 Worlanyo Tashie
Worlanyo Tashie Harry P. de Koning
Harry P. de Koning Nancy O. Duah-Quashie
Nancy O. Duah-Quashie Neils B. Quashie
Neils B. Quashie- 1West African Centre for Cell Biology of Infectious Pathogens, Department of Biochemistry, Cell and Molecular Biology, College of Basic and Applied Sciences, University of Ghana, Accra, Ghana
- 2School of Infection and Immunity, College of Medical, Veterinary and Life Sciences, University of Glasgow, Glasgow, United Kingdom
- 3Department of Epidemiology, Noguchi Memorial Institute for Medical Research, University of Ghana, Accra, Ghana
- 4Centre for Tropical Clinical Pharmacology and Therapeutics, University of Ghana Medical School, Accra, Ghana
Introduction: The increasing resistance of Plasmodium falciparum to existing antimalarial drugs drives the urgent need for novel therapeutic strategies. The purine salvage pathway in P. falciparum is essential for the parasite’s survival due to its complete reliance on host-derived purines for nucleic acid synthesis and other essential processes. Although the purine salvage system has been intensively researched, no purine-based antimalarial drugs have been taken into preclinical development. The current study evaluated the chemotherapeutic potential of some purine nucleobase analogues against P. falciparum.
Methods: In vitro sensitivity assays were conducted using the 72-hour SYBR Green drug assay on laboratory-adapted P. falciparum strains 3D7 and Dd2. The most potent nucleobase analogues were docked into PfENT1 using the PyRx software suite.
Results: The analogues 8-azaguanine, 7-deazaguanine, and 6-thioguanine exhibited average EC50 values of 1.71 µM, 14.9 µM and 15.7 µM, respectively, for 3D7 and 5.2 µM, 16.3 µM and 18.6 µM, respectively, for the Dd2 strain, and subsequently tested against field isolates of P. falciparum. These ex vivo tests showed EC50 values ranging from 0.5 - 4.5 µM for 8-azaguanine, 3.8 - 12.3 µM for 7-deazaguanine, and 4.1 - 15.0 µM for 6-thioguanine. To understand their cellular targeting, molecular docking of the same analogues was performed using the structure of P. falciparum Equilibrative Nucleoside Transporter 1 (PfENT1). This demonstrated that guanine, 8-azaguanine and 7-deazaguanine formed five hydrogen bonds each with the same amino acid residues of PfENT1, whereas 6-thioguanine’s orientation allowed only two hydrogen bonds with PfENT1. The binding pose of inosine was different from these nucleobases.
Discussion: These findings highlight the potential of guanine-based scaffolds, particularly 8-azaguanine and 7-deazaguanine, as promising leads for purine-based antimalarial drug development and the versatility of the PfENT1 transporter in the uptake of purine antimetabolites.
1 Introduction
Malaria remains a major global health challenge, with an estimated 263 million cases and 597,000 deaths reported in 2023 alone (WHO, 2024). Africa bears the highest burden, accounting for approximately 94% of all cases and 95% of deaths due to malaria. Children aged below 5 years accounted for 76% of the deaths due to malaria recorded from Africa in 2023. Among the five Plasmodium species that infect humans, Plasmodium falciparum is the most virulent and is responsible for the majority of malaria-related deaths (Basu and Sahi, 2017; Phillips et al., 2017; WHO, 2024).
While antimalarial resistance is nothing new, the recent emergence of artemisinin partial resistance, which first emerged in South-East Asia and currently in parts of Africa, is particularly alarming and threatens ongoing efforts in the control and elimination of malaria (Balikagala et al., 2021; Ward et al., 2022; Assefa et al., 2024; Rosenthal et al., 2024; WHO, 2024). This necessitates the urgent need for novel therapeutic strategies to tackle the rising threat of drug-resistant malaria (Rosenthal et al., 2024).
One promising strategy lies in targeting the parasite’s absolute dependency on host-derived purines for survival. P. falciparum cannot synthesize purines de novo and relies on salvaging them from host erythrocytes to support essential processes including DNA and RNA synthesis (Baldwin et al., 2007; Frame et al., 2015a). Purine import into the parasite, which is mediated by Equilibrative Nucleoside Transporters (PfENTs), is a critical step in the purine salvage pathway. PfENT1 serves as the primary route for purine uptake in P. falciparum and is essential for the parasite’s intraerythrocytic survival. This transporter has been validated as a promising target for antimalarial drug development (El Bissati et al., 2006; Quashie et al., 2008; Frame et al., 2015b; Zhang et al., 2018).
Purine analogues have demonstrated therapeutic efficacy in treating cancers, viral infections, and protozoan diseases (Muggia et al., 2012; Hulpia et al., 2019b; Pastuch-Gawołek et al., 2019; Natto et al., 2021; Huang et al., 2022; Fiuza et al., 2022). However, despite their broad utility, no purine-based antimalarial drugs have yet reached clinical use. Research has focused on nucleoside analogues including powerful transition state inhibitors of P. falciparum purine nucleoside phosphorylase (PfPNP) such as Immuncillin-H, which displays picomolar Kd for PfPNP but a much lower IC50 for parasite growth (Ting et al., 2005), presumably because of poor uptake of such analogues by the parasite. We prioritized nucleobases over nucleosides due to their direct role in the parasite’s purine metabolism. Oxopurine nucleobases are activated via phosphoribosylation by hypoxanthine-guanine-xanthine-phosphoribosyl transferase (PfHGXPRT), providing a 1-step incorporation into the nucleotide pool. In contrast, nucleosides, if they are to become nucleotides, must first be catabolized into their respective nucleobases by PfPNPs, which introduces an additional and potentially rate-limiting step (Cassera et al., 2011; Ducati et al., 2013; Cheviet et al., 2019). This streamlined activation pathway makes nucleobases promising candidates for therapeutic intervention in P. falciparum. This study evaluated the chemotherapeutic potential of purine nucleobase analogues against P. falciparum as a proof of concept.
2 Materials and methods
2.1 Ethical consideration
The study was approved by the Institutional Review Board of the Noguchi Memorial Institute for Medical Research (NMIMR-IRB CPN: 045/23-24) and the Ethical Review Committee of the Ho Municipal Hospital in Ghana. Before enrolment, the study protocol was thoroughly explained to participants or their parents/guardians, and written informed consent was obtained from each participant and/or their guardian.
2.2 Parasite lines and field isolates
Cryopreserved P. falciparum 3D7 (chloroquine-sensitive) and Dd2 (chloroquine-resistant) laboratory-adapted strains as well as randomly collected P. falciparum clinical isolates were used. The laboratory-adapted strains were acquired from the Department of Epidemiology, Noguchi Memorial Institute for Medical Research, University of Ghana. The clinical isolates were collected from individuals who accessed services at the Municipal Hospital in Ho, Ghana. Participants were randomly recruited based on the detection of P. falciparum mono-infection by microscopy and presented with a parasite density between 1,000 and 250,000 parasites/µL of blood. Individuals on any antimalarial medication at the time of recruitment were excluded. Approximately 2 mL of venous blood was drawn from each participant into Acid Citrate Dextrose vacutainers and immediately transported to the laboratory for analysis.
2.3 Purine analogues
The purine nucleobase analogues evaluated in this study were obtained commercially. These include the following: 1-methylguanine (Fluka Chemika), 2,6-diaminopurine (Sigma-Aldrich), 2,6-dichloropurine (Aldrich Chemistry), 2,8-dimercapto-6-hydroxypurine (Avocado research Chemicals), 2-chlorohypoxanthine (Aldrich Chemistry), 2-hydroxypurine (Sigma-Aldrich), 6-amino-7-deazapurine (Aldrich Chemistry), 6-dimethylaminopurine (Aldrich Chemistry), 6-methoxypurine (Sigma-Aldrich), 6-thioguanine (Thermo Scientific), 7-deazaguanine (Sigma Aldrich), 7-deazahypoxanthine (Alfa Aesar), 8-azaadenine (Sigma-Aldrich), 8-azaguanine (Sigma Aldrich), 8-azahypoxanthine (Sigma-Aldrich), and 9-methylguanine (Fluka Chemika).
2.4 Culturing of P. falciparum
The in vitro continuous culture of P. falciparum (3D7 and Dd2 strains) was carried out using the Trager and Jensen (1976) method with slight modifications. Fresh, non-infected human erythrocytes were infected with P. falciparum parasites and maintained in complete Roswell Park Memorial Institute (RPMI) 1640 media under standard conditions. The culture system consisted of a 5% hematocrit suspension of purified O Rh “D” positive non-infected human erythrocytes in RPMI 1640 media supplemented with the following components: 6 g/L of HEPES (Sigma), 2 g of dextrose (Sigma), 50 mg/mL hypoxanthine (Sigma), 2 g/L of sodium carbonate (NaHCO3; Sigma), and 5 g/L of Albumax II (Sigma). Cultures were incubated at 37°C in an atmosphere of 1% O2, 3% CO2, and 96% N2. The medium was changed daily, and parasitemia levels were monitored through the microscopic examination of Giemsa-stained thin blood smears following protocols described by Moll and colleagues (Moll et al., 2013). Parasite cultures were maintained at parasitemia levels below 5%.
2.5 In vitro drug sensitivity assay
The sensitivities of P. falciparum 3D7 and Dd2 strains to purine nucleobase analogues were assessed using the 72-hour SYBR Green drug assay, as described by Moll and colleagues (Moll et al., 2013), with slight modifications.
2.5.1 Compound preparation
Test compounds were initially dissolved in 100% dimethyl sulfoxide to yield stock concentrations of 20 mM. These stock solutions were diluted in incomplete RPMI 1640 medium to prepare working solutions with final in-well concentrations ranging from 0.39 µM to 100 µM.
2.5.2 Plate set-up
Drug assays were conducted in 96-well opaque, flat-bottomed plates. For each compound, 200 μL of twice the maximum starting concentration (2 × 100 µM) was added to the second column (well) of a row. Serial two-fold dilutions were then carried out across the row from the 2nd to the 10th well to generate a concentration gradient. The 11th well served as the negative control (no drug), while artesunate was used as a positive control.
2.5.3 Parasite culture and inoculation
P. falciparum cultures were adjusted to 1% parasitemia and 1% hematocrit using human erythrocytes. A volume of 100 μL of this parasite suspension was added to each well, resulting in a final volume of 200 μL per well.
2.5.4 Incubation
Plates were placed in a modular incubation chamber gassed with a mixture of 1% O2, 3% CO2, and 96% N2, and incubated at 37°C for 72 hours.
2.5.5 Fluorescence measurement
After incubation, 100 μL of lysis buffer containing SYBR Green was added to each well. The plates were gently mixed, covered with aluminum foil, and incubated at room temperature in the dark for one hour. Fluorescence was measured using a FLUOstar Omega microplate reader (BMG Labtech, USA) with excitation and emission filters set at 485 nm and 520 nm, respectively.
