 Jun-Yan Liu1,2*
Jun-Yan Liu1,2*- 1Center for Nephrology and Metabolomics, Tongji University School of Medicine, Shanghai, China
- 2Division of Nephrology, Shanghai Tenth Peoples Hospital, Tongji University School of Medicine, Shanghai, China
A soluble epoxide hydrolase (sEH) mediates the metabolism of epoxy fatty acids to form the corresponding vicinal diols, which are usually inactive or less active than the epoxide substrates. The sEH enzyme presents in many organs, including but not limited to the liver, heart, spleen, lung, and kidney. Here we summarized the changes in the expression and activity of sEH in multiple renal diseases, such as acute kidney injury (AKI), diabetic nephrology (DN), chronic kidney diseases (CKD), hypertension-mediated renal damage, and other renal dysfunctions. We also discussed the pharmacologic effects and the underlying mechanisms of sEH inhibition by using an inhibitor of sEH and/or the generic deletion of sEH on multiple renal diseases. We believe that sEH is a potential therapeutic target for renal dysfunction although the target disease needs further investigation.
Introduction
A soluble epoxide hydrolase (sEH), a member of epoxide hydrolases (EH) family, presents in almost all living organisms (Newman et al., 2005; Morisseau and Hammock, 2013). In humans, sEH is encoded by the gene EPHX2 (Larsson et al., 1995; Sandberg and Meijer, 1996). The sEH has been well known to mediate the metabolism of epoxides to form the corresponding vicinal diols (Newman et al., 2005). The epoxids that could serve as the substrates for a sEH have been well documented previously, one member of which is epoxy fatty acids, such as the epoxy metabolites of PUFAs, including but not limited to linoleic acid [LA, 18:2 (n-6)], arachidonic acid [ARA, 20:4 (n-6)], alpha-linolenic acid [ALA,18:3 (n-3)], eicosapentaenoic acid [EPA, 20:5 (n-3)], and docosahexaenoic acid [DHA, 22:6 (n-3)] (Figure 1; Newman et al., 2005).
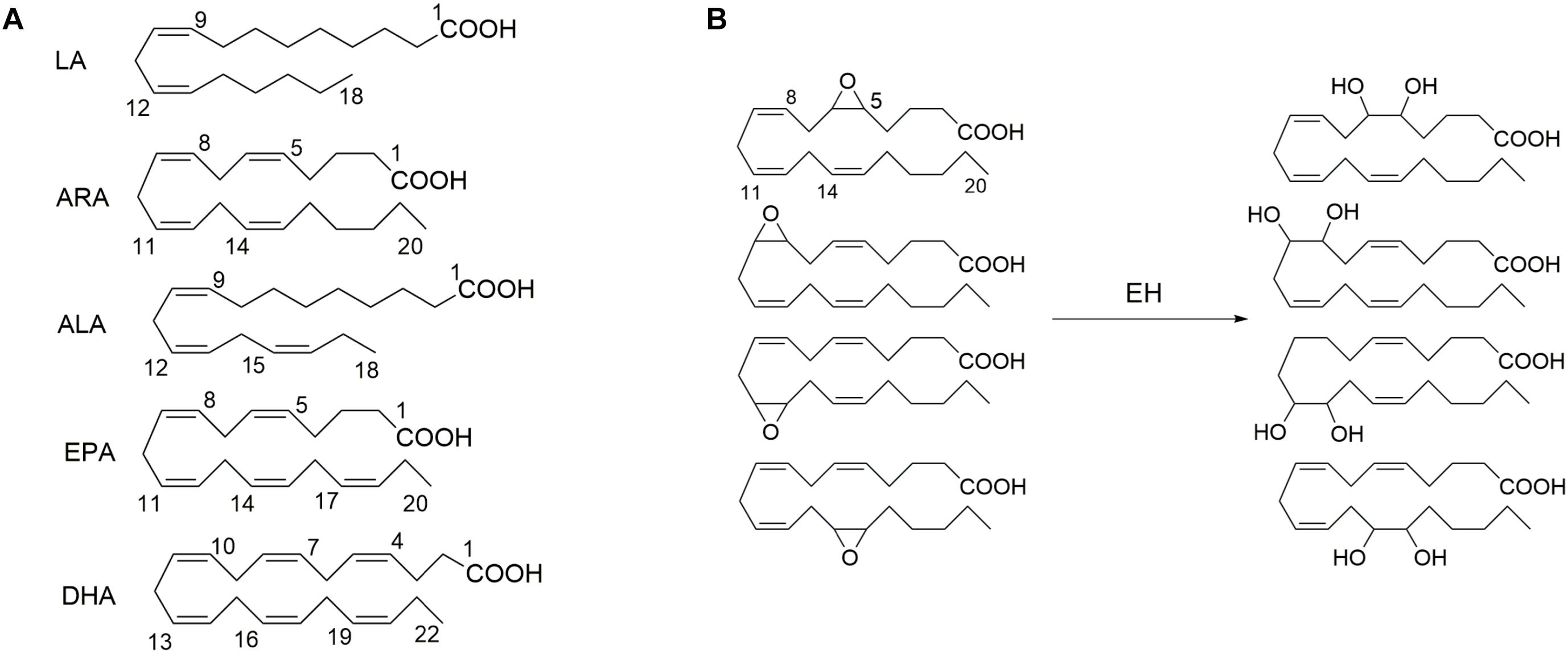
Figure 1. The exemplified polyunsaturated fatty acids (A) that are the common substrates for epoxide hydrolase (EH) and (B) four epoxide regioisomers of ARA that could be metabolized to form the respective diols in the presence of EH such as soluble epoxide hydrolase (sEH) and microsomal epoxide hydrolase (mEH). LA, linoleic acid [18:2 (n–6)]; ARA, arachidonic acid [20:4 (n–6)]; ALA, alpha-linolenic acid [18:3 (n–3)]; EPA, eicosapentaenoic acid [20:5 (n–3)]; and DHA, docosahexaenoic acid [22:6 (n–3)].
Many epoxy fatty acids are multifunctional mediators in vivo and in vitro. For example, EETs, the epoxide metabolites of ARA, are anti-inflammatory (Node et al., 1999), analgesic (Inceoglu et al., 2008), and EDHF (Campbell et al., 1996). However, the sEH-mediated diol metabolites of epoxide fatty acids are usually inactive or less active than their respective epoxide precursors (Newman et al., 2005; Morisseau and Hammock, 2013). The substrate selectivity for the sEH-mediated metabolism of epoxide fatty acids was reported previously (Zeldin et al., 1995; Deng et al., 2017). Although the active epoxy fatty acids could be degraded easily, the circulation and tissue levels of active epoxy fatty acids could be stabilized by both pharmacological interventions of an inhibitor of sEH and target gene disruption of EPHX2. Therefore, sEH inhibitors have been extensively reported to be anti-inflammatory, analgesic, anti-hypertensive, anti-fibrotic, cardioprotective and renoprotective, and other functions in vivo and in vitro (Imig and Hammock, 2009; Morisseau and Hammock, 2013; Sirish et al., 2013; Li et al., 2014; Fan et al., 2015; He et al., 2016).
The sEH presents in almost all mammal organs, such as heart, liver, lung, spleen, intestine, stomach, brain, and urinary and developmental organs1. Kidneys, as the vital part of the urinary and excretory system, also express sEH. The expression and activity were reported upregulated in many kidney diseases for human and animals. Inhibition of sEH was therefore reported to be renoprotective in many renal diseases. As illustrated in Figure 2, the attention to the sEH and kidneys has been consistently rising in the last 10 years. Here we summarized the pathophysiological and pharmacological function of sEH in the onset, prevention, and treatment in multiple kidney-associated diseases. The underlying molecular mechanisms of sEH inhibition on renal diseases were also discussed in this review paper.
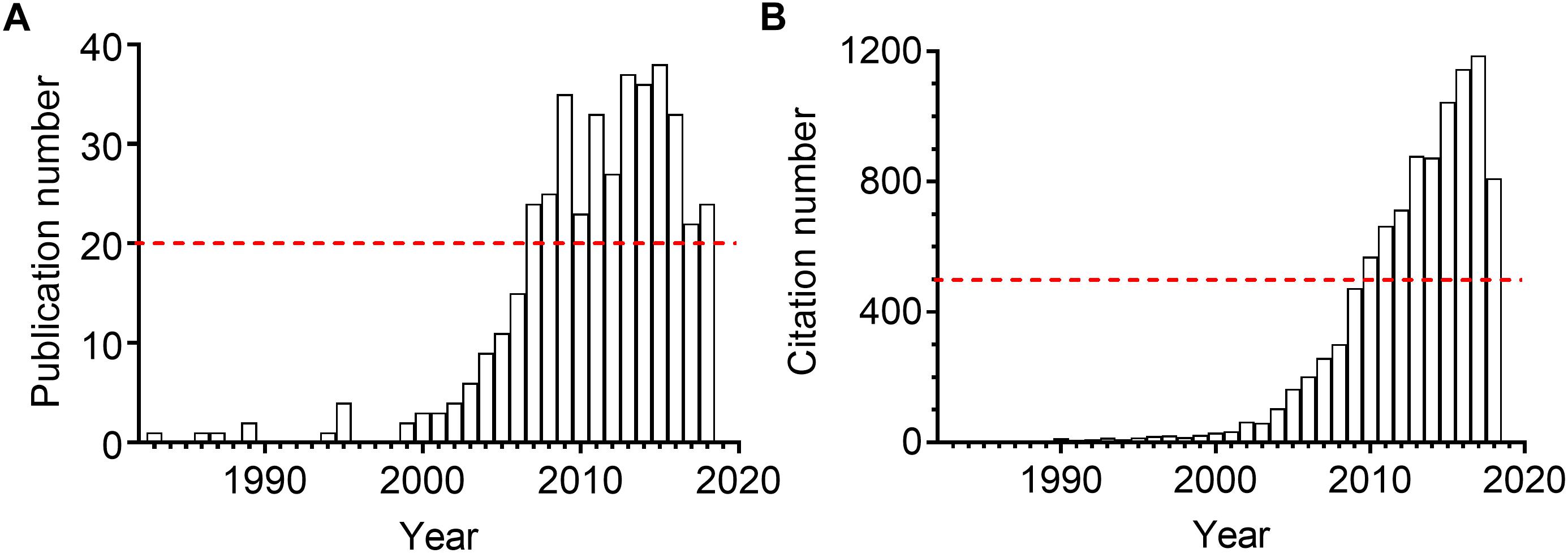
Figure 2. The annual publication numbers (A) and the resulting citation numbers (B) of the scientific paper regarding sEH and kidney. The results were generated by searching the Web of Science with the topic “sEH” combined the topic “kidney or renal” on October 1, 2018.
