 Samudra Prosad Banik1*†
Samudra Prosad Banik1*† Debasis Bagchi2,3,4†
Debasis Bagchi2,3,4† Pradipta Banerjee5†
Pradipta Banerjee5† Sanjoy Chakraborty6†
Sanjoy Chakraborty6† Manashi Bagchi7†
Manashi Bagchi7† Chaitali Bose8†
Chaitali Bose8† Debasmita De8†
Debasmita De8† Sreemoyee Saha1,9†
Sreemoyee Saha1,9† Sudipta Chakraborty1†
Sudipta Chakraborty1†- 1Department of Microbiology, Government General Degree College, Narayangarh, Rathipur, West Bengal, India
- 2Department of Biology, College of Arts and Sciences, Adelphi University, Garden City, NY, United States
- 3Department of Psychology, Gordon F. Derner School of Psychology, College of Arts and Sciences, Adelphi University, Garden City, NY, United States
- 4Department of Pharmaceutical Sciences, College of Pharmacy and Health Sciences, Texas Southern University, Houston, TX, United States
- 5Department of Surgery, University of Pittsburgh, Pittsburgh, PA, United States
- 6Department of Biological Sciences, New York City College of Technology/CUNY5113, Brooklyn, NY, United States
- 7Department of R&D, Dr. Herbs LLC, Concord, CA, United States
- 8Department of Nutrition, Government General Degree College, Narayangarh, Rathipur, West Bengal, India
- 9Department of Microbiology, Maulana Azad College, Kolkata, West Bengal, India
Misfolded proteins have been found to be at the core of an increasing number of cognitive ailments. α-synuclein, a resident chaperone of the neurosynaptic cleft has been implicated in a major share of these neurodegenerative diseases. Over the years, a daunting task for researchers has been the identification of the complex set of conditions which govern the Substantia nigra microenvironment for transformation of α-synuclein from a functional and grossly structureless chaperone to toxic cross-β fibrils. An abundance of Reactive Oxygen Species and a drop in pH of the solvent have been identified to be the key drivers of the fibrillation process which is initiated by Liquid-Liquid phase separation of α-synuclein droplets. Zinc is a significant micronutrient of the human body integral to the proper functioning of the nervous system as well as holistic cognitive development. Many recent studies have deciphered that metal ions including zinc facilitate the fibrillation of α-synuclein by shielding negative charges at the C terminus of the protein. Zinc preferentially binds to Asp121 at the C terminus and His50 at the N terminus to promote fibrillation. On the contrary, zinc has many protective roles to retard fibrillation of the protein at the same time. It downregulates ROS and assists chaperones which prevent non-native aggregation of α-synuclein. The ability of zinc to bind preferentially to α-synuclein coupled with the advent of ultrasensitive detection technologies such as the Surface Enhanced Raman Spectroscopy has led to the prospects of zinc-oxide nanoparticles as effective tools to probe the α-synuclein-based biomarker for early detection of protein aggregates in the body fluid. This review summarizes the significant mechanistic findings which has facilitated our understanding of the fibrillation of α-synuclein, the precise role and mechanism of zinc involved therein and the prospects of using zinc in designing efficient tools for diagnosis of Parkinson’s Disease and other synucleinopathies.
1 Introduction
Amyloids are non-native aggregates formed from cytosolic or membrane associated proteins in neurons which have defined physiological functions but transform into soluble and toxic fibrillar entities under poorly understood altered conditions. Amyloid aggregation is considered to be one of leading causes behind cognitive impairment and onset of several debilitating neurodegenerative diseases (Wells et al., 2021; Wolfe and Cyr, 2011) collectively referred to as amyloidosis (Metkar et al., 2024) Tau proteins representing the characteristic pathology of Alzheimer’s disease (Chen and Yu, 2023) are normally associated with stabilization of neuronal microtubules (Jesu et al., 2004), but give rise to fibrillar tangles along with β-amyloid under a hyperphosphorylated state. Amyloid β protein, on the other hand, is implicated in regulation of synaptic function, improving memory and protection of neurons from oxidative stress (Bishop and Robinson, 2004).
Probably the most significant form of amyloid related neuropathy is associated with α-synuclein, a major neurosynaptic chaperone which mediates release of neurotransmitters (Sulzer and Edwards, 2019). The protein owes its name to its simultaneous localisation in both synaptic vesicle as well as nuclear envelope in a type of stingray (Clayton and George, 1998). In addition to its role as a synaptic chaperone, α-synuclein is also indirectly associated with the regulation of activity of glutamatergic ionotropic and metabotropic receptors activity by virtue of its interaction with other proteins (Calabresi et al., 2023). α-synuclein, one of the major causes of amyloidosis has also been implicated in the highest known numbers of cognitive ailments including Parkinson’s disease, Lewy Body Dementia, Multiple System Atrophy and many other cognitive disorders collective known as α-synucleinopathies (Calabresi et al., 2023). It is a 140-residue protein with three distinct domains; a positively charged N-terminal region (residues 1–60), a hydrophobic non amyloid β component (NAC) region (residues 61–95), and a C-terminal region with amino acids bearing a net negative charge (residues 96–140) (Ohgita et al., 2022). The presence of the two signature features of an aggregation prone protein, namely a hydrophobic region and a sequence with a strong propensity to adopt a beta pleated sheet structure make α-synuclein an ideal candidate for fibrillation; however, the negative charge at the C terminus and its long range non-covalent interaction with the N terminal domain protects the protein from losing its native state and committing to fibrillation. Fibrillation of α-synuclein leads to the formation of a cross beta pleated sheet which causes cellular toxicity by creating pores in the membrane, inducing misfolding of other proteins and eliciting other signalling pathways which results in loss of cellular homeostasis. In spite of a plethora of concerted efforts to understand the molecular basis of fibrillation (Brás and Outeiro, 2021), scientists are yet to decipher the combination of responsible intrinsic and/or extrinsic factors (Longhena et al., 2020; Srinivasan et al., 2021). The available scientific evidences are all equivocal of the fact that synuclein fibrillation consists of two distinct steps a) Nucleation or initiation of fibrillation and b) elongation of fibrillation. Secondary nucleation pathways from dissociated protofibrils are also a prominent part of the process thus resembling a prion like propagation of these generally toxic entities (Coles et al., 2025).
Since the major guiding factor of α-synuclein fibrillation is its charge dependency, bivalent metal ions also play a significant role in governing fibrillation kinetics (Atarod et al., 2022). Particularly, metal ions are believed to mimic the effect of pH by neutralizing the negative charge of the C-terminus, thus increasing the hydrophobicity of the central domain with simultaneous propensity to switch over to the beta pleated sheet (Byrd et al., 2023).
Zinc is a significant trace element with myriad physiological and biochemical functions essential for life. It plays a key part in boosting neuronal health and the micronutrient’s deficiency has been associated with oxidative stress, programmed cell death and necrosis of neurons eventualizing into a plethora of neurodegenerative disorders (Vali et al., 2025). Zinc’s role in Parkinson Disease pathology, in particular, has been intriguing to understand since high zinc concentration promotes fibrillation of α-synuclein (Gao et al., 2022) whereas lower levels assist the cellular chaperones in preventing fibrillation (Al-Harthi et al., 2022). The selective binding of zinc to key regions of α-synuclein has also opened up new avenues for the development of technologies to probe α-synuclein based biosensors for early detection of neuropathies.
The diverse facets of α-synuclein fibrillation render it practically inconceivable to predict the set of extrinsic and intrinsic factors responsible for its phase transition. Metal ions present an additional level of complexity in this regard owing to their differential effect in modulating the fibrillation potential. The primary objective of this review is to present a comprehensive insight of the effect of metal ions, particularly zinc, in modulating α-synuclein aggregation, fibrillation and resultant toxicity analyses along with the technological and scientific challenges involved therein. In addition, the exciting new frontiers of amyloid research, namely, the role of E. coli amyloid curli in shaping the amyloidogenic behaviour of cellular proteins via the gut-brain axis, and the molecular connection between diabetes and neurodegenerative disorders mediated by advanced glycated end products have also been discussed in the perspective of the role of zinc involved therein.
2 α- Synuclein as the chief mediator of synucleinopathies
Parkinson’s disease (PD) is the second most abundant neurological disease after Alzheimer which primarily affects co-ordination in movement. The disease, which was described as Shaking Palsy by its discoverer, James Parkinson in the year 1817 (Goetz, 2011), is caused primarily by death of the dopamine neurons in the Substantia nigra (SN), a region named after the high levels of melanin present in the dopamine neurons in unaffected individuals (Zecca et al., 2003). Loss of dopaminergic neurons results in gross cardinal motor dysfunctions leading to bradykinesia, muscle stiffness, tremors in resting posture, as well as postural and gait impairment (Ramesh and Arachchige, 2023), features which are the hallmarks of PD. Lewy Body Dementia (DLB) is another frequently occurring dementia involving progressive memory loss and hallucinations with cortical depositions of insoluble α-synuclein (Ye et al., 2020). Yet another example of α-synuclein associated cognitive ailment is Multiple System Atrophy which involves depositions of Glial Cytoplasmic Inclusions (GCI) formed of hyperphosphorylated α-synuclein inclusions in oligodendrocytes, better known in the disease perspective as Papp-Lantos bodies (Jellinger, 2014). The characteristic features of MSA include both motor and autonomic disturbances including Parkinsonism, cerebellar ataxia, orthostatic hypotension, urinary dysfunction and cardiovascular issues which often has fatal consequences (Campese et al., 2021). Together, the spectrum of α-synuclein associated neuropathies are referred to as synucleinopathies.
Most of the PD cases are linked to late adult onset and are sporadic in nature. However, in 1997, the first gene associated with familial PD was identified which coded for the presynaptic chaperone protein called α-synuclein (Nussbaum, 2017). The pathology associated with mutant α-synuclein was first detected in the form of characteristic black dots surrounded by vaguely defined halos termed Lewy bodies in histopathological sections of neuronal soma of patient brains affected with this rare inherited form of the disease. Later on, the molecular genetic basis of the disease was linked to a transition mutation in the snca gene A53T which results in the substitution of alanine to threonine in the N terminal 53rd amino acid of the protein leading to its abnormal aggregation (fibrillation) (Lee and Trojanowski, 2006). Subsequently many other mutations were found to be associated with familial forms of PD (Flagmeier et al., 2016) including the A30P mutant aggravating PD by reduced autophagic clearance of protein aggregates (Lei et al., 2019) and E46K mutant which promotes fibrillation of N acetylated α-synuclein by abolishing the long-range electrostatic interactions (Zhao et al., 2020). Lewy bodies are clumps of misfolded/non-native protein aggregates named after their discoverer Frederick Lewy in 1912 (Mueller et al., 2017). Apart from α-synuclein, Lewy bodies also contain many other aggregated cellular proteins, which together constitute an interactome to promote fibrillation of α-synuclein (Lashuel and Novello, 2021). Subsequent clinical cases revealed that α-synuclein pathology is not only restricted to the soma but also extends to neurites which led to the coinage of the term “Lewy neurites” (Braak et al., 1999).
Under normal physiological conditions α-synuclein exists mostly in an N acetylated membrane bound form which results in adoption of an α-helical structure (Runfola et al., 2020). This membrane bound conformation effectively prevents its fibril formation and subsequent toxic consequences (Bell et al., 2022). However, without the acetylation, it is naturally an Intrinsically Disordered Protein lacking any discernible secondary structure (Zhang et al., 2020). Point mutations or gross genetic changes have been known to induce the abnormal conformational changeover of α-synuclein (Waxman and Giasson, 2009). This and many other hitherto uncharacterised factors induce accumulation of pathogenic inclusions differently termed as Lewy bodies in neurons of PD patients (Marotta et al., 2021) or glial cytoplasmic inclusions in oligodendrocytes of Multiple System Atrophy (MSA) patients (Reddy and Dieriks, 2022). Apart from mutations, other significant factors accounting for α-synuclein fibrillation are changes in pH of the microenvironment (Frey et al., 2024), presence of other metal ions temperature (Ariesandi et al., 2013), oxidative stress (Puspita et al., 2017), phosphorylation (Ghanem et al., 2022; Kawahata et al., 2022), molecular crowding (Shtilerman et al., 2002) as well as presence of other proteins in the vicinity (Uversky et al., 2002). Deletion of a stretch of acidic residues along with proline in the C terminal region makes the protein more vulnerable to toxic fibril formation (Farzadfard et al., 2022a) due to loss of long-range electrostatic interactions with the N terminus. The proline residues in the C terminus plausibly function to bring the interacting residues in close vicinity. A particular point mutant of the protein, A53T, associated with familial PD is also known to fibrillate at a significantly higher rate than the normal protein (Coskuner and Wise-Scira, 2013) owing to its increased propensity to form beta fibrils as well as by partially negating the effect of long-range interactions between the N and C terminus. Effect of net charge of the protein on fibrillation is also explained by the observation that pH of the microenvironment is elevated as fibrillation proceeds, along with rise in pKa of the acidic amino acids, in order to prevent association of N and C termini which might block the fibrillation process (Pálmadóttir et al., 2021).
The most acceptable theory to explain the departure of the native α-synuclein into the fibrillation pathway is the Liquid-Liquid phase separation (Ray et al., 2020). This is subsequently converted into a hydrogel-like entity consisting of amyloid oligomers and protofibrils (Mukherjee et al., 2023). The holistic effect of all extrinsic factors accounting for the phase transformation is to shield or neutralize the charges at the N or C terminal ends, as this has been also confirmed by specific site directed mutations of key residues of the protein (Pancoe et al., 2022).
2.1 Liquid-liquid phase separation drives α-synuclein fibrillation
Liquid-liquid phase separation (LLPS) is a physical phenomenon which involves separation of a uniform solution of macromolecules into two distinct liquid phases with different densities of macromolecules, resulting in the formation of coacervate like structures. Phase separation is preceded by accumulation of multiple conformations of α-synuclein monomers some of which are structurally more adapted to develop into protofibrils (Dedmon et al., 2005). Brodie and co-workers used a combination of Molecular Dynamics simulation data substantiated with cross-linking mass spectrometry (MS) and single molecule Förster resonance energy transfer (smFRET) data to generate an ensemble of conformational spaces of such α-synuclein monomers (Brodie et al., 2019). The tendency of α-synuclein to undergo LLPS is facilitated by the presence of low complexity domains (LCDs), one or more sequence stretches with higher abundance of certain amino acids instead of the complete set of 20 amino acids (Mahapatra and Newberry, 2024). These sequences are mostly unstructured representing the IDR regions of proteins and initiate the formation of unique condensates where many molecules of α-synuclein slowly transform into the beta-pleated structure mediated by transient multivalent interactions. This results into a local increase in density of these proteins and marks the initiation of nucleation of the fibres. The process is facilitated by the presence of Polyethylene Glycol, a molecular crowding agent which brings α-synuclein molecules in close proximity by displacing water from the immediate vicinity for enhanced hydrogen bonding, non-covalent interactions and hydrophobic interactions (De Luca et al., 2025). Eventually, further α-synuclein molecules rope in and fibrils are extended leading to the formation of amyloid like hydrogel entities (Mukherjee et al., 2023) (Figure 1). The concentration of α-synuclein in these condensates can reach to as high as 30–40 mM (Dada et al., 2023) which leads to the formation of neurofibrillary tangles or the so-called Lewy bodies. Phase separation of α-synuclein under both cellular and in vitro conditions is guided by the Vesicle Associated Membrane Protein 2 (VAMP2) and its interaction with R-SNARE (Agarwal et al., 2024). The disease pathology is subsequently spread by prion like self-propagation of α-synuclein fibrils (Jan et al., 2021). The indispensable role of the C terminal domain in preventing fibrillation has been demonstrated by an engineered α-synuclein with truncated C terminus which not only displayed accelerated fibrillation kinetics but also promoted the phase separation of WT α-synuclein (Huang et al., 2022). Apart from the intrinsic sequence predisposition and presence of molecular crowding agents, many other extrinsic factors act as mediators of the LLPS; the more significant of these being the pH and the concentration of metal ions in the local microenvironment. At a pH of 7.4, primary nucleation proceeds spontaneously and is the fastest in the absence of other governing factors (Dada et al., 2023) whereas the critical concentration of α-synuclein needed to undergo LLPS is the least at pH 5.5 probably owing to the proximity of the isoelectric point of α-synuclein near that pH assisting separation of proteins from the solution (Hou et al., 2023). A high salt concentration in the medium promotes fibrillation and lowers down the critical concentration of the protein to undergo LLPS by neutralizing the charges at the N and C termini and promoting aggregation of the NAC region (Sawner et al., 2021). In addition to the charge neutralization, it also promotes effective salting out of the protein to induce phase separation.
