 Aude Angelini
Aude Angelini Xinchun Pi
Xinchun Pi Liang Xie
Liang Xie- Department of Medicine-Athero and Lipo, Cardiovascular Research Institute, Baylor College of Medicine, Houston, TX, United States
The heart must consume a significant amount of energy to sustain its contractile activity. Although the fuel demands are huge, the stock remains very low. Thus, in order to supply its daily needs, the heart must have amazing adaptive abilities, which are dependent on dioxygen availability. However, in myriad cardiovascular diseases, “fuel” depletion and hypoxia are common features, leading cardiomyocytes to favor low-dioxygen-consuming glycolysis rather than oxidation of fatty acids. This metabolic switch makes it challenging to distinguish causes from consequences in cardiac pathologies. Finally, despite the progress achieved in the past few decades, medical treatments have not improved substantially, either. In such a situation, it seems clear that much remains to be learned about cardiac diseases. Therefore, in this review, we will discuss how reconciling dioxygen availability and cardiac metabolic adaptations may contribute to develop full and innovative strategies from bench to bedside.
Introduction
In eukaryotes, dioxygen and metabolism are intrinsically bound to energy production. And the major issue of cell metabolism is to supply the tricarboxylic cycle (TCA cycle, also called Krebs cycle or citrate cycle) in acetyl-CoA molecules and to reduce the oxido-reduction coenzymes (FADH2, NADH,H+), in order to continuously sustain the production of ATP by the mitochondrial respiratory chain. In the presence of dioxygen, cell respiration is thus hugely preferred, due to their more positive ATP production efficiency. However, any disturbance, which targets these metabolic pathways, could make the cell metabolism switch into anaerobic process, disrupting the equilibrium between oxidation/reduction processes. During the last decade, modification of both redox state and metabolism imbalance were found to be crucial factors in various physiological pathways from development to aging, and pathological processes including cancer, diabetes mellitus, neurological disorders and cardiovascular diseases (Douglas, 1963; Cunnane et al., 2011; Ma and Li, 2015).
Cardiovascular diseases represent the leading cause of mortality worldwide, while their morbidity is considered as an important economic burden for patients and for their society (Braunwald, 2013, p. 1). Heart failure is a clinical syndrome characterized by the low contractile ability of the myocardium. In the United States, more than 5 million people suffer from heart failure (HF) (Roger et al., 2012). In addition, in spite of significant declines in mortality, the 5-year survival rate is still 50% worse than cancers (Levy et al., 2002; Askoxylakis et al., 2010). Despite advances in our understanding and knowledge of HF, the complexity of the pathophysiology remains a barrier to effective therapeutic strategies. Although there are numerous etiologies that can make a heart fail, alterations of cardiac metabolism and redox-status can be depicted together as a fatal cocktail, dramatically propelling the disease to its lethal outcome (Tsutsui et al., 2011; Wang et al., 2014).
The heart is particularly sensitive to a wide range of environmental parameters, such as glucose and fatty acids (FA) blood levels or blood pressure, which are directly influenced by paracrine, endocrine hormones or neural regulators (Gordan et al., 2015). Hence, any disturbance will be able to affect its function. As a high consumer of energy, the heart must adapt its contractile activity and metabolic function to fit with the environmental availability of fuel sources and dioxygen. However, such a liability has its pros and cons. In this review, we will examine the cardiac metabolism and its involvement in cardiac disease and, conversely, how the metabolic switch from fatty acid oxidation to glycolysis and the alteration of dioxygen availability take part in the pathological progression of HF.
Cardiac Metabolism
Beating 100,000 times every day to pump about 7,500 l of blood, the heart must consume a large amount of energy in order to sustain critical and constant contraction (Abozguia et al., 2009). Although the energy demand is huge, the cardiac stock of fuel is very low, (5 μg of ATP and 8 μg of phosphocreatine per gram of tissue) sufficient for only 10 beats (Beer et al., 2002). Consequently, ATP production must be at a high rate all the time. In the adult human heart, ATP production is estimated at 6 kg per day, representing more than 20–30 times its own mass (Opie, 1968, 1969, 2004; Abozguia et al., 2009; Lopaschuk et al., 2010), demonstrating the enormous capacity of cardiac metabolism.
Under physiological conditions, cardiac metabolism involves three main steps: utilization of substrates; oxidative phosphorylation; ATP transfer and use. Changes in either of these steps may affect the myocardial energy metabolism. In the first step, cardiomyocytes take up and breakdown fuel sources. The heart is an “omnivore” organ, consuming a variety of substances to support its energy needs. A healthy adult heart mainly uses fatty acids (90%) and glucose (10%) as substrates, which are then metabolized by FA β-oxidation (FAO) and glycolysis respectively (Lopaschuk et al., 2010). FA are provided within the cells in the form of triglycerides, which are insoluble lipid complexes associated with lipoproteins or chylomicrons. Cardiomyocyte uptake of fatty acids across the plasmid membrane has been considered a passive diffusion process, but it can be usually facilitated by the fatty acid translocase CD36 (Campbell et al., 2004). Inside the cytosol, free FA are converted into fatty acyl-CoA by fatty acyl-CoA synthase. Then, the second, and most rate limiting, step is the transport of FA across the double mitochondrial membrane into the mitochondrial matrix, when the FAO occurs. For long-chain FA, a carnitine shuttle is required, facilitated by the carnitine-palmitoyl transferase 1 (CPT1) (Saggerson, 1982; Bonnefont et al., 2004). CPT1B is the major isoform located at the outer membrane of the cardiomyocyte mitochondria (Saggerson, 1982; Leij et al., 2002; Hada et al., 2012). In the heart, CPT1B represents a checkpoint that responds to metabolic feedback for FAO (He et al., 2012). Indeed, when citrate TCA intermediates are accumulated within the cells, they can be converted into MalonylCoA molecules, which represses CPT activity in a negative feedback loop, thus modulating efficiently the FAO (Saggerson, 1982; Awan and Saggerson, 1993; Reszko et al., 2004). CPT enzymes belong to the larger family of carnitine acyltransferases, which includes the carnitine O-acetyl transferases (CRATs) and the carnitine octanoyltransferases (COTs) (Hsiao et al., 2006). In this family, the carnitine binding site and the catalytic domain are particularly well-conserved. However, according to their respective ultrastructure, each subfamily would hold specific substrates preferences, these being long-chain FA for CPTs, medium-long FA for COTs and short-chain FA for CRATs (Cox et al., 1998; van der Leij et al., 2000; Hsiao et al., 2006; Seiler et al., 2014). This biochemical data could suggest that the involvement of CRATs and/or COTs in FAO is more substantial than usually thought.
Within mitochondria, fatty acyls are finally included into the β-oxidation breakdown process. FAO is a complex catabolic process comprising a sequence of four reactions: 1- Dehydrogenation, 2- Hydration, 3- Oxidation, 4- Thiolysis. Each sequence leads to the cleavage of a n carbons fatty acyl-CoA into an acetyl-CoA and a (n-2) carbons fatty acyl-CoA. Meanwhile the steps 1 and 3 require the reduction of FAD and NAD+ coenzymes respectively. Hence, finally, in theory, for a 2n carbons fatty acyl, it can be produced n acetyl-CoA, consuming n H2O and reducing back n-1 FADH2 and NADH,H+ coenzymes which will lead to ATP production (Lopaschuk et al., 2010).
Notwithstanding the efficiency of FAO, glycolysis forms an integral part in cardiac metabolism. Glycolysis is able to supply in coenzymes for the TCA cycle in a less-oxygen dependent way, which preserves an equilibrium with the high-oxygen consumer FAO. In addition, beyond to the net metabolic imbalance, glycolysis intermediates can also initiate the production of the indispensable pentoses (riboses and desoxyriboses) within the cardiac cells (Wisneski et al., 1985; Barcia-Vieitez and Ramos-Martínez, 2014). Glucose cell uptake involves specific glucose transporters (GLUT), located at the plasma membrane. In cardiac muscle, GLUT1 and GLUT4 are the most represented transporters and GLUT4 endocytosis depends on insulin (Watson and Pessin, 2001, p. 4; Abel, 2004; Luiken et al., 2004; Aerni-Flessner et al., 2012). Glycolysis is a complex enzymatic process involving cytosolic kinases, isomerases and dehydrogenases (Opie, 2004). Finally, from each molecule of glucose, 2 pyruvates, 2 ATP and 2 NADH,H+ can be produced. Then, pyruvate can cross the double mitochondrial membrane, driven by specific carriers (mitochondrial pyruvate carriers, MPC1 and MPC2 (Bricker et al., 2012). On site within the matrix, pyruvate conversion into acetyl-coA is an oxidative step, which can be catalyzed by the pyruvate dehydrogenase (PDH) (Hansford and Cohen, 1978; Gray et al., 2014; Sun et al., 2015). The PDH represents another key enzyme metabolically feedback-sensitive enzyme (Stanley et al., 1996; Sugden and Holness, 2006), such that a high-amount of acetyl-CoA and NADH,H+ repress its activity, while a bigger pool of CoA and NAD+ is able to boost it (Gray et al., 2014). Finally, both glycolysis and FAO provide acetyl-CoA to fuel the TCA cycle (Barry, 2004). The TCA cycle uses acetyl-CoA as a carbon-pair donor to synthetize citrate from oxaloacetate by aldol condensation. The following steps are oxidoreduction processes, ensuring the reduction of coenzymes QH2 and NAD+/NADH,H+. The net ATP production is based on a proton electrochemical gradient established by the five mitochondrial respiratory chain complexes (complexes I-V), transferring an electron from NADH,H+ to oxygen. The proton uptake across the mitochondrial membrane by the F0-F1 ATP synthase (complex V) ensures the phosphorylation of ADP to ATP. Lastly, to ensure contraction of the heart muscle cells, ATP must be brought into the proper utilization site, the muscle fibers. However, the mitochondrial double membrane is roughly permeable to this molecule. Local mitochondrial creatine kinase initiates the energy shuttle to the cytosol by catalyzing the transfer of a high-energy phosphate from ATP to creatine, releasing ADP and a high-energy phosphocreatine (Ingwall et al., 1985; Wallimann et al., 1998; Schlattner et al., 2006; Figure 1). Due to its smaller size, phosphocreatine easily diffuses from mitochondria to myofibrils, where the muscular creatine kinase converts back energy from phosphocreatine into ATP, releasing creatine (Ingwall et al., 1985; Schlattner et al., 2006; Zervou et al., 2016). In turn, this ATP is used by actin-myosin complexes inside the myofibrils and converted into mechanical force.