The Presence and Localization of sEH in the Kidneys
The enzyme sEH was expressed in all the organs investigated including but not limited to liver, kidney, brain, stomach, intestines, pancreas, prostate, heart, lung, and skin1. The sEH has been reported to present in human and animal kidneys at both transcription and protein levels (Table 1). By using IHC staining, Enayetallah et al. (2004) reported the distribution and expression of sEH in an array of normal human tissues. They found that sEH was frequently expressed in the human kidneys (n = 15) with a high level in the renal proximal tubules but a low level in the renal distal tubules and meager presence in the glomeruli (Enayetallah et al., 2004). In a follow-up study, Enayetallah et al. (2006) found that the sEH is presented in the proximal and cytosolic compartments in hepatocytes and renal proximal tubules. Wang et al. (2013, 2018) also reported that sEH expressed in the renal tubules in the patients with IgAN and other glomerulonephritis. In contrast, Yu et al. (2004) reported the cellular localization of sEH in the human kidneys by examining the biopsies taken from the patients with multiple non-end-stage renal diseases (n = 8) and those without known renal disorders (n = 7). Yu et al. (2004) found that sEH was preferentially expressed in the renal vasculature, mostly in the smooth muscle layers of the arterial wall, while relatively low levels in the surrounding tubules. Also, Yu et al. (2004) reported that sEH expressed in the normal kidneys in a similar pattern to those in the diseased kidneys in the samples investigated. The inconsistent observations among these studies may be due to the different sampling locations of renal biopsies. In addition, the sEH was also reported to be present in the murine and rodent kidneys (Patel et al., 1986; Johansson et al., 1995). The presence of sEH in human and animal kidneys opens a possibility that sEH could be associated by multiple renal diseases.
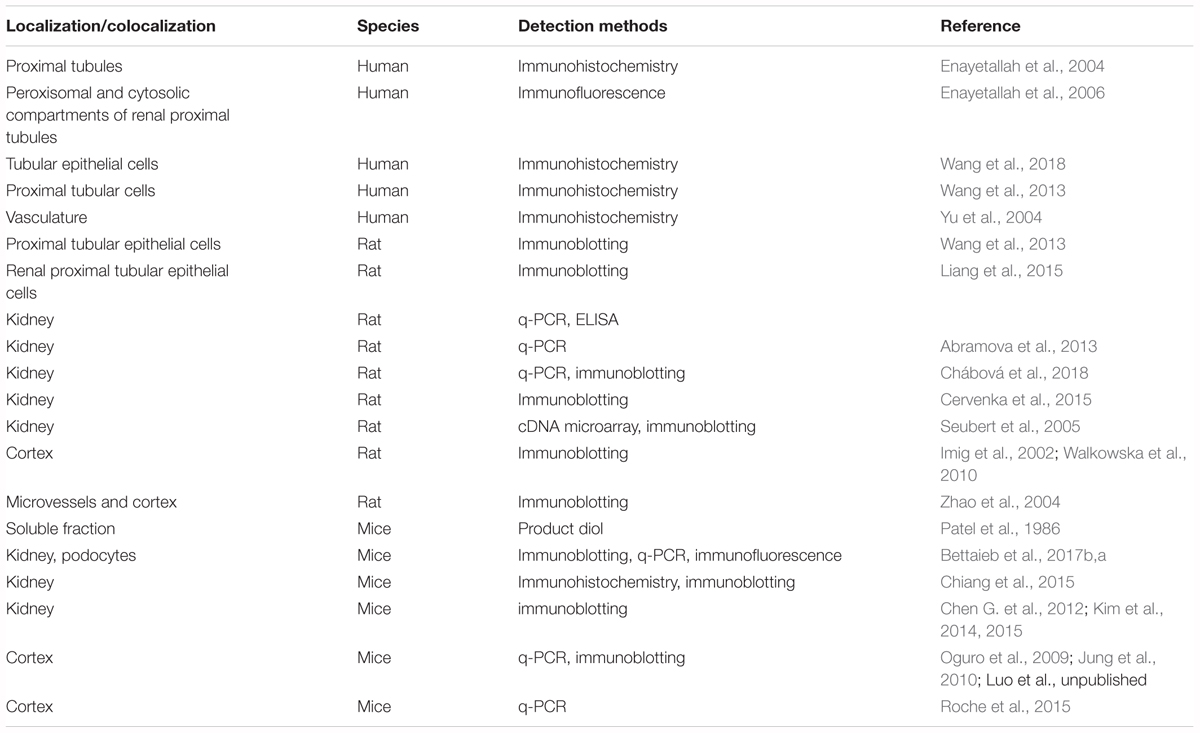
Table 1. The location and co-location of sEH in kidneys from different species.
Preclinical Studies of the Treatment of Renal Dysfunction by Regulation of sEH
Although there has been no drug clinically used as a sEH inhibitor yet, a large amount of pre-clinically experimental evidence supports that sEH may be a potential therapeutic target for several kidney-associated diseases, such as acute kidney injury (AKI), chronic kidney disease (CKD), diabetic nephrology (DN), and hypertension-associated kidney damage.
Regulation of sEH for the Treatment of AKI
AKI is a common fatal disease in hospitals characterized by a sudden and sustained reduction in renal function with the phenotypes of an abrupt decrease in urine output and a dramatic increase in serum creatinine level. The mechanisms underlying the pathogenesis of AKI vary, including but not limited to ischemia/reperfusion, septic shock, toxicant exposure, and inflammation-caused decrease in kidney blood flow, resulting in the damage to renal tissues, and eventually renal dysfunction (Kusch et al., 2013; Persson, 2013; Glodowski and Wagener, 2015; Takaori and Yanagita, 2016), which involve the impairment in glomerulus and renal tubular epithelial cells (Kusch et al., 2013; Prieto-Garcia et al., 2016), and activation of NFκB and GSK-3β (Liu et al., 2012; Kusch et al., 2013; Deng et al., 2017).
Inhibition of sEH has been reported to attenuate the renal injury in multiple murine models of AKI. Although Hashimoto et al. (2015) reported that cisplatin administration decrease the activity and expression of sEH in the kidneys of male ddY mice, Parrish et al. (2009) found that subcutaneous (s.c.) injection of a sEH inhibitor n-butyl ester of 12-(3-adamantan-1-yl-ureido)-dodecanoic acid (nbAUDA, 1, Figure 3), a pro-drug of AUDA (Newman et al., 2005) significantly attenuated the renal injury in a C3H mice model of AKI caused by intraperitoneal (i.p.) injection of cisplatin, which was supported by the blood levels of urea nitrogen (BUN) and histological analysis of kidneys. Also, in a murine model of AKI caused by i.p. injection of cisplatin, Liu et al. reported that oral administration of another sEH inhibitor, 1-adamantan-1-yl-3-(1-methylsulfonyl-piperidin-4-yl-urea) (AR9273, 3) that is structurally different from AUDA, markedly attenuated renal injury, which was supported by the serum levels of urea nitrogen and creatinine, and histological evidence of renal tubular damage and neutrophil infiltration (Liu et al., 2012, 2013). Also, the renoprotective effects of AR9273 were consistent with those in the sEH knockout mice (Liu et al., 2012). It should be noted that the sEH inhibitors were administered 1 day before cisplatin treatment in both studies. Therefore, the renoprotective effects of sEH inhibitors are prophylactic rather than therapeutic effects.
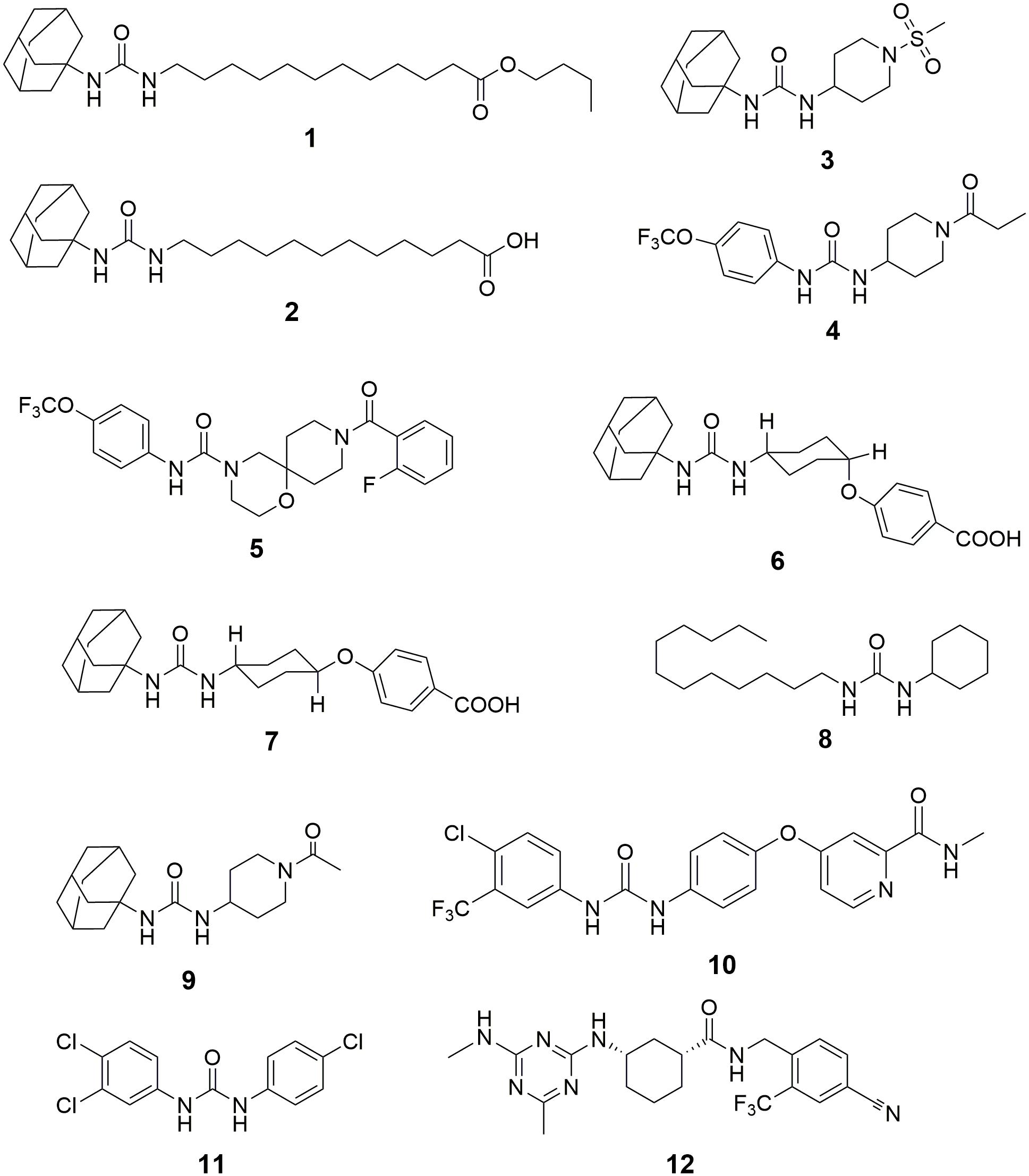
Figure 3. The chemical structures of the inhibitors of sEH summarized in this paper. 1, n-butyl ester of 12-(3-adamantan-1-yl-ureido)-dodecanoic acid (nbAUDA); 2, 12-(3-adamantan-1-yl-ureido)-dodecanoic acid (AUDA); 3, 1-adamantan-1-yl-3-(1-methylsulfonyl-piperidin-4-yl-urea) (AR9273); 4, 1-trifluoromethoxyphenyl-3-(1-propionylpiperidin-4-yl)urea (TPPU); 5, 9-(2-fluorobenzoyl)-N-[4-(trifluoromethoxy)phenyl]-1-oxa-4,9-diazaspiro[5.5]undecane-4-carboxamide; 6, cis-4-[4-(3-adamantan-1-yl-ureido)cyclohexyloxy]benzoic acid (c-AUCB); 7, trans-4-[4-(3-adamantan-1-yl-ureido)cyclohexyloxy]benzoic acid (t-AUCB); 8, 1-cyclohexyl-3-dodecylurea (CDU); 9, 1-(1-acetypiperidin-4-yl)-3-adamantanylurea (AR9281); 10, 4-[4-[[4-chloro-3-(trifluoromethyl)phenyl]carbamoylamino]phenoxy]-N-methyl-pyridine-2-carboxamide (Sorafenib); 11, 3-(4-Chlorophenyl)-1-(3,4-dichlorophenyl)urea (Triclocarban); 12, (1R,3S)-N-[4-cyano-2-(trifluoromethyl)benzyl]-3-[(4-methyl-6-(methylamino)-1,3,5-triazin-2-yl)amino] cyclohexane-1-carboxamide (GSK2256294).
