 Sidharth Tyagi
Sidharth Tyagi Angeles B. Ribera
Angeles B. Ribera Roger A. Bannister
Roger A. Bannister- 1Medical Scientist Training Program, Yale University School of Medicine, New Haven, CT, United States
- 2Department of Physiology and Biophysics, University of Colorado School of Medicine, Aurora, CO, United States
- 3Department of Pathology, University of Maryland School of Medicine, Baltimore, MD, United States
- 4Department of Biochemistry and Molecular Biology, University of Maryland School of Medicine, Baltimore, MD, United States
The P/Q-type CaV2.1 channel regulates neurotransmitter release at neuromuscular junctions (NMJ) and many central synapses. CACNA1A encodes the pore-containing α1A subunit of CaV2.1 channels. In humans, de novo CACNA1A mutations result in a wide spectrum of neurological, neuromuscular, and movement disorders, such as familial hemiplegic migraine type 1 (FHM1), episodic ataxia type 2 (EA2), as well as a more recently discovered class of more severe disorders, which are characterized by ataxia, hypotonia, cerebellar atrophy, and cognitive/developmental delay. Heterologous expression of CaV2.1 channels has allowed for an understanding of the consequences of CACNA1A missense mutations on channel function. In contrast, a mechanistic understanding of how specific CACNA1A mutations lead in vivo to the resultant phenotypes is lacking. In this review, we present the zebrafish as a model to both study in vivo mechanisms of CACNA1A mutations that result in synaptic and behavioral defects and to screen for effective drug therapies to combat these and other CaV2.1 channelopathies.
Introduction
P/Q-type CaV2.1 channels are the predominant voltage-gated Ca2+ channel isoform present at the neuromuscular junction (NMJ) and most central synapses. Since Ca2+ flux via these channels is critical for neurotransmitter release (Llinás et al., 1981; Turner et al., 1992; Uchitel et al., 1992; Dunlap et al., 1994, 1995; Ludwig et al., 1997), mutations in the CaV2.1 α1A subunit would be expected to impact synaptic efficacy. However, as discussed in sections “CaV2.1 Channel Composition” to “The Expanding Spectrum OF CaV2.1-α1A Channelopathies” the direct consequences of mutations on channel function and the resultant neurologic phenotypes vary significantly. For example, two well-studied channelopathies—episodic ataxia type 2 (EA2) and familial hemiplegic migraine type 1 (FHM1)—arise from point mutations in the CACNA1A gene that encodes the α1A subunit (Jen et al., 2007; Pietrobon, 2007, 2010). The mutations that lead to EA2 tend to be loss-of-function mutations, while gain-of-function mutations usually underlie FHM1 (Jen et al., 2001; Tottene et al., 2002; Kaja et al., 2005, 2010; Mantuano et al., 2010; Rajakulendran et al., 2010b; Di Guilmi et al., 2014; Rose et al., 2014; Brusich et al., 2018). However, some ataxic cases have paradoxically been linked to gain-of-channel function mutations (e.g., van den Maagdenberg et al., 2010; Knierim et al., 2011; Gao et al., 2012; Bahamonde et al., 2015; Jiang et al., 2019). These latter examples underscore the diversity of channel dysfunction in this expanding spectrum of ataxic disorders and highlight the need for a model system to rapidly and effectively identify pathological phenotypes.
In this article, we review the: (1) basic information about the CaV2.1 channel heteromultimer; (2) two relatively well-characterized diseases caused by mutation of the CaV2.1 α1A subunit—EA2 and FHM1; (3) the emerging full spectrum of CaV2.1 α1A channelopathies; and (4) the potential that the zebrafish model holds for understanding disease mechanisms and discovering potential therapeutics. Sections “Introduction” to “Familial Hemiplegic Migraine Type 1” are intended to provide sufficient background for the more profound discussion of the more severe neurodevelopmental disorders, which are caused by point mutations in CACNA1A in section “The Expanding Spectrum OF CaV2.1-α1A Channelopathies.” It is important to note that the pathology of this unnamed class of disorders resembles that of spinocerebellar ataxia type 6 (SCA), which is caused by the addition of excess CAG polynucleotide repeats to the CACNA1A transcript (Jodice et al., 1997).
CaV2.1 Channel Composition
High voltage-activated Ca2+ channels, such as the CaV2.1 heteromultimer, are composed minimally of a principal α1 subunit (α1A) and auxiliary β and α2δ subunits (Volsen et al., 1997; Catterall, 2010; Dolphin, 2016). For CaV2.1, an interaction with a γ2 subunit (a.k.a., stargazin) was also reported (Letts et al., 1998; Kang and Campbell, 2003). Like the other nine members of the CaV family, α1A subunits have four transmembrane repeats (I–IV), each with six membrane-spanning α-helices (S1–S6; Mori et al., 1991; please see Figure 1). Of these, the S4 α-helices are thought to be the primary voltage-sensing elements of the channel, a function which is conferred by five to six positively charged amino acids lining a face of the α-helix (Aggarwal and MacKinnon, 1996). The S1–S3 helices form an aqueous conduit that enables passage of the S4 α-helix through the membrane field by facilitating interactions with residues of the “charge transfer center” (formed by conserved negative, polar and hydrophobic residues on the S2 segment and an invariant aspartate residue on the S3 helix; Tao et al., 2010); the S5 and S6 helices line the conventional channel conduction pore (Neely and Hidalgo, 2014; Hering et al., 2018). The relatively long extracellular segment linking the S5 and S6 helices (a.k.a., the P-loop) contains a highly conserved glutamate residue in all four repeats. These four glutamates form the selectivity filter (Yang et al., 1993).
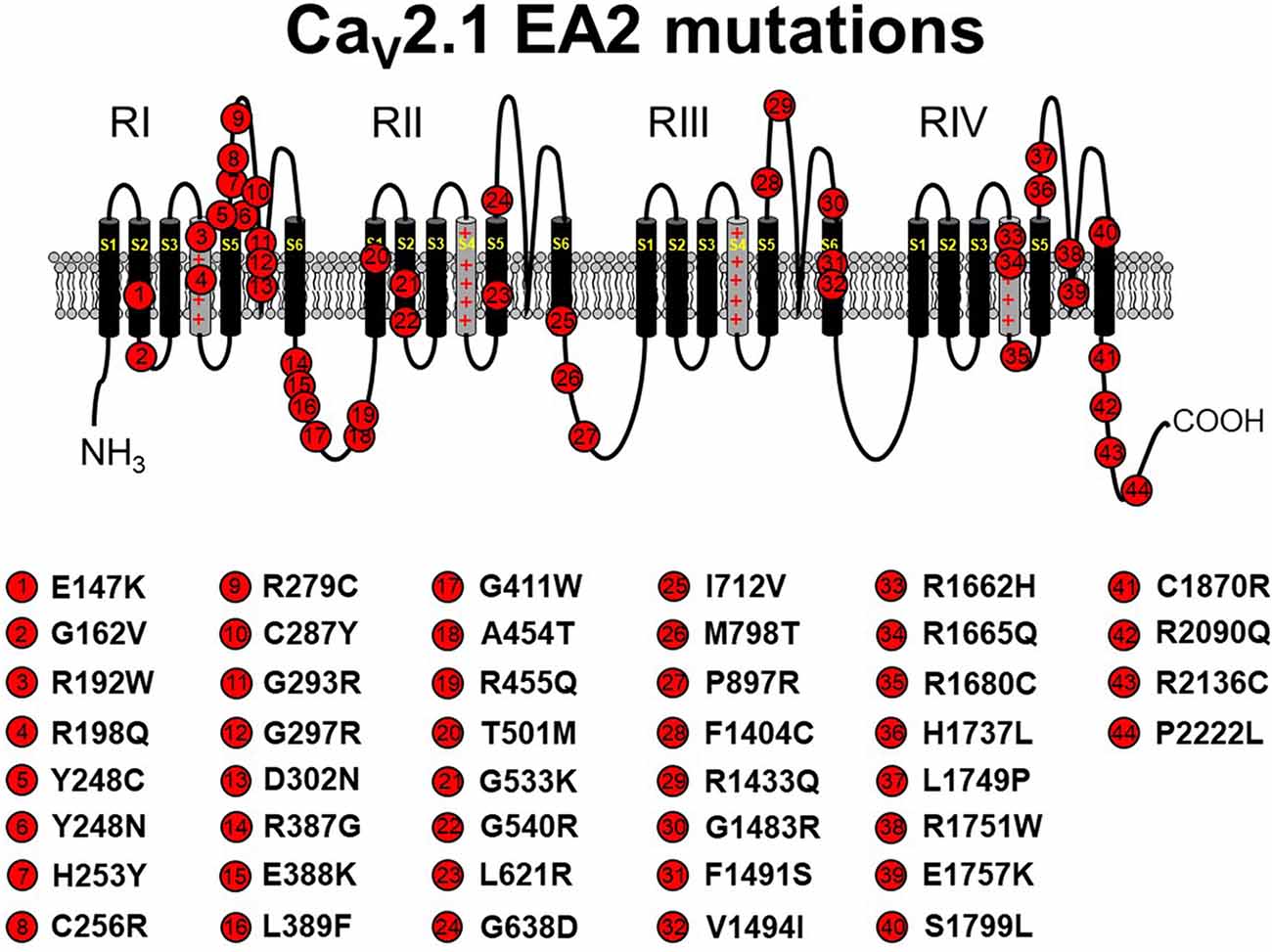
Figure 1. Schematic representation of human CaV2.1 mutations causing episodic ataxia type 2 (EA2). Please note that residue numbering varies between studies due to the existence of multiple CACNA1A splice variants; residue numbers indicated reflect those stated in the original report. Citations to the indicated mutations are listed as follows: E147K—Imbrici et al., 2004; G162V—Maksemous et al. (2016); R192W—Soden et al. (2014); R198Q—Indelicato et al. (2019); Y248C—Zafeiriou et al. (2009); Y248N—Choi et al. (2017); H253Y—van den Maagdenberg et al. (2002); C256R—Mantuano et al. (2004); R279C—Maksemous et al. (2016); C287Y—Jen et al. (2004); G293R—Yue et al. (1997); G297R—Tantsis et al. (2016); D302N—Maksemous et al. (2016); R387G—Maksemous et al. (2016); E388K—Nikaido et al. (2011); L389F—Mantuano et al. (2010); G411W—Maksemous et al. (2016); A454T—Cricchi et al. (2007); R455Q—Isaacs et al. (2017); T501M—Mantuano et al. (2010); G533K—Scoggan et al. (2006); G540R—Rajakulendran et al. (2010a); L621R—Rajakulendran et al. (2010a); G638D—Cuenca-León et al. (2009); I712V—Guerin et al. (2008); M798T—Mantuano et al. (2010); P897R—Mantuano et al. (2010); F1404C—Jen et al. (2001); R1433Q—Pietrobon (2010); G1483R—Mantuano et al. (2004); F1491S—Guida et al. (2001); V1494I—Mantuano et al. (2004); R1662H—Friend et al. (1999); R1665Q—Tonelli et al. (2006); R1680C—Mantuano et al. (2010); H1737L—Spacey et al. (2004); L1749P—Maksemous et al. (2016); R1751W—Bertholon et al. (2009); E1757K—Denier et al. (2001); S1799L—Ohba et al. (2013); C1870R—Mantuano et al. (2010); R2090Q—Melzer et al. (2010); R2136C—Mantuano et al. (2004); P2222L—Sintas et al. (2017). The CaV2.1 schematic was modified from Tyagi et al. (2019) with permission of the authors.
