 Jennifer Dang
Jennifer Dang Jia Yu Liou
Jia Yu Liou Rida Mullah
Rida Mullah Cecilia Giulivi
Cecilia Giulivi- 1Department of Molecular Biosciences, School of Veterinary Medicine, University of California Davis, Davis, CA, United States
- 2MIND Institute, University of California at Davis Medical Center, Sacramento, CA, United States
Introduction
Bruce Ames stands among the most influential biochemists of the past half-century, leaving a profound mark on genetics, toxicology, nutrition, and aging research. Best known for developing the Ames test, a bacterial assay that detects mutagenic chemicals, Ames helped establish the principle that environmental mutagens can be reliably measured and systematically screened (Ames, 1973). This transformed toxicology and cancer prevention, saving countless lives by enabling the regulation of carcinogens. Yet Ames’s legacy extends beyond mutagen detection. In the latter part of his career, he focused on oxidative stress and mitochondrial DNA (mtDNA) mutagenicity as central players in the biology of aging (Ames et al., 1993). While the foundational ideas of oxidative damage and mitochondrial contribution to aging were first proposed by researchers such as Gershman and Harman, Ames contributed by integrating these concepts with experimental observations on mitochondrial decay, nutrition, and disease. He highlighted how reactive oxygen species (ROS) produced during mitochondrial respiration could contribute to cumulative mtDNA damage, potentially impairing mitochondrial function over time. Ames also emphasized the role of micronutrients in modulating this process, proposing that suboptimal nutrition could accelerate oxidative damage and mitochondrial decline through his triage theory (Ames, 2006) (Figure 1). His work helped translate earlier theoretical ideas into a broader framework linking oxidative stress, mitochondrial function, and age-related health outcomes.
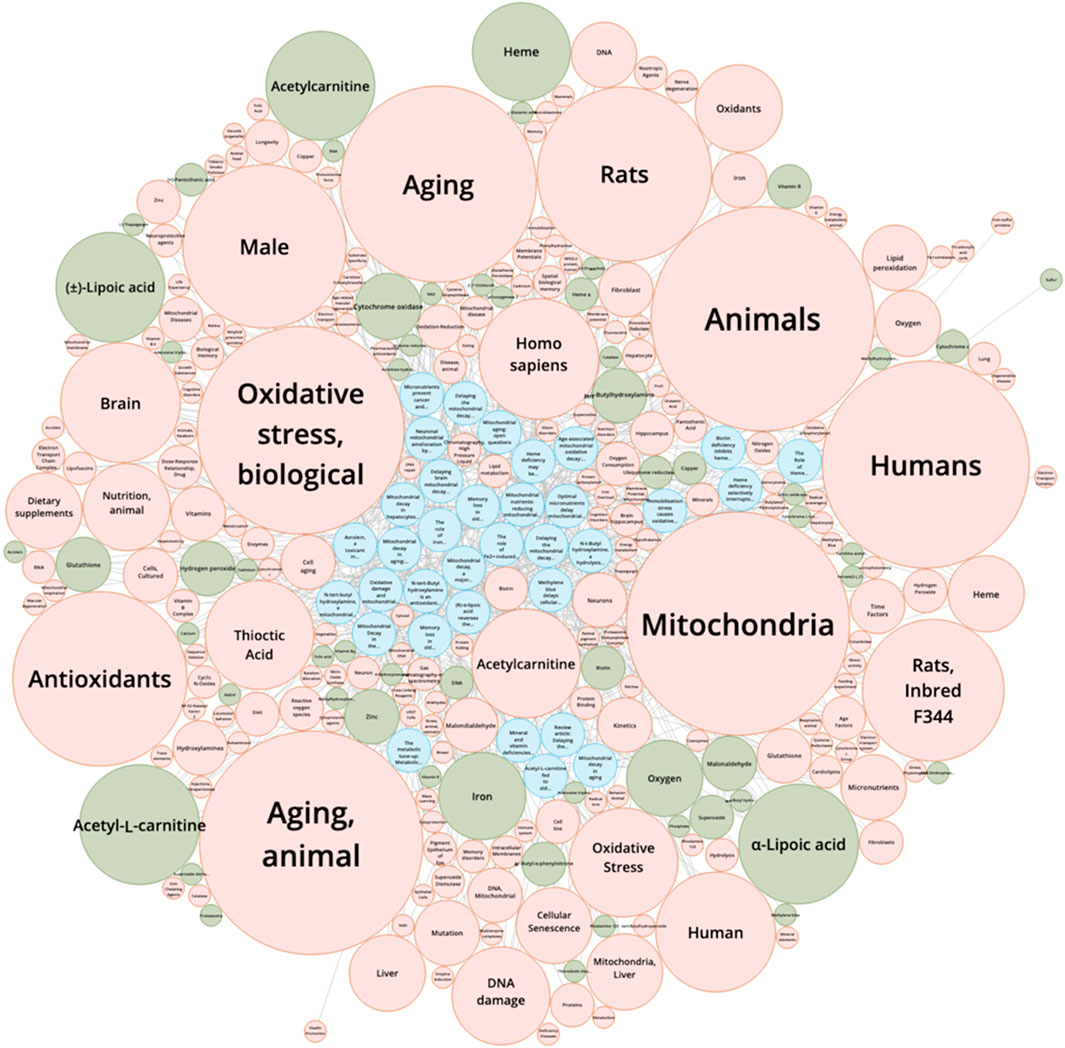
Figure 1. Knowledge Graph Mapping from References. This knowledge graph visualizes the interconnections between key references (articles only), chemical substances, and scientific concepts derived from a SciFinder literature search using the keywords Bruce Ames, mitochondria, and aging. Blue nodes represent primary references, highlighting seminal papers such as Ames (1973, 1993) and subsequent studies on mitochondrial mutagenicity and aging. Green nodes correspond to chemical substances, including antioxidants and micronutrients relevant to Ames’s triage theory and mitochondrial oxidative stress. Orange nodes indicate scientific concepts, such as oxidative stress and aging mechanisms. The edges connecting nodes depict relationships extracted from co-occurrence patterns in the literature, illustrating how Ames’s work intersects with chemical modulators and mechanistic pathways in mitochondrial biology. This figure provides an integrated overview of the interrelationships between studies on Bruce Ames, mitochondrial function, and aging, revealing clusters of high-impact references, commonly studied compounds, and central conceptual themes in the field. Bibliographic and chemical data used to generate this knowledge graph were obtained from SciFinder (CAS, American Chemical Society). The figure was redistributed with permission. Copyright © 2025 the American Chemical Society (ACS). All rights reserved.