In a murine model of AKI caused by ischemia/reperfusion, Deng et al. (2017) reported that administration of a sEH inhibitor, TPPU, 4 attenuated renal injury, which was supported by plasma creatinine, survival time, and histological analysis. In contrast, Zhu et al. (2016) also reported that sEH knockout exacerbated renal injury caused by ischemia/reperfusion in a murine model. Interestingly, Zhu et al. (2016) found that the renal level of 20-HETE and one of its synthetic enzymes, Cyp4a12a, were increased significantly in sEH knockout mice when compared with those of wildtype mice. 20-HETE is a potent nephrotoxic compound, which may abate the renoprotective effects of increased EETs caused by sEH deficiency.
In a murine model of AKI caused by lipopolysaccharide (LPS), Bettaieb et al. (2017a) reported that podocyte-specific sEH deficiency ameliorated the LPS-caused mice renal injury, which was supported by the favorable changes in proteinuria, BUN, and renal mRNA levels and serum levels of inflammatory cytokines. In addition, the in vivo renal protective effect of podocyte-specific sEH knockout was further supported by the in vitro data from the treatment of a sEH inhibitor TPPU to the E11 murine podocytes (Bettaieb et al., 2017a).
Inhibition of sEH for the Treatment of CKD
CKD is one of the top public health problems and leading diseases for all-cause mortality globally (Xie et al., 2018). The putative mechanisms regarding the pathogenesis and progression of CKD have been well documented previously (Fogo, 2007; Malyszko, 2010; Eddy et al., 2012; Yang et al., 2017; Cheung et al., 2018; Mihai et al., 2018; Moradi and Vaziri, 2018; Rossi et al., 2018). Briefly, both generic factor-caused genital abnormality of renal development, and other factors (e.g., inflammation, hypertension, diabetes, dyslipidemia, disorder of cytokines and growth factors, proteinuria, podocyte loss, etc.) can cause irreversible scarring in kidney, resulting in progressive renal fibrosis, and eventually end-stage renal disease, which needs renal replacement therapy, such as hemodialysis and renal transplantation. The progression of CKD has been reported to be associated with the damage to renal glomerulus and renal epithelial cells, accelerating the cell migration and epithelial-mesenchymal transition, and repressing the proliferation of podocytes, which may be mediated by angiotensin-II, PPAR-γ, AMPK, CYPs, and sEH (Fogo, 2007; Malyszko, 2010; Decleves et al., 2011; Kim et al., 2014; Fan et al., 2015).
Inhibition of sEH by both pharmacological interventions with a sEH inhibitor and target gene disruption of sEH has been reported to attenuate the renal injury in several murine and rodent models. Kato et al. (2014) reported that a potent sEH inhibitor, 9-(2-fluorobenzoyl)-N-[4-(trifluoromethoxy)phenyl]-1-oxa-4,9-diazaspiro[5.5]undecane-4-carboxamide (Node et al., 1999), amelio-rated renal injury in a rat model of anti-glomerular basement membrane glomerulonephritis evidenced by time-dependently reducing the rat serum creatinine. Liang et al. (2015) reported that the treatment of AUDA abated the proteinuria-induced epithelial-mesenchymal transition in vivo and in vitro.
By using a rodent model of CKD caused by 5/6 nephrectomy (5/6 NX) in Ren-2 transgenic rats, Kujal et al. (2014) reported that the treatment of a sEH inhibitor, cis-4-[4-(3-adamantan-1-yl-ureido)cyclohexyloxy]benzoic acid (c-AUCB, 6) attenuated the renal and cardiac injuries of the diseased rats by modification of the survival rate, blood pressure, cardiac hypertrophy, proteinuria, degree of glomerular and tubulointerstitial injury, and glomerular volume toward the normative status. In addition, Chábová et al. (2018) reported that co-administration of c-AUCB with a standard renin-angiotensin system (RAS) blockade resulted in additional therapeutic effects in the improvement of rat survival rate, reduce in albuminuria, glomerular and tubulointerstitial injury when compared with the standard RAS blockade alone.
In a murine model of obstructive nephropathy caused by UUO surgery, Kim et al. (2014) reported that the deficiency of sEH abolished the renal interstitial fibrosis and inflammation. Consistently, Chiang et al. (2015) found that renal expression of sEH protein was increased in the UUO-treated mice when compared with the sham mice. In addition, the target gene disruption of sEH abated the UUO-caused renal injury, such as hydronephrosis, renal tubular injury, inflammation, and fibrosis. Also, oral administration of a sEH inhibitor, trans-4-[4-(3-adamantan-1-yl-ureido)cyclohexyloxy]benzoic acid (t -AUCB, 7) resulted in the similar fibro-protective and anti-inflammatory effects to sEH gene knockout (Kim et al., 2014, 2015). Interestingly, Yang et al. (2017) reported that treatment of AUDA resulted in similar results to t-AUCB in a UUO-induced mice model of renal fibrosis.
In addition, Huang et al. (2007) reported that inhibition of sEH by administration of a sEH inhibitor AUDA attenuated the renal injury by regulating the mean arterial pressure, renal vascular resistance, and glomerular filtration rate, and renal blood flow for the obese rats toward normative status. By using a target metabolomic analysis, Luo et al. (unpublished) found that renal sEH was upregulated at both transcription and protein levels time-dependently upon the challenge of high-fat diet (HFD) feeding, which was associated with the progression of renal injury. In addition, inhibition of sEH by TPPU attenuated the HFD-induced renal injury by, at least in part, activation of the Ampk-mediated macro-autophagy and Pax2-mediated chaperone-mediated autophagy.
It is worth noting that not all the study showed the favorite effect of sEH inhibition on kidney diseases. Jung et al. reported in a mice model of chronic renal failure caused by 5/6-nephrectomy that c-TUCB (Inceoglu et al., 2008) failed in lowering blood pressure and even aggravated albuminuria when compared with the placebo controls (El-Sherbeni et al., 2013). The authors thought that this unfavorite effect may be due to the shifts of arachidonic metabolism into lipoxygenase pathway by sEH inhibition.
Inhibition of sEH for the Treatment of Diabetic Nephrology
We separated DN from CKD in the “Inhibition of sEH for the treatment of CKD” since DN is a leading cause of CKD and end-stage renal disease (Lin and Sheu, 2014). DN causes primary renal damage to its microvascular system including glomerular capillaries, influent and affluent arteries at the beginning, and most leading to end-stage renal disease over time (Remuzzi et al., 2006; Afsar and Elsurer, 2017), which may involve the activation of some transcription factors (e.g., activator protein 1, cAMP-response element-binding protein, nuclear factor of activated T cells, NF-κB, stimulating protein 1, and upstream stimulatory factor 1) and may be regulated by some signal pathways such as mTOR, AMPK, GSK-3β, and Deptor 2 (Mariappan, 2012).
Chen G. et al. (2012) reported that sEH deficiency maintained the renal function in a murine model of STZ-induced DN. Compared with the wildtype diabetic mice, sEH-deficient mice resulted in significantly decreased levels of plasma Hb A1c and creatinine, BUN and urinary microalbumin excretion. Bettaieb et al. (2017b) reported that renal sEH protein was upregulated in the mice under HFD and STZ-induced hyperglycemia. In addition, podocyte-specific sEH deficiency preserved renal function in vivo and in vitro via modulation of the renal endoplasmic reticulum (ER) stress, inflammation, fibrosis, and autophagy toward normative conditions, which was further supported by the in vitro data from the pharmacological intervention of sEH with the sEH inhibitor TPPU (Bettaieb et al., 2017b). Katary et al. (2017) reported that in a rodent model of STZ-induced DN, the treatment with t-AUCB attenuated the renal injury by reducing glomerular albumin permeability, albumin, and nephrin excretion levels and restoring the decrease in glomerular α3 integrin and nephrin expression in diabetic rats. However, this beneficial effect of sEH inhibition was unable to be enhanced by co-administration with meloxicam, a cyclooxygenase inhibitor (Katary et al., 2017).