Episodic Ataxia Type 2
EA2 is a rare neurological disease characterized by paroxysmal attacks of ataxia, nystagmus, and vertigo. The majority of CACNA1A mutations that lead to EA2 result in CaV2.1 loss of function by premature termination of the open reading frame, resulting in rapid degradation of truncated protein products (Jen et al., 2001; Pietrobon, 2010; Sintas et al., 2017). Indeed, over 40 pathogenic missense mutations were identified (Pietrobon, 2010; Sintas et al., 2017; see Figure 1). Most of these amino acid substitutions reside in the P-loop or the S5 and S6 helices, themselves, suggesting that impaired ability to form a fully functional channel pore is the likely pathophysiological mechanism of the resultant phenotype for the majority of EA2 missense cases (Jen et al., 2007; Sintas et al., 2017). In some cases, a complete loss of function was observed with missense mutants, likely attributable to ER-associated degradation of the mutant channel and subsequent lack of trafficking to the surface membrane (Page et al., 2004). In addition, some EA2 mutants (e.g., E1761K, F1406C) seem to exert a dominant-negative effect since coexpression of mutant channels with wild-type channels in Xenopus oocytes diminished the amplitude of Ca2+ current elicited by depolarization (Jeng et al., 2006, 2008; Mezghrani et al., 2008). In these latter cases, it was postulated that misfolded mutant channels bound wild-type channels and subsequently induced degradation (Page et al., 2010; Rajakulendran et al., 2012; Dahimene et al., 2016) or competed successfully with the wild-type channel for a limited number of “slots” reserved for CaV2.1 channels at the plasma membrane (Cao et al., 2004; Cao and Tsien, 2010; but see below). In addition, some mutations (e.g., H1736L, A1293D/delY1294, G293R) do not completely abolish channel activity but rather shift the voltage-dependence of CaV2.1 activation to somewhat more positive potentials, thereby decreasing channel open probability (Po; Wappl et al., 2002; Spacey et al., 2004; Pietrobon, 2010).
In a minority of cases, EA2 is precipitated by gain-of-channel function mutations, which suggests that a critical bandwith of Ca2+ flux is required to avoid pathogenicity (e.g., Mantuano et al., 2010; Knierim et al., 2011; Gao et al., 2012; Carreño et al., 2013; Bahamonde et al., 2015). For many yet-to-be characterized Cav2.1 EA2 mutations, whether the mutation produces gain- or loss-of-channel function remains to be seen. Still, these findings underscore the need to resist generalization regarding pathological mechanisms without rigorous investigation of each mutation.
Familial Hemiplegic Migraine Type 1
FHM1 is an inherited migraine condition that results in weakness of half the body for prolonged periods of time. Patients afflicted with FHM1 often display cerebellar degeneration (Elliot et al., 1996). As noted above, FHM1 is most often linked to gain-of-function point mutations in CACNA1A (Tottene et al., 2002; Pietrobon, 2007; see Figure 2). These substitutions occur at a variety of loci within the channel but most commonly in residues thought to line the pore, the S3–S4 or S5–S6 linkers, or the S4 voltage sensor. Even though the locations of the mutations within the channel are variable, analysis in heterologous systems revealed a hyperpolarizing shift in channel activation for most studied mutants (Hans et al., 1999; Tottene et al., 2002, 2005; Adams et al., 2009; Serra et al., 2009). Since these channels open at more hyperpolarizing potentials, channel Po is enhanced, and an FHM1 mutant CaV2.1 channel can carry greater Ca2+ influx than its wild-type counterpart at physiologically relevant membrane potentials. This process may be further facilitated by a reduction in the direct Gβγ-mediated inhibition of presynaptic FHM1 mutant CaV2.1 channels (Melliti et al., 2003; Weiss et al., 2008; Serra et al., 2009; Garza-López et al., 2012, 2013). Mouse knock-in models carrying FHM1-causing CaV2.1 mutations display the migraine aura, cortical spreading depression characteristic of human FHM1 (van den Maagdenberg et al., 2004, 2010). While these gain-of-function biophysical effects of FHM1 mutations are fairly consistent, it is important to state that FHM1 pathology is inarguably a reflection of the balance of the relative manifestation of the mutations between excitatory and inhibitory circuits (Vecchia et al., 2015).
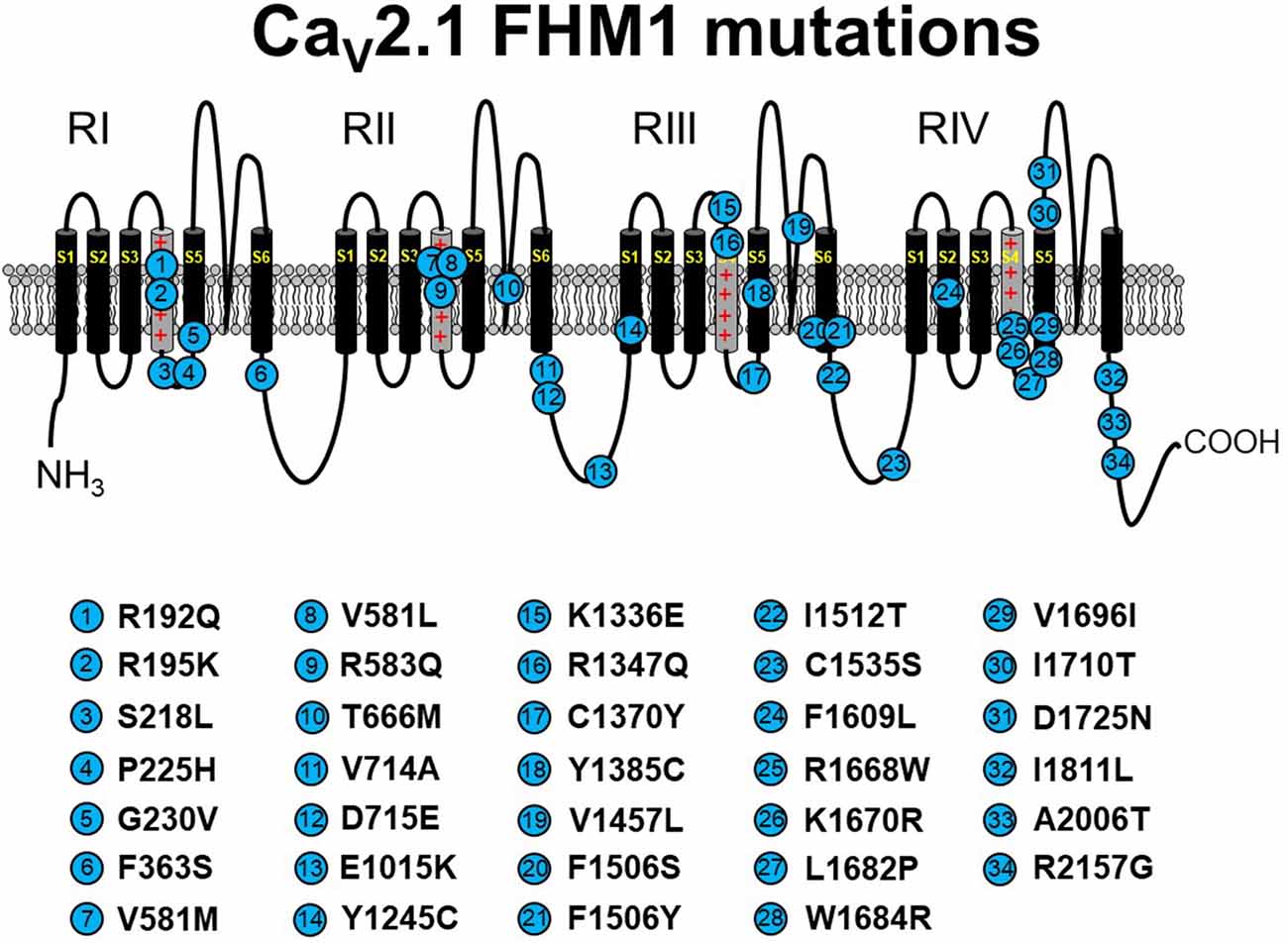
Figure 2. Schematic representation of human CaV2.1 mutations causing familial hemiplegic migraine type 1 (FHM1). Please note that residue numbering varies between studies due to the existence of multiple CACNA1A splice variants; residue numbers indicated reflect those stated in the original report. Citations to the indicated mutations are listed as follows: R192Q—Ophoff et al. (1996); R195K—Ducros et al. (2001); S218L—Kors et al. (2001); P225H—Stuart et al. (2012); G230V—Yang et al. (2014); F363S—Riant et al. (2010); V581M—Cuenca-León et al. (2008); V581L—Freilinger et al. (2011); R583Q—Battistini et al. (1999); T666M—Ophoff et al. (1996); V714A—Ophoff et al. (1996); D715E—Ducros et al. (2001); E1015K—Grieco et al. (2018); Y1245C—Cuenca-León et al. (2008); K1336E—Ducros et al. (2001); R1347Q—Alonso et al. (2004); C1370Y—Thomsen et al. (2007); Y1385C—Vahedi et al. (2000); V1457L—Carrera et al. (1999); F1506S—Riant et al. (2010); F1506Y—Pelzer et al. (2018); I1512T—Grieco et al. (2018); C1535S—Dichgans et al. (2005); F1609L—Pelzer et al. (2018); R1668W—Ducros et al. (2001); K1670R—Riant et al. (2010); L1682P—Weiss et al. (2007); W1684R—Ducros et al. (2001); V1696I—Ducros et al. (2001); I1710T—Kors et al. (2004); D1725N—Riant et al. (2010); I1811L—Ophoff et al. (1996); A2006T—Wilson (2014); R2157G—Grieco et al. (2018). The CaV2.1 schematic was modified from Tyagi et al. (2019) with permission of the authors.
