 Ruoyi Zhou
Ruoyi Zhou Haojie Wang
Haojie Wang- The First Affiliated Hospital of Guangzhou University of Chinese Medicine, Guangzhou, China
Small airways–defined as bronchioles <2 mm in internal diameter that lack cartilaginous support–are frequently involved in the earliest stages of chronic obstructive pulmonary disease (COPD). While COPD is defined per GOLD by persistent post-bronchodilator airflow limitation, small-airway dysfunction can precede spirometric abnormality, motivating earlier, imaging- and physiology-based detection (Agustí et al., 2023). Pathological progression typically begins with loss and stenosis of terminal bronchioles, followed by mucus retention/plugging, fibrotic remodeling, chronic inflammation, microvascular abnormalities, and cellular senescence, ultimately resulting in irreversible impairment of gas exchange. Early diagnosis remains difficult, but a suite of advanced non-invasive modalities–including impulse oscillometry system/forced oscillation techniques (IOS/FOT), single- and multiple-breath washout tests, high-resolution CT with parametric response mapping (PRM), nuclear medicine approaches (e.g., SPECT), dynamic measurements of lung compliance, and Fluorine-19 (19F) MRI–combined with artificial intelligence markedly improve the sensitivity and specificity for detecting small-airway disease. Therapeutic strategies that target cellular senescence and fibrotic pathways–such as senolytics and antifibrotic interventions–are showing promise, particularly approaches that clear senescent cells or block pro-fibrotic signaling. The integration of single-cell omics, high-resolution microvascular imaging, and molecularly targeted therapies is expected to accelerate precision diagnostics and enable personalized early interventions. This review summarizes recent insights into small-airway physiology, key pathophysiological and molecular mechanisms, and current pharmacological strategies, and emphasizes the clinical principle of “early detection, early diagnosis, early intervention” for managing COPD-related small-airway disease.
Background
Small airway disease (SAD) plays a central role in the pathogenesis of chronic obstructive pulmonary disease (COPD), a role that has only recently been fully recognized (2). Small airways, defined as small bronchi with an internal diameter less than 2 mm and no cartilage support, comprise the majority of the distal airway cross-sectional area. Due to their narrow lumen and lack of cartilage support, these airways are extremely sensitive to inhaled noxious particles and irritants, yet are clinically invisible, earning them the nickname “silent zones (3).” Early functional impairment of the small airways often precedes the onset of clinical symptoms, abnormal lung function, or radiographic changes.
Historically, SAD was considered a secondary manifestation of established COPD. However, pathological and high-resolution imaging evidence has changed this understanding: progressive destruction and loss of terminal bronchioles can precede alveolar parenchymal destruction or a significant decline in overall lung function, suggesting that SAD may be the initial step in the disease cascade (4, 5). A multimodal analysis (6) found significant differences in CT small airway/gas trapping indices (including PRM-calibrated fSAD), clinical symptoms, and blood transcriptomic profiles between smokers with COPD, asthma, and ACO. This study linked imaging-defined SAD to molecular phenotypes, supporting the use of SAD as a basis for phenotyping airway diseases and stratifying diagnosis and treatment.
This paradigm shift also explains why patients with similar traditional lung function measures exhibit significant differences in the burden of airway structural damage and clinical outcomes. Research has revealed that SAD reflects the convergence of multiple pathological processes, including chronic inflammation, mucus hypersecretion, epithelial-mesenchymal transition (EMT) and fibrotic remodeling, microvascular rarefaction, epithelial barrier dysfunction, and cellular senescence. Signaling pathways such as TGF-β, PI3K/Akt, and NF-κB play key roles in these processes, collectively regulating inflammation, mucus secretion, EMT, fibrosis, and cellular senescence, driving progressive airway narrowing and ultimately impairing gas exchange. These theoretical and mechanistic advances have revealed several key clinical questions. While conventional pulmonary function testing remains the cornerstone of diagnosing airflow limitation, its sensitivity for early detection of SAD is insufficient (7). Emerging physiological and imaging tools–such as impulse oscillometry system single- and multiple-breath washout tests, high-resolution CT (HRCT) combined with parametric response mapping (PRM), molecular magnetic resonance imaging, and targeted nuclear medicine imaging–when combined with artificial intelligence applications, have the potential to enhance detection capabilities. However, further validation, standardization, and integration into clinical workflows are required. In terms of treatment, anti-aging drugs, anti-fibrosis drugs, and targeted interventions for the above-mentioned signaling pathways have shown potential in preclinical studies, but their translational applications still face challenges such as target specificity, intervention timing, and patient stratification.
Physiological characteristics of the small airway
Small airways are defined as those with a diameter of less than 2 mm and are considered part of the distal airway system, comprising membranous bronchioles, respiratory bronchioles, and alveolar ducts (8). The structures typically originate from the 8th to 15th generations of branches and terminate in the terminal bronchioles. They may range several centimeters in length and exhibit an irregular oval cross-section. The tracheal wall is thin, and contains smooth muscle and a minimal amount of cartilage (9). The inner wall is lined by ciliated columnar epithelial cells. The more distal respiratory bronchioles transition from columnar to cuboidal epithelium and subsequently lead into the alveolar ducts and cavities, which are lined with flat epithelium (10). Additionally, dendritic cells and macrophages residing in the small airways identify and eliminate pathogens, thereby forming an immune barrier. In contrast to the large airways, gas flow in large airways is predominantly turbulent, whereas small airways exhibit predominantly laminar flow, characterized by a lower Reynolds coefficient. Therefore, alterations in gas density have minimal or negligible effects on the resistance of small airways, whereas gas density significantly influences the resistance of large airways (9). Another physiological distinction between the small and large airways is that the liquid lining of the small airways possesses surfactant-like properties, particularly during exhalation. This low surface tension prevents the small airways from collapsing as lung volume decreases (11). These findings suggest that active substances also play a role in regulating gas diffusion distance and lubricating ciliary movement, thereby helping to maintain airway patency. At the end of inspiration, small airways are susceptible to collapse due to a decreased pulling force, leading to dynamic alveolar closure, increased dead space, and reduced gas exchange efficiency. Under normal circumstances, small airways contribute approximately 10% of total airway resistance. However, in the early stages of COPD, small airway resistance significantly increases, reflecting small airway dysfunction (9).
Initiation phase
Epithelial damage–EMT (epithelial-mesenchymal transition)—impaired barrier function–mucus obstruction
The small airway epithelium is the main defense barrier of the respiratory tract against inhaled harmful particles and pathogens. However, prolonged exposure to harmful substances in cigarette smoke and air pollution leads to a cyclical process of repeated damage and continuous repair of the small airway epithelium. The main features of initial epithelial injury are mucociliary dysfunction, goblet cell hyperplasia, and disruption of tight junction proteins, changes that together impair mucociliary clearance and compromise barrier integrity (12). This pathologic progression not only enhances airway hypersensitivity to exogenous stimuli, but also sets the stage for a persistent inflammatory response. In a chronic inflammatory microenvironment, damaged epithelial cells can initiate EMT by activating multiple signaling pathways, such as transforming growth factor-β (TGF-β), Wnt/β-catenin, and the Notch pathway (13). EMT is a process of cellular phenotypic plasticity in which epithelial cells gradually lose polarity and intercellular connections and acquire a mesenchymal-like phenotype (including enhanced migration ability and increased collagen deposition), thereby driving early fibrosis and airway remodeling (14). At the same time, STAT3 activation can amplify pro-fibrotic and pro-inflammatory signals, while dysregulation of the PINK1-Parkin pathway, responsible for mitochondrial quality control, promotes epithelial damage and the progression of EMT by increasing mitochondrial dysfunction and reactive oxygen species (ROS) production. The above signaling pathways synergistically promote epithelial-mesenchymal transition in this stage, laying the molecular foundation for subsequent pathological changes (15). The progression of EMT with fibrotic processes leads to further deterioration of small airway barrier function. Firstly, basement membrane thickening and collagen deposition increase airway stiffness and decrease gas exchange efficiency. Secondly, a compromised barrier promotes infiltration of inflammatory cells and noxious particles into the deeper layers of the airway, thus creating a self-perpetuating cycle of injury and inflammation (16). Although molecular features of EMT (such as downregulation of E-cadherin and upregulation of vimentin) are commonly observed in the small airways of smokers, their exact contribution to airway obstruction remains controversial (17). At the same time, epithelial damage with impaired barrier function promotes the secretion of abnormal mucus in small airways. In patients with COPD, the incidence of small airway mucus plugging is elevated and positively correlates with disease severity (18). Triggered by initial epithelial injury, goblet cell proliferation, mucociliary dysfunction, and up-regulation of mucin (e.g., MUC5AC and MUC5B) expression combine to further impair mucociliary clearance (19–23). Cigarette smoke and air pollution directly induce goblet cell proliferation and promote mucin secretion, which in turn leads to mucus embolism. Simultaneously, secretory immunoglobulin A (SlgA) levels on the surface of small airways are reduced and polymeric immunoglobulin receptor (plgR) expression is decreased. Together, these changes compromise the epithelial antimicrobial barrier and increase susceptibility to infection (24–27). Mucus retention not only impedes airflow on a physical level, but also provides a suitable environment for pathogens to proliferate. This can exacerbate localized inflammation, which in turn creates a self-perpetuating cycle of pathology. This is the initial and critical stage in the progression from small airway disease (SAD) to COPD (5). Therefore, the pathological events from small airway epithelial injury to EMT, barrier damage, and mucus plugging are intertwined, forming a continuous pathological chain: epithelial injury–EMT/TGF-β/STAT3/PINK1-Parkin–barrier disruption–mucus plugging. This is a chain reaction that reveals important molecular mechanisms of early COPD lesions. Future interventions targeting the EMT process (such as TGF-β inhibitors or drugs that regulate oxidative stress) may provide new ideas for the early prevention and treatment of COPD (28).
Expansion Phase
Immune cell recruitment – mediator release – critical pathway activation— transition to senescence
Small airway inflammation is induced by chronic exposure to cigarette smoke and atmospheric particulate matter. These stimuli activate airway epithelial cells and alveolar macrophages, leading to the release of pro-inflammatory mediators (e.g., TNF-α, IL-1β, IL-6) and chemokines (e.g., CXCL8, LTB4) (Figure 1), which recruit neutrophils into the airway wall, ultimately driving the inflammatory response in small airways (29). In the small airways of COPD, neutrophils represent the predominant and earliest infiltrating cells responding to cigarette smoke, followed sequentially by the recruitment of macrophages and CD4+/CD8+ T lymphocytes. These immune cells exacerbate tissue injury through cytotoxicity and the release of inflammatory mediators, and are closely associated with the development of emphysema (30–35). For instance, cigarette smoke and particulate matter can amplify and perpetuate a chronic inflammatory milieu by activating the NF-κB and p38 MAPK signaling pathways, thereby driving the sustained secretion of cytokines such as TNF-α, IL-1β, IL-6, and IL-8. Currently, oxidative stress can activate the PI3K/Akt/mTOR pathway, whose upregulation in the airways of elderly COPD patients has been experimentally demonstrated. This pathway further exacerbates the inflammatory response by suppressing SIRT1/6 activity and enhancing NF-κB pathway activity (31). Approximately 20%–40% of patients with COPD exhibit an inflammatory phenotype characterized by eosinophilic infiltration. This phenotype correlates strongly with small airway dysfunction and often predicts a favorable response to inhaled corticosteroid (ICS) therapy (36, 37). Chronic inflammation also promotes goblet cell hyperplasia and hypertrophy of mucous glands, thereby elevating both the volume and viscosity of secreted mucus. The consequent formation of mucus plugs contributes to the worsening of airflow obstruction (29, 38). Notably, persistent inflammation and oxidative stress can drive DNA damage, telomere shortening, and the accumulation of reactive oxygen species (ROS), thereby inducing cells to enter a state of senescence (39).
