 Robert A. Kozol
Robert A. Kozol Alexander J. Abrams
Alexander J. Abrams David M. James
David M. James Elena Buglo2
Elena Buglo2 Qing Yan
Qing Yan Julia E. Dallman
Julia E. Dallman- 1Department of Biology, University of Miami, Coral Gables, FL, USA
- 2Department of Human Genetics, John P. Hussman Institute for Human Genomics, Dr. John T. Macdonald Foundation, University of Miami, Miami, FL, USA
Zebrafish are a unique cell to behavior model for studying the basic biology of human inherited neurological conditions. Conserved vertebrate genetics and optical transparency provide in vivo access to the developing nervous system as well as high-throughput approaches for drug screens. Here we review zebrafish modeling for two broad groups of inherited conditions that each share genetic and molecular pathways and overlap phenotypically: neurodevelopmental disorders such as Autism Spectrum Disorders (ASD), Intellectual Disability (ID) and Schizophrenia (SCZ), and neurodegenerative diseases, such as Cerebellar Ataxia (CATX), Hereditary Spastic Paraplegia (HSP) and Charcot-Marie Tooth Disease (CMT). We also conduct a small meta-analysis of zebrafish orthologs of high confidence neurodevelopmental disorder and neurodegenerative disease genes by looking at duplication rates and relative protein sizes. In the past zebrafish genetic models of these neurodevelopmental disorders and neurodegenerative diseases have provided insight into cellular, circuit and behavioral level mechanisms contributing to these conditions. Moving forward, advances in genetic manipulation, live imaging of neuronal activity and automated high-throughput molecular screening promise to help delineate the mechanistic relationships between different types of neurological conditions and accelerate discovery of therapeutic strategies.
Introduction
As genetically tractable vertebrates (Streisinger et al., 1981) that share with humans many pathways targeted by FDA approved pharmaceuticals (Renier et al., 2007; Rihel et al., 2010) zebrafish are a powerful model for inherited neurological conditions, both in terms of delineating underlying mechanisms and developing therapeutic strategies. Their small size and optical transparency enable in vivo visualization of cell- and systems-level processes throughout early developmental stages (McLean and Fetcho, 2011; Rasmussen and Sagasti, 2016) while, precocious development of quantifiable behaviors (Brustein et al., 2003) and reduced complexity of the zebrafish nervous system (Goulding, 2009) simplify functional studies of neural circuits. These advantages combined with conserved vertebrate genetics lend themselves to keeping pace with the extraordinary discovery rate of genetic mutations that cause inherited neurological conditions in humans. With the sequencing of the human genome, the formation of worldwide consortia of human geneticists and clinicians and ever-cheaper sequencing technologies, these discoveries have revealed that many inherited disorders with related clinical diagnoses consist of large sets of rare molecular genetic variation (Buxbaum et al., 2012; Gonzalez et al., 2015). Here we focus on these parallel and synergistic frontiers of disease gene discovery and systems-level analyses in zebrafish that promise to yield insight into disease mechanisms and therapies.
To assess zebrafish as a model, we compare studies of two broad classes of inherited neurological conditions: developmental disorders and degenerative diseases, with each class presenting a spectrum of overlapping genotypes and phenotypes. For developmental disorders, we include Autism Spectrum Disorders (ASD), Intellectual Disability (ID) and Schizophrenia (SCZ) that can all affect executive functions, social and overall intellectual abilities (American Psychiatric Association, and DSM-5 Task Force, 2013). Such disorders can co-occur in the same individual (Amaral et al., 2011) supporting overlapping disease etiologies. The second general class we consider are a subset of degenerative diseases including Cerebellar Ataxia (CATX), Hereditary Spastic Paraplegia (HSP), spinal motor atrophy (SMA), amyotrophic lateral sclerosis (ALS) and Charcot-Marie Tooth Disease (CMT) that impair movement due to degeneration of long axon tracts (Züchner and Vance, 2005). In both developmental and degenerative cases there are examples of: (1) distinct clinical features within a given class that result from mutations in the same genes; and (2) shared clinical phenotypes produced by many different types of genetic mutation (Espinós and Palau, 2009; Kaufman et al., 2010; Timmerman et al., 2013; Vissers et al., 2016). For example, ASD affects roughly 1% of the population but even the most commonly mutated genes only account for 1–3% of ASD with hundreds of suspected causal loci (Miles, 2011; De Rubeis and Buxbaum, 2015; Geschwind and State, 2015). It is likely that these two groupings also have phenotypic overlap since age of onset and progression of symptoms varies within each grouping. For example, SCZ that we group with “developmental” disorders also shares symptoms with neurodegenerative conditions. None-the-less, by reviewing the literature on zebrafish models of these two groups of disorders, we hope to highlight the role we feel the zebrafish model has to play in revealing grouped mechanisms of shared clinical features that result from diverse genetic mutations.
Comparing Human and Zebrafish Brains and Genetics
In addition to significant advantages of using zebrafish to model human disease, there are also challenges to modeling disease conditions in zebrafish. For example, in HSP symptoms are associated with degeneration of corticospinal tracts that have no clear homologous cell type in zebrafish. Moreover, for many human brain regions, the baseline studies to determine whether some of these brain regions function similarly to humans still need to be done. Finally, in terms of genetics, zebrafish have retained gene duplicates from a ray-finned fish whole genome duplication (Glasauer and Neuhauss, 2014) that likely provides both advantages (sub-functionalization of pleiotropic phenotypes) and disadvantages (genetic redundancy) for generating disease models.
Conserved Brain Regions?
Many brain regions relevant to human disease show molecular and structural homology in zebrafish (Figure 1). Gene expression patterns that molecularly define large-scale regions of fore-, mid-, hindbrain and spinal cord of the central nervous system (CNS) are generally conserved in vertebrates (Myers et al., 1986; Lumsden and Krumlauf, 1996; Wurst and Bally-Cuif, 2001; McLean et al., 2007; Wullimann et al., 2011). These major regions exhibit both neurochemical identities (e.g., neurotransmitters and receptors; Higashijima et al., 2004; Renier et al., 2007; Jones et al., 2015) and regional connectivity of sub-structures, including the thalamus (Mueller, 2012), optic tectum (Wullimann, 1994), hypothalamic regulatory nuclei (Herget et al., 2014), cerebellum (Hashimoto and Hibi, 2012), medulla oblongata (Kinkhabwala et al., 2011; Koyama et al., 2011), and spinal cord (Higashijima et al., 2004; Wen and Brehm, 2005). Determining homology between human and zebrafish forebrain structures is more challenging because of developmental differences affecting telencephalon topology (eversion vs. invagination) and the elaboration of the mammalian cerebrum (Striedter, 2005). Recent studies have made headway supporting the existence of zebrafish basal ganglia- (Filippi et al., 2014; Wullimann, 2014), cortex- (Ganz et al., 2014), amygdala- (Maximino et al., 2013) and hippocampus-like circuits (O’Connell and Hofmann, 2011; Maximino et al., 2013; Ganz et al., 2014). While these studies support the existence of structurally homologous brain regions, work is still needed to resolve connectivity and functional homology among fore- and midbrain structures. Furthermore some structures in the human brain appear absent in zebrafish including the pons (Wullimann et al., 2011), cortico-thalamic (Mueller, 2012) and cortico-spinal tracts (Babin et al., 2014). With these detailed analyses, we have the necessary anatomical map needed for neurological disease research that is quickly being enriched by functional studies to test the relevance to these brain regions for both zebrafish behaviors and human disorders.
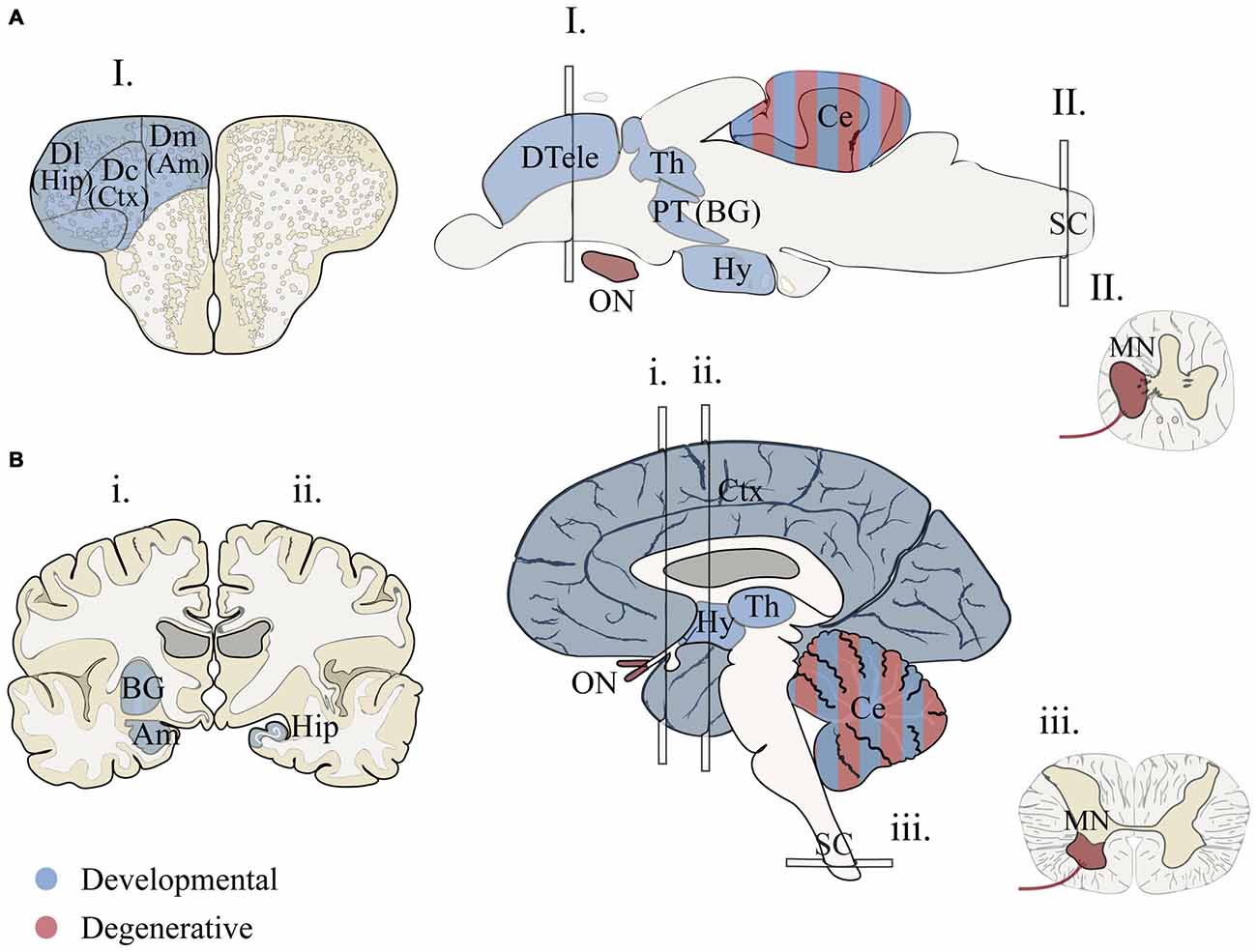
Figure 1. Proposed structural homology between zebrafish and humans for brain regions associated with human neurodevelopmental disorders and neurodegenerative diseases. (A) Adult zebrafish sections for I. telencephalon, brain and II. spinal chord. (B) Adult human sections for i., ii. telencephalon (anterior forebrain), brain and iii. spinal chord (transverse). Two hemi-sections were used (i. and ii.) to illustrate basal ganglia, hippocampus and amygdala. Regions associated with developmental disorders (blue) include cortical and subcortical structures that are vital for language, communication, memory, emotion and intellectual ability (Amaral et al., 2011; Bakhshi and Chance, 2015; Hampson and Blatt, 2015). Homologous forebrain regions for zebrafish are based on models that propose vertebrate structures that may be conserved for aspects of cognition and emotional behavior (Mueller and Wullimann, 2009; Mueller, 2012; Maximino et al., 2013; Filippi et al., 2014; Ganz et al., 2014; Wullimann, 2014). Conserved regions associated with axon degenerative diseases (red) include portions of the motor circuit and optic nerve (De Jonghe et al., 1997; Abrams et al., 2015). Zebrafish brain illustrations were adapted from Wullimann et al. (1996) and Mueller (2012). Am, amygdala; BG, basal ganglia; Ce, cerebellum; Ctx, cortex; Dc, dorsal central pallium; Dl, dorsal lateral pallium; Dm, dorsal medial pallium; DTele, dorsal telencephalon; Hip, hippocampus; Hy, hypothalamus; MN, motor neuron; PT, posterior tuberculum; Th, thalamus; ON, optic nerve.
To address the involvement of brain regions in a particular behavior, advances in functional imaging have been critical. Importantly, these studies focus on larval stages, 5–7 post fertilization, when it is possible to image the entire brain and the larva has already acquired an impressive repertoire of behaviors (Haesemeyer and Schier, 2015). For example, recent studies support functional homology of the zebrafish cerebellum during visual-motor behaviors (Hsieh et al., 2014; Matsui et al., 2014). These findings were made possible by combining behavioral experiments with both genetically encoded calcium imaging to visualize neuronal activity (Matsui et al., 2014) and loose patch recordings from cerebellar Purkinje neurons (Hsieh et al., 2014). These pioneering studies now open the door to sophisticated functional modeling of cerebellar-based neuropathies that can manifest in both neurodevelopmental disorders and neurodegenerative diseases, particularly CATX. Extending these functional technologies to forebrain circuits will be essential to support past studies suggesting similar cognitive and emotional functions exist in subdivisions of the zebrafish forebrain (Northcutt, 2006; O’Connell and Hofmann, 2011; Maximino et al., 2013).
Conserved Genomes?