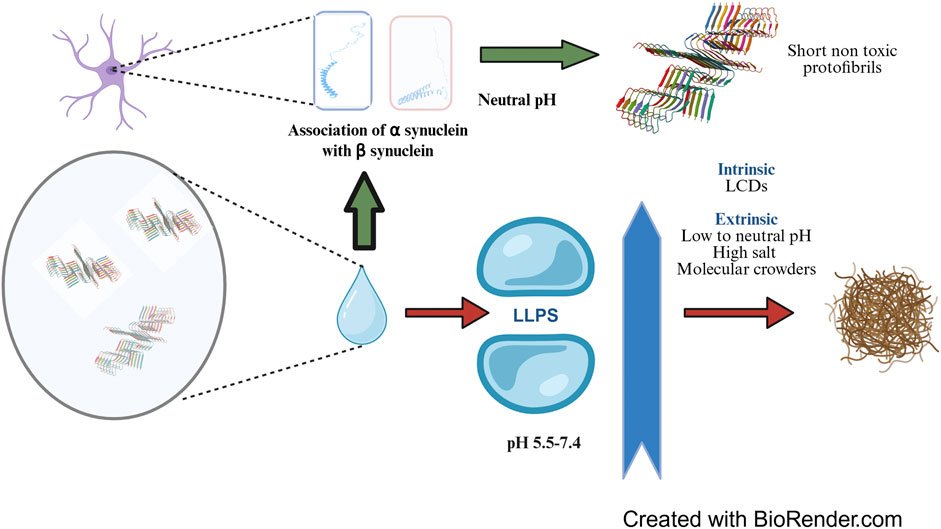
Figure 1. Liquid-Liquid Phase Separation (LLPS) of α-synuclein: LLPS and subsequent fibrillation of α-synuclein is promoted by intrinsic factors such as Low Complexity Domains and solution conditions including low to neutral pH, high salt concentration and presence of molecular crowding agents. LLPS results in local build-up in α-synuclein concentration to as high as 30-40 mM from the physiological concentration of around 50 µM which eventually causes formation of neurofibrillary tangles. In healthy brain, β-synuclein, a homologue of α-synuclein associates with the later at neutral pH and prevents the formation of long, toxic and fibrillar α-synuclein aggregates. The concepts and mechanisms depicted in the figure have been adopted from multiple studies as cited in the text. The protein structure cartoon shown in the figure has been downloaded from RSCB PDB (PDB ID 6OSM, Ni et al. 2019). Figure has been created with BioRender.
Under physiological conditions, LLPS of α-synuclein is prevented by β-synuclein, a related protein of the synuclein family which is present at the presynaptic terminals at a similar concentration as that of α-synuclein (around 50 µM). Several independent reports have indicated that β-synuclein exerts its action by associating with α-synuclein and subsequently a) elongating the lag phase of fibrillation (Jain et al., 2018) and b) arresting secondary nucleation (Brown et al., 2016). β-synuclein has a 78% sequence identity with α-synuclein, differs in few critical aspects from the later including a 11 residue shorter NAC region, absence of cysteine and tryptophan residues and an abundance of proline and acidic residues and forms ordered structure more readily than α-synuclein (Hayashi and Carver, 2022). A higher number of proline residues is probably needed by the protein to make up for the folding flexibility curtailed by the shorter NAC region. A particular point mutant of β-synuclein, P123H, results in pathogenic deposits of Lewy Body Dementia (Psol et al., 2021) Li et al. found that β-synuclein associated with α-synuclein employing electrostatic interactions. In an in vitro microenvironment mimicking presynaptic terminal with equimolar amounts of α and β synuclein, the total mean surface area of α/β-Syn condensates was higher than that of α-Syn alone. Although the association of α/β-Syn condensates apparently promoted LLPS of α-synuclein, its corresponding nucleation and fibrillation was effectively prevented (Li et al., 2024). The pivotal role of NAC region in driving the aggregation is also supported by the observation that the second identified pathogenic point mutation V70M (Valine to Methionine) causes predisposition to a sporadic Lewy Body Dementia. Methionine is less hydrophobic than valine and therefore substitution to methionine should have lessened the aggregation propensity; therefore, this observation is probably a testimonial to the indispensability of Valine at the 70th position for NAC mediated aggregation of β-synuclein and its simultaneous association with α-synuclein.
2.2 Major physiological changes to account for the disease pathology of α-synuclein
Misfolding of α-synuclein has been linked to a plethora of cognitive ailments arising from the death of dopaminergic neurons and collectively referred to as synucleinopathies. The soluble protofibrils formed from α-synuclein exert their cytotoxicity through disruption of mitochondrial function, promotion of autophagy, lysosomal impairment and loss in regulation of calcium homeostasis (Calabresi et al., 2023). Particularly, the protein has been found to be associated with increased production of Reactive Oxygen Species (ROS) and elicitation of inflammatory response (Van Laar et al., 2020), the two primary reasons accounting for loss in cellular homeostasis.
2.3 Augmentation of ROS by α-synuclein and vice versa
As stated earlier, α-synuclein fibrillates by addition of monomers after the initial nucleation; during elongation, smaller fragments of oligomers and protofibrils often gets detached from the primary fibril serving as additional points of nucleation thus aggravating the problem. This avalanche of soluble protofibrils is often believed to be more toxic than the stable full length cross-beta fibrils owing to their membrane permeabilization capability and interaction with various cellular organelles (Winner et al., 2011). Mitochondria represent the most vulnerable site of α-synuclein attack primarily owing to the latter’s interaction with cardiolipin, a major mitochondrial phospholipid with simultaneous formation of transient pores in the membrane (Ghio et al., 2019). The first case of α-synuclein independent PD case linked with mitochondria was reported with respect to consumption of banned drugs contaminated with 1-methyl-4-phenyl-1,2,3,6-tetrabydropyridine (MPTP). The toxic bioactive form of this compound 1-methyl-4-phenylpyridinium (MPP+) blocks the functioning of mitochondrial Complex-1 and NADH-ubiquinone oxidoreductase enzyme resulting in release of free electrons with concomitant ROS generation (Javitch et al., 1985). Creation of transient pores by α-synuclein oligomers induce similar effects of uncoupling electron transport from ATP production, resulting in release of free electrons and cytochrome c and concomitant ROS generation. Apart from directly modulating mitochondrial physiology, α-synuclein oligomers also amplify cytosolic peroxide flux resulting in oxidation of glutathiones (Van Laar et al., 2020). A logical explanation behind this observation comes from the fact that elongation of the polymeric fibrillar structure of α-synuclein requires a constant supply of free protons from the solution in order to mask the C terminal negative charges and prevent the interference of the N and C terminal association during fibrillation. In addition, α-synuclein oligomers can also elicit ER stress and consequent induction of Unfolded Protein Response pathway (Puspita et al., 2017). The consequent rise in intracellular calcium levels leads to further augmentation of ROS induced cellular damage. The diverse mechanisms by which misfolded α-synuclein inflict its cellular toxicity has been summarized in Figure 2.
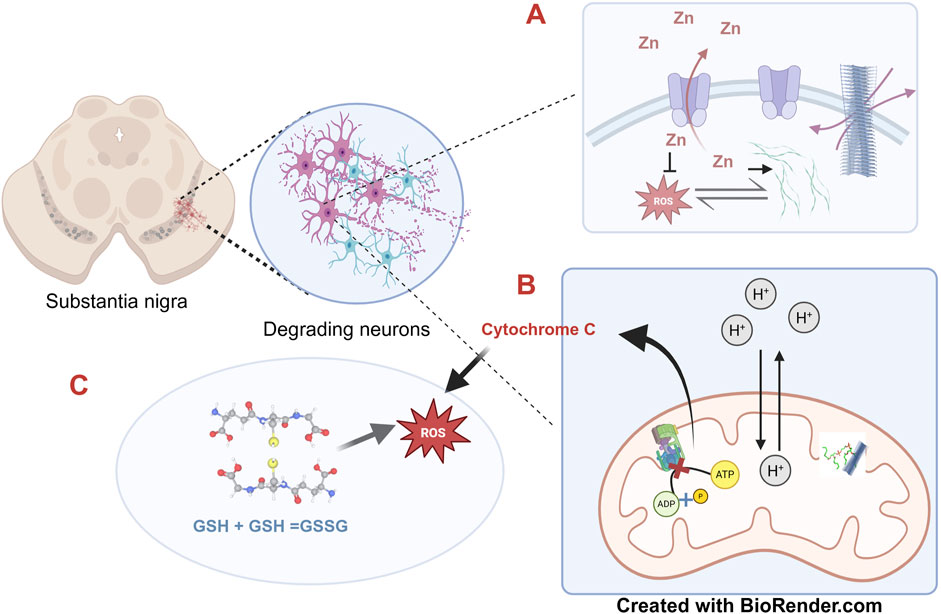
Figure 2. The multiple pathologies of α-synuclein-The substantia nigra in the basal ganglia of the mid brain represents one of the prime sites of α-synuclein mediated pathology where the misfolded protein accounts for the degeneration of the dopaminergic neurons. (A) The synaptic vesicles dissipate the otherwise toxic accumulation of excess zinc ions by the membrane associated ATPase, ATP13A2. A build-up of zinc promotes fibrillation of α-synuclein; however, at the same time also reduces ROS. α-synuclein and ROS augment each other. (B) Neuronal degeneration is accelerated by ATP depletion. Mitochondrial membrane permeability is altered by the toxic cross-beta α-synuclein fibrils through interaction with Cardiolipin, a major mitochondrial membrane lipid, which results in uncoupling of ATP synthesis with simultaneous release of cytochrome c. (C) α-synuclein fibrillation induced oxidative stress leads to accumulation of oxidised glutathione which in turn augments ROS thus self-perpetuating the toxicity. The concepts and mechanisms depicted in the figure have been adopted from multiple studies as cited in the text. Figure has been created with BioRender.
In the reverse perspective, production of cellular ROS is also associated with fibrillation of α-synuclein (Esteves et al., 2008). In a superoxide dismutase 2 (SOD2) transgenic mouse model, development of synucleinopathies was significantly earlier as compared to the control mouse model which proved that a compromise in the capacity to quench free radical led to enhanced fibrillation of α-synuclein (Scudamore and Ciossek, 2018). In related studies, cytochrome c and peroxide radicals also resulted in crosslinking of α-synuclein monomers through the formation 3,3′-dityrosine linkages (Hashimoto et al., 1999; Ruf et al., 2008). Such crosslinking can arrest the formation of conventional cross beta fibrils but at the same time promote off-pathway aggregates as oligomers which are potentially more toxic.
2.4 Effect of pH on α-synuclein fibrillation
Aggregate formation and fibrillation of α-synuclein is favoured at slightly acidic pH, much like the endosomes and the lysosomes. This is explained by the fact that at neutral pH, the protein bears a net negative charge which is nullified on lowering the pH. The C terminal also gets fully protonated and uncharged at this pH which augments the aggregation process. This facilitates the hydrophobic interactions and subsequent aggregation of the central domain of the protein (Moriarty et al., 2017). These results have also been confirmed by single molecule fluorescence studies which has revealed that a slight excess of protons in the solution is conducive towards aggregation of monomeric α-synuclein (Trexler and Rhoades, 2010). However, at a pH below 6, secondary nucleation is favoured than fibril growth thus shifting the kinetics of fibrillation towards formation of toxic protofibrils (Buell et al., 2014). A more recent dissection of the effect of pH on fibrillation has demonstrated that due to the hyperdynamic nature of α-synuclein surface topology, multiple polymorphs of α-synuclein result, each with a different propensity of fibrillation (Frey et al., 2024). The role of the side chain charges in governing the pH susceptibility has also been demonstrated by β-synuclein which develops fibrils at pH 5.8 but not at pH 7.3. As stated in the earlier section, unlike α-synuclein, β-synuclein has a shorter NAC region to readily undergo fibrillation. It was initially thought that the presence of a particular histidine residue at the 63rd position with a pKa of around 6.4 probably serves a critical role in mediating this pH dependent switch of the protein. However, substitution of this histidine with an aspartate residue did not alter the mutant β-synuclein’s behaviour as compared to the wild type with respect to this pH dependent conformational change (Moriarty et al., 2017). Therefore, a more plausible explanation is that at pH 5.8, there are probably enough protons in the solution to shield the significant number of negative charges at the C terminus; however, at pH 7.3, the proton concentration in the physicochemical microenvironment probably falls short to negate these charges. In a LLPS experiment carried out at pH 7.4, α-synuclein in presence of equimolar concentrations of β-synuclein showed a significant increase in turbidity as compared to α-synuclein alone; the corresponding sedimentation experiment indicated that the α/β condensate precipitated readily whereas the β -synuclein was left out in the supernatant justifying β-synuclein’s inability to aggregate at neutral pH (Li et al., 2024).
2.5 Effect of phosphorylation
α-synuclein recovered from Lewy bodies of patients affected with sporadic Parkinson’s disease and LBD as well as from glial cytoplasmic inclusion bodies in oligodendrocytes of patients affected with Multiple System Atrophy has been found to be heavily phosphorylated in the 129th serine residue of the C terminal domain (Ghanem et al., 2022). In contrast, in the brains of healthy adults, only 4% of α-synuclein is constitutively phosphorylated (Fujiwara et al., 2002; Hasegawa et al., 2002). Another specific phosphorylation event at S87 found in the brain of patients with synucleinopathies, and the only one at the NAC region has been found to decrease the protein’s membrane binding affinity and also reduces its fibrillation potential (Paleologou et al., 2010). Several important cellular kinases including the G-protein-coupled receptor kinases (GRKs) (Pronin et al., 2000), the casein kinase II (Sano et al., 2021), polo-like kinases (Inglis et al., 2009), and the leucine-rich repeat kinase 2 (LRRK2) (Greggio et al., 2011) mediate phosphorylation of α-synuclein. This suggests a significant role of phosphorylation in regulating fibril formation of α-synuclein. However, contrasting evidences suggest that the timing of phosphorylation is crucial in governing both the initiation nucleation and fibrillation process. Ghanem and coworkers found that efficiency of phosphorylation at S129 increases manifold when protein aggregation has already been initiated (Ghanem et al., 2022). Using state-of-the-art real time quaking induced conversion (Atarashi et al., 2011) of brain homogenates of PD and DLB patients, the authors found that S129 phosphorylation effectively inhibited subsequent fibril formation as well as toxicity associated with α-synuclein. Therefore, it may be safely said that althoughphosphorylation has been demonstrated to augment the fibrillation process; when it occurs at S129 after initiation of the protein fibrillation, both the aggregation propensity as well as resultant cytotoxicity is decreased (Ghanem et al., 2022). A recent MD simulation study has revealed that phosphorylation brings down the average Solvent Accessible Surface Area of the protein thus promoting its hydrophobic core mediated aggregation (de Bruyn et al., 2024).
2.6 Effect of glycation
The process of attachment of sugar molecules on lysine and arginine residues of proteins via formation of Schiff’s base is referred to as glycation. It occurs obligatorily due to the abundance of carbohydrates as metabolic intermediates; the accumulation of sugar molecules in specific physiological milieus such as the serum, observed in case of diabetic and prediabetic individuals leads to the formation of non-native protein-sugar adducts more popularly known as Advanced Glycated End Products (Rungratanawanich et al., 2021). Many of the AGE adducts of proteins which are apparently non-amyloidogenic, such as serum albumins, show amyloid like properties (Das et al., 2020; Prosad Banik, 2023). Glycation of α-synuclein is mostly concentrated in the N terminus due to the abundance of lysine residues and affects its membrane binding, thus leading to accumulation of toxic oligomers (Vicente Miranda et al., 2017). Additionally, glycation also interferes with the growing end of α-synuclein fibrillation but does not affect the nucleation step (Farzadfard et al., 2022b). Therefore, it can have a toxic effect on α-synuclein fibrillation by increasing the number of oligomeric species and protofibrils rather than full length fibrils (Kumari et al., 2022). This unique behaviour of glycated proteins also explains the strong clinical correlation between chronic hyperglycemia and susceptibility to neurodegenerative disorders.