The Expanding Spectrum of CaV2.1-α1A Channelopathies
EA2 and FHM1 have long been known to be caused primarily by point mutations in CaV2.1 in addition to a few variants that carry deletions or insertions (Jen et al., 2001; Pietrobon, 2007, 2010). However, the biophysical effects of these mutations on channel function are often subtle, and the manifestations of ataxia are paroxysmal (Elliot et al., 1996; Jen et al., 2007; Sintas et al., 2017). With the innovative whole-exome sequencing approach, a new, but yet-to-be-named, class of CaV2.1-linked disorders with developmental components was identified and linked to point mutations in CaV2.1 (Tonelli et al., 2006; Blumkin et al., 2010; Romaniello et al., 2010; Epi4K Consortium and Epilepsy Phenome/Genome Project, 2013; Damaj et al., 2015; Jiang et al., 2016; Weyhrauch et al., 2016; Luo et al., 2017; Travaglini et al., 2017). These disorders represent the far end of the CaV2.1 channelopathy spectrum, which includes FHM1 and EA2. As is the case with spectrum disorders, these more severe disorders often share the characteristics of migraine and ataxia with FHM1 and EA2, respectively. However, the more severe disorders display cognitive deficits, epilepsies, and neurodegeneration that are infrequently observed with FHM1 and EA2 patients. Though similar in presentation, disorders resulting from CaV2.1 missense mutations differ in etiology from SCA6, which is caused by increasing polyglutamine expansions on the channel carboxyl-terminus (Jodice et al., 1997; Frontali, 2001). Moreover, the scattering of mutations within the channel suggests that there are a variety of mechanisms for channel dysfunction underlying this class of disorders (Figure 3). For example, Romaniello et al. (2010) described an A405T substitution in a 12-year-old girl with a family history of CaV2.1 mutation-linked disorders. The patient presented with persistent cerebellar signs (i.e., ataxia, dysmetria, hypotonia) and developmental delay. A405T represents a non-polar to polar substitution in the Repeat I–II linker region of CaV2.1 (Figure 1). The Repeat I–II linker is putatively the site where the auxiliary β subunit interacts with the α1A subunit (Campiglio and Flucher, 2015). A reasonable, but yet-to-be-tested, hypothesis is that the A405T substitution disrupts the α1A-β subunit interaction in much the same way as does an engineered Y392S swap in the I–II loop (Pragnell et al., 1994). Such a disruption would substantially decrease surface expression of the channel by impeding trafficking and, given reduced production of the wild-type protein, would likely result in haploinsufficiency. An alternate explanation is that the A405T substitution that impacts neurotransmitter release, similar to another ataxic variant in the I–II linker, A454T, was demonstrated to curb modulation of CaV2.1 by SNARE proteins via a mechanism involving the β subunit (Cricchi et al., 2007; Serra et al., 2010, 2018).
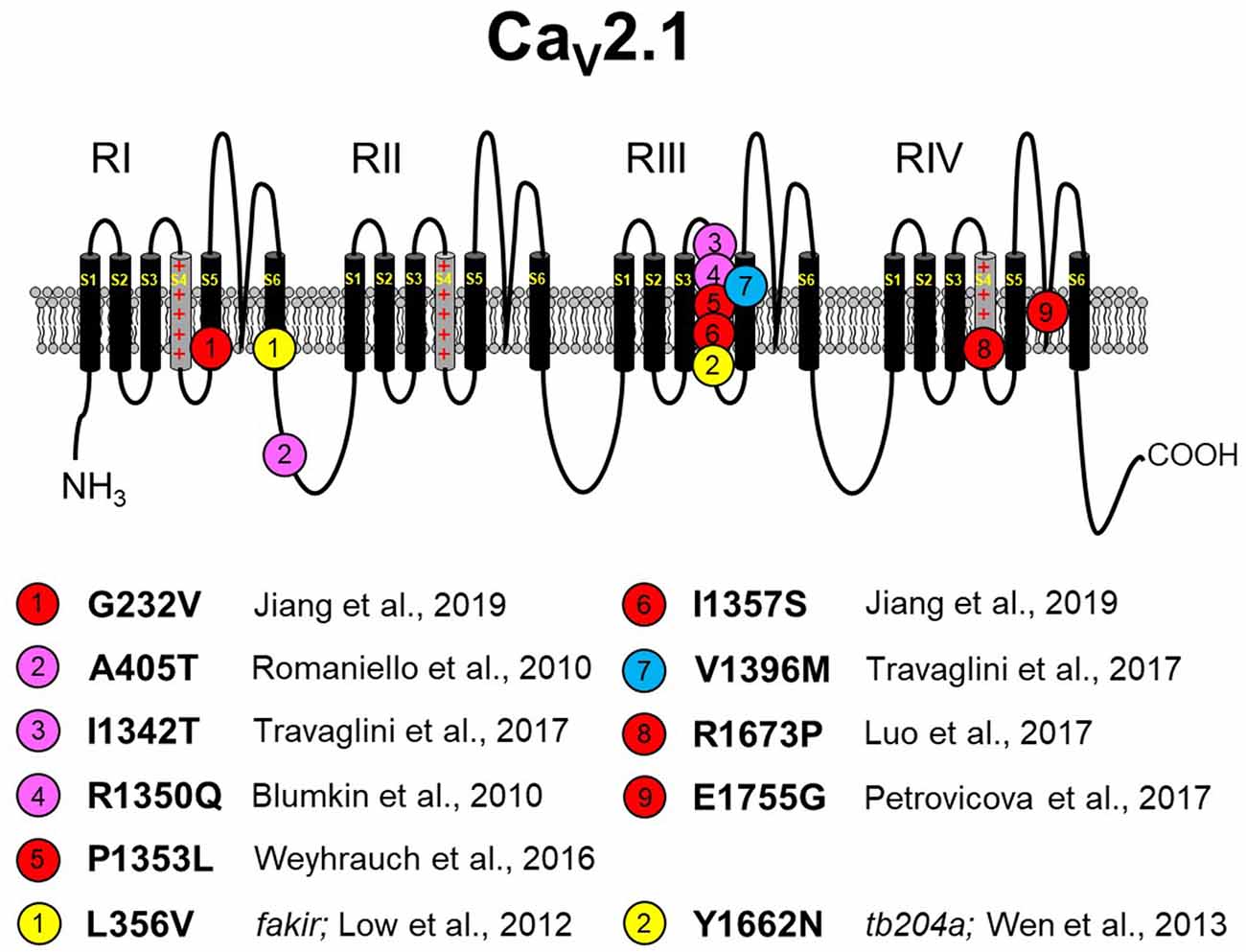
Figure 3. Missense CaV2.1 mutations leading to neurodevelopmental disorders. The zebrafish fakir and tb204a mutants are also depicted as yellow circles. Red circles indicate a loss-of-function human mutation. Blue circles indicate a gain-of-function human mutation. Magenta circles indicate a yet-to-be functionally characterized human mutation. Specific references are indicated below. As in Figures 1, 2, please note that residue numbering varies between studies due to: (1) the existence of multiple known CACNA1A splice variants; and (2) species differences between humans and zebrafish. The CaV2.1 schematic was modified from Tyagi et al. (2019) with permission of the authors. These mutations are discussed in sections “The Expanding Spectrum OF CaV2.1-α1A Channelopathies” and “Zebrafish as a Model SYSTEM for the Study of Severe CaV2.1 Channelopathies.”
Blumkin et al. (2010) reported a R1350Q substitution in a 7-year-old male patient that also presented with cerebellar ataxia, developmental delay, and nonspecific dyskinesia. Although the outward presentation was similar to the patient carrying the A405T substitution, the R1350Q swap inserted a neutral glutamine in place of a basic arginine in the S4 voltage-sensing α-helix of Repeat III (Figure 1). An arginine to glutamine substitution at this position was also reported with a patient exhibiting tremor that was alleviated by a Ca2+ channel blocker (R1345Q in Jiang et al., 2016). Based on the observation that the equivalent substitution in the tottering mutant mouse causes a ~12-mV hyperpolarizing shift in activation (Miki et al., 2008), it is likely that neutralization of this basic residue may have facilitated the movement of the voltage sensor through the membrane field. Such gain of function contrasts with the findings of Weyhrauch et al. (2016), who also described a mutation in the S4 voltage sensor of Repeat III (P1353L) found in a child with developmental delay, gross motor delay, and congenital hypotonia (Figure 1). Electrophysiological analysis of mutant channels expressed heterologously in HEK293 cells revealed near 100% ablation of CaV2.1-mediated Ca2+ current, suggesting that either dominant-negative effects or haploinsufficiency underlies the phenotype. The first possibility was proposed on the basis that mice with only one CACNA1A allele seems normal (Jun et al., 1999). However, the ability of CaV2.1P1353L to out-compete endogenously wild-type channels was not investigated in a neuronal context.
Travaglini et al. (2017) reported a pair of mutations, I1342T and V1396M, in two patients with similar clinical phenotypes involving congenital ataxia, hypotonia, and intellectual disability. The I1342T mutation resides in the extracellular loop between the S3 and S4 helix of α1A in close proximity to the beginning of the Repeat III S4 helix (Figure 1). A reasonable hypothesis for the dysfunction of the I1342T mutant channel is that this substitution alters the conformation of the S4 helix and affects its mobility, though speculation on its relationship to ataxia, hypotonia, and intellectual disability is unfounded without more biophysical information regarding mutant channel dysfunction. The V1396M mutation is found in the proximal S5 pore-forming domain of Repeat III of α1A, a region of the channel that is also predicted to interact with the α2δ subunit on the basis of CaV1.1 cryo-EM structure (Wu et al., 2016). The idea that V1396M facilitates channel expression through an α2δ-mediated mechanism (see Dolphin, 2016, for a review) is particularly intriguing since the current density for the mouse equivalent of Cav2.1 V1396M expressed in HEK293 cells was shown to be nearly double that of wild-type Cav2.1 (Jiang et al., 2019). Though less striking, the introduction of methionine also causes a hyperpolarization in the voltage dependence of activation suggesting the disruption of an inter-helical interaction that restricts voltage-sensor translocation. Three other CaV2.1mutants, which were linked to Lennox–Gastaut epileptic encephalopathy were examined in the same study and were found to have polar effects (Jiang et al., 2019). The A715T mutation at the base of RIIS6 displayed a ~10-mV hyperpolarizing shift in activation, smaller but reminiscent of the ~20-mV hyperpolarizing shift observed in Purkinje cells of CaV2.1 S218L EA2 model mice (Gao et al., 2012). On the other hand, G232V and I1357S, at the bases of RIS5 and RIIS4 helices, respectively, reduced channel plasma membrane expression in both HEK293 and in cortical neurons.
