ, Lim JK. NASH and the gut microbiome: implications for new therapies. Clin Liver Dis (Hoboken). (2022) 19:97–100. doi: 10.1002/cld.1170
24. Zhu L, Baker SS, Gill C, Liu W, Alkhouri R, Baker RD, et al. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: a connection between endogenous alcohol and NASH. Hepatology. (2013) 57:601–9. doi: 10.1002/hep.26093
25. Luedde T, Schwabe RF. NF-kappaB in the liver–linking injury, fibrosis and hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. (2011) 8:108–18. doi: 10.1038/nrgastro.2010.213
26. Jager J, Gremeaux T, Cormont M, Le Marchand-Brustel Y, Tanti JF. Interleukin-1beta-induced insulin resistance in adipocytes through down-regulation of insulin receptor substrate-1 expression. Endocrinology. (2007) 148:241–51. doi: 10.1210/en.2006-0692
27. Rehman K, Akash MS. Mechanisms of inflammatory responses and development of insulin resistance: how are they interlinked? J Biomed Sci. (2016) 23:87. doi: 10.1186/s12929-016-0303-y
28. Garcia-Ruiz C, Fernandez-Checa JC. Mitochondrial oxidative stress and antioxidants balance in fatty liver disease. Hepatol Commun. (2018) 2:1425–39. doi: 10.1002/hep4.1271
29. Marchesini G, Brizi M, Morselli-Labate AM, Bianchi G, Bugianesi E, Mccullough AJ, et al. Association of nonalcoholic fatty liver disease with insulin resistance. Am J Med. (1999) 107:450–5. doi: 10.1016/S0002-9343(99)00271-5
30. Marchesini G, Brizi M, Bianchi G, Tomassetti S, Bugianesi E, Lenzi M, et al. Nonalcoholic fatty liver disease: a feature of the metabolic syndrome. Diabetes. (2001) 50:1844–50. doi: 10.2337/diabetes.50.8.1844
31. Jimba S, Nakagami T, Takahashi M, Wakamatsu T, Hirota Y, Iwamoto Y, et al. Prevalence of non-alcoholic fatty liver disease and its association with impaired glucose metabolism in Japanese adults. Diabet Med. (2005) 22:1141–5. doi: 10.1111/j.1464-5491.2005.01582.x
32. Mitrovic B, Gluvic ZM, Obradovic M, Radunovic M, Rizzo M, Banach M, et al. Non-alcoholic fatty liver disease, metabolic syndrome, and type 2 diabetes mellitus: where do we stand today? Arch Med Sci. (2023) 19:884–94. doi: 10.5114/aoms/150639
33. Chalasani N, Younossi Z, Lavine JE, Diehl AM, Brunt EM, Cusi K, et al. The diagnosis and management of non-alcoholic fatty liver disease: practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology. (2012) 55:2005–23. doi: 10.1002/hep.25762
34. Nascimbeni F, Pais R, Bellentani S, Day CP, Ratziu V, Loria P, et al. From NAFLD in clinical practice to answers from guidelines. J Hepatol. (2013) 59:859–71. doi: 10.1016/j.jhep.2013.05.044
35. Wong VW, Ekstedt M, Wong GL, Hagstrom H. Changing epidemiology, global trends and implications for outcomes of NAFLD. J Hepatol. (2023) 79:842–52. doi: 10.1016/j.jhep.2023.04.036
36. Bugianesi E, Gastaldelli A, Vanni E, Gambino R, Cassader M, Baldi S, et al. Insulin resistance in non-diabetic patients with non-alcoholic fatty liver disease: sites and mechanisms. Diabetologia. (2005) 48:634–42. doi: 10.1007/s00125-005-1682-x
37. Shoelson SE, Herrero L, Naaz A. Obesity, inflammation, and insulin resistance. Gastroenterology. (2007) 132:2169–80. doi: 10.1053/j.gastro.2007.03.059
38. Miyazaki Y, Glass L, Triplitt C, Wajcberg E, Mandarino LJ, Defronzo RA. Abdominal fat distribution and peripheral and hepatic insulin resistance in type 2 diabetes mellitus. Am J Physiol Endocrinol Metab. (2002) 283:E1135–1143. doi: 10.1152/ajpendo.0327.2001
39. Cnop M, Havel PJ, Utzschneider KM, Carr DB, Sinha MK, Boyko EJ, et al. Relationship of adiponectin to body fat distribution, insulin sensitivity and plasma lipoproteins: evidence for independent roles of age and sex. Diabetologia. (2003) 46:459–69. doi: 10.1007/s00125-003-1074-z
40. Bajaj M, Suraamornkul S, Hardies LJ, Pratipanawatr T, Defronzo RA. Plasma resistin concentration, hepatic fat content, and hepatic and peripheral insulin resistance in pioglitazone-treated type II diabetic patients. Int J Obes Relat Metab Disord. (2004) 28:783–9. doi: 10.1038/sj.ijo.0802625
41. Dandona P, Aljada A, Bandyopadhyay A. Inflammation: the link between insulin resistance, obesity and diabetes. Trends Immunol. (2004) 25:4–7. doi: 10.1016/j.it.2003.10.013
42. Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, et al. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest. (2004) 114:1752–61. doi: 10.1172/JCI200421625
43. Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. (2004) 306:457–61. doi: 10.1126/science.1103160
44. Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. (2006) 116:1793–801. doi: 10.1172/JCI29069
45. Summers SA. Ceramides in insulin resistance and lipotoxicity. Prog Lipid Res. (2006) 45:42–72. doi: 10.1016/j.plipres.2005.11.002
46. Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, et al. A central role for JNK in obesity and insulin resistance. Nature. (2002) 420:333–6. doi: 10.1038/nature01137
47. Arkan MC, Hevener AL, Greten FR, Maeda S, Li ZW, Long JM, et al. IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med. (2005) 11:191–8. doi: 10.1038/nm1185
48. Chen Z, Yu R, Xiong Y, Du F, Zhu S. A vicious circle between insulin resistance and inflammation in nonalcoholic fatty liver disease. Lipids Health Dis. (2017) 16:203. doi: 10.1186/s12944-017-0572-9
49. Utzschneider KM, Kahn SE. Review: The role of insulin resistance in nonalcoholic fatty liver disease. J Clin Endocrinol Metab. (2006) 91:4753–61. doi: 10.1210/jc.2006-0587
50. Rhee EJ, Lee WY, Cho YK, Kim BI, Sung KC. Hyperinsulinemia and the development of nonalcoholic Fatty liver disease in nondiabetic adults. Am J Med. (2011) 124:69–76. doi: 10.1016/j.amjmed.2010.08.012
51. Birkenfeld AL, Shulman GI. Nonalcoholic fatty liver disease, hepatic insulin resistance, and type 2 diabetes. Hepatology. (2014) 59:713–23. doi: 10.1002/hep.26672
52. Hudgins LC, Hellerstein M, Seidman C, Neese R, Diakun J, Hirsch J. Human fatty acid synthesis is stimulated by a eucaloric low fat, high carbohydrate diet. J Clin Invest. (1996) 97:2081–91. doi: 10.1172/JCI118645
53. Day CP, Saksena S. Non-alcoholic steatohepatitis: definitions and pathogenesis. J Gastroenterol Hepatol. (2002) 17 Suppl 3:S377–384. doi: 10.1046/j.1440-1746.17.s3.31.x
54. Roden M. Mechanisms of Disease: hepatic steatosis in type 2 diabetes–pathogenesis and clinical relevance. Nat Clin Pract Endocrinol Metab. (2006) 2:335–48. doi: 10.1038/ncpendmet0190
55. Yahagi N, Shimano H, Hasty AH, Matsuzaka T, Ide T, Yoshikawa T, et al. Absence of sterol regulatory element-binding protein-1 (SREBP-1) ameliorates fatty livers but not obesity or insulin resistance in Lep(ob)/Lep(ob) mice. J Biol Chem. (2002) 277:19353–7. doi: 10.1074/jbc.M201584200
56. Koliaki C, Szendroedi J, Kaul K, Jelenik T, Nowotny P, Jankowiak F, et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. (2015) 21:739–46. doi: 10.1016/j.cmet.2015.04.004
57. Hebbard L, George J. Animal models of nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol. (2011) 8:35–44. doi: 10.1038/nrgastro.2010.191
58. Gruben N, Shiri-Sverdlov R, Koonen DP, Hofker MH. Nonalcoholic fatty liver disease: A main driver of insulin resistance or a dangerous liaison? Biochim Biophys Acta. (2014) 1842:2329–43. doi: 10.1016/j.bbadis.2014.08.004
59. Lam TK, Carpentier A, Lewis GF, Van De Werve G, Fantus IG, Giacca A. Mechanisms of the free fatty acid-induced increase in hepatic glucose production. Am J Physiol Endocrinol Metab. (2003) 284:E863–873. doi: 10.1152/ajpendo.00033.2003
60. Gao Z, Zhang X, Zuberi A, Hwang D, Quon MJ, Lefevre M, et al. Inhibition of insulin sensitivity by free fatty acids requires activation of multiple serine kinases in 3T3-L1 adipocytes. Mol Endocrinol. (2004) 18:2024–34. doi: 10.1210/me.2003-0383
61. Otero YF, Stafford JM, Mcguinness OP. Pathway-selective insulin resistance and metabolic disease: the importance of nutrient flux. J Biol Chem. (2014) 289:20462–9. doi: 10.1074/jbc.R114.576355
62. Palma R, Pronio A, Romeo M, Scognamiglio F, Ventriglia L, Ormando VM, et al. The role of insulin resistance in fueling NAFLD pathogenesis: from molecular mechanisms to clinical implications. J Clin Med. (2022) 11:3649. doi: 10.3390/jcm11133649