2.7 Effect of metal ions and their salts on fibrillation
Brain regions affected by neurodegenerative diseases such as PD, Alzheimer’s disease (AD), Amyotrophic Lateral Sclerosis (ALS) and Mass Spectrometry (MS) are found to contain significantly higher concentrations of metal ions such as zinc, iron and copper (Gardner et al., 2017; Miller et al., 2006). The coincidence of cognitive ailments with occupational exposure to these metal ions have been noted on several occasions suggesting a definitive role of the metal ions in disease pathology (Gorell et al., 1997).
α-synuclein fibrillation is strongly affected by metal ions like zinc, copper, iron and calcium (Atarod et al., 2022; Chen et al., 2019). Interestingly, the effect has been significant only in case of divalent/trivalent metal cations (Uversky et al., 2001). Other observations of a high Ferric/ferrous ratio (Zhao et al., 2023) and abundance of oxidised glutathione (Paik et al., 2003) at the SN microenvironment also support the hypothesis that α-synuclein fibrillation is conducive under oxidising conditions. Ferric ions have been shown to bind to a conserved His50 residue in molar equivalents which provides the coordination site for the metal ion. Bound ferric ion decreases lag time and increases rate of fibril formation. However, as reiterated many times in this review, the mechanism of α-synuclein fibrillation is as complex as it can get. Abundance of ferric ions actually halts fibrillation, by inducing formation of spherical oligomers instead of fibrils (Zhao et al., 2023). Probably, the most intriguing effect of metal ions on fibrillation of α-synuclein has been observed with respect to zinc. Unlike the effect of iron, zinc ions are generally known to augment fibrillation. Accumulation of zinc in intracellular milieu has been found to be associated with the death of dopaminergic neurons whereas chelation of zinc reverses the effect (Tamano et al., 2019). However, low concentrations of zinc are actually required by other interacting proteins of α-synuclein to prevent fibrillation (Al-Harthi et al., 2022).
3 Role of zinc in modulation of α-synuclein fibrillation
3.1 Importance of zinc as a micronutrient in the body
In nutrition, any chemical compound that is required in minute quantity (ranges between 1 and 100 mg/day in adults or constitutes 0%–1% of total body weight) is considered as a “trace element” (Stjernswärd et al., 1996). Zinc is an essential trace element which makes up almost 1.5–2.5 gm of human body weight. Following absorption in small intestine, dietary zinc remains stored in skeletal muscles (60%), bones (30%), skin and liver (5%) and the rest is distributed in different organs like brain, heart, pancreas, and kidney or in mammary glands through blood circulation. In blood, zinc either remains bound with albumins which is exchangeable (75%–85%) or with α-2-macroglobulin which is the non-exchangeable form of serum zinc (15%–20%). Urinary or gastrointestinal excretion of zinc occurs depending on its status in body (Willekens and Runnels, 2022). Zinc, the second most copious trace element after iron in humans has crucial structural, catalytic and regulatory roles as it serves as vital component of more than 2,500 proteins encompassing transcriptions factors and over 100 of enzymes (Kiouri et al., 2023). The spectrum of physiological intervention of zinc is as broad as it gets and includes gene expression and regulation, cell signalling, cell proliferation, apoptosis, DNA metabolism and structural stabilization of chromatin (like sperm chromatin and its decondensation as needed), immune responses, defensive action against oxidants and many other vital physiological processes for sustenance of body homeostasis (Björndahl and Kvist, 2014; Costa et al., 2023) (Table 1).
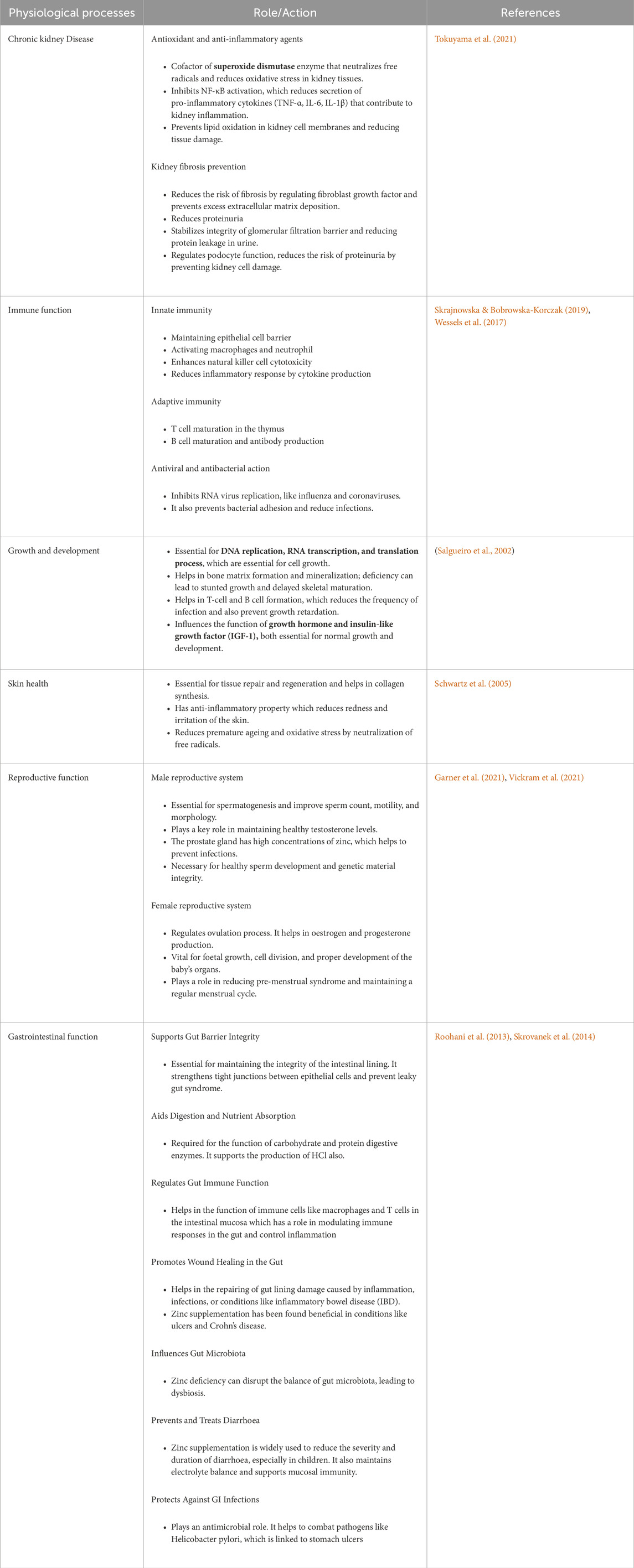
Table 1. The diverse physiological roles of Zinc.
3.2 Role of zinc in minimising ROS and maintaining cellular homeostasis
Zinc serves as co-factor for enzymes involved in defence against oxidative stress. This micro element hinders the activity of pro-oxidant NADPH oxidase, thus maintaining structural stability of the lipid bilayer (Li et al., 2016). Metallothionein (MT) is a cysteine rich protein in cell, which protects from zinc induced toxicity by sequestering the metal. Metallothionine-zinc complex has ROS sequestering activity as it captures and neutralizes the free radicals using cysteine-sulphur ligands; along with its anti-inflammatory and neuroprotective effects (Rodríguez-Menéndez et al., 2018), zinc is also a structural component of the enzyme superoxide dismutase (SOD), found in abundance in the cytoplasm and also a significant enzyme in endogenous antioxidant machineries. SOD contains both copper and zinc and helps to neutralize superoxide radicals thus bringing down the toxicity caused by ROS (Cruz and Soares, 2011). Zinc salts have been also found to be effective in offering protection against lipid peroxidation (Bagchi et al., 1998) by quenching ROS (Bagchi et al., 1997) and also regulates the expression of another cellular antioxidant defence, i.e., glutamate and cysteine ligase, which plays pivotal role in de novo synthesis of glutathione. Therefore, zinc can participate directly in ROS scavenging by glutathione or indirectly by acting as cofactor for the enzyme glutathione peroxidise. Ha et al. has shown that concentration of cellular glutathione can be upregulated by in vitro administration of regulated amount of zinc in epithelial pigment cells of human retina (Tan et al., 2013). Metal Transcription Factor-1 (MTF-1) is dependent on zinc. When the zinc concentration in body rises, it expresses gene for metallothionine and zinc transporter-1 and maintains zinc homeostasis and avert excess metal induced oxidative stress (Günther et al., 2012). MTF-1 also promotes the expression of selenoprotein-1 which binds glutathione and takes part in ROS scavenging. The inertness of zinc towards catalysis of free radical generation reactions additionally boosts its antioxidative efficacy (Oteiza, 2012). Zinc can bind with either thiol or a sulfhydryl group in proteins thus hindering the intra-molecular formation of disulfide (Choi et al., 2018). Zinc alleviates oxidative stress as an anti-inflammatory element by taking part in various molecular signalling pathways which increases the expression of anti-inflammatory protein A20 (otherwise known as TNFAIP3) and downregulates the expression of the pro-inflammatory NF-kB (nuclear factor κB) (Prasad et al., 2010). Zinc has also shown potent effect in attenuation of pro-inflammatory cytokines including Interleukin-6, Tumour Necrosis Factor α (TNF-α), and C-Reactive protein and stimulation of the molecular signalling pathway leading to expression of the A20 protein (Marreiro et al., 2017). Zinc also upregulates the expression of Nrf2, a chief mediator of antioxidant machinery (Olechnowicz et al., 2018).
Zinc has a crucial role in the maintenance of intracellular calcium concentration. Its deficiency causes calcium build-up promoting the release of substance P, a neuropeptide involved in immune response and inflammation resulting in oxidative stress (Weglicki et al., 2011). Deficiency of this micro-element also leads to activation of NADPH oxidase, ROS and reactive nitrogen species (Lee et al., 2021). As zinc is involved in synthesis of the insulin along with its storage and release, it plays a significant role in blood glucose homeostasis with implications in type 2 diabetes, obesity, cardiovascular diseases, altered lipid metabolism and metabolic syndromes (Motamed et al., 2013).
The role of zinc is integral in neuronal health, especially for carrying out routine physiological processes of the Central Nervous System (Li et al., 2023) (Table 2). The hippocampus, amygdala and cortex are the major storage sites of the zinc and a subtly regulated flux of zinc from these regions are vital for carrying out critical functions of the brain including cognition, memory and emotional stability. Scientific evidences have unanimously established the role of zinc ions in modulating glutaminergic synapse transmission (Bertoni-Freddari et al., 2008). The same has been extensively reviewed by Sikora et al. (2020). Physiological concentrations of zinc range between 10 and 50 µM in and around the vicinity of the Substantia nigra (SN) which is typically much higher than that required elsewhere in the body and justifies the diverse integral roles of the metal in the brain. Further rise in concentration of the metal is often met with severe cognitive ailments (Sabouri et al., 2024; Sandstead et al., 2000). Therefore, it is equally essential to control the level of zinc in these cellular micro-niches. ATP13A2 is a P type ATPase generally found in the membranes of acidic vesicles in neurons which, along with the other two more common class of Zinc transporters belonging to the ZnT (SLC30) and ZIP (SLC39) families, is entrusted to serve exactly this role (van Veen et al., 2014). Overexpression of a ATP13A2 orthologue in yeast was shown to impart zinc resistance (Kong et al., 2014); on a different note, an individual with a mutant of ATP13A2 was affected with Kufor-Rakeb syndrome, a disease with shared features of Parkinsonism (van Veen et al., 2014). These observations were subsequently confirmed in a mouse PD model where zinc was shown to increase α-synuclein aggregation; overexpression of ATP13A2 neutralized the effect of zinc thus alleviating α-synuclein pathology whereas its knockout significantly aggravated fibrillation (Gao et al., 2022). It has been confirmed using many independent approaches that the presence of zinc results in increased content of beta structure of α-synuclein.
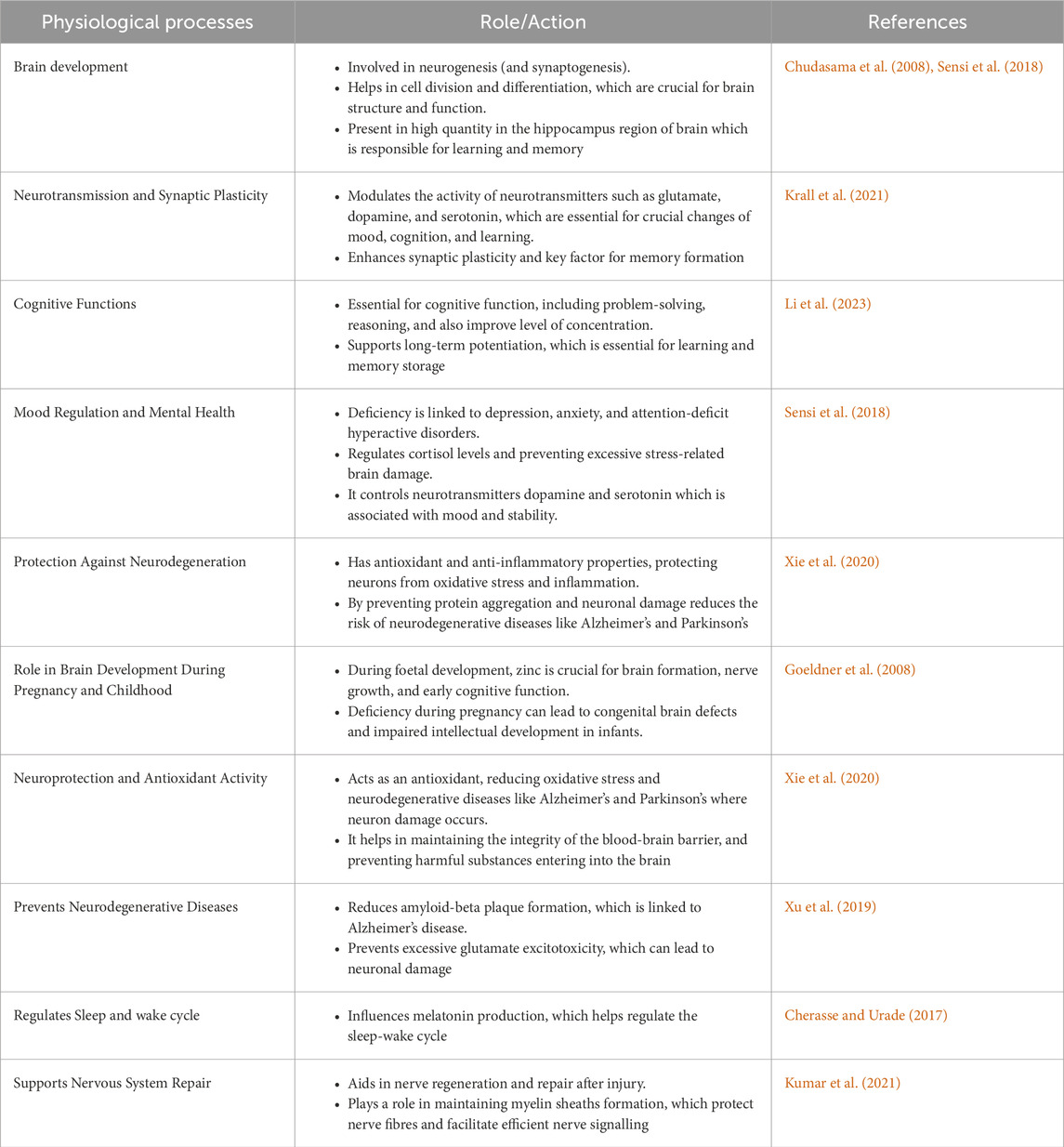
Table 2. Role of zinc in nervous system and cognitive development.