While this concept was groundbreaking, the field has since evolved. Contemporary research has revealed a more complex picture, with some scientists challenging the causal role of mtDNA damage in aging, proposing instead that mitochondrial dysfunction may arise from signaling changes, metabolic imbalances, or programmed processes rather than cumulative mutations (Bratic and Larsson, 2013). This paper will examine Bruce Ames’s contributions, with particular focus on mtDNA mutagenicity, and critically assess how his theories shaped — and continue to spark debate within — modern biogerontology.
The Ames test and the assessment of mutagenicity
In the 1970s, Ames introduced a simple yet powerful tool for detecting chemical mutagens: a bacterial assay using strains of Salmonella typhimurium engineered to be highly sensitive to DNA mutations. If exposure to a chemical restored growth via human or rat liver-activated metabolism in these bacteria (via reversion mutations), the compound was considered mutagenic. Importantly, Ames demonstrated that many environmental chemicals, including industrial pollutants, food additives, and pesticides, scored positive in the assay — and many of these same compounds later proved carcinogenic in animals (Ames et al., 1973).
The test revolutionized toxicology by offering a rapid, inexpensive alternative to long-term rodent studies. Regulatory agencies worldwide adopted it to screen thousands of compounds, dramatically reducing public exposure to carcinogens. The “Ames test” also established a broader principle: genetic mutagenesis underpins carcinogenesis, and therefore, mutagens can be used as predictors of cancer risk. This work reflected Ames’s larger intellectual project: to bridge laboratory assays with real-world human health, from chemical safety to disease prevention.
A shift toward oxidative stress and mtDNA damage
By the 1980s, Ames expanded his focus from exogenous mutagens (environmental chemicals) to endogenous sources of DNA damage within the cell. He became especially interested in mitochondria, the cellular organelles that generate energy but also produce ROS as a byproduct of oxidative phosphorylation (Aliev et al., 2009; Ames, 1983; Ames, 1989; Ames, 2005; Ames, 2010; Ames, 2018; Ames et al., 1991; Ames et al., 1995; Atamna et al., 2002a; Atamna et al., 2001a; Atamna et al., 2007; Atamna et al., 2008; Atamna et al., 2000; Atamna et al., 2001b; Atamna et al., 2002b; Beckman and Ames, 1999; Chen et al., 1995; Gogvadze et al., 2003; Hagen et al., 1999; Hagen et al., 1998; Hagen et al., 2002; Hagen et al., 2000; Hagen et al., 1997; Helbock et al., 1998; Jia et al., 2007; Killilea and Ames, 2008; Killilea et al., 2003; Lal et al., 2008; Liu et al., 2002a; Liu et al., 2002b; Liu et al., 1996; Liu et al., 2000; Long et al., 2009; Lykkesfeldt et al., 1998; Milgram et al., 2007; Shenk et al., 2009; Voloboueva et al., 2007; Voloboueva et al., 2005; Walter et al., 2002; Walter et al., 2013; Yowe and Ames, 1998).
Unlike nuclear DNA, mtDNA is highly vulnerable. It is located near the electron transport chain, where ROS are generated, lacks protective histones, and has more limited repair mechanisms. Ames proposed that cumulative mtDNA mutations lead to a decline in mitochondrial efficiency, greater ROS leakage, and a vicious cycle of escalating damage (Ames et al., 1993). This “mitochondrial decay” hypothesis became central to his later research, with implications for cancer, neurodegeneration, cardiovascular disease, and aging.
Ames further argued that nutrition plays a key role in modulating this process (Aliev et al., 2009; Ames, 1983; Ames, 2010; Ames, 2018; Hagen et al., 1999; Hagen et al., 1998; Hagen et al., 2002; Hagen et al., 2000; Liu et al., 2002b; Long et al., 2009; Milgram et al., 2007; Shenk et al., 2009; Voloboueva et al., 2005). His “triage theory” suggested that when micronutrients (such as vitamins, minerals, and antioxidants) are limited, the body prioritizes short-term survival functions over long-term DNA maintenance. As a result, suboptimal nutrition accelerates oxidative damage, hastening mitochondrial decline and aging (Ames, 2006). An example of how modest micronutrient deficiencies can lead to long-term damage is illustrated in Ames’ work on iron deficiency. Iron, an essential trace mineral for iron-sulfur clusters and heme synthesis, enables complex function in the electron transport chain. Ames posits that when iron is limited, decreased heme and iron-sulfur cluster availability impairs electron transport and causes mitochondrial uncoupling and superoxide release (Walter et al., 2002). If iron deficiency is persistent over time, the resulting ROS leads to ongoing oxidative stress, mtDNA damage, and mitochondrial decline associated with age. Subsequently, if iron deficiency is exacerbated, the preceding symptoms lead to iron deficiency anemia (Pau et al., 2017). This framework, applied to other micronutrients, has elevated nutrition and metabolism as key determinants of genomic stability and longevity.