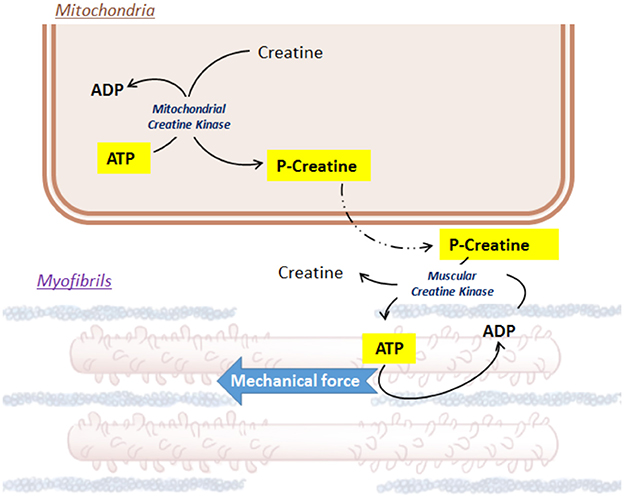
Figure 1. Focus on Creatine/ATP shuttle.
Oxygen Consumption and the Double-edged Redox Signaling in Cardiac Cells
The heart is the highest dioxygen consumer of all organs. Globally, 8–15 mL of dioxygen are perfused per min per 100 g of resting heart, and this rate can increase up to 6–7-fold during exercise, to match closer to ATP needs (Klabunde, 2012). Almost 90% of dioxygen is burnt within the mitochondria as an electron donor for oxidative phosphorylation. However, a lesser amount of dioxygen is used by the oxidase enzymes, mainly NADPH oxidases (Bedard and Krause, 2007; Lassègue et al., 2012) xanthine oxidases (Cantu-Medellin and Kelley, 2013; Battelli et al., 2016) and monoamine oxidases in cardiac cells (Viel et al., 2008; Kaludercic et al., 2014; Vendrov et al., 2015). However, for a minor portion of dioxygen (<2%), reduction is uncompleted, leading to the generation of reactive oxygen species (ROS), and especially the most unstable superoxide anion (O2•−), which can react with itself to produce the non-radical hydrogen peroxide (H2O2), and then OH•− by the Fenton reaction in presence of Fe2+, or OH by Haber-Weiss reaction (Andreyev et al., 2005; Chen et al., 2012). In addition, Nitric oxide synthase can also use O2•− to product nitric oxide (NO•), the precursor of reactive nitrogen species (RNS), leading to ONOO- and ONOOCO2 when they reacted with O2•− and CO2/HCO3- respectively (Augusto et al., 2002).
Thus, ROS/RNS are a common by-product of cellular respiration and metabolism, and their cellular level is tightly regulated by antioxidant systems including enzymatic or non-enzymatic scavenging strategies (β-carotene, ascorbic acid, glutathione-SH, etc.) (Andreyev et al., 2005; Nediani et al., 2010; Chen et al., 2012). Non-exhaustively, enzymatic ROS scavenging system involves the superoxide dismutases that catalyze the O2•− conversion to H2O2. Then, H2O2 can be decomposed in H2O by glutathioneperoxidase-1 and -4 or catalases for a lesser extent. However, an other important scavenging system involves peroxiredoxin-3 (Prx3)/Thioredoxin 2 (Trx2) and Thioredoxin Reductase-2 (TrxR2) axis (Stanley et al., 2011; Cunniff et al., 2014; Li H. et al., 2017). Recent work described TrxR2 as a major controller of H2O2 in mammalian heart mitochondria (Stanley et al., 2011). Interestingly, it has also been demonstrated that energization status of respiring mitochondria directly correlates with Trx2 level, thus suggesting a crucial role of Trx2/TrxR2 and Prx3 axis in mitochondrial function and furthermore in cardiomyocyte ATP production (Stanley et al., 2011).
Hence there are numerous and sophisticated scavenging strategies within healthy cardiomyocytes, and yet ROS remain at a minimal level. In fact, under physiological context, ROS/RNS hold beneficial and protective virtues. For instance, nitric oxide (NO•) regulates endothelium-dependant epicardial and microvascular vasodilation, favoring dioxygen perfusion in response to metabolic stimulation (Quyyumi et al., 1995). Moreover, ROS/RNS are involved as upstream effectors of cell signaling pathways via the now well-known redox signaling (Burgoyne et al., 2007; Nediani et al., 2010). One of the most redox-sensitive pathways is PI3K/AKT that plays a central role in cardiac metabolism and insulin sensitivity (Cook et al., 2002; Sugden, 2003; Morisco et al., 2005; O'Neill et al., 2007). As upstream regulators of MAPK, ERK1/2, p38, and JNK signaling pathways and kinases such as PKC or the histone deacetylates HDAC4 class II, ROS can impact on main transcription factors activity, such as NFAT, MEF2, SRF, and GATA4, which finally orchestrates protein synthesis, cardiomyocyte survival and differentiation, especially by Sabri et al. (1998), Wei et al. (2001), Matsushima et al. (2013), Barajas-Espinosa et al. (2015). In cardiac cells, redox signaling also impacts on main steps of myocyte contractility by targeting Ca2+ exchangers (SERCA2 or the ryanodine receptor) (Sharov et al., 2006; Bellis et al., 2009; Nediani et al., 2010; Tang et al., 2010; Snijder et al., 2011; Simon et al., 2014). Then, ROS have a clear impact on mitochondrial permeability by promoting the opening of the ATP potassium channel and the closure of the mitochondrial permeability transition pore (MPTP) (Cho et al., 2014). Furthermore, RNS (specifically the nitroxyl, HNO donor) can modulate myofilaments Ca2+ sensitivity. In fact, they ensure reversible formation of disulfite bonds between cysteine residues, which improves heart muscle contractility (Gao et al., 2012). Last but not least, according to recent studies, physiological cardiomyocyte stretching leads to an increase of ROS production by NOX2 in a microtubule-dependent manner. This mechano-chemo transduction pathway, called “X-ROS,” favors a surge of Ca2+ release due to a “tuning” of Ryanodine Receptors (Prosser et al., 2011, 2013; Ward et al., 2014). In addition, NOX2-induced oxidation of Ca2+/Calmoduline-dependant kinase, which has been previously described as an intermediary Ca2+ signaling and redox signaling, could be also involved in X-ROS redox signaling (Erickson et al., 2008; Snijder et al., 2011; Ward et al., 2014). Consequently, ROS/RNS are able to act as safeguards of mitochondrial respiratory and metabolic activity, but they remain at the border between physiological and maladaptive cardiac remodeling processes.
Oxidative stress occurs when there is an imbalance between production and scavenging of ROS, else due to an excess of production by the main sources, and/or by a decrease of antioxidant scavenging systems. Contrary to the virtues mentioned previously, under such a context, ROS/RNS become poisonous for the cells when the cellular antioxidant defenses become overwhelmed (Djordjević et al., 2008; Csányi and Miller, 2014). Due its high amount of metabolic activity and dioxygen consumption, the heart is particularly predisposed to excess of ROS. And, since they are particularly unstable and deleterious, ROS can combine with proteins or lipids in a more unspecific and irreversible manner, which can thus disrupt membranes integrity, cytoskeletal organization, metabolic and ionic homeostasis, and enzymatic stoichiometry (Giordano, 2005; Misra et al., 2009; Tsutsui et al., 2011; Csányi and Miller, 2014). During ischemia, myofilaments oxidation by carbonylation leads to a decrease in both Ca2+ sensitivity and myosin Ca2+ ATPase activity, resulting in a decrease in sarcomere contractility (Avner et al., 2012; Balogh et al., 2014). In addition, contrary to physiological process, ROS/RNS can trigger cellular Ca2+ overload, which can reduce Ca2+ sensitivity within the muscle fibers, thus finally triggering the contractility defect and rushing the cardiac cell toward necrosis or apoptosis (Ferrari et al., 2004; Giordano, 2005). Excess of ROS may exacerbate maladaptive process (Richters et al., 2011; Penna et al., 2013). For instance, ERK1/2 and HDAC4 II activation will favor cardiomyocyte hypertrophy, while overactivation of JNK could promote apoptosis (Matsushima et al., 2013; Mutlak and Kehat, 2015; Kanaan and Harper, 2017). Oxidative stress could lead to cysteine oxidation of cGMP-dependant protein kinase (PKG) (Burgoyne et al., 2007). Recent studies demonstrates that expressing of a “redox dead” PKG1αC42S relatively protects the mice from hemodynamic-stress-induced adverse effects (Prysyazhna et al., 2012; Nakamura et al., 2015). By affecting both mitochondrial proteins and phospholipids, ROS also exacerbate the disruption of the mitochondrial respiratory chain and the loss of ATP production. Additional studies described that increase of ROS production by xanthine oxidase is involved in impaired energy metabolism during heart failure and myocardial infarction, and allopurinol (inhibitor of xanthine oxidase) prevents these deleterious effects (Wang Z. et al., 2016; Schuchardt et al., 2017).