Human and zebrafish genomes are highly conserved, with 76–82% of human disease genes present in zebrafish and an average of 20–24% of zebrafish genes duplicated (Howe et al., 2013). In some cases duplicates become sub-functionalized providing an advantage for studying pleiotropic phenotypes (Fleisch et al., 2008; Good et al., 2012; Lagman et al., 2015); in other cases, duplicates have redundant functions providing phenotypic buffering and complicating the generation of disease models (Hinits et al., 2012; Manoli and Driever, 2014). To determine if genes linked to a particular human neurological condition are enriched in gene duplicates we compared gene duplication rates in disease gene orthologs. Because past studies have shown duplicate enrichment in genes associated with neuronal development, signaling pathways and neuronal activity (Howe et al., 2013; Glasauer and Neuhauss, 2014), we hypothesized that neurodevelopmental duplicates would have a higher retention rate compared to neurodegenerative duplicates. Several gene sets showed a high duplicate retention frequency: over 60% (ASD-ID) and 45% (ASD and CMT; Figure 2A). In comparison, ID, ATX and HSP zebrafish orthologs had duplicate retention frequencies of 22–26% similar to the estimated genome-wide (20–24%) duplicate retention rate (Postlethwait et al., 2004; Howe et al., 2013). To determine if neurodevelopmental processes were enriched in sets with high duplicate retention rates we compared gene ortholog Gene Ontology (GO) term enrichments within orthologs sets (Mi et al., 2013). This program compares the frequency of a biological term (e.g., neuron development) with the expected frequency calculated for the genome. ASD (n = 35) and ASD-ID (n = 22) orthologs showed enrichments for GO terms associated with nervous system and neuronal development that confirm large studies analyzing extensive data sets of ASD and ID genes (Parikshak et al., 2013; Pinto et al., 2014). By contrast, CMT orthologs (n = 19) did not show any GO term enrichments overall, but those retained zebrafish CMT duplicates were enriched in genes associated with neuronal development (7/9 genes; Supplementary Material, Table S12). Therefore these genes may be associated with earlier onset cases of CMT, which can occur anywhere between birth and adulthood (De Jonghe et al., 1997). Although these gene sets are small and not statistically powerful, these rates show a general trend for retaining duplicates associated with neural development.
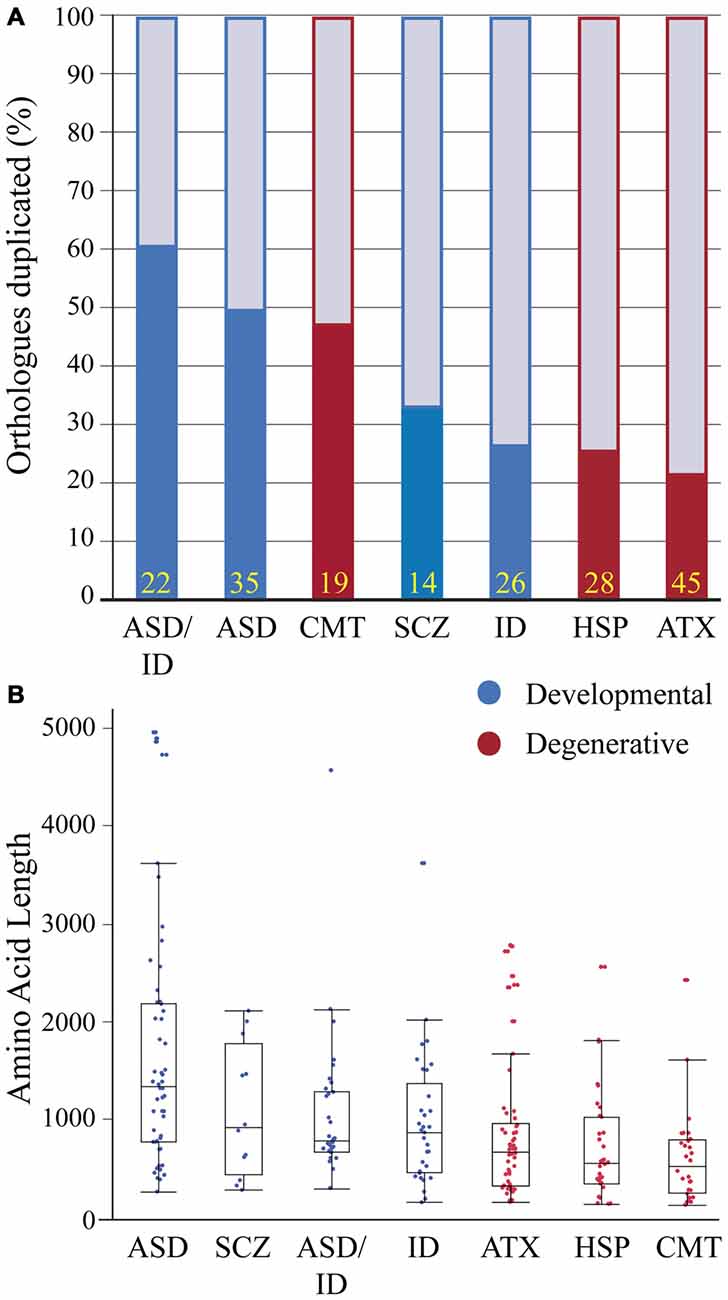
Figure 2. Zebrafish orthologs of human neurological disease genes vary with respect to duplicate retention and average protein size. (A) Gene duplicate retention rates in zebrafish are graphed for neurodevelopmental and neurodegenerative disease groups. Yellow numbers at the base of bars represent sample size. (B) Protein sizes of zebrafish orthologs of human disease genes with floating box plots (median with upper and lower quartile-box and range-whiskers). Note some larger proteins are outliers that fall outside of the calculated range. These gene sets for each human disease only incorporate a small percentage of associated genes and selection criteria varied because of the heterogeneity in genes linked to each disease and an emphasis on producing conservative lists. Each gene set was selected using data from research groups and review articles with the goal of including only the highest confidence disease genes based on either statistical thresholds and/or reoccurrence. Autism spectrum disorders (ASD) genes were chosen from the Simons Foundation Autism Initiative (SFARI.org) “high confidence” and “strong candidate” gene lists, which uses a multi-variable scoring analysis that includes sample size, statistical significance, replication, and functional analysis (Basu et al., 2009). ASD-intellectual disability (ID) genes were found in four separate reviews that provide evidence for reoccurrence in both ASD and ID (Kaufman et al., 2010; Krumm et al., 2014; Srivastava et al., 2014; Vissers et al., 2016). Schizophrenia (SCZ) genes were chosen from a SCZ genetics review, however this list is small and does not provide a confidence level for the disease contribution of each gene (Escudero and Johnstone, 2014). X-linked ID genes (Piton et al., 2013) and Charcot-Marie Tooth Disease (CMT; Timmerman et al., 2014) genes were chosen from recent meta-analyses to which we included a threshold of >5 cases per gene. Autosomal dominant and recessive and X-linked ataxia (ATX; Bird, 2016) and Hereditary Spastic Paraplegia (HSP; Fink, 2014) genes were chosen with well-known inheritance pedigrees. Human proteins for these gene lists were generated using BioMart (Kinsella et al., 2011) and the longest isoforms were used to identify zebrafish orthologs (Supplementary Material, Tables S1–7). Human proteins were then Blasted (Flicek et al., 2014) against the zebrafish proteome and ortholog information was recorded. Zebrafish proteins with low percent coverage, protein identity, e-value and ambiguous gene annotation (e.g., gene-like, LOC1084, etc.) were reciprocally blasted to confirm orthology.
To continue comparing these ortholog classes we looked at the length of the longest protein-coding isoform for each disease gene ortholog. This parameter impacts modeling due to numerous exons and complex splicing. For example SH3 and multiple ankyrin repeat domain 3 (SHANK3) is a large gene and mouse Shank3 knockout models exhibit variable phenotypes depending on whether a mutation is expressed in a predominant isoform (Peça et al., 2011; Zhou et al., 2016). Therefore we compared protein sizes between disease gene orthologs (Figure 2B). Between groups developmental disorders all had larger median protein sizes when compared to degenerative disease orthologs (Figure 2B). However, most notably proteins encoded by ASD genes were more than twice as long when compared to all degenerative disease proteins (Figure 2B). Because ASD genes have been suggested to encompass relatively large genes this result was perhaps not surprising (King et al., 2013; Uddin et al., 2014). Together these results on isoform length and duplicate enrichment suggests that ASD gene modeling in zebrafish will need to grapple with both extensive gene duplication and large complex genes that present challenges for knocking out all protein-coding isoforms and for generating rescue constructs.
Neurodevelopmental and Neurodegenerative Zebrafish Models
Developmental Disorders
Zebrafish models of developmental disorders have benefitted from the accessible embryonic stages and simplified nervous system that reveal an important role for signaling that patterns in the early embryo. Developmental disorders including ASD, ID and SCZ start to manifest phenotypically early in development and include deficits in social, learning and occupational functions (DSM-V). Because brain regions mediating human cognitive symptoms may lack parallels in zebrafish, modeling has focused on embryonic development and disorder comorbidities (such as sensory hypo-/hyper-sensitivity, sleep disruptions, and epilepsy) that still allow testing of etiological theories for these developmental disorders. These studies have paved the way for comprehensive functional assessments that link cellular- and circuit-level phenotypes to changes in behavior.
Molecular to Cellular Mechanisms: Signaling Pathways and Head Size
Mounting evidence links the etiology of neurodevelopmental disorders to embryonic stages. For example, teratogen exposure during gestation can cause developmental disorders (Arndt et al., 2005; Levy, 2011) and embryonic phenotypes in knockout mouse models provide support for an embryonic component underlying neurodevelopmental disorders (Knuesel et al., 2005; Lee et al., 2011; Durak et al., 2015). Zebrafish knockdown models of ASD and ID genes suggest that disrupted patterning of presumptive neural tissue in developmental disorders can occur as early as blastula stages (Yimlamai et al., 2005) and during gastrulation (De Rienzo et al., 2011; Turner et al., 2015). At the molecular level, these disruptions in patterning are likely due to changes in conserved signaling pathways. Several ASD and SCZ zebrafish models have investigated disease genes associated with the Wnt pathway (De Rienzo et al., 2011; Bernier et al., 2014; Brooks et al., 2014). For example, the Wnt interacting protein Chromodomain Helicase DNA binding domain 8 (CHD8) directly affects brain development during gastrulation and increases the size of the optic tectum, mirroring macroencephaly seen in ASD patients carrying CHD8 mutations (Bernier et al., 2014; Sugathan et al., 2014). These genotype-specific features (e.g., macrocephaly) provide a phenotypic screen that can be used to investigate genetic classes within disorders. For example zebrafish potassium channel tetramerization domain containing 13 (kctd13) was shown to have a dose-dependent affect in producing macrocephaly (knockdown) and microcephaly (overexpression) that supports a role for KCTD13 copy number variants causing head size phenotypes (Golzio et al., 2012). Moreover, Disrupted In Schizophrenia 1 (DISC1) interacts with canonical and non-canonical Wnt signaling and zdisc1 morphants and mutants exhibit disorganized axon tracts at larval stages that can be rescued by activating Wnt signaling (De Rienzo et al., 2011). These studies provide clear examples of utilizing zebrafish as an embryonic model to determine molecular and cellular mechanisms that define morphological phenotypes seen in individuals with developmental disorders.
Systems-Level Mechanisms: Disrupting the Balance of Neuronal Activity
Mechanisms affecting neuronal activity can contribute to neurodevelopmental disorders and zebrafish have been used to relate circuit-level changes in activity to behavior (Rubenstein and Merzenich, 2003; Eichler and Meier, 2008; Nelson and Valakh, 2015; Scharf et al., 2015). Circuit-level changes include disrupting the excitatory and inhibitory (E/I) balance, an operational set-point of excitation and inhibition within neural circuits that maintains functional behaviors (Borodinsky et al., 2004; Gatto and Broadie, 2010; Turrigiano, 2012; Vitureira et al., 2012; Davis, 2013). Zebrafish ASD and ID models have looked at E/I balance using transgenic fish lines expressing fluorescent glutamatergic (excitatory) and GABAergic (inhibitory) neurons (Kozol et al., 2015; Hoffman et al., 2016). Recently, Hoffman et al. found that populations of GABAnergic neurons were significantly decreased in contactin associated protein-like 2 (cntnap2ab) mutants, recapitulating a mutant mouse Cntnap2 model and suggesting that in the absence of cntnap2ab larvae fail to maintain inhibitory neuronal populations. This inhibitory decrease was shown to increase seizure susceptibility in cntnap2ab−/− mutants by applying a GABA receptor antagonist (Hoffman et al., 2016). In addition to seizure susceptibility, cntnap2ab−/− mutants had increased nighttime activity providing a circadian disruption for high-throughput drug screening. To identify potential therapies, they screened for drugs that reduced nighttime activity and identified a phytestrogen that restored wild type-like activity states. Like decreased inhibition, increased excitation is also known to contribute to developmental disorders. One well-studied example of this is augmented metabotropic glutamate receptor (mGluR) signaling in Fragile X Syndrome (Scharf et al., 2015). Similar to Fragile x mental retardation 1 (Fmr1) knockout models in mice, a zebrafish fmr1 knockdown model showed behavioral deficits that were ameliorated when treated with an mGluR inhibitor (Tucker et al., 2006). These studies demonstrate how zebrafish genetic models can be used to explore disorder etiology at multiple levels and efficiently test molecular theories for drug discovery.
Systems-Level Mechanisms: Comorbidities and Connecting Cells to Behavior
Individuals with developmental disorders are more likely to have accompanying medical conditions, or comorbidities, than typically developing individuals (Gurney et al., 2006; American Psychiatric Association, and DSM-5 Task Force, 2013; Chen et al., 2013a). Non-cognitive comorbidities such as sensory hypo- or hyper-sensitivity, epilepsy and gastrointestinal (GI) discomfort have revealed cellular-level mechanisms that may underlie behavioral phenotypes in developmental disorders. Several zebrafish knockdowns models of ASD, ID and epilepsy genes have looked at impaired touch sensitivity. Knockdown models of the ASD genes autism susceptibility candidate 2 (auts2) and shank3a exhibit hyposensitivity with concomitant neuronal cell death and morphological changes in skin innervating sensory neurons (Oksenberg et al., 2013; Kozol et al., 2015). Also exploring sensitivity, chromodomain helicase DNA binding protein 2 (chd2) knockdowns and sodium channel, voltage gated, type II, alpha (scn1lab) knockouts display hyper-excitable phenotypes that are characterized by extended or disorganized swimming with epileptiform-like activity in the brain (Baraban et al., 2013; Suls et al., 2013; Galizia et al., 2015). These epileptic swimming bouts provide a stereotyped behavior for high-throughput drug screening. Such a screen in scn1lab mutants identified anti-histamine clemizole as a novel anti-epileptic drug (Baraban et al., 2013). Although more focus has been paid to conditions such as epilepsy, other comorbidities like GI distress in ASD have yet to be investigated comprehensively (Hsiao, 2014; Bresnahan et al., 2015). For instance, chd8 morphants have a decrease in HuC/D positive enteric neurons innervating the gut and have impaired gut motility (Bernier et al., 2014). Again this example provided a cellular to systems level mechanism for GI distress seen in a majority of ASD patients carrying a CHD8 mutation. These examples all show the utility of zebrafish for studying comorbidities that impact the quality of life of large cohorts of patients; therefore a better understanding of the basis for these comorbidities would likely improve patient care.
