 Milan Gerovac
Milan Gerovac Jörg Vogel
Jörg Vogel Alexandre Smirnov
Alexandre Smirnov- 1Institute of Molecular Infection Biology (IMIB), University of Würzburg, Würzburg, Germany
- 2Helmholtz Institute for RNA-based Infection Research (HIRI), Helmholtz Centre for Infection Research (HZI), Würzburg, Germany
- 3UMR 7156—Génétique Moléculaire, Génomique, Microbiologie (GMGM), University of Strasbourg, CNRS, Strasbourg, France
- 4University of Strasbourg Institute for Advanced Study (USIAS), Strasbourg, France
Macromolecular complexes of proteins and RNAs are essential building blocks of cells. These stable supramolecular particles can be viewed as minimal biochemical units whose structural organization, i.e., the way the RNA and the protein interact with each other, is directly linked to their biological function. Whether those are dynamic regulatory ribonucleoproteins (RNPs) or integrated molecular machines involved in gene expression, the comprehensive knowledge of these units is critical to our understanding of key molecular mechanisms and cell physiology phenomena. Such is the goal of diverse complexomic approaches and in particular of the recently developed gradient profiling by sequencing (Grad-seq). By separating cellular protein and RNA complexes on a density gradient and quantifying their distributions genome-wide by mass spectrometry and deep sequencing, Grad-seq charts global landscapes of native macromolecular assemblies. In this review, we propose a function-based ontology of stable RNPs and discuss how Grad-seq and related approaches transformed our perspective of bacterial and eukaryotic ribonucleoproteins by guiding the discovery of new RNA-binding proteins and unusual classes of noncoding RNAs. We highlight some methodological aspects and developments that permit to further boost the power of this technique and to look for exciting new biology in understudied and challenging biological models.
Introduction
When about two decades ago high-throughput approaches in molecular biology became a reality, it all began with parts lists. Genomics shed light on the ensemble of an organism’s genes and provided the first idea about what proteins and noncoding RNAs can in principle be there (Land et al., 2015; Encode Project Consortium et al., 2020). Transcriptomics identified and quantified various kinds of RNAs present in the cell under specific conditions (Wang et al., 2009; Lowe et al., 2017; Hör et al., 2018), and proteomics and metabolomics did the same for proteins and small molecules (Baran et al., 2009; Larance and Lamond, 2015; Omenn et al., 2016). But knowing the pieces does not yet mean playing chess. However essential such catalogs are, understanding what exactly every part does in the cell for a long time required one-by-one characterization, painstaking analysis of interactions with other parts, and rationalization of the ensuing biological effects.
Expectedly, next-generation high-throughput approaches, doing the same in a massively parallel way, soon emerged. Genome-wide functional screens were greatly facilitated by modern mutagenesis tools based on random transposon insertion and CRISPR-mediated gene disruption, silencing, or activation (Ford et al., 2019; Cain et al., 2020; Jiang et al., 2020). They now permit to simultaneously assess the importance of thousands of individual genes, including essential ones, under desired conditions (Langridge et al., 2009; Gilbert et al., 2014; Wang et al., 2014, 2015; Shalem et al., 2015; Peters et al., 2016). Orthogonally, high-throughput phenotyping enables the analysis of genotypes of interest against a wide palette of different conditions, providing insights into the cellular pathways the corresponding genes contribute to Nichols et al. (2011); Kritikos et al. (2017). The molecular mechanisms behind these phenotypes can be attained with interactomic approaches.
Over the last decade, many techniques have been developed to identify partners of specific RNAs and RNA-binding proteins (RBPs) with mass spectrometry-based proteomics and RNA-seq, respectively (Saliba et al., 2017; Giambruno et al., 2018; Lin and Miles, 2019). Among the latter, crosslinking approaches based on the covalent fixation of direct and often transitory RNA–protein associations, such as CLIP-seq, enjoy the widest popularity (Andresen and Holmqvist, 2018; Lee and Ule, 2018; Tree et al., 2018). The wealth of data obtained with these methods connected once-isolated proteins and RNAs into an intricate genome-wide web of interactions, delivering key information for understanding complex phenotypes (Quattrone and Dassi, 2019; Van Nostrand et al., 2020). At the same time, many questions regarding the biological meaning of these associations arose. Which of them take place simultaneously (e.g., on the same transcript) and which are mutually exclusive? Which binding events are just casual handshakes and which represent stable, persistent associations of macromolecules? Do these interactions occur in a simple, one-to-one manner or do they involve more elaborate, multi-subunit complexes? What is the actual diversity of such assemblies? Over the last years, the awareness increased that the virtual edges in RNA-protein interaction networks need to be converted into something more tangible and solid, endowed with clearly defined physical meaning. The time has come to compile the next-level parts list—that of ribonucleoprotein complexes (RNPs).
What Is a Stable RNP?
RNA–protein associations are commonly subdivided in transient and stable. They differ not only in physicochemical properties but also in biologically relevant modes of action. Transiently interacting macromolecules, epitomized by enzyme–substrate complexes, do the best in fugitive encounters: this keeps the enzyme available for further rounds of reaction while leaving the product to go its way (consider RNases or RNA modification factors as examples). In contrast, as we will see below, stable RNPs can only carry their functions out if the partnership between the RNA and the protein persists for a relatively long time. This long-lasting association is a foundation for quite a peculiar kind of molecular behavior which is more characteristic of biological, as opposed to purely chemical, systems. It creates prerequisites for such biologically important properties as structuration, information flow, state maintenance, and switching. Stable complexes are also naturally more amenable to study, and consequently, we know much more about this group of RNPs. The present review will essentially focus on stable RNPs and the methods to characterize them on a global scale. Transient RNA–protein associations, e.g., most enzyme–substrate complexes, temporary low-affinity interactions between RNA chaperones and their clients, and other weak binding events, fall beyond its scope.
What does it take for an RNP to be called stable? Any would-be stable complex must meet at least one of the following requirements: (i) it must have a relatively low dissociation constant, at least in the sub-micromolar range (thermodynamic stability) and/or (ii) its off-rate must be fairly slow, typically <10–4 s–1 (kinetic stability). We refer the reader to the following publications for examples of these parameters among some well-studied RNPs (Yang et al., 2013; Nithin et al., 2019; Licatalosi et al., 2020). It is also instrumental to consider two operational definitions of stability: (i) the “in vivo stability,” when the complex is long-lived within the dynamic cell milieu teeming with thousands of other molecules (which may in various ways influence its integrity), and (ii) the “in vitro stability,” when the complex only needs to be intrinsically robust in (effective) isolation from other cellular components (Helder et al., 2016; Gehring et al., 2017). This distinction is practically very important because it means that we can analyze not only genuinely strong complexes, such as housekeeping RNPs, but also many other, normally dynamic ones which happen to be kinetically trapped, e.g., by dilution into cold buffer upon cell lysis, preventing them from remodeling, disassembly, or degradation (Licatalosi et al., 2020).
Before discussing how this basic physicochemical principle can be converted into working methodology, we will provide an overview of diverse kinds of stable RNPs and explain why they deserve most thorough investigation. Without aspiring to a catch-all classification, we can sort the majority of known stable RNPs in four major classes, based on their biological properties and/or activities and the nature of the interaction between the RNA and the protein components (Figure 1). In fact, the distinction between these classes is sometimes blurred, and some complexes can be well assigned to more than one category. Therefore, it is more appropriate to speak about a continuum of stable RNPs, with four poles typified by relatively “pure” examples and many intermediate cases in between. The main purpose of this classification is to show what kind of stable RNPs fall in the scope of the existing global biochemical approaches, in particular, the complexomic methods described in the second part of this review. We will primarily use examples from bacterial RNA biology, but some particularly interesting cases from eukaryotes will be invoked as well to highlight the general bearing of the proposed ontology.

Figure 1. The world of stable RNPs. The wide diversity of stable RNPs existing in the cell can be presented as a continuum of assemblies with various biochemical and biological properties, roughly partitioned in four distinct classes. Constitutive RNPs (class I) are stable, permanent RNA–protein complexes, mostly with housekeeping roles. Class II comprises dynamic “on-pathway” intermediates, which can be artificially stabilized if deprived of the necessary building blocks. They are subdivided in the active complexes formed by translocating processive enzymes (subclass IIa) and the large RNP assembly intermediates (subclass IIb). Stable-state RNPs (class III) rely on strong facultative RNA–protein interactions where one of the partners influences the stability, the activity, or the localization of the other. In the subclass IIIa, RBPs regulate their RNA ligands by simple binding (often accompanied by the recruitment of other trans-acting factors) or by occlusion of other binding sites. In the subclass IIIb, on the contrary, RNAs regulate their protein partners, often by affecting their activity or localization. Unusually long-lived enzyme–substrate complexes, where the RNA substrate inhibits and traps the protein or RNP enzyme, are separated in the distinct subclass IIIc. Finally, the versatile RNPs, in which the RNA moiety anneals, with the help of its protein partner, to target nucleic acids, form the class IV. This group mainly includes regulatory RNPs but also several types of specialized housekeeping complexes. All classes are illustrated with examples whose structures have been solved (PDB codes: class I—1HQ1, 3Q1Q, 7C79, 3IYQ, 4V60, 7K00, 6D6V, 6G90, 6V4X, 6LQV, and 5GAN; class II—6RH3, 6YAM, 6WD0, 6TQO, 6TNN, 6ZTN, and 6DUQ; class III—6H58, 3ICQ, 5VT0, 4RMO, 2XDD, 2MF1, 2HW8, 1GTF, 1KOG, 2JPP, and 6O1M; class IV—2HVY, 4BY9, 5H9F, 4OO8, 6VRC, 5GUH, 6IFU, 4W5Q, and 4V2S). Proteins are shown in cyan, RNAs in gray, and their base-paired substrates or templates in red (except for RYPER and the 35–40S editosome, for which only low-resolution density maps are available: EMD-5389, EMD-1594; they do not permit to confidently distinguish the RNA and protein components).
Ontology of Stable RNPs
Class I: Constitutive RNPs
Constitutive RNPs are highly organized, permanent RNA-protein associations where one or both partners provide structural and functional support to each other. Much of our current knowledge on the molecular interplay between the RNA and the protein components of ribonucleoproteins comes from the research on constitutive RNPs (Martin and Reiter, 2017). This interplay is remarkably variable. In ribonucleoprotein RNases P, the RNA subunit performs pre-tRNA cleavage, whereas the associated protein(s) assist in its folding and substrate recognition (Guerrier-Takada et al., 1983; Reiter et al., 2010; Wan et al., 2019a; Lan et al., 2020). On the contrary, in the signal recognition particle, the RNA bears the burden of scaffolding and promotes the interaction with the SRP receptor, while a protein subunit works as the enzymatic moiety performing GTP hydrolysis (Peluso et al., 2000). In the ribosome, rRNA usually excels in both duties, serving as the skeleton and the catalytic heart of this giant complex, whereas proteins are relegated to secondary roles (Ban et al., 2000; Muth et al., 2000; Nissen et al., 2000; Routh and Sankaranarayanan, 2017). However, mitochondrial ribosomes often deviate from this rule: in the extreme case of trypanosomatids, the mitochondrial rRNA has suffered such profound structural erosion that the scaffolding task almost entirely falls to the hypertrophied protein shell (Ramrath et al., 2018; Soufari et al., 2020).
Behind all these variations, there is a common architectural theme followed by all constitutive RNPs: both RNA and protein make up a unique and inseparable whole and thereby contribute to the same, shared molecular function. The components of constitutive RNPs are normally bound in an obligate association with each other and only occasionally work outside this context (Pelava et al., 2016; Meyer, 2018; Nakagawa et al., 2018). They are fully hardwired pieces of the cell circuitry. The extreme degree of structural and functional integration earned the most sophisticated of them the qualification of “molecular machines,” and the current physical description of their workings supports this metaphor (Valle et al., 2003; Korostelev et al., 2008; Wilkinson et al., 2020).
As the above-cited examples show, most known constitutive RNPs are housekeeping. The molecular functions of others, like the imposing 13-MDa vault, remain elusive (Tanaka et al., 2009). But can constitutive ribonucleoproteins play regulatory roles? A potential step in this direction is Ro60-Y RNA complexes (Boccitto and Wolin, 2019). In Deinococcus radiodurans, the Ro60-like protein Rsr is tethered via Y RNA to the major bacterial exoribonuclease PNPase and seems to change its substrate specificity by promoting the degradation of structured RNAs (Chen et al., 2013; Sim and Wolin, 2018). This so-called RYPER complex is assembled in response to certain stresses, such as UV irradiation or prolonged stationary phase, and in Salmonella enterica, expression of Y-like RNAs, engaged in a similar kind of RNPs, appears to be confined to certain infection stages (Chen et al., 2013; Westermann et al., 2016), suggesting that they play condition-specific roles in RNA metabolism.
Even more striking cases of regulatory constitutive RNPs are found in the group of scaffolding lncRNAs (Chujo et al., 2016; Smith et al., 2020a). To cite just one telling example, eukaryotic paraspeckles are organized around an architectural lncRNA, NEAT1, stably associated with several RBPs, such as NONO, SFPQ, and FUS (Yamazaki et al., 2019). This core RNP is thought to represent a structurally heterogeneous latticework of RNA-RNA and RNA-protein interactions that nucleate the coalescence of the outer shell components, giving rise to a phase-separated condensate (Fox et al., 2018; Yamazaki et al., 2018). Paraspeckles have been linked with gene expression regulation at various levels, not least by localizing or sequestering specific proteins and transcripts (Chujo et al., 2016; Nakagawa et al., 2018). Interestingly, several families of large and ornately structured noncoding RNAs have been detected in bacteria (Weinberg et al., 2009, 2017; Harris and Breaker, 2018). One of them, OLE, is an abundant, stable transcript, forming membrane-associated RNPs in many Firmicutes to protect bacteria from cold and envelop stress (Puerta-Fernandez et al., 2006; Block et al., 2011; Wallace et al., 2012; Harris et al., 2019; Widner et al., 2020). However, the molecular mechanisms and the biological functions of this and other bacterial lncRNAs still need to be established.
Class II: Pseudo-Stable Intermediates
Subclass IIa: Processive Enzyme–Substrate Complexes
As already said, enzyme–substrate complexes are usually not stable. There are, however, a couple of interesting exceptions. Processive enzymes perform multiple rounds of catalysis without releasing the substrate into solution, like it happens with the elongating RNA polymerase (RNAP), the scanning eukaryotic translation initiation complex, the elongating ribosome, or the translocating transcription termination factor Rho (Hinnebusch, 2011; Gocheva et al., 2015; Kriner et al., 2016; Brito Querido et al., 2020). Of course, such complexes are “stable” only by first approximation, at the compositional level. In reality, they represent an ensemble of discrete short-lived elongation states. Below we will see how this ambivalence can be exploited to glean insight into the mechanics of key gene expression processes.
Translation of an average bacterial ORF, specifying a protein of ∼50 kDa, takes ∼25 s; and during this time, the mRNA and the ribosome are bound to stay together (Johnson et al., 2020). This does not seem particularly long, meaning that processive RNPs are not stable by “in vivo” standards. But this offers a time window generous enough for the experimenter to intervene and leverage their hidden “in vitro stability”. Indeed, many processive RNPs have evolved to firmly hold their template and/or the elongated substrate until a termination signal occurs and enables their dissociation. Yet, elongation depends on physiological temperatures and the regular supply of building blocks (nucleoside triphosphates for RNAP, aminoacyl-tRNAs for the ribosome). If one rapidly cools down the system and dilutes out the building blocks, the bereft processive RNP will stall dead-still and become analyzable. This principle underlies the ribosome profiling, which permits to follow the translational dynamics of all mRNAs in the cell simultaneously (Ingolia, 2016). The same physicochemical basis underlies the sequencing of nascent transcripts (NET-Seq and RNET-seq) and various versions of the transcription run-on assay to create a global snapshot of RNAP activity across the genome (Imashimizu et al., 2015; Jordán-Pla et al., 2019). In either case, the positions of RNA/DNA footprints or toeprints report on the location of the elongating processive RNP on the template.
As seen from these examples, pseudo-stable processive RNPs are deeply involved with core gene expression processes. A singular example of such complexes is the bacterial expressome that physically couples transcription and translation by joining an elongating RNAP and a ribosome via NusG and NusA proteins (O’Reilly et al., 2020; Wang et al., 2020a; Webster et al., 2020). Formation of the expressome and the associated effects on transcription elongation (e.g., antitermination and “pushing” of backtracked RNAP) depend on the relative speed of RNAP and the pioneering ribosome and take place only in those bacteria where the velocities of both molecular machines are properly matched (Burmann et al., 2010; Proshkin et al., 2010; Johnson et al., 2020; Stevenson-Jones et al., 2020).
The inherently variable elongation speed, open to modulation by sequence features and trans-acting factors, creates a fertile ground for regulation. Indeed, genome-wide studies revealed that both RNAP and ribosomes frequently pause, this pausing is often contingent on a specific molecular context or environmental conditions and may profoundly affect the cellular state (Chan et al., 2012; Shalgi et al., 2013; Subramaniam et al., 2014; Richter and Coller, 2015; Choi et al., 2018; Neugebauer, 2019; Yakhnin et al., 2020). The differential speed of RNA polymerase II can influence the pattern of cotranscriptional alternative splicing and thereby effect isoform switching (Muñoz et al., 2009; Ip et al., 2011). In mammalian mitochondria, regulation of RNAP processivity by the transcription elongation factor TEFM is of paramount importance, as it ultimately decides between the whole-genome transcription and the RNA-primed replication of the organellar DNA (Agaronyan et al., 2015; Hillen et al., 2017). Furthermore, the elongation of mitochondria-synthesized polypeptides is moderated to accurately match the incorporation of incoming nuclear-encoded subunits as respiratory complexes are being assembled (Couvillion et al., 2016; Richter-Dennerlein et al., 2016; Wang et al., 2020b).
Bacteria evolved elaborate regulatory strategies operating at the level of transcription elongation, including riboswitches, protein-dependent attenuators, Rho-mediated termination, and a variety of processive antitermination and pausing mechanisms that often involve NusG family proteins (Yakhnin et al., 2006, 2020; Said et al., 2017; Goodson and Winkler, 2018; Kang et al., 2018; Turnbough, 2019; Huang et al., 2020). Additionally, some Hfq-dependent sRNAs, traditionally perceived as posttranscriptional regulators, have been implicated in E. coli RNAP elongation by countering the Rho-dependent termination of the target mRNA (Sedlyarova et al., 2016). One of the most intricate examples of using processive RNPs in regulatory decision-making is leader peptide attenuators. These widespread switch elements elegantly subordinate the transcription of downstream genes to translation elongation on a short upstream ORF (Turnbough, 2019). The two processes are conditionally coupled, depending on the availability of the “sensed” nucleoside triphosphates (Turnbough et al., 1983; Bonekamp et al., 1984; Roland et al., 1988), aminoacyl-tRNAs (Artz and Broach, 1975; Lee and Yanofsky, 1977; Oxender et al., 1979; Johnston et al., 1980), or, in one described case, elongation factor P (Nam et al., 2016). The relative speed and eventual pausing of either machinery determine whether RNAP transcribes all the way down the operon or terminates prematurely (Turnbough, 2019).
Bacteria and eukaryotes also use purely translational regulatory mechanisms relying on the conditional slowdown—up to a complete arrest—of the elongating ribosome (Wilson et al., 2016; Choi et al., 2018). These include the prototypic secMA system, controlling the expression of the protein translocase SecA (Nakatogawa and Ito, 2001; Mitra et al., 2006), and some regulated antibiotic resistance cassettes (Arenz et al., 2014).
Subclass IIb: Assembly Intermediates
A behavior conceptually reminiscent of that of processive RNPs is found in the assembly pathways of complex molecular machines. The ribosome biogenesis proceeds via a series of defined intermediates and is pulled forward by the sequential recruitment of ribosomal proteins and assembly factors, which serve as building blocks (Shajani et al., 2011; Klinge and Woolford, 2019). Assembly intermediates are naturally short-lived in vivo, representing lowly populated states of nascent RNPs. However, some of them exist long enough or become sufficiently abundant (e.g., in exponentially growing bacteria) to enable their capture in the same way as described above, i.e., by snap cooling, dilution, and separation from other cellular components (Chen and Williamson, 2013). This is often achieved by pulling down the intermediate of interest via a unique marker protein or an RNA region (Gupta and Culver, 2014; Wu et al., 2016; Zhang et al., 2016; Barandun et al., 2017; Chaker-Margot et al., 2017; Chen et al., 2017b; Cheng et al., 2017, 2019, 2020; Heuer et al., 2017; Hunziker et al., 2019). But the most decisive—and impressive—implementation of this “in vitro stabilization” workaround has been attained in cryo-EM studies relying on advanced particle classification from heterogeneous samples. These works brought about unique data on the architecture of assembly intermediates and biogenesis factors for ribosomes and spliceosomes—and this without perturbing the natural assembly pathways (Sashital et al., 2014; Brown et al., 2017; Ameismeier et al., 2018; Wan et al., 2019b; Itoh et al., 2020; Jaskolowski et al., 2020; Soufari et al., 2020). Alternatively, intermediates can be enriched by “starving” the assembly for one of the building blocks. This approach is widely adopted as it permits to generate higher amounts of analyzable complexes, which is particularly instrumental in studying very short-lived ribosome and spliceosome assembly stages (Li et al., 2013; Jomaa et al., 2014; Davis et al., 2016; Ni et al., 2016; Sun et al., 2017; Zeng et al., 2018; Razi et al., 2019; Seffouh et al., 2019; Du et al., 2020; Rabuck-Gibbons et al., 2020; Rai et al., 2021). However, this comes at a risk of altering the native assembly route and accumulating off-pathway, dead-end particles.
Class III: Stable-State RNPs
Stable-state RNPs form the most numerous and actively studied RNP class. Like class I RNPs, they rely on strong RNA-protein interactions. Contrary to the former group, however, these interactions are not obligate and rather represent one of two (or sometimes more) distinct alternative states, each associated with a (dramatically) different outcome for the partners. Usually very long-lived in vitro, such complexes may be dynamic in vivo as other, competing molecules eventually drive one of the partners out and thereby switch the state of the system. Such properties open endless possibilities for gene expression control. Depending on which partner suffers (or enjoys) the consequences of such an interaction and whether it involves simple RNA binding or an enzymatic event, class III can be subdivided in “protein–controls–RNA” or “RNA–controls–protein”-type RNPs and inhibitory enzyme–substrate complexes.
Subclass IIIa: “Protein–Controls–RNA”-Type RNPs
This is likely the most widespread kind of stable-state RNPs: a protein binds a transcript and influences its stability, localization or, in the case of mRNAs, translation. In eukaryotes, most of such regulation converges at the level of the extensive 3′-UTR harboring recognition sites for multiple RBPs and miRNAs (Mayya and Duchaine, 2019). This enables combinatorial control of prodigious complexity where diverse trans-acting factors boost, mitigate, or override the effects of each other, thereby achieving highly nuanced outcomes (Iadevaia and Gerber, 2015; Dassi, 2017). Bacteria also set great store by regulating their mRNAs but do this usually via the 5′-UTR, near the translation start site, where most bacterial RBPs and sRNAs bind (Meyer, 2017; Holmqvist and Vogel, 2018b). The paradigm of such mechanisms is the broadly conserved protein CsrA that strongly binds to ANGGA motifs so frequent in Shine-Dalgarno sequences (Schubert et al., 2007; Holmqvist et al., 2016; Potts et al., 2017; Romeo and Babitzke, 2018). This preference makes CsrA family proteins potent translational repressors whose high-affinity binding can only be overcome by a dedicated protein antagonist, such as FliW or CesT, or a special group of “sponge” RNAs, which will be described in the next section (Mukherjee et al., 2011, 2016; Altegoer et al., 2016; Dugar et al., 2016; Katsowich et al., 2017; Sowa et al., 2017; Ye et al., 2018; Oshiro et al., 2019). In rare cases, CsrA upregulates the expression of its target through mRNA stabilization or translational activation (Yakhnin et al., 2013; Renda et al., 2020).
A beautiful example of translational feedback control is the Bacillus subtilis undecameric RBP TRAP that, under tryptophan-replete conditions, binds to the 5′-UTR of tryptophan-related mRNAs and prevents their translation (Merino et al., 1995; Du et al., 1997; Du and Babitzke, 1998; Babitzke, 2004; Yakhnin et al., 2004). Another widespread mechanism of translational repression is the autoregulation of ribosomal protein operons: some r-proteins interact with a specific region in the 5′-UTR of their own mRNA that mimics their native binding site within the ribosome, thereby inhibiting their own translation as well as the production of other r-proteins encoded on the same polycistronic message (Meyer, 2018). A similar molecular mimicry case is the autoregulation of the E. coli threonyl-tRNA synthetase, which recognizes a tRNAThr-like element in its own 5′-UTR (Romby et al., 1996; Torres-Larios et al., 2002). Interestingly, the RNA chaperone Hfq, more known as an sRNA cofactor (see Class IV), can itself serve as a translational inhibitor of certain mRNAs, including its own (Večerek et al., 2005; Desnoyers and Massé, 2012; Chen and Gottesman, 2017; Azam and Vanderpool, 2018; Morita and Aiba, 2019). In Pseudomonas, Hfq joins forces with another global regulator, Crc, to shut down the translation of numerous mRNAs (Moreno et al., 2015; Sonnleitner et al., 2018; Pei et al., 2019). Recent data indicate that RBP association with target mRNAs in bacteria may occur cotranscriptionally and, similar to eukaryotes, support combinatorial regulatory modes (Kambara et al., 2018; Gebhardt et al., 2020; Melamed et al., 2020).