Seminal work from Richard Tsien’s laboratory in the early 1990s revealed that four highly conserved glutamate residues within the P-loop are the structural basis of Ca2+ selectivity among all CaV channels (Yang et al., 1993). Two such mutations in α1A are known to occur at the same glutamate in Repeat IV. Mutation of this residue to glycine causes ataxia and cognitive deficits running through three generations of the Slovak family (E1755G in Petrovicova et al., 2017), and as noted above, a reversal of charge via substitution of a lysine for the glutamate causes EA2 (E1761K in Denier et al., 2001). The glutamate to lysine mutation ablates inward Ba2+ flux via the channel in Xenopus oocytes (Jeng et al., 2006). Since coexpression of the CaV2.1 E1761K mutant with the wild-type channel reduced the amplitude of the current in an RNA dose-dependent manner, the authors postulated that the E1761K resulted in a dominant-negative effect. While this mechanism could certainly underlie this particular channelopathy, conversion of any one of the glutamates in the selectivity filter to lysine effectively transforms CaV channels into non-specific monovalent ion channels that are subject to block by divalent ions (Yang et al., 1993). In this regard, Jeng et al. (2006) used a concentration of Ba2+ (40 mM) in their experiments showing the ablation of inward current via E1761K channels, which most likely would have blocked the mutant channel. At more physiological divalent ion concentrations (i.e., <2 mM Ca2+), currents carried by Na+ and K+ might be visible and pathogenic. Indeed, aberrant Na+ and K+ flux via CaV1.2 Repeat III glutamate to lysine mutant channels can prolong action potential duration in cardiac-like iPSCs (Ye et al., 2019), while the equivalent mutation in CaV1.1 is postulated to cause K+ accumulation in the transverse tubules (Beqollari et al., 2018) and to accelerate muscle fatigue in mice (Lee et al., 2015). Thus, the possibility that the E1761K mutation augments neurotransmitter release by prolonging neuronal action potential duration is not unreasonable, nor is the idea that excessive K+ secretion into restricted extracellular compartments may excite neighboring neurons or vascular smooth muscle cells (see Filosa et al., 2006).
Recently, Luo et al. (2017) described an 8-year-old female patient with congenital ataxia, hypotonia, cerebellar atrophy, and global developmental delay. The trio-based exome sequencing of this patient revealed a de novo missense mutation (R1673P) in the gene for CaV2.1. The mutation resulted in an arginine to proline substitution within the Repeat IV S4 voltage-sensing helix of CaV2.1. The R1673P mutation was predicted to be “probably damaging” by PolyPhen-2, a protein structure prediction software. As a means to identify the molecular mechanism by which R1673P precipitates the clinical phenotype, transgenic flies expressing the Drosophila equivalent of wild-type CaV2.1 and CaV2.1 R1673P in a CaV2.1-deficient Drosophila (i.e., cacophony mutants) background were generated. In these experiments, the mutant CaV2.1 R1673P was able to rescue the photoreceptor response in 3-day-old larvae to a greater extent than the wild-type channel suggesting a gain-of-function effect. At 30 days, the rescue of the electroretinogram had dissipated, but substantial photoreceptor degeneration was observed in the R1673P line but not in wild-type or CaV2.1-deficient flies. It is possible that the early effects of gain-of-function Ca2+channel activity triggered neurodegeneration secondary to Ca2+ toxicity. In contrast, however, voltage-clamp experiments showed that the R1673P mutation causes a profound loss-of-function for channels expressed heterologously in tsA-201 cells (Tyagi et al., 2019). Specifically, the rat ortholog of R1673P (R1624P) displayed a ~25-mV depolarizing shift in activation and resultant weak activation at physiologically relevant membrane potentials. Further work is needed to understand how the loss of function at the molecular level leads to neurodegeneration at the systemic level.
Zebrafish as a Model System for the Study of Severe CaV2.1 Channelopathies
Heterologous expression systems are the industry standard for the identification of pathogenic channel dysfunction. However, it is often difficult to extrapolate information gleaned using this approach to neurological dysfunction in patients. To bridge this gap, animal models are employed. Mice carrying FHM1 or EA2 mutations were very useful in understanding the pathophysiology underlying these disorders. However, no mouse line yet exists that models the more severe developmental disorders discussed above. The paucity of such models may be due to the uncertain viability or breeding capability of mice with grave developmental defects and the monetary risk associated with this endeavor. By contrast, simpler organisms like Drosophila have rapid propagation, are relatively easy to manipulate genetically, and lack the burden of cost. The obvious shortcoming of Drosophila is that insects are both phylogenetically and physiologically far removed from humans. A notable shortcoming is that Drosophila lack a true CaV2.1 channel (Smith et al., 1996).
Zebrafish—Danio rerio—offers a unique complement to the strengths of flies and mice as models for the study of severe CaV2.1 channelopathies. The zebrafish is useful to investigate mechanisms because of the conservation of most fundamental physiology processes (e.g., neurotransmitter release) with mammals with a reduced risk of embryonic lethality. Similar to many zebrafish genes, the gene encoding the CaV2.1 α-subunit is duplicated, yielding cacna1aa and cacna1ab. Two zebrafish loss-of-function cacna1ab mutants, tb204a (Wen et al., 2013) and fakir (Low et al., 2012), were studied previously. For both mutations, the loss-of-channel function was sizable, but incomplete. The tb204a mutation results in a tyrosine-to-asparagine substitution (Y1662N) within the carboxyl terminus of CaV2.1a and a depolarizing shift in channel activation, similar to what was found for the rat cognate of CaV2.1 R1673P (Tyagi et al., 2019). Homozygous cacna1abtb204a−/− larvae were viable and had reduced motility. Moreover, there was an increased incidence of synaptic failure at the NMJ due to reduced Ca2+ flux into the presynaptic NMJ, as detected by imaging of presynaptic intracellular Ca2+ (Wen et al., 2013). While this defect accurately predicted reduced motor function, neither sensory nor central effects of the mutation were assessed so their potential contribution to the behavioral phenotype cannot be excluded. Interestingly, both swimming behavior and NMJ synaptic transmission were rescued in cacna1abtb204a−/− larvae by 3,4-diaminopyridine (a K+ channel blocker) and Roscovitine (a P/Q-type channel agonist; Yan et al., 2002; Buraei et al., 2007; Tarr et al., 2013).
The fakir cacna1ab mutation results in a L356V substitution in the S6 helix of Repeat I (Figure 1). Like the tb204a larvae, fakir mutants display reduced locomotor behavior compared to wild-type siblings. In addition, heterologously expressed fakir and tb204 mutant channels had reductions in current amplitude and similar depolarizing shifts in channel activation properties (Low et al., 2012; Wen et al., 2013). a priori, L356V would appear to be a conservative amino acid change. However, L356 (located at the cytoplasmic side of S6 in RI) is highly conserved across species. Interestingly, the tb204a mutation (Y1662N) resides in an analogous location in S6 of RIV. While no disease-causing mutations have yet been identified in RIS6, human pathogenic point mutations were detected in the S6 helices of Repeats II–IV (Figures 1–3). Two of the mutations in S6 domains, V1494I and I1811L, would, similar to fakir, also be considered to be conservative substitutions. Overall, despite the identification of several S6 mutations, how L356V or other S6 mutations lead to perturbed channel function remains unknown. However, the fact that this is a highly conserved region across species suggests that mutations, even conservative ones, would be of consequence.
Despite the somewhat similar effects on channel activity produced by the two different cacna1ab mutations, substantially different mechanisms were proposed for how channel dysfunction leads to abnormal locomotor behavior. Consistent with the behavioral immotility, Low et al. (2012) found that rigorous swimming could be evoked in wild-type, but not fakir mutant, slow-twitch muscle by tactile stimulation. However, examination of responses to direct application of acetylcholine as well as miniature end plate current properties revealed little differences in transmission between motor neurons and slow-twitch fibers in fakir vs. wild-type larvae, nor were defects detected in evoked transmission between CaP motor neuron and fast-twitch muscle fibers. On this basis and consistent with the initial identification of fakir as a reduced touch-sensitive mutant (Granato et al., 1996), Low et al. (2012) proposed that fakir mutants have defective swimming responses to tactile stimulation because the relevant sensory neuron Rohon–Beard cell required cacna1ab for function. However, this hypothesis was not tested directly by recording from Rohon–Beard neurons or their post-synaptic partners. In contrast, a study of the tb204 allele provided strong evidence to support defective transmission at the NMJ (Wen et al., 2013). Supporting evidence was provided by paired recordings between one type of motor neuron, CaP, and its fast-muscle target cell. Whether similar transmission defects occur at the NMJs formed between other motor neurons and muscle targets has not been studied. Thus, the mechanistic bases for the reduced motility defects of fakir and tb204a mutants have not been resolved.
Despite this impasse, the viability of both the fakir and the tb204 mutant lines bodes well for the potential usefulness of zebrafish larvae carrying missense mutations corresponding to those which cause severe human CaV2.1 channelopathies (e.g., CaV2.1 R1673P). The generation of such models through CRISPR-Cas9 technology would enable the study of individual mutations with approaches encompassing the molecular, systemic, and behavioral levels. In particular, via paired CaP motor neuron—muscle recordings and imaging of depolarization-induced Ca2+ flux into presynaptic terminals allow assessment of whether impairments in locomotor function result from NMJ defects.
Since zebrafish were successfully used to screen for compounds for the treatment of Dravet syndrome, a SCNA1ANa+ channelopathy (Griffin et al., 2017), one can envision that this approach could be used to identify and/or refine small molecules to combat both CaV2.1 gain- and loss-of-function disorders. Compounds that partially counteract channel gain of function, notably gabapentin and pregabalin, were available for clinical use for sometime (Sills, 2006). However, a need for alternatives arose as both the aforementioned compounds were shown to have some addictive capability (Bonnet et al., 2018; Althobaiti et al., 2019). In regard to loss-of-function disorders, 3,4-diaminopyridine was approved for acute treatment of Lambert—Eaton syndrome, a condition secondary to an aggressive lung cancer in which autoantibodies to CaV2.1 are generated (García and Beam, 1996; Maddison, 2012). Unfortunately, the arrhythmogenic potential of this compound precludes its long-term use in other contexts including the neurodevelopmental disorders discussed above. By contrast, derivatives of Roscovitine, such as those pioneered by the Meriney group, are logical candidates for further development (Tarr et al., 2013; Wu et al., 2018). Another possibility, which may not be a stretch given nascent cryo-EM images and the increasingly frequent implementation of deep learning approaches, is the modification of the L-type channel agonist (-)Bay K 8644 for use as a specific P/Q-type channel agonist (Zhao et al., 2019).
Despite these advantages, the zebrafish model system does pose some challenges. The fact that gene duplication endowed teleosts with two cacna1a genes can be problematic, even though the characterization of the tb204a mutant revealed that cacna1aa channel isoform makes little, if any, contribution to neurotransmission at the NMJ (Wen et al., 2013). However, sequence similarity between the isoforms may complicate knockdown experiments using antisense strategies and the production of reliable antibodies. Finally, zebrafish, like flies and mice, are not human. Nonetheless, the flexibility of the fish model makes it potentially useful as a first-line indicator of individual mutations and a vehicle for the development of personalized therapies.
Conclusions
Whole-exome sequencing is bringing new CaV2.1 mutations out of the woodwork (see Damaj et al., 2015; Jiang et al., 2016; Weyhrauch et al., 2016; Luo et al., 2017; Travaglini et al., 2017). Many of the syndromes caused by these point mutations are more severe than the typical EA2 and FHM1 in that they present with not only ataxia or migraine but also with neurodevelopmental delay, nystagmus, epilepsy, cerebellar degeneration, hypotonia, and cognitive dysfunction. Modeling these more severe disorders is problematic because of the heterogeneous effects on channel function and the limitations intrinsic to flies and mice. Although not without some disadvantages, zebrafish present a useful model system for the timely characterization of pathological phenotypes and pharmacological correction.