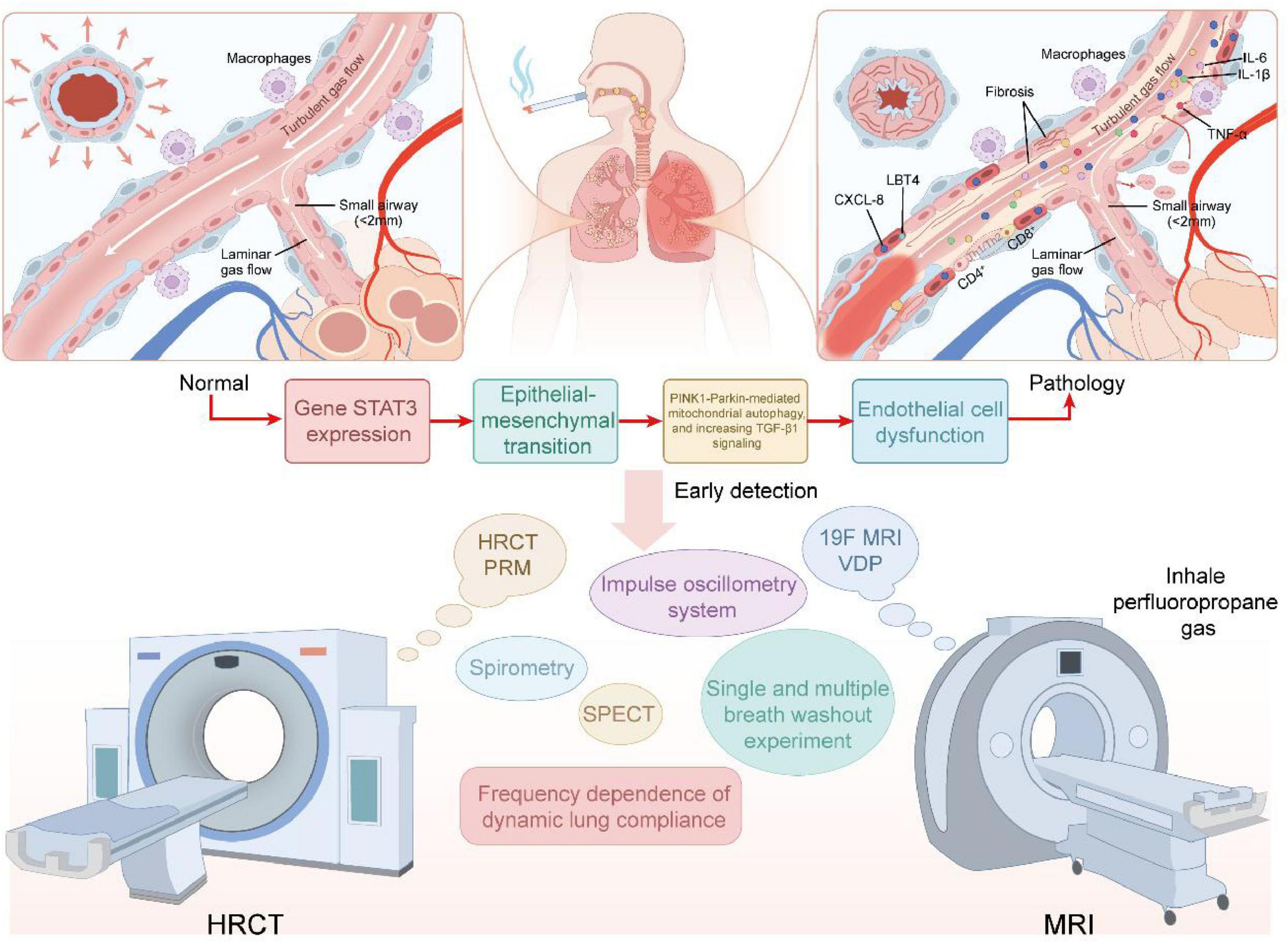
Figure 1. Top panels show enlarged small-airway cross-sections (left: near-normal; right: pathological changes including mucus plugging, epithelial injury, inflammatory-cell infiltration, and focal fibrosis). The middle panel depicts a molecular–cellular cascade–exposure (e.g., cigarette smoke or air pollution) → STAT3/TGF-β-driven epithelial-to-mesenchymal transition (EMT), PINK1–Parkin-related mitochondrial/autophagy dysfunction → endothelial dysfunction–illustrating the principal sequence Initiation → Expansion → Remodeling. The lower panel lists imaging and physiological modalities for early detection and phenotyping (HRCT-PRM, CT arterial pruning, 19F-MRI ventilation defect percentage (VDP), impulse oscillometry system (IOS), multiple-breath washout [MBW], SPECT, and conventional spirometry), emphasizing the complementary roles of imaging and functional assays in identifying individuals with functional small-airways disease (fSAD) and gas trapping despite preserved spirometry (pre-COPD/early SAD). This diagram is conceptual; several mechanistic links remain to be confirmed by longitudinal and mechanistic studies.
Cellular senescence, the senescence-associated secretory phenotype (SASP), miR-34a, and sirtuins (SIRT1/6)
Chronic inflammation and oxidative stress induce epithelial cells, fibroblasts, and endothelial cells into a senescent phenotype through the ATM/ATR-p53/p21 pathway (39). Senescent cells exhibit permanent growth arrest and secrete a group of pro-inflammatory and pro-fibrotic factors, the senescence-associated secretory phenotype (SASP), which mainly includes IL-6, IL-8, MMP-9, MMP-12, etc. These factors can amplify inflammation, promote ECM remodeling and induce functional decline of adjacent cells in a paracrine manner (40, 41). Meanwhile, microRNA-34a (miR-34a) has been reported to be upregulated in the regulation of aging and oxidative stress, and promotes the formation of aging phenotypes and the persistence of SASP by downregulating SIRT1/6 (key deacetylases involved in antioxidant and repair responses) (42–44). Within fibroblasts, SASP factors stimulate the accumulation of collagen and laminin, resulting in subepithelial fibrosis and consequent stiffening of the airway wall. Additionally, dysregulated iron metabolism amplifies ROS production through the Fenton reaction, which in turn drives both fibroblast senescence and extracellular matrix (ECM) remodeling (45). Short peptides produced after ECM is cleaved by proteases can act as damage-associated molecular patterns (DAMPs) or direct chemokines, activating receptors such as TLRs, inducing cytokine release and promoting neutrophil recruitment. Studies in the lungs have shown that these short peptides can prevent immune cell infiltration and affect the process of repairing fibrosis in chronic severe lung cancer (46). Furthermore, emerging evidence indicates that substance P, a neuropeptide, may mediate neurogenic inflammation in response to epithelial irritation and damage caused by cigarette smoke. This neurogenic response exacerbates mucus hypersecretion and inflammatory cell infiltration in the small airways (47). Moreover, studies have indicated a potential role for substance P in mediating repair mechanisms after epithelial damage in the small airways. Nevertheless, aberrant overexpression of this neuropeptide may promote the development of fibrosis (48). Treatment with a neurokinin 1 (NK1) receptor antagonist attenuates airway inflammation and bronchial hyperresponsiveness, leading to concomitant improvement in lung function (49). A study found that the migration of airway smooth muscle cells in asthma model rats was significantly increased. The NK1 receptor antagonist WIN62577 can inhibit this migration and reduce the expression of α-tubulin, suggesting that it can alleviate airway remodeling (50). Another study showed that in a bleomycin-induced rat pulmonary fibrosis model, the NK1R antagonist aprepitant significantly reduced the activity of the TGF-β/Smad3 signaling pathway and the expression of inflammatory cytokines (such as TNF-α and IFN-α), while increasing the level of antioxidants, exerting anti-fibrotic and anti-inflammatory effects (51). These results suggest that NK1R blockade may alleviate disease progression by reducing airway inflammation, fibrosis, and remodeling. However, the current relevant mechanism research and clinical evidence are still in the preliminary stage.
In summary, the inflammation-senescence axis drives the progression of small airway disease toward COPD, forming a vicious cycle. In this cycle, chronic inflammation promotes the release of pro-inflammatory mediators and chemokines, activates key signaling pathways, and elevates ROS production, collectively culminating in cellular senescence. Senescent cells release the senescence-associated secretory phenotype (SASP), which exacerbates inflammation, promotes extracellular matrix (ECM) remodeling, and induces secondary senescence in neighboring cells. This process establishes a self-perpetuating cycle that serves as a persistent mechanism underlying small airway structural damage and progressive airflow limitation, thereby driving disease worsening. This process thereby establishes the pathological foundation for subsequent airway structural remodeling. Emerging evidence suggests that the NLRP3 inflammasome acts as a pivotal amplifier of inflammation and is strongly implicated in the acute exacerbations of chronic respiratory diseases. Upon stimulation by reactive oxygen species (ROS), extracellular ATP, or lysosomal destabilization, NLRP3 activation drives the maturation and release of pro-inflammatory cytokines such as IL-1β and IL-18, which in turn promote neutrophil recruitment and tissue injury. This feed-forward mechanism establishes a ROS–IL-1β amplification loop, thereby perpetuating airway inflammation and contributing to recurrent exacerbations. Notably, experimental data indicate that pharmacological or genetic inhibition of NLRP3 can effectively interrupt this cycle, dampening the inflammatory cascade within the small airways. Furthermore, a combinatorial approach targeting both NLRP3 activation and oxidative stress has been proposed as a promising therapeutic strategy, with the potential to restore small airway function, mitigate structural injury, and ultimately prevent disease progression (52).
Remodeling phase
Proliferation of smooth muscle cells–Fibrosis–Vascular Pruning
Airway smooth muscle (ASM) in COPD small airway pathology commonly exhibits cellular hyperplasia and hypertrophy, leading to airway wall thickening, increased airway contractility, and directly contributing to small airway stenosis and airflow limitation (53). Epithelial and immune cells release various pro-proliferative/pro-fibrotic factors (such as TGF-β and PDGF) when epithelial injury and chronic inflammatory microenvironment occur. These factors stimulate ASM proliferation and induce phenotypic transformation by acting through the PI3K/Akt/mTOR and MAPK signaling pathways. Additionally, tobacco-induced Oxidative stress is able to amplify these pro-proliferative signals and further promote ASM hyperplasia (53, 54). Concomitant with ASM hyperplasia, local epithelium may undergo epithelial-mesenchymal transition (EMT). Meanwhile, recruited or activated fibroblasts differentiate into α-SMA-positive myofibroblasts, which synthesize large amounts of type I/III collagen and laminin, resulting in excessive sub-basement membrane ECM deposition and solidification of the airway wall, thereby reducing lung compliance and exacerbating irreversible stenosis (5, 54). In this process, the imbalance between Matrix Metalloproteinases (MMPs) and Tissue Inhibitors of Metalloproteinases (TIMPs) shows a “bidirectional effect”. On one side, ECM degradation mediated by MMP leads to elastic fiber rupture and emphysematous changes. On the other side, ECM fragments and pro-fibrotic signals trigger fibroblast activation and collagen deposition, ultimately resulting in spatially heterogeneous remodeling characterized by the coexistence of destruction and deposition (54, 55). The molecular mechanisms of how STAT3 activation and PINK1-Parkin regulation of mitochondria bridge epithelial damage and fibrosis have been described in detail in the initial section and will not be repeated here. From a biomechanical perspective, ASM hyperplasia and ECM deposition alter local tissue tension and generate mechanical compression, promoting distal capillary dysfunction and structural loss (capillary rarefaction). Concurrently, chronic inflammation, endothelial injury, and intermittent hypoxia also drive arteriolar intima-media thickening (arterial remodeling). These two types of vascular changes (arterial remodeling and capillary rarefaction) negatively affect clinical outcome (FEV1, DLCO, etc.) by increasing pulmonary vascular resistance or decreasing gas diffusion capacity, respectively (56, 57). Further evidence from the COPDGene cohort demonstrated that CT-based showed that “Arterial pruning”–decrease in distal small arterial vessel volume–was associated with emphysema progression, pulmonary function deterioration, and adverse outcomes, suggesting that vascular pruning is not only a concomitant change but also serves as an independent driving factor of disease progression (56, 58). Longitudinal study evidence from the COPDGene cohort indicates that, after excluding the influence of baseline lung parenchymal destruction, baseline arterial pruning can predict accelerated emphysema progression and decline in lung function (56); And in smokers without obvious COPD, the early presence of arterial pruning suggests its pathogenic role in increasing pulmonary vascular resistance and promoting right ventricular remodeling (58). Although the pruning process is regulated by the inflammatory reaction (e.g., cigarette smoke-induced endothelial-mesenchymal transition) (59, 60), and may be exacerbated by fibrosis compression (61), its independent impact on the disease course and clinical outcome highlights its core importance in the pathogenesis. Arterial remodeling and capillary rarefaction are two distinct angiopathies, each having unique physiological impacts. Arterial remodeling is manifested by structural alterations in muscular arteries, including intimal thickening and reduction in lumen diameter. These changes can increase pulmonary vascular resistance, induce pulmonary hypertension, lead to right ventricular dysfunction, and consequently cause hemodynamic impairment and limited motor capabilities (62, 63). In contrast, capillary rarefaction refers to decrease in capillary density. Gas exchange is directly impaired by reducing diffusing capacity (DLCO) and disrupting ventilation-perfusion matching, ultimately resulting in hypoxemia and ventilation/perfusion ratio (V/Q) imbalance (64–66). It is important that there is a synergistic effect between these two lesions: hypertension secondary to arterial remodeling exacerbates capillary loss, while capillary rarefaction further increases vascular resistance, collectively contributing to clinical deterioration in COPD patients.
In summary, ASM proliferation, ECM fibrosis, and vascular pruning are spatiotemporally coupled. molecular signals driven by epithelium/immune cells (TGF-β, STAT3, ROS, miRNA, etc.) not only directly promote the functional activation of smooth muscle and fibroblasts that leads to fibrosis, but also exacerbate tissue hypoxia and progressive injury by altering tissue mechanics and microvascular blood supply. Therefore, multi-target comprehensive treatment for this axis holds significant translational potential and merits further exploration in clinical trials (15, 55, 56).