2.5.6 Data analysis
Fluorescence readings were exported to Microsoft Excel and analyzed in GraphPad Prism version 9.0 (GraphPad Software, USA). Dose-response curves were generated using a four-parameter logistic regression model (log[test compound] versus fluorescence) to determine EC50 values. Each assay was conducted in duplicate to ensure reproducibility.
2.6 Ex vivo drug sensitivity assay technique
The sensitivity of P. falciparum field isolates to purine nucleobase analogues was assessed using the 72-hour SYBR Green drug assay, essentially as described in Section 2.5. Field isolates were diluted 20-fold by mixing 1 part of the isolate with 19 parts of complete RPMI 1640 medium. The final-well concentrations of the test compounds ranged from 0.39 µM to 100 µM. All other steps, including the preparation of test compounds, plate set-up, incubation conditions, SYBR Green staining, fluorescence measurement and data analysis were identical to the in vitro assay protocol described in Section 2.5. Each compound was tested in duplicate to ensure reproducibility, and artesunate was included as an internal control.
2.7 Cytotoxicity assay
The cytotoxic effects of the potent purine analogues were evaluated on uninfected erythrocytes using a modified version of the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)-based colorimetric assay, as described by Appiah-Opong and colleagues (Appiah-Opong et al., 2022; Appiah-Opong et al., 2011).
Briefly, 100 µL of each purine analogue, at concentrations ranging from 6.25 µM to 100 µM, was dispensed into separate wells of a 96-well microtiter plate. Subsequently, 100 µL of uninfected erythrocytes at 2% hematocrit was added to each well. Control wells containing 100 µL of each purine analogue in 100 µL of complete RPMI 1640 medium were set up in parallel for background signal correction.
The plates were incubated at 37°C for 72 hours in a modular incubation chamber gassed with a mixture of 1% O2, 3% CO2, and 96% N2. After incubation, 40 µL of 2.5 mg/mL MTT solution (prepared in phosphate-buffered saline) was added to each well in the dark. The plates were gently mixed and incubated for an additional 2 hours at 37°C.
At the end of the incubation period, absorbance was measured at 570 nm using the FLUOstar Omega microplate reader. All experiments were performed in triplicate. The 50% cytotoxic concentrations (CC50) of each compound were determined by non-linear regression analysis. Selectivity indices were calculated as the ratio of CC50 to EC50.
2.8 Molecular docking studies
The structure-activity relationship of potent purine nucleobase analogues identified through in vitro sensitivity testing was investigated using molecular docking. Docking studies were conducted with PyRx-v0.8, a virtual screening tool which has an in-built AutoDock-Vina for docking simulations (Dallakyan and Olson, 2015). Within PyRx, AutoDock Vina was used to compute binding affinities and generate docking poses of ligands in the defined binding pocket of PfENT1. The tool automates several steps, including file format conversion, energy minimization, and execution of the Vina engine with user-specified parameters.
2.8.1 Preparation of protein target and ligands for docking studies
The cryo-electron microscopy (cryo-EM)-resolved 3D structure of PfENT1 in complex with nanobody 48 and inosine (PDB code: 7WN1) (Wang et al., 2023) was retrieved from the RCSB-PDB database in PDB format. The chemical structures of the ligands were drawn using ChemDraw (version 23.1.1) and subsequently imported into Chem3D (version 23.1.1) for 3D optimization. The structures were then saved in Structure-Data File format. Preparation and refining of the PfENT1 protein, such as removal of native ligand and assigning hydrogen polarities and charges were performed using UCSF ChimeraX, adhering to the protocol described by Meng and colleagues (Meng et al., 2023). This was followed by energy minimization and geometry optimization of the proteins’ structures using PyRx-v0.8. Using the in-built Open Babel in PyRx, all the ligand structures were converted to Autodock suitable PDBQT format, and their energy minimized using Universal Forcefield (UFF). The 2D structures of the most potent guanine derivatives identified in this study are shown in Figure 1.
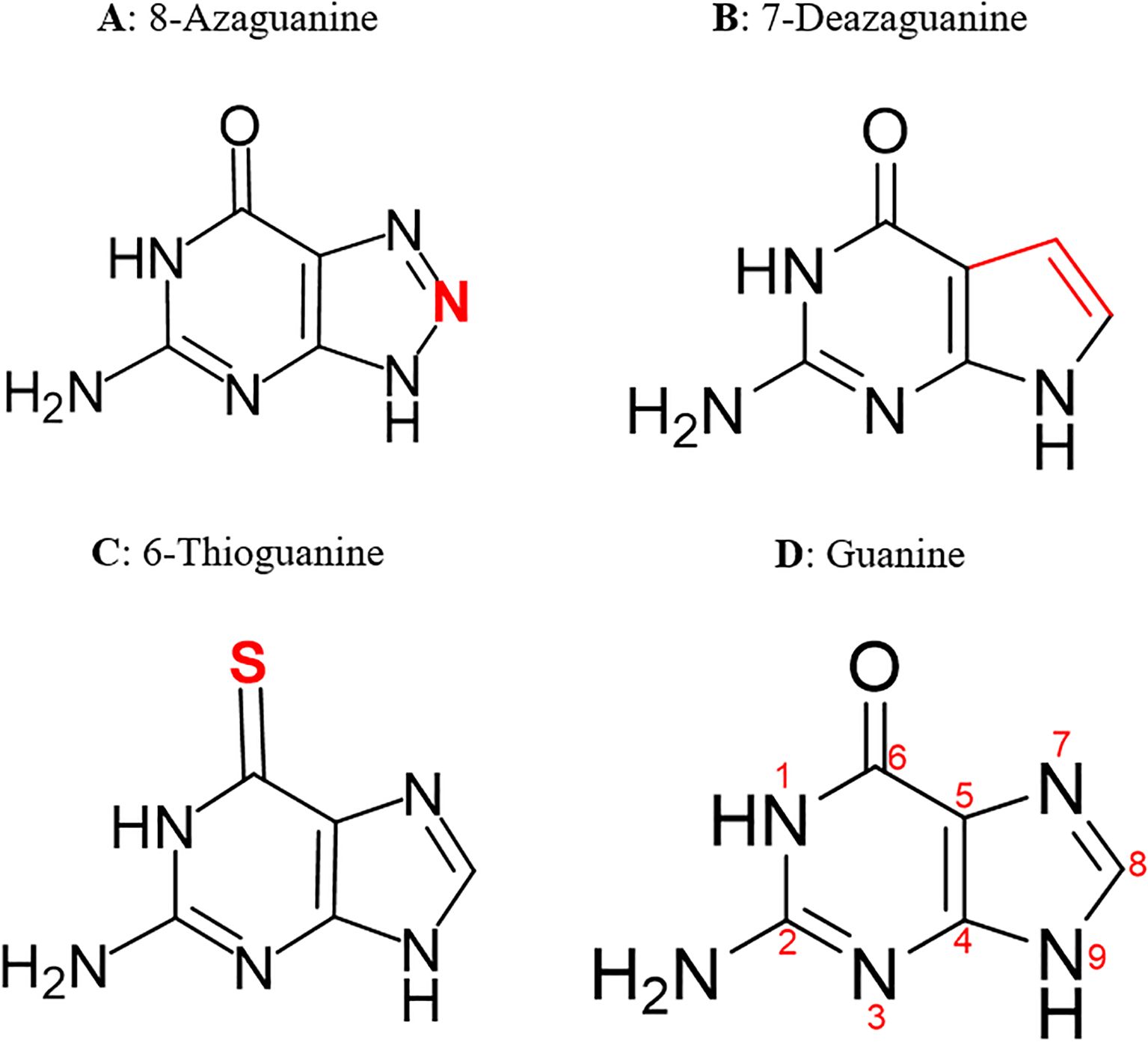
Figure 1. 2D structures of the most potent guanine derivatives. Structural modifications to the guanine scaffold are highlighted in red.
2.8.2 Molecular docking of PfENT1 with guanine derivatives
Before proceeding with ligand docking, a redocking procedure was performed to validate the docking protocol by reproducing the original ligand poses observed in the cryo-EM structure. Each ligand was individually docked into the binding site of the prepared PfENT1 protein. The binding site was predicted using the COACH-D server (Wu et al., 2018), and a grid box was defined with the following parameters, chosen to be sufficiently large to include the entire protein-binding site of PfENT1: a center of 117.515×119.996×116.537 Å, and dimensions of 69.9708×48.6471×53.2414 Å. Docking was performed using AutoDock Vina within PyRx, with the “exhaustiveness” parameter set to 8 to improve search thoroughness. All other parameters were maintained at their default settings Docking results were clustered based on the root mean square deviation criterion. Complexes with a root mean square deviation of 0.0 were selected for further analysis. The best protein-ligand poses were visualized using Discovery Studio 2021 (BIOVIA). Key interactions within the active site were analyzed to identify significant contacts between the ligands and critical amino acid residues anchoring the compounds.
3 Results
3.1 In vitro sensitivity assay of 3D7 and Dd2 strains using purine analogues
All test compounds were initially screened in vitro using the 72-hour SYBR Green assay. Analogues that demonstrated inhibitory activity against P. falciparum were subsequently validated in biologically independent replicate experiments. The EC50 values obtained from these replicates exhibited minimal variation, with no statistically significant differences observed between experiments (p > 0.05).
Among the tested purine nucleobase analogues, only four - 8-azaguanine, 7-deazaguanine, 6-thioguanine, and 2,6-dichloropurine - demonstrated activity against the in vitro growth of P. falciparum parasites. Their EC50 values were 1.71 µM, 14.9 µM, 15.7 µM and 45.5 µM, respectively, for the 3D7 strain, and 5.2 µM, 16.3 µM, 18.6 µM and 36.19 µM, respectively, for the Dd2 strain. There was a borderline significant difference (p = 0.025) in the sensitivities of the 3D7 and Dd2 strains to 8-azaguanine but not to the other purine nucleobases (Table 1) and we conclude that purine derivates are not cross-resistant with chloroquine. The graphical representation of the EC50 of the potent guanine derivatives obtained from the in vitro experiments is shown in Figure 2.
