Abstract
The inability of individual neurons to compensate for aging-related damage leads to a gradual loss of functional plasticity in the brain accompanied by progressive impairment in learning and memory. Whereas this loss in neuroplasticity is gradual during normal aging, in neurodegenerative diseases such as Alzheimer’s disease (AD), this loss is accelerated dramatically, leading to the incapacitation of patients within a decade of onset of cognitive symptoms. The mechanisms that underlie this accelerated loss of neuroplasticity in AD are still not completely understood. While the progressively increasing proteinopathy burden, such as amyloid β (Aβ) plaques and tau tangles, definitely contribute directly to a neuron’s functional demise, the role of non-neuronal cells in controlling neuroplasticity is slowly being recognized as another major factor. These non-neuronal cells include astrocytes, microglia, and oligodendrocytes, which through regulating brain homeostasis, structural stability, and trophic support, play a key role in maintaining normal functioning and resilience of the neuronal network. It is believed that chronic signaling from these cells affects the homeostatic network of neuronal and non-neuronal cells to an extent to destabilize this harmonious milieu in neurodegenerative diseases like AD. Here, we will examine the experimental evidence regarding the direct and indirect pathways through which astrocytes and microglia can alter brain plasticity in AD, specifically as they relate to the development and progression of tauopathy. In this review article, we describe the concepts of neuroplasticity and glial plasticity in healthy aging, delineate possible mechanisms underlying tau-induced plasticity dysfunction, and discuss current clinical trials as well as future disease-modifying approaches.
Introduction
Neuroplasticity Is Essential for Healthy Brain Function
In the late 1800s and early 1900s, the scientist Santiago Ramón y Cajal set down his Neuron doctrine which defined the neuron as the physiologic, metabolic, anatomic, and genetic basis of the nervous system (Ramón y Cajal, 1899/1904; Shepherd, 1991; Jones, 1994; Mateos-Aparicio and Rodríguez-Moreno, 2019). He also proposed that the brain can improve its function by improving the connections between neurons, establishing a paradigm that is now known as neuroplasticity (Ramón y Cajal, 1892, 1894a,b,c; Stahnisch and Nitsch, 2002). In 1949, Hebb (1949) formally defined neuroplasticity to refer to increased neuronal activity leading to stronger neuronal connections. Indeed, a simplified way to illustrate the Hebbian definition of neuroplasticity is through the statement: “cells that fire together, wire together” (Shatz, 1992). It follows that plasticity can be thought of as a continuum of neuronal activity that results in improved brain function through an iterative and cooperative process.
Plasticity can be assessed at different levels (Figure 1). Synaptic plasticity can be measured structurally at the level of altered dendritic spine morphology, which is modulated by synaptic activity and determines synapse strength (Figure 1A; Harris and Kater, 1994; Gipson and Olive, 2017). Experimental evidence in rats, mice, birds, squirrels, organotypic brain slices, and neuronal cultures have revealed that increases in neuronal activity lead to spine induction (Yuste and Bonhoeffer, 2001). Also, the size of a dendritic spine head correlates with the number of postsynaptic receptors and the number of presynaptic vesicles ready for release (Yuste and Bonhoeffer, 2001). The functional readouts of synaptic plasticity can be captured through long-term potentiation (LTP) and long-term depression (LTD), which are processes through which synaptic transmission is either strengthened or weakened, respectively (Figure 1B; Bear and Malenka, 1994). During LTP, synaptic activity is increased, unused dendritic spines are pruned, activated spines are stabilized and new spines are made (De Roo et al., 2008). The opposite happens in LTD: synaptic activity is reduced, synapses are weakened, and dendritic spines shrink or are lost completely (Gipson and Olive, 2017). Synaptic plasticity within a group of neurons is modulated through homeostatic scaling, a process in which neurons keep their output activity within a physiologic range to normalize overall signaling (Figure 1B; Chowdhury and Hell, 2018). Viewed at the level of large neural networks and the brain as an organ, plasticity affects phenomena such as cognition, learning, memory, and resilience (Figure 1C; Stampanoni Bassi et al., 2019). The brain is thus a very adaptable, self-regulating organ that is capable of rewiring itself, if necessary. However, the capacity of the brain to accomplish these tasks efficiently and effectively is impeded by aging and even more so in conditions like injury and neurodegenerative disease.
Figure 1
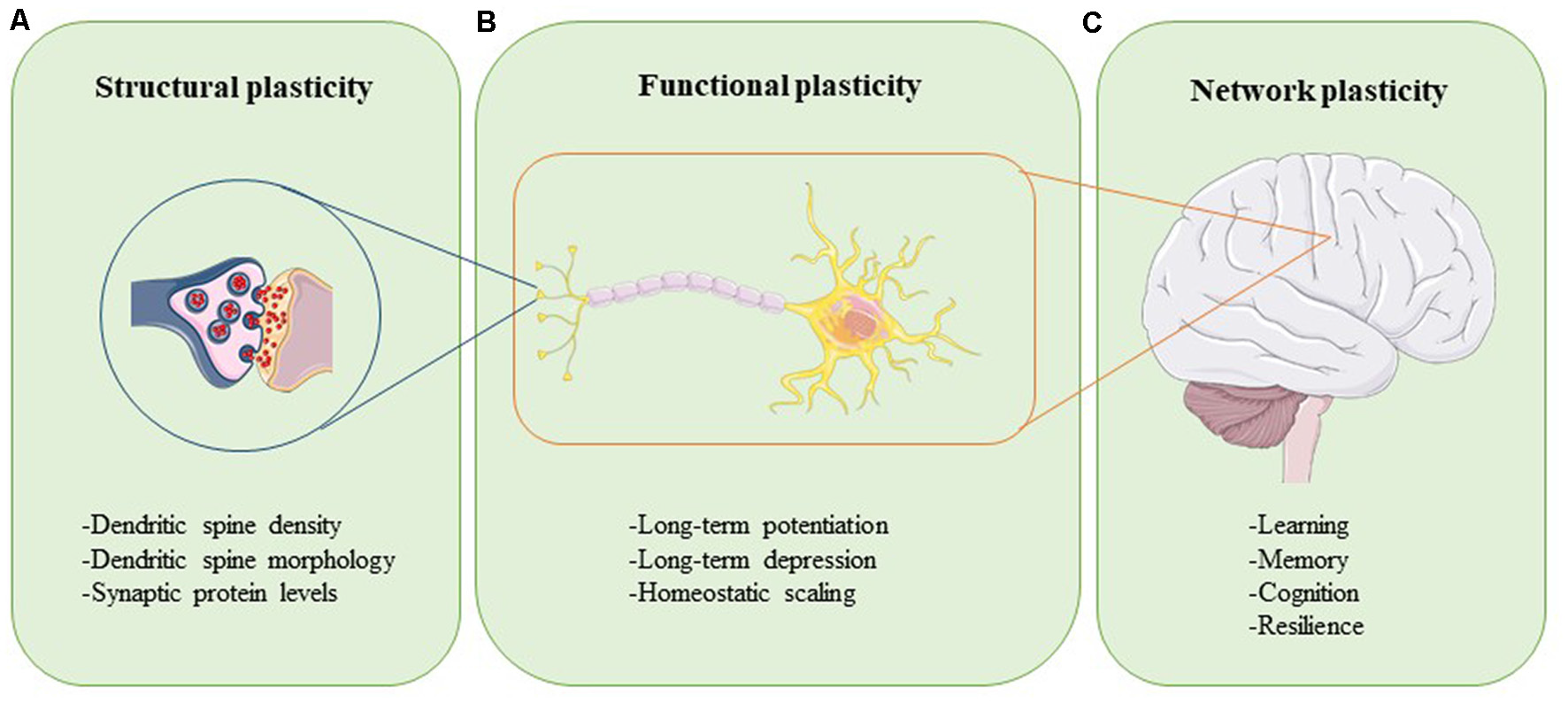
Neuroplasticity can be assessed at different levels. Structural plasticity refers to the morphological changes that occur at the synapse, such as altered dendritic spine density, dendritic spine shape, and synaptic protein profiles (A). Functional plasticity affects neuronal circuit regulation and includes processes such as long-term potentiation (LTP), long-term depression (LTD), and homeostatic scaling (B). At the organ level, network plasticity modulates learning, memory, cognition, and resilience (C). Proper function and careful integration of all levels of plasticity are required for healthy brain function.
Glia Are Integrally Involved in Neuroplasticity
The healthy functioning of neuronal networks in the brain requires non-neuronal cells as well (Allen and Lyons, 2018). Glial cells, such as astrocytes and microglia, are constantly interfacing with neurons to modulate and refine signaling besides providing trophic support (Allen and Lyons, 2018). The “tripartite” synapse model was established to describe the interactions between the presynaptic compartment, the post-synaptic compartment, and the perisynaptic astrocytic processes that surround them (Perea et al., 2009). As more information regarding the importance of microglia in neuronal signaling emerged, a “quadripartite” synapse model was proposed that includes perisynaptic processes from microglia along with the other components (Schafer et al., 2013). Due to their proximity to the synapse, glial cells are in a critical position to manipulate synaptic activity. Indeed, glial cells respond rapidly to changes in their environment in a matter of seconds (Nimmerjahn et al., 2005). Such stimulus-driven responses of these non-neuronal cells aim to preserve the homeostasis of the neuronal and synaptic milieu (Vainchtein and Molofsky, 2020). This quick response of the microglia and astrocytes to compensate or adjust their function can be thought of as glial plasticity. It can be hypothesized that the rapid alterations in glial plasticity may have a long-term and protracted effect on neuronal health and neuroplasticity. The mechanisms by which these non-neuronal cells regulate synaptic activity and plasticity are of immense interest because of the enormous impact these processes have on cognition and healthy brain function.
Neuroplasticity Loss in AD and Related Tauopathies
Alzheimer’s disease (AD) is a neurodegenerative disease characterized clinically by progressive memory loss, loss of executive function, confusion, disorientation, and aphasia (Alzheimer’s Association, 2020). Neuropathologically, AD is characterized by intracellular neurofibrillary tangles (NFTs) and extracellular amyloid β (Aβ) plaques (Braak and Braak, 1991). The pathophysiology of AD follows a stereotypical spatio-temporal pattern: extracellular Aβ plaque pathology appears first in the frontal lobes, even before the appearance of any clinical symptoms, followed by the appearance of NFT deposits in the transentorhinal cortex and hippocampus (Braak and Braak, 1991). Imaging studies investigating connectivity in these brain regions have shown that, even in preclinical or pre-pathological stages, subtle functional connectivity disturbances can be detected (Grieder et al., 2018; Tijms et al., 2018). For example, the default mode network (DMN) is affected even before robust Aβ plaque pathology sets in and presages the emergence of Aβ plaques (Palmqvist et al., 2017). This network includes hubs such as the medial prefrontal cortex, posterior cingulate cortex, retrosplenial cortex, basal forebrain, and thalamus (Alves et al., 2019). Aβ deposition leads to hypometabolism in brain regions connected by the DMN and this hypometabolism may ultimately lead to cognitive deterioration (Pascoal et al., 2019). It should also be mentioned that Aβ deposits are directly correlated with increasing microglial and astrocyte engagement (Selkoe and Hardy, 2016). It is possible, that since the healthy brain is fairly plastic, especially in younger ages, it can compensate for changes in neuronal connectivity, neuroinflammation, and initial Aβ proteinopathy, until a threshold is reached at which the compensatory alterations fail (Burke and Barnes, 2006; Figure 2A).
Figure 2
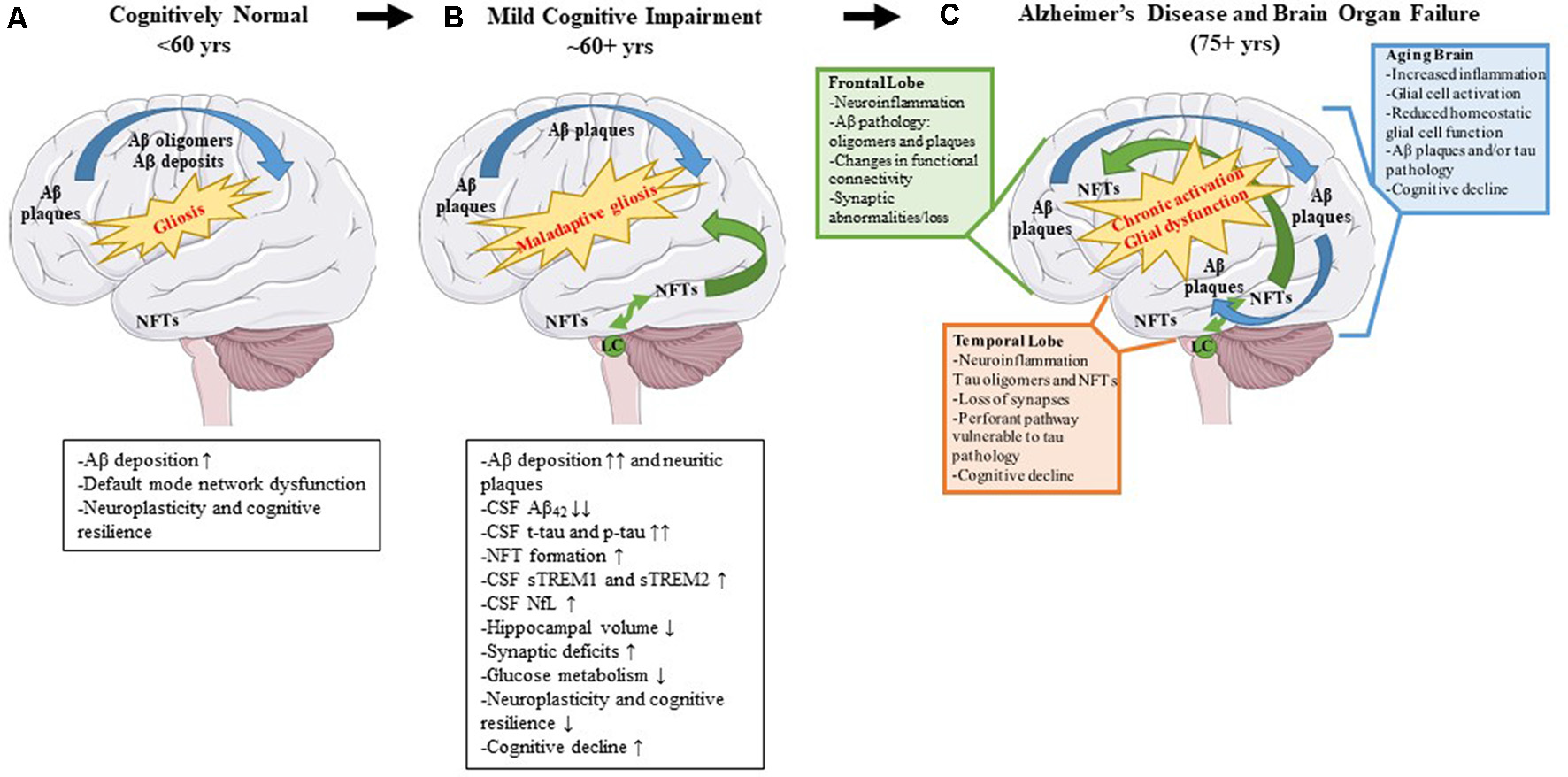
Pathological changes and biomarker alterations observed in the development of Alzheimer’s disease (AD). Moving left to right: schematic depicting brain alterations in a cognitively normal individual (A), in an mild cognitive impairment (MCI) individual (B), and in an end-stage AD patient (C). Amyloid β (Aβ) pathology begins in the frontal cortex and spreads throughout the brain (blue arrows). Neurofibrillary tangle (NFT) pathology begins in the medial temporal cortex and locus coeruleus and spreads to the rest of the brain (green arrows). Gliosis increases with age, and the onset of the disease leads to maladaptive gliosis and eventual chronic glial activation and dysfunction. Biomarker alterations corresponding to each stage are listed under (A,B) in black boxes. Abnormal changes in the frontal and temporal lobes as well as changes observed in an aged brain are listed in (C) in green, orange, and blue boxes, respectively.
NFTs are complex intracellular deposits that appear decades after initial Aβ emergence. These intracellular inclusions are composed primarily of hyperphosphorylated forms of the protein tau which is a microtubule-binding protein that stabilizes microtubules (Iqbal et al., 2016). NFTs first appear in the entorhinal cortex and propagate to the hippocampus, the center of learning, and memory (Braak and Braak, 1991) initiating a toxic cascade of events leading ultimately to impaired cognition (DeKosky and Scheff, 1990). Several studies have shown that the severity of cognitive impairment correlates with intracellular NFT burden and neuritic plaque pathology and less with Aβ plaque pathology (Arriagada et al., 1992; Giannakopoulos et al., 2003; Figure 2B). During this time, the glial response is hypothesized to shift to a more toxic neurodegenerative phenotype characterized by a unique gene expression signature (reviewed in Deczkowska et al., 2018). This loss of glial plasticity, where these glial cells lose their protective function and assume a neurotoxic function, may be related to loss of neuroplasticity leading to neurodegeneration and cognitive failure in the end stage of the disease (Figure 2C). A key factor to consider is that many elderly people have Aβ deposits or even tau pathology in their brains and retain high levels of cognition (Price and Morris, 1999; Knopman et al., 2003; Crary et al., 2014). Because the brain is very adaptable, it can compensate for synaptic dysfunction or synaptic loss in the early phases by altering the properties of remaining synapses (Figure 2A). However, at some point, the ability of the brain to compensate is exceeded and efforts to stave off neuronal loss and cognitive decline are no longer viable (DeKosky and Scheff, 1990; Scheff et al., 1990; Figures 2B,C).
Since one of the hallmarks of AD is NFTs, AD is included under the umbrella of neurodegenerative diseases called tauopathies. In AD and other tauopathies, tau becomes hyperphosphorylated, dissociates from microtubules, and accumulates as intracellular inclusions of misfolded, non-functional protein. Such intracellular inclusions of aggregated tau protein are referred to as NFTs (Iqbal et al., 2016). The majority of tauopathies are sporadic, but familial frontotemporal dementia (FTD) has been linked to mutations in the gene that encodes tau (microtubule-associated protein tau, MAPT, Ghetti et al., 2015). FTD patients display severe frontal and temporal lobe atrophy and dementia (Ghetti et al., 2015). Tau-positron emission tomography (PET) tracing and 18F FDG (fluorodeoxyglucose)-PET studies show overlap between hypometabolism of glucose and tau deposition in the brains of FTD patients (Su et al., 2020). Also, iPSC neurons derived from FTD patients show abnormally increased activity when chronically depolarized (Sohn et al., 2019). While studies like these have begun to reveal altered synaptic function in FTD patients, pre-clinical models of tauopathy have provided us with mechanistic insight into how neuronal plasticity is affected by tau pathology. Reduction in spine density, a common feature of tauopathy mouse models, is associated with loss of function (Crimins et al., 2012; Sydow et al., 2016). It follows then that many mice expressing mutated forms of human tau display reduced levels of pre- and post-synaptic proteins (Schindowski et al., 2006; Yoshiyama et al., 2007; Crimins et al., 2012; de Calignon et al., 2012; Van der Jeugd et al., 2012). Interestingly, some of these changes are observed before the formation of NFTs (Yoshiyama et al., 2007). Some tauopathy models are characterized by electrophysiological abnormalities (Schindowski et al., 2006; Sydow et al., 2011; Crimins et al., 2012; Levenga et al., 2013; Polydoro et al., 2014) and others show non-convulsive epileptiform activity (Decker et al., 2016; Maeda et al., 2016). Mouse models deficient for tau have also provided information concerning synaptic plasticity. These studies complicate the picture since it seems that tau is involved in the breakdown of synaptic plasticity (as discussed above); however, it also seems to be necessary for maintaining these processes. A recent study found that Mapt−/− mice display hyperactivity, altered LTP, and impaired memory, while a partial deletion of mouse tau (Mapt+/−) was able to partially rescue this phenotype (Biundo et al., 2018). Other studies have found similar results (Ikegami et al., 2000; Ahmed et al., 2014; Ma et al., 2014). Tau knockout mice also show both reduced LTD (Kimura et al., 2014; Regan et al., 2015) and impaired α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) internalization (Regan et al., 2015). Finally, an underlying factor that is common across many tau transgenic models is increased neuroinflammation in the form of microgliosis and astrocytosis (Schindowski et al., 2006; Yoshiyama et al., 2007; de Calignon et al., 2012; Maeda et al., 2016). Indeed, a network-based analysis of neurodegenerative diseases based on prion models paired with cell-type classification showed that, during the development of the disease, the majority of the upregulated genes were expressed by microglia (Vincenti et al., 2015). These studies highlight the importance of understanding how tau-mediated changes to synaptic plasticity are intimately connected with glial plasticity alterations.