Hereditary Neurodegenerative Disorders
Some common cellular mechanisms underlying degenerative diseases have been elucidated through gene discovery and zebrafish modeling of rare hereditary diseases. The degeneration of axon tracts in the central and peripheral nervous system are a clinical feature in neurodegenerative disorders such as Charcot-Marie-Tooth disease type 2 (CMT2), HSP, SMA or spinal muscle atrophy (SMA), ALS, as well as some forms of CATX which have phenotypic and mechanistic overlap (Züchner and Vance, 2005; Timmerman et al., 2013; Bargiela et al., 2015; Burté et al., 2015). The early development of zebrafish peripheral, motor and sensory neurons provide a foundation that has been used to dissect molecular mechanisms at both the cellular and systems level especially in models of SMA and ALS (McGown et al., 2013; Wiley et al., 2014). Using an innovative strategy to develop SMA therapies, one study used a high-throughput synthetic genetic array (SGA) screen in fission yeast to identify gene networks that when targeted with drugs reversed motor axon outgrowth deficits in a zebrafish SMA model (Wiley et al., 2014). Zebrafish models of these neurodegenerative diseases have also focused on molecular mechanisms such as axonal transport, mitochondrial dynamics, and autophagy, while also measuring morphological changes at the systems level such as alterations at the neuromuscular junction, degeneration of motor and sensory neurons, and disruptions of Purkinje cell (PC) development.
Cellular Level Mechanisms: Axonal Transport
A subset of the causative genes in these disorders are directly involved in axonal transport processes (Timmerman et al., 2013). The optical transparency of zebrafish and transgenic lines available make zebrafish an ideal model to study the relationship between axonal transport and axon degeneration in vivo (Plucińska et al., 2012; O’Donnell et al., 2014). Mutations in Kinesin Family member 5A (KIF5A), a molecular motor for transporting microtubule-mediated cargo, have been reported in both CMT2 (Crimella et al., 2012), and HSP patients (Reid et al., 2002; Fichera et al., 2004). A kif5a mutant zebrafish shows decreased touch response, and defective sensory neuronal maintenance all within the larval stages of development (Campbell et al., 2014). Furthermore the authors found that kif5a specifically affects the transport and distribution of mitochondria in neurons, but not lysosomes or presynaptic vesicles. Dominant mutations in Atlastin GTPase 1 (ATL1) encoding atlastin-1 cause an early onset form of HSP (Dürr et al., 2004). Morpholino knockdown of atl1 in zebrafish causes decreased mobility in larval fish and specifically disrupts axon tracts of spinal motor neurons (Fassier et al., 2010). Fassier et al. (2010) further demonstrated that the phenotype is the result of altered BMP signaling and that atlastin may play a role in BMP receptor trafficking. This link to BMP receptor trafficking suggested blocking BMP receptors as a therapeutic strategy that indeed ameliorated both cellular and behavioral phenotypes in the atl1 morphant zebrafish model. These zebrafish models support axonal transport as a cellular mechanism that could explain why long axons in CMT2 and HSP are primarily affected by genetic mutations in genes associated with transport processes.
Cellular Level Mechanisms: Mitochondrial Neuropathies
Mitochondrial dysfunction is another common mechanism in neurodegeneration. Dominant mutations in Mitofusin 2 (MFN2) are the primary cause of axonal degeneration in Charcot-Marie-Tooth Neuropathy (CMT2), and MFN2 has been implicated in the fusion and transport of mitochondria in neurons (Chen et al., 2003; Züchner et al., 2004; Baloh et al., 2007). Murine Mfn2 knockout and knock-in models are embryonic or postnatal lethal and do not develop a peripheral neuropathy, however a conditional knockout model did produce cerebellar degeneration and neonatal lethality (Chen et al., 2003, 2007; Strickland et al., 2014). In contrast, a stable mfn2L285X loss-of-function zebrafish model does recapitulate the motor neuron degenerative phenotype showing progressive loss of swimming ability, loss of neuromuscular junctions (NMJs), and early lethality by 1 year of age (Chapman et al., 2013). The authors found that the transport of mitochondria is disrupted in cultured motor neurons from the homozygous mfn2L285X at 24 hpf suggesting that a primary transport defect occurs before the onset of symptoms. In addition to axon degeneration of motor neurons a portion of patients with MFN2 mutations also develop optic atrophy (Züchner et al., 2006). The optic nerve seems to be particularly susceptible to mitochondrial dysfunction and is often affected in clinical spectrum phenotypes classified as mitochondrial optic neuropathies (Yu-Wai-Man et al., 2009). Recessive mutations in the nuclear encoded mitochondrial gene Optic Atrophy 3 (OPA3) cause a spectrum disorder classified as Costeff syndrome and includes optic atrophy, ataxia, extra pyramidal dysfunction, and increased urinary excretion of 3-methylglutaconic acid (MGC; Costeff et al., 1989). Zebrafish opa3 null mutants show increased MGC at both 5 dpf and at 2–5 months (Pei et al., 2010). At 1 year they show decreased optic nerve thickness and retinal ganglion cell density. Mutants have detectable changes in movement behaviors at larval stages and adults show loss of horizontal swimming. The authors speculate that the swimming phenotype can be attributed to ataxia, however TUNEL and histological staining of the cerebellum did not reveal any abnormalities. A third disease gene linked to mitochondrial dynamics and a spectrum of degenerative neurological conditions that include optic atrophy, CMT and cerebellar degeneration is Solute Carrier family 25, member 46 SLC25A46; Abrams et al., 2015). Studies in zebrafish and patient stem cells linked disruption of slc25a46 to reduced mitochondrial fission, altered distribution of mitochondria in motor neurons, and defective maintenance of neuronal processes. Even though cellular phenotypes were dramatic, swimming deficits in slc25a46 morphants were mild.
Cellular Level Mechanisms: Cerebellar Purkinje Neurons
Ataxia and associated sensory and motor phenotypes result from genetic mutations that affect various cell types within the spinocerebellar circuit. Cerebellar PCs are commonly affected and appear especially sensitive to peroxisomal dysfunction associated with PC loss or cerebellar atrophy (Akbar and Ashizawa, 2015). Consistent with this, zebrafish ataxia models, such as sorting nexin 14 (snx14) morphants show decreases in PC progenitors while cwf19-like 1 (cwf1911) morphants show disruptions in overall hindbrain morphology (Burns et al., 2014; Akizu et al., 2015). Alternatively, one group has focused on primary motor neurons for functional studies of Potassium Channel, voltage gated shaw related subfamily c, member 3 (KCN3) mutations that cause Spinocerebellar ataxia type 13 (Waters et al., 2006). They found that zebrafish kcn3a is expressed in the primary motor neurons and overexpression of a dominant negative version of this potassium channel decreases in neuronal excitability during fictive swimming (Issa et al., 2012). To follow up this study, Issa et al. also investigated the affect of KCN3 mutations associated with infant onset ataxia. Overexpression of two human KCN3 mutations demonstrated axonal defects that were only found in a mutation associated with infantile onset of SCA13. Given that neurodegenerative phenotypes become more severe with age, there has been doubt as to whether modeling these diseases in zebrafish larvae would be informative. The studies reviewed above indicate that indeed it is possible to gain functional insight into the basic biology of these disease genes from modeling in the larva. As a newer model, however, approaches in zebrafish are not quite as standardized as those in longer established models like Drosophila and mouse resulting in the diversity of modeling strategies employed by different researchers to model these inherited neurological conditions.
Lessons Learned From Modeling Inheritable Disease Genes
Homeostatic Plasticity: Unlinking Cellular and Systems Level Phenotypes
In some less common examples, genetic mutations cause cellular- and molecular-level phenotypes without leading to behavioral phenotypes. In zebrafish where’s waldo and strumpy mutants, neuromuscular synaptogenesis is defected but the mutants exhibit normal motility (Hutson and Chien, 2002; Panzer et al., 2005). Similar phenomena have been observed in mouse hypoxanthine-guanine phosporibosyltransferase (HPRT) and HPRT-adenine phosporibosyltransferase (APRT) mutant models of Lesch-Nyhan syndrome genes that produced an expected drop in dopamine but lacked self mutilation behaviors (Kuehn et al., 1987; Engle et al., 1996; Jinnah et al., 1999). These observations indicate that molecular- and cellular-level phenotype does not always correspond to behavioral phenotype, suggesting the existence of compensatory mechanisms at the systems-level. Besides genetic compensation from other genes, the nervous system also has remarkable capacity to stabilize its functions via homeostatic plasticity. Compensatory homeostatic plasticity operates on multiple levels, regulating synaptogenesis, synaptic strength and intrinsic excitability to stabilize neural circuit output in the context of genetic and/or environmental perturbations (Turrigiano, 2012; Vitureira et al., 2012; Davis, 2013). In zebrafish strumpyp37er mutants have enlarged NMJ acetylcholine receptor clusters are compared to wild-type (Panzer et al., 2005), but lack motor phenotypes, indicating homeostatic plasticity. In addition, defective homeostatic plasticity has been associated to a variety of human neurological diseases, including ASD, ID and Fragile X Syndrome (Rubenstein and Merzenich, 2003; Eichler and Meier, 2008; Gatto and Broadie, 2010; Yizhar et al., 2011; Wondolowski and Dickman, 2013; Nelson and Valakh, 2015). Mutant animal models with cellular but not behavioral phenotype have the potential to shed light on mechanisms of homeostatic plasticity at the systems-level. Given the diversity of molecular genetic pathways that contribute to developmental and degenerative disorders, a potential therapeutic target is to boost compensatory mechanisms that act to re-establish systems-level function.
Genetic Buffering: Molecular Compensation for Genetic Lesions
It is also common, though not necessarily well-represented in the literature, for knockout models to lack phenotypes. In a zebrafish study that generated mutant lines with 32 distinct lesions in 24 genes, most of the mutants exhibit a wild-type phenotype (Kok et al., 2015). Rather than a unique characteristic of zebrafish, such phenotypic buffering is found across single-celled to multi-cellular organisms. For example, >70% in fission yeast and >80% in bakers yeast, of genes can be individually mutated with little effect on haploid viability in the laboratory setting (Kim et al., 2010)1. Also in mice, though it is hard to accurately estimate the proportion of knockout mice without detectable phenotypes due to a lack of publications, the number is considerable (Barbaric et al., 2007). This phenomenon can be explained by the recent finding that other genes in a regulatory network can provide genetic compensation in mutants (Rossi et al., 2015). Such compensation can vary depending on genetic background (Gerlai, 1996; Pearson, 2002). Another recent study in zebrafish shows that the oncogenic B-RAF proto-oncogene (BRAFV600E) mutations rarely covert carrier cells into cancer cells unless in a p53 mutant background (Kaufman et al., 2016). The importance of genetic background for gene regulatory compensation likely contributes to disease penetrance and expressivity in humans as well (Zlotogora, 2003; Andersen and Al-Chalabi, 2011; Cooper et al., 2013; Persico and Napolioni, 2013), and suggests the importance to move from analyzing single gene to systems-level analysis of gene regulatory networks in disease models (Döhr et al., 2005; Barabási et al., 2011; Chen et al., 2014; Wiley et al., 2014; Marbach et al., 2016).
Broad Gene Expression; Local Pathology
Both developmental and degenerative diseases are associated with dysfunction in specific regions of the CNS (Figure 1). None-the-less most genes have heterogeneous spatial and temporal expression patterns that extend well beyond the windows of time and specific neural circuits associated with the disorder. Therefore we did a limited meta-analysis of mRNA expression of zebrafish disease orthologs. Unfortunately a zebrafish gene expression atlas does not exist and the most complete data sets only span embryonic development. Therefore we chose to compile previously published in situ hybridization data and determine the relative enrichment of gene expression in brain regions and spinal cord (Figure 3; Suplementary Material, Tables S13–18). During the first day of development, both gene sets show enriched expression in the hindbrain with the developmental set having enriched expression in the forebrain. By the second and third day, expression patterns become broader showing similar enrichment throughout the brain at these stages of morphogenesis and circuit formation. Broad CNS expression patterns suggest that genes play functional roles throughout development and across the nervous system despite being associated with symptoms that disrupt specific circuits during particular times of life.