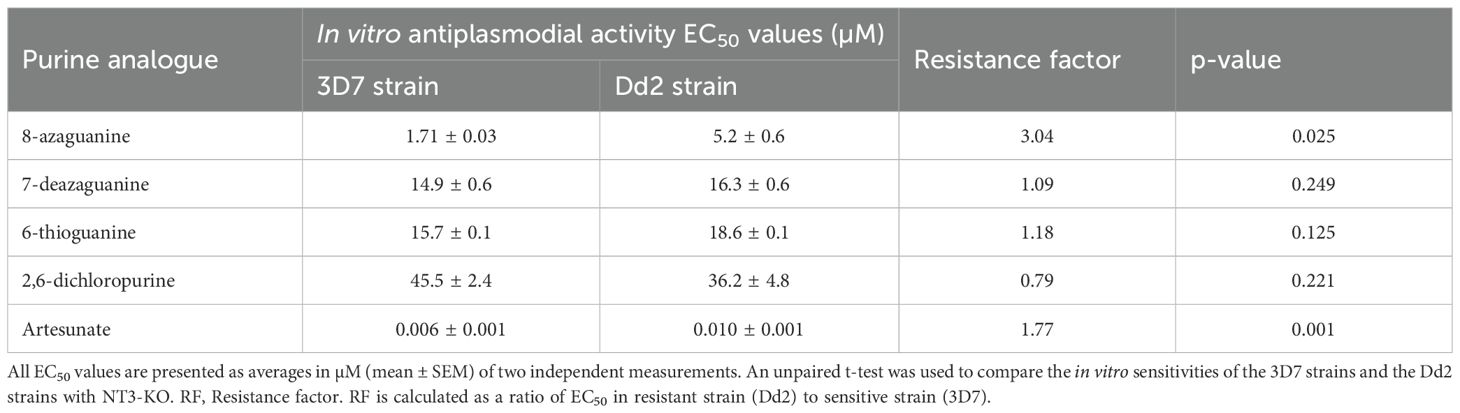
Table 1. Effect of test purine nucleobases on 3D7 and Dd2 strains of P. falciparum in vitro.
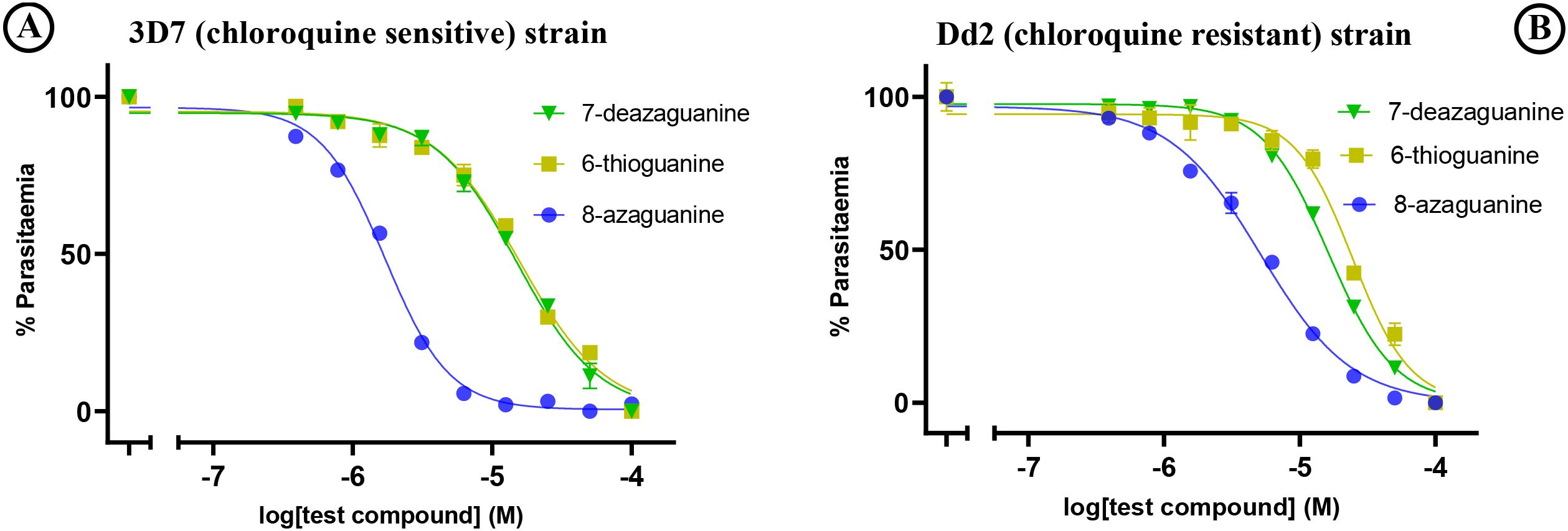
Figure 2. In vitro sensitivity testing of (A) 3D7 and (B) Dd2 strains against test purine nucleobase analogues. The graphs show a single representative experiment in duplicate; error bars are SEM.
3.2 Ex vivo testing of field isolates of P. falciparum
Following the initial screening of purine nucleobase analogues against 3D7 and Dd2 strains, only four compounds inhibited the in vitro growth of the parasites at low micromolar concentrations: 8-azaguanine, 6-thioguanine, 7-deazaguanine, and 2,6-dichloropurine. Notably, three of these active compounds shared a guanine scaffold and exhibited the most potent inhibitory effects. Therefore, based on these observations, the guanine derivatives were selected for further testing against four P. falciparum field isolates. 8-Azaguanine was the most potent analogue from the ex vivo testing, with EC50 values ranging from 0.5 - 4.5 µM across all isolates. This was followed by 7-deazaguanine, which had EC50 values ranging from 3.8 - 12.3 µM, whereas 6-thioguanine had a larger range of EC50 values on the field isolates, with EC50 values from 4.1 - 15.0 µM with a single isolate being slightly more sensitive to this analogue than to 7-deazaguanine. The ex vivo testing confirmed the efficacy of these three analogues against field isolates of P. falciparum and confirmed 8-azaguanine as the most potent of the analogues tested (Figure 3; Table 2).
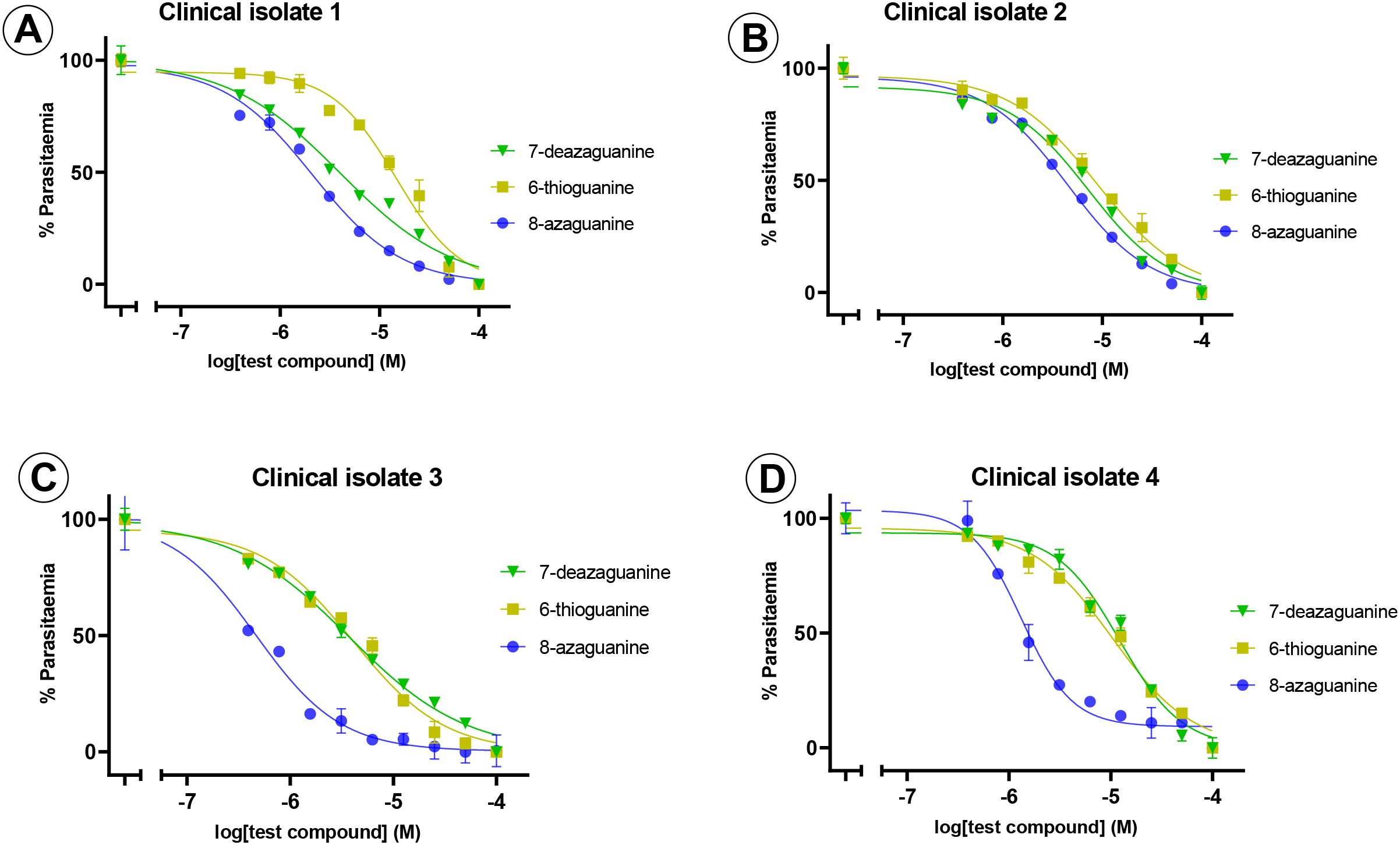
Figure 3. Ex vivo sensitivity testing of field isolates of P. falciparum against purine nucleobase analogues. The graphs show a single representative experiment in duplicate; error bars are SEM. Frames (A–D) show clinical isolates 1–4.
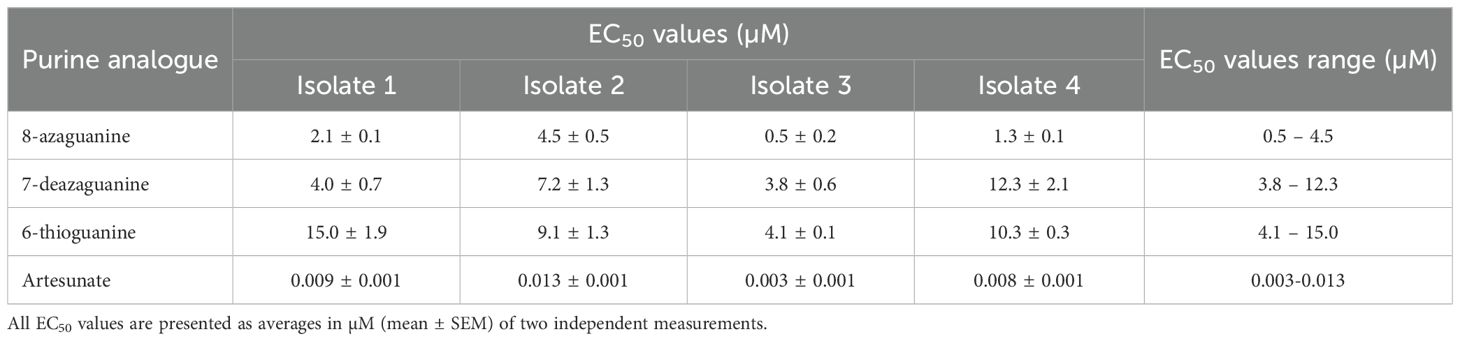
Table 2. Ex vivo results of the effect of purine nucleobases on P. falciparum field isolates.
