 Gamze Kocak1
Gamze Kocak1 Mylla M. Dimas2
Mylla M. Dimas2 Luiz F. S. E. Silva1,3,4
Luiz F. S. E. Silva1,3,4 Danyelle Silva-Amaral5
Danyelle Silva-Amaral5 Congxin Sun1
Congxin Sun1 Sophie Cruddas1
Sophie Cruddas1 Timothy Barrett1,6
Timothy Barrett1,6 Daniel Martins-de-Souza5,7,8,9
Daniel Martins-de-Souza5,7,8,9 Edecio Cunha-Neto3,4
Edecio Cunha-Neto3,4 Patricia S. Brocardo2
Patricia S. Brocardo2 Tetsushi Kataura10
Tetsushi Kataura10 Viktor I. Korolchuk11
Viktor I. Korolchuk11 Sovan Sarkar1*
Sovan Sarkar1*- 1Department of Cancer and Genomic Sciences, School of Medical Sciences, College of Medicine and Health, University of Birmingham, Birmingham, United Kingdom
- 2Department of Morphological Sciences, Centre of Biological Sciences, Federal University of Santa Catarina, Florianopolis, Santa Catarina, Brazil
- 3Laboratory of Immunology, Heart Institute (InCor), and Division of Clinical Immunology and Allergy, Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo, São Paulo, Brazil
- 4Institute for Investigation in Immunology (III), Institutos Nacionais de Ciências e Tecnologias (INCT), São Paulo, Brazil
- 5Laboratory of Neuroproteomics, Institute of Biology, University of Campinas, Campinas, Brazil
- 6Department of Endocrinology, Birmingham Women’s and Children’s Hospital, Birmingham, United Kingdom
- 7Experimental Medicine Research Cluster (EMRC), University of Campinas, Campinas, Brazil
- 8D’Or Institute for Research and Education (IDOR), São Paulo, Brazil
- 9INCT in Modelling Human Complex Diseases With 3D Platforms (Model3D), Conselho Nacional de Desenvolvimento Científico e Tecnológico, São Paulo, Brazil
- 10Department of Neurology, Institute of Medicine, University of Tsukuba, Tsukuba, Japan
- 11Biosciences Institute, Faculty of Medical Sciences, Newcastle University, Newcastle upon Tyne, United Kingdom
Autophagy is an evolutionarily conserved catabolic process that plays a central role in maintaining cellular homeostasis by degrading and recycling damaged or surplus proteins, organelles, and other cellular macromolecules and components. A growing body of evidence highlights a bidirectional relationship between autophagy and nicotinamide adenine dinucleotide (NAD+), a vital metabolic cofactor involved in numerous cellular processes, including energy metabolism, genomic maintenance, stress resistance, and cell survival. Autophagy supports NAD+ homeostasis by recycling metabolic precursors, while NAD+-dependent enzymes such as sirtuins and PARPs regulate autophagy initiation and lysosomal function. Disruption of this autophagy–NAD+ axis has emerged as a common feature in several neurodegenerative diseases, where impaired cellular clearance and metabolic dysfunction contribute to neuronal vulnerability. In this review, we summarize the advances of the molecular links between autophagy and NAD+ metabolism, with a particular focus on their roles in mitochondrial quality control, bioenergetic regulation, and cellular resilience. We also discuss the therapeutic potential of targeting the autophagy–NAD+ axis to promote neuroprotection in neurodegenerative disease.
1 Introduction
Maintaining cellular homeostasis is essential to the physiology of an organism. Macroautophagy (henceforth, autophagy) is among the main catabolic processes by which recycling of cellular macromolecules and organelles is carried out. This process involves a double-membraned vesicle called autophagosome engulfing the autophagic cargo, and its subsequent fusion with the lysosome to mediate cargo digestion (Yamamoto et al., 2023). Increasingly, dysregulation of autophagy is being linked to ageing and the pathology in a myriad of human diseases, including several neurodegenerative and lysosomal storage diseases (Seranova et al., 2017; 2020; Aman et al., 2021; Nixon and Rubinsztein, 2024). Post-mitotic neuronal cells are particularly vulnerable to autophagy malfunction due to their inability to clear toxic protein aggregates and damaged mitochondria, and thus, failure to maintain autophagy-mediated cellular homeostasis in neurons contributes to neurodegeneration (Palmer et al., 2025).
Autophagy is a multistep, tightly regulated pathway initiated by the formation of a phagophore, a cup-shaped membrane that elongates and engulfs cargo destined for degradation (Figure 1). This nascent autophagosome requires the coordinated action of autophagy-related (ATG) proteins, including the ULK1 initiation complex, the class III phosphatidylinositol 3-kinase (PI3K) complex, and two ubiquitin-like conjugation systems that mediate lipidation of ATG8/LC3 proteins onto the growing membrane (Yamamoto et al., 2023). Autophagosomes can capture bulk cytosolic material, but they also selectively target damaged or superfluous organelles, aggregated proteins, or invading pathogens, a process often mediated by selective autophagy receptors such as p62/SQSTM1, NBR1, NDP52, and OPTN, which bridge ubiquitinated cargo to LC3 (Vargas et al., 2023). Once sealed, the mature autophagosome traffics along the cytoskeleton and fuses with lysosomes, generating autolysosomes in which the cargo is degraded by acidic hydrolases. The resulting macromolecular building blocks, including amino acids, fatty acids, and nucleotides, are recycled back into the cytoplasm to support biosynthesis and energy production. Autophagy is under dynamic regulation by nutrient and stress signalling pathways, with mTORC1 acting as a key negative regulator and AMPK as an additional modulator, thereby coupling autophagy induction to cellular energy status (Rabanal-Ruiz et al., 2017). In neurons, selective forms of autophagy such as mitophagy play a particularly critical role in preserving organelle quality and bioenergetic competence (Nixon and Rubinsztein, 2024). Thus, autophagy integrates degradative capacity with metabolic flexibility, and its impairment not only compromises proteostasis and organelle integrity but also undermines cellular adaptation to stress, contributing to the pathogenesis of age-related and neurodegenerative disorders (Palmer et al., 2025).
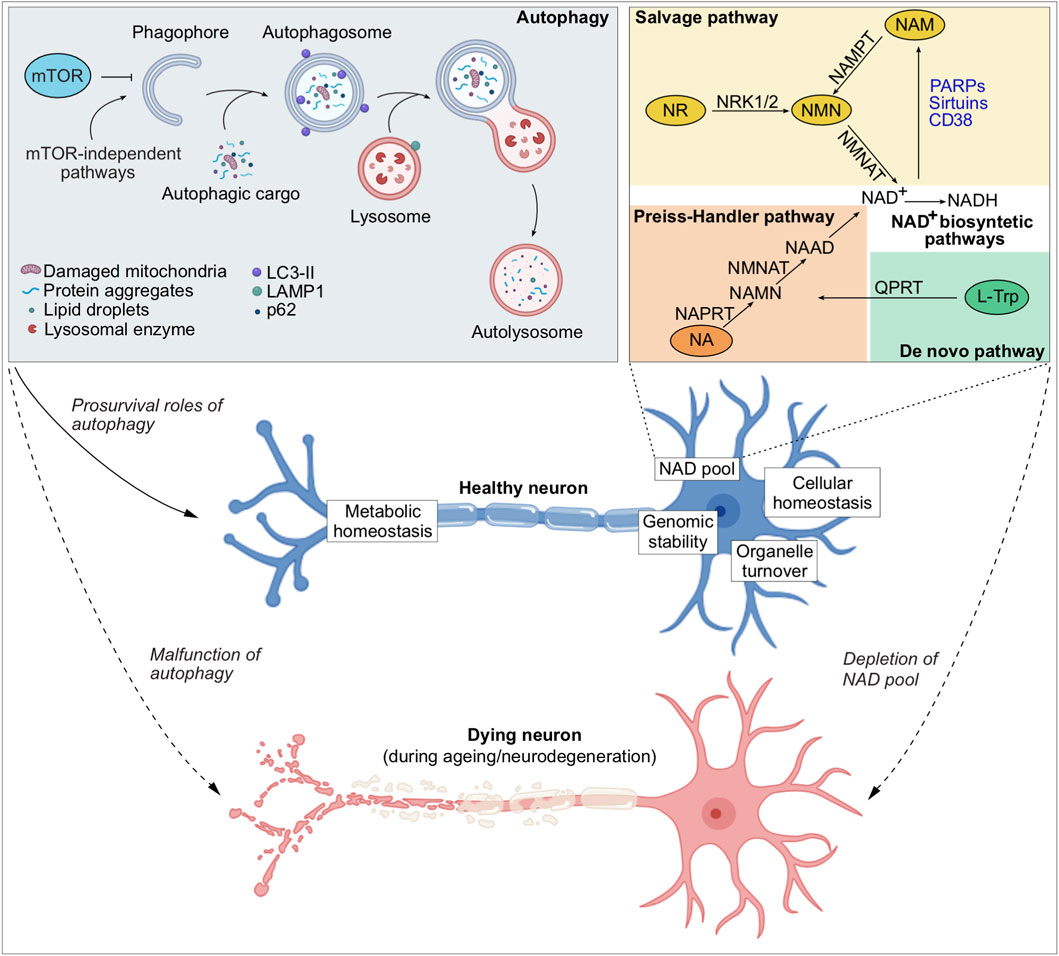
Figure 1. Autophagy and NAD+ metabolism in neuronal health and disease. Schematic overview illustrating the interplay between autophagy and NAD+ metabolism in maintaining neuronal homeostasis. Autophagy can be initiated through mTOR-dependent or mTOR-independent pathways, driving the formation of phagophores that elongate and capture autophagic cargo (such as damaged organelles, protein aggregates, lipid droplets) to form autophagosomes. The autophagosomes subsequently fuse with the lysosomes to degrade the autophagic cargo, thereby ensuring organelle turnover and recycling of the macromolecular breakdown products to aid metabolic homeostasis. Apart from cellular macromolecules and organelles, autophagy maintains intracellular NAD pool. NAD+ levels are sustained through multiple biosynthesis pathways, including the salvage, the Preiss–Handler, and de novo pathways. Among these, the salvage pathway predominates, recycling NAM (a by-product of NAD+-consuming enzymes such as sirtuins, PARPs, and CD38) back into NAD+. A healthy neuron is maintained by adequate autophagy and intracellular NAD+ levels, maintaining metabolic and cellular homeostasis, genomic stability, and efficient organelle turnover. In contrast, malfunction of autophagy or depletion of the NAD pool during ageing and neurodegeneration leads to neuronal cell death.
Beyond its canonical degradative function, autophagy has emerged as a central regulator of cellular metabolism linked to cell survival. Recent studies have identified a pro-survival role of autophagy by maintaining intracellular levels of a metabolic cofactor, nicotinamide adenine dinucleotide (NAD+). Loss of autophagy leads to depletion of NAD+ that mediates cytotoxicity in an evolutionarily conserved mechanism from yeast cells to human neurons (Kataura et al., 2022; Sun et al., 2023). Consistent with these findings, NAD+ functions as a cofactor for a wide array of redox and non-redox enzymes that facilitate energy metabolism, DNA repair, chromatin remodelling, and other biological processes, thereby playing a key role in maintaining genomic stability and cellular health (Covarrubias et al., 2021). In cells, NAD+ biosynthesis occurs via three main pathways: the de novo pathway from tryptophan, the Preiss-Handler pathway from nicotinic acid, and the salvage pathway from nicotinamide (Figure 1); the latter being most utilized by the cells (Katsyuba et al., 2020; Covarrubias et al., 2021). In the salvage pathway, the NAD+ biosynthetic enzymes include nicotinamide phosphoribosyl transferase (NAMPT) and nicotinamide mononucleotide adenylyl transferases (NMNATs) (Cambronne and Kraus, 2020). NAMPT converts NAM to NMN, which is then converted to NAD+ by NMNATs. The NMNAT isoforms—NMNAT1, NMNAT2 and NMNAT3—are respectively localised in the nucleus, cytoplasm and mitochondria, and enable subcellular control of NAD+ synthesis (Berger et al., 2005). The localisation of NAMPT and NMNAT3 in mitochondria indicates the existence of an intrinsic mitochondrial NAD+-salvage pathway critical for sustaining mitochondrial NAD+ levels and bioenergetic function (Berger et al., 2005; Nikiforov et al., 2011; Wang X. et al., 2019). Apart from the roles of NAD+ biosynthetic enzymes, NAD+-dependent enzymes (PARPs, SIRTs, CD38/CD157, and SARM1) can directly regulate autophagy and mitochondrial quality control in a context-dependent manner, linking metabolic state to degradative pathway regulation (Fang et al., 2016; Wilson et al., 2023).
Together, multiple lines of evidence delineate an intricate bidirectional interplay between autophagy and NAD+ metabolism that ensures cellular homeostasis under metabolic stress. Moreover, both autophagy and NAD+ levels are necessary for mitochondrial turnover and function, which are crucial for neuronal homeostasis and survival (Fang et al., 2016; Kataura et al., 2024). Of biomedical relevance, pharmacological strategies enhancing autophagy or boosting NAD+ levels show therapeutic benefits in neurodegenerative disease models (Menzies et al., 2017; Lautrup et al., 2019). Therefore, the autophagy–NAD+ axis represents a promising therapeutic avenue for ageing and neurodegenerative diseases that are characterized by progressive decline in autophagic activity and NAD+ levels (Wilson et al., 2023). In this review, we focus on the significance of the bidirectional relationship between autophagy and NAD+, with a particular emphasis on the role of autophagy–NAD+ axis in cellular, mitochondrial and bioenergetic homeostasis in neurons, and its implications and therapeutic opportunities in neurodegenerative diseases.
2 The link between autophagy and NAD+ in neurons
Autophagy or NAD+ homeostasis have been shown to independently support cellular health across species, from yeast to mice (Katsyuba et al., 2020; Aman et al., 2021; Covarrubias et al., 2021; Wilson et al., 2023; Yamamoto et al., 2023). Growing evidence suggests a bidirectional relationship between autophagy and NAD+, where NAD+ replenishment can normalize cellular function by modulating autophagy (Fang et al., 2014; 2016; 2019a; Kataura et al., 2024), whilst activation or restoring autophagy can improve NAD+ levels across various cell types (Desquiret-Dumas et al., 2013; Alshawi and Agius, 2019; Zhang et al., 2020; Kataura et al., 2024). Our recent findings reveal that autophagy plays a vital role in maintaining intracellular NAD pool (Kataura et al., 2022; Sun et al., 2023). The mechanisms underlying this reciprocal relationship, especially in neuronal homeostasis, are not fully understood. We will further discuss about this link in the context of mitochondrial and bioenergetic homeostasis, as well as overall cellular homeostasis, in neurons.
2.1 Autophagy–NAD+ axis in maintaining cellular homeostasis in neurons
Autophagy and NAD+ are vital for neuronal homeostasis and survival, whereas their deficits are associated with ageing and neurodegeneration (Sedlackova and Korolchuk, 2020; Schmauck-Medina et al., 2022; Wilson et al., 2023) (Figure 1). Studies on the molecular roles of NAD+ in cellular physiology highlight autophagy as a key regulator—by preventing DNA damage leading to excessive NAD+ consumption through mitochondrial quality control, and by mitigating nutrient stress via the recycling of amino acids, lipids, and nucleosides (Sedlackova and Korolchuk, 2020). We recently demonstrated that autophagy plays a crucial role in supporting cellular survival by maintaining intracellular NAD+ levels (Kataura et al., 2022; Sun et al., 2023). This is pertinent for post-mitotic neurons which rely on functional autophagy for clearing undesirable cellular materials like protein aggregates and damaged mitochondria (Stavoe and Holzbaur, 2019). Genetic studies in mice have shown that inducible knockout of essential autophagy-related genes (Atg5 or Atg7) in the brain leads to accumulation of protein aggregates and neurodegeneration (Hara et al., 2006; Komatsu et al., 2006), implying that basal autophagy is crucial for neuronal survival.
Emerging evidence highlights a crucial role for autophagy in maintaining neuronal homeostasis through the regulation of NAD+ metabolism. Studies employing autophagy-deficient models, including ATG5−/− human embryonic stem cells (hESC)-derived neurons and mouse embryonic fibroblasts, have revealed that loss of autophagy disrupts cellular metabolism and NAD+ homeostasis, leading to mitochondrial dysfunction, disrupted proteostasis, and increased cell death (Kataura et al., 2022; Sun et al., 2023). Mechanistically, these effects are driven by hyperactivation of NAD+-consuming enzymes (NADases) such as poly-ADP ribose polymerases (PARPs) and sirtuins (SIRTs), which deplete the total NAD pool. Notably, depletion of NAD+ and NADH triggered a cytotoxic cascade involving mitochondrial depolarization and oxidative stress, culminating in cell death (Kataura et al., 2022; Sun et al., 2023). This phenomenon was evolutionarily conserved from yeast to mammals (Kataura et al., 2022). In these autophagy-deficient models, boosting NAD+ levels by supplementation with NAD+ precursors such as nicotinamide (NAM), nicotinamide riboside (NR) or nicotinamide mononucleotide (NMN), or by preventing NAD+ consumption with PARP and SIRT inhibitors, restored mitochondrial function and bioenergetics, and improved cell viability (Kataura et al., 2022; Sun et al., 2023). Interestingly, NAD+ boosters also suppressed the buildup of aggresomes (accumulation of misfolded protein aggregates) that are typically observed in autophagy-deficient cells (Sun et al., 2023), highlighting a potential interplay between mitochondrial and protein homeostasis (Katsyuba et al., 2018; Ruan et al., 2020; Nowicka et al., 2021; Romani et al., 2021). These findings suggest the importance of the autophagy–NAD+ axis in neuronal survival.
