 Alejandro Soto
Alejandro Soto Francesco Mambretti
Francesco Mambretti Emanuele Locatelli
Emanuele Locatelli Iliya D. Stoev
Iliya D. Stoev- 1Institute of Biological and Chemical Systems - Biological Information Processing, Karlsruhe Institute of Technology, Karlsruhe, Germany
- 2Istituto Italiano di Tecnologia, Atomistic Simulations Group, Genoa, Italy
- 3Department of Physics and Astronomy, University of Padua, Padua, Italy
- 4Istituto Nazionale di Fisica Nucleare (INFN), Sezione di Padova, Padua, Italy
In this work, we investigate the dependence of the melting temperature of low-valency DNA constructs on the length of non-inert dangling ends, controlling their sequence composition. We compare two computational models to evaluate their effectiveness and limitations in predicting the melting behavior of DNA oligomers (bivalent linkers) and more complex structures (trivalent nanostars), benchmarking the results against experimental spectroscopic data. Our results suggest that the length of non-inert dangling ends has minimal impact on the melting point of the DNA duplex for the duplexes we studied, informing the future design of DNA supramolecular constructs.
1 Introduction
Beyond its primary use as genetic material, DNA has been exploited as a building block for nanoscale technological applications for almost 40 years (Seeman and Sleiman, 2017; Jonoska and Winfree, 2023). DNA nanotechnology employs the same physics and chemistry as DNA’s biological function, using Watson-Crick base-pairing rules of complementarity to achieve high-yield self-assembly of complex structures (Rothemund, 2006; Dey et al., 2021; Liu et al., 2024). As practical applications for DNA-based nanosystems are explored, precise thermodynamic control over the hierarchical self-assembly of complex higher-order structures is paramount (Biffi et al., 2013; Conrad et al., 2019; Dong et al., 2020; Conrad et al., 2022; Stoev et al., 2018; 2020). A deep understanding of structure-property relationships is critical for the advancement of DNA-based nanotechnology, enabling innovative applications, such as environmental monitoring, biomolecular interactions detection and identification of pathogenic biomarkers (Shen et al., 2021; Huang et al., 2021; Can et al., 2025).
The supramolecular assembly of DNA hydrogels and other high-order DNA materials follows a hierarchical, step-by-step strategy, based on complex DNA “building blocks”, constructs that are rather stable at intermediate temperatures and that bind together through short complementary single-stranded sequences called “sticky ends”. This strategy exploits the dependence of the melting temperature on the duplex length: the design allows the building blocks to remain stable in a target thermodynamic region where dynamic formation and breaking of bonds is taken into account, shaping the overall phase diagram (Biffi et al., 2013; Rovigatti et al., 2014; Locatelli et al., 2017; Chen et al., 2020; Abraham et al., 2024).
However, the presence of unpaired bases also influences the stability of the construct. As previously reported (Di Michele et al., 2014), a moderate increase in melting temperature
Here we obtain the melting temperatures as a function of