Single-cell omics unveils molecular lineages and remodeling mechanisms of the small airways
In small airway stenosis, fibroblast-driven ECM remodeling is the core of structural stenosis, while persistent inflammation dominated by neutrophils and macrophages provides the cellular and enzymatic basis for this process (such as MMPs and ROS), promoting the degradation and redeposition of ECM (67, 68). Recent single-cell and multimodal omics studies have revealed, at both molecular and cellular levels, how distinct cell populations aggregate early and orchestrate extracellular matrix (ECM) remodeling. Single-cell atlases of terminal bronchioles demonstrate that, in the early stages of COPD (GOLD I–II), M1-like macrophages and neutrophils preferentially accumulate at alveolar attachment sites, where their presence correlates with elastic fiber degradation. These findings indicate that immune cell infiltration and ECM injury occur in close temporal and spatial proximity, synergistically driving the initiation of tissue destruction (69). Multiple single-cell studies, including global scRNA-seq analyses of COPD alveolar regions and airway terminals, have uncovered a complex network of ligand–receptor interactions among basal cells, resident fibroblasts, and immune cells. These signaling pairs drive the coordinated expression of inflammatory mediators and matrix-regulating factors, thereby exacerbating ECM metabolic imbalance within the local microenvironment and accelerating irreversible remodeling processes (70, 71). A recent study integrating macro- and single-cell RNA-seq data further demonstrated that significant expression changes of RNA methylation regulatory factors and autophagy pathways were present in immune cells of COPD patients, and that methylation regulatory factors and autophagy-related genes were upregulated in T cells and macrophages. Furthermore, such epigenetic and autophagy signals were positively correlated with inflammatory factor levels, suggesting that epigenetic modifications and cellular autophagy may serve as upstream regulatory points linking environmental exposure to persistent immune homeostasis imbalance and tissue damage (72).
Spirometry
In the absence of a non-invasive gold standard for diagnosing small airway obstruction (SAD), traditional pulmonary function tests remain the most commonly used screening tool in clinical practice, mainly because they are easy to operate, reproducible and low-cost (73). In 1972, the maximum mid-expiratory flow (MMEF) was proposed as the best vital capacity parameter for identifying small airway obstruction (SAO) (74). MMEF is commonly referred to as the mean forced expiratory flow between 25% and 75% of forced vital capacity (FEF 25%–75%), which represents the average expiratory flow during this portion of forced vital capacity. It represents the most sensitive measure of airflow in peripheral airways and is indicative of airflow obstruction, which is reduced in the early stages of SAD (75) and it is considered more accurate in detecting small airway disease than the forced expiratory volume in 1 s (FEV1) (76). Its use is grounded in the assumption that the middle and posterior portions of the FVC reflect airflow in small airways, which are susceptible to collapse at the end of expiration due owing to the absence of cartilage support. However, the clinical application of MMEF has remained controversial because of its insufficient sensitivity and specificity for assessing small airways, as well as its dependence on accurate measurement of the FVC (77). To overcome this limitation, some researchers have proposed the ratio of forced expiratory volume in 3 s (FEV3) to forced expiratory volume in 6 s (FEV6) as an alternative indicator for assessing small airway obstruction. The theoretical basis lies in the fact that FEV3 can capture more expiratory volume than FEV1, thereby including gas from the small airways (78, 79). Additionally, the use of FEV6 eliminates the requirement for precise measurement of FVC (80). Currently, the FEV3/FEV6 ratio has been demonstrated to be highly sensitivity for early small airway disease associated with airflow obstruction (3). In the multicenter COPD Genetic Epidemiology (COPD Gene) study, which included former and current smokers, a low FEV3/FEV6 (below the lower limit of normal [LLN]) was linked to the presence of gas trapping on CT imaging and a worse quality of life. This suggests its potential value in clinical screening, especially for early risk identification of former or current smokers. However, it remains uncertain whether these findings translate into increased mortality (81). In addition, studies from large cohorts (e.g., SPIROMICS) have shown that when FEV3/FEV6 is below the lower limit (LLN), HRCT/PRM-defined gas trapping and functional small airways disease (fSAD) can be detected even in smokers with normal FEV1/FVC, suggesting that this simple and easily obtainable respiratory function indicator can help identify potential early small airways lesions (82). Large cohort analyses from the COPDGene cohort also showed that fSAD quantified by CT-PRM was common in subjects who had not yet developed airflow limitation diagnosed by spirometry and was associated with accelerated FEV1 decline and adverse clinical outcomes during follow-up, which reinforced the value of imaging fSAD in early identification and risk stratification (83). Therefore, using FEV3/FEV6 as a preliminary screening in high-risk smoking populations combined with imaging methods such as PRM can help to early identify and stratify the management of individuals with subclinical small airway disease. Owing to its simplicity and low cost, spirometry remains the most commonly utilized diagnostic method for patients with or at risk of developing COPD in clinical management and epidemiological studies (84).
Single and multiple breath washout experiments
Increased ventilation heterogeneity is a characteristic physiological abnormality observed in respiratory diseases such as asthma and COPD (85, 86). The single-breath washout (SBW) maneuver involves a rapid exhalation to residual volume (RV), followed by inhalation of 100% oxygen to total lung capacity (TLC), and a subsequent exhalation from TLC back to RV, during which inert gas is washed out from the lungs. The most commonly assessed parameter is the slope of phase III (SIII), which reflects the homogeneity of gas clearance across lung regions and helps detect areas of hypoventilation and gas trapping. The operation is relatively simple and can reflect the closure of the small airways (87), but it requires the patient to cooperate with maximal breathing. The results are easily affected by factors such as age and obesity, and the sensitivity is relatively limited. The single-breath washout (SBW) test is suitable for patients capable of performing forced vital capacity maneuvers, such as adults and older children. It requires only one deep inhalation followed by a forced exhalation, making it simple to administer and highly feasible for assessing obstructive diseases such as COPD (88, 89). Studies have shown that the completion rate of SBWZ among children is as high as over 92%, which is significantly faster than MBW (83%) (89). Its limitations include the need for a high level of patient cooperation, low sensitivity in detecting mild small airway lesions, and the strong dependence of single washout parameters (such as SIII) on the subject’s effort. Multiple-breath washout (MBW) evaluates pulmonary gas mixing and clearance over repeated inspiratory and expiratory cycles, offering a comprehensive assessment of ventilation heterogeneity. Because it requires only quiet tidal breathing–without breath-holding or forced exhalation–it is particularly well suited for young children, infants, and patients unable to perform spirometry (90). Key parameters include the lung clearance index (LCI), Scond, Sacin, and functional residual capacity (FRC), which respectively reflect overall ventilation heterogeneity, convection-dependent heterogeneity in the conducting airways, diffusion-dependent heterogeneity in the peripheral airways and alveolar regions, and theresting end-expiratory lung volume (91). Over the past two decades, a large body of literature has emerged on the LCI, demonstrating that it is more sensitive than spirometry in detecting early obstructive lung disease in children with cystic fibrosis (CF) (92–94). MBW testing is time-consuming (average 2–5 min per test for healthy subjects (90), typically requires three or more repeats to ensure data reliability (90), and requires specialized gas analysis equipment and strict quality management. Nevertheless, MBW is widely applied in children and in the screening of early-stage conditions such as cystic fibrosis and primary ciliary dyskinesia, as it requires minimal patient coordination and is highly sensitive to subclinical small airway disease (95–97). Clinically, the two can be combined to evaluate small airway lesions, or combined with other examinations for a more accurate diagnosis.
Impulse oscillometry system
Forced oscillations are pressure or flow signals, or waves, that originate below the human hearing range, typically spanning 5–37 Hz, and are superimposed on normal tidal breathing, providing information about resistance and reactance on the basis of respiratory mechanics, i.e., airflow obstructions both inside and outside the bronchi. The use of multiple wave frequencies enables the exploration of the impedance frequency dependence to more precisely describe the location of airway obstruction. However, recent studies have shown that the impedance frequency dependence increases with disease severity or is influenced by the patient’s chest wall stiffness (87). This method tends to underestimate the true condition of lung mechanics (98–103). Currently, the latest pulse oscillation method is employed to assess small airway size by analyzing the heterogeneity of low-frequency resistance at 5–20 Hz (R5-R20), low-frequency reactance at 5 Hz (X5), or the area under the reactance curve between 5 Hz and the resonant frequency (100). This is because, at high oscillation frequencies (20 Hz), the signal is dominated by the characteristics of the upper airways, whereas at low frequencies (below 10 Hz), the signal reflects the entire tracheobronchial tree, including small airway resistance. At lower oscillation frequencies, reactance reflects the elastic properties of the parenchyma, airways, and chest wall, whereas at higher frequencies, inertial forces become predominant. At the resonant frequency (8 Hz), the elastic and inertial forces are equal and opposite, at which point they cancel each other out, and the pressure-flow relationship at this frequency reflects only the resistance of the system (104). Compared with spirometry, this method does not require much effort from the patient. It can also accurately detect small airway disease in all age groups (3, 105). In addition, impulse oscillometry has been shown to be more sensitive than spirometry in detecting small airway disease. Spirometry and impulse oscillomerty are recommended in combination for hospitalized patients to assess small airway function comprehensively and enable timely intervention (106). The use of an impulse oscillome try system has been more extensively studied in obstructive airway diseases. Recent studies have demonstrated that impulse oscillometry can be utilized to detect early manifestations of chronic obstructive pulmonary disease (COPD) (107). It is more useful than emphysema in the assessment of disease and can identify small airway dysfunction in patients with normal pulmonary function but early symptoms of chronic obstructive pulmonary disease (COPD) (76, 108). Moreover, the incidence of respiratory symptoms has been shown to be significantly greater in patients with small airway disease (SAD), as defined by impulse oscillometry (76). Correlations between impulse oscillometry (IOS) parameters and clinical indicators, including the severity of bronchiectasis and the presence of potential pathogenic microorganisms in sputum (109, 110). Park demonstrated that impulse oscillometry (IOS) may be a more sensitive method for predicting disease progression and prognosis than traditional lung function tests are. It can also differentiate between small airway disease (SAD) and chronic obstructive pulmonary disease (COPD), serving as an effective tool for assessing the risk of acute exacerbations in both asthma and COPD patients. In the clinical management of elderly patients with chronic obstructive pulmonary disease (COPD), impulse oscillometry (IOS) offers several advantages (111). A prospective study demonstrated that patients with small airway disease (SAD) defined by impulse oscillometry (IOS) experienced a more rapid decline in lung function and a greater risk of acute exacerbations (112). However, the consistency of results from oscillometric and other small airway detection methods in diagnosing small airway dysfunction remains a topic of ongoing discussion.
Frequency dependence of dynamic lung compliance
Dynamic lung compliance (Cdyn) refers to the ratio of changes in lung volume to the corresponding changes in airway pressure during respiration. Unlike static compliance, which purely reflects the elasticity of lung tissue and is not affected by airway resistance, dynamic compliance reflects the combined effects of lung tissue elastic resistance and airway resistance. In clinical practice, pulmonary function tests are frequently used to measure end-expiratory and end-inspiratory lung volumes and pressures in real time. Dynamic compliance (Cdyn) is calculated by dividing tidal volume by the difference between peak inspiratory pressure and end-expiratory pressure (113). Dynamic compliance includes the resistance to airflow through the airways in addition to the elasticity of lung tissue and is therefore usually lower than static compliance (114). Dynamic compliance reflects the combined influence of lung tissue elasticity and airway resistance and is commonly used in clinical practice to evaluate their interactive effects. Under normal conditions, the time constants of each alveolar unit are closely aligned, causing dynamic compliance to remain nearly constant as the respiratory rate increases from a resting state (≈8–16 breaths/minute) to an active state (≈60–120 breaths/minute). In other words, the normal lung time constant (τ = R⋅C) is both uniform and brief, ensuring synchronized inflation and exhalation of each lung area and thereby maintaining a constant Cdyn value regardless of changes in respiratory rate. This phenomenon demonstrates that, in healthy individuals, the respiratory rate and dynamic compliance are largely independent (104). When small airway obstruction or dysfunction develops, this uniformity is disrupted, resulting in the progressive appearance of frequency-dependent dynamic compliance (115). An early study by Woolcock (116) et al. found that even if the routine lung function indicators of patients with chronic bronchitis or asthma were close to normal, their dynamic compliance was significantly reduced at higher respiratory rates, while there was no such change in the healthy control group. These results suggest that airway obstruction leads to uneven ventilation, preventing some lung regions from being adequately deflated during rapid breathing, thereby reducing overall compliance. Thus, in patients with small airways disease, dynamic compliance typically decreases with increasing respiratory rate, disrupting the normal rate independence. The frequency dependence of dynamic compliance provides a sensitive indicator for the early diagnosis of small airway disease. As early as 1969, Woolcock et al. discovered latent airway obstruction through compliance testing at different respiratory rates (116). In recent years, with growing attention to COPD and early small airway disease, researchers have sought to integrate traditional compliance measurements with novel respiratory impedance techniques, such as oscillometry. The oscillometric low-frequency reactivity (AX) is closely related to the elasticity of peripheral lung units and is considered to reflect small airway patency and lung compliance (108). Teixeira et al. (117) used oscillometry to measure the Cdyn of patients with both chronic bronchitis and COPD, which was significantly lower than that of the healthy control group. This indicates that the decrease in dynamic compliance is related to the severity of airflow limitation. In the future, we can try to integrate static compliance and oscillometry to further improve the sensitivity of early detection of small airway disease and provide more basis for early intervention of COPD.