Cardiac metabolism and adaptation are thus widely dependent on redox balance within the cells of the myocardium. Meanwhile, ROS level is deeply sensitive to the energy performance within cardiomyocytes. Recently, it has been demonstrated that oxygen consumption and H2O2 production are directly proportional to level of long-chain palmitoyl-CoA concentration (Cortassa et al., 2017) which perfectly matches with this concept. This also underlines the significant issues that drive heart metabolic adaptions. Consequently, linked together, this amount of knowledge emphasizes ambiguous roles of ROS in cardiac physiology and pathology.
Lability and Physiological Adaptation within the Heart
When the heart is correctly perfused with adequate dioxygen concentration, up to 95% of the ATP production is due to oxidative phosphorylation of FA inside the mitochondria, while the remaining ~5% is provided by glycolysis and to a lesser extent the TCA cycle (<1%) (Scolletta and Biagioli, 2010). For 1 mole of an unsaturated long-chain fatty acids like palmitate, the net energy production is estimated at 128 ATP, whereas this rate reaches at only 38 ATP for 1 mole of glucose (Stanley et al., 1997). However, in comparison to glycolysis, FAO uses 12% more dioxygen and thus FAO is considered less dioxygen-efficient (Mjos, 1971; Grynberg and Demaison, 1996; Afanasiev et al., 2016). Consequently, even a major component of cardiac energy production, FAO is thus particularly sensitive to dioxygen concentration, and a tight equilibrium must be kept between availability of fuel sources. Dioxygen must be available during ATP production, and ATP is consumed during contraction. Therefore, the heart requires some flexibility and adaptability, metabolically switching between FAO and glycolysis when needed for optimal functionality. For almost five decades now, it has been well established that a reciprocal imbalance links FAO and glycolysis. This competitive process, historically described as the “Randle cycle” (Neely et al., 1972; Randle, 1998), is such that an increase of availability for one fuel source will favor its use, repressing the use of the others. For instance, an excess in acetyl-CoA production from FA breakdown inhibits PDH, thus reducing the glycolytic rate (Stanley et al., 1996; Sharma et al., 2005). By contrast, an activation of glycolysis favors the activity of acetyl-CoA carboxylase, which in turns increases the level of malonyl-CoA, thus reducing CPT1-dependant FA shuttle and β-oxidation (Awan and Saggerson, 1993; Reszko et al., 2004).
However, additional regulatory processes participate in this cardiac metabolic flexibility, beyond whatever substrate is available. These processes involve a wide range of biomolecular and biochemical parameters including protein activity and translocation as well as allosteric modulation of enzymes but also gene expression (nuclear and mitochondrial gene program). Among them, PPARs and PGC-1α represent powerful regulators of cardiac metabolism (Barger and Kelly, 2000; Campbell et al., 2002; Duncan and Finck, 2008; Finck et al., 2008; Rowe et al., 2010). PPAR isoforms (α, β, δ) can bind to Retinoid X Receptor (RXR) transcriptional factor, whereas PGC-1α secondly acts as a co-activator, increasing the transcriptional activity of the RXR-PPAR heterocomplex. PPARα is the major isoform in the cardiac muscle, whereas PGC-1α is more ubiquitous but particularly sensitive to a wide number of extracellular or physiological stimuli such as starvation and exercise in the striated muscles (Lopaschuk and Spafford, 1990; Miura et al., 2008; Chinsomboon et al., 2009). In intracellular compartment, PGC-1α is also a downstream effector of AMPK as well as cAMP and calcineurin signaling pathway (Schaeffer et al., 2004; Jäger et al., 2007; Miura et al., 2008; Chinsomboon et al., 2009). PPARα/RXR/PGC-1α heterocomplex is mostly dedicated to FA activation, transport and breakdown, through the expression of fatty acyl-CoA synthase, CPT1B, and long-chain fatty acyl-CoA dehydrogenase, but also PDH (Barger and Kelly, 2000; Hopkins et al., 2003; Duncan and Finck, 2008). By contrast, targets of PPAR β/δ are involved in both FAO (CPT1B and malonyl-CoA decarboxylase) and Glycolysis (activating especially GLUT4 receptor and Phosphofructokinase) (Burkart et al., 2007; Risérus et al., 2008).
Also contributing to the metabolic homeostatis, endocrine hormones (insulin or glucagon-like peptides) and neurohormones (catecholamines) can adapt rapidly several parameters such as blood FA or glucose levels, heartbeat or blood pressure, thus connecting metabolic adaptation of the heart to the whole organism and reciprocally (Gordan et al., 2015). Whereas during normal homeostasis the maintenance of cardiac metabolism is relatively subtle, the heart's energy needs can evolve suddenly and drastically. For example, athletic exercise can increase cardiac output up to 6-fold from resting values, thus requiring a proportional increase in ATP production (Warburton et al., 2002). Under such a context, cardiac metabolism reaches peak activity (Figure 2A). Increases in myocardial workload typically boost FA uptake and FAO. But the glycolysis is also stimulated by exercise in human, or by other stressors such as β-adrenergic or workload stimulation in the hearts of both large animals and rats (Andersson et al., 1991). In addition, exercise increases lactate blood level, which leads to elevated lactate use and oxidation by the cardiac muscle (Kaijser and Berglund, 1992; Jeffrey et al., 1995; Goodwin and Taegtmeyer, 2000), notably by decreasing fatty acids uptake and oxidation (Schönekess, 1997). By contrast, during prolonged exercise of more than 30 min, fatty acids are released by the adipose tissue in the blood circulation, favoring their absorption by the heart (Martin et al., 1993). Meanwhile, it has been demonstrated that the level of malonyl-CoA decreases after 15–30 min of high pacing-induced stimulation in the hearts of pigs and rats, thus facilitating the removal of CPT1 inhibition (Goodwin et al., 1998; Winder, 1998; Zhou et al., 2008). According to this data, longer-term exercise tolerance seems to be associated with the myocardial re-uptake and breakdown of FA. However, an abrupt increase of workload in pigs leads also to a 2.5-fold increase of FAO, although the level of both malonyl-CoA or AMPK remains unchanged (King et al., 2005). Seemingly contradictory, these studies suggest that acute adaptation involves different pathways that remain poorly investigated.
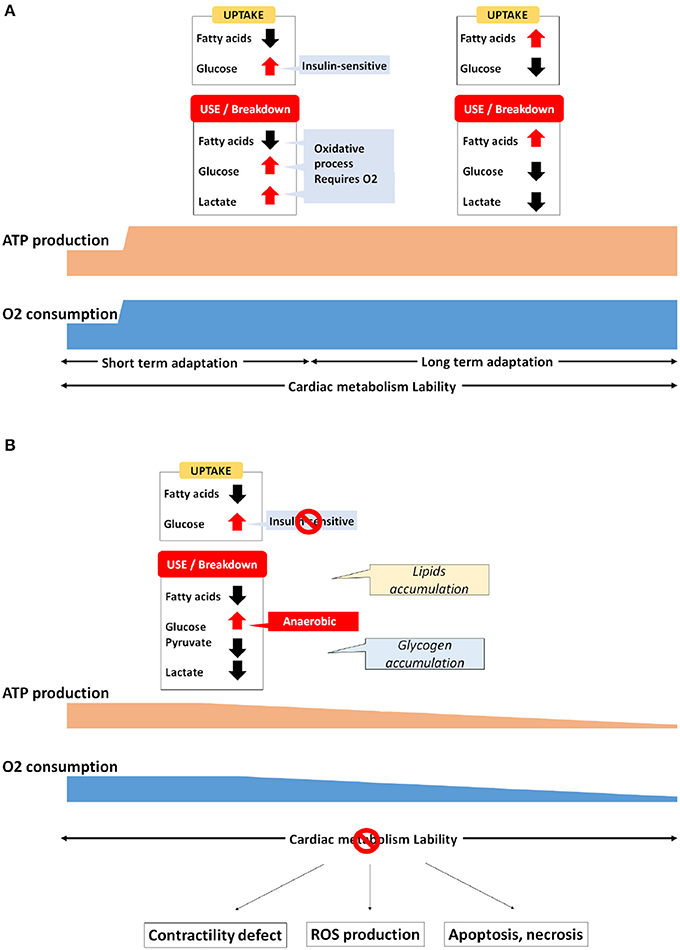
Figure 2. Metabolic adaptation in the cardiac muscle (A) Physiological, (B) Pathological.
Metabolic Adaptations, Beyond Good and Evil in Cardiac Diseases
In cardiovascular diseases, regardless the etiology, energetic starvation is described as a fairly common feature. However, specific metabolic alterations can vary widely from one patient to another (Neubauer, 2007; Turer et al., 2010). This illustrates how the whole complexity of cardiac metabolism under pathological context remains fairly comprehended.