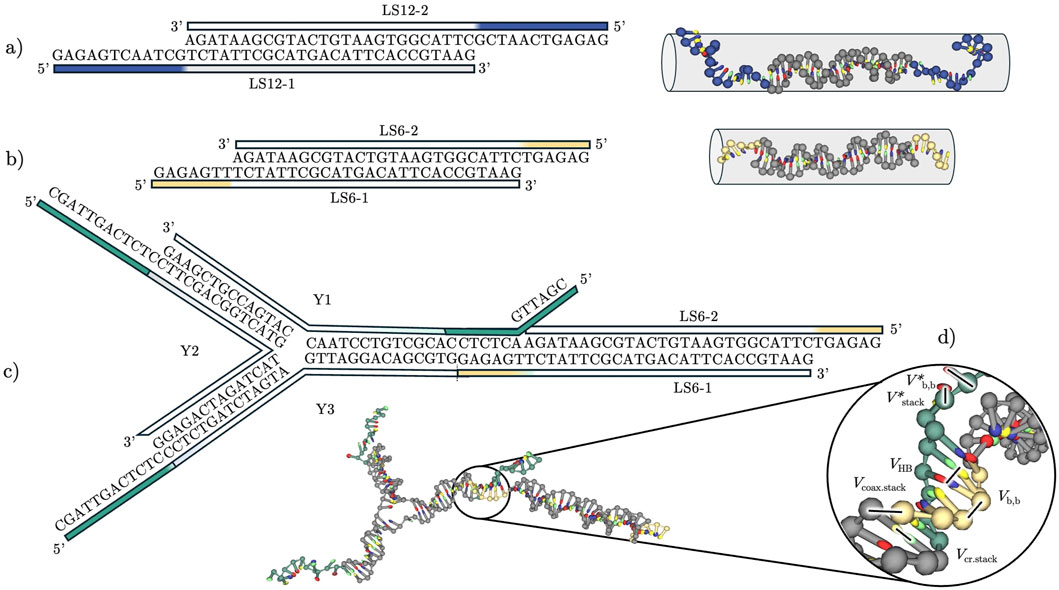
Figure 1. Schematic of the building blocks employed in this study: (a) Linker “LS12” with a 12-nucleotide-long sticky end. Linear sequence representation and three-dimensional representation showing the approximate volume the construct occupies. (b) Linker “LS6” with a 6-nucleotide-long sticky end. As in (a), we show a linear sequence and three-dimensional representations giving the approximate volume the construct occupies. (c) A trivalent nanostar (Y-shape) joined with a double-stranded linker LS6, justifying the rational design of our non-inert sequences, although here we performed simulations and experiments only on the individual building blocks. (d) Potential energy contributions included in the oxDNA model, used to calculate intra- and inter-strand interactions.
In the following text, we give information on the short DNA constructs employed in this study, specifically bivalent “linkers” (L, Figures 1a,b) and trivalent “Y-shapes” (Y, Figure 1c). We start by briefly presenting experimental methods and theoretical models, NUPACK and oxDNA. We then discuss the experimental ground truth and expose a limitation of NUPACK that, upon inclusion of sticky ends, shows a qualitative discrepancy with experiments, which however can be corrected by using melting curve normalization or a method based on first derivatives to extract melting temperature values (cf. Supplementary Figure S4). Instead, results of direct sampling from Molecular Dynamics simulations with the oxDNA model qualitatively reproduce both the literature results (Di Michele et al., 2014) and the experimental data (Can et al., 2025). We conclude with a discussion of our results from a thermodynamic standpoint, critically reviewing and pointing to specific strengths and limitations of each approach.
2 Methods
2.1 System description
Linkers are bivalent constructs consisting of a 26-base-long double-stranded DNA (dsDNA) core with single-stranded DNA (ssDNA) ends (dangling ends or tails) on both sides. The length of the tails
Finally, to facilitate comparison with thermodynamic information on shorter oligos, we included in our analysis dimers used in a preceding work (Di Michele et al., 2014) (see SI).
2.2 Experimental methods
We ordered the custom-made DNA oligonucleotides in Table 1 from biomers.net GmbH. The oligonucleotides were HPLC-purified, freeze-dried and subsequently dissolved in 10 mM phosphate buffer saline (PBS) supplemented with 100 mM NaCl (pH 7.6), following an established protocol (Stoev et al., 2018; 2020). All oligo concentrations were then measured using NanoDrop 1000 and the hybridisation behaviour verified using polyacrylamide gel electrophoresis and UV-visible spectroscopy. Y-shaped (trivalent) nanostars are formed by the designed single strands Y1, Y2 and Y3, while bivalent DNA linkers are formed by linker strands LS-1 and LS-2. The linkers LS

Table 1. DNA strands comprising Y-shapes and linkers, with labelled sticky ends (black-green) and hybridising segments between Y1 and Y2, Y1 and Y3, Y2 and Y3 (shades of grey), LSX-1 and LSX-2 (matching colour). All synthesised DNA strands were purified via HPLC before use.