We do not fully understand how different factors, be they genetic or environmental, synergize, and predispose the brain to develop inexorable neurodegenerative diseases, such as tauopathies. Also, still unknown is how two seemingly disparate pathological proteinopathies, Aβ and tau, synergize and lead to loss of neuroplasticity observed in AD. A clear picture of how neuroplasticity and glial plasticity result in brain organ failure through a complex interaction between neurons, synapses, and non-neuronal cell function is only now emerging. Here we will review the known roles of non-neuronal cells—astrocytes, oligodendrocytes, and microglia—as they pertain to neuronal health and synaptic plasticity in AD and relate this to disrupted glial plasticity.
Early Neuroplasticity Loss in AD and in Mouse Models Is Reversible
Synaptic dysregulation has been suggested to begin in the prodromal phase of AD, even before Aβ plaque formation (Walsh and Selkoe, 2007), perhaps triggered by oligomeric Aβ or transient soluble Aβ species that are neurotoxic (Heiss et al., 2017). Interestingly, not only does Aβ affect synaptic plasticity, other cleavage products of the amyloid precursor protein (APP), the parent molecule of Aβ, can also have neurotoxic functions. For example, the APP intracellular domain (AICD), produced as a by-product of the amyloidogenic processing of the APP protein, has been shown to regulate synaptic function and may also play a role in synaptic plasticity (Konietzko, 2012; Pousinha et al., 2017; Skaper et al., 2017). It is also noteworthy that soluble APPα (the most common proteolytic cleavage product of APP in healthy brains) has been suggested to play a role in synaptic growth and plasticity, but whether APPα is helpful or damaging remains highly debated (Corbett and Hooper, 2018).
The majority of studies seeking to improve synaptic plasticity have been conducted using either transgenic rodent models of Aβ deposition or injection models of Aβ (Hao et al., 2011; Bachstetter et al., 2012; Chen et al., 2012; Duffy and Hölscher, 2013; Wu et al., 2013; Deibel et al., 2016; Iaccarino et al., 2016; Ahmad et al., 2017; Wilkaniec et al., 2018; Liang et al., 2019; Martorell et al., 2019), while very few have been executed in tau transgenic models (Martorell et al., 2019), leading to a relative lack of understanding of tau-related abnormalities in neuroplasticity and glial plasticity. In wild type mice or human APP transgenic mice, various inhibitors, agonists, or small molecules as well as brain stimulation and environmental enrichment have been tested (Supplementary Table S1). In most cases, these manipulations resulted in improved performance in cognitive paradigms, increased synaptic function, increased the density of dendritic spines and increased levels of synaptic proteins (Hao et al., 2011; Bachstetter et al., 2012; Chen et al., 2012; Wu et al., 2013; Ahmad et al., 2017; Liang et al., 2019; Martorell et al., 2019). Many of these treatments reduced neuroinflammation (Hao et al., 2011; Bachstetter et al., 2012; Chen et al., 2012; Wu et al., 2013; Ahmad et al., 2017; Liang et al., 2019) and a few studies also reported improved neuronal survival (Chen et al., 2012; Ahmad et al., 2017). This suggests some form of synergy between processes connecting brain immunity, neuroplasticity, and neuronal survival. However, other studies involving cholinergic depletion and optogenetic stimulation did not result in improved neuroplasticity and sometimes even decreased cognitive performance and increased neuroinflammation (Deibel et al., 2016; Iaccarino et al., 2016). Effects on Aβ and tau neuropathology were mixed, with some studies showing reduced burden (Hao et al., 2011; Chen et al., 2012; Wu et al., 2013; Iaccarino et al., 2016; Ahmad et al., 2017; Martorell et al., 2019), while others reported no effect (Bachstetter et al., 2012; Deibel et al., 2016; Liang et al., 2019), and these changes did not always correspond to the studies that showed improved synaptic plasticity.
Synaptic plasticity has also been modulated in non-AD mouse models with various agents, neuropeptides, and exercise (Soto et al., 2015; Liu, Yi et al., 2018; Luo et al., 2019; Zhou et al., 2019). These studies showed improvements in synaptic protein levels, LTP, and cognitive performance as well as reduced neuroinflammation and, when assessed, neurodegeneration (Soto et al., 2015; Liu, Yi et al., 2018; Luo et al., 2019; Zhou et al., 2019). These studies are only some representative examples of findings in the field, but what can be observed is that much of the research on synaptic plasticity in AD has been conducted in rodent models that only accumulate Aβ plaque pathology (Pozueta et al., 2013). They do not take into account the effects of tau pathology on synaptic dysfunction and synapse loss, which are the major determinates of cognitive decline in AD.
Chronic Glial Signaling Counteracts Glial Plasticity in Neurodegenerative Conditions
Neuroinflammation increases with age and is considered a normal part of brain aging (Cribbs et al., 2012). Aged brains are characterized by increased activation of glial cells and elevated levels of pro-inflammatory molecules and reduced anti-inflammatory mediators (Di Benedetto et al., 2017). Glial cells in an aged brain are primed for inflammatory responses and they are slow in returning to baseline following acute insults (Di Benedetto et al., 2017). Also, senescent glial cells have a reduced ability to perform their homeostatic functions (Di Benedetto et al., 2017), thus, increasing the vulnerability of the aging brain to proteinopathy-related injuries. Indeed, neurodegenerative disease is accompanied by progressively exacerbated neuroinflammation marked by astrogliosis and microgliosis (Serrano-Pozo et al., 2011; Di Benedetto et al., 2017). Topics of critical interest in the field are how does abnormal proteinopathy impact glial cell functionality, how does dysregulated glial function alter intraneuronal proteostasis, and finally, how do these alterations contribute to the progression of disease?
Glia can directly regulate short-term neuroplasticity in the synaptic cleft, either through homosynaptic connections or by modulating complex synaptic loops—thus they support neuronal activity at the cellular and network level (Perea et al., 2009; Schafer et al., 2013). Glia also possesses immense plasticity of their own as they can react within seconds to environmental threats to rebalance network homeostasis (Nimmerjahn et al., 2005). Since glial plasticity processes are critical in supporting and complementing neuronal health and function, glial plasticity and neuronal plasticity are integrated processes. In diseases such as AD, or even during aging, chronic gliosis can lead to loss of function (Hickman et al., 2013) whereby glia are unable to respond to challenges resulting in dyshomeostasis, morphological changes, and disrupted signaling (Bellamy et al., 2015). In the next sections of this review, we will discuss how tau accumulation in AD is related to loss of brain function.
Experimental Models Show That NFT Pathology Correlates With Gliosis and Cognitive Decline
Relative to the effects of Aβ pathology, cognitive decline and neurodegenerative features are closely associated with NFT pathology in AD (Arriagada et al., 1992; Gómez-Isla et al., 1997; Giannakopoulos et al., 2003). Until recently, it was widely believed that NFTs deposited as intracellular inclusions are the toxic form of tau, but recent research has provided evidence that even soluble forms of phosphorylated tau may have a pathogenic effect on neuroplasticity (Fá et al., 2016; Ghag et al., 2018). The questions remain though, how do different forms of Aβ and tau affect neuroplasticity and glial plasticity, and what is the spatio-temporal relationship between these proteinopathies and dysfunctional plasticity? In this review, we will focus on tauopathy and its effects on neuroplasticity and glial plasticity.
In AD, tau pathology originates mostly in the trans-entorhinal and hippocampal regions of the brain and then spreads out of the medial temporal lobe to other brain areas as the disease progresses (Mudher et al., 2017). This has been recapitulated in rodent transgenic models of tauopathy where the spread of tau pathology out of the medial temporal lobe involves the propagation of soluble tau species along synaptically connected neuronal networks (de Calignon et al., 2012; Calafate et al., 2015; DeVos et al., 2018). Another mechanism of tau spread involves the dispersal of tau pathology via glial cells (Asai et al., 2015; Hopp et al., 2018). Gliosis closely follows the development of tauopathy in transgenic mouse models (Ramsden et al., 2005; Schindowski et al., 2006; Yoshiyama et al., 2007; de Calignon et al., 2012; Maeda et al., 2016). We developed several mouse models of tauopathy in our lab to understand how gliosis is differentially regulated in the presence of intracellular phosphorylated tau and NFT inclusions (Koller et al., 2019). These models express different human mutant tau constructs following neonatal delivery of recombinant adeno-associated viruses (AAV). We observed a dramatic induction of gliosis, especially of hypertrophic microglia in rodents with NFT pathology but not in mice with somatodendritic hyperphosphorylated tau (Figure 3). Microglia present in NFT-rich regions display considerably hypertrophic morphology (Figure 3K) when compared to microglia in a mouse brain with soluble phosphorylated tau but no NFTs (Figure 3G) or microglia in control cohorts (Figure 3C). Astrocytes display similar changes (Figures 3D,H,L). This might reflect a continuum of glial functionality accompanying the spectrum of tau pathogenesis, starting from accumulation of relatively soluble hyperphosphorylated tau in the somatodendritic compartment towards more neurotoxic NFT deposits.
Figure 3
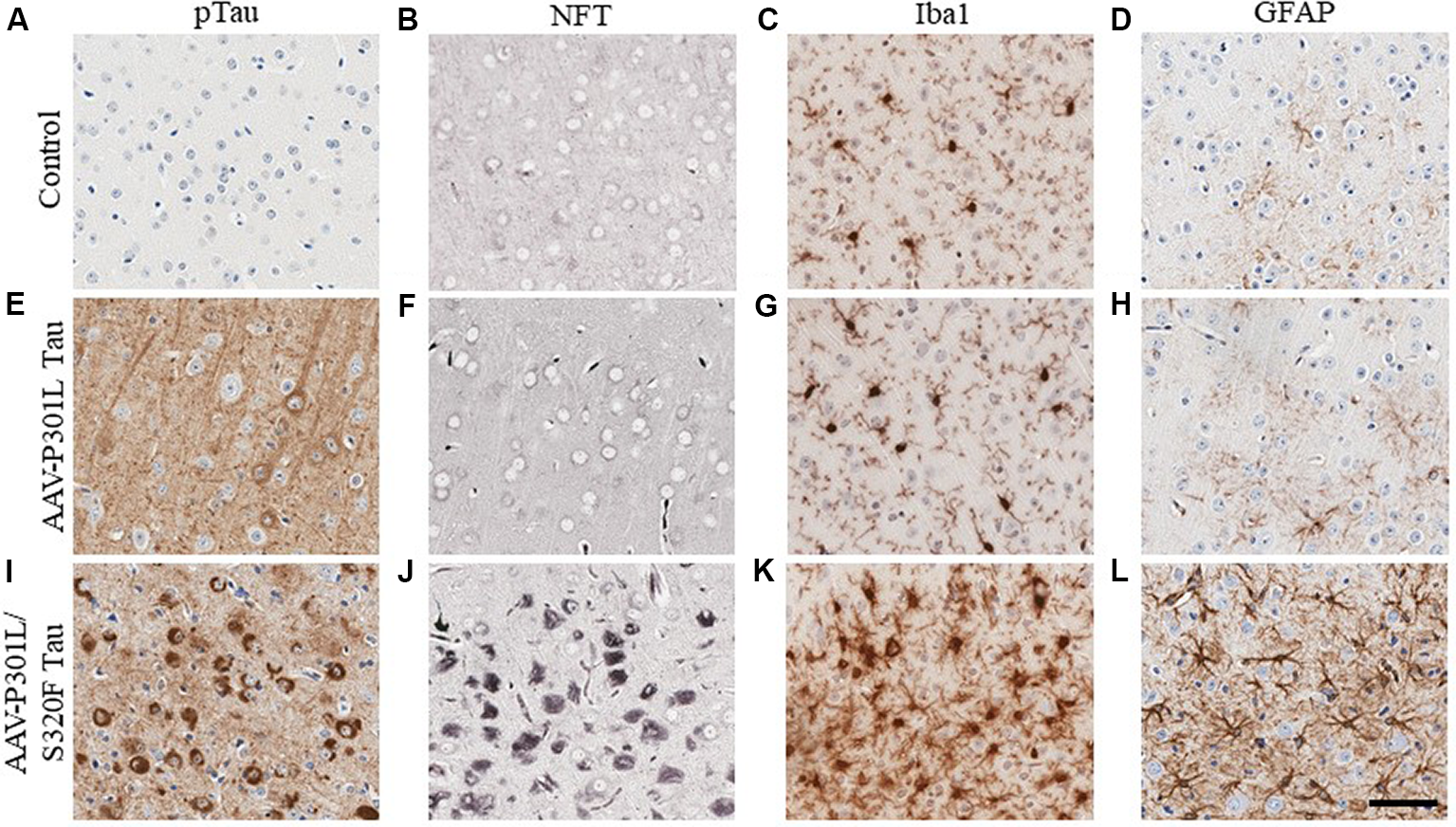
Phosphorylated tau vs. neurofibrillary tangle (NFT) tau species determine microglial and astroglial phenotype. Immunohistochemical detection of phosphorylated tau (CP13 antibody, pSer202 tau epitope), NFT tau (histological Gallyas silver stain), microglia (Iba1 antibody), and astrocytes (GFAP antibody) in the cortex of 3-month-old nontransgenic (C57/Bl6) mice that received neonatal intracerebroventricular delivery of adeno-associated virus (AAV) expressing P301L tau or P301L/S320F tau compared to age-matched naive C57/Bl6 control mice. AAV-P301L tau expressing mice accumulate intracellularly and neuropil phosphorylated tau (ptau; E), while in the AAV-P301L/S320F tau expressing mice, ptau is concentrated in the somatodendritic compartment (I). No ptau is observed in control (A). Histological Gallyas silver stain indicates frank NFT pathology only in the AAV-P301L/S320F tau mice (J), and not in the AAV-P301L tau mice (F) or control group (B). Detection of microglia (Iba1 antibody) and astrocytes (GFAP antibody) reveals increased hypertrophy of microglia and astrocytes in the AAV-P301L/S320F tau mice (K,L) compared to the AAV-P301L tau (G,H) and control mice (C,D). Scale bar: 50 μm.
In vivo imaging studies of rTg4510 mice have shown that both pathological tau and neurodegeneration correlate with microglial activation (Sahara et al., 2018). Also, the elimination of senescent microglia and astrocytes can block the cognitive impairment and neurodegeneration observed in tauopathy mouse models (Bussian et al., 2018). However, glial cell responses are not always detrimental and are needed to protect the brain. In some instances, astrocytes can form protective scars (Sofroniew, 2009) and microglia can mount a rapid protective phagocytic response to neurotoxic proteinaceous species (Meyer-Luehmann et al., 2008; Sarlus and Heneka, 2017). These are reactive events designed to provide some resilience to the homeostasis of the neural network, but emerging evidence suggests that the glial response to progressive neuropathological disruption in neurons, as observed through morphological or functional changes, may be indicative of some sort of loss of glial plasticity when this reactive process leads to progressive neuro-glial toxicity rather than a compensatory healing outcome (Alibhai et al., 2018). Indeed, the presence of soluble forms of neurotoxic tau can trigger microglial senescence, reduce microglial surveillance, abort successful phagocytosis and clearance, and worsen neuro-glial homeostasis (Streit et al., 2009; Sanchez-Mejias et al., 2016).
Consistent with reports from other laboratories (Ramsden et al., 2005; Schindowski et al., 2006), we observed that mice with accelerated NFT deposits also developed rapid memory impairment (Koller et al., 2019). While our results were consistent with human data (reviewed in Nelson et al., 2012), other preclinical studies have shown that tau conformers, independent of NFT, underlie memory deficits and network alterations (Santacruz et al., 2005; Bolós et al., 2017; Green et al., 2019). The effects of accumulating proteinopathy on dendritic spine morphology and strength of synaptic connectivity may also be dependent on heterogeneous age-progressive spatial and temporally relevant factors, not just on the intracellular proteinopathy burden. How glial cell function and their evolving plasticity across the spectrum of age-progressive proteostasis contribute to such feedback or feedforward mechanisms to determine neuroplasticity and neurodegeneration remain of interest.
Role of Non-Neuronal Cells in AD and Related Tauopathies
Age and Neuroinflammation Increase the Vulnerability of the Brain to AD Pathologies
Following the development of Aβ deposits in the frontal cortices, tau pathology starts accumulating in areas distal to Aβ deposits, such as in the entorhinal cortex and hippocampus (Braak and Braak, 1991). The molecular determinants that trigger tau hyperphosphorylation and the development of NFTs in the entorhinal cortex and then the hippocampus are unknown. The amyloid cascade hypothesis posits that cellular changes downstream of the development of Aβ pathology initiate tau pathogenesis (Hardy and Higgins, 1992). However, since ptau has been identified in brainstem nuclei of individuals as young as 30 years of age, subtle changes in tau physiology may begin much earlier in life than had been previously thought (Braak and Tredici, 2011). The hippocampus is the site of early synaptic loss in AD (Scheff and Price, 2006; Scheff et al., 2006; Bastin et al., 2020) and it is this synaptic loss and the development of NFTs that correlate with the eventual cognitive decline (Terry et al., 1991; Arriagada et al., 1992; Giannakopoulos et al., 2003). The entorhinal cortex and the hippocampus are selectively vulnerable to aging-related changes, and the major synaptic pathway connecting these two brain regions, the perforant pathway, is especially vulnerable to tau pathology (García-Sierra et al., 2000, 2001; Thal et al., 2000). Additionally, the extent of tau pathology and synaptic loss in this pathway correlates with the degree of dementia, especially in the oldest old (García-Sierra et al., 2000; Thal et al., 2000; Robinson et al., 2014), consistent with the observation that aging is the major risk factor for AD type neurodegenerative changes.
A key factor to consider is that many elderly people have Aβ plaques in their brains and retain high levels of cognition (Price and Morris, 1999; Knopman et al., 2003). Also, while many people who accumulate tau pathology in their brain develop various tauopathies, other people have been shown to accumulate tau pathology during aging, with only a little or only mild cognitive decline (clinically diagnosed as primary age-related tauopathy or PART; Crary et al., 2014). Thus, we do not fully understand how different predisposing factors synergize to develop neurodegenerative diseases and how some people show greater resilience to proteinopathy and age successfully. Additionally, we also still do not fully understand how glial dysfunction and altered neuroplasticity and glial plasticity fit into the neurodegenerative cascade in AD. In the next few sections, we will explore the individual role of glial cells (astrocytes, microglia, and oligodendrocytes) as they pertain to neuronal health and synaptic plasticity in AD in more granular details (Figure 4).