The work of Dr. Ames can be cross-referenced with that of our team, which has also extensively studied mitochondrial dysfunction in the context of aging and neurodegeneration. However, our focus is more disease-specific. While Dr. Ames emphasized systemic mitochondrial decay and the role of oxidative damage in aging and age-related diseases (Ames et al., 1993), our research explores mitochondrial bioenergetics, redox balance, and apoptotic pathways in long-lived or post-mitotic cells, particularly neurons, in conditions such as Huntington’s disease, fragile X-associated tremor/ataxia syndrome, and schizophrenia (Giulivi et al., 2010; Napoli et al., 2016; Song et al., 2016; Napoli et al., 2013). Together, our works highlight the centrality of mitochondria in both age-related and disease-specific cellular decline. Ames’s systemic and nutritional perspective complements our team’s mechanistic and cell-specific insights, suggesting that interventions aimed at preserving mitochondrial function could benefit both healthy aging and the mitigation of neurodegenerative disorders.
Early theoretical and experimental links between mtDNA damage and aging
The idea that mitochondrial DNA mutations contribute to aging did not originate solely with Bruce Ames. Earlier work laid critical foundations. In the 1950s, Rebecca Gershman was the first to suggest that oxygen- and nitrogen-centered free radicals could play a role in biological damage, a radical departure from the prevailing view that oxygen was purely beneficial (Gerschman et al., 1954). Building on this, Denham Harman proposed the Free Radical Theory of Aging in 1956, later refining it in 1972 into the Mitochondrial Free Radical Theory of Aging (MFRTA). Harman argued that mitochondria are both generators and victims of reactive oxygen species, and that cumulative oxidative damage leads to cellular dysfunction and senescence (Harman, 1972). Ames and his colleagues played a significant role in developing and advancing the theory. In the mid-90s, Ames published research showing that mitochondrial function declines with age and that this decay could be mitigated in rats with certain micronutrients or antioxidants such as acetyl-L-carnitine, α-lipoic acid, and N-tert-butylhydroxylamine (Atamna et al., 2001b; Hagen et al., 2002; Shigenaga et al., 1994). While not the originator of the idea, Ames’s work provided crucial experimental evidence and added significant detail to the understanding of mitochondrial decline in aging.
Further experimental support for these ideas began to emerge in the 1980s. Pikó and colleagues (1988) provided some of the earliest evidence by reporting increased deletions in the mtDNA of aged rodents, linking mitochondrial genomic instability to aging phenotypes (Piko et al., 1988). This strengthened the hypothesis that mtDNA integrity was a critical factor in age-related decline.
More decisive evidence arrived in the early 2000s with the development of mtDNA mutator mice (Trifunovic et al., 2004). These mice carried mutations in the mitochondrial DNA polymerase gamma (Polg) that caused an accelerated accumulation of mtDNA mutations. The animals exhibited a progeroid syndrome — premature aging features such as hair loss, osteoporosis, and reduced lifespan. This was widely interpreted as proof that increased mtDNA mutation burden could drive aging, offering direct experimental validation of earlier theoretical models (Trifunovic et al., 2004).
Ames’s later work on oxidative stress and mitochondrial decay fit squarely within this trajectory. While Gershman and Harman framed the initial conceptual theories and studies, like Pikó’s and the mutator mice, provided experimental support, Ames helped integrate these ideas into a broader picture of mutagenesis, nutrition, and public health, making the subject accessible across disciplines.
Mitochondrial mutagenesis and the biology of aging
The idea that mtDNA damage drives aging resonated deeply within biogerontology. For decades, the “oxidative stress theory of aging” was one of the most widely accepted models of senescence. Numerous studies have appeared to support this: aged tissues exhibit higher levels of mtDNA mutations, dysfunctional mitochondria, and oxidative stress biomarkers. In animal models, interventions that boosted antioxidant defenses or enhanced mitochondrial function sometimes extended lifespan or delayed age-related decline (Shigenaga et al., 1994).
Ames’s work thus provided both a mechanistic hypothesis and a therapeutic rationale: reduce oxidative damage, protect mtDNA, and thereby slow aging. This vision helped catalyze entire subfields focused on antioxidants, caloric restriction mimetics, and mitochondrial-targeted therapies.
Challenges and current understanding of mitochondria in aging
Despite the compelling evidence that oxidative stress and mtDNA mutations contribute to cellular decline, more recent research has revealed that the relationship between mitochondria and aging is far more complex than initially proposed. For example, interventions with antioxidants, while effective at reducing oxidative markers, have generally failed to extend lifespan in both human or animal studies due to antioxidants disrupting crucial oxidant signaling (Ristow and Schmeisser, 2011). This suggests that reactive oxygen species (ROS) are not simply damaging agents; at low levels, they may play important signaling roles that promote cellular adaptation and survival, a concept now referred to as mitohormesis (Ristow and Schmeisser, 2011).
Although mtDNA mutations do accumulate with age, the actual mutation burden in many tissues remains relatively low (Larsson, 2010) and often insufficient to explain widespread age-related physiological decline. Some aged individuals maintain robust mitochondrial function despite the presence of detectable mtDNA mutations, suggesting that mitochondrial DNA damage alone is not the sole driver of aging (Larsson, 2010). Mouse models with artificially accelerated mtDNA mutations, such as Polg mutator mice, display progeroid phenotypes (Trifunovic et al., 2004). Still, the levels of mutation in these models far exceed those found in natural aging, indicating that these experimental systems may exaggerate the impact of mtDNA mutations (Trifunovic et al., 2004).
Current research has shifted toward a more nuanced view of mitochondrial aging, emphasizing that dysfunction arises from multiple interconnected mechanisms. Epigenetic regulation, altered signaling (e.g., NAD+/sirtuins), proteostasis, and changes in mitochondrial dynamics all contribute to aging more broadly than just impaired ATP production (Sun et al., 2016; Budinger and Chandel, 2025). With age, the balance between mitochondrial fission and fusion is disrupted, and defective mitochondria that should be cleared by mitophagy accumulate, driving cellular dysfunction. While mitochondrial function may decline in sedentary individuals, evidence suggests this decline is not inevitable, as maintaining an active lifestyle can preserve mitochondrial efficiency and energy metabolism.