In hypertrophic remodeling, increases in hemodynamic pressure and workload urge the heart to reinforce its contractile strength by increasing its muscular mass. Contrary to most somatic cell types, cardiomyocytes are no longer able to proliferate (Ahuja et al., 2007; Zebrowski and Engel, 2013). Hence, they must increase their size and their stock of both sarcomeres and mitochondria to sustain the upgraded myocardial needs. Cardiomyocyte hypertrophy also requires some metabolic adaptation. Contrary to physiological remodeling, pathological hypertrophy is characterized by a return to fetal gene program that favors anaerobic glycolysis over that of FA, pyruvate and lactate oxidative breakdown (Christe and Rodgers, 1994; Sack and Kelly, 1998; Barger and Kelly, 1999; van der Vusse et al., 2000; Lehman and Kelly, 2002a,b; Ingwall, 2009). In addition, data in both rat and human has validated the switch toward anaerobic glycolysis in pathological cardiac hypertrophy (Ritchie and Delbridge, 2006; Ma and Li, 2015). In hypertrophied hearts, the level of PGC-1α and PPAR is also decreased (Barger and Kelly, 2000; Duncan and Finck, 2008). Conversely, the same maladaptive fetal metabolic patterns are observed in the heart of transgenic mice lacking PPARα, and these are associated with a significant increase in malonyl-CoA (Campbell et al., 2002; Finck et al., 2008). Moreover, an overexpression of PPARα in neonatal rat cardiomyocytes is able to counteract the hypertrophic remodeling process, which suppresses protein synthesis and mitigates CPT1 reduction Cardiac-specific disruption of PPARβ/δ is also associated with cardiac hypertrophy development and a decrease of FA oxidation (Cheng et al., 2004). Above-mentioned activation of PPAR/PGC-1α pathway is sufficient to prevent the decline of FAO, thus maintaining FA oxidation rate. Moreover, increased cardiomyocyte size is associated with a decrease in oxygen tension per area, stabilizing the hypoxia-inducible factor 1α (HIF1α) (Des Tombe et al., 2002; Chu et al., 2012; He et al., 2016). HIF1α takes part in the subtle readjustment of anaerobic glycolysis by favoring fetal lactate dehydrogenase (LDH-A) and pyruvate dehydrogenase kinase (PDK1) activity along with hypoxia-induced GLUT1 expression (Brahimi-Horn et al., 2007). Initially, the fact that GLUT1 expression now supersedes GLUT4 favors insulin resistance. Then, despite glucose uptake being particularly enhanced, pyruvate and lactate oxidation remains low, because their activity is drastically repressed by the increase of PDK1 and the replacement by LDH-A, respectively. In addition, it also well known that hypoxia is able to repress FA metabolism gene expression (Brahimi-Horn et al., 2007). This further validates the relationship between HIF1α, PPAR expression, and cardiac metabolism, thus accordingly reinforcing the notion of the Randle cycle switch in heart failure scenarios (Figure 2B).
Pathological hypertrophy can progress quickly toward HF. Although etiology of HF is particularly complex, metabolic disruption is clearly involved in this progression (Ingwall and Weiss, 2004; Doenst et al., 2013; Abdurrachim et al., 2015). In mild-to-moderate HF, it is generally thought that phosphocreatine level decreases whereas upstream cardiac metabolism toggles from FAO toward glucose/pyruvate oxidation (Sack et al., 1996; Ye et al., 2001; Phillips et al., 2010). As mentioned previously, phosphocreatine is the substrate of creatine kinases that actively take part in ATP buffering. Gupta et al. demonstrated that ATP flux and myocardial contractility are directly related to myocardial CK (M-CK) expression and activity (Gupta et al., 2012). In fact, while overexpression of M-CK in mouse keeps higher ATP flux and better cardiac function, especially at 4 weeks following thoracic aortic constriction; this beneficial impact is lost when M-CK is raised back to the normal level (Gupta et al., 2012). Hence the failing heart is definitively an engine “out of fuel” (Neubauer, 2007), and this is directly related to M-CK activity (Gupta et al., 2012). As long as it can be supplied with adequate fuel, the heart is able to maintain its contractile activity, albeit at a lower output. However in end-stage HF, oxidation of FA, pyruvate and lactate is shunted, favoring the lower dioxygen-consuming glycolytic pathway (Ussher, 2014). In human idiopathic dilated cardiomyopathy, a common precursor to HF, FA uptake and oxidation are substantially decreased in patients (Dávila-Román et al., 2002). Furthermore, metabolic assays performed in rat and human hearts (by mass spectrometry or by 14C and 3H radioactivity measurements) strongly ascribed a correlation between FA deregulation and HF severity (Turer et al., 2009, 2010; Doenst et al., 2010). In addition, during pacing stress, the over-activation of glucose uptake, usually occurring in control groups, is disrupted in both human DCM patients and canine models of DCM (Nikolaidis et al., 2004a,b; Bergman et al., 2009). Taken together, this amount of data points out a lack of metabolic-related responsiveness as a common feature of HF.
In addition, in such a context of failing heart, dioxygen level (availability or starvation) represents another critical point in occurring maladaptive process. Consequently, signaling pathways that contribute to metabolic switching in the heart differ significantly between non-ischemic HF (when dioxygen remains perfused) and ischemic HF (when heart becomes dioxygen-straved).
Metabolic disturbances, in non-ischemic HF, remain close to those leading to HF, such as the switch from FA to glucose utilization. For instance, in an experiment on pressure overload, using mice, transverse aortic constriction (TAC) is followed by an acute increase of myocardial glucose use in the day following the experiment, and this rate reached at higher level 7 days after TAC (Zhong et al., 2013). Additional data demonstrated that the metabolic switch is still preserved 6–8 weeks after TAC in mice (Kolwicz et al., 2012; Pereira et al., 2013, 2014), and this has been also validated in other animal models of pressure overload or volume overload or HF in rats, rabbits and dogs (Taegtmeyer and Overturf, 1988; Allard et al., 1994; Christe and Rodgers, 1994; Christian et al., 1998; Labinskyy et al., 2007). Another study, this time in patients suffering from non-ischemic DCM, right ventricle glucose uptake had been monitored by PET imaging (using the radioactive isotope [18F] fluodeoxyglucose) (Wang L. et al., 2016). Conclusions of these experiments demonstrated a correlation between increased right ventricle glucose uptake and dysfunction (Wang L. et al., 2016). In addition, a recent paper established the metabolomics profile of non-ischemic HF in patients (Mueller-Hennessen et al., 2017). In this clinical study, plasma metabolite profile has shown to widely differ between HF patients and controls, even during resting time, while cardiopulmonary exercise testing clearly exacerbated these differences (Mueller-Hennessen et al., 2017). More precisely, in this article, it has been demonstrated a clear decrease of complex lipids and FA, counterbalanced by an increase in glutamate and purine byproducts, whereas glycolysis was impaired (which is also established as a marker of pre-diabetes) (Mueller-Hennessen et al., 2017). However, in ischemic HF, inefficient blood supply by coronary arteries leads to oxygen starvation within the myocardium, impairing both contractile activity and metabolic imbalance. Not surprisingly, it stands to reason that ischemia compromises all the oxidative steps involved in cardiac metabolism. It reduces the ATP production from both FA and pyruvate but increases anaerobic glycolysis, in proportion with the degree of ischemia (Ferrari et al., 2004; Turer et al., 2009). In such a low oxygen context, the remaining part of oxidation always favors FAO rather than pyruvate use. By contrast, pyruvate is mostly converted into lactate, in order to fuel anaerobic glycolysis in NADH,H+ coenzyme (Josan et al., 2014; Chen et al., 2015; Mariotti et al., 2016). Consequently, to avoid a massive acidification inside the failing cardiomyocytes, an extended part of this newly-produced ATP is dedicated to ionic exchange and homeostasis. The accumulation of reducing coenzymes (NADH,H+ and FADH2) shunts the activity of several metabolic enzymes, such as acyl-CoA dehydrogenase or 3-hydroxylacyl-CoA dehydrogenase, which are directly sensitive to redox state (Nocito et al., 2015). Thus, FA-derived intermediate metabolites accumulate rapidly within the cardiac cells. Fatty acyl CoA favor mitochondrial storage, whereas fatty acyl-carnitines may be accumulated in both mitochondria and other cellular compartments (Kler et al., 1991; Fukushima et al., 2015). Such a build-up of fatty acyl esters and carnitine triggers disturbances in mitochondrial morphology (irregular cristae, amorphous densities) which in turn impact on metabolic function (Kuzmicic et al., 2014; Elezaby et al., 2015). In the failing heart, mitochondrial dysfunction is, indeed, a well-established common feature (Huss and Kelly, 2005; Banović and Ristić, 2016; Goh et al., 2016). At first, their morphology is usually affected, since they are described as smaller in size with membrane disruption and matrix depletion. In these morphologically altered mitochondria, the complex integrity of the respiratory chain is abrogated, ATP production is reduced, and the level of ROS dramatically increased (Banović and Ristić, 2016; Sverdlov et al., 2016). As previously mentioned, excess of ROS will lead to protein degradation, loss of membrane integrity, Ca2+ overload, thus propelling the cardiomyocytes to necrosis and apoptosis (Giordano, 2005; Misra et al., 2009; Sanada et al., 2011; Tsutsui et al., 2011; Csányi and Miller, 2014). While hypoxic conditions