Figure 4
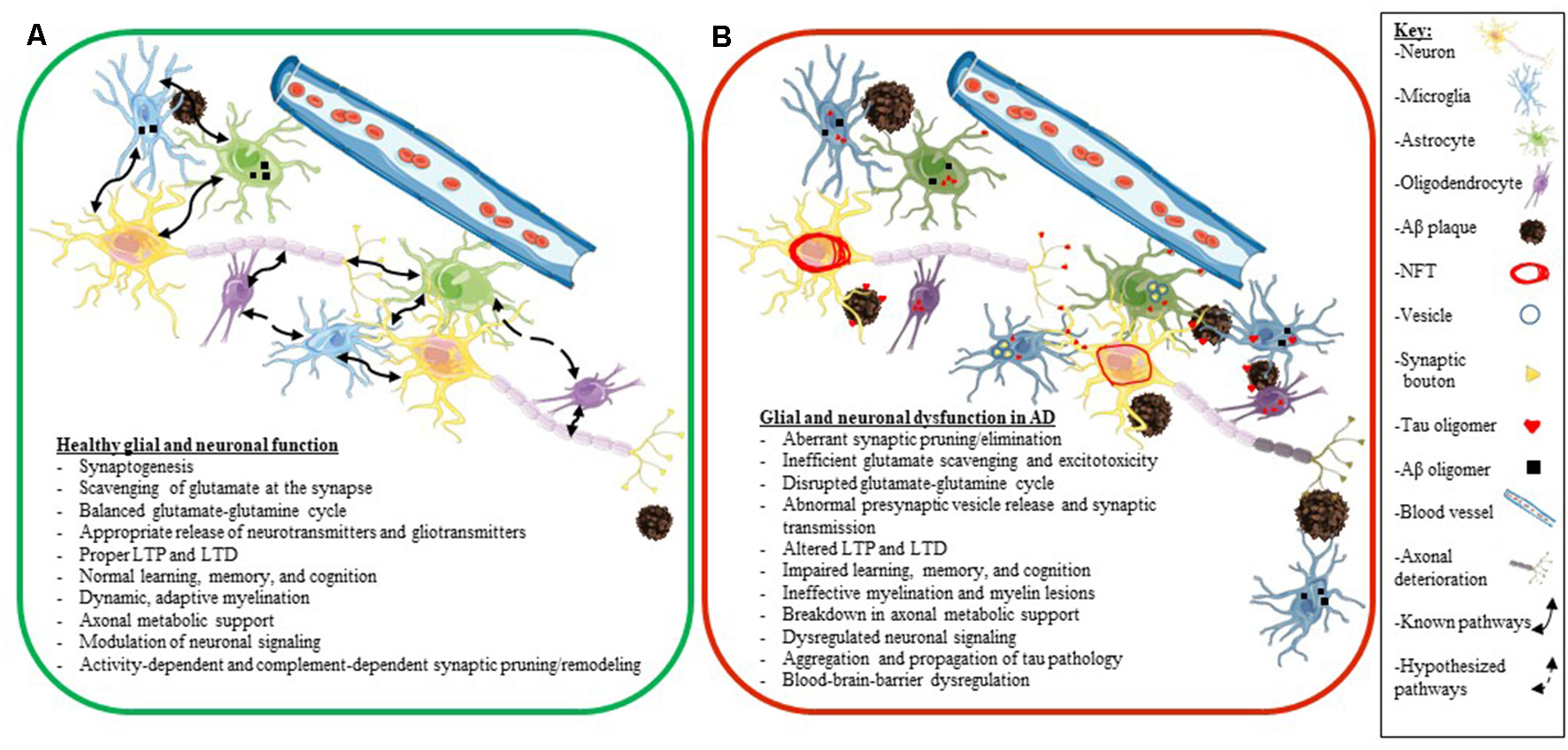
Glial and neuronal function and plasticity are dysregulated in AD. Schematics depicting healthy glial and neuronal function (A) compared to AD-induced glial and neuronal dysfunction (B). Normal communication and support processes occur between neurons, astrocytes, microglia, and oligodendrocytes even in the presence of Aβ plaques and oligomers (A). In AD, healthy communication and function break down between neurons, astrocytes, microglia, and oligodendrocytes (B). Astrocytes and microglia take up tau, Aβ, and synaptic boutons; tau oligomers spread across synapses and travel between different cell types (neurons, astrocytes, microglia, and oligodendrocytes); neurons develop NFT deposits, and Aβ plaques convert to neuritic plaques and cause the death of neurites (B). Listed below the schematics are dysregulated processes seen in a normally functioning brain (A) compared to an AD brain (B). The key for the schematics is located on the right side of the figure.
Astrocytes Are Not Just Supporting Cells in the CNS
Astrocytes are often described as the supporting cells of the nervous system. Emerging research has shown that they are not just the “glue” of the central nervous system (CNS), but that they are integral to many functional and physiological aspects of the nervous system (Allen and Lyons, 2018) including synaptogenesis (Chung et al., 2015). They facilitate the development of synapses through direct contact as well as through secreted factors such as thrombospondins, SPARCL1/Hevin, and cholesterol. They also regulate the localization of postsynaptic glutamate receptors through the release of glypicans (Allen and Eroglu, 2017). A new study showed that astrocytes are critical during the integration of adult-born hippocampal neurons into established neuronal circuits and the formation of dendritic spines on these neurons (Sultan et al., 2015). On the other hand, astrocytes also regulate synapse elimination (Allen and Eroglu, 2017). They can directly phagocytose synapses through Mertk and Megf10 receptors (Chung et al., 2013), or they can release cues such as SPARC and block synapse formation (Kucukdereli et al., 2011). They also can release TGFβ, which elicits increased C1q in neurons and marks synapses for phagocytosis by microglia (Schafer et al., 2012; Bialas and Stevens, 2013). Thus, astrocytes are dynamically related to synaptic homeostasis.
Astrocytes are not only involved in the structural maintenance of synapses; they also control their function as part of the tripartite synapse. Astrocytes regulate levels of glutamate at the synapse by taking up excess glutamate through their glutamate transporters (Stogsdill and Eroglu, 2017). Once they have removed glutamate from the synapse, astrocytes convert it to glutamine, which they provide to neurons to convert back to glutamate to be used for excitatory neurotransmission (the glutamate-glutamine cycle; Tani et al., 2014). This is a dynamic process, as high levels of neuronal activity are accompanied by increased glutamate transporters on astrocytic processes, while low activity levels result in a reduced number of glutamate transporters (Allen, 2014). Additionally, neuronal uptake of glutamate is also important for producing γ-aminobutyric acid (GABA) in GABAergic neurons and facilitating the function of inhibitory neurons (Schousboe et al., 2004). Therefore, astrocytes are indispensable in both excitatory and inhibitory signaling. Additionally, homeostatic synaptic scaling, a process for tuning neuronal network signaling, is regulated by TNFα secreted from glial cells (Stellwagen and Malenka, 2006).
Astrocytes directly participate in neuronal signaling by releasing gliotransmitters such as d-serine, glutamate, and adenosine triphosphate (ATP), each of which can affect neuronal activity and change synaptic transmission (Halassa and Haydon, 2010). In particular, d-serine is a co-agonist for N-methyl-D-aspartate receptors (NMDARs); therefore, the release of d-serine from astrocytes has the potential to modulate LTP (Henneberger et al., 2010). Providing further evidence for bi-directional communication between astrocytes and neurons, neurotransmitter release from neurons has been shown to cause local intracellular calcium increases in astrocytic processes (Di Castro et al., 2011) and astrocytes have been shown to regulate spike timing-dependent depression, a form of LTD (Min and Nevian, 2012), as well as neuronal excitatory postsynaptic currents (Pascual et al., 2012). Since LTP and LTD are important for learning and memory (Bliss and Collingridge, 1993; Abel and Lattal, 2001), this illustrates the critical role astrocytes in neuroplasticity. Indeed, astrocytic activation has been shown to increase hippocampal LTP, augment memory acquisition, and improve cognitive performance (Adamsky et al., 2018). Also, gene expression alterations in the hippocampus following learning indicate that there are changes in the energy metabolism of both astrocytes and neurons, further supporting the idea that memory formation requires concerted actions of neurons and glia (Tadi et al., 2015).
Oligodendrocytes Provide Trophic Support to Axons and Modulate Neuronal Signaling
Oligodendrocytes are glial cells that wrap around neuronal axons and produce the myelin sheath, which acts as an insulator around the axon and facilitates rapid signal conduction. Although not the only contributing factor, it has been suggested that neuronal activity plays a role in myelination and that adaptive myelination is a form of plasticity since it allows for the fine-tuning of neuronal circuits and modulation of neuronal signaling (Foster et al., 2019). Aspects of myelination that can be modified include the thickness of the myelin, the size of the internodes, and the length of myelination between internodes (Waxman, 1980; Wu et al., 2012; Ford et al., 2015). Increased neuronal activity in the brain and spinal tracts has been shown to increase the differentiation of oligodendrocyte progenitor cells (OPCs) into mature myelinating oligodendrocytes and increased myelination affects neuronal circuitry and signal conduction in the CNS regions affected (Li et al., 2010; Gibson et al., 2014; Nagy et al., 2017; Mitew et al., 2018). Interestingly, it has been found that learning a new task enhances oligodendrogenesis in adult mice while inhibiting new oligodendrocyte production is detrimental to learning (Almeida and Lyons, 2017). Also, social isolation results in impaired myelination in mice resulting in deficits in sociability and working memory tasks (Almeida and Lyons, 2017). These studies highlight the importance of neuronal activity in myelination and demonstrate the necessity of dynamic myelination in learning and memory.
Another key aspect of the oligodendrocyte-neuron relationship involves metabolic coupling. Oligodendrocytes produce lactate and, using the MCT1 transporter, provide it to neuronal axons as an energy source (Fünfschilling et al., 2012; Lee et al., 2012). It has been suggested that this process is regulated by a neuronal activity because glutamate released by myelinated axons during action potentials activates NMDA receptors and increases Ca2+ in the surrounding myelin (Micu et al., 2016). Oligodendrocytic NMDA receptor activation leads to augmented trafficking of glucose transporter to the myelin sheath, increasing glucose uptake and allowing for the production of more lactate to support the energy needs of the axon (Krasnow and Attwell, 2016; Saab et al., 2016). This evidence provides further support that oligodendrocytes are instrumental in activity-dependent neuroplasticity.
Microglia Are Dynamic Glial Cells That Regulate CNS Immunity and Directly Influence Neuroplasticity
Microglia are the innate immune cells of the brain. They constantly scan their microenvironment with their highly dynamic processes, phagocytose debris, and react to injuries or perceived threats (Nimmerjahn et al., 2005). In addition to their immune function, microglia play a critical role in processes that affect neuronal plasticity. For example, microglia have been shown to sense signals indicative of high neuronal firing rates and make contact with those active neurons to modulate their activity (Li et al., 2012). Also, ATP released by microglia causes astrocytes to release glutamate, activating neuronal metabotropic glutamate receptors and resulting in increased frequency of postsynaptic currents (Pascual et al., 2012). Microglia have even been shown to modulate neuronal circuit synchronization (Akiyoshi et al., 2018). Therefore, microglia surveil the brain not only to find debris and clear them away but also to correct abnormal signaling and prevent circuit failure (Akiyoshi et al., 2018). Based on the evidence that microglia interact at the level of synapses, they were added to the “tripartite synapse” model, modifying the model to include microglia as part of the “quadripartite synapse” (Schafer et al., 2013). As we have seen with astrocytes and oligodendrocytes, plasticity is a two-way street in microglia as well, as shown by multiple experimental models. Microglial motility is at least partially dependent upon signaling through their purinergic receptors activated by ATP/ADP (Koizumi et al., 2013).
A notable microglial receptor important for plasticity is the CX3C chemokine receptor 1 (CX3CR1). Hippocampal slices from Cx3cr1 knockout mice display LTP deficits compared to hippocampi from wild type littermates (Rogers et al., 2011). Also, Cx3cr1 knockout mice perform poorly in the Morris water maze and contextual fear conditioning paradigms, showing that signaling through CX3CR1 is important for plasticity and cognition (Rogers et al., 2011). Another study found that microglia support synapse formation during BDNF-dependent learning (Parkhurst et al., 2013). They showed that the post-synaptic density increased following learning of a motor task, and when microglia were ablated, new synapses were not established and mice did not learn the motor task (Parkhurst et al., 2013).
Microglia also directly affect the structure of synapses. During development, microglial synaptic pruning occurs to eliminate excess connections which are needed for healthy brain development (Paolicelli et al., 2011). Delayed synaptic pruning due to a reduction in microglia results in structural and electrophysiological abnormalities, which may play a role in developmental disorders (Paolicelli et al., 2011). It has been shown that synaptic pruning is mediated by the complement system (Fourgeaud and Boulanger, 2007; Stevens et al., 2007; Schafer et al., 2012; Stephan et al., 2012; Sierra et al., 2013) and is sensory- and activity-dependent (Tremblay et al., 2010; Schafer et al., 2012; Sierra et al., 2013). Also, a recent study found that triggering receptor expressed on myeloid cells 2 (TREM2) is necessary for synaptic pruning during development (Filipello et al., 2018), which is especially interesting since TREM2 variants have been linked to increased risk for AD and FTD (Guerreiro et al., 2013a,b; Filipello et al., 2018). We will discuss the function of TREM2 in more specific details in later sections.
In adulthood, synapses continue to undergo pruning (although to a lesser extent than during development) and remain dynamic, facilitating learning and memory as well as responding to injury (Zuo et al., 2005; Lohmann and Kessels, 2014). Interestingly, a recent study postulated that “synaptic pruning” may be too simple of a term for synapse removal as the researchers observed selective trogocytosis, or partial elimination, of the presynaptic compartment, but not the post-synaptic compartment in the hippocampus (Weinhard et al., 2018). They also observed the initiation of spine head filopodia at sites of microglia-synapse contact (Weinhard et al., 2018). Therefore, microglia have the capacity for extremely elegant synaptic remodeling and play a crucial role in regulating neuronal circuitry and plasticity (Weinhard et al., 2018). Another study has shown that intra-cortical injection of heat-killed bacteria leads to a process of “synaptic stripping” (Trapp et al., 2007), similar to that observed previously in the facial nucleus axotomy model (Blinzinger and Kreutzberg, 1968). This is a process mediated by microglia that removes synaptic connections of neurons by closely apposing the neuronal perikarya. Because there was no neurodegeneration, this process of synaptic elimination, where the microglia ensheathe the neurons, is considered a neuroprotective response. Microglia, when under the influence of glucocorticoid-induced stress, can also reduce spine density in a combinatorial mouse model of tau and Aβ (Pedrazzoli et al., 2019), suggesting a multi-system control of neuronal plasticity in the diseased brain. Additionally, in injury conditions, microglial function leads to a broader brain plasticity phenomenon, defined as “plasticity of plasticity” which is related to their response to modifying their morphology, function, and transcriptional identity in response to environmental threats (Banati, 2002). Such changes in microglia may induce neuroplasticity on a cellular and physiological scale. Overall, one of the major questions in the field exists around whether synaptic pruning, while usually a tightly regulated process, becomes dysregulated and if this aberrant pruning plays a role in neurodegenerative diseases (Stevens et al., 2007). Thus, more studies examining the spatio-temporal context of synaptic pruning by microglia are needed to improve our understanding of this process in both heathy and disease states.
Glial Homeostasis and Plasticity Are Lost During Aging
Aging Alters the Capacity of the Brain for Synaptic Plasticity
Age is the primary risk factor for neurodegenerative diseases such as AD (Alzheimer’s Association, 2020). The brain undergoes major changes just during the process of aging. Neurons of the aged brain display less complex dendritic arborization, shorter dendrites, and fewer dendritic spines (Dickstein et al., 2007). Also, cognitive domains such as processing speed, working memory, inhibition, and long-term memory tend to decline with age, while other processes such as knowledge storage and implicit memory are relatively preserved (Park et al., 2002; Park and Reuter-Lorenz, 2009). However, aging is a process that varies from person to person, with some people aging more “successfully” than others (Morrison and Baxter, 2012; Harada et al., 2013) and possessing higher levels of cognitive reserve even with an unfavorable proteinopathy burden (Harada et al., 2013). One method that our highly adaptive brains use to meet the challenges presented by aging is employing compensatory or alternative circuitry to overcome existing dysfunctional connections (Park and Reuter-Lorenz, 2009). This idea is supported by imaging studies that have shown over-activation and greater bilateral activation of the prefrontal cortex of aged adults (Park et al., 2001; Cabeza, 2002; Reuter-Lorenz, 2002). The brain does reach a threshold, however, at which the need for circuit reorganization exceeds its ability to adapt, at which point cognitive problems manifest (Burke and Barnes, 2006), especially since the aging brain is less efficient at compensating than the brain of a young person (Park and Reuter-Lorenz, 2009). Therefore, a large part of cognitive decline seen with aging can be attributed to the reduced ability of the brain to undergo synaptic plasticity (Morrison and Baxter, 2012). As age is the primary risk factor for neurodegenerative diseases like AD, much interest exists in exploring the effect of age on synaptic and glial plasticity in the brain.
Aged Glial Cells Have Altered Inflammatory Profiles and Reduced Functionality
A recent transcriptomic study done in mice found that astrocytes take on a more reactive phenotype in normal aging and that this occurs in a region-specific manner in the brain: hippocampal and striatal astrocytes preferentially display large gene expression changes during aging (Clarke et al., 2018). Microglia also display age-dependent regional heterogeneity (Lawson et al., 1990; Grabert et al., 2016). A recent genome-wide analysis found that microglia in the young adult brain show a unique “immune-vigilant” phenotype in the hippocampus and the cerebellum, whereas, in the aged adult brain, these two brain regions diverge in their immunogenic phenotype: microglia in the cerebellum become increasingly more immune-vigilant while those in the hippocampus display the opposite effect (Grabert et al., 2016). Such age-dependent regional changes in microglial and astrocytic function may probably be an underlying factor in triggering regional vulnerability to lesions or injuries in a selective fashion observed in different neurodegenerative diseases.
Resting microglia in aged animals show a decreased capacity to survey their microenvironment and they display slower response to acute injury as well as a sustained, long-lasting response to chronic injury (Damani et al., 2011). Also, the “sensome,” or the collection of genes that encode the proteins used by microglia to surveille their surroundings, undergoes massive downregulation in aged mice compared to young adult mice (Hickman et al., 2013). Aged astrocytes are also deficient in their maintenance of brain homeostasis and function. Reactive astrocytes in an aged brain are deficient in supporting neuronal survival and neuronal function such as debris clearance or synaptogenesis (Liddelow et al., 2017). This reactive astrocytosis can be induced by the release of cytokines such as Il-1α, TNF, and C1q originating from microglia (Liddelow et al., 2017). Additionally this reactive astrocytic environment has been shown to not only be toxic to mature oligodendrocytes but also impair the differentiation of immature oligodendrocytes (Liddelow et al., 2017). These studies show that aged glial cells do not possess the capacity to perform their respective functions as effectively as those found in a young brain and this may make the brain more susceptible to injury or disease. These data also demonstrate the paramount importance of probing the neuron-astrocyte-microglia-oligodendrocyte nexus in aging and neurodegenerative disease to better understand the effect of neuroplasticity and glial plasticity on brain organ function.
Glial Plasticity Is Altered in Neurodegenerative Disease
Several Microglial Genes Increase the Risk of Developing AD and Other Tauopathies
Genome-wide association studies have identified risk variants for AD in several proteins expressed by microglia (such as TREM2, TREM1, APOE, PLCG2, ABI3, CR1, and CD33), further linking microglial function to AD and suggesting a role in disease pathogenesis (Sims et al., 2017; Hansen et al., 2018; Henstridge et al., 2019; McQuade and Blurton-Jones, 2019). Many of these genes are connected to Aβ metabolism, but how they affect tauopathy remains unanswered. Specifically, here we will discuss the role of TREM2 and TREM1 as well as their relationship to Aβ, tau, and cognitive decline in neurodegenerative proteinopathies.
TREM2 variants have been associated with increased risk of developing AD (Sims et al., 2017; Carmona et al., 2018) as well as FTD and other neurodegenerative diseases (Guerreiro et al., 2013a,b; Yeh et al., 2017). For example, AD patients with the R47H mutation in TREM2 display earlier age of onset and shorter disease duration (Slattery et al., 2014; Korvatska et al., 2015). Also, (R47H) TREM2 patient brains display fewer microglia around Aβ plaques, indicating altered microglial function and increased axonal dystrophy (Yuan et al., 2016). In the cerebrospinal fluid (CSF) of AD patients, soluble TREM2 (sTREM2) levels are elevated when compared to healthy controls (Brosseron et al., 2018) and increase with disease progression (Suárez-Calvet et al., 2016a). Although this elevation is not reliable enough for AD diagnosis, it may be indicative of microglial activity (Suárez-Calvet et al., 2016b; Brosseron et al., 2018). New data suggests that increased CSF sTREM2 is correlated with tau-related neurodegeneration, while decreased CSF levels of sTREM2 are associated with Aβ pathology (Suárez-Calvet et al., 2019). While the (R47H) TREM2 mutation is not linked with FTD, biallelic mutations in TREM2 have been associated with FTD (Guerreiro et al., 2013a; Slattery et al., 2014). These findings warrant more research into the association between TREM2 function, neuroplasticity, and tau abnormalities.
Pre-clinical studies exploring the effect of TREM2 in tauopathy have not been entirely conclusive. One group showed that tau transgenic mice lacking Trem2 display reduced inflammation and neurodegeneration with no alterations in tau phosphorylation or solubility (Leyns et al., 2017), while another group observed that tau phosphorylation was increased in tau transgenic mice deficient for Trem2 (Bemiller et al., 2017). Of note, these studies were conducted in different tauopathy mouse models and the mice were of different ages, which may suggest an age-dependent effect of Trem2, which has been observed when assessing the effect of Trem2 deficiency on Aβ pathology (Jay et al., 2017). Additionally, in a TREM2 overexpression model, tau mice showed attenuated tau hyperphosphorylation, neuronal death, and synaptic loss as well as improved spatial cognitive performance (Jiang et al., 2016a). The authors attributed these results to the reduced neuroinflammation that they observed in these mice (Jiang et al., 2016a). How TREM2-mediated glial activity is mechanistically related to neuroplasticity remains entirely unexplored.