63. Odegaard JI, Ricardo-Gonzalez RR, Red Eagle A, Vats D, Morel CR, Goforth MH, et al. Alternative M2 activation of Kupffer cells by PPARdelta ameliorates obesity-induced insulin resistance. Cell Metab. (2008) 7:496–507. doi: 10.1016/j.cmet.2008.04.003
64. Sica A, Invernizzi P, Mantovani A. Macrophage plasticity and polarization in liver homeostasis and pathology. Hepatology. (2014) 59:2034–42. doi: 10.1002/hep.26754
65. Backhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, et al. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A. (2004) 101:15718–23. doi: 10.1073/pnas.0407076101
66. Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, et al. Diversity of the human intestinal microbial flora. Science. (2005) 308:1635–8. doi: 10.1126/science.1110591
67. Mazmanian SK, Liu CH, Tzianabos AO, Kasper DL. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell. (2005) 122:107–18. doi: 10.1016/j.cell.2005.05.007
68. Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. (2006) 444:1027–31. doi: 10.1038/nature05414
69. Sabio G, Das M, Mora A, Zhang Z, Jun JY, Ko HJ, et al. A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Science. (2008) 322:1539–43. doi: 10.1126/science.1160794
70. Turnbaugh PJ, Backhed F, Fulton L, Gordon JI. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe. (2008) 3:213–23. doi: 10.1016/j.chom.2008.02.015
71. Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A. (2005) 102:11070–5. doi: 10.1073/pnas.0504978102
72. Sonnenburg JL, Angenent LT, Gordon JI. Getting a grip on things: how do communities of bacterial symbionts become established in our intestine? Nat Immunol. (2004) 5:569–73. doi: 10.1038/ni1079
73. Rabot S, Membrez M, Bruneau A, Gerard P, Harach T, Moser M, et al. Germ-free C57BL/6J mice are resistant to high-fat-diet-induced insulin resistance and have altered cholesterol metabolism. FASEB J. (2010) 24:4948–59. doi: 10.1096/fj.10.164921
74. Kolodziejczyk AA, Zheng D, Shibolet O, Elinav E. The role of the microbiome in NAFLD and NASH. EMBO Mol Med. (2019) 11. doi: 10.15252/emmm.201809302
75. Boursier J, Mueller O, Barret M, Machado M, Fizanne L, Araujo-Perez F, et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology. (2016) 63:764–75. doi: 10.1002/hep.28356
76. Lee G, You HJ, Bajaj JS, Joo SK, Yu J, Park S, et al. Distinct signatures of gut microbiome and metabolites associated with significant fibrosis in non-obese NAFLD. Nat Commun. (2020) 11:4982. doi: 10.1038/s41467-020-18754-5
77. De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, et al. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci U S A. (2010) 107:14691–6. doi: 10.1073/pnas.1005963107
78. Ou J, Carbonero F, Zoetendal EG, Delany JP, Wang M, Newton K, et al. Diet, microbiota, and microbial metabolites in colon cancer risk in rural Africans and African Americans. Am J Clin Nutr. (2013) 98:111–20. doi: 10.3945/ajcn.112.056689
79. Moran-Ramos S, Cerqueda-Garcia D, Lopez-Contreras B, Larrieta-Carrasco E, Villamil-Ramirez H, Molina-Cruz S, et al. A metagenomic study identifies a Prevotella copri enriched microbial profile associated with non-alcoholic steatohepatitis in subjects with obesity. J Gastroenterol Hepatol. (2023) 38:791–9. doi: 10.1111/jgh.16147
80. Iljazovic A, Roy U, Gálvez EJC, Lesker TR, Zhao B, Gronow A, et al. Perturbation of the gut microbiome by Prevotella spp. enhances host susceptibility to mucosal inflammation. Mucosal Immunol. (2021) 14:113–24. doi: 10.1038/s41385-020-0296-4
81. Larsen JM. The immune response to Prevotella bacteria in chronic inflammatory disease. Immunology. (2017) 151:363–74. doi: 10.1111/imm.12760
82. Jumpertz R, Le DS, Turnbaugh PJ, Trinidad C, Bogardus C, Gordon JI, et al. Energy-balance studies reveal associations between gut microbes, caloric load, and nutrient absorption in humans. Am J Clin Nutr. (2011) 94:58–65. doi: 10.3945/ajcn.110.010132
83. Kau AL, Ahern PP, Griffin NW, Goodman AL, Gordon JI. Human nutrition, the gut microbiome and the immune system. Nature. (2011) 474:327–36. doi: 10.1038/nature10213
84. Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med. (2013) 19:576–85. doi: 10.1038/nm.3145
85. Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science. (2013) 341:1241214. doi: 10.1126/science.1241214
86. Sharon G, Garg N, Debelius J, Knight R, Dorrestein PC, Mazmanian SK. Specialized metabolites from the microbiome in health and disease. Cell Metab. (2014) 20:719–30. doi: 10.1016/j.cmet.2014.10.016
87. Neis EP, Dejong CH, Rensen SS. The role of microbial amino acid metabolism in host metabolism. Nutrients. (2015) 7:2930–46. doi: 10.3390/nu7042930
88. Levy M, Kolodziejczyk AA, Thaiss CA, Elinav E. Dysbiosis and the immune system. Nat Rev Immunol. (2017) 17:219–32. doi: 10.1038/nri.2017.7
89. Festi D, Colecchia A, Sacco T, Bondi M, Roda E, Marchesini G. Hepatic steatosis in obese patients: clinical aspects and prognostic significance. Obes Rev. (2004) 5:27–42. doi: 10.1111/j.1467-789X.2004.00126.x
90. Compare D, Coccoli P, Rocco A, Nardone OM, De Maria S, Carteni M, et al. Gut–liver axis: the impact of gut microbiota on non alcoholic fatty liver disease. Nutr Metab Cardiovasc Dis. (2012) 22:471–6. doi: 10.1016/j.numecd.2012.02.007
91. Le Roy T, Llopis M, Lepage P, Bruneau A, Rabot S, Bevilacqua C, et al. Intestinal microbiota determines development of non-alcoholic fatty liver disease in mice. Gut. (2013) 62:1787–94. doi: 10.1136/gutjnl-2012-303816
92. Loomba R, Seguritan V, Li W, Long T, Klitgord N, Bhatt A, et al. Gut microbiome-based metagenomic signature for non-invasive detection of advanced fibrosis in human nonalcoholic fatty liver disease. Cell Metab. (2017) 25:1054–62. doi: 10.1016/j.cmet.2017.04.001
93. Quevrain E, Maubert MA, Michon C, Chain F, Marquant R, Tailhades J, et al. Identification of an anti-inflammatory protein from Faecalibacterium prausnitzii, a commensal bacterium deficient in Crohn's disease. Gut. (2016) 65:415–25. doi: 10.1136/gutjnl-2014-307649
94. Zeng H, Liu J, Jackson MI, Zhao FQ, Yan L, Combs GFJr. Fatty liver accompanies an increase in lactobacillus species in the hind gut of C57BL/6 mice fed a high-fat diet. J Nutr. (2013) 143:627–31. doi: 10.3945/jn.112.172460
95. Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. (2007) 56:1761–72. doi: 10.2337/db06-1491
96. De Wit N, Derrien M, Bosch-Vermeulen H, Oosterink E, Keshtkar S, Duval C, et al. Saturated fat stimulates obesity and hepatic steatosis and affects gut microbiota composition by an enhanced overflow of dietary fat to the distal intestine. Am J Physiol Gastrointest Liver Physiol. (2012) 303:G589–599. doi: 10.1152/ajpgi.00488.2011
97. Taira T, Yamaguchi S, Takahashi A, Okazaki Y, Yamaguchi A, Sakaguchi H, et al. Dietary polyphenols increase fecal mucin and immunoglobulin A and ameliorate the disturbance in gut microbiota caused by a high fat diet. J Clin Biochem Nutr. (2015) 57:212–6. doi: 10.3164/jcbn.15-15
98. Bisanz JE, Upadhyay V, Turnbaugh JA, Ly K, Turnbaugh PJ. Meta-Analysis Reveals Reproducible Gut Microbiome Alterations in Response to a High-Fat Diet. Cell Host Microbe. (2019) 26:265–72. doi: 10.1016/j.chom.2019.06.013
99. Singh RP, Halaka DA, Hayouka Z, Tirosh O. High-fat diet induced alteration of mice microbiota and the functional ability to utilize fructooligosaccharide for ethanol production. Front Cell Infect Microbiol. (2020) 10:376. doi: 10.3389/fcimb.2020.00376
100. Munukka E, Wiklund P, Pekkala S, Volgyi E, Xu L, Cheng S, et al. Women with and without metabolic disorder differ in their gut microbiota composition. Obesity. (2012) 20:1082–7. doi: 10.1038/oby.2012.8
101. Parks BW, Nam E, Org E, Kostem E, Norheim F, Hui ST, et al. Genetic control of obesity and gut microbiota composition in response to high-fat, high-sucrose diet in mice. Cell Metab. (2013) 17:141–52. doi: 10.1016/j.cmet.2012.12.007
102. Murphy EA, Velazquez KT, Herbert KM. Influence of high-fat diet on gut microbiota: a driving force for chronic disease risk. Curr Opin Clin Nutr Metab Care. (2015) 18:515–20. doi: 10.1097/MCO.0000000000000209
103. Islam MR, Arthur S, Haynes J, Butts MR, Nepal N, Sundaram U. The role of gut microbiota and metabolites in obesity-associated chronic gastrointestinal disorders. Nutrients. (2022) 14:624. doi: 10.3390/nu14030624