In contrast to toxic effects of zinc reported by most studies, the metal has been shown to mitigate oxidative stress (Méndez-Álvarez et al., 2002) as well as prevent toxicity of dopaminergic neurons (Ajjimaporn et al., 2008). The physiological role and requirement of zinc is evident from the fact that zinc is stored in within synaptic vesicles at the terminals of glutaminergic neurons especially in the forebrain region. It is released along with glutamate and plays an integral role in mediating transmission of synapse through NMDA and GABA receptors (Anderson et al., 2017; Vergnano et al., 2014). Pathological consequences start occurring when an excess of synaptic zinc leaks in to the post synaptic neurons through the ion channels (Sensi et al., 2011).
3.3 Understanding the basis of the “zinc effect” on α-synuclein
As stated previously, many divalent metal cations have the potential to augment the fibrillation of α-synuclein by a common mechanism of shielding the negative charge density on the protein. The effect is more prominent in case of the trivalent lanthanides which can additionally bind to the central NAC region and accelerate fibril formation (Bai et al., 2016). A closer look reveals that different cations vary with regards to their binding affinity to α-synuclein and subsequent effect on the kinetics of nucleation and elongation.
Native nano-electrospray ionisation and ion mobility-mass spectrometry (nESI(-IM)-MS) can differentiate conformations of the protein based on their charged states, relying on the principle that denatured proteins are more extended and therefore are comparatively more protonated than native compact states with a lower Solvent Accessible Surface Area. Therefore, denatured conformations of proteins can readily be differentiated from the native conformers based on their status of protonation. Due to the inherently Intrinsically Disordered nature of α-synuclein, an ensemble of native state monomers of the proteins can be distinguished with a charge ranging between +5 and +18. Using this technique, the binding of zinc to the protein was probed quantitatively and at equimolar ratios of metal ion: protein, zinc was found to exhibit significant binding to α-synuclein along with sodium, copper and lanthanum (Moons et al., 2020). In a subsequent study conducted to understand the effect of zinc on α-synuclein fibrillation, the metal ion was found to decrease the lag time of aggregation (tlag) with a simultaneous increase in apparent rate constant (kapp). However, the fibrils formed were significantly shorter than those formed in the presence of copper, calcium and magnesium (Atarod et al., 2022). FTIR analysis of amide I vibrational spectrum also confirmed that in presence zinc, additional regions of the protein take up the beta pleated sheet conformation (Li et al., 2022). A combination of single residue resolution level NMR spectroscopy and truncated versions of α-synuclein mutants have revealed the binding sites of zinc. In presence of 100 µM zinc, the most significant binding site has been deciphered to be the Asp121 and Asp135 in the C terminus; however, no additional binding interaction was noted due to the presence of other negatively charged residues in the vicinity including Glu130, Glu131, Glu137 and Glu139. This clearly indicated that the affinity of zinc for the C terminal domain was not solely governed by only electrostatic interactions but was primarily a result of the unique conformation of the protein centred around the Asp121 residue. Binding at a threefold excess molar concentration of zinc revealed the second binding site of the metal at His50 in the N terminus but no additional affinity was noted for the C terminus. The trend of zinc binding to α-synuclein is in unison with the observed effects; at low physiological concentrations, zinc serves a twofold function of both neutralizing the negative charge density at the C terminal of α-synuclein and assists other chaperones which work in a concerted manner to prevent aggregation of α-synuclein, whereas at higher concentrations, zinc promotes fibrillation by binding to His50 at the N terminus and lowering its pKa, much like that observed in case of iron (Valiente-Gabioud et al., 2012). Given the fact that physiological concentrations of zinc seldom exceed 50 μM at the substantia nigra micromilieu, it is apparent that in vivo, zinc induced α-synuclein toxicity is not an issue of concern. Interestingly enough, the shorter fibrils formed in presence of zinc were not cytotoxic unlike the longer ones formed in presence of the other cations (Atarod et al., 2022). This observation came as a surprise given the fact that oligomers of α-synuclein impart more toxic effects than the fully mature fibrils due to their stronger ability to induce pore formation in the membrane (Zhang et al., 2013). One of the several possible answers to explain this may lie in the fact that zinc ions serve as metal activators of certain chaperonin proteins which bind and neutralize synuclein oligomers (Al-Harthi et al., 2022). Another clue to explain the apparent harmlessness of zinc induced synuclein oligomers comes from a related study in tau proteins implicated in disease pathology of AD. Zinc induced oligomerization of tau occurred in a temperature dependent manner without the necessity of additional cofactors such as heparin. However, these oligomers did not eventually develop into amyloid fibrils and were dissociated on lowering the temperature (Roman et al., 2019) indicating the reversibility in the formation of the oligomers. The results if extrapolatable in case of α-synuclein will imply that oligomers of α-synuclein formed in the presence of zinc are also transient unstable entities not capable of inducing pores in lipid membranes. A similar study conducted by Kim et al. much earlier, had indicated that heat induced aggregation of recombinant α-synuclein was facilitated in presence of zinc (Kim et al., 2000). A possible explanation of these findings is the fact that increasing temperature augments hydrophobic interaction at the NAC region of α-synuclein; however, at the same time temperature lowers the pKa of the acidic side chains in the C terminus thus facilitating the opposite effect of keeping fibrillation at bay. Zinc masks these negative charges even at elevated temperatures to promote fibrillation. As soon as the temperature drops, the hydrophobicity driven factor is neutralized, rendering the proteins into random aggregates which lack a structured fibrillar component and therefore incapable of effecting membrane damage. Zinc is also known to induce liquid-liquid phase separation of proteins implicated in neurodegenerative disorders (Singh et al., 2020; Yatoui et al., 2022). The metal was shown to induce phase separation in tau, the amyloid like protein associated with AD pathology and shift the equilibrium phase boundary of LLPS towards the region of lower protein concentration (Singh et al., 2020). However, there has been little or no study on the effect of zinc in the LLPS of α-synuclein. The dual role of zinc on aggregation and fibrillation of α-synuclein has been summarised in Figure 3.
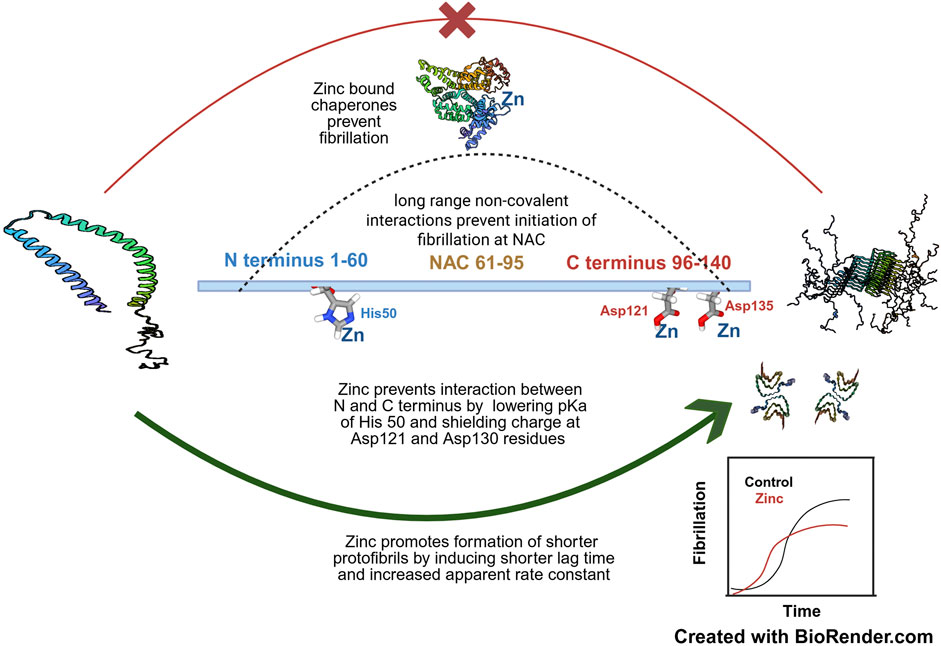
Figure 3. The concentration dependent effect of zinc on α-synuclein fibrillation- Zinc at moderate to high concentrations (around 100 µM) promotes aggregation of NAC domain by shielding the C terminal negative charges of Asp121 and Asp135 and at even higher concentrations also lowers the pKa of His50 at the N terminus-the cumulative effect is to prevent the long range non-covalent interaction of the N and C terminus which prevents initiation of fibrillation. Zinc induces formation of short fibrils by decreasing lag time and increasing association constant as represented in the inset. However, lower concentrations of zinc helps to prevent fibrillation by assisting in the chaperonin activity of proteins which keeps α-synuclein in its physiological conformation. The PDB files of α-synuclein, namely, 1XQ8 (Ulmer et al. 2005), 2N0A (Tuttle et al. 2016) and 6OSM (Ni et al. 2019) and HSA, 6YG9 (Mishra et al. 2020) have been used in this figure. The concepts and mechanisms depicted in the figure have been adopted from multiple studies as cited in the text. Figure has been created with BioRender.
3.4 Role of other micronutrients in shaping amyloid fibrillation: iodine and selenium in regulating α-synuclein fibrillation
Apart from zinc, other physiologically significant non-metal micronutrients such as iodine and selenium have also been demonstrated to possess anti-amyloidogenic properties. Iodine is an acclaimed chaotropic agent capable of modulating the conformational stability of a plethora of proteins by reorienting the water structure around the protein’s microenvironment. Iodine has also been shown to interact with amyloid fibrils, amyloid polymorphs and form complex with mature insulin amyloid fibrils (Hiramatsu et al., 2020; Takekiyo et al., 2022). By using a combination of biophysical techniques, Taketiyo et al. demonstrated that ethylammonium iodide (EAI) can inhibit the formation of α-synuclein amyloid aggregate via interaction between I3− ions and Lys residues in the N-terminal domain of the protein (Takekiyo et al., 2022).
Selenium and selenoproteins have also been implicated in modulating α-synuclein fibrillation. Free Selenium can interact with α-Synuclein and induce the formation of protein aggregates (Maass et al., 2020). SelS and SelP are the two major selenoproteins of the body implicated in antioxidant defence and selenium transport respectively (Lu and Holmgren, 2009). Additionally, SelS has been described as the regulator of the protein aggregation; it probably acts by mediating transcriptional downregulation of aggregation prone proteins; however, the exact mechanism is yet to be deciphered. A recent study on PD patients has suggested that serum concentrations of Se and SelP can potentially be used as a biomarker for monitoring α-Synuclein aggregation (Salaramoli et al., 2024).
3.5 Leads towards zinc based therapeutic strategies to counteract synucleinopathies
There are currently very few safe therapeutic strategies to directly degrade and clear amyloid fibrils. One of these is the bacterial enzyme nattokinase which is a serine protease capable of efficiently removing fibrinolytic clots (Ni et al., 2023). A new enzyme from earthworm termed lumbrokinase has been reported by Girigoswami et al. with the potential to degrade fibrin clots (Metkar et al., 2017). The polysaccharide carrageenan isolated from certain species of red algae possess the potential to degrade amyloid fibrils (Makshakova et al., 2023). Girigoswami et al. has recently developed a nanoformulated carrageenan with enhanced degradation potential as compared to the conventional one (Udayakumar et al., 2024).
The ability of zinc to arrest fibrillation by assisting chaperones and to induce formation of non-toxic protofibrils and oligomers have led to the prospects for the development of zinc oxide nanoparticle-based biomarkers. In a study carried out by Asthana et al., zinc oxide nanoparticles showed significant binding affinity with α-synuclein fibrils via a) enthalpy driven interaction with the amphipathic “KA: TKE/QGV” repeats and b) enthalpy-driven interaction with the central NAC region as well as the C terminus; this led to arrest of subsequent fibrillation (Asthana et al., 2020). Based on this observation, a zinc oxide nanoparticle coated aluminium microelectrode has been designed as an efficient tool for early detection of α-synuclein fibrillation (Adam et al., 2021a). A related study based on Surface Enhanced Raman Spectroscopy reported that zinc oxide nanoparticles can effectively bind and sequester α-synuclein oligomers provided they have not been exposed to physiological temperatures (37°C) which would lead to irreversible formation of amyloid fibrils (Slekiene et al., 2022). Further refinement in this regard has been achieved with surface moderated zinc oxide nanoparticle corona which is able to effectively remove flocs or amorphous aggregates of α-synuclein (Jena et al., 2025). In order to maximize the surface area for enhanced sensitivity in detection of amyloids, a zinc nanoflower-based on nano-silver thin film has been developed (Akhtar et al., 2017) and subsequently upgraded to degrade the amyloid fibrils (Girigoswami et al., 2019). As discussed before, Thioflavin-T positive glycated proteins in the bloodstream of diabetic or prediabetic patients can elicit amyloid like complicacies. Similar studies exploring the effect of zinc nanoparticles on glycated proteins need to be conducted to determine whether zinc-oxide nanoparticles can also trap these protein-sugar adducts before they develop into toxic amyloids capable of developing cognitive ailments and other complicacies.
4 Discussion: bottlenecks in current technology and future of research in alpha-synucleinopathies
The discovery of the strong clinical correlation between Type 2 diabetes and neurodegeneration and the molecular crosstalks involved therein has opened up new avenues of research in elucidating the molecular and physiological basis of cognitive decline, but at the same time it has also seriously complicated our understanding of complex neurodegenerative diseases such as PD and other alpha-synucleinopathies. As already stated, serum proteins existing in a high sugar milieu for a long period develop into Advanced Glycated End products (AGEs) via Amadori reaction and subsequent Maillard rearrangement. Many of these adducts have shown Thioflavin-T positive amyloidogenic behaviour (Hsu et al., 2019). Several other proteins irrespective of the state of glycation, show resemblance to amyloids and collectively constitute the “amyloid proteome” (Gottwald and Röcken, 2021). Although the nature and fibril forming mechanism of most of these proteins have been delineated in detail, the same is not true for the glycated adducts. The burden of glycated proteins can be estimated from the fact that diabetic individuals are more than 50% susceptible in developing cognitive disorders as compared to non-diabetic persons (Cao et al., 2024). The management of glycated proteins is further escalated by the fact that they are recalcitrant to ubiquitin mediated degradation owing to selective modification of lysine residues via glycation. Therefore, efficient and early detection of glycation is pivotal for avoiding many diseases.
Previously, people used immunological techniques such as ELISA and immunoblotting for detection of glycated protein adducts which frequently led to overestimation of AGEs due to conversion of Amadori products into carboxy methyl lysine or pentosidines (Suzuki et al., 2022). Thereafter, improvisations on fluorescence-based methods such as measurement of skin autofluorescence to detect skin AGEs (Koetsier et al., 2010) has facilitated detection of subcutaneous deposition of AGEs. In spite of the intrinsically challenging task of ionisation of glycated proteins and peptides owing to their charged status (Bae et al., 2012), detection of glycation has also been made possible by the development of advanced proteomic tools like stable isotopic dilution analysis liquid chromatography-tandem mass spectrometry and stable isotope labelling with amino acids in cell culture (SILAC) high resolution mass spectrometry (Rabbani et al., 2016). It must, however, be reinstated that our understanding of the role of glycation in mediating neurodegenerative disorders is still at rudimentary level, and preliminary studies have indicated that although metal ions like zinc and iron inhibit glycation of amyloidogenic proteins (Baraka-Vidot et al., 2014). Another potential but less explored avenue on amyloid plaque formation research is the role of bacterial Curli system on cellular amyloid proteins such as α-synuclein and amyloid protein A. Curli is itself an amyloid like protein and an integral component of biofilm produced by some strains of Escherichia coli and several Salmonella sp. (Oh et al., 2012). Inside human body, curli, produced by resident E. coli of gut can be transported to the brain via the Gut-Brain axis (Zheng et al., 2023). Curli can both augment or inhibit the fibrillation of human amyloids depending on the type of protein and/or cellular conditions (Christensen et al., 2019; Ivanova et al., 2021). Zinc is an essential constituent of Curli biogenesis in E. coli and while in gut, the bacterium draws its requisite zinc from the physiological reserve. YkgM and ZinT proteins maintains the optimal zinc concentration required for Curli biogenesis and assembly (Lim et al., 2011). However, it is imperative to state that more oriented studies need to be conducted to understand the role of zinc and other metals in mediating the fibrillation microenvironment of bacterial curl as well as other its effect on other human amyloids. A schematic representation of amyloids resulting from glycated adducts as well as the mode of transport of E. coli curli via the vagus nerve to the brain is presented in Figure 4.