Importantly, the effects of aging on mitochondrial function are highly cell-type and tissue-specific. A 2024 study (Ehinger et al., 2024) found only minor, largely insignificant changes in mitochondrial respiration in blood cells across the human lifespan. In contrast, other research examining long-lived, differentiated, or non-dividing cells—including neurons, cardiomyocytes, and skeletal muscle fibers—has documented substantial declines in mitochondrial function, including reduced ATP production, altered membrane potential, and impaired oxidative capacity (Chubanava et al., 2025; Peterson et al., 2012; Boengler et al., 2017; Johnson et al., 2013; Seo et al., 2016; Tocchi et al., 2015; Marzetti et al., 2013; Short et al., 2005; Sagar and Gustafsson, 2023; Li et al., 2023; Jeong et al., 2024; Chen et al., 2023; Grevendonk et al., 2021; Harper et al., 2021; Kamarulzaman and Makpol, 2025; Nuccio et al., 2024; Tepp et al., 2016; Wang et al., 2025; Zhang et al., 2025; Springer-Sapp et al., 2025; Hepple, 2014). These observations indicate that mitochondrial aging is not uniform throughout the body, and specific post-mitotic cells may be particularly susceptible to dysfunction.
Lifestyle factors, particularly physical activity, also play an essential role in modulating mitochondrial aging. Evidence suggests that many of the declines observed in sedentary individuals reflect reduced activity rather than inevitable aging itself. This distinction is underscored by findings in animal models, where dietary restrictions and moderate physical exercise enhanced mitochondrial activity and decreased ROS formation (Ristow and Schmeisser, 2011).
Taken together, these findings challenge the traditional view that oxidative damage and mtDNA mutations alone are the primary drivers of aging. Instead, mitochondrial dysfunction appears to be multifactorial, involving disrupted signaling, imbalanced dynamics, impaired quality control, lifestyle influences, and tissue- and cell-type specific vulnerabilities. This contemporary understanding complements and refines Ames’s original hypotheses, highlighting the complexity of mitochondrial biology and emphasizing that interventions targeting signaling pathways, network dynamics, and lifestyle factors may be more effective than strategies that focus exclusively on preventing oxidative damage or mutagenesis.
The enduring influence of Professor Bruce Ames
Even with these challenges, Ames’s influence remains profound. His work continues to shape scientific and public discourse in several ways:
• Methodological impact: The Ames test remains a staple in toxicology, underscoring its lasting contribution to public health.
• Conceptual legacy: By linking mutagenesis, mitochondrial biology, and nutrition, Ames helped frame aging as a mechanistic, molecular process rather than an inevitable, mysterious decline.
• Nutritional insights: His triage theory broadened how scientists think about micronutrient deficiencies, suggesting that even marginal deficits can accelerate long-term genomic instability (Ames, 2006).
• Catalyst for new theories: Even as the oxidative stress model is revised, the debates it inspired have fueled breakthroughs in mitohormesis, mitochondrial signaling, and metabolic regulation.
In this sense, the critiques of Ames’s model are not rejections but evolutions — refining the questions he first posed about damage, repair, and aging.
Conclusion
Bruce Ames’s career illustrates how a scientist’s influence can transcend specific findings. His Ames test forever changed the field of chemical safety and mutagen screening. At the same time, his later work on oxidative stress and mtDNA mutagenicity provided a powerful, albeit imperfect, framework for understanding aging. Although modern evidence challenges the view that accumulated mtDNA mutations are the central drivers of aging, Ames’s theories catalyzed decades of research that continue to shape biogerontology. Today, as scientists explore mitochondrial signaling, metabolic pathways, and the complex interplay between genetics and environment, they build on the foundation Ames helped create. His legacy is therefore not tied to a single test or theory, but to a larger vision: that careful mechanistic science, applied to the most fundamental processes of biology, can illuminate the path to better health and longer life.
Author contributions
JD: Writing – review and editing. JL: Writing – review and editing. RM: Writing – review and editing. CG: Writing – original draft, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. The study was supported by NIH (NS128751) and discretionary funds (CG). JD and RM are supported by the Bridges to Baccalaureate Program (5T34GM150440-02).
Conflict of interest
CG serves as an Editorial Board Member of Scientific Reports. She has received compensation as a Field Chief Editor for Frontiers in Molecular Biosciences and honoraria for participating in NIH peer review meetings.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that Generative AI was used in the creation of this manuscript. Artificial intelligence technology was used to improve readability and language (Grammarly).