Inhibition of sEH for the Attenuation of Hypertension-Associated Kidney Disorders
Hypertension is the second leading cause of end-stage renal disease after diabetes in the United States (U.S. Renal Data System, 2009). Although the susceptibility to hypertension-associated renal injury differs significantly in various populations, a consensus is that hypertension causes damage to glomerular arteries and capillary, and endothelial cells, leading to the injuries to glomerular filtration barrier and podocytes, eventually renal dysfunction, which may be manipulated by renin-angiotensin–aldosterone system, reactive oxidative species, endothelial dysfunction, and genetic and epigenetic factors (Mennuni et al., 2014).
Hypertension is a risk factor for the development of renal dysfunction. The renal sEH was upregulated in hypertensive status. By using a gene microarray analysis, Seubert et al. (2005) reported that sEH was significantly upregulated in the kidneys of spontaneously hypertensive rats (SHR) when compared with the non-hypertensive Wistar-Kyoto (WKY) rats. Also, Abramova et al. (2013, 2017) found that the renal mRNA level and protein concentration of sEH were increased at in the inherited stress-induced arterial hypertension (ISIAH) rats when compared with the normative Wistar Albino Glaxo (WAG) rats. Tain et al. (2015) reported that renal sEH was upregulated in dexamethasone- and a high fructose-induced rodent model of programmed hypertension. These data suggest the possibility that inhibition of sEH could protect the kidney from injury associated with hypertension.
Zhao et al. (2004) reported that chronic administration of a sEH inhibitor 1-cyclohexyl-3-dodecyl urea (CDU, 8) significantly attenuated renal injury in an angiotensin-induced rodent model of hypertension, which was supported by the observation that CDU treatment maintained the renal vasculatures and glomerulus toward normative status. Imig et al. (2005) reported that administration of AUDA significantly lowered blood pressure and protected renal damage by decreasing the urinary microalbumin excretion in the rodent models of hypertension induced by both normal-salt angiotensin and high-salt angiotensin. Imig et al. also reported that another sEH inhibitor AR9281 attenuated glomeruli injury and reduced renal inflammation in the angiotensin-induced hypertensive rats (Imig et al., 2009). Olearczyk et al. (2009) reported that treatment with AUDA protected kidneys from glomerular and tubular damage in spontaneously diabetic Goto-Kakizaki rats induced by angiotensin II with high salt diet. Sporkova et al. (2011) reported that treatment of c-AUCB improved renal function by maintaining the renal blood flow toward normative status while exhibiting an anti-hypertensive effect in the 2-kidney 1 clip hypertensive rats. Imig et al. (2012) reported that t-AUCB treatment reversed the increase in urinary levels of albumin and kidney injury marker-1 (KIM-1) in the spontaneously hypertensive obese rats. In addition, co-administration of t-AUCB with rosiglitazone, an agonist of PPARγ, resulted in additive reno-protection (Imig et al., 2012). Cervenka et al. (2015) reported that in an aorto-caval fistula-induced Ren-2 transgenic hypertensive rats model of congestive heart failure (CHF), c-AUCB treatment improved rats survival rate and increased renal blood flow, glomerular filtration rate and fractional sodium excretion.
Association of sEh Polymorphisms With Kidney-Associated Diseases
Several sEH polymorphisms, such as Lys55Arg, rs41507953 (K55R), rs751141 (R287Q), and rs1042032, were reported to correlate with several renal-associated diseases. Based on current data, lower sEH activity is associated with the improved renal function. Shuey et al. (2017) reported that sEH Lys55Arg polymorphism was associated with an increased incidence of AKI following cardiac surgery in patients without preexisting CKD. Also, the sEH activity that was characterized by the ratio of DHOMEs to EpOMEs was increased in sEH 55Arg variant carriers when compared with the investigated wildtype carriers.
Lee et al. (2011) investigated the association of three single nucleotide polymorphisms [SNPs, rs41507953 (K55R), rs751141 (R287Q), and rs1042032] of sEH with IgAN progression in a retrospective cohort including 401 IgAN patients and 402 normal healthy controls. They reported that the patients carrying the variant allele (A) of rs751141 (R287Q) were associated with a better kidney survival (P < 0.001) and a lower sEH activity (P < 0.05) than those with the wildtype allele (G) (Lee et al., 2011). Also, Wang et al. (2013, 2018) reported that the level of sEH expression correlated with proteinuria and infiltration of macrophages positively in the IgAN patients and other glomerulonephritis.
Dreisbach et al. (2005) conducted a case-control study to investigate the possible correlation of multiple SNPs in sEH (sEH-Q and sEH-RR) and CYP 2C8, 2C9, and 2J2 with an increased risk of hypertension in African American individuals. They found that these SNPs are not associated with the increased risk of hypertension in the African American population (Dreisbach et al., 2005). Ma et al. (2018) reported that the A allele of an exonic polymorphism in sEH rs751141 correlated with the incidence of DN in the Chinese T2D population negatively, which could be modulated by homocysteine level status.
Gervasini et al. (2015) reported that the renal transplant recipients who carried the rs1042032GG genotype in sEH were associated with the significantly lower estimated glomerular filtration rate and higher serum creatinine levels on year after grafting compared with those with wildtype A-allele. This was consistent with the study by Lee et al. (2008), in which the presence of the rs1042032 AA genotype in sEH was reported to correlate with a protective role for allograft function for the kidney transplant recipients.
Molecular Mechanisms Underlying the Regulation of sEH for Reno-Protection
The sEH has been reported to be upregulated by both endogenous and exogenous factors at both mRNA and protein levels. Wang et al. (2013, 2018) reported that the expression of sEH was increased in renal tissues in the patients with glomerulonephritis when compared with the control renal tissues by IHC analysis. Both mRNA and protein levels of sEH were found higher in the kidneys of SHR than those of the control WKY rats (Yu et al., 2000; Koeners et al., 2011). The sEH was also upregulated in the renal cortex by angiotensin-II, two-kidney-one-clip, and a HFD (Imig et al., 2002; Zhao et al., 2004; Kopkan et al., 2012; Luo et al., unpublished).
An sEH can be down-regulated by generic target gene disruption of Ephx2 and chemical knockdown by various sEH inhibitors. In most cases, sEH is believed to be inhibited at protein level primarily due to the formation of the hydrogen bonds between the inhibitors and the active sites Tyr461 and Tyr385 in both human and mammal sEH enzymes (Argiriadi et al., 1999, 2000; Gomez et al., 2004). Recently, Luo et al. (unpublished) found that an sEH inhibitor TPPU could inhibit murine sEH transcriptionally.
The mechanisms underlying the reno-protection of sEH inhibition are summarized in Figure 4. The renoprotective effects of sEH inhibition are mostly believed to be ascribed to the increased renal levels of EETs resulted from pharmacological interventions with a sEH inhibitor or genetic disruption of sEH based on two main facts. The one is that the renal and/or circulation levels EETs or the intrarenal ratio of EETs to DHETs were hypothesized and observed to be increased following sEH inhibition (Zhao et al., 2004; Huang et al., 2007; Kim et al., 2014, 2015; Kujal et al., 2014; Deng et al., 2017; Luo et al., unpublished). The other is that EETs have been investigated to be the active mediators, which could result in the protection of kidneys from multiple injuries. In addition to inhibition of sEH, the metabolic pathway of EETs, upregulation of CYP2C and/or 2J, the biosynthetic pathways of EETs, is an alternative approach to stabilize the EETs level in vivo. Upregulation of Cyp2J was previously reported to protect the kidneys function in the 5/6-nephrectomized rat (Zhao et al., 2012), which supports that the reno-protection of EETs.
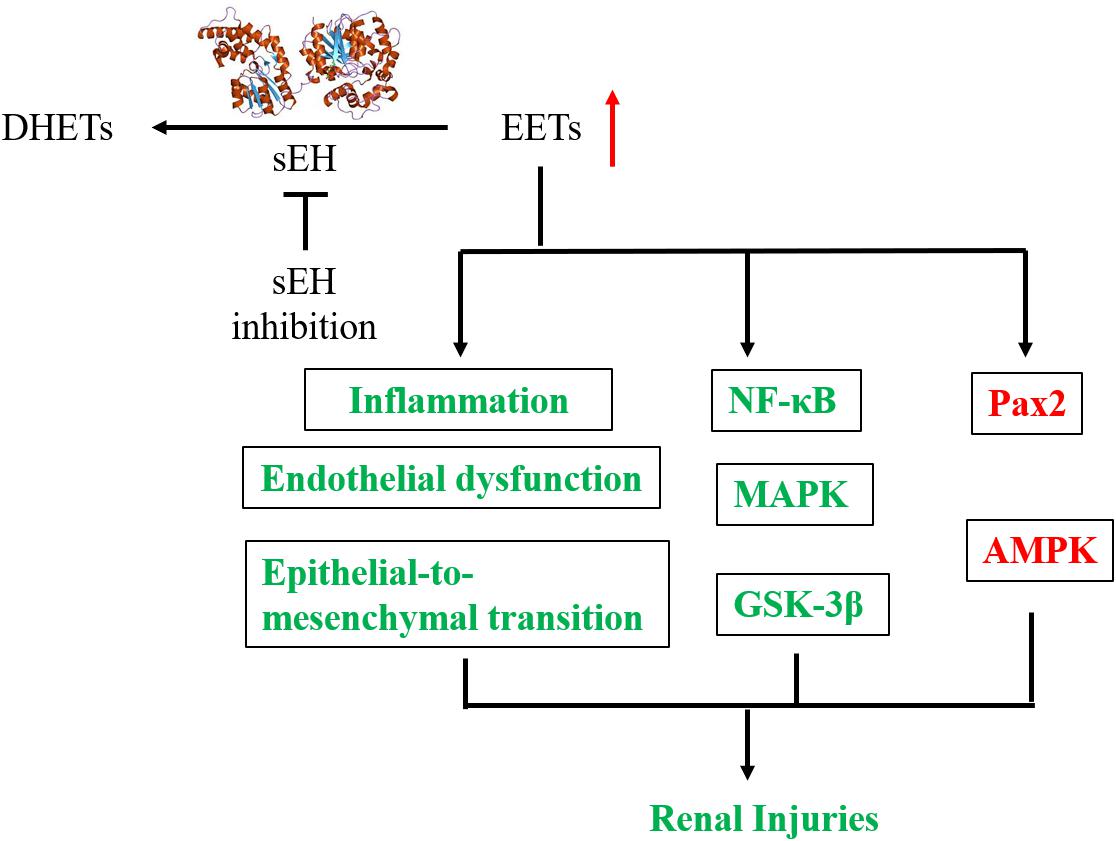
Figure 4. A putative mechanic summary of reno-protection of sEH inhibition. The items highlighted with red mean increased while those with green mean decreased caused by sEH inhibition with a chemical inhibitor or generic disruption.