UV-visible absorption spectra were then recorded at a wavelength of 260 nm by Cary 3,500 Multicell Peltier UV-visible spectrophotometer (Agilent Technologies). At this wavelength, we used the differential absorbance of ssDNA and dsDNA to confirm the formation of the Y-shapes and linkers, alongside obtaining information about their melting temperatures. All temperature ramps (heating and cooling) were performed between
2.3 Statistical mechanics approach: NUPACK
The NUPACK tool (Zadeh et al., 2011) was designed to construct and analyse nucleic acid systems, predicting DNA secondary structure from the sequences, i.e., the base-pairs of a set of DNA strands. For each candidate secondary structure, NUPACK algorithms add an empirical estimate of the free energy parameters (Ke et al., 2024), with a strand association penalty energy and stacking contributions (Fornace et al., 2023). The tool allows identification of the Minimum Free Energy (MFE) structure under given conditions, viz., temperature, salt concentration and other thermodynamic parameters. NUPACK advantages include open accessibility, ease of use, and the ability to provide estimates of equilibrium and thermodynamic properties, such as melting profiles, even for systems involving multiple interacting strands (Zadeh et al., 2011). On the other hand, the tool is limited in the properties it can predict for small constructs and may propose highly improbable configurations in the presence of dangling ends (Buterez, 2021). Another issue stems from NUPACK’s inability to distinguish between broken base pairs and single bases belonging to tails, which leads to severe underestimation of bonded fractions at low temperatures for short oligomers. In this analysis, we used the energy parameter set “dna04”, with 100 mM
2.4 Computational approach with oxDNA
OxDNA (Ouldridge et al., 2010a; Šulc et al., 2012; Doye et al., 2013) is a widely used physical model that enables in-silico studies of DNA origami and other types of all-DNA fluids, including their self-assembly kinetics. In addition to its effectiveness in reproducing the mechanics and thermodynamics of DNA constructs (Ouldridge et al., 2011; Srinivas et al., 2013; Skoruppa et al., 2018, oxDNA is arguably the most accessible simulation code for DNA, providing introductory material (Sengar et al., 2021; Poppleton et al., 2023), supporting a number of web tools (Poppleton et al., 2021; Bohlin et al., 2022; Poppleton et al., 2020; Suma et al., 2019) and allowing extensions (Kaufhold et al., 2022).