The examples just quoted give class IIIa RNPs a uniquely regulatory flair. However, their housekeeping roles are not to be underestimated: in eukaryotes, such complexes punctuate the ages of the normal life cycle of many cellular transcripts (Müller-McNicoll and Neugebauer, 2013). Numerous RBPs govern the intracellular localization of transcripts—a feature present not only in morphologically complex eukaryotic but also in simpler bacterial cells (Nevo-Dinur et al., 2011; Moffitt et al., 2016; Eliscovich and Singer, 2017; Béthune et al., 2019; Mahbub et al., 2020). The cap-binding complex, poly(A)-binding protein, and the associated factors serve as tokens of stability and translatability of eukaryotic mRNAs (Müller-McNicoll and Neugebauer, 2013; Rissland, 2017). It is likely that a somewhat analogous role is played in bacteria by Hfq, which stably associates with the intrinsic terminators of both coding and noncoding RNAs, protecting them from 3′-to-5′ exoribonucleases (Sauer and Weichenrieder, 2011; Holmqvist et al., 2016).
In a few special cases, regulatory proteins form stable-state complexes with entire class I RNPs, like it happens with the ribosome-associated inhibitor RaiA, that stabilizes bacterial ribosomes in an inactive 70S form, or the dormancy factors HPF and RMF, which induce ribosome dimerization into inert 100S particles under stress, stationary phase, and sometimes even normal growth conditions (Kato et al., 2010; Polikanov et al., 2012; Matzov et al., 2017). Similar mechanisms have been described in eukaryotes (Ben-Shem et al., 2011; Brown et al., 2018; Barandun et al., 2019). There exist also examples of regulatory RBPs that inhibit ribosomes translating specific mRNAs (Darnell et al., 2011; Chen et al., 2014).
Subclass IIIb: “RNA–Controls–Protein”-Type RNPs
In this scenario, opposite to the previous one, the RNA rules over its protein partner. This rule is not necessarily a repressive one: for example, many lncRNAs simply recruit specific proteins to the sites where their activity is required (Kopp and Mendell, 2018). Nevertheless, it is more common to showcase “RNA–controls–protein” complexes with so-called sequestration mechanisms. In classical regulation by sequestration, the decoy RNA lures the cognate RBP into a tight but unproductive association, effectively debarring the protein from acting on its bona fide RNA targets. Although protein sequestration by RNA is found in eukaryotes too (Kino et al., 2010; Lee et al., 2016; Egloff et al., 2018), the finest examples thereof arguably come from bacterial RNA biology.
The E. coli small RNA decoy GlmY mimics its mate sRNA GlmZ in all respects but the presence of the base-pairing region employed by GlmZ to activate the translation of the glmS mRNA (Reichenbach et al., 2008; Urban and Vogel, 2008). GlmS makes glucosamine-6-phosphate (GlcN6P) required for the bacterial cell wall synthesis (Milewski, 2002). When GlcN6P is abundant, a regulatory RBP, RapZ, captures GlmZ and delivers it to RNase E for cleavage; this prevents unnecessary GlmS production (Göpel et al., 2013, 2016; Durica-Mitic et al., 2020). But when GlcN6P is scarce, GlmY jumps in and titers RapZ out by chaining it in stable—and completely inert—class IIIb RNPs (Gonzalez et al., 2017a; Khan et al., 2020). GlmZ thereby escapes degradation and activates glmS translation (Göpel et al., 2013).
RapZ is a very specific protein and its sequestration has only local regulatory consequences. How much more far-reaching could be the effect of detaining a globally acting RBP! This is exactly what happens to the Pseudomonas Hfq protein sponged by the abundant CrcZ sRNA (Sonnleitner and Bläsi, 2014; Pusic et al., 2016; Sonnleitner et al., 2017). The currently best-understood case of such control, from both functional and structural perspectives, is provided by the widespread noncoding RNAs that sequester members of the already mentioned CsrA/Rsm protein family (Babitzke and Romeo, 2007). These decoy sRNAs are basically a concatenation of numerous GGA motifs specifically recognized by CsrA-like proteins. Most of these potential binding sites are low (micromolar) affinity, when considered in isolation. But together they show high cooperativity, so that the initial fleeting and seemingly innocuous encounter with the target protein turns the decoy RNA into a molecular “black hole” avidly absorbing multiple CsrA dimers (Duss et al., 2014). As a result, CsrA is no longer available for repressing its mRNA targets, and an entire large regulon gets activated (Romeo and Babitzke, 2018).
Subclass IIIc: Inhibitory Enzyme–Substrate RNPs
We have seen above that enzyme–substrate complexes sometimes conceal remarkable in vitro stability, which can be accessed by relatively simple experimental means. In contrast to those class IIa RNPs, inhibitory enzyme–substrate complexes are long-lived in vivo. They arise as a special kind of stable-state RNPs formed in an unusual, indeed conspicuous, manner. An enzyme, which is fully active on its regular RNA substrates, eventually encounters an unusual one, which it cannot process normally, and both partners freeze in a stable complex. The simplest realization of this scenario is found in type III toxin–antitoxin systems used to abort phage infection or stabilize plasmids (Goeders et al., 2016). Here an endoribonuclease toxin is physiologically inactivated by the cognate pseudoknotted RNA antitoxin, with which it forms a highly stable, closed RNP (Blower et al., 2011; Samson et al., 2013; Short et al., 2013; Rao et al., 2015). This inhibitory complex forms via a processing reaction performed by the toxin on the antitoxin precursor: the enzyme inadvertently snares itself as it cleaves the “poisoned” substrate. This prevents other RNAs from accessing the toxin, rendering the latter perfectly harmless for the host, at least under normal circumstances (Short et al., 2013, 2018).
In another mechanism broadly conserved among bacteria, the housekeeping form of RNAP is fooled into binding to 6S RNA that structurally imitates a molten DNA duplex (Wassarman, 2018). The resulting complex is extremely stable and effectively sequesters RNAP from σ70-dependent promoters, tilting the balance in favor of alternative σ-factors, such as σS, insensitive to 6S RNA (Wassarman and Storz, 2000; Gildehaus et al., 2007; Chen et al., 2017a). In this stable state (typically observed when bacteria enter the stationary phase), transcription undergoes global remodeling (Cavanagh et al., 2008). However, when new resources permit to resume growth, the system must be reset. RNAP cannot simply “spit out” the 6S RNA and is forced to disentangle itself from the problematic substrate by using it as a template to synthesize a short product RNA (pRNA) (Wassarman and Saecker, 2006; Gildehaus et al., 2007). The conformational change operating during this weird transcriptional act forces the 6S RNA-pRNA duplex out (Wurm et al., 2010; Beckmann et al., 2012; Chen et al., 2017a). An analogous system has been reported in mammalian cells: the SINE-encoded B2 RNA competitively inhibits RNA polymerase II and, just like 6S RNA, exploits self-templated transcription as a release mechanism (Espinoza et al., 2004; Yakovchuk et al., 2009; Wagner et al., 2013).
Inhibitory enzyme–substrate complexes may form accidentally, e.g., when RNAP encounters a DNA damage site or a ribosome translates an aberrant mRNA. Their resolution is ensured by dedicated cellular mechanisms, including the Mfd-mediated disassembly of the transcription elongation complex (Selby and Sancar, 1993; Shi et al., 2020; Kang et al., 2021) and the ribosome rescue by trans-translation and no-stop/no-go-mediated pathways (Keiler, 2015; Simms et al., 2017).
Class IV: Matchmaking RNPs
Matchmaking RNPs can be counted among the most advanced evolutionary inventions in the domain of nucleic acid recognition. Their tremendous success in all three branches of life is largely due to their unique labor division scheme, radically different from what RBPs typically do (Liu et al., 2020). The latter recognize and act upon their targets with the help of elaborate binding sites that are intrinsically predisposed to accept more-or-less well-defined short RNA sequences embedded in a particular kind of 3D structure (Jankowsky and Harris, 2015). With some exceptions (Hall, 2016), the RNA recognition code used by RBPs is notoriously complex and not in the least universal. By contrast, within matchmaking RNPs, the task of target discrimination is entrusted to an RNA moiety that relies on a simple and universal set of base-pairing rules and can operate on longer sequences and not always in the best structural context. The protein subunit of such RNPs assumes the role of protector and presenter, ensuring that the RNA is displayed in a conformation optimal for rapid querying of potential targets; it also facilitates formation of a stable duplex, if the match is deemed satisfactory, and sometimes performs further molecular acts (Gorski et al., 2017).
Beside protein-based regulators, the class IV RNPs stand out as truly versatile RNA binders – and not only because of a more flexible target recognition strategy. Their key advantage is that they work as programmable devices where an RNA-“instruction” is plugged into the universal protein-“player” to chase and manipulate one specific kind of targets. The same “player,” however, can be reprogrammed with another RNA, which imparts a new specificity to hunt down different molecular preys. As a corollary, matchmaking RNPs, aptly dubbed “search engines” in a recent review (Dendooven and Luisi, 2017), can – via a huge number of possible guides – target practically any gene in the cell. Because such guiding RNA moieties are much easier to evolve than the equivalent number of target-specific RBPs, entire suites of new regulatory RNAs emerge around the same “player” protein, forming new functional classes within the noncoding transcriptome (Dutcher and Raghavan, 2018; Jose et al., 2019). The protein in its turn accedes to the status of “hub” and tremendously increases its regulatory reach and physiological importance (Vidal et al., 2011).
Because of the “target/programming” lingo, matchmaking RNPs are naturally associated with regulation. Indeed, in bacteria and eukaryotes special small RNA subtypes have been implicated in gene expression control. The widely conserved bacterial homohexameric Sm-like RNA chaperone Hfq interacts with over a hundred sRNAs, primarily engaging their intrinsic terminators (Holmqvist et al., 2016; Updegrove et al., 2016). This interaction is required for their stability and target recognition (Moll et al., 2003; Otaka et al., 2011; Ishikawa et al., 2012; Dimastrogiovanni et al., 2014). The latter is achieved with the help of the single-stranded base-pairing module, or seed sequence, typically situated on the 5′-end of sRNAs (Balbontin et al., 2010; Papenfort et al., 2010; Sauer et al., 2012; Fröhlich et al., 2013; Dimastrogiovanni et al., 2014). When the Hfq-sRNA complex encounters a true mRNA target, the protein catalyzes the duplex formation between the two transcripts (Santiago-Frangos and Woodson, 2018). Depending on the position of the duplex with respect to the translation initiation site, this RNA-RNA interaction may lead to translational inhibition, activation, or a change in the mRNA stability (Wagner and Romby, 2015). Although the sRNA-Hfq complexes are thermodynamically stable in vitro (with typical Kds in the low-nanomolar range), they show rapid exchange dynamics in vivo, where many different RNAs incessantly chase each other from the limited number Hfq hexamers (Wagner, 2013). At the systems level, this means that the population of Hfq-sRNPs is never the same and changes in function of the transcriptional profile which, in its turn, adapts to environmental conditions (Chao et al., 2012; Chihara et al., 2019). By this means, the Hfq-dependent regulon is constantly remodeled and stays flexible to provide rapid and adequate regulatory responses (Wagner and Romby, 2015). Very similar targeting mechanisms are employed in eukaryotes by Argonaute-associated small RNAs (Salomon et al., 2015; Sheu-Gruttadauria and MacRae, 2017).
Other types of matchmaking RNPs are involved in housekeeping processes. Probably the most ancient realization of the RNA-guided recognition principle can be found in the ribosome: bacterial 30S subunits typically find ORFs within “target” mRNAs with the help of the anti-Shine-Dalgarno sequence at the 3′-end of 16S rRNA (Shine and Dalgarno, 1974; Steitz and Jakes, 1975). Furthermore, the U1, U2, and U6 snRNPs (built around Sm-family protein oligomers akin to Hfq) exploit RNA-RNA base-pairing rules to locate the 5′-splice site and the branch point in pre-mRNAs (Papasaikas and Valcárcel, 2016). Other similar cases include U3 snoRNP, that chaperones critical pre-18S rRNA regions during SSU assembly (Barandun et al., 2018), and U7 snRNP (another Sm-ring complex), that guides the 3′-processing of histone mRNAs (Sun et al., 2020). However, in all these examples, guiding RNA moieties are stably integrated and cannot be exchanged to target different suites of transcripts. Much closer to the bespoke matchmakers described in the previous paragraph are snoRNPs and scaRNPs, that employ invariant protein scaffolds and different RNA guides to direct 2′-O-methylation or pseudouridylation at specific sites of archeal and eukaryotic rRNAs and snRNAs (Dupuis-Sandoval et al., 2015; Bohnsack and Sloan, 2018). Similarly, the RNA editosome edits several kinetoplast messengers in trypanosomatids using an ∼40-protein scaffold and ∼1,200 distinct guide RNAs (Göringer, 2012).
Special classes of matchmaking RNPs are employed as genome defense agents. For instance, siRNA-programmed AGO2 is used to combat viral infection in fungi, plants and invertebrates (Ding, 2010). PIWI-interacting RNAs repress transposons via a “ping-pong” cycle of base-pairing events in the germ line of most animals (Ozata et al., 2019). Finally, the large universe of CRISPR systems provides a number of fascinating mechanisms by which crRNA-guided endonucleases, either embedded in exuberantly complex RNPs or, on the contrary, stunningly simplistic, seek and destroy parasitic nucleic acids in prokaryotes (Mohanraju et al., 2016).
Why a High-Throughput Approach to Stable RNPs?
Our knowledge of this mesmerizing diversity and mechanisms of stable RNPs is the fruit of decades of research relying on diverse biochemical, structural, and molecular biology techniques. While many of these methods continue to play a critical role by laying a solid ground for our understanding of these tiniest biological units, the next-generation, high-throughput approaches are knocking ever louder on the door. The impetus for their development is not only down-to-earth pragmatic (i.e., massively parallel RNP characterization). A comprehensive, genome-wide approach actually becomes a necessity when big, transversal questions are raised. One such question is ontological: what stable RNPs exist in the studied biological system under given conditions? Here the profiling nature of a high-throughput technique by far outperforms the serendipity of the traditional “by-chance” discovery. Another advantage of a “bird’s-eye” view on the cellular RNP ensemble is the access to systems-level information: what is the functional state of the cell? How are resources allocated among various RNP-based processes, such as transcription, RNA processing, translation, or degradation? How are the ongoing regulatory programs implemented on the level of persistent RNA-protein associations? Regarding the interactions themselves, genome-wide techniques provide unique biochemical data on the “in vitro” (affinity distribution, specificity) and “in vivo” (occupancy, competition, interconversion) parameters of RNPs (Campbell and Wickens, 2015; Jankowsky and Harris, 2015). And if one wishes to recast all this in a form of defined, tangible physical entities, as advocated here, a complexomic approach will be an obvious option.
Profiling Cellular Complexes With Complexomic Techniques
The logic of the complexomic approach is radically different from that of the traditional bait-prey interactomics (Smirnov et al., 2017a). In complexomics, there are no baits nor preys, no need to tag or catch molecules. In fact, this group of methods does not directly profile interactions; what it looks at is macromolecular complexes, their composition, and physical properties. In a typical complexomic experiment (Figure 2A), biological material (cultured cells or a tissue) is lysed and resolved by one or several biochemical techniques that partition complexes according to a certain physicochemical parameter (size, shape, charge, hydrophobicity etc.). Upon fractionation, the content of each fraction is analyzed quantitatively, and the profiles of individual macromolecules across the whole separation range are reconstructed computationally. Comparison of their distributions (“correlation profiling”), often in conjunction with the physical information gleaned from the separation principle applied, permits to assign macromolecules to distinct complexes, evaluate the composition of known assemblies, and even propose new ones.
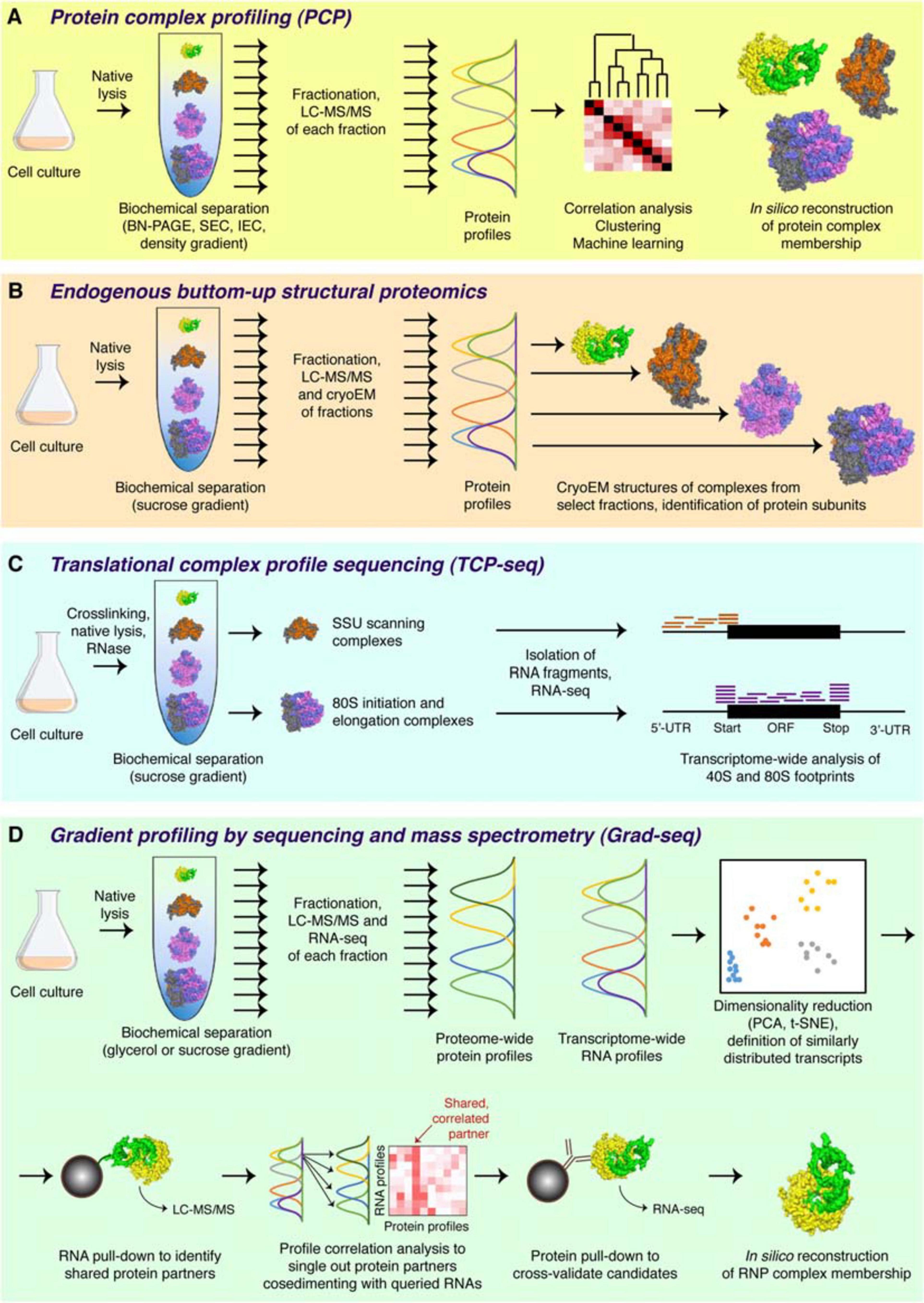
Figure 2. Major complexomic approaches. (A) In protein complex profiling (PCP), biological material is lysed under native conditions and resolved by one or several biochemical techniques (here velocity sedimentation in a density gradient). Each fraction is then analyzed by quantitative mass spectrometry, and protein profiles are cross-correlated to infer their involvement in the same or different complexes. (B) In endogenous bottom-up structural proteomics, the cell lysate is resolved on a sucrose gradient and profiled by mass spectrometry, like in PCP. In addition, individual fractions are subjected to cryoEM, followed by unsupervised, model-free 3D particle classification to determine structures of individual complexes and directly identify their protein constituents. (C) In the ribosome profiling family of approaches, represented here by TCP-seq, the lysate is chemically crosslinked, treated with an RNase to digest unprotected mRNA, and subjected to sucrose gradient centrifugation. Only the 40S and the 80S fractions are collected for subsequent analysis by RNA-seq to visualize the footprints of scanning 40S subunits or translating 80S ribosomes. (D) In Grad-seq, density gradient centrifugation is used to resolve RNPs up to the size of a monosome. Both the protein and the RNA components of each fraction are profiled by mass spectrometry and RNA-seq, respectively. The RNA profiles are clustered to identify cohorts of similarly behaving transcripts (likely forming the same kind of RNPs). Select members of each cohort are used as baits in pull-down assays to identify their protein partners. Then, the sedimentation profiles of the enriched proteins are correlated with those of the RNA baits, and the most consistent and recurrent candidates are cross-validated by RIP-seq or CLIP-seq. If the mutual interaction is confirmed, the RNP membership of the analyzed cohort can be considered established.
Each step of this standard pipeline can be played differently: the last two decades witnessed a flurry of studies employing a wide diversity of complexomic approaches tailored to various biological systems and research questions. For example, Blue-Native (BN) PAGE is traditionally used in microbial and mitochondrial complexomics as it permits to analyze large (up to 30 MDa), soluble or membrane-embedded protein complexes, which cannot be satisfactorily resolved by other techniques (Schägger and Von Jagow, 1991; Strecker et al., 2010). In earlier 2D implementations, BN-PAGE gels were stained, and individual protein spots were identified by western blotting or mass spectrometry (Camacho-Carvajal et al., 2004; Farhoud et al., 2005; Pyndiah et al., 2007; Klodmann et al., 2011). Such an approach was naturally limited in scope as it was restricted to abundant proteins (or those for which specific antibodies were available). Therefore, it has been supplanted by 1D BN-PAGE with regular gel slicing and systematic LC-MS/MS analysis of each slice (Wessels et al., 2009). This extremely successful method enabled unbiased profiling of hundreds of proteins across one gel, assignment of molecular weights to the complexes they form, and the possibility to compare such profiles proteome-wide to infer protein complex memberships in many species (Helbig et al., 2009; Heide et al., 2012; Schwenk et al., 2012; Wessels et al., 2013; de Almeida et al., 2016; Müller et al., 2016; Schimo et al., 2017; Senkler et al., 2017; Vidoni et al., 2017; Rugen et al., 2019; Páleníková et al., 2021b).
Other separation techniques, such as clear native PAGE, sucrose gradient centrifugation, and size exclusion chromatography, have been successfully used to partition complexes by size and shape (Andersen et al., 2003; Peltier et al., 2006; Hartman et al., 2007; Chen and Williamson, 2013; Kirkwood et al., 2013; Larance et al., 2016; Páleníková et al., 2021a). However, the majority of complexomic studies now rely on a combination of orthogonal fractionation methods, thus decreasing the probability of chance co-elution for proteins involved in closely migrating but otherwise unrelated assemblies. Such fractionation schemes can be quite complex, include both parallel and sequential steps, and yield hundreds-to-thousands of fractions (Dong et al., 2008; Menon et al., 2009; Havugimana et al., 2012; Gordon et al., 2013; Wan et al., 2015; Gazestani et al., 2016; Shatsky et al., 2016). As a result, researchers have access to a wider variety of physical information about each complex and, based on richer complexome profiling datasets, can predict with higher confidence the involvement of each protein in macromolecular assemblies.
In one stunning methodological development, the sucrose gradient fractionation of the cell lysate is coupled to both mass spectrometry and cryo-electron microscopy—to directly classify and visualize complexes present in select fractions, solve their structures, and thereby identify the constituent proteins (Figure 2B). Pioneered by studies of bacterial ribosome assembly (Sashital et al., 2014; Davis et al., 2016), the technique has been recently extended to cover virtually any macromolecular complex in the cell (Ho et al., 2020). With this bottom-up endogenous structural proteomics approach, complexomics achieves its most visual expression and promises a wealth of exciting data in the years to come.
Because complexomic techniques do not require genetic manipulation of the biological system and provide a large amount of data on the physical organization of the proteome, they have been used to map complexomes not only of model but also of some traditionally “difficult” organisms, including numerous bacteria, archaea, protists, algae, plants, animals, and human subjects (Peltier et al., 2006; Kristensen et al., 2012; Chatzispyrou et al., 2017; Moutaoufik et al., 2019; Van Strien et al., 2019 and the references above). The biological insights brought about by these studies are impressive: from the organization of giant respiratory supercomplexes, through the fine structural coupling of metabolic pathways, to unexpected properties and compositions of gene expression machines, to new hints at possible functions of “orphan” proteins and molecular mechanisms of human diseases. From this “protein-only” complexomic perspective only few steps were remaining to the vast RNP world (Figure 3).