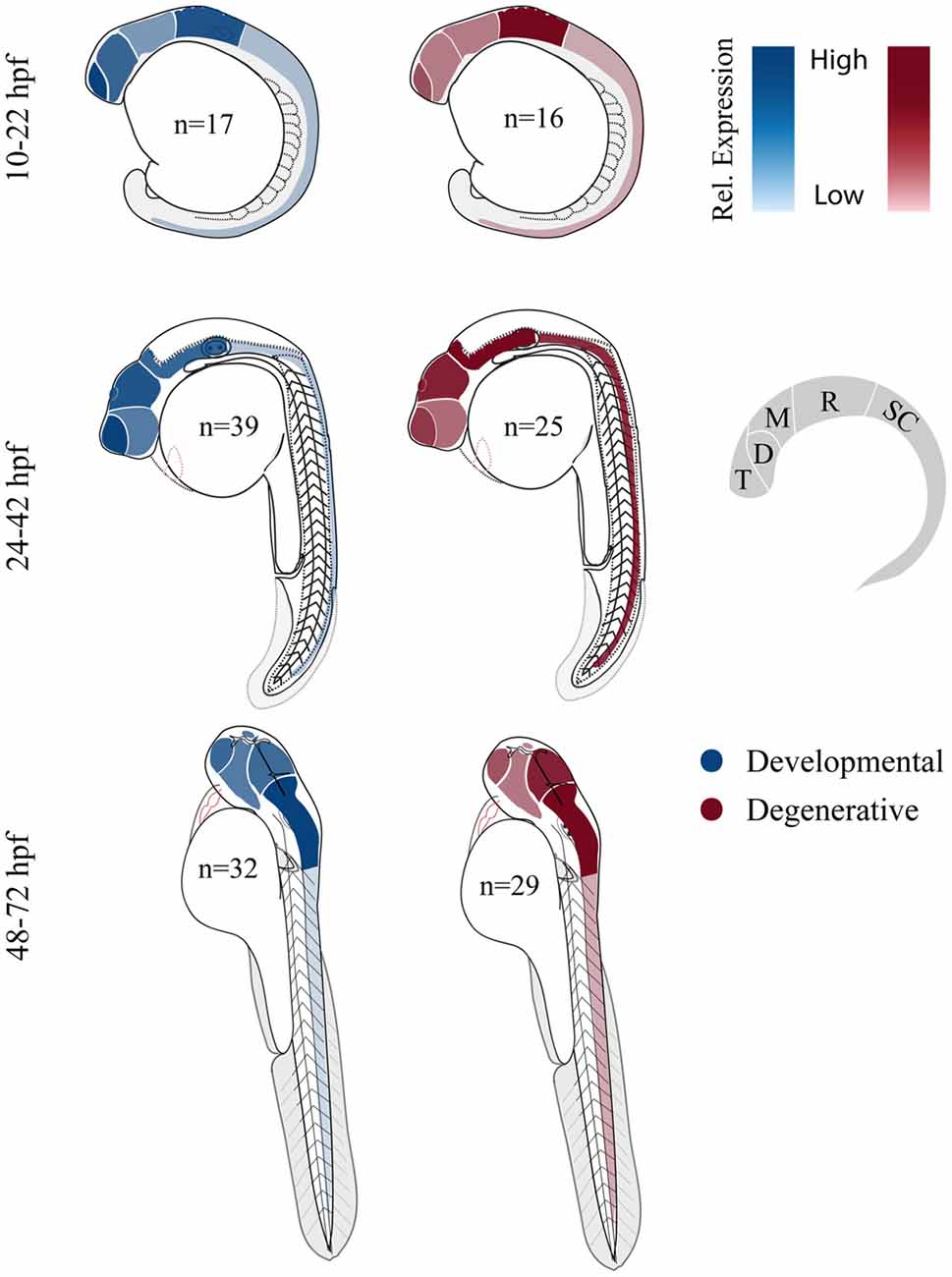
Figure 3. Comparing relative central nervous system (CNS) gene expression for developmental and degenerative disease gene orthologs in zebrafish. Previously published in situ hybridization data was collected from the zebrafish model organism database (ZFIN.org). Expression level was scored for each gene proportionally from 0 to 3; 0, no expression; 1, expression; 2, moderate expression; and 3, high level of expression, (i.e., gene x showed expression in the telencephalon (1) at 12 hpf and comparatively high expression in the telencephalon (3) at 36 hpf). Zebrafish larvae illustrations were adapted from Kimmel et al. (1995). D, diencephalon; M, mesencephalon; R, rhombencephalon; T, telencephalon and SC, spinal cord (De Jonghe et al., 1997; Kudoh et al., 2001; Thisse et al., 2001; Wurst and Bally-Cuif, 2001; Groth et al., 2002; Rauch et al., 2003; Thisse and Thisse, 2004, 2005; Croushore et al., 2005; Imamura and Kishi, 2005; Meyer et al., 2005; Thompson et al., 2005; Yimlamai et al., 2005; Liu et al., 2006; Mendelsohn et al., 2006; Meyer and Smith, 2006; Cheng et al., 2007; George et al., 2007; Goruppi et al., 2007; Katsuyama et al., 2007; Patten et al., 2007; Anichtchik et al., 2008; Stuebe et al., 2008; Sun et al., 2008; Yoshida and Mishina, 2008; Zhou et al., 2008; Emond et al., 2009; Ferrante et al., 2009; Monnich et al., 2009; Patten and Ali, 2009; Titus et al., 2009; Wood et al., 2009; Appelbaum et al., 2010; Davey et al., 2010; Fassier et al., 2010; Rissone et al., 2010; Takada and Appel, 2010; Mapp et al., 2011; Yeh et al., 2011; Artuso et al., 2012; Dresner et al., 2012; Gomez et al., 2012; Imai et al., 2012; Mueller, 2012; Pujol-Martí et al., 2012; Xing et al., 2012; Yanicostas et al., 2012; Baraban et al., 2013; Campbell and Marlow, 2013; Haug et al., 2013; Ng et al., 2013; Recher et al., 2013; Suls et al., 2013; Vatine et al., 2013; Bernier et al., 2014; Garbarino et al., 2014; Housley et al., 2014; Hsieh et al., 2014; Galizia et al., 2015; Kozol et al., 2015; Wakayama et al., 2015).
Looking Forward: Neural Circuits, Behavior, and Therapy
Because of the large number of rare mutations linked to inherited nervous system diseases, an important frontier for disease modeling is strategies that leverage to make stable F0 mutant models of inherited neurological disorders (Jao et al., 2013; Shah et al., 2015). Such models enable the rapid screening of candidate disease genes for whether they produce disease relevant phenotypes in the zebrafish model. To this end, many groups currently augment their MO studies by demonstrating similar phenotypes in F0 mutants (Bernier et al., 2014; Aspatwar et al., 2015; Bögershausen et al., 2015; O’Rawe et al., 2015; Wheeler et al., 2015; Xing et al., 2015). Still others have found mismatches between morphant and stable mutant phenotypes (Kok et al., 2015; Rossi et al., 2015; Stainier et al., 2015). Clearly in some cases these mismatches ascribed to non-specific effects of the morpholino (Kok et al., 2015) can also be explained by compensatory mechanisms masking the phenotype in the stable mutant (Rossi et al., 2015). To address the challenges of gene duplicates and multiple mutation causes of disease (Shah et al., 2015), several labs have further pioneered a strategy to pool guides targeting multiple genes and inject them together to efficiently screen multiple mutations in the F0 generation. Combined with a large repertoire of behaviors that develop within 5 days of fertilization and diverse transgenic lines for rapid screening of cellular phenotypes, F0 CRISPR zebrafish mutagenesis promises to contribute significantly to our understanding of genetic variation linked to nervous system disorders.
To model specific patient missense mutations and to better understand the basic biology of disease genes by tagging them in vivo or create conditional mutant alleles, several groups have also recently pioneered the use CRISPR/Cas9 for more sophisticated genome engineering. One novel strategy effectively “enhancer traps” a gene of interest by replacing the last exon with an engineered last exon encoding the C-terminal end of the coding sequence in frame with a cleavable p2A sequence followed by a fluorescent reporter (Li et al., 2015). In this way, the spatial and temporal expression dynamics of the protein can be captured. Also recently, Hoshijima et al. (2016) have developed streamlined strategies to precisely edit the genome and generate conditional mutant alleles flanked by LoxP sites. Such conditional mutant alleles have been used to great effect in mouse models to test when the mutation acts to produce different disease phenotypes. For example, in a mouse Shank3 autism model, rescuing the mutant Shank3 protein in the adult was sufficient to rescue social interactions and excessive grooming but not anxiety and repetitive motor behaviors (Mei et al., 2016).
The development of approaches that enable monitoring of behavior-relevant neural circuits in the intact larvae will be a boon for modeling inherited neurological disease (Ahrens et al., 2013; Fosque et al., 2015; Randlett et al., 2015; Dunn et al., 2016). Such approaches have the significant advantage of being unbiased. While many of these systems-level analyses use light-sheet or high-end microscopy to capture data (Ahrens et al., 2013; Fosque et al., 2015; Dunn et al., 2016), others use standard confocal microscopy to identify relevant brain circuits (Randlett et al., 2015) that are often spatially distributed across the nervous system. Several groups have also made concerted efforts towards establishing brain atlases to structurally and functionally annotate the brain (Ronneberger et al., 2012; Turner et al., 2014; Randlett et al., 2015) which is crucial since the ability to interpret imaging data is only as good as our understanding of brain regions (Arrenberg and Driever, 2013). Once these circuits are identified, they can then be studied in greater depth using in vivo calcium imaging with genetically encoded calcium indicators- (GECIs; Chen et al., 2013b), laser or enzymatic ablation of parts of the circuit (Liu and Fetcho, 1999; Tabor et al., 2014; Chen et al., 2016), electrophysiological recordings (Koyama et al., 2011; Baraban, 2013; Johnston et al., 2013; Wen et al., 2013), and optogenetics (Wyart et al., 2009; Kimura et al., 2013) to dissect circuit properties. Most of these approaches have yet to be broadly applied to zebrafish models of disease but once applied more broadly they promise to significantly contribute to our understanding of systems-level neural circuit mechanisms that contribute to symptoms of inherited neurological disorders.
In addition to genetic screens, due to their small size and their tendency to absorb drugs added directly to the water, zebrafish larvae are uniquely amenable for high-throughput drug screens (Rihel et al., 2010; Rihel and Schier, 2013; Bruni et al., 2014). High-throughput behavioral screens in zebrafish have enabled the classification of neuro-active drugs with respect to their impact on whole organism behavior. The ability to screen compounds in this manner is crucial since neuroactive drug discovery is still more empirical—a matter of what works—rather than rational—a matter of what makes sense based on chemistry and known molecular targets (Bruni et al., 2014). As highlighted in neurodevelopmental and neurodegenerative sections above, several disease models have made great use of the ability to use drugs to enhance or suppress mutant phenotypes as a means to identify therapeutic strategies (Fassier et al., 2010; Baraban et al., 2013; Hoffman et al., 2016).
Conclusion
The continued expertise and innovations of zebrafish genetic and developmental tools will continue to make zebrafish an attractive neurological disease model. Going forward, combining standard assays that allow comparisons across models with newer approaches would be ideal to enable a better understanding of the molecular, cellular, and systems-level groupings of these neurological conditions. Finally, zebrafish will certainly contribute to consortia of research groups that use multiple animal models for discovering essential molecular to circuit level mechanisms underlying neurological disease.
Author Contributions
RAK conducted all meta-analyses, made all figures and wrote introductory and developmental disorder sections and conducted extensive edits to coordinate sections. AJA wrote the bulk of the neurodegenerative section with the exception of the ataxia section that was written by EB. DMJ wrote comorbidity section that centered on GI distress in developmental disorders. QY wrote the lessons learned section. JED conceived the scope of the review, wrote frontiers section and helped RAK to conduct extensive edits to coordinate sections.
Funding
This work was supported by support from the National Institutes of Health Institute of Mental Health R03MH103857 to JED and from the Institute of General Medicine, an IMSD graduate fellowship from parent grant R25GM076419 to DMJ.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to acknowledge our human genetics colleagues Stephan Züchner, Rebecca Schüle, Margaret Pericak-Vance, and Joseph Buxbaum without whom we would never have started modeling human genetic disorders and diseases in zebrafish.
Footnotes
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fnmol.2016.00055/abstract
References
Abrams, A. J., Hufnagel, R. B., Rebelo, A., Zanna, C., Patel, N., Gonzalez, M. A., et al. (2015). Mutations in SLC25A46, encoding a UGO1-like protein, cause an optic atrophy spectrum disorder. Nat. Genet. 47, 926–932. doi: 10.1038/ng.3354
Ahrens, M. B., Orger, M. B., Robson, D. N., Li, J. M., and Keller, P. J. (2013). Whole-brain functional imaging at cellular resolution using light-sheet microscopy. Nat. Methods 10, 413–420. doi: 10.1038/nmeth.2434
Akbar, U., and Ashizawa, T. (2015). Ataxia. Neurol. Clin. 33, 225–248. doi: 10.1016/j.ncl.2014.09.004
Akizu, N., Cantagrel, V., Zaki, M. S., Al-Gazali, L., Wang, X., Rosti, R. O., et al. (2015). Biallelic mutations in SNX14 cause a syndromic form of cerebellar atrophy and lysosome-autophagosome dysfunction. Nat. Genet. 47, 528–534. doi: 10.1038/ng.3256
Amaral, D. G., Dawson, G., and Geschwind, D. H. (2011). Autism Spectrum Disorders. New York, NY: Oxford University Press, Inc.
American Psychiatric Association, and DSM-5 Task Force. (2013). Diagnostic and Statistical Manual of Mental Disorders : DSM-5. Washington, D.C.: American Psychiatric Association.
Andersen, P. M., and Al-Chalabi, A. (2011). Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat. Rev. Neurol. 7, 603–615. doi: 10.1038/nrneurol.2011.150
Anichtchik, O., Diekmann, H., Fleming, A., Roach, A., Goldsmith, P., and Rubinsztein, D. C. (2008). Loss of PINK1 function affects development and results in neurodegeneration in zebrafish. J. Neurosci. 28, 8199–8207. doi: 10.1523/jneurosci.0979-08.2008
Appelbaum, L., Wang, G., Yokogawa, T., Skariah, G. M., Smith, S. J., Mourrain, P., et al. (2010). Circadian and homeostatic regulation of structural synaptic plasticity in hypocretin neurons. Neuron 68, 87–98. doi: 10.1016/j.neuron.2010.09.006
Arndt, T. L., Stodgell, C. J., and Rodier, P. M. (2005). The teratology of autism. Int. J. Dev. Neurosci. 23, 189–199. doi: 10.1016/j.ijdevneu.2004.11.001
Arrenberg, A. B., and Driever, W. (2013). Integrating anatomy and function for zebrafish circuit analysis. Front. Neural Circuits 7:74. doi: 10.3389/fncir.2013.00074
Artuso, L., Romano, A., Verri, T., Domenichini, A., Argenton, F., Santorelli, F. M., et al. (2012). Mitochondrial DNA metabolism in early development of zebrafish (Danio rerio). Biochim. Biophys. Acta 1817, 1002–1011. doi: 10.1016/j.bbabio.2012.03.019
Aspatwar, A., Tolvanen, M. E., Ojanen, M. J., Barker, H. R., Saralahti, A. K., Bäuerlein, C. A., et al. (2015). Inactivation of ca10a and ca10b genes leads to abnormal embryonic development and alters movement pattern in zebrafish. PLoS One 10:e0134263. doi: 10.1371/journal.pone.0134263
Babin, P. J., Goizet, C., and Raldua, D. (2014). Zebrafish models of human motor neuron diseases: advantages and limitations. Prog. Neurobiol. 118, 36–58. doi: 10.1016/j.pneurobio.2014.03.001
Bakhshi, K., and Chance, S. A. (2015). The neuropathology of schizophrenia: a selective review of past studies and emerging themes in brain structure and cytoarchitecture. Neuroscience 303, 82–102. doi: 10.1016/j.neuroscience.2015.06.028
Baloh, R. H., Schmidt, R. E., Pestronk, A., and Milbrandt, J. (2007). Altered axonal mitochondrial transport in the pathogenesis of Charcot-Marie-Tooth disease from mitofusin 2 mutations. J. Neurosci. 27, 422–430. doi: 10.1523/jneurosci.4798-06.2007
Baraban, S. C. (2013). Forebrain electrophysiological recording in larval zebrafish. J. Vis. Exp. 17:50104. doi: 10.3791/50104
Baraban, S. C., Dinday, M. T., and Hortopan, G. A. (2013). Drug screening in Scn1a zebrafish mutant identifies clemizole as a potential Dravet syndrome treatment. Nat. Commun. 4:2410. doi: 10.1038/ncomms3410
Barabási, A. L., Gulbahce, N., and Loscalzo, J. (2011). Network medicine: a network-based approach to human disease. Nat. Rev. Genet. 12, 56–68. doi: 10.1038/nrg2918
Barbaric, I., Miller, G., and Dear, T. N. (2007). Appearances can be deceiving: phenotypes of knockout mice. Brief. Funct. Genomic. Proteomic. 6, 91–103. doi: 10.1093/bfgp/elm008
Bargiela, D., Shanmugarajah, P., Lo, C., Blakely, E. L., Taylor, R. W., Horvath, R., et al. (2015). Mitochondrial pathology in progressive cerebellar ataxia. Cerebellum Ataxias 2:16. doi: 10.1186/s40673-015-0035-x
Basu, S. N., Kollu, R., and Banerjee-Basu, S. (2009). AutDB: a gene reference resource for autism research. Nucleic Acids Res. 37, D832–D836. doi: 10.1093/nar/gkn835
Bernier, R., Golzio, C., Xiong, B., Stessman, H. A., Coe, B. P., Penn, O., et al. (2014). Disruptive CHD8 mutations define a subtype of autism early in development. Cell 158, 263–276. doi: 10.1016/j.cell.2014.06.017
Bird, T. D. (2016). Hereditary Ataxia Overview. Gene Reviews. Seattle, WA: University of Washington.