In a nutshell, oxDNA is a top-down, nucleotide-level coarse-grained model that accounts for different potential energy contributions between base pairs (Figure 1d) (Sengar et al., 2021). The oxDNA code supports both Monte Carlo (MC), with native support for Umbrella Sampling (US), and Molecular Dynamics (MD) simulations. Notable for our work is the GPU acceleration of oxDNA, which enables scaling up in simulations of larger systems (Rovigatti et al., 2015).
We performed standard MD simulations, also termed here “unbiased sampling”, and MC simulations with Umbrella Sampling (cf. SI). We employed the oxDNA2 model (Snodin et al., 2015), which explicitly includes the effect of screened electrostatic interactions, and set the Na+ concentration to
3 Results and discussion
3.1 Validation on reference system
First, we briefly validated our chosen numerical methods on a reference system (Di Michele et al., 2014) (details in the SI). We observed that both unbiased MD and US with oxDNA qualitatively retrieve the reported results. Notably, while we reliably captured a difference in melting temperature
3.2 Experimental results
Next, we investigated experimentally through UV-visible spectroscopy a set of duplexes with non-inert tails (Section 2.1), thereby providing ground truth for comparison with the two in-silico approaches we selected. We show normalised transmittance at 260 nm in Figure 2a. At this wavelength, single-stranded DNA absorbs more strongly than double-stranded DNA, which yields a sigmoidal curve. The mid-point of the sigmoidal curve–equivalent to the temperature at which the fraction of bonded bases equals 0.5 (a value that closely matches the temperature corresponding to the maximum of the first derivative, as shown in Supplementary Figure S3)–represents a transient dynamic state between a fully assembled and fully dissociated duplex, where on average half of all hydrogen bonds are formed (during cooling) or broken (during heating). Notably, the melting temperature displays little to no variation as we deleted bases from the sticky ends of the double-stranded linkers. However, the Y-shapes display a notably different melting temperature from the linkers, with
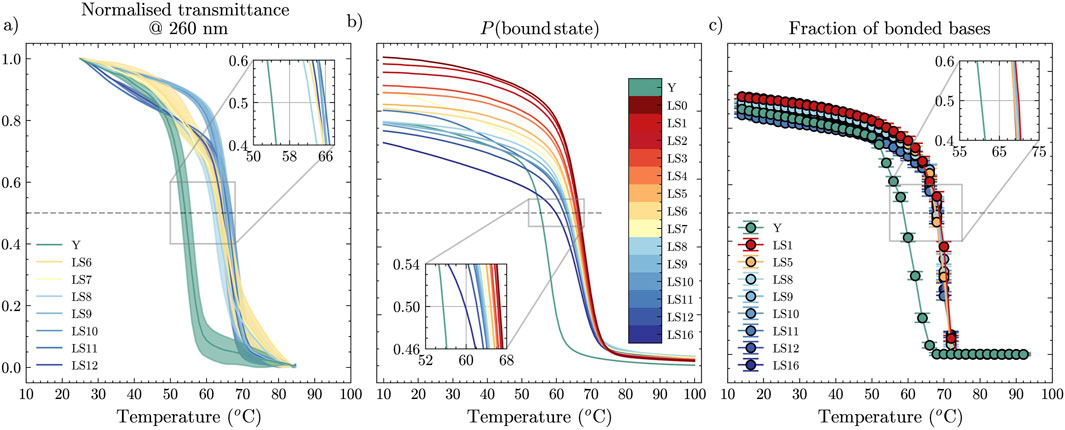
Figure 2. Melting curves as a function of temperature, obtained from (a) UV-visible absorbance experiments, (b) NUPACK statistical mechanics treatment, and (c) oxDNA MD unbiased simulations for dimers with dangling ends of different length and for a Y-shape. Error bars in (a) are obtained from the values measured in heating and cooling ramps.
In Figure 3, we included a summary of these experimental melting-temperature results, shown as square symbols, where we indeed observed that
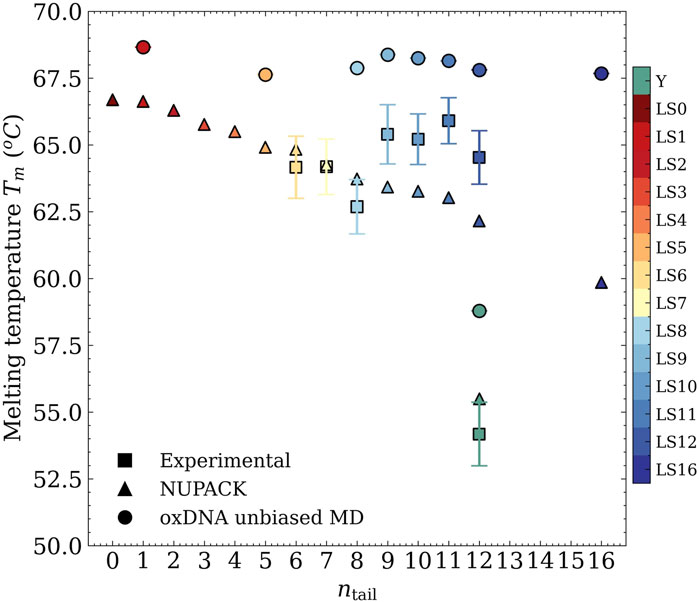
Figure 3. Melting temperature as a function of the length of the sticky ends from UV-visible absorbance experiments (squares), NUPACK statistical treatment (triangles), and oxDNA MD unbiased simulations (circles).
