 Li-Tung Huang1,2*
Li-Tung Huang1,2*- 1Department of Pediatrics, Kaohsiung Chang Gung Memorial Hospital, Chang Gung University College of Medicine, Kaohsiung, Taiwan
- 2Department of Traditional Chinese Medicine, Chang Gung University, Linkou, Taiwan
Early-life stress includes prenatal, postnatal, and adolescence stress. Early-life stress can affect the development of the hypothalamic-pituitary-adrenal (HPA) axis, and cause cellular and molecular changes in the developing hippocampus that can result in neurobehavioral changes later in life. Epidemiological data implicate stress as a cause of seizures in both children and adults. Emerging evidence indicates that both prenatal and postnatal stress can prime the developing brain for seizures and an increase in epileptogenesis. This article reviews the cellular and molecular changes encountered during prenatal and postnatal stress, and assesses the possible link between these changes and increases in seizure occurrence and epileptogenesis in the developing hippocampus. In addititon, the priming effect of prenatal and postnatal stress for seizures and epileptogenesis is discussed. Finally, the roles of epigenetic modifications in hippocampus and HPA axis programming, early-life stress, and epilepsy are discussed.
Introduction
The early-life environment is one of the most important factors affecting life-long health (Anand, 2000; van den Bergh et al., 2005; Lupien et al., 2009; Boksa, 2010; Strüber et al., 2014). In humans, early-life stress is associated with a preterm birth and a low birth weight, and can prime the neonate for further complications later in life that include psychiatric disorders, aged-related cognitive dysfunction, obesity, and hypertension (Barker et al., 1989; Fowden et al., 2005; Lemaire et al., 2006; Lahiri et al., 2009; Strüber et al., 2014). Animal studies also suggest that exposure to stressors or steroids during early-life alter the programming of the hypothalamic-pituitary-adrenal (HPA) axis, neurobehavior, and neuroimmune systems (Matthews, 2000; Mueller and Bale, 2008; Lupien et al., 2009; Brunton and Russell, 2010; Chen and Zhang, 2011; Lai and Huang, 2011; Strüber et al., 2014). Epigenetic modification has gained increasing attention in recent years because of its connection with early-life adversities (Weaver et al., 2004; Meaney et al., 2007; Mueller and Bale, 2008; Chen and Zhang, 2011; McClelland et al., 2011a,b; Murgatroyd and Spengler, 2011; Lucassen et al., 2013; Rabbe and Spengler, 2013). On the other hand, stress during development can have a significant epigenetic impact on the brain, and this relationship is bidirectional (Hunter, 2012).
Early-life stressors include prenatal, postnatal, and adolescence stress (Lupien et al., 2009; Schmidt, 2010). For example, in humans, early-life stress can include prenatal stressors such as exposure to exogenous glucocorticoids, maternal infection (King et al., 2005; Sørensen et al., 2009; Jenkins, 2013), and birth complications, as well as postnatal stressors such as exposure to exogenous glucocorticoids, maternal postpartum depression, loss of a parent, exposure to family conflict and violence, neglect, or physical maltreatment (De Bellis, 2002; King et al., 2005; Frodl et al., 2010). Both prenatal and postnatal stress can increase the likelihood of seizures in early life (Joels, 2009; Koe et al., 2009) and epileptogenesis in later life. This article focuses only on the influences of prenatal stress and postnatal stress.
Hippocampal and HPA Axis Development
The hippocampus develops primarily during the fetal period in both rodents and primates (Seress et al., 2001; Khalaf-Nazzal and Francis, 2013). The limbic system, which includes the hippocampus, amygdala, and anterior cingulate cortex are already formed during the third and fourth month. Dentate gyrus forms at late stages of embryogenesis, however small numbers of dentate gyrus cells are formed from mid-embyrogenesis making temporal matching and connectivity of cells from other hippocampal subfields (Deguchi et al., 2011). Rodents and primates differ in the timing at which the majority of the dentate granule cells are produced; however, both rodents and primates produce ~85% postnatally (Bayer, 1980a; Rakic and Nowakowski, 1981). A similar percentage of cornus ammonis (CA) 1–3 subfield neurons are produced during the last days of gestation in rodents, and during the first half of pregnancy in primates (Bayer, 1980b; Rakic and Nowakowski, 1981). The hippocampal subfields can be recognized with distinct molecular markers from embryonic stages (Khalaf-Nazzal and Francis, 2013).
In the rodent, maturation and full differentiation of the hippocampal formation takes place during early postnatal periods (Avishai-Eliner et al., 2002). During the first postnatal weeks, neuronal birth, differentiation, and migration are ongoing (Altman and Bayer, 1990; Gould and Cameron, 1996). Neurogenesis of granule cells peaks during the second week of life in rodents (Bayer, 1980a), and during the third month in humans (Seress et al., 2001). In addition, synaptogenesis and the establishment of enduring connectivity patterns continue for weeks in the rodent, and for years in humans (Avishai-Eliner et al., 2002).
Glucocorticoids are released from the adrenal glands in response to stress, readily cross the blood-brain barrier, and activate hippocampal glucocorticoids receptors (McEwen, 1998). Glucocorticoids interact with their receptors in multiple target tissues, especially the HPA axis. Glucocorticoids act via two intracellular receptors, the glucocorticoid receptor (GR) and the mineralocorticoid receptor (MR) to regulate gene transcription. In addition, glucocorticoids can change neural function via rapid nongenomic actions. GR and MR differ in ligand affinity and distribution (de Kloet et al., 2005): GR has a lower affinity than MR has, and therefore are more frequently occupied when corticosterone levels increase (de Kloet et al., 2005). The actions of glucocorticoids depend on the functionality of the balance between GR and MR in the brain (de Kloet et al., 2005).
There is a distinct ontogenic profile for GR and MR in the fetal rat brain (Diaz et al., 1998). GR mRNA is present in the anterior hypothalamus, hippocampus, and pituitary by gestational day 13 (Diaz et al., 1998), whereas MR mRNA is present in the hippocampus by gestational day 16 and the hypothalamus by day 17 (Diaz et al., 1998). GR and MR in the rat fetal brain are low throughout gestation, but increase rapidly after birth, consistent with the postnatal development of the brain in the rat (Diaz et al., 1998).
During pregnancy, the mother's HPA axis undergoes major changes (Lindsay and Nieman, 2005). Cortisol secretion increases steadily through gestation (Jung et al., 2011); thus, the normal physiological responses to stressors and the cortisol awakening response (i.e., basal HPA activity) are attenuated (Lindsay and Nieman, 2005). For most of the pregnancy, the baby and mother share a common corticotrophin-releasing hormone (CRH)-adrenocorticotropic hormone (ACTH)-cortisol axis (McLean et al., 1995).
By the end of the first week of life (Bohn et al., 1994; Vazquez et al., 1998), the number of MRs reaches adult levels. The number of GRs present during the first few week of life, however, is ~30% of adult levels, but approach adult levels after ~30 days of life. Both GR and MR are highly expressed in the developing brain, and have different and complex ontogenies that allow intricate brain development.
Between postnatal day 4 and 14, neonatal rat pups have low basal corticosterone levels and the corticosterone response to stressors is blunted, which constitutes the so-called stress hyporesponsive period (SHRP) (Levine, 2005). However, disruption of normal maternal behavior in rat during the SHRP can influence HPA axis development. In humans, the HPA axis is highly reactive and labile during early infancy, but organizes between 2 and 6 months of age through interactions between the infant and caregiver. The quality of caregiving that the infant receives predicts the infant's ability to self-regulate later in life. Sensitive caregiving is associated with better self-regulatory abilities and optimal functioning of the child's HPA system (Gunnar and Cheatham, 2002; Gunnar and Donzella, 2002).
Effects of Pre-/Post-Natal Stress on Seizure Susceptibility and Epileptogenesis
Epileptogenesis is a process through which the normal brain develops epilepsy, and the hippocampus is implicated in the pathogenesis of both the initiation and propagation phases (Pitkänen and Lukasiuk, 2011). Mesial temporal lobe epilepsy (MTLE), the most common focal intractable epilepsy, is thought to be a multi-stage process of increasing epileptogenesis commencing in early life. The ongoing process of epileptogenesis and the course of epilepsy might be negatively influenced by the stress associated with the disease itself (Joels, 2009; Sawyer and Escayg, 2010). As a result, a negative loop might occur in which stress promotes epileptogenesis in predisposed individuals or lowers seizure threshold in epilepsy patients, thereby increasing the likelihood of exposure to stress, which in turn exacerbates the disease. Epidemiological data implicate stress in the cause of epilepsy and seizures in both children and adults (Temkin and Davis, 1984; Swinkels et al., 1998; Bosnjak et al., 2002).
Stress is a natural factor that may exacerbate or trigger seizures (Novakova et al., 2013; van Campen et al., 2013). HPA-related stress hormones, especially glucocorticoid and CRH, can affect excitatory and inhibitory processes in brain areas that are critically involved in seizure generation. Glucocorticoid exposure can alter plasticity in the hippocampus through increasing extracellular glutamate levels and calcium conductance (either voltage- or ligand-gated), alter expression of N-methyl-D-aspartate (NMDA) receptor subunits, and reduce glial uptake of glutamate, and thus, facilitate epileptiform discharges and seizures in animals. Glucocorticoids facilitate epileptiform discharges and seizures in animals. CRH is expressed in interneurons in both the developing and adult hippocampus and is released during stress (Sakanaka et al., 1987; Chen et al., 2001). Both glucocorticoids and CRH are important hormones that regulate the stress response and may contribute to seizure-induced loss of neurons, dendritic spines, and branching if it persists for a prolonged period (Ribak and Baram, 1996; Chen et al., 2012).
Negative life events and stress sensitivity are linked with childhood epilepsy (van Campen et al., 2012, 2013). In addition, epidemiological data implicate stress in the causation of epilepsy and seizures in children (Bosnjak et al., 2002). Specifically, early-life stress might create an enduring vulnerability to limbic epilepsy through altering glucocorticoids (Kumar et al., 2007), HPA axis (Joels, 2009), CRH (Baram and Hatalski, 1998), inflammation (Vezzani et al., 2013), membrane receptors such as gamma-aminobutyric acid (GABA) (Reddy, 2013), NMDA (Olney et al., 1991), and 2-amino-3-(3-hydroxy-5-methylisoxazol-4-yl) propionic acid (AMPA) receptors and neurotransmission (Rogawski, 2013), cellular electrophysiology, such as long-term potentiation (LTP) and long-term depression (Blaise et al., 2008), limbic area structures (Wong and Guo, 2013), and neuronal cell proliferation and neurogenesis (McCabe et al., 2001).
Physiological Mechanisms by Which Pre-/Post-Natal Stress Affects the Developing Hippocampus
Prenatal Stress
Glucocorticoid hormones
During pregnancy, women have naturally elevated levels of cortisol. In general, normal glucocorticoid concentrations are essential for the development of several organs, including the central nervous system. Prenatal stress or synthetic glucocorticoid administration exposes the fetus to high glucocorticoid levels, which leads to downregulation of GR in the hippocampus, attenuation of negative feedback for the HPA axis, and enhanced HPA axis activity (Reul and de Kloet, 1985; Harris and Seckl, 2011).
Placental CRH
In humans, placental CRH activity is modulated by the maternal HPA axis (Wadhwa et al., 1998). Placental CRH concentration is a significant predictor of spontaneous preterm birth (Glynn et al., 2001; Sandman et al., 2006) and intrauterine growth restriction (IUGR) (Wadhwa et al., 2004), and can influence hippocampal development in the fetus. Prenatal stress activates the maternal HPA axis, which increases placental CRH production and its subsequent release into the bloodstream. A positive feed-forward loop between cortisol and placental CRH indicates that prenatal stress leads to progressively higher fetal plasma CRH levels. Placental CRH may penetrate the blood-brain barrier of the fetus, and subsequently influence both the function and the integrity of the hippocampus (Kastin and Akerstrom, 2002), presumably by activating CRH receptors (Sandman et al., 1999; Wadhwa et al., 2001).
Placental 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2)
The placenta is an effective barrier between the maternal and fetal hormonal environments in humans, being rich in 11β-HSD2, which converts cortisol to inactive cortisone (Benediktsson et al., 1997). Downregulation of placental 11β-HSD 2 increases glucocorticoid exposure for the placenta and fetus. Maternal stress not only increases her own circulating cortisol, it also reduces the expression and activity of 11β-HSD 2 in the placenta, leaving the fetus less protected (Avishai-Eliner et al., 2002; Mairesse et al., 2007). Moreover, inhibition of 11β-HSD2 might contribute to low birth weight, IUGR, and pregnancy disorders such as preterm birth and preeclampsia (Causevic and Mohaupt, 2007; Michael and Papageorghiou, 2008).