Bögershausen, N., Tsai, I. C., Pohl, E., Kiper, P. Ö., Beleggia, F., Percin, E. F., et al. (2015). RAP1-mediated MEK/ERK pathway defects in Kabuki syndrome. J. Clin. Invest. 125, 3585–3599. doi: 10.1172/jci80102
Borodinsky, L. N., Root, C. M., Cronin, J. A., Sann, S. B., Gu, H. L., and Spitzer, N. C. (2004). Activity-dependent homeostatic specification of transmitter expression in embryonic neurons. Nature 429, 523–530. doi: 10.1038/nature02518
Bresnahan, M., Hornig, M., Schultz, A. F., Gunnes, N., Hirtz, D., Lie, K. K., et al. (2015). Association of maternal report of infant and toddler gastrointestinal symptoms with autism: evidence from a prospective birth cohort. JAMA Psychiatry 72, 466–474. doi: 10.1001/jamapsychiatry.2014.3034
Brooks, S. S., Wall, A. L., Golzio, C., Reid, D. W., Kondyles, A., Willer, J. R., et al. (2014). A novel ribosomopathy caused by dysfunction of RPL10 disrupts neurodevelopment and causes X-linked microcephaly in humans. Genetics 198, 723–733. doi: 10.1534/genetics.114.168211
Bruni, G., Lakhani, P., and Kokel, D. (2014). Discovering novel neuroactive drugs through high-throughput behavior-based chemical screening in the zebrafish. Front. Pharmacol. 5:153. doi: 10.3389/fphar.2014.00153
Brustein, E., Saint-Amant, L., Buss, R. R., Chong, M., McDearmid, J. R., and Drapeau, P. (2003). Steps during the development of the zebrafish locomotor network. J. Physiol. Paris 97, 77–86. doi: 10.1016/j.jphysparis.2003.10.009
Burns, R., Majczenko, K., Xu, J., Peng, W., Yapici, Z., Dowling, J. J., et al. (2014). Homozygous splice mutation in CWF19L1 in a Turkish family with recessive ataxia syndrome. Neurology 83, 2175–2182. doi: 10.1212/wnl.0000000000001053
Burté, F., Carelli, V., Chinnery, P. F., and Yu-Wai-Man, P. (2015). Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat. Rev. Neurol. 11, 11–24. doi: 10.1038/nrneurol.2014.228
Buxbaum, J. D., Daly, M. J., Devlin, B., Lehner, T., Roeder, K., State, M. W., et al. (2012). The autism sequencing consortium: large-scale, high-throughput sequencing in autism spectrum disorders. Neuron 76, 1052–1056. doi: 10.1016/j.neuron.2012.12.008
Campbell, P. D., and Marlow, F. L. (2013). Temporal and tissue specific gene expression patterns of the zebrafish kinesin-1 heavy chain family, kif5s, during development. Gene Expr. Patterns 13, 271–279. doi: 10.1016/j.gep.2013.05.002
Campbell, P. D., Shen, K., Sapio, M. R., Glenn, T. D., Talbot, W. S., and Marlow, F. L. (2014). Unique function of Kinesin Kif5A in localization of mitochondria in axons. J. Neurosci. 34, 14717–14732. doi: 10.1523/jneurosci.2770-14.2014
Chapman, A. L., Bennett, E. J., Ramesh, T. M., De Vos, K. J., and Grierson, A. J. (2013). Axonal transport defects in a mitofusin 2 loss of function model of charcot-marie-tooth disease in zebrafish. PLoS One 8:e67276. doi: 10.1371/journal.pone.0067276
Chen, H., Detmer, S. A., Ewald, A. J., Griffin, E. E., Fraser, S. E., and Chan, D. C. (2003). Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 160, 189–200. doi: 10.1083/jcb.200211046
Chen, H., McCaffery, J. M., and Chan, D. C. (2007). Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell 130, 548–562. doi: 10.1016/j.cell.2007.06.026
Chen, J. C., Alvarez, M. J., Talos, F., Dhruv, H., Rieckhof, G. E., Iyer, A., et al. (2014). Identification of causal genetic drivers of human disease through systems-level analysis of regulatory networks. Cell 159, 402–414. doi: 10.1016/j.cell.2014.09.021
Chen, M. H., Su, T. P., Chen, Y. S., Hsu, J. W., Huang, K. L., Chang, W. H., et al. (2013a). Comorbidity of allergic and autoimmune diseases among patients with ADHD: a nationwide population-based study. J. Atten. Disord. doi: 10.1177/1087054712474686 [Epub ahead of print].
Chen, T. W., Wardill, T. J., Sun, Y., Pulver, S. R., Renninger, S. L., Baohan, A., et al. (2013b). Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499, 295–300. doi: 10.1038/nature12354
Chen, S., Chiu, C. N., McArthur, K. L., Fetcho, J. R., and Prober, D. A. (2016). TRP channel mediated neuronal activation and ablation in freely behaving zebrafish. Nat. Methods 13, 147–150. doi: 10.1038/nmeth.3691
Cheng, L., Chong, M., Fan, W., Guo, X., Zhang, W., Yang, X., et al. (2007). Molecular cloning, characterization and developmental expression of foxp1 in zebrafish. Dev. Genes Evol. 217, 699–707. doi: 10.1007/s00427-007-0177-9
Cooper, D. N., Krawczak, M., Polychronakos, C., Tyler-Smith, C., and Kehrer-Sawatzki, H. (2013). Where genotype is not predictive of phenotype: towards an understanding of the molecular basis of reduced penetrance in human inherited disease. Hum. Genet. 132, 1077–1130. doi: 10.1007/s00439-013-1331-2
Costeff, H., Gadoth, N., Apter, N., Prialnic, M., and Savir, H. (1989). A familial syndrome of infantile optic atrophy, movement disorder and spastic paraplegia. Neurology 39, 595–597. doi: 10.1212/wnl.39.4.595
Crimella, C., Baschirotto, C., Arnoldi, A., Tonelli, A., Tenderini, E., Airoldi, G., et al. (2012). Mutations in the motor and stalk domains of KIF5A in spastic paraplegia type 10 and in axonal Charcot-Marie-Tooth type 2. Clin. Genet. 82, 157–164. doi: 10.1111/j.1399-0004.2011.01717.x
Croushore, J. A., Blasiole, B., Riddle, R. C., Thisse, C., Thisse, B., Canfield, V. A., et al. (2005). Ptena and ptenb genes play distinct roles in zebrafish embryogenesis. Dev. Dyn. 234, 911–921. doi: 10.1002/dvdy.20576
Davey, C., Tallafuss, A., and Washbourne, P. (2010). Differential expression of neuroligin genes in the nervous system of zebrafish. Dev. Dyn. 239, 703–714. doi: 10.1002/dvdy.22195
Davis, G. W. (2013). Homeostatic signaling and the stabilization of neural function. Neuron 80, 718–728. doi: 10.1016/j.neuron.2013.09.044
De Jonghe, P., Timmerman, V., Nelis, E., Martin, J. J., and Van Broeckhoven, C. (1997). Charcot-Marie-Tooth disease and related peripheral neuropathies. J. Peripher. Nerv. Syst. 2, 370–387.
De Rienzo, G., Bishop, J. A., Mao, Y., Pan, L., Ma, T. P., Moens, C. B., et al. (2011). Disc1 regulates both beta-catenin-mediated and noncanonical Wnt signaling during vertebrate embryogenesis. FASEB J. 25, 4184–4197. doi: 10.1096/fj.11-186239
De Rubeis, S., and Buxbaum, J. D. (2015). Genetics and genomics of autism spectrum disorder: embracing complexity. Hum. Mol. Genet. 24, R24–R31. doi: 10.1093/hmg/ddv273
Döhr, S., Klingenhoff, A., Maier, H., Hrabé de Angelis, M., Werner, T., and Schneider, R. (2005). Linking disease-associated genes to regulatory networks via promoter organization. Nucleic Acids Res. 33, 864–872. doi: 10.1093/nar/gki230
Dresner, E., Malishkevich, A., Arviv, C., Leibman Barak, S., Alon, S., Ofir, R., et al. (2012). Novel evolutionary-conserved role for the activity-dependent neuroprotective protein (ADNP) family that is important for erythropoiesis. J. Biol. Chem. 287, 40173–40185. doi: 10.1074/jbc.m112.387027
Dunn, T. W., Mu, Y., Narayan, S., Randlett, O., Naumann, E. A., Yang, C. T., et al. (2016). Brain-wide mapping of neural activity controlling zebrafish exploratory locomotion. Elife 5:e12741. doi: 10.7554/elife.12741
Durak, O., de Anda, F. C., Singh, K. K., Leussis, M. P., Petryshen, T. L., Sklar, P., et al. (2015). Ankyrin-G regulates neurogenesis and Wnt signaling by altering the subcellular localization of beta-catenin. Mol. Psychiatry 20, 388–397. doi: 10.1038/mp.2014.42
Dürr, A., Camuzat, A., Colin, E., Tallaksen, C., Hannequin, D., Coutinho, P., et al. (2004). Atlastin1 mutations are frequent in young-onset autosomal dominant spastic paraplegia. Arch. Neurol. 61, 1867–1872. doi: 10.1001/archneur.61.12.1867
Eichler, S. A., and Meier, J. C. (2008). E-I balance and human diseases–from molecules to networking. Front. Mol. Neurosci. 1:2. doi: 10.3389/neuro.02.002.2008
Emond, M. R., Biswas, S., and Jontes, J. D. (2009). Protocadherin-19 is essential for early steps in brain morphogenesis. Dev. Biol. 334, 72–83. doi: 10.1016/j.ydbio.2009.07.008
Engle, S. J., Womer, D. E., Davies, P. M., Boivin, G., Sahota, A., Simmonds, H. A., et al. (1996). HPRT-APRT-deficient mice are not a model for lesch-nyhan syndrome. Hum. Mol. Genet. 5, 1607–1610. doi: 10.1093/hmg/5.10.1607
Escudero, I., and Johnstone, M. (2014). Genetics of schizophrenia. Curr. Psychiatry Rep. 16:502. doi: 10.1007/s11920-014-0502-8
Espinós, C., and Palau, F. (2009). Genetics and pathogenesis of inherited ataxias and spastic paraplegias. Adv. Exp. Med. Biol. 652, 263–296. doi: 10.1007/978-90-481-2813-6_18
Fassier, C., Hutt, J. A., Scholpp, S., Lumsden, A., Giros, B., Nothias, F., et al. (2010). Zebrafish atlastin controls motility and spinal motor axon architecture via inhibition of the BMP pathway. Nat. Neurosci. 13, 1380–1387. doi: 10.1038/nn.2662
Ferrante, M. I., Romio, L., Castro, S., Collins, J. E., Goulding, D. A., Stemple, D. L., et al. (2009). Convergent extension movements and ciliary function are mediated by ofd1, a zebrafish orthologue of the human oral-facial-digital type 1 syndrome gene. Hum. Mol. Genet. 18, 289–303. doi: 10.1093/hmg/ddn356
Fichera, M., Lo Giudice, M., Falco, M., Sturnio, M., Amata, S., Calabrese, O., et al. (2004). Evidence of kinesin heavy chain (KIF5A) involvement in pure hereditary spastic paraplegia. Neurology 63, 1108–1110. doi: 10.1212/01.wnl.0000138731.60693.d2
Filippi, A., Mueller, T., and Driever, W. (2014). vglut2 and gad expression reveal distinct patterns of dual GABAergic versus glutamatergic cotransmitter phenotypes of dopaminergic and noradrenergic neurons in the zebrafish brain. J. Comp. Neurol. 522, 2019–2037. doi: 10.1002/cne.23524
Fink, J. K. (2014). Herditary Spastic Paraplegia Overview. GeneReviews. Seattle, WA: University of Washington.