104. Mitsou EK, Kakali A, Antonopoulou S, Mountzouris KC, Yannakoulia M, Panagiotakos DB, et al. Adherence to the Mediterranean diet is associated with the gut microbiota pattern and gastrointestinal characteristics in an adult population. Br J Nutr. (2017) 117:1645–55. doi: 10.1017/S0007114517001593
105. Garcia-Mantrana I, Selma-Royo M, Alcantara C, Collado MC. Shifts on gut microbiota associated to mediterranean diet adherence and specific dietary intakes on general adult population. Front Microbiol. (2018) 9:890. doi: 10.3389/fmicb.2018.00890
106. Nagpal R, Shively CA, Appt SA, Register TC, Michalson KT, Vitolins MZ, et al. Gut microbiome composition in non-human primates consuming a western or mediterranean diet. Front Nutr. (2018) 5:28. doi: 10.3389/fnut.2018.00028
107. Forlano R, Mullish BH, Roberts LA, Thursz MR, Manousou P. The intestinal barrier and its dysfunction in patients with metabolic diseases and non-alcoholic fatty liver disease. Int J Mol Sci. (2022) 23:662. doi: 10.3390/ijms23020662
108. Soppert J, Brandt EF, Heussen NM, Barzakova E, Blank LM, Kuepfer L, et al. Blood endotoxin levels as biomarker of nonalcoholic fatty liver disease: a systematic review and meta-analysis. Clin Gastroenterol Hepatol. (2023) 21:2746–58. doi: 10.1016/j.cgh.2022.11.030
109. Fialho A, Fialho A, Thota P, Mccullough AJ, Shen B. Small intestinal bacterial overgrowth is associated with non-alcoholic fatty liver disease. J Gastrointestin Liver Dis. (2016) 25:159–65. doi: 10.15403/jgld.2014.1121.252.iwg
110. Shanab AA, Scully P, Crosbie O, Buckley M, O'mahony L, Shanahan F, et al. Small intestinal bacterial overgrowth in nonalcoholic steatohepatitis: association with toll-like receptor 4 expression and plasma levels of interleukin 8. Dig Dis Sci. (2011) 56:1524–34. doi: 10.1007/s10620-010-1447-3
111. Gkolfakis P, Tziatzios G, Leite G, Papanikolaou IS, Xirouchakis E, Panayiotides IG, et al. Prevalence of small intestinal bacterial overgrowth syndrome in patients with non-alcoholic fatty liver disease/non-alcoholic steatohepatitis: a cross-sectional study. Microorganisms. (2023) 11:723. doi: 10.3390/microorganisms11030723
112. Fang J, Yu CH, Li XJ, Yao JM, Fang ZY, Yoon SH, et al. Gut dysbiosis in nonalcoholic fatty liver disease: pathogenesis, diagnosis, and therapeutic implications. Front Cell Infect Microbiol. (2022) 12:997018. doi: 10.3389/fcimb.2022.997018
113. Hildebrandt MA, Hoffmann C, Sherrill-Mix SA, Keilbaugh SA, Hamady M, Chen YY, et al. High-fat diet determines the composition of the murine gut microbiome independently of obesity. Gastroenterology. (2009) 137:1716–24. doi: 10.1053/j.gastro.2009.08.042
114. Loomba R, Seguritan V, Li W, Long T, Klitgord N, Bhatt A, et al. Gut microbiome-based metagenomic signature for non-invasive detection of advanced fibrosis in human nonalcoholic fatty liver disease. Cell Metab. (2019) 30:607. doi: 10.1016/j.cmet.2019.08.002
115. Li Q, Rempel JD, Yang J, Minuk GY. The effects of pathogen-associated molecular patterns on peripheral blood monocytes in patients with non-alcoholic fatty liver disease. J Clin Exp Hepatol. (2022) 12:808–17. doi: 10.1016/j.jceh.2021.11.011
116. Pettinelli P, Arendt BM, Schwenger KJP, Sivaraj S, Bhat M, Comelli EM, et al. Relationship between hepatic gene expression, intestinal microbiota, and inferred functional metagenomic analysis in NAFLD. Clin Transl Gastroenterol. (2022) 13:e00466. doi: 10.14309/ctg.0000000000000466
117. Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. (2012) 482:179–85. doi: 10.1038/nature10809
118. Ikeda T, Nishida A, Yamano M, Kimura I. Short-chain fatty acid receptors and gut microbiota as therapeutic targets in metabolic, immune, and neurological diseases. Pharmacol Ther. (2022) 239:108273. doi: 10.1016/j.pharmthera.2022.108273
119. Liu M, Lu Y, Xue G, Han L, Jia H, Wang Z, et al. Role of short-chain fatty acids in host physiology. Animal Model Exp Med. (2024) 7:641–52. doi: 10.1002/ame2.12464
120. Kondo T, Kishi M, Fushimi T, Kaga T. Acetic acid upregulates the expression of genes for fatty acid oxidation enzymes in liver to suppress body fat accumulation. J Agric Food Chem. (2009) 57:5982–6. doi: 10.1021/jf900470c
121. Morrison DJ, Preston T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes. (2016) 7:189–200. doi: 10.1080/19490976.2015.1134082
122. Liu W, Luo X, Tang J, Mo Q, Zhong H, Zhang H, et al. A bridge for short-chain fatty acids to affect inflammatory bowel disease, type 1 diabetes, and non-alcoholic fatty liver disease positively: by changing gut barrier. Eur J Nutr. (2021) 60:2317–30. doi: 10.1007/s00394-020-02431-w
123. Park JH, Kotani T, Konno T, Setiawan J, Kitamura Y, Imada S, et al. Promotion of intestinal epithelial cell turnover by commensal bacteria: role of short-chain fatty acids. PLoS ONE. (2016) 11:e0156334. doi: 10.1371/journal.pone.0156334
124. Aoki R, Onuki M, Hattori K, Ito M, Yamada T, Kamikado K, et al. Commensal microbe-derived acetate suppresses NAFLD/NASH development via hepatic FFAR2 signalling in mice. Microbiome. (2021) 9:188. doi: 10.1186/s40168-021-01125-7
125. Zhou D, Pan Q, Xin FZ, Zhang RN, He CX, Chen GY, et al. Sodium butyrate attenuates high-fat diet-induced steatohepatitis in mice by improving gut microbiota and gastrointestinal barrier. World J Gastroenterol. (2017) 23:60–75. doi: 10.3748/wjg.v23.i1.60
126. Zhou D, Chen YW, Zhao ZH, Yang RX, Xin FZ, Liu XL, et al. Sodium butyrate reduces high-fat diet-induced non-alcoholic steatohepatitis through upregulation of hepatic GLP-1R expression. Exp Mol Med. (2018) 50:1–12. doi: 10.1038/s12276-018-0183-1
127. Baumann A, Jin CJ, Brandt A, Sellmann C, Nier A, Burkard M, et al. Oral supplementation of sodium butyrate attenuates the progression of non-alcoholic steatohepatitis. Nutrients. (2020) 12:951. doi: 10.3390/nu12040951
128. Yang T, Yang H, Heng C, Wang H, Chen S, Hu Y, et al. Amelioration of non-alcoholic fatty liver disease by sodium butyrate is linked to the modulation of intestinal tight junctions in db/db mice. Food Funct. (2020) 11:10675–89. doi: 10.1039/D0FO01954B
129. Chen J, Vitetta L. Gut Microbiota Metabolites in NAFLD Pathogenesis and Therapeutic Implications. Int J Mol Sci. (2020) 21. doi: 10.3390/ijms21155214
130. Hartmann P, Chen WC, Schnabl B. The intestinal microbiome and the leaky gut as therapeutic targets in alcoholic liver disease. Front Physiol. (2012) 3:402. doi: 10.3389/fphys.2012.00402
131. Engstler AJ, Aumiller T, Degen C, Durr M, Weiss E, Maier IB, et al. Insulin resistance alters hepatic ethanol metabolism: studies in mice and children with non-alcoholic fatty liver disease. Gut. (2016) 65:1564–71. doi: 10.1136/gutjnl-2014-308379
132. Chen J, Thomsen M, Vitetta L. Interaction of gut microbiota with dysregulation of bile acids in the pathogenesis of nonalcoholic fatty liver disease and potential therapeutic implications of probiotics. J Cell Biochem. (2019) 120:2713–20. doi: 10.1002/jcb.27635
133. Miyata M, Funaki A, Fukuhara C, Sumiya Y, Sugiura Y. Taurine attenuates hepatic steatosis in a genetic model of fatty liver disease. J Toxicol Sci. (2020) 45:87–94. doi: 10.2131/jts.45.87
134. Alvarez-Sola G, Uriarte I, Latasa MU, Fernandez-Barrena MG, Urtasun R, Elizalde M, et al. Fibroblast growth factor 15/19 (FGF15/19) protects from diet-induced hepatic steatosis: development of an FGF19-based chimeric molecule to promote fatty liver regeneration. Gut. (2017) 66:1818–28. doi: 10.1136/gutjnl-2016-312975
135. Mouzaki M, Comelli EM, Arendt BM, Bonengel J, Fung SK, Fischer SE, et al. Intestinal microbiota in patients with nonalcoholic fatty liver disease. Hepatology. (2013) 58:120–7. doi: 10.1002/hep.26319
136. Del Chierico F, Nobili V, Vernocchi P, Russo A, De Stefanis C, Gnani D, et al. Gut microbiota profiling of pediatric nonalcoholic fatty liver disease and obese patients unveiled by an integrated meta-omics-based approach. Hepatology. (2017) 65:451–64. doi: 10.1002/hep.28572
137. Tacke F, Zimmermann HW. Macrophage heterogeneity in liver injury and fibrosis. J Hepatol. (2014) 60:1090–6. doi: 10.1016/j.jhep.2013.12.025