2.2 Mitochondrial homeostasis in neuronal health
Beyond energy production, mitochondria are pivotal regulators of neuronal signalling and plasticity (Rangaraju et al., 2019). Maintaining mitochondrial homeostasis—or mitostasis, which encompasses mitochondrial function, distribution, and quality—is essential for neuronal health due to their unique morphology, extensive architecture, and high metabolic demands (Misgeld and Schwarz, 2017). A key aspect of mitostasis is mitophagy, which selectively eliminates damaged mitochondria (Pickles et al., 2018), thereby preventing oxidative stress and sustaining mitochondrial and cellular NAD+ levels (Sedlackova and Korolchuk, 2020; Kataura et al., 2024). The PINK1–Parkin pathway is a well-characterized mechanism in this process; PINK1 senses mitochondrial damage, while Parkin tags impaired mitochondria for autophagic clearance (Narendra and Youle, 2024).
Neurons mainly generate ATP through oxidative phosphorylation (OXPHOS), in contrast to glial cells that rely mainly on glycolysis (Zheng et al., 2016). Although the brain constitute only about 2% of body mass, it consumes about 20% of body’s oxygen to support energy-intensive functions, including action potentials, neurotransmission, synaptic vesicle recycling, and axonal transport (Raichle and Gusnard, 2002; Howarth et al., 2012). Glucose is first metabolized through glycolysis to pyruvate, which enters the tricarboxylic acid cycle (TCA) via pyruvate dehydrogenase. The TCA cycle generates NADH and FADH2, which fuel the mitochondrial electron transport chain (ETC) and ATP synthase (Martínez-Reyes and Chandel, 2020). Notably, neuronal metabolism is spatially compartmentalized: somata preferentially use aerobic glycolysis to limit ROS, whereas axon terminals depend on OXPHOS, preserving nuclear and cytoplasmic integrity (Wei et al., 2023).
Central to these mitochondrial functions is NAD+, cycles between oxidised (NAD+) and reduced (NADH) forms to drive electron transport and ATP production. NAD+ exists in compartmentalised pools—approximately 250 µM in the mitochondria, 100 µM in the cytoplasm, and in the nucleus—regulated by NAD+-consuming enzymes that modulate turnover in a compartment-specific manner (Cambronne et al., 2016). The NAD+ biosynthetic enzyme NAMPT, which progressively declines with ageing and neurodegeneration, is essential for maintaining mitochondrial and neuronal health; its loss disrupts mitochondrial homeostasis and causes neurodegeneration in mice (Wang et al., 2017; Lautrup et al., 2019; Shen et al., 2023; Chen et al., 2024). NAD+ precursor supplementation can rescue mitochondrial dysfunction, mitophagy, and alleviate disease-related phenotypes (Yoshino et al., 2018; Dölle and Tzoulis, 2025). These findings highlight the neuroprotective roles of NAD+, suggesting that its effects may be mediated through the enhancement of mitochondrial biogenesis, function and mitophagy.
Altogether, NAD+ availability is closely intertwined with mitochondrial health. A decline in NAD+ levels impair mitochondrial function, diminishes ATP production, and jeopardises neuronal viability (Lautrup et al., 2019; Wilson et al., 2023). Moreover, the PINK1–Parkin pathway and mitophagy intersect with NAD+-dependent signalling, underscoring the bidirectional crosstalk between mitochondria quality control and NAD+ homeostasis (Fang et al., 2016; Kataura et al., 2024). This precise spatial and metabolic regulation positions NAD+ as a central hub integrating mitochondrial homeostasis with autophagy-dependent neuronal survival.
2.3 NAD+-dependent enzymes in neuronal autophagy
Key NAD+-consuming enzymes—PARPs, SIRTs, CD38/CD157, and SARM1—coordinately regulate neuronal physiology and autophagy (Zhang D.-X. et al., 2016; Covarrubias et al., 2021; Wilson et al., 2023). Among these, SIRTs are NAD+-dependent deacetylases and mono-ADP-ribosyltransferases that control transcriptional activities and various cellular processes including DNA repair, mitochondrial biogenesis, and autophagy (Houtkooper et al., 2012; Ng and Tang, 2013; Wu et al., 2022; Baeken, 2024). SIRT1, SIRT2, and SIRT3 are prominent in central nervous system (CNS). SIRT1, mainly nuclear, promotes mitophagy and mitochondrial biogenesis via deacetylation of LC3, FOXO3 and PGC-1α (Ng and Tang, 2013; Baeken, 2024). Its deficiency impairs autophagy and energy homeostasis, whereas its overexpression induces autophagy even under nutrient-rich conditions (Araki et al., 2004; Lee et al., 2008). SIRT3, the mitochondrial isoform of the sirtuins family, preserves mitochondrial integrity by directly regulating key enzymes of the TCA cycle and antioxidant defense systems, particularly during oxidative stress (Sidorova-Darmos et al., 2018). SIRT3 has been also shown to modulate mitophagy in specific disease contexts (Yu et al., 2017; Wang C. et al., 2019), and its overexpression alleviates neurological and mitochondrial dysfunction (Yang et al., 2024).
PARPs, particularly PARP1, are activated by DNA damage to catalyze ADP-ribosylation reactions that consume NAD+ and facilitate repair, thereby maintaining genomic integrity (Ray Chaudhuri and Nussenzweig, 2017). While moderate PARP activation supports cell survival, chronic or excessive activity depletes NAD+, causes mitochondrial depolarization, inhibits autophagy, and ultimately triggers cell death (Virág et al., 2013; Bai, 2015). PARP1 also modulates autophagy through AMPK–mTOR pathway and FOXO3a transcription factor (Muñoz-Gámez et al., 2009; Meng et al., 2018; Wang et al., 2018). In neurons, PARP-induced NAD+ depletion contributes to the pathogenesis of several neurodegenerative diseases (Mao and Zhang, 2022). Moreover, excessive PARP activation suppresses sirtuin activity either by depleting NAD+ or through transcriptional repression, thereby aggravating mitochondrial dysfunction (Cantó et al., 2013). Collectively, these findings underscore the therapeutic potential of targeting the PARP–SIRT interplay to preserve NAD+ homeostasis and protect against neurodegeneration.
CD38, an NADase whose activity increases with ageing, degrades NAD+ into ADP-ribose and cyclic ADP-ribose, making it a major contributor to the age-related decline in brain NAD+ levels (Camacho-Pereira et al., 2016; Tarragó et al., 2018). CD38-mediated NAD+ depletion reduces SIRT3 activity and impairs mitophagy, establishing a feed-forward loop that exacerbates mitochondrial dysfunction and energy failure (Camacho-Pereira et al., 2016). In neurons, another NADase, SARM1 (Sterile alpha and TIR motif-containing 1), acts as a metabolic sensor activated by axonal injury, cleaving NAD+ into nicotinamide and ADP-ribose to trigger axonal degeneration (Gerdts et al., 2015; Figley et al., 2021). This process contributes to pathological axonal pruning and neurodegeneration, whilst pharmacological inhibition of SARM1 confers neuroprotection in models of peripheral neuropathy and traumatic brain injury (Gerdts et al., 2015; Figley et al., 2021).
Collectively, these enzymes not only consume NAD+ but also modulate key metabolic and autophagy regulators such as AMPK and mTOR (Covarrubias et al., 2021; Wilson et al., 2023). Restoring NAD+ levels, frequently via precursors such as NMN or NR, enhance autophagic flux, sustain mitochondrial health, and improve neuronal survival in models of neurodegeneration (Yoshino et al., 2018; Lautrup et al., 2019; Xie et al., 2020). In summary, NAD+-consuming enzymes function as critical metabolic sensors that regulate neuronal autophagy, linking cellular energy metabolism to stress responses, protein turnover, and organelle maintenance—processes essential for neuronal function and survival.
3 Autophagy and NAD+ in neuronal homeostasis and neurodegenerative diseases
Defective autophagy is increasingly recognized as a major driver of neurodegeneration because impaired clearance of macromolecules (Menzies et al., 2015; Seranova et al., 2017; 2020; Palhegyi et al., 2019; Nixon and Rubinsztein, 2024) or damaged organelles (Narendra and Youle, 2024; Antico et al., 2025) disrupt neuronal homeostasis. Recent demonstration that the loss of autophagy depletes intracellular NAD+ levels suggests a role of autophagy in the maintenance of cellular NAD pool (Kataura et al., 2022; Sun et al., 2023). Decline in autophagic activity and NAD+ levels has been suggested as contributing factors to ageing and age-related neurodegenerative diseases that are marked by progressive neuronal cell death (Sedlackova and Korolchuk, 2020; Schmauck-Medina et al., 2022; Wilson et al., 2023; Kolotyeva et al., 2024) (Figure 1). In this section, we will discuss the interplay between autophagy and NAD+, and the therapeutic benefits of targeting the autophagy–NAD+ axis, in neurodegenerative diseases that are characterized by autophagy and NAD+ deficits (Table 1).
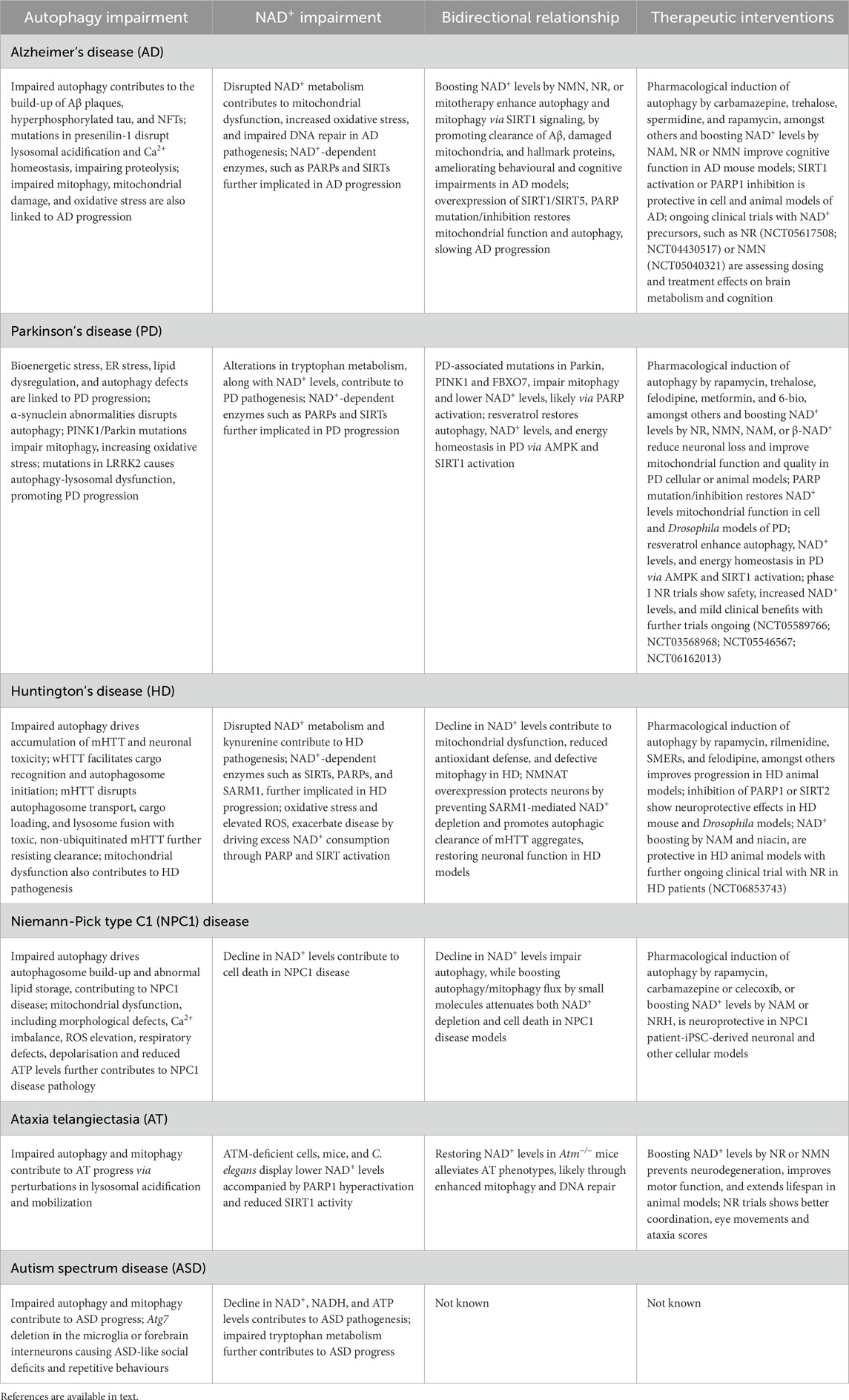
Table 1. Implications of autophagy-NAD+ axis in neurodegenerative diseases.
3.1 Alzheimer’s disease
Alzheimer’s disease (AD) is the most prevalent neurodegenerative disorder. It is associated with cognitive impairment and behavioral symptoms due to progressive neuronal loss (Knopman et al., 2021; Zhang et al., 2024). Familial AD is driven by autosomal dominant mutations in amyloid precursor protein (APP) or presenilin (PSEN1 or PSEN2) genes, which lead to increased β-amyloid (Aβ) production. In contrast, sporadic AD that accounts for over 95% of cases, arises from a multifactorial interplay of genetic risk factors, ageing, metabolic stress, and mitochondrial dysfunction (Dorszewska et al., 2016). AD is pathologically defined by the accumulation of Aβ plaques and tau neurofibrillary tangles (NFTs) in the brain (Knopman et al., 2021; Zhang et al., 2024). Increasing evidence suggests crosstalk between Aβ and tau that contributes to cellular dysfunctions and neurotoxicity (Zhang et al., 2021). A complex interplay of disruption in synaptic homeostasis and cellular clearance pathways like autophagy has been suggested in AD pathogenesis (Knopman et al., 2021; Zhang et al., 2021; 2024).
Autophagy dysfunction is considered a major contributing factor in neuronal vulnerability to genetic and environmental factors underlying AD. Autophagy has been suggested to play a role in the generation of Aβ, via processing of the amyloid precursor protein (APP) by presenilin-1 inside the autophagosomes. Aβ either undergoes autophagic degradation or is secreted into extracellular space by autophagy (Yu W. H. et al., 2005; Nilsson et al., 2013; Tian et al., 2013). Moreover, tau is also degraded by autophagy (Wang et al., 2009; Krüger et al., 2012; Lee et al., 2013; Neu et al., 2025). Defective autophagic clearance leads to the accumulation of Aβ and hyperphosphorylated tau, contributing to synaptic dysfunction and neuronal cell death (Nixon, 2024; Nixon and Rubinsztein, 2024). Failure in autophagy has been evidenced by the accumulation of autophagic vacuoles in AD patient brains, dystrophic neurites in mouse models and iPSC-derived cortical neurons, indicative of a block in autophagic flux that is suggested to arise from impaired lysosomal proteolysis (Boland et al., 2008; Nixon and Yang, 2011; Piras et al., 2016; Hung and Livesey, 2018). Mutations in presenilin-1 mediate autophagic dysfunction by impairing lysosomal acidification and Ca2+ homeostasis, thereby disrupting lysosomal proteolytic activity (Lee et al., 2010; 2015; Lee J.-H.et al., 2020). Mutant tau can also cause buildup of autophagosomes by preventing axonal transport via destabilization of microtubules (Rodríguez-Martín et al., 2013; Butzlaff et al., 2015; Cario and Berger, 2023). Furthermore, impairment of mitophagy has been demonstrated in AD patient brain and iPSC-derived neurons, and in cell and animal models (Cummins et al., 2019; Fang et al., 2019b; Mary et al., 2023). This could lead to the accumulation of damaged mitochondria and oxidative stress that are involved in the progression of AD (Wang et al., 2020; Ionescu-Tucker and Cotman, 2021). In addition, multiple lines of evidence suggest disruption in NAD+ metabolism as a contributing factor in AD (Lautrup et al., 2019; Wang et al., 2021). Decreased NAD+ and NADH levels, observed in both ageing and AD mouse brains and neuronal cultures, are linked to mitochondrial dysfunction and oxidative stress (Abeti et al., 2011; Liu D. et al., 2013; Hou et al., 2018; 2021; Lautrup et al., 2019; van der Velpen et al., 2021). NAD+-consuming enzymes, such as PARPs and SIRTs, are also implicated in AD—although SIRT activity is reduced in AD, PARP is activated that could drive NAD+ depletion (Strosznajder et al., 2012; Mehramiz et al., 2023). Together, these findings highlight a pathological link between autophagy and mitochondrial dysfunction, along with NAD+ deficits, in AD pathogenesis (Bhatia and Sharma, 2021).