3.3 NUPACK predictions
For the same oligomers, we computed a prediction of the melting temperature using NUPACK. Figure 2b shows melting curves for different lengths of the dangling ends,
3.4 oxDNA results
Approaching the melting-point determination task using unbiased MD with the oxDNA model leads to the results reported in Figure 2c, where we again provide the curves for different values of
4 Conclusion
We investigated the effect of adding non-inert nucleotides to the sticky end attached to a duplex core in DNA constructs. We compared NUPACK and oxDNA models with an experimental reference; oxDNA was used in the context of Molecular Dynamics simulations, both unbiased and with Umbrella Sampling. In the SI, we summarise additional attempts at using advanced sampling techniques, discussing the challenges and potential pitfalls one encounters along the way. US promotes quicker convergence, but becomes impractical for strands containing even a few tens of bases since the required simulation time increases significantly and it becomes non-trivial to estimate the bias weights. Unbiased MD is cheaper to run and capable of handling larger systems, even with tens of molecules owing to GPU-acceleration; on the other hand, its uniform exploration of states in the melting region implies longer simulation times, since fluctuations become large and the system requires more time to reach steady state. One significant disadvantage of explicit counting of hydrogen bonds in unbiased MD is that it requires careful handling of statistical results by running multiple independent simulations for extended periods. For the long duplexes, the equilibration time was estimated to be approximately
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author contributions
AS: Writing – review and editing, Methodology, Formal Analysis, Software, Writing – original draft, Investigation, Visualization. FM: Writing – original draft, Formal Analysis, Software, Visualization, Resources, Methodology, Investigation, Writing – review and editing, Validation. EL: Writing – original draft, Writing – review and editing, Software, Formal Analysis, Investigation, Validation, Methodology. IS: Writing – review and editing, Validation, Funding acquisition, Project administration, Supervision, Data curation, Writing – original draft, Resources, Conceptualization, Investigation.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. AS and IDS kindly acknowledge financial help from the Karlsruhe Institute of Technology Excellence Strategy via the Young Investigator Group Preparation Program. We acknowledge funding by the Carl-Zeiss-Foundation through the Seed Funding Program of the Center for Synthetic Genomics.
Acknowledgments
FM acknowledges CINECA through ISCRA initiative, in detail IsCc4 project (ID: HP10CSR1I8). FM also gratefully acknowledges the Data Science and Computation Facility and its Support Team for their support and assistance on the IIT High Performance Computing Infrastructure.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2025.1646428/full#supplementary-material
References
Abraham, G. R., Chaderjian, A. S., Nguyen, A. B., Wilken, S., and Saleh, O. A. (2024). Nucleic acid liquids. Rep. Prog. Phys. 87, 066601. doi:10.1088/1361-6633/ad4662
Biffi, S., Cerbino, R., Bomboi, F., Paraboschi, E. M., Asselta, R., Sciortino, F., et al. (2013). Phase behavior and critical activated dynamics of limited-valence dna nanostars. Proc. Natl. Acad. Sci. 110, 15633–15637. doi:10.1073/pnas.1304632110
Bohlin, J., Matthies, M., Poppleton, E., Procyk, J., Mallya, A., Yan, H., et al. (2022). Design and simulation of dna, rna and hybrid protein–nucleic acid nanostructures with oxview. Nat. Protoc. 17, 1762–1788. doi:10.1038/s41596-022-00688-5