HRCT/CT and PRM
High-resolution computed tomography (HRCT) is the most effective imaging modality for assessing small airway disease (Figure 1). Direct signs of small airway disease observed on high-resolution computed tomography (HRCT) scans result from alterations in the airway wall or lumen. On HRCT scans, abnormal small airways may present as tubular, nodular, or branching linear structures. Indirect signs of small airway disease result from alterations in the lung parenchyma distal to the affected small airways and include air trapping, subsegmental atelectasis, centrilobular emphysema, and nodules (118). However, the spatial resolution of HRCT is limited (approximately 2 mm), making it generally impossible to directly visualize the smaller terminal bronchioles. As a result, early small airway lesions are often difficult to detect on conventional HRCT images. In contrast, parametric response mapping (PRM) is a quantitative imaging technique based on the registration of paired inspiratory and expiratory scans (119). This method classifies each registered lung voxel according to the HU value of the inspiratory and expiratory CT: it not only identifies the low-density area caused by emphysema, but also marks the emphysematous gas retention area of the lung (functional small airway disease, fSAD), and generates the corresponding 3D map (120). Compared with traditional spirometry, PRM has the advantages of spatial visualization and numerical reproducibility. Many studies have used the volume percentage of fSAD in PRM representing non-emphysematous gas retention (PRMfSAD) as an indicator for evaluating small airways (83, 121–124), including analyses from large cohorts such as COPDGene. Small airway disease is a transitional stage in the progression from normal airways to emphysema in severe disease, as demonstrated by the follow-up Parametric Response Mapping (PRM) study in the SPIROMICS cohort, which used inspiratory and expiratory HRCT scans to assess small airway disease (120, 125). A previous study compared 78 COPD patients at different stages of SAD and demonstrated that small airway stenosis and obstruction appear earlier than emphysema, so HRCT has advantages in the early diagnosis of COPD (126). The advantage of PRM is that it provides spatially resolved functional imaging and reproducible quantitative indicators, which go beyond the capabilities of traditional lung function tests (which can only give average results of the whole lung) (120, 127). PRM analysis can clearly visualize the spatial distribution of gas trapping and differentiate the causes of gas trapping (small airway disease or emphysema), facilitating the differential diagnosis of different COPD phenotypes (120). Compared with simple lung function measurement, PRM also has better reproducibility and objectivity (127). Therefore, in smokers or other high-risk groups, when screening, grading, or follow-up of early small airway lesions is required, PRM can be considered to obtain more functional information than conventional HRCT. However, PRM also has limitations. First, it requires two scans, one for inspiration and one for expiration, which increases examination time, cost, and radiation exposure (119), and therefore has not yet become a routine examination method. Secondly, PRM results rely on accurate image registration, and its measurements are based on changes in lung parenchymal density, which may be interfered with by changes in pulmonary vascular or other tissue density, affecting the relevance of small airway disease detection (127). Furthermore, PRM reflects functional gas trapping rather than direct structural imaging, so anatomical details such as bronchial wall thickening and mucus plugging require HRCT assessment. Overall, HRCT and PRM are complementary examination methods, capable of visualizing changes in lung structure and assessing small airway dysfunction Therefore, combining patient information and HRCT to assess SAD still has good predictive value. At the same time, some studies have found that PRMfSAD may indicate early alveolar attachment loss. Although previous studies support the relationship between PRM and alveolar attachment (121), this deserves further exploration. Currently, recent studies have shown that generative AI techniques can be used to reliably assess small airway disease using deep learning in inspiratory chest CT, without the need for additional respiratory CT scans. The fSAD obtained from inspiratory CT is strongly correlated with PRMfSAD and is significantly associated with a decrease in FEV1, while providing higher reproducibility (128).
SPECT
Quantitative single-photon emission computed tomography (SPECT/CT) can objectively quantify regional heterogeneity in human ventilation and is more sensitive than chest CT and lung function tests in detecting early airway changes in COPD patients. The coefficient of variation (CV) of the distribution of radioactive tracer values inhaled during the test can generate heterogeneity maps and density curves for small lung regions (129, 130). The area under the coefficient of variation (CV) curve (AUC-CV) at a predetermined threshold serves as a marker of ventilation in homogeneity and may have clinical value in assessing the severity and distribution of lung disease. Studies have shown that AUC-CV values are sensitive to the presence of COPD, asthma, and airflow obstruction, and are correlated with even mild abnormalities in pulmonary function tests (PFTs), including in otherwise healthy individuals. Therefore, measuring the AUC-CV value may serve as an early marker of airway disease in active smokers at risk for COPD (131, 132). A study by Juneau demonstrated that the AUC-CV40% can serve not only as a marker for peripheral airway disease but also as a clinical indicator for respiratory symptoms. It can also serve as an auxiliary tool for clinicians to assess smokers with respiratory symptoms. However, whether this marker can serve as a prognostic indicator for COPD requires further investigation (133). In fact, although Technegas technology has benefited more than 4.4 million patients in more than 60 countries since it was first used in Australia in the 1980s, it has only recently been approved for use in some regions (such as the United States) (134). In addition, current guidelines mainly recommend SPECT/CT for evaluation of pulmonary embolism rather than COPD screening (134). Current guidelines mainly recommend SPECT/CT for the evaluation of pulmonary embolism, rather than COPD screening (135). Therefore, SPECT ventilation imaging is currently mostly used in special occasions such as scientific research and preoperative evaluation of pulmonary surgery, and has not yet been included in the routine diagnostic process of COPD (136, 137). In summary, although SPECT/CT ventilation imaging can help detect airway function changes early, it is still mainly a research tool due to the high complexity of its equipment and operation and strict requirements for tracers, and its clinical practicality and maturity are limited.
Perfluoropropane-based 19F MRI and ventilation defect percentage (VDP)
Perfluoropropane 19F MRI is emerging lung ventilation imaging technique that uses inert fluorinated gases, which are detectable in MRI (Figure 1), to depict lung ventilation distribution. After subjects inhale a gas mixture containing approximately 79% perfluoropropane and undergo MRI scanning, the spatial distribution of the 19F signal during lung inflation is obtained and quantified as the ventilation defect percentage (VDP). Pippard and Neal developed a novel technique to assess the ventilation defect percentage (VDP), which reflects air trapping in the small airways. The subjects inhaled perfluoropropane gas and underwent fluorine-19 (19F) MRI scans. Inspiratory fluorine-19 (19F) MR images revealed regions of poor lung ventilation, as indicated by the heterogeneity in the distribution of perfluoropropane. VDP was calculated by dividing the volume of aerated lungs displayed on the inspiratory fluorine-19 (19F) MRI scan by the volume determined on the proton MRI scan (excluding the trachea and large airways). In their study, after bronchodilator inhalation, the VDP of asthma patients decreased by an average of 33%, whereas that of COPD patients decreased by an average of 14%. The FEV and FVC reflect airway disease, whereas VDP provides insights into regional air trapping and dead space, particularly in the small airways. Unlike FEV1 and FVC, which mainly reflect global airflow limitation, VDP can reflect local gas trapping and dead space, and is more sensitive in small airway lesions. Compared with other detection methods, such as hyperpolarized MRI or helium (He) MRI, perfluoropropane 19F MRI technology is straightforward, cost-effective, and capable of accurately depicting the distribution of lung gas during both inhalation and exhalation. It also demonstrates good tolerance in test subjects and maintains high clinical safety. Furthermore, perfluoropropane is an inert gas that is not metabolized and is rapidly cleared through exhalation, making this method safer for lung imaging and ensuring that it does not cause lasting effects on the body. Overall, this is a promising, investigational approach that can sensitively detect regional ventilation defects, but its availability is currently limited and has not yet been widely used in routine clinical practice. This imaging technology aids in detecting the early stages of COPD and asthma and may facilitate early intervention to enhance outcomes and more effectively evaluate treatment effects (138, 139). A review pointed out that although lung ventilation imaging is the most common case of 19F MRI in clinical practice, it is still relatively rare overall. In addition, the operation requires the subject to hold their breath for a short time to obtain clear images, which poses a tolerance challenge for patients with severe COPD. To this end, the research team is developing fast imaging and reconstruction algorithms to shorten scanning time and reduce the need for breath holding (138).
Although conventional spirometry is widely used and cost-effective, its sensitivity for detecting early small airway disease (SAD) remains limited. Many patients with pathological changes may still present with preserved FEV1 and FVC values, thereby delaying the recognition of early or pre-COPD phenotypes. Emerging techniques such as multiple-breath washout (MBW) and impulse oscillometry (IOS) allow non-invasive assessment of ventilation heterogeneity and peripheral airway resistance, offering superior sensitivity for the early detection of SAD. Imaging-based approaches, including parametric response mapping (PRM), single-photon emission computed tomography (SPECT), and 19F magnetic resonance imaging (19F-MRI), provide high-resolution insights into regional small airway obstruction and remodeling. However, their clinical application remains constrained by high costs, limited availability, and the need for further validation. Overall, these methods complement spirometry by bridging the diagnostic gap between pre-COPD and early COPD, thereby facilitating earlier intervention strategies and potentially improving patient outcomes (Table 1).
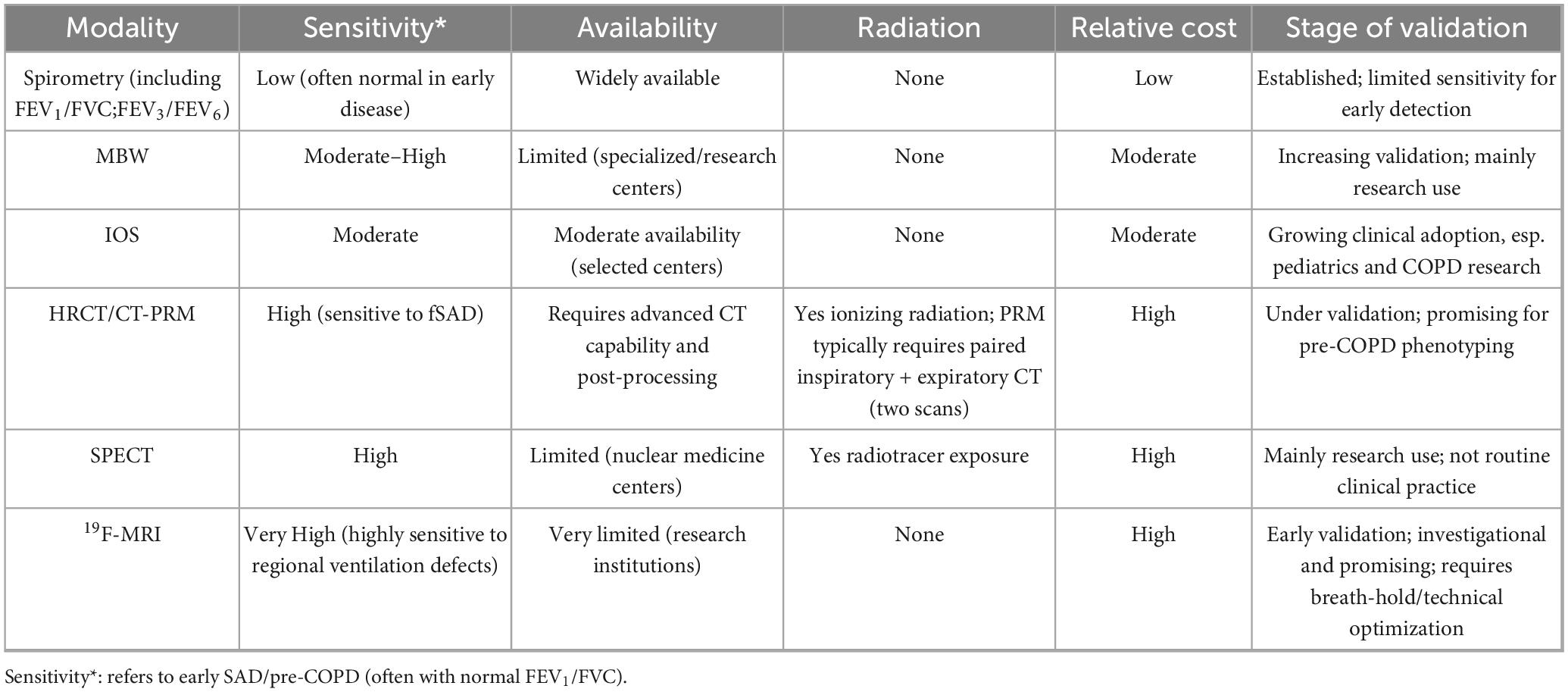
Table 1. Comparison of different pairs of assays.