Therapeutic strategies involving upregulation of autophagy and NAD+ levels have been shown to improve neuronal health and cognitive outcomes in AD animal models (Menzies et al., 2017; Dölle and Tzoulis, 2025). Pharmacological autophagy inducers, such as carbamazepine, trehalose, spermidine, and rapamycin or its analog temsirolimus, amongst others, have been shown to enhance the clearance of Aβ plaques and tau tangles, and ameliorate disease phenotypes including cognitive deficits, in various AD mouse models (Caccamo et al., 2010; Rodríguez-Navarro et al., 2010; Spilman et al., 2010; Majumder et al., 2011; Schaeffer et al., 2012; Du et al., 2013; Li et al., 2013; Ozcelik et al., 2013; Jiang et al., 2014a; 2014b; Portbury et al., 2017; Freitag et al., 2022). Supplementation with NAD+ precursors, such as NAM, NR or NMN, restores NAD+ levels and also prevents neurodegenerative pathology and cognitive decline in AD mouse models (Green et al., 2008; Gong et al., 2013; Liu D. et al., 2013; Yao et al., 2017; Hou et al., 2018; 2021; Rehman et al., 2021; Ma et al., 2024; Xiong et al., 2024). Interestingly, some studies have revealed potential crosstalk between these pathways. For instance, NMN promotes the clearance of hyperphosphorylated tau by enhancing autophagy (Ma et al., 2024), also lowers Aβ plaques (Yao et al., 2017), to ameliorate behavioural and cognitive impairments in AD mice. Likewise, NMN and NR prevent cognitive decline and proteotoxicity in C. elegans models of AD by inducing mitophagy (Sorrentino et al., 2017; Fang et al., 2019b). Beyond NAD+ replenishment, mitotherapy—introducing healthy mitochondria into defective neuronal cells—has been shown to activate autophagy via the NAD+-dependent SIRT1 signalling pathway to enhance the clearance of Aβ and damaged mitochondria, and improve cognitive function, in a mouse model of AD (Yang et al., 2023). Additionally, overexpression of SIRT1 or SIRT5, genetic mutation of Parp, or inhibition of PARP1 is protective in cell and animal models of AD (Qin et al., 2006; Min et al., 2010; Abeti et al., 2011; Martire et al., 2016; Wu et al., 2021; Yu et al., 2021).
There have been multiple clinical trials with NAD+ precursors in AD (Dölle and Tzoulis, 2025). Oral administration of NR in healthy individuals elevates neuronal NAD+ levels and lowers biomarkers related to neurodegenerative pathology (Vreones et al., 2023). However, clinical trials in AD with NR or NAM have not yielded any major cognitive improvements (Orr et al., 2024; Grill et al., 2025). Ongoing trials are evaluating the optimal dose and treatment period on brain metabolism and cognitive function, amongst other parameters, with NR (NCT05617508; NCT04430517) or NMN (NCT05040321). Overall, the interplay between autophagy and NAD+ metabolism, and their convergence on key cellular processes such as mitochondrial homeostasis and neuronal survival, highlight the therapeutic promise of targeting the autophagy–NAD+ axis in AD.
3.2 Parkinson’s disease
Parkinson’s disease (PD) is the second most prevalent neurodegenerative disease worldwide. It is clinically characterized by motor symptoms including resting tremor, bradykinesia, rigidity, and postural instability, as well as a broad spectrum of non-motor features including cognitive changes (Poewe et al., 2017; Bloem et al., 2021). Pathologically, PD is marked by the progressive degeneration of dopaminergic neurons in the substantia nigra pars compacta and the accumulation of Lewy bodies that are mainly composed of aggregated α-synuclein (Kalia and Kalia, 2015; Poewe et al., 2017). Lewy body pathology in PD not only contributes to dopaminergic neuronal loss but also disrupts dopamine neurotransmission by impairing intracellular transport (Kalia and Kalia, 2015; Aarsland et al., 2021; Calabresi et al., 2023). The selective vulnerability of dopaminergic neurons in PD has been linked to mitochondrial dysfunction (Borsche et al., 2021; Henrich et al., 2023). Multiple cellular stress pathways aid to mitochondrial abnormalities, including bioenergetic disruption, ER stress, altered lipid metabolism, and autophagy malfunction (Lynch-Day et al., 2012; Van Laar and Berman, 2013; Gómez-Suaga et al., 2018; Zambon et al., 2019; Flores-Leon and Outeiro, 2023).
Impairment in autophagy, including chaperone-mediated autophagy (CMA), in PD contributes to α-synuclein aggregation, mitochondrial dysfunction, and neuronal loss (Cuervo et al., 2004; Lynch-Day et al., 2012; Menzies et al., 2015; Hou et al., 2020). PD is a multigenic disorder, and mutations in multiple PD-associated genes deregulate autophagy at distinct stages, including the selective autophagy process of mitophagy as well as CMA (Lynch-Day et al., 2012; Gan-Or et al., 2015). α-synuclein gene multiplication inhibits autophagosome biogenesis (Winslow et al., 2010; Decressac et al., 2013), Lewy bodies containing α-synuclein prevent autophagosome maturation (Tanik et al., 2013), whilst α-synuclein point mutations (A53T or A30P) impair CMA (Cuervo et al., 2004; Martinez-Vicente et al., 2008). Mutations in mitophagy-related genes, encoding for PINK1 and Parkin, further compromise mitochondrial clearance. Loss-of-function mutations in PINK1 and Parkin impair the recognition and degradation of damaged mitochondria, thereby increasing oxidative stress and energy failure (Pickrell and Youle, 2015; Narendra and Youle, 2024). Mutations in LRRK2 is suggested to cause autophagy and lysosomal dysfunction, as well as disruption of CMA (Orenstein et al., 2013; Madureira et al., 2020).
Alterations in metabolic pathways also contribute to the pathogenesis of PD. One such example is the kynurenine pathway of tryptophan metabolism, which influences NAD+ levels and is implicated in PD (Lim et al., 2017; Castro-Portuguez and Sutphin, 2020). Multiple studies in genetic and neurotoxin-induced models have demonstrated impairment in NAD+ metabolism and decrease in NAD+ levels in PD, along with the role of NAD+ depletion in dopaminergic neurodegeneration (Schöndorf et al., 2018; Lundt and Ding, 2021; Pérez et al., 2021). For instance, PD-associated mutations in Parkin, PINK1 and FBXO7, all of which impair mitophagy, reduce cellular NAD+ levels that is suggested via PARP activation (Burchell et al., 2013; Lehmann et al., 2016; 2017; Delgado-Camprubi et al., 2017). Moreover, NAD+-consuming enzymes such as PARPs and SIRTs have also been implicated in PD, further underscoring the involvement of NAD+-dependent mechanisms (He et al., 2022; Lengyel-Zhand et al., 2022).
Therapeutic strategies involving autophagy induction and NAD+ restoration in PD models have demonstrated promising neuroprotective effects, including amelioration of motor deficits and neuropathological phenotypes (Menzies et al., 2017; Pérez et al., 2021; Sanchez-Mirasierra et al., 2022; Dölle and Tzoulis, 2025). Pharmacological agents inducing autophagy, such as rapamycin, trehalose, felodipine, metformin, and 6-Bio, amongst others, have been shown to alleviate disease phenotypes in mouse models of PD (Malagelada et al., 2010; Liu K. et al., 2013; Bai et al., 2015; Suresh et al., 2017; Ryu et al., 2018; Pupyshev et al., 2019; Siddiqi et al., 2019). Likewise, supplementation with NAD+ precursors, such as NR, NMN, NAM, and β-NAD+, have been shown to improve mitochondrial bioenergetic function and exert cytoprotective effects in cellular and animal models of PD (Jia et al., 2008; Lu et al., 2014; Lehmann et al., 2016; 2017; Zou et al., 2016; Schöndorf et al., 2018; Shan et al., 2019; Pérez et al., 2021). For instance, NR administration rescues mitochondrial dysfunction in PD patient iPSC-derived neurons and prevents the decline of motor ability and age-dependent loss of dopaminergic neurons in GBA-PD Drosophila model by increasing NAD+ levels (Schöndorf et al., 2018). Moreover, modulating the activity of NAD+-consuming enzyme, such as by PARP inhibition or mutation, restores NAD+ levels and rescues mitochondrial dysfunction in cell and Drosophila models of PD (Lehmann et al., 2016; 2017; Delgado-Camprubi et al., 2017). Amongst others, resveratrol has been shown to improve autophagy, NAD+ levels and energy homeostasis in PD patient fibroblasts via activation of AMPK and SIRT1 (Ferretta et al., 2014). Randomised phase I clinical trials with NR in 20–30 PD patients have reported no safety concerns, elevation in brain and blood NAD+ levels, along with mild clinical improvement (Brakedal et al., 2022; Berven et al., 2023). Clinical trials involving larger patient cohort with NR (NCT05589766; NCT03568968; NCT05546567; NCT06162013) are currently ongoing. Overall, these studies highlight the therapeutic potential of targeting the autophagy–NAD+ axis in PD.
3.3 Huntington’s disease
Huntington’s disease (HD) is a progressive, autosomal dominant neurodegenerative disorder caused by a CAG trinucleotide expansion in the Huntingtin (HTT) gene (Bates et al., 2015). This expansion results in an abnormally extended polyglutamine (PolyQ) tract in the huntingtin protein (HTT), producing a mutant form (mHTT) with toxic gain-of-function properties (Zoghbi and Orr, 2000). The accumulation of mHTT aggregates within neurons is a pathological hallmark of HD and contributes to widespread neuronal dysfunction and degeneration (Yamamoto et al., 2000). The number of CAG repeats is strongly associated with disease characteristic—larger expansions correlate with earlier onset and increased severity. Alleles with more than 40 CAG repeats are fully penetrant and lead to HD development, and typically with more than 60 CAG repeats in early-onset. Intermediate alleles (36–39 CAG repeats) show reduced penetrance, while alleles with fewer than 36 CAG repeats are considered non-pathological (Zoghbi and Orr, 2000; Bates et al., 2015). Clinically, HD presents with a triad of progressive motor dysfunction, cognitive decline, and psychiatric disturbances, progressively worsening into dementia (Peavy et al., 2010; Roos, 2010). The motor symptoms are primarily attributed to selective degeneration of medium spiny neurons in the striatum, which precedes cortical neuronal loss (Zoghbi and Orr, 2000; Bates et al., 2015; Oh et al., 2022).
A critical cellular process disrupted in HD is autophagy, which is the predominant intracellular clearance route for mHTT (Sarkar and Rubinsztein, 2008; Menzies et al., 2015). Wild-type HTT functions as a scaffold protein, facilitating cargo recognition and autophagosome initiation (Ochaba et al., 2014; Rui et al., 2015). In contrast, mHTT impairs autophagy at multiple stages—disrupting autophagosome transport, hindering cargo loading, and blocking fusion with lysosomes (Martinez-Vicente et al., 2010; Wong and Holzbaur, 2014; Oh et al., 2022; Pircs et al., 2022). Studies in various HD models have elucidated how dysregulated autophagy contributes to mHTT accumulation and neuronal toxicity (Martin et al., 2015; Menzies et al., 2015). Moreover, mHTT can undergo conformation-dependent recognition by selective autophagy that correlates with their degradation and toxicity. A toxic mHTT protein lacking Lys63 polyubiquitination and p62 interaction, which are essential for aggrephagy (degradation of protein aggregates by autophagy), slow autophagic clearance due to its resistance to this selective autophagy process (Fu et al., 2017).
Apart from defective autophagy, mitochondrial dysfunction and disrupted NAD+ metabolism are pathological features in HD that reflect impairment in cellular energy homeostasis (Lundt and Ding, 2021; Sharma et al., 2021). This decline in NAD+ levels in HD is linked to mitochondrial dysfunction, weakened antioxidant defence, and aberrant mitophagy (Xie et al., 2020), which could be arising due to autophagy defect. Hyperactivation of kynurenine pathway as well as NAD+-consuming enzymes, such as SIRTs, PARPs, and SARM1, are also implicated in the pathogenesis of HD (Lloret and Beal, 2019; Xie et al., 2020; Lundt and Ding, 2021). Oxidative stress and elevated ROS, commonly reported in HD (Kim et al., 2021; Sharma et al., 2021), can further activate PARPs and SIRTs that accelerate NAD+ consumption. As a potential therapeutic intervention, pharmacological inhibition of PARP1 or SIRT2 demonstrate neuroprotective effects in HD mouse and Drosophila models (Pallos et al., 2008; Cardinale et al., 2015; Paldino et al., 2017). In a study involving injury-induced axonal degeneration where NAD+ levels were decreased, overexpression of the NAD+ biosynthetic enzyme NMNAT1 is shown to protect mouse dorsal root ganglion neurons against axonal damage by preventing SARM1-mediated NAD+ depletion (Sasaki et al., 2016). Nmnat overexpression also restores neuronal function in a Drosophila model of HD where the neuroprotective effects are attributed to autophagic removal of mHTT aggregates (Zhu et al., 2019).
Given the interdependence between autophagy, mitochondrial integrity, and NAD+ metabolism, numerous studies have explored therapeutic strategies targeting these pathways. Pharmacological inducers of autophagy, such as rapamycin, rilmenidine, SMERs, and felodipine, amongst others, have been shown to enhance autophagic flux, facilitate mHTT clearance, and alleviate disease phenotypes in various HD animal models (Ravikumar et al., 2004; Sarkar et al., 2007; Williams et al., 2008; Rose et al., 2010; Menzies et al., 2017; Siddiqi et al., 2019). Likewise, supplementation with NAD+ precursors, such as NAM and niacin, are protective in HD animal models (Pallos et al., 2008; Hathorn et al., 2011; Xie et al., 2020; Dölle and Tzoulis, 2025). Recently, a clinical trial has been launched to study the efficacy and safety of NR in HD patients (NCT06853743). Overall, targeting the autophagy–NAD+ axis has demonstrated therapeutic benefits in HD models.
3.4 Niemann-Pick type C1 disease
Niemann-Pick Type C1 (NPC1) disease is a rare autosomal recessive lysosomal storage disorder primarily caused by mutations in the NPC1 gene, which encodes a polytopic transmembrane cholesterol transporter located on the lysosomal membrane (Li et al., 2017). Loss of NPC1 function impairs intracellular lipid trafficking, leading to the accumulation of lipids, particularly cholesterol and sphingolipids, in the late endosomes and lysosomes (Lloyd-Evans et al., 2008; Patterson et al., 2017). Clinical presentations of NPC1 disease are highly heterogenous and depend on the age of onset, although the disease is typically characterised by progressive neurodegenerative dementia and hepatosplenomegaly (Vanier et al., 1996; Las Heras et al., 2023). The NPC1 gene is highly conserved across eukaryotes, and thus various disease models have provided key insights into the pathophysiology of NPC1 disease (Fog and Kirkegaard, 2019; Lee S.-E. et al., 2020), showing that defective lipid homeostasis causes cellular degeneration through mechanisms involving impaired autophagy, mitochondrial dysfunction, and more recently, NAD+ decline (Sarkar et al., 2013; Maetzel et al., 2014; Vilaça et al., 2014; Kataura et al., 2022; 2024).
Impaired autophagic flux in NPC1 disease is characterised by the accumulation of autophagosomes due to defective SNARE machinery, which is required for the fusion of autophagosomes with late-endosomes or lysosomes (Sarkar et al., 2013). Additionally, reduced VEGF levels leading to abnormal sphingosine accumulation have been implicated in autophagy dysfunction and neuronal loss in NPC1 disease (Lee et al., 2014). A recent study identified that lysosomal cholesterol accumulation leads to hyperactivation of mTORC1 (Davis et al., 2021), which can repress autophagy by inhibiting the autophagy initiator ULK1 complex. These findings indicate that multiple molecular mechanisms underlie autophagy dysfunction in NPC1 disease. Nonetheless, stimulating autophagy with rapamycin, carbamazepine or celecoxib restore autophagic flux and improve cell viability in NPC1 patient iPSC-derived neurons (Maetzel et al., 2014; Kuo et al., 2015; Kataura et al., 2024).
Mitochondrial dysfunction is also evident in NPC1 disease. In NPC1-deficient cells, cholesterol is significantly elevated in mitochondrial membranes, which is associated with various mitochondrial abnormalities such as morphological defects, elevated Ca2+ levels, increased ROS production, reduced respiratory capacity, mitochondrial depolarisation and decreased ATP levels (Yu W. et al., 2005; Woś et al., 2016; Casas et al., 2023; Kataura et al., 2024). Mitochondrial dysfunction can trigger apoptosis and influence other regulated cell death pathways including necroptosis, both of which have been implicated in NPC1 pathogenesis (Erickson and Bernard, 2002; Cougnoux et al., 2016; Zareba et al., 2024). Given that autophagy plays a critical role in mitochondrial quality control via mitophagy, impaired autophagy and aberrant mitochondrial cholesterol may act synergistically to disrupt mitochondrial homeostasis and promote cell death in NPC1 disease.