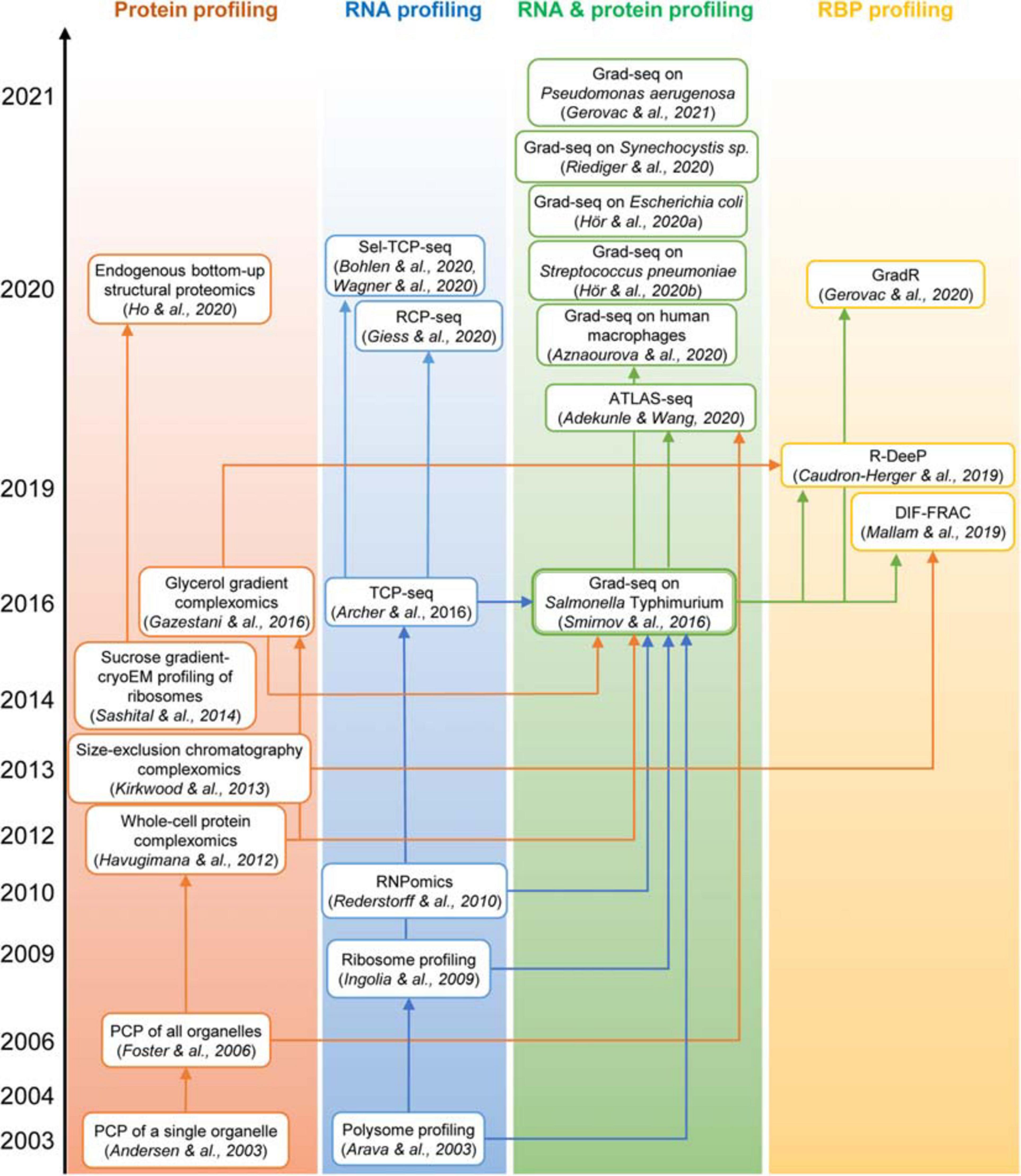
Figure 3. The “family tree” of principal complexomic approaches and the conceptual links between them. High-throughput techniques initially interrogating only the protein or only the RNA dimension of stable macromolecular complexes (the first two columns) inspired the development of the Grad-seq family of methods that profile both components simultaneously to infer stable RNA–protein associations (the third column). Adaptations of the latter technique have recently enabled the systematic identification of proteins involved in stable RNPs, be they genuine RBPs or indirectly associated subunits (the last column).
Incorporating RNA in the Complexome Landscape
Though largely focused on proteins, some complexomic studies had already reserved a place of honor to major RNPs, most often ribosomes (Wessels et al., 2013; Gazestani et al., 2016; Larance et al., 2016; Chatzispyrou et al., 2017; Van Strien et al., 2019; Páleníková et al., 2021a). These works resulted in several interesting findings, including details of bacterial and mitochondrial ribosome assembly (Chen and Williamson, 2013; Sashital et al., 2014; Davis et al., 2016; Rugen et al., 2019) and some of the first observations of CRISPR ribonucleoproteins (Menon et al., 2009). However, because the RNA composition of fractions was not explicitly interrogated, the interpretation of a wide diversity of observed RBP distributions, e.g., those of mitochondrial PPR proteins (Wessels et al., 2013; Senkler et al., 2017; Rugen et al., 2019), numerous RNA-related complexes in chloroplasts (Peltier et al., 2006), or a potential RNA chaperone in archaea (Menon et al., 2009), was precluded.
First attempts to visualize the RNA component of RNPs relied on low-throughput northern blotting (e.g., Wassarman and Storz, 2000). The polysome and ribosome profiling, later joined by the translation complex profile sequencing (TCP-seq), the selective TCP-seq (Sel-TCP-seq) and the ribosome complex profiling (RCP-seq) (Figure 2C), marked a decisive turn in handling the RNA dimension by introducing the unbiased and comprehensive quantification of mRNAs isolated from defined sucrose gradient fractions by microarray and later RNA-seq (Arava et al., 2003; Ingolia et al., 2009; Archer et al., 2016; Bohlen et al., 2020; Giess et al., 2020; Wagner et al., 2020). Such approaches have revolutionized our view of dynamic translation but are naturally limited to specific types of class IIa (elongating or scanning ribosomes), class IIIc (stalled ribosomes), and sometimes lncRNA-ribosome complexes (van Heesch et al., 2014; Carlevaro-Fita et al., 2016). Another transcriptome-wide technique, RNPomics, that similarly resolves cellular complexes in a gradient, sequences the constituent RNAs, and focuses instead on the sedimentation range 10S–30S, thereby excluding ribosome-bound transcripts (Rederstorff et al., 2010). This procedure enriches well for RBP-bound noncoding RNAs, but because all RNP-containing fractions are ultimately pulled and analyzed together, it does not really profile them and thus cannot provide further information about the diversity and the composition of these RNPs. Such a feat has become a reality only with the advent of gradient profiling by sequencing and mass spectrometry (Grad-seq).
Profiling Stable RNPs With Grad-Seq
General Pipeline
Grad-seq was introduced in 2016 as a new technique to comprehensively chart the landscape of an organism’s stable RNA and protein complexes without resorting to tagging or enrichment of cellular components (Smirnov et al., 2016, 2017a). This hybrid method marries the logic of a traditional complexomic approach with the power of RNA-seq to quantitatively profile transcripts of all expressed genes (Figure 3). The Grad-seq pipeline (Figure 2D) begins with the native lysis of biological material (bacterial or human cell culture) at low temperature to stabilize dynamic complexes. The cleared lysate is then loaded onto a linear density gradient and subjected to velocity centrifugation to resolve cellular complexes across the size range of interest (typically up to monosomes). Upon centrifugation, the gradient is split in a series of equal-volume fractions, and their RNA and protein components are isolated. The distributions of selected macromolecules across the gradient can be analyzed with conventional methods, i.e., gel electrophoresis, staining, western and northern blotting. To profile proteins and transcripts genome-wide, each fraction is subjected to label-free LC-MS/MS and RNA-seq. The obtained data are normalized with the help of added spike-in molecules and the bona fide in-gradient distributions of individual macromolecules revealed with conventional methods. The result is a complete collection of sedimentation profiles of potentially all expressed proteins and RNAs present in the studied biological model.
Peaks in each distribution indicate the stable assemblies of profiled molecules. Calculation of the sedimentation coefficient for each fraction, based on the behavior of known complexes or standards, enables access to molecular weight estimates for any detected complex (Erickson, 2009). With this “sedimentation ruler” one can make reasonable assumptions regarding the complexity of each observed particle, e.g., evidence for oligomerization or the degree of heterogeneity. Besides this physical information, one can now compare profiles between them, and this is where the genuine power of the approach reveals itself. Like classical complexomics, Grad-seq permits to evaluate and either confirm or reject proposed stable associations between proteins and RNAs, based on the similarity or dissimilarity of their sedimentation profiles. When the profiles are compared with each other globally, e.g., with the help of such analytical tools as PCA and t-SNE, one can go even further and forward hypotheses about novel complexes between similarly distributed proteins and transcripts (Smirnov et al., 2016; Hör et al., 2020a,b).
Additionally, if uncharacterized transcripts or proteins co-segregate with other components, whose molecular role has been determined (e.g., RNAP, ribosomal subunits, RNA chaperones, or other established RBPs), one can leverage guilt-by-association and predict their involvement in a similar kind of processes or assemblies. Such hypotheses can be verified with the help of accessory techniques, such as RNA and protein pull-downs, to robustly establish the composition of the observed RNPs (Figure 2D). Therefore, Grad-seq bridges the biochemical profiling of cellular complexes with functional information, which makes it a versatile functional genomics approach.
Case Study: Comparative Analysis of Bacterial Stable RNPs
To date, Grad-seq has been used to profile stable RNPs in several model bacteria, such as the γ-proteobacteria S. Typhimurium (Smirnov et al., 2016; Gerovac et al., 2020; Venturini et al., 2020), Escherichia coli (Hör et al., 2020a), and Pseudomonas aeruginosa (Gerovac et al., 2021), the firmicute Streptococcus pneumoniae (Hör et al., 2020b), the cyanobacterium Synechocystis sp. (Riediger et al., 2020), and even in human cells (Aznaourova et al., 2020). Additionally, the RNPs formed by P. aeruginosa RNAs have been analyzed along with phage ΦKZ transcripts during viral infection in what can be considered the first “dual Grad-seq” experiment (Gerovac et al., 2021). These known biological models provided the first touchstone and an essential benchmark for this new technique. Figures 4–6 show back-to-back select examples of stable RNPs detected in the three proteobacteria and the evolutionarily distant S. pneumoniae. Let us consider them in further detail to see what kind of functional insights can be gleaned from such analysis.
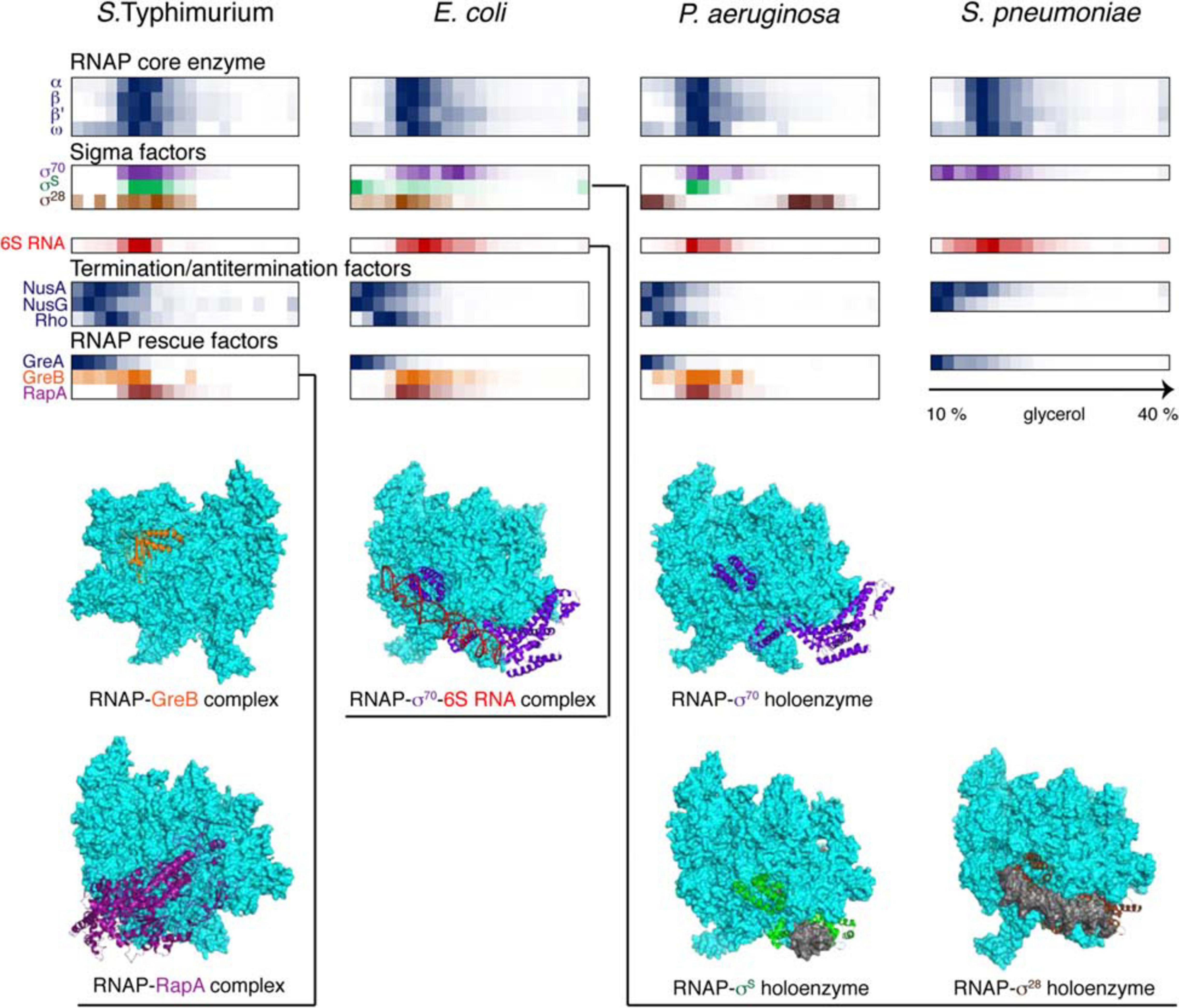
Figure 4. Back-to-back view of the transcriptional machinery from S. Typhimurium, E. coli, P. aeruginosa, and S. pneumoniae profiled by Grad-seq. The Grad-seq distributions of select proteins and RNAs, forming the RNAP holoenzyme or involved in transcription regulation and rescue, are shown as heat-maps reflecting the abundance of each macromolecule along the gradient. Some of the profiles highlighted with color are illustrated with the corresponding structures below (PDB codes: 6RIN, 4S20, 6P1K, 5IPL, 6PMI, and 5VT0). Note that S. pneumoniae, in contrast to the proteobacteria, has only one Gre factor.
The core transcriptional machinery (Figure 4) is well assembled in all examined species. RNAP is abundantly present as the holoenzyme, as can be judged by the cosedimentation of σ-factors with the core subunits in the 15-18S range, corresponding to transcription initiation or early elongation events. However, some elongation accidents have obviously taken place in the proteobacterial species, as can be attested by the recruitment of the elongation factor GreB, stimulating RNA cleavage and reactivation of deeply backtracked RNAP (Abdelkareem et al., 2019), and the ATPase RapA, forcing the translocation of stalled RNAP (Liu et al., 2015). Interestingly, GreA, specialized on very small backtracks usually caused by nucleotide misincorporation, is not recruited, highlighting the division of labor between the two paralogues present in enterobacteria (Borukhov et al., 1993). Similarly, the termination factors NusA, NusG, and Rho are at best only marginally associated with RNAP; indeed, Rho shows a peak of ∼10S, which agrees well with the 10.4S reported for a free hexamer (Geiselmann et al., 1992).
The translational machinery (Figure 5) features well-defined 30 and 50S subunits recognizable by their rRNA and r-protein markers. At the bottom of the gradient, 70S ribosomes engaged in translation are prominently represented; this is where most mRNAs naturally peak. Interestingly, endogenous P. aeruginosa mRNAs were observed mostly in low-molecular-weight complexes under these conditions. By contrast, during phage infection, viral messengers accumulated in ribosome-rich fractions, suggesting that they were effectively overtaking the translational apparatus of the cell (Gerovac et al., 2021).
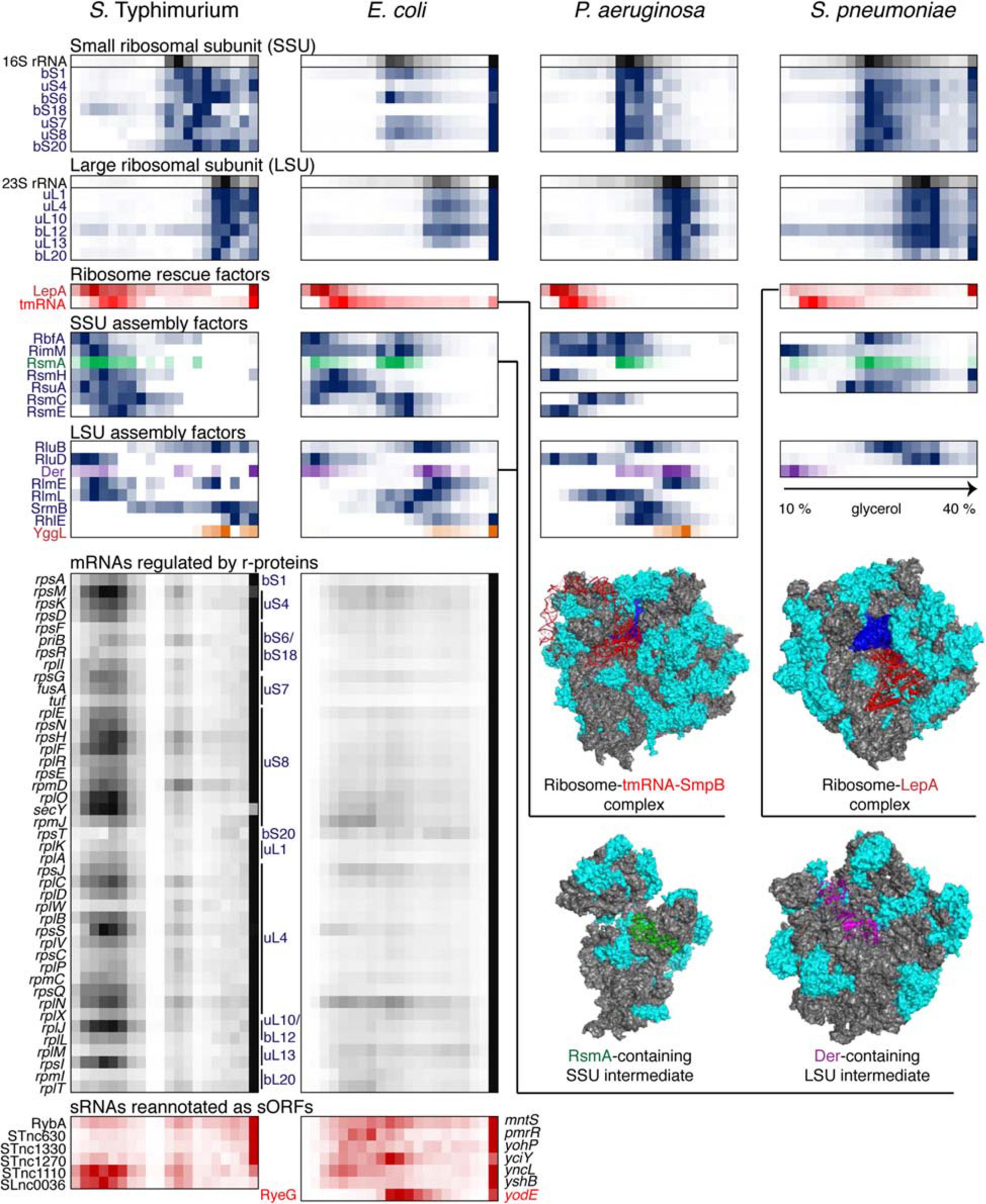
Figure 5. Back-to-back view of the translational machinery and the associated RNAs from S. Typhimurium, E. coli, P. aeruginosa, and S. pneumoniae profiled by Grad-seq. The Grad-seq distributions of select proteins and RNAs involved in the ribosome structure, assembly of rescue, are depicted in the same way as in Figure 4, and some of the complexes are illustrated with solved cryo-EM structures on the right (PDB codes: 6Q95, 5J8B, 4ADV, and 3J8G). Additionally, the Grad-seq profiles of autoregulated r-protein-encoding mRNAs are provided below to showcase their involvement in low molecular weight RNPs (left side of the profile) or association with translating ribosomes (last fraction). The r-proteins negatively regulating each cistron are specified on the right of the profiles. Certain enterobacterial sRNAs, shown in red at the bottom of the figure, have been re-annotated as short mRNAs due to the presence of a confirmed ORF and association with translating ribosomes. Their old sRNA identifiers are given on the left of each profile and the new approved protein-coding gene names are provided on the right.
Like transcription, translation elongation sometimes seems to go awry. In S. pneumoniae, the LepA GTPase, also known as elongation factor 4, is recruited to the ribosomes, suggesting inefficient translocation (Heller et al., 2017). In E. coli, the presence in the same fraction of tmRNA (in addition to the free tmRNA-SmpB complex sedimenting at 11–12S) betrays the accumulation of no-stop or no-go translation elongation complexes awaiting resolution (Keiler, 2015). In S. Typhimurium, both these aberrant states are clearly present. Similar to earlier observations (Chen and Williamson, 2013), several ribosome assembly intermediates appear to be abundant and long-lived enough to permit their detection by Grad-seq. These include the broadly conserved RsmA/KsgA and RbfA stages of 30S biogenesis, readily observable in all four bacteria (Datta et al., 2007; Boehringer et al., 2012). Association of other assembly factors with immature ribosomal subunits shows a great degree of species- and likely condition-specificity.
In this regard, it is appealing to compare in more detail the two enterobacterial datasets, given that S. Typhimurium and E. coli are genetically extremely close, and both were harvested during the transition between the exponential and the stationary phases of growth (Smirnov et al., 2016; Hör et al., 2020a). In both species, 6S RNA is already beginning to engage the housekeeping (σ70) form of RNAP, and the recruitment of σ28 marks the commitment of both cultures to foster motility at the wake of the exponential phase (Barembruch and Hengge, 2007). However, the E. coli σS, unlike its Salmonella counterpart, does not yet have a privileged access to the transcriptional machinery (Figure 4). Moreover, most ribosomal proteins and mRNAs in E. coli sharply peak at the bottom of the gradient, indicating active translation (Figure 5). Considerably higher amounts of ribosome assembly intermediates are visible in E. coli, as compared to the S. Typhimurium culture, suggesting actively ongoing ribosome biogenesis (Chen and Williamson, 2013). In S. Typhimurium, but not E. coli, r-protein-encoding mRNAs appear to be significantly involved in low-molecular-weight complexes reminiscent of the already mentioned autoregulatory class IIIa RNPs (Meyer, 2018). The corresponding r-proteins are also more abundant in extraribosomal fractions in S. Typhimurium, suggesting that they may indeed engage in inhibitory complexes with their own messengers, as would be expected when the ribosome production is curtailed (Figure 5). Altogether, these observations suggest that, at the moment of harvest, the E. coli population was physiologically considerably more exponential than the S. Typhimurium one, which had already begun to show some stationary-phase hallmarks. This little exercise illustrates how analysis of stable complexes by Grad-seq can inform on the functional state of the studied system.
This state is to a large extent governed by class III and IV RNPs formed by key posttranscriptional regulators CsrA and Hfq which had their fair share in the three proteobacterial gradients (Figure 6). CsrA homologues are expectedly involved in class IIIb complexes with their sRNA decoys (Babitzke and Romeo, 2007). However, in S. Typhimurium and P. aeruginosa, a significant proportion of CsrA/Rsm proteins appears to have reached some of their mRNA targets, such as flhDC, glgC and nhaR in enterobacteria (Baker et al., 2002; Pannuri et al., 2012; Yakhnin et al., 2013) and pslA, PA0081, and PA4492 in P. aeruginosa (Brencic and Lory, 2009; Irie et al., 2010). The majority of Hfq-dependent sRNAs in S. Typhimurium form complexes of ∼350 kDa, and even larger assemblies are visible in P. aeruginosa. Given the ∼67 kDa of one Hfq hexamer plus an average of ∼40 kDa sRNA, this suggests the existence in vivo of rather more complex RNPs than traditionally believed (Dimastrogiovanni et al., 2014; Obregon et al., 2015; Bandyra et al., 2016; Caillet et al., 2019). Interestingly, several Salmonella Hfq-dependent sRNAs, including ArcZ, DsrA, and RprA, co-migrate with RNAP, which is reminiscent of their antitermination activity during transcription of the σS-encoding rpoS mRNA as cells enter the stationary phase (Sedlyarova et al., 2016). Remarkably, only part of Hfq-dependent sRNAs cosediment with the low-molecular-weight pool of Hfq in E. coli, whereas the majority are found in the pellet associated with translating ribosomes (Hör et al., 2020a). It appears that in this case most Hfq-dependent sRNAs follow their mRNA targets, while the free Hfq pool is usurped by a few high-affinity binders that are less inclined to interact with actively translated messengers.
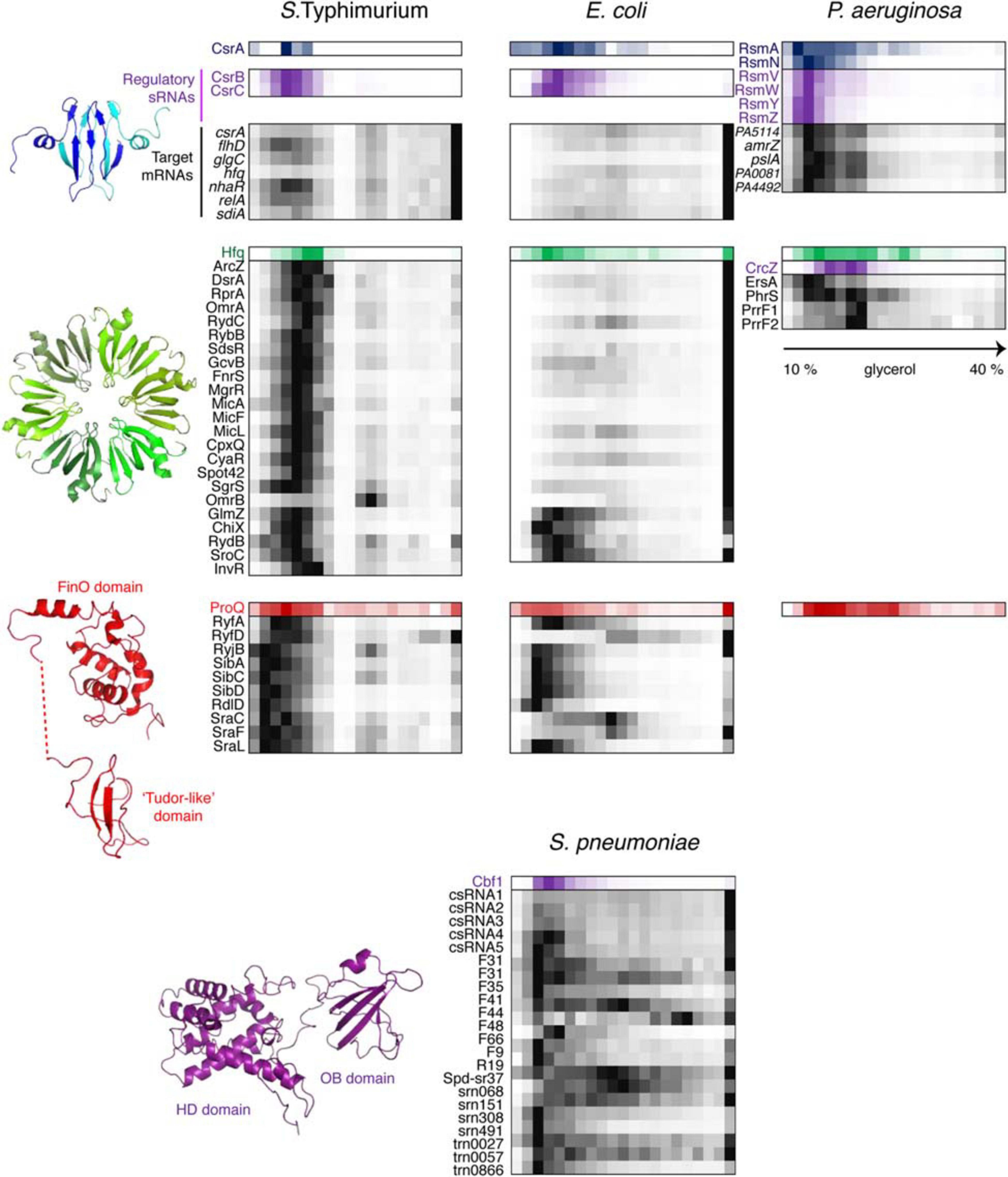
Figure 6. Back-to-back view of global regulatory RBPs and the associated RNAs from S. Typhimurium, E. coli, P. aeruginosa, and S. pneumoniae profiled by Grad-seq. The Grad-seq distributions of three major proteobacterial RBPs, CsrA, Hfq, and ProQ, and the S. pneumoniae exoribonuclease Cbf1 are shown along with select profiles of their sRNA and mRNA ligands (except for the P. aeruginosa ProQ, for which in vivo ligands have not been studied as for now). Decoy sRNAs in proteobacteria are highlighted with violet. The CsrA, Hfq, and ProQ structures—PDB codes: 1Y00, 2YLB, 5NB9, and 5NBB. The 3D structure of Cbf1 is predicted by RaptorX (Xu, 2019). The P. aeruginosa ProQ protein lacks the “Tudor-like” domain (Gerovac et al., 2021).