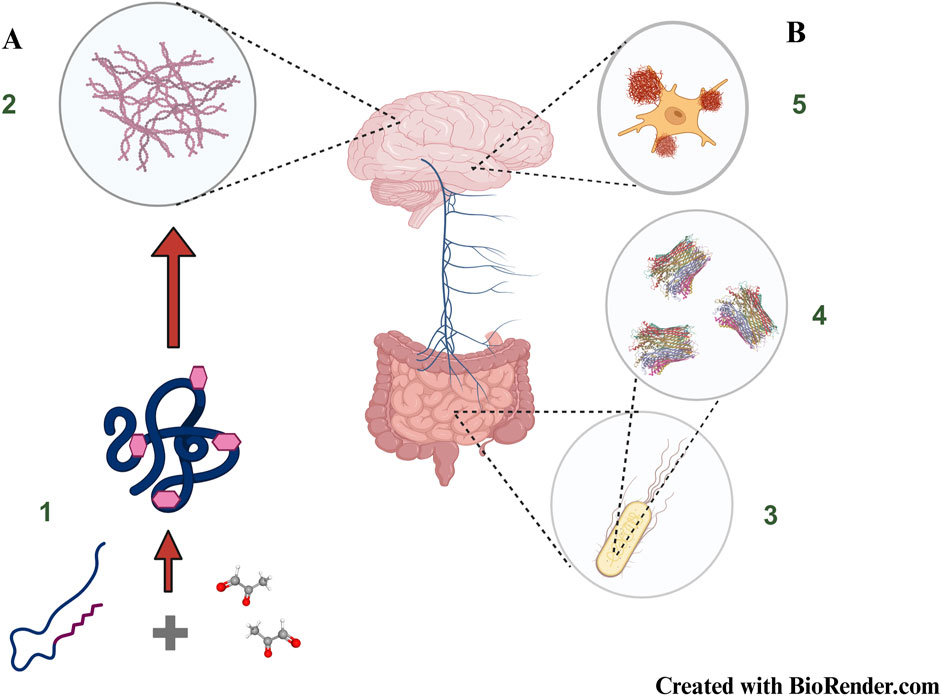
Figure 4. New frontiers in brain amyloid formation: (A) Non-enzymatic glycation of proteins resulting in the formation of Advanced Glycation End Products (1) mimic amyloid like behaviour (2) and constitute one of the major causes to account for diabetes induced neurodegeneration (B) Gut resident Escherichia coli (3) produce bacterial amyloids known as curli (4) which can potentially be transported to the brain via the vagus nerve (Gut-Brain Axis) and influence abnormal behaviour of resident amyloidogenic proteins (5). The cartoon structure of Curli has been taken from PDB ID 6LQH, Zhang et al. 2020.The concepts and mechanisms depicted in the figure have been adopted from multiple studies as cited in the text. Figure has been created with BioRender.
5 Conclusion
Management of α-synucleinopathies has been an insurmountable hurdle for both the scientific as well as the biomedical community. The delineation of almost all structural and physicochemical facets of α-synuclein has facilitated the development of biomarkers to detect protein aggregates associated with cognitive ailments such as synucleinopathies (Bagree et al., 2023). Particularly, significant progress has been made possible through the use of Surface Enhanced Raman Spectroscopy (SERS), a technique which achieves detection of miniscule analytes through amplification of Raman scattering and thus can diagnose a cognitive problem before the actual onset of the symptoms (Colniță et al., 2023). However, we are yet to reach the desirable Technology Readiness Level to adopt these tools in a real-life clinical setting (Ganguly et al., 2021). In this regard, the role of zinc has emerged on a dual perspective in both prevention and cure of these complex multifactorial ailments. The intrinsically strong zinc-α-synuclein interaction has serious prospects for circumventing the current technological bottlenecks. A gold nano-rod incorporated zinc-oxide nanocomposite has stepped a foot forward by discriminating between native and fibrillated α-synuclein (Adam et al., 2021b) and seems to hold a lot of promise in identification of the chemical microenvironment conducive to fibrillation. The high affinity of zinc especially towards the C-terminal acidic domain of α-synuclein has facilitated the development of zinc-oxide nanocomposites to probe the deposition of α-synuclein aggregates (Di Mari et al., 2024). More importantly, considering the fact that many neurodegenerative ailments have been traced to have roots in lifestyle diseases, proper management of our diet including adoption of food processing techniques is of paramount importance to ensure the requisite absorption and bioavailability of micronutrients including zinc.
Author contributions
SB: Conceptualization, Investigation, Writing – original draft, Writing – review and editing. DB: Conceptualization, Formal Analysis, Investigation, Resources, Supervision, Validation, Writing – review and editing. PB: Formal Analysis, Investigation, Validation, Writing – review and editing. SaC: Investigation, Validation, Writing – review and editing. MB: Conceptualization, Formal Analysis, Writing – review and editing. CB: Conceptualization, Investigation, Writing – original draft. DD: Formal Analysis, Validation, Writing – original draft. SS: Formal Analysis, Validation, Writing – original draft. SuC: Validation, Writing – original draft.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
The authors are grateful to Officer-in-Charge, Government General Degree College, Narayangarh and Principal, Maulana Azad College, Kolkata for extending all kinds of support to complete this review in time. Sincere acknowledgement is due to Krishnananda Chattopadhay, Chief Scientist, Structural Biology and Bioinformatics Division, CSIR-Indian Institute of Chemical Biology, Kolkata, India for kindly helping with some of the experiments of our lab cited in this work.
Conflict of interest
Author MB was employed by Dr. Herbs LLC.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Adam, H., Gopinath, S. C. B., Arshad, M. K. M., Parmin, N. A., and Hashim, U. (2021a). Distinguishing normal and aggregated alpha-synuclein interaction on gold nanorod incorporated zinc oxide nanocomposite by electrochemical technique. Int. J. Biol. Macromol. 171, 217–224. doi:10.1016/j.ijbiomac.2021.01.014
Adam, H., Gopinath, S. C. B., and Hashim, U. (2021b). Integration of aluminium interdigitated electrodes with zinc oxide as nanocomposite for selectively detect alpha-synuclein for Parkinson’s disease diagnosis. J. Phys. Conf. Ser. 2129 (1), 012094. doi:10.1088/1742-6596/2129/1/012094
Agarwal, A., Chandran, A., Raza, F., Ungureanu, I. M., Hilcenko, C., Stott, K., et al. (2024). VAMP2 regulates phase separation of α-synuclein. Nat. Cell Biol. 26 (8), 1296–1308. doi:10.1038/s41556-024-01451-6
Ajjimaporn, A., Shavali, S., Ebadi, M., and Govitrapong, P. (2008). Zinc rescues dopaminergic SK–N–SH cell lines from methamphetamine-induced toxicity. Brain Res. Bull. 77 (6), 361–366. doi:10.1016/j.brainresbull.2008.09.006
Akhtar, N., Metkar, S. K., Girigoswami, A., and Girigoswami, K. (2017). ZnO nanoflower based sensitive nano-biosensor for amyloid detection. Mater. Sci. Eng. C 78, 960–968. doi:10.1016/j.msec.2017.04.118
Al-Harthi, S., Kharchenko, V., Mandal, P., Gourdoupis, S., and Jaremko, Ł. (2022). Zinc ions prevent α-synuclein aggregation by enhancing chaperone function of human serum albumin. Int. J. Biol. Macromol. 222, 2878–2887. doi:10.1016/j.ijbiomac.2022.10.066
Anderson, C. T., Kumar, M., Xiong, S., and Tzounopoulos, T. (2017). Cell-specific gain modulation by synaptically released zinc in cortical circuits of audition. ELife 6, e29893. doi:10.7554/eLife.29893
Ariesandi, W., Chang, C. F., Chen, T. E., and Chen, Y. R. (2013). Temperature-dependent structural changes of Parkinson’s alpha-synuclein reveal the role of pre-existing oligomers in alpha-synuclein fibrillization. PLoS ONE 8 (1), e53487. doi:10.1371/journal.pone.0053487
Asthana, S., Bhattacharyya, D., Kumari, S., Nayak, P. S., Saleem, M., Bhunia, A., et al. (2020). Interaction with zinc oxide nanoparticle kinetically traps α-synuclein fibrillation into off-pathway non-toxic intermediates. Int. J. Biol. Macromol. 150, 68–79. doi:10.1016/j.ijbiomac.2020.01.269
Atarashi, R., Kazunori, S., Katsuya, S., and Nishida, N. (2011). Real-time quaking-induced conversion. Prion 5 (3), 150–153. doi:10.4161/pri.5.3.16893
Atarod, D., Mamashli, F., Ghasemi, A., Moosavi-Movahedi, F., Pirhaghi, M., Nedaei, H., et al. (2022). Bivalent metal ions induce formation of α-synuclein fibril polymorphs with different cytotoxicities. Sci. Rep. 12 (1), 11898. doi:10.1038/s41598-022-15472-4
Bae, Y. J., Shin, Y. S., Moon, J. H., and Kim, M. S. (2012). Degree of ionization in MALDI of peptides: thermal explanation for the gas-phase ion formation. J. Am. Soc. Mass Spectrom. 23 (8), 1326–1335. doi:10.1007/s13361-012-0406-y
Bagchi, D., Bagchi, M., and Stohs, S. J. (1997). Comparative in vitro oxygen radical scavenging ability of zinc methionine and selected zinc salts and antioxidants. General Pharmacol. Vasc. Syst. 28 (1), 85–91. doi:10.1016/S0306-3623(96)00178-4
Bagchi, D., Vuchetich, P. J., Bagchi, M., Tran, M. X., Krohn, R. L., Ray, S. D., et al. (1998). Protective effects of zinc salts on TPA-Induced hepatic and brain lipid peroxidation, glutathione depletion, DNA damage and peritoneal macrophage activation in mice. General Pharmacol. Vasc. Syst. 30 (1), 43–50. doi:10.1016/S0306-3623(97)00072-4
Bagree, G., De Silva, O., Liyanage, P. D., Ramarathinam, S. H., Sharma, S. K., Bansal, V., et al. (2023). α-synuclein as a promising biomarker for developing diagnostic tools against neurodegenerative synucleionopathy disorders. TrAC Trends Anal. Chem. 159, 116922. doi:10.1016/j.trac.2023.116922
Bai, J., Zhang, Z., Liu, M., and Li, C. (2016). α-synuclein-lanthanide metal ions interaction: binding sites, conformation and fibrillation. BMC Biophys. 9 (1), 1. doi:10.1186/s13628-016-0026-1
Baraka-Vidot, J., Navarra, G., Leone, M., Bourdon, E., Militello, V., and Rondeau, P. (2014). Deciphering metal-induced oxidative damages on glycated albumin structure and function. Biochimica Biophysica Acta (BBA) - General Subj. 1840 (6), 1712–1724. doi:10.1016/j.bbagen.2013.12.017
Bell, R., Thrush, R. J., Castellana-Cruz, M., Oeller, M., Staats, R., Nene, A., et al. (2022). N-Terminal acetylation of α-Synuclein slows down its aggregation process and alters the morphology of the resulting aggregates. Biochemistry 61 (17), 1743–1756. doi:10.1021/acs.biochem.2c00104
Bertoni-Freddari, C., Fattoretti, P., Casoli, T., Di Stefano, G., Giorgetti, B., and Balietti, M. (2008). Brain aging: the zinc connection. Exp. Gerontol. 43 (5), 389–393. doi:10.1016/j.exger.2007.11.001
Bishop, G. M., and Robinson, S. R. (2004). Physiological roles of Amyloid-β and implications for its removal in Alzheimer’s disease. Drugs and Aging 21 (10), 621–630. doi:10.2165/00002512-200421100-00001
Björndahl, L., and Kvist, U. (2014). “Structure of chromatin in spermatozoa,” in Genetic damage in human spermatozoa. Editors E. Baldi, and M. Muratori (New York: Springer), 1–11. doi:10.1007/978-1-4614-7783-9_1
Braak, H., Sandmann-Keil, D., Gai, W., and Braak, E. (1999). Extensive axonal lewy neurites in Parkinson’s disease: a novel pathological feature revealed by α-synuclein immunocytochemistry. Neurosci. Lett. 265 (1), 67–69. doi:10.1016/S0304-3940(99)00208-6
Brás, I. C., and Outeiro, T. F. (2021). Alpha-synuclein: mechanisms of release and pathology progression in synucleinopathies. Cells 10 (2), 375–19. doi:10.3390/cells10020375
Brodie, N. I., Popov, K. I., Petrotchenko, E. V., Dokholyan, N. V., and Borchers, C. H. (2019). Conformational ensemble of native α-synuclein in solution as determined by short-distance crosslinking constraint-guided discrete molecular dynamics simulations. PLOS Comput. Biol. 15 (3), e1006859. doi:10.1371/journal.pcbi.1006859
Brown, J. W. P., Buell, A. K., Michaels, T. C. T., Meisl, G., Carozza, J., Flagmeier, P., et al. (2016). β-Synuclein suppresses both the initiation and amplification steps of α-synuclein aggregation via competitive binding to surfaces. Sci. Rep. 6 (1), 36010. doi:10.1038/srep36010
Buell, A. K., Galvagnion, C., Gaspar, R., Sparr, E., Vendruscolo, M., Knowles, T. P. J., et al. (2014). Solution conditions determine the relative importance of nucleation and growth processes in α-synuclein aggregation. Proc. Natl. Acad. Sci. U. S. A. 111 (21), 7671–7676. doi:10.1073/pnas.1315346111
Byrd, E. J., Wilkinson, M., Radford, S. E., and Sobott, F. (2023). Taking charge: metal ions accelerate amyloid aggregation in sequence variants of α-Synuclein. J. Am. Soc. Mass Spectrom. 34 (3), 493–504. doi:10.1021/jasms.2c00379
Calabresi, P., Di Lazzaro, G., Marino, G., Campanelli, F., and Ghiglieri, V. (2023). Advances in understanding the function of alpha-synuclein: implications for Parkinson’s disease. Brain 146 (9), 3587–3597. doi:10.1093/brain/awad150
Campese, N., Fanciulli, A., Stefanova, N., Haybaeck, J., Kiechl, S., and Wenning, G. K. (2021). Neuropathology of multiple system atrophy: kurt Jellinger`s legacy. J. Neural Transm. 128 (10), 1481–1494. doi:10.1007/s00702-021-02383-3
Cao, F., Yang, F., Li, J., Guo, W., Zhang, C., Gao, F., et al. (2024). The relationship between diabetes and the dementia risk: a meta-analysis. Diabetology Metabolic Syndrome 16 (1), 101. doi:10.1186/s13098-024-01346-4
Chen, B., Wen, X., Jiang, H., Wang, J., Song, N., and Xie, J. (2019). Interactions between iron and α-synuclein pathology in Parkinson’s disease. Free Radic. Biol. Med. 141, 253–260. doi:10.1016/j.freeradbiomed.2019.06.024
Chen, Y., and Yu, Y. (2023). Tau and neuroinflammation in Alzheimer’s disease: interplay mechanisms and clinical translation. J. Neuroinflammation 20 (1), 165. doi:10.1186/s12974-023-02853-3
Cherasse, Y., and Urade, Y. (2017). Dietary zinc acts as a sleep modulator. Int. J. Mol. Sci. 18 (11), 2334. doi:10.3390/ijms18112334
Choi, S., Liu, X., and Pan, Z. (2018). Zinc deficiency and cellular oxidative stress: prognostic implications in cardiovascular diseases. Acta Pharmacol. Sin. 39 (7), 1120–1132. doi:10.1038/aps.2018.25