3.3 Cytotoxicity and selectivity indices of potent purine analogues
All three potent guanine derivatives exhibited CC50 values above 50 µM on uninfected erythrocytes at the highest tested analogue concentration of 100 µM, suggesting acceptable safety profiles. 7-Deazaguanine demonstrated the lowest cytotoxicity, i.e. the highest CC50, 125.7 ± 2.7 µM (n=3). 8-Azaguanine showed the highest overall selectivity, with selectivity indices of 33.59 against the 3D7 strain and 11.05 against Dd2 strain, indicating strong antiplasmodial activity relative to its cytotoxicity. All three analogues demonstrated selectivity indices above the recommended threshold of 2 (Appiah-Opong et al., 2022) which indicates a favourable therapeutic potential (Table 3).
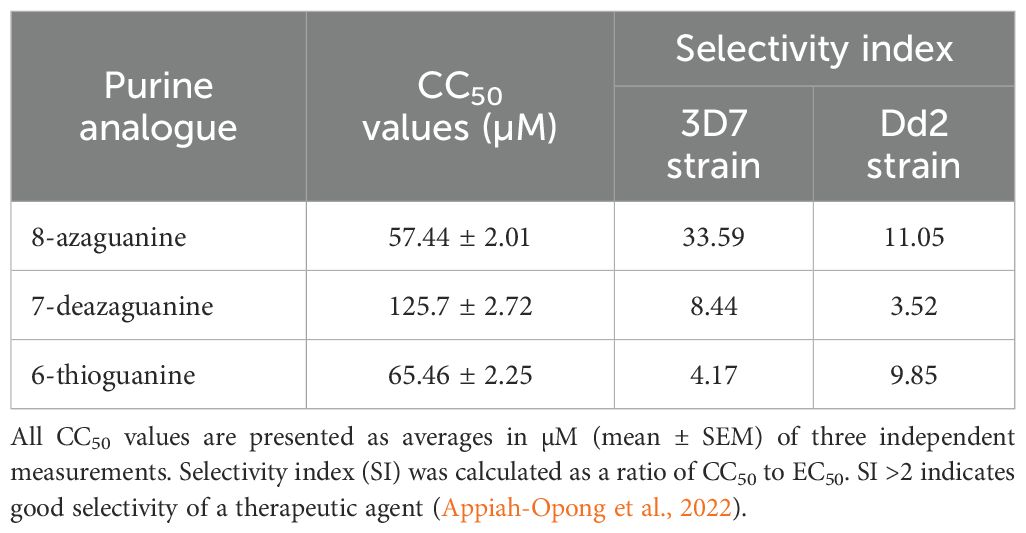
Table 3. Cytotoxicity and selectivity indices of potent purine analogues.
3.4 Molecular docking studies
To understand the protein-ligand interactions, the potent guanine derivatives identified from the in vitro drug sensitivity testing were further docked into PfENT1 protein.
3.4.1 Redocking to validate the docking procedure
Redocking was performed to confirm that the docking protocol could be used to accurately replicate the experimental poses of the ligand in the binding site, thereby validating the method for subsequent docking experiments with purine nucleobases. This process assessed whether the docking program could recreate the original cryo-EM resolved 3D structure poses of the ligands using PyRx.
Figure 4A illustrates the protein-ligand interactions in the cryo-EM structure of the PfENT1-inosine complex, retrieved from the RCSB-PDB database (Wang et al., 2023). Wang and colleagues (Wang et al., 2023) reported that Gln135 of PfENT1 forms a hydrogen bond pair with the inosine keto group and N1(H), while Trp53 and Asp287, form hydrogen bonds with the 5’ and 3’ hydroxy groups, respectively, of the ribose moiety, and Arg291 further engages the 3’ and/or 2’ hydroxy moieties. Other residues observed by Wang and colleagues (Wang et al., 2023) to be involved in nucleoside recognition include Phe283 and Leu393. Additionally, the authors reported the following residues to be within 6 angstroms of the inosine’s ribose group: Asn250, Thr253, Asn354, and Asn358. The redocking procedure confirmed these interactions. As shown in Figure 4B, Gln135 of PfENT1 formed the same pair of hydrogen bonds with the 6-keto and N1(H) positions of inosine. Asp287 also engaged 3’-OH in the redocking, and Arg291 forms a hydrogen bond with 2’-OH. The only significant difference between the cryo-EM structure and the redocking is that in the latter Trp53 appears to engage in π-stacking with the purine ring rather than a hydrogen bond with the 5’-OH of ribose; this difference depends on the orientation of this tryptophan’s indole ring system within the binding pocket, which in turn would depend on whether the energy gain from the 5’-OH hydrogen bond or the π-π interaction is stronger.
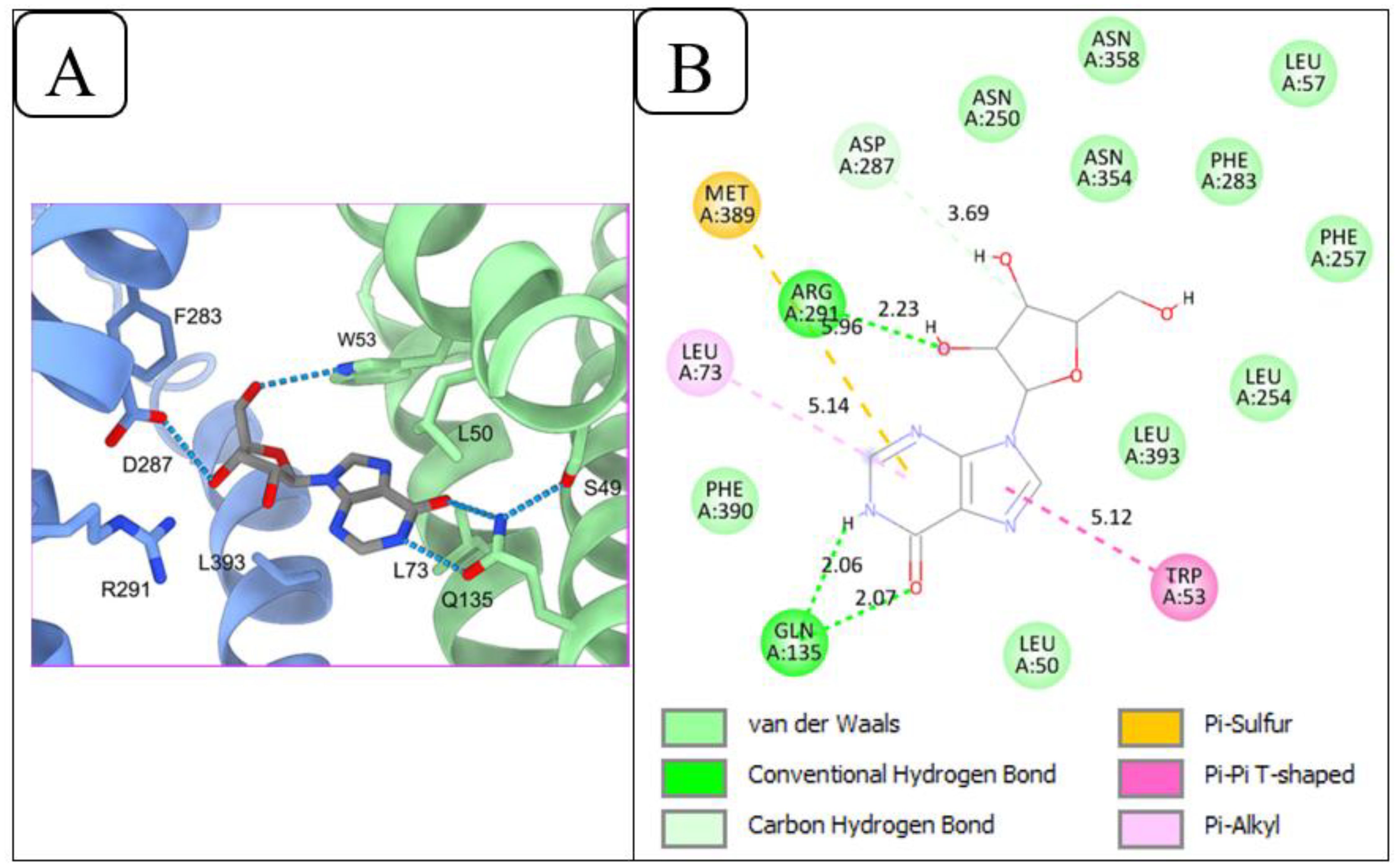
Figure 4. Redocking of PfENT1 and inosine to validate the docking procedure. (A) Inosine binding sites of PfENT1 from the cryo-EM solved 3D structure of the PfENT1-inosine complex, retrieved from the RCSB-PDB database. (B) Protein-ligand interactions of the redocked PfENT1 and inosine, performed using PyRx.
These findings show that the redocking process yields outputs close to the experimentally observed poses, validating the docking procedure for further protein-ligand interaction studies.
3.4.2 Molecular docking results of the guanine scaffolds
The interactions of 8-azaguanine and 7-deazaguanine with PfENT1 were similar. PfENT1 formed five (Rosenthal et al., 2024) hydrogen bonds with both 8-azaguanine and 7-deazaguanine via the following amino acid residues: Asn54, Ile157, Gly158, Gln284 and Asp287 (Figures 5A, B). Both ligands further made Van der Waals contact with PfENT1 through the amino acid residues Leu50, Trp53, Ile159, Ser160, Val162, Phe283, Gln285, and Phe288. Docking guanine into PfENT1 yielded protein-ligand interactions identical to the binding of 8-azaguanine and 7-deazaguanine (Figure 5C). Thus, the two most potent nucleobase analogues interacted identically with PfENT1, yet in a very different orientation from inosine, illustrated perhaps most clearly by the fact that Asp287 interacts with 3’-OH of inosine but with N(9)H in the guanine analogues, and Gln135, the only residue contributing H-bonds with the oxopurine base moiety in inosine (to 6-keto and N(1)H), is not involved at all in binding the guanine analogues. Instead the 6-keto group now forms a hydrogen bond with Asn54 and N(1)H with Ile157 – neither being involved in the binding of inosine. Interestingly, the 2-amine moiety of the guanine analogues, which does not exist in inosine, contributes two strong H-bonds, with Gly158 and Gln284, which also are not engaged in inosine binding. Clearly, the two oxopurine nucleobase analogues interact with PfENT1 very differently from the oxopurine nucleoside, with scarce overlap between the binding positions.