The quick look at what Grad-seq permitted to reveal about known RNA-protein complexes inspired confidence in its ability to go beyond this canonical purview and interrogate new biology that had eluded the attention of researchers. Unexpected discoveries were not slow to appear.
New RNPs Discovered by Grad-Seq
Class IIa: Translated Small ORFs in Presumed sRNAs
A common problem in the automatic de novo annotation of protein-coding genes is the default cut-off of 100 amino acids below which the genome sequence is typically not queried to avoid false positives (Harrison et al., 2002). This means that, in the absence of additional experimental evidence, key proteins such as CsrA (61 aa), RpoZ (91 aa), and many ribosomal proteins would have been simply missed—rather a disconcerting prospect. Molding the annotation from a related species by homology search improves the situation but does not sort the issue entirely out, since preexisting knowledge is still required, and certain ORFs—albeit functional—are too short and insufficiently conserved to be robustly detected (Storz et al., 2014). Therefore, direct experimental evidence of the polypeptide production is necessary. This can be achieved either by the identification of the molecule in question (by MS or western blotting) or by demonstrating the involvement of the corresponding mRNA in translation (Makarewich and Olson, 2017). The former approach has been recently used to support the existence of 170 small proteins, including 89 uncharacterized ones, in a S. Typhimurium Grad-seq dataset (Venturini et al., 2020). Their detection has become possible thanks to a higher sensitivity offered by Grad-seq protein profiling which, by fractionating the cell lysate, considerably decreases the complexity of analyzed samples. Moreover, 82 of the uncharacterized small proteins were found to form complexes with other molecules, providing first evidence of their functionality.
The first Grad-seq analysis unveiled the fundamental biochemical dichotomy between coding and noncoding RNAs in Salmonella: the former expectedly tended to cosediment with 30S and 70S ribosomes whereas the latter preferred small RNPs (Smirnov et al., 2016). The flagrant association of a few sRNAs with ribosomal particles urged to reconsider their coding potential and, with support of complementary experimental and comparative genomic evidence, re-annotate them as small mRNAs (Figure 5) (Hemm et al., 2008, 2010; Waters et al., 2011; Kato et al., 2012; Smirnov et al., 2016; Hör et al., 2020a). For example, the E. coli Grad-seq dataset exposed the interesting case of the prophage-encoded sRNA RyeG strongly associated with 30S and 70S ribosomes (Bak et al., 2015; Hör et al., 2020a). The existence within this transcript of a short yodE ORF, encoding a 48-aa bacteriostatic peptide, has been corroborated in vitro by 30S toeprinting and in vivo by modified ribosome profiling and functional assays, indicating that RyeG is in fact a minute mRNA (Weaver et al., 2019; Hör et al., 2020a).
Class IIb: New Ribosome Assembly Factors
Earlier studies showed that many proteins involved in ribosome biogenesis stably associate with free 30S and 50S subunits during gradient centrifugation, which can be exploited to predict new ribosome assembly factors (Chen and Williamson, 2013). The bacterial Grad-seq datasets revealed a few proteins cosedimenting with either subunit, including several uncharacterized ones (Smirnov et al., 2016; Hör et al., 2020a,b; Gerovac et al., 2021). One of them, the 12-kDa DUF469 protein YggL found in many γ- and β-proteobacteria, showed pronounced association with 50S and 70S particles (Figure 5). Given that yggL is mostly expressed in the exponential phase, and its deletion has a strong effect on the 50S levels, the protein likely functions in late LSU assembly (Chen and Williamson, 2013; Smirnov et al., 2016; Hör et al., 2020a). This specific example provides a modus operandi to study other candidate ribosome biogenesis factors spotted by Grad-seq, especially in less studied species (Hör et al., 2020b; Riediger et al., 2020).
Class IIIb: A Scaffolding lncRNA to Organize the Immune Response to Bacterial Infection
Grad-seq has been recently adapted to eukaryotic cells to study lncRNPs produced by immune cells in response to bacterial infection (Aznaourova et al., 2020). Human macrophages, activated by Salmonella lipopolysaccharide, were lysed and resolved on a glycerol gradient to comprehensively profile subribosomal RNPs. Focusing on the transcripts upregulated by infection-relevant stimuli, the authors noticed that the uncharacterized cytoplasmic lncRNA MaIL1 cosedimented with components of the ubiquitin-proteasome system. Its main partner, identified by RNA affinity purification and confirmed by Grad-seq profiling, is the ubiquitin-adapter optineurin (OPTN), which platforms the TBK1 kinase within the TLR4-TRIF pathway to induce type I interferon expression (Gleason et al., 2011; Munitic et al., 2013). MaIL1 is strictly required for the stability and the ubiquitin-dependent aggregation of OPTN. The emerging model posits that MaIL1 scaffolds the assembly of the ubiquitin-associated OPTN platform to enable type I interferon production and thereby activate antibacterial defense mechanisms (Aznaourova et al., 2020). A large repertoire of distinct lncRNPs revealed by Grad-seq in this study promises further surprises in this dynamically evolving research area (Walther and Schulte, 2020).
Class IIIc: A Trapped RNase to Protect sRNAs?
While sRNAs are omnipresent in bacteria, their associated proteins have much patchier phylogenetic distributions, and ample evidence indicates that, for instance, Hfq, important as it is in proteobacteria, does not play as pervasive a role in other bacterial clades (Zheng et al., 2016; Santiago-Frangos et al., 2017). Defining the organizational principles of regulatory sRNPs in Gram-positive bacteria has become a big challenge in microbial RNA biology over the last years. Consequently, performing Grad-seq in the model Firmicute Streptococcus pneumoniae, lacking all presently known global sRNA-binding RBPs, promised to be an exciting endeavor (Hör et al., 2020b). Gratifyingly, the 141 currently annotated pneumococcal sRNAs showed massive involvement in small RNPs of varying complexity. Some of them showed remarkably similar sedimentation patterns, suggesting shared RNP architecture (Figure 6). Capturing their protein partners from the cell lysate identified several candidate RBPs. One of them, the conserved 3′–5′ exoribonuclease Cbf1/YhaM (Oussenko et al., 2002; Lécrivain et al., 2018), strongly interacted with the five csRNAs, involved in competence control (Schnorpfeil et al., 2013; Laux et al., 2015), but not with differently distributed control transcripts. UV CLIP-seq corroborated this finding and revealed a number of additional sRNA and mRNA ligands, indicating that Cbf1 is in fact a global RNA binder. Importantly, the in-gradient distributions of many Cbf1-associated sRNAs turned out to be highly correlated with the Cbf1 profile, strongly supporting their involvement in the same stable sRNPs (Figure 6). The deletion of this unusual RNase, composed of an HD-domain (Histidine/Aspartate-containing metal-dependent phosphohydrolase) and an Oligonucleotide-Binding (OB)-fold, resulted in csRNA destabilization, supporting its protective role, similar to Hfq in Gram-negative bacteria (Oussenko et al., 2002; Hör et al., 2020b).
How can an RNase stably associate with—and even protect—RNA instead of destroying it? Cbf1 crosslinks tend to occur on the 3′-ends of transcripts, where this exoribonuclease normally cleaves (Hör et al., 2020b). However, the role of Cbf1 in cellular RNA turnover is at best modest (Oussenko et al., 2005; Lécrivain et al., 2018). In fact, in vitro assays and recent in vivo data revealed that Cbf1 trims the single-stranded 3′-oligo(U)-tail of its ligands by ∼3 nucleotides but fails to digest any further (Lécrivain et al., 2018; Hör et al., 2020b). The enzyme does not seem to be halted by a stable secondary structure, as no such feature has been observed around trimmed sites in vivo (Lécrivain et al., 2018), nor impeded by another RBP sitting on the 3′-end of the target RNA, since the same behavior has been recapitulated in a minimal system on protein-free RNA (Hör et al., 2020b). This lack of processivity may be an intrinsic feature of Cbf1 itself, which is reminiscent of the behavior of another HD-domain protein acting on the RNA 3′-end, the CCA-adding enzyme (Cca). Unlike template-dependent polymerases, Cca stably anchors to its tRNA substrate and does not translocate upon addition of nucleotides, which makes the catalyzed reaction self-limiting—it necessarily stops after addition of the 3-nucleotide CCA-tail (Shi et al., 1998; Cho et al., 2006; Kuhn et al., 2015). Whatever the exact mechanics of the Cbf1 cleavage is, current biochemical and functional evidence converges on a model where this exoribonuclease forms class IIIc RNPs (inhibited enzyme-substrate complexes) with its RNA ligands and thereby shields them from cellular degradative enzymes (Hör et al., 2020b).
Cbf1 and the associated csRNAs are involved in competence control in S. pneumoniae by repressing the comC mRNA (Schnorpfeil et al., 2013; Laux et al., 2015; Aprianto et al., 2018; Hör et al., 2020b). However, the large number of identified Cbf1 ligands and phenotypic data suggest that its physiological roles may go way beyond this small regulon (van Opijnen and Camilli, 2012; Lécrivain et al., 2018; Hör et al., 2020b). The other pneumococcal sRNPs uncovered by Grad-seq also await detailed characterization, promising identification of additional RBPs involved in riboregulation in Gram-positive bacteria (Zheng et al., 2017).
Classes IIIa and IV: ProQ, a New Global RNA Chaperone Acting on mRNAs and sRNAs
When Grad-seq entered the stage (Smirnov et al., 2016), CsrA and Hfq largely dominated the panorama of posttranscriptional regulation in enterobacteria (Van Assche et al., 2015). However, Grad-seq analysis of the RNPs formed by Salmonella sRNAs uncovered a surprising heterogeneity of profiles: while Hfq-binding sRNAs constituted a well-defined biochemical class of complexes (Figure 6), they accounted for only ∼30% of sRNA distributions, the other two thirds of noncoding RNAs being involved in different kinds of assemblies (Smirnov et al., 2016, 2017a). A group of such unusual transcripts clearly clustered together and were selected for RNA pull-downs to identify their shared protein partners. This analysis zeroed in on ProQ, an RNA chaperone of unknown functional role involved in osmoregulation (Milner and Wood, 1989; Kunte et al., 1999; Chaulk et al., 2011).
ProQ RIP-seq, later supported by CLIP-seq experiments, identified the same sRNAs and several hundred other transcripts, both coding and noncoding, as specific ProQ ligands (Smirnov et al., 2016; Holmqvist et al., 2018a). The ProQ-associated sRNAs are mostly Hfq-independent and, unlike classical Hfq-binding riboregulators, highly structured. Their in-gradient profiles correlated well with that of ProQ, indicating the formation of small (100-150 kDa) stable RNPs (Figure 6). The abundance and the stability of most ProQ-binding sRNAs and at least some mRNAs depend on ProQ which, similar to Hfq, tends to bind its ligands close to the 3′-end, protecting them from cellular RNases (Smirnov et al., 2016; Holmqvist et al., 2018a). RNA-seq analyses of ΔproQ strains uncovered a profound impact of this prolific RNA binder on gene expression in enterobacteria, making it, along with Hfq and CsrA, the third global regulatory RBP in this group (Smirnov et al., 2016; Melamed et al., 2020).
The ProQ research is now booming: from an obscure protein (unfairly) commanding only passing interest it has grown into a major topic of bacterial RNA biology (Olejniczak and Storz, 2017; Holmqvist et al., 2020). ProQ homologues have been found in a number of proteobacteria (Attaiech et al., 2016; Smirnov et al., 2016) and implicated in key physiological processes, including competence (Sexton and Vogel, 2004; Attaiech et al., 2016; Durieux et al., 2019), virulence, chemotaxis, motility and biofilm formation (Sheidy and Zielke, 2013; Westermann et al., 2019), metabolism (Kunte et al., 1999; Smirnov et al., 2016), stress resistance, plasmid and core genome maintenance (Smirnov et al., 2016; Bauriedl et al., 2020; Gerovac et al., 2020). Many known base-pairing sRNAs repressing their targets in-cis or in-trans turned out to be avid ProQ binders, but only in a handful of cases the role of this protein in regulation has been solved mechanistically (Smirnov et al., 2017b; Silva et al., 2019; Westermann et al., 2019), and an even larger number of ProQ-dependent sRNAs remain totally uncharacterized. Pioneering biochemical studies highlighted generic RNA chaperone properties of ProQ homologs (Chaulk et al., 2010, 2011). Their role as new RNA matchmakers, mechanistically distinct from Hfq, is being intensively studied, but the molecular details of the interplay between ProQ and its natural RNA ligands only begin to emerge (Melamed et al., 2020). Structural and functional studies highlighted the importance of the conserved N-terminal FinO-like domain, which harbors most of the RNA-binding activity and places ProQ into a larger family of FinO-like RNA chaperones (Chaulk et al., 2010, 2011; Glover et al., 2015; Gonzalez et al., 2017b; Eidelpes et al., 2020; Gerovac et al., 2020, 2021; Immer et al., 2020; Pandey et al., 2020; Stein et al., 2020). However, the role of the C-terminal “Tudor-like” domain (Figure 6), that contributes to the RNA chaperone activities of ProQ (Chaulk et al., 2011; Gonzalez et al., 2017b), does not cease to intrigue researchers. Recent comparative analysis uncovered striking structural and functional similarities between several small β-barrel (SSB) folds, such as Sm, OB, cold-shock and Tudor domains, and proposed a unifying concept of “urfold” highlighting their biological relatedness (Youkharibache et al., 2019). The remarkable frequency of the SSB urfold among bacterial global RBPs, including Hfq, ribosomal protein S1, Cbf1, ProQ, PNPase, RNases E and R, suggests that it may constitute a recurrent signature of pleiotropic RNA-binding regulators.
Beyond Grad-Seq
The basic Grad-seq pipeline has recently seen two interesting adaptations, marking new important developments in RNA-protein complexomics (Figure 3). In one case, ATLAS-seq brings the complex profiling logic to the cell biology level: entire organelles from mildly lysed cells are resolved on a sucrose gradient and systematically profiled for their protein and RNA contents (Foster et al., 2006; Adekunle and Wang, 2020). Applied to mouse liver, this method revealed genome-wide distributions of transcripts across diverse cellular microenvironments, identified cases of differential localization for mRNA isoforms, and visualized RBPs following their targets (Adekunle and Wang, 2020).
Since Grad-seq studies broadly illustrated the dependence of RBP sedimentation on their RNA ligands (Figures 4–6), three independently developed approaches have leveraged this principle to systematically profile the protein components of stable RNPs (Caudron-Herger et al., 2019; Mallam et al., 2019; Gerovac et al., 2020). They do so by comparing global MS-derived protein profiles in lysates treated or not with an RNase, as destruction of the RNA component typically shifts the distribution of the associated proteins to lighter fractions or causes their disassembly. Thus, R-Deep employed the sucrose gradient fractionation of HeLa S3 cells and identified 1,784 proteins whose distributions changed upon RNase treatment, of which only ∼70% were known to be direct RNA binders (Caudron-Herger et al., 2019). Similarly, DIF-FRAC resolved complexes by size-exclusion chromatography and found >1,000 RNA-dependent proteins in HEK293T and mouse embryonic stem cells, of which ∼40% lacked RBP annotation. Based on the effect of the RNase treatment on the RNP behavior, all RNA-protein complexes were broadly classified into apo-stable (i.e., persisting even in the absence of RNA), structural (fully dependent on the RNA component), and compositional (with only some protein subunits contingent on the presence of RNA) (Mallam et al., 2019). Finally, GradR profiled Salmonella protein complexes on glycerol gradients and revealed a number of RNA-dependent proteins, including Hfq, CsrA, and ProQ, while also spotting an uncharacterized plasmid-encoded FinO/ProQ homolog, FopA, as a potential RBP. RIP-seq and in vitro assays confirmed its specific binding to the Inc sRNA and showed that FopA dramatically accelerates the base-pairing between Inc and the cis-encoded repZ mRNA involved in plasmid replication (Gerovac et al., 2020). This study brought to three the number of ProQ/FinO-like chaperones simultaneously residing in the same bacterial cell (ProQ, FinO, FopA), which is an exceptional case with potentially interesting evolutionary implications. All in all, these approaches beautifully complement the existing RBP discovery techniques, such as RNA interactome capture and the new crosslinking-based methods OOPS, PTex, TRAPP, and XRNAX (Hentze et al., 2018; Queiroz et al., 2019; Shchepachev et al., 2019; Trendel et al., 2019; Urdaneta et al., 2019; Smith et al., 2020b), by adding those RNP components which do not interact with RNA directly or crosslink poorly yet contribute to the structure and the functionality of the complexes. Like the classical Grad-seq, they provide important biochemical data on the organization and the RNA dependency of cellular RNPs.
Outlook
Considering Grad-seq warts-and-all, next to the advances it brought about in six model species, the method still must face up to a few challenging issues. The most important one is profile matching. A recent critical appraisal exposed a surprisingly high false discovery rate of global complexomic analyses, when complex membership is called by direct matching of protein distributions (Shatsky et al., 2016). This happens despite the ever more sophisticated bioinformatic pipelines used for the analysis of complexomic data (Dong et al., 2008; Havugimana et al., 2012; Kristensen et al., 2012; Giese et al., 2014; Gazestani et al., 2016; Páleníková et al., 2021a). The problem comes, on the one side, from natural spurious overlapping of unrelated profiles, and on the other, from the general analytical caveat of matching algorithms: most statistical measures are inherently tailored to look for differences, not similarities (Motulsky, 2010). For the moment, Grad-seq suffers from the same limitation: while clustering algorithms and especially dimensionality reduction methods (PCA, t-SNE) could be applied to classify and compare profiles, predicting complexes based solely on such profile similarities is dangerous. Therefore, Grad-seq prudently incorporates an RNA pull-down step to prioritize protein candidates and only then performs profile matching to call RNPs – expectedly with a much higher success rate (Figure 2D). It is desirable, however, to develop in future more elaborate similarity metrics and information-rich profile descriptors to enable automated detection of RNPs from Grad-seq datasets with acceptable accuracy and sensitivity.
Data mining is another issue. The Grad-seq publications highlighted in this review primarily reported on a few particularly striking findings illustrating the power of the method. But those are merely the tip of the iceberg, and the RNA and especially the protein Grad-seq datasets await deeper exploration to unearth new potentially interesting macromolecular assemblies. This is particularly important if we want to approach the functions of understudied proteins and transcripts.
We contemplate many possible avenues for further development of RNP complexomics. It will be exciting to apply Grad-seq family approaches to more exotic and recalcitrant biological systems, such as newly discovered microbes, evolutionarily diverged eukaryotes, and semi-autonomous organelles, like mitochondria and chloroplasts, in search for novel RNA biology (Castelle and Banfield, 2018; Adl et al., 2019). We foresee that RNP complexomics will become increasingly comparative, confronting different species or assessing how the ensemble of stable complexes within the same organism changes between conditions or in time. Experimental settings and statistical tools have definitely matured to carry out such complex studies (Heide et al., 2012; Kristensen et al., 2012; Moutaoufik et al., 2019; Van Strien et al., 2019; Gerovac et al., 2021; Páleníková et al., 2021a,b). Finally, albeit the identification of RNA from cryoEM structures remains challenging (Greber et al., 2014), the expansion of the bottom-up structural proteomics (Figure 2B, Ho et al., 2020) to the RNP world is now as tantalizing a perspective as never before.
Author Contributions
AS designed the study, analyzed data, wrote the original draft, and prepared the figures. MG and JV contributed to the data and reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work and its open access publication were funded by CNRS and the University of Strasbourg through the Programme Investissement d’Avenir (ANR-10-IDEX-0002-02), University of Strasbourg Insitute for Advanced Study (USIAS-2020-021), and LabEx “MitoCross”.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
AS acknowledges the continuous support from the University of Strasbourg and CNRS. The authors apologize to all colleagues whose studies could not be cited due to space limitations.