Buterez, D. (2021). Scaling up dna digital data storage by efficiently predicting dna hybridisation using deep learning. Sci. Rep. 11, 20517. doi:10.1038/s41598-021-97238-y
Can, A. E., Ali, A. W. U., Oelschlaeger, C., Willenbacher, N., and Stoev, I. D. (2025). Mechanically tunable dna hydrogels as prospective biosensing modules. Macromol. Rapid Commun. n/a, 2500149. doi:10.1002/marc.202500149
Chen, J., Zhu, Y., Liu, H., and Wang, L. (2020). Tailoring dna self-assembly to build hydrogels. Top. Curr. Chem. 378, 32. doi:10.1007/s41061-020-0295-7
Conrad, N., Kennedy, T., Fygenson, D. K., and Saleh, O. A. (2019). Increasing valence pushes dna nanostar networks to the isostatic point. Proc. Natl. Acad. Sci. 116, 7238–7243. doi:10.1073/pnas.1819683116
Conrad, N., Chang, G., Fygenson, D. K., and Saleh, O. A. (2022). Emulsion imaging of a dna nanostar condensate phase diagram reveals valence and electrostatic effects. J. Chem. Phys. 157, 234203. doi:10.1063/5.0130808
Dey, S., Fan, C., Gothelf, K. V., Li, J., Lin, C., Liu, L., et al. (2021). Dna origami. Nat. Rev. Methods Prim. 1, 13. doi:10.1038/s43586-020-00009-8
Di Michele, L., Mognetti, B. M., Yanagishima, T., Varilly, P., Ruff, Z., Frenkel, D., et al. (2014). Effect of inert tails on the thermodynamics of dna hybridization. J. Am. Chem. Soc. 136, 6538–6541. doi:10.1021/ja500027v
Dong, Y., Yao, C., Zhu, Y., Yang, L., Luo, D., and Yang, D. (2020). Dna functional materials assembled from branched dna: design, synthesis, and applications. Chem. Rev. 120, 9420–9481. doi:10.1021/acs.chemrev.0c00294
Doye, J. P., Ouldridge, T. E., Louis, A. A., Romano, F., Šulc, P., Matek, C., et al. (2013). Coarse-graining dna for simulations of dna nanotechnology. Phys. Chem. Chem. Phys. 15, 20395–20414. doi:10.1039/c3cp53545b
Fornace, M. E., Huang, J., Newman, C. T., Porubsky, N. J., Pierce, M. B., and Pierce, N. A. (2023). NUPACK Analysis Des. Nucleic Acid Struct. Devices, Syst doi:10.26434/chemrxiv-2022-xv98l
Huang, Z., Qiu, L., Zhang, T., and Tan, W. (2021). Integrating dna nanotechnology with aptamers for biological and biomedical applications. Matter 4, 461–489. doi:10.1016/j.matt.2020.11.002
Jonoska, N., and Winfree, E. (2023). Visions of DNA nanotechnology at 40 for the next 40: a tribute to nadrian C. Seeman. Springer Nature.
Kaufhold, W. T., Pfeifer, W., Castro, C. E., and Di Michele, L. (2022). Probing the mechanical properties of dna nanostructures with metadynamics. ACS nano 16, 8784–8797. doi:10.1021/acsnano.1c08999
Ke, Y., Sharma, E., Wayment-Steele, H. K., Becker, W. R., Ho, A., Marklund, E., et al. (2024). High-throughput dna melt measurements enable improved models of dna folding thermodynamics. BioRxiv, 2024–01. doi:10.1038/s41467-025-60455-4
Liu, H., Matthies, M., Russo, J., Rovigatti, L., Narayanan, R. P., Diep, T., et al. (2024). Inverse design of a pyrochlore lattice of dna origami through model-driven experiments. Science 384, 776–781. doi:10.1126/science.adl5549
Locatelli, E., Handle, P. H., Likos, C. N., Sciortino, F., and Rovigatti, L. (2017). Condensation and demixing in solutions of dna nanostars and their mixtures. ACS nano 11, 2094–2102. doi:10.1021/acsnano.6b08287
Ouldridge, T. E., Louis, A. A., and Doye, J. P. (2010a). Dna nanotweezers studied with a coarse-grained model of dna. Phys. Rev. Lett. 104, 178101. doi:10.1103/PhysRevLett.104.178101
Ouldridge, T. E., Louis, A. A., and Doye, J. P. (2010b). Extracting bulk properties of self-assembling systems from small simulations. J. Phys. Condens. Matter 22, 104102. doi:10.1088/0953-8984/22/10/104102
Ouldridge, T. E., Louis, A. A., and Doye, J. P. (2011). Structural, mechanical, and thermodynamic properties of a coarse-grained dna model. J. Chem. Phys. 134, 085101. doi:10.1063/1.3552946
Poppleton, E., Bohlin, J., Matthies, M., Sharma, S., Zhang, F., and Šulc, P. (2020). Design, optimization and analysis of large dna and rna nanostructures through interactive visualization, editing and molecular simulation. Nucleic acids Res. 48, e72. doi:10.1093/nar/gkaa417