Author Contributions
ST, AR, and RB wrote the article. All authors read and approved the final manuscript.
Funding
This work was supported by National Institute of Neurological Disorders and Stroke/National Institute of Health (NIH) grants NS103777 and NS086839 to RB and AR, respectively. ST was supported by the Boettcher Foundation.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Adams, P. J., Garcia, E., David, L. S., Mulatz, K. J., Spacey, S. D., and Snutch, T. P. (2009). CaV2.1 P/Q-type calcium channel alternative splicing affects the functional impact of familial hemiplegic migraine mutations: implications for calcium channelopathies. Channels 3, 110–121. doi: 10.4161/chan.3.2.7932
Aggarwal, S. K., and MacKinnon, R. (1996). Contribution of the S4 segment to gating charge in the Shaker K+ channel. Neuron 16, 1169–1177. doi: 10.1016/s0896-6273(00)80143-9
Alonso, I., Barros, J., Tuna, A., Seixas, A., Coutinho, P., Sequeiros, J., et al. (2004). A novel R1347Q mutation in the predicted voltage sensor segment of the P/Q-type calcium-channel α-subunit in a family with progressive cerebellar ataxia and hemiplegic migraine. Clin. Genet. 65, 70–72. doi: 10.1111/j‥2004.00187.x
Althobaiti, Y. S., Almalki, A., Alsaab, H., Alsanie, W., Gaber, A., Alhadidi, Q., et al. (2019). Pregabalin: potential for addiction and a possible glutamatergic mechanism. Sci. Rep. 9:15136. doi: 10.1038/s41598-019-51556-4
Bahamonde, M. I., Serra, S. A., Drechsel, O., Rahman, R., Marcé-Grau, A., Prieto, M., et al. (2015). A single amino acid deletion (ΔF1502) in the S6 segment of CaV2.1 domain III associated with congenital ataxia increases channel activity and promotes Ca2+ influx. PLoS One 10:e0146035. doi: 10.1371/journal.pone.0146035
Battistini, S., Stenirri, S., Piatti, M., Gelfi, C., Righetti, P. G., Rocchi, R., et al. (1999). A new CACNA1A gene mutation in acetazolamide-responsive familial hemiplegic migraine and ataxia. Neurology 53, 38–43. doi: 10.1212/wnl.53.1.38
Beqollari, D., Dockstader, K., and Bannister, R. A. (2018). A skeletal muscle L-type Ca2+ channel with a single mutation in the selectivity filter (CaV1.1 E1014K) conducts K+. J. Biol. Chem. 293, 3126–3133. doi: 10.1074/jbc.M117.812446
Bertholon, P., Chabrier, S., Riant, F., Tournier-Lasserve, E., and Peyron, R. (2009). Episodic ataxia type 2: unusual aspects in clinical and genetic presentation. J. Neurol. Neurosurg. Psychiatry 80, 1289–1292. doi: 10.1136/jnnp.2008.159103
Blumkin, L., Michelson, M., Leshinsky-Silver, E., Kivity, S., Lev, D., and Lerman-Sagie, T. (2010). Congenital ataxia, mental retardation and dyskinesia associated with a novel CACNA1A mutation. J. Child Neurol. 25, 892–897. doi: 10.1177/0883073809351316
Bonnet, U., Richter, E. L., Isbruch, K., and Scherbaum, N. (2018). On the addictive power of gabapentinoids: a mini-review. Psychiatr. Danub. 30, 142–149. doi: 10.24869/psyd.2018.142
Brusich, D. J., Spring, A. M., James, T. D., Yeates, C. J., Helms, T. H., and Frank, C. A. (2018). Drosophila CaV2 channels harboring human migraine mutations cause synapse hyperexcitability that can be suppressed by inhibition of a Ca2+ store release pathway. PLoS Genet. 14:e1007577. doi: 10.1371/journal.pgen.1007577
Buraei, Z., Schofield, G., and Elmslie, K. S. (2007). Roscovitine differentially affects CaV2 and KV channels by binding to the open state. Neuropharmacology 52, 883–894. doi: 10.1016/j.neuropharm.2006.10.006
Campiglio, M., and Flucher, B. E. (2015). The role of auxiliary subunits for the functional diversity of voltage-gated calcium channels. J. Cell. Physiol. 230, 2019–2031. doi: 10.1002/jcp.24998
Cao, Y. Q., Piedras-Rentería, E. S., Smith, G. B., Chen, G., Harata, N. C., and Tsien, R. W. (2004). Presynaptic Ca2+ channels compete for channel type-preferring slots in altered neurotransmission arising from Ca2+ channelopathy. Neuron 43, 387–400. doi: 10.1016/j.neuron.2004.07.014
Cao, Y. Q., and Tsien, R. W. (2010). Different relationship of N- and P/Q-type Ca2+ channels to channel-interacting slots in controlling neurotransmission at cultured hippocampal synapses. J. Neurosci. 30, 4536–4546. doi: 10.1523/jneurosci.5161-09.2010
Carreño, O., Corominas, R., Serra, S. A., Sintas, C., Fernández-Castillo, N., Vila-Pueyo, M., et al. (2013). Screening of CACNA1A and ATP1A2 genes in hemiplegic migraine: clinical, genetic, and functional studies. Mol. Genet. Genomic Med. 1, 206–222. doi: 10.1002/mgg3.24
Carrera, P., Piatti, M., Stenirri, S., Grimaldi, L. M., Marchioni, E., Curcio, M., et al. (1999). Genetic heterogeneity in Italian families with familial hemiplegic migraine. Neurology 53, 26–33. doi: 10.1212/wnl.53.1.26
Catterall, W. A. (2010). Ion channel voltage sensors: structure, function, and pathophysiology. Neuron 67, 915–928. doi: 10.1016/j.neuron.2010.08.021
Choi, K. D., Kim, J. S., Kim, H. J., Jung, I., Jeong, S.-H., Lee, S.-H., et al. (2017). Genetic variants associated with episodic ataxia in Korea. Sci. Rep. 7:13855. doi: 10.1038/s41598-017-14254-7
Cricchi, F., Di Lorenzo, C., Grieco, G. S., Rengo, C., Cardinale, A., Racaniello, M., et al. (2007). Early-onset progressive ataxia associated with the first CACNA1A mutation identified within the I-II loop. J. Neurol. Sci. 254, 69–71. doi: 10.1016/j.jns.2007.01.008
Cuenca-León, E., Banchs, I., Serra, S. A., Latorre, P., Fernàndez-Castillo, N., Corominas, R., et al. (2009). Late-onset episodic ataxia type 2 associated with a novel loss-of-function mutation in the CACNA1A gene. J. Neurol. Sci. 280, 10–14. doi: 10.1016/j.jns.2009.01.005
Cuenca-León, E., Corominas, R., Fernàndez-Castillo, N., Volpini, V., Del Toro, M., Roig, M., et al. (2008). Genetic analysis of 27 Spanish patients with hemiplegic migraine, basilar-type migraine and childhood periodic syndromes. Cephalalgia 28, 1039–1047. doi: 10.1111/j.1468-2982.2008.01645.x
Dahimene, S., Page, K. M., Nieto-Rostro, M., Pratt, W. S., D’Arco, M., and Dolphin, A. C. (2016). A CaV2.1 N-terminal fragment relieves the dominant-negative inhibition by an episodic ataxia 2 mutant. Neurobiol. Dis. 93, 243–256. doi: 10.1016/j.nbd.2016.05.020
Damaj, L., Lupien-Meilleur, A., Lortie, A., Riou, É., Ospina, L. H., Gagnon, L., et al. (2015). CACNA1A haploinsufficiency causes cognitive impairment, autism and epileptic encephalopathy with mild cerebellar symptoms. Eur. J. Hum. Genet. 23, 1505–1512. doi: 10.1038/ejhg.2015.21
Denier, C., Ducros, A., Durr, A., Eymard, B., Chassande, B., and Tournier-Lasserve, E. (2001). Missense CACNA1A mutation causing episodic ataxia type 2. Arch. Neurol. 58, 292–295. doi: 10.1001/archneur.58.2.292
Di Guilmi, M. N., Wang, T., Inchauspe, C. G., Forsythe, I. D., Ferrari, M. D., van den Maagdenberg, A. M., et al. (2014). Synaptic gain-of-function effects of mutant CaV2.1 channels in a mouse model of familial hemiplegic migraine are due to increased basal [Ca2+]i. J. Neurosci. 34, 7047–7058. doi: 10.1523/JNEUROSCI.2526-13.2014
Dichgans, M., Herzog, J., Freilinger, T., Wilke, M., and Auer, D. P. (2005). 1H-MRS alterations in the cerebellum of patients with familial hemiplegic migraine type 1. Neurology 64, 608–613. doi: 10.1212/01.wnl.0000151855.98318.50
Dolphin, A. C. (2016). Voltage-gated calcium channels and their auxiliary subunits: physiology and pathophysiology and pharmacology. J. Physiol. 594, 5369–5390. doi: 10.1113/jp272262
Ducros, A., Denier, C., Joutel, A., Cecillon, M., Lescoat, C., Vahedi, K., et al. (2001). The clinical spectrum of familial hemiplegic migraine associated with mutations in a neuronal calcium channel. N. Engl. J. Med. 345, 17–24. doi: 10.1056/nejm200107053450103
Dunlap, K., Luebke, J. I., and Turner, T. J. (1994). Identification of calcium channels that control neurosecretion. Science 266, 828–830. doi: 10.1126/science.266.5186.828-a
Dunlap, K., Luebke, J. I., and Turner, T. J. (1995). Exocytotic Ca2+ channels in mammalian central neurons. Trends Neurosci. 18, 89–98. doi: 10.1016/0166-2236(95)80030-6
Elliot, M. A., Peroutka, S. J., Welch, S., and May, E. F. (1996). Familial hemiplegic migraine, nystagmus, and cerebellar atrophy. Ann. Neurol. 39, 100–106. doi: 10.1002/ana.410390115
Epi4K Consortium and Epilepsy Phenome/Genome Project. (2013). De novo mutations in epileptic encephalopathies. Nature 501, 217–221. doi: 10.1038/nature12439
Filosa, J. A., Bonev, A. D., Straub, S. V., Meredith, A. L., Wilkerson, M. K., Aldrich, R. W., et al. (2006). Local potassium signaling couples neuronal activity to vasodilation in the brain. Nat. Neurosci. 9, 1397–1403. doi: 10.1038/nn1779
Freilinger, T., Ackl, N., Ebert, A., Schmidt, C., Rautenstrauss, B., Dichgans, M., et al. (2011). A novel mutation in CACNA1A associated with hemiplegic migraine, cerebellar dysfunction and late-onset cognitive decline. J. Neurol. Sci. 300, 160–163. doi: 10.1016/j.jns.2010.09.032
Friend, K. L., Crimmins, D., Phan, T. G., Sue, C. M., Colley, A., Fung, V. S., et al. (1999). Detection of a novel missense mutation and second recurrent mutation in the CACNA1A gene in individuals with EA-2 and FHM. Hum. Genet. 105, 261–265. doi: 10.1007/s004390051099
Frontali, M. (2001). Spinocerebellar ataxia type 6: channelopathy or glutamine repeat disorder? Brain Res. Bull. 56, 227–231. doi: 10.1016/s0361-9230(01)00574-3
Gao, Z., Todorov, B., Barrett, C. F., van Dorp, S., Ferrari, M. D., van den Maagdenberg, A. M., et al. (2012). Cerebellar ataxia by enhanced CaV2.1 currents is alleviated by Ca2+-dependent K+-channel activators in CACNA1A(S218L) mutant mice. J. Neurosci. 32, 15533–15546. doi: 10.1523/JNEUROSCI.2454-12.2012
García, K. D., and Beam, K. G. (1996). Reduction of calcium currents by Lambert-Eaton syndrome sera: motoneurons are preferentially affected, and L-type currents are spared. J. Neurosci. 16, 4903–4913. doi: 10.1523/jneurosci.16-16-04903.1996
Garza-López, E., González-Ramírez, R., Gandini, M. A., Sandoval, A., and Felix, R. (2013). The familial hemiplegic migraine type 1 mutation K1336E affects direct G protein-mediated regulation of neuronal P/Q-type Ca2+ channels. Cephalalgia 33, 398–407. doi: 10.1177/0333102412475236
Garza-López, E., Sandoval, A., González-Ramírez, R., Gandini, M. A., van den Maagdenberg, A., De Waard, M., et al. (2012). Familial hemiplegic migraine type 1 mutations W1684R and V1696I alter G protein-mediated regulation of CaV2.1 voltage-gated calcium channels. Biochim. Biophys. Acta. 1822, 1238–1246. doi: 10.1016/j.bbadis.2012.04.008
Granato, M., van Eeden, F. J., Schach, U., Trowe, T., Brand, M., Furutani-Seiki, M., et al. (1996). Genes controlling and mediating locomotion behavior of the zebrafish embryo and larva. Development 123, 399–413.