Recently, TREM1 variants have also been associated with AD. This variant is associated with increased Aβ burden as well as exacerbated cognitive decline in humans with AD (Replogle et al., 2015; Liu et al., 2020). A study of Aβ-PET imaging in cognitively normal individuals, MCI individuals, and AD patients that was conducted in the ADNI cohort showed that the TREM1 variant investigated was associated with MCI and was even more strongly linked with AD (Liu et al., 2020). Interestingly, assessment of the plasma of AD patients led to the finding that soluble TREM1 (sTREM1) levels were elevated, and concentrations of sTREM1 correlated to disease progression in AD patients (Jiang et al., 2019). Additionally, plasma sTREM1 levels also correlated positively with the amount of total tau found in AD plasma and this relationship was stronger in severe AD cases (Jiang et al., 2019).
Pre-clinical studies of TREM1 have mainly focused on Aβ pathology. An in vitro study found increased Aβ burden was likely due to impaired Aβ phagocytosis (Jiang et al., 2016b). These findings were supported by evidence showing that TREM1 deficiency in APP/PSEN1 mice leads to increased Aβ42 levels and overall Aβ burden (Jiang et al., 2016b). Also, TREM1 replacement by microglial overexpression as well as activation of TREM1 signaling reversed Aβ pathology and ameliorated cognitive deficits (Jiang et al., 2016b). The relationship between TREM1 and tau pathology has yet to be explored in-depth, although, one study found that TREM1 is elevated in cortical lysates from the h-tau tauopathy mouse model (Garwood et al., 2010). In the light of these results, the importance of TREM2 and TREM1 in AD therapies is being increasingly recognized as timely, spurring research into these targets (Saadipour, 2017; Carmona et al., 2018).
Microglia Phagocytose Disease-Associated Synapses and Propagate Tau Pathology
Tau associates with presynaptic vesicles via its amino-terminal domain and hinders vesicle release (Zhou et al., 2017). Moreover, the complement protein C1q is found extracellularly adjacent to the post-synaptic density in older tau transgenic mice (Dejanovic et al., 2018) and helps microglia engulf synaptic material in a C1q-dependent manner (Dejanovic et al., 2018). Treatment with an anti-C1q antibody inhibited microglial engulfment of synaptic material, prevented the loss of synapses, and also boosted synaptic density (Dejanovic et al., 2018). This study highlights the idea that tau-induced microglial phagocytosis of synapses leads to synaptic loss (Dejanovic et al., 2018). Another study showed that complement-dependent microglial phagocytosis can destroy synapses between engram cells, thus destroying encoded remote memories (Wang et al., 2020). These newly emerging studies form the foundation of our recent understanding of how microglial function is related to synaptic function and learning and memory.
Phagocytosis of tau-laden synapses may also have a downside, as additional evidence has shown that microglia are involved in the propagation of tau. We know that tau can spread from neuron to neuron along with synaptic connections (de Calignon et al., 2012; Calafate et al., 2015; DeVos et al., 2018) and is released in synaptic vesicles (Pooler et al., 2013). Tau can also be transmitted through exosomes (Saman et al., 2012; Polanco et al., 2016), ectosomes (Dujardin et al., 2014), or tunneling nanotubes (Abounit et al., 2016; Tardivel et al., 2016). It has also been shown that microglia phagocytize tau and secrete it in exosomes that then can be taken up by neurons, thereby spreading tau to neurons that may not be synaptically connected (Asai et al., 2015). Another study found that tau contained in microglia does indeed have “seeding” ability and can induce tau accumulation in recipient cells (Hopp et al., 2018). Further supporting the importance of microglia in tau transmission, microglial ablation following tau expression in the entorhinal cortex resulted in reduced tau phosphorylation and propagation to the dentate gyrus (Asai et al., 2015). This effect may be based on the amount of microglia that are removed from the brain, as partial microglial depletion (~30%) did not alter tauopathy in rTg4510 mice (Bennett et al., 2018a). A better understanding of whether microglial phagocytosis of tau-laden synapses contributes to the propagation of tau pathology could lead to the discovery of new modes for targeting tauopathy at the level of synapses.
Astrocytes Accumulate Tau Pathology in Aging and Neurodegenerative Disease
Astrocytes also undergo morphological and functional changes in response to tauopathy. Indeed, 50% of humans aged 75 years and older show tau accumulated in the cell bodies and processes of astrocytes (Schultz et al., 2004). Glial tau pathology is not usually associated with AD, therefore, it has been suggested that this effect is likely due to aging (such as that found in aging-related tau astrogliopathy or ARTAG), although other tauopathies are characterized by glial tau pathology (Schultz et al., 2004; Lace et al., 2012; Kovacs et al., 2016). Types of inclusions in other tauopathies include astrocytic plaques in corticobasal degeneration (CBD), tufted astrocytes in progressive supranuclear palsy (PSP), and globular astrocytic inclusions in globular glial tauopathy (Kovacs, 2015). Even though the human brain expresses six different isoforms of tau (Sergeant et al., 1997), certain isoforms accumulate selectively in astrocytes compared to neurons in these different tauopathies (Ferrer et al., 2014). Also, tufted astrocytes in PSP and astrocytic plaques in CBD are labeled by antibodies specific to the carboxyl-terminus and the middle portion of the tau protein but are not detected by antibodies against the amino-terminus, suggesting that a portion of the amino-terminus of the tau protein may be missing in astrocytic tau pathology (Ferrer, 2018). On the other hand, in the thorn-shaped astrocytes found in ARTAG, tau is labeled at the carboxy-terminus, the middle portion of tau, and the amino-terminus (Ferrer et al., 2018). This raises questions about whether different types of astrocytic tau pathology are composed of different isoforms or truncations of tau protein.
Another unknown is whether the tau found in astrocytic inclusions originates from an external source, such as neurons, or if astrocytes express tau under some circumstances (reviewed in Kovacs, 2020). However, as only a couple studies have found evidence of astrocytes expressing tau (Shin et al., 1991; Miyazono et al., 1993), it is fairly widely accepted that astrocytes do not express tau, or if they do, it is expressed at very low levels. We have already discussed that astrocytes can phagocytose synapses (Kucukdereli et al., 2011; Chung et al., 2013; Allen and Eroglu, 2017) and we know that tau can be released from neurons (Pooler et al., 2013). Therefore, the proximity of astrocytes to synapses allows them to uptake extracellular tau released from neurons as shown by some groups (Perea et al., 2019). Also, astrocytic tau is seed-competent, as sarkosyl-insoluble brain homogenate from ARTAG brains was shown to seed tau pathology in neurons, astrocytes, and oligodendrocytes in the hippocampus of wild type mice (Ferrer et al., 2018). Therefore, astrocytes, as well as microglia, can spread tau in the brain. Additionally, while AD brain homogenate only seeded intraneuronal tau pathology, CBD and PSP brain homogenate led to oligodendrocytic, astrocytic and neuronal tau pathology, supporting the idea that different strains of the tau protein found in different tauopathies have unique cell-type specific pathogenic properties (Narasimhan et al., 2017). This is supported by another study that reported similar results, showing that injection of brain extracts from several different tauopathies (AD, tangle-only dementia, PSP, CBD, PiD, and argyrophilic grain disease) into ALZ17 mice or non-transgenic mice led to the formation of silver-positive tau inclusions and that the type of tau inclusion formed differed depending upon the type of brain extract injected (Clavaguera et al., 2013). Finally, another study showed an injection of CBD or AD brain extracts in young PS19 mice led to the development of tau pathology specifically in oligodendrocytes and neurons, respectively, and this cell-type specificity was noticed even in brain regions distal from the site of injection indicating that the biological nature of the original seeds was preserved during transmission (Boluda et al., 2015). Interestingly, the astrocytic inclusions found in PSP and CBD have been preferentially observed near blood vessels (Shibuya et al., 2011). In a study conducted in the GFAP-tau transgenic model that accumulates astrocytic tau, tau was observed in astrocytic end-feet abutting the blood-brain-barrier (Forman et al., 2005). As neurodegenerative diseases have been associated with a blood-brain-barrier breakdown (Zhao et al., 2015), this is a decidedly interesting observation with wide-reaching implications regarding the impact of tauopathy on the integrity of the blood-brain barrier (Bennett et al., 2018b).
Breakdown of Astrocytic Support and Synaptic Modulation in Tauopathy
In AD, brain glucose uptake is impaired as shown by PET studies (Bischof et al., 2016). A recent study found that astrocytes comprise a significant part of the in vivo18F-FDG PET signal (Carter et al., 2019). In a longitudinal study of familial AD patients, they showed that decreased astrocyte function, as assessed by 11C-deuterium-L-deprenyl (11C-DED) binding, aligned with reduced 18F-FDG PET uptake representative of glucose metabolism (Carter et al., 2019). Also, in a study of post-mortem brains, it was found that cognitively normal individuals with AD pathology exhibited enhanced expression of GLT-1 compared to cognitively impaired individuals with AD pathology, suggesting a protective role of glutamate transport in the brain (Kobayashi et al., 2018). Other studies also showed that AD patient brains display reduced glutamate transporters (GLAST and GLT-1) and vesicular glutamate transporter (VGLUT1; Masliah et al., 1996; Jacob et al., 2007). This reduction in glutamate transporters was associated with increased excitotoxicity and neurodegeneration, which suggests that deficient clearance of glutamate from the synapse leads to excessive activation of glutamate receptors (Masliah et al., 1996). AD brains also display lowered levels of enzymes involved in the metabolism of glutamate and GABA as well as those involved in the inter-conversion of glutamate to glutamine (Burbaeva et al., 2014). Also, the PS19 tauopathy model, which expresses FTD-associated P301S mutant tau, shows abnormal levels of glutamate and glutamine in the parahippocampal and hippocampal regions of the brain, respectively, in an age-dependent manner (Vemula et al., 2019). In a mouse model of astrocytic tauopathy (the GFAP-tau model), mice exhibited impaired motor function along with the accumulation of astrocytic tau and reduced expression of GLT-1, which aligns with what is observed in human CBD brains (Dabir et al., 2006). These studies show the dramatic dysregulation of the glutamate-glutamine cycle, functionally linking neurons and astrocytes in AD and other tauopathies. It may follow then that neuronal function and plasticity are directly modified by loss of astrocytic plasticity. In vitro experiments demonstrated that astrocytes that internalize tau oligomers display altered spontaneous intracellular Ca2+ transients and reduced release of gliotransmitters, including ATP (Piacentini et al., 2017). This causes lowered presynaptic vesicle release and depressed synaptic transmission in adjacent neurons (Piacentini et al., 2017). Altered synaptic transmission has implications for learning, memory, and cognition. Indeed, in AD, although LTD is relatively stable, LTP is impaired (Koch et al., 2012). Therefore, irregular glutamate-glutamine metabolism and transport paired with abnormal levels of neurotransmitters result in altered neuronal health and signaling that adversely impacts synaptic plasticity.
Oligodendrocytes Accumulate Tau Pathology and Display Tau-Related Dysfunction
Some tauopathies are characterized by oligodendrocytic tau pathology in the form of coiled bodies or threads (Ferrer et al., 2014). Interestingly, Braak observed that the pattern for myelination during development inversely correlates to the pattern of NFT development and spread in AD (Braak and Braak, 1996). This observation has been corroborated by more recent findings indicating that white matter degeneration in AD follows the pattern of myelogenesis in reverse (Stricker et al., 2009). These studies suggest that those neurons that were last myelinated may be more susceptible to pathological changes in AD (Benitez et al., 2014).
Additionally, tau is transported in the white matter of the brain as well as the gray matter (Ferrer et al., 2019). Unilateral injections of brain fractions isolated from several different human tauopathy brains into the corpus callosum resulted in coiled body and thread tau pathology in both the ipsilateral and contralateral corpus callosum, indicating seeding and spread of tau pathology in oligodendrocytes (Ferrer et al., 2019). Also, increased levels of hyperphosphorylated tau in the cortex of AD brains predicted the severity of white matter hyperintensities seen in magnetic resonance imaging (MRI), which denote lesions usually caused by demyelination and/or axonal loss (McAleese et al., 2015). White matter hyperintensities have been shown to correlate with cognitive decline in patients with MCI (Tosto et al., 2014) as well as in those with dementia in general (Brickman et al., 2014). Since we know that myelination is important for neuronal plasticity and learning, these white matter lesions have great potential to disrupt neuronal signaling and negatively influence synaptic plasticity.
Also, inflammatory conditions, such as those seen in an AD brain, are sufficient to impair remyelination and stop OPCs from differentiating into mature oligodendrocytes (Miron et al., 2013). It is not surprising then that several genetic risk factors for AD are expressed in oligodendrocytes or OPCs such as CLU, BIN1, ZCWPW1, and EPHA1 (Henstridge et al., 2019). Also, oligodendrocytes are especially vulnerable to glutamate excitotoxicity, which is a feature of lost astrocytic homeostasis (Matute et al., 2007). Also, it has been shown that regenerated oligodendrocytes require astrocytes to form new myelin (Liu, Yiting et al., 2018). These studies strongly argue for a multi-system loss of plasticity in AD-type neurodegenerative diseases. These findings once more emphasize the importance of the elegant integration of different cell types in the CNS and illustrate how easily the loss of homeostasis in one cell type can drastically harm the other cell types, which in turn, has consequences for brain organ homeostasis.
Therapeutic Approaches to Targeting Synaptic Plasticity
Throughout this review, we have discussed glial contributions to neuroplasticity and the elegant interwoven functions of astrocytes, microglia, and oligodendrocytes in the healthy brain. We have also discussed how these systems are altered by age and by tau proteinopathy, leading to a loss of overall plasticity. Currently, multiple clinical trials are underway to assess new therapeutic approaches targeting tau, though there are no disease-modifying therapies or prophylactics for tauopathies yet. Some treatments, such as monoclonal tau antibodies and active tau vaccines are showing early promise (Cummings et al., 2019; Jadhav et al., 2019), but an effective tau treatment or a cure for AD and related dementias remain elusive.
All the treatments currently used in the clinic for AD are neuroplasticity modulators while few address the underlying proteinopathy (Weller and Budson, 2018). It is possible that since glial homeostasis and synaptic plasticity are intimately connected, new therapeutics should target glial plasticity to combat proteinopathy and improve synaptic plasticity, cognition, and quality of life. Interestingly, as described in the next sections, some neuroplasticity modulators also affect glial function, which highlights potential cooperative interactions between neuronal and glial plasticity. Described below are a selection of some of the current and past clinical trials targeting synaptic plasticity and cognition.
Brain Stimulation and Cognitive Training Improve Cognitive Function
Several types of brain stimulation including transcranial Direct Current Stimulation (tDCS) and Transcranial Magnetic Stimulation (TMS) have been shown to improve cognition in MCI and AD patients (Supplementary Table S2). tDCS has been associated with modulation of cortical excitability and synaptic connections and has been suggested as a potential therapy in multiple diseases and disorders because it is non-invasive and painless and has minimal or mild side effects (Stagg and Nitsche, 2011; Elder and Taylor, 2014). tDCS was shown to improve cognitive performance in healthy elderly individuals and reduce hyperexcitability in the stimulated brain regions (Meinzer et al., 2013). Although a single session of tDCS does not cause long-lasting alterations, it has been found that repeated tDCS sessions result in progressive and long-lasting improvement (Reis et al., 2009; Stagg and Nitsche, 2011). Also, fMRI assessment of the brains of these older individuals undergoing treatment showed that the treatment reversed the connectivity pattern to that matching younger brain performance characteristics (Meinzer et al., 2013). Acute assessment of tDCS in MCI patients revealed that MCI individuals were able to reach a similar performance level as healthy controls in a semantic word-retrieval task, a task in which performance worsens early on in dementia, accompanied by restoration of resting-state connectivity (Meinzer et al., 2015). As AD is associated with the altered function of several brain networks (DMN, control network, dorsal attention network, sensor-motor network, and salience network, among others; Brier et al., 2012), tDCS could be a potential mechanism to improve cognitive functioning in AD and MCI on a global scale (Flöel, 2014).
Another method of brain stimulation is TMS. TMS can be conducted either with a single pulse, paired-pulse, or repetitive stimulation (Elder and Taylor, 2014). The most common side effect of TMS is transient pain, but TMS is non-invasive and has been assessed to be safe (Elder and Taylor, 2014). TMS has been used in older adults to improve different types of memory and to ameliorate neuropsychiatric conditions such as depression (Elder and Taylor, 2014). There are several clinical trials underway to assess the efficacy of different types of TMS in AD, and repetitive TMS has been shown to result in improved performance during a naming task in AD patients (Cotelli et al., 2008; Supplementary Table S2). Brain stimulation is an effective intervention for improving cognition as it has minimal side effects and does not involve an invasive procedure. Additionally, clinical trials are attempting to enhance the cognitive outcomes of brain stimulation by pairing it with various cognitive training tasks.
Cognitive training can include computer-based training, the learning of particular tasks, or musical training, among others, and these interventions have already shown promise in MCI and AD (Supplementary Table S2). For example, a study in MCI patients found that a 12-week computer-based training program targeting memory and attention resulted in improved performance on recognition and recall tasks and found that this improvement in functioning lasted for 6 months post-training (Herrera et al., 2012). Another study assessing hippocampal activation as well as verbal memory in MCI patients showed that cognitive training involving auditory-verbal perception improved the patients’ ability to remember and their overall hippocampal function (Rosen et al., 2011). To improve experience-induced neuronal plasticity, older adults at risk for developing AD participated in 13 weeks of cognitive training targeting attention and working memory (Leung et al., 2015). These two cognitive domains were improved following the training period, while other cognitive domains, such as verbal or visual-spatial memory were not affected, suggesting experience-dependent plasticity (Leung et al., 2015). In a study assessing mild and moderate AD patients, individuals were trained on a structure-from-motion task. Both groups were impaired on the task, but the mild AD group was able to improve their performance after several training sessions, while the moderate AD group was unable to improve (Kim and Park, 2010). These results suggest that several cognitive domains that decline in MCI and AD remain plastic and can be re-trained; however, these studies also show that some domains can change only during the early stages of the disease, after which they cannot be rescued (Kim and Park, 2010).
Several clinical trials are assessing whether brain stimulation alone or in combination with cognitive training could be used to treat MCI and AD (Supplementary Table S2). For example, in one ongoing study, older individuals with mild neurocognitive disorder due to AD will be subjected to tDCS with working memory training or with tDCS alone (Cheng et al., 2015). The hypothesis is that the modulatory effect of tDCS will have an enhancing effect on working memory performance in the adaptive N-back task (Cheng et al., 2015). As this study and the other clinical trials combining brain stimulation and cognitive training are ongoing, the results remain to be determined.
Physical Activity and Cognitive Training Prevent Cognitive Decline and Improve Cognition
Physical fitness training, aquatic exercise, “exergames,” as well as dancing have been suggested as potential therapeutics for AD. In a 6 month study, a resistance training exercise in women with probable MCI led to improved attention, memory, and brain plasticity (Nagamatsu et al., 2012). In a different study with both aquatic- and land-based exercise that was conducted for 16 weeks in elderly women, plasma Aβ and HSP27 levels were increased and vascular health was improved in these women (Kim et al., 2018). As cognitive training has also been shown to improve cognitive functioning, some studies have combined physical activity and cognitive exercises to enhance functional outcomes. For example, a trial using “exergames” (virtual reality–enhanced exercise) in MCI patients posited that since exercise can lead to cognitive improvement, a regimen of stationary cycling paired with virtual reality tours (cyber cycling) would increase participation and enhance cognition (Anderson-Hanley et al., 2012). The results of this study revealed that cyber cycling improves executive functioning and reduces the risk of MCI progression (Anderson-Hanley et al., 2012). Another study that combined physical training and cognitive training over 8 weeks found that this training reduced delta, theta, and beta rhythms, induced functional plasticity, and improved cognitive scores (Styliadis et al., 2015). Therefore, combinatorial therapies pairing exercise with cognitive training show promise for modifying cognitive decline in patients at risk for dementia.
Pharmacological Interventions Leading to Improved Cognitive Function
One method for drug discovery is to test treatments that have already been approved for use in other diseases. A glucagon-like peptide 1 (GLP-1) analog called Liraglutide has been used to treat obesity and diabetes and is being assessed in an ongoing clinical trial to improve brain glucose metabolism as well as cognition in patients with mild AD (Femminella et al., 2019). A previous clinical trial had evaluated the efficacy of daily administration of Liraglutide in AD patients over 6 months and found improved glucose metabolism, but no change in cognitive scores or amyloid load (Gejl et al., 2016). The researchers suggested further study of this compound and cited the small number of participants and the short treatment window (Gejl et al., 2016). An ongoing Liraglutide trial has incorporated an extended treatment window of 12 months and recruited a substantially larger cohort of study participants (Femminella et al., 2019).