Poppleton, E., Romero, R., Mallya, A., Rovigatti, L., and Šulc, P. (2021). Oxdna. Org: a public webserver for coarse-grained simulations of dna and rna nanostructures. Nucleic acids Res. 49, W491–W498. doi:10.1093/nar/gkab324
Poppleton, E., Matthies, M., Mandal, D., Romano, F., Šulc, P., and Rovigatti, L. (2023). Oxdna: coarse-grained simulations of nucleic acids made simple. J. Open Source Softw. 8, 4693. doi:10.21105/joss.04693
Rothemund, P. W. (2006). Folding dna to create nanoscale shapes and patterns. Nature 440, 297–302. doi:10.1038/nature04586
Rovigatti, L., Bomboi, F., and Sciortino, F. (2014). Accurate phase diagram of tetravalent dna nanostars. J. Chem. Phys. 140. doi:10.1063/1.4870467
Rovigatti, L., Šulc, P., Reguly, I. Z., and Romano, F. (2015). A comparison between parallelization approaches in molecular dynamics simulations on gpus. J. Comput. Chem. 36, 1–8. doi:10.1002/jcc.23763
Santa Lucia, H., and Hicks, D. (2004). Thermodynamics and kinetics of dna self-assembly. Annu. Rev. Biophysics 33, 415–440. doi:10.1146/annurev.biophys.32.110601.141800
Seeman, N. C., and Sleiman, H. F. (2017). Dna nanotechnology. Nat. Rev. Mater. 3, 17068–23. doi:10.1038/natrevmats.2017.68
Sengar, A., Ouldridge, T. E., Henrich, O., Rovigatti, L., and Šulc, P. (2021). A primer on the oxdna model of dna: when to use it, how to simulate it and how to interpret the results. Front. Mol. Biosci. 8, 693710. doi:10.3389/fmolb.2021.693710
Shen, L., Wang, P., and Ke, Y. (2021). Dna nanotechnology-based biosensors and therapeutics. Adv. Healthc. Mater. 10, 2002205. doi:10.1002/adhm.202002205
Skoruppa, E., Nomidis, S. K., Marko, J. F., and Carlon, E. (2018). Bend-induced twist waves and the structure of nucleosomal dna. Phys. Rev. Lett. 121, 088101. doi:10.1103/PhysRevLett.121.088101
Snodin, B. E., Randisi, F., Mosayebi, M., Šulc, P., Schreck, J. S., Romano, F., et al. (2015). Introducing improved structural properties and salt dependence into a coarse-grained model of dna. J. Chem. Phys. 142, 234901. doi:10.1063/1.4921957
Srinivas, N., Ouldridge, T. E., Šulc, P., Schaeffer, J. M., Yurke, B., Louis, A. A., et al. (2013). On the biophysics and kinetics of toehold-mediated dna strand displacement. Nucleic acids Res. 41, 10641–10658. doi:10.1093/nar/gkt801
Stoev, I. D., Caciagli, A., Xing, Z., and Eiser, E. (2018). Using single-beam optical tweezers for the passive microrheology of complex fluids. Opt. Trapp. Opt. Micromanipulation XV (SPIE) 10723, 251–262. doi:10.1117/12.2318019
Stoev, I. D., Cao, T., Caciagli, A., Yu, J., Ness, C., Liu, R., et al. (2020). On the role of flexibility in linker-mediated dna hydrogels. Soft Matter 16, 990–1001. doi:10.1039/c9sm01398a
Šulc, P., Romano, F., Ouldridge, T. E., Rovigatti, L., Doye, J. P., and Louis, A. A. (2012). Sequence-dependent thermodynamics of a coarse-grained dna model. J. Chem. Phys. 137, 135101. doi:10.1063/1.4754132
Suma, A., Poppleton, E., Matthies, M., Šulc, P., Romano, F., Louis, A. A., et al. (2019). Tacoxdna: a user-friendly web server for simulations of complex dna structures, from single strands to origami. J. Comput. Chem. 40, 2586–2595. doi:10.1002/jcc.26029
Keywords: DNA nanotechnology, melting curves, oxDNA, NUPACK, molecular dynamics, umbrella sampling
Citation: Soto A, Mambretti F, Locatelli E and Stoev ID (2025) Modelling the melting of DNA oligomers with non-inert dangling ends. Front. Mol. Biosci. 12:1646428. doi: 10.3389/fmolb.2025.1646428
Received: 13 June 2025; Accepted: 28 July 2025;
Published: 13 August 2025.
Edited by:
Li-Tang Yan, Tsinghua University, ChinaCopyright © 2025 Soto, Mambretti, Locatelli and Stoev. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Iliya D. Stoev, aWxpeWEuc3RvZXZAa2l0LmVkdQ==