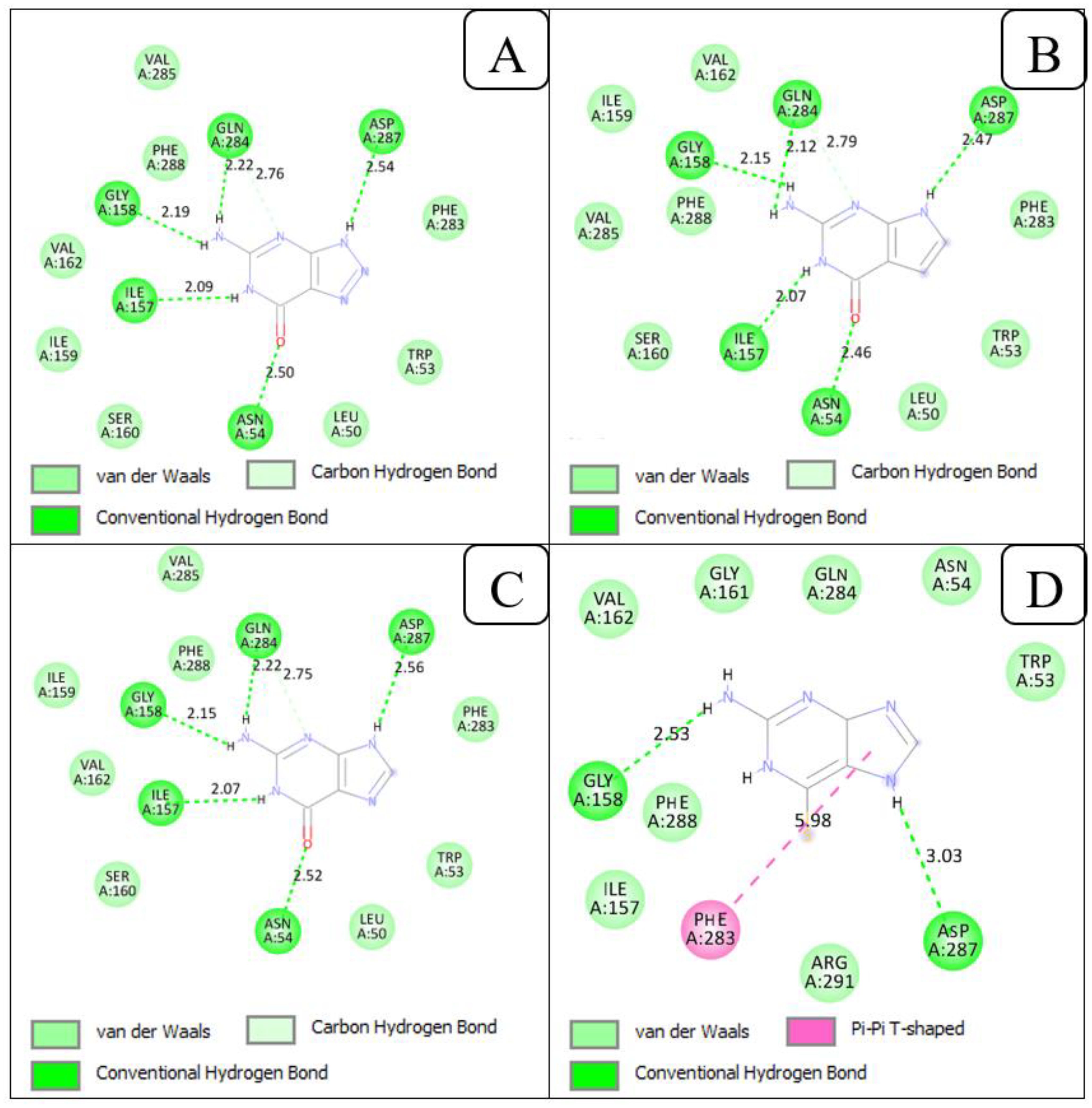
Figure 5. 2D view of the interactions of PfENT1 protein with the guanine scaffolds. (A) Protein-ligand interactions of PfENT1 and 8-azaguanine. (B) Protein-ligand interactions of PfENT1 and 7-deazaguanine. (C) Protein-ligand interactions of PfENT1 and guanine. (D) Protein-ligand interactions of PfENT1 and 6-thioguanine.
The orientation of 6-thioguanine has shifted again (Figure 5D), highlighting the importance of the Asn54 H-bond with the 6-keto oxygen for the positioning of guanine nucleobases in this transporter, as the thio moiety does not form strong hydrogen bonds (Civcir, 2001). The positional shift is further enabled by the dominant tautomeric form of 6-thioguanine including the protonation of N7 rather than N9 as is the case for 7-deazaguanine and 8-azaguanine (Contreras and Madariaga, 1998; Maciejczyk and Pyrka, 2023), resulting in the strong Asp287 H-bond with N (7)H in 6-thioguanine rather than N (Baldwin et al., 2007)H. The change in position results in the further loss of the H-bonds by Ile157 (to N(1)H) and Gln284 to the 2-amine but the binding pose is stabilized by a new π-π-hydrophobic interaction with the Phe283 side chain, just as adenine is stabilized by Phe aromatic interactions in the binding pocket of the Trypanosoma brucei TbAT1/P2 transporter (Munday et al., 2015), and Gly158 maintains H-bond with the 2-amine. The contribution of the 2-amine moiety in the binding of guanine and its analogues is consistent with our previous observation of approximately 3-fold higher affinity by PfENT1 for guanine than to hypoxanthine, just like the bonding by five strong H-bonds (distance between 2.56 and 2.07 Å) is consistent with the extraordinarily low Ki value for guanine of 0.11 ± 0.01 µM (Quashie et al., 2008), corresponding to a Gibbs free energy of interaction (ΔG0) of −39.7 kJ/mol. In addition to the hydrogen bonds, the amino acid residues, Trp53, Asn54, Ile157, Gly161, Val162, Gln284, Phe288 and Arg291 increased the strength of the interaction between PfENT1 and 6-thioguanine through Van der Waals interactions.
4 Discussion
Purine analogues have been utilized in clinical settings for the treatment of cancers and viral infections (Muggia et al., 2012; Jordheim et al., 2013; Pastuch-Gawołek et al., 2019), and are under development against certain protozoan diseases (Hulpia et al., 2019b; Fiuza et al., 2022). Despite significant efforts to target the purine transport system in the development of antimalarial drugs (Quashie et al., 2010; Frame et al., 2015a; Zhang et al., 2018; Sosa et al., 2019), no purine-based antimalarial drugs are currently available in clinical practice. Here, we evaluated the chemotherapeutic potential of some purine nucleobase analogues against P. falciparum isolates and use them to explore the issues of purine drug uptake in Plasmodium.
Our findings revealed that guanine-derived analogues, specifically 8-azaguanine, 7-deazaguanine, and 6-thioguanine, inhibited the in vitro growth of P. falciparum at low micromolar concentrations. The potent activity of these guanine derivatives against P. falciparum is likely mediated through one of several mechanisms. These may include inhibition of PfENTs, or disruption of key enzymes involved in purine salvage metabolism within the parasite, or these compounds may act as “subversive” substrates for the parasite’s purine salvage enzymes, resulting in incorporation into DNA or RNA (Galmarini et al., 2002; Pruijssers and Denison, 2019; Rehan et al., 2019; Thomson and Lamont, 2019; Borbone et al., 2021; Kamzeeva et al., 2023). This pathway would commence with the conversion to monophosphate nucleotides by PfHGXPRT followed by further enzymatic steps to the analogues of (2’-deoxy)ATP and/or (2’-deoxy)GTP (Cassera et al., 2011; Ducati et al., 2013; Cheviet et al., 2019), which would eventually impair nucleic acid synthesis and/or structure and ultimately lead to parasite death.
The analogue 8-azaguanine was the most potent analogue identified in this study. 8-Azaguanine is an azapurine, and previous studies have demonstrated that azapurines are important analogues with a broad spectrum of anti-infective and antineoplastic activity (Hahn, 1979; Giorgi and Scartoni, 2009; Wang et al., 2016; Hou et al., 2020), acting by incorporation into RNA (Nelson et al., 1975). For instance, 8-azapurines possess a broad spectrum of biological activity and are studied for their interactions with many enzymes and receptors for their antitumour and antiviral activity (Giorgi and Scartoni, 2009; Wang et al., 2016; Kim et al., 2019). Despite the observed potency of 8-azaguanine and 6-thioguanine against P. falciparum parasites, a possible challenge with their direct usage as antimalarial drugs is their reported toxicities to human cells when used in cancer treatments (Berman et al., 1985; Wang et al., 2016; Kim et al., 2019; Tseligka et al., 2023). 8-Azaguanine and 6-thioguanine are approved drugs that have been previously used in the treatment of acute leukaemias (Colsky et al., 1955; Wang et al., 2016; Kim et al., 2019). However, both analogues are toxic to human cells, posing a challenge to their continuous usage in cancer treatment (Berman et al., 1985; Wang et al., 2016; Kim et al., 2019; Tseligka et al., 2023). 8-Azaguanine produces toxicity to human cells through its incorporation into RNA, whereas 6-thioguanine produces toxicity to human cells through its incorporation into DNA (Keough et al., 2006; Kim et al., 2019). It is, however, worth noting that the short-term administration of these potent guanine analogues for severe malaria cases is less likely to cause the toxicities associated with extended use, as seen with prolonged cancer treatment regimens. Moreover, an effective antimalarial dosage may stay well below what is required in cancer chemotherapy, especially when used in combination with another antimalarial agent. Thus, these potent guanine analogues against P. falciparum infection remain promising, especially as the starting point for further drug discovery efforts.
Cytotoxicity testing of the analogues on human erythrocytes indicated acceptable safety profiles, with 8-azaguanine exhibiting the highest selectivity and 7-deazaguanine demonstrating the lowest cytotoxicity. Thus, with a favourable toxicity profile, the 7-deazaguanine scaffold could become a preferred candidate for the development of purine-based antimalarial drugs just like 7-substituted, 7-deazapurine nucleosides have provided excellent scaffolds against cancer and viral infections (Perlíková and Hocek, 2017), and against both human and animal forms of African and American trypanosomiasis (Hulpia et al., 2019a; Hulpia et al., 2020; Fiuza et al., 2022; Mabille et al., 2022). In contrast to these other cell types, P. falciparum only incorporates nucleobases into their nucleotide pool, through PfHGXPRT, with nucleosides first degraded to oxopurine nucleobases (Cassera et al., 2011), hence the decision to focus on the nucleobase moieties in this study. However, given that the 7-deazanucleosides are also believed to be active in their nucleotide form, for instance through phosphorylation by adenosine kinase in kinetoplastid protozoa or mammalian cells (Perlíková and Hocek, 2017; Hulpia et al., 2019b), the nucleoside and and nucleobase prodrugs ultimately converge to the same active compound in cellulo.