The biochemical functions of EETs have been well documented in several review papers (Spector et al., 2004; Spector and Norris, 2007; Wang and DuBois, 2012; Imig, 2015). EETs were reported to be anti-inflammatory via modulation of the NFκB/IκBK signaling pathway (Node et al., 1999). Reno-protective effects of sEH inhibition, such as anti-inflammatory and fibro-protective, were reported to be involved in the modulation of NFκB signaling pathway and cytokine orchestra toward inflammation resolution in several animal studies (Liu et al., 2012; Kim et al., 2014, 2015; Bettaieb et al., 2017a). Also, EETs are potent EDHF (Campbell et al., 1996; Archer et al., 2003). Inhibition of sEH was reported to protect the kidney from the multiple injuries associated with the improvement of the renal microvessel dilation and renal blood flowing (Sporkova et al., 2011; Cervenka et al., 2015).
Epoxyeicosatrienoic acids have been reported to induce cell growth and inhibit the cell apoptosis (Dhanasekaran et al., 2006; Nieves and Moreno, 2007). EETs were found to protect pig kidney proximal tubule LLC-PK1 cells from cisplatin-induced p38 MAPK activation, and Bax mitochondrial trafficking (Liu et al., 2013), which was further supported by that inhibition of sEH by AR9273 treatment and sEH-deficiency significantly attenuated cisplatin-induced murine renal injury via inhibition of the p38 MAPK phosphorylation (Liu et al., 2013). The renal podocyte-specific sEH deficiency ameliorated murine kidney injury associated with decreased LPS-induced NF-κB and MAPK activation and attenuated endoplasmic reticulum stress (Bettaieb et al., 2017a). EETs were also reported to inhibit the apoptosis of renal tubular epithelial cells via, at least in part, the modulation of the phosphorylation of GSK-3β, which was also supported by the in vivo data from TPPU treatment in a murine model of AKI (Deng et al., 2017). Also, AUDA treatment was reported to attenuate renal injury by modulation of the PI3K-Akt-GSK-3β signaling pathway (Liang et al., 2015). Also, sEH gene inhibition and exogenous EETs significantly protected HK-2 cells from TNF-induced apoptosis in vitro associated with activation of the PI3K-Akt-NOS3 and AMPK signaling cascades (Chen G. et al., 2012).
Recently, Luo et al. (unpublished) found that EETs protected the murine renal mesangial cells and tubular epithelial cells from palmitic acid-induced injury by activation of Ampk and Pax2, which was supported by the in vivo data from the TPPU treatment to the mice feeding on a HFD.
In addition to EETs, other epoxy fatty acids were also reported to contribute to the renoprotective role of sEH inhibition. 19(20)-epoxydocosapentaenoic acid, the principal epoxy metabolite of DHA that could also be stabilized by sEH inhibition, was found to mitigate the renal fibrosis in a mouse model of experimental UUO-induced renal fibrosis by decreasing renal epithelial-to-mesenchymal transition (Sharma et al., 2016).
Besides sEH, microsomal EH (mEH), encoded by EPHX1, has the similar function to sEH in mediating the hydrolysis of epoxy fatty acids (Newman et al., 2005; Rawal et al., 2009). However, mEH was found to modified slightly while sEH was up-regulated significantly in renal microvessels by the treatment of angiotensin-II infusion (Zhao et al., 2004). Recently, mEH was found to be decreased significantly while sEH altered non-significantly at mRNA level in the blood cells of the uremic patients when compared with healthy controls (Hu et al., 2018). The pathophysiological and pharmacological role of mEH in kidney diseases and other disorders needs further investigation.
Clinical Studies of sEH Inhibitors
As far as we know, there is no clinical drug used as a sEH inhibitor. However, some sEH chemical inhibitors failed in, or are under, or will be tested in clinical trials. In addition, some clinically used drugs or human used agents were found to be the potent sEH inhibitors although they have their specific mode of action other than sEH inhibition. For example, sorafenib (Imig and Hammock, 2009) is a clinical drug for the treatment of advanced renal cell carcinoma, advanced hepatocellular carcinoma, and radioactive iodine resistant advanced thyroid carcinoma as an inhibitor of multi-kinase, such as VEGFR, PDGFR, and Raf family kinases (Ng and Chen, 2006; Wilhelm et al., 2008; Smalley et al., 2009; Iyer et al., 2010). However, since sorafenib has the N,N′-disubstituted substructure that is similar to many potent sEH inhibitors (Figure 3), it was found to be a potent sEH inhibitor (Liu et al., 2009). Also, triclocarban (He et al., 2016) has been widely used as an anti-microbial agent in various personal care products (Brausch and Rand, 2011; Bu et al., 2013), is also a potent inhibitor of sEH (Liu et al., 2011).
In addition, two sEH inhibitors, AR9281 and GSK2256294, finished the phase I clinical trials. AR9281 showed well-tolerated at a single oral dose (10–1000 mg) and multiple doses (100–400 mg every 8 h) during the test period (1 week) and dose-dependent blood drug concentration and activity to inhibit the blood sEH with 90% or greater inhibition. This study suggested a twice-daily or thrice-daily dosing regimen for the patients (Chen D. et al., 2012). GSK2256294 was also well-tolerated at the tested doses. Plasma drug concentration increased at a dose-related manner with a half-life of 25–43 h. The activity of GSK2256294 to inhibit sEH was dose-dependent, from an average of 41.9% (2 mg) to 99.8% (20 mg) (Lazaar et al., 2016).
Wang et al. (2018) reported that renal sEH expression was increased in patients with IgA nephrology. Before that, Wang et al. (2013) reported that renal sEH expression was upregulated in the patients with glomerulonephritis including IgA nephrology. These human data suggest that sEH may be a therapeutic target for some renal diseases like IgA nephrology. However, Hu et al. (2018) reported that the mEH (EPHX1) was downregulated while sEH was changed slightly in the whole blood cells of the uremic patients when compared with healthy controls. Therefore, further studies are needed to optimize a target renal disease for the follow-up clinical study of a sEH inhibitor.
Conclusion
In summary, sEH may be a promising therapeutic target for the prevention and treatment of renal disorders. Also, various small molecular sEH inhibitors were synthesized in several laboratories with favorable pharmacokinetics and pharmacodynamics in multiple animals and humans. To move sEH inhibitors toward clinical use, the next step should focus more on the functional investigation of sEH in the pathology and pathophysiology of the renal dysfunction by using human samples to target a renal disease for systemic evaluation of the pharmacological and toxicological effects of sEH inhibitors.
Author Contributions
J-YL designed and wrote the paper.
Funding
This study was supported by the National Natural Science Foundation of China (NSFC) grant 81470588.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
AKI, acute kidney injury; ALA, alpha-linolenic acid; AR9273, 1-adamantan-1-yl-3-(1-methylsulfonyl-piperidin-4-yl-urea); ARA, arachidonic acid; AUDA, 12-(3-adamantan-1-yl-ureido)-dodecanoic acid; BUN, blood urea nitrogen; c-AUCB, cis-4-[4-(3-adamantan-1-yl-ureido)cyclohexyloxy]benzoic acid; CDU, 1-cyclohexyl-3-dodecyl urea; CKD, chronic kidney disease; CYP, cytochrome P450; DHA, docosahexaenoic acid; DHOME, dihydroxyoctadecanoic acid; DN, diabetic nephrology; EDHF, endothelium-derived hyperpolarizing factor; EET, epoxyeicosatrienoic acid; EH, soluble epoxide hydrolase; EPA, eicosapentaenoic acid; EpOME, epoxyoctadecanoic acid; GBM, glomerular basement membrane; 20-HETE, 20-hydroxyeicosatetraenoic acid; IgAN, IgA nephropathy; IHC, immunohistochemical or immunohistochemistry; LA, linoleic acid; nbAUDA, n-butyl ester of 12-(3-adamantan-1-yl-ureido)-dodecanoic acid; PUFA, polyunsaturated fatty acid; SNP, single nucleotide polymorphism; STZ, streptozotocin; t-AUCB, trans-AUCB; TPPU, 1-trifluoromethoxyphenyl-3-(1-propionylpiperidin-4-yl)urea; UUO, unilateral ureteral obstruction.