Recently, declining NAD+ levels have been identified as a key mediator of cell death in NPC1 disease and are closely associated with autophagy dysfunction (Kataura et al., 2022; 2024). In NPC1-deficient cells, both NAD+ and NADH levels are significantly reduced whilst boosting NAD+ levels by supplementing NAD+ precursors, such as NAM or NRH, restores cell survival. Notably, enhancing autophagy/mitophagy flux using small molecules attenuates both NAD+ depletion and cell death, suggesting that NAD+ loss occurs downstream of impaired autophagy and serves as a trigger for cell death––consistent with the previous findings in ATG5−/− autophagy-deficient cells (Kataura et al., 2022; 2024; Sun et al., 2023). These results support the concept that the autophagy–NAD+ axis could be a new therapeutic target in NPC1 disease.
Currently, two drugs—Miglustat and Arimoclomol—have been approved for NPC1 treatment. Miglustat, the first approved drug, is a glucosylceramide synthase inhibitor that reduces glycosphingolipids accumulation and remained the only available treatment for 15 years (Pineda et al., 2018) Similarly, the cholesterol-lowering agent, 2-Hydroxypropyl-β-cyclodextrin (HPβCD) has shown promise in clinical trials for NPC1, although further validation is needed (Hastings et al., 2022). In 2024, the FDA approved Arimoclomol as part of a combination therapy with Miglustat. Arimoclomol is thought to enhance lysosomal function by upregulating heat shock proteins and promote cell survival under lysosomal stress (Mengel et al., 2021). As this drug was effective in patients who received Miglustat as their background treatment (Mengel et al., 2021), combination therapies that act via multiple mechanisms appear promising for NPC1 disease. Notably, stimulating autophagy, which is sufficient to rescue cell death in NPC1-deficient cells, was unable to alleviate cholesterol accumulation. Likewise, HPβCD did not improve autophagic function (Sarkar et al., 2013). Taken together, it is possible that a dual approach—combining lipid-lowering agents with drugs targeting the autophagy–NAD+ axis—may offer an optimal disease-modifying strategy for NPC1 disease.
3.5 Other neurological diseases
Ataxia-telangiectasia (A-T) is a rare, autosomal recessive genetic disorder, characterized by progressive cerebellar ataxia, telangiectasia and immunodeficiency (Collyer and Rajan, 2024). A-T is caused by mutations in ataxia telangiectasia-mutated (ATM) gene, which encodes for ATM kinase that regulates of double-strand DNA break (DSB) signalling and stress responses (Lee and Paull, 2021). ATM deficiency is associated with impairment in DNA damage response, mitochondrial dysfunction and oxidative stress (Valentin-Vega et al., 2012; Fang et al., 2016; Stagni et al., 2018; Lee and Paull, 2021; Leeson et al., 2024). ATM is suggested to undergo autophagic degradation and aid in autolysosome formation, whereas its loss deregulates autophagy and mitophagy possibly via perturbations in lysosomal acidification and mobilization (Fang et al., 2016; Stagni et al., 2020; Sunderland et al., 2020; Cheng et al., 2021; Hwang et al., 2023). Additionally, ATM-deficient cells, mice and C. elegans display lower NAD+ levels (Fang et al., 2016; Yang et al., 2021). This was found to be associated with increased PARP1 and reduced SIRT1 activity in ATM-deficient cells (Fang et al., 2016). Boosting NAD+ levels via supplementation with NAD+ precursors, such as NR or NMN, prevents neurodegeneration, improves motor function, and extends lifespan in animal models (Fang et al., 2016; Yang et al., 2021). Alleviation of A-T phenotypes by restoring NAD+ levels in Atm−/− mice are suggested to be via upregulation of mitophagy and DNA repair (Fang et al., 2016; Yang et al., 2021). Clinical trials with NR in a small cohort of A-T patients have shown improvements in motor coordination, eye movements and ataxia scores (Veenhuis et al., 2021; Steinbrücker et al., 2023; Presterud et al., 2024). Overall, these findings suggest NAD+ supplementation as a promising therapeutic strategy for A-T.
Autism spectrum disorder (ASD) is a multifaceted neurodevelopmental condition characterised by difficulties in social communication and repetitive behaviours (Lord et al., 2020). Autophagy and mitochondrial dysfunction, along with oxidative stress, have been reported in ASD (Essa et al., 2013b; Deng et al., 2021). Impairment in autophagy, suggested to be arising from mTOR activation, is seen in autistic children as well as in rat, mouse and cellular experimental models of ASD (Tang et al., 2014; Huber et al., 2015; Zhang J. et al., 2016; Lieberman et al., 2020; Deri et al., 2025; Ham et al., 2025). Perturbations in the expression and phosphorylation of several autophagy-related proteins has been found via multi-omics approach in ASD mouse brain (Deri et al., 2025). Moreover, deletion of Atg7, an essential autophagy gene, in the microglia or forebrain interneurons in mice leads to ASD-like social behavioural impairments and repetitive behaviours (Kim et al., 2017; Hui et al., 2019). In addition, decreased levels of NAD+, NADH and ATP are found in patients with autism (Adams et al., 2011; Essa et al., 2013a; Launay et al., 2023). Tryptophan metabolism, which generates NAD+ and several neuroactive intermediates, is reported to be impaired in ASD, as evidenced by low tryptophan levels in autistic children (Kałuzna-Czaplinska et al., 2010; Essa et al., 2013b), whilst other studies found no change (Almulla et al., 2023; Launay et al., 2023). More studies are warranted to determine the therapeutic benefits of autophagy–NAD+ axis in experimental models of ASD.
4 Conclusions and prospects
Autophagy and NAD+ play a pivotal role in the regulation of numerous cellular processes, including energy metabolism, genomic maintenance, stress resistance, cell survival. Although emerging evidence have underscored the importance of the reciprocal relationship between autophagy and NAD+ metabolism, their crosstalk in the progression of neurodegenerative diseases by maintaining cellular, mitochondrial and bioenergetic homeostasis is not completely understood. To bridge this knowledge gap, future research should aim to elucidate the underlying mechanisms of this bidirectional interaction and its impact on neuronal health and the neurodegenerative process. Importantly, most current findings have been derived from conventional in vitro and animal models, which often fail to fully recapitulate human neuronal complexity and disease pathology. Therefore, there is an urgent need to employ more physiologically relevant systems—such as iPSC-derived cerebral organoids—to better model the intricate dynamics of the autophagy–NAD+ relationship in a disease-specific context.
From a therapeutic standpoint, pharmacological induction of autophagy and enhancement of NAD+ levels using precursors such as NAM, NMN, NR, and NRH have individually demonstrated neuroprotective effects in preclinical studies. These interventions have been associated with improved mitochondrial function, reduced protein aggregation, and enhanced neuronal survival. Future therapeutic interventions could focus on modulating NAD+ metabolism and autophagy to restore cellular function and mitigate the effects of neurodegeneration.
Author contributions
GK: Conceptualization, Visualization, Writing – original draft, Writing – review and editing. MD: Writing – original draft, Writing – review and editing. LS: Writing – original draft, Writing – review and editing. DS-A: Writing – original draft, Writing – review and editing. CS: Writing – original draft, Writing – review and editing. SC: Writing – original draft, Writing – review and editing. TB: Funding acquisition, Writing – review and editing. DM-S: Funding acquisition, Writing – review and editing. EC-N: Funding acquisition, Writing – review and editing. PB: Funding acquisition, Writing – review and editing. TK: Funding acquisition, Writing – original draft, Writing – review and editing. VK: Funding acquisition, Writing – original draft, Writing – review and editing. SS: Conceptualization, Funding acquisition, Supervision, Writing – original draft, Writing – review and editing.
Funding
The authors declare that financial support was received for the research and/or publication of this article. This work is supported grants from Medical Research Council (MRC) (MR/Z504488/1) to SS and VK; Action Medical Research/LifeArc (GN3049), LifeArc (Philanthropic Fund P2019-0004, and Pathfinder Award), Wellcome Trust (109626/Z/15/Z), Wolfram Syndrome UK, and Birmingham Fellowship to SS; University of Birmingham Brazil Institute (UBBI) Visiting Fellowship to LS and SS; UBBI–UNICAMP Seed Fund to SS and DM-S.; UBBI–USP Seed Fund to SS and EC-N; JSPS (20K22912; 25K18725), AMED (JP24gm6710024) and Nippon Shinyaku to TK (the funder Nippon Shinyaku was not involved in the study design, collection, analysis, interpretation of data, the writing of this article, or the decision to submit it for publication); FAPESP (São Paulo State Research Funding Agency) Brazil (2022/00758-0) to EC-N; FAPESP (2023/00841-7, 2019/00098-7, 2017/25588-1, and 2024/03869-2) and CAPES (Coordination of Superior Level Staff Improvement) Brazil (88887.495565/2020–00) to DM-S; FAPESP (2024/22132-0) to DS-A; CNPq Productivity Research Fellowship (309675/2025-1) to PB; MRC DPFS (MR/P007732/1) to TB. Both SS and VK are Former Fellows for life at Hughes Hall, University of Cambridge, United Kingdom.
Conflict of interest
Authors SS and TB are scientific advisors for NMN Bio Ltd. Author VK is a scientific advisor for Longaevus Technologies. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The authors declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aarsland, D., Batzu, L., Halliday, G. M., Geurtsen, G. J., Ballard, C., Ray Chaudhuri, K., et al. (2021). Parkinson disease-associated cognitive impairment. Nat. Rev. Dis. Primer 7, 7. doi:10.1038/s41572-021-00280-3
Abeti, R., Abramov, A. Y., and Duchen, M. R. (2011). β-amyloid activates PARP causing astrocytic metabolic failure and neuronal death. Brain J. Neurol. 134, 1658–1672. doi:10.1093/brain/awr104
Adams, J. B., Audhya, T., McDonough-Means, S., Rubin, R. A., Quig, D., Geis, E., et al. (2011). Nutritional and metabolic status of children with autism vs. neurotypical children, and the association with autism severity. Nutr. Metab. 8, 34. doi:10.1186/1743-7075-8-34
Almulla, A. F., Thipakorn, Y., Tunvirachaisakul, C., and Maes, M. (2023). The tryptophan catabolite or kynurenine pathway in autism spectrum disorder; a systematic review and meta-analysis. Autism Res. 16, 2302–2315. doi:10.1002/aur.3044
Alshawi, A., and Agius, L. (2019). Low metformin causes a more oxidized mitochondrial NADH/NAD redox state in hepatocytes and inhibits gluconeogenesis by a redox-independent mechanism. J. Biol. Chem. 294, 2839–2853. doi:10.1074/jbc.RA118.006670
Aman, Y., Schmauck-Medina, T., Hansen, M., Morimoto, R. I., Simon, A. K., Bjedov, I., et al. (2021). Autophagy in healthy aging and disease. Nat. Aging 1, 634–650. doi:10.1038/s43587-021-00098-4
Antico, O., Thompson, P. W., Hertz, N. T., Muqit, M. M. K., and Parton, L. E. (2025). Targeting mitophagy in neurodegenerative diseases. Nat. Rev. Drug Discov. 24, 276–299. doi:10.1038/s41573-024-01105-0
Araki, T., Sasaki, Y., and Milbrandt, J. (2004). Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science 305, 1010–1013. doi:10.1126/science.1098014
Baeken, M. W. (2024). Sirtuins and their influence on autophagy. J. Cell. Biochem. 125, e30377. doi:10.1002/jcb.30377
Bai, P. (2015). Biology of Poly(ADP-Ribose) polymerases: the factotums of cell maintenance. Mol. Cell 58, 947–958. doi:10.1016/j.molcel.2015.01.034
Bai, X., Wey, M. C.-Y., Fernandez, E., Hart, M. J., Gelfond, J., Bokov, A. F., et al. (2015). Rapamycin improves motor function, reduces 4-hydroxynonenal adducted protein in brain, and attenuates synaptic injury in a mouse model of synucleinopathy. Pathobiol. Aging Age Relat. Dis. 5, 28743. doi:10.3402/pba.v5.28743
Bates, G. P., Dorsey, R., Gusella, J. F., Hayden, M. R., Kay, C., Leavitt, B. R., et al. (2015). Huntington disease. Nat. Rev. Dis. Primer 1, 15005. doi:10.1038/nrdp.2015.5
Berger, F., Lau, C., Dahlmann, M., and Ziegler, M. (2005). Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J. Biol. Chem. 280, 36334–36341. doi:10.1074/jbc.M508660200
Berven, H., Kverneng, S., Sheard, E., Søgnen, M., Af Geijerstam, S. A., Haugarvoll, K., et al. (2023). NR-SAFE: a randomized, double-blind safety trial of high dose nicotinamide riboside in Parkinson’s disease. Nat. Commun. 14, 7793. doi:10.1038/s41467-023-43514-6
Bhatia, V., and Sharma, S. (2021). Role of mitochondrial dysfunction, oxidative stress and autophagy in progression of Alzheimer’s disease. J. Neurol. Sci. 421, 117253. doi:10.1016/j.jns.2020.117253
Bloem, B. R., Okun, M. S., and Klein, C. (2021). Parkinson’s disease. Lancet lond. Engl. 397, 2284–2303. doi:10.1016/S0140-6736(21)00218-X
Boland, B., Kumar, A., Lee, S., Platt, F. M., Wegiel, J., Yu, W. H., et al. (2008). Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease. J. Neurosci. 28, 6926–6937. doi:10.1523/JNEUROSCI.0800-08.2008
Borsche, M., Pereira, S. L., Klein, C., and Grünewald, A. (2021). Mitochondria and Parkinson’s disease: clinical, molecular, and translational aspects. J. Park. Dis. 11, 45–60. doi:10.3233/JPD-201981
Brakedal, B., Dölle, C., Riemer, F., Ma, Y., Nido, G. S., Skeie, G. O., et al. (2022). The NADPARK study: a randomized phase I trial of nicotinamide riboside supplementation in Parkinson’s disease. Cell Metab. 34, 396–407.e6. doi:10.1016/j.cmet.2022.02.001
Burchell, V. S., Nelson, D. E., Sanchez-Martinez, A., Delgado-Camprubi, M., Ivatt, R. M., Pogson, J. H., et al. (2013). The Parkinson’s disease-linked proteins Fbxo7 and Parkin interact to mediate mitophagy. Nat. Neurosci. 16, 1257–1265. doi:10.1038/nn.3489
Butzlaff, M., Hannan, S. B., Karsten, P., Lenz, S., Ng, J., Voßfeldt, H., et al. (2015). Impaired retrograde transport by the Dynein/Dynactin complex contributes to Tau-induced toxicity. Hum. Mol. Genet. 24, 3623–3637. doi:10.1093/hmg/ddv107
Caccamo, A., Majumder, S., Richardson, A., Strong, R., and Oddo, S. (2010). Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. J. Biol. Chem. 285, 13107–13120. doi:10.1074/jbc.M110.100420
Calabresi, P., Mechelli, A., Natale, G., Volpicelli-Daley, L., Di Lazzaro, G., and Ghiglieri, V. (2023). Alpha-synuclein in Parkinson’s disease and other synucleinopathies: from overt neurodegeneration back to early synaptic dysfunction. Cell Death Dis. 14, 176. doi:10.1038/s41419-023-05672-9