Impaired uterine blood flow
The impact of maternal anxiety on fetal blood flow can be determined by using ultrasound to measure the blood flow pattern in the uterine arteries. Sjostrom et al. found that, at 37–40 gestational weeks, mothers with high-trait anxiety scores had fetuses with higher indices of blood flow in the umbilical artery, and lower values in the fetal middle cerebral artery, suggesting a change in blood distribution that favored brain circulation (Sjöström et al., 1997).
Postnatal Stress
CRH
CRH is expressed in hippocampal interneurons and is released from axon terminals during stress. CRH is produced in several populations of cells in the developing hippocampus, such as Cajal-Retzius cells, and is involved in the maturation of hippocampal circuitry (Chen et al., 2001).
Chronic early-life stress, which was imposed by creating “simulated poverty” in the cage, resulted in cognitive problems and dendritic atrophy with loss of dendritic spines and synapses (Brunson et al., 2005). Many of the persistent effects of early-life stress are reversible with subsequent treatment with a CRH receptor 1 (CRHR1) antagonist (Fenoglio et al., 2005). Adult mice lacking CRHR1 in the forebrain were relatively resistant to the deleterious effects of chronic stress of social defeat (Wang et al., 2011a). Interestingly, the local deletion of CRHR1 also protected adult mice from the adverse effects of chronic early-life stress on learning and memory (Wang et al., 2011b). Infusion of CRHR1 antagonists immediately following this early-life stress prevented the learning and memory deficits, rescued LTP, and restored the integrity of the dendritic structure (Ivy et al., 2010). These findings provide direct evidence for a need for CRH-CRHR1 signaling in the persistent effects of chronic early-life stress on hippocampal synapses. In this regard, Karsten and Baram propose that early-life experience can result in persistently altered regulation of CRH expression, which provides the neurobiological substrate to subsequent stress and some adult psychopathology (Karsten and Baram, 2013). In line with the preclinical data, single-nucleotide polymorphisms in the CRHR1 gene protect against depression in individuals exposed to childhood maltreatment (Tyrka et al., 2009).
Glucocorticoid hormones
Glucocorticoids are released from the adrenal glands in response to stress, readily cross the blood-brain barrier, and activate hippocampal glucocorticoids receptors (McEwen, 1998). Schmidt et al. demonstrated that glucocorticoid excess during the SHRP has only limited consequences on the adult behavioral phenotype (Schmidt et al., 2002). In addition, glucocorticoid administration early in life does not reproduce the effects of stress on hippocampal function and integrity when given in a non-stressful manner (Leverenz et al., 1999). Together, glucocorticoids play a minor role, and other factors may contribute more to the mechanisms by which early-life stress influences hippocampal development and function throughout life.
Prenatal Stress
Prenatal stress is an important programming factor in brain development and function. A recent cross-sectional study indicated that 6% of pregnant women reported high levels of psychological stress during their pregnancies that resulted from conditions including depression, panic disorder, or domestic violence (Woods et al., 2009). Talge et al. reviewed several prospective studies related to prenatal maternal stress, and found a substantial number of emotional/behavioral problems in children, including attention deficit hyperactivity disorder, anxiety, and language delay, that were attributed to prenatal stress or anxiety in ~15% of the subjects (Talge et al., 2007).
Cellular and Molecular Alterations in the Developing Hippocampus that May Link Prenatal Stress to Seizure and Epileptogenesis
Glucocorticoid and CRH and HPA Axis
The density of hippocampal GRs was lower by ~50% in prenatal stress female offspring; however, no difference was observed between prenatally stressed and control males (Szuran et al., 2000). This female-specific decrease in hippocampal GRs was also shown by Weinstock et al. (1992).
Szuran et al. restrained pregnant rat dams for 30 min/day during gestational days 15–19. Prenatally stressed females had higher basal corticosterone levels (Szuran et al., 2000). Exposure to exogenous glucocorticoids during the last week of gestation increased basal and stress-induced plasma corticosterone levels in adult rats (Seckl, 2004) and attenuated the HPA axis response (Seckl, 2004; Welberg and Seckl, 2001). Endogenous glucocorticoids mediated some of the changes in HPA responsiveness in prenatally stressed offspring, both in rodents and primates (Matthews, 2000).
Inflammation
Restrained pregnant mice dam offspring showed increased interleukin-1β and tumor necrosis factor-α level in the hippocampus, increased interleukin-1β immunoreactive microglial cells, and increased activated microglia. In addition, systemic administration of lipopolysaccharide induced a significant increase in tumor necrosis factor-α in the hippocampus of only prenatally stressed mice but not non-stressed animals (Diz-Chaves et al., 2012, 2013).
Membrane Receptors and Neurotransmitter
Maternal immune activation caused reduced basal neurotransmission of dopamine and glutamate, as well as reduced levels of the inhibitory transmitter GABA, within the hippocampus (Bitanihirwe et al., 2010). Prenatal stress also reduced the expression and activity of metabotropic glutamate receptor 5, which is implicated in the regulation of synaptic plasticity and neurogenesis in the hippocampus of male rats (Morley-Fletcher et al., 2011).
Cellular Electrophysiology
A significant downregulation of hippocampal genes also was reported in 23-day-old female rats whose mothers were stressed from gestational days 17–21 (Bogoch et al., 2007). This included presynaptic voltage-gated Ca2+ type P/Q and several K+ channels that regulate the neuron membrane potential and suggests a potential decrease in the excitability of newly formed synapses.
Spine and Dendrite and Cell Morphology
Hayashi and colleagues reported that rats exposed to prenatal stress had a significant 32% reduction in synaptic density within the hippocampal CA3 area, as measured on postnatal day 35 (Hayashi et al., 1998). Lemaire et al. (2000) reported a reduction in the number of granule cells within the hippocampal dentate gyrus of prenatally stressed rats measured 28 days postnatally.
Neuronal Cell Proliferation and Neurogenesis
In male mice, prolonged prenatal stress decreased cell proliferation in the hippocampus by 60% on postnatal day 10 (Kawamura et al., 2006). In another experimental paradigm, daily maternal restraint during the last week of gestation resulted in deficits of hippocampal neurogenesis (Lemaire et al., 2006). The relationship between prenatal stress and neurogenesis is complicated and depends on the stressor type, sex, and environment. Prenatal stress seems to have both enhancing and suppressing effects on the development of hippocampal neurons in a stressor intensity-dependent manner (Fujioka et al., 2006). Fujioka et al. reported that short-lasting (i.e., 30 min, once daily, between gestation days 15–17) and mild prenatal stress seemed to enhance neonatal neurogenesis, facilitate LTP, and the differentiation of processes of hippocampal neurons, whereas long-lasting (i.e., 240 min, once daily, between gestation days 15–17) and severe prenatal stress impaired their morphology.
Effects of Prenatal Stress on Seizure Susceptibility and Epileptogenesis
Beck and Gavin treated pregnant mice with beta-2-thienylalanine solvent or a sham injection on gestational days 10–12. Audiogenic seizures were tested on postnatal day 23. An increase in audiogenic seizure frequencies were observed in injected mice, irrespective of the nature of the injected substance. This finding suggested that the act of manipulation, rather than the test substance, caused stress and increased seizure propensity (Beck and Gavin, 1976). Frye and Bayon exposed rats to 20 min of restraint stress toward the end of their pregnancy (Frye and Bayon, 1999). They found that the prenatally stressed offspring had more partial seizures and tonic-clonic seizures with long durations than did control rats. Edwards et al. examined how stress exposure at different times during gestation might affect later limbic system excitability and the propensity to develop epilepsy (Edwards et al., 2002). Pregnant dams were restrained under bright light for 45 min, three times a day during either early gestation (gestational days 5–12) or mid-late gestation (gestational days 12–20). Offspring of the stressed dams were then tested as an infant at postnatal day 10 or as adults, and were compared with offspring from non-stressed dams. Outcome measures assessed were the stimulation-induced seizure threshold, after-discharge threshold, and the rate of seizure development using electrical hippocampal kindling. Both prenatal stressors significantly lowered after-discharge threshold in pups, but this effect appeared to diminish by adulthood in the early gestational stress group. In addition, mid to late gestational stress accelerated kindling rates in all infant offspring and in adult males, but had no effect in adult female rats. Notably, Young et al. administered dexamethasone or betamethasone on gestational days 15–18, and tested the seizure threshold and kindling parameters (Young et al., 2006). They found prenatal betamethasone treatment increased seizure threshold for both models. Prenatal dexamethasone treatment increased kindling threshold, but not seizure threshold. Kindling rate was unaffected by either glucocorticoid treatment (Young et al., 2006). Velisek showed prenatal exposure to betamethasone decreased postnatal susceptibility to flurothyl-induced clonic seizures but not to kainic acid-induced seizures. Prenatal hydrocortisone decreased postnatal weight but did not affect seizure susceptibility (Velíšek, 2011). In their subsequent work, Yum et al. demonstrated that prenatal restraint stress (2 × 45 min) in rats on gestational day 15 would increase susceptibility to spasms on postnatal day 15 (Yum et al., 2012).
Shang et al. showed an association between the onset risk of infantile spasms and the degree of maternal stress (Shang et al., 2010). However, in a population-based cohort study in Denmark, Li et al. studied children who were hospitalized because of epilepsy and born to women who had lost a close relative during pregnancy 1 year before pregnancy (Li et al., 2008). In this study, no association was found between this particular form of prenatal stress and the risk of epilepsy.
Indirect evidence links prenatal stress and an increased likelihood of childhood seizures in children with autistic disorder. Minshew et al. pointed out that epilepsy is found in about one-third of patients with autistic disorder, a disorder related to prenatal stress (Kinney et al., 2008), compared with a prevalence of only 2–3% in the general population (Minshew et al., 2005). Table 1 summarizes the current rodent studies regarding the impact of prenatal stress on seizure occurrence and epileptogenesis.
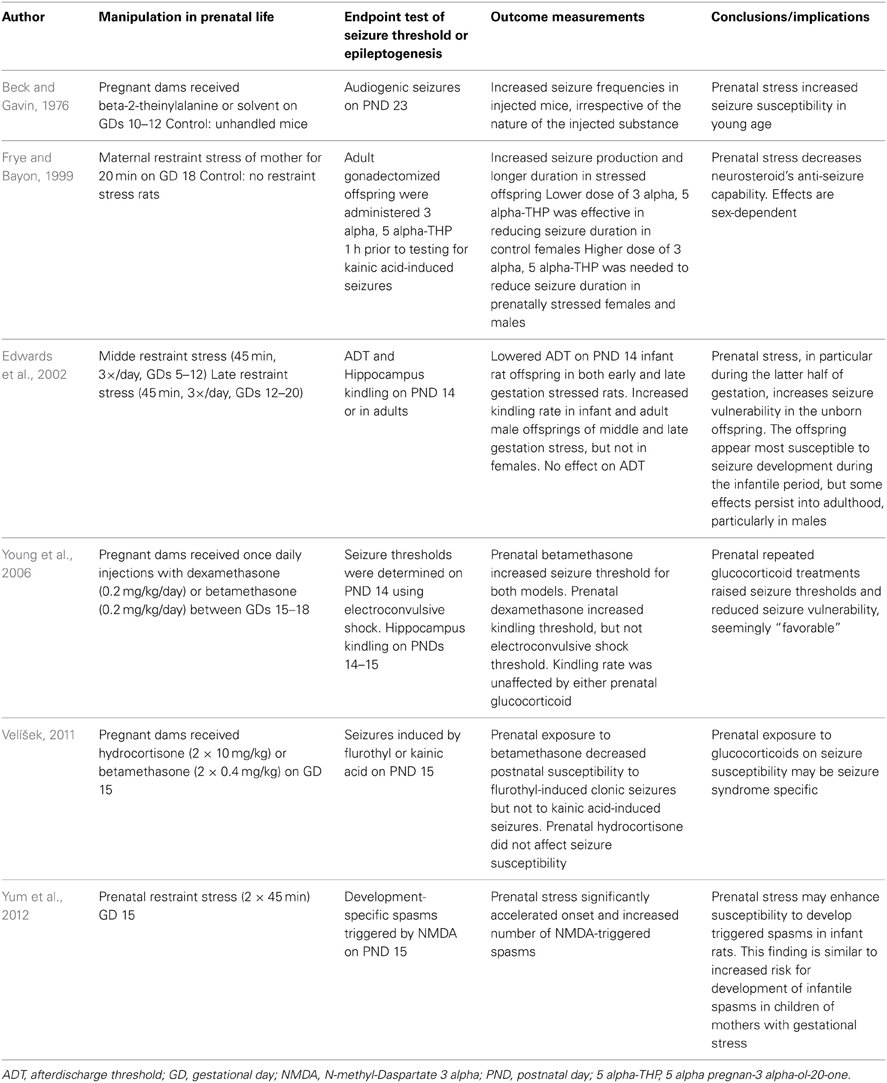
Table 1. Summary of rodent studies investigating effects of prenatal stress in rodent models of epilepsy/epileptogenesis.