Fleisch, V. C., Schonthaler, H. B., von Lintig, J., and Neuhauss, S. C. (2008). Subfunctionalization of a retinoid-binding protein provides evidence for two parallel visual cycles in the cone-dominant zebrafish retina. J. Neurosci. 28, 8208–8216. doi: 10.1523/jneurosci.2367-08.2008
Flicek, P., Amode, M. R., Barrell, D., Beal, K., Billis, K., Brent, S., et al. (2014). Ensembl 2014. Nucleic Acids Res. 42, D749–D755. doi: 10.1093/nar/gkt1196
Fosque, B. F., Sun, Y., Dana, H., Yang, C. T., Ohyama, T., Tadross, M. R., et al. (2015). Neural circuits. Labeling of active neural circuits in vivo with designed calcium integrators. Science 347, 755–760. doi: 10.1126/science.1260922
Galizia, E. C., Myers, C. T., Leu, C., de Kovel, C. G., Afrikanova, T., Cordero-Maldonado, M. L., et al. (2015). CHD2 variants are a risk factor for photosensitivity in epilepsy. Brain 138, 1198–1207. doi: 10.1093/brain/awv052
Ganz, J., Kroehne, V., Freudenreich, D., Machate, A., Geffarth, M., Braasch, I., et al. (2014). Subdivisions of the adult zebrafish pallium based on molecular marker analysis. F1000Res. 3:308. doi: 10.12688/f1000research.5595.2
Garbarino, G., Costa, S., Pestarino, M., and Candiani, S. (2014). Differential expression of synapsin genes during early zebrafish development. Neuroscience 280, 351–367. doi: 10.1016/j.neuroscience.2014.09.015
Gatto, C. L., and Broadie, K. (2010). Genetic controls balancing excitatory and inhibitory synaptogenesis in neurodevelopmental disorder models. Front. Synaptic Neurosci. 2:4. doi: 10.3389/fnsyn.2010.00004
George, B., Datar, R. H., Wu, L., Cai, J., Patten, N., Beil, S. J., et al. (2007). p53 gene and protein status: the role of p53 alterations in predicting outcome in patients with bladder cancer. J. Clin. Oncol. 25, 5352–5358. doi: 10.1200/jco.2006.10.4125
Gerlai, R. (1996). Gene-targeting studies of mammalian behavior: is it the mutation or the background genotype? Trends Neurosci. 19, 177–181. doi: 10.1016/s0166-2236(96)20020-7
Geschwind, D. H., and State, M. W. (2015). Gene hunting in autism spectrum disorder: on the path to precision medicine. Lancet Neurol. 14, 1109–1120. doi: 10.1016/S1474-4422(15)00044-7
Glasauer, S. M., and Neuhauss, S. C. (2014). Whole-genome duplication in teleost fishes and its evolutionary consequences. Mol. Genet. Genomics 289, 1045–1060. doi: 10.1007/s00438-014-0889-2
Golzio, C., Willer, J., Talkowski, M. E., Oh, E. C., Taniguchi, Y., Jacquemont, S., et al. (2012). KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature 485, 363–367. doi: 10.1038/nature11091
Gomez, G., Lee, J. H., Veldman, M. B., Lu, J., Xiao, X., and Lin, S. (2012). Identification of vascular and hematopoietic genes downstream of etsrp by deep sequencing in zebrafish. PLoS One 7:e31658. doi: 10.1371/journal.pone.0031658
Gonzalez, M., Falk, M. J., Gai, X., Postrel, R., Schüle, R., and Zuchner, S. (2015). Innovative genomic collaboration using the GENESIS (GEM.app) platform. Hum. Mutat. 36, 950–956. doi: 10.1002/humu.22836
Good, S., Yegorov, S., Martijn, J., Franck, J., and Bogerd, J. (2012). New insights into ligand-receptor pairing and coevolution of relaxin family peptides and their receptors in teleosts. Int. J. Evol. Biol. 2012:310278. doi: 10.1155/2012/310278
Goruppi, S., Patten, R. D., Force, T., and Kyriakis, J. M. (2007). Helix-loop-helix protein p8, a transcriptional regulator required for cardiomyocyte hypertrophy and cardiac fibroblast matrix metalloprotease induction. Mol. Cell. Biol. 27, 993–1006. doi: 10.1128/mcb.00996-06
Goulding, M. (2009). Circuits controlling vertebrate locomotion: moving in a new direction. Nat. Rev. Neurosci. 10, 507–518. doi: 10.1038/nrn2608
Groth, C., Nornes, S., McCarty, R., Tamme, R., and Lardelli, M. (2002). Identification of a second presenilin gene in zebrafish with similarity to the human Alzheimer’s disease gene presenilin2. Dev. Genes Evol. 212, 486–490. doi: 10.1007/s00427-002-0269-5
Gurney, J. G., McPheeters, M. L., and Davis, M. M. (2006). Parental report of health conditions and health care use among children with and without autism: national survey of children’s health. Arch. Pediatr. Adolesc. Med. 160, 825–830. doi: 10.1001/archpedi.160.8.825
Haesemeyer, M., and Schier, A. F. (2015). The study of psychiatric disease genes and drugs in zebrafish. Curr. Opin. Neurobiol. 30, 122–130. doi: 10.1016/j.conb.2014.12.002
Hampson, D. R., and Blatt, G. J. (2015). Autism spectrum disorders and neuropathology of the cerebellum. Front. Neurosci. 9:420. doi: 10.3389/fnins.2015.00420
Hashimoto, M., and Hibi, M. (2012). Development and evolution of cerebellar neural circuits. Dev. Growth Differ. 54, 373–389. doi: 10.1111/j.1440-169x.2012.01348.x
Haug, M. F., Gesemann, M., Mueller, T., and Neuhauss, S. C. (2013). Phylogeny and expression divergence of metabotropic glutamate receptor genes in the brain of zebrafish (Danio rerio). J. Comp. Neurol. 521, 1533–1560. doi: 10.1002/cne.23240
Herget, U., Wolf, A., Wullimann, M. F., and Ryu, S. (2014). Molecular neuroanatomy and chemoarchitecture of the neurosecretory preoptic-hypothalamic area in zebrafish larvae. J. Comp. Neurol. 522, 1542–1564. doi: 10.1002/cne.23480
Higashijima, S., Masino, M. A., Mandel, G., and Fetcho, J. R. (2004). Engrailed-1 expression marks a primitive class of inhibitory spinal interneuron. J. Neurosci. 24, 5827–5839. doi: 10.1523/JNEUROSCI.5342-03.2004
Hinits, Y., Pan, L., Walker, C., Dowd, J., Moens, C. B., and Hughes, S. M. (2012). Zebrafish Mef2ca and Mef2cb are essential for both first and second heart field cardiomyocyte differentiation. Dev. Biol. 369, 199–210. doi: 10.1016/j.ydbio.2012.06.019
Hoffman, E. J., Turner, K. J., Fernandez, J. M., Cifuentes, D., Ghosh, M., Ijaz, S., et al. (2016). Estrogens suppress a behavioral phenotype in zebrafish mutants of the autism risk gene, CNTNAP2. Neuron 89, 725–733. doi: 10.1016/j.neuron.2015.12.039
Hoshijima, K., Jurynec, M. J., and Grunwald, D. J. (2016). Precise editing of the zebrafish genome made simple and efficient. Dev. Cell 36, 654–667. doi: 10.1016/j.devcel.2016.02.015
Housley, M. P., Reischauer, S., Dieu, M., Raes, M., Stainier, D. Y., and Vanhollebeke, B. (2014). Translational profiling through biotinylation of tagged ribosomes in zebrafish. Development 141, 3988–3993. doi: 10.1242/dev.111849
Howe, K., Clark, M. D., Torroja, C. F., Torrance, J., Berthelot, C., Muffato, M., et al. (2013). The zebrafish reference genome sequence and its relationship to the human genome. Nature 496, 498–503. doi: 10.1038/nature12111
Hsiao, E. Y. (2014). Gastrointestinal issues in autism spectrum disorder. Harv. Rev. Psychiatry 22, 104–111. doi: 10.1097/HRP.0000000000000029
Hsieh, J. Y., Ulrich, B., Issa, F. A., Wan, J., and Papazian, D. M. (2014). Rapid development of Purkinje cell excitability, functional cerebellar circuit and afferent sensory input to cerebellum in zebrafish. Front. Neural Circuits 8:147. doi: 10.3389/fncir.2014.00147
Hutson, L. D., and Chien, C. B. (2002). Wiring the zebrafish: axon guidance and synaptogenesis. Curr. Opin. Neurobiol. 12, 87–92. doi: 10.1016/s0959-4388(02)00294-5
Imai, H., Oomiya, Y., Kikkawa, S., Shoji, W., Hibi, M., Terashima, T., et al. (2012). Dynamic changes in the gene expression of zebrafish Reelin receptors during embryogenesis and hatching period. Dev. Growth Differ. 54, 253–263. doi: 10.1111/j.1440-169x.2012.01327.x
Imamura, S., and Kishi, S. (2005). Molecular cloning and functional characterization of zebrafish ATM. Int. J. Biochem. Cell Biol. 37, 1105–1116. doi: 10.1016/j.biocel.2004.10.015
Issa, F. A., Mock, A. F., Sagasti, A., and Papazian, D. M. (2012). Spinocerebellar ataxia type 13 mutation that is associated with disease onset in infancy disrupts axonal pathfinding during neuronal development. Dis. Model. Mech. 5, 921–929. doi: 10.1242/dmm.010157
Jao, L. E., Wente, S. R., and Chen, W. (2013). Efficient multiplex biallelic zebrafish genome editing using a CRISPR nuclease system. Proc. Natl. Acad. Sci. U S A 110, 13904–13909. doi: 10.1073/pnas.1308335110
Jinnah, H. A., Jones, M. D., Wojcik, B. E., Rothstein, J. D., Hess, E. J., Friedmann, T., et al. (1999). Influence of age and strain on striatal dopamine loss in a genetic mouse model of Lesch-Nyhan disease. J. Neurochem. 72, 225–229. doi: 10.1046/j.1471-4159.1999.0720225.x
Johnston, L., Ball, R. E., Acuff, S., Gaudet, J., Sornborger, A., and Lauderdale, J. D. (2013). Electrophysiological recording in the brain of intact adult zebrafish. J. Vis. Exp. 81:e51065. doi: 10.3791/51065
Jones, L. J., McCutcheon, J. E., Young, A. M., and Norton, W. H. (2015). Neurochemical measurements in the zebrafish brain. Front. Behav. Neurosci. 9:246. doi: 10.3389/fnbeh.2015.00246
Katsuyama, Y., Oomiya, Y., Dekimoto, H., Motooka, E., Takano, A., Kikkawa, S., et al. (2007). Expression of zebrafish ROR alpha gene in cerebellar-like structures. Dev. Dyn. 236, 2694–2701. doi: 10.1002/dvdy.21275
Kaufman, L., Ayub, M., and Vincent, J. B. (2010). The genetic basis of non-syndromic intellectual disability: a review. J. Neurodev. Disord. 2, 182–209. doi: 10.1007/s11689-010-9055-2
Kaufman, C. K., Mosimann, C., Fan, Z. P., Yang, S., Thomas, A. J., Ablain, J., et al. (2016). A zebrafish melanoma model reveals emergence of neural crest identity during melanoma initiation. Science 351:aad2197. doi: 10.1126/science.aad2197
Kim, D. U., Hayles, J., Kim, D., Wood, V., Park, H. O., Won, M., et al. (2010). Analysis of a genome-wide set of gene deletions in the fission yeast Schizosaccharomyces pombe. Nat. Biotechnol. 28, 617–623. doi: 10.1038/nbt.1628
Kimmel, C. B., Ballard, W. W., Kimmel, S. R., Ullmann, B., and Schilling, T. F. (1995). Stages of embryonic development of the zebrafish. Dev. Dyn. 203, 253–310. doi: 10.1002/aja.1002030302
Kimura, Y., Satou, C., Fujioka, S., Shoji, W., Umeda, K., Ishizuka, T., et al. (2013). Hindbrain V2a neurons in the excitation of spinal locomotor circuits during zebrafish swimming. Curr. Biol. 23, 843–849. doi: 10.1016/j.cub.2013.03.066
King, I. F., Yandava, C. N., Mabb, A. M., Hsiao, J. S., Huang, H. S., Pearson, B. L., et al. (2013). Topoisomerases facilitate transcription of long genes linked to autism. Nature 501, 58–62. doi: 10.1038/nature12504
Kinkhabwala, A., Riley, M., Koyama, M., Monen, J., Satou, C., Kimura, Y., et al. (2011). A structural and functional ground plan for neurons in the hindbrain of zebrafish. Proc. Natl. Acad. Sci. U S A 108, 1164–1169. doi: 10.1073/pnas.1012185108
Kinsella, R. J., Kähäri, A., Haider, S., Zamora, J., Proctor, G., Spudich, G., et al. (2011). Ensembl BioMarts: a hub for data retrieval across taxonomic space. Database (Oxford) 2011:bar030. doi: 10.1093/database/bar030
Knuesel, I., Elliott, A., Chen, H. J., Mansuy, I. M., and Kennedy, M. B. (2005). A role for synGAP in regulating neuronal apoptosis. Eur. J. Neurosci. 21, 611–621. doi: 10.1111/j.1460-9568.2005.03908.x