Further exploration of the structure-activity relationships of the antimalarial activity of both compounds by medicinal chemistry, aided by mechanism of action and structural docking studies, should be able to improve the antimalarial activities of these potent guanine analogues and potentially reduce human cell toxicity: note, for instance, that 7-deazaadenosine (tubercidin) is toxic to human cells but most 7-substituted tubercidin analogues (Hulpia et al., 2019a) and 7-deazainosine derivatives (Hulpia et al., 2020) are not. Tubercidin and the 7-substituted tubercidin sangivamycine also display potent antiplasmodial activity but are not selective (Coomber et al., 1994). Thus, a more promising starting point for medicinal chemistry exploration may be to combine the active modifications to guanine, for instance, 6-thio,8-azaguanine, 6-thio,7-deazaguanine and 7-deaza,8-azaguanine, like combining the tubercidin and cordycepin scaffolds yielded 3’-deoxy,7-deazaadenosine, a highly potent trypanocide that is curative in vivo with low dose oral administration, even in models of cerebral sleeping sickness (Hulpia et al., 2019b). Importantly, it has already been established that 8-azaguanine and 6-thioguanine are very good substrates of PfHGXPRT, which shows 336 and 19.6-fold higher catalytic efficiency (kcat/Km), respectively, than the human HGPRT for these analogues, although it has >5-fold lower catalytic efficiency for guanine itself (Keough et al., 2006). This clearly shows that PfHGXPRT, performing the critical phosphoribosylation step from guanine (analogue) to GMP (analogue), can easily accommodate changes at oxopurine nucleobase position 6, and the 8-aza modification. Moreover, 7-deaza,8-azaguanine is the guanine analogue of allopurinol (7-deaza,8-azahypoxanthine), which is in use as an antileishmanial agent and not toxic to humans – it is widely used as treatment for gout – because it is not incorporated into the nucleotide pool by human purine metabolic enzymes (Day et al., 2007), but is incorporated into RNA in Leishmania and Trypanosoma species (Looker et al., 1986). Allopurinol is also an excellent substrates for protozoan nucleoside transporters (Al-Salabi et al., 2003; Natto et al., 2005). In Plasmodium, a combination with allopurinol improved the efficacy of quinine in acute complicated malaria (Sarma et al., 1998).
The limited amino acid sequence similarity between the PfENTs and hENTs (Arora et al., 2016) further provides the possibility of optimizing these potent analogues to improve their efficacy and selectivity for PfENTs over the hENTs. More important than the sequence dissimilarity, ENT transporters have been widely documented to have highly different and exclusive substrate selectivities even within the same organism. For example, in Leishmania species, the NT1.1 and NT1.2 transporters are selective for adenosine and uridine, whereas NT2 recognises only the oxopurine nucleosides inosine and guanosine (Aldfer et al., 2022), despite 32% identity/52% similarity on amino acid sequence. Indeed, a single amino acid replacement can alter the substrate or inhibitor preferences of a transport protein as shown for instance for the UapA nucleobase transporter of Aspergillus nidulans (Koukaki et al., 2005) and for the T. brucei pentamidine transporter TbAQP2 (Alghamdi et al., 2020).
To gain further insight into the interaction modes of the potent guanine derivatives with PfENTs, molecular docking studies were carried out. The docking of a cryo-EM-resolved 3D structure of PfENT1 (Wang et al., 2023) with the potent guanine derivatives identified in this study revealed that guanine, 8-azaguanine, 7-deazaguanine all formed five hydrogen bonds with the same amino acid residues of PfENT1. Hydrogen bonds, due to their specificity, are critical in biomolecular interactions and contribute significantly to the stability and selectivity of target-ligand complexes (Murali et al., 2023). The binding mode of guanine by PfENT1, then, appears to rely primarily on H-bonds with the 6-keto group, the 2-amine moiety and N1(H), i.e. the pyrimidine half of the purine ring, in addition to the H-bond between Asp287 and N (Baldwin et al., 2007)H. The fact that this is identical to the reported binding mode of the facilitative nucleobase transporter hFNT1 present in the human erythrocyte membrane (Wallace et al., 2002), apart from the N (Baldwin et al., 2007)H interaction, is a significant advantage in the design of effectively targeted guanine analogues against intra-erythrocytic P. falciparum. It contrasts the binding mode of the nucleobase transporters of the kinetoplastid species T. brucei (TbH2 (Wallace et al., 2002),, T. cruzi (TcrNT2 (Aldfer et al., 2024), and Leishmania major (LmajNBT1 (Al-Salabi et al., 2003),, all of which form very strong hydrogen bonds with N7.
The protein-ligand interactions of PfENT1 with these guanine derivatives highlighted some specific positions on the guanine ring that can be exploited in designing purine-based antimalarial drugs that are specific and selective for the parasite. With positions 1, 2, 6 and 9 in 8-azaguanine and 7-deazaguanine identified as essential for high affinity towards PfENT1, positions 3, 7, and 8 offer flexibility for potential structural optimization. These findings align with the observations of Wang and colleagues (Wang et al., 2023), who demonstrated that positions 1 and 6 of inosine’s nucleobase moiety are critical for binding, while positions 3, 7, and 8 exhibit flexibility for further optimization. For 6-thioguanine, which assumes a different orientation in the PfENT1 binding pocket, positions 1, 3, 6 and 9 are potentially available for modifications. Interestingly, inosine, guanine and 6-thioguanine each interact with a distinct set of amino acid residues in the binding pocket, showing an extraordinary versatility of substrate for an ENT-family transporter. Our data are completely consistent with the reports by multiple research groups that this carrier can accommodate both nucleosides and nucleobases, and oxopurines as well as the aminopurine nucleoside adenosine (Carter et al., 2000; Parker et al., 2000; Quashie et al., 2008). The structural studies presented here and by Wang and colleagues (Wang et al., 2023) provide the clearest rationale to date for these observations. For instance, the glutamine residue binding inosine with a pair of H-bonds with 6-keto and N1(H) would equally be able to bond with the 6-amine/N1 motif of adenosine. And although inosine binding involves H-bonds to the ribose hydroxy moieties, which would be expected to correlate with low affinity for nucleobases, this is compensated for by a shift of the purine ring position allowing the formation of several new H-bonds. The single substitution of the guanine keto group with a thio moiety initiated yet another orientation for energy optimization. All this demonstrates that PfENT1 is quintessentially suited as a conduit or target for purine antimetabolites, consistent with the observations of Frame and colleagues (Frame et al., 2015b).
While recent reports show that 7-deaza-7-substituted nucleosides possess extraordinarily potent and selective and antiprotozoal activity (Hulpia et al., 2019a; Hulpia et al., 2020; Natto et al., 2021; Fiuza et al., 2022; Lin et al., 2022), the antimalarial effects and/or selectivity of these nucleosides has been disappointing (Serge Van Calenbergh and Guy Caljon, unpublished). However, we stress that this is most likely because, uniquely in Plasmodium, nucleosides must first be broken down by PfPNPs before being phosphoribosylated into the nucleoside pool as nucleotides (Cheviet et al., 2019). This adds the necessity to accommodate the structure-activity relationships of additional enzymes to the compound design, and the different structure-activity relationships may be mutually exclusive. Thus, our results would justify an exploration of the antimalarial activity of 7-deaza,7-substituted oxopurine nucleobases for antimalarial activity similar to those of the corresponding nucleosides against many other protozoa.
In conclusion, the guanine derivatives 7-deazaguanine, 6-thioguanine, and particularly 8-azaguanine demonstrated significant inhibitory activity against the in vitro growth of P. falciparum at low micromolar concentrations. Our findings suggest that these guanine derivatives hold promise as potential starting points for the development of purine-based antimalarial drugs. Employing medicinal chemistry approaches to optimize these analogues could enhance their antimalarial efficacy and their selectivity for PfENT1. Further studies are necessary to elucidate the mechanisms through which these potent guanine derivatives and/or their metabolites inhibit P. falciparum growth. In addition, site-directed mutagenesis of PfENT1 could be used to validate the predicted binding interactions identified in this study and provide deeper insights into the structure-activity relationships of these analogues.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Institutional Review Board of the Noguchi Memorial Institute for Medical Research. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
WT: Methodology, Investigation, Writing – original draft, Formal Analysis, Data curation. HK: Writing – original draft, Supervision, Conceptualization, Resources, Methodology, Formal Analysis, Writing – review & editing. ND-Q: Supervision, Writing – review & editing, Formal Analysis, Validation. NQ: Supervision, Conceptualization, Writing – original draft, Funding acquisition, Project administration, Resources.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. Worlanyo Tashie was supported by a WACCBIP-World Bank ACE PhD fellowship (WACCBIP+NCDs: Awandare).
Acknowledgments
We extend our sincere gratitude to the members of Lab 121 at the Department of Epidemiology, Noguchi Memorial Institute for Medical Research - Mr. Felix Zoiku, Mr. Prince A. Fordjour, Ms. Nana Aba Ennuson, Ms. Selassie Bruku, Mr. Philip Opoku-Agyeman, Mr. Prince Donkor, Dr. Sena Matrevi, and Dr. Kwesi Tandoh - for their insightful discussions, supportive environment, and for sharing their expertise in various laboratory techniques. We are also grateful to members of the Harry De Koning Laboratory at the University of Glasgow, especially Mr. Hamza A. A. Elati, Mr. Jamal I. Asseri, Ms. Tahani A. AlSiari, Ms. Maha Aloraini, and Mr. Mustafa M. Aldfer, for generously sharing their expertise in various laboratory techniques. We gratefully acknowledge the West African Centre for Cell Biology of Infectious Pathogens (WACCBIP) for funding this research through the WACCBIP-World Bank African Centres of Excellence PhD Fellowship (WACCBIP+NCDs: Awandare).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
Cryo-EM, Cryo electron microscopy; PfENT, P. falciparum equilibrative nucleoside transporters; PfHGXPRT, P. falciparum hypoxanthine-guanine-xanthine-phosphoribosyl transferase; PfPNP, P. falciparum purine nucleoside phosphorylases; RPMI, Roswell Park Memorial Institute; SI, Selectivity index; CC50, 50% cytotoxic concentrations.