Another clinical trial assessed the effectiveness of the dopamine D2 agonist, rotigotine (RTG), in improving synaptic plasticity in patients with mild AD by altering cortical excitability and cholinergic transmission (Martorana et al., 2013). A paired-pulse paradigm was used to investigate the effect of RTG on cortical excitability by assessing intracortical inhibition, and facilitation and short-latency afferent inhibition was used to evaluate the effect of RTG on cholinergic transmission. Cortical excitability increased and cholinergic transmission returned to the baseline seen in the control subjects (Martorana et al., 2013). A similar RTG study evaluated the effect of RTG on cortical plasticity using transcranial stimulation with theta-burst stimulation (TBS) to induce LTP or LTD in mild AD patients (Koch et al., 2014). Cortical plasticity was improved and an additional assessment with short-latency afferent inhibition revealed increased cholinergic activity as well (Koch et al., 2014).
A study targeting the cholinergic system using Donepezil, an acetylcholinesterase (AChE) inhibitor, along with cognitive training has been done in AD patients (Gonzalez Rothi et al., 2009). AChE inhibitors increase the amount of acetylcholine at the synapse and improve cholinergic signaling. This tends to slow the decline of cognition in AD, but not necessarily reverse the cognitive impairment (Gonzalez Rothi et al., 2009). In this study, the premise was that if cognitive training was paired with increased cholinergic signaling, then cognition might improve and not just remain stable (Gonzalez Rothi et al., 2009). Assessment of performance revealed that half of the participants showed improvement on the verbal naming task, showing that this combined treatment is only partially effective (Gonzalez Rothi et al., 2009).
Multimodal Therapies or Changes in Lifestyle Could Improve Overall Brain and Bodily Function
A few studies have combined more than two modes of treating cognition and synaptic plasticity and have implemented a lifestyle change regimen. A study involving a dietary regimen, physical activity, cognitive intervention, and a specific dietary supplement (epigallocatechin gallate) is underway to assess changes in function and structural connectivity in the brain. Another study has found that aerobic and resistance training, cognitive training, and vitamin D supplement have potential as a synergistic intervention to enhance mobility as well as cognition in older adults, especially those with MCI (Montero-Odasso et al., 2018). If these trials do establish the efficacy of a combined approach for slowing the loss of cognitive function or even reversing cognitive decline, then this would be an exciting step in the direction of using multimodal treatments to overcome the challenges presented by age-related dementia, MCI, and AD.
Neuroplasticity Targeted Therapies Resolve Gliosis in Mouse Models
If neuronal and glial plasticity are connected, possibly these neuroplasticity targeted therapies could also ameliorate glial dysfunction or at least stabilize their function to support overall maintenance of brain organ homeostasis. Such a scenario would have a translational impact, when combined with promising therapies targeting the underlying proteinopathies in AD and related tauopathies (https://www.alzforum.org/therapeutics/aducanumab and https://www.alzforum.org/therapeutics/aci-35; retrieved June 8, 2020). In mouse models of AD-type amyloidosis, most of the environmental enrichment strategies, such as physical exercise and brain stimulation, have resulted in reduced Aβ and neuroinflammation (Iaccarino et al., 2016; Xu et al., 2016; Adaikkan et al., 2019; Martorell et al., 2019). Even in models that do not deposit Aβ, manipulating microglia could rescue effective behavior and induce adult neurogenesis following environmental enrichment (Ortega-Martinez et al., 2019). Interestingly, in wild type mice, such interventions have resulted in sustained effects on glial activation by recruiting microglia and upregulating NF-KB/MAPK pathways (Garza et al., 2020), suggesting that these manipulations can alter glial plasticity independent of their effects on AD-type proteinopathy. Due to the paucity of research on tauopathy using these manipulations, the relationship between these therapeutics and tau-mediated alterations in neuronal and glial plasticity remains unknown. Pharmacological interventions such as GLP-1 can also restore neurite complexity, dendritic spine morphogenesis, and spine development in non-AD experimental models (Yoon et al., 2020). Its effect on brain plasticity in Aβ models mostly show positive regulation (Cai et al., 2014; McClean et al., 2015), though the effect on tauopathy-related brain dysfunction remains unclear.
Conclusion
Here, we have broadly described the impact of glial dysregulation on synaptic plasticity in AD and highlight the concept of “plasticity of plasticity” in the context of neurodegenerative dementias such as AD and related tauopathies. Experimental models have shown that synaptic plasticity, as a stand-alone paradigm, is necessary to maintain healthy brain function and its dysregulation in AD leads to progressive and inexorable brain organ failure. The essential role of glia and their plastic functioning in regulating synaptic plasticity in homeostatic and diseased conditions remain uncertain, though their role in CNS injury is well-documented. Glial cells are not just supporting cells in the CNS, but they are integral players in processes such as synaptic pruning, glutamate scavenging, the glutamate-glutamine cycle, gliotransmitter and neurotransmitter release, LTP/LTD, myelination, metabolic support, synaptic transmission, and learning and memory. In AD, the disrupted proteostasis can fundamentally alter the capacity of glial cells to perform their functions, having detrimental effects on neuronal function and plasticity and resulting in a feed-forward toxicity cycle.
So far, there are no effective disease-modifying therapies for AD. Many of the available drugs used in the clinic as symptomatic therapies in AD are neuroplasticity modulators with a possible role in altering neuroinflammation. More research needs to be done in terms of utilizing these manipulations to illuminate the concept of glia-mediated “plasticity of plasticity” phenomenon and its possible therapeutic contribution in neurodegenerative dementias. Clinical trials that can target both glial plasticity as well as synaptic plasticity should be encouraged, but the optimal therapeutic or combination of therapeutics has yet to be determined. As with any treatment or therapy, the optimal time of intervention is a key factor to be considered, especially while attempting to re-instate synaptic plasticity by rebalancing glial function and plasticity in a milieu of neurodegenerative proteinopathy.
Statements
Author contributions
PC and EK conceived the original idea and wrote the manuscript. EK drafted the manuscript and designed the tables and figures and performed PubMed. PC reviewed PubMed searches. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Institutes of Health (R01 AG055798 and T32 AG061892).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2020.00151/full#supplementary-material.
- AAV
adeno-associated viruses
- Aβ
Amyloid β
- AD
Alzheimer’s disease
- AICD
APP intracellular domain
- AMPAR
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor
- APOE
Apolipoprotein E
- APP
amyloid precursor protein
- ARTAG
aging-related tau astrogliopathy
- ATP
adenosine triphosphate
- CBD
corticobasal degeneration
- CNS
central nervous system
- CSF
cerebrospinal fluid
- Cx3CR1
CX3C chemokine receptor 1
- C1q
complement component 1q
- DBS
deep brain stimulation
- DMN
default mode network
- FDG-PET
fluorodeoxyglucose-positron emission tomography
- FTD
frontotemporal dementia
- GABA
γ-aminobutyric acid
- GFAP
glial fibrillary acidic protein
- LTD
long-term depression
- LTP
long-term potentiation
- MAPT
microtubule associated protein tau
- MCI
mild cognitive impairment
- NFT
neurofibrillary tangle
- NMDAR
N-Methyl-d-aspartic acid or N-Methyl-d-aspartate receptor
- OPC
oligodendrocyte progenitor cells
- PART
primary age-related tauopathy
- PET
positron emission tomography
- PSP
progressive supranuclear palsy
- tDCS
transcranial Direct Current Stimulation
- Tg
transgenic
- TMS
transcranial magnetic stimulation
- TREM
triggering receptor expressed on myeloid cells.
Abbreviations
References
1
Abel T. Lattal K. M. (2001). Molecular mechanisms of memory acquisition, consolidation and retrieval. Curr. Opin. Neurobiol.11, 180–187. 10.1016/s0959-4388(00)00194-x
2
Abounit S. Wu J. W. Duff K. Victoria G. S. Zurzolo C. (2016). Tunneling nanotubes: a possible highway in the spreading of tau and other prion-like proteins in neurodegenerative diseases. Prion10, 344–351. 10.1080/19336896.2016.1223003
3
Adaikkan C. Middleton S. J. Marco A. Pao P. C. Mathys H. Kim D. N. et al . (2019). Gamma entrainment binds higher-order brain regions and offers neuroprotection. Neuron102, 929.e8–943.e8. 10.1016/j.neuron.2019.04.011
4
Adamsky A. Kol A. Kreisel T. Doron A. Ozeri-Engelhard N. Melcer T. et al . (2018). Astrocytic activation generates de novo neuronal potentiation and memory enhancement. Cell174, 59.e14–71.e14. 10.1016/j.cell.2018.05.002
5
Ahmad A. Ali T. Park H. Y. Badshah H. Rehman S. U. Kim M. O. (2017). Neuroprotective effect of fisetin against amyloid-β-induced cognitive/synaptic dysfunction, neuroinflammation and neurodegeneration in adult mice. Mol. Neurobiol.54, 2269–2285. 10.1007/s12035-016-9795-4
6
Ahmed T. Van der Jeugd A. Blum D. Galas M. C. D’Hooge R. Buee L. et al . (2014). Cognition and hippocampal synaptic plasticity in mice with a homozygous tau deletion. Neurobiol. Aging35, 2474–2478. 10.1016/j.neurobiolaging.2014.05.005
7
Akiyoshi R. Wake H. Kato D. Horiuchi H. Ono R. Ikegami A. et al . (2018). Microglia enhance synapse activity to promote local network synchronization. eNeuro5:ENEURO.0088-18.2018. 10.1523/ENEURO.0088-18.2018
8
Alibhai J. D. Diack A. B. Manson J. C. (2018). Unravelling the glial response in the pathogenesis of Alzheimer’s disease. FASEB J.32, 5766–5777. 10.1096/fj.201801360r
9
Allen N. J. (2014). Astrocyte regulation of synaptic behavior. Annu. Rev. Cell. Dev. Biol.30, 439–463. 10.1146/annurev-cellbio-100913-013053
10
Allen N. J. Eroglu C. (2017). Cell biology of astrocyte-synapse interactions. Neuron96, 697–708. 10.1016/j.neuron.2017.09.056
11
Allen N. J. Lyons D. A. (2018). Glia as architects of central nervous system formation and function. Science362, 181–185. 10.1126/science.aat0473
12
Almeida R. G. Lyons D. A. (2017). On myelinated axon plasticity and neuronal circuit formation and function. J. Neurosci.37, 10023–10034. 10.1523/jneurosci.3185-16.2017
13
Alves P. N. Foulon C. Karolis V. Bzdok D. Margulies D. S. Volle E. et al . (2019). An improved neuroanatomical model of the default-mode network reconciles previous neuroimaging and neuropathological findings. Commun. Biol.2:370. 10.1038/s42003-019-0611-3
14
Alzheimer’s Association . (2020). 2020 Alzheimer’s disease facts and figures. Alzheimers Dement. [Epub ahead of print]. 10.1002/alz.12068
15
Anderson-Hanley C. Arciero P. J. Brickman A. M. Nimon J. P. Okuma N. Westen S. C. et al . (2012). Exergaming and older adult cognition: a cluster randomized clinical trial. Am. J. Prev. Med.42, 109–119. 10.1016/j.amepre.2011.10.016
16
Arriagada P. V. Growdon J. H. Hedley-Whyte E. T. Hyman B. T. (1992). Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology42, 631–639. 10.1212/wnl.42.3.631
17
Asai H. Ikezu S. Tsunoda S. Medalla M. Luebke J. Haydar T. et al . (2015). Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci.18, 1584–1593. 10.1038/nn.4132
18
Bachstetter A. D. Norris C. M. Sompol P. Wilcock D. M. Goulding D. Neltner J. H. et al . (2012). Early stage drug treatment that normalizes proinflammatory cytokine production attenuates synaptic dysfunction in a mouse model that exhibits age-dependent progression of Alzheimer’s disease-related pathology. J. Neurosci.32, 10201–10210. 10.1523/jneurosci.1496-12.2012
19
Banati R. B. (2002). Brain plasticity and microglia: is transsynaptic glial activation in the thalamus after limb denervation linked to cortical plasticity and central sensitisation?J. Physiol.96, 289–299. 10.1016/s0928-4257(02)00018-9
20
Bastin C. Bahri M. A. Meyer F. Manard M. Delhaye E. Plenevaux A. et al . (2020). In vivo imaging of synaptic loss in Alzheimer’s disease with [18F]UCB-H positron emission tomography. Eur. J. Nucl. Med. Mol. Imaging47, 390–402. 10.1007/s00259-019-04461-x
21
Bear M. F. Malenka R. C. (1994). Synaptic plasticity: LTP and LTD. Curr. Opin. Neurobiol.4, 389–399. 10.1016/0959-4388(94)90101-5
22
Bellamy T. C. Dunaevsky A. Parri H. R. (2015). Glial plasticity. Neural Plast.2015:723891. 10.1155/2015/723891
23
Bemiller S. M. McCray T. J. Allan K. Formica S. V. Xu G. Wilson G. et al . (2017). TREM2 deficiency exacerbates tau pathology through dysregulated kinase signaling in a mouse model of tauopathy. Mol. Neurodegener.12:74. 10.1186/s13024-017-0216-6
24
Benitez A. Fieremans E. Jensen J. H. Falangola M. F. Tabesh A. Ferris S. H. et al . (2014). White matter tract integrity metrics reflect the vulnerability of late-myelinating tracts in Alzheimer’s disease. Neuroimage Clin.4, 64–71. 10.1016/j.nicl.2013.11.001
25
Bennett R. E. Bryant A. Hu M. Robbins A. B. Hopp S. C. Hyman B. T. (2018a). Partial reduction of microglia does not affect tau pathology in aged mice. J. Neuroinflammation15:311. 10.1186/s12974-018-1348-5
26
Bennett R. E. Robbins A. B. Hu M. Cao X. Betensky R. A. Clark T. et al . (2018b). Tau induces blood vessel abnormalities and angiogenesis-related gene expression in P301L transgenic mice and human Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A115, E1289–E1298. 10.1073/pnas.1710329115
27
Bialas A. R. Stevens B. (2013). TGF-β signaling regulates neuronal C1q expression and developmental synaptic refinement. Nat. Neurosci.16, 1773–1782. 10.1038/nn.3560
28
Bischof G. N. Jessen F. Fliessbach K. Dronse J. Hammes J. Neumaier B. et al . (2016). Impact of tau and amyloid burden on glucose metabolism in Alzheimer’s disease. Ann. Clin. Transl. Neurol.3, 934–939. 10.1002/acn3.339
29
Biundo F. Del Prete D. Zhang H. Arancio O. D’Adamio L. (2018). A role for tau in learning, memory and synaptic plasticity. Sci. Rep.8:3184. 10.1038/s41598-018-21596-3
30
Blinzinger K. Kreutzberg G. (1968). Displacement of synaptic terminals from regenerating motor neurons by microglial cells. Z. Zellforsch. Mikrosk. Anat.85, 145–157. 10.1007/bf00325030
31
Bliss T. V. Collingridge G. L. (1993). A synaptic model of memory: long-term potentiation in the hippocampus. Nature361, 31–39. 10.1038/361031a0
32
Bolós M. Pallas-Bazarra N. Terreros-Roncal J. Perea J. R. Jurado-Arjona J. Ávila J. et al . (2017). Soluble Tau has devastating effects on the structural plasticity of hippocampal granule neurons. Transl. Psychiatry7:1267. 10.1038/s41398-017-0013-6
33
Boluda S. Iba M. Zhang B. Raible K. M. Lee V. M. Trojanowski J. Q. (2015). Differential induction and spread of tau pathology in young PS19 tau transgenic mice following intracerebral injections of pathological tau from Alzheimer’s disease or corticobasal degeneration brains. Acta Neuropathol.129, 221–237. 10.1007/s00401-014-1373-0
34
Braak H. Braak E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol.82, 239–259. 10.1007/BF00308809
35
Braak H. Braak E. (1996). Development of Alzheimer-related neurofibrillary changes in the neocortex inversely recapitulates cortical myelogenesis. Acta Neuropathol.92, 197–201. 10.1007/s004010050508
36
Braak H. Tredici K. D. (2011). The pathological process underlying Alzheimer’s disease in individuals under thirty. Acta Neuropathol.121, 171–181. 10.1007/s00401-010-0789-4
37
Brickman A. M. Schupf N. Manly J. J. Stern Y. Luchsinger J. A. Provenzano F. A. et al . (2014). APOE ε4 and risk for Alzheimer’s disease: do regionally distributed white matter hyperintensities play a role?Alzheimers Dement.10, 619–629. 10.1016/j.jalz.2014.07.155
38
Brier M. R. Thomas J. B. Snyder A. Z. Benzinger T. L. Zhang D. Raichle M. E. et al . (2012). Loss of intranetwork and internetwork resting state functional connections with Alzheimer’s disease progression. J. Neurosci.32, 8890–8899. 10.1523/JNEUROSCI.5698-11.2012