Postnatal Stress
Early-life adversity (childhood abuse and neglect, loss of parents, or extreme poverty) occurs worldwide and are all too common in the lives of children (Jones, 2008; Sandberg and Rutter, 2008). In the Dunedin Study birth cohort of 1037 children, followed prospectively for 32 years, maltreatment includes maternal rejection, harsh discipline, sexual abuse, physical abuse, and disruptive caregiver changes (Danese et al., 2009). For each child, the cumulative index counts the number of maltreatment indicators experienced during the first decade of life; 63.7% of children experienced no maltreatment, 26.7% experienced one form of maltreatment, and 9.6% experienced two or more forms of maltreatment (Danese et al., 2009). Clinical evidence from life-course epidemiology study points to the importance of early life experiences in shaping adult disease (Poulton et al., 2010).
Cellular and Molecular Alterations in the Developing Hippocampus that May Link Postnatal Stress to Seizure and Epileptogenesis
Glucocorticoid and CRH and HPA Axis
A 24-h maternal separation paradigm in 11-day-old rat pups can lead to a decrease in the expression of GR and MR mRNA in the hippocampus (van Oers et al., 1998). Likewise, expression levels of GR and MR are down regulated in the hippocampus of maternally separated mice on postnatal day 9 (Schmidt et al., 2002). In addition, neonatal infection in mice led to altered hippocampal GR and MR mRNA, as well as proteins, following a subsequent adult infection (Wynne et al., 2011).
Wang et al. demonstrated that early postnatal life stress impairs hippocampus-dependent spatial learning and memory in adult mice, and is associated with physiological, morphological, and molecular abnormalities in the hippocampus (Wang et al., 2011a,b). Impairments of spatial learning and memory in early postnatal life stress are recapitulated by forebrain CRH overexpression and attenuated by forebrain CRHR1 inactivation. This suggests the forebrain CRH-CRHR1 system is crucial for modulating and programming cognitive functions by early-life stress (Wang et al., 2011a,b).
Inflammation
In rat, maternal separation on postnatal day 9 caused increased hippocampal interleukin-1 receptor in male offspring (Viviani et al., 2014). In the hippocampus, a decrease in BDNF mRNA and an increase in interleukin-1β mRNA were observed in rats with a neonatal infection and an immune challenge in adults (Bilbo et al., 2008).
Membrane Receptors and Neurotransmitter
Maternal separation on postnatal day 9 decreased the levels of the AMPA receptor GluA1 and GluA2 subunits, altered NMDA receptor subunits GluN2B to GluN2A ratio, and increased interleukin-1 receptor interactions with GluN2B at the synapse of male hippocampal neurons (Viviani et al., 2014). This mechanism is part of a complex re-organization of the excitatory glutamatargic synapses. Hsu et al. reported two episodes of handling with maternal separation during early postnatal development resulted in long-term changes in postsynaptic GABA receptor function and subunit expression in hippocampal dentate gyrus (Hsu et al., 2003).
Cellular Electrophysiology
Maternal separation prevented the stress-induced transformation from early to late LTP in the dentate gyrus of adult male rats (Wang et al., 2013b). However, maternal separation for 24h on postnatal day 3 facilitated LTP in the dentate gyrus after an acute stress (Oomen et al., 2010).
Spine and Dendrite and Cell Morphology
An altered granule cell dendritic morphology (Oomen et al., 2010), a lower number of hippocampal neurons and glia (Leventopoulos et al., 2007; Fabricius et al., 2008), and a reduced mossy fiber density (Hout et al., 2002) have been reported following maternal separation (Rodenas-Ruano et al., 2012). Wang et al. demonstrated that postnatally stressed adult mice had decreased hippocampal nectin-3 levels and dendritic spine loss via CRH mechanism (Wang et al., 2013a).
Neuronal Cell Proliferation and Neurogenesis
Maternal separation for 180 min leads to an increase in cell proliferation on postnatal day 21 (Nair et al., 2007); however, in 2- to 7-month-old rats, cell proliferation was reduced (Mirescu et al., 2004; Oomen et al., 2010; Hulshof et al., 2011).
Maternal separation for 24h on postnatal day 3 increases hippocampal neurogenesis (Oomen et al., 2009). Similar to cell proliferation, early stress is associated with distinct consequences on hippocampal neurogenesis that manifest in a temporally regulated manner, i.e., enhanced in young adulthood and impaired in middle-aged (Suri et al., 2013).
Effects of Postnatal Stress on Seizure Susceptibility and Epileptogenesis
Edwards et al. investigated the effects of maternal separation on kindling epileptogenesis utilizing a relatively benign separation protocol that included 60 min on postnatal days 4 and 5 (Edwards et al., 2002). The comparison group included the other littermates, which were briefly handled but not removed from the mother. This postnatal manipulation had no effect on after-discharge threshold or rapid hippocampal kindling rates when assessed at 2 weeks of age.
To investigate the effects of maternal separation on the long-term consequences of early-life status epilepticus, Lai et al. tested whether maternal separation for 1h affected the long-term sequelae of emotional disorders following seizure early in life (Lai et al., 2006). Lai et al. used maternal separation that involved 1h of isolation daily during postnatal days 2 and 9, and used lithium-pilocarpine-induced status epilepticus on postnatal day 10 rats. As adults, anxiety-related behavior was assessed using the elevated plus maze test and seizure susceptibility was assessed by pentylenetetrazol-induced seizures. Rats exposed to maternal separation and seizures demonstrated a reduced pentylenetetrazol threshold for seizure induction compared to non-handled rats or rats exposed to isolation or seizure alone. Metyrapone (a corticosterone synthesis inhibitor) treatment prior to seizure did not reverse this enhanced excitability, indicating a partial role of glucocorticoids in this context. Salzberg et al. examined the effects of maternal separation on limbic excitability and the development of amygdala kindling (Salzberg et al., 2007). Postnatal stress was induced by separating pups from their mothers for 180 min daily from postnatal days 2–14. The comparison condition was mother and pup separation for 15 min per day over the same period, an exposure referred to as early handling. At 8 weeks of age, equivalent to young adult life, rats were tested for the after-discharge threshold and subjected to rapid amygdala kindling. Rats exposed to early-life stress exhibited significantly lower seizure thresholds and an accelerated rate of kindling, compared to early handled rats. These effects on limbic excitability and epileptogenesis were specifically observed in female rats, whereas males did not demonstrate changes in epilepsy outcomes, despite demonstrating increases in anxiety-like behavior. Using the rat amygdala-kindling model, Kumar et al. demonstrated that early-life stress induced by maternal separation accelerates the progression of focal limbic seizures to secondary generalized convulsive seizures in adult rats (Kumar et al., 2011). Desgent et al. used a two-hit model of TLE characterized by two early-life insults: a freeze lesion-induced cortical malformation on postnatal day 1, and a prolonged hyperthermic seizure on postnatal day 10 (Desgent et al., 2012). They demonstrated that after both insults, females did not develop MTLE while all males did. This correlated with a rise in corticosterone levels on postnatal day 1 following the lesion, but only in males. Their data demonstrated sexual dimorphism in the long-term vulnerability for developing epilepsy in the lesion plus hyperthermia animal model of MTLE, and suggested that the response to early-life stress at postnatal day 1 contributed significantly to epileptogenesis in a sex-specific manner (Desgent et al., 2012). Ali et al. demonstrated changes in firing patterns in thalamocortical and hippocampal regions resulting from both maternal separation and amygdala kindling, which might reflect cellular changes underlying the enhanced vulnerability to kindling in rats that had been exposed to early-life stress (Ali et al., 2013).
Similarly, Leussis and Heinrichs cross-fostered El pups to CD-1 dams because CD-1 dams exhibit a higher quality of maternal care than El dams. El pups raised by CD-1 dams experienced delayed seizure onset and reduced seizure frequency, suggesting that early-life environment can play an important role in shaping the adult seizure phenotype (Leussis and Heinrichs, 2009). It should be noted the El mouse model has not been verified for its effect on early-life stress. In addition, El pups raised in a biparental environment with both the El dam and sire attending the pups received more parental attention than El pups raised by only the El dam, yet they showed an earlier development of seizures (Orefice and Heinrichs, 2008). Together, early-life environment can interact with a genetic predisposition to shape the future seizure phenotype.
Van Campen et al. studied the effect of stress on seizure frequency in childhood epilepsy. They found stress sensitivity was reported in half of the children with epilepsy. They suggested that experiencing negative life events might cause a larger response to daily stressors, thereby increasing the likelihood to induce epileptic activity in childhood (van Campen et al., 2012). Table 2 summarizes the current rodent studies regarding the impacts of postnatal stress upon seizure occurrence and epileptogenesis.
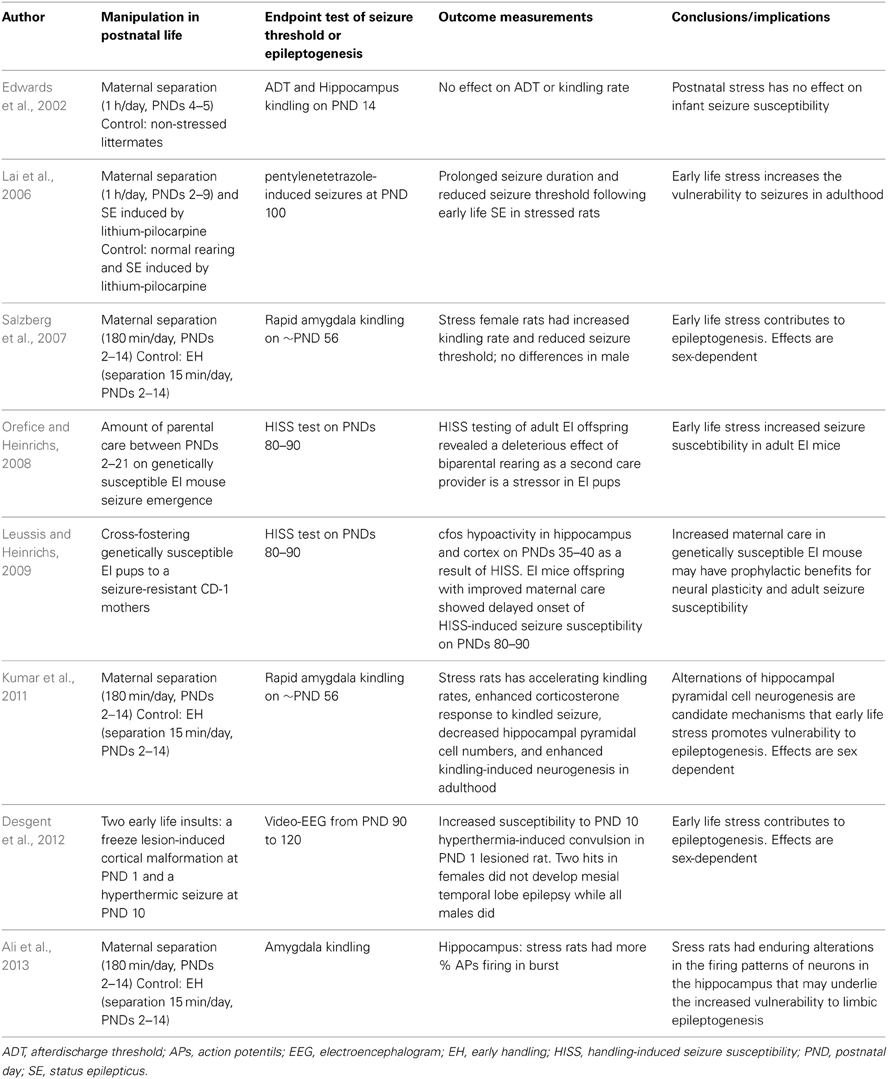
Table 2. Summary of rodent studies investigating effects of postnatal stress in rodent models of epilepsy/epileptogenesis.