Camacho-Pereira, J., Tarragó, M. G., Chini, C. C. S., Nin, V., Escande, C., Warner, G. M., et al. (2016). CD38 dictates age-related NAD decline and mitochondrial dysfunction through an SIRT3-Dependent mechanism. Cell Metab. 23, 1127–1139. doi:10.1016/j.cmet.2016.05.006
Cambronne, X. A., and Kraus, W. L. (2020). Location, location, location: compartmentalization of NAD+ synthesis and functions in mammalian cells. Trends biochem. Sci. 45, 858–873. doi:10.1016/j.tibs.2020.05.010
Cambronne, X. A., Stewart, M. L., Kim, D., Jones-Brunette, A. M., Morgan, R. K., Farrens, D. L., et al. (2016). Biosensor reveals multiple sources for mitochondrial NAD+. Science 352, 1474–1477. doi:10.1126/science.aad5168
Cantó, C., Sauve, A. A., and Bai, P. (2013). Crosstalk between poly(ADP-ribose) polymerase and sirtuin enzymes. Mol. Asp. Med. 34, 1168–1201. doi:10.1016/j.mam.2013.01.004
Cardinale, A., Paldino, E., Giampà, C., Bernardi, G., and Fusco, F. R. (2015). PARP-1 inhibition is neuroprotective in the R6/2 Mouse model of Huntington’s disease. PLoS One 10, e0134482. doi:10.1371/journal.pone.0134482
Cario, A., and Berger, C. L. (2023). Tau, microtubule dynamics, and axonal transport: new paradigms for neurodegenerative disease. BioEssays News Rev. Mol. Cell. Dev. Biol. 45, e2200138. doi:10.1002/bies.202200138
Casas, M., Murray, K. D., Hino, K., Vierra, N. C., Simó, S., Trimmer, J. S., et al. (2023). NPC1-dependent alterations in KV2.1-CaV1.2 nanodomains drive neuronal death in models of Niemann-Pick Type C disease. Nat. Commun. 14, 4553. doi:10.1038/s41467-023-39937-w
Castro-Portuguez, R., and Sutphin, G. L. (2020). Kynurenine pathway, NAD+ synthesis, and mitochondrial function: targeting tryptophan metabolism to promote longevity and healthspan. Exp. Gerontol. 132, 110841. doi:10.1016/j.exger.2020.110841
Chen, C., Wang, T., Gao, T.-Y., Chen, Y.-L., Lu, Y.-B., and Zhang, W.-P. (2024). Ablation of NAMPT in dopaminergic neurons leads to neurodegeneration and induces Parkinson’s disease in mouse. Brain Res. Bull. 218, 111114. doi:10.1016/j.brainresbull.2024.111114
Cheng, A., Tse, K.-H., Chow, H.-M., Gan, Y., Song, X., Ma, F., et al. (2021). ATM loss disrupts the autophagy-lysosomal pathway. Autophagy 17, 1998–2010. doi:10.1080/15548627.2020.1805860
Collyer, J., and Rajan, D. S. (2024). Ataxia telangiectasia. Semin. Pediatr. Neurol. 52, 101169. doi:10.1016/j.spen.2024.101169
Cougnoux, A., Cluzeau, C., Mitra, S., Li, R., Williams, I., Burkert, K., et al. (2016). Necroptosis in Niemann-Pick disease, type C1: a potential therapeutic target. Cell Death Dis. 7, e2147. doi:10.1038/cddis.2016.16
Covarrubias, A. J., Perrone, R., Grozio, A., and Verdin, E. (2021). NAD+ metabolism and its roles in cellular processes during ageing. Nat. Rev. Mol. Cell Biol. 22, 119–141. doi:10.1038/s41580-020-00313-x
Cuervo, A. M., Stefanis, L., Fredenburg, R., Lansbury, P. T., and Sulzer, D. (2004). Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 305, 1292–1295. doi:10.1126/science.1101738
Cummins, N., Tweedie, A., Zuryn, S., Bertran-Gonzalez, J., and Götz, J. (2019). Disease-associated tau impairs mitophagy by inhibiting Parkin translocation to mitochondria. EMBO J. 38, e99360. doi:10.15252/embj.201899360
Davis, O. B., Shin, H. R., Lim, C.-Y., Wu, E. Y., Kukurugya, M., Maher, C. F., et al. (2021). NPC1-mTORC1 signaling couples cholesterol sensing to organelle homeostasis and is a targetable pathway in Niemann-Pick type C. Dev. Cell 56, 260–276.e7. doi:10.1016/j.devcel.2020.11.016
Decressac, M., Mattsson, B., Weikop, P., Lundblad, M., Jakobsson, J., and Björklund, A. (2013). TFEB-mediated autophagy rescues midbrain dopamine neurons from α-synuclein toxicity. Proc. Natl. Acad. Sci. U. S. A. 110, E1817–E1826. doi:10.1073/pnas.1305623110
Delgado-Camprubi, M., Esteras, N., Soutar, M. P., Plun-Favreau, H., and Abramov, A. Y. (2017). Deficiency of Parkinson’s disease-related gene Fbxo7 is associated with impaired mitochondrial metabolism by PARP activation. Cell Death Differ. 24, 120–131. doi:10.1038/cdd.2016.104
Deng, Z., Zhou, X., Lu, J.-H., and Yue, Z. (2021). Autophagy deficiency in neurodevelopmental disorders. Cell Biosci. 11, 214. doi:10.1186/s13578-021-00726-x
Deri, E., Kumar Ojha, S., Kartawy, M., Khaliulin, I., and Amal, H. (2025). Multi-omics study reveals differential expression and phosphorylation of autophagy-related proteins in autism spectrum disorder. Sci. Rep. 15, 10878. doi:10.1038/s41598-025-95860-8
Desquiret-Dumas, V., Gueguen, N., Leman, G., Baron, S., Nivet-Antoine, V., Chupin, S., et al. (2013). Resveratrol induces a mitochondrial complex I-dependent increase in NADH oxidation responsible for sirtuin activation in liver cells. J. Biol. Chem. 288, 36662–36675. doi:10.1074/jbc.M113.466490
Dölle, C., and Tzoulis, C. (2025). NAD augmentation as a disease-modifying strategy for neurodegeneration. Trends Endocrinol. Metab. Tem. S1043-2760 (25), 00070. doi:10.1016/j.tem.2025.03.013
Dorszewska, J., Prendecki, M., Oczkowska, A., Dezor, M., and Kozubski, W. (2016). Molecular basis of familial and sporadic alzheimer’s disease. Curr. Alzheimer Res. 13, 952–963. doi:10.2174/1567205013666160314150501
Du, J., Liang, Y., Xu, F., Sun, B., and Wang, Z. (2013). Trehalose rescues Alzheimer’s disease phenotypes in APP/PS1 transgenic mice. J. Pharm. Pharmacol. 65, 1753–1756. doi:10.1111/jphp.12108
Erickson, R. P., and Bernard, O. (2002). Studies on neuronal death in the mouse model of Niemann-Pick C disease. J. Neurosci. Res. 68, 738–744. doi:10.1002/jnr.10257
Essa, M. M., Braidy, N., Waly, M. I., Al-Farsi, Y. M., Al-Sharbati, M., Subash, S., et al. (2013a). Impaired antioxidant status and reduced energy metabolism in autistic children. Res. Autism Spectr. Disord. 7, 557–565. doi:10.1016/j.rasd.2012.12.006
Essa, M. M., Subash, S., Braidy, N., Al-Adawi, S., Lim, C. K., Manivasagam, T., et al. (2013b). Role of NAD(+), oxidative stress, and tryptophan metabolism in autism spectrum disorders. Int. J. Tryptophan Res. IJTR 6, 15–28. doi:10.4137/IJTR.S11355
Fang, E. F., Scheibye-Knudsen, M., Brace, L. E., Kassahun, H., SenGupta, T., Nilsen, H., et al. (2014). Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell 157, 882–896. doi:10.1016/j.cell.2014.03.026
Fang, E. F., Kassahun, H., Croteau, D. L., Scheibye-Knudsen, M., Marosi, K., Lu, H., et al. (2016). NAD+ replenishment improves lifespan and healthspan in Ataxia Telangiectasia models via mitophagy and DNA repair. Cell Metab. 24, 566–581. doi:10.1016/j.cmet.2016.09.004
Fang, E. F., Hou, Y., Lautrup, S., Jensen, M. B., Yang, B., SenGupta, T., et al. (2019a). NAD+ augmentation restores mitophagy and limits accelerated aging in Werner syndrome. Nat. Commun. 10, 5284. doi:10.1038/s41467-019-13172-8
Fang, E. F., Hou, Y., Palikaras, K., Adriaanse, B. A., Kerr, J. S., Yang, B., et al. (2019b). Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 22, 401–412. doi:10.1038/s41593-018-0332-9
Ferretta, A., Gaballo, A., Tanzarella, P., Piccoli, C., Capitanio, N., Nico, B., et al. (2014). Effect of resveratrol on mitochondrial function: implications in parkin-associated familiar Parkinson’s disease. Biochim. Biophys. Acta 1842, 902–915. doi:10.1016/j.bbadis.2014.02.010
Figley, M. D., Gu, W., Nanson, J. D., Shi, Y., Sasaki, Y., Cunnea, K., et al. (2021). SARM1 is a metabolic sensor activated by an increased NMN/NAD+ ratio to trigger axon degeneration. Neuron 109, 1118–1136.e11. doi:10.1016/j.neuron.2021.02.009
Flores-Leon, M., and Outeiro, T. F. (2023). More than meets the eye in Parkinson’s disease and other synucleinopathies: from proteinopathy to lipidopathy. Acta Neuropathol. (Berl.) 146, 369–385. doi:10.1007/s00401-023-02601-0
Fog, C. K., and Kirkegaard, T. (2019). Animal models for Niemann-Pick type C: implications for drug discovery and development. Expert Opin. Drug Discov. 14, 499–509. doi:10.1080/17460441.2019.1588882
Freitag, K., Sterczyk, N., Wendlinger, S., Obermayer, B., Schulz, J., Farztdinov, V., et al. (2022). Spermidine reduces neuroinflammation and soluble amyloid beta in an Alzheimer’s disease mouse model. J. Neuroinflammation 19, 172. doi:10.1186/s12974-022-02534-7
Fu, Y., Wu, P., Pan, Y., Sun, X., Yang, H., Difiglia, M., et al. (2017). A toxic mutant huntingtin species is resistant to selective autophagy. Nat. Chem. Biol. 13, 1152–1154. doi:10.1038/nchembio.2461
Gan-Or, Z., Dion, P. A., and Rouleau, G. A. (2015). Genetic perspective on the role of the autophagy-lysosome pathway in Parkinson disease. Autophagy 11, 1443–1457. doi:10.1080/15548627.2015.1067364
Gerdts, J., Brace, E. J., Sasaki, Y., DiAntonio, A., and Milbrandt, J. (2015). SARM1 activation triggers axon degeneration locally via NAD+ destruction. Science 348, 453–457. doi:10.1126/science.1258366
Gómez-Suaga, P., Bravo-San Pedro, J. M., González-Polo, R. A., Fuentes, J. M., and Niso-Santano, M. (2018). ER-mitochondria signaling in Parkinson’s disease. Cell Death Dis. 9, 337. doi:10.1038/s41419-017-0079-3
Gong, B., Pan, Y., Vempati, P., Zhao, W., Knable, L., Ho, L., et al. (2013). Nicotinamide riboside restores cognition through an upregulation of proliferator-activated receptor-γ coactivator 1α regulated β-secretase 1 degradation and mitochondrial gene expression in Alzheimer’s mouse models. Neurobiol. Aging 34, 1581–1588. doi:10.1016/j.neurobiolaging.2012.12.005
Green, K. N., Steffan, J. S., Martinez-Coria, H., Sun, X., Schreiber, S. S., Thompson, L. M., et al. (2008). Nicotinamide restores cognition in Alzheimer’s disease transgenic mice via a mechanism involving sirtuin inhibition and selective reduction of Thr231-phosphotau. J. Neurosci. 28, 11500–11510. doi:10.1523/JNEUROSCI.3203-08.2008
Grill, J. D., Tam, S., Thai, G., Vides, B., Pierce, A. L., Green, K., et al. (2025). Phase 2A proof-of-concept Double-Blind, randomized, placebo-controlled trial of nicotinamide in early Alzheimer disease. Neurology 104, e210152. doi:10.1212/WNL.0000000000210152
Ham, A., Chang, A. Y., Li, H., Bain, J. M., Goldman, J. E., Sulzer, D., et al. (2025). Impaired macroautophagy confers substantial risk for intellectual disability in children with autism spectrum disorders. Mol. Psychiatry 30, 810–824. doi:10.1038/s41380-024-02741-z
Hara, T., Nakamura, K., Matsui, M., Yamamoto, A., Nakahara, Y., Suzuki-Migishima, R., et al. (2006). Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885–889. doi:10.1038/nature04724
Hastings, C., Liu, B., Hurst, B., Cox, G. F., and Hrynkow, S. (2022). Intravenous 2-hydroxypropyl-β-cyclodextrin (Trappsol® CycloTM) demonstrates biological activity and impacts cholesterol metabolism in the central nervous system and peripheral tissues in adult subjects with Niemann-Pick disease type C1: results of a phase 1 trial. Mol. Genet. Metab. 137, 309–319. doi:10.1016/j.ymgme.2022.10.004
Hathorn, T., Snyder-Keller, A., and Messer, A. (2011). Nicotinamide improves motor deficits and upregulates PGC-1α and BDNF gene expression in a mouse model of Huntington’s disease. Neurobiol. Dis. 41, 43–50. doi:10.1016/j.nbd.2010.08.017
He, L., Wang, J., Yang, Y., Li, J., and Tu, H. (2022). Mitochondrial sirtuins in Parkinson’s disease. Neurochem. Res. 47, 1491–1502. doi:10.1007/s11064-022-03560-w
Henrich, M. T., Oertel, W. H., Surmeier, D. J., and Geibl, F. F. (2023). Mitochondrial dysfunction in Parkinson’s disease - a key disease hallmark with therapeutic potential. Mol. Neurodegener. 18, 83. doi:10.1186/s13024-023-00676-7
Hou, Y., Lautrup, S., Cordonnier, S., Wang, Y., Croteau, D. L., Zavala, E., et al. (2018). NAD+ supplementation normalizes key Alzheimer’s features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency. Proc. Natl. Acad. Sci. U. S. A. 115, E1876–E1885. doi:10.1073/pnas.1718819115
Hou, X., Watzlawik, J. O., Fiesel, F. C., and Springer, W. (2020). Autophagy in Parkinson’s disease. J. Mol. Biol. 432, 2651–2672. doi:10.1016/j.jmb.2020.01.037
Hou, Y., Wei, Y., Lautrup, S., Yang, B., Wang, Y., Cordonnier, S., et al. (2021). NAD+ supplementation reduces neuroinflammation and cell senescence in a transgenic mouse model of Alzheimer’s disease via cGAS-STING. Proc. Natl. Acad. Sci. U. S. A. 118, e2011226118. doi:10.1073/pnas.2011226118
Houtkooper, R. H., Pirinen, E., and Auwerx, J. (2012). Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol. 13, 225–238. doi:10.1038/nrm3293
Howarth, C., Gleeson, P., and Attwell, D. (2012). Updated energy budgets for neural computation in the neocortex and cerebellum. J. Cereb. Blood Flow. Metab. 32, 1222–1232. doi:10.1038/jcbfm.2012.35
Huber, K. M., Klann, E., Costa-Mattioli, M., and Zukin, R. S. (2015). Dysregulation of Mammalian target of Rapamycin signaling in mouse models of autism. J. Neurosci. 35, 13836–13842. doi:10.1523/JNEUROSCI.2656-15.2015
Hui, K. K., Takashima, N., Watanabe, A., Chater, T. E., Matsukawa, H., Nekooki-Machida, Y., et al. (2019). GABARAPs dysfunction by autophagy deficiency in adolescent brain impairs GABAA receptor trafficking and social behavior. Sci. Adv. 5, eaau8237. doi:10.1126/sciadv.aau8237
Hung, C. O. Y., and Livesey, F. J. (2018). Altered γ-Secretase processing of APP disrupts lysosome and autophagosome function in monogenic alzheimer’s disease. Cell Rep. 25, 3647–3660.e2. doi:10.1016/j.celrep.2018.11.095
Hwang, M., Jun, D. W., Song, B. R., Shim, H., Lee, C.-H., and Kim, S. (2023). Ataxia-telangiectasia mutated is involved in autolysosome Formation. Biomol. Ther. 31, 559–565. doi:10.4062/biomolther.2023.003
Ionescu-Tucker, A., and Cotman, C. W. (2021). Emerging roles of oxidative stress in brain aging and Alzheimer’s disease. Neurobiol. Aging 107, 86–95. doi:10.1016/j.neurobiolaging.2021.07.014
Jia, H., Li, X., Gao, H., Feng, Z., Li, X., Zhao, L., et al. (2008). High doses of nicotinamide prevent oxidative mitochondrial dysfunction in a cellular model and improve motor deficit in a Drosophila model of Parkinson’s disease. J. Neurosci. Res. 86, 2083–2090. doi:10.1002/jnr.21650
Jiang, T., Yu, J.-T., Zhu, X.-C., Tan, M.-S., Wang, H.-F., Cao, L., et al. (2014a). Temsirolimus promotes autophagic clearance of amyloid-β and provides protective effects in cellular and animal models of Alzheimer’s disease. Pharmacol. Res. 81, 54–63. doi:10.1016/j.phrs.2014.02.008
Jiang, T., Yu, J.-T., Zhu, X.-C., Zhang, Q.-Q., Cao, L., Wang, H.-F., et al. (2014b). Temsirolimus attenuates tauopathy in vitro and in vivo by targeting tau hyperphosphorylation and autophagic clearance. Neuropharmacology 85, 121–130. doi:10.1016/j.neuropharm.2014.05.032
Kalia, L. V., and Kalia, S. K. (2015). α-Synuclein and Lewy pathology in Parkinson’s disease. Curr. Opin. Neurol. 28, 375–381. doi:10.1097/WCO.0000000000000215
Kałuzna-Czaplinska, J., Michalska, M., and Rynkowski, J. (2010). Determination of tryptophan in urine of autistic and healthy children by gas chromatography/mass spectrometry. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 16, CR488–CR492.