138. Caligiuri A, Gentilini A, Marra F. Molecular pathogenesis of NASH. Int J Mol Sci. (2016) 17. doi: 10.3390/ijms17091575
139. Yuan F, Cai JN, Dai M, Lv X. Inhibition of P2Y(6) receptor expression in Kupffer cells alleviates alcoholic steatohepatitis in mice. Int Immunopharmacol. (2022) 109:108909. doi: 10.1016/j.intimp.2022.108909
140. Liaskou E, Wilson DV, Oo YH. Innate immune cells in liver inflammation. Mediators Inflamm. (2012) 2012:949157. doi: 10.1155/2012/949157
141. Zimmermann HW, Tacke F. Modification of chemokine pathways and immune cell infiltration as a novel therapeutic approach in liver inflammation and fibrosis. Inflamm Allergy Drug Targets. (2011) 10:509–36. doi: 10.2174/187152811798104890
142. Bedossa P. Diagnosis of non-alcoholic fatty liver disease/non-alcoholic steatohepatitis: Why liver biopsy is essential. Liver Int. (2018) 38 Suppl 1:64–6. doi: 10.1111/liv.13653
143. Zhang P. Influence of foods and nutrition on the gut microbiome and implications for intestinal health. Int J Mol Sci. (2022) 23. doi: 10.3390/ijms23179588
144. Yang C, Wu J, Yang L, Hu Q, Li L, Yang Y, et al. Altered gut microbial profile accompanied by abnormal short chain fatty acid metabolism exacerbates nonalcoholic fatty liver disease progression. Sci Rep. (2024) 14:22385. doi: 10.1038/s41598-024-72909-8
145. Suk KT, Kim DJ. Gut microbiota: novel therapeutic target for nonalcoholic fatty liver disease. Expert Rev Gastroenterol Hepatol. (2019) 13:193–204. doi: 10.1080/17474124.2019.1569513
146. Albillos A, De Gottardi A, Rescigno M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J Hepatol. (2020) 72:558–77. doi: 10.1016/j.jhep.2019.10.003
147. Han TR, Yang WJ, Tan QH, Bai S, Zhong H, Tai Y, et al. Gut microbiota therapy for nonalcoholic fatty liver disease: Evidence from randomized clinical trials. Front Microbiol. (2022) 13:1004911. doi: 10.3389/fmicb.2022.1004911
148. Kumari N, Kumari R, Dua A, Singh M, Kumar R, Singh P, et al. From gut to hormones: unraveling the role of gut microbiota in (phyto)estrogen modulation in health and disease. Mol Nutr Food Res. (2024) 68:2300688. doi: 10.1002/mnfr.202300688
149. Schwenger KJ, Clermont-Dejean N, Allard JP. The role of the gut microbiome in chronic liver disease: the clinical evidence revised. JHEP Rep. (2019) 1:214–26. doi: 10.1016/j.jhepr.2019.04.004
150. Zhang C, Yang M, Ericsson AC. The potential gut microbiota-mediated treatment options for liver cancer. Front Oncol. (2020) 10:524205. doi: 10.3389/fonc.2020.524205
151. Huang W, Kong D. The intestinal microbiota as a therapeutic target in the treatment of NAFLD and ALD. Biomed Pharmacother. (2021) 135:111235. doi: 10.1016/j.biopha.2021.111235
152. Li M, Liu T, Yang T, Zhu J, Zhou Y, Wang M, et al. Gut microbiota dysbiosis involves in host non-alcoholic fatty liver disease upon pyrethroid pesticide exposure. Environ Sci Ecotechnol. (2022) 11:100185. doi: 10.1016/j.ese.2022.100185
153. Dua A, Nigam A, Saxena A, Dhingra GG, Kumar R. Microbial bioproduction of antiaging molecules. In:Singh SP, Upadhyay SK, , editors. Microbial Bioreactors for Industrial Molecules. Wiley (2023). p. 512. doi: 10.1002/9781119874096.ch22
154. Smeuninx B, Boslem E, Febbraio MA. Current and future treatments in the fight against non-alcoholic fatty liver disease. Cancers. (2020) 12. doi: 10.3390/cancers12071714
155. Gao X, Zhu Y, Wen Y, Liu G, Wan C. Efficacy of probiotics in non-alcoholic fatty liver disease in adult and children: a meta-analysis of randomized controlled trials. Hepatol Res. (2016) 46:1226–33. doi: 10.1111/hepr.12671
156. Kumar R, Sood U, Gupta V, Singh M, Scaria J, Lal R. Recent advancements in the development of modern probiotics for restoring human gut microbiome dysbiosis. Indian J Microbiol. (2020) 60:12–25. doi: 10.1007/s12088-019-00808-y
157. Choi HH, Cho YS. Fecal microbiota transplantation: current applications, effectiveness, and future perspectives. Clin Endosc. (2016) 49:257–65. doi: 10.5946/ce.2015.117
158. Kumar R, Sood U, Kaur J, Anand S, Gupta V, Patil KS, et al. The rising dominance of microbiology: what to expect in the next 15 years? Microb Biotechnol. (2022) 15:110–28. doi: 10.1111/1751-7915.13953
159. Li Z, Yang S, Lin H, Huang J, Watkins PA, Moser AB, et al. Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease. Hepatology. (2003) 37:343–50. doi: 10.1053/jhep.2003.50048
160. Mencarelli A, Cipriani S, Renga B, Bruno A, D'amore C, Distrutti E, et al. VSL#3 resets insulin signaling and protects against NASH and atherosclerosis in a model of genetic dyslipidemia and intestinal inflammation. PLoS ONE. (2012) 7:e45425. doi: 10.1371/journal.pone.0045425
161. Lynch SV, Pedersen O. The human intestinal microbiome in health and disease. N Engl J Med. (2016) 375:2369–79. doi: 10.1056/NEJMra1600266
162. Xie C, Halegoua-Demarzio D. Role of probiotics in non-alcoholic fatty liver disease: does gut microbiota matter? Nutrients. (2019) 11:2837. doi: 10.3390/nu11112837
163. Okubo H, Sakoda H, Kushiyama A, Fujishiro M, Nakatsu Y, Fukushima T, et al. Lactobacillus casei strain Shirota protects against nonalcoholic steatohepatitis development in a rodent model. Am J Physiol Gastrointest Liver Physiol. (2013) 305:G911–918. doi: 10.1152/ajpgi.00225.2013
164. Wongkuna S, Ambat A, Ghimire S, Mattiello Samara P, Maji A, Kumar R, et al. Identification of a microbial sub-community from the feral chicken gut that reduces Salmonella colonization and improves gut health in a gnotobiotic chicken model. Microbiology Spectrum. (2024) 12:e01621–01623. doi: 10.1128/spectrum.01621-23
165. Binda S, Hill C, Johansen E, Obis D, Pot B, Sanders ME, et al. Criteria to qualify microorganisms as “probiotic” in foods and dietary supplements. Front Microbiol. (2020) 11:1662. doi: 10.3389/fmicb.2020.01662
166. Mohamad Nor MH, Ayob N, Mokhtar NM, Raja Ali RA, Tan GC, Wong Z, et al. The effect of probiotics (MCP((R)) BCMC((R)) Strains) on hepatic steatosis, small intestinal mucosal immune function, and intestinal barrier in patients with non-alcoholic fatty liver disease. Nutrients. (2021) 13:3192. doi: 10.3390/nu13093192
167. Wong VW, Won GL, Chim AM, Chu WC, Yeung DK, Li KC, et al. Treatment of nonalcoholic steatohepatitis with probiotics. A proof-of-concept study. Ann Hepatol. (2013) 12:256–62. doi: 10.1016/S1665-2681(19)31364-X
168. Duseja A, Acharya SK, Mehta M, Chhabra S, Shalimar Rana S, Das A, et al. High potency multistrain probiotic improves liver histology in non-alcoholic fatty liver disease (NAFLD): a randomised, double-blind, proof of concept study. BMJ Open Gastroenterol. (2019) 6:e000315. doi: 10.1136/bmjgast-2019-000315
169. Ahn SB, Jun DW, Kang BK, Lim JH, Lim S, Chung MJ. Randomized, double-blind, placebo-controlled study of a multispecies probiotic mixture in nonalcoholic fatty liver disease. Sci Rep. (2019) 9:5688. doi: 10.1038/s41598-019-42059-3
170. Ma YY, Li L, Yu CH, Shen Z, Chen LH, Li YM. Effects of probiotics on nonalcoholic fatty liver disease: a meta-analysis. World J Gastroenterol. (2013) 19:6911–8. doi: 10.3748/wjg.v19.i40.6911
171. Eslamparast T, Poustchi H, Zamani F, Sharafkhah M, Malekzadeh R, Hekmatdoost A. Synbiotic supplementation in nonalcoholic fatty liver disease: a randomized, double-blind, placebo-controlled pilot study. Am J Clin Nutr. (2014) 99:535–42. doi: 10.3945/ajcn.113.068890
172. Sepideh A, Karim P, Hossein A, Leila R, Hamdollah M, Mohammad EG, et al. Effects of multistrain probiotic supplementation on glycemic and inflammatory indices in patients with nonalcoholic fatty liver disease: a double-blind randomized clinical trial. J Am Coll Nutr. (2016) 35:500–5. doi: 10.1080/07315724.2015.1031355
173. Kobyliak N, Abenavoli L, Mykhalchyshyn G, Kononenko L, Boccuto L, Kyriienko D, et al. A Multi-strain probiotic reduces the fatty liver index, cytokines and aminotransferase levels in NAFLD patients: evidence from a randomized clinical trial. J Gastrointestin Liver Dis. (2018) 27:41–9. doi: 10.15403/jgld.2014.1121.271.kby
174. Sharpton SR, Maraj B, Harding-Theobald E, Vittinghoff E, Terrault NA. Gut microbiome-targeted therapies in nonalcoholic fatty liver disease: a systematic review, meta-analysis, and meta-regression. Am J Clin Nutr. (2019) 110:139–49. doi: 10.1093/ajcn/nqz042