Epigenetic Modifications in Development Programming and the Effects of Stress
Epigenetic modifications regulate gene expression without altering the DNA sequence. Epigenetic changes involve DNA methylation at cytosine-guanine sequences-CpG sites, histone posttranslational modifications (histone methylation, acetylation, phosphorylation, ubiquitylation, sumoylation, and propionylation), and microRNAs (Gräff et al., 2011). Epigenetic mechanisms control nucleosome spacing and how they are condensed, which subsequently determines gene activity. Briefly, chromatin exists in an inactivated and condensed state (heterochromatin) that prevents gene transcription, but when activated to an open state (euchromatin), genes can be transcribed.
It is now clear in both humans and animals that glucocorticois and stress have a significant epigenetic impact, and the relationship between the stress response and epigenetics in the brain is bidirectional (Hunter, 2012). Epigenetic alterations have become especially attractive to researchers in recent years, as increasing evidence indicates that they can be induced by physical and social exposure early in life (Meaney et al., 2007). For some neurobiological disorders, exposure to environmental agents during early developmental stages can epigenetically disturb gene regulation in a long-term manner and cause significant pathological manifestations later in life. This process is the latent early-life associated regulation model by Lahiri et al. (2009).
Epigenetic dysregulation has been associated with prenatal IUGR and disease in both humans and rodents (Baserga et al., 2007, 2010; Friso et al., 2008). Prenatal stress can cause increased DNA methylation in the frontal cortex and hippocampus (Mychasiuk et al., 2011; Matrisciano et al., 2013) and a lower DNA methyltransferase 3a immunoreactivity in the dentate gyrus in offspring (Sierksma et al., 2013).
Variations in maternal care in the rat result in differences in hippocampal development and synaptic plasticity in the offspring (Macrì and Würbel, 2006). Observational studies provide evidence for two forms of maternal behaviors during the first week of lactation: licking/grooming (LG) and the arched-back nursing (ABN) posture (Liu et al., 1997; Francis et al., 1999). In the rat, the adult offspring of high LG-ABN mothers show increased hippocampal GR expression and enhanced glucocorticoid feedback sensitivity compared to animals reared by low LG-ABN mothers (Liu et al., 1997; Francis et al., 1999). In addition, adult offspring of high LG-ABN mothers exhibited modest HPA stress responses compared to animals reared by low LG-ABN mothers (Menard and Hakvoort, 2007). In hippocampus, offspring from high LG-ABN mothers had hypomethylation of CpG dinucleotides in the exon 17 GR promoter sequence, and increased histone acetylation that might account for higher transcription of the GR gene (Weaver et al., 2004). The maternal effect is mediated by enhanced serotonergic activity and an increased expression of NGFI-A, which binds the exon 17 GR promoter sequence (Weaver et al., 2007). Cross-fostering experiments showed a causal relationship between maternal care and changes in the exon 17 GR promoter methylation (Weaver et al., 2005, 2006).
Epigenetic Modification is a Shared Pathogenic Substrate of Both Early-Life Stress and Epilepsy
As stated above, epigenetic modifications underpin the programming effects of early-life stress. Interestingly, a wealth of evidence indicates that dysregulation of epigenetic mechanisms occurs in several human epilepsy syndromes. Epigenetic mechanisms can influence the acute deployment of genes resulting from seizures themselves or can have gradual effects on the steady-state expression profile of candidate genes that persist into epilepsy. Epigenetic modifications can affect seizure and epilepsy in several ways (Lubin, 2012; Roopra et al., 2012).
Firstly, histone acetylation is involved in epileptogenesis in human epilepsy patients. Seizure activity results in gene expression changes, including alterations in mRNA levels for glutamate receptor 2 and BDNF, the two well-characterized epileptogenesis-related genes. Of interest, histone acetyltransferase-mediated increases in histone acetylation levels at the promoter regions of the glutamate receptor 2 and BDNF genes have been shown to correlate with their gene expression changes following seizures in an experimental animal model (Huang et al., 2002b).
Secondly, DNA methylation has been highlighted as a component of the methylation hypothesis of epileptogenesis (Kobow and Blumcke, 2011). DNA methyltransferase enzymes 1 and 3a specifically, were increased in neurons from the temporal neocortices of 25 MTLE patients (Zhu et al., 2012). Using a rat model of MTLE, Williams-Karnesky et al. identified an increase in hippocampal DNA methylation that correlates with an increased DNA methyltransferase activity, disruption of adenosine homeostasis, and spontaneous recurrent seizures. To test the effects of adenosine, they used bioengineered silk implants to deliver a defined dose of adenosine over 10 days to the brains of epileptic rats (Williams-Karnesky et al., 2013). Adenosine implants reversed DNA hypermethylation seen in the epileptic brain, inhibited sprouting of mossy fibers in the hippocampus, and prevented the progression of epilepsy for at least 3 months (Williams-Karnesky et al., 2013).
Thirdly, transcription factors are involved in epileptogenesis in human epilepsy patients. Repressor element-1 silencing transcription factor and neuronal restrictive silencer factor serve to repress gene expression through dynamic recruitment of epigenetic complexes (Qureshi and Mehler, 2009). Of interest, repressor element-1 silencing transcription has been implicated in the regulation of several epileptogenesis specific factors, including growth factors, neurotransmitter receptors, ion channels, circuit excitability, and neurogenesis (Huang et al., 1999; McClelland et al., 2011a,b; Roopra et al., 2012).
Fourthly, methyl-CpG-binding protein 2 can regulate neuronal activity and is itself controlled by activity (Roopra et al., 2012).
Taken together, early-life stress can prime seizure occurrence and increases epileptogenesis. In addition, epigenetic modification is a shared pathogenic substrate of early-life stress and epilepsy.
Coexistence of Early-Life Stress and Early-Life Seizures
Seizure is one of the most common pediatric emergencies, with the highest incidence in the first year of life. Animal studies have demonstrated early-life seizures differ from adult seizures by the seizure behaviors, the electroencephalogram features, and their consequences. Notwithstanding the higher susceptibility to seizures, the immature brain is less vulnerable to seizure-induced injuries than the mature brain (Dube et al., 2001; Holmes and Ben-Ari, 2001; Huang et al., 2012). However, under some circumstances seizure in the immature brain can cause permanent brain damage (Dube et al., 2006).
For humans, most early-life seizures occur in premature and sick neonates (Scher et al., 1993; Miller et al., 2002; Scher, 2003) who are hospitalized and separated from their mothers, and thus, under stress (Field, 1994; Anand, 2000). Reciprocally, early-life stress may prime the occurrence of seizures and act via glucocorticoids, thereby potentiating the excitotoxic effects of concurrent neurological insults (Sapolsky, 1996), such as seizure (Huang et al., 2002a; Lai et al., 2006).
As stated above, early-life stress can prime the seizure occurrence and subsequent epileptogenesis. Currently, more attention is being paid to the effect of early-life stress on adult-onset seizure; however, little work has focused on the effect of early-life stress on the early-life seizure (Beck and Gavin, 1976; Edwards et al., 2002; Lai et al., 2006; Young et al., 2006; Velíšek, 2011; Yum et al., 2012). Indeed, to study the coexistence of early-life stress and early-life seizures is of both experimental and clinical importance.
The Concept of Development Origins of Health and Disease (DOHaD)
Barker et al. noted that low birth weight was associated with an increased risk of adverse outcomes in adulthood, such as coronary heart disease, stroke, high blood pressure, and type 2 diabetes (Barker et al., 1989). Gluckman et al. proposed the concept of DOHaD by observing the enduring effects of the fetal environment on physical health and disease in adulthood. The process of fetal programming or developmental plasticity is one of the core assumptions of DOHaD (Gluckman et al., 2007). Gluckman et al. use the concept of predictive adaptive responses to describe the developing organism by making phenotypic responses during development to obtain an adaptive advantage (Gluckman et al., 2005). The fetus will predict and make adaptive responses to a broad range of environmental cues to aid fitness and survival in later life. If the prediction is correct, then there will be a good match between the phenotype adopted and the environment in which the organism will later live. If the prediction is poor, there will be a mismatch between the environment experienced and the phenotype induced. The authors propose that developmental mismatch triggers or exacerbates certain diseases and provide a useful explanation for the DOHaD phenomenon (Gluckman et al., 2005, 2007). Furthermore, the notion of epigenetic modifications is applied to the DOHaD approach (Waterland and Michels, 2007). The DOHaD approach has become so popular that an international society has been formed, and this society is actively promoting research and collaboration in this area.
Figure 1 depicts the path form early-life adversity to long-term neuropsychiatric disorders, along with the underlying molecular and cellular mechanisms and epigenetic modifications, with a match or mismatch adaptation that leads to the final outcome.
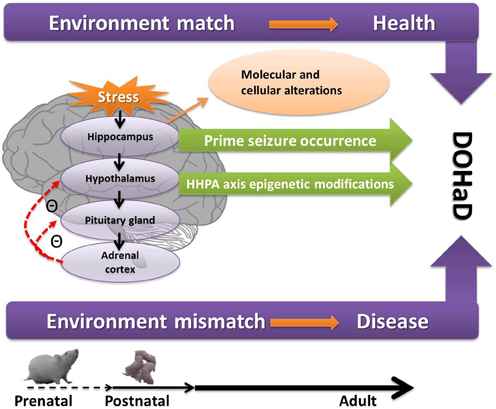
Figure 1. A conceptual diagram of how prenatal and postnatal stress act on the hippocampus and HPA axis and lead to neuropsychiatric disorders through epigenetic modifications, the so-called DOHaD as presently understood. If the early programming environment matches the later adult environment, the adults are healthy. If mismatch occurs, the adults are more likely to have diseases. Figure 1 also shows the increase seizure propensity in the context of early-life stress. See text for details. DOHaD, development origins of health and disease; HHPA, hippocampus and hypothalamic-pituitary-adrenal.
Conclusions and Perspectives
Early-life stress can elicit detrimental effects on hippocampal development by altering the HPA axis, neuroplasticity, and behavior. Developmental plasticity allows an organism to adapt to environmental changes in the critical stages of early life. As highlighted in this review, early-life stress programs the development of the HPA axis, exerts profound effects on neural plasticity, primes seizure occurrence, and increases epileptogenesis. Epigenetic modifications play an important role in both early-life stress and epilepsy.
A number of important points made throughout the manuscript are reinforced here. Reducing damage done by prenatal and postnatal stress may help reduce the cost of treating adult diseases. Protecting pregnant mothers from harmful stress exposure and supporting programs to reduce stress or anxiety during pregnancy might lead to improvements in the health and well-being of their children later in life. Ideally, intervention and prevention should be achieved before pregnancy begins. In terms of postnatal stress, psychosocial interventions in early life can affect brain development and thereby benefit children at risk. Other perinatal adversities such as perinatal infection, nutritional disorders, and toxin exposures must be cautiously avoided and treated. The potential therapeutic value of pharmacological agents, such as CRHR1 antagonists, MR and GR antagonists should be explored.
Recently, an increasing number of studies have shown that early-life stress primes seizure occurrence and increases epileptogenesis. An increased understanding of the link between early-life stress and epilepsy could improve the care and treatment of patients with epilepsy, while also allowing better management of other stress-related neurological disorders.
In the future, we will need to better determine the developmental windows during which preventative or therapeutic interventions can reverse the adverse effects of developmental programming. It will also be important to better understand stress biomarkers, especially epigenetic biomarkers. An increasing number of studies have provided clues as to how early-life stress induces changes at the cellular, molecular, and epigenetic levels. Continued progress on these fronts will provide great insight into disease mechanisms, in turn leading to the potential identification of novel targets for therapy and prevention.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grant CMRPG8B0112 from Chang Gung Hospital, Kaohsiung, Taiwan and NMRPD1C0731 (NSC102-2314-B-182-025-MY3) from National Science Council, Taiwan to Dr. Li-Tung Huang.