Kataura, T., Sedlackova, L., Otten, E. G., Kumari, R., Shapira, D., Scialo, F., et al. (2022). Autophagy promotes cell survival by maintaining NAD levels. Dev. Cell 57, 2584–2598.e11. doi:10.1016/j.devcel.2022.10.008
Kataura, T., Sedlackova, L., Sun, C., Kocak, G., Wilson, N., Banks, P., et al. (2024). Targeting the autophagy-NAD axis protects against cell death in Niemann-Pick type C1 disease models. Cell Death Dis. 15, 382. doi:10.1038/s41419-024-06770-y
Katsyuba, E., Mottis, A., Zietak, M., De Franco, F., van der Velpen, V., Gariani, K., et al. (2018). De novo NAD+ synthesis enhances mitochondrial function and improves health. Nature 563, 354–359. doi:10.1038/s41586-018-0645-6
Katsyuba, E., Romani, M., Hofer, D., and Auwerx, J. (2020). NAD+ homeostasis in health and disease. Nat. Metab. 2, 9–31. doi:10.1038/s42255-019-0161-5
Kim, H.-J., Cho, M.-H., Shim, W. H., Kim, J. K., Jeon, E.-Y., Kim, D.-H., et al. (2017). Deficient autophagy in microglia impairs synaptic pruning and causes social behavioral defects. Mol. Psychiatry 22, 1576–1584. doi:10.1038/mp.2016.103
Kim, A., Lalonde, K., Truesdell, A., Gomes Welter, P., Brocardo, P. S., Rosenstock, T. R., et al. (2021). New avenues for the treatment of Huntington’s disease. Int. J. Mol. Sci. 22, 8363. doi:10.3390/ijms22168363
Knopman, D. S., Amieva, H., Petersen, R. C., Chételat, G., Holtzman, D. M., Hyman, B. T., et al. (2021). Alzheimer disease. Nat. Rev. Dis. Primer 7, 33. doi:10.1038/s41572-021-00269-y
Kolotyeva, N. A., Groshkov, A. A., Rozanova, N. A., Berdnikov, A. K., Novikova, S. V., Komleva, Y. K., et al. (2024). Pathobiochemistry of aging and neurodegeneration: deregulation of NAD+ metabolism in brain cells. Biomolecules 14, 1556. doi:10.3390/biom14121556
Komatsu, M., Waguri, S., Chiba, T., Murata, S., Iwata, J., Tanida, I., et al. (2006). Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441, 880–884. doi:10.1038/nature04723
Krüger, U., Wang, Y., Kumar, S., and Mandelkow, E.-M. (2012). Autophagic degradation of tau in primary neurons and its enhancement by trehalose. Neurobiol. Aging 33, 2291–2305. doi:10.1016/j.neurobiolaging.2011.11.009
Kuo, S.-Y., Castoreno, A. B., Aldrich, L. N., Lassen, K. G., Goel, G., Dančík, V., et al. (2015). Small-molecule enhancers of autophagy modulate cellular disease phenotypes suggested by human genetics. Proc. Natl. Acad. Sci. U. S. A. 112, E4281–E4287. doi:10.1073/pnas.1512289112
Las Heras, M., Szenfeld, B., Ballout, R. A., Buratti, E., Zanlungo, S., Dardis, A., et al. (2023). Understanding the phenotypic variability in Niemann-Pick disease type C (NPC): a need for precision medicine. NPJ Genomic Med. 8, 21. doi:10.1038/s41525-023-00365-w
Launay, J.-M., Delorme, R., Pagan, C., Callebert, J., Leboyer, M., and Vodovar, N. (2023). Impact of IDO activation and alterations in the kynurenine pathway on hyperserotonemia, NAD+ production, and AhR activation in autism spectrum disorder. Transl. Psychiatry 13, 380. doi:10.1038/s41398-023-02687-w
Lautrup, S., Sinclair, D. A., Mattson, M. P., and Fang, E. F. (2019). NAD+ in brain aging and neurodegenerative disorders. Cell Metab. 30, 630–655. doi:10.1016/j.cmet.2019.09.001
Lee, J.-H., and Paull, T. T. (2021). Cellular functions of the protein kinase ATM and their relevance to human disease. Nat. Rev. Mol. Cell Biol. 22, 796–814. doi:10.1038/s41580-021-00394-2
Lee, I. H., Cao, L., Mostoslavsky, R., Lombard, D. B., Liu, J., Bruns, N. E., et al. (2008). A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. U. S. A. 105, 3374–3379. doi:10.1073/pnas.0712145105
Lee, J.-H., Yu, W. H., Kumar, A., Lee, S., Mohan, P. S., Peterhoff, C. M., et al. (2010). Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 141, 1146–1158. doi:10.1016/j.cell.2010.05.008
Lee, M. J., Lee, J. H., and Rubinsztein, D. C. (2013). Tau degradation: the ubiquitin-proteasome system versus the autophagy-lysosome system. Prog. Neurobiol. 105, 49–59. doi:10.1016/j.pneurobio.2013.03.001
Lee, H., Lee, J. K., Park, M. H., Hong, Y. R., Marti, H. H., Kim, H., et al. (2014). Pathological roles of the VEGF/SphK pathway in Niemann-Pick type C neurons. Nat. Commun. 5, 5514. doi:10.1038/ncomms6514
Lee, J.-H., McBrayer, M. K., Wolfe, D. M., Haslett, L. J., Kumar, A., Sato, Y., et al. (2015). Presenilin 1 maintains lysosomal Ca(2+) homeostasis via TRPML1 by regulating vATPase-mediated lysosome acidification. Cell Rep. 12, 1430–1444. doi:10.1016/j.celrep.2015.07.050
Lee, J.-H., Wolfe, D. M., Darji, S., McBrayer, M. K., Colacurcio, D. J., Kumar, A., et al. (2020a). β2-adrenergic agonists rescue lysosome acidification and function in PSEN1 deficiency by reversing defective ER-to-lysosome delivery of ClC-7. J. Mol. Biol. 432, 2633–2650. doi:10.1016/j.jmb.2020.02.021
Lee, S.-E., Shin, N., Kook, M. G., Kong, D., Kim, N. G., Choi, S. W., et al. (2020b). Human iNSC-derived brain organoid model of lysosomal storage disorder in Niemann-Pick disease type C. Cell Death Dis. 11, 1059. doi:10.1038/s41419-020-03262-7
Leeson, H. C., Aguado, J., Gómez-Inclán, C., Chaggar, H. K., Fard, A. T., Hunter, Z., et al. (2024). Ataxia Telangiectasia patient-derived neuronal and brain organoid models reveal mitochondrial dysfunction and oxidative stress. Neurobiol. Dis. 199, 106562. doi:10.1016/j.nbd.2024.106562
Lehmann, S., Costa, A. C., Celardo, I., Loh, S. H. Y., and Martins, L. M. (2016). Parp mutations protect against mitochondrial dysfunction and neurodegeneration in a PARKIN model of Parkinson’s disease. Cell Death Dis. 7, e2166. doi:10.1038/cddis.2016.72
Lehmann, S., Loh, S. H. Y., and Martins, L. M. (2017). Enhancing NAD+ salvage metabolism is neuroprotective in a PINK1 model of Parkinson’s disease. Biol. Open 6, bio.022186–147. doi:10.1242/bio.022186
Lengyel-Zhand, Z., Puentes, L. N., and Mach, R. H. (2022). PARkinson’s: from cellular mechanisms to potential therapeutics. Pharmacol. Ther. 230, 107968. doi:10.1016/j.pharmthera.2021.107968
Li, L., Zhang, S., Zhang, X., Li, T., Tang, Y., Liu, H., et al. (2013). Autophagy enhancer carbamazepine alleviates memory deficits and cerebral amyloid-β pathology in a mouse model of Alzheimer’s disease. Curr. Alzheimer Res. 10, 433–441. doi:10.2174/1567205011310040008
Li, X., Lu, F., Trinh, M. N., Schmiege, P., Seemann, J., Wang, J., et al. (2017). 3.3 Å structure of Niemann-Pick C1 protein reveals insights into the function of the C-terminal luminal domain in cholesterol transport. Proc. Natl. Acad. Sci. U. S. A. 114, 9116–9121. doi:10.1073/pnas.1711716114
Lieberman, O. J., Cartocci, V., Pigulevskiy, I., Molinari, M., Carbonell, J., Broseta, M. B., et al. (2020). mTOR suppresses macroautophagy during striatal postnatal development and is hyperactive in mouse models of autism spectrum disorders. Front. Cell. Neurosci. 14, 70. doi:10.3389/fncel.2020.00070
Lim, C. K., Fernández-Gomez, F. J., Braidy, N., Estrada, C., Costa, C., Costa, S., et al. (2017). Involvement of the kynurenine pathway in the pathogenesis of Parkinson’s disease. Prog. Neurobiol. 155, 76–95. doi:10.1016/j.pneurobio.2015.12.009
Liu, D., Pitta, M., Jiang, H., Lee, J.-H., Zhang, G., Chen, X., et al. (2013a). Nicotinamide forestalls pathology and cognitive decline in Alzheimer mice: evidence for improved neuronal bioenergetics and autophagy procession. Neurobiol. Aging 34, 1564–1580. doi:10.1016/j.neurobiolaging.2012.11.020
Liu, K., Shi, N., Sun, Y., Zhang, T., and Sun, X. (2013b). Therapeutic effects of rapamycin on MPTP-induced Parkinsonism in mice. Neurochem. Res. 38, 201–207. doi:10.1007/s11064-012-0909-8
Lloret, A., and Beal, M. F. (2019). PGC-1α, sirtuins and PARPs in Huntington’s disease and other neurodegenerative conditions: NAD+ to rule them all. Neurochem. Res. 44, 2423–2434. doi:10.1007/s11064-019-02809-1
Lloyd-Evans, E., Morgan, A. J., He, X., Smith, D. A., Elliot-Smith, E., Sillence, D. J., et al. (2008). Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat. Med. 14, 1247–1255. doi:10.1038/nm.1876
Lord, C., Brugha, T. S., Charman, T., Cusack, J., Dumas, G., Frazier, T., et al. (2020). Autism spectrum disorder. Nat. Rev. Dis. Primer 6, 5. doi:10.1038/s41572-019-0138-4
Lu, L., Tang, L. E., Wei, W., Hong, Y., Chen, H., Ying, W., et al. (2014). Nicotinamide mononucleotide improves energy activity and survival rate in an in vitro model of Parkinson’s disease. Exp. Ther. Med. 8, 943–950. doi:10.3892/etm.2014.1842
Lundt, S., and Ding, S. (2021). NAD+ metabolism and diseases with motor dysfunction. Genes 12, 1776. doi:10.3390/genes12111776
Lynch-Day, M. A., Mao, K., Wang, K., Zhao, M., and Klionsky, D. J. (2012). The role of autophagy in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2, a009357. doi:10.1101/cshperspect.a009357
Ma, R.-Y., Li, L., Yang, H., Zou, B., Ma, R.-X., Zhang, Y., et al. (2024). Therapeutic effect of nicotinamide mononucleotide on Alzheimer’s disease through activating autophagy and anti-oxidative stress. Biomed. Pharmacother. Biomedecine Pharmacother. 178, 117199. doi:10.1016/j.biopha.2024.117199
Madureira, M., Connor-Robson, N., and Wade-Martins, R. (2020). LRRK2: autophagy and lysosomal activity. Front. Neurosci. 14, 498. doi:10.3389/fnins.2020.00498
Maetzel, D., Sarkar, S., Wang, H., Abi-Mosleh, L., Xu, P., Cheng, A. W., et al. (2014). Genetic and chemical correction of cholesterol accumulation and impaired autophagy in hepatic and neural cells derived from Niemann-Pick Type C patient-specific iPS cells. Stem Cell Rep. 2, 866–880. doi:10.1016/j.stemcr.2014.03.014
Majumder, S., Richardson, A., Strong, R., and Oddo, S. (2011). Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS One 6, e25416. doi:10.1371/journal.pone.0025416
Malagelada, C., Jin, Z. H., Jackson-Lewis, V., Przedborski, S., and Greene, L. A. (2010). Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson’s disease. J. Neurosci. 30, 1166–1175. doi:10.1523/JNEUROSCI.3944-09.2010
Mao, K., and Zhang, G. (2022). The role of PARP1 in neurodegenerative diseases and aging. FEBS J. 289, 2013–2024. doi:10.1111/febs.15716
Martin, D. D. O., Ladha, S., Ehrnhoefer, D. E., and Hayden, M. R. (2015). Autophagy in Huntington disease and huntingtin in autophagy. Trends Neurosci. 38, 26–35. doi:10.1016/j.tins.2014.09.003
Martínez-Reyes, I., and Chandel, N. S. (2020). Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 11, 102. doi:10.1038/s41467-019-13668-3
Martinez-Vicente, M., Talloczy, Z., Kaushik, S., Massey, A. C., Mazzulli, J., Mosharov, E. V., et al. (2008). Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J. Clin. Invest. 118, 777–788. doi:10.1172/JCI32806
Martinez-Vicente, M., Talloczy, Z., Wong, E., Tang, G., Koga, H., Kaushik, S., et al. (2010). Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat. Neurosci. 13, 567–576. doi:10.1038/nn.2528
Martire, S., Fuso, A., Mosca, L., Forte, E., Correani, V., Fontana, M., et al. (2016). Bioenergetic impairment in animal and cellular models of Alzheimer’s disease: PARP-1 inhibition rescues metabolic dysfunctions. J. Alzheimers Dis. JAD 54, 307–324. doi:10.3233/JAD-151040
Mary, A., Eysert, F., Checler, F., and Chami, M. (2023). Mitophagy in Alzheimer’s disease: molecular defects and therapeutic approaches. Mol. Psychiatry 28, 202–216. doi:10.1038/s41380-022-01631-6
Mehramiz, M., Porter, T., O’Brien, E. K., Rainey-Smith, S. R., and Laws, S. M. (2023). A potential role for Sirtuin-1 in Alzheimer’s disease: reviewing the biological and environmental evidence. J. Alzheimers Dis. Rep. 7, 823–843. doi:10.3233/ADR-220088
Meng, Y. Y., Wu, C. W., Yu, B., Li, H., Chen, M., and Qi, G. X. (2018). PARP-1 involvement in autophagy and their roles in apoptosis of vascular smooth muscle cells under oxidative stress. Folia Biol. (Praha) 64, 103–111. doi:10.14712/fb2018064030103
Mengel, E., Patterson, M. C., Da Riol, R. M., Del Toro, M., Deodato, F., Gautschi, M., et al. (2021). Efficacy and safety of arimoclomol in Niemann-Pick disease type C: results from a double-blind, randomised, placebo-controlled, multinational phase 2/3 trial of a novel treatment. J. Inherit. Metab. Dis. 44, 1463–1480. doi:10.1002/jimd.12428
Menzies, F. M., Fleming, A., and Rubinsztein, D. C. (2015). Compromised autophagy and neurodegenerative diseases. Nat. Rev. Neurosci. 16, 345–357. doi:10.1038/nrn3961
Menzies, F. M., Fleming, A., Caricasole, A., Bento, C. F., Andrews, S. P., Ashkenazi, A., et al. (2017). Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron 93, 1015–1034. doi:10.1016/j.neuron.2017.01.022
Min, S.-W., Cho, S.-H., Zhou, Y., Schroeder, S., Haroutunian, V., Seeley, W. W., et al. (2010). Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 67, 953–966. doi:10.1016/j.neuron.2010.08.044
Misgeld, T., and Schwarz, T. L. (2017). Mitostasis in neurons: maintaining mitochondria in an extended cellular Architecture. Neuron 96, 651–666. doi:10.1016/j.neuron.2017.09.055
Muñoz-Gámez, J. A., Rodríguez-Vargas, J. M., Quiles-Pérez, R., Aguilar-Quesada, R., Martín-Oliva, D., de Murcia, G., et al. (2009). PARP-1 is involved in autophagy induced by DNA damage. Autophagy 5, 61–74. doi:10.4161/auto.5.1.7272
Narendra, D. P., and Youle, R. J. (2024). The role of PINK1-Parkin in mitochondrial quality control. Nat. Cell Biol. 26, 1639–1651. doi:10.1038/s41556-024-01513-9
Neu, M., Parfentev, I., Potts, G. K., Stöberl, N., Korffmann, J., Striebinger, A., et al. (2025). A role for the autophagy receptor NBR1 in the degradation of tau aggregates. Neurobiol. Dis. 214, 107060. doi:10.1016/j.nbd.2025.107060
Ng, F., and Tang, B. L. (2013). Sirtuins’ modulation of autophagy. J. Cell. Physiol. 228, 2262–2270. doi:10.1002/jcp.24399
Nikiforov, A., Dölle, C., Niere, M., and Ziegler, M. (2011). Pathways and subcellular compartmentation of NAD biosynthesis in human cells: from entry of extracellular precursors to mitochondrial NAD generation. J. Biol. Chem. 286, 21767–21778. doi:10.1074/jbc.M110.213298
Nilsson, P., Loganathan, K., Sekiguchi, M., Matsuba, Y., Hui, K., Tsubuki, S., et al. (2013). Aβ secretion and plaque formation depend on autophagy. Cell Rep. 5, 61–69. doi:10.1016/j.celrep.2013.08.042
Nixon, R. A. (2024). Autophagy-lysosomal-associated neuronal death in neurodegenerative disease. Acta Neuropathol. (Berl.) 148, 42. doi:10.1007/s00401-024-02799-7
Nixon, R. A., and Rubinsztein, D. C. (2024). Mechanisms of autophagy-lysosome dysfunction in neurodegenerative diseases. Nat. Rev. Mol. Cell Biol. 25, 926–946. doi:10.1038/s41580-024-00757-5
Nixon, R. A., and Yang, D.-S. (2011). Autophagy failure in Alzheimer’s disease--locating the primary defect. Neurobiol. Dis. 43, 38–45. doi:10.1016/j.nbd.2011.01.021
Nowicka, U., Chroscicki, P., Stroobants, K., Sladowska, M., Turek, M., Uszczynska-Ratajczak, B., et al. (2021). Cytosolic aggregation of mitochondrial proteins disrupts cellular homeostasis by stimulating the aggregation of other proteins. eLife 10, e65484. doi:10.7554/eLife.65484
Ochaba, J., Lukacsovich, T., Csikos, G., Zheng, S., Margulis, J., Salazar, L., et al. (2014). Potential function for the Huntingtin protein as a scaffold for selective autophagy. Proc. Natl. Acad. Sci. U. S. A. 111, 16889–16894. doi:10.1073/pnas.1420103111
Oh, Y. M., Lee, S. W., Kim, W. K., Chen, S., Church, V. A., Cates, K., et al. (2022). Age-related Huntington’s disease progression modeled in directly reprogrammed patient-derived striatal neurons highlights impaired autophagy. Nat. Neurosci. 25, 1420–1433. doi:10.1038/s41593-022-01185-4
Orenstein, S. J., Kuo, S.-H., Tasset, I., Arias, E., Koga, H., Fernandez-Carasa, I., et al. (2013). Interplay of LRRK2 with chaperone-mediated autophagy. Nat. Neurosci. 16, 394–406. doi:10.1038/nn.3350