175. Huang Y, Wang X, Zhang L, Zheng K, Xiong J, Li J, et al. Effect of probiotics therapy on nonalcoholic fatty liver disease. Comput Math Methods Med. (2022) 2022:7888076. doi: 10.1155/2022/7888076
176. Cai J, Dong J, Chen D, Ye H. The effect of synbiotics in patients with NAFLD: a systematic review and meta-analysis. Therap Adv Gastroenterol. (2023) 16:17562848231174299. doi: 10.1177/17562848231174299
177. Cao C, Shi M, Wang X, Yao Y, Zeng R. Effects of probiotics on non-alcoholic fatty liver disease: a review of human clinical trials. Front Nutr. (2023) 10:1155306. doi: 10.3389/fnut.2023.1155306
178. Song Y, Liu S, Zhang L, Zhao W, Qin Y, Liu M. The effect of gut microbiome-targeted therapies in nonalcoholic fatty liver disease: a systematic review and network meta-analysis. Front Nutr. (2024) 11:1470185. doi: 10.3389/fnut.2024.1470185
179. Liu L, Li P, Liu Y, Zhang Y. Efficacy of probiotics and synbiotics in patients with nonalcoholic fatty liver disease: a meta-analysis. Dig Dis Sci. (2019) 64:3402–12. doi: 10.1007/s10620-019-05699-z
180. Brunner JD, Chia N. Metabolic model-based ecological modeling for probiotic design. Elife. (2024) 13:e83690. doi: 10.7554/eLife.83690
181. Cho MS, Kim SY, Suk KT, Kim BY. Modulation of gut microbiome in nonalcoholic fatty liver disease: pro-, pre-, syn-, and antibiotics. J Microbiol. (2018) 56:855–67. doi: 10.1007/s12275-018-8346-2
182. Vallianou N, Stratigou T, Christodoulatos GS, Dalamaga M. Understanding the role of the gut microbiome and microbial metabolites in obesity and obesity-associated metabolic disorders: current evidence and perspectives. Curr Obes Rep. (2019) 8:317–32. doi: 10.1007/s13679-019-00352-2
183. Rezende ESV, Lima GC, Naves MMV. Dietary fibers as beneficial microbiota modulators: a proposed classification by prebiotic categories. Nutrition. (2021) 89:111217. doi: 10.1016/j.nut.2021.111217
184. Duarte FND, Rodrigues JB, Da Costa Lima M, Lima MDS, Pacheco MTB, Pintado MME, et al. Potential prebiotic properties of cashew apple (Anacardium occidentale L.) agro-industrial byproduct on Lactobacillus species. J Sci Food Agric. (2017) 97:3712–9. doi: 10.1002/jsfa.8232
185. Sun Y, Hu J, Zhang S, He H, Nie Q, Zhang Y, et al. Prebiotic characteristics of arabinogalactans during in vitro fermentation through multi-omics analysis. Food Chem Toxicol. (2021) 156:112522. doi: 10.1016/j.fct.2021.112522
186. Bomhof MR, Parnell JA, Ramay HR, Crotty P, Rioux KP, Probert CS, et al. Histological improvement of non-alcoholic steatohepatitis with a prebiotic: a pilot clinical trial. Eur J Nutr. (2019) 58:1735–45. doi: 10.1007/s00394-018-1721-2
187. Stachowska E, Portincasa P, Jamiol-Milc D, Maciejewska-Markiewicz D, Skonieczna-Zydecka K. The relationship between prebiotic supplementation and anthropometric and biochemical parameters in patients with NAFLD-A systematic review and meta-analysis of randomized controlled trials. Nutrients. (2020) 12. doi: 10.3390/nu12113460
188. Malaguarnera M, Vacante M, Antic T, Giordano M, Chisari G, Acquaviva R, et al. Bifidobacterium longum with fructo-oligosaccharides in patients with non alcoholic steatohepatitis. Dig Dis Sci. (2012) 57:545–53. doi: 10.1007/s10620-011-1887-4
189. Safavi M, Farajian S, Kelishadi R, Mirlohi M, Hashemipour M. The effects of synbiotic supplementation on some cardio-metabolic risk factors in overweight and obese children: a randomized triple-masked controlled trial. Int J Food Sci Nutr. (2013) 64:687–93. doi: 10.3109/09637486.2013.775224
190. Jafarpour D, Shekarforoush SS, Ghaisari HR, Nazifi S, Sajedianfard J, Eskandari MH. Protective effects of synbiotic diets of Bacillus coagulans, Lactobacillus plantarum and inulin against acute cadmium toxicity in rats. BMC Complement Altern Med. (2017) 17:291. doi: 10.1186/s12906-017-1803-3
191. Janssen AWF, Houben T, Katiraei S, Dijk W, Boutens L, Van Der Bolt N, et al. Modulation of the gut microbiota impacts nonalcoholic fatty liver disease: a potential role for bile acids. J Lipid Res. (2017) 58:1399–416. doi: 10.1194/jlr.M075713
192. Bergheim I, Weber S, Vos M, Kramer S, Volynets V, Kaserouni S, et al. Antibiotics protect against fructose-induced hepatic lipid accumulation in mice: role of endotoxin. J Hepatol. (2008) 48:983–92. doi: 10.1016/j.jhep.2008.01.035
193. Brown E, Hydes T, Hamid A, Cuthbertson DJ. Emerging and established therapeutic approaches for nonalcoholic fatty liver disease. Clin Ther. (2021) 43:1476–504. doi: 10.1016/j.clinthera.2021.07.013
194. Gangarapu V, Ince AT, Baysal B, Kayar Y, Kilic U, Gok O, et al. Efficacy of rifaximin on circulating endotoxins and cytokines in patients with nonalcoholic fatty liver disease. Eur J Gastroenterol Hepatol. (2015) 27:840–5. doi: 10.1097/MEG.0000000000000348
195. Mahana D, Trent CM, Kurtz ZD, Bokulich NA, Battaglia T, Chung J, et al. Antibiotic perturbation of the murine gut microbiome enhances the adiposity, insulin resistance, and liver disease associated with high-fat diet. Genome Med. (2016) 8:48. doi: 10.1186/s13073-016-0297-9
196. Ebrahimi F, Simon TG, Hagstrom H, Sun J, Bergman D, Forss A, et al. Antibiotic use and development of nonalcoholic fatty liver disease: a population-based case-control study. Liver Int. (2023) 43:2186–97. doi: 10.1111/liv.15663
197. Safari Z, Gerard P. The links between the gut microbiome and non-alcoholic fatty liver disease (NAFLD). Cell Mol Life Sci. (2019) 76:1541–58. doi: 10.1007/s00018-019-03011-w
198. Vrieze A, Van Nood E, Holleman F, Salojarvi J, Kootte RS, Bartelsman JF, et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology. (2012) 143:913–6. doi: 10.1053/j.gastro.2012.06.031
199. Kelly CR, Kahn S, Kashyap P, Laine L, Rubin D, Atreja A, et al. Update on Fecal Microbiota Transplantation 2015: Indications, Methodologies, Mechanisms, and Outlook. Gastroenterology. (2015) 149:223–37. doi: 10.1053/j.gastro.2015.05.008
200. Walker AW, Parkhill J. Microbiology. Fighting obesity with bacteria. Science. (2013) 341:1069–70. doi: 10.1126/science.1243787
201. Anderson JL, Edney RJ, Whelan K. Systematic review: faecal microbiota transplantation in the management of inflammatory bowel disease. Aliment Pharmacol Ther. (2012) 36:503–16. doi: 10.1111/j.1365-2036.2012.05220.x
202. He Z, Cui BT, Zhang T, Li P, Long CY, Ji GZ, et al. Fecal microbiota transplantation cured epilepsy in a case with Crohn's disease: the first report. World J Gastroenterol. (2017) 23:3565–8. doi: 10.3748/wjg.v23.i19.3565
203. Kootte RS, Levin E, Salojarvi J, Smits LP, Hartstra AV, Udayappan SD, et al. Improvement of insulin sensitivity after lean donor feces in metabolic syndrome is driven by baseline intestinal microbiota composition. Cell Metab. (2017) 26:611–9. doi: 10.1016/j.cmet.2017.09.008
204. Cai TT, Ye XL, Yong HJ, Song B, Zheng XL, Cui BT, et al. Fecal microbiota transplantation relieve painful diabetic neuropathy: a case report. Medicine. (2018) 97:e13543. doi: 10.1097/MD.0000000000013543
205. De Groot P, Scheithauer T, Bakker GJ, Prodan A, Levin E, Khan MT, et al. Donor metabolic characteristics drive effects of faecal microbiota transplantation on recipient insulin sensitivity, energy expenditure and intestinal transit time. Gut. (2020) 69:502–12. doi: 10.1136/gutjnl-2019-318320
206. Zhou D, Pan Q, Shen F, Cao HX, Ding WJ, Chen YW, et al. Total fecal microbiota transplantation alleviates high-fat diet-induced steatohepatitis in mice via beneficial regulation of gut microbiota. Sci Rep. (2017) 7:1529. doi: 10.1038/s41598-017-01751-y
207. Zhang X, Coker OO, Chu ES, Fu K, Lau HCH, Wang YX, et al. Dietary cholesterol drives fatty liver-associated liver cancer by modulating gut microbiota and metabolites. Gut. (2021) 70:761–74. doi: 10.1136/gutjnl-2019-319664
208. Gupta M, Krishan P, Kaur A, Arora S, Trehanpati N, Singh TG, et al. Mechanistic and physiological approaches of fecal microbiota transplantation in the management of NAFLD. Inflamm Res. (2021) 70:765–76. doi: 10.1007/s00011-021-01480-z
209. Michailidis L, Currier AC, Le M, Flomenhoft DR. Adverse events of fecal microbiota transplantation: a meta-analysis of high-quality studies. Ann Gastroenterol. (2021) 34:802–14. doi: 10.20524/aog.2021.0655