References
Ali, I., O'Brien, P., Kumar, G., Zheng, T., Jones, N. C., Pinault, D. et al. (2013). Enduring effects of early life stress on firing patterns of hippocampal and thalamocortical neurons in rats: implications for limbic epilepsy. PLoS ONE 8:e66962. doi: 10.1371/journal.pone.0066962
Altman, J., and Bayer, S. A. (1990). Migration and distribution of two populations of hip- pocampal granule cell precursors during the perinatal and postnatal periods. J. Comp. Neurol. 301, 365–381. doi: 10.1002/cne.903010304
Anand, K. J. (2000). Effects of perinatal pain and stress. Prog. Brain Res. 122, 117–129. doi: 10.1016/S0079-6123(08)62134-2
Avishai-Eliner, S., Brunson, K. L., Sandman, C. A., and Baram, T. Z. (2002). Stressed out? or in (utero). Trends Neurosci. 25, 518–524. doi: 10.1016/S0166-2236(02)02241-5
Baram, T. Z., and Hatalski, C. G. (1998). Neuropeptide-mediated excitability a key triggering mechanism for seizure generation in the developing brain. Trends Neurosci. 21, 471–476. doi: 10.1016/S0166-2236(98)01275-2
Barker, D. J., Winter, P. D., Osmond, C., Margetts, B., and Simmonds, S. J. (1989). Weight in infancy and death from ischaem heart disease. Lancet 2, 577–580.
Baserga, M., Hale, M. A., Wang, Z. M., Yu, X., and Callaway, C. W. (2007). Uteroplacental insufficiency alters nephrogenesis and downregulates cyclooxygenase-2 expression in a model of IUGR with adult-onset hypertension. Am. J. Physiol. Regul. Integr. Comp. Physiol. 292, R1943–R1955. doi: 10.1152/ajpregu.00558.2006
Baserga, M., Kaur, R., Hale, M. A., Bares, A., and Yu, X. (2010). Fetal growth restriction alters transcription factor binding and epigenetic mechanisms of renal 11beta-hydroxysteroid dehydrogenase type 2 in a sex-specific manner. Am. J. Physiol. Regul. Integr. Comp. Physiol. 299, R334–R342. doi: 10.1152/ajpregu.00122.2010
Bayer, S. A. (1980a). Development of the hippocampal region in the rat. I. Neurogenesis examined with 3H-thymidine autoradiography. J. Comp. Neurol. 190, 87–114. doi: 10.1002/cne.901900107
Bayer, S. A. (1980b). Development of the hippocampal region in the rat. II. Morphogenesis during embryonic and early postnatal life. J. Comp. Neurol. 190, 115–134. doi: 10.1002/cne.901900108
Beck, S. L., and Gavin, D. L. (1976). Suceptibility of mice to audiogenic seizures is increased by handling their dams during gestation. Science 193, 427–428. doi: 10.1126/science.945614
Benediktsson, R., Calder, A. A., Edwards, C. R., and Seckl, J. R. (1997). Placental 11β-hydroxysteroid dehydrogenase: a key regulator of fetal glucocorticoid exposure. Clin. Endocrinol. 46, 161–166. doi: 10.1046/j.1365-2265.1997.1230939.x
Bilbo, S. D., Barrientos, R. M., Eads, A. S., Northcutt, A., Watkins, L. R., Rudy, J. W., et al. (2008). Early-life infection leads to altered BDNF and IL-1beta mRNA expression in rat hippocampus following learning in adulthood. Brain Behav. Immun. 22, 451–455. doi: 10.1016/j.bbi.2007.10.003
Bitanihirwe, B. K., Peleg-Raibstein, D., Mouttet, F., Feldon, J., and Meyer, U. (2010). Late prenatal immune activation in mice leads to behavioral and neurochemical abnormalities relevant to the negative symptoms of schizophrenia. Neuropsychopharmacology 35, 2462–2478. doi: 10.1038/npp.2010.129
Blaise, J. H., Koranda, J. L., Chow, U., Haines, K. E., and Dorward, E. C. (2008). Neonatal isolation stress alters bidirectional long-term synaptic plasticity in amygdalo-hippocampal synapses in freely behaving adult rats. Brain Res. 1193, 25–33. doi: 10.1016/j.brainres.2007.11.049
Bogoch, Y., Biala, Y. N., Linial, M., and Weinstock, M. (2007). Anxiety induced by prenatal stress is associated with suppression of hippocampal genes involved in synaptic function. J. Neurochem. 101, 1018–1030. doi: 10.1111/j.1471-4159.2006.04402.x
Bohn, M. C., Dean, D., Hussain, S., and Giuliano, R. (1994). Development of mRNAs for glucocorticoid and mineralocorticoid receptors in rat hippocampus. Dev. Brain Res. 77, 157–162. doi: 10.1016/0165-3806(94)90192-9
Boksa, P. (2010). Effects of prenatal infection on brain development and behavior: a review of findings from animal models. Brain Behav. Immun. 24, 881–897. doi: 10.1016/j.bbi.2010.03.005
Bosnjak, J., Vukovic-Bobic, M., and Mejaski-Bosnjak, V. (2002). Effect of war on the occurrence of epileptic seizures in children. Epilepsy Behav. 3, 502–509. doi: 10.1016/S1525-5050(02)00602-9
Brunson, K. L., Kramar, E., Lin, B., Chen, Y., Colgin, L. L., Yanagihara, T. K., et al. (2005). Mechanisms of late-onset cognitive decline after early-life stress. J. Neurosci. 25, 9328–9338. doi: 10.1523/JNEUROSCI.2281-05.2005
Brunton, P. J., and Russell, J. A. (2010). Prenatal social stress in the rat programmes neuroendocrine and behavioural responses to stress in the adult offspring: sex specific effects. J. Neuroendocrinol. 22, 258–271. doi: 10.1111/j.1365-2826.2010.01969.x
Causevic, M., and Mohaupt, M. (2007). 11[beta]-Hydroxysteroid dehydrogenase type 2 in pregnancy and preeclampsia. Mol. Aspects Med. 28, 220–226. doi: 10.1016/j.mam.2007.04.003
Chen, M., and Zhang, L. (2011). Epigenetic mechanisms in developmental programming of adult disease. Drug Discov. Today 16, 1007–1018. doi: 10.1016/j.drudis.2011.09.008
Chen, Y., Andres, A. L., Frotscher, M., and Baram, T. Z. (2012). Tuning synaptic transmission in the hippocampus by stress: the CRH system. Front. Cell. Neurosci. 6:13. doi: 10.3389/fncel.2012.00013
Chen, Y., Bender, R. A., Frotscher, M., and Baram, T. Z. (2001). Novel and transient populations of corticotropin-releasing hormone-expressing neurons in developing hippocampus suggest unique functional roles: a quantitative spatiotemporal analysis. J. Neurosci. 21, 7171–7181.
Danese, A., Moffitt, T. E., Harrington, H., Milne, B. J., Polanczyk, G., Pariante, C. M., et al. (2009). Adverse childhood experiences and adult risk factors for age-related disease: depression, inflammation, and clustering of metabolic risk markers. Arch. Pediatr. Adolesc. Med. 163, 1135–1143. doi: 10.1001/archpediatrics.2009.214
De Bellis, M. D. (2002). Developmental traumatology: a contributory mechanism for alcohol and substance use disorders. Psychoneuroendocrinology 27, 155–170. doi: 10.1016/S0306-4530(01)00042-7
Deguchi, Y., Donato, F., Galimberti, I., Cabuy, E., and Caroni, P. (2011). Temporally matched subpopulations of selectively interconnected principal neurons in the hippocampus. Nat. Neurosci. 14, 495–504. doi: 10.1038/nn.2768
de Kloet, E. R., Joels, M., and Holsboer, F. (2005). Stress and the brain: from adaptation to disease. Nat. Rev. Neurosci. 6, 463–475. doi: 10.1038/nrn1683
Desgent, S., Duss, S., Sanon, N. T., Lema, P., Lévesque, M., Hébert, D., et al. (2012). Early-life stress is associated with gender-based vulnerability to epileptogenesis in rat pups. PLoS ONE 7:e42622. doi: 10.1371/journal.pone.0042622
Diaz, R., Brown, R. W., and Seckl, J. R. (1998). Distinct ontogeny of glucocorticoid and mineralocorticoid receptor and 11[beta]-hydroxysteroid dehydrogenase types I and II mRNAs in the fetal rat brain suggest a complex control of glucocorticoid actions. J. Neurosci. 18, 2570–2580.
Diz-Chaves, Y., Astiz, M., Bellini, M. J., and Garcia-Segura, L. M. (2013). Prenatal stress increases the expression of proinflammatory cytokines and exacerbates the inflammatory response to LPS in the hippocampal formation of adult male mice. Brain Behav. Immun. 28, 196–206. doi: 10.1016/j.bbi.2012.11.013
Diz-Chaves, Y., Pernia, O., Carrero, P., and Garcia-Segura, L. M. (2012). Prenatal stress causes alterations in the morphology of microglia and the inflammatory response of the hippocampus of adult female mice. J. Neuroinflammation 9, 71. doi: 10.1186/1742-2094-9-71
Dube, C., Boyet, S., Marescaux, C., and Nehlig, A. (2001). Relationship between neuronal loss and interictal glucose metabolism during the chronic phase of the lithium-pilocarpine model of epilepsy in the immature and adult rat. Exp. Neurol. 167, 227–241. doi: 10.1006/exnr.2000.7561
Dube, C., Richichi, C., Bender, R. A., Chung, G., Litt, B., Baram, T. Z., et al. (2006). Temporal lobe epilepsy after experimental prolonged febrile seizures: prospective analysis. Brain 129, 911–922. doi: 10.1093/brain/awl018
Edwards, H. E., Dortok, D., Tam, J., Won, D., and Burnham, W. M. (2002). Prenatal stress alters seizure thresholds and the development of kindled seizures in infant and adult rats. Horm. Behav. 42, 437–447. doi: 10.1006/hbeh.2002.1839
Fabricius, K., Wörtwein, G., and Pakkenberg, B. (2008). The impact of maternal separation on adult mouse behaviour and on the total neuron number in the mouse hippocampus. Brain Struct. Funct. 212, 403–416. doi: 10.1007/s00429-007-0169-6
Fenoglio, K. A., Brunson, K. L., Avishai-Eliner, S., Stone, B. A., Kapadia, B. J., Baram, T. Z., et al. (2005). Enduring, handling-evoked enhancement of hippocampal memory function and glucocorticoid receptor expression involves activation of the corticotropin-releasing factor type 1 receptor. Endocrinology 146, 4090–4096. doi: 10.1210/en.2004-1285
Field, T. (1994). The effects of mother's physical and emotional unavailability on emotion regulation. Monogr. Soc. Res. Child Dev. 59, 208–227. doi: 10.2307/1166147
Fowden, A. L., Giussani, D. A., and Forhead, A. J. (2005). Endocrine and metabolic programming during intrauterine development. Early Hum. Dev. 81, 723–734. doi: 10.1016/j.earlhumdev.2005.06.007
Francis, D. J., Diorio, J., Liu, D., and Meaney, M. J. (1999). Nongenomic transmission across generations of maternal behavior and stress responses in the rat. Science 286, 1155–1158. doi: 10.1126/science.286.5442.1155
Friso, S., Pizzolo, F., Choi, S. W., Guarini, P., and Castagna, A. (2008). Epigenetic control of 11 beta-hydroxysteroid dehydrogenase 2 gene promoter is related to human hypertension. Atherosclerosis 199, 323–327. doi: 10.1016/j.atherosclerosis.2007.11.029
Frodl, T., Reinhold, E., Koutsouleris, N., Reiser, M., and Meisenzahl, E. M. (2010). Interaction of childhood stress with hippocampus and prefrontal cortex volume reduction in major depression. J. Psychiatr. Res. 44, 799–807. doi: 10.1016/j.jpsychires.2010.01.006
Frye, C. A., and Bayon, L. E. (1999). Prenatal stress reduces the effectiveness of the neurosteroid 3 alpha, 5 alpha-THP to block kainic-acid-induced seizures. Dev. Psychobiol. 34, 227–234. doi: 10.1002/(SICI)1098-2302(199904)34:3<227::AID-DEV7>3.0.CO;2-H
Fujioka, A., Fujioka, T., Ishida, Y., Maekawa, T., and Nakamura, S. (2006). Differential effects of prenatal stress on the morphological maturation of hippocampal neurons. Neuroscience 141, 907–915. doi: 10.1016/j.neuroscience.2006.04.046
Gluckman, P. D., Hanson, M. A., and Beedle, A. S. (2007). Early life events and their consequences for later disease: a life history and evolutionary perspective. Am. J. Hum. Biol. 19, 1–19. doi: 10.1002/ajhb.20590
Gluckman, P. D., Hanson, M. A., and Spencer, H. G. (2005). Predictive adaptive responses and human evolution. Trends Ecol. Evol. 20, 527–533. doi: 10.1016/j.tree.2005.08.001
Glynn, L. M., Wadhwa, P. D., Dunkel Schetter, C., Chicz-Demet, A., and Sandman, C. A. (2001). When stress happens matters: effects of earthquake timing on stress responsivity in pregnancy. Am. J. Obstet. Gynecol. 184, 637–642. doi: 10.1067/mob.2001.111066
Gould, E., and Cameron, H. A. (1996). Regulation of neuronal birth, migration and death in the rat dentate gyrus. Dev. Neurosci. 18, 22–35. doi: 10.1159/000111392
Gräff, J., Kim, D., Dobbin, M. M., and Tsai, L. H. (2011). Epigenetic regulation of gene expression in physiological and pathological brain processes. Physiol. Rev. 91, 603–649. doi: 10.1152/physrev.00012.2010
Gunnar, M. R., and Cheatham, C. L. (2002). Brain and behavior interfaces: stress and the developing brain. Infant Ment. Health J. 24, 195–211. doi: 10.1002/imhj.10052
Gunnar, M. R., and Donzella, B. (2002). Social regulation of the cortisol levels in early human development. Psychoneuroendocrinology 27, 199–220. doi: 10.1016/S0306-4530(01)00045-2
Harris, A., and Seckl, J. (2011). Glucocorticoids, prenatal stress and the programming of disease. Horm. Behav. 59, 279–289. doi: 10.1016/j.yhbeh.2010.06.007
Hayashi, A., Nagaoka, M., Yamada, K., Ichitani, Y., Miake, Y., Okado, N., et al. (1998). Maternal stress induces synaptic loss and developmental disabilities of offspring. Int. J. Dev. Neurosci. 16, 209–219. doi: 10.1016/S0736-5748(98)00028-8
Holmes, G. L., and Ben-Ari, Y. (2001). The neurobiology and consequences of epilepsy in the developing brain. Pediatr. Res. 49, 320–325. doi: 10.1203/00006450-200103000-00004
Hout, R. L., Plotsky, P. M., Lenox, R. H., and McNamara, R. K. (2002). Neonatal maternal separation reduces hippocampal mossy fiber density in adult long evans rats. Brain Res. 950, 52–63. doi: 10.1016/S0006-8993(02)02985-2
Hsu, F. C., Zhang, G. J., Raol, Y. S., Valentino, R. J., Coulter, D. A., Brooks-Kayal, A. R., et al. (2003). Repeated neonatal handling with maternal separation permanently alters hippocampal GABAA receptors and behavioral stress responses. Proc. Natl. Acad. Sci. U.S.A. 100, 12213–12218. doi: 10.1073/pnas.2131679100
Huang, L. T., Holmes, G. L., Lai, M. C., Huang, P. L., Wang, C. L., Wang, T. J., et al. (2002a). Maternal deprivation stress exacerbates cognitive deficits in immature rats with recurrent seizures. Epilepsia 43, 1141–1148. doi: 10.1046/j.1528-1157.2002.14602.x
Huang, Y., Doherty, J. J., and Dingledine, R. (2002b). Altered histone acetylation at glutamate receptor 2 and brain-derived neurotrophic factor genes is an early event triggered by status epilepticus. J. Neurosci. 22, 8422–8428.