References
Aldfer M. M., AlSiari T. A., Elati H. A. A., Natto M. J., Alfayez I. A., Campagnaro G. D., et al. (2022). Nucleoside transport and nucleobase uptake null mutants in leishmania mexicana for the routine expression and characterization of purine and pyrimidine transporters. Int. J. Mol. Sci. 23, 8139. doi: 10.3390/ijms23158139
Aldfer M. M., Hulpia F., van Calenbergh S., and De Koning H. P. (2024). Mapping the transporter-substrate interactions of the Trypanosoma cruzi NB1 nucleobase transporter reveals the basis for its high affinity and selectivity for hypoxanthine and guanine and lack of nucleoside uptake. Mol. Biochem. Parasitol. 258, 111616. doi: 10.1016/j.molbiopara.2024.111616
Alghamdi A. H., Munday J. C., Campagnaro G. D., Gurvic D., Svensson F., Okpara C. E., et al. (2020). Positively selected modifications in the pore of TbAQP2 allow pentamidine to enter Trypanosoma brucei. Elife. 9, e56416. doi: 10.7554/elife.56416
Al-Salabi M. I., Wallace L. J. M., and de Koning H. P. (2003). A Leishmania major nucleobase transporter responsible for allopurinol uptake is a functional homolog of the Trypanosoma brucei H2 transporte. R. Mol. Pharmacol. 63, 814–820. doi: 10.1124/mol.63.4.814
Appiah-Opong R., Agyemang K., Dotse E., Atchoglo P., Owusu K. B., Aning A., et al. (2022). Anti-plasmodial, cytotoxic and antioxidant activities of selected Ghanaian medicinal plants. J. Evid Based Integr. Med., 27, 1–8. doi: 10.1177/2515690X211073709
Appiah-Opong R., Nyarko A. K., Dodoo D., Gyang F. N., Koram K. A., and Ayisi N. K. (2011). Antiplasmodial activity of extracts of Tridax procumbens and Phyllanthus amarus in in vitro Plasmodium falciparum culture systems. Ghana Med. J. 45, 143–150.
Arora A., Deniskin R., Sosa Y., Nishtala S. N., Henrich P. P., Kumar T. R., et al. (2016). Substrate and inhibitor specificity of the plasmodium berghei equilibrative nucleoside transporter type 1. Mol. Pharmacol. 89, 678–685. doi: 10.1124/mol.115.101386
Assefa A., Fola A. A., and Tasew G. (2024). Emergence of Plasmodium falciparum strains with artemisinin partial resistance in East Africa and the Horn of Africa: is there a need to panic? Malaria J. 23, 34. doi: 10.1186/s12936-024-04848-8
Baldwin S. A., McConkey G. A., Cass C. E., and Young J. D. (2007). Nucleoside transport as a potential target for chemotherapy in malaria. Curr. Pharm. Des. 13, 569–580. doi: 10.2174/138161207780162845
Balikagala B., Fukuda N., Ikeda M., Katuro O. T., Tachibana S. I., Yamauchi M., et al. (2021). Evidence of artemisinin-resistant malaria in africa. N Engl. J. Med. 385, 1163–1171. doi: 10.1056/nejmoa2101746
Basu S. and Sahi P. K. (2017). Malaria: an update. Indian J. Pediatrics. 84, 521–528. doi: 10.1007/s12098-017-2332-2
Berman J. J., Tong C., and Williams G. M. (1985). Toxicity of 6-thioguanine and 8-azaguanine to non-dividing liver cell cultures. Cell Biol. Toxicol. 1, 67–73. doi: 10.1007/bf00717792
Borbone N., Piccialli G., Roviello G. N., and Oliviero G. (2021). Nucleoside analogs and nucleoside precursors as drugs in the fight against SARS-coV-2 and other coronaviruses. Molecules. 26, 986. doi: 10.3390/molecules26040986
Carter N. S., Ben Mamoun C., Liu W., Silva E. O., Landfear S. M., Goldberg D. E., et al. (2000). Isolation and functional characterization of the PfNT1 nucleoside transporter gene from Plasmodium falciparum. J. Biol. Chem. 275, 10683–10691. doi: 10.1074/jbc.275.14.10683
Cassera B. M., Zhang Y., Hazleton Z. K., and Schramm L. V. (2011). Purine and pyrimidine pathways as targets in plasmodium falciparum. Curr. Topics Medicinal Chem. 11, 2103–2115. doi: 10.2174/156802611796575948
Cheviet T., Lefebvre-Tournier I., Wein S., and Peyrottes S. (2019). Plasmodium purine metabolism and its inhibition by nucleoside and nucleotide analogues. J. Med. Chem. 62, 8365–8391. doi: 10.1021/acs.jmedchem.9b00182
Civcir P. U. (2001). Tautomerism of 6-thioguanine in the gas and aqueous phases using AM1 and PM3 methods. J. Mol. Structure: THEOCHEM. 536, 161–171. doi: 10.1016/s0166-1280(00)00636-9
Colsky J., Meiselas L. E., Rosen S. J., and Schulman I. (1955). Response of patients with leukemia to 8-azaguanine. Blood. 10, 482–492. doi: 10.1182/blood.v10.5.482.482
Contreras J. G. and Madariaga S. T. (1998). Azaguanine: A theoretical study of its tautomerism and protonation in the gas phase and aqueous solution. Bioorganic Chem. 26, 345–355. doi: 10.1006/bioo.1998.1113
Coomber D. W., O’Sullivan W. J., and Gero A. M. (1994). Adenosine analogues as antimetabolites against Plasmodium falciparum malaria. Int. J. Parasitol. 24, 357–365. doi: 10.1016/0020-7519(94)90083-3
Dallakyan S. and Olson A. J. (2015). Small-molecule library screening by docking with PyRx. Methods Mol. Biol. 1263, 243–250. doi: 10.1007/978-1-4939-2269-7_19
Day R. O., Graham G. G., Hicks M., McLachlan A. J., Stocker S. L., and Williams K. M. (2007). Clinical pharmacokinetics and pharmacodynamics of allopurinol and oxypurinol. Clin. Pharmacokinet. 46, 623–644. doi: 10.2165/00003088-200746080-00001
Ducati R. G., Namanja-Magliano H. A., and Schramm V. L. (2013). Transition-state inhibitors of purine salvage and other prospective enzyme targets in malaria. Future Medicinal Chem. 5, 1341–1360. doi: 10.4155/fmc.13.51
El Bissati K., Zufferey R., Witola W. H., Carter N. S., Ullman B., and Ben Mamoun C. (2006). The plasma membrane permease PfNT1 is essential for purine salvage in the human malaria parasite Plasmodium falciparum. Proc. Natl. Acad. Sci. 103, 9286–9291. doi: 10.1073/pnas.0602590103
Fiuza L. F. d. A., Batista D. G. J., Girão R. D., Hulpia F., Finamore-Araújo P., Aldfer M. M., et al. (2022). Phenotypic Evaluation of Nucleoside Analogues against Trypanosoma cruzi Infection: In Vitro and In Vivo Approaches. Molecules. 27, 8087. doi: 10.3390/molecules27228087
Frame I. J., Deniskin R., Arora A., and Akabas M. H. (2015a). Purine import into malaria parasites as a target for antimalarial drug development. Ann. N Y Acad. Sci. 1342, 19–28. doi: 10.1111/nyas.12568
Frame I. J., Deniskin R., Rinderspacher A., Katz F., Deng S. X., Moir R. D., et al. (2015b). Yeast-based high-throughput screen identifies Plasmodium falciparum equilibrative nucleoside transporter 1 inhibitors that kill malaria parasites. ACS Chem. Biol. 10, 775–783. doi: 10.1021/cb500981y
Galmarini C. M., Mackey J. R., and Dumontet C. (2002). Nucleoside analogues and nucleobases in cancer treatment. Lancet Oncol. 3, 415–424. doi: 10.1016/s1470-2045(02)00788-x
Giorgi I. and Scartoni V. (2009). 8-Azapurine nucleus: A versatile Scaffold for different targets. Mini-Reviews Medicinal Chem. 9, 1367–1378. doi: 10.2174/138955709789957440
Hahn F. E. (1979). “Mechanism of action of antieukaryotic and antiviral compounds.” in Antibiotics;, ed. Hahn F. E. (Heidelberg: Springer-Verlag Berlin), 472. doi: 10.1007/978-3-642-46407-2
Hou F., Wan Y., Gan Q., Xian M., and Huang W. (2020). Identification of 8-azaguanine biosynthesis-related genes provides insight into the enzymatic and non-enzymatic biosynthetic pathway for 1,2,3-triazole. Front. Bioeng Biotechnol. 8, 603514. doi: 10.3389/fbioe.2020.603514
Huang S., Bian Y., Huang C., and Miao L. (2022). Is monitoring of the intracellular active metabolite levels of nucleobase and nucleoside analogs ready for precision medicine applications? Eur. J. Drug Metab. Pharmacokinet. 47, 761–775. doi: 10.1007/s13318-022-00786-5
Hulpia F., Bouton J., Campagnaro G., Alfayez I., Mabille D., Maes L., et al. (2020). C6–O-alkylated 7-deaza inosine nucleoside analogues: Discovery of potent and selective anti-sleeping sickness agents. Eur. J. Medicinal Chem. 188, 112018. doi: 10.1016/j.ejmech.2019.112018
Hulpia F., Campagnaro G. D., Scortichini M., Van Hecke K., Maes L., de Koning H. P., et al. (2019a). Revisiting tubercidin against kinetoplastid parasites: Aromatic substitutions at position 7 improve activity and reduce toxicity. Eur. J. Medicinal Chem. 164, 689–705. doi: 10.1016/j.ejmech.2018.12.050
Hulpia F., Mabille D., Campagnaro G. D., Schumann G., Maes L., Roditi I., et al. (2019b). Combining tubercidin and cordycepin scaffolds results in highly active candidates to treat late-stage sleeping sickness. Nat. Commun. 10, 5564. doi: 10.1038/s41467-019-13522-6
Jordheim L. P., Durantel D., Zoulim F., and Dumontet C. (2013). Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat. Rev. Drug Discovery. 12, 447–464. doi: 10.1038/nrd4010
Kamzeeva P. N., Aralov A. V., Alferova V. A., and Korshun V. A. (2023). Recent advances in molecular mechanisms of nucleoside antivirals. Curr. Issues Mol. Biol. 45, 6851–6879. doi: 10.3390/cimb45080433
Keough D. T., Skinner-Adams T., Jones M. K., Ng A., Brereton I. M., Guddat L. W., et al. (2006). Lead compounds for antimalarial chemotherapy: Purine base analogs discriminate between human and P. Falciparum 6-oxopurine phosphoribosyltransferases. J. Medicinal Chem. 49, 7479–7486. doi: 10.1021/jm061012j
Kim N., Choi J., Song A., Choi W., Park H., Park S., et al. (2019). Direct potentiation of NK cell cytotoxicity by 8-azaguanine with potential antineoplastic activity. Int. Immunopharmacology. 67, 152–159. doi: 10.1016/j.intimp.2018.12.020
Koukaki M., Vlanti A., Goudela S., Pantazopoulou A., Gioule H., Tournaviti S., et al. (2005). The nucleobase-ascorbate transporter (NAT) signature motif in UapA defines the function of the purine translocation pathway. J. Mol. Biol. 350, 499–513. doi: 10.1016/j.jmb.2005.04.076
Lin C., Karalic I., Matheeussen A., Feijens P.-B., Hulpia F., Maes L., et al. (2022). Exploration of 6-methyl-7-(Hetero)Aryl-7-Deazapurine ribonucleosides as antileishmanial agents. Eur. J. Medicinal Chem. 237, 114367. doi: 10.1016/j.ejmech.2022.114367
Looker D. L., Marr J. J., and Berens R. L. (1986). Mechanisms of action of pyrazolopyrimidines in Leishmania donovani. J. Biol. Chem. 261, 9412–9415. doi: 10.1016/s0021-9258(18)67670-7
Mabille D., Ilbeigi K., Hendrickx S., Ungogo M. A., Hulpia F., Lin C., et al. (2022). Nucleoside analogues for the treatment of animal trypanosomiasis. Int. J. Parasitol. Drugs Drug Resist. 19, 21–30. doi: 10.1016/j.ijpddr.2022.05.001
Maciejczyk M. and Pyrka M. (2023). Tautomeric equilibrium and spectroscopic properties of 8-azaguanine revealed by quantum chemistry methods. Eur. Biophys. J. 52, 545–557. doi: 10.1007/s00249-023-01672-x
Meng E. C., Goddard T. D., Pettersen E. F., Couch G. S., Pearson Z. J., Morris J. H., et al. (2023). UCSF ChimeraX: Tools for structure building and analysis. Protein Science. 32, e4792. doi: 10.1002/pro.4792
Moll K., Kaneko A., Scherf A., and Wahlgren M. (2013). Methods in malaria research. 6 ed (UK: EVIMalaR Glasgow).