Christensen, L. F. B., Jensen, K. F., Nielsen, J., Vad, B. S., Christiansen, G., and Otzen, D. E. (2019). Reducing the amyloidogenicity of functional amyloid protein FapC increases its ability to inhibit α-Synuclein fibrillation. ACS Omega 4 (2), 4029–4039. doi:10.1021/acsomega.8b03590
Chudasama, Y., Wright, K. S., and Murray, E. A. (2008). Hippocampal lesions in rhesus monkeys disrupt emotional responses but not reinforcer devaluation effects. Biol. Psychiatry 63 (11), 1084–1091. doi:10.1016/j.biopsych.2007.11.012
Clayton, D. F., and George, J. M. (1998). The synucleins: a family of proteins involved in synaptic function, plasticity, neurodegeneration and disease. Trends Neurosci. 21 (6), 249–254. doi:10.1016/S0166-2236(97)01213-7
Coles, N. P., Elsheikh, S., Quesnel, A., Butler, L., Jennings, C., Tarzi, C., et al. (2025). Molecular insights into α-Synuclein fibrillation: a raman spectroscopy and machine learning approach. ACS Chem. Neurosci. 16, 687–698. doi:10.1021/acschemneuro.4c00726
Colniță, A., Toma, V. A., Brezeştean, I. A., Tahir, M. A., and Dina, N. E. (2023). A review on integrated ZnO-Based SERS biosensors and their potential in detecting biomarkers of neurodegenerative diseases. Biosensors 13 (5), 499. doi:10.3390/bios13050499
Coskuner, O., and Wise-Scira, O. (2013). Structures and free energy landscapes of the A53T mutant-type α-synuclein protein and impact of A53T mutation on the structures of the wild-type α-synuclein protein with dynamics. ACS Chem. Neurosci. 4 (7), 1101–1113. doi:10.1021/cn400041j
Costa, M. I., Sarmento-Ribeiro, A. B., and Gonçalves, A. C. (2023). Zinc: from biological functions to therapeutic potential. Int. J. Mol. Sci. 24 (5), 4822. doi:10.3390/ijms24054822
Dada, S. T., Hardenberg, M. C., Toprakcioglu, Z., Mrugalla, L. K., Cali, M. P., McKeon, M. O., et al. (2023). Spontaneous nucleation and fast aggregate-dependent proliferation of α-synuclein aggregates within liquid condensates at neutral pH. Proc. Natl. Acad. Sci. 120 (9), e2208792120. doi:10.1073/pnas.2208792120
Das, A., Basak, P., Pramanik, A., Majumder, R., Ghosh, A., Hazra, S., et al. (2020). Ribosylation induced structural changes in bovine serum albumin: understanding high dietary sugar induced protein aggregation and amyloid formation. Heliyon 6 (9), e05053. doi:10.1016/j.heliyon.2020.e05053
de Bruyn, E., Dorn, A. E., Rossetti, G., Fernandez, C., Outeiro, T. F., Schulz, J. B., et al. (2024). Impact of phosphorylation on the physiological form of human alpha-Synuclein in aqueous solution. J. Chem. Inf. Model. 64, 8215–8226. doi:10.1021/acs.jcim.4c01172
Dedmon, M. M., Lindorff-Larsen, K., Christodoulou, J., Vendruscolo, M., and Dobson, C. M. (2005). Mapping long-range interactions in α-Synuclein using spin-label NMR and ensemble molecular dynamics simulations. J. Am. Chem. Soc. 127 (2), 476–477. doi:10.1021/ja044834j
De Luca, G., Malacrida, L., Vetri, V., and Sancataldo, G. (2025). Unveiling water ordering in liquid–liquid phase separation using bovine serum albumin-polyethylene glycol systems. J. Mol. Liq. 433, 127865. doi:10.1016/j.molliq.2025.127865
Di Mari, G. M., Scuderi, M., Lanza, G., Salluzzo, M. G., Salemi, M., Caraci, F., et al. (2024). Pain-free alpha-synuclein detection by low-cost hierarchical nanowire based electrode. Nanomaterials 14 (2), 170. doi:10.3390/nano14020170
Esteves, A. R., Arduíno, D. M., Swerdlow, R. H., Oliveira, C. R., and Cardoso, S. M. (2008). Oxidative stress involvement in α-Synuclein oligomerization in Parkinson’s disease cybrids. Antioxidants and Redox Signal. 11 (3), 439–448. doi:10.1089/ars.2008.2247
Farzadfard, A., König, A., Petersen, S. V., Nielsen, J., Vasili, E., Dominguez-Meijide, A., et al. (2022a). Glycation modulates alpha-synuclein fibrillization kinetics: a sweet spot for inhibition. J. Biol. Chem. 298 (5), 101848. doi:10.1016/j.jbc.2022.101848
Farzadfard, A., Pedersen, J. N., Meisl, G., Somavarapu, A. K., Alam, P., Goksøyr, L., et al. (2022b). The C-terminal tail of α-synuclein protects against aggregate replication but is critical for oligomerization. Commun. Biol. 5 (1), 123. doi:10.1038/s42003-022-03059-8
Flagmeier, P., Meisl, G., Vendruscolo, M., Knowles, T. P. J., Dobson, C. M., Buell, A. K., et al. (2016). Mutations associated with familial Parkinson’s disease alter the initiation and amplification steps of α-synuclein aggregation. Proc. Natl. Acad. Sci. 113 (37), 10328–10333. doi:10.1073/pnas.1604645113
Frey, L., Ghosh, D., Qureshi, B. M., Rhyner, D., Guerrero-Ferreira, R., Pokharna, A., et al. (2024). On the pH-dependence of α-synuclein amyloid polymorphism and the role of secondary nucleation in seed-based amyloid propagation. ELife 12. doi:10.7554/elife.93562
Fujiwara, H., Hasegawa, M., Dohmae, N., Kawashima, A., Masliah, E., Goldberg, M. S., et al. (2002). alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 4 (2), 160–164. doi:10.1038/ncb748
Ganguly, U., Singh, S., Pal, S., Prasad, S., Agrawal, B. K., Saini, R. V., et al. (2021). Alpha-synuclein as a biomarker of Parkinson’s disease: good, but not good enough. Front. Aging Neurosci. 13, 702639. doi:10.3389/fnagi.2021.702639
Gao, H., Sun, H., Yan, N., Zhao, P., Xu, H., Zheng, W., et al. (2022). ATP13A2 declines zinc-induced accumulation of α-Synuclein in a Parkinson’s disease model. Int. J. Mol. Sci. 23 (14), 8035. doi:10.3390/ijms23148035
Gardner, B., Dieriks, B. V., Cameron, S., Mendis, L. H. S., Turner, C., Faull, R. L. M., et al. (2017). Metal concentrations and distributions in the human olfactory bulb in Parkinson’s disease. Sci. Rep. 7 (1), 10454. doi:10.1038/s41598-017-10659-6
Garner, T. B., Hester, J. M., Carothers, A., and Diaz, F. J. (2021). Role of zinc in female reproduction. Biol. Reproduction 104 (5), 976–994. doi:10.1093/biolre/ioab023
Ghanem, S. S., Majbour, N. K., Vaikath, N. N., Ardah, M. T., Erskine, D., Jensen, N. M., et al. (2022). α-Synuclein phosphorylation at serine 129 occurs after initial protein deposition and inhibits seeded fibril formation and toxicity. Proc. Natl. Acad. Sci. 119 (15), e2109617119. doi:10.1073/pnas.2109617119
Ghio, S., Camilleri, A., Caruana, M., Ruf, V. C., Schmidt, F., Leonov, A., et al. (2019). Cardiolipin promotes pore-forming activity of alpha-synuclein oligomers in mitochondrial membranes. ACS Chem. Neurosci. 10 (8), 3815–3829. doi:10.1021/acschemneuro.9b00320
Girigoswami, A., Ramalakshmi, M., Akhtar, N., Metkar, S. K., and Girigoswami, K. (2019). ZnO nanoflower petals mediated amyloid degradation - an in vitro electrokinetic potential approach. Mater. Sci. Eng. C 101, 169–178. doi:10.1016/j.msec.2019.03.086
Goeldner, C., Reiss, D., Wichmann, J., Meziane, H., Kieffer, B. L., and Ouagazzal, A. M. (2008). Nociceptin receptor impairs recognition memory via interaction with NMDA receptor-dependent mitogen-activated protein kinase/extracellular signal-regulated kinase signaling in the hippocampus. J. Neurosci. 28 (9), 2190–2198. doi:10.1523/JNEUROSCI.3711-07.2008
Goetz, C. G. (2011). The history of Parkinson’s disease: early clinical descriptions and neurological therapies. Cold Spring Harb. Perspect. Med. 1 (1), a008862. doi:10.1101/cshperspect.a008862
Gorell, J. M., Johnson, C. C., Rybicki, B. A., Peterson, E. L., Kortsha, G. X., Brown, G. G., et al. (1997). Occupational exposures to metals as risk factors for Parkinson’s disease. Neurology 48 (3), 650–658. doi:10.1212/WNL.48.3.650
Gottwald, J., and Röcken, C. (2021). The amyloid proteome: a systematic review and proposal of a protein classification system. Crit. Rev. Biochem. Mol. Biol. 56 (5), 526–542. doi:10.1080/10409238.2021.1937926
Greggio, E., Bisaglia, M., Civiero, L., and Bubacco, L. (2011). Leucine-rich repeat kinase 2 and alpha-synuclein: intersecting pathways in the pathogenesis of Parkinson’s disease? Mol. Neurodegener. 6 (1), 6. doi:10.1186/1750-1326-6-6
Günther, V., Lindert, U., and Schaffner, W. (2012). The taste of heavy metals: gene regulation by MTF-1. Biochimica Biophysica Acta (BBA) - Mol. Cell Res. 1823 (9), 1416–1425. doi:10.1016/j.bbamcr.2012.01.005
Hasegawa, M., Fujiwara, H., Nonaka, T., Wakabayashi, K., Takahashi, H., Lee, V. M.-Y., et al. (2002). Phosphorylated α-Synuclein is ubiquitinated in α-Synucleinopathy lesions. J. Biol. Chem. 277 (50), 49071–49076. doi:10.1074/jbc.M208046200
Hashimoto, M., Takeda, A., Hsu, L. J., Takenouchi, T., and Masliah, E. (1999). Role of cytochrome c as a stimulator of α-synuclein aggregation in lewy body disease. J. Biol. Chem. 274 (41), 28849–28852. doi:10.1074/jbc.274.41.28849
Hayashi, J., and Carver, J. A. (2022). β-Synuclein: an enigmatic protein with diverse functionality. Biomolecules 12 (1), 142. doi:10.3390/biom12010142
Hiramatsu, T., Yamamoto, N., Ha, S., Masuda, Y., Yasuda, M., Ishigaki, M., et al. (2020). Iodine staining as a useful probe for distinguishing insulin amyloid polymorphs. Sci. Rep. 10 (1), 16741. doi:10.1038/s41598-020-73460-y
Hou, K., Liu, T., Li, J., Xian, M., Sun, L., and Wei, J. (2023). Liquid-liquid phase separation regulates alpha-synuclein aggregate and mitophagy in Parkinson’s disease. Front. Neurosci. 17, 1250532. doi:10.3389/fnins.2023.1250532
Hsu, Y. H., Chen, Y. W., Wu, M. H., and Tu, L. H. (2019). Protein glycation by glyoxal promotes amyloid formation by islet amyloid polypeptide. Biophysical J. 116 (12), 2304–2313. doi:10.1016/j.bpj.2019.05.013
Huang, S., Mo, X., Wang, J., Ye, X., Yu, H., and Liu, Y. (2022). α-Synuclein phase separation and amyloid aggregation are modulated by C-terminal truncations. FEBS Lett. 596 (11), 1388–1400. doi:10.1002/1873-3468.14361
Inglis, K. J., Chereau, D., Brigham, E. F., Chiou, S.-S., Schöbel, S., Frigon, N. L., et al. (2009). Polo-like kinase 2 (PLK2) phosphorylates α-Synuclein at serine 129 in central nervous system. J. Biol. Chem. 284 (5), 2598–2602. doi:10.1074/jbc.C800206200
Ivanova, M. I., Lin, Y., Lee, Y.-H., Zheng, J., and Ramamoorthy, A. (2021). Biophysical processes underlying cross-seeding in amyloid aggregation and implications in amyloid pathology. Biophys. Chem. 269, 106507. doi:10.1016/j.bpc.2020.106507
Jain, M. K., Singh, P., Roy, S., and Bhat, R. (2018). Comparative analysis of the conformation, aggregation, interaction, and fibril morphologies of human α-β-and γ-Synuclein proteins. Biochemistry 57 (26), 3830–3848. doi:10.1021/acs.biochem.8b00343
Jan, A., Gonçalves, N. P., Vaegter, C. B., Jensen, P. H., and Ferreira, N. (2021). The prion-like spreading of alpha-synuclein in Parkinson's disease: update on models and hypotheses. Int. J. Mol. Sci. 22 (15), 8338. doi:10.3390/ijms22158338
Javitch, J. A., D’Amato, R. J., Strittmatter, S. M., and Snyder, S. H. (1985). Parkinsonism-inducing neurotoxin, N-methyl-4-phenyl-1,2,3,6 -tetrahydropyridine: uptake of the metabolite N-methyl-4-phenylpyridine by dopamine neurons explains selective toxicity. Proc. Natl. Acad. Sci. 82 (7), 2173–2177. doi:10.1073/pnas.82.7.2173
Jellinger, K. A. (2014). Neuropathology of multiple system atrophy: new thoughts about pathogenesis. Mov. Disord. 29 (14), 1720–1741. doi:10.1002/mds.26052
Jena, S., Subham, K., Kalra, H., and Jha, S. (2025). Multimeric interacting interface of biologically synthesized zinc oxide nanoparticle corona efficiently sequesters α-synuclein against protein fibrillation. Biomaterials Sci. 13, 3336–3353. doi:10.1039/D5BM00143A
Jesu, J., Avila, J., Lucas, J. J., and Hernandez, F. (2004). Role of tau protein in both physiological and pathological conditions. Physioal Rev. 84, 361–384. doi:10.1152/physrev.00024.2003
Kawahata, I., Finkelstein, D. I., and Fukunaga, K. (2022). Pathogenic impact of α-Synuclein phosphorylation and its kinases in α-Synucleinopathies. Int. J. Mol. Sci. 23 (11), 6216. doi:10.3390/ijms23116216
Kim, T. D., Paik, S. R., Yang, C.-H., and Kim, J. (2000). Structural changes in α-synuclein affect its chaperone-like activity in vitro. Protein Sci. 9 (12), 2489–2496. doi:10.1110/ps.9.12.2489
Kiouri, D. P., Tsoupra, E., Peana, M., Perlepes, S. P., Stefanidou, M. E., and Chasapis, C. T. (2023). Multifunctional role of zinc in human health: an update. EXCLI J. 22, 809–827. doi:10.17179/excli2023-6335