leads cardiomyocytes to drastic adaptations, during reperfusion phase following ischemia, cells suffer this time from a sudden burst of dioxygen. Under such an acute re-oxygenation context, a high and uncontrolled release of ROS occurs, due to functional remaining mitochondria and re-activated xanthine oxidase, and this reveals as particularly deleterious (Sanada et al., 2011; Raedschelders et al., 2012; Zuo et al., 2013). Myocardial infarction in mice is notably characterized by a higher level of myosin and actin carbonylation, resulting in a decrease in Ca2+ sensitivity and myocardial dysfunction (Dalle-Donne et al., 2001; Castro et al., 2013; Balogh et al., 2014). In addition, actin oxidation, which is NOX-dependent phenomenon, reorganizes the thin filaments, which favor cell survival in yeast (Farah et al., 2011; Wilson et al., 2016). In the case of failing cardiomyocytes, we can suggest that myofilament oxidation could prevent depolymerization and stabilize actin-myosin complex, which finally reduces ATP needs. Hence, resulting sarcomeres can sustain their contractility, even in a slower, weaker but less energy-consuming fashion. While long-term ischemia following by reperfusion is clearly a deleterious cocktail for the heart, surprisingly, acute but repetitive periods of ischemia is beneficial and cardioprotective (Eltzschig and Eckle, 2011; Frank et al., 2012; Zuo et al., 2013). In fact, this “warm up” effect (called ischemic preconditioning, IPC) is able to improve recovery after a myocardial infarction and to reduce the severity of arrhythmia (Yang et al., 2010). A wide number of research works was led in order to elucidate the mechanisms involved in this surprising phenomenon. Briefly, it has been demonstrated that IPC positively impacts on sarcolemmal and mitochondrial ATP K+ channels. This prevents deleterious ischemia-induced Ca2+ overload and could favor ATP production by impacting on adenylate cyclase and muscular creatine kinase (Turrell et al., 2011; Zuo et al., 2013; Donato et al., 2017). In addition, within mitochondrial matrix, influx of K+ inhibits mitochondrial respiratory chain (especially Complex II) and favors the matrix swelling, which was described as an efficient enhancer of FAO and ATP production (Kaasik et al., 2007; Zuo et al., 2013; Donato et al., 2017; Javadov et al., in press). Due to the rapid switch between oxygen starvation/reperfusion during this phenomenon, the role of ROS was rapidly suspected in IPC (Bolli et al., 1989; Qiu et al., 1997). An acute increase in ROS was indeed taken in evidence when short periods of ischemia were challenged, resulting in K+ ATP channels activation (Gross et al., 2007). Furthermore, beneficial effects of IPC are critically blunted when antioxidants are administrated, validating crucial and beneficial impact of ROS in IPC (Skyschally et al., 2003; Khanna et al., 2008). More specifically, a recent research study demonstrated that a surge of O2•− arises within the first minute following ischemia (Zhu and Zuo, 2013). In particular, this early burst of ROS is due to myoglobin oxidation, releasing O2•− (Zhu and Zuo, 2013). Myoglobin (Mb) is an oxygen- and iron-binding hemoprotein that ensures buffering dioxygen in striated muscle cells. Beyond this traditional function, myoglobin is also an oxygen-sensitive safeguard of NO homeostasis, since oxygenated Mb acts as NO scavenger in normoxia, while Mb oxidation favors NO generation during hypoxia (Rassaf et al., 2007; Totzeck et al., 2014). NO, generated from myoglobin oxidation reduces cardiac oxygen consumption and energy status, which helps to prevent myocardial damages during short phases of hypoxia or ischemia/reperfusion (Hendgen-Cotta et al., 2017). Moreover, based on its oxidation status, myoglobin can bind to FA (especially with long chain oleate and palmitate) (Götz et al., 1994; Shih et al., 2014, 2015; Jue et al., 2016). A recent work (using Mb −/− mice) demonstrated that myoglobin has a new crucial role in FAO by preventing lipid accumulation and alleviating FAO in cardiomyocytes, which preserves cardiac function (Hendgen-Cotta et al., 2017). Such an amount of data reinforces the idea that dioxygen availability and metabolism are deeply bounded in cardiac adaptation, and myoglobin oxidation seems to actively take part in this process as being a ROS/RNS/metabolic sensor in cardiomyocyte. More broadly, while ROS/RNS have been described in a Manichean viewpoint for years, ischemia/reperfusion and IPC provide evidences that they definitively work as a double-edged sword, based on other critical parameters influencing cardiomyocyte needs (especially dioxygen and fuel).
This is the reason why therapeutic strategies are still such challenging in cardiac diseases.
Discussion: Therapeutic Strategies
Cardiac metabolism attracted closer attention in the last decade, appearing as a promising therapeutic target. However, despite the range of accumulated data and technical skills dedicated to assessing this topic (Taegtmeyer et al., 2016), the whole understanding suffers from numerous gaps which remain poorly explored. Meanwhile, although our knowledge of cardiac diseases is expanding, most treatments are still only moderately effective. Hence, many strategies have been envisaged for which we will discuss below the pros and cons in the view of the most recent publications.
Antioxidant Strategy
In the first instance, antioxidant agents demonstrated a therapeutic appeal (for an exhaustive review and discussion about this topic, refer to Goszcz et al., 2015). Briefly, two main strategies have been developed in the past decades: else supply in natural antioxidants, or inhibit the main deleterious sources of ROS.
Primarily, several trials have used vitamins and enzymes, due to their ROS scavenging properties. In particular, it has been demonstrated that addition of retinoic acid and catalase in the culture medium inhibits Angiotensine II/TNF-α-induced hypertrophy in neonatal rat ventricular cardiomyocytes (Nakamura et al., 1998). Following these promising results, a larger-scale clinical study was launched in order to investigate the clear impact of antioxidant treatment in HF. Called HOPE (Heart Outcomes Prevention Evaluation), this study resulted in disappointing results, taken in evidence that higher dose of retinoic acid is pro-oxidant, worsening of the cardiac defect (Sleight, 2000; Gerstein, 2001). In addition, some studies trend to suggest vitamin E and vitamin C can be cardioprotective in patients, even if these virtues remain slightly debated (Hemilä and Kaprio, 2009, 2011).
Instead of focusing on ROS buffering strategies, other studies tried to act more specifically on enzymatic sources of ROS. One of them is the pharmacological inhibition of Xantine Oxidase, by allopurinol. First, in both rat and mouse models of dilated cardiomyopathy, administration of allopurinol is able to decrease the level of ROS, which contributes to slow down the disease (Engberding et al., 2004; Wang Z. et al., 2016). Interestingly, in a clinical study led on HF patients, allopurinol was able to restore M-CK activity, hence to sustain the transfer of energy from mitochondria to sarcomere (Hirsch et al., 2012). More recent study revealed an improvement of walk effort resistance in patients suffering from left ventricular dysfunction, even with higher administrated dose (300 mg daily for 1 week then 600 mg daily for 3 months) (Ansari-Ramandi et al., 2017). However, in patients undergoing percutaneous intervention, allopurinol pretreatment revealed as not effective enough to modify cardiac biomarkers, and especially the level of creatine kinase or troponin-T (Alemzadeh-Ansari et al., 2017; Ansari-Ramandi et al., 2017). This last work may lead to suggest the idea that relevance of such a treatment may depend on numerous parameters.
More recently, ubiquinone (coQ10), another natural antioxidant, has received attention, because it takes part in the redox balance through the mitochondrial respiratory chain, hence playing a critical role in OXPHOS. In addition, it has been demonstrated that HF severity is proportional to the depletion of coQ10, suggesting that a supply of this antioxidant could really help in slowing down the fatal evolution of the disease (Belardinelli et al., 2005). Consequently, many clinical trials have been performed, with large patient cohorts. However, although promising, comparison of the results was challenging due to cohort heterogeneity and lack of randomization. Finally from 41 relevant studies, the most recent meta-analysis identified only 13 accurate studies (Fotino et al., 2013). From this amount of data, the beneficial effect of coQ10 on ejection fraction was validated, but the impact on long-term survival rate following the treatment remained unclear (Fotino et al., 2013). A more recent randomized study including 420 HF patients for 2 years shed new light concerning the coQ10 therapeutic strategy (Mortensen et al., 2014). Called, Q-Symbio, this large-scale study has strongly established a reduction of mortality due to cardiovascular causes as well as a decrease of hospital admission for HF among patients treated with coQ10 (Mortensen et al., 2014).
Finally, antioxidant strategy has carried out promising clinical trials, but its whole efficiency remains mixed. These drugs might mainly act as “redox sponges,” favoring ROS scavenging in a certain extent, without addressing the pathogenesis. This makes sense since deleterious oxidative stress is both a cause and a consequence in cardiovascular diseases, especially in HF. Consequently, therapeutic use of antioxidants would be a good complementary to any other strategy.
By contrast, a growing number of clues suggest that restoring the supply of “cardiac fuel” can bring sustainable solutions (Fukushima et al., 2015; Salzman et al., 2017).