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aliev, G., Liu, J., Shenk, J. C., Fischbach, K., Pacheco, G. J., Chen, S. G., et al. (2009). Neuronal mitochondrial amelioration by feeding acetyl-L-carnitine and lipoic acid to aged rats. J. Cell Mol. Med. 13 (2), 320–333. doi:10.1111/j.1582-4934.2008.00324.x
Ames, B. N. (1973). Carcinogens are mutagens: their detection and classification. Environ. Health Perspect. 6, 115–118. doi:10.1289/ehp.7306115
Ames, B. N. (1983). Dietary carcinogens and anticarcinogens. Oxygen radicals and degenerative diseases. Science. 221 (4617), 1256–1264. doi:10.1126/science.6351251
Ames, B. N. (1989). Endogenous oxidative DNA damage, aging, and cancer. Free Radic. Res. Commun. 7 (3-6), 121–128. doi:10.3109/10715768909087933
Ames, B. N. (2005). Increasing longevity by tuning up metabolism. To maximize human health and lifespan, scientists must abandon outdated models of micronutrients. EMBO Rep. 6 (Suppl. 1), S20–S24. doi:10.1038/sj.embor.7400426
Ames, B. N. (2006). Low micronutrient intake may accelerate the degenerative diseases of aging through allocation of scarce micronutrients by triage. Proc. Natl. Acad. Sci. U. S. A. 103 (47), 17589–17594. doi:10.1073/pnas.0608757103
Ames, B. N. (2010). Optimal micronutrients delay mitochondrial decay and age-associated diseases. Mech. Ageing Dev. 131 (7-8), 473–479. doi:10.1016/j.mad.2010.04.005
Ames, B. N. (2018). Prolonging healthy aging: longevity vitamins and proteins. Proc. Natl. Acad. Sci. U. S. A. 115 (43), 10836–10844. doi:10.1073/pnas.1809045115
Ames, B. N., Durston, W. E., Yamasaki, E., and Lee, F. D. (1973). Carcinogens are mutagens: a simple test system combining liver homogenates for activation and bacteria for detection. Proc. Natl. Acad. Sci. U. S. A. 70 (8), 2281–2285. doi:10.1073/pnas.70.8.2281
Ames, B. N., and Gold, L. S. (1991). Endogenous mutagens and the causes of aging and cancer. Mutat. Res. 250 (1-2), 3–16. doi:10.1016/0027-5107(91)90157-j
Ames, B. N., Shigenaga, M. K., and Hagen, T. M. (1993). Oxidants, antioxidants, and the degenerative diseases of aging. Proc. Natl. Acad. Sci. U. S. A. 90 (17), 7915–7922. doi:10.1073/pnas.90.17.7915
Ames, B. N., Shigenaga, M. K., and Hagen, T. M. (1995). Mitochondrial decay in aging. Biochim. Biophys. Acta 1271 (1), 165–170. doi:10.1016/0925-4439(95)00024-x
Atamna, H., Paler-Martinez, A., and Ames, B. N. (2000). N-t-butyl hydroxylamine, a hydrolysis product of alpha-phenyl-N-t-butyl nitrone, is more potent in delaying senescence in human lung fibroblasts. J. Biol. Chem. 275 (10), 6741–6748. doi:10.1074/jbc.275.10.6741
Atamna, H., Liu, J., and Ames, B. N. (2001a). Heme deficiency selectively interrupts assembly of mitochondrial complex IV in human fibroblasts: revelance to aging. J. Biol. Chem. 276 (51), 48410–48416. doi:10.1074/jbc.M108362200
Atamna, H., Robinson, C., Ingersoll, R., Elliott, H., and Ames, B. N. (2001b). N-t-Butyl hydroxylamine is an antioxidant that reverses age-related changes in mitochondria in vivo and in vitro. FASEB J. 15 (12), 2196–2204. doi:10.1096/fj.01-0134com
Atamna, H., Killilea, D. W., Killilea, A. N., and Ames, B. N. (2002a). Heme deficiency may be a factor in the mitochondrial and neuronal decay of aging. Proc. Natl. Acad. Sci. U. S. A. 99 (23), 14807–14812. doi:10.1073/pnas.192585799
Atamna, H., Walter, P. B., and Ames, B. N. (2002b). The role of heme and iron-sulfur clusters in mitochondrial biogenesis, maintenance, and decay with age. Arch. Biochem. Biophys. 397 (2), 345–353. doi:10.1006/abbi.2001.2671
Atamna, H., Newberry, J., Erlitzki, R., Schultz, C. S., and Ames, B. N. (2007). Biotin deficiency inhibits heme synthesis and impairs mitochondria in human lung fibroblasts. J. Nutr. 137 (1), 25–30. doi:10.1093/jn/137.1.25
Atamna, H., Nguyen, A., Schultz, C., Boyle, K., Newberry, J., Kato, H., et al. (2008). Methylene blue delays cellular senescence and enhances key mitochondrial biochemical pathways. FASEB J. 22 (3), 703–712. doi:10.1096/fj.07-9610com
Beckman, K. B., and Ames, B. N. (1999). Endogenous oxidative damage of mtDNA. Mutat. Res. 424 (1-2), 51–58. doi:10.1016/s0027-5107(99)00007-x
Boengler, K., Kosiol, M., Mayr, M., Schulz, R., and Rohrbach, S. (2017). Mitochondria and ageing: role in heart, skeletal muscle and adipose tissue. J. Cachexia Sarcopenia Muscle 8 (3), 349–369. doi:10.1002/jcsm.12178
Bratic, A., and Larsson, N. G. (2013). The role of mitochondria in aging. J. Clin. Invest 123 (3), 951–957. doi:10.1172/JCI64125
Budinger, G. R. S., and Chandel, N. S. (2025). Mitochondria dysfunction: cause or consequence of physiologic aging? Genes Dev. 39 (15-16), 917–919. doi:10.1101/gad.353106.125
Chen, Q., Fischer, A., Reagan, J. D., Yan, L. J., and Ames, B. N. (1995). Oxidative DNA damage and senescence of human diploid fibroblast cells. Proc. Natl. Acad. Sci. U. S. A. 92 (10), 4337–4341. doi:10.1073/pnas.92.10.4337