Huang, L. T., Tain, Y. L., Lai, M. C., and Yang, S. N. (2012). Neonatal seizures: dialogues between clinic and bench. J. Formos. Med. Assoc. 111, 239–244. doi: 10.1016/j.jfma.2011.12.007
Huang, Y., Myers, S. J., and Dingledine, R. (1999). Transcriptional repression by REST: recruitment of Sin3A and histone deacetylase to neuronal genes. Nat. Neurosci. 2, 867–872. doi: 10.1038/13165
Hulshof, H. J., Novati, A., Sgoifo, A., Luiten, P. G. M., den Boer, J. A., Meerlo, P., et al. (2011). Maternal separation decreases adult hippocampal cell proliferation and impairs cognitive performance but has little effect on stress sensitivity and anxiety in adult Wistar rats. Behav. Brain Res. 216, 552–560. doi: 10.1016/j.bbr.2010.08.038
Hunter, R. G. (2012). Epigenetic effects of stress and corticosteroids in the brain. Front. Cell. Neurosci. 6:18. doi: 10.3389/fncel.2012.00018
Ivy, A. S., Rex, C. S., Chen, Y., Dubé, C., Maras, P. M., Grigoriadis, D. E., et al. (2010). Hippocampal dysfunction and cognitive impairments provoked by chronic early-life stress involve excessive activation of CRH receptors. J. Neurosci. 30, 13005–13015. doi: 10.1523/JNEUROSCI.1784-10.2010
Jenkins, T. A. (2013). Perinatal complications and schizophrenia: involvement of the immune system. Front. Neurosci. 7:110. doi: 10.3389/fnins.2013.00110
Joels, M. (2009). Stress, the hippocampus, and epilepsy. Epilepsia 50, 586–597. doi: 10.1111/j.1528-1167.2008.01902.x
Jones, D. (2008). “Childhood maltreatment,” in Rutter's Child and Adolescent Psychiatry, eds M. Rutter, D. Bishop, D. Pine, S. Scott, J. Stevenson, E. Taylor, et al. (Malden, MA: Blackwell Publishing), 421–439. doi: 10.1002/9781444300895.ch28
Jung, C., Ho, J. T., Torpy, D. J., Rogers, A., Doogue, M., Lewis, J. G. et al. (2011). A longitudinal study of plasma and urinary cortisol in pregnancy and postpartum. J. Clin. Endocrinol. Metab. 96, 1533–1540. doi: 10.1210/jc.2010-2395
Karsten, C. A., and Baram, T. Z. (2013). How does a deuron “know” to modulate its epigenetic machinery in response to early-life environment/experience? Front. Psychiatry 4:89. doi: 10.3389/fpsyt.2013.00089
Kastin, A. J., and Akerstrom, V. (2002). Differential interactions of urocortin/corticotropin-releasing hormone peptides with the blood-brain barrier. Neuroendocrinology 75, 367–374. doi: 10.1159/000059433
Kawamura, T., Chen, J., Takahashi, T., Ichitani, Y., and Nakahara, D. (2006). Prenatal stress suppresses cell proliferation in the early developing brain. Neuroreport 17, 1515–1518. doi: 10.1097/01.wnr.0000236849.53682.6d
Khalaf-Nazzal, R., and Francis, F. (2013). Hippocampal development—old and new findings. Neuroscience 248C, 225–242. doi: 10.1016/j.neuroscience.2013.05.061
King, S., Laplante, D., and Joober, R. (2005). Understanding putative risk factors for schizophrenia: retrospective and prospective studies. J. Psychiatry Neurosci. 30, 342–348.
Kinney, D. K., Miller, A. M., Crowley, D. J., Huang, E., and Gerber, E. (2008). Autism prevalence following prenatal exposure to hurricanes and tropical storms in Louisiana. J. Autism Dev. Disord. 38, 481–488. doi: 10.1007/s10803-007-0414-0
Kobow, K., and Blumcke, I. (2011). The methylation hypothesis: do epigenetic chromatin modifications play a role in epileptogenesis? Epilepsia 52(Suppl. 4), 15–19. doi: 10.1111/j.1528-1167.2011.03145.x
Koe, A. S., Jones, N. C., and Salzberg, M. R. (2009). Early life stress as an influence on limbic epilepsy: an hypothesis whose time has come? Front. Behav. Neurosci. 3:24. doi: 10.3389/neuro.08.024.2009
Kumar, G., Couper, A., O'Brien, T. J., Salzberg, M. R., Jones, N. C., Rees, S. M., et al. (2007). The acceleration of amygdala kindling epileptogenesis by chronic low-dose corticosterone involves both mineralocorticoid and glucocorticoid receptors. Psychoneuroendocrinology 32, 834–842. doi: 10.1016/j.psyneuen.2007.05.011
Kumar, G., Jones, N. C., Morris, M. J., Rees, S., O'Brien, T. J., Salzberg, M. R., et al. (2011). Early life stress enhancement of limbic epileptogenesis in adult rats: mechanistic insights. PLoS ONE 6:e24033. doi: 10.1371/journal.pone.0024033
Lahiri, D. K., Maloney, B., and Zawia, N. H. (2009). The LEARn model: an epigenetic explanation for idiopathic neurobiological diseases. Mol. Psychiatry 14, 992–1003. doi: 10.1038/mp.2009.82
Lai, M. C., Holmes, G. L., Lee, K. H., Yang, S. N., Wang, C. A., Wu, C. L., et al. (2006). Effect of neonatal isolation on outcome following neonatal seizure in rats-the role of corticosterone. Epilepsy Res. 68, 123–136. doi: 10.1016/j.eplepsyres.2005.10.005
Lai, M. C., and Huang, L. T. (2011). Effects of early life stress on neuroendocrine and neurobehavior: mechanisms and implications. Pediatr. Neonatol. 52, 122–129. doi: 10.1016/j.pedneo.2011.03.008
Lemaire, V., Koehl, M., LeMoal, M., and Abrous, D. N. (2000). Prenatal stressproduces learning deficits associated with an inhibition of neurogensis in the hippocampus. Proc. Natl. Acad. Sci. U.S.A. 97, 11032–11037. doi: 10.1073/pnas.97.20.11032
Lemaire, V., Lamarque, S., LeMoal, M., Piazza, P. V., and Abrous, D. N. (2006). Postnatal stimulation of the pups counteracts prenatal stress-induced deficits in hippocampal neurogenesis. Biol. Psychiatry 59, 786–792. doi: 10.1016/j.biopsych.2005.11.009
Leussis, M. P., and Heinrichs, S. C. (2009). Quality of rearing guides expression of behavioral and neural seizure phenotypes in el mice. Brain Res. 1260, 84–93. doi: 10.1016/j.brainres.2009.01.007
Leventopoulos, M., Ruedi-Bettschen, D., Knuesel, I., Feldon, J., Pryce, C. R., Opacka-Juffry, J., et al. (2007). Long-term effects of early life deprivation on brain glia in Fischer rats. Brain Res. 1142, 119–126. doi: 10.1016/j.brainres.2007.01.039
Leverenz, J. B., Wilkinson, C. W., Wamble, M., Corbin, S., Grabber, J. E., Raskind, M,. A., et al. (1999). Effect of chronic high-dose exogenous cortisol on hippocampal neuronal number in aged nonhuman primates. J. Neurosci. 19, 2356–2361.
Levine, S. (2005). Developmental determinants of sensitivity and resistance to stress. Psychoneuroendocrinology 30, 939–946. doi: 10.1016/j.psyneuen.2005.03.013
Li, J., Vestergaard, M., Obel, C., Precht, D. H., Christensen, J., Lu, M., et al. (2008). Prenatal stress and epilepsy in later life: a nationwide follow-up study in Denmark. Epilepsy Res. 81, 52–57. doi: 10.1016/j.eplepsyres.2008.04.014
Lindsay, J. R., and Nieman, L. K. (2005). The hypothalamic-pituitary-adrenal axis in pregnancy: challenges in disease detection and treatment. Endocr. Rev. 26, 775–799. doi: 10.1210/er.2004-0025
Liu, D., Diorio, J., Tannenbaum, B., Caldji, C., Francis, D., and Freed-man, A. (1997). Maternal care, hippocampal glucocorticoid receptors, and hypothalamic-pituitaryadrenal response to stress. Science 277, 1659–1662. doi: 10.1126/science.277.5332.1659
Lubin, F. D. (2012). Epileptogenesis: can the science of epigenetics give us answers? Epilepsy Curr. 12, 105–110. doi: 10.5698/1535-7511-12.3.105
Lucassen, P. J., Naninck, E. F., van Goudoever, J. B., Fitzsimons, C., Joels, M., Korosi, A., et al. (2013). Perinatal programming of adult hippocampal structure and function; emerging roles of stress, nutrition and epigenetics. Trends Neurosci. 36, 621–631. doi: 10.1016/j.tins.2013.08.002
Lupien, S. J., McEwen, B. S., Gunnar, M. R., and Heim, C. (2009). Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nat. Rev. Neurosci. 10, 434–445. doi: 10.1038/nrn2639
Macrì, S., and Würbel, H. (2006). Developmental plasticity of HPA and fear responses in rats: a critical review of the maternal mediation hypothesis. Horm. Behav. 50, 667–680. doi: 10.1016/j.yhbeh.2006.06.015
Mairesse, J., Lesage, J., Breton, C., Breant, B., Hahn, T., Darnaudery, M., et al. (2007). Maternal stress alters endocrine function of the feto-placental unit in rats. Am. J. Physiol. Endocrinol. Metab. 292, E1526–E1533. doi: 10.1152/ajpendo.00574.2006
Matrisciano, F., Tueting, P., Dalal, I., Kadriu, B., Grayson, D. R., Davis, J. M., et al. (2013). Epigenetic modifications of GABAergic interneurons are associated with the schizophrenia-like phenotype induced by prenatal stress in mice. Neuropharmacology 68, 184–194. doi: 10.1016/j.neuropharm.2012.04.013
Matthews, S. G. (2000). Antenatal glucocorticoids and programming of the developing CNS. Pediatr. Res. 47, 291–300. doi: 10.1203/00006450-200003000-00003
McCabe, B. K., Silveira, D. C., Cilio, M. R., Cha, B. H., Liu, X., Sogawa, Y., et al. (2001). Reduced neurogenesis after neonatal seizures. J. Neurosci. 21, 2094–2103.