Muggia F., Diaz I., and Peters G. J. (2012). Nucleoside and nucleobase analogs in cancer treatment: not only sapacitabine, but also gemcitabine. Expert Opin. Investigational Drugs 21, 403–408. doi: 10.1517/13543784.2012.666236
Munday J. C., Tagoe D. N., Eze A. A., Krezdorn J. A., Rojas López K. E., Alkhaldi A. A., et al. (2015). Functional analysis of drug resistance-associated mutations in the Trypanosoma brucei adenosine transporter 1 (TbAT1) and the proposal of a structural model for the protein. Mol. Microbiol. 96, 887–900. doi: 10.1111/mmi.12979
Murali M., Nair B., Vishnu V. R., Aneesh T. P., and Nath L. R. (2023). 2,4-Dihydroxycinnamic acid as spike ACE2 inhibitor and apigenin as RdRp inhibitor in Nimbamritadi Panchatiktam Kashayam against COVID-19: an in silico and in vitro approach. Mol. Diversity. 27, 2353–2363. doi: 10.1007/s11030-022-10552-z
Natto M. J., Hulpia F., Kalkman E. R., Baillie S., Alhejeli A., Miyamoto Y., et al. (2021). Deazapurine nucleoside analogues for the treatment of trichomonas vaginalis. ACS Infect. Dis. 7, 1752–1764. doi: 10.1021/acsinfecdis.1c00075
Natto M. J., Wallace L. J. M., Candlish D., Al-Salabi M. I., Coutts S. E., and de Koning H. P. (2005). Trypanosoma brucei: expression of multiple purine transporters prevents the development of allopurinol resistance. Exp. Parasitology. 109, 80–86. doi: 10.1016/j.exppara.2004.11.004
Nelson J. A., Carpenter J. W., Rose L. M., and Adamson D. J. (1975). Mechanisms of action of 6-thioguanine, 6-mercaptopurine, and 8-azaguanine. Cancer Res. 35, 2872–2878.
Parker M. D., Hyde R. J., Yao S. Y. M., Mcrobert L., Cass C. E., Young J. D., et al. (2000). Identification of a nucleoside/nucleobase transporter from Plasmodium falciparum, a novel target for anti-malarial chemotherapy. Biochem. J. 349, 67–75. doi: 10.1042/bj3490067
Pastuch-Gawołek G., Gillner D., Król E., Walczak K., and Wandzik I. (2019). Selected nucleos(t)ide-based prescribed drugs and their multi-target activity. Eur. J. Pharmacol. 865, 172747. doi: 10.1016/j.ejphar.2019.172747
Perlíková P. and Hocek M. (2017). Pyrrolo[2,3-d]pyrimidine (7-deazapurine) as a privileged scaffold in design of antitumor and antiviral nucleosides. Med. Res. Rev. 37, 1429–1460. doi: 10.1002/med.21465
Phillips M. A., Burrows J. N., Manyando C., van Huijsduijnen R. H., Van Voorhis W. C., and Wells T. N. C. (2017). Malaria. Nat. Rev. Dis. Primers. 3, 17050. doi: 10.1038/nrdp.2017.50
Pruijssers A. J. and Denison M. R. (2019). Nucleoside analogues for the treatment of coronavirus infections. Curr. Opin. Virol. 35, 57–62. doi: 10.1016/j.coviro.2019.04.002
Quashie N., Dorin-Semblat D., Bray P., Biagini G., Doerig C., Ranford-Cartwright L., et al. (2008). A comprehensive model of purine uptake by the malaria parasite Plasmodium falciparum: Identification of four purine transport activities in intraerythrocytic parasites. Biochem. J. 411, 287–295. doi: 10.1042/bj20071460
Quashie N. B., Ranford-Cartwright L. C., and de Koning H. P. (2010). Uptake of purines in Plasmodium falciparum-infected human erythrocytes is mostly mediated by the human equilibrative nucleoside transporter and the human facilitative nucleobase transporter. Malaria J. 9, 36. doi: 10.1186/1475-2875-9-s2-p66
Rehan S., Shahid S., Salminen T. A., Jaakola V., and Paavilainen V. O. (2019). Current progress on equilibrative nucleoside transporter function and inhibitor design. SLAS Discovery. 24, 953–968. doi: 10.1177/2472555219870123
Rosenthal P. J., Asua V., Bailey J. A., Conrad M. D., Ishengoma D. S., Kamya M. R., et al. (2024). The emergence of artemisinin partial resistance in Africa: how do we respond? Lancet Infect. Dis. 24, e591–e600. doi: 10.1016/s1473-3099(24)00141-5
Sarma P. S., Mandal A. K., and Khamis H. J. (1998). Allopurinol as an additive to quinine in the treatment of acute complicated falciparum malaria. Am. J. Trop. Med. Hyg. 58, 454–457. doi: 10.4269/ajtmh.1998.58.454
Sosa Y., Deniskin R., Frame I. J., Steiginga M. S., Bandyopadhyay D., Graybill T. L., et al. (2019). Identification via a Parallel Hit Progression Strategy of Improved Small Molecule Inhibitors of the Malaria Purine Uptake Transporter that Inhibit Plasmodium falciparum Parasite Proliferation. ACS Infect. Diseases. 5, 1738–1753. doi: 10.1021/acsinfecdis.9b00168
Thomson J. M. and Lamont I. L. (2019). Nucleoside analogues as antibacterial agents. Front. Microbiol. 10. doi: 10.3389/fmicb.2019.00952
Ting L., Shi W., Lewandowicz A., Singh V., Mwakingwe A., Birck M. R., et al. (2005). Targeting a novel plasmodium falciparum purine recycling pathway with specific immucillins. J. Biol. Chem. 280, 9547–9554. doi: 10.1074/jbc.m412693200
Trager W. and Jensen J. B. (1976). Human malaria parasites in continuous culture. Science. 193, 673–675. doi: 10.1126/science.781840
Tseligka E. D., Conzelmann S., Cambet Y., Schaer T., Negro F., and Clément S. (2023). Identification of selective hepatitis delta virus ribozyme inhibitors by high-throughput screening of small molecule libraries. JHEP Rep. 5, 100652. doi: 10.1016/j.jhepr.2022.100652
Wallace L. J. M., Candlish D., and De Koning H. (2002). Different Substrate Recognition Motifs of Human and Trypanosome Nucleobase Transporters: Selective uptake of purine antimetabolites. J. Biol. Chem. 277, 26149–26156. doi: 10.1074/jbc.m202835200
Wang S., Liu J. C., Kim D., Datti A., and Zacksenhaus E. (2016). Targeted Pten deletion plus p53-R270H mutation in mouse mammary epithelium induces aggressive claudin-low and basal-like breast cancer. Breast Cancer Res. 18, 9. doi: 10.1186/s13058-015-0668-y
Wang C., Yu L., Zhang J., Zhou Y., Sun B., Xiao Q., et al. (2023). Structural basis of the substrate recognition and inhibition mechanism of Plasmodium falciparum nucleoside transporter PfENT1. Nat. Commun. 14, 1727. doi: 10.1038/s41467-023-37411-1
Ward K. E., Fidock D. A., and Bridgford J. L. (2022). Plasmodium falciparum resistance to artemisinin-based combination therapies. Curr. Opin. Microbiol. 69, 102193. doi: 10.1016/j.mib.2022.102193
Wu Q., Peng Z., Zhang Y., and Yang J. (2018). COACH-D: improved protein-ligand binding sites prediction with refined ligand-binding poses through molecular docking. Nucleic Acids Res. 46, W438–Ww42. doi: 10.1093/nar/gky439
Keywords: Plasmodium falciparum, purine salvage system, purine analogues, in vitro sensitivity testing, PfENT1 protein, drug discovery, nucleotide metabolism, purine antimetabolites
Citation: Tashie W, de Koning HP, Duah-Quashie NO and Quashie NB (2025) Guanine derivatives as promising candidates for the development of purine-based antimalarial drugs. Front. Parasitol. 4:1634209. doi: 10.3389/fpara.2025.1634209
Received: 23 May 2025; Accepted: 14 July 2025;
Published: 30 July 2025.
Edited by:
Alena Zikova, Institute of Parasitology, CzechiaReviewed by:
Brajesh Kumar Singh, Brown University, United StatesBarbara Castro-Pimentel Figueiredo, Federal University of Bahia (UFBA), Brazil
Sumaira Javaid, University of Karachi, Pakistan
Copyright © 2025 Tashie, de Koning, Duah-Quashie and Quashie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Neils B. Quashie, TlF1YXNoaWVAbm9ndWNoaS51Zy5lZHUuZ2g=