Facilitate FA Uptake and Oxidation
One approach involves refueling the heart, else by the stabilization or by the increase of FA uptake and/or FAO. However, this strategy seems to be dangerously close to maladaptive processes, regarding the level of dioxygen available that may not be matched. Consequently, there have been some ambiguous and contradictory data, resulting from this approach. For instance, high fat diet feeding to mice worsens TAC-induced left ventricular remodeling and this is associated with insulin resistance (Raher et al., 2008). By contrast, in hypertensive rat model, the same high fat diet treatment seems to be beneficial, as it is associated with reduction of the hypertrophic remodeling process, mostly by improving cardiac contractility (Okere et al., 2005). Equivalent results were obtained after lipid administration on the hearts of arctic ground squirrels, protecting them against adverse effects of ischemia/reperfusion (Salzman et al., 2017). All these results match with several puzzling observations of human patients (usually called the “obesity paradox”). Indeed, the risk for developing cardiovascular disease is higher in obese people, but the survival rate is also higher in these overweight patients compared to normal or low-weight patients (Oreopoulos et al., 2008; Clark et al., 2011; Nagarajan et al., 2016). By contrast, clinical trials demonstrated that polyunsaturated fatty acids administration (PUFAs) are beneficial for preventing or delaying the progression of HF, whereas monounsaturated FA supply in patients suffering from end-stage HF trigger the decline of cardiac function (Mozaffarian et al., 2005; Xin et al., 2012; Imamura et al., 2013; Masson et al., 2013).
Decrease FA Uptake and Oxidation
As a mirror effect, a preferred alternative consists in the reduction of the FA uptake. This approach may avoid a toggling of the metabolic machinery, hence matching better with oxygen availability. Moreover, in accordance with the Randle cycle, a decrease of FA uptake and oxidation would be able to increase proportionally glucose use and breakdown.
As a key checkpoint in mitochondrial FA uptake, CPT1 represents a prime target in order to reduce FAO, thus, several CPT1 inhibitors have been designed. Among them, etomoxir and perhexeline received previously closer attention. Several studies in both human patients and rodent models validated some beneficial effects, and particularly their anti-ischemic virtues, by favoring the switch to glucose oxidation (Fillmore and Lopaschuk, 2013). A recent study focused on Mitochondrial Trifunctional Protein Deficiency (MTFD), a genetic disease of which the most severe neonatal form leads to cardiomyopathy and hepatic dysfunction. In this study, etomoxir-treated MTFD fibroblasts exhibited a clearing of cytotoxic fatty-acyls carnitine and improved mitochondrial respiration rate and ATP production (Lefort et al., 2017). Due to its ability to boost glucose utilization, etomoxir was previously used as an anti-diabetic agent. In addition, a 3-month treatment, led in a small group of patients, demonstrated an improvement of LV ejection fraction and exercise tolerance as well as a better overall medical condition (Schmidt-Schweda and Holubarsch, 2000). Beyond cardiac metabolism, the treatment with etomoxir can improve Ca2+ handling by increasing SERCA2a expression (Zarain-Herzberg et al., 1996; Rupp and Vetter, 2000). However, a larger-scale study (Etomoxir for the Recovery of Glucose Oxidation, ERGO) had been hastily stopped because HF patients rapidly exhibited a disturbing increase of transaminase blood level, suggesting a severe hepatotoxicity (Holubarsch et al., 2007). Thus, the irreversible impact of etomoxir on the hepatic tissue seems to supersede its potential beneficial effects on cardiac function.
New challenging approaches carry on more specifically targeting CPT1B, the more cardiac specific isoform. In such a purpose, oxfenicine (Sepa-Kishi et al., 2016) or perhexeline (ex anti-angina agent from the 1970's) (Chong et al., 2016) hold promising data both in vitro and in animal studies.
While high doses of these agents were associated with neurotoxicity and hepatotoxicity (Shah et al., 1982), more interestingly, clinical trials demonstrated that a lower dose of perhexeline plasma level specifically inhibits cardiac CPT1B (Kennedy et al., 1996). Although perhexiline virtues on LV function were demonstrated decades ago in an ex vivo canine model (Ono et al., 1982), there is a recent growth of interest for this metabolic agent (Chong et al., 2016). Notably, it has been demonstrated that mice fed with perhexiline preserve partly the cardiac function, reducing FAO and increasing glucose use (Yin et al., 2013). Several clinical trials in patients also validate a positive impact in electrophysiological parameters. However, recent studies trend to demonstrate that this metabolic agent also exhibits many beneficial side effects on other main effectors involved in maladaptive cardiac remodeling, such as NOX2 complex (Liberts et al., 2007; Gatto et al., 2013), Kruppel-like factor 14 (Guo et al., 2015) and mTORC1 (Balgi et al., 2009), thus, reducing inflammation, lipid metabolism and autophagy.
In addition, as a natural feedback inhibitor of CPT1, MalonylCoA can be considered as another way of slowing down FAO. Usually, this TCA byproduct is converted to AcetylCoA, and this reaction is catalyzed by MalonylCoA Decarboxylase (MCD) (Hamilton and Saggerson, 2000). Several recent data trend to validate MCD inhibition as promising therapeutic strategy. First, in a swine model of HF, administration of MCD inhibitors improves cardiac function and upregulates glycolysis (Dyck et al., 2004). More recently, MCD gene silencing in both mouse and rat are associated with a better-preserved ejection fraction following surgical induction of myocardial infarction (Masoud et al., 2014; Wu et al., 2014). In such a situation, it has been demonstrated that the preservation of cardiac function is coupled with an upholding of ATP production. However, since the experiments that yielded this information are particularly recent, this strategy has not been tried yet on HF patients.
As the terminal enzyme of the β-oxidation cycle, 3-KetoAcylCoAThiolase represents another interesting therapeutic target. Its catalytic activity can be partly inhibited by the trimetazidine. Usually used as an anti-angina treatment in more than 90 countries through the world, trimetazidine is efficient in decreasing FA oxidation, thus increasing glucose oxidation. This drug holds beneficial impact on myocardial infarction and HF (D'hahan et al., 1997; Belardinelli, 2000; Saeedi et al., 2005; Belardinelli et al., 2008; Marazzi et al., 2009; Zhou and Chen, 2014). Trimetazidine is also associated with cardioprotective effects in pressure-overload cardiac hypertrophy, where FA oxidation is already disrupted. In fact, data suggest that trimetazidine modulates the production of H+ and regulates the Ca2+ handling (by acting on SERCA catalytic activity), thus preserving ionic exchange and homeostasis inside the cardiac cells, which contributes to an improvement of the cardiac function (Meng et al., 2006). Ranolazine and Dichloroacetate are two other anti-angina agents which act on cardiac metabolism by increasing PDH activity, thus removing the FA-associated negative feedback upon glucose oxidation (Scirica and Morrow, 2007; Scirica et al., 2007; Gutierrez et al., 2015). Research and clinical trials demonstrated that their beneficial effects are quite equivalent to trimetazidine (Clarke et al., 1993; Gralinski et al., 1996; Battiprolu and Rodnick, 2014; Caminiti et al., 2016; Coppini et al., 2017).
Modulate AMPK Signaling Pathway
In cardiac metabolism, the AMP-activated protein kinase (AMPK) acts as a “fuel gauge” due to its sensitivity to AMP/ATP and creatine/phosphocreatine ratios (Hardie, 2004; Shirwany and Zou, 2014). AMPK mediates cell catabolism and ATP production by different strategies. First, AMPK positively impacts on GLUT1 expression, GLUT4 endocytosis, and phosphofructose kinase 2 activity, thus promoting glucose uptake (Zhao et al., 2017). AMPK can also increase FAO by decreasing acetylCoA carboxylases activity, which consequently decreases the level of malonyl CoA and thus release the CPT1 inhibition. In addition, AMPK can inhibit mTORC1, which reduces protein synthesis and favors autophagy. Meanwhile AMPK reversely activates FoxOs, which contributes to glycolysis by regulating PGC1-α and Glucose-6-Phosphatase thus boosting both FAO and glycolysis (Jäger et al., 2007; Zhu et al., 2010; Kousteni, 2012; Eijkelenboom and Burgering, 2013). Notably, AMPK pathways are also involved in oxidative stress; AMPK/PKC-2α can regulate NOX2 activity (Balteau et al., 2014), while AMPK/FOXO3 activates Thrx2 in endothelial cells (Hou et al., 2010). Lastly, AMPK works in a reverse crosstalk with AKT signaling that usually favors anabolism and cell survival (Zhao et al., 2017). Hence, AMPK activation is foreseen as an up-and-coming therapeutic strategy (Beauloye et al., 2011). Among them, metformin, a common drug for the treatment of Type 2 diabetes, has caught careful attention. Indeed, metformin induced-AMPK activation can protect from myocardial injury and cell apoptosis following ischemia (Kim et al., 2011; Yin et al., 2011) and it improves cardiac function (and oxidative metabolism) in rodent models of HF (Benes et al., 2011; Fu et al., 2011). Yet, long-term activation of AMPK is deleterious for cardiac function, due to negative feedback on CPT1 expression and activity, thus reducing FA use and metabolism. Cardioprotective effects of metformin were also demonstrated in rat model of chronic HF (Wang et al., 2011). However, a recent meta-analysis validates with caution this beneficial impact only on experimental myocardial infarction (Hesen et al., 2017). And this is the line with observations led in clinical trials. In fact, in non-diabetic patients who underwent acute myocardial infarction, clinical studies about metformin treatment remain disappointing or contradictory (Lexis et al., 2014). Moreover, although beneficial effects were suggested in a clinical trial focusing in HF with preserved ejection fraction (Shah et al., 2010), metformin seems to exacerbate cardiac disruption in end-stage HF by increasing lactate acidosis (Wong et al., 2009; Hou et al., 2010; Doenst et al., 2013). This data validates the “Janus double-headed” role of AMPK in cardiac metabolism. Thus, although interesting, AMPK treatment seems to be case-by-case-dependent. Consequently, caution remains needed concerning the use of AMPK activators in clinical trials.