References
Abdelkareem, M., Saint-André, C., Takacs, M., Papai, G., Crucifix, C., Guo, X., et al. (2019). Structural basis of transcription: RNA polymerase backtracking and its reactivation. Mol. Cell 75, 298–309e4. doi: 10.1016/j.molcel.2019.04.029
Adekunle, D. A., and Wang, E. T. (2020). Transcriptome-wide organization of subcellular microenvironments revealed by ATLAS-Seq. Nucleic Acids Res. 48, 5859–5872. doi: 10.1093/nar/gkaa334
Adl, S. M., Bass, D., Lane, C. E., Lukeš, J., Schoch, C. L., Smirnov, A., et al. (2019). Revisions to the classification, nomenclature, and diversity of eukaryotes. J. Eukaryot. Microbiol. 66, 4–119. doi: 10.1111/jeu.12691
Agaronyan, K., Morozov, Y. I., Anikin, M., and Temiakov, D. (2015). Mitochondrial biology. Replication-transcription switch in human mitochondria. Science 347, 548–551. doi: 10.1126/science.aaa0986
Altegoer, F., Rensing, S. A., and Bange, G. (2016). Structural basis for the CsrA-dependent modulation of translation initiation by an ancient regulatory protein. Proc. Natl. Acad. Sci. U.S.A. 113, 10168–10173. doi: 10.1073/pnas.1602425113
Ameismeier, M., Cheng, J., Berninghausen, O., and Beckmann, R. (2018). Visualizing late states of human 40S ribosomal subunit maturation. Nature 558, 249–253. doi: 10.1038/s41586-018-0193-0
Andersen, J. S., Wilkinson, C. J., Mayor, T., Mortensen, P., Nigg, E. A., and Mann, M. (2003). Proteomic characterization of the human centrosome by protein correlation profiling. Nature 426, 570–574. doi: 10.1038/nature02166
Andresen, L., and Holmqvist, E. (2018). CLIP-seq in bacteria: global recognition patterns of bacterial RNA-binding proteins. Methods Enzymol. 612, 127–145. doi: 10.1016/bs.mie.2018.08.008
Aprianto, R., Slager, J., Holsappel, S., and Veening, J. W. (2018). High-resolution analysis of the pneumococcal transcriptome under a wide range of infection-relevant conditions. Nucleic Acids Res. 46, 9990–10006. doi: 10.1093/nar/gky750
Arava, Y., Wang, Y., Storey, J. D., Liu, C. L., Brown, P. O., and Herschlag, D. (2003). Genome-wide analysis of mRNA translation profiles in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 100, 3889–3894. doi: 10.1073/pnas.0635171100
Archer, S. K., Shirokikh, N. E., Beilharz, T. H., and Preiss, T. (2016). Dynamics of ribosome scanning and recycling revealed by translation complex profiling. Nature 535, 570–574. doi: 10.1038/nature18647
Arenz, S., Meydan, S., Starosta, A. L., Berninghausen, O., Beckmann, R., Vázquez-Laslop, N., et al. (2014). Drug sensing by the ribosome induces translational arrest via active site perturbation. Mol. Cell 56, 446–452. doi: 10.1016/j.molcel.2014.09.014
Artz, S. W., and Broach, J. R. (1975). Histidine regulation in Salmonella Typhimurium: an activator attenuator model of gene regulation. Proc. Natl. Acad. Sci. U.S.A. 72, 3453–3457. doi: 10.1073/pnas.72.9.3453
Attaiech, L., Boughammoura, A., Brochier-Armanet, C., Allatif, O., Peillard-Fiorente, F., Edwards, R. A., et al. (2016). Silencing of natural transformation by an RNA chaperone and a multitarget small RNA. Proc. Natl. Acad. Sci. U.S.A. 113, 8813–8818. doi: 10.1073/pnas.1601626113
Azam, M. S., and Vanderpool, C. K. (2018). Translational regulation by bacterial small RNAs via an unusual Hfq-dependent mechanism. Nucleic Acids Res. 46, 2585–2599. doi: 10.1093/nar/gkx1286
Aznaourova, M., Janga, H., Sefried, S., Kaufmann, A., Dorna, J., Volkers, S. M., et al. (2020). Noncoding RNA MaIL1 is an integral component of the TLR4-TRIF pathway. Proc. Natl. Acad. Sci. U.S.A. 117, 9042–9053. doi: 10.1073/pnas.1920393117
Babitzke, P. (2004). Regulation of transcription attenuation and translation initiation by allosteric control of an RNA-binding protein: the Bacillus subtilis TRAP protein. Curr. Opin. Microbiol. 7, 132–139. doi: 10.1016/j.mib.2004.02.003
Babitzke, P., and Romeo, T. (2007). CsrB sRNA family: sequestration of RNA-binding regulatory proteins. Curr. Opin. Microbiol. 10, 156–163. doi: 10.1016/j.mib.2007.03.007
Bak, G., Lee, J., Suk, S., Kim, D., Young Lee, J., Kim, K. S., et al. (2015). Identification of novel sRNAs involved in biofilm formation, motility, and fimbriae formation in Escherichia coli. Sci. Rep. 5, 15287. doi: 10.1038/srep15287
Baker, C. S., Morozov, I., Suzuki, K., Romeo, T., and Babitzke, P. (2002). CsrA regulates glycogen biosynthesis by preventing translation of glgC in Escherichia coli. Mol. Microbiol. 44, 1599–1610. doi: 10.1046/j.1365-2958.2002.02982.x
Balbontin, R., Fiorini, F., Figueroa-Bossi, N., Casadesús, J., and Bossi, L. (2010). Recognition of heptameric seed sequence underlies multi-target regulation by RybB small RNA in Salmonella enterica. Mol. Microbiol. 78, 380–394. doi: 10.1111/j.1365-2958.2010.07342.x
Ban, N., Nissen, P., Hansen, J., Moore, P. B., and Steitz, T. A. (2000). The complete atomic structure of the large ribosomal subunit at 2.4 A resolution. Science 289, 905–920. doi: 10.1126/science.289.5481.905
Bandyra, K. J., Sinha, D., Syrjanen, J., Luisi, B. F., and De Lay, N. R. (2016). The ribonuclease polynucleotide phosphorylase can interact with small regulatory RNAs in both protective and degradative modes. RNA 22, 360–372. doi: 10.1261/rna.052886.115
Baran, R., Reindl, W., and Northen, T. R. (2009). Mass spectrometry based metabolomics and enzymatic assays for functional genomics. Curr. Opin. Microbiol. 12, 547–552. doi: 10.1016/j.mib.2009.07.004
Barandun, J., Chaker-Margot, M., Hunziker, M., Molloy, K. R., Chait, B. T., and Klinge, S. (2017). The complete structure of the small-subunit processome. Nat. Struct. Mol. Biol. 24, 944–953. doi: 10.1038/nsmb.3472
Barandun, J., Hunziker, M., and Klinge, S. (2018). Assembly and structure of the SSU processome-a nucleolar precursor of the small ribosomal subunit. Curr. Opin. Struct. Biol. 49, 85–93. doi: 10.1016/j.sbi.2018.01.008
Barandun, J., Hunziker, M., Vossbrinck, C. R., and Klinge, S. (2019). Evolutionary compaction and adaptation visualized by the structure of the dormant microsporidian ribosome. Nat. Microbiol. 4, 1798–1804. doi: 10.1038/s41564-019-0514-6
Barembruch, C., and Hengge, R. (2007). Cellular levels and activity of the flagellar sigma factor FliA of Escherichia coli are controlled by FlgM-modulated proteolysis. Mol. Microbiol. 65, 76–89. doi: 10.1111/j.1365-2958.2007.05770.x
Bauriedl, S., Gerovac, M., Heidrich, N., Bischler, T., Barquist, L., Vogel, J., et al. (2020). The minimal meningococcal ProQ protein has an intrinsic capacity for structure-based global RNA recognition. Nat. Commun. 11, 2823. doi: 10.1038/s41467-020-16650-6
Beckmann, B. M., Hoch, P. G., Marz, M., Willkomm, D. K., Salas, M., and Hartmann, R. K. (2012). A pRNA-induced structural rearrangement triggers 6S-1 RNA release from RNA polymerase in Bacillus subtilis. EMBO J. 31, 1727–1738. doi: 10.1038/emboj.2012.23
Ben-Shem, A., Garreau De Loubresse, N., Melnikov, S., Jenner, L., Yusupova, G., and Yusupov, M. (2011). The structure of the eukaryotic ribosome at 3.0 A resolution. Science 334, 1524–1529. doi: 10.1126/science.1212642
Béthune, J., Jansen, R. P., Feldbrügge, M., and Zarnack, K. (2019). Membrane-associated RNA-binding proteins orchestrate organelle-coupled translation. Trends Cell Biol. 29, 178–188. doi: 10.1016/j.tcb.2018.10.005
Block, K. F., Puerta-Fernandez, E., Wallace, J. G., and Breaker, R. R. (2011). Association of OLE RNA with bacterial membranes via an RNA-protein interaction. Mol. Microbiol. 79, 21–34. doi: 10.1111/j.1365-2958.2010.07439.x
Blower, T. R., Pei, X. Y., Short, F. L., Fineran, P. C., Humphreys, D. P., Luisi, B. F., et al. (2011). A processed noncoding RNA regulates an altruistic bacterial antiviral system. Nat. Struct. Mol. Biol. 18, 185–190. doi: 10.1038/nsmb.1981
Boccitto, M., and Wolin, S. L. (2019). Ro60 and Y RNAs: structure, functions, and roles in autoimmunity. Crit. Rev. Biochem. Mol. Biol. 54, 133–152. doi: 10.1080/10409238.2019.1608902
Boehringer, D., O’Farrell, H. C., Rife, J. P., and Ban, N. N. (2012). Structural insights into methyltransferase KsgA function in 30S ribosomal subunit biogenesis. J. Biol. Chem. 287, 10453–10459. doi: 10.1074/jbc.M111.318121
Bohlen, J., Fenzl, K., Kramer, G., Bukau, B., and Teleman, A. A. (2020). Selective 40S footprinting reveals cap-tethered ribosome scanning in human cells. Mol. Cell 79, 561–574e5. doi: 10.1016/j.molcel.2020.06.005
Bohnsack, M. T., and Sloan, K. E. (2018). Modifications in small nuclear RNAs and their roles in spliceosome assembly and function. Biol. Chem. 399, 1265–1276. doi: 10.1515/hsz-2018-0205
Bonekamp, F., Clemmesen, K., Karlström, O., and Jensen, K. F. (1984). Mechanism of UTP-modulated attenuation at the pyrE gene of Escherichia coli: an example of operon polarity control through the coupling of translation to transcription. EMBO J. 3, 2857–2861. doi: 10.1002/j.1460-2075.1984.tb02220.x
Borukhov, S., Sagitov, V., and Goldfarb, A. (1993). Transcript cleavage factors from E. coli. Cell 72, 459–466. doi: 10.1016/0092-8674(93)90121-6
Brencic, A., and Lory, S. (2009). Determination of the regulon and identification of novel mRNA targets of Pseudomonas aeruginosa RsmA. Mol. Microbiol. 72, 612–632. doi: 10.1111/j.1365-2958.2009.06670.x
Brito Querido, J., Sokabe, M., Kraatz, S., Gordiyenko, Y., Skehel, J. M., Fraser, C. S., et al. (2020). Structure of a human 48S translational initiation complex. Science 369, 1220–1227. doi: 10.1126/science.aba4904
Brown, A., Baird, M. R., Yip, M. C., Murray, J., and Shao, S. (2018). Structures of translationally inactive mammalian ribosomes. eLife 7, e40486. doi: 10.7554/eLife.40486
Brown, A., Rathore, S., Kimanius, D., Aibara, S., Bai, X. C., Rorbach, J., et al. (2017). Structures of the human mitochondrial ribosome in native states of assembly. Nat. Struct. Mol. Biol. 24, 866–869. doi: 10.1038/nsmb.3464
Burmann, B. M., Schweimer, K., Luo, X., Wahl, M. C., Stitt, B. L., Gottesman, M. E., et al. (2010). A NusE:NusG complex links transcription and translation. Science 328, 501–504. doi: 10.1126/science.1184953
Caillet, J., Baron, B., Boni, I. V., Caillet-Saguy, C., and Hajnsdorf, E. (2019). Identification of protein-protein and ribonucleoprotein complexes containing Hfq. Sci. Rep. 9, 14054. doi: 10.1038/s41598-019-50562-w
Cain, A. K., Barquist, L., Goodman, A. L., Paulsen, I. T., Parkhill, J., and Van Opijnen, T. (2020). A decade of advances in transposon-insertion sequencing. Nat. Rev. Genet. 21, 526–540. doi: 10.1038/s41576-020-0244-x
Camacho-Carvajal, M. M., Wollscheid, B., Aebersold, R., Steimle, V., and Schamel, W. W. (2004). Two-dimensional Blue native/SDS gel electrophoresis of multi-protein complexes from whole cellular lysates: a proteomics approach. Mol. Cell Proteomics. 3, 176–182. doi: 10.1074/mcp.T300010-MCP200
Campbell, Z. T., and Wickens, M. (2015). Probing RNA-protein networks: biochemistry meets genomics. Trends Biochem. Sci. 40, 157–164. doi: 10.1016/j.tibs.2015.01.003
Carlevaro-Fita, J., Rahim, A., Guigó, R., Vardy, L. A., and Johnson, R. (2016). Cytoplasmic long noncoding RNAs are frequently bound to and degraded at ribosomes in human cells. RNA 22, 867–882. doi: 10.1261/rna.053561.115
Castelle, C. J., and Banfield, J. F. (2018). Major new microbial groups expand diversity and alter our understanding of the tree of life. Cell 172, 1181–1197. doi: 10.1016/j.cell.2018.02.016
Caudron-Herger, M., Rusin, S. F., Adamo, M. E., Seiler, J., Schmid, V. K., Barreau, E., et al. (2019). R-DeeP: proteome-wide and quantitative identification of RNA-dependent proteins by density gradient ultracentrifugation. Mol. Cell 75, 184–199e10. doi: 10.1016/j.molcel.2019.04.018
Cavanagh, A. T., Klocko, A. D., Liu, X., and Wassarman, K. M. (2008). Promoter specificity for 6S RNA regulation of transcription is determined by core promoter sequences and competition for region 4.2 of σ70. Mol. Microbiol. 67, 1242–1256. doi: 10.1111/j.1365-2958.2008.06117.x
Chaker-Margot, M., Barandun, J., Hunziker, M., and Klinge, S. (2017). Architecture of the yeast small subunit processome. Science 355, eaal1880. doi: 10.1126/science.aal1880
Chan, C. T. Y., Pang, Y. L. J., Deng, W., Babu, I. R., Dyavaiah, M., Begley, T. J., et al. (2012). Reprogramming of tRNA modifications controls the oxidative stress response by codon-biased translation of proteins. Nat. Commun. 3, 937. doi: 10.1038/ncomms1938
Chao, Y., Papenfort, K., Reinhardt, R., Sharma, C. M., and Vogel, J. (2012). An atlas of Hfq-bound transcripts reveals 3′ UTRs as a genomic reservoir of regulatory small RNAs. EMBO J. 31, 4005–4019. doi: 10.1038/emboj.2012.229
Chatzispyrou, I. A., Alders, M., Guerrero-Castillo, S., Zapata Perez, R., Haagmans, M. A., Mouchiroud, L., et al. (2017). A homozygous missense mutation in ERAL1, encoding a mitochondrial rRNA chaperone, causes Perrault syndrome. Hum. Mol. Genet. 26, 2541–2550. doi: 10.1093/hmg/ddx152
Chaulk, S., Lu, J., Tan, K., Arthur, D. C., Edwards, R. A., Frost, L. S., et al. (2010). N. meningitidis 1681 is a member of the FinO family of RNA chaperones. RNA Biol. 7, 812–819. doi: 10.4161/rna.7.6.13688
Chaulk, S. G., Smith Frieday, M. N. S., Arthur, D. C., Culham, D. E., Edwards, R. A., Soo, P., et al. (2011). ProQ is an RNA chaperone that controls ProP levels in Escherichia coli. Biochemistry 50, 3095–3106. doi: 10.1021/bi101683a
Chen, E., Sharma, M. R., Shi, X., Agrawal, R. K., and Joseph, S. (2014). Fragile X mental retardation protein regulates translation by binding directly to the ribosome. Mol. Cell 54, 407–417. doi: 10.1016/j.molcel.2014.03.023
Chen, J., and Gottesman, S. (2017). Hfq links translation repression to stress-induced mutagenesis in E. coli. Genes Dev. 31, 1382–1395. doi: 10.1101/gad.302547.117
Chen, J., Wassarman, K. M., Feng, S., Leon, K., Feklistov, A., Winkelman, J. T., et al. (2017a). 6S RNA mimics B-form DNA to regulate Escherichia coli RNA polymerase. Mol. Cell. 68, 388–397e6. doi: 10.1016/j.molcel.2017.09.006
Chen, S. S., and Williamson, J. R. (2013). Characterization of the ribosome biogenesis landscape in E. coli using quantitative mass spectrometry. J. Mol. Biol. 425, 767–779. doi: 10.1016/j.jmb.2012.11.040
Chen, W., Xie, Z., Yang, F., and Ye, K. (2017b). Stepwise assembly of the earliest precursors of large ribosomal subunits in yeast. Nucleic Acids Res. 45, 6837–6847. doi: 10.1093/nar/gkx254
Chen, X., Taylor, D. W., Fowler, C. C., Galan, J. E., Wang, H. W., and Wolin, S. L. (2013). An RNA degradation machine sculpted by Ro autoantigen and noncoding RNA. Cell 153, 166–177. doi: 10.1016/j.cell.2013.02.037
Cheng, J., Baßler, J., Fischer, P., Lau, B., Kellner, N., Kunze, R., et al. (2019). Thermophile 90S pre-ribosome structures reveal the reverse order of co-transcriptional 18S rRNA subdomain integration. Mol. Cell 75, 1256–1269e7. doi: 10.1016/j.molcel.2019.06.032
Cheng, J., Kellner, N., Berninghausen, O., Hurt, E., and Beckmann, R. (2017). 3.2-Å-resolution structure of the 90S preribosome before A1 pre-rRNA cleavage. Nat. Struct. Mol. Biol. 24, 954–964. doi: 10.1038/nsmb.3476
Cheng, J., Lau, B., La Venuta, G., Ameismeier, M., Berninghausen, O., Hurt, E., et al. (2020). 90S pre-ribosome transformation into the primordial 40S subunit. Science 369, 1470–1476. doi: 10.1126/science.abb4119
Chihara, K., Bischler, T., Barquist, L., Monzon, V. A., Noda, N., Vogel, J., et al. (2019). Conditional Hfq association with small noncoding RNAs in Pseudomonas aeruginosa revealed through comparative UV cross-linking immunoprecipitation followed by high-throughput sequencing. mSystems 4, e590–e519. doi: 10.1128/mSystems.00590-19
Cho, H. D., Chen, Y., Varani, G., and Weiner, A. M. (2006). A model for C74 addition by CCA-adding enzymes: C74 addition, like C75 and A76 addition, does not involve tRNA translocation. J. Biol. Chem. 281, 9801–9811. doi: 10.1074/jbc.M512603200
Choi, J., Grosely, R., Prabhakar, A., Lapointe, C. P., Wang, J., and Puglisi, J. D. (2018). How messenger RNA and nascent chain sequences regulate translation elongation. Annu Rev Biochem 87, 421–449. doi: 10.1146/annurev-biochem-060815-014818
Chujo, T., Yamazaki, T., and Hirose, T. (2016). Architectural RNAs (arcRNAs): a class of long noncoding RNAs that function as the scaffold of nuclear bodies. Biochim Biophys Acta 1859, 139–146. doi: 10.1016/j.bbagrm.2015.05.007
Couvillion, M. T., Soto, I. C., Shipkovenska, G., and Churchman, L. S. (2016). Synchronized mitochondrial and cytosolic translation programs. Nature 533, 499–503. doi: 10.1038/nature18015
Darnell, J. C., Van Driesche, S. J., Zhang, C., Hung, K. Y., Mele, A., Fraser, C. E., et al. (2011). FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261. doi: 10.1016/j.cell.2011.06.013
Dassi, E. (2017). Handshakes and fights: the regulatory interplay of RNA-binding proteins. Front Mol Biosci 4:67. doi: 10.3389/fmolb.2017.00067
Datta, P. P., Wilson, D. N., Kawazoe, M., Swami, N. K., Kaminishi, T., Sharma, M. R., et al. (2007) Structural aspects of RbfA action during small ribosomal subunit assembly. Mol. Cell 28, 434–445. doi: 10.1016/j.molcel.2007.08.026
Davis, J. H., Tan, Y. Z., Carragher, B., Potter, C. S., Lyumkis, D., and Williamson, J. R. (2016). Modular assembly of the bacterial large ribosomal subunit. Cell 167, 1610–1622e5. doi: 10.1016/j.cell.2016.11.020
de Almeida, N. M., Wessels, H. J. C. T., De Graaf, R. M., Ferousi, C., Jetten, M. S. M., Keltjens, J. T., et al. (2016). Membrane-bound electron transport systems of an anammox bacterium: a complexome analysis. Biochim Biophys Acta 1857, 1694–1704. doi: 10.1016/j.bbabio.2016.07.006
Dendooven, T., and Luisi, B. F. (2017). RNA search engines empower the bacterial intranet. Biochem Soc Trans 45, 987–997. doi: 10.1042/BST20160373
Desnoyers, G., and Massé, E. (2012). Noncanonical repression of translation initiation through small RNA recruitment of the RNA chaperone Hfq. Genes Dev. 26, 726–739. doi: 10.1101/gad.182493.111
Dimastrogiovanni, D., Fröhlich, K. S., Bandyra, K. J., Bruce, H. A., Hohensee, S., Vogel, J., et al. (2014). Recognition of the small regulatory RNA RydC by the bacterial Hfq protein. eLife 3, e05375. doi: 10.7554/eLife.05375
Dong, M., Yang, L. L., Williams, K., Fisher, S. J., Hall, S. C., Biggin, M. D., et al. (2008). A “tagless” strategy for identification of stable protein complexes genome-wide by multidimensional orthogonal chromatographic separation and iTRAQ reagent tracking. J Proteome Res 7, 1836–1849. doi: 10.1021/pr700624e
Du, H., and Babitzke, P. (1998). trp RNA-binding attenuation protein-mediated long distance RNA refolding regulates translation of trpE in Bacillus subtilis. J. Biol. Chem. 273, 20494–20503. doi: 10.1074/jbc.273.32.20494
Du, H., Tarpey, R., and Babitzke, P. (1997). The trp RNA-binding attenuation protein regulates TrpG synthesis by binding to the trpG ribosome binding site of Bacillus subtilis. J Bacteriol 179, 2582–2586. doi: 10.1128/jb.179.8.2582-2586.1997
Du, Y., An, W., Zhu, X., Sun, Q., Qi, J., and Ye, K. (2020). Cryo-EM structure of 90S small ribosomal subunit precursors in transition states. Science 369, 1477–1481. doi: 10.1126/science.aba9690
Dugar, G., Svensson, S. L., Bischler, T., Wäldchen, S., Reinhardt, R., Sauer, M., et al. (2016). The CsrA-FliW network controls polar localization of the dual-function flagellin mRNA in Campylobacter jejuni. Nat. Commun. 7, 11667. doi: 10.1038/ncomms11667
Dupuis-Sandoval, F., Poirier, M., and Scott, M. S. (2015). The emerging landscape of small nucleolar RNAs in cell biology. Wiley Interdiscip Rev RNA 6, 381–397. doi: 10.1002/wrna.1284
Durica-Mitic, S., Göpel, Y., Amman, F., and Görke, B. (2020). Adaptor protein RapZ activates endoribonuclease RNase E by protein-protein interaction to cleave a small regulatory RNA. RNA 26, 1198–1215. doi: 10.1261/rna.074047.119
Durieux, I., Ginevra, C., Attaiech, L., Picq, K., Juan, P. A., Jarraud, S., et al. (2019). Diverse conjugative elements silence natural transformation in Legionella species. Proc. Natl. Acad. Sci. U.S.A. 116, 18613–18618. doi: 10.1073/pnas.1909374116
Duss, O., Michel, E., Yulikov, M., Schubert, M., Jeschke, G., and Allain, F. H. T. (2014). Structural basis of the non-coding RNA RsmZ acting as a protein sponge. Nature 509, 588–592. doi: 10.1038/nature13271
Dutcher, H. A., and Raghavan, R. (2018). Origin, evolution, and loss of bacterial small RNAs. Microbiol Spectr 6, doi: 10.1128/microbiolspec.RWR-0004-2017 ∗∗∗P∗∗∗,
Eidelpes, R., Kim, H. J., Glover, J. N. M., and Tollinger, M. (2020). NMR resonance assignments of the FinO-domain of the RNA chaperone RocC. Biomol NMR Assign doi: 10.1007/s12104-020-09983-2 [Epub ahead of print].
Eliscovich, C., and Singer, R. H. (2017). RNP transport in cell biology: the long and winding road. Curr Opin Cell Biol 45, 38–46. doi: 10.1016/j.ceb.2017.02.008
Encode Project Consortium, Snyder, M. P., Gingeras, T. R., Moore, J. E., Weng, Z., Gerstein, M. B., et al. (2020). Perspectives on ENCODE. Nature 583, 693–698. doi: 10.1038/s41586-020-2449-8
Egloff, S., Studniarek, C., and Kiss, T. (2018). 7SK small nuclear RNA, a multifunctional transcriptional regulatory RNA with gene-specific features. Transcription 9, 95–101. doi: 10.1080/21541264.2017.1344346
Erickson, H. P. (2009). Size and shape of protein molecules at the nanometer level determined by sedimentation, gel filtration, and electron microscopy. Biol Proced Online 11, 32–51. doi: 10.1007/s12575-009-9008-x
Espinoza, C. A., Allen, T. A., Hieb, A. R., Kugel, J. F., and Goodrich, J. A. (2004). B2 RNA binds directly to RNA polymerase II to repress transcript synthesis. Nat. Struct. Mol. Biol. 11, 822–829. doi: 10.1038/nsmb812
Farhoud, M. H., Wessels, H. J. C. T., Steenbakkers, P. J. M., Mattijssen, S., Wevers, R. A., Van Engelen, B. G., et al. (2005). Protein complexes in the archaeon Methanothermobacter thermautotrophicus analyzed by blue native/SDS-PAGE and mass spectrometry. Mol. Cell Proteomics. 4, 1653–1663. doi: 10.1074/mcp.M500171-MCP200
Ford, K., Mcdonald, D., and Mali, P. (2019). Functional genomics via CRISPR-Cas. J Mol Biol 431, 48–65. doi: 10.1016/j.jmb.2018.06.034
Foster, L. J., De Hoog, C. L., Zhang, Y., Zhang, Y., Xie, X., Mootha, V. K., et al. (2006). A mammalian organelle map by protein correlation profiling. Cell 125, 187–199. doi: 10.1016/j.cell.2006.03.022
Fox, A. H., Nakagawa, S., Hirose, T., and Bond, C. S. (2018). Paraspeckles: where long noncoding RNA meets phase separation. Trends Biochem. Sci. 43, 124–135. doi: 10.1016/j.tibs.2017.12.001
Fröhlich, K. S., Papenfort, K., Fekete, A., and Vogel, J. (2013). A small RNA activates CFA synthase by isoform-specific mRNA stabilization. EMBO J. 32, 2963–2979. doi: 10.1038/emboj.2013.222
Gazestani, V. H., Nikpour, N., Mehta, V., Najafabadi, H. S., Moshiri, H., Jardim, A., et al. (2016). A protein complex map of Trypanosoma brucei. PLoS Negl. Trop. Dis. 10:e0004533. doi: 10.1371/journal.pntd.0004533
Gebhardt, M. J., Kambara, T. K., Ramsey, K. M., and Dove, S. L. (2020). Widespread targeting of nascent transcripts by RsmA in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U.S.A. 117, 10520–10529. doi: 10.1073/pnas.1917587117
Gehring, N. H., Wahle, E., and Fischer, U. (2017). Deciphering the mRNP code: RNA-bound determinants of post-transcriptional gene regulation. Trends Biochem. Sci. 42, 369–382. doi: 10.1016/j.tibs.2017.02.004
Geiselmann, J., Yager, T. D., Gill, S. C., Calmettes, P., and Von Hippel, P. H. (1992). Physical properties of the Escherichia coli transcription termination factor rho. 1. Association states and geometry of the rho hexamer. Biochemistry 31, 111–121. doi: 10.1021/bi00116a017
Gerovac, M., El Mouali, Y., Kuper, J., Kisker, C., Barquist, L., and Vogel, J. (2020). Global discovery of bacterial RNA-binding proteins by RNase-sensitive gradient profiles reports a new FinO domain protein. RNA 26, 1448–1463. doi: 10.1261/rna.076992.120
Gerovac, M., Wicke, L., Chihara, K., Schneider, C., Lavigne, R., and Vogel, J. (2021). A Grad-seq view of RNA and protein complexes in Pseudomonas aeruginosa under standard and bacteriophage predation conditions. mBio 12, e3454–20. doi: 10.1128/mBio.03454-20
Giambruno, R., Mihailovich, M., and Bonaldi, T. (2018). Mass spectrometry-based proteomics to unveil the non-coding RNA world. Front Mol Biosci 5:90. doi: 10.3389/fmolb.2018.00090
Giese, H., Ackermann, J., Heide, H., Bleier, L., Dröse, S., Wittig, I., et al. (2014). NOVA: a software to analyze complexome profiling data. Bioinformatics 31, 440–441. doi: 10.1093/bioinformatics/btu623
Giess, A., Torres Cleuren, Y. N., Tjeldnes, H., Krause, M., Bizuayehu, T. T., Hiensch, S., et al. (2020). Profiling of small ribosomal subunits reveals modes and regulation of translation initiation. Cell Rep 31, 107534. doi: 10.1016/j.celrep.2020.107534
Gilbert, L. A., Horlbeck, M. A., Adamson, B., Villalta, J. E., Chen, Y., Whitehead, E. H., et al. (2014). Genome-scale CRISPR-mediated control of gene repression and activation. Cell 159, 647–661. doi: 10.1016/j.cell.2014.09.029
Gildehaus, N., Neußer, T., Wurm, R., and Wagner, R. (2007). Studies on the function of the riboregulator 6S RNA from E. coli: RNA polymerase binding, inhibition of in vitro transcription and synthesis of RNA-directed de novo transcripts. Nucleic Acids Res. 35, 1885–1896. doi: 10.1093/nar/gkm085
Gleason, C. E., Ordureau, A., Gourlay, R., Arthur, J. S., and Cohen, P. (2011). Polyubiquitin binding to optineurin is required for optimal activation of TANK-binding kinase 1 and production of interferon beta. J. Biol. Chem. 286, 35663–35674. doi: 10.1074/jbc.M111.267567
Glover, J. N. M., Chaulk, S. G., Edwards, R. A., Arthur, D., Lu, J., and Frost, L. S. (2015). The FinO family of bacterial RNA chaperones. Plasmid 78, 79–87. doi: 10.1016/j.plasmid.2014.07.003
Gocheva, V., Le Gall, A., Boudvillain, M., Margeat, E., and Nollmann, M. (2015). Direct observation of the translocation mechanism of transcription termination factor Rho. Nucleic Acids Res. 43, 2367–2377. doi: 10.1093/nar/gkv085
Goeders, N., Chai, R., Chen, B., Day, A., and Salmond, G. P. (2016). Structure, evolution, and functions of bacterial type III toxin-antitoxin systems. Toxins (Basel) 8, 282. doi: 10.3390/toxins8100282
Gonzalez, G. M., Durica-Mitic, S., Hardwick, S. W., Moncrieffe, M. C., Resch, M., Neumann, P., et al. (2017a). Structural insights into RapZ-mediated regulation of bacterial amino-sugar metabolism. Nucleic Acids Res. 45, 10845–10860. doi: 10.1093/nar/gkx732
Gonzalez, G. M., Hardwick, S. W., Maslen, S. L., Skehel, J. M., Holmqvist, E., Vogel, J., et al. (2017b). Structure of the Escherichia coli ProQ RNA-binding protein. RNA 23, 696–711. doi: 10.1261/rna.060343.116
Goodson, J. R., and Winkler, W. C. (2018). Processive antitermination. Microbiol Spectr. 6, doi: 10.1128/microbiolspec.RWR-0031-2018.