Therapeutic perspectives
Chronic airway inflammation represents the core pathological basis and primary driving factor in the progression of chronic obstructive pulmonary disease (COPD) and various small airway diseases. It is characterized by inflammatory cell infiltration and excessive production of proinflammatory cytokines, which contribute to airway remodeling, airflow limitation, and acute exacerbations of the disease (140, 141). Given this underlying mechanism, anti-inflammatory therapy has emerged as a central strategy in the management of these conditions, aiming to reduce inflammation, alleviate symptoms, and slow disease progression (140, 141). Anti-inflammatory drugs primarily function by targeting key inflammatory signaling pathways–such as nuclear factor-κB (NF-κB) and mitogen-activated protein kinase (MAPK)–modulating the balance between proinflammatory and anti-inflammatory mediators, inhibiting the activation and migration of inflammatory cells, and ameliorating oxidative stress and protease/antiprotease imbalance (141–143). These agents can block the activation of NF-κB and MAPK pathways, thereby reducing the transcription and release of proinflammatory cytokines including interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α). Additionally, they can modulate the redox state via histone deacetylase 1 (HDAC1)-dependent mechanisms, thereby attenuating oxidative damage in lung tissue (141, 142, 144). Inhaled glucocorticoids (ICS) are a cornerstone of anti-inflammatory treatment in COPD. They exert their effects by binding to the glucocorticoid receptor (GR), inhibiting the transcription of proinflammatory genes (e.g., TNF-α, IL-6), and activating anti-inflammatory genes. COPD patients with elevated blood eosinophil counts (BECs) show a better response to ICS treatment, an effect associated with the suppression of type 2 inflammatory pathways rather than direct reduction in eosinophil numbers (145). Antifibrotic agents constitute a class of compounds that interfere with the fibrotic process. Their mechanisms include inhibiting the activation and proliferation of fibroblasts and myofibroblasts, regulating extracellular matrix (ECM) synthesis, and suppressing profibrotic inflammatory responses. Specific actions involve blocking signaling pathways such as transforming growth factor-β (TGF-β) and cyclic adenosine monophosphate/protein kinase A (cAMP/PKA), reducing ECM synthesis by inhibiting synthases, and modulating inflammatory mediator secretion to alleviate inflammatory responses (146, 147). Studies indicate that targeting the TGF-β pathway can inhibit fibroblast-to-myofibroblast differentiation and reduce ECM deposition (148). For instance, pirfenidone inhibits TGF-β-induced Smad3, p38, and protein kinase B (Akt) phosphorylation, thereby suppressing fibroblast proliferation and collagen production (146). Epigallocatechin gallate (EGCG), a green tea polyphenol, demonstrates antifibrotic potential in respiratory diseases by inhibiting the TGF-β1 pathway, downregulating α-smooth muscle actin (α-SMA) and collagen expression, and reducing ECM deposition. In a bleomycin-induced pulmonary fibrosis model, EGCG reduced hydroxyproline content and enhanced antioxidant enzyme activity, mitigating pulmonary fibrosis (149). BI 1015550, a phosphodiesterase 4B (PDE4B) inhibitor, exerts antifibrotic effects by inhibiting TGF-β-stimulated myofibroblast transformation and ECM protein expression in fibroblasts from idiopathic pulmonary fibrosis (IPF) patients (150). Nintedanib, a tyrosine kinase inhibitor, inhibits fibroblast proliferation and exhibits synergistic effects with BI 1015550 (150, 151). Moreover, nintedanib inhibits collagen fiber assembly, representing a novel antifibrotic mechanism (151). Senolytics are small molecules or biological agents that selectively induce apoptosis in senescent cells (SCs) without affecting non-senescent cells (152). These drugs were initially identified via a “network targeting” strategy: senescent cells accumulate with age or disease and upregulate anti-apoptotic pathways (SCAPs) such as B-cell lymphoma-2/extra large (BCL-2/BCL-XL), phosphatidylinositol 3-kinase/protein kinase B (PI3K/AKT), hypoxia-inducible factor-1α (HIF-1α), and receptor/tyrosine kinase-dependent pathways, counteracting the pro-apoptotic effects of the senescence-associated secretory phenotype (SASP) (152, 153). Senolytics eliminate senescent cells by transiently inhibiting SCAPs, inducing mitochondrial outer membrane permeabilization and caspase-mediated cell death, while sparing healthy cells (153). In a bleomycin-induced mouse model of pulmonary fibrosis (simulating IPF), the combination of dasatinib and quercetin (D + Q) reduced senescent fibroblasts and epithelial cells, decreased expression of fibrotic factors such as type I collagen α1 chain (Col1a1) and TGF-β, and improved lung compliance (154). Multiple in vivo and in vitro studies have shown that senolytics (such as D + Q) can reduce the burden of senescent cells in the respiratory epithelium or mouse models and improve inflammatory indicators, but efficacy and safety data at the population level are still limited and no consensus has been reached. Some small sample or early trials have shown positive signals, but data from larger-scale, randomized controlled trials are still scarce, so its clinical feasibility and long-term risks need to be carefully evaluated (155, 156). In a cigarette smoke-induced lung injury model using p16-3MR mice, ganciclovir (GCV) eliminated p16+ senescent cells, inhibited neutrophil inflammation mediated by the C-X-C chemokine ligand 1-keratinocyte chemoattractant (CXCL1-KC) axis, and restored mitochondrial function. This intervention reduced senescence-associated β-galactosidase-positive (SA-β-Gal+) cells, reversed lung aging, diminished neutrophil infiltration in bronchoalveolar lavage fluid, and ultimately attenuated smoke-induced alveolar enlargement, highlighting its potential value in early COPD intervention (157).
Conclusion
Small airway disease (SAD) is present throughout the course of COPD but plays a particularly critical role in its early development. Three interrelated pathological mechanisms drive injury and remodeling of the small airways. First, chronic inflammation–primarily mediated by neutrophils and macrophages–directly damages the epithelium and extracellular matrix (ECM) through proteases (e.g., MMPs, elastase), chemokines, and reactive oxygen species, thereby promoting structural destruction and airway narrowing. Second, fibrosis and epithelial–mesenchymal transition (EMT), activated by profibrotic pathways such as TGF-β, induce fibroblast-to-myofibroblast transdifferentiation and excessive ECM deposition. Third, the senescence–SASP axis, whereby senescent cells amplify local inflammation and propagate paracrine senescence through the secretion of SASP factors (e.g., IL-6, IL-8, PAI-1, MMPs), synergistically accelerates remodeling and functional decline. These mechanisms do not act in isolation but continuously interact within the small airway microenvironment, collectively driving the progression from SAD to overt COPD (55, 158, 159).
To achieve “early detection, early diagnosis, and early treatment,” the combination of HRCT and PRM has been shown to differentiate functional small airway disease (fSAD) from emphysema at the voxel level and predict subsequent functional and structural progression. The latest deep learning methods can even approximate PRM information using only inspiratory CT, significantly reducing the need for additional respiratory scans (119, 125). MBW and IOS are more sensitive for detecting airway malfunction and peripheral airway resistance when spirometry is normal (97, 160), and can be considered for use in outpatient clinics or longitudinal follow-up. Furthermore, single-cell omics has revealed the spatiotemporal landscape of small airway cell subsets and immune-stromal interactions, laying the foundation for linking imaging phenotypes with molecular mechanisms. These complementary approaches enhance the sensitivity of small airway detection.
Future therapeutic strategies for COPD should focus on disrupting key pathogenic circuits of small airway disease. Promising approaches include: senolytics or senomorphics to attenuate SASP-driven inflammation and paracrine senescence; microRNA- or exosome-based precision interventions to restore anti-senescence pathways; antifibrotic, anti-TGF-β, and antioxidant therapies to limit ECM deposition and oxidative damage; and targeting immune regulatory axes, such as neutrophil chemotaxis/activation or PI3Kδ signaling, to reduce protease-mediated injury (55, 156, 159, 161).
Author contributions
RZ: Writing – original draft. HW: Writing – review & editing, Supervision. YZ: Supervision, Writing – review & editing. JM: Writing – review & editing. LY: Funding acquisition, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work supported by the National Natural Science Foundation of China, China, 82474411.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2025.1648612/full#supplementary-material
Abbreviations
SA, small airways; IOS, impulse oscillometry; PRM, parameter response mapping; ASM, airway smooth muscle; CS, cigarette smoke; MMPS, matrix metalloproteinases; EMT, epithelial-mesenchymal transition; NK1, neurokinin-1 receptor; CXCL8 C-X-C, chemokine ligand 8; IL-17A, interleukin-17A; TNF-α, tumor necrosis factor-α; IL-1β, interleukin-1β; SlgA, secretory immunoglobulin; plgR, polymeric Ig receptor; SASP, senescence-associated secretory phenotype; PAI-1, plasminogen activator inhibitor-1; D + Q, Dasatinib + Quercetin; Antagomir, antisense oligonucleotides; MMEF, the maximum mid-expiratory flow; LLN, below the lower limit of normal; ROS, reactive oxygen species; LTB4, leukotriene B4; QCT, quantitative computed tomography; DLCO, the diffusing capacity of the lung for carbon monoxide; ECM, extracellular matrix; SAO, small airway obstruction; FEV1, the forced expiratory volume in 1 s; FVC, the forced vital capacity; RV, the residual volume; SBW, single-breath washout; MBW, multiple-breath washout; TLC, total lung capacity; SIII, the third-phase slope; LCI, the lung clearance index; FRC, functional residual capacity; CF, cystic fibrosis; SAD, small airway disease; Cdyn, dynamic lung compliance; HRCT, high-resolution computed tomography; HU, Hounsfield units; CV, coefficient of variation; VDP, the ventilation defect percentage; ICS, inhaled glucocorticoids; NF-κB, nuclear factor-κB; MAPK, mitogen-activated protein kinase; IL-6, interleukin-6; TNF-α, tumor necrosis factor-α; HDAC1, histone deacetylase 1; ICS, inhaled glucocorticoids; GR, glucocorticoid receptor; BECs, blood eosinophil counts; TGF-β, transforming growth factor-β; cAMP, cyclic adenosine monophosphate; PKA, protein kinase A; EGCG, epigallocatechin gallate; α-SMA, α-smooth muscle actin; PDE4B, phosphodiesterase 4B; IPF, idiopathic pulmonary fibrosis; SCs, senescent cells; BCL-2, B-cell lymphoma-2; BCL-XL, B-cell lymphoma-extra large; PI3K, phosphatidylinositol 3-kinase; HIF-1α, hypoxia-inducible factor-1α; Col1a1 type I, collagen α1 chain; GCV, ganciclovir; CXCL1-KC C-X-C, chemokine ligand 1-keratinocyte chemoattractant; SA-β-Gal+, senescence-associated β-galactosidase-positive.