Chen, X., Ji, Y., Liu, R., Zhu, X., Wang, K., Yang, X., et al. (2023). Mitochondrial dysfunction: roles in skeletal muscle atrophy. J. Transl. Med. 21 (1), 503. doi:10.1186/s12967-023-04369-z
Chubanava, S., Karavaeva, I., Ehrlich, A. M., Justicia, R. M., Basse, A. L., Kulik, I., et al. (2025). NAD depletion in skeletal muscle does not compromise muscle function or accelerate aging. Cell Metab. 37 (7), 1460–1481.e17. doi:10.1016/j.cmet.2025.04.002
Ehinger, J. K., Westerlund, E., Frostner, E. A., Karlsson, M., Paul, G., Sjovall, F., et al. (2024). Mitochondrial function in peripheral blood cells across the human lifespan. NPJ Aging 10 (1), 10. doi:10.1038/s41514-023-00130-4
Gerschman, R., Gilbert, D. L., Nye, S. W., Dwyer, P., and Fenn, W. O. (1954). Oxygen poisoning and x-irradiation: a mechanism in common. Science 119 (3097), 623–626. doi:10.1126/science.119.3097.623
Giulivi, C., Zhang, Y. F., Omanska-Klusek, A., Ross-Inta, C., Wong, S., Hertz-Picciotto, I., et al. (2010). Mitochondrial dysfunction in autism. JAMA 304 (21), 2389–2396. doi:10.1001/jama.2010.1706
Gogvadze, V., Walter, P. B., and Ames, B. N. (2003). The role of Fe2+-induced lipid peroxidation in the initiation of the mitochondrial permeability transition. Arch. Biochem. Biophys. 414 (2), 255–260. doi:10.1016/s0003-9861(02)00750-6
Grevendonk, L., Connell, N. J., McCrum, C., Fealy, C. E., Bilet, L., Bruls, Y. M. H., et al. (2021). Impact of aging and exercise on skeletal muscle mitochondrial capacity, energy metabolism, and physical function. Nat. Commun. 12 (1), 4773. doi:10.1038/s41467-021-24956-2
Hagen, T. M., Yowe, D. L., Bartholomew, J. C., Wehr, C. M., Do, K. L., Park, J. Y., et al. (1997). Mitochondrial decay in hepatocytes from old rats: membrane potential declines, heterogeneity and oxidants increase. Proc. Natl. Acad. Sci. U. S. A. 94 (7), 3064–3069. doi:10.1073/pnas.94.7.3064
Hagen, T. M., Ingersoll, R. T., Wehr, C. M., Lykkesfeldt, J., Vinarsky, V., Bartholomew, J. C., et al. (1998). Acetyl-L-carnitine fed to old rats partially restores mitochondrial function and ambulatory activity. Proc. Natl. Acad. Sci. U. S. A. 95 (16), 9562–9566. doi:10.1073/pnas.95.16.9562
Hagen, T. M., Ingersoll, R. T., Lykkesfeldt, J., Liu, J., Wehr, C. M., Vinarsky, V., et al. (1999). (R)-alpha-lipoic acid-supplemented old rats have improved mitochondrial function, decreased oxidative damage, and increased metabolic rate. FASEB J. 13 (2), 411–418. doi:10.1096/fasebj.13.2.411
Hagen, T. M., Vinarsky, V., Wehr, C. M., and Ames, B. N. (2000). (R)-alpha-lipoic acid reverses the age-associated increase in susceptibility of hepatocytes to tert-butylhydroperoxide both in vitro and in vivo. Antioxid. Redox Signal 2 (3), 473–483. doi:10.1089/15230860050192251
Hagen, T. M., Liu, J., Lykkesfeldt, J., Wehr, C. M., Ingersoll, R. T., Vinarsky, V., et al. (2002). Feeding acetyl-L-carnitine and lipoic acid to old rats significantly improves metabolic function while decreasing oxidative stress. Proc. Natl. Acad. Sci. U. S. A. 99 (4), 1870–1875. doi:10.1073/pnas.261708898
Harman, D. (1972). The biologic clock: the mitochondria? J. Am. Geriatr. Soc. 20 (4), 145–147. doi:10.1111/j.1532-5415.1972.tb00787.x
Harper, C., Gopalan, V., and Goh, J. (2021). Exercise rescues mitochondrial coupling in aged skeletal muscle: a comparison of different modalities in preventing sarcopenia. J. Transl. Med. 19 (1), 71. doi:10.1186/s12967-021-02737-1
Helbock, H. J., Beckman, K. B., Shigenaga, M. K., Walter, P. B., Woodall, A. A., Yeo, H. C., et al. (1998). DNA oxidation matters: the HPLC-electrochemical detection assay of 8-oxo-deoxyguanosine and 8-oxo-guanine. Proc. Natl. Acad. Sci. U. S. A. 95 (1), 288–293. doi:10.1073/pnas.95.1.288
Hepple, R. T. (2014). Mitochondrial involvement and impact in aging skeletal muscle. Front. Aging Neurosci. 6, 211. doi:10.3389/fnagi.2014.00211
Jeong, I., Cho, E. J., Yook, J. S., Choi, Y., Park, D. H., Kang, J. H., et al. (2024). Mitochondrial adaptations in aging skeletal muscle: implications for resistance exercise training to treat sarcopenia. Life (Basel) 14 (8), 962. doi:10.3390/life14080962
Jia, L., Liu, Z., Sun, L., Miller, S. S., Ames, B. N., Cotman, C. W., et al. (2007). Acrolein, a toxicant in cigarette smoke, causes oxidative damage and mitochondrial dysfunction in RPE cells: protection by (R)-alpha-lipoic acid. Invest Ophthalmol. Vis. Sci. 48 (1), 339–348. doi:10.1167/iovs.06-0248
Johnson, M. L., Robinson, M. M., and Nair, K. S. (2013). Skeletal muscle aging and the mitochondrion. Trends Endocrinol. Metab. 24 (5), 247–256. doi:10.1016/j.tem.2012.12.003
Kamarulzaman, N. T., and Makpol, S. (2025). The link between mitochondria and sarcopenia. J. Physiol. Biochem. 81 (1), 1–20. doi:10.1007/s13105-024-01062-7