Kok, F. O., Shin, M., Ni, C. W., Gupta, A., Grosse, A. S., van Impel, A., et al. (2015). Reverse genetic screening reveals poor correlation between morpholino-induced and mutant phenotypes in zebrafish. Dev. Cell 32, 97–108. doi: 10.1016/j.devcel.2014.11.018
Koyama, M., Kinkhabwala, A., Satou, C., Higashijima, S., and Fetcho, J. (2011). Mapping a sensory-motor network onto a structural and functional ground plan in the hindbrain. Proc. Natl. Acad. Sci. U S A 108, 1170–1175. doi: 10.1073/pnas.1012189108
Kozol, R. A., Cukier, H. N., Zou, B., Mayo, V., De Rubeis, S., Cai, G., et al. (2015). Two knockdown models of the autism genes SYNGAP1 and SHANK3 in zebrafish produce similar behavioral phenotypes associated with embryonic disruptions of brain morphogenesis. Hum. Mol. Genet. 24, 4006–4023. doi: 10.1093/hmg/ddv138
Krumm, N., O’Roak, B. J., Shendure, J., and Eichler, E. E. (2014). A de novo convergence of autism genetics and molecular neuroscience. Trends Neurosci. 37, 95–105. doi: 10.1016/j.tins.2013.11.005
Kudoh, T., Tsang, M., Hukriede, N. A., Chen, X., Dedekian, M., Clarke, C. J., et al. (2001). A gene expression screen in zebrafish embryogenesis. Genome Res. 11, 1979–1987. doi: 10.1101/gr.209601
Kuehn, M. R., Bradley, A., Robertson, E. J., and Evans, M. J. (1987). A potential animal model for Lesch-Nyhan syndrome through introduction of HPRT mutations into mice. Nature 326, 295–298. doi: 10.1038/326295a0
Lagman, D., Callado-Pérez, A., Franzén, I. E., Larhammar, D., and Abalo, X. M. (2015). Transducin duplicates in the zebrafish retina and pineal complex: differential specialisation after the teleost tetraploidisation. PLoS One 10:e0121330. doi: 10.1371/journal.pone.0121330
Lee, F. H., Fadel, M. P., Preston-Maher, K., Cordes, S. P., Clapcote, S. J., Price, D. J., et al. (2011). Disc1 point mutations in mice affect development of the cerebral cortex. J. Neurosci. 31, 3197–3206. doi: 10.1523/JNEUROSCI.4219-10.2011
Levy, Y. (2011). Developmental delay revisited. Dev. Disabil. Res. Rev. 17, 180–184. doi: 10.1002/ddrr.1112
Li, J., Zhang, B. B., Ren, Y. G., Gu, S. Y., Xiang, Y. H., and Du, J. L. (2015). Intron targeting-mediated and endogenous gene integrity-maintaining knockin in zebrafish using the CRISPR/Cas9 system. Cell Res. 25, 634–637. doi: 10.1038/cr.2015.43
Liu, Q., Duff, R. J., Liu, B., Wilson, A. L., Babb-Clendenon, S. G., Francl, J., et al. (2006). Expression of cadherin10, a type II classic cadherin gene, in the nervous system of the embryonic zebrafish. Gene Expr. Patterns 6, 703–710. doi: 10.1016/j.modgep.2005.12.009
Liu, K. S., and Fetcho, J. R. (1999). Laser ablations reveal functional relationships of segmental hindbrain neurons in zebrafish. Neuron 23, 325–335. doi: 10.1016/s0896-6273(00)80783-7
Lumsden, A., and Krumlauf, R. (1996). Patterning the vertebrate neuraxis. Science 274, 1109–1115. doi: 10.1126/science.274.5290.1109
Manoli, M., and Driever, W. (2014). nkx2.1 and nkx2.4 genes function partially redundant during development of the zebrafish hypothalamus, preoptic region and pallidum. Front. Neuroanat. 8:145. doi: 10.3389/fnana.2014.00145
Mapp, O. M., Walsh, G. S., Moens, C. B., Tada, M., and Prince, V. E. (2011). Zebrafish Prickle1b mediates facial branchiomotor neuron migration via a farnesylation-dependent nuclear activity. Development 138, 2121–2132. doi: 10.1242/dev.060442
Marbach, D., Lamparter, D., Quon, G., Kellis, M., Kutalik, Z., and Bergmann, S. (2016). Tissue-specific regulatory circuits reveal variable modular perturbations across complex diseases. Nat. Methods 13, 366–370. doi: 10.1038/nmeth.3799
Matsui, H., Namikawa, K., Babaryka, A., and Köster, R. W. (2014). Functional regionalization of the teleost cerebellum analyzed in vivo. Proc. Natl. Acad. Sci. U S A 111, 11846–11851. doi: 10.1073/pnas.1403105111
Maximino, C., Lima, M. G., Oliveira, K. R., Batista Ede, J., and Herculano, A. M. (2013). “Limbic associative” and “autonomic” amygdala in teleosts: a review of the evidence. J. Chem. Neuroanat. 48–49, 1–13. doi: 10.1016/j.jchemneu.2012.10.001
McGown, A., McDearmid, J. R., Panagiotaki, N., Tong, H., Al Mashhadi, S., Redhead, N., et al. (2013). Early interneuron dysfunction in ALS: insights from a mutant sod1 zebrafish model. Ann. Neurol. 73, 246–258. doi: 10.1002/ana.23780
McLean, D. L., Fan, J., Higashijima, S., Hale, M. E., and Fetcho, J. R. (2007). A topographic map of recruitment in spinal cord. Nature 446, 71–75. doi: 10.1038/nature05588
McLean, D. L., and Fetcho, J. R. (2011). Movement, technology and discovery in the zebrafish. Curr. Opin. Neurobiol. 21, 110–115. doi: 10.1016/j.conb.2010.09.011
Mei, Y., Monteiro, P., Zhou, Y., Kim, J. A., Gao, X., Fu, Z., et al. (2016). Adult restoration of Shank3 expression rescues selective autistic-like phenotypes. Nature 530, 481–484. doi: 10.1038/nature16971
Mendelsohn, B. A., Yin, C., Johnson, S. L., Wilm, T. P., Solnica-Krezel, L., and Gitlin, J. D. (2006). Atp7a determines a hierarchy of copper metabolism essential for notochord development. Cell Metab. 4, 155–162. doi: 10.1016/j.cmet.2006.05.001
Meyer, M. P., and Smith, S. J. (2006). Evidence from in vivo imaging that synaptogenesis guides the growth and branching of axonal arbors by two distinct mechanisms. J. Neurosci. 26, 3604–3614. doi: 10.1523/JNEUROSCI.0223-06.2006
Meyer, M. P., Trimmer, J. S., Gilthorpe, J. D., and Smith, S. J. (2005). Characterization of zebrafish PSD-95 gene family members. J. Neurobiol. 63, 91–105. doi: 10.1002/neu.20118
Mi, H., Muruganujan, A., Casagrande, J. T., and Thomas, P. D. (2013). Large-scale gene function analysis with the PANTHER classification system. Nat. Protoc. 8, 1551–1566. doi: 10.1038/nprot.2013.092
Miles, J. H. (2011). Autism spectrum disorders—a genetics review. Genet. Med. 13, 278–294. doi: 10.1097/GIM.0b013e3181ff67ba
Monnich, M., Banks, S., Eccles, M., Dickinson, E., and Horsfield, J. (2009). Expression of cohesin and condensin genes during zebrafish development supports a non-proliferative role for cohesin. Gene Expr. Patterns 9, 586–594. doi: 10.1016/j.gep.2009.08.004
Mueller, T., and Wullimann, M. F. (2009). An evolutionary interpretation of teleostean forebrain anatomy. Brain Behav. Evol. 74, 30–42. doi: 10.1159/000229011
Mueller, T. (2012). What is the thalamus in zebrafish? Front. Neurosci. 6:64. doi: 10.3389/fnins.2012.00064
Myers, P. Z., Eisen, J. S., and Westerfield, M. (1986). Development and axonal outgrowth of identified motoneurons in the zebrafish. J. Neurosci. 6, 2278–2289.
Nelson, S. B., and Valakh, V. (2015). Excitatory/inhibitory balance and circuit homeostasis in autism spectrum disorders. Neuron 87, 684–698. doi: 10.1016/j.neuron.2015.07.033
Ng, M. C., Yang, Y. L., and Lu, K. T. (2013). Behavioral and synaptic circuit features in a zebrafish model of fragile X syndrome. PLoS One 8:e51456. doi: 10.1371/journal.pone.0051456
Northcutt, R. G. (2006). Connections of the lateral and medial divisions of the goldfish telencephalic pallium. J. Comp. Neurol. 494, 903–943. doi: 10.1002/cne.20853
O’Connell, L. A., and Hofmann, H. A. (2011). The vertebrate mesolimbic reward system and social behavior network: a comparative synthesis. J. Comp. Neurol. 519, 3599–3639. doi: 10.1002/cne.22735
O’Donnell, K. C., Lulla, A., Stahl, M. C., Wheat, N. D., Bronstein, J. M., and Sagasti, A. (2014). Axon degeneration and PGC-1alpha-mediated protection in a zebrafish model of alpha-synuclein toxicity. Dis. Model. Mech. 7, 571–582. doi: 10.1242/dmm.013185
Oksenberg, N., Stevison, L., Wall, J. D., and Ahituv, N. (2013). Function and regulation of AUTS2, a gene implicated in autism and human evolution. PLoS Genet. 9:e1003221. doi: 10.1371/journal.pgen.1003221
O’Rawe, J. A., Wu, Y., Dorfel, M. J., Rope, A. F., Au, P. Y., Parboosingh, J. S., et al. (2015). TAF1 variants are associated with dysmorphic features, intellectual disability and neurological manifestations. Am. J. Hum. Genet. 97, 922–932. doi: 10.1016/j.ajhg.2015.11.005
Panzer, J. A., Gibbs, S. M., Dosch, R., Wagner, D., Mullins, M. C., Granato, M., et al. (2005). Neuromuscular synaptogenesis in wild-type and mutant zebrafish. Dev. Biol. 285, 340–357. doi: 10.1016/j.ydbio.2005.06.027
Parikshak, N. N., Luo, R., Zhang, A., Won, H., Lowe, J. K., Chandran, V., et al. (2013). Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell 155, 1008–1021. doi: 10.1016/j.cell.2013.10.031
Patten, S. A., and Ali, D. W. (2009). PKCgamma-induced trafficking of AMPA receptors in embryonic zebrafish depends on NSF and PICK1. Proc. Natl. Acad. Sci. U S A 106, 6796–6801. doi: 10.1073/pnas.0811171106
Patten, S. A., Sihra, R. K., Dhami, K. S., Coutts, C. A., and Ali, D. W. (2007). Differential expression of PKC isoforms in developing zebrafish. Int. J. Dev. Neurosci. 25, 155–164. doi: 10.1016/j.ijdevneu.2007.02.003
Peça, J., Feliciano, C., Ting, J. T., Wang, W., Wells, M. F., Venkatraman, T. N., et al. (2011). Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 472, 437–442. doi: 10.1038/nature09965
Pei, W., Kratz, L. E., Bernardini, I., Sood, R., Yokogawa, T., Dorward, H., et al. (2010). A model of Costeff Syndrome reveals metabolic and protective functions of mitochondrial OPA3. Development 137, 2587–2596. doi: 10.1242/dev.043745
Persico, A. M., and Napolioni, V. (2013). Autism genetics. Behav. Brain Res. 251, 95–112. doi: 10.1016/j.bbr.2013.06.012
Pinto, D., Delaby, E., Merico, D., Barbosa, M., Merikangas, A., Klei, L., et al. (2014). Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am. J. Hum. Genet. 94, 677–694. doi: 10.1016/j.ajhg.2014.03.018
Piton, A., Redin, C., and Mandel, J. L. (2013). XLID-causing mutations and associated genes challenged in light of data from large-scale human exome sequencing. Am. J. Hum. Genet. 93, 368–383. doi: 10.1016/j.ajhg.2013.06.013
Plucińska, G., Paquet, D., Hruscha, A., Godinho, L., Haass, C., Schmid, B., et al. (2012). In vivo imaging of disease-related mitochondrial dynamics in a vertebrate model system. J. Neurosci. 32, 16203–16212. doi: 10.1523/JNEUROSCI.1327-12.2012
Postlethwait, J., Amores, A., Cresko, W., Singer, A., and Yan, Y. L. (2004). Subfunction partitioning, the teleost radiation and the annotation of the human genome. Trends Genet. 20, 481–490. doi: 10.1016/j.tig.2004.08.001
Pujol-Martí, J., Zecca, A., Baudoin, J. P., Faucherre, A., Asakawa, K., Kawakami, K., et al. (2012). Neuronal birth order identifies a dimorphic sensorineural map. J. Neurosci. 32, 2976–2987. doi: 10.1523/JNEUROSCI.5157-11.2012
Randlett, O., Wee, C. L., Naumann, E. A., Nnaemeka, O., Schoppik, D., Fitzgerald, J. E., et al. (2015). Whole-brain activity mapping onto a zebrafish brain atlas. Nat Methods 12, 1039–1046. doi: 10.1038/nmeth.3581
Rasmussen, J. P., and Sagasti, A. (2016). Learning to swim, again: axon regeneration in fish. Exp. Neurol. doi: 10.1016/j.expneurol.2016.02.022 [Epub ahead of print].