References
1. Agustí A, Celli B, Criner G, Halpin D, Anzueto A, Barnes P, et al. Global initiative for chronic obstructive lung disease 2023 report: gold executive summary. Am J Respir Crit Care Med. (2023) 207:819–37. doi: 10.1164/rccm.202301-0106PP
2. McDonough J, Yuan R, Suzuki M, Seyednejad N, Elliott W, Sanchez P, et al. Small-airway obstruction and emphysema in chronic obstructive pulmonary disease. N Engl J Med. (2011) 365:1567–75. doi: 10.1056/NEJMoa1106955
3. McNulty W, Usmani O. Techniques of assessing small airways dysfunction. Eur Clin Respir J. (2014) 1:1. doi: 10.3402/ecrj.v1.25898
4. Capron T, Bourdin A, Perez T, Chanez P. COPD beyond proximal bronchial obstruction: phenotyping and related tools at the bedside. Eur Respir Rev. (2019) 28:190010. doi: 10.1183/16000617.0010-2019
5. Higham A, Quinn A, Cançado J, Singh D. The pathology of small airways disease in COPD: historical aspects and future directions. Respir Res. (2019) 20:49. doi: 10.1186/s12931-019-1017-y
6. Fangal V, Saferali A, Castaldi P, Hersh C, Weiss S. Distinct physiological, transcriptomic, and imaging characteristics of asthma-COPD overlap compared to asthma and COPD in smokers. EBioMedicine. (2024) 110:105453. doi: 10.1016/j.ebiom.2024.105453
7. Stockley J, Cooper B, Stockley R, Sapey E. Small airways disease: time for a revisit? Int J Chron Obstruct Pulmon Dis. (2017) 12:2343–53. doi: 10.2147/COPD.S138540
8. Allen T. Pathology of small airways disease. Arch Pathol Lab Med. (2010) 134:702–18. doi: 10.5858/134.5.702
9. Macklem P. The physiology of small airways. Am J Respir Crit Care Med. (1998) 157:S181–3. doi: 10.1164/ajrccm.157.5.rsaa-2
10. Berg K, Wright J. The pathology of chronic obstructive pulmonary disease: progress in the 20th and 21st Centuries. Arch Pathol Lab Med. (2016) 140:1423–8. doi: 10.5858/arpa.2015-0455-RS
11. Macklem P, Proctor D, Hogg J. The stability of peripheral airways. Respir Physiol. (1970) 8:191–203. doi: 10.1016/0034-5687(70)90015-0
12. Heijink I, Brandenburg S, Postma D, van Oosterhout A. Cigarette smoke impairs airway epithelial barrier function and cell-cell contact recovery. Eur Respir J. (2012) 39:419–28. doi: 10.1183/09031936.00193810
13. Mottais A, Riberi L, Falco A. Epithelial-mesenchymal transition mechanisms in chronic airway diseases: a common process to target? Int J Mol Sci. (2023) 24:12412. doi: 10.3390/ijms241512412
14. Sohal S, Walters E. Role of epithelial mesenchymal transition (EMT) in chronic obstructive pulmonary disease (COPD). Respir Res. (2013) 14:120. doi: 10.1186/1465-9921-14-120
15. Wei Y, Li Q, He K, Liao G, Cheng L, Li M, et al. Mechanism of cigarette smoke in promoting small airway remodeling in mice via STAT 3 / PINK 1-Parkin / EMT. Free Radic Biol Med. (2024) 224:447–56. doi: 10.1016/j.freeradbiomed.2024.08.036
16. Sohal S, Walters E. Epithelial mesenchymal transition (EMT) in small airways of COPD patients. Thorax. (2013) 68:783–4. doi: 10.1136/thoraxjnl-2013-203373
17. Milara J, Peiró T, Serrano A, Cortijo J. Epithelial to mesenchymal transition is increased in patients with COPD and induced by cigarette smoke. Thorax. (2013) 68:410–20. doi: 10.1136/thoraxjnl-2012-201761
18. Hogg J, Chu F, Utokaparch S. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. (2004) 350:2645–53. doi: 10.1056/NEJMoa032158
19. Kirkham S, Kolsum U, Rousseau K, Singh D, Vestbo J, Thornton D. MUC5B is the major mucin in the gel phase of sputum in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2008) 178:1033–9. doi: 10.1164/rccm.200803-391OC
20. Kesimer M, Ford A, Ceppe A. Airway mucin concentration as a marker of chronic bronchitis. N Engl J Med. (2017) 377:911–22. doi: 10.1056/NEJMoa1701632
21. Hessel J, Heldrich J, Fuller J, Staudt M, Radisch S, Hollmann C, et al. Intraflagellar transport gene expression associated with short cilia in smoking and COPD. PLoS One. (2014) 9:e85453. doi: 10.1371/journal.pone.0085453
22. Yaghi A, Zaman A, Cox G, Dolovich M. Ciliary beating is depressed in nasal cilia from chronic obstructive pulmonary disease subjects. Respir Med. (2012) 106:1139–47. doi: 10.1016/j.rmed.2012.04.001
23. Fang Z, Wang Z, Chen Z, Peng Y, Fu Y, Yang Y, et al. Fine particulate matter contributes to COPD-like pathophysiology: experimental evidence from rats exposed to diesel exhaust particles. Respir Res. (2024) 25:14. doi: 10.1186/s12931-023-02623-y
24. Pilette C, Godding V, Kiss R, Delos M, Verbeken E, Decaestecker C, et al. Reduced epithelial expression of secretory component in small airways correlates with airflow obstruction in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2001) 163:185–94. doi: 10.1164/ajrccm.163.1.9912137
25. Polosukhin V, Richmond B, Du R, Cates J, Wu P, Nian H, et al. Secretory IgA deficiency in individual small airways is associated with persistent inflammation and remodeling. Am J Respir Crit Care Med. (2017) 195:1010–21. doi: 10.1164/rccm.201604-0759OC
26. Ladjemi M, Lecocq M, Weynand B, Bowen H, Gould H, Van Snick J, et al. Increased IgA production by B-cells in COPD via lung epithelial interleukin-6 and TACI pathways. Eur Respir J. (2015) 45:980–93. doi: 10.1183/09031936.00063914
27. Du R, Richmond B, Blackwell T, Cates J, Massion P, Ware L, et al. Secretory IgA from submucosal glands does not compensate for its airway surface deficiency in chronic obstructive pulmonary disease. Virchows Arch. (2015) 467:657–65. doi: 10.1007/s00428-015-1854-0
28. Brandsma C, de Vries M, Costa R, Woldhuis R, Königshoff M, Timens W. Lung ageing and COPD: is there a role for ageing in abnormal tissue repair? Eur Respir Rev. (2017) 26:170073. doi: 10.1183/16000617.0073-2017
29. Shaykhiev R. Emerging biology of persistent mucous cell hyperplasia in COPD. Thorax. (2019) 74:4–6. doi: 10.1136/thoraxjnl-2018-212271
30. Cosio M, Guerassimov A. Chronic obstructive pulmonary disease. Inflammation of small airways and lung parenchyma. Am J Respir Crit Care Med. (1999) 160:S21–5. doi: 10.1164/ajrccm.160.supplement_1.7
31. Aghali A, Koloko Ngassie M, Pabelick C. Cellular senescence in aging lungs and diseases. Cells. (2022) 11:1781. doi: 10.3390/cells11111781
32. Yadav A, Gu W, Zhang T, Xu X, Yu L. Current perspectives on biological therapy for COPD. COPD. (2023) 20:197–209. doi: 10.1080/15412555.2023.2187210
33. Vlahos R, Bozinovski S. Role of alveolar macrophages in chronic obstructive pulmonary disease. Front Immunol. (2014) 5:435. doi: 10.3389/fimmu.2014.00435
34. Williams M, Todd I, Fairclough L. The role of CD8 + T lymphocytes in chronic obstructive pulmonary disease: a systematic review. Inflamm Res. (2021) 70:11–8. doi: 10.1007/s00011-020-01408-z
35. Wu J, Zhao X, Xiao C, Xiong G, Ye X, Li L, et al. The role of lung macrophages in chronic obstructive pulmonary disease. Respir Med. (2022) 205:107035. doi: 10.1016/j.rmed.2022.107035
36. Maetani T, Tanabe N, Sato A, Shiraishi Y, Sakamoto R, Ogawa E, et al. Association between blood eosinophil count and small airway eosinophils in smokers with and without COPD. ERJ Open Res. (2023) 9:235–2023. doi: 10.1183/23120541.00235-2023
37. David B, Bafadhel M, Koenderman L, De Soyza A. Eosinophilic inflammation in COPD: from an inflammatory marker to a treatable trait. Thorax. (2021) 76:188–95. doi: 10.1136/thoraxjnl-2020-215167
38. Haswell L, Hewitt K, Thorne D, Richter A, Gaça M. Cigarette smoke total particulate matter increases mucous secreting cell numbers in vitro: a potential model of goblet cell hyperplasia. Toxicol In Vitro. (2010) 24:981–7. doi: 10.1016/j.tiv.2009.12.019
39. Rodier F, Coppé J, Patil C, Hoeijmakers W, Muñoz D, Raza S, et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. (2009) 11:973–9. doi: 10.1038/ncb1909
40. Barnes P, Baker J, Donnelly L. Cellular senescence as a mechanism and target in chronic lung diseases. Am J Respir Crit Care Med. (2019) 200:556–64. doi: 10.1164/rccm.201810-1975TR
41. Coppé J, Patil C, Rodier F, Sun Y, Muñoz D, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. (2008) 6:2853–68. doi: 10.1371/journal.pbio.0060301
42. Yao H, Chung S, Hwang J, Rajendrasozhan S, Sundar I, Dean D, et al. SIRT1 protects against emphysema via FOXO3-mediated reduction of premature senescence in mice. J Clin Invest. (2012) 122:2032–45. doi: 10.1172/JCI60132
43. Houssaini A, Breau M, Kebe K, Abid S, Marcos E, Lipskaia L, et al. mTOR pathway activation drives lung cell senescence and emphysema. JCI Insight. (2018) 3:e93203. doi: 10.1172/jci.insight.93203
44. Baker J, Vuppusetty C, Colley T, Papaioannou A, Fenwick P, Donnelly L, et al. Oxidative stress dependent microRNA-34a activation via PI3Kα reduces the expression of sirtuin-1 and sirtuin-6 in epithelial cells. Sci Rep. (2016) 6:35871. doi: 10.1038/srep35871
45. Baker J, Fenwick P, Donnelly L, Barnes P, Cloonan S. Altered iron metabolism and elevated cellular senescence in COPD small airway epithelial cells. Eur Respiratory J. (2020) 56:3707. doi: 10.1183/13993003.congress-2020.3707
46. Gaggar A, Weathington N. Bioactive extracellular matrix fragments in lung health and disease. J Clin Invest. (2016) 126:3176–84. doi: 10.1172/JCI83147
47. De Swert K, Bracke K, Demoor T, Brusselle G, Joos G. Role of the tachykinin NK1 receptor in a murine model of cigarette smoke-induced pulmonary inflammation. Respir Res. (2009) 10:37. doi: 10.1186/1465-9921-10-37
48. Redkiewicz P. The Regenerative Potential of Substance P. Int J Mol Sci. (2022) 23:750. doi: 10.3390/ijms23020750
49. Advenier C, Lagente V, Boichot E. The role of tachykinin receptor antagonists in the prevention of bronchial hyperresponsiveness, airway inflammation and cough. Eur Respir J. (1997) 10:1892–906. doi: 10.1183/09031936.97.10081892
50. Wei B, Sun M, Shang Y, Zhang C, Jiao X. Neurokinin 1 receptor promotes rat airway smooth muscle cell migration in asthmatic airway remodelling by enhancing tubulin expression. J Thorac Dis. (2018) 10:4849–57. doi: 10.21037/jtd.2018.07.114
51. Mohamed M, Abed El Baky MF, Ali ME, Hafez HM. Aprepitant exerts anti-fibrotic effect via inhibition of TGF-β/Smad3 pathway in bleomycin-induced pulmonary fibrosis in rats. Environ Toxicol Pharmacol. (2022) 95:103940. doi: 10.1016/j.etap.2022.103940
52. Leszczyńska K, Jakubczyk D, Górska S. The NLRP3 inflammasome as a new target in respiratory disorders treatment. Front Immunol. (2022) 13:1006654. doi: 10.3389/fimmu.2022.1006654
53. Yan F, Gao H, Zhao H, Bhatia M, Zeng Y. Roles of airway smooth muscle dysfunction in chronic obstructive pulmonary disease. J Transl Med. (2018) 16:262. doi: 10.1186/s12967-018-1635-z
54. Karakioulaki M, Papakonstantinou E, Stolz D. Extracellular matrix remodelling in COPD. Eur Respir Rev. (2020) 29:190124. doi: 10.1183/16000617.0124-2019
55. Christopoulou M, Papakonstantinou E, Stolz D. Matrix metalloproteinases in chronic obstructive pulmonary disease. Int J Mol Sci. (2023) 24:3786. doi: 10.3390/ijms24043786
56. Pistenmaa C, Nardelli P, Ash S, Come C, Diaz A, Rahaghi F, et al. Pulmonary arterial pruning and longitudinal change in percent emphysema and lung function: the genetic epidemiology of COPD study. Chest. (2021) 160:470–80. doi: 10.1016/j.chest.2021.01.084
57. Blanco I, Piccari L, Barberà J. Pulmonary vasculature in COPD: the silent component. Respirology. (2016) 21:984–94. doi: 10.1111/resp.12772
58. Washko G, Nardelli P, Ash S, Vegas Sanchez-Ferrero G, Rahaghi FN, Come CE, et al. Arterial vascular pruning, right ventricular size, and clinical outcomes in chronic obstructive pulmonary disease. A longitudinal observational study. Am J Respir Crit Care Med. (2019) 200:454–61. doi: 10.1164/rccm.201811-2063OC
59. Bhattarai P, Lu W, Hardikar A, Dey S, Gaikwad A, Shahzad A, et al. Endothelial to mesenchymal transition is an active process in smokers and patients with early COPD contributing to pulmonary arterial pathology. ERJ Open Res. (2024) 10:00767–2024. doi: 10.1183/23120541.00767-2023