McClelland, S., Flynn, C., Dube, C., Richichi, C., Zha, Q., Ghestem, A., et al. (2011a). Neuron-restrictive silencer factor-mediated hyperpolarization-activated cyclic nucleotide gated channelopathy in experimental temporal lobe epilepsy. Ann. Neurol. 70, 454–464. doi: 10.1002/ana.22479
McClelland, S., Korosi, A., Cope, J., Ivy, A., and Baram, T. Z. (2011b). Emerging roles of epigenetic mechanisms in the enduring effects of early-life stress and experience on learning and memory. Neurobiol. Learn. Mem. 96, 79–88. doi: 10.1016/j.nlm.2011.02.008
McEwen, B. S. (1998). Protective and damaging effects of stress mediators. N. Engl. J. Med. 338, 171–179. doi: 10.1056/NEJM199801153380307
McLean, M., Bisits, A., Davies, J., Woods, R., Lowry, P., Smith, R., et al. (1995). A placental clock controlling the length of human pregnancy. Nat. Med. 1, 460–463. doi: 10.1038/nm0595-460
Meaney, M. J., Szyf, M., and Seckl, J. R. (2007). Epigenetic mechanisms of perinatal programming of hypothalamic-pituitary-adrenal function and health. Trends Mol. Med. 13, 269–277. doi: 10.1016/j.molmed.2007.05.003
Menard, J. L., and Hakvoort, R. M. (2007). Variations of maternal care alter offspring levels of behavioural defensiveness in adulthood: evidence for a threshold model. Behav. Brain Res. 176, 302–313. doi: 10.1016/j.bbr.2006.10.014
Michael, A. E., and Papageorghiou, A. T. (2008). Potential significance of physiological and pharmacological glucocorticoids in early pregnancy. Hum. Reprod. Update 14, 497–517. doi: 10.1093/humupd/dmn021
Miller, S. P., Weiss, J., Barnwell, A., Ferriero, D. M., Latal-Hajnal, B., Ferrer-Rogers, A., et al. (2002). Seizure-associated brain injury in term newborns with perinatal asphyxia. Neurology 58, 542–548. doi: 10.1212/WNL.58.4.542
Minshew, N., Sweeney, J., Bauman, M., and Webb, S. J. (2005). “Neurological aspects of autism,” in Handbook of Autism and Pervasive Developmental Disorders, 3rd Edn., eds F. R. Volkmar, R. Paul, A. Klin, and D. Cohen (Hoboken,NJ: Wiley), 473–514.
Mirescu, C., Peters, J. D., and Gould, E. (2004). Early life experience alters response of adult neurogenesis to stress. Nat. Neurosci. 7, 841–846. doi: 10.1038/nn1290
Morley-Fletcher, S., Mairesse, J., Soumier, A., Banasr, M., Fagioli, F., Gabriel, C., et al. (2011). Chronic agomelatine treatment corrects behavioral, cellular, and biochemical abnormalities induced by prenatal stress in rats. Psychopharmacology 217, 301–313. doi: 10.1007/s00213-011-2280-x
Mueller, B. R., and Bale, T. L. (2008). Sex-specific programming of offspring emotionality after stress early in pregnancy. J. Neurosci. 28, 9055–9065. doi: 10.1523/JNEUROSCI.1424-08.2008
Murgatroyd, C., and Spengler, D. (2011). Epigenetics of early child development. Front. Psychiatry 2:16. doi: 10.3389/fpsyt.2011.00016
Mychasiuk, R., Gibb, R., and Kolb, B. (2011). Prenatal bystander stress induces neuroanatomical changes in the prefrontal cortex and hippocampus of developing rat offspring. Brain Res. 1412, 55–62. doi: 10.1016/j.brainres.2011.07.023
Nair, A., Vadodaria, K. C., Banerjee, S. B., Benekareddy, M., Dias, B. G., Duman, R. S., et al. (2007). Stressor-specific regulation of distinct brain-derived neurotrophic factor transcripts and cyclic AMP response element-binding protein expression in the postnatal and adult rat hippocampus. Neuropsychopharmacology 32, 1504–1519. doi: 10.1038/sj.npp.1301276
Novakova, B., Harris, P. R., Ponnusamy, A., and Reuber, M. (2013). The role of stress as a trigger for epileptic seizures: a narrative review of evidence from human and animal studies. Epilepsia 54, 1866–1876. doi: 10.1111/epi.12377
Olney, J.W., Labruyere, J., Wang, G., Wozniak, D. F., Price, M.T., and Sesma, M.A. (1991). NMDA antagonist neurotoxicity: mechanism and prevention. Science 254, 1515–1518. doi: 10.1126/science.1835799
Oomen, C. A., Girardi, C. E., Cahyadi, R., Verbeek, E. C., Krugers, H., Joels, M., et al. (2009). Opposite effects of early maternal deprivation on neurogenesis in male versus female rats. PLoS ONE 4:e3675. doi: 10.1371/journal.pone.0003675
Oomen, C. A., Soeters, H., Audureau, N., Vermunt, L., van Hasselt, F. N., Manders, E. M., et al. (2010). Severe early life stress hampers spatial learning and neurogenesis, but improves hippocampal synaptic plasticity and emotional learning under high-stress conditions in adulthood. J. Neurosci. 30, 6635–6645. doi: 10.1523/JNEUROSCI.0247-10.2010
Orefice, L. L., and Heinrichs, S. C. (2008). Paternal care paradoxically increases offspring seizure susceptibility in the El mouse model of epilepsy. Epilepsy Behav. 12, 234–241. doi: 10.1016/j.yebeh.2007.09.004
Pitkänen, A., and Lukasiuk, K. (2011). Mechanisms of epileptogenesis and potential treatment targets. Lancet Neurol. 10, 173–186. doi: 10.1016/S1474-4422(10)70310-0
Poulton, R., Caspi, A., Milne, B. J., Thomson, W. M., Taylor, A., Sears, M. R., et al. (2010). Association between children's experience of socioeconomic disadvantage and adult health: a life-course study. Lancet 360, 1640–1645. doi: 10.1016/S0140-6736(02)11602-3
Qureshi, I. A., and Mehler, M. F. (2009). Regulation of non-coding RNA networks in the nervous system-what is the REST of the story? Neurosci. Lett. 466, 73–80. doi: 10.1016/j.neulet.2009.07.093
Rabbe, F. J., and Spengler, D. (2013). Epigenetic risk factors in PTSD and depression. Front. Psychiatry 4:80. doi: 10.3389/fpsyt.2013.00080
Rakic, P., and Nowakowski, R. S. (1981). The time of origin of neurons in the hippocampal region of the rhesus monkey. J. Comp. Neurol. 196, 99–128. doi: 10.1002/cne.901960109
Reddy, D. S. (2013). Role of hormones and neurosteroids in epileptogenesis. Front. Cell. Neurosci. 7:115. doi: 10.3389/fncel.2013.00115
Reul, J. M., and de Kloet, E. R. (1985). Two receptor systems for corticosterone in rat brain: microdistribution and differential occupation. Endocrinology 117, 2505–2511. doi: 10.1210/endo-117-6-2505
Ribak, C. E., and Baram, T. Z. (1996). Selective death of hippocampal CA3 pyramidal cells with mossy fiber afferents after CRH-induced status epilepticus in infant rats. Dev. Brain Res. 91, 245–251. doi: 10.1016/0165-3806(95)00183-2
Rodenas-Ruano, A., Chavez, A. E., Cossio, M. J., Castillo, P. E., and Zukin, R. S. (2012). REST-dependent epigenetic remodeling promotes the developmental switch in synaptic NMDA receptors. Nat. Neurosci. 15, 1382–1390. doi: 10.1038/nn.3214
Rogawski, M. A. (2013). AMPA receptors as a molecular target in epilepsy therapy. Acta Neurol. Scand. Suppl. 197, 9–18. doi: 10.1111/ane.12099
Roopra, A., Dingledine, R., and Hsieh, J. (2012). Epigenetics and epilepsy. Epilepsia 53(Suppl. 9) 2–10. doi: 10.1111/epi.12030
Sakanaka, M., Shibasaki, T., and Lederis, K. (1987). Corticotropin releasing factor-like 960 immunoreactivity in the rat brain as revealed by a modified cobalt-glucose 961oxidase-diaminobenzidine method. J. Comp. Neurol. 260, 256–298. doi: 10.1002/cne.902600209
Salzberg, M., Kumar, G., Supit, L., Jones, N. C., Morris, M. J., Rees, S., et al. (2007). Early postnatal stress confers enduring vulnerability to limbic epileptogenesis. Epilepsia 48, 2079–2085. doi: 10.1111/j.1528-1167.2007.01246.x
Sandberg, S., and Rutter, M. (2008). “Acute life stresses,” in Rutter's Child and Adolescent Psychiatry, eds M. Rutter, D. Bishop, D. Pine, S. Scott, J. Stevenson, E. Taylor, et al. (Malden, MA: Blackwell Publishing), 392–406. doi: 10.1002/9781444300895.ch26
Sandman, C. A., Glynn, L., Schetter, C. D., Wadhwa, P., Garite, T., Chicz-DeMet, A., et al. (2006). Elevated maternal cortisol early in pregnancy predicts third trimester levels of placental corticotropin releasing hormone (CRH): priming the placental clock. Peptides 27, 1457–1463. doi: 10.1016/j.peptides.2005.10.002
Sandman, C. A., Wadhwa, P. D., Glynn, L., Chicz-Demet, A., Porto, M., Garite, T. J., et al. (1999). Corticotrophin-releasing hormone and fetal responses in human pregnancy. Ann. N.Y. Acad. Sci. 897, 66–75. doi: 10.1111/j.1749-6632.1999.tb07879.x
Sapolsky, M. (1996). Stress, glucocorticoids, and damage to the nervous system: the current state of confusion. Stress 1, 1–19. doi: 10.3109/10253899609001092
Sawyer, N. T., and Escayg, A. (2010). Stress and epilepsy: multiple models, multiple outcomes. J. Clin. Neurophysiol. 27, 424–452. doi: 10.1097/WNP.0b013e3181fe0573
Scher, M. S. (2003). Neonatal seizures and brain damage. Pediatr. Neurol. 29, 381–390. doi: 10.1016/S0887-8994(03)00399-0
Scher, M. S., Hamid, M. Y., Steppe, D. A., Beggarly, M. E., and Painter, M. J. (1993). Ictal and interictal electrographic seizure durations in preterm and term neonates. Epilepsia 34, 284–288. doi: 10.1111/j.1528-1157.1993.tb02412.x
Schmidt, M. V. (2010). Molecular mechanisms of early life stress-lessons from mouse models. Neurosci. Biobehav. Rev. 34, 845–852. doi: 10.1016/j.neubiorev.2009.05.002
Schmidt, M. V., Oitzl, M. S., Levine, S., and De Kloet, E. R. (2002). The HPA system during the postnatal development of CD1 mice and the effects of maternal deprivation. Dev. Brain Res. 139, 39–49. doi: 10.1016/S0165-3806(02)00519-9
Seckl, J. R. (2004). Prenatal glucocorticoids and long-term programming. Eur. J. Endocrinol. 151, U49–U62. doi: 10.1530/eje.0.151U049
Seress, L., Abraham, H., Tornoczky, T., and Kosztolanyi, G. (2001). Cell formation in the human hippocampal formation from mid-gestation to the late postnatal period. Neuroscience 105, 831–843. doi: 10.1016/S0306-4522(01)00156-7
Shang, N. X., Zou, L. P., Zhao, J. B., Zhang, F., and Li, H. (2010). Association between prenatal stress and infantile spasms: a case-control study in China. Pediatr. Neurol. 42, 181–186. doi: 10.1016/j.pediatrneurol.2009.09.003
Sierksma, A. S., Prickaerts, J., Chouliaras, L., Rostamian, S., Delbroek, L., Rutten, B. P., et al. (2013). Behavioral and neurobiological effects of prenatal stress exposure in male and female APPswe/PS1dE9 mice. Neurobiol. Aging 34, 319–337. doi: 10.1016/j.neurobiolaging.2012.05.012
Sjöström, K., Valentin, L., Thelin T., and Marsál, K. (1997). Maternal anxiety in late pregnancy and fetal hemodynamics. Eur. J. Obstet. Gynecol. Reprod. Biol. 74, 49–55. doi: 10.1016/S0301-2115(97)00100-0
Sørensen, H. J., Mortensen, E. L., Reinisch, J. M., and Mednick, S. A. (2009). Association between prenatal exposure to bacterial infection and risk of schizophrenia. Schizophr. Bull. 35, 631–637. doi: 10.1093/schbul/sbn121
Strüber, N., Strüber, D., and Roth, G. (2014). Impact of early adversity on glucocorticoid regulation and later mental disorders. Neurosci. Biobehav. Rev. 38, 17–37. doi: 10.1016/j.neubiorev.2013.10.015
Suri, D., Veenit, V., Sarkar, A., Thiagarajan, D., Kumar, A., Nestler, E. J., et al. (2013). Early stress evokes age-dependent biphasic changes in hippocampal neurogenesis, BDNF expression, and cognition. Biol. Psychiatry 73, 658–666. doi: 10.1016/j.biopsych.2012.10.023
Swinkels, W. A., Engelsman, M., Kasteleijn-Nolst Trenite, D. G., Baal, M. G., de Haan, G. J., Oosting, J., et al. (1998). Influence of an evacuation in February 1995 in the placeNetherlands on the seizure frequency in patients with epilepsy: a controlled study. Epilepsia 39, 1203–1207. doi: 10.1111/j.1528-1157.1998.tb01312.x
Szuran, T. F., Pliška, V., Pokorny, J., and Welzl, H. (2000). Prenatal stress in rats: effects on plasma corticosterone, hippocampal glucocorticoid receptors, and maze performance. Physiol. Behav. 71, 353–362. doi: 10.1016/S0031-9384(00)00351-6
Talge, N. M., Neal, C., and Glover, V. (2007). Antenatal maternal stress and long-term effects on child neurodevelopment: how and why? J. Child Psychol. Psychiatry 48, 245–261. doi: 10.1111/j.1469-7610.2006.01714.x
Temkin, N. R., and Davis, G. R. (1984). Stress as a risk factor for seizures among adults with epilepsy. Epilepsia 25, 450–456. doi: 10.1111/j.1528-1157.1984.tb03442.x
Tyrka, A. R., Price, L. H., Gelernter, J., Schepker, C., Anderson, G. M., Carpenter, L. L., et al. (2009). Interaction of childhood maltreatment with the corticotropin-releasing hormone receptor gene: effects on hypothalamic-pituitary-adrenal axis reactivity. Biol. Psychiatry 66, 681–685. doi: 10.1016/j.biopsych.2009.05.012
van Campen, J. S., Jansen, F. E., de Graan, P. N., Braun, K. P., and Joels, M. (2013). Early life stress in epilepsy: a seizure precipitant and risk factor for epileptogenesis. Epilepsy Behav. doi: 10.1016/j.yebeh.2013.09.029. [Epub ahead of print].