Grieco, G. S., Gagliardi, S., Ricca, I., Pansarasa, O., Neri, M., Gualandi, F., et al. (2018). New CACNA1A deletions are associated to migraine phenotypes. J. Headache Pain 19:75. doi: 10.1186/s10194-018-0891-x
Griffin, A., Hamling, K. R., Knupp, K., Hong, S., Lee, L. P., and Baraban, S. C. (2017). Clemizole and modulators of serotonin signalling suppress seizures in Dravet syndrome. Brain 140, 669–683. doi: 10.1093/brain/aww342
Guerin, A. A., Feigenbaum, A., Donner, E. J., and Yoon, G. (2008). Stepwise developmental regression associated with novel CACNA1A mutation. Pediatr. Neurol. 39, 363–364. doi: 10.1016/j.pediatrneurol.2008.07.030
Guida, S., Trettel, F., Pagnutti, S., Mantuano, E., Tottene, A., Veneziano, L., et al. (2001). Complete loss of P/Q calcium channel activity caused by a CACNA1A missense mutation carried by patients with episodic ataxia type 2. Am. J. Hum. Genet. 68, 759–764. doi: 10.1086/318804
Hans, M., Luvisetto, S., Williams, M. E., Spagnolo, M., Urrutia, A., Tottene, A., et al. (1999). Functional consequences of mutations in the human α1A calcium channel subunit linked to familial hemiplegic migraine. J. Neurosci. 19, 1610–1619. doi: 10.1523/JNEUROSCI.19-05-01610.1999
Hering, S., Zangerl-Plessl, E. M., Beyl, S., Hohaus, A., Andranovits, S., and Timin, E. N. (2018). Calcium channel gating. Pflugers Arch. 470, 1291–1309. doi: 10.1007/s00424-018-2163-7
Imbrici, P., Jaffe, S. L., Eunson, L. H., Davies, N. P., Herd, C., Robertson, R., et al. (2004). Dysfunction of the brain calcium channel CaV2.1 in absence epilepsy and episodic ataxia. Brain 127, 2682–2692. doi: 10.1093/brain/awh301
Indelicato, E., Nachbauer, W., Karner, E., Eigentler, A., Wagner, M., Unterberger, I., et al. (2019). The neuropsychiatric phenotype in CACNA1A mutations: a retrospective single center study and review of the literature. Eur. J. Neurol. 26, 66.e1–66.e7. doi: 10.1111/ene.13765
Isaacs, D. A., Bradshaw, M. J., Brown, K., and Hedera, P. (2017). Case report of novel CACNA1A gene mutation causing episodic ataxia type 2. SAGE Open Med. Case Rep. 5:2050313X17706044. doi: 10.1177/2050313x17706044
Jen, J. C., Graves, T. D., Hess, E. J., Hanna, M. G., Griggs, R. C., and Baloh, R. W. (2007). Primary episodic ataxias: diagnosis, pathogenesis and treatment. Brain 130, 2484–2493. doi: 10.1093/brain/awm126
Jen, J., Kim, G. W., and Baloh, R. W. (2004). Clinical spectrum of episodic ataxia type 2. Neurology 62, 17–22. doi: 10.1212/01.wnl.0000101675.61074.50
Jen, J., Wan, J., Graves, M., Yu, H., Mock, A. F., Coulin, C. J., et al. (2001). Loss-of-function EA2 mutations are associated with impaired neuromuscular transmission. Neurology 57, 1843–1848. doi: 10.1212/wnl.57.10.1843
Jeng, C. J., Chen, Y. T., Chen, Y. W., and Tang, C. Y. (2006). Dominant-negative effects of human P/Q-type Ca2+channel mutations associated with episodic ataxia type 2. Am. J. Physiol. Cell Physiol. 290, C1209–C1220. doi: 10.1152/ajpcell.00247.2005
Jeng, C. J., Sun, M. C., Chen, Y. W., and Tang, C. Y. (2008). Dominant-negative effects of episodic ataxia type 2 mutations involve disruption of membrane trafficking of human P/Q-type Ca2+ channels. J. Cell. Physiol. 214, 422–433. doi: 10.1002/jcp.21216
Jiang, X., Raju, P. K., D’Avanzo, N., Lachance, M., Pepin, J., Dubeau, F., et al. (2019). Both gain-of-function and loss-of-function de novo CACNA1A mutations cause severe developmental epileptic encephalopathies in the spectrum of Lennox-Gastaut syndrome. Epilepsia 60, 1881–1894. doi: 10.1111/epi.16316
Jiang, H. S., Wang, D. M., Wang, Q., Yang, M., Wang, W., Pan, S. Y., et al. (2016). Missense mutation R1345Q in CACNA1A gene causes a new type of ataxia with episodic tremor: clinical features, genetic analysis and treatment in a familial case. Nan Fang Yi Ke Da Xue Xue Bao 36, 883–886.
Jodice, C., Mantuano, E., Veneziano, L., Trettel, F., Sabbadini, G., Calandriello, L., et al. (1997). Episodic ataxia type 2 (EA2) and spinocerebellar ataxia type 6 (SCA6) due to CAG repeat expansion in the CACNA1A gene on chromosome 19p. Hum. Mol. Genet. 6, 1973–1978. doi: 10.1093/hmg/6.11.1973
Jun, K., Piedras-Rentería, E. S., Smith, S. M., Wheeler, D. B., Lee, S. B., Lee, T. G., et al. (1999). Ablation of P/Q-type Ca2+ channel currents, altered synaptic transmission and progressive ataxia in mice lacking the α1A-subunit. Proc. Natl. Acad. Sci. U S A 96, 15245–15250. doi: 10.1073/pnas.96.26.15245
Kaja, S., Van de Ven, R. C., Broos, L. A., Frants, R. R., Ferrari, M. D., Van den Maagdenberg, A. M., et al. (2010). Severe and progressive neurotransmitter release aberrations in familial hemiplegic migraine type 1 CACNA1A S218L knock-in mice. J. Neurophysiol. 104, 1445–1455. doi: 10.1152/jn.00012.2010
Kaja, S., van de Ven, R. C., Broos, L. A., Veldman, H., van Dijk, J. G., Verschuuren, J. J., et al. (2005). Gene dosage-dependent transmitter release changes at neuromuscular synapses of CACNA1A R192Q knockin mice are non-progressive and do not lead to morphological changes or muscle weakness. Neuroscience 135, 81–95. doi: 10.1016/j.neuroscience.2005.04.069
Kang, M. G., and Campbell, K. P. (2003). γ subunit of voltage-activated calcium channels. J. Biol. Chem. 278, 21315–21318. doi: 10.1074/jbc.R300004200
Knierim, E., Leisle, L., Wagner, C., Weschke, B., Lucke, B., Bohner, G., et al. (2011). Recurrent stroke due to a novel voltage sensor mutation in CaV2.1 responds to verapamil. Stroke 42, e14–e17. doi: 10.1161/STROKEAHA.110.600023
Kors, E. E., Melberg, A., Vanmolkot, K. R., Kumlien, E., Haan, J., Raininko, R., et al. (2004). Childhood epilepsy, familial hemiplegic migraine, cerebellar ataxia and a new CACNA1A mutation. Neurology 63, 1136–1137. doi: 10.1212/01.wnl.0000138571.48593.fc
Kors, E. E., Terwindt, G. M., Vermeulen, F. L., Fitzsimons, R. B., Jardine, P. E., Heywood, P., et al. (2001). Delayed cerebral edema and fatal coma after minor head trauma: role of the CACNA1A calcium channel subunit gene and relationship with familial hemiplegic migraine. Ann. Neurol. 49, 753–760. doi: 10.1002/ana.1031
Lee, C. S., Dagnino-Acosta, A., Yarotskyy, V., Hanna, A., Lyfenko, A., Knoblauch, M., et al. (2015). Ca2+ permeation and/or binding to CaV1.1 fine-tunes skeletal muscle Ca2+ signaling to sustain muscle function. Skelet. Muscle. 5:4. doi: 10.1186/s13395-014-0027-1
Letts, V. A., Felix, R., Biddlecome, G. H., Arikkath, J., Mahaffey, C. L., Valenzuela, A., et al. (1998). The mouse stargazer gene encodes a neuronal Ca2+-channel γ subunit. Nat. Genet. 19, 340–347. doi: 10.1038/1228
Llinás, R., Steinberg, I. Z., and Walton, K. (1981). Presynaptic calcium currents in squid giant synapse. Biophys. J. 33, 289–321. doi: 10.1016/s0006-3495(81)84898-9
Low, S. E., Woods, I. G., Lachance, M., Ryan, J., Schier, A. F., and Saint-Amant, L. (2012). Touch responsiveness in zebrafish requires voltage-gated calcium channel 2.1b. J. Neurophysiol. 108, 148–159. doi: 10.1152/jn.00839.2011
Ludwig, A., Flockerzi, V., and Hofmann, F. (1997). Regional expression and cellular localization of the α1 and βsubunit of high voltage-activated calcium channels in rat brain. J. Neurosci. 17, 1339–1349. doi: 10.1523/JNEUROSCI.17-04-01339.1997
Luo, X., Rosenfeld, J. A., Yamamoto, S., Harel, T., Zuo, Z., Hall, M., et al. (2017). Clinically severe CACNA1A alleles affect synaptic function and neurodegeneration differentially. PLoS Genet. 13:e1006905. doi: 10.1371/journal.pgen.1006905
Maddison, P. (2012). Treatment in Lambert-Eaton myasthenic syndrome. Ann. N Y Acad. Sci. 1275, 78–84. doi: 10.1111/j.1749-6632.2012.06769.x
Maksemous, N., Roy, B., Smith, R. A., and Griffiths, L. R. (2016). Next-generation sequencing identifies novel CACNA1A gene mutations in episodic ataxia type 2. Mol. Genet. Genomic Med. 4, 211–222. doi: 10.1002/mgg3.196
Mantuano, E., Romano, S., Veneziano, L., Gellera, C., Castellotti, B., Caimi, S., et al. (2010). Identification of novel and recurrent CACNA1A gene mutations in fifteen patients with episodic ataxia type 2. J. Neurol. Sci. 291, 30–36. doi: 10.1016/j.jns.2010.01.010
Mantuano, E., Veneziano, L., Spadaro, M., Giunti, P., Guida, S., Leggio, M. G., et al. (2004). Clusters of non-truncating mutations of P/Q type Ca2+ channel subunit CaV2.1 causing episodic ataxia 2. J. Med. Genet. 41:e82. doi: 10.1136/jmg.2003.015396