Orr, M. E., Kotkowski, E., Ramirez, P., Bair-Kelps, D., Liu, Q., Brenner, C., et al. (2024). A randomized placebo-controlled trial of nicotinamide riboside in older adults with mild cognitive impairment. GeroScience 46, 665–682. doi:10.1007/s11357-023-00999-9
Ozcelik, S., Fraser, G., Castets, P., Schaeffer, V., Skachokova, Z., Breu, K., et al. (2013). Rapamycin attenuates the progression of tau pathology in P301S tau transgenic mice. PLoS One 8, e62459. doi:10.1371/journal.pone.0062459
Paldino, E., Cardinale, A., D’Angelo, V., Sauve, I., Giampà, C., and Fusco, F. R. (2017). Selective sparing of striatal interneurons after poly (ADP-Ribose) polymerase 1 inhibition in the R6/2 mouse model of Huntington’s disease. Front. Neuroanat. 11, 61. doi:10.3389/fnana.2017.00061
Palhegyi, A. M., Seranova, E., Dimova, S., Hoque, S., and Sarkar, S. (2019). Biomedical implications of autophagy in macromolecule storage disorders. Front. Cell Dev. Biol. 7, 179. doi:10.3389/fcell.2019.00179
Pallos, J., Bodai, L., Lukacsovich, T., Purcell, J. M., Steffan, J. S., Thompson, L. M., et al. (2008). Inhibition of specific HDACs and sirtuins suppresses pathogenesis in a Drosophila model of Huntington’s disease. Hum. Mol. Genet. 17, 3767–3775. doi:10.1093/hmg/ddn273
Palmer, J. E., Wilson, N., Son, S. M., Obrocki, P., Wrobel, L., Rob, M., et al. (2025). Autophagy, aging, and age-related neurodegeneration. Neuron 113, 29–48. doi:10.1016/j.neuron.2024.09.015
Patterson, M. C., Clayton, P., Gissen, P., Anheim, M., Bauer, P., Bonnot, O., et al. (2017). Recommendations for the detection and diagnosis of Niemann-Pick disease type C: an update. Neurol. Clin. Pract. 7, 499–511. doi:10.1212/CPJ.0000000000000399
Peavy, G. M., Jacobson, M. W., Goldstein, J. L., Hamilton, J. M., Kane, A., Gamst, A. C., et al. (2010). Cognitive and functional decline in Huntington’s disease: dementia criteria revisited. Mov. Disord. 25, 1163–1169. doi:10.1002/mds.22953
Pérez, M. J., Baden, P., and Deleidi, M. (2021). Progresses in both basic research and clinical trials of NAD+ in Parkinson’s disease. Mech. Ageing Dev. 197, 111499. doi:10.1016/j.mad.2021.111499
Pickles, S., Vigié, P., and Youle, R. J. (2018). Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr. Biol. CB 28, R170–R185. doi:10.1016/j.cub.2018.01.004
Pickrell, A. M., and Youle, R. J. (2015). The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 85, 257–273. doi:10.1016/j.neuron.2014.12.007
Pineda, M., Walterfang, M., and Patterson, M. C. (2018). Miglustat in Niemann-Pick disease type C patients: a review. Orphanet J. Rare Dis. 13, 140. doi:10.1186/s13023-018-0844-0
Piras, A., Collin, L., Grüninger, F., Graff, C., and Rönnbäck, A. (2016). Autophagic and lysosomal defects in human tauopathies: analysis of post-mortem brain from patients with familial Alzheimer disease, corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol. Commun. 4, 22. doi:10.1186/s40478-016-0292-9
Pircs, K., Drouin-Ouellet, J., Horváth, V., Gil, J., Rezeli, M., Garza, R., et al. (2022). Distinct subcellular autophagy impairments in induced neurons from patients with Huntington’s disease. Brain J. Neurol. 145, 3035–3057. doi:10.1093/brain/awab473
Poewe, W., Seppi, K., Tanner, C. M., Halliday, G. M., Brundin, P., Volkmann, J., et al. (2017). Parkinson disease. Nat. Rev. Dis. Primer 3, 17013. doi:10.1038/nrdp.2017.13
Portbury, S. D., Hare, D. J., Sgambelloni, C., Perronnes, K., Portbury, A. J., Finkelstein, D. I., et al. (2017). Trehalose improves cognition in the transgenic Tg2576 mouse model of Alzheimer’s disease. J. Alzheimers Dis. JAD 60, 549–560. doi:10.3233/JAD-170322
Presterud, R., Deng, W. H., Wennerström, A. B., Burgers, T., Gajera, B., Mattsson, K., et al. (2024). Long-term nicotinamide riboside use improves coordination and eye movements in Ataxia Telangiectasia. Mov. Disord. Off. J. Mov. Disord. Soc. 39, 360–369. doi:10.1002/mds.29645
Pupyshev, A. B., Tikhonova, M. A., Akopyan, A. A., Tenditnik, M. V., Dubrovina, N. I., and Korolenko, T. A. (2019). Therapeutic activation of autophagy by combined treatment with rapamycin and trehalose in a mouse MPTP-induced model of Parkinson’s disease. Pharmacol. Biochem. Behav. 177, 1–11. doi:10.1016/j.pbb.2018.12.005
Qin, W., Yang, T., Ho, L., Zhao, Z., Wang, J., Chen, L., et al. (2006). Neuronal SIRT1 activation as a novel mechanism underlying the prevention of Alzheimer disease amyloid neuropathology by calorie restriction. J. Biol. Chem. 281, 21745–21754. doi:10.1074/jbc.M602909200
Rabanal-Ruiz, Y., Otten, E. G., and Korolchuk, V. I. (2017). mTORC1 as the main gateway to autophagy. Essays Biochem. 61, 565–584. doi:10.1042/EBC20170027
Raichle, M. E., and Gusnard, D. A. (2002). Appraising the brain’s energy budget. Proc. Natl. Acad. Sci. U. S. A. 99, 10237–10239. doi:10.1073/pnas.172399499
Rangaraju, V., Lewis, T. L., Hirabayashi, Y., Bergami, M., Motori, E., Cartoni, R., et al. (2019). Pleiotropic mitochondria: the influence of mitochondria on neuronal development and disease. J. Neurosci. 39, 8200–8208. doi:10.1523/JNEUROSCI.1157-19.2019
Ravikumar, B., Vacher, C., Berger, Z., Davies, J. E., Luo, S., Oroz, L. G., et al. (2004). Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 36, 585–595. doi:10.1038/ng1362
Ray Chaudhuri, A., and Nussenzweig, A. (2017). The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 18, 610–621. doi:10.1038/nrm.2017.53
Rehman, I. U., Ahmad, R., Khan, I., Lee, H. J., Park, J., Ullah, R., et al. (2021). Nicotinamide ameliorates amyloid beta-induced oxidative stress-mediated neuroinflammation and neurodegeneration in adult Mouse brain. Biomedicines 9, 408. doi:10.3390/biomedicines9040408
Rodríguez-Martín, T., Cuchillo-Ibáñez, I., Noble, W., Nyenya, F., Anderton, B. H., and Hanger, D. P. (2013). Tau phosphorylation affects its axonal transport and degradation. Neurobiol. Aging 34, 2146–2157. doi:10.1016/j.neurobiolaging.2013.03.015
Rodríguez-Navarro, J. A., Rodríguez, L., Casarejos, M. J., Solano, R. M., Gómez, A., Perucho, J., et al. (2010). Trehalose ameliorates dopaminergic and tau pathology in parkin deleted/tau overexpressing mice through autophagy activation. Neurobiol. Dis. 39, 423–438. doi:10.1016/j.nbd.2010.05.014
Romani, M., Sorrentino, V., Oh, C.-M., Li, H., de Lima, T. I., Zhang, H., et al. (2021). NAD+ boosting reduces age-associated amyloidosis and restores mitochondrial homeostasis in muscle. Cell Rep. 34, 108660. doi:10.1016/j.celrep.2020.108660
Roos, R. A. C. (2010). Huntington’s disease: a clinical review. Orphanet J. Rare Dis. 5, 40. doi:10.1186/1750-1172-5-40
Rose, C., Menzies, F. M., Renna, M., Acevedo-Arozena, A., Corrochano, S., Sadiq, O., et al. (2010). Rilmenidine attenuates toxicity of polyglutamine expansions in a mouse model of Huntington’s disease. Hum. Mol. Genet. 19, 2144–2153. doi:10.1093/hmg/ddq093
Ruan, L., Wang, Y., Zhang, X., Tomaszewski, A., McNamara, J. T., and Li, R. (2020). Mitochondria-associated proteostasis. Annu. Rev. Biophys. 49, 41–67. doi:10.1146/annurev-biophys-121219-081604
Rui, Y.-N., Xu, Z., Patel, B., Chen, Z., Chen, D., Tito, A., et al. (2015). Huntingtin functions as a scaffold for selective macroautophagy. Nat. Cell Biol. 17, 262–275. doi:10.1038/ncb3101
Ryu, Y.-K., Park, H.-Y., Go, J., Choi, D.-H., Kim, Y.-H., Hwang, J. H., et al. (2018). Metformin inhibits the development of L-DOPA-Induced dyskinesia in a murine model of parkinson’s disease. Mol. Neurobiol. 55, 5715–5726. doi:10.1007/s12035-017-0752-7
Sanchez-Mirasierra, I., Ghimire, S., Hernandez-Diaz, S., and Soukup, S.-F. (2022). Targeting macroautophagy as a therapeutic opportunity to treat parkinson’s disease. Front. Cell Dev. Biol. 10, 921314. doi:10.3389/fcell.2022.921314
Sarkar, S., and Rubinsztein, D. C. (2008). Huntington’s disease: degradation of mutant huntingtin by autophagy. FEBS J. 275, 4263–4270. doi:10.1111/j.1742-4658.2008.06562.x
Sarkar, S., Perlstein, E. O., Imarisio, S., Pineau, S., Cordenier, A., Maglathlin, R. L., et al. (2007). Small molecules enhance autophagy and reduce toxicity in Huntington’s disease models. Nat. Chem. Biol. 3, 331–338. doi:10.1038/nchembio883
Sarkar, S., Carroll, B., Buganim, Y., Maetzel, D., Ng, A. H. M., Cassady, J. P., et al. (2013). Impaired autophagy in the lipid-storage disorder Niemann-Pick type C1 disease. Cell Rep. 5, 1302–1315. doi:10.1016/j.celrep.2013.10.042
Sasaki, Y., Nakagawa, T., Mao, X., DiAntonio, A., and Milbrandt, J. (2016). NMNAT1 inhibits axon degeneration via blockade of SARM1-mediated NAD+ depletion. eLife 5, e19749. doi:10.7554/eLife.19749
Schaeffer, V., Lavenir, I., Ozcelik, S., Tolnay, M., Winkler, D. T., and Goedert, M. (2012). Stimulation of autophagy reduces neurodegeneration in a mouse model of human tauopathy. Brain J. Neurol. 135, 2169–2177. doi:10.1093/brain/aws143
Schmauck-Medina, T., Molière, A., Lautrup, S., Zhang, J., Chlopicki, S., Madsen, H. B., et al. (2022). New hallmarks of ageing: a 2022 Copenhagen ageing meeting summary. Aging 14, 6829–6839. doi:10.18632/aging.204248
Schöndorf, D. C., Ivanyuk, D., Baden, P., Sanchez-Martinez, A., De Cicco, S., Yu, C., et al. (2018). The NAD+ precursor nicotinamide riboside rescues mitochondrial defects and neuronal loss in iPSC and fly models of parkinson’s disease. Cell Rep. 23, 2976–2988. doi:10.1016/j.celrep.2018.05.009
Sedlackova, L., and Korolchuk, V. I. (2020). The crosstalk of NAD, ROS and autophagy in cellular health and ageing. Biogerontology 21, 381–397. doi:10.1007/s10522-020-09864-0
Seranova, E., Connolly, K. J., Zatyka, M., Rosenstock, T. R., Barrett, T., Tuxworth, R. I., et al. (2017). Dysregulation of autophagy as a common mechanism in lysosomal storage diseases. Essays Biochem. 61, 733–749. doi:10.1042/EBC20170055
Seranova, E., Palhegyi, A. M., Verma, S., Dimova, S., Lasry, R., Naama, M., et al. (2020). Human induced pluripotent stem cell models of neurodegenerative disorders for studying the biomedical implications of autophagy. J. Mol. Biol. 432, 2754–2798. doi:10.1016/j.jmb.2020.01.024
Shan, C., Gong, Y.-L., Zhuang, Q.-Q., Hou, Y.-F., Wang, S.-M., Zhu, Q., et al. (2019). Protective effects of β-nicotinamide adenine dinucleotide against motor deficits and dopaminergic neuronal damage in a mouse model of Parkinson’s disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 94, 109670. doi:10.1016/j.pnpbp.2019.109670
Sharma, A., Behl, T., Sharma, L., Aelya, L., and Bungau, S. (2021). Mitochondrial dysfunction in Huntington’s disease: pathogenesis and therapeutic opportunities. Curr. Drug Targets 22, 1637–1667. doi:10.2174/1389450122666210224105945
Shen, C., Chen, C., Wang, T., Gao, T.-Y., Zeng, M., Lu, Y.-B., et al. (2023). The depletion of NAMPT disturbs mitochondrial homeostasis and causes neuronal degeneration in Mouse Hippocampus. Mol. Neurobiol. 60, 1267–1280. doi:10.1007/s12035-022-03142-5
Siddiqi, F. H., Menzies, F. M., Lopez, A., Stamatakou, E., Karabiyik, C., Ureshino, R., et al. (2019). Felodipine induces autophagy in mouse brains with pharmacokinetics amenable to repurposing. Nat. Commun. 10, 1817. doi:10.1038/s41467-019-09494-2
Sidorova-Darmos, E., Sommer, R., and Eubanks, J. H. (2018). The role of SIRT3 in the brain under physiological and pathological conditions. Front. Cell. Neurosci. 12, 196. doi:10.3389/fncel.2018.00196
Sorrentino, V., Romani, M., Mouchiroud, L., Beck, J. S., Zhang, H., D’Amico, D., et al. (2017). Enhancing mitochondrial proteostasis reduces amyloid-β proteotoxicity. Nature 552, 187–193. doi:10.1038/nature25143
Spilman, P., Podlutskaya, N., Hart, M. J., Debnath, J., Gorostiza, O., Bredesen, D., et al. (2010). Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer’s disease. PLoS One 5, e9979. doi:10.1371/journal.pone.0009979
Stagni, V., Cirotti, C., and Barilà, D. (2018). Ataxia-Telangiectasia mutated kinase in the control of oxidative stress, Mitochondria, and autophagy in cancer: a maestro with a large orchestra. Front. Oncol. 8, 73. doi:10.3389/fonc.2018.00073
Stagni, V., Ferri, A., Cirotti, C., and Barilà, D. (2020). ATM kinase-dependent regulation of autophagy: a key player in senescence? Front. Cell Dev. Biol. 8, 599048. doi:10.3389/fcell.2020.599048
Stavoe, A. K. H., and Holzbaur, E. L. F. (2019). Autophagy in neurons. Annu. Rev. Cell Dev. Biol. 35, 477–500. doi:10.1146/annurev-cellbio-100818-125242
Steinbrücker, K., Tiefenthaler, E., Schernthaner, E.-M., Jungwirth, J., and Wortmann, S. B. (2023). Nicotinamide riboside for Ataxia Telangiectasia: a report of an early treated individual. Neuropediatrics 54, 78–81. doi:10.1055/a-1959-9404
Strosznajder, J. B., Czapski, G. A., Adamczyk, A., and Strosznajder, R. P. (2012). Poly(ADP-ribose) polymerase-1 in amyloid beta toxicity and Alzheimer’s disease. Mol. Neurobiol. 46, 78–84. doi:10.1007/s12035-012-8258-9
Sun, C., Seranova, E., Cohen, M. A., Chipara, M., Roberts, J., Astuti, D., et al. (2023). NAD depletion mediates cytotoxicity in human neurons with autophagy deficiency. Cell Rep. 42, 112372. doi:10.1016/j.celrep.2023.112372
Sunderland, P., Augustyniak, J., Lenart, J., Bużańska, L., Carlessi, L., Delia, D., et al. (2020). ATM-deficient neural precursors develop senescence phenotype with disturbances in autophagy. Mech. Ageing Dev. 190, 111296. doi:10.1016/j.mad.2020.111296
Suresh, S. N., Chavalmane, A. K., Dj, V., Yarreiphang, H., Rai, S., Paul, A., et al. (2017). A novel autophagy modulator 6-Bio ameliorates SNCA/α-synuclein toxicity. Autophagy 13, 1221–1234. doi:10.1080/15548627.2017.1302045
Tang, G., Gudsnuk, K., Kuo, S.-H., Cotrina, M. L., Rosoklija, G., Sosunov, A., et al. (2014). Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 83, 1131–1143. doi:10.1016/j.neuron.2014.07.040
Tanik, S. A., Schultheiss, C. E., Volpicelli-Daley, L. A., Brunden, K. R., and Lee, V. M. Y. (2013). Lewy body-like α-synuclein aggregates resist degradation and impair macroautophagy. J. Biol. Chem. 288, 15194–15210. doi:10.1074/jbc.M113.457408
Tarragó, M. G., Chini, C. C. S., Kanamori, K. S., Warner, G. M., Caride, A., de Oliveira, G. C., et al. (2018). A potent and specific CD38 inhibitor ameliorates age-related metabolic dysfunction by reversing tissue NAD+ decline. Cell Metab. 27, 1081–1095.e10. doi:10.1016/j.cmet.2018.03.016
Tian, Y., Chang, J. C., Fan, E. Y., Flajolet, M., and Greengard, P. (2013). Adaptor complex AP2/PICALM, through interaction with LC3, targets Alzheimer’s APP-CTF for terminal degradation via autophagy. Proc. Natl. Acad. Sci. U. S. A. 110, 17071–17076. doi:10.1073/pnas.1315110110
Valentin-Vega, Y. A., Maclean, K. H., Tait-Mulder, J., Milasta, S., Steeves, M., Dorsey, F. C., et al. (2012). Mitochondrial dysfunction in ataxia-telangiectasia. Blood 119, 1490–1500. doi:10.1182/blood-2011-08-373639
van der Velpen, V., Rosenberg, N., Maillard, V., Teav, T., Chatton, J.-Y., Gallart-Ayala, H., et al. (2021). Sex-specific alterations in NAD+ metabolism in 3xTg Alzheimer’s disease mouse brain assessed by quantitative targeted LC-MS. J. Neurochem. 159, 378–388. doi:10.1111/jnc.15362
Van Laar, V. S., and Berman, S. B. (2013). The interplay of neuronal mitochondrial dynamics and bioenergetics: implications for Parkinson’s disease. Neurobiol. Dis. 51, 43–55. doi:10.1016/j.nbd.2012.05.015
Vanier, M. T., Duthel, S., Rodriguez-Lafrasse, C., Pentchev, P., and Carstea, E. D. (1996). Genetic heterogeneity in Niemann-Pick C disease: a study using somatic cell hybridization and linkage analysis. Am. J. Hum. Genet. 58, 118–125.