39
Brosseron F. Traschütz A. Widmann C. N. Kummer M. P. Tacik P. Santarelli F. et al . (2018). Characterization and clinical use of inflammatory cerebrospinal fluid protein markers in Alzheimer’s disease. Alzheimers Res. Ther.10:25. 10.1186/s13195-018-0353-3
40
Burbaeva G. S. H. Boksha I. S. Tereshkina E. B. Savushkina O. K. Prokhorova T. A. Vorobyeva E. A. (2014). Glutamate and GABA-metabolizing enzymes in post-mortem cerebellum in Alzheimer’s disease: phosphate-activated glutaminase and glutamic acid decarboxylase. Cerebellum13, 607–615. 10.1007/s12311-014-0573-4
41
Burke S. N. Barnes C. A. (2006). Neural plasticity in the ageing brain. Nat. Rev. Neurosci.7, 30–40. 10.1038/nrn1809
42
Bussian T. J. Aziz A. Meyer C. F. Swenson B. L. van Deursen J. M. Baker D. J. (2018). Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature562, 578–582. 10.1038/s41586-018-0543-y
43
Cabeza R. (2002). Hemispheric asymmetry reduction in older adults: the HAROLD model. Psychol. Aging17, 85–100. 10.1037/0882-7974.17.1.85
44
Cai H. Y. Hölscher C. Yue X. H. Zhang S. X. Wang X. H. Qiao F. et al . (2014). Lixisenatide rescues spatial memory and synaptic plasticity from amyloid β protein-induced impairments in rats. Neuroscience277, 6–13. 10.1016/j.neuroscience.2014.02.022
45
Calafate S. Buist A. Miskiewicz K. Vijayan V. Daneels G. de Strooper B. et al . (2015). Synaptic contacts enhance cell-to-cell tau pathology propagation. Cell Rep.11, 1176–1183. 10.1016/j.celrep.2015.04.043
46
Carmona S. Zahs K. Wu E. Dakin K. Bras J. Guerreiro R. (2018). The role of TREM2 in Alzheimer’s disease and other neurodegenerative disorders. Lancet Neurol.17, 721–730. 10.1016/s1474-4422(18)30232-1
47
Carter S. F. Chiotis K. Nordberg A. Rodriguez-Vieitez E. (2019). Longitudinal association between astrocyte function and glucose metabolism in autosomal dominant Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging46, 348–356. 10.1007/s00259-018-4217-7
48
Chen R. Zhang J. Wu Y. Wang D. Feng G. Tang Y. P. et al . (2012). Monoacylglycerol lipase is a therapeutic target for Alzheimer’s disease. Cell Rep.2, 1329–1339. 10.1016/j.celrep.2012.09.030
49
Cheng C. P. Chan S. S. Mak A. D. Chan W. C. Cheng S. T. Shi L. et al . (2015). Would transcranial direct current stimulation (tDCS) enhance the effects of working memory training in older adults with mild neurocognitive disorder due to Alzheimer’s disease: study protocol for a randomized controlled trial. Trials16:479. 10.1186/s13063-015-0999-0
50
Chowdhury D. Hell J. W. (2018). Homeostatic synaptic scaling: molecular regulators of synaptic AMPA-type glutamate receptors. F1000Res.7:234. 10.12688/f1000research.13561.1
51
Chung W. S. Allen N. J. Eroglu C. (2015). Astrocytes control synapse formation, function and elimination. Cold Spring Harb. Perspect. Biol.7:a020370. 10.1101/cshperspect.a020370
52
Chung W. S. Clarke L. E. Wang G. X. Stafford B. K. Sher A. Chakraborty C. et al . (2013). Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature504, 394–400. 10.1038/nature12776
53
Clarke L. E. Liddelow S. A. Chakraborty C. Münch A. E. Heiman M. Barres B. A. (2018). Normal aging induces A1-like astrocyte reactivity. Proc. Natl. Acad. Sci. U S A115, E1896–E1905. 10.1073/pnas.1800165115
54
Clavaguera F. Akatsu H. Fraser G. Crowther R. A. Frank S. Hench J. et al . (2013). Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc. Natl. Acad. Sci. U S A110, 9535–9540. 10.1073/pnas.1301175110
55
Corbett N. J. Hooper N. M. (2018). Soluble amyloid precursor protein α: friend or foe?Adv. Exp. Med. Biol.1112, 177–183. 10.1007/978-981-13-3065-0_13
56
Cotelli M. Manenti R. Cappa S. F. Zanetti O. Miniussi C. (2008). Transcranial magnetic stimulation improves naming in Alzheimer disease patients at different stages of cognitive decline. Eur. J. Neurol.15, 1286–1292. 10.1111/j.1468-1331.2008.02202.x
57
Crary J. F. Trojanowski J. Q. Schneider J. A. Abisambra J. F. Abner E. L. Alafuzoff I. et al . (2014). Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol.128, 755–766. 10.1007/s00401-014-1349-0
58
Cribbs D. H. Berchtold N. C. Perreau V. Coleman P. D. Rogers J. Tenner A. J. et al . (2012). Extensive innate immune gene activation accompanies brain aging, increasing vulnerability to cognitive decline and neurodegeneration: a microarray study. J. Neuroinflammation9:179. 10.1186/1742-2094-9-179
59
Crimins J. L. Rocher A. B. Luebke J. I. (2012). Electrophysiological changes precede morphological changes to frontal cortical pyramidal neurons in the rTg4510 mouse model of progressive tauopathy. Acta Neuropathol.124, 777–795. 10.1007/s00401-012-1038-9
60
Cummings J. Lee G. Ritter A. Sabbagh M. Zhong K. (2019). Alzheimer’s disease drug development pipeline: 2019. Alzheimers Dement.5, 272–293. 10.1016/j.trci.2019.05.008
61
Dabir D. V. Robinson M. B. Swanson E. Zhang B. Trojanowski J. Q. Lee V. M. et al . (2006). Impaired glutamate transport in a mouse model of tau pathology in astrocytes. J. Neurosci.26, 644–654. 10.1523/jneurosci.3861-05.2006
62
Damani M. R. Zhao L. Fontainhas A. M. Amaral J. Fariss R. N. Wong W. T. (2011). Age-related alterations in the dynamic behavior of microglia. Aging Cell10, 263–276. 10.1111/j.1474-9726.2010.00660.x
63
de Calignon A. Polydoro M. Suarez-Calvet M. William C. Adamowicz D. H. Kopeikina K. J. et al . (2012). Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron73, 685–697. 10.1016/j.neuron.2011.11.033
64
Decker J. M. Krüger L. Sydow A. Dennissen F. J. Siskova Z. Mandelkow E. et al . (2016). The Tau/A152T mutation, a risk factor for frontotemporal-spectrum disorders, leads to NR2B receptor-mediated excitotoxicity. EMBO Rep.17, 552–569. 10.15252/embr.201541439
65
Deczkowska A. Keren-Shaul H. Weiner A. Colonna M. Schwartz M. Amit I. (2018). Disease-associated microglia: a universal immune sensor of neurodegeneration. Cell173, 1073–1081. 10.1016/j.cell.2018.05.003
66
Deibel S. H. Weishaupt N. Regis A. M. Hong N. S. Keeley R. J. Balog R. J. et al . (2016). Subtle learning and memory impairment in an idiopathic rat model of Alzheimer’s disease utilizing cholinergic depletions and β-amyloid. Brain Res.1646, 12–24. 10.1016/j.brainres.2016.05.033
67
Dejanovic B. Huntley M. A. De Maziere A. Meilandt W. J. Wu T. Srinivasan K. et al . (2018). Changes in the synaptic proteome in tauopathy and rescue of tau-induced synapse loss by C1q antibodies. Neuron100, 1322.e7–1336.e7. 10.1016/j.neuron.2018.10.014
68
DeKosky S. T. Scheff S. W. (1990). Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann. Neurol.27, 457–464. 10.1002/ana.410270502
69
De Roo M. Klauser P. Muller D. (2008). LTP promotes a selective long-term stabilization and clustering of dendritic spines. PLoS Biol.6:e219. 10.1371/journal.pbio.0060219
70
DeVos S. L. Corjuc B. T. Oakley D. H. Nobuhara C. K. Bannon R. N. Chase A. et al . (2018). Synaptic tau seeding precedes tau pathology in human Alzheimer’s disease brain. Front. Neurosci.12:267. 10.3389/fnins.2018.00267
71
Di Benedetto S. Müller L. Wenger E. Düzel S. Pawelec G. (2017). Contribution of neuroinflammation and immunity to brain aging and the mitigating effects of physical and cognitive interventions. Neurosci. Biobehav. Rev.75, 114–128. 10.1016/j.neubiorev.2017.01.044
72
Di Castro M. A. Chuquet J. Liaudet N. Bhaukaurally K. Santello M. Bouvier D. et al . (2011). Local Ca2+ detection and modulation of synaptic release by astrocytes. Nat. Neurosci.14, 1276–1284. 10.1038/nn.2929
73
Dickstein D. L. Kabaso D. Rocher A. B. Luebke J. I. Wearne S. L. Hof P. R. (2007). Changes in the structural complexity of the aged brain. Aging Cell6, 275–284. 10.1111/j.1474-9726.2007.00289.x
74
Duffy A. M. Hölscher C. (2013). The incretin analogue D-Ala2GIP reduces plaque load, astrogliosis and oxidative stress in an APP/PS1 mouse model of Alzheimer’s disease. Neuroscience228, 294–300. 10.1016/j.neuroscience.2012.10.045
75
Dujardin S. Bégard S. Caillierez R. Lachaud C. Delattre L. Carrier S. et al . (2014). Ectosomes: a new mechanism for non-exosomal secretion of tau protein. PLoS One9:e100760. 10.1371/journal.pone.0100760
76
Elder G. J. Taylor J. P. (2014). Transcranial magnetic stimulation and transcranial direct current stimulation: treatments for cognitive and neuropsychiatric symptoms in the neurodegenerative dementias?Alzheimers Res. Ther.6:74. 10.1186/s13195-014-0074-1
77
Fá M. Puzzo D. Piacentini R. Staniszewski A. Zhang H. Baltrons M. A. et al . (2016). Extracellular tau oligomers produce an immediate impairment of LTP and memory. Sci. Rep.6:19393. 10.1038/srep19393
78
Femminella G. D. Frangou E. Love S. B. Busza G. Holmes C. Ritchie C. et al . (2019). Evaluating the effects of the novel GLP-1 analogue liraglutide in Alzheimer’s disease: study protocol for a randomised controlled trial (ELAD study). Trials20:191. 10.1186/s13063-019-3259-x
79
Ferrer I. (2018). Astrogliopathy in tauopathies. Neuroglia1, 126–150. 10.3390/neuroglia1010010
80
Ferrer I. Aguiló García M. Carmona M. Andrés-Benito P. Torrejón-Escribano B. Garcia-Esparcia P. et al . (2019). Involvement of oligodendrocytes in tau seeding and spreading in tauopathies. Front. Aging Neurosci.11:112. 10.3389/fnagi.2019.00112
81
Ferrer I. García M. A. González I. L. Lucena D. D. Villalonga A. R. Tech M. C. et al . (2018). Aging-related tau astrogliopathy (ARTAG): not only tau phosphorylation in astrocytes. Brain Pathol.28, 965–985. 10.1111/bpa.12593
82
Ferrer I. López-González I. Carmona M. Arregui L. Dalfó E. Torrejón-Escribano B. et al . (2014). Glial and neuronal tau pathology in tauopathies: characterization of disease-specific phenotypes and tau pathology progression. J. Neuropathol. Exp. Neurol.73, 81–97. 10.1097/nen.0000000000000030
83
Filipello F. Morini R. Corradini I. Zerbi V. Canzi A. Michalski B. et al . (2018). The microglial innate immune receptor TREM2 is required for synapse elimination and normal brain connectivity. Immunity48, 979.e8–991.e8. 10.1016/j.immuni.2018.04.016
84
Flöel A. (2014). tDCS-enhanced motor and cognitive function in neurological diseases. NeuroImage85, 934–947. 10.1016/j.neuroimage.2013.05.098
85
Ford M. C. Alexandrova O. Cossell L. Stange-Marten A. Sinclair J. Kopp-Scheinpflug C. et al . (2015). Tuning of Ranvier node and internode properties in myelinated axons to adjust action potential timing. Nat. Commun.6:8073. 10.1038/ncomms9073
86
Forman M. S. Lal D. Zhang B. Dabir D. V. Swanson E. Lee V. M. et al . (2005). Transgenic mouse model of tau pathology in astrocytes leading to nervous system degeneration. J. Neurosci.25, 3539–3550. 10.1523/jneurosci.0081-05.2005
87
Foster A. Y. Bujalka H. Emery B. (2019). Axoglial interactions in myelin plasticity: evaluating the relationship between neuronal activity and oligodendrocyte dynamics. Glia67, 2038–2049. 10.1002/glia.23629
88
Fourgeaud L. Boulanger L. M. (2007). Synapse remodeling, compliments of the complement system. Cell131, 1034–1036. 10.1016/j.cell.2007.11.031
89
Fünfschilling U. Supplie L. M. Mahad D. Boretius S. Saab A. S. Edgar J. et al . (2012). Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature485, 517–521. 10.1038/nature11007
90
García-Sierra F. Hauw J. J. Duyckaerts C. Wischik C. M. Luna-Muñoz J. Mena R. (2000). The extent of neurofibrillary pathology in perforant pathway neurons is the key determinant of dementia in the very old. Acta Neuropathol.100, 29–35. 10.1007/s004010051189
91
García-Sierra F. Wischik C. M. Harrington C. R. Luna-Muñoz J. Mena R. (2001). Accumulation of C-terminally truncated tau protein associated with vulnerability of the perforant pathway in early stages of neurofibrillary pathology in Alzheimer’s disease. J. Chem. Neuroanat.22, 65–77. 10.1016/s0891-0618(01)00096-5
92
Garwood C. J. Cooper J. D. Hanger D. P. Noble W. (2010). Anti-inflammatory impact of minocycline in a mouse model of tauopathy. Front. Psychiatry1:136. 10.3389/fpsyt.2010.00136
93
Garza K. M. Zhang L. Borron B. Wood L. B. Singer A. C. (2020). Gamma visual stimulation induces a neuroimmune signaling profile distinct from acute neuroinflammation. J. Neurosci.40, 1211–1225. 10.1523/jneurosci.1511-19.2019
94
Gejl M. Gjedde A. Egefjord L. Møller A. Hansen S. B. Vang K. et al . (2016). In Alzheimer’s disease, 6-month treatment with GLP-1 analog prevents decline of brain glucose metabolism: randomized, placebo-controlled, double-blind clinical trial. Front. Aging Neurosci.8:108. 10.3389/fnagi.2016.00108
95
Ghag G. Bhatt N. Cantu D. V. Guerrero-Munoz M. J. Ellsworth A. Sengupta U. et al . (2018). Soluble tau aggregates, not large fibrils, are the toxic species that display seeding and cross-seeding behavior. Protein Sci.27, 1901–1909. 10.1002/pro.3499
96
Ghetti B. Oblak A. L. Boeve B. F. Johnson K. A. Dickerson B. C. Goedert M. (2015). Invited review: frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: a chameleon for neuropathology and neuroimaging. Neuropathol. Appl. Neurobiol.41, 24–46. 10.1111/nan.12213
97
Giannakopoulos P. Herrmann F. R. Bussière T. Bouras C. Kövari E. Perl D. P. et al . (2003). Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology60, 1495–1500. 10.1212/01.wnl.0000063311.58879.01
98
Gibson E. M. Purger D. Mount C. W. Goldstein A. K. Lin G. L. Wood L. S. et al . (2014). Neuronal activity promotes oligodendrogenesis and adaptive myelination in the mammalian brain. Science344:1252304. 10.1126/science.1252304
99
Gipson C. D. Olive M. F. (2017). Structural and functional plasticity of dendritic spines - root or result of behavior?Genes Brain Behav.16, 101–117. 10.1111/gbb.12324
100
Gómez-Isla T. Hollister R. West H. Mui S. Growdon J. H. Petersen R. C. et al . (1997). Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann. Neurol.41, 17–24. 10.1002/ana.410410106
101
Gonzalez Rothi L. J. Fuller R. Leon S. A. Kendall D. Moore A. Wu S. S. et al . (2009). Errorless practice as a possible adjuvant to donepezil in Alzheimer’s disease. J. Int. Neuropsychol. Soc.15, 311–322. 10.1017/s1355617709090201
102
Grabert K. Michoel T. Karavolos M. H. Clohisey S. Baillie J. K. Stevens M. P. et al . (2016). Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nat. Neurosci.19, 504–516. 10.1038/nn.4222
103
Green C. Sydow A. Vogel S. Anglada-Huguet M. Wiedermann D. Mandelkow E. et al . (2019). Functional networks are impaired by elevated tau-protein but reversible in a regulatable Alzheimer’s disease mouse model. Mol. Neurodegener.14:13. 10.1186/s13024-019-0316-6
104
Grieder M. Wang D. J. J. Dierks T. Wahlund L. O. Jann K. (2018). Default mode network complexity and cognitive decline in mild Alzheimer’s disease. Front. Neurosci.12:770. 10.3389/fnins.2018.00770
105
Guerreiro R. J. Lohmann E. Brás J. M. Gibbs J. R. Rohrer J. D. Gurunlian N. et al . (2013a). Using exome sequencing to reveal mutations in TREM2 presenting as a frontotemporal dementia-like syndrome without bone involvement. JAMA Neurol.70, 78–84. 10.1001/jamaneurol.2013.579
106
Guerreiro R. Wojtas A. Bras J. Carrasquillo M. Rogaeva E. Majounie E. et al . (2013b). TREM2 variants in Alzheimer’s disease. N. Engl. J. Med.368, 117–127. 10.1056/NEJMoa1211851
107
Halassa M. M. Haydon P. G. (2010). Integrated brain circuits: astrocytic networks modulate neuronal activity and behavior. Annu. Rev. Physiol.72, 335–355. 10.1146/annurev-physiol-021909-135843
108
Hansen D. V. Hanson J. E. Sheng M. (2018). Microglia in Alzheimer’s disease. J. Cell Biol.217, 459–472. 10.1083/jcb.201709069
109
Hao W. Liu Y. Liu S. Walter S. Grimm M. O. Kiliaan A. J. et al . (2011). Myeloid differentiation factor 88-deficient bone marrow cells improve Alzheimer’s disease-related symptoms and pathology. Brain134, 278–292. 10.1093/brain/awq325
110
Harada C. N. Natelson Love M. C. Triebel K. L. (2013). Normal cognitive aging. Clin. Geriatr. Med.29, 737–752. 10.1016/j.cger.2013.07.002
111
Hardy J. A. Higgins G. A. (1992). Alzheimer’s disease: the amyloid cascade hypothesis. Science256, 184–185. 10.1126/science.1566067
112
Harris K. M. Kater S. B. (1994). Dendritic spines: cellular specializations imparting both stability and flexibility to synaptic function. Annu. Rev. Neurosci.17, 341–371. 10.1146/annurev.ne.17.030194.002013
113
Hebb D. O. (1949). The Organization of Behavior: A Neurophychological Theory.New York, NY: Wiley.