Göpel, Y., Khan, M. A., and Görke, B. (2016). Domain swapping between homologous bacterial small RNAs dissects processing and Hfq binding determinants and uncovers an aptamer for conditional RNase E cleavage. Nucleic Acids Res. 44, 824–837. doi: 10.1093/nar/gkv1161
Göpel, Y., Papenfort, K., Reichenbach, B., Vogel, J., and Görke, B. (2013). Targeted decay of a regulatory small RNA by an adaptor protein for RNase E and counteraction by an anti-adaptor RNA. Genes Dev. 27, 552–564. doi: 10.1101/gad.210112.112
Gordon, S. M., Deng, J., Tomann, A. B., Shah, A. S., Lu, L. J., and Davidson, W. S. (2013). Multi-dimensional co-separation analysis reveals protein-protein interactions defining plasma lipoprotein subspecies. Mol. Cell Proteomics. 12, 3123–3134. doi: 10.1074/mcp.M113.028134
Göringer, H. U. (2012). ‘Gestalt,’ composition and function of the Trypanosoma brucei editosome. Annu Rev Microbiol 66, 65–82. doi: 10.1146/annurev-micro-092611-150150
Gorski, S. A., Vogel, J., and Doudna, J. A. (2017). RNA-based recognition and targeting: sowing the seeds of specificity. Nat Rev Mol Cell Biol 18, 215–228. doi: 10.1038/nrm.2016.174
Greber, B. J., Boehringer, D., Leibundgut, M., Bieri, P., Leitner, A., Schmitz, N., et al. (2014). The complete structure of the large subunit of the mammalian mitochondrial ribosome. Nature 515, 283–286. doi: 10.1038/nature13895
Guerrier-Takada, C., Gardiner, K., Marsh, T., Pace, N., and Altman, S. (1983). The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell 35, 849–857. doi: 10.1016/0092-8674(83)90117-4
Gupta, N., and Culver, G. M. (2014). Multiple in vivo pathways for Escherichia coli small ribosomal subunit assembly occur on one pre-rRNA. Nat. Struct. Mol. Biol. 21, 937–943. doi: 10.1038/nsmb.2887
Hall, T. M. (2016). De-coding and re-coding RNA recognition by PUF and PPR repeat proteins. Curr. Opin. Struct. Biol. 36, 116–121. doi: 10.1016/j.sbi.2016.01.010
Harris, K. A., and Breaker, R. R. (2018). Large noncoding RNAs in bacteria. Microbiol Spectr 6, doi: 10.1128/microbiolspec.RWR-0005-2017.
Harris, K. A., Odzer, N. B., and Breaker, R. R. (2019). Disruption of the OLE ribonucleoprotein complex causes magnesium toxicity in Bacillus halodurans. Mol. Microbiol. 112, 1552–1563. doi: 10.1111/mmi.14379
Harrison, P. M., Kumar, A., Lang, N., Snyder, M., and Gerstein, M. (2002). A question of size: the eukaryotic proteome and the problems in defining it. Nucleic Acids Res. 30, 1083–1090. doi: 10.1093/nar/30.5.1083
Hartman, N. T., Sicilia, F., Lilley, K. S., and Dupree, P. (2007). Proteomic complex detection using sedimentation. Anal Chem 79, 2078–2083. doi: 10.1021/ac061959t
Havugimana, P. C., Hart, G. T., Nepusz, T., Yang, H., Turinsky, A. L., Li, Z., et al. (2012). A census of human soluble protein complexes. Cell 150, 1068–1081. doi: 10.1016/j.cell.2012.08.011
Heide, H., Bleier, L., Steger, M., Ackermann, J., Dröse, S., Schwamb, B., et al. (2012). Complexome profiling identifies TMEM126B as a component of the mitochondrial complex I assembly complex. Cell Metab 16, 538–549. doi: 10.1016/j.cmet.2012.08.009
Helbig, A. O., De Groot, M. J. L., Van Gestel, R. A., Mohammed, S., De Hulster, E. A., Luttik, M. A., et al. (2009). A three-way proteomics strategy allows differential analysis of yeast mitochondrial membrane protein complexes under anaerobic and aerobic conditions. Proteomics 9, 4787–4798. doi: 10.1002/pmic.200800951
Helder, S., Blythe, A. J., Bond, C. S., and Mackay, J. P. (2016). Determinants of affinity and specificity in RNA-binding proteins. Curr. Opin. Struct. Biol. 38, 83–91. doi: 10.1016/j.sbi.2016.05.005
Heller, J. L. E., Kamalampeta, R., and Wieden, H. J. (2017). Taking a step back from back-translocation: an integrative view of LepA/EF4’s cellular function. Mol Cell Biol 37, e653–e616. doi: 10.1128/MCB.00653-16
Hemm, M. R., Paul, B. J., Miranda-Ríos, J., Zhang, A., Soltanzad, N., and Storz, G. (2010). Small stress response proteins in Escherichia coli: proteins missed by classical proteomic studies. J Bacteriol 192, 46–58. doi: 10.1128/JB.00872-09
Hemm, M. R., Paul, B. J., Schneider, T. D., Storz, G., and Rudd, K. E. (2008). Small membrane proteins found by comparative genomics and ribosome binding site models. Mol. Microbiol. 70, 1487–1501. doi: 10.1111/j.1365-2958.2008.06495.x
Hentze, M. W., Castello, A., Schwarzl, T., and Preiss, T. (2018). A brave new world of RNA-binding proteins. Nat Rev Mol Cell Biol 19, 327–341. doi: 10.1038/nrm.2017.130
Heuer, A., Thomson, E., Schmidt, C., Berninghausen, O., Becker, T., Hurt, E., et al. (2017). Cryo-EM structure of a late pre-40S ribosomal subunit from Saccharomyces cerevisiae. Elife 6, e30189. doi: 10.7554/eLife.30189
Hillen, H. S., Parshin, A. V., Agaronyan, K., Morozov, Y. I., Graber, J. J., Chernev, A., et al. (2017). Mechanism of transcription anti-termination in human mitochondria. Cell 171, 1082–1093e13. doi: 10.1016/j.cell.2017.09.035
Hinnebusch, A. G. (2011). Molecular mechanism of scanning and start codon selection in eukaryotes. Microbiol Mol Biol Rev 75, 434–467. doi: 10.1128/MMBR.00008-11
Ho, C. M., Li, X., Lai, M., Terwilliger, T. C., Beck, J. R., Wohlschlegel, J., et al. (2020). Bottom-up structural proteomics: cryoEM of protein complexes enriched from the cellular milieu. Nat Methods 17, 79–85. doi: 10.1038/s41592-019-0637-y
Holmqvist, E., Berggren, S., and Rizvanovic, A. (2020). RNA-binding activity and regulatory functions of the emerging sRNA-binding protein ProQ. Biochim Biophys Acta Gene Regul Mech 1863, 194596. doi: 10.1016/j.bbagrm.2020.194596
Holmqvist, E., Li, L., Bischler, T., Barquist, L., and Vogel, J. (2018a). Global maps of ProQ binding in vivo reveal target recognition via RNA structure and stability control at mRNA 3′ ends. Mol. Cell 70, e976. doi: 10.1016/j.molcel.2018.04.017
Holmqvist, E., and Vogel, J. (2018b). RNA-binding proteins in bacteria. Nat Rev Microbiol 16, 601–615. doi: 10.1038/s41579-018-0049-5
Holmqvist, E., Wright, P. R., Li, L., Bischler, T., Barquist, L., Reinhardt, R., et al. (2016). Global RNA recognition patterns of post-transcriptional regulators Hfq and CsrA revealed by UV crosslinking in vivo. EMBO J. 35, 991–1011. doi: 10.15252/embj.201593360
Hör, J., Di Giorgio, S., Gerovac, M., Venturini, E., Förstner, K. U., and Vogel, J. (2020a). Grad-seq shines light on unrecognized RNA and protein complexes in the model bacterium Escherichia coli. Nucleic Acids Res. 48, 9301–9319. doi: 10.1093/nar/gkaa676
Hör, J., Garriss, G., Di Giorgio, S., Hack, L. M., Vanselow, J. T., Förstner, K. U., et al. (2020b). Grad-seq in a Gram-positive bacterium reveals exonucleolytic sRNA activation in competence control. EMBO J. 39, e103852. doi: 10.15252/embj.2019103852
Hör, J., Gorski, S. A., and Vogel, J. (2018). Bacterial RNA biology on a genome scale. Mol. Cell 70, 785–799. doi: 10.1016/j.molcel.2017.12.023
Huang, Y. H., Hilal, T., Loll, B., Bürger, J., Mielke, T., Böttcher, C., et al. (2020). Structure-based mechanisms of a molecular RNA polymerase/chaperone machine required for ribosome biosynthesis. Mol. Cell 79, 1024–1036e5. doi: 10.1016/j.molcel.2020.08.010
Hunziker, M., Barandun, J., Buzovetsky, O., Steckler, C., Molina, H., and Klinge, S. (2019). Conformational switches control early maturation of the eukaryotic small ribosomal subunit. Elife 8, e45185. doi: 10.7554/eLife.45185
Iadevaia, V., and Gerber, A. P. (2015). Combinatorial control of mRNA fates by RNA-binding proteins and non-coding RNAs. Biomolecules 5, 2207–2222. doi: 10.3390/biom5042207
Imashimizu, M., Takahashi, H., Oshima, T., Mcintosh, C., Bubunenko, M., Court, D. L., et al. (2015). Visualizing translocation dynamics and nascent transcript errors in paused RNA polymerases in vivo. Genome Biol 16, 98. doi: 10.1186/s13059-015-0666-5
Immer, C., Hacker, C., and Wohnert, J. (2020). Solution structure and RNA-binding of a minimal ProQ-homolog from Legionella pneumophila (Lpp1663). RNA 26, 2031–2043. doi: 10.1261/rna.077354.120
Ingolia, N. T. (2016). Ribosome footprint profiling of translation throughout the genome. Cell 165, 22–33. doi: 10.1016/j.cell.2016.02.066
Ingolia, N. T., Ghaemmaghami, S., Newman, J. R., and Weissman, J. S. (2009). Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324, 218–223. doi: 10.1126/science.1168978
Ip, J. Y., Schmidt, D., Pan, Q., Ramani, A. K., Fraser, A. G., Odom, D. T., et al. (2011). Global impact of RNA polymerase II elongation inhibition on alternative splicing regulation. Genome Res 21, 390–401. doi: 10.1101/gr.111070.110
Irie, Y., Starkey, M., Edwards, A. N., Wozniak, D. J., Romeo, T., and Parsek, M. R. (2010). Pseudomonas aeruginosa biofilm matrix polysaccharide Psl is regulated transcriptionally by RpoS and post-transcriptionally by RsmA. Mol. Microbiol. 78, 158–172. doi: 10.1111/j.1365-2958.2010.07320.x
Ishikawa, H., Otaka, H., Maki, K., Morita, T., and Aiba, H. (2012). The functional Hfq-binding module of bacterial sRNAs consists of a double or single hairpin preceded by a U-rich sequence and followed by a 3′ poly(U) tail. RNA 18, 1062–1074. doi: 10.1261/rna.031575.111
Itoh, Y., Naschberger, A., Mortezaei, N., Herrmann, J. M., and Amunts, A. (2020). Analysis of translating mitoribosome reveals functional characteristics of translation in mitochondria of fungi. Nat. Commun. 11, 5187. doi: 10.1038/s41467-020-18830-w
Jankowsky, E., and Harris, M. E. (2015). Specificity and nonspecificity in RNA-protein interactions. Nat Rev Mol Cell Biol 16, 533–544. doi: 10.1038/nrm4032
Jaskolowski, M., Ramrath, D. J. F., Bieri, P., Niemann, M., Mattei, S., Calderaro, S., et al. (2020). Structural insights into the mechanism of mitoribosomal large subunit biogenesis. Mol. Cell 79, 629–644e4. doi: 10.1016/j.molcel.2020.06.030
Jiang, W., Oikonomou, P., and Tavazoie, S. (2020). Comprehensive genome-wide perturbations via CRISPR adaptation reveal complex genetics of antibiotic sensitivity. Cell 180, 1002–1017e31. doi: 10.1016/j.cell.2020.02.007
Johnson, G. E., Lalanne, J. B., Peters, M. L., and Li, G. W. (2020). Functionally uncoupled transcription-translation in Bacillus subtilis. Nature 585, 124–128. doi: 10.1038/s41586-020-2638-5
Johnston, H. M., Barnes, W. M., Chumley, F. G., Bossi, L., and Roth, J. R. (1980). Model for regulation of the histidine operon of Salmonella. Proc. Natl. Acad. Sci. U.S.A. 77, 508–512. doi: 10.1073/pnas.77.1.508
Jomaa, A., Jain, N., Davis, J. H., Williamson, J. R., Britton, R. A., and Ortega, J. (2014). Functional domains of the 50S subunit mature late in the assembly process. Nucleic Acids Res. 42, 3419–3435. doi: 10.1093/nar/gkt1295
Jordán-Pla, A., Pérez-Martínez, M. E., and Pérez-Ortín, J. E. (2019). Measuring RNA polymerase activity genome-wide with high-resolution run-on-based methods. Methods 159-160, 177–182. doi: 10.1016/j.ymeth.2019.01.017
Jose, B. R., Gardner, P. P., and Barquist, L. (2019). Transcriptional noise and exaptation as sources for bacterial sRNAs. Biochem Soc Trans 47, 527–539. doi: 10.1042/BST20180171
Kambara, T. K., Ramsey, K. M., and Dove, S. L. (2018). Pervasive targeting of nascent transcripts by Hfq. Cell Rep 23, 1543–1552. doi: 10.1016/j.celrep.2018.03.134
Kang, J. Y., Llewellyn, E., Chen, J., Olinares, P. D. B., Brewer, J., Chait, B. T., et al. (2021). Structural basis for transcription complex disruption by the Mfd translocase. eLife 10, e62117. doi: 10.7554/eLife.62117
Kang, J. Y., Mooney, R. A., Nedialkov, Y., Saba, J., Mishanina, T. V., Artsimovitch, I., et al. (2018). Structural basis for transcript elongation control by NusG family universal regulators. Cell 173, 1650–1662e14. doi: 10.1016/j.cell.2018.05.017
Kato, A., Chen, H. D., Latifi, T., and Groisman, E. A. (2012). Reciprocal control between a bacterium’s regulatory system and the modification status of its lipopolysaccharide. Mol. Cell 47, 897–908. doi: 10.1016/j.molcel.2012.07.017
Kato, T., Yoshida, H., Miyata, T., Maki, Y., Wada, A., and Namba, K. (2010). Structure of the 100S ribosome in the hibernation stage revealed by electron cryomicroscopy. Structure 18, 719–724. doi: 10.1016/j.str.2010.02.017
Katsowich, N., Elbaz, N., Pal, R. R., Mills, E., Kobi, S., Kahan, T., et al. (2017). Host cell attachment elicits posttranscriptional regulation in infecting enteropathogenic bacteria. Science 355, 735–739. doi: 10.1126/science.aah4886
Keiler, K. C. (2015). Mechanisms of ribosome rescue in bacteria. Nat Rev Microbiol 13, 285–297. doi: 10.1038/nrmicro3438
Khan, M. A., Durica-Mitic, S., Göpel, Y., Heermann, R., and Görke, B. (2020). Small RNA-binding protein RapZ mediates cell envelope precursor sensing and signaling in Escherichia coli. EMBO J. 39, e103848. doi: 10.15252/embj.2019103848
Kino, T., Hurt, D. E., Ichijo, T., Nader, N., and Chrousos, G. P. (2010). Noncoding RNA Gas5 is a growth arrest- and starvation-associated repressor of the glucocorticoid receptor. Sci Signal 3, ra8. doi: 10.1126/scisignal.2000568
Kirkwood, K. J., Ahmad, Y., Larance, M., and Lamond, A. I. (2013). Characterization of native protein complexes and protein isoform variation using size-fractionation-based quantitative proteomics. Mol. Cell Proteomics. 12, 3851–3873. doi: 10.1074/mcp.M113.032367
Klinge, S., and Woolford, J. L. Jr. (2019). Ribosome assembly coming into focus. Nat Rev Mol Cell Biol 20, 116–131. doi: 10.1038/s41580-018-0078-y
Klodmann, J., Senkler, M., Rode, C., and Braun, H. P. (2011). Defining the protein complex proteome of plant mitochondria. Plant Physiol 157, 587–598. doi: 10.1104/pp.111.182352
Kopp, F., and Mendell, J. T. (2018). Functional classification and experimental dissection of long noncoding RNAs. Cell 172, 393–407. doi: 10.1016/j.cell.2018.01.011
Korostelev, A., Ermolenko, D. N., and Noller, H. F. (2008). Structural dynamics of the ribosome. Curr Opin Chem Biol 12, 674–683. doi: 10.1016/j.cbpa.2008.08.037
Kriner, M. A., Sevostyanova, A., and Groisman, E. A. (2016). Learning from the leaders: gene regulation by the transcription termination factor Rho. Trends Biochem. Sci. 41, 690–699. doi: 10.1016/j.tibs.2016.05.012
Kristensen, A. R., Gsponer, J., and Foster, L. J. (2012). A high-throughput approach for measuring temporal changes in the interactome. Nat Methods 9, 907–909. doi: 10.1038/nmeth.2131
Kritikos, G., Banzhaf, M., Herrera-Dominguez, L., Koumoutsi, A., Wartel, M., Zietek, M., et al. (2017). A tool named Iris for versatile high-throughput phenotyping in microorganisms. Nat. Microbiol. 2, 17014. doi: 10.1038/nmicrobiol.2017.14
Kuhn, C. D., Wilusz, J. E., Zheng, Y., Beal, P. A., and Joshua-Tor, L. (2015). On-enzyme refolding permits small RNA and tRNA surveillance by the CCA-adding enzyme. Cell 160, 644–658. doi: 10.1016/j.cell.2015.01.005
Kunte, H. J., Crane, R. A., Culham, D. E., Richmond, D., and Wood, J. M. (1999). Protein ProQ influences osmotic activation of compatible solute transporter ProP in Escherichia coli K-12. J Bacteriol 181, 1537–1543. doi: 10.1128/JB.181.5.1537-1543.1999
Lan, P., Zhou, B., Tan, M., Li, S., Cao, M., Wu, J., et al. (2020). Structural insight into precursor ribosomal RNA processing by ribonuclease MRP. Science 369, 656–663. doi: 10.1126/science.abc0149
Land, M., Hauser, L., Jun, S. R., Nookaew, I., Leuze, M. R., Ahn, T. H., et al. (2015). Insights from 20 years of bacterial genome sequencing. Funct Integr Genomics 15, 141–161. doi: 10.1007/s10142-015-0433-4
Langridge, G. C., Phan, M. D., Turner, D. J., Perkins, T. T., Parts, L., Haase, J., et al. (2009). Simultaneous assay of every Salmonella Typhi gene using one million transposon mutants. Genome Res 19, 2308–2316. doi: 10.1101/gr.097097.109
Larance, M., Kirkwood, K. J., Tinti, M., Brenes Murillo, A., Ferguson, M. A. J., and Lamond, A. I. (2016). Global membrane protein interactome analysis using in vivo crosslinking and mass spectrometry-based protein correlation profiling. Mol. Cell Proteomics. 15, 2476–2490. doi: 10.1074/mcp.O115.055467
Larance, M., and Lamond, A. I. (2015). Multidimensional proteomics for cell biology. Nat Rev Mol Cell Biol 16, 269–280. doi: 10.1038/nrm3970
Laux, A., Sexauer, A., Sivaselvarajah, D., Kaysen, A., and Brückner, R. (2015). Control of competence by related non-coding csRNAs in Streptococcus pneumoniae R6. Front Genet 6:246. doi: 10.3389/fgene.2015.00246
Lécrivain, A. L., Le Rhun, A., Renault, T. T., Ahmed-Begrich, R., Hahnke, K., and Charpentier, E. (2018). In vivo 3′-to-5′ exoribonuclease targetomes of Streptococcus pyogenes. Proc. Natl. Acad. Sci. U.S.A. 115, 11814–11819. doi: 10.1073/pnas.1809663115
Lee, F., and Yanofsky, C. (1977). Transcription termination at the trp operon attenuators of Escherichia coli and Salmonella Typhimurium: RNA secondary structure and regulation of termination. Proc. Natl. Acad. Sci. U.S.A. 74, 4365–4369. doi: 10.1073/pnas.74.10.4365
Lee, F. C. Y., and Ule, J. (2018). Advances in CLIP technologies for studies of protein-RNA interactions. Mol. Cell 69, 354–369. doi: 10.1016/j.molcel.2018.01.005
Lee, S., Kopp, F., Chang, T. C., Sataluri, A., Chen, B., Sivakumar, S., et al. (2016). Noncoding RNA NORAD regulates genomic stability by sequestering PUMILIO proteins. Cell 164, 69–80. doi: 10.1016/j.cell.2015.12.017
Li, N., Chen, Y., Guo, Q., Zhang, Y., Yuan, Y., Ma, C., et al. (2013). Cryo-EM structures of the late-stage assembly intermediates of the bacterial 50S ribosomal subunit. Nucleic Acids Res. 41, 7073–7083. doi: 10.1093/nar/gkt423
Licatalosi, D. D., Ye, X., and Jankowsky, E. (2020). Approaches for measuring the dynamics of RNA-protein interactions. Wiley Interdiscip Rev RNA 11, e1565. doi: 10.1002/wrna.1565
Lin, C., and Miles, W. O. (2019). Beyond CLIP: advances and opportunities to measure RBP-RNA and RNA-RNA interactions. Nucleic Acids Res. 47, 5490–5501. doi: 10.1093/nar/gkz295
Liu, B., Zuo, Y., and Steitz, T. A. (2015). Structural basis for transcription reactivation by RapA. Proc. Natl. Acad. Sci. U.S.A. 112, 2006–2010. doi: 10.1073/pnas.1417152112
Liu, S., Li, B., Liang, Q., Liu, A., Qu, L., and Yang, J. (2020). Classification and function of RNA-protein interactions. Wiley Interdiscip Rev RNA 11, e1601. doi: 10.1002/wrna.1601
Lowe, R., Shirley, N., Bleackley, M., Dolan, S., and Shafee, T. (2017). Transcriptomics technologies. PLoS Comput Biol 13:e1005457. doi: 10.1371/journal.pcbi.1005457
Mahbub, M., Hemm, L., Yang, Y., Kaur, R., Carmen, H., Engl, C., et al. (2020). mRNA localization, reaction centre biogenesis and thylakoid membrane targeting in cyanobacteria. Nat Plants 6, 1179–1191. doi: 10.1038/s41477-020-00764-2
Makarewich, C. A., and Olson, E. N. (2017). Mining for micropeptides. Trends Cell Biol. 27, 685–696. doi: 10.1016/j.tcb.2017.04.006
Mallam, A. L., Sae-Lee, W., Schaub, J. M., Tu, F., Battenhouse, A., Jang, Y. J., et al. (2019). Systematic discovery of endogenous human ribonucleoprotein complexes. Cell Rep 29, 1351–1368e5. doi: 10.1016/j.celrep.2019.09.060
Martin, W. J., and Reiter, N. J. (2017). Structural roles of noncoding RNAs in the heart of enzymatic complexes. Biochemistry 56, 3–13. doi: 10.1021/acs.biochem.6b01106
Matzov, D., Aibara, S., Basu, A., Zimermann, E., Bashan, A., Yap, M. N., et al. (2017). The cryo-EM structure of hibernating 100S ribosome dimer from pathogenic Staphylococcus aureus. Nat. Commun. 8, 723. doi: 10.1038/s41467-017-00753-8
Mayya, V. K., and Duchaine, T. F. (2019). Ciphers and executioners: how 3′-untranslated regions determine the fate of messenger RNAs. Front Genet 10:6. doi: 10.3389/fgene.2019.00006
Melamed, S., Adams, P. P., Zhang, A., Zhang, H., and Storz, G. (2020). RNA-RNA interactomes of ProQ and Hfq reveal overlapping and competing roles. Mol. Cell 77, 411–425e7. doi: 10.1016/j.molcel.2019.10.022
Menon, A. L., Poole, F. L. II, Cvetkovic, A., Trauger, S. A., Kalisiak, E., Scott, J. W., et al. (2009). Novel multiprotein complexes identified in the hyperthermophilic archaeon Pyrococcus furiosus by non-denaturing fractionation of the native proteome. Mol. Cell Proteomics. 8, 735–751. doi: 10.1074/mcp.M800246-MCP200
Merino, E., Babitzke, P., and Yanofsky, C. (1995). trp RNA-binding attenuation protein (TRAP)-trp leader RNA interactions mediate translational as well as transcriptional regulation of the Bacillus subtilis trp operon. J Bacteriol 177, 6362–6370. doi: 10.1128/jb.177.22.6362-6370.1995
Meyer, M. M. (2017). The role of mRNA structure in bacterial translational regulation. Wiley Interdiscip Rev RNA 8, e1370. doi: 10.1002/wrna.1370
Meyer, M. M. (2018). rRNA mimicry in RNA regulation of gene expression. Microbiol Spectr 6, doi: 10.1128/microbiolspec.RWR-0006-2017.