Footnotes
References
Abramova, T. O., Redina, O. E., Smolenskaya, S. E., and Markel, A. L. (2013). Elevated expression of the Ephx2 mRNA in the kidney of hypertensive ISIAH rats. Mol. Biol. 47, 821–826. doi: 10.1134/S0026893313060022
Abramova, T. O., Ryazanova, M. A., Antonov, E. V., Redina, O. E., and Markel, A. L. (2017). Increase in the concentration of sEH protein in renal medulla of ISIAH rats with inherited stress-induced arterial hypertension. Mol. Biol. 51, 389–392. doi: 10.7868/S0026898417020021
Afsar, B., and Elsurer, R. (2017). Increased renal resistive index in type 2 diabetes: clinical relevance, mechanisms and future directions. Diabetes Metab. Syndr. 11, 291–296. doi: 10.1016/j.dsx.2016.08.019
Archer, S. L., Gragasin, F. S., Wu, X. C., Wang, S. H., McMurtry, S., Kim, D. H., et al. (2003). Endothelium-derived hyperpolarizing factor in human internal mammary artery is 11,12-epoxyeicosatrienoic acid and causes relaxation by activating smooth muscle BKCa channels. Circulation 107, 769–776. doi: 10.1161/01.CIR.0000047278.28407.C2
Argiriadi, M. A., Morisseau, C., Goodrow, M. H., Dowdy, D. L., Hammock, B. D., and Christianson, D. W. (2000). Binding of alkylurea inhibitors to epoxide hydrolase implicates active site tyrosines in substrate activation. J. Biol. Chem. 275, 15265–15270. doi: 10.1074/jbc.M000278200
Argiriadi, M. A., Morisseau, C., Hammock, B. D., and Christianson, D. W. (1999). Detoxification of environmental mutagens and carcinogens: structure, mechanism, and evolution of liver epoxide hydrolase. Proc. Natl. Acad. Sci. U.S.A. 96, 10637–10642. doi: 10.1073/pnas.96.19.10637
Bettaieb, A., Koike, S., Chahed, S., Zhao, Y., Bachaalany, S., Hashoush, N., et al. (2017a). Podocyte-specific soluble epoxide hydrolase deficiency in mice attenuates acute kidney injury. FEBS J. 284, 1970–1986. doi: 10.1111/febs.14100
Bettaieb, A., Koike, S., Hsu, M. F., Ito, Y., Chahed, S., Bachaalany, S., et al. (2017b). Soluble epoxide hydrolase in podocytes is a significant contributor to renal function under hyperglycemia. Biochim. Biophys. Acta Gen. Subj. 1861, 2758–2765. doi: 10.1016/j.bbagen.2017.07.021
Brausch, J. M., and Rand, G. M. (2011). A review of personal care products in the aquatic environment: environmental concentrations and toxicity. Chemosphere 82, 1518–1532. doi: 10.1016/j.chemosphere.2010.11.018
Bu, Q., Wang, B., Huang, J., Deng, S., and Yu, G. (2013). Pharmaceuticals and personal care products in the aquatic environment in China: a review. J. Hazard. Mater. 262, 189–211. doi: 10.1016/j.jhazmat.2013.08.040
Campbell, W. B., Gebremedhin, D., Pratt, P. F., and Harder, D. R. (1996). Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circ. Res. 78, 415–423. doi: 10.1161/01.RES.78.3.415
Cervenka, L., Melenovsky, V., Huskova, Z., Skaroupkova, P., Nishiyama, A., and Sadowski, J. (2015). Inhibition of soluble epoxide hydrolase counteracts the development of renal dysfunction and progression of congestive heart failure in Ren-2 transgenic hypertensive rats with aorto-caval fistula. Clin. Exp. Pharmacol. Physiol. 42, 795–807. doi: 10.1111/1440-1681.12419
Chábová, V. C., Kujal, P., Skaroupkova, P., Varnourkova, Z., Vackova, S., Huskova, Z., et al. (2018). Combined inhibition of soluble epoxide hydrolase and renin-angiotensin system exhibits superior renoprotection to renin-angiotensin system blockade in 5/6 nephrectomized Ren-2 transgenic hypertensive rats with established chronic kidney disease. Kidney Blood Press. Res. 43, 329–349. doi: 10.1159/000487902
Chen, D., Whitcomb, R., MacIntyre, E., Tran, V., Do, Z. N., Sabry, J., et al. (2012). Pharmacokinetics and pharmacodynamics of AR9281, an inhibitor of soluble epoxide hydrolase, in single- and multiple-dose studies in healthy human subjects. J. Clin. Pharmacol. 52, 319–328. doi: 10.1177/0091270010397049
Chen, G., Xu, R., Wang, Y., Wang, P., Zhao, G., Xu, X., et al. (2012). Genetic disruption of soluble epoxide hydrolase is protective against streptozotocin-induced diabetic nephropathy. Am. J. Physiol. Endocrinol. Metab. 303,E563–E575. doi: 10.1152/ajpendo.00591.2011
Cheung, K. L., Zakai, N. A., Callas, P. W., Howard, G., Mahmoodi, B. K., Peralta, C. A., et al. (2018). Mechanisms and mitigating factors for venous thromboembolism in chronic kidney disease: the REGARDS study. J. Thromb. Haemost. 16, 1743–1752. doi: 10.1111/jth.14235
Chiang, C. W., Lee, H. T., Tarng, D. C., Kuo, K. L., Cheng, L. C., and Lee, T. S. (2015). Genetic deletion of soluble epoxide hydrolase attenuates inflammation and fibrosis in experimental obstructive nephropathy. Mediat. Inflamm. 2015:693260. doi: 10.1155/2015/693260
Decleves, A. E., Mathew, A. V., Cunard, R., and Sharma, K. (2011). AMPK mediates the initiation of kidney disease induced by a high-fat diet. J. Am. Soc. Nephrol. 22, 1846–1855. doi: 10.1681/ASN.2011010026
Deng, B. Q., Luo, Y., Kang, X., Li, C. B., Morisseau, C., Yang, J., et al. (2017). Epoxide metabolites of arachidonate and docosahexaenoate function conversely in acute kidney injury involved in GSK3 beta signaling. Proc. Natl. Acad. Sci. U.S.A. 114, 12608–12613. doi: 10.1073/pnas.1705615114
Dhanasekaran, A., Gruenloh, S., Jacobs, E., and Medhora, M. (2006). Epoxyeicosatrienoic acids (EETs) protect cardiovascular cells from apoptosis mediated by caspase 3-dependent pathways. FASEB J. 20, A123–A123.
Dreisbach, A. W., Japa, S., Sigel, A., Parenti, M. B., Hess, A. E., Srinouanprachanh, S. L., et al. (2005). The prevalence of CYP2C8, 2C9, 2J2, and soluble epoxide hydrolase polymorphisms in African Americans with hypertension. Am. J. Hypertens. 18, 1276–1281. doi: 10.1016/j.amjhyper.2005.04.019
Eddy, A. A., Lopez-Guisa, J. M., Okamura, D. M., and Yamaguchi, I. (2012). Investigating mechanisms of chronic kidney disease in mouse models. Pediatr. Nephrol. 27, 1233–1247. doi: 10.1007/s00467-011-1938-2
El-Sherbeni, A. A., Aboutabl, M. E., Zordoky, B. N. M., Anwar-Mohamed, A., and El-Kadi, A. O. S. (2013). Determination of the dominant arachidonic acid cytochrome P450 monooxygenases in rat heart, lung, kidney, and liver: protein expression and metabolite kinetics. AAPS J. 15, 112–122. doi: 10.1208/s12248-012-9425-7
Enayetallah, A. E., French, R. A., Barber, M., and Grant, D. F. (2006). Cell-specific subcellular localization of soluble epoxide hydrolase in human tissues. J. Histochem. Cytochem. 54, 329–335. doi: 10.1369/jhc.5A6808.2005
Enayetallah, A. E., French, R. A., Thibodeau, M. S., and Grant, D. F. (2004). Distribution of soluble epoxide hydrolase and of cytochrome P4502C8, 2C9, and 2J2 in human tissues. J. Histochem. Cytochem. 52, 447–454. doi: 10.1177/002215540405200403
Fan, F., Muroya, Y., and Roman, R. J. (2015). Cytochrome P450 eicosanoids in hypertension and renal disease. Curr. Opin. Nephrol. Hypertens. 24, 37–46. doi: 10.1097/MNH.0000000000000088
Fogo, A. B. (2007). Mechanisms of progression of chronic kidney disease. Pediatr. Nephrol. 22, 2011–2022. doi: 10.1007/s00467-007-0524-0
Gervasini, G., Garcia-Cerrada, M., Coto, E., Vergara, E., Garcia-Pino, G., Alvarado, R., et al. (2015). A 3′ -UTR polymorphism in soluble epoxide hydrolase gene is associated with acute rejection in renal transplant recipients. PLoS One 10:e0133563. doi: 10.1371/journal.pone.0133563
Glodowski, S. D., and Wagener, G. (2015). New insights into the mechanisms of acute kidney injury in the intensive care unit. J. Clin. Anesth. 27, 175–180. doi: 10.1016/j.jclinane.2014.09.011
Gomez, G. A., Morisseau, C., Hammock, B. D., and Christianson, D. W. (2004). Structure of human epoxide hydrolase reveals mechanistic inferences on bifunctional catalysis in epoxide and phosphate ester hydrolysis. Biochemistry 43, 4716–4723. doi: 10.1021/bi036189j
Hashimoto, T., Fang, Y. I., Ohata, H., and Honda, K. (2015). Change in soluble epoxide hydrolase (sEH) during cisplatin-induced acute renal failure in mice. J. Toxicol. Sci. 40, 451–457. doi: 10.2131/jts.40.451
He, J. L., Wang, C. J., Zhu, Y., and Ai, D. (2016). Soluble epoxide hydrolase: a potential target for metabolic diseases. J. Diabetes 8, 305–313. doi: 10.1111/1753-0407.12358
Hu, D. Y., Luo, Y., Li, C. B., Zhou, C. Y., Li, X. H., Peng, A., et al. (2018). Oxylipin profiling of human plasma reflects the renal dysfunction in uremic patients. Metabolomics 14:104. doi: 10.1007/s11306-018-1402-4
Huang, H., Morisseau, C., Wang, J. F., Yang, T. X., Falck, J. R., Hammock, B. D., et al. (2007). Increasing or stabilizing renal epoxyeicosatrienoic acid production attenuates abnormal renal function and hypertension in obese rats. Am. J. Physiol. Renal Physiol. 293, F342–F349. doi: 10.1152/ajprenal.00004.2007
Imig, J. D. (2015). Epoxyeicosatrienoic acids, hypertension, and kidney injury. Hypertension 65, 476–482. doi: 10.1161/HYPERTENSIONAHA.114.03585
Imig, J. D., Carpenter, M. A., and Shaw, S. (2009). The soluble epoxide hydrolase inhibitor AR9281 decreases blood pressure, ameliorates renal injury and improves vascular function in hypertension. Pharmaceuticals 2, 217–227. doi: 10.3390/ph2030217
Imig, J. D., and Hammock, B. D. (2009). Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat. Rev. Drug Discov. 8, 794–805. doi: 10.1038/nrd2875