Melliti, K., Grabner, M., and Seabrook, G. R. (2003). The familial hemiplegic migraine mutation R192Q reduces G-protein-mediated inhibition of P/Q-type (CaV2.1) calcium channels expressed in human embryonic kidney cells. J. Physiol. 546, 337–347. doi: 10.1113/jphysiol.2002.026716
Melzer, N., Classen, J., Reiners, K., and Buttmann, M. (2010). Fluctuating neuromuscular transmission defects and inverse acetazolamide response in episodic ataxia type 2 associated with the novel CaV2.1 single amino acid substitution R2090Q. J. Neurol. Sci. 296, 104–106. doi: 10.1016/j.jns.2010.06.024
Mezghrani, A., Monteil, A., Watschinger, K., Sinnegger-Brauns, M. J., Barrère, C., Bourinet, E., et al. (2008). A destructive interaction mechanism accounts for dominant-negative effects of misfolded mutants of voltage-gated calcium channels. J. Neurosci. 28, 4501–4511. doi: 10.1523/JNEUROSCI.2844-07.2008
Miki, T., Zwingman, T. A., Wakamori, M., Lutz, C. M., Cook, S. A., Hosford, D. A., et al. (2008). Two novel alleles of tottering with distinct CaV2.1 calcium channel neuropathologies. Neuroscience 155, 31–44. doi: 10.1016/j.neuroscience.2008.05.028
Mori, Y., Friedrich, T., Kim, M. S., Mikami, A., Nakai, J., Ruth, P., et al. (1991). Primary structure and functional expression from complementary DNA of a brain calcium channel. Nature 350, 398–402. doi: 10.1038/350398a0
Neely, A., and Hidalgo, P. (2014). Structure-function of proteins interacting with the α1 pore-forming subunit of high-voltage-activated calcium channels. Front. Physiol. 5:209. doi: 10.3389/fphys.2014.00209
Nikaido, K., Tachi, N., Ohya, K., Wada, T., and Tsutsumi, H. (2011). New mutation of CACNA1A gene in episodic ataxia type 2. Pediatr. Int. 53, 415–416. doi: 10.1111/j.1442-200x.2011.03390.x
Ohba, C., Osaka, H., Iai, M., Yamashita, S., Suzuki, Y., Aida, N., et al. (2013). Diagnostic utility of whole exome sequencing in patients showing cerebellar and/or vermis atrophy in childhood. Neurogenetics 14, 225–232. doi: 10.1007/s10048-013-0375-8
Ophoff, R. A., Terwindt, G. M., Vergouwe, M. N., van Eijk, R., Oefner, P. J., Hoffman, S. M., et al. (1996). Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell 87, 543–552. doi: 10.1016/s0092-8674(00)81373-2
Page, K. M., Heblich, F., Davies, A., Butcher, A. J., Leroy, J., Bertaso, F., et al. (2004). Dominant-negative calcium channel suppression by truncated constructs involves a kinase implicated in the unfolded protein response. J. Neurosci. 24, 5400–5409. doi: 10.1523/jneurosci.0553-04.2004
Page, K. M., Heblich, F., Margas, W., Pratt, W. S., Nieto-Rostro, M., Chaggar, K., et al. (2010). N terminus is key to the dominant negative suppression of CaV2 calcium channels. J. Biol. Chem. 285, 835–844. doi: 10.1074/jbc.M109.065045
Pelzer, N., Haan, J., Stam, A. H., Vijfhuizen, L. S., Koelewijn, S. C., Smagge, A., et al. (2018). Clinical spectrum of hemiplegic migraine and chances of finding a pathogenic mutation. Neurology 90, e575–e582. doi: 10.1212/wnl.0000000000004966
Petrovicova, A., Brozman, M., Kurca, E., Gobo, T., Dluha, J., and Kalmarova, K. (2017). Novel missense variant of CACNA1A gene in a Slovak family with episodic ataxia type 2. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czech Repub. 161, 107–110. doi: 10.5507/bp.2016.066
Pietrobon, D. (2007). Familial hemiplegic migraine. Neurotherapeutics 4, 274–284. doi: 10.1016/j.nurt.2007.01.008
Pietrobon, D. (2010). CaV2.1 channelopathies. Pflugers Arch. 460, 375–393. doi: 10.1007/s00424-010-0802-8
Pragnell, M., De Waard, M., Mori, Y., Tanabe, T., Snutch, T. P., and Campbell, K. P. (1994). Calcium channel β-subunit binds to a conserved motif in the I-II cytoplasmic linker of the α 1-subunit. Nature 368, 67–70. doi: 10.1038/368067a0
Rajakulendran, S., Graves, T. D., Labrum, R. W., Kotzadimitriou, D., Eunson, L., Davis, M. B., et al. (2010a). Genetic and functional characterisation of the P/Q calcium channel in episodic ataxia with epilepsy. J. Physiol. 588, 1905–1913. doi: 10.1113/jphysiol.2009.186437
Rajakulendran, S., Schorge, S., Kullmann, D. M., and Hanna, M. G. (2010b). Dysfunction of the CaV2.1 calcium channel in cerebellar ataxias. F1000 Biol. Rep. 2:4. doi: 10.3410/b2-4
Rajakulendran, S., Kaski, D., and Hanna, M. G. (2012). Neuronal P/Q-type calcium channel dysfunction in inherited disorders of the CNS. Nat. Rev. Neurol. 8, 86–96. doi: 10.1038/nrneurol.2011.228
Riant, F., Ducros, A., Ploton, C., Barbance, C., Depienne, C., and Tournier-Lasserve, E. (2010). De novo mutations in ATP1A2 and CACNA1A are frequent in early-onset sporadic hemiplegic migraine. Neurology 75, 967–972. doi: 10.1212/wnl.0b013e3181f25e8f
Romaniello, R., Zucca, C., Tonelli, A., Bonato, S., Baschirotto, C., Zanotta, N., et al. (2010). A wide spectrum of clinical, neurophysiological and neuroradiological abnormalities in a family with a novel CACNA1A mutation. J. Neurol. Neurosurg. Psychiatry 81, 840–843. doi: 10.1136/jnnp.2008.163402
Rose, S. J., Kriener, L. H., Heinzer, A. K., Fan, X., Raike, R. S., van den Maagdenberg, A. M., et al. (2014). The first knockin mouse model of episodic ataxia type 2. Exp. Neurol. 261, 553–562. doi: 10.1016/j.expneurol.2014.08.001
Scoggan, K. A., Friedman, J. H., and Bulman, D. E. (2006). CACNA1A mutation in a EA-2 patient responsive to acetazolamide and valproic acid. Can. J. Neurol. Sci. 33, 68–72. doi: 10.1017/s0317167100004728
Serra, S. A., Cuenca-León, E., Llobet, A., Rubio-Moscardo, F., Plata, C., Carreño, O., et al. (2010). A mutation in the first intracellular loop of CACNA1A prevents P/Q channel modulation by SNARE proteins and lowers exocytosis. Proc. Natl. Acad. Sci. U S A 107, 1672–1677. doi: 10.1073/pnas.0908359107
Serra, S., Fernàndez-Castillo, N., Macaya, A., Cormand, B., Valverde, B., and Fernández-Fernández, J. M. (2009). The hemiplegic migraine-associated Y1245C mutation in CACNA1A results in a gain of channel function due to its effect on the voltage sensor and G-protein-mediated inhibition. Pflügers Arch. 458, 489–502. doi: 10.1007/s00424-009-0637-3
Serra, S. A., Gené, G. G., Elorza-Vidal, X., and Fernández-Fernández, J. M. (2018). Cross talk between β subunits, intracellular Ca2+ signaling, and SNAREs in the modulation of CaV2.1 channel steady-state inactivation. Physiol. Rep. 6:e13557. doi: 10.14814/phy2.13557
Sills, G. J. (2006). The mechanisms of action of gabapentin and pregabalin. Curr. Opin. Pharmacol. 6, 108–113. doi: 10.1016/j.coph.2005.11.003
Sintas, C., Carreño, O., Fernàndez-Castillo, N., Corominas, R., Vila-Pueyo, M., Toma, C., et al. (2017). Mutation spectrum in the CACNA1A gene in 49 patients with episodic ataxia. Sci. Rep. 7:2514. doi: 10.1038/s41598-017-02554-x
Smith, L. A., Wang, X., Peixoto, A. A., Neumann, E. K., Hall, L. M., and Hall, J. C. (1996). A Drosophila calcium channel α1 subunit gene maps to a genetic locus associated with behavioral and visual defects. J. Neurosci. 16, 7868–7879. doi: 10.1523/JNEUROSCI.16-24-07868.1996
Soden, S. E., Saunders, C. J., Willig, L. K., Farrow, E. G., Smith, L. D., Petrikin, J. E., et al. (2014). Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci. Transl. Med. 265:265ra168. doi: 10.1126/scitranslmed.3010076
Spacey, S. D., Hildebrand, M. E., Materek, L. A., Bird, T. D., and Snutch, T. P. (2004). Functional implications of a novel EA2 mutation in the P/Q-type calcium channel. Ann. Neurol. 56, 213–220. doi: 10.1002/ana.20169
Stuart, S., Roy, B., Davies, G., Maksemous, N., Smith, R., and Griffiths, L. R. (2012). Detection of a novel mutation in the CACNA1A gene. Twin Res. Hum. Genet. 15, 120–125. doi: 10.1375/twin.15.1.120
Tantsis, E. M., Gill, D., Griffiths, L., Lawson, J., Maksemous, N., Ouvrier, R., et al. (2016). Eye movement disorders are an early manifestation of CACNA1A mutations in children. Dev. Med. Child Neurol. 58, 639–644. doi: 10.1111/dmcn.13033
Tao, X., Lee, A., Limapichat, W., Dougherty, D. A., and MacKinnon, R. (2010). A gating charge transfer center in voltage sensors. Science 328, 67–73. doi: 10.1126/science.1185954
Tarr, T. B., Malick, W., Liang, M., Valdomir, G., Frasso, M., Lacomis, D., et al. (2013). Evaluation of a novel calcium channel agonist for therapeutic potential in Lambert-Eaton myasthenic syndrome. J. Neurosci. 33, 10559–10567. doi: 10.1523/JNEUROSCI.4629-12.2013