210. Amorim R, Soares P, Chavarria D, Benfeito S, Cagide F, Teixeira J, et al. Decreasing the burden of non-alcoholic fatty liver disease: From therapeutic targets to drug discovery opportunities. Eur J Med Chem. (2024) 277:116723. doi: 10.1016/j.ejmech.2024.116723
211. Arellano-Garcia L, Portillo MP, Martinez JA, Milton-Laskibar I. Usefulness of probiotics in the management of NAFLD: evidence and involved mechanisms of action from preclinical and human models. Int J Mol Sci. (2022) 23:3167. doi: 10.3390/ijms23063167
212. Craven L, Rahman A, Nair Parvathy S, Beaton M, Silverman J, Qumosani K, et al. Allogenic fecal microbiota transplantation in patients with nonalcoholic fatty liver disease improves abnormal small intestinal permeability: a randomized control trial. Am J Gastroenterol. (2020) 115:1055–65. doi: 10.14309/ajg.0000000000000661
213. Xue L, Deng Z, Luo W, He X, Chen Y. Effect of fecal microbiota transplantation on non-alcoholic fatty liver disease: a randomized clinical trial. Front Cell Infect Microbiol. (2022) 12:759306. doi: 10.3389/fcimb.2022.759306
214. Ianiro G, Puncochar M, Karcher N, Porcari S, Armanini F, Asnicar F, et al. Variability of strain engraftment and predictability of microbiome composition after fecal microbiota transplantation across different diseases. Nat Med. (2022) 28:1913–23. doi: 10.1038/s41591-022-01964-3
215. Defilipp Z, Bloom PP, Torres Soto M, Mansour MK, Sater MRA, Huntley MH, et al. Drug-resistant E. coli bacteremia transmitted by fecal microbiota transplant. N Engl J Med. (2019) 381:2043–50. doi: 10.1056/NEJMoa1910437
216. Bashiardes S, Shapiro H, Rozin S, Shibolet O, Elinav E. Non-alcoholic fatty liver and the gut microbiota. Mol Metab. (2016) 5:782–94. doi: 10.1016/j.molmet.2016.06.003
217. Almeqdadi M, Gordon FD. Farnesoid X receptor agonists: a promising therapeutic strategy for gastrointestinal diseases. Gastro Hep Adv. (2024) 3:344–52. doi: 10.1016/j.gastha.2023.09.013
218. Zhao J, Li B, Zhang K, Zhu Z. The effect and safety of obeticholic acid for patients with nonalcoholic steatohepatitis: a systematic review and meta-analysis of randomized controlled trials. Medicine. (2024) 103:e37271. doi: 10.1097/MD.0000000000037271
219. Neuschwander-Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet. (2015) 385:956–65. doi: 10.1016/S0140-6736(14)61933-4
220. Shen WJ, Azhar S, Kraemer FB. SR-B1: a unique multifunctional receptor for cholesterol influx and efflux. Annu Rev Physiol. (2018) 80:95–116. doi: 10.1146/annurev-physiol-021317-121550
221. Patel K, Harrison SA, Elkhashab M, Trotter JF, Herring R, Rojter SE, et al. Cilofexor, a nonsteroidal FXR agonist, in patients with noncirrhotic NASH: a phase 2 randomized controlled trial. Hepatology. (2020) 72:58–71. doi: 10.1002/hep.31205
222. Tully DC, Rucker PV, Chianelli D, Williams J, Vidal A, Alper PB, et al. Discovery of tropifexor (LJN452), a highly potent non-bile acid FXR agonist for the treatment of cholestatic liver diseases and nonalcoholic steatohepatitis (NASH). J Med Chem. (2017) 60:9960–73. doi: 10.1021/acs.jmedchem.7b00907
223. Ratziu V, Harrison SA, Loustaud-Ratti V, Bureau C, Lawitz E, Abdelmalek M, et al. Hepatic and renal improvements with FXR agonist vonafexor in individuals with suspected fibrotic NASH. J Hepatol. (2023) 78:479–92. doi: 10.1016/j.jhep.2022.10.023
224. Fu ZD, Cai XL, Yang WJ, Zhao MM, Li R, Li YF. Novel glucose-lowering drugs for non-alcoholic fatty liver disease. World J Diabetes. (2021) 12:84–97. doi: 10.4239/wjd.v12.i1.84
225. Meng W, Ellsworth BA, Nirschl AA, Mccann PJ, Patel M, Girotra RN, et al. Discovery of dapagliflozin: a potent, selective renal sodium-dependent glucose cotransporter 2 (SGLT2) inhibitor for the treatment of type 2 diabetes. J Med Chem. (2008) 51:1145–9. Available online at: https://pubs.acs.org/doi/10.1021/jm701272q
226. Androutsakos T, Nasiri-Ansari N, Bakasis AD, Kyrou I, Efstathopoulos E, Randeva HS, et al. SGLT-2 inhibitors in NAFLD: expanding their role beyond diabetes and cardioprotection. Int J Mol Sci. (2022) 23:3107. doi: 10.3390/ijms23063107
227. Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology. (2007) 132:2131–57. doi: 10.1053/j.gastro.2007.03.054
228. An H, Jang Y, Choi J, Hur J, Kim S, Kwon Y. New insights into AMPK, as a potential therapeutic target in metabolic dysfunction-associated steatotic liver disease and hepatic fibrosis. Biomol Ther. (2025) 33:18–38. doi: 10.4062/biomolther.2024.188
229. Zhao P, Sun X, Chaggan C, Liao Z, In Wong K, He F, et al. An AMPK-caspase-6 axis controls liver damage in nonalcoholic steatohepatitis. Science. (2020) 367:652–60. doi: 10.1126/science.aay0542
230. Fullerton MD, Galic S, Marcinko K, Sikkema S, Pulinilkunnil T, Chen ZP, et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat Med. (2013) 19:1649–54. doi: 10.1038/nm.3372
231. Bhattacharya D, Basta B, Mato JM, Craig A, Fernandez-Ramos D, Lopitz-Otsoa F, et al. Aramchol downregulates stearoyl CoA-desaturase 1 in hepatic stellate cells to attenuate cellular fibrogenesis. JHEP Rep. (2021) 3:100237. doi: 10.1016/j.jhepr.2021.100237
232. Feldstein AE, Canbay A, Angulo P, Taniai M, Burgart LJ, Lindor KD, et al. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology. (2003) 125:437–43. doi: 10.1016/S0016-5085(03)00907-7
233. Perseghin G, Petersen K, Shulman GI. Cellular mechanism of insulin resistance: potential links with inflammation. Int J Obes Relat Metab Disord. (2003) 27 Suppl 3:S6–11. doi: 10.1038/sj.ijo.0802491
234. Hotamisligil GS. Role of endoplasmic reticulum stress and c-Jun NH2-terminal kinase pathways in inflammation and origin of obesity and diabetes. Diabetes. (2005) 54 Suppl 2:S73–78. doi: 10.2337/diabetes.54.suppl_2.S73
235. Liu Q, Bengmark S, Qu S. The role of hepatic fat accumulation in pathogenesis of non-alcoholic fatty liver disease (NAFLD). Lipids Health Dis. (2010) 9:42. doi: 10.1186/1476-511X-9-42
236. Sanyal AJ, Chalasani N, Kowdley KV, Mccullough A, Diehl AM, Bass NM, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med. (2010) 362:1675–85. doi: 10.1056/NEJMoa0907929
237. Van Der Windt DJ, Sud V, Zhang H, Tsung A, Huang H. The effects of physical exercise on fatty liver disease. Gene Expr. (2018) 18:89–101. doi: 10.3727/105221617X15124844266408
238. Targher G, Bertolini L, Padovani R, Rodella S, Tessari R, Zenari L, et al. Prevalence of nonalcoholic fatty liver disease and its association with cardiovascular disease among type 2 diabetic patients. Diabetes Care. (2007) 30:1212–8. doi: 10.2337/dc06-2247
239. Leite NC, Villela-Nogueira CA, Cardoso CR, Salles GF. Non-alcoholic fatty liver disease and diabetes: from physiopathological interplay to diagnosis and treatment. World J Gastroenterol. (2014) 20:8377–92. doi: 10.3748/wjg.v20.i26.8377
240. Williams KH, Burns K, Constantino M, Shackel NA, Prakoso E, Wong J, et al. An association of large-fibre peripheral nerve dysfunction with non-invasive measures of liver fibrosis secondary to non-alcoholic fatty liver disease in diabetes. J Diabetes Complications. (2015) 29:1240–7. doi: 10.1016/j.jdiacomp.2015.06.015
241. Byrne CD, Targher G. NAFLD as a driver of chronic kidney disease. J Hepatol. (2020) 72:785–801. doi: 10.1016/j.jhep.2020.01.013
242. Lombardi R, Airaghi L, Targher G, Serviddio G, Maffi G, Mantovani A, et al. Liver fibrosis by FibroScan((R)) independently of established cardiovascular risk parameters associates with macrovascular and microvascular complications in patients with type 2 diabetes. Liver Int. (2020) 40:347–54. doi: 10.1111/liv.14274
243. Mantovani A, Dalbeni A, Beatrice G, Cappelli D, Gomez-Peralta F. Non-alcoholic fatty liver disease and risk of macro- and microvascular complications in patients with type 2 diabetes. J Clin Med. (2022) 11:968. doi: 10.3390/jcm11040968
244. Wang TY, Wang RF, Bu ZY, Targher G, Byrne CD, Sun DQ, et al. Association of metabolic dysfunction-associated fatty liver disease with kidney disease. Nat Rev Nephrol. (2022) 18:259–68. doi: 10.1038/s41581-021-00519-y
245. Sumida Y, Yoneda M. Current and future pharmacological therapies for NAFLD/NASH. J Gastroenterol. (2018) 53:362–76. doi: 10.1007/s00535-017-1415-1