60. Sohal S. Endothelial to mesenchymal transition (EndMT): an active process in chronic obstructive pulmonary disease (COPD)? Respir Res. (2016) 17:20. doi: 10.1186/s12931-016-0337-4
61. Gaikwad A, Lu W, Dey S, Bhattarai P, Chia C, Larby J, et al. Vascular remodelling in idiopathic pulmonary fibrosis patients and its detrimental effect on lung physiology: potential role of endothelial-to-mesenchymal transition. ERJ Open Res. (2022) 8:00571–2021. doi: 10.1183/23120541.00571-2021
62. Bhattarai P, Lu W, Gaikwad A, Dey S, Chia C, Larby J, et al. Arterial remodelling in smokers and in patients with small airway disease and COPD: implications for lung physiology and early origins of pulmonary hypertension. ERJ Open Res. (2022) 8:254–2022. doi: 10.1183/23120541.00254-2022
63. Chaouat A, Naeije R, Weitzenblum E. Pulmonary hypertension in COPD. Eur Respir J. (2008) 32:1371–85. doi: 10.1183/09031936.00015608
64. Peinado V, Pizarro S, Barberà J. Pulmonary vascular involvement in COPD. Chest. (2008) 134:808–14. doi: 10.1378/chest.08-0820
65. Santos S, Peinado V, Ramírez J, Melgosa T, Roca J, Rodriguez-Roisin R, et al. Characterization of pulmonary vascular remodelling in smokers and patients with mild COPD. Eur Respir J. (2002) 19:632–8. doi: 10.1183/09031936.02.00245902
66. Rodríguez-Roisin R, Drakulovic M, Rodríguez D, Roca J, Barberà J, Wagner P. Ventilation-perfusion imbalance and chronic obstructive pulmonary disease staging severity. J Appl Physiol. (2009) 106:1902–8. doi: 10.1152/japplphysiol.00085.2009
67. Hoenderdos K, Condliffe A. The neutrophil in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. (2013) 48:531–9. doi: 10.1165/rcmb.2012-0492TR
68. Mercer P, Shute J, Bhowmik A, Donaldson G, Wedzicha J, Warner JA. MMP-9, TIMP-1 and inflammatory cells in sputum from COPD patients during exacerbation. Respir Res. (2005) 6:151. doi: 10.1186/1465-9921-6-151
69. Booth S, Hsieh A, Mostaco-Guidolin L, Koo H, Wu K, Aminazadeh F, et al. A single-cell atlas of small airway disease in chronic obstructive pulmonary disease: a cross-sectional study. Am J Respir Crit Care Med. (2023) 208:472–86. doi: 10.1164/rccm.202303-0534OC
70. Sauler M, McDonough J, Adams T, Kothapalli N, Barnthaler T, Werder R, et al. Characterization of the COPD alveolar niche using single-cell RNA sequencing. Nat Commun. (2022) 13:494. doi: 10.1038/s41467-022-28062-9
71. Huang Q, Wang Y, Zhang L, Qian W, Shen S, Wang J, et al. Single-cell transcriptomics highlights immunological dysregulations of monocytes in the pathobiology of COPD. Respir Res. (2022) 23:367. doi: 10.1186/s12931-022-02293-2
72. Liao S, Zhang L, Shi L, Hu H, Gu Y, Wang T, et al. Integrating bulk and single-cell RNA sequencing data: unveiling RNA methylation and autophagy-related signatures in chronic obstructive pulmonary disease patients. Sci Rep. (2025) 15:4005. doi: 10.1038/s41598-025-87437-2
73. Konstantinos Katsoulis K, Kostikas K, Kontakiotis T. Techniques for assessing small airways function: possible applications in asthma and COPD. Respir Med. (2016) 119:e2–9. doi: 10.1016/j.rmed.2013.05.003
74. McFadden E, Linden DA. A reduction in maximum mid-expiratory flow rate. A spirographic manifestation of small airway disease. Am J Med. (1972) 52:725–37. doi: 10.1016/0002-9343(72)90078-2
75. Kwon D, Choi Y, Kim T, Byun M, Cho J, Kim H, et al. FEF25-75% values in patients with normal lung function can predict the development of chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. (2020) 15:2913–21. doi: 10.2147/COPD.S261732
76. Chiu H, Hsiao Y, Su K, Lee Y, Ko H, Perng D. Small airway dysfunction by impulse oscillometry in symptomatic patients with preserved pulmonary function. J Allergy Clin Immunol Pract. (2020) 8:229–35.e3. doi: 10.1016/j.jaip.2019.06.035
77. Knox-Brown B, Mulhern O, Feary J, Amaral A. Spirometry parameters used to define small airways obstruction in population-based studies: systematic review. Respir Res. (2022) 23:67. doi: 10.1186/s12931-022-01990-2
78. Quanjer P, Stanojevic S, Cole T, Baur X, Hall G, Culver B, et al. Multi-ethnic reference values for spirometry for the 3-95-yr age range: the global lung function 2012 equations. Eur Respir J. (2012) 40:1324–43. doi: 10.1183/09031936.00080312
79. Quanjer P, Weiner D, Pretto J, Brazzale D, Boros P. Measurement of FEF25-75% and FEF75% does not contribute to clinical decision making. Eur Respir J. (2014) 43:1051–8. doi: 10.1183/09031936.00128113
80. Hansen J, Sun X, Wasserman K. Discriminating measures and normal values for expiratory obstruction. Chest. (2006) 129:369–77. doi: 10.1378/chest.129.2.369
81. Dilektasli A, Porszasz J, Casaburi R, Stringer W, Bhatt S, Pak Y, et al. A novel spirometric measure identifies mild COPD unidentified by standard criteria. Chest. (2016) 150:1080–90. doi: 10.1016/j.chest.2016.06.047
82. Yee N, Markovic D, Buhr R, Fortis S, Arjomandi M, Couper D, et al. Significance of FEV3/FEV6 in recognition of early airway disease in smokers at risk of development of COPD: analysis of the SPIROMICS cohort. Chest. (2022) 161:949–59. doi: 10.1016/j.chest.2021.10.046
83. Bhatt S, Soler X, Wang X, Murray S, Anzueto A, Beaty T, et al. Association between functional small airway disease and FEV1 decline in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2016) 194:178–84. doi: 10.1164/rccm.201511-2219OC
84. Solomon D. Are small airways tests helpful in the detection of early airflow obstruction? Chest. (1978) 74:567–9. doi: 10.1378/chest.74.5.567
85. Zimmermann S, Tonga K, Thamrin C. Dismantling airway disease with the use of new pulmonary function indices. Eur Respir Rev. (2019) 28:180122. doi: 10.1183/16000617.0122-2018
86. King G. Cutting edge technologies in respiratory research: lung function testing. Respirology. (2011) 16:883–90. doi: 10.1111/j.1440-1843.2011.02013.x
87. Lapp N. Physiological approaches to detection of small airway disease. Environ Res. (1973) 6:253–64. doi: 10.1016/0013-9351(73)90038-8
88. Vontetsianos A, Chynkiamis N, Anagnostopoulou C. Small airways dysfunction and lung hyperinflation in long COVID-19 Patients as potential mechanisms of persistent dyspnoea. Adv Respir Med. (2024) 92:329–37. doi: 10.3390/arm92050031
89. Singer F, Stern G, Thamrin C, Abbas C, Casaulta C, Frey U, et al. A new double-tracer gas single-breath washout to assess early cystic fibrosis lung disease. Eur Respir J. (2013) 41:339–45. doi: 10.1183/09031936.00044312
90. Subbarao P, Milla C, Aurora P, Davies J, Davis S, Hall G, et al. Multiple-breath washout as a lung function test in cystic fibrosis. A cystic fibrosis foundation workshop report. Ann Am Thorac Soc. (2015) 12:932–9. doi: 10.1513/AnnalsATS.201501-021FR
91. Robinson P, Latzin P, Verbanck S, Hall G, Horsley A, Gappa M, et al. Consensus statement for inert gas washout measurement using multiple- and single- breath tests. Eur Respir J. (2013) 41:507–22. doi: 10.1183/09031936.00069712
92. Ramsey K, Rosenow T, Turkovic L, Skoric B, Banton G, Adams A, et al. Lung clearance index and structural lung disease on computed tomography in early cystic fibrosis. Am J Respir Crit Care Med. (2016) 193:60–7. doi: 10.1164/rccm.201507-1409OC
93. Stanojevic S, Bowerman C, Robinson P. Multiple breath washout: measuring early manifestations of lung pathology. Breathe. (2021) 17:210016. doi: 10.1183/20734735.0016-2021
94. Frauchiger B, Binggeli S, Yammine S, Spycher B, Krüger L, Ramsey K, et al. Longitudinal course of clinical lung clearance index in children with cystic fibrosis. Eur Respir J. (2021) 58:2002686. doi: 10.1183/13993003.02686-2020
95. Ramsey K, Foong R, Grdosic J, Harper A, Skoric B, Clem C, et al. Multiple-breath washout outcomes are sensitive to inflammation and infection in children with cystic fibrosis. Ann Am Thorac Soc. (2017) 14:1436–42. doi: 10.1513/AnnalsATS.201611-935OC
96. Downing B, Irving S, Bingham Y, Fleming L, Bush A, Saglani S. Feasibility of lung clearance index in a clinical setting in pre-school children. Eur Respir J. (2016) 48:1074–80. doi: 10.1183/13993003.00374-2016
97. Bell A, Lawrence P, Singh D, Horsley A. Feasibility and challenges of using multiple breath washout in COPD. Int J Chron Obstruct Pulmon Dis. (2018) 13:2113–9. doi: 10.2147/COPD.S164285
98. Galant S, Morphew T. Adding oscillometry to spirometry in guidelines better identifies uncontrolled asthma, future exacerbations, and potential targeted therapy. Ann Allergy Asthma Immunol. (2024) 132:21–9. doi: 10.1016/j.anai.2023.08.011
99. Cottini M, Lombardi C, Berti A, Comberiati P. Small-airway dysfunction in paediatric asthma. Curr Opin Allergy Clin Immunol. (2021) 21:128–34. doi: 10.1097/ACI.0000000000000728
100. Cottini M, Licini A, Lombardi C, Berti A. Clinical characterization and predictors of ios-defined small-airway dysfunction in asthma. J Allergy Clin Immunol Pract. (2020) 8: 997–1004.e2. doi: 10.1016/j.jaip.2019.10.040
101. Sly P, Hantos Z. The international collaboration to improve respiratory health in children (INCIRCLE) ERS clinical research collaboration. Eur Respir J. (2018) 52:1801867. doi: 10.1183/13993003.01867-2018
102. Bates J, Irvin C, Farré R, Hantos Z. Oscillation mechanics of the respiratory system. Compr Physiol. (2011) 1:1233–72. doi: 10.1002/cphy.c100058
103. Dubois A, Brody A, Lewis D, Burgess B. Oscillation mechanics of lungs and chest in man. J Appl Physiol. (1956) 8:587–94. doi: 10.1152/jappl.1956.8.6.587
104. Hogg J, Paré P, Hackett T. The contribution of small airway obstruction to the pathogenesis of chronic obstructive pulmonary disease. Physiol Rev. (2017) 97:529–52. doi: 10.1152/physrev.00025.2015
105. King G, Chung L, Usmani O, Nilsen K, Thompson B. Improving asthma outcomes: clinicians’ perspectives on peripheral airways. J Allergy Clin Immunol Glob. (2024) 3:100228. doi: 10.1016/j.jacig.2024.100228
106. Mou T, Wang Y, Fu Y, Wang Y, Li G. Analysis of the correlations and inconsistencies between spirometry and impulse oscillometry in the diagnosis of small-airway dysfunction. BMC Pulm Med. (2024) 24:619. doi: 10.1186/s12890-024-03420-z
107. Frantz S, Nihlén U, Dencker M, Engström G, Löfdahl C, Wollmer P. Impulse oscillometry may be of value in detecting early manifestations of COPD. Respir Med. (2012) 106:1116–23. doi: 10.1016/j.rmed.2012.04.010
108. Li L, Yan T, Yang J, Li Y, Fu L, Lan L, et al. Impulse oscillometry for detection of small airway dysfunction in subjects with chronic respiratory symptoms and preserved pulmonary function. Respir Res. (2021) 22:68. doi: 10.1186/s12931-021-01662-7
109. Guan W, Gao Y, Xu G, Lin Z, Tang Y, Li H, et al. Impulse oscillometry in adults with bronchiectasis. Ann Am Thorac Soc. (2015) 12:657–65. doi: 10.1513/AnnalsATS.201406-280OC
110. Guan W, Yuan J, Gao Y, Li H, Zheng J, Chen R, et al. Impulse oscillometry and spirometry small-airway parameters in mild to moderate bronchiectasis. Respir Care. (2016) 61:1513–22. doi: 10.4187/respcare.04710
111. Park H, Lee H, Lee H, Park T, Heo E, Kim D, et al. Diagnosis and evaluation of small airway disease and COPD using impulse oscillometry. Sci Rep. (2024) 14:28030. doi: 10.1038/s41598-024-79818-w
112. Lu L, Wu F, Tang G, Wan Q, Deng Z, Peng J, et al. Associations of small airway dysfunction assessed by impulse oscillometry with lung function decline and exacerbations in participants with chronic obstructive pulmonary disease: a prospective cohort study in China. Respir Med. (2025) 241:108075. doi: 10.1016/j.rmed.2025.108075
113. Sundaram M, Karthika M. Respiratory mechanics: to balance the mechanical breaths!! Indian J Crit Care Med. (2021) 25:10–1. doi: 10.5005/jp-journals-10071-23700
114. Desai J, Moustarah F. Pulmonary Compliance. ineligible companies. Disclosure: Fady Moustarah declares no relevant financial relationships with ineligible companies. Treasure Island, FL: StatPearls (2025).