Increase Blood Glucose Level and Release
In cardiac disease, insulin tolerance or resistance is a crucial parameter. Upstream insulin and glycolysis, glucagon-like peptide-1 (GLP-1) is a gut-secreted incretin (metabolic hormone) which can promote the release of insulin after meals to activate cell glucose uptake from blood (Nadkarni et al., 2014). In experiments using dogs and rats, GLP-1 infusion can increase glucose uptake and improve cardiac function (Nikolaidis et al., 2004a; Sokos et al., 2006; Poornima et al., 2008). Regarding its short half-life in plasma, pharmacological agonists of GLP-1 have been developed with promising but sometimes contradictory results (Liu et al., 2014; Margulies et al., 2014; Trujillo et al., 2015). GLP-1 receptor blockage abolished IPC cardioprotective effects in a rat model of myocardial infarction, suggesting a deep impact of GLP-1 on pathways involved in IPC (Basalay et al., 2016). Then, recent studies in H9c2 myoblasts demonstrated, in particular, that GLP-1 and analogs can decrease oxidative stress and also hypoxia/reoxygenation-induced apoptosis via activation of PI3K/AKT pathway (Chang et al., 2014; Jiang et al., 2016; Petersen et al., 2016). Another approach demonstrated that GLP-1 can also activate AMPK, which reduces hyperglycemia-induced NOX2 activity in adult rat cardiomyocytes (Balteau et al., 2014). Beyond glycemic control, GLP-1 and analogs exhibit side anti-inflammatory and anti-atherogenic effects (Anagnostis et al., 2011; Petersen et al., 2016). Such a pleiotropic action has thus very recently led them to greater and promising interest from bench to bedside.
Myocardial Stimulation by Catecholamines
Last but not least, neurotransmitters can be viewed as other way of positively impacting in cardiac metabolism (Slatton and Eichhorn, 1996; Bouzamondo et al., 2001; Lechat, 2004). Indeed, sympathetic and parasympathetic stimulations by catecholamines are crucial in cardiac adaptive processes. As previously described, metabolic demands increase during rigorous exercise. It is well described that short-term β-adrenergic stimulation increases glucose uptake and oxidation (Morisco et al., 2005; Nagoshi et al., 2011). Stimulation by the use of an agonist (epinephrine) can also favor Ca2+ handling cycle, with potential beneficial in myocardial contractility (Collins-Nakai et al., 1994; Briston et al., 2014; Grimm et al., 2015; Ho et al., 2016). Moreover, recent study demonstrated that a selective β3-adrenergic receptor agonist (BRL37344) favors left ventricular relaxation in Langendorff-perfused rat hearts via the NO/cGMP/PKG axis, thus by activating specific redox signaling and favoring beneficial adaptive process (Angelone et al., 2008; Cannavo and Koch, 2017). NMR spectroscopy in rat heart also demonstrated that acute β-adrenergic receptor stimulation by isoprotenerol leads to a sudden lactate production from glycogen stock (Khemtong et al., 2015). However, long-term activation leads to insulin resistance, making therapy challenging (Morisco et al., 2005; Ciccarelli et al., 2011). In fact, β-adrenergic receptor stimulation overactivates cAMP/PKA signaling pathway, leading to a decrease in insulin-dependent GLUT4 membrane translocation and expression, and thereby to a disruption of glucose uptake (Mangmool et al., 2016).
By contrast, long-term blockade with a non-selective β-adrenergic and α1-adrenergic antagonist (carvedilol) reduces FA oxidation by favoring glucose use and breakdown in HF patients, which may provide longer-term and more efficient therapeutic benefits (Eichhorn et al., 1990; Eichhorn, 1998, 2000). In fact, more recent studies demonstrated that carvedilol encloses a bunch of beneficial side-effects including antioxidant, anti-inflammatory and anti-apoptotic virtues, notably by favoring ROS scavenging systems and by activating specific miRNA involved in cardiomyocyte survival (miR-125b-5, miR-199-3p, miR-214) (Bayoumi et al., 2017, p. 125; Park et al., 2016). Network meta-analyses remain ambivalent concerning carvedilol superiority on mortality benefits, when compared with specific β1-adrenergic receptor blockers, but several bias (sampling, dose administration) made these studies to be taken with caution (Kveiborg et al., 2007; DiNicolantonio et al., 2013; Zhang et al., 2016; Li J. et al., 2017).
Although non-exhaustive, these studies underline an updated interest for β-blockers. Combined administration of carvedilol with antioxidant could be an interesting therapeutic strategy in cardiac diseases, and first clinical trials have shown encouraging potential (Budni et al., 2013; El-Shitany and El-Desoky, 2016).
Conclusions
Cardiac metabolism works in a finely tuned fashion: with enough fuel and dioxygen, cardiomyocytes can rapidly and efficiently generate ATP to convert into mechanical force. The current body of research now clearly demonstrates that maladaptive metabolic behaviors as well as ROS/RNS double-edged effects are intimately involved in both the genesis as well as the progression of cardiac diseases. As a result, though many therapies have been developed for years, none of them can be considered as a panacea, since the fact to reconcile fuel sources and oxidative capacity is the toughest challenge. For now, the best therapeutic approach may be a combination of drugs with complementary virtues. On the bedside of patients, a case-by-case approach is therefore necessary and based on specific patient clinical history.
Author Contributions
AA, XP, and LX wrote the manuscript. AA prepared the figures.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work is supported NIH grants R01 HL122736 (to LX), NIH grants R01 HL061656 and HL112890 (to XP). We wish to thank W.H.D Towley-Tilson and J. Comer for proofreading this manuscript and B. de Latte for comments.
Abbreviations
AMPK, AMP-activated protein kinase; CK, Creatine Kinase; CRATs: Carnitine O-acetyl transferases; COTs, Carnitine octanoyltransferases; CPT1, Carnitine palmitoyl transferase 1; DCM, Dilated cardiomyopathy; FA, Fatty acids; FAO, Fatty acids β-oxidation; FACS, Fatty acyl CoA Synthase; G6P, Glucose 6-Phosphate; GLP-1, Glucagon-like peptide-1; GLUT, Glucose transporter; HF, Heart failure; HIF1α, Hypoxia-inducible factor 1α; IPC, Ischemic preconditioning; JNK, (Stress-activated kinase)/c-jun N-terminal kinase; LDH, Lactate dehydrogenase; MAPK, Mitogen-associated protein kinase; PDH, Pyruvate dehydrogenase; PDK1, Pyruvate deshydrogenase kinase 1; PGC-1α, Peroxisome proliferator-activated receptor γ coactivator-1α; PPAR, Peroxisome proliferator-activated Receptor; ROS, Reactive oxygen species; RXR, Retinoid X receptor; SERCA, Sarcoplasmic reticulum Ca2+ ATPase; TAC, Transversal aortic constriction; TCA cycle, Tricarboxylic cycle.
References
Abdurrachim, D., Luiken, J. J. F. P., Nicolay, K., Glatz, J. F. C., Prompers, J. J., and Nabben, M. (2015). Good and bad consequences of altered fatty acid metabolism in heart failure: evidence from mouse models. Cardiovasc. Res. 106, 194–205. doi: 10.1093/cvr/cvv105
Abel, E. D. (2004). Glucose transport in the heart. Front. Biosci. J. Virtual Libr. 9, 201–215. doi: 10.2741/1216
Abozguia, K., Shivu, G. N., Ahmed, I., Phan, T. T., and Frenneaux, M. P. (2009). The heart metabolism: pathophysiological aspects in ischaemia and heart failure. Curr. Pharm. Des. 15, 827–835. doi: 10.2174/138161209787582101
Aerni-Flessner, L., Abi-Jaoude, M., Koenig, A., Payne, M., and Hruz, P. W. (2012). GLUT4, GLUT1, and GLUT8 are the dominant GLUT transcripts expressed in the murine left ventricle. Cardiovasc. Diabetol. 11:63. doi: 10.1186/1475-2840-11-63
Afanasiev, S. A., Egorova, M. V., Kutsykova, T. V., and Popov, S. V. (2016). Influence of fatty acids on oxygen consumption in isolated cardiomyocytes of rats with ischemic or diabetic heart disease. Vestn. Ross. Akad. Med. Nauk 2, 97–101. doi: 10.15690/vramn612
Ahuja, P., Sdek, P., and MacLellan, W. R. (2007). Cardiac myocyte cell cycle control in development, disease and regeneration. Physiol. Rev. 87, 521–544. doi: 10.1152/physrev.00032.2006
Alemzadeh-Ansari, M. J., Hosseini, S. K., Talasaz, A. H., Mohammadi, M., Tokaldani, M. L., Jalali, A., et al. (2017). Effect of high-dose allopurinol pretreatment on cardiac biomarkers of patients undergoing elective percutaneous coronary intervention: a randomized clinical trial. Am. J. Ther. 24, e723–e729. doi: 10.1097/MJT.0000000000000411
Allard, M. F., Schönekess, B. O., Henning, S. L., English, D. R., and Lopaschuk, G. D. (1994). Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. Am. J. Physiol. 267, H742–H750.