114
Heiss J. K. Barrett J. Yu Z. Haas L. T. Kostylev M. A. Strittmatter S. M. (2017). Early activation of experience-independent dendritic spine turnover in a mouse model of Alzheimer’s disease. Cereb. Cortex27, 3660–3674. 10.1093/cercor/bhw188
115
Henneberger C. Papouin T. Oliet S. H. Rusakov D. A. (2010). Long-term potentiation depends on release of D-serine from astrocytes. Nature463, 232–236. 10.1038/nature08673
116
Henstridge C. M. Hyman B. T. Spires-Jones T. L. (2019). Beyond the neuron-cellular interactions early in Alzheimer disease pathogenesis. Nat. Rev. Neurosci.20, 94–108. 10.1038/s41583-018-0113-1
117
Herrera C. Chambon C. Michel B. F. Paban V. Alescio-Lautier B. (2012). Positive effects of computer-based cognitive training in adults with mild cognitive impairment. Neuropsychologia50, 1871–1881. 10.1016/j.neuropsychologia.2012.04.012
118
Hickman S. E. Kingery N. D. Ohsumi T. K. Borowsky M. L. Wang L. C. Means T. K. et al . (2013). The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci.16, 1896–1905. 10.1038/nn.3554
119
Hopp S. C. Lin Y. Oakley D. Roe A. D. DeVos S. L. Hanlon D. et al . (2018). The role of microglia in processing and spreading of bioactive tau seeds in Alzheimer’s disease. J. Neuroinflammation15:269. 10.1186/s12974-018-1309-z
120
Iaccarino H. F. Singer A. C. Martorell A. J. Rudenko A. Gao F. Gillingham T. Z. et al . (2016). Gamma frequency entrainment attenuates amyloid load and modifies microglia. Nature540, 230–235. 10.1038/nature20587
121
Ikegami S. Harada A. Hirokawa N. (2000). Muscle weakness, hyperactivity and impairment in fear conditioning in tau-deficient mice. Neurosci. Lett.279, 129–132. 10.1016/s0304-3940(99)00964-7
122
Iqbal K. Liu F. Gong C. X. (2016). Tau and neurodegenerative disease: the story so far. Nat. Rev. Neurol.12, 15–27. 10.1038/nrneurol.2015.225
123
Jacob C. P. Koutsilieri E. Bartl J. Neuen-Jacob E. Arzberger T. Zander N. et al . (2007). Alterations in expression of glutamatergic transporters and receptors in sporadic Alzheimer’s disease. J. Alzheimers Dis.11, 97–116. 10.3233/jad-2007-11113
124
Jadhav S. Avila J. Schöll M. Kovacs G. G. Kövari E. Skrabana R. et al . (2019). A walk through tau therapeutic strategies. Acta Neuropathol. Commun.7:22. 10.1186/s40478-019-0664-z
125
Jay T. R. Hirsch A. M. Broihier M. L. Miller C. M. Neilson L. E. Ransohoff R. M. et al . (2017). Disease progression-dependent effects of TREM2 deficiency in a mouse model of Alzheimer’s disease. J. Neurosci.37, 637–647. 10.1523/JNEUROSCI.2110-16.2016
126
Jiang T. Gong P. Y. Tan M. S. Xue X. Huang S. Zhou J. S. et al . (2019). Soluble TREM1 concentrations are increased and positively correlated with total tau levels in the plasma of patients with Alzheimer’s disease. Aging Clin. Exp. Res.31, 1801–1805. 10.1007/s40520-019-01122-9
127
Jiang T. Zhang Y. D. Chen Q. Gao Q. Zhu X. C. Zhou J. S. et al . (2016a). TREM2 modifies microglial phenotype and provides neuroprotection in P301S tau transgenic mice. Neuropharmacology105, 196–206. 10.1016/j.neuropharm.2016.01.028
128
Jiang T. Zhang Y. D. Gao Q. Zhou J. S. Zhu X. C. Lu H. et al . (2016b). TREM1 facilitates microglial phagocytosis of amyloid β. Acta Neuropathol.132, 667–683. 10.1007/s00401-016-1622-5
129
Jones E. G. (1994). The neuron doctrine 1891. J. Hist. Neurosci.3, 3–20. 10.1080/09647049409525584
130
Kim J. H. Jung Y. S. Kim J. W. Ha M. S. Ha S. M. Kim D. Y. (2018). Effects of aquatic and land-based exercises on amyloid β, heat shock protein 27 and pulse wave velocity in elderly women. Exp. Gerontol.108, 62–68. 10.1016/j.exger.2018.03.024
131
Kim N. G. Park J. H. (2010). Learning to perceive structure from motion and neural plasticity in patients with Alzheimer’s disease. Neuropsychologia48, 1464–1471. 10.1016/j.neuropsychologia.2010.01.016
132
Kimura T. Whitcomb D. J. Jo J. Regan P. Piers T. Heo S. et al . (2014). Microtubule-associated protein tau is essential for long-term depression in the hippocampus. Philos. Trans. R. Soc. Lond. B Biol. Sci.369:20130144. 10.1098/rstb.2013.0144
133
Knopman D. S. Parisi J. E. Salviati A. Floriach-Robert M. Boeve B. F. Ivnik R. J. et al . (2003). Neuropathology of cognitively normal elderly. J. Neuropathol. Exp. Neurol.62, 1087–1095. 10.1093/jnen/62.11.1087
134
Kobayashi E. Nakano M. Kubota K. Himuro N. Mizoguchi S. Chikenji T. et al . (2018). Activated forms of astrocytes with higher GLT-1 expression are associated with cognitive normal subjects with Alzheimer pathology in human brain. Sci. Rep.8:1712. 10.1038/s41598-018-19442-7
135
Koch G. Di Lorenzo F. Bonnì S. Giacobbe V. Bozzali M. Caltagirone C. et al . (2014). Dopaminergic modulation of cortical plasticity in Alzheimer’s disease patients. Neuropsychopharmacology39, 2654–2661. 10.1038/npp.2014.119
136
Koch G. Di Lorenzo F. Bonnì S. Ponzo V. Caltagirone C. Martorana A. (2012). Impaired LTP- but not LTD-like cortical plasticity in Alzheimer’s disease patients. J. Alzheimers Dis.31, 593–599. 10.3233/jad-2012-120532
137
Koizumi S. Ohsawa K. Inoue K. Kohsaka S. (2013). Purinergic receptors in microglia: functional modal shifts of microglia mediated by P2 and P1 receptors. Glia61, 47–54. 10.1002/glia.22358
138
Koller E. J. De La Cruz E. G. Machula T. Ibanez K. R. Lin W. L. Williams T. et al . (2019). Combining P301L and S320F tau variants produces a novel accelerated model of tauopathy. Hum. Mol. Genet.28, 3255–3269. 10.1093/hmg/ddz151
139
Konietzko U. (2012). AICD nuclear signaling and its possible contribution to Alzheimer’s disease. Curr. Alzheimer Res.9, 200–216. 10.2174/156720512799361673
140
Korvatska O. Leverenz J. B. Jayadev S. McMillan P. Kurtz I. Guo X. et al . (2015). R47H Variant of TREM2 associated with Alzheimer disease in a large late-onset family: clinical, genetic, and neuropathological study. JAMA Neurol.72, 920–927. 10.1001/jamaneurol.2015.0979
141
Kovacs G. G. (2015). Invited review: neuropathology of tauopathies: principles and practice. Neuropathol. Appl. Neurobiol.41, 3–23. 10.1111/nan.12208
142
Kovacs G. G. (2020). Astroglia and tau: new perspectives. Front. Aging Neurosci.12:96. 10.3389/fnagi.2020.00096
143
Kovacs G. G. Ferrer I. Grinberg L. T. Alafuzoff I. Attems J. Budka H. et al . (2016). Aging-related tau astrogliopathy (ARTAG): harmonized evaluation strategy. Acta Neuropathol.131, 87–102. 10.1007/s00401-015-1509-x
144
Krasnow A. M. Attwell D. (2016). NMDA receptors: power switches for oligodendrocytes. Neuron91, 3–5. 10.1016/j.neuron.2016.06.023
145
Kucukdereli H. Allen N. J. Lee A. T. Feng A. Ozlu M. I. Conatser L. M. et al . (2011). Control of excitatory CNS synaptogenesis by astrocyte-secreted proteins Hevin and SPARC. Proc. Natl. Acad. Sci. U S A108, E440–449. 10.1073/pnas.1104977108
146
Lace G. Ince P. G. Brayne C. Savva G. M. Matthews F. E. de Silva R. et al . (2012). Mesial temporal astrocyte tau pathology in the MRC-CFAS ageing brain cohort. Dement. Geriatr. Cogn. Disord.34, 15–24. 10.1159/000341581
147
Lawson L. J. Perry V. H. Dri P. Gordon S. (1990). Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience39, 151–170. 10.1016/0306-4522(90)90229-w
148
Lee Y. Morrison B. M. Li Y. Lengacher S. Farah M. H. Hoffman P. N. et al . (2012). Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature487, 443–448. 10.1038/nature11314
149
Leung N. T. Tam H. M. Chu L. W. Kwok T. C. Chan F. Lam L. C. et al . (2015). Neural plastic effects of cognitive training on aging brain. Neural Plast.2015:535618. 10.1155/2015/535618
150
Levenga J. Krishnamurthy P. Rajamohamedsait H. Wong H. Franke T. F. Cain P. et al . (2013). Tau pathology induces loss of GABAergic interneurons leading to altered synaptic plasticity and behavioral impairments. Acta Neuropathol. Commun.1:34. 10.1186/2051-5960-1-34
151
Leyns C. E. G. Ulrich J. D. Finn M. B. Stewart F. R. Koscal L. J. Remolina Serrano J. et al . (2017). TREM2 deficiency attenuates neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy. Proc. Natl. Acad. Sci. U S A114, 11524–11529. 10.1073/pnas.1710311114
152
Li Q. Brus-Ramer M. Martin J. H. McDonald J. W. (2010). Electrical stimulation of the medullary pyramid promotes proliferation and differentiation of oligodendrocyte progenitor cells in the corticospinal tract of the adult rat. Neurosci. Lett.479, 128–133. 10.1016/j.neulet.2010.05.043
153
Li Y. Du X. F. Liu C. S. Wen Z. L. Du J. L. (2012). Reciprocal regulation between resting microglial dynamics and neuronal activity in vivo. Dev. Cell23, 1189–1202. 10.1016/j.devcel.2012.10.027
154
Liang F. Yang S. Zhang Y. Hao T. (2019). Social housing promotes cognitive function through enhancing synaptic plasticity in APP/PS1 mice. Behav. Brain Res.368:111910. 10.1016/j.bbr.2019.111910
155
Liddelow S. A. Guttenplan K. A. Clarke L. E. Bennett F. C. Bohlen C. J. Schirmer L. et al . (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature541, 481–487. 10.1038/nature21029
156
Liu Y. Zhang Y. Zheng X. Fang T. Yang X. Luo X. et al . (2018). Galantamine improves cognition, hippocampal inflammation, and synaptic plasticity impairments induced by lipopolysaccharide in mice. J. Neuroinflammation15:112. 10.1186/s12974-018-1141-5
157
Liu Y. Given K. S. Owens G. P. Macklin W. B. Bennett J. L. (2018). Distinct patterns of glia repair and remyelination in antibody-mediated demyelination models of multiple sclerosis and neuromyelitis optica. Glia66, 2575–2588. 10.1002/glia.23512
158
Liu Y. S. Yan W. J. Tan C. C. Li J. Q. Xu W. Cao X. P. et al . (2020). Common variant in TREM1 influencing brain amyloid deposition in mild cognitive impairment and Alzheimer’s disease. Neurotox. Res.37, 661–668. 10.1007/s12640-019-00105-y
159
Lohmann C. Kessels H. W. (2014). The developmental stages of synaptic plasticity. J. Physiol.592, 13–31. 10.1113/jphysiol.2012.235119
160
Luo H. Xiang Y. Qu X. Liu H. Liu C. Li G. et al . (2019). Apelin-13 suppresses neuroinflammation against cognitive deficit in a streptozotocin-induced rat model of Alzheimer’s disease through activation of BDNF-TrkB signaling pathway. Front. Pharmacol.10:395. 10.3389/fphar.2019.00395
161
Ma Q. L. Zuo X. Yang F. Ubeda O. J. Gant D. J. Alaverdyan M. et al . (2014). Loss of MAP function leads to hippocampal synapse loss and deficits in the Morris Water Maze with aging. J. Neurosci.34, 7124–7136. 10.1523/JNEUROSCI.3439-13.2014
162
Maeda S. Djukic B. Taneja P. Yu G. Q. Lo I. Davis A. et al . (2016). Expression of A152T human tau causes age-dependent neuronal dysfunction and loss in transgenic mice. EMBO Rep.17, 530–551. 10.15252/embr.201541438
163
Martorana A. Di Lorenzo F. Esposito Z. Lo Giudice T. Bernardi G. Caltagirone C. et al . (2013). Dopamine D2-agonist rotigotine effects on cortical excitability and central cholinergic transmission in Alzheimer’s disease patients. Neuropharmacology64, 108–113. 10.1016/j.neuropharm.2012.07.015
164
Martorell A. J. Paulson A. L. Suk H. J. Abdurrob F. Drummond G. T. Guan W. et al . (2019). Multi-sensory gamma stimulation ameliorates Alzheimer’s-associated pathology and improves cognition. Cell177, 256.e22–271.e22. 10.1016/j.cell.2019.02.014
165
Masliah E. Alford M. DeTeresa R. Mallory M. Hansen L. (1996). Deficient glutamate transport is associated with neurodegeneration in Alzheimer’s disease. Ann. Neurol.40, 759–766. 10.1002/ana.410400512
166
Mateos-Aparicio P. Rodríguez-Moreno A. (2019). The impact of studying brain plasticity. Front. Cell. Neurosci.13:66. 10.3389/fncel.2019.00066
167
Matute C. Alberdi E. Domercq M. Sánchez-Gómez M. V. Pérez-Samartín A. Rodríguez-Antigüedad A. et al . (2007). Excitotoxic damage to white matter. J. Anat.210, 693–702. 10.1111/j.1469-7580.2007.00733.x
168
McAleese K. E. Firbank M. Dey M. Colloby S. J. Walker L. Johnson M. et al . (2015). Cortical tau load is associated with white matter hyperintensities. Acta Neuropathol. Commun.3:60. 10.1186/s40478-015-0240-0
169
McClean P. L. Jalewa J. Hölscher C. (2015). Prophylactic liraglutide treatment prevents amyloid plaque deposition, chronic inflammation and memory impairment in APP/PS1 mice. Behav. Brain Res.293, 96–106. 10.1016/j.bbr.2015.07.024
170
McQuade A. Blurton-Jones M. (2019). Microglia in Alzheimer’s disease: exploring how genetics and phenotype influence risk. J. Mol. Biol.431, 1805–1817. 10.1016/j.jmb.2019.01.045
171
Meinzer M. Lindenberg R. Antonenko D. Flaisch T. Flöel A. (2013). Anodal transcranial direct current stimulation temporarily reverses age-associated cognitive decline and functional brain activity changes. J. Neurosci.33, 12470–12478. 10.1523/JNEUROSCI.5743-12.2013
172
Meinzer M. Lindenberg R. Phan M. T. Ulm L. Volk C. Flöel A. (2015). Transcranial direct current stimulation in mild cognitive impairment: behavioral effects and neural mechanisms. Alzheimers Dement.11, 1032–1040. 10.1016/j.jalz.2014.07.159
173
Meyer-Luehmann M. Spires-Jones T. L. Prada C. Garcia-Alloza M. de Calignon A. Rozkalne A. et al . (2008). Rapid appearance and local toxicity of amyloid-β plaques in a mouse model of Alzheimer’s disease. Nature451, 720–724. 10.1038/nature06616
174
Micu I. Plemel J. R. Lachance C. Proft J. Jansen A. J. Cummins K. et al . (2016). The molecular physiology of the axo-myelinic synapse. Exp. Neurol.276, 41–50. 10.1016/j.expneurol.2015.10.006
175
Min R. Nevian T. (2012). Astrocyte signaling controls spike timing-dependent depression at neocortical synapses. Nat. Neurosci.15, 746–753. 10.1038/nn.3075
176
Miron V. E. Boyd A. Zhao J. W. Yuen T. J. Ruckh J. M. Shadrach J. L. et al . (2013). M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat. Neurosci.16, 1211–1218. 10.1038/nn.3469
177
Mitew S. Gobius I. Fenlon L. R. McDougall S. J. Hawkes D. Xing Y. L. et al . (2018). Pharmacogenetic stimulation of neuronal activity increases myelination in an axon-specific manner. Nat. Commun.9:306. 10.1038/s41467-017-02719-2
178
Miyazono M. Iwaki T. Kitamoto T. Shin R. W. Fukui M. Tateishi J. (1993). Widespread distribution of tau in the astrocytic elements of glial tumors. Acta Neuropathol.86, 236–241. 10.1007/bf00304137
179
Montero-Odasso M. Almeida Q. J. Burhan A. M. Camicioli R. Doyon J. Fraser S. et al . (2018). SYNERGIC TRIAL (SYNchronizing exercises, remedies in gait and cognition) a multi-Centre randomized controlled double blind trial to improve gait and cognition in mild cognitive impairment. BMC Geriatr.18:93. 10.1186/s12877-018-0782-7
180
Morrison J. H. Baxter M. G. (2012). The ageing cortical synapse: hallmarks and implications for cognitive decline. Nat. Rev. Neurosci.13, 240–250. 10.1038/nrn3200
181
Mudher A. Colin M. Dujardin S. Medina M. Dewachter I. Alavi Naini S. M. et al . (2017). What is the evidence that tau pathology spreads through prion-like propagation?Acta Neuropathol. Commun.5:99. 10.1186/s40478-017-0488-7
182
Nagamatsu L. S. Handy T. C. Hsu C. L. Voss M. Liu-Ambrose T. (2012). Resistance training promotes cognitive and functional brain plasticity in seniors with probable mild cognitive impairment. Arch. Intern. Med.172, 666–668. 10.1001/archinternmed.2012.379
183
Nagy B. Hovhannisyan A. Barzan R. Chen T. J. Kukley M. (2017). Different patterns of neuronal activity trigger distinct responses of oligodendrocyte precursor cells in the corpus callosum. PLoS Biol.15:e2001993. 10.1371/journal.pbio.2001993
184
Narasimhan S. Guo J. L. Changolkar L. Stieber A. McBride J. D. Silva L. V. et al . (2017). Pathological tau strains from human brains recapitulate the diversity of tauopathies in nontransgenic mouse brain. J. Neurosci.37, 11406–11423. 10.1523/JNEUROSCI.1230-17.2017
185
Nelson P. T. Alafuzoff I. Bigio E. H. Bouras C. Braak H. Cairns N. J. et al . (2012). Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J. Neuropathol. Exp. Neurol.71, 362–381. 10.1097/NEN.0b013e31825018f7
186
Nimmerjahn A. Kirchhoff F. Helmchen F. (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science308, 1314–1318. 10.1126/science.1110647
187
Ortega-Martinez S. Palla N. Zhang X. Lipman E. Sisodia S. S. (2019). Deficits in enrichment-dependent neurogenesis and enhanced anxiety behaviors mediated by expression of Alzheimer’s disease-linked Ps1 variants are rescued by microglial depletion. J. Neurosci.39, 6766–6780. 10.1523/JNEUROSCI.0884-19.2019
188
Palmqvist S. Schöll M. Strandberg O. Mattsson N. Stomrud E. Zetterberg H. et al . (2017). Earliest accumulation of β-amyloid occurs within the default-mode network and concurrently affects brain connectivity. Nat. Commun.8:1214. 10.1038/s41467-017-01150-x
189
Paolicelli R. C. Bolasco G. Pagani F. Maggi L. Scianni M. Panzanelli P. et al . (2011). Synaptic pruning by microglia is necessary for normal brain development. Science333, 1456–1458. 10.1126/science.1202529
190
Park D. C. Lautenschlager G. Hedden T. Davidson N. S. Smith A. D. Smith P. K. (2002). Models of visuospatial and verbal memory across the adult life span. Psychol. Aging17, 299–320. 10.1037/0882-7974.17.2.299
191
Park D. C. Polk T. A. Mikels J. A. Taylor S. F. Marshuetz C. (2001). Cerebral aging: integration of brain and behavioral models of cognitive function. Dialogues Clin. Neurosci.3, 151–165.
192
Park D. C. Reuter-Lorenz P. (2009). The adaptive brain: aging and neurocognitive scaffolding. Annu. Rev. Psychol.60, 173–196. 10.1146/annurev.psych.59.103006.093656
193
Parkhurst C. N. Yang G. Ninan I. Savas J. N. Yates J. R. Lafaille J. J. et al . (2013). Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell155, 1596–1609. 10.1016/j.cell.2013.11.030
194
Pascoal T. A. Mathotaarachchi S. Kang M. S. Mohaddes S. Shin M. Park A. Y. et al . (2019). Aβ-induced vulnerability propagates via the brain’s default mode network. Nat. Commun.10:2353. 10.1038/s41467-019-10217-w
195
Pascual O. Ben Achour S. Rostaing P. Triller A. Bessis A. (2012). Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proc. Natl. Acad. Sci. U S A109, E197–E205. 10.1073/pnas.1111098109
196
Pedrazzoli M. Losurdo M. Paolone G. Medelin M. Jaupaj L. Cisterna B. et al . (2019). Glucocorticoid receptors modulate dendritic spine plasticity and microglia activity in an animal model of Alzheimer’s disease. Neurobiol. Dis.132:104568. 10.1016/j.nbd.2019.104568
197
Perea J. R. López E. Díez-Ballesteros J. C. Ávila J. Hernández F. Bolós M. (2019). Extracellular monomeric tau is internalized by astrocytes. Front. Neurosci.13:442. 10.3389/fnins.2019.00442
198
Perea G. Navarrete M. Araque A. (2009). Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci.32, 421–431. 10.1016/j.tins.2009.05.001
199
Piacentini R. Li Puma D. D. Mainardi M. Lazzarino G. Tavazzi B. Arancio O. et al . (2017). Reduced gliotransmitter release from astrocytes mediates tau-induced synaptic dysfunction in cultured hippocampal neurons. Glia65, 1302–1316. 10.1002/glia.23163
200
Polanco J. C. Scicluna B. J. Hill A. F. Götz J. (2016). Extracellular vesicles isolated from the brains of rTg4510 mice seed tau protein aggregation in a threshold-dependent manner. J. Biol. Chem.291, 12445–12466. 10.1074/jbc.m115.709485
201
Polydoro M. Dzhala V. I. Pooler A. M. Nicholls S. B. McKinney A. P. Sanchez L. et al . (2014). Soluble pathological tau in the entorhinal cortex leads to presynaptic deficits in an early Alzheimer’s disease model. Acta Neuropathol.127, 257–270. 10.1007/s00401-013-1215-5
202
Pooler A. M. Phillips E. C. Lau D. H. Noble W. Hanger D. P. (2013). Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep.14, 389–394. 10.1038/embor.2013.15
203
Pousinha P. A. Mouska X. Raymond E. F. Gwizdek C. Dhib G. Poupon G. et al . (2017). Physiological and pathophysiological control of synaptic GluN2B-NMDA receptors by the C-terminal domain of amyloid precursor protein. eLife6:e25659. 10.7554/eLife.25659
204
Pozueta J. Lefort R. Shelanski M. L. (2013). Synaptic changes in Alzheimer’s disease and its models. Neuroscience251, 51–65. 10.1016/j.neuroscience.2012.05.050
205
Price J. L. Morris J. C. (1999). Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Ann. Neurol.45, 358–368. 10.1002/1531-8249(199903)45:3<358::aid-ana12>3.0.co;2-x
206
Ramón y Cajal S. (1892). El nuevo concepto de la histología en los centros nerviosos. Rev. Ciencias. Med. Barcelona18, 361–376, 457–476, 505–520, 529–541.
207
Ramón y Cajal S. (1894a). Consideraciones Generales Sobre la Morfología de la Célula Nerviosa.Madrid: Moya.
208
Ramón y Cajal S. (1894b). Consideraciones generales sobre la morfología de la célula nerviosa. Veter. Espa.37, 257–260, 273–275, 289–291.
209
Ramón y Cajal S. (1894c). The Croonian lecture: la fine structure des centres nerveux. Proc. R. Soc. Land55, 444–468. 10.1098/rspl.1894.0063
210
Ramón y Cajal S. (1899/1904). Textura del Sistema Nervioso del Hombre y de Los Vertebrados.Madrid: Moya.