Vargas, J. N. S., Hamasaki, M., Kawabata, T., Youle, R. J., and Yoshimori, T. (2023). The mechanisms and roles of selective autophagy in mammals. Nat. Rev. Mol. Cell Biol. 24, 167–185. doi:10.1038/s41580-022-00542-2
Veenhuis, S. J. G., van Os, N. J. H., Janssen, A. J. W. M., van Gerven, M. H. J. C., Coene, K. L. M., Engelke, U. F. H., et al. (2021). Nicotinamide riboside improves ataxia scores and immunoglobulin levels in ataxia Telangiectasia. Mov. Disord. 36, 2951–2957. doi:10.1002/mds.28788
Vilaça, R., Silva, E., Nadais, A., Teixeira, V., Matmati, N., Gaifem, J., et al. (2014). Sphingolipid signalling mediates mitochondrial dysfunctions and reduced chronological lifespan in the yeast model of Niemann-Pick type C1. Mol. Microbiol. 91, 438–451. doi:10.1111/mmi.12470
Virág, L., Robaszkiewicz, A., Rodriguez-Vargas, J. M., and Oliver, F. J. (2013). Poly(ADP-ribose) signaling in cell death. Mol. Asp. Med. 34, 1153–1167. doi:10.1016/j.mam.2013.01.007
Vreones, M., Mustapic, M., Moaddel, R., Pucha, K. A., Lovett, J., Seals, D. R., et al. (2023). Oral nicotinamide riboside raises NAD+ and lowers biomarkers of neurodegenerative pathology in plasma extracellular vesicles enriched for neuronal origin. Aging Cell 22, e13754. doi:10.1111/acel.13754
Wang, Y., Martinez-Vicente, M., Krüger, U., Kaushik, S., Wong, E., Mandelkow, E.-M., et al. (2009). Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum. Mol. Genet. 18, 4153–4170. doi:10.1093/hmg/ddp367
Wang, X., Zhang, Q., Bao, R., Zhang, N., Wang, Y., Polo-Parada, L., et al. (2017). Deletion of nampt in projection neurons of adult mice leads to motor dysfunction, neurodegeneration, and death. Cell Rep. 20, 2184–2200. doi:10.1016/j.celrep.2017.08.022
Wang, C., Xu, W., Zhang, Y., Zhang, F., and Huang, K. (2018). PARP1 promote autophagy in cardiomyocytes via modulating FoxO3a transcription. Cell Death Dis. 9, 1047. doi:10.1038/s41419-018-1108-6
Wang, C., Yang, Y., Zhang, Y., Liu, J., Yao, Z., and Zhang, C. (2019a). Protective effects of metformin against osteoarthritis through upregulation of SIRT3-mediated PINK1/Parkin-dependent mitophagy in primary chondrocytes. Biosci. Trends 12, 605–612. doi:10.5582/bst.2018.01263
Wang, X., Zhang, Z., Zhang, N., Li, H., Zhang, L., Baines, C. P., et al. (2019b). Subcellular NAMPT-mediated NAD+ salvage pathways and their roles in bioenergetics and neuronal protection after ischemic injury. J. Neurochem. 151, 732–748. doi:10.1111/jnc.14878
Wang, W., Zhao, F., Ma, X., Perry, G., and Zhu, X. (2020). Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: recent advances. Mol. Neurodegener. 15, 30. doi:10.1186/s13024-020-00376-6
Wang, X., He, H.-J., Xiong, X., Zhou, S., Wang, W.-W., Feng, L., et al. (2021). NAD+ in Alzheimer’s disease: molecular mechanisms and systematic therapeutic evidence obtained in vivo. Front. Cell Dev. Biol. 9, 668491. doi:10.3389/fcell.2021.668491
Wei, Y., Miao, Q., Zhang, Q., Mao, S., Li, M., Xu, X., et al. (2023). Aerobic glycolysis is the predominant means of glucose metabolism in neuronal somata, which protects against oxidative damage. Nat. Neurosci. 26, 2081–2089. doi:10.1038/s41593-023-01476-4
Williams, A., Sarkar, S., Cuddon, P., Ttofi, E. K., Saiki, S., Siddiqi, F. H., et al. (2008). Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat. Chem. Biol. 4, 295–305. doi:10.1038/nchembio.79
Wilson, N., Kataura, T., Korsgen, M. E., Sun, C., Sarkar, S., and Korolchuk, V. I. (2023). The autophagy-NAD axis in longevity and disease. Trends Cell Biol. 33, 788–802. doi:10.1016/j.tcb.2023.02.004
Winslow, A. R., Chen, C.-W., Corrochano, S., Acevedo-Arozena, A., Gordon, D. E., Peden, A. A., et al. (2010). α-Synuclein impairs macroautophagy: implications for Parkinson’s disease. J. Cell Biol. 190, 1023–1037. doi:10.1083/jcb.201003122
Wong, Y. C., and Holzbaur, E. L. F. (2014). The regulation of autophagosome dynamics by huntingtin and HAP1 is disrupted by expression of mutant huntingtin, leading to defective cargo degradation. J. Neurosci. 34, 1293–1305. doi:10.1523/JNEUROSCI.1870-13.2014
Woś, M., Szczepanowska, J., Pikuła, S., Tylki-Szymańska, A., Zabłocki, K., and Bandorowicz-Pikuła, J. (2016). Mitochondrial dysfunction in fibroblasts derived from patients with Niemann-Pick type C disease. Arch. Biochem. Biophys. 593, 50–59. doi:10.1016/j.abb.2016.02.012
Wu, S., Wei, Y., Li, J., Bai, Y., Yin, P., and Wang, S. (2021). SIRT5 represses neurotrophic pathways and Aβ production in Alzheimer’s disease by targeting autophagy. ACS Chem. Neurosci. 12, 4428–4437. doi:10.1021/acschemneuro.1c00468
Wu, Q.-J., Zhang, T.-N., Chen, H.-H., Yu, X.-F., Lv, J.-L., Liu, Y.-Y., et al. (2022). The sirtuin family in health and disease. Signal Transduct. Target. Ther. 7, 402. doi:10.1038/s41392-022-01257-8
Xie, N., Zhang, L., Gao, W., Huang, C., Huber, P. E., Zhou, X., et al. (2020). NAD+ metabolism: pathophysiologic mechanisms and therapeutic potential. Signal Transduct. Target. Ther. 5, 227. doi:10.1038/s41392-020-00311-7
Xiong, X., Hou, J., Zheng, Y., Jiang, T., Zhao, X., Cai, J., et al. (2024). NAD+-boosting agent nicotinamide mononucleotide potently improves mitochondria stress response in Alzheimer’s disease via ATF4-dependent mitochondrial UPR. Cell Death Dis. 15, 744. doi:10.1038/s41419-024-07062-1
Yamamoto, A., Lucas, J. J., and Hen, R. (2000). Reversal of neuropathology and motor dysfunction in a conditional model of Huntington’s disease. Cell 101, 57–66. doi:10.1016/S0092-8674(00)80623-6
Yamamoto, H., Zhang, S., and Mizushima, N. (2023). Autophagy genes in biology and disease. Nat. Rev. Genet. 24, 382–400. doi:10.1038/s41576-022-00562-w
Yang, B., Dan, X., Hou, Y., Lee, J.-H., Wechter, N., Krishnamurthy, S., et al. (2021). NAD+ supplementation prevents STING-induced senescence in ataxia telangiectasia by improving mitophagy. Aging Cell 20, e13329. doi:10.1111/acel.13329
Yang, X., Zhou, P., Zhao, Z., Li, J., Fan, Z., Li, X., et al. (2023). Improvement effect of mitotherapy on the cognitive ability of alzheimer’s disease through NAD+/SIRT1-Mediated autophagy. Antioxid. Basel Switz. 12, 2006. doi:10.3390/antiox12112006
Yang, J., Yu, Z., Jiang, Y., Zhang, Z., Tian, Y., Cai, J., et al. (2024). SIRT3 alleviates painful diabetic neuropathy by mediating the FoxO3a-PINK1-Parkin signaling pathway to activate mitophagy. CNS Neurosci. Ther. 30, e14703. doi:10.1111/cns.14703
Yao, Z., Yang, W., Gao, Z., and Jia, P. (2017). Nicotinamide mononucleotide inhibits JNK activation to reverse Alzheimer disease. Neurosci. Lett. 647, 133–140. doi:10.1016/j.neulet.2017.03.027
Yoshino, J., Baur, J. A., and Imai, S.-I. (2018). NAD+ intermediates: the biology and therapeutic potential of NMN and NR. Cell Metab. 27, 513–528. doi:10.1016/j.cmet.2017.11.002
Yu, W., Gong, J.-S., Ko, M., Garver, W. S., Yanagisawa, K., and Michikawa, M. (2005a). Altered cholesterol metabolism in Niemann-Pick type C1 mouse brains affects mitochondrial function. J. Biol. Chem. 280, 11731–11739. doi:10.1074/jbc.M412898200
Yu, W. H., Cuervo, A. M., Kumar, A., Peterhoff, C. M., Schmidt, S. D., Lee, J.-H., et al. (2005b). Macroautophagy--a novel beta-amyloid peptide-generating pathway activated in Alzheimer’s disease. J. Cell Biol. 171, 87–98. doi:10.1083/jcb.200505082
Yu, W., Gao, B., Li, N., Wang, J., Qiu, C., Zhang, G., et al. (2017). Sirt3 deficiency exacerbates diabetic cardiac dysfunction: role of Foxo3A-Parkin-mediated mitophagy. Biochim. Biophys. Acta Mol. Basis Dis. 1863, 1973–1983. doi:10.1016/j.bbadis.2016.10.021
Yu, Y., Fedele, G., Celardo, I., Loh, S. H. Y., and Martins, L. M. (2021). Parp mutations protect from mitochondrial toxicity in Alzheimer’s disease. Cell Death Dis. 12, 651. doi:10.1038/s41419-021-03926-y
Zambon, F., Cherubini, M., Fernandes, H. J. R., Lang, C., Ryan, B. J., Volpato, V., et al. (2019). Cellular α-synuclein pathology is associated with bioenergetic dysfunction in Parkinson’s iPSC-derived dopamine neurons. Hum. Mol. Genet. 28, 2001–2013. doi:10.1093/hmg/ddz038
Zareba, J., Cattaneo, E. F., Villani, A., Othman, A., Streb, S., and Peri, F. (2024). NPC1 links cholesterol trafficking to microglial morphology via the gastrosome. Nat. Commun. 15, 8638. doi:10.1038/s41467-024-52874-6
Zhang, D.-X., Zhang, J.-P., Hu, J.-Y., and Huang, Y.-S. (2016a). The potential regulatory roles of NAD(+) and its metabolism in autophagy. Metabolism 65, 454–462. doi:10.1016/j.metabol.2015.11.010
Zhang, J., Zhang, J.-X., and Zhang, Q.-L. (2016b). PI3K/AKT/mTOR-mediated autophagy in the development of autism spectrum disorder. Brain Res. Bull. 125, 152–158. doi:10.1016/j.brainresbull.2016.06.007
Zhang, Z., Xu, H. N., Li, S., Jr, A. D., Chellappa, K., Davis, J. G., et al. (2020). Rapamycin maintains NAD+/NADH redox homeostasis in muscle cells. Aging 12, 17786–17799. doi:10.18632/aging.103954
Zhang, H., Wei, W., Zhao, M., Ma, L., Jiang, X., Pei, H., et al. (2021). Interaction between Aβ and Tau in the pathogenesis of alzheimer’s disease. Int. J. Biol. Sci. 17, 2181–2192. doi:10.7150/ijbs.57078
Zhang, J., Zhang, Y., Wang, J., Xia, Y., Zhang, J., and Chen, L. (2024). Recent advances in Alzheimer’s disease: mechanisms, clinical trials and new drug development strategies. Signal Transduct. Target. Ther. 9, 211. doi:10.1038/s41392-024-01911-3
Zheng, X., Boyer, L., Jin, M., Mertens, J., Kim, Y., Ma, L., et al. (2016). Metabolic reprogramming during neuronal differentiation from aerobic glycolysis to neuronal oxidative phosphorylation. eLife 5, e13374. doi:10.7554/eLife.13374
Zhu, Y., Li, C., Tao, X., Brazill, J. M., Park, J., Diaz-Perez, Z., et al. (2019). Nmnat restores neuronal integrity by neutralizing mutant Huntingtin aggregate-induced progressive toxicity. Proc. Natl. Acad. Sci. U. S. A. 116, 19165–19175. doi:10.1073/pnas.1904563116
Zoghbi, H. Y., and Orr, H. T. (2000). Glutamine repeats and neurodegeneration. Annu. Rev. Neurosci. 23, 217–247. doi:10.1146/annurev.neuro.23.1.217
Keywords: autophagy, NAD+, NAD+ precursor, NAD+-dependent enzyme, neuroprotection, neuronal cell death, autophagy inducer, NAD+ supplementation
Citation: Kocak G, Dimas MM, Silva LFSE, Silva-Amaral D, Sun C, Cruddas S, Barrett T, Martins-de-Souza D, Cunha-Neto E, Brocardo PS, Kataura T, Korolchuk VI and Sarkar S (2025) Autophagy–NAD+ axis: emerging insights into neuronal homeostasis and neurodegenerative diseases. Front. Mol. Biosci. 12:1695486. doi: 10.3389/fmolb.2025.1695486
Received: 29 August 2025; Accepted: 04 November 2025;
Published: 26 November 2025.
Edited by:
Francescopaolo Iavarone, Telethon Institute of Genetics and Medicine (TIGEM), ItalyReviewed by:
Wei Zhou, National Neuroscience Institute (NNI), SingaporeQuanjiang Zhang, University of California, Los Angeles, United States
Copyright © 2025 Kocak, Dimas, Silva, Silva-Amaral, Sun, Cruddas, Barrett, Martins-de-Souza, Cunha-Neto, Brocardo, Kataura, Korolchuk and Sarkar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sovan Sarkar, cy5zYXJrYXJAYmhhbS5hYy51aw==