246. Egan M, Prasad S. PURLs: statins for patients with nonalcoholic fatty liver? J Fam Pract. (2011) 60:536–8.
247. Ayada I, Van Kleef LA, Zhang H, Liu K, Li P, Abozaid YJ, et al. Dissecting the multifaceted impact of statin use on fatty liver disease: a multidimensional study. EBioMedicine. (2023) 87:104392. doi: 10.1016/j.ebiom.2022.104392
248. Younossi ZM, Zelber-Sagi S, Henry L, Gerber LH. Lifestyle interventions in nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol. (2023) 20:708–22. doi: 10.1038/s41575-023-00800-4
249. St George A, Bauman A, Johnston A, Farrell G, Chey T, George J. Effect of a lifestyle intervention in patients with abnormal liver enzymes and metabolic risk factors. J Gastroenterol Hepatol. (2009) 24:399–407. doi: 10.1111/j.1440-1746.2008.05694.x
250. Promrat K, Kleiner DE, Niemeier HM, Jackvony E, Kearns M, Wands JR, et al. Randomized controlled trial testing the effects of weight loss on nonalcoholic steatohepatitis. Hepatology. (2010) 51:121–9. doi: 10.1002/hep.23276
251. Sun WH, Song MQ, Jiang CQ, Xin YN, Ma JL, Liu YX, et al. Lifestyle intervention in non-alcoholic fatty liver disease in Chengyang District, Qingdao, China. World J Hepatol. (2012) 4:224–30. doi: 10.4254/wjh.v4.i7.224
252. Eckard C, Cole R, Lockwood J, Torres DM, Williams CD, Shaw JC, et al. Prospective histopathologic evaluation of lifestyle modification in nonalcoholic fatty liver disease: a randomized trial. Therap Adv Gastroenterol. (2013) 6:249–59. doi: 10.1177/1756283X13484078
253. Wong VW, Chan RS, Wong GL, Cheung BH, Chu WC, Yeung DK, et al. Community-based lifestyle modification programme for non-alcoholic fatty liver disease: a randomized controlled trial. J Hepatol. (2013) 59:536–42. doi: 10.1016/j.jhep.2013.04.013
254. Gepner Y, Shelef I, Komy O, Cohen N, Schwarzfuchs D, Bril N, et al. The beneficial effects of Mediterranean diet over low-fat diet may be mediated by decreasing hepatic fat content. J Hepatol. (2019) 71:379–88. doi: 10.1016/j.jhep.2019.04.013
255. Plauth M, Bernal W, Dasarathy S, Merli M, Plank LD, Schutz T, et al. ESPEN guideline on clinical nutrition in liver disease. Clin Nutr. (2019) 38:485–521. doi: 10.1016/j.clnu.2018.12.022
256. Marti-Aguado D, Clemente-Sanchez A, Bataller R. Cigarette smoking and liver diseases. J Hepatol. (2022) 77:191–205. doi: 10.1016/j.jhep.2022.01.016
257. Zelber-Sagi S, Moore JB. Practical lifestyle management of nonalcoholic fatty liver disease for busy clinicians. Diabetes Spectr. (2024) 37:39–47. doi: 10.2337/dsi23-0009
258. Papamiltiadous ES, Roberts SK, Nicoll AJ, Ryan MC, Itsiopoulos C, Salim A, et al. A randomised controlled trial of a Mediterranean Dietary Intervention for Adults with Non Alcoholic Fatty Liver Disease (MEDINA): study protocol. BMC Gastroenterol. (2016) 16:14. doi: 10.1186/s12876-016-0426-3
259. Mendonca N, Gregorio MJ, Salvador C, Henriques AR, Canhao H, Rodrigues AM. Low adherence to the mediterranean diet is associated with poor socioeconomic status and younger age: a cross-sectional analysis of the EpiDoC cohort. Nutrients. (2022) 14. doi: 10.3390/nu14061239
260. George ES, Forsyth AK, Reddy A, Itsiopoulos C, Roberts SK, Nicoll AJ, et al. A Mediterranean and low-fat dietary intervention in non-alcoholic fatty liver disease patients: Exploring participant experience and perceptions about dietary change. J Hum Nutr Diet. (2023) 36:592–602. doi: 10.1111/jhn.13069
261. Shema A, Nouha T, Nesrine K, Asma M, Emna BM, Leila M, et al. Adherence and barriers to Mediterranean diet in Tunisian patients with nonalcoholic fatty liver disease. Future Sci OA. (2024) 10:FSO919. doi: 10.2144/fsoa-2023-0140
262. Haigh L, Bremner S, Houghton D, Henderson E, Avery L, Hardy T, et al. Barriers and facilitators to mediterranean diet adoption by patients with nonalcoholic fatty liver disease in Northern Europe. Clin Gastroenterol Hepatol. (2019) 17:1364–71. doi: 10.1016/j.cgh.2018.10.044
263. Keating SE, Chawla Y, De A, George ES. Lifestyle intervention for metabolic dysfunction-associated fatty liver disease: a 24-h integrated behavior perspective. Hepatol Int. (2024) 18:959–76. doi: 10.1007/s12072-024-10663-9
264. Fernandez T, Vinuela M, Vidal C, Barrera F. Lifestyle changes in patients with non-alcoholic fatty liver disease: a systematic review and meta-analysis. PLoS One. (2022) 17:e0263931. doi: 10.1371/journal.pone.0263931
265. Stine JG, Long MT, Corey KE, Sallis RE, Allen AM, Armstrong MJ, et al. American College of Sports Medicine (ACSM) International Multidisciplinary Roundtable report on physical activity and nonalcoholic fatty liver disease. Hepatol Commun. (2023) 7:108. doi: 10.1097/HC9.0000000000000108
266. Kim D, Murag S, Cholankeril G, Cheung A, Harrison SA, Younossi ZM, et al. Physical activity, measured objectively, is associated with lower mortality in patients with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. (2021) 19:1240–47. doi: 10.1016/j.cgh.2020.07.023
267. Kim D, Konyn P, Cholankeril G, Ahmed A. Physical activity is associated with nonalcoholic fatty liver disease and significant fibrosis measured by FibroScan. Clin Gastroenterol Hepatol. (2022) 20:e1438–55. doi: 10.1016/j.cgh.2021.06.029
268. Chun HS, Lee M, Lee HA, Oh SY, Baek HJ, Moon JW, et al. Association of physical activity with risk of liver fibrosis, sarcopenia, and cardiovascular disease in nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. (2023) 21:358–69. doi: 10.1016/j.cgh.2021.12.043
269. Francque SM, Marchesini G, Kautz A, Walmsley M, Dorner R, Lazarus JV, et al. Non-alcoholic fatty liver disease: a patient guideline. JHEP Rep. (2021) 3:100322. doi: 10.1016/j.jhepr.2021.100322
270. Al-Ozairi E, Irshad M, Alkandari J, Mashankar A, Alroudhan D, Le Roux CW. Liver fibrosis and liver stiffness in patients with obesity and type 1 diabetes. Diabetes Obes Metab. (2024) 26:4052–9. doi: 10.1111/dom.15760
271. Kakegawa T, Sugimoto K, Saito K, Yunaiyama D, Araki Y, Wada T, et al. Favorable liver and skeletal muscle changes in patients with MASLD and T2DM receiving glucagon-like peptide-1 receptor agonist: a prospective cohort study. Medicine (Baltimore). (2024) 103:e38444. doi: 10.1097/MD.0000000000038444
272. Quinn CE, Hamilton PK, Lockhart CJ, Mcveigh GE. Thiazolidinediones: effects on insulin resistance and the cardiovascular system. Br J Pharmacol. (2008) 153:636–45. doi: 10.1038/sj.bjp.0707452
273. Lee M, Hong S, Cho Y, Rhee H, Yu MH, Bae J, et al. Synergistic benefit of thiazolidinedione and sodium-glucose cotransporter 2 inhibitor for metabolic dysfunction-associated steatotic liver disease in type 2 diabetes: a 24-week, open-label, randomized controlled trial. BMC Med. (2025) 23:266. doi: 10.1186/s12916-025-04017-x
274. Michalopoulou E, Thymis J, Lampsas S, Pavlidis G, Katogiannis K, Vlachomitros D, et al. The triad of risk: linking MASLD, cardiovascular disease and type 2 diabetes; from pathophysiology to treatment. J Clin Med. (2025) 14:428. doi: 10.3390/jcm14020428
275. Pompili S, Vetuschi A, Gaudio E, Tessitore A, Capelli R, Alesse E, et al. Long-term abuse of a high-carbohydrate diet is as harmful as a high-fat diet for development and progression of liver injury in a mouse model of NAFLD/NASH. Nutrition. (2020) 2020:110782. doi: 10.1016/j.nut.2020.110782
276. Sukkar SG, Muscaritoli M. A clinical perspective of low carbohydrate ketogenic diets: a narrative review. Front Nutr. (2021) 8:642628. doi: 10.3389/fnut.2021.642628
277. Torres-Pena JD, Arenas-De Larriva AP, Alcala-Diaz JF, Lopez-Miranda J, Delgado-Lista J. Different dietary approaches, non-alcoholic fatty liver disease and cardiovascular disease: a literature review. Nutrients. (2023) 15:1483. doi: 10.3390/nu15061483
278. Nseir W, Hellou E, Assy N. Role of diet and lifestyle changes in nonalcoholic fatty liver disease. World J Gastroenterol. (2014) 20:9338−44.
279. Romero-Gomez M, Zelber-Sagi S, Trenell M. Treatment of NAFLD with diet, physical activity and exercise. J Hepatol. (2017) 67:829–46. doi: 10.1016/j.jhep.2017.05.016
280. Erten M. Visfatin as a promising marker of cardiometabolic risk. Acta Cardiol Sin. (2021) 37:464–72. doi: 10.6515/ACS.202109_37(5).20210323B
281. Semmler G, Datz C, Reiberger T, Trauner M. Diet and exercise in NAFLD/NASH: beyond the obvious. Liver Int. (2021) 41:2249–68. doi: 10.1111/liv.15024
282. Manco M, Putignani L, Bottazzo GF. Gut microbiota, lipopolysaccharides, and innate immunity in the pathogenesis of obesity and cardiovascular risk. Endocr Rev. (2010) 31:817–44. doi: 10.1210/er.2009-0030
283. Krajmalnik-Brown R, Ilhan ZE, Kang DW, Dibaise JK. Effects of gut microbes on nutrient absorption and energy regulation. Nutr Clin Pract. (2012) 27:201–14. doi: 10.1177/0884533611436116
284. Mach N, Fuster-Botella D. Endurance exercise and gut microbiota: a review. J Sport Health Sci. (2017) 6:179–97. doi: 10.1016/j.jshs.2016.05.001
285. Severino A, Tohumcu E, Tamai L, Dargenio P, Porcari S, Rondinella D, et al. The microbiome-driven impact of western diet in the development of noncommunicable chronic disorders. Best Pract Res Clin Gastroenterol. (2024) 72:101923. doi: 10.1016/j.bpg.2024.101923
286. Lazo M, Solga SF, Horska A, Bonekamp S, Diehl AM, Brancati FL, et al. Effect of a 12-month intensive lifestyle intervention on hepatic steatosis in adults with type 2 diabetes. Diabetes Care. (2010) 33:2156–63. doi: 10.2337/dc10-0856
287. Sullivan S, Kirk EP, Mittendorfer B, Patterson BW, Klein S. Randomized trial of exercise effect on intrahepatic triglyceride content and lipid kinetics in nonalcoholic fatty liver disease. Hepatology. (2012) 55:1738–45. doi: 10.1002/hep.25548
288. Thyfault JP, Rector RS. Exercise combats hepatic steatosis: potential mechanisms and clinical implications. Diabetes. (2020) 69:517–24. doi: 10.2337/dbi18-0043
289. Keating SE, Sabag A, Hallsworth K, Hickman IJ, Macdonald GA, Stine JG, et al. Exercise in the management of metabolic-associated fatty liver disease (MAFLD) in adults: a position statement from exercise and sport science Australia. Sports Med. (2023) 53:2347–71. doi: 10.1007/s40279-023-01918-w
290. Galaviz KI, Narayan KMV, Lobelo F, Weber MB. Lifestyle and the prevention of type 2 diabetes: a status report. Am J Lifestyle Med. (2018) 12:4–20. doi: 10.1177/1559827615619159
291. Syeda USA, Battillo D, Visaria A, Malin SK. The importance of exercise for glycemic control in type 2 diabetes. Am J Med Open. (2023) 9:100031. doi: 10.1016/j.ajmo.2023.100031
292. Patel R, Sina RE, Keyes D. Lifestyle Modification for Diabetes and Heart Disease Prevention. Treasure Island, FL: StatPearls. (2024).