Milewski, S. (2002). Glucosamine-6-phosphate synthase–the multi-facets enzyme. Biochim Biophys Acta 1597, 173–192. doi: 10.1016/s0167-4838(02)00318-7
Milner, J. L., and Wood, J. M. (1989). Insertion proQ220::Tn5 alters regulation of proline porter II, a transporter of proline and glycine betaine in Escherichia coli. J Bacteriol 171, 947–951. doi: 10.1128/jb.171.2.947-951.1989
Mitra, K., Schaffitzel, C., Fabiola, F., Chapman, M. S., Ban, N., and Frank, J. (2006). Elongation arrest by SecM via a cascade of ribosomal RNA rearrangements. Mol. Cell 22, 533–543. doi: 10.1016/j.molcel.2006.05.003
Moffitt, J. R., Pandey, S., Boettiger, A. N., Wang, S., and Zhuang, X. (2016). Spatial organization shapes the turnover of a bacterial transcriptome. eLife 5, e13065. doi: 10.7554/eLife.13065
Mohanraju, P., Makarova, K. S., Zetsche, B., Zhang, F., Koonin, E. V., and Van Der Oost, J. (2016). Diverse evolutionary roots and mechanistic variations of the CRISPR-Cas systems. Science 353, aad5147. doi: 10.1126/science.aad5147
Moll, I., Afonyushkin, T., Vytvytska, O., Kaberdin, V. R., and Bläsi, U. (2003). Coincident Hfq binding and RNase E cleavage sites on mRNA and small regulatory RNAs. RNA 9, 1308–1314. doi: 10.1261/rna.5850703
Moreno, R., Hernández-Arranz, S., La Rosa, R., Yuste, L., Madhushani, A., Shingler, V., et al. (2015). The Crc and Hfq proteins of Pseudomonas putida cooperate in catabolite repression and formation of ribonucleic acid complexes with specific target motifs. Environ Microbiol 17, 105–118. doi: 10.1111/1462-2920.12499
Morita, T., and Aiba, H. (2019). Mechanism and physiological significance of autoregulation of the Escherichia coli hfq gene. RNA 25, 264–276. doi: 10.1261/rna.068106.118
Motulsky, H. (2010). Intuitive biostatistics: a nonmathematical guide to statistical thinking. New York, NY: Oxford University Press.
Moutaoufik, M. T., Malty, R., Amin, S., Zhang, Q., Phanse, S., Gagarinova, A., et al. (2019). Rewiring of the human mitochondrial interactome during neuronal reprogramming reveals regulators of the respirasome and neurogenesis. iScience 19, 1114–1132. doi: 10.1016/j.isci.2019.08.057
Mukherjee, S., Oshiro, R. T., Yakhnin, H., Babitzke, P., and Kearns, D. B. (2016). FliW antagonizes CsrA RNA binding by a noncompetitive allosteric mechanism. Proc. Natl. Acad. Sci. U.S.A. 113, 9870–9875. doi: 10.1073/pnas.1602455113
Mukherjee, S., Yakhnin, H., Kysela, D., Sokoloski, J., Babitzke, P., and Kearns, D. B. (2011). CsrA-FliW interaction governs flagellin homeostasis and a checkpoint on flagellar morphogenesis in Bacillus subtilis. Mol. Microbiol. 82, 447–461. doi: 10.1111/j.1365-2958.2011.07822.x
Müller, C. S., Bildl, W., Haupt, A., Ellenrieder, L., Becker, T., Hunte, C., et al. (2016). Cryo-slicing Blue Native-Mass Spectrometry (csBN-MS), a novel technology for high resolution complexome profiling. Mol. Cell Proteomics. 15, 669–681. doi: 10.1074/mcp.M115.054080
Müller-McNicoll, M., and Neugebauer, K. M. (2013). How cells get the message: dynamic assembly and function of mRNA-protein complexes. Nat. Rev. Genet. 14, 275–287. doi: 10.1038/nrg3434
Munitic, I., Giardino Torchia, M. L., Meena, N. P., Zhu, G., Li, C. C., and Ashwell, J. D. (2013). Optineurin insufficiency impairs IRF3 but not NF-kappaB activation in immune cells. J Immunol 191, 6231–6240. doi: 10.4049/jimmunol.1301696
Muñoz, M. J., Pérez Santangelo, M. S., Paronetto, M. P., De La Mata, M., Pelisch, F., Boireau, S., et al. (2009). DNA damage regulates alternative splicing through inhibition of RNA polymerase II elongation. Cell 137, 708–720. doi: 10.1016/j.cell.2009.03.010
Muth, G. W., Ortoleva-Donnelly, L., and Strobel, S. A. (2000). A single adenosine with a neutral pKa in the ribosomal peptidyl transferase center. Science 289, 947–950. doi: 10.1126/science.289.5481.947
Nakagawa, S., Yamazaki, T., and Hirose, T. (2018). Molecular dissection of nuclear paraspeckles: towards understanding the emerging world of the RNP milieu. Open Biol 8, 180150. doi: 10.1098/rsob.180150
Nakatogawa, H., and Ito, K. (2001). Secretion monitor, SecM, undergoes self-translation arrest in the cytosol. Mol. Cell 7, 185–192. doi: 10.1016/s1097-2765(01)00166-6
Nam, D., Choi, E., Shin, D., and Lee, E. J. (2016). tRNAPro-mediated downregulation of elongation factor P is required for mgtCBR expression during Salmonella infection. Mol. Microbiol. 102, 221–232. doi: 10.1111/mmi.13454
Neugebauer, K. M. (2019). Nascent RNA and the coordination of splicing with transcription. Cold Spring Harb Perspect Biol 11, a032227. doi: 10.1101/cshperspect.a032227
Nevo-Dinur, K., Nussbaum-Shochat, A., Ben-Yehuda, S., and Amster-Choder, O. (2011). Translation-independent localization of mRNA in E. coli. Science 331, 1081–1084. doi: 10.1126/science.1195691
Ni, X., Davis, J. H., Jain, N., Razi, A., Benlekbir, S., Mcarthur, A. G., et al. (2016). YphC and YsxC GTPases assist the maturation of the central protuberance, GTPase associated region and functional core of the 50S ribosomal subunit. Nucleic Acids Res. 44, 8442–8455. doi: 10.1093/nar/gkw678
Nichols, R. J., Sen, S., Choo, Y. J., Beltrao, P., Zietek, M., Chaba, R., et al. (2011). Phenotypic landscape of a bacterial cell. Cell 144, 143–156. doi: 10.1016/j.cell.2010.11.052
Nissen, P., Hansen, J., Ban, N., Moore, P. B., and Steitz, T. A. (2000). The structural basis of ribosome activity in peptide bond synthesis. Science 289, 920–930. doi: 10.1126/science.289.5481.920
Nithin, C., Mukherjee, S., and Bahadur, R. P. (2019). A structure-based model for the prediction of protein-RNA binding affinity. RNA 25, 1628–1645. doi: 10.1261/rna.071779.119
Obregon, K. A., Hoch, C. T., and Sukhodolets, M. V. (2015). Sm-like protein Hfq: composition of the native complex, modifications, and interactions. Biochim Biophys Acta 1854, 950–966. doi: 10.1016/j.bbapap.2015.03.016
Olejniczak, M., and Storz, G. (2017). ProQ/FinO-domain proteins: another ubiquitous family of RNA matchmakers? Mol. Microbiol. 104, 905–915. doi: 10.1111/mmi.13679
Omenn, G. S., Lane, L., Lundberg, E. K., Beavis, R. C., Overall, C. M., and Deutsch, E. W. (2016). Metrics for the Human Proteome Project 2016: progress on identifying and characterizing the human proteome, including post-translational modifications. J Proteome Res 15, 3951–3960. doi: 10.1021/acs.jproteome.6b00511
O’Reilly, F. J., Xue, L., Graziadei, A., Sinn, L., Lenz, S., Tegunov, D., et al. (2020). In-cell architecture of an actively transcribing-translating expressome. Science 369, 554–557. doi: 10.1126/science.abb3758
Oshiro, R. T., Rajendren, S., Hundley, H. A., and Kearns, D. B. (2019). Robust stoichiometry of FliW-CsrA governs flagellin homeostasis and cytoplasmic organization in Bacillus subtilis. mBio 10, e533–19. doi: 10.1128/mBio.00533-19
Otaka, H., Ishikawa, H., Morita, T., and Aiba, H. (2011). PolyU tail of rho-independent terminator of bacterial small RNAs is essential for Hfq action. Proc. Natl. Acad. Sci. U.S.A. 108, 13059–13064. doi: 10.1073/pnas.1107050108
Oussenko, I. A., Abe, T., Ujiie, H., Muto, A., and Bechhofer, D. H. (2005). Participation of 3′-to-5′ exoribonucleases in the turnover of Bacillus subtilis mRNA. J Bacteriol 187, 2758–2767. doi: 10.1128/JB.187.8.2758-2767.2005
Oussenko, I. A., Sanchez, R., and Bechhofer, D. H. (2002). Bacillus subtilis YhaM, a member of a new family of 3′-to-5′ exonucleases in gram-positive bacteria. J Bacteriol 184, 6250–6259. doi: 10.1128/jb.184.22.6250-6259.2002
Oxender, D. L., Zurawski, G., and Yanofsky, C. (1979). Attenuation in the Escherichia coli tryptophan operon: role of RNA secondary structure involving the tryptophan codon region. Proc. Natl. Acad. Sci. U.S.A. 76, 5524–5528. doi: 10.1073/pnas.76.11.5524
Ozata, D. M., Gainetdinov, I., Zoch, A., O’carroll, D., and Zamore, P. D. (2019). PIWI-interacting RNAs: small RNAs with big functions. Nat. Rev. Genet. 20, 89–108. doi: 10.1038/s41576-018-0073-3
Páleníková, P., Harbour, M. E., Ding, S., Fearnley, I. M., Van Haute, L., Rorbach, J., et al. (2021a). Quantitative density gradient analysis by mass spectrometry (qDGMS) and complexome profiling analysis (ComPrAn) R package for the study of macromolecular complexes. Biochim Biophys Acta Bioenerg 1862, 148399. doi: 10.1016/j.bbabio.2021.148399
Páleníková, P., Harbour, M. E., Prodi, F., Minczuk, M., Zeviani, M., Ghelli, A., et al. (2021b). Duplexing complexome profiling with SILAC to study human respiratory chain assembly defects. Biochim Biophys Acta Bioenerg 1862, 148395. doi: 10.1016/j.bbabio.2021.148395
Pandey, S., Gravel, C. M., Stockert, O. M., Wang, C. D., Hegner, C. L., Leblanc, H., et al. (2020). Genetic identification of the functional surface for RNA binding by Escherichia coli ProQ. Nucleic Acids Res. 48, 4507–4520. doi: 10.1093/nar/gkaa144
Pannuri, A., Yakhnin, H., Vakulskas, C. A., Edwards, A. N., Babitzke, P., and Romeo, T. (2012). Translational repression of NhaR, a novel pathway for multi-tier regulation of biofilm circuitry by CsrA. J Bacteriol 194, 79–89. doi: 10.1128/JB.06209-11
Papasaikas, P., and Valcárcel, J. (2016). The spliceosome: the ultimate RNA chaperone and sculptor. Trends Biochem. Sci. 41, 33–45. doi: 10.1016/j.tibs.2015.11.003
Papenfort, K., Bouvier, M., Mika, F., Sharma, C. M., and Vogel, J. (2010). Evidence for an autonomous 5′ target recognition domain in an Hfq-associated small RNA. Proc. Natl. Acad. Sci. U.S.A. 107, 20435–20440. doi: 10.1073/pnas.1009784107
Pei, X. Y., Dendooven, T., Sonnleitner, E., Chen, S., Bläsi, U., and Luisi, B. F. (2019). Architectural principles for Hfq/Crc-mediated regulation of gene expression. Elife 8, e43158. doi: 10.7554/eLife.43158
Pelava, A., Schneider, C., and Watkins, N. J. (2016). The importance of ribosome production, and the 5S RNP-MDM2 pathway, in health and disease. Biochem Soc Trans 44, 1086–1090. doi: 10.1042/BST20160106
Peltier, J. B., Cai, Y., Sun, Q., Zabrouskov, V., Giacomelli, L., Rudella, A., et al. (2006). The oligomeric stromal proteome of Arabidopsis thaliana chloroplasts. Mol. Cell Proteomics. 5, 114–133. doi: 10.1074/mcp.M500180-MCP200
Peluso, P., Herschlag, D., Nock, S., Freymann, D. M., Johnson, A. E., and Walter, P. (2000). Role of 4.5S RNA in assembly of the bacterial signal recognition particle with its receptor. Science 288, 1640–1643. doi: 10.1126/science.288.5471.1640
Peters, J. M., Colavin, A., Shi, H., Czarny, T. L., Larson, M. H., Wong, S., et al. (2016). A comprehensive, CRISPR-based functional analysis of essential genes in bacteria. Cell 165, 1493–1506. doi: 10.1016/j.cell.2016.05.003
Polikanov, Y. S., Blaha, G. M., and Steitz, T. A. (2012). How hibernation factors RMF, HPF, and YfiA turn off protein synthesis. Science 336, 915–918. doi: 10.1126/science.1218538
Potts, A. H., Vakulskas, C. A., Pannuri, A., Yakhnin, H., Babitzke, P., and Romeo, T. (2017). Global role of the bacterial post-transcriptional regulator CsrA revealed by integrated transcriptomics. Nat. Commun. 8, 1596. doi: 10.1038/s41467-017-01613-1
Proshkin, S., Rahmouni, A. R., Mironov, A., and Nudler, E. (2010). Cooperation between translating ribosomes and RNA polymerase in transcription elongation. Science 328, 504–508. doi: 10.1126/science.1184939
Puerta-Fernandez, E., Barrick, J. E., Roth, A., and Breaker, R. R. (2006). Identification of a large noncoding RNA in extremophilic eubacteria. Proc. Natl. Acad. Sci. U.S.A. 103, 19490–19495. doi: 10.1073/pnas.0607493103
Pusic, P., Tata, M., Wolfinger, M. T., Sonnleitner, E., Häussler, S., and Bläsi, U. (2016). Cross-regulation by CrcZ RNA controls anoxic biofilm formation in Pseudomonas aeruginosa. Sci. Rep. 6, 39621. doi: 10.1038/srep39621
Pyndiah, S., Lasserre, J. P., Ménard, A., Claverol, S., Prouzet-Mauléon, V., Mégraud, F., et al. (2007). Two-dimensional blue native/SDS gel electrophoresis of multiprotein complexes from Helicobacter pylori. Mol. Cell Proteomics. 6, 193–206. doi: 10.1074/mcp.M600363-MCP200
Quattrone, A., and Dassi, E. (2019). The architecture of the human RNA-binding protein regulatory network. iScience 21, 706–719. doi: 10.1016/j.isci.2019.10.058
Queiroz, R. M. L., Smith, T., Villanueva, E., Marti-Solano, M., Monti, M., Pizzinga, M., et al. (2019). Comprehensive identification of RNA-protein interactions in any organism using orthogonal organic phase separation (OOPS). Nat Biotechnol 37, 169–178. doi: 10.1038/s41587-018-0001-2
Rabuck-Gibbons, J. N., Popova, A. M., Greene, E. M., Cervantes, C. F., Lyumkis, D., and Williamson, J. R. (2020). SrmB rescues trapped ribosome assembly intermediates. J Mol Biol 432, 978–990. doi: 10.1016/j.jmb.2019.12.013
Rai, J., Parker, M. D., Huang, H., Choy, S., Ghalei, H., Johnson, M. C., et al. (2021). An open interface in the pre-80S ribosome coordinated by ribosome assembly factors Tsr1 and Dim1 enables temporal regulation of Fap7. RNA 27, 221–233. doi: 10.1261/rna.077610.120
Ramrath, D. J. F., Niemann, M., Leibundgut, M., Bieri, P., Prange, C., Horn, E. K., et al. (2018). Evolutionary shift toward protein-based architecture in trypanosomal mitochondrial ribosomes. Science 362, eaau7735. doi: 10.1126/science.aau7735
Rao, F., Short, F. L., Voss, J. E., Blower, T. R., Orme, A. L., Whittaker, T. E., et al. (2015). Co-evolution of quaternary organization and novel RNA tertiary interactions revealed in the crystal structure of a bacterial protein-RNA toxin-antitoxin system. Nucleic Acids Res. 43, 9529–9540. doi: 10.1093/nar/gkv868
Razi, A., Davis, J. H., Hao, Y., Jahagirdar, D., Thurlow, B., Basu, K., et al. (2019). Role of Era in assembly and homeostasis of the ribosomal small subunit. Nucleic Acids Res. 47, 8301–8317. doi: 10.1093/nar/gkz571
Rederstorff, M., Bernhart, S. H., Tanzer, A., Zywicki, M., Perfler, K., Lukasser, M., et al. (2010). RNPomics: defining the ncRNA transcriptome by cDNA library generation from ribonucleo-protein particles. Nucleic Acids Res. 38, e113. doi: 10.1093/nar/gkq057
Reichenbach, B., Maes, A., Kalamorz, F., Hajnsdorf, E., and Görke, B. (2008). The small RNA GlmY acts upstream of the sRNA GlmZ in the activation of glmS expression and is subject to regulation by polyadenylation in Escherichia coli. Nucleic Acids Res. 36, 2570–2580. doi: 10.1093/nar/gkn091
Reiter, N. J., Osterman, A., Torres-Larios, A., Swinger, K. K., Pan, T., and Mondragón, A. (2010). Structure of a bacterial ribonuclease P holoenzyme in complex with tRNA. Nature 468, 784–789. doi: 10.1038/nature09516
Renda, A., Poly, S., Lai, Y. J., Pannuri, A., Yakhnin, H., Potts, A. H., et al. (2020). CsrA-mediated translational activation of ymdA expression in Escherichia coli. mBio 11, e849–20. doi: 10.1128/mBio.00849-20
Richter, J. D., and Coller, J. (2015). Pausing on polyribosomes: make way for elongation in translational control. Cell 163, 292–300. doi: 10.1016/j.cell.2015.09.041
Richter-Dennerlein, R., Oeljeklaus, S., Lorenzi, I., Ronsör, C., Bareth, B., Schendzielorz, A. B., et al. (2016). Mitochondrial protein synthesis adapts to influx of nuclear-encoded protein. Cell 167, 471–483e10. doi: 10.1016/j.cell.2016.09.003
Riediger, M., Spät, P., Bliger, R., Voigt, K., Maček, B., and Hess, W. R. (2020). Analysis of a photosynthetic cyanobacterium rich in internal membrane systems via gradient profiling by sequencing (Grad-seq). Plant Cell doi: 10.1093/plcell/koaa017 [Epub ahead of print].
Rissland, O. S. (2017). The organization and regulation of mRNA-protein complexes. Wiley Interdiscip Rev RNA 8, e1369. doi: 10.1002/wrna.1369
Roland, K. L., Liu, C. G., and Turnbough, C. L. Jr. (1988). Role of the ribosome in suppressing transcriptional termination at the pyrBI attenuator of Escherichia coli K-12. Proc. Natl. Acad. Sci. U.S.A. 85, 7149–7153. doi: 10.1073/pnas.85.19.7149
Romby, P., Caillet, J., Ebel, C., Sacerdot, C., Graffe, M., Eyermann, F., et al. (1996). The expression of E. coli threonyl-tRNA synthetase is regulated at the translational level by symmetrical operator-repressor interactions. EMBO J. 15, 5976–5987. doi: 10.1002/j.1460-2075.1996.tb00984.x
Romeo, T., and Babitzke, P. (2018). Global regulation by CsrA and its RNA antagonists. Microbiol Spectr 6, doi: 10.1128/microbiolspec.RWR-0009-2017.
Routh, S. B., and Sankaranarayanan, R. (2017). Mechanistic insights into catalytic RNA-protein complexes involved in translation of the genetic code. Adv Protein Chem Struct Biol 109, 305–353. doi: 10.1016/bs.apcsb.2017.04.002
Rugen, N., Straube, H., Franken, L. E., Braun, H. P., and Eubel, H. (2019). Complexome profiling reveals association of PPR proteins with ribosomes in the mitochondria of plants. Mol. Cell Proteomics. 18, 1345–1362. doi: 10.1074/mcp.RA119.001396
Said, N., Krupp, F., Anedchenko, E., Santos, K. F., Dybkov, O., Huang, Y. H., et al. (2017). Structural basis for lambdaN-dependent processive transcription antitermination. Nat. Microbiol. 2, 17062. doi: 10.1038/nmicrobiol.2017.62
Saliba, A. E., Santos, S. C., and Vogel, J. (2017). New RNA-seq approaches for the study of bacterial pathogens. Curr. Opin. Microbiol. 35, 78–87. doi: 10.1016/j.mib.2017.01.001
Salomon, W. E., Jolly, S. M., Moore, M. J., Zamore, P. D., and Serebrov, V. (2015). Single-molecule imaging reveals that Argonaute reshapes the binding properties of its nucleic acid guides. Cell 162, 84–95. doi: 10.1016/j.cell.2015.06.029
Samson, J. E., Spinelli, S., Cambillau, C., and Moineau, S. (2013). Structure and activity of AbiQ, a lactococcal endoribonuclease belonging to the type III toxin-antitoxin system. Mol. Microbiol. 87, 756–768. doi: 10.1111/mmi.12129
Santiago-Frangos, A., Jeliazkov, J. R., Gray, J. J., and Woodson, S. A. (2017). Acidic C-terminal domains autoregulate the RNA chaperone Hfq. Elife 6, e27049. doi: 10.7554/eLife.27049
Santiago-Frangos, A., and Woodson, S. A. (2018). Hfq chaperone brings speed dating to bacterial sRNA. Wiley Interdiscip Rev RNA 9, e1475. doi: 10.1002/wrna.1475
Sashital, D. G., Greeman, C. A., Lyumkis, D., Potter, C. S., Carragher, B., and Williamson, J. R. (2014). A combined quantitative mass spectrometry and electron microscopy analysis of ribosomal 30S subunit assembly in E. coli. Elife 3, e04491. doi: 10.7554/eLife.04491
Sauer, E., Schmidt, S., and Weichenrieder, O. (2012). Small RNA binding to the lateral surface of Hfq hexamers and structural rearrangements upon mRNA target recognition. Proc. Natl. Acad. Sci. U.S.A. 109, 9396–9401. doi: 10.1073/pnas.1202521109
Sauer, E., and Weichenrieder, O. (2011). Structural basis for RNA 3′-end recognition by Hfq. Proc. Natl. Acad. Sci. U.S.A. 108, 13065–13070. doi: 10.1073/pnas.1103420108
Schägger, H., and Von Jagow, G. (1991). Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal Biochem 199, 223–231. doi: 10.1016/0003-2697(91)90094-a
Schimo, S., Wittig, I., Pos, K. M., and Ludwig, B. (2017). Cytochrome c oxidase biogenesis and metallochaperone interactions: steps in the assembly pathway of a bacterial complex. PLoS One 12:e0170037. doi: 10.1371/journal.pone.0170037
Schnorpfeil, A., Kranz, M., Kovács, M., Kirsch, C., Gartmann, J., Brunner, I., et al. (2013). Target evaluation of the non-coding csRNAs reveals a link of the two-component regulatory system CiaRH to competence control in Streptococcus pneumoniae R6. Mol. Microbiol. 89, 334–349. doi: 10.1111/mmi.12277
Schubert, M., Lapouge, K., Duss, O., Oberstrass, F. C., Jelesarov, I., Haas, D., et al. (2007). Molecular basis of messenger RNA recognition by the specific bacterial repressing clamp RsmA/CsrA. Nat. Struct. Mol. Biol. 14, 807–813. doi: 10.1038/nsmb1285
Schwenk, J., Harmel, N., Brechet, A., Zolles, G., Berkefeld, H., Müller, C. S., et al. (2012). High-resolution proteomics unravel architecture and molecular diversity of native AMPA receptor complexes. Neuron 74, 621–633. doi: 10.1016/j.neuron.2012.03.034
Sedlyarova, N., Shamovsky, I., Bharati, B. K., Epshtein, V., Chen, J., Gottesman, S., et al. (2016). sRNA-mediated control of transcription termination in E. coli. Cell 167, 111–121e3. doi: 10.1016/j.cell.2016.09.004
Seffouh, A., Jain, N., Jahagirdar, D., Basu, K., Razi, A., Ni, X., et al. (2019). Structural consequences of the interaction of RbgA with a 50S ribosomal subunit assembly intermediate. Nucleic Acids Res. 47, 10414–10425. doi: 10.1093/nar/gkz770
Selby, C. P., and Sancar, A. (1993). Molecular mechanism of transcription-repair coupling. Science 260, 53–58. doi: 10.1126/science.8465200
Senkler, J., Senkler, M., Eubel, H., Hildebrandt, T., Lengwenus, C., Schertl, P., et al. (2017). The mitochondrial complexome of Arabidopsis thaliana. Plant J 89, 1079–1092. doi: 10.1111/tpj.13448
Sexton, J. A., and Vogel, J. P. (2004). Regulation of hypercompetence in Legionella pneumophila. J Bacteriol 186, 3814–3825. doi: 10.1128/JB.186.12.3814-3825.2004
Shajani, Z., Sykes, M. T., and Williamson, J. R. (2011). Assembly of bacterial ribosomes. Annu Rev Biochem 80, 501–526. doi: 10.1146/annurev-biochem-062608-160432
Shalem, O., Sanjana, N. E., and Zhang, F. (2015). High-throughput functional genomics using CRISPR-Cas9. Nat. Rev. Genet. 16, 299–311. doi: 10.1038/nrg3899
Shalgi, R., Hurt, J. A., Krykbaeva, I., Taipale, M., Lindquist, S., and Burge, C. B. (2013). Widespread regulation of translation by elongation pausing in heat shock. Mol. Cell 49, 439–452. doi: 10.1016/j.molcel.2012.11.028
Shatsky, M., Dong, M., Liu, H., Yang, L. L., Choi, M., Singer, M. E., et al. (2016). Quantitative tagless copurification: a method to validate and identify protein-protein interactions. Mol. Cell Proteomics. 15, 2186–2202. doi: 10.1074/mcp.M115.057117
Shchepachev, V., Bresson, S., Spanos, C., Petfalski, E., Fischer, L., Rappsilber, J., et al. (2019). Defining the RNA interactome by total RNA-associated protein purification. Mol Syst Biol 15, e8689. doi: 10.15252/msb.20188689
Sheidy, D. T., and Zielke, R. A. (2013). Analysis and expansion of the role of the Escherichia coli protein ProQ. PLoS One 8:e79656. doi: 10.1371/journal.pone.0079656
Sheu-Gruttadauria, J., and MacRae, I. J. (2017). Structural foundations of RNA silencing by Argonaute. J Mol Biol 429, 2619–2639. doi: 10.1016/j.jmb.2017.07.018
Shi, J., Wen, A., Zhao, M., Jin, S., You, L., Shi, Y., et al. (2020). Structural basis of Mfd-dependent transcription termination. Nucleic Acids Res. 48, 11762–11772. doi: 10.1093/nar/gkaa904
Shi, P. Y., Maizels, N., and Weiner, A. M. (1998). CCA addition by tRNA nucleotidyltransferase: polymerization without translocation? EMBO J. 17, 3197–3206. doi: 10.1093/emboj/17.11.3197
Shine, J., and Dalgarno, L. (1974). The 3′-terminal sequence of Escherichia coli 16S ribosomal RNA: complementarity to nonsense triplets and ribosome binding sites. Proc. Natl. Acad. Sci. U.S.A. 71, 1342–1346. doi: 10.1073/pnas.71.4.1342
Short, F. L., Akusobi, C., Broadhurst, W. R., and Salmond, G. P. C. (2018). The bacterial Type III toxin-antitoxin system, ToxIN, is a dynamic protein-RNA complex with stability-dependent antiviral abortive infection activity. Sci. Rep. 8, 1013. doi: 10.1038/s41598-017-18696-x
Short, F. L., Pei, X. Y., Blower, T. R., Ong, S. L., Fineran, P. C., Luisi, B. F., et al. (2013). Selectivity and self-assembly in the control of a bacterial toxin by an antitoxic noncoding RNA pseudoknot. Proc. Natl. Acad. Sci. U.S.A. 110, E241–E249. doi: 10.1073/pnas.1216039110
Silva, I. J., Barahona, S., Eyraud, A., Lalaouna, D., Figueroa-Bossi, N., Massé, E., et al. (2019). SraL sRNA interaction regulates the terminator by preventing premature transcription termination of rho mRNA. Proc. Natl. Acad. Sci. U.S.A. 116, 3042–3051. doi: 10.1073/pnas.1811589116
Sim, S., and Wolin, S. L. (2018). Bacterial Y RNAs: gates, tethers, and tRNA mimics. Microbiol Spectr 6, doi: 10.1128/microbiolspec.RWR-0023-2018.