Imig, J. D., Walsh, K. A., Khan, M. A. H., Nagasawa, T., Cherian-Shaw, M., Shaw, S. M., et al. (2012). Soluble epoxide hydrolase inhibition and peroxisome proliferator activated receptor gamma agonist improve vascular function and decrease renal injury in hypertensive obese rats. Exp. Biol. Med. 237, 1402–1412. doi: 10.1258/ebm.2012.012225
Imig, J. D., Zhao, X. Y., Capdevila, J. H., Morisseau, C., and Hammock, B. D. (2002). Soluble epoxide hydrolase inhibition lowers arterial blood pressure in angiotensin II hypertension. Hypertension 39, 690–694. doi: 10.1161/hy0202.103788
Imig, J. D., Zhao, X. Y., Zaharis, C. Z., Olearczyk, J. J., Pollock, D. M., Newman, J. W., et al. (2005). An orally active epoxide hydrolase inhibitor lowers blood pressure and provides renal protection in salt-sensitive hypertension. Hypertension 46, 975–981. doi: 10.1161/01.HYP.0000176237.74820.75
Inceoglu, B., Jinks, S. L., Ulu, A., Hegedus, C. M., Georgi, K., Schmelzer, K. R., et al. (2008). Soluble epoxide hydrolase and epoxyeicosatrienoic acids modulate two distinct analgesic pathways. Proc. Natl. Acad. Sci. U.S.A. 105, 18901–18906. doi: 10.1073/pnas.0809765105
Iyer, R., Fetterly, G., Lugade, A., and Thanavala, Y. (2010). Sorafenib: a clinical and pharmacologic review. Expert Opin. Pharmacother. 11, 1943–1955. doi: 10.1517/14656566.2010.496453
Johansson, C., Stark, A., Sandberg, M., Ek, B., Rask, L., and Meijer, J. (1995). Tissue-specific basal expression of soluble murine epoxide hydrolase and effects of clofibrate on the messenger-rna levels in extrahepatic tissues and liver. Arch. Toxicol. 70, 61–63. doi: 10.1007/s002040050250
Jung, O., Jansen, F., Mieth, A., Barbosa-Sicard, E., Pliquett, R. U., Babelova, A., et al. (2010). Inhibition of the soluble epoxide hydrolase promotes albuminuria in mice with progressive renal disease. PLoS One 5:e11979. doi: 10.1371/journal.pone.0011979
Katary, M. M., Pye, C., and Elmarakby, A. A. (2017). Meloxicam fails to augment the reno-protective effects of soluble epoxide hydrolase inhibition in streptozotocin-induced diabetic rats via increased 20-HETE levels. Prostaglandins Other Lipid Mediat. 132, 3–11. doi: 10.1016/j.prostaglandins.2016.08.004
Kato, Y., Fuchi, N., Nishimura, Y., Watanabe, A., Yagi, M., Nakadera, Y., et al. (2014). Discovery of 1-oxa-4,9-diazaspiro[5.5]undecane-based trisubstituted urea derivatives as highly potent soluble epoxide hydrolase inhibitors and orally active drug candidates for treating of chronic kidney diseases. Bioorg. Med. Chem. Lett. 24, 565–570. doi: 10.1016/j.bmcl.2013.12.020
Kim, J., Imig, J. D., Yang, J., Hammock, B. D., and Padanilam, B. J. (2014). Inhibition of soluble epoxide hydrolase prevents renal interstitial fibrosis and inflammation. Am. J. Physiol. Renal Physiol. 307, F971–F980. doi: 10.1152/ajprenal.00256.2014
Kim, J., Yoon, S. P., Toews, M. L., Imig, J. D., Hwang, S. H., Hammock, B. D., et al. (2015). Pharmacological inhibition of soluble epoxide hydrolase prevents renal interstitial fibrogenesis in obstructive nephropathy. Am. J. Physiol. Renal Physiol. 308, F131–F139. doi: 10.1152/ajprenal.00531.2014
Koeners, M. P., Wesseling, S., Ulu, A., Sepulveda, R. L., Morisseau, C., Braam, B., et al. (2011). Soluble epoxide hydrolase in the generation and maintenance of high blood pressure in spontaneously hypertensive rats. Am. J. Physiol. Endocrinol. Metab. 300, E691–E698. doi: 10.1152/ajpendo.00710.2010
Kopkan, L., Huskova, Z., Sporkova, A., Varcabova, S., Honetschlagerova, Z., Hwang, S. H., et al. (2012). Soluble epoxide hydrolase inhibition exhibits antihypertensive actions independently of nitric oxide in mice with renovascular hypertension. Kidney Blood Press. Res. 35, 595–607. doi: 10.1159/000339883
Kujal, P., Chabova, V. C., Skaroupkova, P., Huskova, Z., Vernerova, Z., Kramer, H. J., et al. (2014). Inhibition of soluble epoxide hydrolase is renoprotective in 5/6 nephrectomized Ren-2 transgenic hypertensive rats. Clin. Exp. Pharmacol. Physiol. 41, 227–237. doi: 10.1111/1440-1681.12204
Kusch, A., Hoff, U., Bubalo, G., Zhu, Y., Fechner, M., Schmidt-Ullrich, R., et al. (2013). Novel signalling mechanisms and targets in renal ischaemia and reperfusion injury. Acta Physiol. 208, 25–40. doi: 10.1111/apha.12089
Larsson, C., White, I., Johansson, C., Stark, A., and Meijer, J. (1995). Localization of the human soluble epoxide hydrolase gene (Ephx2) to chromosomal region 8p21-P12. Hum. Genet. 95, 356–358. doi: 10.1007/BF00225209
Lazaar, A. L., Yang, L., Boardley, R. L., Goyal, N. S., Robertson, J., Baldwin, S. J., et al. (2016). Pharmacokinetics, pharmacodynamics and adverse event profile of GSK2256294, a novel soluble epoxide hydrolase inhibitor. Br. J. Clin. Pharmacol. 81, 971–979. doi: 10.1111/bcp.12855
Lee, J. P., Yang, S. H., Kim, D. K., Lee, H., Kim, B., Cho, J. Y., et al. (2011). In vivo activity of epoxide hydrolase according to sequence variation affects the progression of human IgA nephropathy. Am. J. Physiol. Renal Physiol. 300, F1283–F1290. doi: 10.1152/ajprenal.00733.2010
Lee, S. H., Lee, J., Cha, R., Park, M. H., Ha, J. W., Kim, S., et al. (2008). Genetic variations in soluble epoxide hydrolase and graft function in kidney transplantation. Transplant. Proc. 40, 1353–1356. doi: 10.1016/j.transproceed.2008.03.137
Li, L. J., Li, N., Pang, W., Zhang, X., Hammock, B. D., Ai, D., et al. (2014). Opposite effects of gene deficiency and pharmacological inhibition of soluble epoxide hydrolase on cardiac fibrosis. PLoS One 9:e94092. doi: 10.1371/journal.pone.0094092
Liang, Y. X., Jing, Z. Y., Deng, H., Li, Z. Q., Zhuang, Z., Wang, S., et al. (2015). Soluble epoxide hydrolase inhibition ameliorates proteinuria-induced epithelial-mesenchymal transition by regulating the PI3K-Akt-GSK-3 beta signaling pathway. Biochem. Biophys. Res. Commun. 463, 70–75. doi: 10.1016/j.bbrc.2015.05.020
Lin, Y. F., and Sheu, W. H. H. (2014). From sugar to kidney: a never-ending battle. J. Diabetes Investig. 5, 482–483. doi: 10.1111/jdi.12230
Liu, J. Y., Park, S. H., Morisseau, C., Hwang, S. H., Hammock, B. D., and Weiss, R. H. (2009). Sorafenib has soluble epoxide hydrolase inhibitory activity, which contributes to its effect profile in vivo. Mol. Cancer Ther. 8, 2193–2203. doi: 10.1158/1535-7163.MCT-09-0119
Liu, J. Y., Qiu, H., Morisseau, C., Hwang, S. H., Tsai, H. J., Ulu, A., et al. (2011). Inhibition of soluble epoxide hydrolase contributes to the anti-inflammatory effect of antimicrobial triclocarban in a murine model. Toxicol. Appl. Pharmacol. 255, 200–206. doi: 10.1016/j.taap.2011.06.017
Liu, Y. M., Lu, X. D., Nguyen, S., Olson, J. L., Webb, H. K., and Kroetz, D. L. (2013). Epoxyeicosatrienoic acids prevent cisplatin-induced renal apoptosis through a p38 mitogen-activated protein kinase-regulated mitochondrial pathway. Mol. Pharmacol. 84, 925–934. doi: 10.1124/mol.113.088302
Liu, Y. M., Webb, H. K., Fukushima, H., Micheli, J., Markova, S., Olson, J. L., et al. (2012). Attenuation of cisplatin-induced renal injury by inhibition of soluble epoxide hydrolase involves nuclear factor kappa B signaling. J. Pharmacol. Exp. Ther. 341, 725–734. doi: 10.1124/jpet.111.191247
Ma, L., Yan, M., Kong, X., Jiang, Y., Zhao, T., Zhao, H., et al. (2018). Association of EPHX2 R287Q polymorphism with diabetic nephropathy in Chinese type 2 diabetic patients. J. Diabetes Res. 2018:6. doi: 10.1155/2018/2786470
Malyszko, J. (2010). Mechanism of endothelial dysfunction in chronic kidney disease. Clin. Chim. Acta 411, 1412–1420. doi: 10.1016/j.cca.2010.06.019
Mariappan, M. M. (2012). Signaling mechanisms in the regulation of renal matrix metabolism in diabetes. Exp. Diabetes Res. 2012:749812. doi: 10.1155/2012/749812
Mennuni, S., Rubattu, S., Pierelli, G., Tocci, G., Fofi, C., and Volpe, M. (2014). Hypertension and kidneys: unraveling complex molecular mechanisms underlying hypertensive renal damage. J. Hum. Hypertens. 28, 74–79. doi: 10.1038/jhh.2013.55
Mihai, S., Codrici, E., Popescu, I. D., Enciu, A. M., Albulescu, L., Necula, L. G., et al. (2018). Inflammation-related mechanisms in chronic kidney disease prediction, progression, and outcome. J. Immunol. Res. 2018:2180373. doi: 10.1155/2018/2180373
Moradi, H., and Vaziri, N. D. (2018). Molecular mechanisms of disorders of lipid metabolism in chronic kidney disease. Front. Biosci. 23, 146–161. doi: 10.2741/4585
Morisseau, C., and Hammock, B. D. (2013). Impact of soluble epoxide hydrolase and epoxyeicosanoids on human health. Annu. Rev. Pharmacol. Toxicol. 53, 37–58. doi: 10.1146/annurev-pharmtox-011112-140244
Newman, J. W., Morisseau, C., and Hammock, B. D. (2005). Epoxide hydrolases: their roles and interactions with lipid metabolism. Prog. Lipid Res. 44, 1–51. doi: 10.1016/j.plipres.2004.10.001
Ng, R., and Chen, E. X. (2006). Sorafenib (BAY 43-9006): review of clinical development. Curr. Clin. Pharmacol. 1, 223–228. doi: 10.2174/157488406778249325
Nieves, D., and Moreno, J. J. (2007). Epoxyeicosatrienoic acids induce growth inhibition and calpain/caspase-12 dependent apoptosis in PDGF cultured 3T6 fibroblast. Apoptosis 12, 1979–1988. doi: 10.1007/s10495-007-0123-3
Node, K., Huo, Y. Q., Ruan, X. L., Yang, B. C., Spiecker, M., Ley, K., et al. (1999). Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science 285, 1276–1279. doi: 10.1126/science.285.5431.1276