211
Ramsden M. Kotilinek L. Forster C. Paulson J. McGowan E. SantaCruz K. et al . (2005). Age-dependent neurofibrillary tangle formation, neuron loss, and memory impairment in a mouse model of human tauopathy (P301L). J. Neurosci.25, 10637–10647. 10.1523/JNEUROSCI.3279-05.2005
212
Regan P. Piers T. Yi J. H. Kim D. H. Huh S. Park S. J. et al . (2015). Tau phosphorylation at serine 396 residue is required for hippocampal LTD. J. Neurosci.35, 4804–4812. 10.1523/JNEUROSCI.2842-14.2015
213
Reis J. Schambra H. M. Cohen L. G. Buch E. R. Fritsch B. Zarahn E. et al . (2009). Noninvasive cortical stimulation enhances motor skill acquisition over multiple days through an effect on consolidation. Proc. Natl. Acad. Sci. U S A106, 1590–1595. 10.1073/pnas.0805413106
214
Replogle J. M. Chan G. White C. C. Raj T. Winn P. A. Evans D. A. et al . (2015). A TREM1 variant alters the accumulation of Alzheimer-related amyloid pathology. Ann. Neurol.77, 469–477. 10.1002/ana.24337
215
Reuter-Lorenz P. (2002). New visions of the aging mind and brain. Trends Cogn. Sci.6:394. 10.1016/s1364-6613(02)01957-5
216
Robinson J. L. Molina-Porcel L. Corrada M. M. Raible K. Lee E. B. Lee V. M. et al . (2014). Perforant path synaptic loss correlates with cognitive impairment and Alzheimer’s disease in the oldest-old. Brain137, 2578–2587. 10.1093/brain/awu190
217
Rogers J. T. Morganti J. M. Bachstetter A. D. Hudson C. E. Peters M. M. Grimmig B. A. et al . (2011). CX3CR1 deficiency leads to impairment of hippocampal cognitive function and synaptic plasticity. J. Neurosci.31, 16241–16250. 10.1523/JNEUROSCI.3667-11.2011
218
Rosen A. C. Sugiura L. Kramer J. H. Whitfield-Gabrieli S. Gabrieli J. D. (2011). Cognitive training changes hippocampal function in mild cognitive impairment: a pilot study. J. Alzheimers Dis.26, 349–357. 10.3233/jad-2011-0009
219
Saab A. S. Tzvetavona I. D. Trevisiol A. Baltan S. Dibaj P. Kusch K. et al . (2016). Oligodendroglial NMDA receptors regulate glucose import and axonal energy metabolism. Neuron91, 119–132. 10.1016/j.neuron.2016.05.016
220
Saadipour K. (2017). TREM1: a potential therapeutic target for Alzheimer’s disease. Neurotox. Res.32, 14–16. 10.1007/s12640-017-9716-y
221
Sahara N. Maeda J. Ishikawa A. Tokunaga M. Suhara T. Higuchi M. (2018). Microglial activation during pathogenesis of tauopathy in rTg4510 mice: implications for the early diagnosis of tauopathy. J. Alzheimers Dis.64, S353–S359. 10.3233/jad-179933
222
Saman S. Kim W. Raya M. Visnick Y. Miro S. Jackson B. et al . (2012). Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J. Biol. Chem.287, 3842–3849. 10.1074/jbc.m111.277061
223
Sanchez-Mejias E. Navarro V. Jimenez S. Sanchez-Mico M. Sanchez-Varo R. Nuñez-Diaz C. et al . (2016). Soluble phospho-tau from Alzheimer’s disease hippocampus drives microglial degeneration. Acta Neuropathol.132, 897–916. 10.1007/s00401-016-1630-5
224
Santacruz K. Lewis J. Spires T. Paulson J. Kotilinek L. Ingelsson M. et al . (2005). Tau suppression in a neurodegenerative mouse model improves memory function. Science309, 476–481. 10.1126/science.1113694
225
Sarlus H. Heneka M. T. (2017). Microglia in Alzheimer’s disease. J. Clin. Invest.127, 3240–3249. 10.1172/JCI90606
226
Schafer D. P. Lehrman E. K. Kautzman A. G. Koyama R. Mardinly A. R. Yamasaki R. et al . (2012). Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron74, 691–705. 10.1016/j.neuron.2012.03.026
227
Schafer D. P. Lehrman E. K. Stevens B. (2013). The “quad-partite” synapse: microglia-synapse interactions in the developing and mature CNS. Glia61, 24–36. 10.1002/glia.22389
228
Scheff S. W. DeKosky S. T. Price D. A. (1990). Quantitative assessment of cortical synaptic density in Alzheimer’s disease. Neurobiol. Aging11, 29–37. 10.1016/0197-4580(90)90059-9
229
Scheff S. W. Price D. A. (2006). Alzheimer’s disease-related alterations in synaptic density: neocortex and hippocampus. J. Alzheimers Dis.9, 101–115. 10.3233/jad-2006-9s312
230
Scheff S. W. Price D. A. Schmitt F. A. Mufson E. J. (2006). Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol. Aging27, 1372–1384. 10.1016/j.neurobiolaging.2005.09.012
231
Schindowski K. Bretteville A. Leroy K. Bégard S. Brion J. P. Hamdane M. et al . (2006). Alzheimer’s disease-like tau neuropathology leads to memory deficits and loss of functional synapses in a novel mutated tau transgenic mouse without any motor deficits. Am. J. Pathol.169, 599–616. 10.2353/ajpath.2006.060002
232
Schousboe A. Sarup A. Bak L. K. Waagepetersen H. S. Larsson O. M. (2004). Role of astrocytic transport processes in glutamatergic and GABAergic neurotransmission. Neurochem. Int.45, 521–527. 10.1016/j.neuint.2003.11.001
233
Schultz C. Ghebremedhin E. Del Tredici K. Rüb U. Braak H. (2004). High prevalence of thorn-shaped astrocytes in the aged human medial temporal lobe. Neurobiol. Aging25, 397–405. 10.1016/s0197-4580(03)00113-1
234
Selkoe D. J. Hardy J. (2016). The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med.8, 595–608. 10.15252/emmm.201606210
235
Sergeant N. David J. P. Goedert M. Jakes R. Vermersch P. Buée L. et al . (1997). Two-dimensional characterization of paired helical filament-tau from Alzheimer’s disease: demonstration of an additional 74-kDa component and age-related biochemical modifications. J. Neurochem.69, 834–844. 10.1046/j.1471-4159.1997.69020834.x
236
Serrano-Pozo A. Mielke M. L. Gómez-Isla T. Betensky R. A. Growdon J. H. Frosch M. P. et al . (2011). Reactive glia not only associates with plaques but also parallels tangles in Alzheimer’s disease. Am. J. Pathol.179, 1373–1384. 10.1016/j.ajpath.2011.05.047
237
Shatz C. J. (1992). The developing brain. Sci. Am.267, 60–67. 10.1038/scientificamerican0992-60
238
Shepherd G. M. (1991). Foundations of the Neuron Doctrine.New York, NY: Oxford University Press.
239
Shibuya K. Yagishita S. Nakamura A. Uchihara T. (2011). Perivascular orientation of astrocytic plaques and tuft-shaped astrocytes. Brain Res.1404, 50–54. 10.1016/j.brainres.2011.06.014
240
Shin R. W. Iwaki T. Kitamoto T. Tateishi J. (1991). Hydrated autoclave pretreatment enhances tau immunoreactivity in formalin-fixed normal and Alzheimer’s disease brain tissues. Lab. Invest.64, 693–702.
241
Sierra A. Abiega O. Shahraz A. Neumann H. (2013). Janus-faced microglia: beneficial and detrimental consequences of microglial phagocytosis. Front. Cell. Neurosci.7:6. 10.3389/fncel.2013.00006
242
Sims R. van der Lee S. J. Naj A. C. Bellenguez C. Badarinarayan N. Jakobsdottir J. et al . (2017). Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat. Genet.49, 1373–1384. 10.1038/ng.3916
243
Skaper S. D. Facci L. Zusso M. Giusti P. (2017). Synaptic plasticity, dementia and alzheimer disease. CNS Neurol. Disord Drug Targets16, 220–233. 10.2174/1871527316666170113120853
244
Slattery C. F. Beck J. A. Harper L. Adamson G. Abdi Z. Uphill J. et al . (2014). R47H TREM2 variant increases risk of typical early-onset alzheimer’s disease but not of prion or frontotemporal dementia. Alzheimers Dement.10, 602.e4–608.e4. 10.1016/j.jalz.2014.05.1751
245
Sofroniew M. V. (2009). Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci.32, 638–647. 10.1016/j.tins.2009.08.002
246
Sohn P. D. Huang C. T. Yan R. Fan L. Tracy T. E. Camargo C. M. et al . (2019). Pathogenic tau impairs axon initial segment plasticity and excitability homeostasis. Neuron104, 458.e5–470.e5. 10.1016/j.neuron.2019.08.008
247
Soto I. Graham L. C. Richter H. J. Simeone S. N. Radell J. E. Grabowska W. et al . (2015). APOE stabilization by exercise prevents aging neurovascular dysfunction and complement induction. PLoS Biol.13:e1002279. 10.1371/journal.pbio.1002279
248
Stagg C. J. Nitsche M. A. (2011). Physiological basis of transcranial direct current stimulation. Neuroscientist17, 37–53. 10.1177/1073858410386614
249
Stahnisch F. W. Nitsch R. (2002). Santiago Ramon y Cajal’s concept of neuronal plasticity: the ambiguity lives on. Trends Neurosci.25, 589–591. 10.1016/s0166-2236(02)02251-8
250
Stampanoni Bassi M. Iezzi E. Gilio L. Centonze D. Buttari F. (2019). Synaptic plasticity shapes brain connectivity: implications for network topology. Int. J. Mol. Sci.20:6193. 10.3390/ijms20246193
251
Stellwagen D. Malenka R. C. (2006). Synaptic scaling mediated by glial TNF-α. Nature440, 1054–1059. 10.1038/nature04671
252
Stephan A. H. Barres B. A. Stevens B. (2012). The complement system: an unexpected role in synaptic pruning during development and disease. Annu. Rev. Neurosci.35, 369–389. 10.1146/annurev-neuro-061010-113810
253
Stevens B. Allen N. J. Vazquez L. E. Howell G. R. Christopherson K. S. Nouri N. et al . (2007). The classical complement cascade mediates CNS synapse elimination. Cell131, 1164–1178. 10.1016/j.cell.2007.10.036
254
Stogsdill J. A. Eroglu C. (2017). The interplay between neurons and glia in synapse development and plasticity. Curr. Opin. Neurobiol.42, 1–8. 10.1016/j.conb.2016.09.016
255
Streit W. J. Braak H. Xue Q. S. Bechmann I. (2009). Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathol.118, 475–485. 10.1007/s00401-009-0556-6
256
Stricker N. H. Schweinsburg B. C. Delano-Wood L. Wierenga C. E. Bangen K. J. Haaland K. Y. et al . (2009). Decreased white matter integrity in late-myelinating fiber pathways in Alzheimer’s disease supports retrogenesis. NeuroImage45, 10–16. 10.1016/j.neuroimage.2008.11.027
257
Styliadis C. Kartsidis P. Paraskevopoulos E. Ioannides A. A. Bamidis P. D. (2015). Neuroplastic effects of combined computerized physical and cognitive training in elderly individuals at risk for dementia: an eLORETA controlled study on resting states. Neural Plast.2015:172192. 10.1155/2015/172192
258
Su Y. Fu J. Yu J. Zhao Q. Guan Y. Zuo C. et al . (2020). Tau PET imaging with [18F]PM-PBB3 in frontotemporal dementia with MAPT mutation. J. Alzheimers Dis.76, 149–157. 10.3233/jad-200287
259
Suárez-Calvet M. Araque Caballero M. Kleinberger G. Bateman R. J. Fagan A. M. Morris J. C. et al . (2016a). Early changes in CSF sTREM2 in dominantly inherited Alzheimer’s disease occur after amyloid deposition and neuronal injury. Sci. Transl. Med.8:369ra178. 10.1126/scitranslmed.aag1767
260
Suárez-Calvet M. Kleinberger G. Araque Caballero M. Brendel M. Rominger A. Alcolea D. et al . (2016b). sTREM2 cerebrospinal fluid levels are a potential biomarker for microglia activity in early-stage Alzheimer’s disease and associate with neuronal injury markers. EMBO Mol. Med.8, 466–476. 10.15252/emmm.201506123
261
Suárez-Calvet M. Morenas-Rodríguez E. Kleinberger G. Schlepckow K. Araque Caballero M. Franzmeier N. et al . (2019). Early increase of CSF sTREM2 in Alzheimer’s disease is associated with tau related-neurodegeneration but not with amyloid-β pathology. Mol. Neurodegener.14:1. 10.1186/s13024-018-0301-5
262
Sultan S. Li L. Moss J. Petrelli F. Cassé F. Gebara E. et al . (2015). Synaptic integration of adult-born hippocampal neurons is locally controlled by astrocytes. Neuron88, 957–972. 10.1016/j.neuron.2015.10.037
263
Sydow A. Hochgräfe K. Könen S. Cadinu D. Matenia D. Petrova O. et al . (2016). Age-dependent neuroinflammation and cognitive decline in a novel Ala152Thr-Tau transgenic mouse model of PSP and AD. Acta Neuropathol. Commun.4:17. 10.1186/s40478-016-0281-z
264
Sydow A. Van der Jeugd A. Zheng F. Ahmed T. Balschun D. Petrova O. et al . (2011). Reversibility of Tau-related cognitive defects in a regulatable FTD mouse model. J. Mol. Neurosci.45, 432–437. 10.1007/s12031-011-9604-5
265
Tadi M. Allaman I. Lengacher S. Grenningloh G. Magistretti P. J. (2015). Learning-induced gene expression in the hippocampus reveals a role of neuron -astrocyte metabolic coupling in long term memory. PLoS One10:e0141568. 10.1371/journal.pone.0141568
266
Tani H. Dulla C. G. Farzampour Z. Taylor-Weiner A. Huguenard J. R. Reimer R. J. (2014). A local glutamate-glutamine cycle sustains synaptic excitatory transmitter release. Neuron81, 888–900. 10.1016/j.neuron.2013.12.026
267
Tardivel M. Bégard S. Bousset L. Dujardin S. Coens A. Melki R. et al . (2016). Tunneling nanotube (TNT)-mediated neuron-to neuron transfer of pathological Tau protein assemblies. Acta Neuropathol. Commun.4:117. 10.1186/s40478-016-0386-4
268
Terry R. D. Masliah E. Salmon D. P. Butters N. DeTeresa R. Hill R. et al . (1991). Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann. Neurol.30, 572–580. 10.1002/ana.410300410
269
Thal D. R. Holzer M. Rüb U. Waldmann G. Günzel S. Zedlick D. et al . (2000). Alzheimer-related tau-pathology in the perforant path target zone and in the hippocampal stratum oriens and radiatum correlates with onset and degree of dementia. Exp. Neurol.163, 98–110. 10.1006/exnr.2000.7380
270
Tijms B. M. Ten Kate M. Gouw A. A. Borta A. Verfaillie S. Teunissen C. E. et al . (2018). Gray matter networks and clinical progression in subjects with predementia Alzheimer’s disease. Neurobiol. Aging61, 75–81. 10.1016/j.neurobiolaging.2017.09.011
271
Tosto G. Zimmerman M. E. Carmichael O. T. Brickman A. M. Alzheimer’s Disease Neuroimaging Initiative . (2014). Predicting aggressive decline in mild cognitive impairment: the importance of white matter hyperintensities. JAMA Neurol.71, 872–877. 10.1001/jamaneurol.2014.667
272
Trapp B. D. Wujek J. R. Criste G. A. Jalabi W. Yin X. Kidd G. J. et al . (2007). Evidence for synaptic stripping by cortical microglia. Glia55, 360–368. 10.1002/glia.20462
273
Tremblay M. Lowery R. L. Majewska A. K. (2010). Microglial interactions with synapses are modulated by visual experience. PLoS Biol.8:e1000527. 10.1371/journal.pbio.1000527
274
Vainchtein I. D. Molofsky A. V. (2020). Astrocytes and microglia: in sickness and in health. Trends Neurosci.43, 144–154. 10.1016/j.tins.2020.01.003
275
Van der Jeugd A. Hochgräfe K. Ahmed T. Decker J. M. Sydow A. Hofmann A. et al . (2012). Cognitive defects are reversible in inducible mice expressing pro-aggregant full-length human Tau. Acta Neuropathol.123, 787–805. 10.1007/s00401-012-0987-3
276
Vemula P. Jing Y. Zhang H. Hunt J. B. Sandusky-Beltran L. A. Lee D. C. et al . (2019). Altered brain arginine metabolism in a mouse model of tauopathy. Amino Acids51, 513–528. 10.1007/s00726-018-02687-x
277
Vincenti J. E. Murphy L. Grabert K. McColl B. W. Cancellotti E. Freeman T. C. et al . (2015). Defining the microglia response during the time course of chronic neurodegeneration. J. Virol.90, 3003–3017. 10.1128/jvi.02613-15
278
Walsh D. M. Selkoe D. J. (2007). A β oligomers—a decade of discovery. J. Neurochem.101, 1172–1184. 10.1111/j.1471-4159.2006.04426.x
279
Wang C. Yue H. Hu Z. Shen Y. Ma J. Li J. et al . (2020). Microglia mediate forgetting via complement-dependent synaptic elimination. Science367, 688–694. 10.1126/science.aaz2288
280
Waxman S. G. (1980). Determinants of conduction velocity in myelinated nerve fibers. Muscle Nerve3, 141–150. 10.1002/mus.880030207
281
Weinhard L. di Bartolomei G. Bolasco G. Machado P. Schieber N. L. Neniskyte U. et al . (2018). Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nat. Commun.9:1228. 10.1038/s41467-018-03566-5
282
Weller J. Budson A. (2018). Current understanding of Alzheimer’s disease diagnosis and treatment. F1000Res.7:1161. 10.12688/f1000research.14506.1
283
Wilkaniec A. Gąssowska-Dobrowolska M. Strawski M. Adamczyk A. Czapski G. A. (2018). Inhibition of cyclin-dependent kinase 5 affects early neuroinflammatory signalling in murine model of amyloid β toxicity. J. Neuroinflammation15:1. 10.1186/s12974-017-1027-y
284
Wu J. Bie B. Yang H. Xu J. J. Brown D. L. Naguib M. (2013). Activation of the CB2 receptor system reverses amyloid-induced memory deficiency. Neurobiol. Aging34, 791–804. 10.1016/j.neurobiolaging.2012.06.011
285
Wu L. M. Williams A. Delaney A. Sherman D. L. Brophy P. J. (2012). Increasing internodal distance in myelinated nerves accelerates nerve conduction to a flat maximum. Curr. Biol.22, 1957–1961. 10.1016/j.cub.2012.08.025
286
Xu H. Gelyana E. Rajsombath M. Yang T. Li S. Selkoe D. (2016). Environmental enrichment potently prevents microglia-mediated neuroinflammation by human amyloid β-protein oligomers. J. Neurosci.36, 9041–9056. 10.1523/JNEUROSCI.1023-16.2016
287
Yeh F. L. Hansen D. V. Sheng M. (2017). TREM2, microglia, and neurodegenerative diseases. Trends Mol. Med.23, 512–533. 10.1016/j.molmed.2017.03.008
288
Yoon G. Kim Y. K. Song J. (2020). Glucagon-like peptide-1 suppresses neuroinflammation and improves neural structure. Pharmacol. Res.152:104615. 10.1016/j.phrs.2019.104615
289
Yoshiyama Y. Higuchi M. Zhang B. Huang S. M. Iwata N. Saido T. C. et al . (2007). Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron53, 337–351. 10.1016/j.neuron.2007.01.010
290
Yuan P. Condello C. Keene C. D. Wang Y. Bird T. D. Paul S. M. et al . (2016). TREM2 haplodeficiency in mice and humans impairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy. Neuron90, 724–739. 10.1016/j.neuron.2016.05.003
291
Yuste R. Bonhoeffer T. (2001). Morphological changes in dendritic spines associated with long-term synaptic plasticity. Annu. Rev. Neurosci.24, 1071–1089. 10.1146/annurev.neuro.24.1.1071
292
Zhao Z. Nelson A. R. Betsholtz C. Zlokovic B. V. (2015). Establishment and dysfunction of the blood-brain barrier. Cell163, 1064–1078. 10.1016/j.cell.2015.10.067
293
Zhou L. Flores J. Noël A. Beauchet O. Sjöström P. J. LeBlanc A. C. (2019). Methylene blue inhibits Caspase-6 activity and reverses Caspase-6-induced cognitive impairment and neuroinflammation in aged mice. Acta Neuropathol. Commun.7:210. 10.1186/s40478-019-0856-6
294
Zhou L. McInnes J. Wierda K. Holt M. Herrmann A. G. Jackson R. J. et al . (2017). Tau association with synaptic vesicles causes presynaptic dysfunction. Nat. Commun.8:15295. 10.1038/ncomms15295
295
Zuo Y. Lin A. Chang P. Gan W. B. (2005). Development of long-term dendritic spine stability in diverse regions of cerebral cortex. Neuron46, 181–189. 10.1016/j.neuron.2005.04.001
Summary
Keywords
synaptic plasticity, glial plasticity, Alzheimer’s disease, tauopathy, therapeutics
Citation
Koller EJ and Chakrabarty P (2020) Tau-Mediated Dysregulation of Neuroplasticity and Glial Plasticity. Front. Mol. Neurosci. 13:151. doi: 10.3389/fnmol.2020.00151
Received
09 June 2020
Accepted
20 July 2020
Published
21 August 2020
Volume
13 - 2020
Edited by
Isabel Lastres-Becker, Autonomous University of Madrid, Spain
Reviewed by
Michiyo Iba, National Institute on Aging (NIH), United States; Gustavo Fernando Pigino, Medical Research Institute Mercedes and Martín Ferreyra (INIMEC), Argentina
Updates
Copyright
© 2020 Koller and Chakrabarty.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paramita Chakrabarty pchakrabarty@ufl.edu
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.