Killilea, D. W., and Ames, B. N. (2008). Magnesium deficiency accelerates cellular senescence in cultured human fibroblasts. Proc. Natl. Acad. Sci. U. S. A. 105 (15), 5768–5773. doi:10.1073/pnas.0712401105
Killilea, D. W., Atamna, H., Liao, C., and Ames, B. N. (2003). Iron accumulation during cellular senescence in human fibroblasts in vitro. Antioxid. Redox Signal 5 (5), 507–516. doi:10.1089/152308603770310158
Lal, A., Atamna, W., Killilea, D. W., Suh, J. H., and Ames, B. N. (2008). Lipoic acid and acetyl-carnitine reverse iron-induced oxidative stress in human fibroblasts. Redox Rep. 13 (1), 2–10. doi:10.1179/135100008X259150
Larsson, N. G. (2010). Somatic mitochondrial DNA mutations in mammalian aging. Annu. Rev. Biochem. 79 (1), 683–706. doi:10.1146/annurev-biochem-060408-093701
Li, A., Shami, G. J., Griffiths, L., Lal, S., Irving, H., and Braet, F. (2023). Giant mitochondria in cardiomyocytes: cellular architecture in health and disease. Basic Res. Cardiol. 118 (1), 39. doi:10.1007/s00395-023-01011-3
Liu, J., Yeo, H. C., Overvik-Douki, E., Hagen, T., Doniger, S. J., Chyu, D. W., et al. (2000). Chronically and acutely exercised rats: biomarkers of oxidative stress and endogenous antioxidants. J. Appl. Physiol. 89 (1), 21–28. doi:10.1152/jappl.2000.89.1.21
Liu, J., Wang, X., Shigenaga, M. K., Yeo, H. C., Mori, A., and Ames, B. N. (1996). Immobilization stress causes oxidative damage to lipid, protein, and DNA in the brain of rats. FASEB J. 10 (13), 1532–1538. doi:10.1096/fasebj.10.13.8940299
Liu, J., Head, E., Gharib, A. M., Yuan, W., Ingersoll, R. T., Hagen, T. M., et al. (2002a). Memory loss in old rats is associated with brain mitochondrial decay and RNA/DNA oxidation: partial reversal by feeding acetyl-L-carnitine and/or R-alpha -lipoic acid. Proc. Natl. Acad. Sci. U. S. A. 99 (4), 2356–2361. doi:10.1073/pnas.261709299
Liu, J., Killilea, D. W., and Ames, B. N. (2002b). Age-associated mitochondrial oxidative decay: improvement of carnitine acetyltransferase substrate-binding affinity and activity in brain by feeding old rats acetyl-L- carnitine and/or R-alpha -lipoic acid. Proc. Natl. Acad. Sci. U. S. A. 99 (4), 1876–1881. doi:10.1073/pnas.261709098
Long, J., Gao, F., Tong, L., Cotman, C. W., Ames, B. N., and Liu, J. (2009). Mitochondrial decay in the brains of old rats: ameliorating effect of alpha-lipoic acid and acetyl-L-carnitine. Neurochem. Res. 34 (4), 755–763. doi:10.1007/s11064-008-9850-2
Lykkesfeldt, J., Hagen, T. M., Vinarsky, V., and Ames, B. N. (1998). Age-associated decline in ascorbic acid concentration, recycling, and biosynthesis in rat hepatocytes--reversal with (R)-alpha-lipoic acid supplementation. FASEB J. 12 (12), 1183–1189. doi:10.1096/fasebj.12.12.1183
Marzetti, E., Calvani, R., Cesari, M., Buford, T. W., Lorenzi, M., Behnke, B. J., et al. (2013). Mitochondrial dysfunction and sarcopenia of aging: from signaling pathways to clinical trials. Int. J. Biochem. Cell Biol. 45 (10), 2288–2301. doi:10.1016/j.biocel.2013.06.024
Milgram, N. W., Araujo, J. A., Hagen, T. M., Treadwell, B. V., and Ames, B. N. (2007). Acetyl-L-carnitine and alpha-lipoic acid supplementation of aged beagle dogs improves learning in two landmark discrimination tests. FASEB J. 21 (13), 3756–3762. doi:10.1096/fj.07-8531com
Napoli, E., Wong, S., Hung, C., Ross-Inta, C., Bomdica, P., and Giulivi, C. (2013). Defective mitochondrial disulfide relay system, altered mitochondrial morphology and function in Huntington's disease. Hum. Mol. Genet. 22 (5), 989–1004. doi:10.1093/hmg/dds503
Napoli, E., Song, G., Wong, S., Hagerman, R., and Giulivi, C. (2016). Altered bioenergetics in primary dermal fibroblasts from adult carriers of the FMR1 premutation before the onset of the neurodegenerative disease fragile X-Associated Tremor/Ataxia syndrome. Cerebellum 15 (5), 552–564. doi:10.1007/s12311-016-0779-8
Nuccio, A., Nogueira-Ferreira, R., Moreira-Pais, A., Attanzio, A., Duarte, J. A., Luparello, C., et al. (2024). The contribution of mitochondria to age-related skeletal muscle wasting: a sex-specific perspective. Life Sci. 336, 122324. doi:10.1016/j.lfs.2023.122324
Paul, B. T., Manz, D. H., Torti, F. M., and Torti, S. V. (2017). Mitochondria and Iron: current questions. Expert Rev. Hematol. 10 (1), 65–79. doi:10.1080/17474086.2016.1268047
Peterson, C. M., Johannsen, D. L., and Ravussin, E. (2012). Skeletal muscle mitochondria and aging: a review. J. Aging Res. 2012, 194821. doi:10.1155/2012/194821
Piko, L., Hougham, A. J., and Bulpitt, K. J. (1988). Studies of sequence heterogeneity of mitochondrial DNA from rat and mouse tissues: evidence for an increased frequency of deletions/additions with aging. Mech. Ageing Dev. 43 (3), 279–293. doi:10.1016/0047-6374(88)90037-1
Ristow, M., and Schmeisser, S. (2011). Extending life span by increasing oxidative stress. Free Radic. Biol. Med. 51 (2), 327–336. doi:10.1016/j.freeradbiomed.2011.05.010