Rauch, G. J., Lyons, D. A., Middendorf, I., Friedlander, B., Arana, N., Reyes, T., et al. (2003). Submission and curation of gene expression data. ZFIN Direct Data Submission. Available online at: http://zfin.org
Recher, G., Jouralet, J., Brombin, A., Heuze, A., Mugniery, E., Hermel, J. M., et al. (2013). Zebrafish midbrain slow-amplifying progenitors exhibit high levels of transcripts for nucleotide and ribosome biogenesis. Development 140, 4860–4869. doi: 10.1242/dev.099010
Reid, E., Kloos, M., Ashley-Koch, A., Hughes, L., Bevan, S., Svenson, I. K., et al. (2002). A kinesin heavy chain (KIF5A) mutation in hereditary spastic paraplegia (SPG10). Am. J. Hum. Genet. 71, 1189–1194. doi: 10.1086/344210
Renier, C., Faraco, J. H., Bourgin, P., Motley, T., Bonaventure, P., Rosa, F., et al. (2007). Genomic and functional conservation of sedative-hypnotic targets in the zebrafish. Pharmacogenet. Genomics 17, 237–253. doi: 10.1097/fpc.0b013e3280119d62
Rihel, J., Prober, D. A., Arvanites, A., Lam, K., Zimmerman, S., Jang, S., et al. (2010). Zebrafish behavioral profiling links drugs to biological targets and rest/wake regulation. Science 327, 348–351. doi: 10.1126/science.1183090
Rihel, J., and Schier, A. F. (2013). Sites of action of sleep and wake drugs: insights from model organisms. Curr. Opin. Neurobiol. 23, 831–840. doi: 10.1016/j.conb.2013.04.010
Rissone, A., Sangiorgio, L., Monopoli, M., Beltrame, M., Zucchi, I., Bussolino, F., et al. (2010). Characterization of the neuroligin gene family expression and evolution in zebrafish. Dev. Dyn. 239, 688–702. doi: 10.1002/dvdy.22196
Ronneberger, O., Liu, K., Rath, M., Rueβ, D., Mueller, T., Skibbe, H., et al. (2012). ViBE-Z: a framework for 3D virtual colocalization analysis in zebrafish larval brains. Nat. Methods 9, 735–742. doi: 10.1038/nmeth.2076
Rossi, A., Kontarakis, Z., Gerri, C., Nolte, H., Hölper, S., Krüger, M., et al. (2015). Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature 524, 230–233. doi: 10.1038/nature14580
Rubenstein, J. L., and Merzenich, M. M. (2003). Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2, 255–267. doi: 10.1034/j.1601-183x.2003.00037.x
Scharf, S. H., Jaeschke, G., Wettstein, J. G., and Lindemann, L. (2015). Metabotropic glutamate receptor 5 as drug target for Fragile X syndrome. Curr. Opin. Pharmacol. 20, 124–134. doi: 10.1016/j.coph.2014.11.004
Shah, A. N., Davey, C. F., Whitebirch, A. C., Miller, A. C., and Moens, C. B. (2015). Rapid reverse genetic screening using CRISPR in zebrafish. Nat. Methods 12, 535–540. doi: 10.1038/nmeth.3360
Srivastava, S., Cohen, J., Pevsner, J., Aradhya, S., McKnight, D., Butler, E., et al. (2014). A novel variant in GABRB2 associated with intellectual disability and epilepsy. Am. J. Med. Genet. A 164A, 2914–2921. doi: 10.1002/ajmg.a.36714
Stainier, D. Y., Kontarakis, Z., and Rossi, A. (2015). Making sense of anti-sense data. Dev. Cell 32, 7–8. doi: 10.1016/j.devcel.2014.12.012
Streisinger, G., Walker, C., Dower, N., Knauber, D., and Singer, F. (1981). Production of clones of homozygous diploid zebra fish (Brachydanio rerio). Nature 291, 293–296. doi: 10.1038/291293a0
Strickland, A. V., Rebelo, A. P., Zhang, F., Price, J., Bolon, B., Silva, J. P., et al. (2014). Characterization of the mitofusin 2 R94W mutation in a knock-in mouse model. J. Peripher. Nerv. Syst. 19, 152–164. doi: 10.1111/jns5.12066
Stuebe, S., Wieland, T., Kraemer, E., Stritzky, A., Schroeder, D., Seekamp, S., et al. (2008). Sphingosine-1-phosphate and endothelin-1 induce the expression of rgs16 protein in cardiac myocytes by transcriptional activation of the rgs16 gene. Naunyn Schmiedebergs Arch. Pharmacol. 376, 363–373. doi: 10.1007/s00210-007-0214-2
Sugathan, A., Biagioli, M., Golzio, C., Erdin, S., Blumenthal, I., Manavalan, P., et al. (2014). CHD8 regulates neurodevelopmental pathways associated with autism spectrum disorder in neural progenitors. Proc. Natl. Acad. Sci. U S A 111, E4468–E4477. doi: 10.1073/pnas.1405266111
Suls, A., Jaehn, J. A., Kecskes, A., Weber, Y., Weckhuysen, S., Craiu, D. C., et al. (2013). De novo loss-of-function mutations in CHD2 cause a fever-sensitive myoclonic epileptic encephalopathy sharing features with Dravet syndrome. Am. J. Hum. Genet. 93, 967–975. doi: 10.1016/j.ajhg.2013.09.017
Sun, X. J., Xu, P. F., Zhou, T., Hu, M., Fu, C. T., Zhang, Y., et al. (2008). Genome-wide survey and developmental expression mapping of zebrafish SET domain-containing genes. PLoS One 3:e1499. doi: 10.1371/journal.pone.0001499
Tabor, K. M., Bergeron, S. A., Horstick, E. J., Jordan, D. C., Aho, V., Porkka-Heiskanen, T., et al. (2014). Direct activation of the Mauthner cell by electric field pulses drives ultrarapid escape responses. J. Neurophysiol. 112, 834–844. doi: 10.1152/jn.00228.2014
Takada, N., and Appel, B. (2010). Identification of genes expressed by zebrafish oligodendrocytes using a differential microarray screen. Dev. Dyn. 239, 2041–2047. doi: 10.1002/dvdy.22338
Thisse, B., Pflumio, S., Furthauer, M., Loppin, B., Heyer, V., Degrave, A., et al. (2001). Expression of the zebrafish genome during embryogenesis. ZFIN Direct Data Submission. Available online at: http://zfin.org
Thisse, B., and Thisse, C. (2004). Fast release clones: a high throughput expression analysis. ZFIN Direct Data Submission. Available online at: http://zfin.org
Thisse, C., and Thisse, B. (2005). High throughput expression analysis of ZF models consortium clones. ZFIN Direct Data Submission. Available online at: http://zfin.org
Thompson, C. M., Davis, E., Carrigan, C. N., Cox, H. D., Bridges, R. J., and Gerdes, J. M. (2005). Inhibitor of the glutamate vesicular transporter (VGLUT). Curr. Med. Chem. 12, 2041–2056. doi: 10.2174/0929867054637635
Timmerman, V., Clowes, V. E., and Reid, E. (2013). Overlapping molecular pathological themes link Charcot-Marie-Tooth neuropathies and hereditary spastic paraplegias. Exp. Neurol. 246, 14–25. doi: 10.1016/j.expneurol.2012.01.010
Timmerman, V., Strickland, A. V., and Zuchner, S. (2014). Genetics of Charcot-Marie-Tooth (CMT) disease within the frame of the human genome project success. Genes (Basel) 5, 13–32. doi: 10.3390/genes5010013
Titus, T. A., Yan, Y. L., Wilson, C., Starks, A. M., Frohnmayer, J. D., Bremiller, R. A., et al. (2009). The fanconi anemia/BRCA gene network in zebrafish: embryonic expression and comparative genomics. Mutat. Res. 668, 117–132. doi: 10.1016/j.mrfmmm.2008.11.017
Tucker, B., Richards, R. I., and Lardelli, M. (2006). Contribution of mGluR and Fmr1 functional pathways to neurite morphogenesis, craniofacial development and fragile X syndrome. Hum. Mol. Genet. 15, 3446–3458. doi: 10.1093/hmg/ddl422
Turner, K. J., Bracewell, T. G., and Hawkins, T. A. (2014). Anatomical dissection of zebrafish brain development. Methods Mol. Biol. 1082, 197–214. doi: 10.1007/978-1-62703-655-9_14
Turner, T. N., Sharma, K., Oh, E. C., Liu, Y. P., Collins, R. L., Sosa, M. X., et al. (2015). Loss of δ-catenin function in severe autism. Nature 520, 51–56. doi: 10.1038/nature14186
Turrigiano, G. (2012). Homeostatic synaptic plasticity: local and global mechanisms for stabilizing neuronal function. Cold Spring Harb. Perspect. Biol. 4:a005736. doi: 10.1101/cshperspect.a005736
Uddin, M., Tammimies, K., Pellecchia, G., Alipanahi, B., Hu, P., Wang, Z., et al. (2014). Brain-expressed exons under purifying selection are enriched for de novo mutations in autism spectrum disorder. Nat. Genet. 46, 742–747. doi: 10.1038/ng.2980
Vatine, G. D., Zada, D., Lerer-Goldshtein, T., Tovin, A., Malkinson, G., Yaniv, K., et al. (2013). Zebrafish as a model for monocarboxyl transporter 8-deficiency. J. Biol. Chem. 288, 169–180. doi: 10.1074/jbc.M112.413831
Vissers, L. E., Gilissen, C., and Veltman, J. A. (2016). Genetic studies in intellectual disability and related disorders. Nat. Rev. Genet. 17, 9–18. doi: 10.1038/nrg3999
Vitureira, N., Letellier, M., and Goda, Y. (2012). Homeostatic synaptic plasticity: from single synapses to neural circuits. Curr. Opin. Neurobiol. 22, 516–521. doi: 10.1016/j.conb.2011.09.006
Wakayama, Y., Fukuhara, S., Ando, K., Matsuda, M., and Mochizuki, N. (2015). Cdc42 mediates Bmp-induced sprouting angiogenesis through Fmnl3-driven assembly of endothelial filopodia in zebrafish. Dev. Cell 32, 109–122. doi: 10.1016/j.devcel.2014.11.024
Waters, B. M., Chu, H. H., Didonato, R. J., Roberts, L. A., Eisley, R. B., Lahner, B., et al. (2006). Mutations in Arabidopsis yellow stripe-like1 and yellow stripe-like3 reveal their roles in metal ion homeostasis and loading of metal ions in seeds. Plant Physiol. 141, 1446–1458. doi: 10.1104/pp.106.082586
Wen, H., and Brehm, P. (2005). Paired motor neuron-muscle recordings in zebrafish test the receptor blockade model for shaping synaptic current. J. Neurosci. 25, 8104–8111. doi: 10.1523/jneurosci.2611-05.2005
Wen, H., Linhoff, M. W., Hubbard, J. M., Nelson, N. R., Stensland, D., Dallman, J., et al. (2013). Zebrafish calls for reinterpretation for the roles of P/Q calcium channels in neuromuscular transmission. J. Neurosci. 33, 7384–7392. doi: 10.1523/JNEUROSCI.5839-12.2013
Wheeler, N. A., Lister, J. A., and Fuss, B. (2015). The autotaxin-lysophosphatidic acid axis modulates histone acetylation and gene expression during oligodendrocyte differentiation. J. Neurosci. 35, 11399–11414. doi: 10.1523/JNEUROSCI.0345-15.2015
Wiley, D. J., Juan, I., Le, H., Cai, X., Baumbach, L., Beattie, C., et al. (2014). Yeast augmented network analysis (YANA): a new systems approach to identify therapeutic targets for human genetic diseases. F1000Res 3:121. doi: 10.12688/f1000research.4188.1
Wondolowski, J., and Dickman, D. (2013). Emerging links between homeostatic synaptic plasticity and neurological disease. Front. Cell. Neurosci. 7:223. doi: 10.3389/fncel.2013.00223
Wood, J. D., Bonath, F., Kumar, S., Ross, C. A., and Cunliffe, V. T. (2009). Disrupted-in-schizophrenia 1 and neuregulin 1 are required for the specification of oligodendrocytes and neurones in the zebrafish brain. Hum. Mol. Genet. 18, 391–404. doi: 10.1093/hmg/ddn361
Wullimann, M. F. (1994). The teleostean torus longitudinalis: a short review on its structure, histochemistry, connectivity, possible function and phylogeny. Eur. J. Morphol. 32, 235–242.
Wullimann, M. F., Rupp, B., and Reichert, H. (1996). Neuroanatomy of the Zebrafish Brain: A Topological Atlas. Basel, CHE: Birkhauser.
Wullimann, M. F. (2014). Ancestry of basal ganglia circuits: new evidence in teleosts. J. Comp. Neurol. 522, 2013–2018. doi: 10.1002/cne.23525
Wullimann, M. F., Mueller, T., Distel, M., Babaryka, A., Grothe, B., and Koster, R. W. (2011). The long adventurous journey of rhombic lip cells in jawed vertebrates: a comparative developmental analysis. Front. Neuroanat. 5:27. doi: 10.3389/fnana.2011.00027
Wurst, W., and Bally-Cuif, L. (2001). Neural plate patterning: upstream and downstream of the isthmic organizer. Nat. Rev. Neurosci. 2, 99–108. doi: 10.1038/35053516
Wyart, C., Del Bene, F., Warp, E., Scott, E. K., Trauner, D., Baier, H., et al. (2009). Optogenetic dissection of a behavioural module in the vertebrate spinal cord. Nature 461, 407–410. doi: 10.1038/nature08323
Xing, L., Hoshijima, K., Grunwald, D. J., Fujimoto, E., Quist, T. S., Sneddon, J., et al. (2012). Zebrafish foxP2 zinc finger nuclease mutant has normal axon pathfinding. PLoS One 7:e43968. doi: 10.1371/journal.pone.0043968
Xing, L., Son, J. H., Stevenson, T. J., Lillesaar, C., Bally-Cuif, L., Dahl, T., et al. (2015). A serotonin circuit acts as an environmental sensor to mediate midline axon crossing through EphrinB2. J. Neurosci. 35, 14794–14808. doi: 10.1523/JNEUROSCI.1295-15.2015
Yanicostas, C., Barbieri, E., Hibi, M., Brice, A., Stevanin, G., and Soussi-Yanicostas, N. (2012). Requirement for zebrafish ataxin-7 in differentiation of photoreceptors and cerebellar neurons. PLoS One 7:e50705. doi: 10.1371/journal.pone.0050705
Yeh, C. M., Liu, Y. C., Chang, C. J., Lai, S. L., Hsiao, C. D., and Lee, S. J. (2011). Ptenb mediates gastrulation cell movements via Cdc42/AKT1 in zebrafish. PLoS One 6:e18702. doi: 10.1371/journal.pone.0018702
Yimlamai, D., Konnikova, L., Moss, L. G., and Jay, D. G. (2005). The zebrafish down syndrome cell adhesion molecule is involved in cell movement during embryogenesis. Dev. Biol. 279, 44–57. doi: 10.1016/j.ydbio.2004.12.001
Yizhar, O., Fenno, L. E., Prigge, M., Schneider, F., Davidson, T. J., O’Shea, D. J., et al. (2011). Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature 477, 171–178. doi: 10.1038/nature10360
Yoshida, T., and Mishina, M. (2008). Zebrafish orthologue of mental retardation protein IL1RAPL1 regulates presynaptic differentiation. Mol. Cell Neurosci. 39, 218–228. doi: 10.1016/j.mcn.2008.06.013
Yu-Wai-Man, P., Griffiths, P. G., Hudson, G., and Chinnery, P. F. (2009). Inherited mitochondrial optic neuropathies. J. Med. Genet. 46, 145–158. doi: 10.1136/jmg.2007.054270
Zhou, W., Horstick, E. J., Hirata, H., and Kuwada, J. Y. (2008). Identification and expression of voltage-gated calcium channel beta subunits in Zebrafish. Dev. Dyn. 237, 3842–3852. doi: 10.1002/dvdy.21776
Zhou, Y., Kaiser, T., Monteiro, P., Zhang, X., Van der Goes, M. S., Wang, D., et al. (2016). Mice with Shank3 Mutations associated with ASD and schizophrenia display both shared and distinct defects. Neuron 89, 147–162. doi: 10.1016/j.neuron.2015.11.023
Zlotogora, J. (2003). Penetrance and expressivity in the molecular age. Genet. Med. 5, 347–352. doi: 10.1097/01.gim.0000086478.87623.69
Züchner, S., De Jonghe, P., Jordanova, A., Claeys, K. G., Guergueltcheva, V., Cherninkova, S., et al. (2006). Axonal neuropathy with optic atrophy is caused by mutations in mitofusin 2. Ann. Neurol. 59, 276–281. doi: 10.1002/ana.20797
Züchner, S., Mersiyanova, I. V., Muglia, M., Bissar-Tadmouri, N., Rochelle, J., Dadali, E. L., et al. (2004). Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 36, 449–451. doi: 10.1038/ng1341
Keywords: zebrafish, disease modeling, autism spectrum disorder, intellectual disability, schizophrenia, ataxia, Charcot-Marie tooth, hereditary spastic paraplegia
Citation: Kozol RA, Abrams AJ, James DM, Buglo E, Yan Q and Dallman JE (2016) Function Over Form: Modeling Groups of Inherited Neurological Conditions in Zebrafish. Front. Mol. Neurosci. 9:55. doi: 10.3389/fnmol.2016.00055
Received: 05 April 2016; Accepted: 23 June 2016;
Published: 07 July 2016.
Edited by:
Robert J. Harvey, UCL School of Pharmacy, UKCopyright © 2016 Kozol, Abrams, James, Buglo, Yan and Dallman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert A. Kozol, cm9ia296b2xAYmlvLm1pYW1pLmVkdQ==
Julia E. Dallman, SmRhbGxtYW5AYmlvLm1pYW1pLmVkdQ==