van Campen, J. S., Jansen, F. E., Steinbusch, L. C., Joëls. M., and Braun, K. P. (2012). Stress sensitivity of childhood epilepsy is related to experienced negative life events. Epilepsia 53, 1554–1562. doi: 10.1111/j.1528-1167.2012.03566.x
van den Bergh, B. R., Mulder, E. J., Mennes, M., and Glover, V. (2005). Antenatal maternal anxiety and stress and the neurobehavioural development of the fetus and child: links and possible mechanisms a review. Neurosci. Biobehav. Rev. 29, 237–258. doi: 10.1016/j.neubiorev.2004.10.007
van Oers, H. J., De Kloet, E. R., Whelan, T., and Levine, S. (1998). Maternal deprivation effect on the infant's neural stress markers is reversed by tactile stimulation and feeding but not by suppressing corticosterone. J. Neurosci. 18, 10171–10179.
Vazquez, D. M., Lopez, J. F., Morano, M. I., Kwak, S. P., Watson, S. J., Akil, H., et al. (1998). Alpha, beta, and gamma mineralocorticoid receptor messenger ribonucleic acid splice variants: differential expression and rapid regulation in the developing hippocampus. Endocrinology 139, 3165–3177.
Velíšek, L. (2011). Prenatal corticosteroid exposure alters early developmental seizures and behavior. Epilepsy Res. 95, 9–19. doi: 10.1016/j.eplepsyres.2011.01.019
Vezzani, A., Friedman, A., and Dingledine, R. J. (2013). The role of inflammation in epileptogenesis. Neuropharmacology 69, 16–24. doi: 10.1016/j.neuropharm.2012.04.004
Viviani, B., Boraso, M., Valero, M., Gardoni, F., Marco, E. M., Llorente, R., et al. (2014). Early maternal deprivation immunologically primes hippocampal synapses by redistributing interleukin-1 receptor type I in a sex dependent manner. Brain Behav. Immun. 35, 135–143. doi: 10.1016/j.bbi.2013.09.008
Wadhwa, P. D., Garite, T. J., Porto, M., Glynn, L., Chicz-DeMet, A., Dunkel-Schetter, C., et al. (2004). Placental corticotropin-releasing hormone (CRH), spontaneous preterm birth, and fetal growth restriction: a prospective investigation. Am. J. Obstet. Gynecol. 191, 1063–1069. doi: 10.1016/j.ajog.2004.06.070
Wadhwa, P. D., Porto, M., Chicz-DeMet, A., and Sandman, C. A. (1998). Maternal CRH levels in early third trimester predict length of gestation in human pregnancy. Am. J. Obstet. Gynecol. 179, 1079–1085. doi: 10.1016/S0002-9378(98)70219-4
Wadhwa, P. D., Sandman, C. A., and Garite, T. J. (2001). The neurobiology of stress in human pregnancy: implications for prematurity and development of the fetal central nervous system. Prog. Brain Res. 133, 131–142. doi: 10.1016/S0079-6123(01)33010-8
Wang, X. D., Chen, Y., Wolf, M., Wagner, K. V., Liebl, C., Scharf, S. H., et al. (2011a). Forebrain CRHR1 deficiency attenuates chronic stress-induced cognitive deficits and dendritic remodeling. Neurobiol. Dis. 42, 300–310. doi: 10.1016/j.nbd.2011.01.020
Wang, X. D., Rammes, G., Kraev, I., Wolf, M., Liebl, C., Scharf, S. H., et al. (2011b). Forebrain CRF1 modulates early-life stress-programmed cognitive deficits. J. Neurosci. 31, 13625–13634. doi: 10.1523/JNEUROSCI.2259-11.2011
Wang, X. D., Su, Y. A., Wagner, K. V., Avrabos, C., Scharf, S. H., Hartmann, J., et al. (2013a). Nectin-3 links CRHR1 signaling to stress-induced memory deficits and spine loss. Nat. Neurosci. 16, 706–713. doi: 10.1038/nn.3395
Wang, H., Meyer, K., and Korz, V. (2013b). Stress induced hippocampal mineralocorticoid and estrogen receptor β gene expression and long-term potentiation in male adult rats is sensitive to early-life stress experience. Psychoneuroendocrinology 38, 250–262. doi: 10.1016/j.psyneuen.2012.06.004
Waterland, R. A., and Michels, K. B. (2007). Epigenetic epidemiology of the developmental origins hypothesis. Annu. Rev. Nutr. 27, 63–388. doi: 10.1146/annurev.nutr.27.061406.093705
Weaver, I. C., Cervoni, N., Champagne, F. A., D'Alessio, A. C., Sharma, S., Seckl, J. R., et al. (2004). Epigenetic programming by maternal behavior. Nat. Neurosci. 7, 847–854. doi: 10.1038/nn1276
Weaver, I. C., Champagne, F. A., Brown, S. E., Dymov, S., Sharma, S., Meaney, M. J., et al. (2005). Reversal of maternal programming of stress responses in adult offspring through methyl supplementation: altering epigenetic marking later in life. J. Neurosci. 25, 11045–11054. doi: 10.1523/JNEUROSCI.3652-05.2005
Weaver, I. C., D'Alessio, A. C., Brown, S. E., Hellstrom, I. C., Dymov, S., Sharma, S., et al. (2007). The transcription factor nerve growth factor-inducible protein a mediates epigenetic programming: altering epigenetic marks by immediate-early genes. J. Neurosci. 27, 1756–1768. doi: 10.1523/JNEUROSCI.4164-06.2007
Weaver, I. C., Meaney, M. J., and Szyf, M. (2006). Maternal care effects on the hippocampal transcriptome and anxiety-mediated behaviors in the offspring that are reversible in adulthood. Proc. Natl. Acad. Sci. U.S.A. 103, 3480–3485. doi: 10.1073/pnas.0507526103
Weinstock, M., Matlina, E., Maor, G. I., Rosen, H., and McEwen, B. S. (1992). Prenatal stress selectivity alters the reactivity of the hypothalamic-pituitary adrenal system in the female rat. Brain Res. 595, 195–200. doi: 10.1016/0006-8993(92)91049-K
Welberg, L. A., and Seckl, J. R. (2001). Prenatal stress, glucocorticoids and the programming of the brain. J. Neuroendocrinol. 13, 113–128. doi: 10.1111/j.1365-2826.2001.00601.x
Williams-Karnesky, R. L., Sandau, U. S., Lusardi, T. A., Lytle, N. K., Farrell, J. M., Pritchard, E. M., et al. (2013). Epigenetic changes induced by adenosine augmentation therapy prevent epileptogenesis. J. Clin. Invest. 123, 3552–3563. doi: 10.1172/JCI65636
Wong, M., and Guo, D. (2013). Dendritic spine pathology in epilepsy: cause or consequence? Neuroscience 251, 141–150. doi: 10.1016/j.neuroscience.2012.03.048
Woods, S. M., Melville, J. L., Guo, Y., Fan, M. Y., and Gavin, A. (2009). Psychosocial stress during pregnancy. Am. J. Obstet. Gynecol. 202, e1–e7. doi: 10.1016/j.ajog.2009.07.041
Wynne, O., Horvat, J. C., Kim, R. Y., Ong, L. K., Smith, R., Hansbro, P. M., et al. (2011). Neonatal respiratory infection and adult re-infection: effect on glucocorticoid and mineralocorticoid receptors in the hippocampus in BALB/c mice. Brain Behav. Immun. 25, 1214–1222. doi: 10.1016/j.bbi.2011.03.014
Young, N. A., Teskey, G. C., Henry, L. C., and Edwards, H. E. (2006). Exogenous antenatal glucocorticoid treatment reduces susceptibility for hippocampal kindled and maximal electroconvulsive seizures in infant rats. Exp. Neurol. 198, 303–312. doi: 10.1016/j.expneurol.2005.11.013
Yum, M. S., Chachua, T., Velíšková, J., and Velíšek, L. (2012). Prenatal stress promotes development of spasms in infant rats. Epilepsia 53, e46–e49. doi: 10.1111/j.1528-1167.2011.03357.x
Keywords: early-life stress, epigenetic, epileptogenesis, hippocampus, hypothalamic-pituitary-adrenal axis, prenatal stress, postnatal stress, seizure
Citation: Huang L-T (2014) Early-life stress impacts the developing hippocampus and primes seizure occurrence: cellular, molecular, and epigenetic mechanisms. Front. Mol. Neurosci. 7:8. doi: 10.3389/fnmol.2014.00008
Received: 29 November 2013; Accepted: 21 January 2014;
Published online: 10 February 2014.
Edited by:
Xiao-Dong Wang, Zhejiang University, ChinaReviewed by:
Mathias V. Schmidt, Max Planck Institute of Psychiatry, GermanyXiao-Dong Wang, Zhejiang University, China
Baojin Ding, University of Massachusetts Medical School, USA
Copyright © 2014 Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li-Tung Huang, Department of Pediatrics, Kaohsiung Chang Gung Memorial Hospital, 123 Ta-Pei Road, Niausung, Kaohsiung 833, Taiwan e-mail:aHVhbmdfbGlAcGllLmNvbS50dw==;bGl0dW5nLmh1YW5nQGdtYWlsLmNvbQ==