Oguro, A., Fujita, N., and Imaoka, S. (2009). Regulation of soluble epoxide hydrolase (sEH) in mice with diabetes: high glucose suppresses sEH expression. Drug Metab. Pharmacokinet. 24, 438–445. doi: 10.2133/dmpk.24.438
Olearczyk, J. J., Quigley, J. E., Mitchell, B. C., Yamamoto, T., Kim, I. H., Newman, J. W., et al. (2009). Administration of a substituted adamantyl urea inhibitor of soluble epoxide hydrolase protects the kidney from damage in hypertensive Goto-Kakizaki rats. Clin. Sci. 116, 61–70. doi: 10.1042/CS20080039
Parrish, A. R., Chen, G., Burghardt, R. C., Watanabe, T., Morisseau, C., and Hammock, B. D. (2009). Attenuation of cisplatin nephrotoxicity by inhibition of soluble epoxide hydrolase. Cell Biol. Toxicol. 25, 217–225. doi: 10.1007/s10565-008-9071-0
Patel, B. N., Mackness, M. I., Nwosu, V., and Connock, M. J. (1986). Subcellular-localization of epoxide hydrolase in mouse-liver and kidney. Biochem. Pharmacol. 35, 231–235. doi: 10.1016/0006-2952(86)90519-8
Persson, P. B. (2013). Mechanisms of acute kidney injury. Acta Physiol. 207, 430–431. doi: 10.1111/apha.12063
Prieto-Garcia, L., Pericacho, M., Sancho-Martinez, S. M., Sanchez, A., Martinez-Salgado, C., Lopez-Novoa, J. M., et al. (2016). Mechanisms of triple whammy acute kidney injury. Pharmacol. Ther. 167, 132–145. doi: 10.1016/j.pharmthera.2016.07.011
Rawal, S., Morisseau, C., Hammock, B. D., and Shivachar, A. C. (2009). Differential subcellular distribution and colocalization of the microsomal and soluble epoxide hydrolases in cultured neonatal rat brain cortical astrocytes. J. Neurosci. Res. 87, 218–227. doi: 10.1002/jnr.21827
Remuzzi, G., Benigni, A., and Remuzzi, A. (2006). Mechanisms of progression and regression of renal lesions of chronic nephropathies and diabetes. J. Clin. Invest. 116, 288–296. doi: 10.1172/JCI27699
Roche, C., Guerrot, D., Harouki, N., Duflot, T., Besnier, M., Remy-Jouet, I., et al. (2015). Impact of soluble epoxide hydrolase inhibition on early kidney damage in hyperglycemic overweight mice. Prostaglandins Other Lipid Mediat. 120, 148–154. doi: 10.1016/j.prostaglandins.2015.04.011
Rossi, G. P., Seccia, T. M., Barton, M., Danser, A. H. J., de Leeuw, P. W., Dhaun, N., et al. (2018). Endothelial factors in the pathogenesis and treatment of chronic kidney disease Part I: general mechanisms: a joint consensus statement from the European society of hypertension working group on endothelin and endothelial factors and the Japanese society of hypertension. J. Hypertens. 36, 451–461. doi: 10.1097/HJH.0000000000001599
Sandberg, M., and Meijer, J. (1996). Structural characterization of the human soluble epoxide hydrolase gene (EPHX2). Biochem. Biophys. Res. Commun. 221, 333–339. doi: 10.1006/bbrc.1996.0596
Seubert, J. M., Xu, F., Graves, J. P., Collins, J. B., Sieber, S. O., Paules, R. S., et al. (2005). Differential renal gene expression in prehypertensive and hypertensive spontaneously hypertensive rats. Am. J. Physiol. Renal Physiol. 289, F552–F561. doi: 10.1152/ajprenal.00354.2004
Sharma, A., Hye Khan, M. A., Levick, S. P., Lee, K. S., Hammock, B. D., and Imig, J. D. (2016). Novel Omega-3 fatty acid epoxygenase metabolite reduces kidney fibrosis. Int. J. Mol. Sci. 17:E751. doi: 10.3390/ijms17050751
Shuey, M. M., Billings, F. T., Wei, S. Z., Milne, G. L., Nian, H., Yu, C., et al. (2017). Association of gain-of-function EPHX2 polymorphism Lys55Arg with acute kidney injury following cardiac surgery. PLoS One 12:e0175292. doi: 10.1371/journal.pone.0175292
Sirish, P., Li, N., Liu, J. Y., Lee, K. S. S., Hwang, S. H., Qiu, H., et al. (2013). Unique mechanistic insights into the beneficial effects of soluble epoxide hydrolase inhibitors in the prevention of cardiac fibrosis. Proc. Natl. Acad. Sci. U.S.A. 110, 5618–5623. doi: 10.1073/pnas.1221972110
Smalley, K. S., Xiao, M., Villanueva, J., Nguyen, T. K., Flaherty, K. T., Letrero, R., et al. (2009). CRAF inhibition induces apoptosis in melanoma cells with non-V600E BRAF mutations. Oncogene 28, 85–94. doi: 10.1038/onc.2008.362
Spector, A. A., Fang, X., Snyder, G. D., and Weintraub, N. L. (2004). Epoxyeicosatrienoic acids (EETs): metabolism and biochemical function. Prog. Lipid Res. 43, 55–90. doi: 10.1016/S0163-7827(03)00049-3
Spector, A. A., and Norris, A. W. (2007). Action of epoxyeicosatrienoic acids on cellular function. Am. J. Physiol. Cell Physiol. 292, C996–C1012. doi: 10.1152/ajpcell.00402.2006
Sporkova, A., Kopkan, L., Varcabova, S., Huskova, Z., Hwang, S. H., Hammock, B. D., et al. (2011). Role of cytochrome P-450 metabolites in the regulation of renal function and blood pressure in 2-kidney 1-clip hypertensive rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 300, R1468–R1475. doi: 10.1152/ajpregu.00215.2010
Tain, Y. L., Huang, L. T., Chan, J. Y. H., and Lee, C. T. (2015). Transcriptome analysis in rat kidneys: importance of genes involved in programmed hypertension. Int. J. Mol. Sci. 16, 4744–4758. doi: 10.3390/ijms16034744
Takaori, K., and Yanagita, M. (2016). Insights into the mechanisms of the acute kidney injury-to-chronic kidney disease continuum. Nephron 134, 172–176. doi: 10.1159/000448081
U.S. Renal Data System (2009). USRDS 2009 Annual Data Report: Atlas of Chronic Kidney Disease and End-Stage Renal Disease in the United States. Bethesda, MD: National Institute of Diabetes and Digestive and Kidney Diseases.
Walkowska, A., Skaroupkova, P., Huskova, Z., Vanourkova, Z., Chabova, V. C., Tesar, V., et al. (2010). Intrarenal cytochrome P-450 metabolites of arachidonic acid in the regulation of the nonclipped kidney function in two-kidney, one-clip Goldblatt hypertensive rats. J. Hypertens. 28, 582–593. doi: 10.1097/HJH.0b013e328334dfd4
Wang, D. Z., and DuBois, R. N. (2012). Epoxyeicosatrienoic acids: a double-edged sword in cardiovascular diseases and cancer. J. Clin. Invest. 122, 19–22. doi: 10.1172/JCI61453
Wang, Q., Liang, Y., Qiao, Y., Zhao, X., Yang, Y., Yang, S., et al. (2018). Expression of soluble epoxide hydrolase in renal tubular epithelial cells regulates macrophage infiltration and polarization in IgA nephropathy. Am. J. Physiol. Renal Physiol. 315, F915–F926. doi: 10.1152/ajprenal.00534.2017
Wang, Q., Pang, W., Cui, Z., Shi, J. B., Liu, Y., Liu, B., et al. (2013). Upregulation of soluble epoxide hydrolase in proximal tubular cells mediated proteinuria-induced renal damage. Am. J. Physiol. Renal Physiol. 304, F168–F176. doi: 10.1152/ajprenal.00129.2012
Wilhelm, S. M., Adnane, L., Newell, P., Villanueva, A., Llovet, J. M., and Lynch, M. (2008). Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol. Cancer Ther. 7, 3129–3140. doi: 10.1158/1535-7163.MCT-08-0013
Xie, Y., Bowe, B., Mokdad, A. H., Xian, H., Yan, Y., Li, T. T., et al. (2018). Analysis of the Global Burden of Disease study highlights the global, regional, and national trends of chronic kidney disease epidemiology from 1990 to 2016. Kidney Int. 94, 567–581. doi: 10.1016/j.kint.2018.04.011
Yang, S. H., Kim, Y. C., An, J. N., Kim, J. H., Lee, J., Lee, H. Y., et al. (2017). Active maintenance of endothelial cells prevents kidney fibrosis. Kidney Res. Clin. Pract. 36, 329–341. doi: 10.23876/j.krcp.2017.36.4.329
Yu, Z. G., Davis, B. B., Morisseau, C., Hammock, B. D., Olson, J. L., Kroetz, D. L., et al. (2004). Vascular localization of soluble epoxide hydrolase in the human kidney. Am. J. Physiol. Renal Physiol. 286, F720–F726. doi: 10.1152/ajprenal.00165.2003
Yu, Z. G., Xu, F. Y., Huse, L. M., Morisseau, C., Draper, A. J., Newman, J. W., et al. (2000). Soluble epoxide hydrolase regulates hydrolysis of vasoactive epoxyeicosatrienoic acids. Circ. Res. 87, 992–998. doi: 10.1161/01.RES.87.11.992
Zeldin, D. C., Wei, S. Z., Falck, J. R., Hammock, B. D., Snapper, J. R., and Capdevila, J. H. (1995). Metabolism of epoxyeicosatrienoic acids by cytosolic epoxide hydrolase - substrate structural determinants of asymmetric catalysis. Arch. Biochem. Biophys. 316, 443–451. doi: 10.1006/abbi.1995.1059
Zhao, G., Tu, L., Li, X. G., Yang, S. L., Chen, C., Xu, X. Z., et al. (2012). Delivery of AAV2-CYP2J2 protects remnant kidney in the 5/6-nephrectomized rat via inhibition of apoptosis and fibrosis. Hum. Gene Ther. 23, 688–699. doi: 10.1089/hum.2011.135
Zhao, X. Y., Yamamoto, T., Newman, J. W., Kim, I. H., Watanabe, T., Hammock, B. D., et al. (2004). Soluble epoxide hydrolase inhibition protects the kidney from hypertension-induced damage. J. Am. Soc. Nephrol. 15, 1244–1253.
Keywords: chronic kidney disease, acute kidney disease, soluble epoxide hydrolase, epoxyeicosatrienoic acid, renal dysfunction
Citation: Liu J-Y (2019) Inhibition of Soluble Epoxide Hydrolase for Renal Health. Front. Pharmacol. 9:1551. doi: 10.3389/fphar.2018.01551
Received: 20 October 2018; Accepted: 19 December 2018;
Published: 10 January 2019.
Edited by:
John D. Imig, Medical College of Wisconsin, United StatesReviewed by:
John M. Seubert, University of Alberta, CanadaMd Abdul Hye Khan, Medical College of Wisconsin, United States
Craig R. Lee, University of North Carolina at Chapel Hill, United States
Copyright © 2019 Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun-Yan Liu, jyliu@tongji.edu.cn orcid.org/0000-0002-3018-0335