293. Houghton D, Thoma C, Hallsworth K, Cassidy S, Hardy T, Burt AD, et al. Exercise reduces liver lipids and visceral adiposity in patients with nonalcoholic steatohepatitis in a randomized controlled trial. Clin Gastroenterol Hepatol. (2017) 15:96–102. doi: 10.1016/j.cgh.2016.07.031
294. Marek RJ, Coulon SM, Brown JD, Lydecker JA, Marek S, Malcolm R, et al. Characteristics of weight loss trajectories in a comprehensive lifestyle intervention. Obesity. (2017) 25:2062–7. doi: 10.1002/oby.21942
295. Zhong F, Zhou X, Xu J, Gao L. Rodent models of nonalcoholic fatty liver disease. Digestion. (2020) 101:522–35. doi: 10.1159/000501851
296. Karimkhanloo H, Keenan SN, Bayliss J, De Nardo W, Miotto PM, Devereux CJ, et al. Mouse strain-dependent variation in metabolic associated fatty liver disease (MAFLD): a comprehensive resource tool for pre-clinical studies. Sci Rep. (2023) 13:4711. doi: 10.1038/s41598-023-32037-1
297. Leclercq IA, Farrell GC, Schriemer R, Robertson GR. Leptin is essential for the hepatic fibrogenic response to chronic liver injury. J Hepatol. (2002) 37:206–13. doi: 10.1016/S0168-8278(02)00102-2
298. Paz-Filho G, Mastronardi C, Delibasi T, Wong ML, Licinio J. Congenital leptin deficiency: diagnosis and effects of leptin replacement therapy. Arq Bras Endocrinol Metabol. (2010) 54:690–7. doi: 10.1590/S0004-27302010000800005
299. Lee UE, Friedman SL. Mechanisms of hepatic fibrogenesis. Best Pract Res Clin Gastroenterol. (2011) 25:195–206. doi: 10.1016/j.bpg.2011.02.005
300. Bissig-Choisat B, Alves-Bezerra M, Zorman B, Ochsner SA, Barzi M, Legras X, et al. A human liver chimeric mouse model for non-alcoholic fatty liver disease. JHEP Rep. (2021) 3:100281. doi: 10.1016/j.jhepr.2021.100281
301. Ding Y, Dai X, Bao M, Xing Y, Liu J, Zhao S, et al. Hepatic transcriptome signatures in mice and humans with nonalcoholic fatty liver disease. Animal Model Exp Med. (2023) 6:317–28. doi: 10.1002/ame2.12338
302. Vacca M, Kamzolas I, Harder LM, Oakley F, Trautwein C, Hatting M, et al. An unbiased ranking of murine dietary models based on their proximity to human metabolic dysfunction-associated steatotic liver disease (MASLD). Nat Metab. (2024) 6:1178−96. doi: 10.1038/s42255-024-01043-6
303. Lau JK, Zhang X, Yu J. Animal models of non-alcoholic fatty liver disease: current perspectives and recent advances. J Pathol. (2017) 241:36–44. doi: 10.1002/path.4829
304. Chua D, Low ZS, Cheam GX, Ng AS, Tan NS. Utility of human relevant preclinical animal models in navigating NAFLD to MAFLD paradigm. Int J Mol Sci. (2022) 23. doi: 10.3390/ijms232314762
305. Barrow F, Khan S, Fredrickson G, Wang H, Dietsche K, Parthiban P, et al. Microbiota-driven activation of intrahepatic B cells aggravates NASH through innate and adaptive signaling. Hepatology. (2021) 74:704–22. doi: 10.1002/hep.31755
306. Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol. (2013) 31:563–604. doi: 10.1146/annurev-immunol-020711-074950
307. Haas JT, Vonghia L, Mogilenko DA, Verrijken A, Molendi-Coste O, Fleury S, et al. Transcriptional network analysis implicates altered hepatic immune function in NASH development and resolution. Nat Metab. (2019) 1:604–14. doi: 10.1038/s42255-019-0076-1
308. Deczkowska A, David E, Ramadori P, Pfister D, Safran M, Li B, et al. XCR1(+) type 1 conventional dendritic cells drive liver pathology in non-alcoholic steatohepatitis. Nat Med. (2021) 27:1043–54. doi: 10.1038/s41591-021-01344-3
309. Luo XY, Takahara T, Kawai K, Fujino M, Sugiyama T, Tsuneyama K, et al. IFN-gamma deficiency attenuates hepatic inflammation and fibrosis in a steatohepatitis model induced by a methionine- and choline-deficient high-fat diet. Am J Physiol Gastrointest Liver Physiol. (2013) 305:G891–899. doi: 10.1152/ajpgi.00193.2013
310. Bahcecioglu IH, Yalniz M, Ataseven H, Ilhan N, Ozercan IH, Seckin D, et al. Levels of serum hyaluronic acid, TNF-alpha and IL-8 in patients with nonalcoholic steatohepatitis. Hepatogastroenterology. (2005) 52:1549–53.
311. Wolf MJ, Adili A, Piotrowitz K, Abdullah Z, Boege Y, Stemmer K, et al. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell. (2014) 26:549–64. doi: 10.1016/j.ccell.2014.09.003
312. Lo JC, Wang Y, Tumanov AV, Bamji M, Yao Z, Reardon CA, et al. Lymphotoxin beta receptor-dependent control of lipid homeostasis. Science. (2007) 316:285–8. doi: 10.1126/science.1137221
313. Syn WK, Agboola KM, Swiderska M, Michelotti GA, Liaskou E, Pang H, et al. NKT-associated hedgehog and osteopontin drive fibrogenesis in non-alcoholic fatty liver disease. Gut. (2012) 61:1323–9. doi: 10.1136/gutjnl-2011-301857
314. Coombes JD, Choi SS, Swiderska-Syn M, Manka P, Reid DT, Palma E, et al. Osteopontin is a proximal effector of leptin-mediated non-alcoholic steatohepatitis (NASH) fibrosis. Biochim Biophys Acta. (2016) 1862:135–44. doi: 10.1016/j.bbadis.2015.10.028
315. Malehmir M, Pfister D, Gallage S, Szydlowska M, Inverso D, Kotsiliti E, et al. Platelet GPIbalpha is a mediator and potential interventional target for NASH and subsequent liver cancer. Nat Med. (2019) 25:641–55. doi: 10.1055/s-0038-1677172
316. Soehnlein O, Steffens S, Hidalgo A, Weber C. Neutrophils as protagonists and targets in chronic inflammation. Nat Rev Immunol. (2017) 17:248–61. doi: 10.1038/nri.2017.10
317. Remmerie A, Martens L, Thone T, Castoldi A, Seurinck R, Pavie B, et al. Osteopontin expression identifies a subset of recruited macrophages distinct from Kupffer cells in the fatty liver. Immunity. (2020) 53:641–57. doi: 10.1016/j.immuni.2020.08.004
318. Barrow F, Khan S, Wang H, Revelo XS. The emerging role of B cells in the pathogenesis of NAFLD. Hepatology. (2021) 74:2277–86. doi: 10.1002/hep.31889
319. Pan Z, Chan WK, Eslam M. The role of B cells in metabolic (dysfunction)-associated fatty liver disease. Hepatobiliary Surg Nutr. (2021) 10:875–7. doi: 10.21037/hbsn-21-404
320. Pinto AT, Lukacs-Kornek V. The role of dendritic cells in MASH: friends or foes? Front Immunol. (2024) 15:1379225. doi: 10.3389/fimmu.2024.1379225
321. Michail S, Lin M, Frey MR, Fanter R, Paliy O, Hilbush B, et al. Altered gut microbial energy and metabolism in children with non-alcoholic fatty liver disease. FEMS Microbiol Ecol. (2015) 91:1–9. doi: 10.1093/femsec/fiu002
322. Affo S, Morales-Ibanez O, Rodrigo-Torres D, Altamirano J, Blaya D, Dapito DH, et al. CCL20 mediates lipopolysaccharide induced liver injury and is a potential driver of inflammation and fibrosis in alcoholic hepatitis. Gut. (2014) 63:1782–92. doi: 10.1136/gutjnl-2013-306098
323. De Minicis S, Rychlicki C, Agostinelli L, Saccomanno S, Candelaresi C, Trozzi L, et al. Dysbiosis contributes to fibrogenesis in the course of chronic liver injury in mice. Hepatology. (2014) 59:1738–49. doi: 10.1002/hep.26695
324. Omori K, Nakamura A, Miyoshi H, Takahashi K, Kitao N, Nomoto H, et al. Effects of dapagliflozin and/or insulin glargine on beta cell mass and hepatic steatosis in db/db mice. Metabolism. (2019) 98:27–36. doi: 10.1016/j.metabol.2019.06.006
325. Meng Z, Liu X, Li T, Fang T, Cheng Y, Han L, et al. The SGLT2 inhibitor empagliflozin negatively regulates IL-17/IL-23 axis-mediated inflammatory responses in T2DM with NAFLD via the AMPK/mTOR/autophagy pathway. Int Immunopharmacol. (2021) 94:107492. doi: 10.1016/j.intimp.2021.107492
326. Nasiri-Ansari N, Nikolopoulou C, Papoutsi K, Kyrou I, Mantzoros CS, Kyriakopoulos G, et al. Empagliflozin attenuates non-alcoholic fatty liver disease (NAFLD) in high fat diet fed ApoE((-/-)) mice by activating autophagy and reducing ER stress and apoptosis. Int J Mol Sci. (2021) 22:818. doi: 10.3390/ijms22020818
327. Blaslov K, Bulum T, Zibar K, Duvnjak L. Incretin based therapies: a novel treatment approach for non-alcoholic fatty liver disease. World J Gastroenterol. (2014) 20:7356–65. doi: 10.3748/wjg.v20.i23.7356
Keywords: MASLD, MASH, NAFLD, NASH, microbiome, dysbiosis, metabolic syndrome
Citation: Dua A, Kumari R, Singh M, Kumar R, Pradeep S, Ojesina AI and Kumar R (2025) Metabolic Dysfunction-Associated Steatotic Liver Disease (MASLD): the interplay of gut microbiome, insulin resistance, and diabetes. Front. Med. 12:1618275. doi: 10.3389/fmed.2025.1618275
Received: 25 April 2025; Accepted: 18 July 2025;
Published: 14 August 2025.
Edited by:
Zhengyang Bao, Wuxi Maternity and Child Health Care Hospital, ChinaReviewed by:
Jinwei Zhang, University of Exeter, United KingdomErnest Saenz, Heidelberg University, Germany
Copyright © 2025 Dua, Kumari, Singh, Kumar, Pradeep, Ojesina and Kumar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roshan Kumar, cm9zaGFuemhjQGdtYWlsLmNvbQ==; Akinyemi I. Ojesina, YW9qZXNpbmFAbWN3LmVkdQ==