Thomsen, L. L., Kirchmann, M., Bjornsson, A., Stefansson, H., Jensen, R. M., Fasquel, A. C., et al. (2007). The genetic spectrum of a population-based sample of familial hemiplegic migraine. Brain 130, 346–356. doi: 10.1093/brain/awl334
Tonelli, A., D’Angelo, M. G., Salati, R., Villa, L., Germinasi, C., Frattini, T., et al. (2006). Early onset, non-fluctuating spinocerebellar ataxia and a novel missense mutation in CACNA1A gene. J. Neurol. Sci. 241, 13–17. doi: 10.1016/j.jns.2005.10.007
Tottene, A., Fellini, T., Pagnutti, S., Luvisetto, S., Striessnig, J., Fletcher, C., et al. (2002). Familial hemiplegic migraine mutations increase Ca2+ influx through single human CaV2.1 channels and decrease maximal CaV2.1 current density in neurons. Proc. Natl. Acad. Sci. U S A 99, 13284–13289. doi: 10.1073/pnas.192242399
Tottene, A., Pivotto, F., Fellin, T., Cesetti, T., van den Maagdenberg, A. M., and Pietrobon, D. (2005). Specific kinetic alterations of human CaV2.1 calcium channels produced by mutation S218L causing familial hemiplegic migraine and delayed cerebral edema and coma after minor head trauma. J. Biol. Chem. 280, 17678–17686. doi: 10.1074/jbc.m501110200
Travaglini, L., Nardella, M., Bellacchio, E., D’Amico, A., Capuano, A., Frusciante, R., et al. (2017). Missense mutations of CACNA1A are a frequent cause of autosomal dominant nonprogressive congenital ataxia. Eur. J. Paediatr. Neurol. 221, 450–456. doi: 10.1016/j.ejpn.2016.11.005
Turner, T. J., Adams, M. E., and Dunlap, K. (1992). Calcium channels coupled to glutamate release identified by ω-Aga-IVA. Science 258, 310–313. doi: 10.1126/science.1357749
Tyagi, S., Bendrick, T. R., Filipova, D., Papadopoulos, S., and Bannister, R. A. (2019). A mutation in CaV2.1 linked to a severe neurodevelopmental disorder impairs channel gating. J. Gen. Physiol. 151, 850–859. doi: 10.1085/jgp.201812237
Uchitel, O. D., Protti, D. A., Sanchez, V., Cherksey, B. D., Sugimori, M., and Llinás, R. (1992). P-type voltage-dependent calcium channel mediates presynaptic calcium influx and transmitter release in mammalian synapses. Proc. Natl. Acad. Sci. U S A 89, 3330–3333. doi: 10.1073/pnas.89.8.3330
Vahedi, K., Denier, C., Ducros, A., Bousson, V., Levy, C., Chabriat, H., et al. (2000). CACNA1A gene de novo mutation causing hemiplegic migraine, coma and cerebellar atrophy. Neurology 55, 1040–1042. doi: 10.1212/wnl.55.7.1040
van den Maagdenberg, A. M., Kors, E. E., Brunt, E. R., Pascual, J., Ravine, D., Keeling, S., et al. (2002). Episodic ataxia type 2. Three novel truncating mutations and one novel missense mutation in the CACNA1A gene. J. Neurol. 249, 1515–1519. doi: 10.1007/s00415-002-0860-8
van den Maagdenberg, A. M., Pietrobon, D., Pizzorusso, T., Kaja, S., Broos, L. A. M., Cesetti, T., et al. (2004). A CACNA1A knockin migraine mouse model with increased susceptibility to cortical spreading depression. Neuron 41, 701–710. doi: 10.1016/s0896-6273(04)00085-6
van den Maagdenberg, A. M., Pizzorusso, T., Kaja, S., Terpolilli, N., Shapovalova, M., Hoebeek, F. E., et al. (2010). High cortical spreading depression susceptibility and migraine-associated symptoms in CaV2.1 S218L mice. Ann. Neurol. 67, 85–98. doi: 10.1002/ana.21815
Vecchia, D., Tottene, A., van den Maagdenberg, A. M., and Pietrobon, D. (2015). Abnormal cortical synaptic transmission in CaV2.1 knockin mice with the S218L missense mutation which causes a severe familial hemiplegic migraine syndrome in humans. Front. Cell. Neurosci. 9:8. doi: 10.3389/fncel.2015.00008
Volsen, S. G., Day, N. C., McCormack, A. L., Smith, W., Craig, P. J., Beattie, R. E., et al. (1997). The expression of voltage-dependent calcium channel β subunits in human cerebellum. Neuroscience 80, 161–174. doi: 10.1016/s0306-4522(97)00115-2
Wappl, E., Koschak, A., Poteser, M., Sinnegger, M. J., Walter, D., Eberhart, A., et al. (2002). Functional consequences of P/Q-type Ca2+ channel CaV2.1 missense mutations associated with episodic ataxia type 2 and progressive ataxia. J. Biol. Chem. 277, 6960–6966. doi: 10.1074/jbc.M110948200
Weiss, N., Sandoval, A., Felix, R., van den Maagdenberg, A., and De Waard, M. (2008). The S218L familial hemiplegic migraine mutation promotes deinhibition of CaV2.1 calcium channels during direct G-protein regulation. Pflugers Arch. 457, 315–326. doi: 10.1007/s00424-008-0541-2
Weiss, N., Tournier-Lasserve, E., and De Waard, M. (2007). Role of P/Q calcium channel in familial hemiplegic migraine. Med. Sci. 23, 53–63. doi: 10.1051/medsci/200723153
Wen, H., Linhoff, M. W., Hubbard, J. M., Nelson, N. R., Stensland, D., Dallman, J., et al. (2013). Zebrafish calls for reinterpretation for the roles of P/Q calcium channels in neuromuscular transmission. J. Neurosci. 33, 7384–7392. doi: 10.1523/JNEUROSCI.5839-12.2013
Weyhrauch, D. L., Ye, D., Boczek, N. J., Tester, D. J., Gavrilova, R. H., Patterson, M. C., et al. (2016). Whole exome sequencing and heterologous cellular electrophysiology studies elucidate a novel loss-of-function mutation in the CACNA1A-encoded neuronal P/Q-type calcium channel in a child with congenital hypotonia and developmental delay. Pediatr. Neurol. 55, 46–51. doi: 10.1016/j.pediatrneurol.2015.10.014
Wilson, G. N. (2014). Exome analysis of connective tissue dysplasia: death and rebirth of clinical genetics? Am. J. Med. Genet. A 164A, 1209–1212. doi: 10.1002/ajmg.a.36463
Wu, M., White, B. H. V., Boehm, A., Meriney, C. J., Kerrigan, K., Frasso, M., et al. (2018). New CaV2 calcium channel gating modifiers with agonist activity and therapeutic potential to treat neuromuscular disease. Neuropharmacology 131, 176–189. doi: 10.1016/j.neuropharm.2017.12.022
Wu, J., Yan, Z., Li, Z., Qian, X., Lu, S., Dong, M., et al. (2016). Structure of the voltage-gated calcium channel CaV1.1 at 3.6 Å resolution. Nature 537, 191–196. doi: 10.1038/nature19321
Yan, Z., Chi, P., Bibb, J. A., Ryan, T. A., and Greengard, P. (2002). Roscovitine: a novel regulator of P/Q-type calcium channels and transmitter release in central neurons. J. Physiol. 540, 761–770. doi: 10.1113/jphysiol.2001.013376
Yang, J., Ellinor, P. T., Sather, W. A., Zhang, J. F., and Tsien, R. W. (1993). Molecular determinants of Ca2+ selectivity and ion permeation in L-type Ca2+ channels. Nature 366, 158–161. doi: 10.1038/366158a0
Yang, Y., Muzny, D. M., Xia, F., Niu, Z., Person, R., Ding, Y., et al. (2014). Molecular findings among patients referred for clinical whole-exome sequencing. JAMA 312, 1870–1879. doi: 10.1001/jama.2014.14601
Ye, D., Tester, D. J., Zhou, W., Papagiannis, J., and Ackerman, M. J. (2019). A pore-localizing CACNA1C-E1115K missense mutation, identified in a patient with idiopathicQT prolongation, bradycardia and autism spectrum disorder, converts the L-type calcium channel into a hybrid nonselective monovalent cation channel. Heart Rhythm. 16, 270–278. doi: 10.1016/j.hrthm.2018.08.030
Yue, Q., Jen, J. C., Nelson, S. F., and Baloh, R. W. (1997). Progressive ataxia due to a missense mutation in a calcium-channel gene. Am. J. Hum. Genet. 61, 1078–1087. doi: 10.1086/301613
Zafeiriou, D. I., Lehmann-Horn, F., Vargiami, E., Teflioudi, E., Ververi, A., and Jurkat-Rott, K. (2009). Episodic ataxia type 2 showing ictal hyperhidrosis with hypothermia and interictal chronic diarrhea due to a novel CACNA1A mutation. Eur. J. Paediatr. Neurol. 13, 191–193. doi: 10.1016/j.ejpn.2008.02.011
Keywords: CaV2.1, α1A, P/Q-type, channelopathy, familial hemiplegic migraine type 1, episodic ataxia type 2, vertebrate models, zebrafish
Citation: Tyagi S, Ribera AB and Bannister RA (2020) Zebrafish as a Model System for the Study of Severe CaV2.1 (α1A) Channelopathies. Front. Mol. Neurosci. 12:329. doi: 10.3389/fnmol.2019.00329
Received: 14 October 2019; Accepted: 23 December 2019;
Published: 07 February 2020.
Edited by:
Baojin Ding, University of Louisiana at Lafayette, United StatesReviewed by:
Paul Brehm, Oregon Health & Science University, United StatesJose M. Fernandez-Fernandez, Pompeu Fabra University, Spain
Noèlia Fernàndez-Castillo, Center for Biomedical Research in the Network of Rare Diseases (CIBERER), Spain
Copyright © 2020 Tyagi, Ribera and Bannister. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roger A. Bannister, cmJhbm5pc3RlckBzb20udW1hcnlsYW5kLmVkdQ==