Simms, C. L., Thomas, E. N., and Zaher, H. S. (2017). Ribosome-based quality control of mRNA and nascent peptides. Wiley Interdiscip Rev RNA 8, doi: 10.1002/wrna.1366.
Smirnov, A., Förstner, K. U., Holmqvist, E., Otto, A., Günster, R., Becher, D., et al. (2016). Grad-seq guides the discovery of ProQ as a major small RNA-binding protein. Proc. Natl. Acad. Sci. U.S.A. 113, 11591–11596. doi: 10.1073/pnas.1609981113
Smirnov, A., Schneider, C., Hör, J., and Vogel, J. (2017a). Discovery of new RNA classes and global RNA-binding proteins. Curr. Opin. Microbiol. 39, 152–160. doi: 10.1016/j.mib.2017.11.016
Smirnov, A., Wang, C., Drewry, L. L., and Vogel, J. (2017b). Molecular mechanism of mRNA repression in trans by a ProQ-dependent small RNA. EMBO J. 36, 1029–1045. doi: 10.15252/embj.201696127
Smith, K. P., Hall, L. L., and Lawrence, J. B. (2020a). Nuclear hubs built on RNAs and clustered organization of the genome. Curr Opin Cell Biol 64, 67–76. doi: 10.1016/j.ceb.2020.02.015
Smith, T., Villanueva, E., Queiroz, R. M. L., Dawson, C. S., Elzek, M., Urdaneta, E. C., et al. (2020b). Organic phase separation opens up new opportunities to interrogate the RNA-binding proteome. Curr Opin Chem Biol 54, 70–75. doi: 10.1016/j.cbpa.2020.01.009
Sonnleitner, E., and Bläsi, U. (2014). Regulation of Hfq by the RNA CrcZ in Pseudomonas aeruginosa carbon catabolite repression. PLoS Genet 10:e1004440. doi: 10.1371/journal.pgen.1004440
Sonnleitner, E., Prindl, K., and Bläsi, U. (2017). The Pseudomonas aeruginosa CrcZ RNA interferes with Hfq-mediated riboregulation. PLoS One 12:e0180887. doi: 10.1371/journal.pone.0180887
Sonnleitner, E., Wulf, A., Campagne, S., Pei, X. Y., Wolfinger, M. T., Forlani, G., et al. (2018). Interplay between the catabolite repression control protein Crc, Hfq and RNA in Hfq-dependent translational regulation in Pseudomonas aeruginosa. Nucleic Acids Res. 46, 1470–1485. doi: 10.1093/nar/gkx1245
Soufari, H., Waltz, F., Parrot, C., Durrieu-Gaillard, S., Bochler, A., Kuhn, L., et al. (2020). Structure of the mature kinetoplastids mitoribosome and insights into its large subunit biogenesis. Proc. Natl. Acad. Sci. U.S.A. 117, 29851–29861. doi: 10.1073/pnas.2011301117
Sowa, S. W., Gelderman, G., Leistra, A. N., Buvanendiran, A., Lipp, S., Pitaktong, A., et al. (2017). Integrative FourD omics approach profiles the target network of the carbon storage regulatory system. Nucleic Acids Res. 45, 1673–1686. doi: 10.1093/nar/gkx048
Stein, E. M., Kwiatkowska, J., Basczok, M. M., Gravel, C. M., Berry, K. E., and Olejniczak, M. (2020). Determinants of RNA recognition by the FinO domain of the Escherichia coli ProQ protein. Nucleic Acids Res. 48, 7502–7519. doi: 10.1093/nar/gkaa497
Steitz, J. A., and Jakes, K. (1975). How ribosomes select initiator regions in mRNA: base pair formation between the 3′ terminus of 16S rRNA and the mRNA during initiation of protein synthesis in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 72, 4734–4738. doi: 10.1073/pnas.72.12.4734
Stevenson-Jones, F., Woodgate, J., Castro-Roa, D., and Zenkin, N. (2020). Ribosome reactivates transcription by physically pushing RNA polymerase out of transcription arrest. Proc. Natl. Acad. Sci. U.S.A. 117, 8462–8467. doi: 10.1073/pnas.1919985117
Storz, G., Wolf, Y. I., and Ramamurthi, K. S. (2014). Small proteins can no longer be ignored. Annu Rev Biochem 83, 753–777. doi: 10.1146/annurev-biochem-070611-102400
Strecker, V., Wumaier, Z., Wittig, I., and Schägger, H. (2010). Large pore gels to separate mega protein complexes larger than 10 MDa by blue native electrophoresis: isolation of putative respiratory strings or patches. Proteomics 10, 3379–3387. doi: 10.1002/pmic.201000343
Subramaniam, A. R., Zid, B. M., and O’shea, E. K. (2014). An integrated approach reveals regulatory controls on bacterial translation elongation. Cell 159, 1200–1211. doi: 10.1016/j.cell.2014.10.043
Sun, Q., Zhu, X., Qi, J., An, W., Lan, P., Tan, D., et al. (2017). Molecular architecture of the 90S small subunit pre-ribosome. Elife 6, e22086. doi: 10.7554/eLife.22086
Sun, Y., Zhang, Y., Aik, W. S., Yang, X. C., Marzluff, W. F., Walz, T., et al. (2020). Structure of an active human histone pre-mRNA 3′-end processing machinery. Science 367, 700–703. doi: 10.1126/science.aaz7758
Tanaka, H., Kato, K., Yamashita, E., Sumizawa, T., Zhou, Y., Yao, M., et al. (2009). The structure of rat liver vault at 3.5 angstrom resolution. Science 323, 384–388. doi: 10.1126/science.1164975
Torres-Larios, A., Dock-Bregeon, A. C., Romby, P., Rees, B., Sankaranarayanan, R., Caillet, J., et al. (2002). Structural basis of translational control by Escherichia coli threonyl tRNA synthetase. Nat Struct Biol 9, 343–347. doi: 10.1038/nsb789
Tree, J. J., Gerdes, K., and Tollervey, D. (2018). Transcriptome-wide analysis of protein-RNA and RNA-RNA interactions in pathogenic bacteria. Methods Enzymol 612, 467–488. doi: 10.1016/bs.mie.2018.08.009
Trendel, J., Schwarzl, T., Horos, R., Prakash, A., Bateman, A., Hentze, M. W., et al. (2019). The human RNA-binding proteome and its dynamics during translational arrest. Cell 176, 391–403. doi: 10.1016/j.cell.2018.11.004
Turnbough, C. L. Jr. (2019). Regulation of bacterial gene expression by transcription attenuation. Microbiol Mol Biol Rev 83, e00019–e19. doi: 10.1128/MMBR.00019-19
Turnbough, C. L. Jr., Hicks, K. L., and Donahue, J. P. (1983). Attenuation control of pyrBI operon expression in Escherichia coli K-12. Proc. Natl. Acad. Sci. U.S.A. 80, 368–372. doi: 10.1073/pnas.80.2.368
Updegrove, T. B., Zhang, A., and Storz, G. (2016). Hfq: the flexible RNA matchmaker. Curr. Opin. Microbiol. 30, 133–138. doi: 10.1016/j.mib.2016.02.003
Urban, J. H., and Vogel, J. (2008). Two seemingly homologous noncoding RNAs act hierarchically to activate glmS mRNA translation. PLoS Biol 6:e64. doi: 10.1371/journal.pbio.0060064
Urdaneta, E. C., Vieira-Vieira, C. H., Hick, T., Wessels, H. H., Figini, D., Moschall, R., et al. (2019). Purification of cross-linked RNA-protein complexes by phenol-toluol extraction. Nat. Commun. 10, 990. doi: 10.1038/s41467-019-08942-3
Valle, M., Zavialov, A., Sengupta, J., Rawat, U., Ehrenberg, M., and Frank, J. (2003). Locking and unlocking of ribosomal motions. Cell 114, 123–134. doi: 10.1016/s0092-8674(03)00476-8
Van Assche, E., Van Puyvelde, S., Vanderleyden, J., and Steenackers, H. P. (2015). RNA-binding proteins involved in post-transcriptional regulation in bacteria. Front Microbiol 6:141. doi: 10.3389/fmicb.2015.00141
van Heesch, S., Van Iterson, M., Jacobi, J., Boymans, S., Essers, P. B., De Bruijn, E., et al. (2014). Extensive localization of long noncoding RNAs to the cytosol and mono- and polyribosomal complexes. Genome Biol 15, R6. doi: 10.1186/gb-2014-15-1-r6
Van Nostrand, E. L., Freese, P., Pratt, G. A., Wang, X., Wei, X., Xiao, R., et al. (2020). A large-scale binding and functional map of human RNA-binding proteins. Nature 583, 711–719. doi: 10.1038/s41586-020-2077-3
van Opijnen, T., and Camilli, A. (2012). A fine scale phenotype-genotype virulence map of a bacterial pathogen. Genome Res 22, 2541–2551. doi: 10.1101/gr.137430.112
Van Strien, J., Guerrero-Castillo, S., Chatzispyrou, I. A., Houtkooper, R. H., Brandt, U., and Huynen, M. A. (2019). COmplexome Profiling ALignment (COPAL) reveals remodeling of mitochondrial protein complexes in Barth syndrome. Bioinformatics 35, 3083–3091. doi: 10.1093/bioinformatics/btz025
Večerek, B., Moll, I., and Bläsi, U. (2005). Translational autocontrol of the Escherichia coli hfq RNA chaperone gene. RNA 11, 976–984. doi: 10.1261/rna.2360205
Venturini, E., Svensson, S. L., Maaß, S., Gelhausen, R., Eggenhofer, F., Li, L., et al. (2020). A global data-driven census of Salmonella small proteins and their potential functions in bacterial virulence. microLife 1, uqaa002. doi: 10.1093/femsml/uqaa002
Vidal, M., Cusick, M. E., and Barabási, A. L. (2011). Interactome networks and human disease. Cell 144, 986–998. doi: 10.1016/j.cell.2011.02.016
Vidoni, S., Harbour, M. E., Guerrero-Castillo, S., Signes, A., Ding, S., Fearnley, I. M., et al. (2017). MR-1S interacts with PET100 and PET117 in module-based assembly of human cytochrome c oxidase. Cell Rep 18, 1727–1738. doi: 10.1016/j.celrep.2017.01.044
Wagner, E. G. H., and Romby, P. (2015). Small RNAs in bacteria and archaea: who they are, what they do, and how they do it. Adv Genet 90, 133–208. doi: 10.1016/bs.adgen.2015.05.001
Wagner, S., Herrmannová, A., Hronová, V., Gunišová, S., Sen, N. D., Hannan, R. D., et al. (2020). Selectve translation complex profiling reveals staged initiation and co-translational assembly of initiation factor complexes. Mol. Cell 79, 546–560e7. doi: 10.1016/j.molcel.2020.06.004
Wagner, S. D., Yakovchuk, P., Gilman, B., Ponicsan, S. L., Drullinger, L. F., Kugel, J. F., et al. (2013). RNA polymerase II acts as an RNA-dependent RNA polymerase to extend and destabilize a non-coding RNA. EMBO J. 32, 781–790. doi: 10.1038/emboj.2013.18
Wallace, J. G., Zhou, Z., and Breaker, R. R. (2012). OLE RNA protects extremophilic bacteria from alcohol toxicity. Nucleic Acids Res. 40, 6898–6907. doi: 10.1093/nar/gks352
Walther, K., and Schulte, L. N. (2020). The role of lncRNAs in innate immunity and inflammation. RNA Biol. doi: 10.1080/15476286.2020.1845505 [Epub ahead of print].
Wan, C., Borgeson, B., Phanse, S., Tu, F., Drew, K., Clark, G., et al. (2015). Panorama of ancient metazoan macromolecular complexes. Nature 525, 339–344. doi: 10.1038/nature14877
Wan, F., Wang, Q., Tan, J., Tan, M., Chen, J., Shi, S., et al. (2019a). Cryo-electron microscopy structure of an archaeal ribonuclease P holoenzyme. Nat. Commun. 10, 2617. doi: 10.1038/s41467-019-10496-3
Wan, R., Bai, R., and Shi, Y. (2019b). Molecular choreography of pre-mRNA splicing by the spliceosome. Curr. Opin. Struct. Biol. 59, 124–133. doi: 10.1016/j.sbi.2019.07.010
Wang, C., Molodtsov, V., Firlar, E., Kaelber, J. T., Blaha, G., Su, M., et al. (2020a). Structural basis of transcription-translation coupling. Science 369, 1359–1365. doi: 10.1126/science.abb5317
Wang, C., Richter-Dennerlein, R., Pacheu-Grau, D., Liu, F., Zhu, Y., Dennerlein, S., et al. (2020b). MITRAC15/COA1 promotes mitochondrial translation in a ND2 ribosome-nascent chain complex. EMBO Rep 21, e48833. doi: 10.15252/embr.201948833
Wang, T., Birsoy, K., Hughes, N. W., Krupczak, K. M., Post, Y., Wei, J. J., et al. (2015). Identification and characterization of essential genes in the human genome. Science 350, 1096–1101. doi: 10.1126/science.aac7041
Wang, T., Wei, J. J., Sabatini, D. M., and Lander, E. S. (2014). Genetic screens in human cells using the CRISPR-Cas9 system. Science 343, 80–84. doi: 10.1126/science.1246981
Wang, Z., Gerstein, M., and Snyder, M. (2009). RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63. doi: 10.1038/nrg2484
Wassarman, K. M. (2018). 6S RNA, a global regulator of transcription. Microbiol Spectr 6, doi: 10.1128/microbiolspec.RWR-0019-2018.
Wassarman, K. M., and Saecker, R. M. (2006). Synthesis-mediated release of a small RNA inhibitor of RNA polymerase. Science 314, 1601–1603. doi: 10.1126/science.1134830
Wassarman, K. M., and Storz, G. (2000). 6S RNA regulates E. coli RNA polymerase activity. Cell 101, 613–623. doi: 10.1016/s0092-8674(00)80873-9
Waters, L. S., Sandoval, M., and Storz, G. (2011). The Escherichia coli MntR miniregulon includes genes encoding a small protein and an efflux pump required for manganese homeostasis. J Bacteriol 193, 5887–5897. doi: 10.1128/JB.05872-11
Weaver, J., Mohammad, F., Buskirk, A. R., and Storz, G. (2019). Identifying small proteins by ribosome profiling with stalled initiation complexes. mBio 10, e2819–18. doi: 10.1128/mBio.02819-18
Webster, M. W., Takacs, M., Zhu, C., Vidmar, V., Eduljee, A., Abdelkareem, M., et al. (2020). Structural basis of transcription-translation coupling and collision in bacteria. Science 369, 1355–1359. doi: 10.1126/science.abb5036
Weinberg, Z., Lunse, C. E., Corbino, K. A., Ames, T. D., Nelson, J. W., Roth, A., et al. (2017). Detection of 224 candidate structured RNAs by comparative analysis of specific subsets of intergenic regions. Nucleic Acids Res. 45, 10811–10823. doi: 10.1093/nar/gkx699
Weinberg, Z., Perreault, J., Meyer, M. M., and Breaker, R. R. (2009). Exceptional structured noncoding RNAs revealed by bacterial metagenome analysis. Nature 462, 656–659. doi: 10.1038/nature08586
Wessels, H. J., Vogel, R. O., Lightowlers, R. N., Spelbrink, J. N., Rodenburg, R. J., Van Den Heuvel, L. P., et al. (2013). Analysis of 953 human proteins from a mitochondrial HEK293 fraction by complexome profiling. PLoS One 8:e68340. doi: 10.1371/journal.pone.0068340
Wessels, H. J., Vogel, R. O., Van Den Heuvel, L., Smeitink, J. A., Rodenburg, R. J., Nijtmans, L. G., et al. (2009). LC-MS/MS as an alternative for SDS-PAGE in blue native analysis of protein complexes. Proteomics 9, 4221–4228. doi: 10.1002/pmic.200900157
Westermann, A. J., Förstner, K. U., Amman, F., Barquist, L., Chao, Y., Schulte, L. N., et al. (2016). Dual RNA-seq unveils noncoding RNA functions in host-pathogen interactions. Nature 529, 496–501. doi: 10.1038/nature16547
Westermann, A. J., Venturini, E., Sellin, M. E., Förstner, K. U., Hardt, W. D., and Vogel, J. (2019). The major RNA-binding protein ProQ impacts virulence gene expression in Salmonella enterica serovar Typhimurium. mBio 10, e2504–18. doi: 10.1128/mBio.02504-18
Widner, D. L., Harris, K. A., Corey, L., and Breaker, R. R. (2020). Bacillus halodurans OapB forms a high-affinity complex with the P13 region of the noncoding RNA OLE. J. Biol. Chem. 295, 9326–9334. doi: 10.1074/jbc.RA120.012676
Wilkinson, M. E., Charenton, C., and Nagai, K. (2020). RNA splicing by the spliceosome. Annu Rev Biochem 89, 359–388. doi: 10.1146/annurev-biochem-091719-064225
Wilson, D. N., Arenz, S., and Beckmann, R. (2016). Translation regulation via nascent polypeptide-mediated ribosome stalling. Curr. Opin. Struct. Biol. 37, 123–133. doi: 10.1016/j.sbi.2016.01.008
Wu, S., Tutuncuoglu, B., Yan, K., Brown, H., Zhang, Y., Tan, D., et al. (2016). Diverse roles of assembly factors revealed by structures of late nuclear pre-60S ribosomes. Nature 534, 133–137. doi: 10.1038/nature17942
Wurm, R., Neusser, T., and Wagner, R. (2010). 6S RNA-dependent inhibition of RNA polymerase is released by RNA-dependent synthesis of small de novo products. Biol. Chem. 391, 187–196. doi: 10.1515/BC.2010.018
Xu, J. (2019). Distance-based protein folding powered by deep learning. Proc. Natl. Acad. Sci. U.S.A. 116, 16856–16865. doi: 10.1073/pnas.1821309116
Yakhnin, A. V., Baker, C. S., Vakulskas, C. A., Yakhnin, H., Berezin, I., Romeo, T., et al. (2013). CsrA activates flhDC expression by protecting flhDC mRNA from RNase E-mediated cleavage. Mol. Microbiol. 87, 851–866. doi: 10.1111/mmi.12136
Yakhnin, A. V., Fitzgerald, P. C., Mcintosh, C., Yakhnin, H., Kireeva, M., Turek-Herman, J., et al. (2020). NusG controls transcription pausing and RNA polymerase translocation throughout the Bacillus subtilis genome. Proc. Natl. Acad. Sci. U.S.A. 117, 21628–21636. doi: 10.1073/pnas.2006873117
Yakhnin, A. V., Yakhnin, H., and Babitzke, P. (2006). RNA polymerase pausing regulates translation initiation by providing additional time for TRAP-RNA interaction. Mol. Cell 24, 547–557. doi: 10.1016/j.molcel.2006.09.018
Yakhnin, H., Zhang, H., Yakhnin, A. V., and Babitzke, P. (2004). The trp RNA-binding attenuation protein of Bacillus subtilis regulates translation of the tryptophan transport gene trpP (yhaG) by blocking ribosome binding. J Bacteriol 186, 278–286. doi: 10.1128/jb.186.2.278-286.2004
Yakovchuk, P., Goodrich, J. A., and Kugel, J. F. (2009). B2 RNA and Alu RNA repress transcription by disrupting contacts between RNA polymerase II and promoter DNA within assembled complexes. Proc. Natl. Acad. Sci. U.S.A. 106, 5569–5574. doi: 10.1073/pnas.0810738106
Yamazaki, T., Nakagawa, S., and Hirose, T. (2019). Architectural RNAs for membraneless nuclear body formation. Cold Spring Harb Symp Quant Biol 84, 227–237. doi: 10.1101/sqb.2019.84.039404
Yamazaki, T., Souquere, S., Chujo, T., Kobelke, S., Chong, Y. S., Fox, A. H., et al. (2018). Functional domains of NEAT1 architectural lncRNA induce paraspeckle assembly through phase separation. Mol. Cell 70, 1038–1053e7. doi: 10.1016/j.molcel.2018.05.019
Yang, X., Li, H., Huang, Y., and Liu, S. (2013). The dataset for protein-RNA binding affinity. Protein Sci 22, 1808–1811. doi: 10.1002/pro.2383
Ye, F., Yang, F., Yu, R., Lin, X., Qi, J., Chen, Z., et al. (2018). Molecular basis of binding between the global post-transcriptional regulator CsrA and the T3SS chaperone CesT. Nat. Commun. 9, 1196. doi: 10.1038/s41467-018-03625-x
Youkharibache, P., Veretnik, S., Li, Q., Stanek, K. A., Mura, C., and Bourne, P. E. (2019). The small β-barrel domain: a survey-based structural analysis. Structure 27, 6–26. doi: 10.1016/j.str.2018.09.012
Zeng, R., Smith, E., and Barrientos, A. (2018). Yeast mitoribosome large subunit assembly proceeds by hierarchical incorporation of protein clusters and modules on the inner membrane. Cell Metab 27, 645–656e7. doi: 10.1016/j.cmet.2018.01.012
Zhang, L., Wu, C., Cai, G., Chen, S., and Ye, K. (2016). Stepwise and dynamic assembly of the earliest precursors of small ribosomal subunits in yeast. Genes Dev. 30, 718–732. doi: 10.1101/gad.274688.115
Zheng, A., Panja, S., and Woodson, S. A. (2016). Arginine patch predicts the RNA annealing activity of Hfq from Gram-negative and Gram-positive bacteria. J Mol Biol 428, 2259–2264. doi: 10.1016/j.jmb.2016.03.027
Keywords: ribonucleoprotein, complexomics, Grad-seq, RNA-binding protein, noncoding RNA, sRNA, ProQ, FinO domain
Citation: Gerovac M, Vogel J and Smirnov A (2021) The World of Stable Ribonucleoproteins and Its Mapping With Grad-Seq and Related Approaches. Front. Mol. Biosci. 8:661448. doi: 10.3389/fmolb.2021.661448
Received: 30 January 2021; Accepted: 04 March 2021;
Published: 07 April 2021.
Edited by:
Björn Voß, University of Stuttgart, GermanyReviewed by:
Jens Georg, University of Freiburg, GermanyElena Evguenieva-Hackenberg, University of Giessen, Germany
Jai Justin Tree, University of New South Wales, Australia
Copyright © 2021 Gerovac, Vogel and Smirnov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexandre Smirnov, YWxleGFuZHJlc21pcm5vdkB1bmlzdHJhLmZy