Sagar, S., and Gustafsson, A. B. (2023). Cardiovascular aging: the mitochondrial influence. J. Cardiovasc Aging 3 (3), 33. doi:10.20517/jca.2023.22
Seo, D. Y., Lee, S. R., Kim, N., Ko, K. S., Rhee, B. D., and Han, J. (2016). Age-related changes in skeletal muscle mitochondria: the role of exercise. Integr. Med. Res. 5 (3), 182–186. doi:10.1016/j.imr.2016.07.003
Shenk, J. C., Liu, J., Fischbach, K., Xu, K., Puchowicz, M., Obrenovich, M. E., et al. (2009). The effect of acetyl-L-carnitine and R-alpha-lipoic acid treatment in ApoE4 mouse as a model of human Alzheimer's disease. J. Neurol. Sci. 283 (1-2), 199–206. doi:10.1016/j.jns.2009.03.002
Shigenaga, M. K., Hagen, T. M., and Ames, B. N. (1994). Oxidative damage and mitochondrial decay in aging. Proc. Natl. Acad. Sci. U. S. A. 91 (23), 10771–10778. doi:10.1073/pnas.91.23.10771
Short, K. R., Bigelow, M. L., Kahl, J., Singh, R., Coenen-Schimke, J., Raghavakaimal, S., et al. (2005). Decline in skeletal muscle mitochondrial function with aging in humans. Proc. Natl. Acad. Sci. U. S. A. 102 (15), 5618–5623. doi:10.1073/pnas.0501559102
Song, G., Napoli, E., Wong, S., Hagerman, R., Liu, S., Tassone, F., et al. (2016). Altered redox mitochondrial biology in the neurodegenerative disorder fragile X-tremor/ataxia syndrome: use of antioxidants in precision medicine. Mol. Med. 22, 548–559. doi:10.2119/molmed.2016.00122
Springer-Sapp, C. B., Ogbara, O., Canellas Da Silva, M., Henderson, A., Liu, Y., Prior, S. J., et al. (2025). Age and sex-specific changes in mitochondrial quality control in skeletal and cardiac muscle. Front. Aging 6, 1606110. doi:10.3389/fragi.2025.1606110
Sun, N., Youle, R. J., and Finkel, T. (2016). The mitochondrial basis of aging. Mol. Cell. 61 (5), 654–666. doi:10.1016/j.molcel.2016.01.028
Tepp, K., Timohhina, N., Puurand, M., Klepinin, A., Chekulayev, V., Shevchuk, I., et al. (2016). Bioenergetics of the aging heart and skeletal muscles: modern concepts and controversies. Ageing Res. Rev. 28, 1–14. doi:10.1016/j.arr.2016.04.001
Tocchi, A., Quarles, E. K., Basisty, N., Gitari, L., and Rabinovitch, P. S. (2015). Mitochondrial dysfunction in cardiac aging. Biochim. Biophys. Acta 1847 (11), 1424–1433. doi:10.1016/j.bbabio.2015.07.009
Trifunovic, A., Wredenberg, A., Falkenberg, M., Spelbrink, J. N., Rovio, A. T., Bruder, C. E., et al. (2004). Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429 (6990), 417–423. doi:10.1038/nature02517
Voloboueva, L. A., Liu, J., Suh, J. H., Ames, B. N., and Miller, S. S. (2005). (R)-alpha-lipoic acid protects retinal pigment epithelial cells from oxidative damage. Invest Ophthalmol. Vis. Sci. 46 (11), 4302–4310. doi:10.1167/iovs.04-1098
Voloboueva, L. A., Killilea, D. W., Atamna, H., and Ames, B. N. (2007). N-tert-butyl hydroxylamine, a mitochondrial antioxidant, protects human retinal pigment epithelial cells from iron overload: relevance to macular degeneration. FASEB J. 21 (14), 4077–4086. doi:10.1096/fj.07-8396com
Walter, P. B., Knutson, M. D., Paler-Martinez, A., Lee, S., Xu, Y., Viteri, F. E., et al. (2002). Iron deficiency and iron excess damage mitochondria and mitochondrial DNA in rats. Proc. Natl. Acad. Sci. U. S. A. 99 (4), 2264–2269. doi:10.1073/pnas.261708798
Walter, P. B., Porter, J., Evans, P., Kwiatkowski, J. L., Neufeld, E. J., Coates, T., et al. (2013). Increased leucocyte apoptosis in transfused beta-thalassaemia patients. Br. J. Haematol. 160 (3), 399–403. doi:10.1111/bjh.12076
Wang, K., Gan, M., Lei, Y., Liao, T., Li, J., Niu, L., et al. (2025). Perspectives on mitochondrial dysfunction in the regeneration of aging skeletal muscle. Cell Mol. Biol. Lett. 30 (1), 94. doi:10.1186/s11658-025-00771-1
Yowe, D. L., and Ames, B. N. (1998). Quantitation of age-related mitochondrial DNA deletions in rat tissues shows that their pattern of accumulation differs from that of humans. Gene 209 (1-2), 23–30. doi:10.1016/s0378-1119(97)00628-8
Keywords: mtDNA mutagenesis, oxidative stress, Ames bacterial mutagenicity test, triage theory, mitochondrial dysfunction, aging, nutrition
Citation: Dang J, Liou JY, Mullah R and Giulivi C (2025) The legacy of Bruce Ames and mitochondrial DNA mutagenicity: integrating oxidative stress, aging, and modern perspectives. Front. Mol. Biosci. 12:1710944. doi: 10.3389/fmolb.2025.1710944
Received: 22 September 2025; Accepted: 09 October 2025;
Published: 29 October 2025.
Edited by:
Giuseppe Valacchi, North Carolina State University, United StatesReviewed by:
Enrique Cadenas, University of Southern California, United StatesCopyright © 2025 Dang, Liou, Mullah and Giulivi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cecilia Giulivi, Y2dpdWxpdmlAdWNkYXZpcy5lZHU=
†These authors have contributed equally to this work to the work