Koetsier, M., Nur, E., Chunmao, H., Lutgers, H. L., Links, T. P., Smit, A. J., et al. (2010). Skin color independent assessment of aging using skin autofluorescence. Opt. Express 18 (14), 14416–14429. doi:10.1364/OE.18.014416
Kong, S. M. Y., Chan, B. K. K., Park, J.-S., Hill, K. J., Aitken, J. B., Cottle, L., et al. (2014). Parkinson’s disease-linked human PARK9/ATP13A2 maintains zinc homeostasis and promotes α-Synuclein externalization via exosomes. Hum. Mol. Genet. 23 (11), 2816–2833. doi:10.1093/hmg/ddu099
Krall, R. F., Tzounopoulos, T., and Aizenman, E. (2021). The function and regulation of zinc in the brain. Neuroscience 457, 235–258. doi:10.1016/j.neuroscience.2021.01.010
Kumar, V., Kumar, A., Singh, K., Avasthi, K., and Kim, J.-J. (2021). Neurobiology of zinc and its role in neurogenesis. Eur. J. Nutr. 60 (1), 55–64. doi:10.1007/s00394-020-02454-3
Kumari, M., Kumar, B., Bisht, K. S., and Maiti, T. K. (2022). Glycation renders ɑ-synuclein oligomeric strain and modulates microglia activation. BioRxiv. doi:10.1101/2022.01.15.476311
Lashuel, H. A., and Novello, S. (2021). Lewy body-associated proteins: victims, instigators, or innocent bystanders? The case of AIMP2 and alpha-synuclein. Neurobiol. Dis. 156, 105417. doi:10.1016/j.nbd.2021.105417
Lee, S. H., Lee, M. W., Ko, D. G., Choi, B. Y., and Suh, S. W. (2021). The role of nadph oxidase in neuronal death and neurogenesis after acute neurological disorders. Antioxidants 10 (5), 739. doi:10.3390/antiox10050739
Lee, V. M. Y., and Trojanowski, J. Q. (2006). Mechanisms of Parkinson’s disease linked to pathological α-Synuclein: new targets for drug discovery. Neuron 52 (1), 33–38. doi:10.1016/j.neuron.2006.09.026
Lei, Z., Cao, G., and Wei, G. (2019). A30P mutant α-synuclein impairs autophagic flux by inactivating JNK signaling to enhance ZKSCAN3 activity in midbrain dopaminergic neurons. Cell Death Dis. 10 (2), 133. doi:10.1038/s41419-019-1364-0
Li, M. S., Adesina, S. E., Ellis, C. L., Gooch, J. L., Hoover, R. S., and Williams, C. R. (2016). NADPH oxidase-2 mediates zinc deficiency-induced oxidative stress and kidney damage. Am. J. Physiology-Cell Physiology 312 (1), C47–C55. doi:10.1152/ajpcell.00208.2016
Li, X., Yu, L., Liu, X., Shi, T., Zhang, Y., Xiao, Y., et al. (2024). β-synuclein regulates the phase transitions and amyloid conversion of α-synuclein. Nat. Commun. 15 (1), 8748. doi:10.1038/s41467-024-53086-8
Li, Y., Yu, Y., and Ma, G. (2022). Modulation effects of Fe3+, Zn2+, and Cu2+ ions on the amyloid fibrillation of α-Synuclein: insights from a FTIR investigation. Molecules 27 (23), 8383. doi:10.3390/molecules27238383
Li, Z., Liu, Y., Wei, R., Yong, V. W., and Xue, M. (2023). The important role of zinc in neurological diseases. Biomolecules 13 (1), 28. doi:10.3390/biom13010028
Lim, J., Lee, K.-M., Kim, S. H., Kim, Y., Kim, S.-H., Park, W., et al. (2011). YkgM and ZinT proteins are required for maintaining intracellular zinc concentration and producing curli in enterohemorrhagic Escherichia coli (EHEC) O157:H7 under zinc deficient conditions. Int. J. Food Microbiol. 149 (2), 159–170. doi:10.1016/j.ijfoodmicro.2011.06.017
Longhena, F., Faustini, G., and Bellucci, A. (2020). Study of alpha-synuclein fibrillation: state of the art and expectations. Neural Regen. Res. 15 (1), 59–60. doi:10.4103/1673-5374.264453
Lu, J., and Holmgren, A. (2009). Selenoproteins. J. Biol. Chem. 284 (2), 723–727. doi:10.1074/jbc.R800045200
Maass, F., Michalke, B., Willkommen, D., Schulte, C., Tönges, L., Boerger, M., et al. (2020). Selenium speciation analysis in the cerebrospinal fluid of patients with Parkinson’s disease. J. Trace Elem. Med. Biol. 57, 126412. doi:10.1016/j.jtemb.2019.126412
Mahapatra, A., and Newberry, R. W. (2024). Liquid–liquid phase separation of α-synuclein is highly sensitive to sequence complexity. Protein Sci. 33 (4), e4951. doi:10.1002/pro.4951
Makshakova, O., Bogdanova, L., Faizullin, D., Khaibrakhmanova, D., Ziganshina, S., Ermakova, E., et al. (2023). The ability of some polysaccharides to disaggregate lysozyme amyloid fibrils and renature the protein. Pharmaceutics 15 (2), 624. doi:10.3390/pharmaceutics15020624
Marotta, N. P., Ara, J., Uemura, N., Lougee, M. G., Meymand, E. S., Zhang, B., et al. (2021). Alpha-synuclein from patient lewy bodies exhibits distinct pathological activity that can be propagated in vitro. Acta Neuropathol. Commun. 9 (1), 188. doi:10.1186/s40478-021-01288-2
Marreiro, D. N., Cruz, K. J. C., Morais, J. B. S., Beserra, J. B., Severo, J. S., and Soares de Oliveira, A. R. (2017). Zinc and oxidative stress: current mechanisms. Antioxidants 6 (2), 24. doi:10.3390/antiox6020024
Méndez-Álvarez, E., Soto-Otero, R., Hermida-Ameijeiras, Á., López-Real, A. M., and Labandeira-Garcia, J. L. (2002). Effects of aluminum and zinc on the oxidative stress caused by 6-hydroxydopamine autoxidation: relevance for the pathogenesis of Parkinson’s disease. Biochimica Biophysica Acta (BBA) - Mol. Basis Dis. 1586 (2), 155–168. doi:10.1016/S0925-4439(01)00077-1
Metkar, S. K., Girigoswami, A., Murugesan, R., and Girigoswami, K. (2017). Lumbrokinase for degradation and reduction of amyloid fibrils associated with amyloidosis. J. Appl. Biomed. 15 (2), 96–104. doi:10.1016/j.jab.2017.01.003
Metkar, S. K., Udayakumar, S., Girigoswami, A., and Girigoswami, K. (2024). Amyloidosis-history and development, emphasis on insulin and prion amyloids. Brain Disord. 13, 100106. doi:10.1016/j.dscb.2023.100106
Miller, L. M., Wang, Q., Telivala, T. P., Smith, R. J., Lanzirotti, A., and Miklossy, J. (2006). Synchrotron-based infrared and X-ray imaging shows focalized accumulation of Cu and Zn co-localized with β-amyloid deposits in Alzheimer’s disease. J. Struct. Biol. 155 (1), 30–37. doi:10.1016/j.jsb.2005.09.004
Mishra, A., Castañeda, T. R., Bader, B., Elshorst, S., Cummings, P., Scherer, D. S., et al. (2020). Triantennary GalNAc molecular imaging probes for monitoring hepatocyte function in a rat model of nonalcoholic steatohepatitis. Adv. Sci. 7, 2002997. doi:10.1002/advs.202002997
Moons, R., Konijnenberg, A., Mensch, C., Van Elzen, R., Johannessen, C., Maudsley, S., et al. (2020). Metal ions shape α-synuclein. Sci. Rep. 10 (1), 16293. doi:10.1038/s41598-020-73207-9
Moriarty, G. M., Olson, M. P., Atieh, T. B., Janowska, M. K., Khare, S. D., and Baum, J. (2017). A pH-dependent switch promotes β-synuclein fibril formation via glutamate residues. J. Biol. Chem. 292 (39), 16368–16379. doi:10.1074/jbc.M117.780528
Motamed, S., Ebrahimi, M., Safarian, M., Ghayour-Mobarhan, M., Mouhebati, M., Azarpazhouh, M., et al. (2013). Micronutrient intake and the presence of the metabolic syndrome. North Am. J. Med. Sci. 5 (6), 377–385. doi:10.4103/1947-2714.114171
Mueller, C., Ballard, C., Corbett, A., and Aarsland, D. (2017). Historical landmarks in dementia with lewy bodies. Lancet Neurology 16 (5), 348. doi:10.1016/S1474-4422(17)30089-3
Mukherjee, S., Sakunthala, A., Gadhe, L., Poudyal, M., Sawner, A. S., Kadu, P., et al. (2023). Liquid-liquid phase separation of α-Synuclein: a new mechanistic insight for α-Synuclein aggregation associated with Parkinson’s disease pathogenesis. J. Mol. Biol. 435 (1), 167713. doi:10.1016/j.jmb.2022.167713
Ni, A., Li, H., Wang, R., Sun, R., and Zhang, Y. (2023). Degradation of amyloid β-peptides catalyzed by nattokinase in vivo and in vitro. Food Sci. Hum. Wellness 12 (5), 1905–1916. doi:10.1016/j.fshw.2023.02.042
Ni, X., McGlinchey, R. P., Jiang, J., and Lee, J. C. (2019). Structural insights into α-Synuclein fibril polymorphism: effects of parkinson’s disease-related c-terminal truncations. J. Mol. Biol. 431 (19), 3913–3919. doi:10.1016/j.jmb.2019.07.001
Nussbaum, R. L. (2017). The identification of alpha-synuclein as the first parkinson disease gene. J. Parkinson’s Dis. 7 (1), S43–S49. doi:10.3233/JPD-179003
Oh, Y. J., Cui, Y., Kim, H., Li, Y., Hinterdorfer, P., and Park, S. (2012). Characterization of curli A production on living bacterial surfaces by scanning probe microscopy. Biophysical J. 103 (8), 1666–1671. doi:10.1016/j.bpj.2012.09.004
Ohgita, T., Namba, N., Kono, H., Shimanouchi, T., and Saito, H. (2022). Mechanisms of enhanced aggregation and fibril formation of Parkinson’s disease-related variants of α-synuclein. Sci. Rep. 12 (1), 6770. doi:10.1038/s41598-022-10789-6
Olechnowicz, J., Tinkov, A., Skalny, A., and Suliburska, J. (2018). Zinc status is associated with inflammation, oxidative stress, lipid, and glucose metabolism. J. Physiological Sci. 68 (1), 19–31. doi:10.1007/s12576-017-0571-7
Oteiza, P. I. (2012). Zinc and the modulation of redox homeostasis. Free Radic. Biol. Med. 53 (9), 1748–1759. doi:10.1016/j.freeradbiomed.2012.08.568
Paik, S. R., Lee, D., Cho, H.-J., Lee, E.-N., and Chang, C.-S. (2003). Oxidized glutathione stimulated the amyloid formation of α-synuclein. FEBS Lett. 537 (1–3), 63–67. doi:10.1016/S0014-5793(03)00081-4
Paleologou, K. E., Oueslati, A., Shakked, G., Rospigliosi, C. C., Kim, H.-Y., Lamberto, G. R., et al. (2010). Phosphorylation at S87 is enhanced in synucleinopathies, inhibits α-Synuclein oligomerization, and influences synuclein-membrane interactions. J. Neurosci. 30 (9), 3184–3198. doi:10.1523/JNEUROSCI.5922-09.2010
Pálmadóttir, T., Malmendal, A., Leiding, T., Lund, M., and Linse, S. (2021). Charge regulation during amyloid formation of α-Synuclein. J. Am. Chem. Soc. 143 (20), 7777–7791. doi:10.1021/jacs.1c01925
Pancoe, S. X., Wang, Y. J., Shimogawa, M., Perez, R. M., Giannakoulias, S., and Petersson, E. J. (2022). Effects of mutations and post-translational modifications on α-Synuclein in vitro aggregation: Mutation/PTM effects on α-Synuclein aggregation. J. Mol. Biol. 434 (23), 167859. doi:10.1016/j.jmb.2022.167859
Prasad, A., Bao, B., Beck, F., and Sarkar, F. (2010). Zinc-suppressed inflammatory cytokines by induction of A20-mediated inhibition of nuclear factor-κB. Nutr. (Burbank, Los Angel. Cty. Calif.) 27, 816–823. doi:10.1016/j.nut.2010.08.010
Pronin, A. N., Morris, A. J., Surguchov, A., and Benovic, J. L. (2000). Synucleins are a novel class of substrates for G protein-coupled receptor kinases. J. Biol. Chem. 275 (34), 26515–26522. doi:10.1074/jbc.M003542200
Prosad Banik, S. (2023). Glycation-induced amyloid formation in proteins: an emerging perspective to explore diabetes associated onset of neurodegenerative symptoms. Curr. Nutr. Food Sci. 20 (1), 2–7. doi:10.2174/1573401319666230224094812
Psol, M., Darvas, S. G., Leite, K., Mahajani, S. U., Bähr, M., and Kügler, S. (2021). Dementia with lewy bodies—associated ß-synuclein mutations V70M and P123H cause mutation-specific neuropathological lesions. Hum. Mol. Genet. 30 (3–4), 247–264. doi:10.1093/hmg/ddab036
Puspita, L., Chung, S. Y., and Shim, J. (2017). Oxidative stress and cellular pathologies in Parkinson’s disease. Mol. Brain 10 (1), 53. doi:10.1186/s13041-017-0340-9
Rabbani, N., Ashour, A., and Thornalley, P. J. (2016). Mass spectrometric determination of early and advanced glycation in biology. Glycoconj. J. 33 (4), 553–568. doi:10.1007/s10719-016-9709-8
Ramesh, S., and Arachchige, A. S. P. M. (2023). Depletion of dopamine in Parkinson’s disease and relevant therapeutic options: a review of the literature. AIMS Neurosci. 10 (3), 200–231. doi:10.3934/NEUROSCIENCE.2023017
Ray, S., Singh, N., Kumar, R., Patel, K., Pandey, S., Datta, D., et al. (2020). α-Synuclein aggregation nucleates through liquid–liquid phase separation. Nat. Chem. 12 (8), 705–716. doi:10.1038/s41557-020-0465-9
Reddy, K., and Dieriks, B. V. (2022). Multiple system atrophy: α-synuclein strains at the neuron-oligodendrocyte crossroad. Mol. Neurodegener. 17 (1), 77. doi:10.1186/s13024-022-00579-z
Rodríguez-Menéndez, S., García, M., Fernández, B., Álvarez, L., Fernández-Vega-Cueto, A., Coca-Prados, M., et al. (2018). The zinc-metallothionein redox system reduces oxidative stress in retinal pigment epithelial cells. Nutrients 10 (12), 1874. doi:10.3390/nu10121874
Roman, A.Yu., Devred, F., Byrne, D., La Rocca, R., Ninkina, N. N., Peyrot, V., et al. (2019). Zinc induces temperature-dependent reversible self-assembly of tau. J. Mol. Biol. 431 (4), 687–695. doi:10.1016/j.jmb.2018.12.008
Roohani, N., Hurrell, R., Kelishadi, R., and Schulin, R. (2013). Zinc and its importance for human health: an integrative review. J. Res. Med. Sci. 18 (144), 144–157.