115. Chiang S. Distribution of ventilation and frequency-dependence of dynamic lung compliance. Thorax. (1971) 26:721–6. doi: 10.1136/thx.26.6.721
116. Woolcock A, Vincent N, Macklem P. Frequency dependence of compliance as a test for obstruction in the small airways. J Clin Invest. (1969) 48:1097–106. doi: 10.1172/JCI106066
117. Teixeira E, Ribeiro C, Lopes A, de Melo P. Respiratory oscillometry and functional performance in different COPD phenotypes. Int J Chron Obstruct Pulmon Dis. (2024) 19:667–82. doi: 10.2147/COPD.S446085
118. Teel GS, Engeler CE, Tashijian JH. Imaging of small airways disease. Radiographics. (1996) 16:27-41. doi: 10.1148/radiographics.16.1.27
119. Chen B, Liu Z, Lu J, Li Z, Kuang K, Yang J, et al. Deep learning parametric response mapping from inspiratory chest CT scans: a new approach for small airway disease screening. Respir Res. (2023) 24:299. doi: 10.1186/s12931-023-02611-2
120. Galbán C, Han M, Boes J, Chughtai K, Meyer C, Johnson T, et al. Computed tomography-based biomarker provides unique signature for diagnosis of COPD phenotypes and disease progression. Nat Med. (2012) 18:1711–5. doi: 10.1038/nm.2971
121. Vasilescu D, Martinez F, Marchetti N, Galbán C, Hatt C, Meldrum C, et al. Noninvasive imaging biomarker identifies small airway damage in severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2019) 200:575–81. doi: 10.1164/rccm.201811-2083OC
122. Bodduluri S, Nakhmani A, Reinhardt J, Wilson C, McDonald M, Rudraraju R, et al. Deep neural network analyses of spirometry for structural phenotyping of chronic obstructive pulmonary disease. JCI Insight. (2020) 5:e132781. doi: 10.1172/jci.insight.132781
123. Maselli D, Yen A, Wang W. Small airway disease and emphysema are associated with future exacerbations in smokers with CT-derived bronchiectasis and COPD: Results from the COPDGene cohort. Radiology. (2021) 300:706–14. doi: 10.1148/radiol.2021204052
124. Trivedi A, Hall C, Goss C. Quantitative CT characteristics of cluster phenotypes in the severe asthma research program cohorts. Radiology. (2022) 304:450–9. doi: 10.1148/radiol.210363
125. Boes J, Hoff B, Bule M, Johnson T, Rehemtulla A, Chamberlain R, et al. Parametric response mapping monitors temporal changes on lung CT scans in the subpopulations and intermediate outcome measures in COPD Study (SPIROMICS). Acad Radiol. (2015) 22:186–94. doi: 10.1016/j.acra.2014.08.015
126. McDonough J, Yuan R, Suzuki M, et al. Small-airway obstruction and emphysema in chronic obstructive pulmonary disease. N Engl J Med. (2011) 365:1567–75. doi: 10.1056/NEJMoa1106955
127. Chen B, Gao P, Yang Y, Ma Z, Sun Y, Lu J, et al. Discordant definitions of small airway dysfunction between spirometry and parametric response mapping: the HRCT-based study. Insights Imaging. (2024) 15:233. doi: 10.1186/s13244-024-01819-0
128. Chaudhary M, Awan H, Gerard S, Bodduluri S, Comellas A, Barjaktarevic I, et al. Deep learning estimation of small airway disease from inspiratory chest computed tomography: clinical validation, repeatability, and associations with adverse clinical outcomes in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2025) 211:1185–95. doi: 10.1164/rccm.202409-1847OC
129. Norberg P, Persson H, Carlsson G, Bake B, Kentson M, Sandborg M, et al. Quantitative lung SPECT applied on simulated early COPD and humans with advanced COPD. EJNMMI Res. (2013) 3:28. doi: 10.1186/2191-219X-3-28
130. Norberg P, Persson H, Schmekel B, Carlsson G, Wahlin K, Sandborg M, et al. Does quantitative lung SPECT detect lung abnormalities earlier than lung function tests? Results of a pilot study. EJNMMI Res. (2014) 4:39. doi: 10.1186/s13550-014-0039-1
131. Suga K, Kawakami Y, Koike H, Iwanaga H, Tokuda O, Okada M, et al. Lung ventilation-perfusion imbalance in pulmonary emphysema: assessment with automated V/Q quotient SPECT. Ann Nucl Med. (2010) 24:269–77. doi: 10.1007/s12149-010-0369-7
132. Nagao M, Murase K, Ichiki T, Sakai S, Yasuhara Y, Ikezoe J. Quantitative analysis of technegas SPECT: evaluation of regional severity of emphysema. J Nucl Med. (2000) 41:590–5.
133. Juneau D, Leblond A, Chatta R. SPECT/CT to quantify early small airway disease and its relationship to clinical symptoms in smokers with normal lung function: a pilot study. Front Physiol. (2024) 15:1417463. doi: 10.3389/fphys.2024.1417463
134. Le Roux P, Schafer W, Blanc-Beguin F, Tulchinsky M. Ventilation scintigraphy with radiolabeled carbon nanoparticulate aerosol (Technegas): state-of-the-art review and diagnostic applications to pulmonary embolism during COVID-19 pandemic. Clin Nucl Med. (2023) 48:8–17. doi: 10.1097/RLU.0000000000004426
135. Bajc M, Schümichen C, Grüning T, Lindqvist A, Le Roux P, Alatri A, et al. EANM guideline for ventilation/perfusion single-photon emission computed tomography (SPECT) for diagnosis of pulmonary embolism and beyond. Eur J Nucl Med Mol Imaging. (2019) 46:2429–51. doi: 10.1007/s00259-019-04450-0
136. Myc L, Shim Y, Laubach V, Dimastromatteo J. Role of medical and molecular imaging in COPD. Clin Transl Med. (2019) 8:12. doi: 10.1186/s40169-019-0231-z
137. Milne S, King G. Advanced imaging in COPD: insights into pulmonary pathophysiology. J Thorac Dis. (2014) 6:1570–85. doi: 10.3978/j.issn.2072-1439.2014.11.30
138. Pippard BJ, Neal MA, Holland CW. Assessing lung ventilation and bronchodilator response in asthma and chronic obstructive pulmonary disease with(19)F MRI. Radiology. (2024) 313:e240949. doi: 10.1148/radiol.240949
139. Unger EC. Role of imaging in diagnosis of small airway disease. Radiology. (2024) 313:e243461. doi: 10.1148/radiol.243461
140. Yousuf A, Brightling C. Biologic drugs: a new target therapy in COPD? COPD. (2018) 15:99–107. doi: 10.1080/15412555.2018.1437897
141. Wang C, Zhou J, Wang J, Li S, Fukunaga A, Yodoi J, et al. Progress in the mechanism and targeted drug therapy for COPD. Signal Transduct Target Ther. (2020) 5:248. doi: 10.1038/s41392-020-00345-x
142. Zhao Z, Tong Y, Kang Y, Qiu Z, Li Q, Xu C, et al. Sodium butyrate (SB) ameliorated inflammation of COPD induced by cigarette smoke through activating the GPR43 to inhibit NF-κB/MAPKs signaling pathways. Mol Immunol. (2023) 163:224–34. doi: 10.1016/j.molimm.2023.10.007
143. Hao D, Li Y, Shi J, Jiang J. Baicalin alleviates chronic obstructive pulmonary disease through regulation of HSP72-mediated JNK pathway. Mol Med. (2021) 27:53. doi: 10.1186/s10020-021-00309-z
144. Deng M, Tong R, Bian Y, Hou G. Astaxanthin attenuates cigarette smoking-induced oxidative stress and inflammation in a sirtuin 1-dependent manner. Biomed Pharmacother. (2023) 159:114230. doi: 10.1016/j.biopha.2023.114230
145. Lea S, Higham A, Beech A, Singh D. How inhaled corticosteroids target inflammation in COPD. Eur Respir Rev. (2023) 32:230084. doi: 10.1183/16000617.0084-2023
146. Conte E, Gili E, Fagone E, Fruciano M, Iemmolo M, Vancheri C. Effect of pirfenidone on proliferation, TGF-β-induced myofibroblast differentiation and fibrogenic activity of primary human lung fibroblasts. Eur J Pharm Sci. (2014) 58:13–9. doi: 10.1016/j.ejps.2014.02.014
147. Tannheimer S, Wright C, Salmon M. Combination of roflumilast with a beta-2 adrenergic receptor agonist inhibits proinflammatory and profibrotic mediator release from human lung fibroblasts. Respir Res. (2012) 13:28. doi: 10.1186/1465-9921-13-28
148. Györfi A, Matei A, Distler J. Targeting TGF-β signaling for the treatment of fibrosis. Matrix Biol. (2018) 68-69:8–27. doi: 10.1016/j.matbio.2017.12.016
149. Mokra D, Adamcakova J, Mokry J. Green tea polyphenol (-)-Epigallocatechin-3-Gallate (EGCG): a time for a new player in the treatment of respiratory diseases? Antioxidants. (2022) 11:1566. doi: 10.3390/antiox11081566
150. Herrmann F, Hesslinger C, Wollin L, et al. BI 1015550 is a PDE4B inhibitor and a clinical drug candidate for the oral treatment of idiopathic pulmonary fibrosis. Front Pharmacol. (2022) 13:838449. doi: 10.3389/fphar.2022.838449
151. Knüppel L, Ishikawa Y, Aichler M, Heinzelmann K, Hatz R, Behr J, et al. A novel antifibrotic mechanism of nintedanib and pirfenidone. Inhibition of collagen fibril assembly. Am J Respir Cell Mol Biol. (2017) 57:77–90. doi: 10.1165/rcmb.2016-0217OC
152. Zhu Y, Doornebal E, Pirtskhalava T, Giorgadze N, Wentworth M, Fuhrmann-Stroissnigg H, et al. New agents that target senescent cells: the flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463. Aging. (2017) 9:955–63. doi: 10.18632/aging.101202
153. Kirkland J, Tchkonia T. Senolytic drugs: from discovery to translation. J Intern Med. (2020) 288:518–36. doi: 10.1111/joim.13141
154. Schafer M, White T, Iijima K, Haak A, Ligresti G, Atkinson E, et al. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun. (2017) 8:14532. doi: 10.1038/ncomms14532
155. Nambiar A, Kellogg D, Justice J, Goros M, Gelfond J, Pascual R, et al. Senolytics dasatinib and quercetin in idiopathic pulmonary fibrosis: results of a phase I, single-blind, single-center, randomized, placebo-controlled pilot trial on feasibility and tolerability. EBioMedicine. (2023) 90:104481. doi: 10.1016/j.ebiom.2023.104481
156. Baker J, Daly L, Hassibi S, et al. Senolytic therapy reduces inflammation in epithelial cells from COPD patients and in smoke-exposure mice. Front Med. (2025) 12:1451056. doi: 10.3389/fmed.2025.1451056
157. Kaur G, Muthumalage T, Rahman I. Clearance of senescent cells reverts the cigarette smoke-induced lung senescence and airspace enlargement in p16-3MR mice. Aging Cell. (2023) 22:e13850. doi: 10.1111/acel.13850
158. Subedi S, Guntipally M, Suwal N, Thapa R, Bashyal S, Panth N, et al. Cellular senescence in chronic obstructive pulmonary disease: molecular mechanisms and therapeutic interventions. Ageing Res Rev. (2025) 110:102813. doi: 10.1016/j.arr.2025.102813
159. Saito A, Horie M, Nagase T. TGF-β signaling in lung health and disease. Int J Mol Sci. (2018) 19:2460. doi: 10.3390/ijms19082460
160. Porojan-Suppini N, Fira-Mladinescu O, Marc M, Tudorache E, Oancea C. Lung function assessment by impulse oscillometry in adults. Ther Clin Risk Manag. (2020) 16:1139–50. doi: 10.2147/TCRM.S275920
Keywords: COPD, small airways, lung mechanics, early diagnosis of COPD, small airway disease
Citation: Zhou R, Wang H, Zhang Y, Mai J and Yang L (2025) Small airway disease as a key factor in COPD: new perspectives and insights. Front. Med. 12:1648612. doi: 10.3389/fmed.2025.1648612
Received: 08 July 2025; Accepted: 11 September 2025;
Published: 26 September 2025.
Edited by:
Matthew Brendan O’Rourke, University of Technology Sydney, AustraliaReviewed by:
Yan Sun, First Affiliated Hospital of Harbin Medical University, ChinaGuansong Wang, Third Military Medical University, China
Vrushali D. Fangal, Channing Division of Network Medicine, Brigham and Women’s Hospital, United States
Copyright © 2025 Zhou, Wang, Zhang, Mai and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liuliu Yang, eWxsMTAzMUAxMjYuY29t