Ruf, R. A. S., Lutz, E. A., Zigoneanu, I. G., and Pielak, G. J. (2008). Alpha-synuclein conformation affects its tyrosine-dependent oxidative aggregation. Biochemistry 47 (51), 13604–13609. doi:10.1021/bi801884z
Runfola, M., De Simone, A., Vendruscolo, M., Dobson, C. M., and Fusco, G. (2020). The N-terminal acetylation of α-Synuclein changes the affinity for lipid membranes but not the structural properties of the bound state. Sci. Rep. 10 (1), 204. doi:10.1038/s41598-019-57023-4
Rungratanawanich, W., Qu, Y., Wang, X., Essa, M. M., and Song, B.-J. (2021). Advanced glycation end products (AGEs) and other adducts in aging-related diseases and alcohol-mediated tissue injury. Exp. and Mol. Med. 53 (2), 168–188. doi:10.1038/s12276-021-00561-7
Sabouri, S., Rostamirad, M., and Dempski, R. E. (2024). Unlocking the brain’s zinc code: implications for cognitive function and disease. Front. Biophysics 2, 1406868. doi:10.3389/frbis.2024.1406868
Salaramoli, S., Joshaghani, H. R., Shoeibi, A., and Hashemy, S. I. (2024). Selenium and selenoproteins role in Parkinson’s disease: is there a link between selenoproteins and accumulated alpha-synuclein? J. Trace Elem. Med. Biol. 81, 127344. doi:10.1016/j.jtemb.2023.127344
Salgueiro, M. J., Zubillaga, M. B., Lysionek, A. E., Caro, R. A., Weill, R., and Boccio, J. R. (2002). The role of zinc in the growth and development of children. Nutrition 18 (6), 510–519. doi:10.1016/S0899-9007(01)00812-7
Sandstead, H. H., Frederickson, C. J., and Penland, J. G. (2000). History of zinc as related to brain function. J. Nutr. 130 (2), 496S–502S. doi:10.1093/jn/130.2.496S
Sano, K., Iwasaki, Y., Yamashita, Y., Irie, K., Hosokawa, M., Satoh, K., et al. (2021). Tyrosine 136 phosphorylation of α-synuclein aggregates in the lewy body dementia brain: involvement of serine 129 phosphorylation by casein kinase 2. Acta Neuropathol. Commun. 9 (1), 182. doi:10.1186/s40478-021-01281-9
Sawner, A. S., Ray, S., Yadav, P., Mukherjee, S., Panigrahi, R., Poudyal, M., et al. (2021). Modulating α-Synuclein liquid–liquid phase separation. Biochemistry 60 (48), 3676–3696. doi:10.1021/acs.biochem.1c00434
Schwartz, J. R., Marsh, R. G., and Draelos, Z. D. (2005). Zinc and skin health: overview of physiology and pharmacology. Dermatol. Surg. 31 (1), 837–847. doi:10.1111/j.1524-4725.2005.31729
Scudamore, O., and Ciossek, T. (2018). Increased oxidative stress exacerbates α-Synuclein aggregation in vivo. J. Neuropathology and Exp. Neurology 77 (6), 443–453. doi:10.1093/jnen/nly024
Sensi, S. L., Granzotto, A., Siotto, M., and Squitti, R. (2018). Copper and zinc dysregulation in Alzheimer’s disease. Trends Pharmacol. Sci. 39 (12), 1049–1063. doi:10.1016/j.tips.2018.10.001
Sensi, S. L., Paoletti, P., Koh, J.-Y., Aizenman, E., Bush, A. I., and Hershfinkel, M. (2011). The neurophysiology and pathology of brain zinc. J. Neurosci. 31 (45), 16076–16085. doi:10.1523/JNEUROSCI.3454-11.2011
Shtilerman, M. D., Ding, T. T., and Lansbury, P. T. (2002). Molecular crowding accelerates fibrillization of α-Synuclein: could an increase in the cytoplasmic protein concentration induce Parkinson’s disease? Biochemistry 41 (12), 3855–3860. doi:10.1021/bi0120906
Sikora, J., Kieffer, B. L., Paoletti, P., and Ouagazzal, A.-M. (2020). Synaptic zinc contributes to motor and cognitive deficits in 6-hydroxydopamine mouse models of Parkinson’s disease. Neurobiol. Dis. 134, 104681. doi:10.1016/j.nbd.2019.104681
Singh, V., Xu, L., Boyko, S., Surewicz, K., and Surewicz, W. K. (2020). Zinc promotes liquid–liquid phase separation of tau protein. J. Biol. Chem. 295 (18), 5850–5856. doi:10.1074/jbc.AC120.013166
Skrajnowska, D., and Bobrowska-Korczak, B. (2019). Role of zinc in immune system and anti-cancer defense mechanisms. Nutrients 11 (10), 2273. doi:10.3390/nu11102273
Skrovanek, S., DiGuilio, K., Bailey, R., Huntington, W., Urbas, R., Mayilvaganan, B., et al. (2014). Zinc and gastrointestinal disease. World J. Gastrointest. Pathophysiol. 5 (4), 496–513. doi:10.4291/wjgp.v5.i4.496
Slekiene, N., Snitka, V., Bruzaite, I., and Ramanavicius, A. (2022). Influence of TiO2 and ZnO nanoparticles on α-Synuclein and β-Amyloid aggregation and formation of protein fibrils. Materials 15 (21), 7664. doi:10.3390/ma15217664
Srinivasan, E., Chandrasekhar, G., Chandrasekar, P., Anbarasu, K., Vickram, A. S., Karunakaran, R., et al. (2021). Alpha-synuclein aggregation in Parkinson’s disease. Front. Med. 8, 736978. doi:10.3389/fmed.2021.736978
Stjernswärd, J., Colleau, S. M., and Ventafridda, V. (1996). The world health organization cancer pain and palliative care program past, present, and future. J. Pain Symptom Manag. 12 (2), 65–72. doi:10.1016/0885-3924(96)00109-1
Sulzer, D., and Edwards, R. H. (2019). The physiological role of α-synuclein and its relationship to Parkinson’s disease. J. Neurochem. 150 (5), 475–486. doi:10.1111/jnc.14810
Suzuki, A., Yabu, A., and Nakamura, H. (2022). Advanced glycation end products in musculoskeletal system and disorders. Methods 203, 179–186. doi:10.1016/j.ymeth.2020.09.012
Takekiyo, T., Yamada, N., Amo, T., Asano, A., and Yoshimura, Y. (2022). Triiodide ion-induced inhibition of amyloid aggregate formation: a case study of α-synuclein. J. Mol. Liq. 360, 119446. doi:10.1016/j.molliq.2022.119446
Tamano, H., Morioka, H., Nishio, R., Takeuchi, A., and Takeda, A. (2019). Blockade of rapid influx of extracellular Zn2+ into nigral dopaminergic neurons overcomes paraquat-induced Parkinson’s disease in rats. Mol. Neurobiol. 56 (6), 4539–4548. doi:10.1007/s12035-018-1398-9
Tan, S. M., Stefanovic, N., Tan, G., Wilkinson-Berka, J. L., and de Haan, J. B. (2013). Lack of the antioxidant glutathione Peroxidase-1 (GPx1) exacerbates retinopathy of prematurity in mice. Investigative Ophthalmol. and Vis. Sci. 54 (1), 555–562. doi:10.1167/iovs.12-10685
Tokuyama, A., Kanda, E., Itano, S., Kondo, M., Wada, Y., Kadoya, H., et al. (2021). Effect of zinc deficiency on chronic kidney disease progression and effect modification by hypoalbuminemia. PLoS One 16 (5), e0251554. doi:10.1371/journal.pone.0251554
Trexler, A. J., and Rhoades, E. (2010). Single molecule characterization of α-synuclein in aggregation-prone states. Biophysical J. 99 (9), 3048–3055. doi:10.1016/j.bpj.2010.08.056
Tuttle, M., Comellas, G., Nieuwkoop, A. J., Covell, D. J., Berthold, D. A., Kloepper, K. D., et al. (2016). Solid-state NMR structure of a pathogenic fibril of full-length human α-synuclein. Nat. Struct. Mol. Biol. 23, 409–415. doi:10.1038/nsmb.3194
Udayakumar, S., Metkar, S. K., Girigoswami, A., Deepika, B., Janani, G., Kanakaraj, L., et al. (2024). Exploring the amyloid degradation potential of nanoformulated carrageenan-bridging in vitro and in vivo perspectives. Int. J. Biol. Macromol. 279, 134814. doi:10.1016/j.ijbiomac.2024.134814
Ulmer, T. S., Bax, A., Cole, N. B., and Nussbaum, R. L. (2005). Structure and dynamics of micelle-bound human alpha-synuclein. J. Biol. Chem. 280 (10), 9595–9603. doi:10.1074/jbc.M411805200
Uversky, V. N., Li, J., and Fink, A. L. (2001). Metal-triggered structural transformations, aggregation, and fibrillation of human α-synuclein: a possible molecular link between parkinson’s disease and heavy metal exposure. J. Biol. Chem. 276 (47), 44284–44296. doi:10.1074/jbc.M105343200
Uversky, V. N., Li, J., Souillac, P., Millett, I. S., Doniach, S., Jakes, R., et al. (2002). Biophysical properties of the synucleins and their propensities to fibrillate: inhibition of α-synuclein assembly by β- and γ-synucleins. J. Biol. Chem. 277 (14), 11970–11978. doi:10.1074/jbc.M109541200
Vali, R., Shirvanian, K., Farkhondeh, T., Aschner, M., Samini, F., and Samarghandian, S. (2025). A review study on the effect of zinc on oxidative stress-related neurological disorders. J. Trace Elem. Med. Biol. 88, 127618. doi:10.1016/j.jtemb.2025.127618
Valiente-Gabioud, A. A., Torres-Monserrat, V., Molina-Rubino, L., Binolfi, A., Griesinger, C., and Fernández, C. O. (2012). Structural basis behind the interaction of Zn²⁺ with the protein α-synuclein and the Aβ peptide: a comparative analysis. J. Inorg. Biochem. 117, 334–341. doi:10.1016/j.jinorgbio.2012.06.011
Van Laar, V. S., Chen, J., Zharikov, A. D., Bai, Q., Di Maio, R., Dukes, A. A., et al. (2020). α-Synuclein amplifies cytoplasmic peroxide flux and oxidative stress provoked by mitochondrial inhibitors in CNS dopaminergic neurons in vivo. Redox Biol. 37, 101695. doi:10.1016/j.redox.2020.101695
van Veen, S., Sørensen, D. M., Holemans, T., Holen, H. W., Palmgren, M. G., and Vangheluwe, P. (2014). Cellular function and pathological role of ATP13A2 and related P-type transport ATPases in Parkinson’s disease and other neurological disorders. Front. Mol. Neurosci. 7, 48. doi:10.3389/fnmol.2014.00048
Vergnano, A. M., Rebola, N., Savtchenko, L. P., Pinheiro, P. S., Casado, M., Kieffer, B. L., et al. (2014). Zinc dynamics and action at excitatory synapses. Neuron 82 (5), 1101–1114. doi:10.1016/j.neuron.2014.04.034
Vicente Miranda, H., Szegő, É. M., Oliveira, L. M. A., Breda, C., Darendelioglu, E., de Oliveira, R. M., et al. (2017). Glycation potentiates α-synuclein-associated neurodegeneration in synucleinopathies. Brain 140 (5), 1399–1419. doi:10.1093/brain/awx056
Vickram, S., Rohini, K., Srinivasan, S., Veenakumari, D. N., Archana, K., Anbarasu, K., et al. (2021). Role of zinc (Zn) in human reproduction: a journey from initial spermatogenesis to childbirth. Int. J. Mol. Sci. 22 (4), 2188–16. doi:10.3390/ijms22042188
Waxman, E. A., and Giasson, B. I. (2009). Molecular mechanisms of α-synuclein neurodegeneration. Biochimica Biophysica Acta - Mol. Basis Dis. 1792 (7), 616–624. doi:10.1016/j.bbadis.2008.09.013
Weglicki, W. B., Chmielinska, J. J., Kramer, J. H., and Mak, I. T. (2011). Cardiovascular and intestinal responses to oxidative and nitrosative stress during prolonged magnesium deficiency. Am. J. Med. Sci. 342 (2), 125–128. doi:10.1097/MAJ.0b013e318222e88c
Wells, C., Brennan, S., Keon, M., and Ooi, L. (2021). The role of amyloid oligomers in neurodegenerative pathologies. Int. J. Biol. Macromol. 181, 582–604. doi:10.1016/j.ijbiomac.2021.03.113
Wessels, I., Maywald, M., and Rink, L. (2017). Zinc as a gatekeeper of immune function. Nutrients 9 (12), 1286. doi:10.3390/nu9121286
Willekens, J., and Runnels, L. W. (2022). Impact of zinc transport mechanisms on embryonic and brain development. Nutrients 14 (12), 2526. doi:10.3390/nu14122526
Winner, B., Jappelli, R., Maji, S. K., Desplats, P. A., Boyer, L., Aigner, S., et al. (2011). In vivo demonstration that α-synuclein oligomers are toxic. Proc. Natl. Acad. Sci. U. S. A. 108 (10), 4194–4199. doi:10.1073/pnas.1100976108
Wolfe, K. J., and Cyr, D. M. (2011). Amyloid in neurodegenerative diseases: friend or foe? Seminars Cell Dev. Biol. 22 (5), 476–481. doi:10.1016/j.semcdb.2011.03.011
Xie, Z., Wu, H., and Zhao, J. (2020). Multifunctional roles of zinc in Alzheimer’s disease. NeuroToxicology 80, 112–123. doi:10.1016/j.neuro.2020.07.003
Xu, Y., Xiao, G., Liu, L., and Lang, M. (2019). Zinc transporters in Alzheimer’s disease. Mol. Brain 12 (1), 106. doi:10.1186/s13041-019-0528-2
Yatoui, D., Tsvetkov, P. O., La Rocca, R., Baksheeva, V. E., Allegro, D., Breuzard, G., et al. (2022). Binding of two zinc ions promotes liquid-liquid phase separation of Tau. Int. J. Biol. Macromol. 223, 1223–1229. doi:10.1016/j.ijbiomac.2022.11.060
Ye, R., Touroutoglou, A., Brickhouse, M., Katz, S., Growdon, J. H., Johnson, K. A., et al. (2020). Topography of cortical thinning in the lewy body diseases. NeuroImage Clin. 26, 102196. doi:10.1016/j.nicl.2020.102196
Zecca, L., Zucca, F. A., Wilms, H., and Sulzer, D. (2003). Neuromelanin of the substantia nigra: a neuronal black hole with protective and toxic characteristics. Trends Neurosci. 26 (11), 578–580. doi:10.1016/j.tins.2003.08.009
Zhang, H., Griggs, A., Rochet, J. C., and Stanciu, L. A. (2013). In vitro study of α-synuclein protofibrils by Cryo-EM suggests a Cu2+-dependent aggregation pathway. Biophysical J. 104 (12), 2706–2713. doi:10.1016/j.bpj.2013.04.050
Zhang, M., Shi, H., Zhang, X., Zhang, X., and Huang, Y. (2020). Cryo-EM structure of the nonameric CsgG-CsgF complex and its implications for controlling curli biogenesis in Enterobacteriaceae. PLoS Biol. 18 (6), e3000748. doi:10.1371/journal.pbio.3000748
Zhang, P., Park, H.-J., Zhang, J., Junn, E., Andrews, R. J., Velagapudi, S. P., et al. (2020). Translation of the intrinsically disordered protein α-synuclein is inhibited by a small molecule targeting its structured mRNA. Proc. Natl. Acad. Sci. 117 (3), 1457–1467. doi:10.1073/pnas.1905057117
Zhao, K., Li, Y., Liu, Z., Long, H., Zhao, C., Luo, F., et al. (2020). Parkinson’s disease associated mutation E46K of α-synuclein triggers the formation of a distinct fibril structure. Nat. Commun. 11 (1), 2643. doi:10.1038/s41467-020-16386-3
Zhao, Q., Tao, Y., Zhao, K., Ma, Y., Xu, Q., Liu, C., et al. (2023). Structural insights of Fe3+ induced α-synuclein fibrillation in Parkinson’s disease. J. Mol. Biol. 435 (1), 167680. doi:10.1016/j.jmb.2022.167680
Keywords: neurodegenerative disorders, α-synuclein, synucleinopathies, Parkinson’s disease, liquid-liquid phase separation, zinc as micronutrient, ZnO nanoparticle
Citation: Banik SP, Bagchi D, Banerjee P, Chakraborty S, Bagchi M, Bose C, De D, Saha S and Chakraborty S (2025) Subtle concentration changes in zinc hold the key to fibrillation of α-synuclein: an updated insight on the micronutrient’s role in prevention of neurodegenerative disorders. Front. Mol. Biosci. 12:1603364. doi: 10.3389/fmolb.2025.1603364
Received: 31 March 2025; Accepted: 16 June 2025;
Published: 10 July 2025.
Edited by:
Yuan-Xiang Pan, University of Illinois at Urbana-Champaign, United StatesReviewed by:
Joshua Emmerson, Washington University in St. Louis, United StatesAgnishwar Girigoswami, Chettinad Academy of Research and Education (CARE), India
Copyright © 2025 Banik, Bagchi, Banerjee, Chakraborty, Bagchi, Bose, De, Saha and Chakraborty. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Samudra Prosad Banik, c2FtdWRyYXBiQGdtYWlsLmNvbQ==
†ORCID: Samudra Prosad Banik, orcid.org/0000-0003-0075-7508. Debasis Bagchi, orcid.org/0000-0001-9478-6427. Pradipta Banerjee, orcid.org/0000-0002-2609-8849. Sanjoy Chakrobarty, orcid.org/0000-0002-6213-5483. Manashi Bagchi, orcid.org/0000-0002-1527-6419. Chaitali Bose, orcid.org/0000-0003-1981-631X. Debasmita De, orcid.org/0009-0007-4811-5113. Sreemoyee Saha, orcid.org/0009-0006-0348-7274. Sudipta Chakrobarty, orcid.org/0009-0008-0167-0678