Anagnostis, P., Athyros, V. G., Adamidou, F., Panagiotou, A., Kita, M., Karagiannis, A., et al. (2011). Glucagon-like peptide-1-based therapies and cardiovascular disease: looking beyond glycaemic control. Diabetes Obes. Metab. 13, 302–312. doi: 10.1111/j.1463-1326.2010.01345.x
Andersson, B., Blomström-Lundqvist, C., Hedner, T., and Waagstein, F. (1991). Exercise hemodynamics and myocardial metabolism during long-term beta-adrenergic blockade in severe heart failure. J. Am. Coll. Cardiol. 18, 1059–1066. doi: 10.1016/0735-1097(91)90767-4
Andreyev, A. Y., Kushnareva, Y. E., and Starkov, A. A. (2005). Mitochondrial metabolism of reactive oxygen species. Biochem. Mosc. 70, 200–214. doi: 10.1007/s10541-005-0102-7
Angelone, T., Filice, E., Quintieri, A. M., Imbrogno, S., Recchia, A., Pulerà, E., et al. (2008). Beta3-adrenoceptors modulate left ventricular relaxation in the rat heart via the NO-cGMP-PKG pathway. Acta Physiol. Oxf. Engl. 193, 229–239. doi: 10.1111/j.1748-1716.2008.01838.x
Ansari-Ramandi, M. M., Maleki, M., Alizadehasl, A., Amin, A., Taghavi, S., Alemzadeh-Ansari, M. J., et al. (2017). Safety and effect of high dose allopurinol in patients with severe left ventricular systolic dysfunction. J. Cardiovasc. Thorac. Res. 9, 102–107. doi: 10.15171/jcvtr.2017.17
Askoxylakis, V., Thieke, C., Pleger, S. T., Most, P., Tanner, J., Lindel, K., et al. (2010). Long-term survival of cancer patients compared to heart failure and stroke: a systematic review. BMC Cancer 10:105. doi: 10.1186/1471-2407-10-105
Augusto, O., Bonini, M. G., Amanso, A. M., Linares, E., Santos, C. C. X., and De Menezes, S. L. (2002). Nitrogen dioxide and carbonate radical anion: two emerging radicals in biology. Free Radic. Biol. Med. 32, 841–859. doi: 10.1016/S0891-5849(02)00786-4
Avner, B. S., Shioura, K. M., Scruggs, S. B., Grachoff, M., Geenen, D. L., Helseth, D. L., et al. (2012). Myocardial infarction in mice alters sarcomeric function via post-translational protein modification. Mol. Cell. Biochem. 363, 203–215. doi: 10.1007/s11010-011-1172-z
Awan, M. M., and Saggerson, E. D. (1993). Malonyl-CoA metabolism in cardiac myocytes and its relevance to the control of fatty acid oxidation. Biochem. J. 295(Pt 1), 61–66. doi: 10.1042/bj2950061
Balgi, A. D., Fonseca, B. D., Donohue, E., Tsang, T. C. F., Lajoie, P., Proud, C. G., et al. (2009). Screen for chemical modulators of autophagy reveals novel therapeutic inhibitors of mTORC1 signaling. PLoS ONE 4:e7124. doi: 10.1371/journal.pone.0007124
Balogh, A., Santer, D., Pásztor, E. T., Tóth, A., Czuriga, D., Podesser, B. K., et al. (2014). Myofilament protein carbonylation contributes to the contractile dysfunction in the infarcted LV region of mouse hearts. Cardiovasc. Res. 101, 108–119. doi: 10.1093/cvr/cvt236
Balteau, M., Van Steenbergen, A., Timmermans, A. D., Dessy, C., Behets-Wydemans, G., Tajeddine, N., et al. (2014). AMPK activation by glucagon-like peptide-1 prevents NADPH oxidase activation induced by hyperglycemia in adult cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 307, H1120–H1133. doi: 10.1152/ajpheart.00210.2014
Banović, M. D., and Ristić, A. D. (2016). The role of mitochondrial dysfunction in heart failure and potential therapeutic targets. Curr. Pharm. Des. 22, 4752–4762. doi: 10.2174/1381612822666160614081853
Barajas-Espinosa, A., Basye, A., Angelos, M. G., and Chen, C.-A. (2015). Modulation of p38 kinase by DUSP4 is important in regulating cardiovascular function under oxidative stress. Free Radic. Biol. Med. 89, 170–181. doi: 10.1016/j.freeradbiomed.2015.07.013
Barcia-Vieitez, R., and Ramos-Martínez, J. I. (2014). The regulation of the oxidative phase of the pentose phosphate pathway: new answers to old problems. IUBMB Life 66, 775–779. doi: 10.1002/iub.1329
Barger, P. M., and Kelly, D. P. (1999). Fatty acid utilization in the hypertrophied and failing heart: molecular regulatory mechanisms. Am. J. Med. Sci. 318, 36–42. doi: 10.1016/S0002-9629(15)40570-1
Barger, P. M., and Kelly, D. P. (2000). PPAR signaling in the control of cardiac energy metabolism. Trends Cardiovasc. Med. 10, 238–245. doi: 10.1016/S1050-1738(00)00077-3
Barry, W. H. (2004). Heart physiology from cell to circulation, 4th ed. Circulation 110, e313–e313. doi: 10.1161/01.CIR.0000143724.99618.62
Basalay, M. V., Mastitskaya, S., Mrochek, A., Ackland, G. L., Del Arroyo, A. G., Sanchez, J., et al. (2016). Glucagon-like peptide-1 (GLP-1) mediates cardioprotection by remote ischaemic conditioning. Cardiovasc. Res. 112, 669–676. doi: 10.1093/cvr/cvw216
Battelli, M. G., Polito, L., Bortolotti, M., and Bolognesi, A. (2016). Xanthine oxidoreductase-derived reactive species: physiological and pathological effects. Oxid. Med. Cell. Longev. 2016:3527579. doi: 10.1155/2016/3527579
Battiprolu, P. K., and Rodnick, K. J. (2014). Dichloroacetate selectively improves cardiac function and metabolism in female and male rainbow trout. Am. J. Physiol. Heart Circ. Physiol. 307, H1401–H1411. doi: 10.1152/ajpheart.00755.2013
Bayoumi, A. S., Park, K.-M., Wang, Y., Teoh, J.-P., Aonuma, T., Tang, Y., et al. (2017). A carvedilol-responsive microRNA, miR-125b-5p protects the heart from acute myocardial infarction by repressing pro-apoptotic bak1 and klf13 in cardiomyocytes. J. Mol. Cell. Cardiol. 114, 72–82. doi: 10.1016/j.yjmcc.2017.11.003
Beauloye, C., Bertrand, L., Horman, S., and Hue, L. (2011). AMPK activation, a preventive therapeutic target in the transition from cardiac injury to heart failure. Cardiovasc. Res. 90, 224–233. doi: 10.1093/cvr/cvr034
Bedard, K., and Krause, K.-H. (2007). The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev. 87, 245–313. doi: 10.1152/physrev.00044.2005
Beer, M., Seyfarth, T., Sandstede, J., Landschütz, W., Lipke, C., Köstler, H., et al. (2002). Absolute concentrations of high-energy phosphate metabolites in normal, hypertrophied, and failing human myocardium measured noninvasively with (31)P-SLOOP magnetic resonance spectroscopy. J. Am. Coll. Cardiol. 40, 1267–1274. doi: 10.1016/S0735-1097(02)02160-5
Belardinelli, R. (2000). Trimetazidine and the contractile response of dysfunctional myocardium in ischaemic cardiomyopathy. Rev. Port. Cardiol. Orgao Of. Soc. Port. Cardiol. Port. J. Cardiol. Off. J. Port. Soc. Cardiol. 19(Suppl. 5), V35–V39. doi: 10.1053/euhj.2001.2653
Belardinelli, R., Cianci, G., Gigli, M., Mazzanti, M., and Lacalaprice, F. (2008). Effects of trimetazidine on myocardial perfusion and left ventricular systolic function in type 2 diabetic patients with ischemic cardiomyopathy. J. Cardiovasc. Pharmacol. 51, 611–615. doi: 10.1097/FJC.0b013e31817bdd66
Belardinelli, R., Muçaj, A., Lacalaprice, F., Solenghi, M., Principi, F., Tiano, L., et al. (2005). Coenzyme Q10 improves contractility of dysfunctional myocardium in chronic heart failure. BioFactors Oxf. Engl. 25, 137–145. doi: 10.1002/biof.5520250115
Bellis, A., Castaldo, D., Trimarco, V., Monti, M. G., Chivasso, P., Sadoshima, J., et al. (2009). Cross-talk between PKA and Akt protects endothelial cells from apoptosis in the late ischemic preconditioning. Arterioscler. Thromb. Vasc. Biol. 29, 1207–1212. doi: 10.1161/ATVBAHA.109.184135
Benes, J., Kazdova, L., Drahota, Z., Houstek, J., Medrikova, D., Kopecky, J., et al. (2011). Effect of metformin therapy on cardiac function and survival in a volume-overload model of heart failure in rats. Clin. Sci. Lond. Engl. 121, 29–41. doi: 10.1042/CS20100527
Bergman, B. C., Tsvetkova, T., Lowes, B., and Wolfel, E. E. (2009). Myocardial FFA metabolism during rest and atrial pacing in humans. Am. J. Physiol. Endocrinol. Metab. 296, E358–E366. doi: 10.1152/ajpendo.90747.2008
Bolli, R., Jeroudi, M. O., Patel, B. S., DuBose, C. M., Lai, E. K., Roberts, R., et al. (1989). Direct evidence that oxygen-derived free radicals contribute to postischemic myocardial dysfunction in the intact dog. Proc. Natl. Acad. Sci. U.S.A. 86, 4695–4699. doi: 10.1073/pnas.86.12.4695
Bonnefont, J.-P., Djouadi, F., Prip-Buus, C., Gobin, S., Munnich, A., and Bastin, J. (2004). Carnitine palmitoyltransferases 1 and 2: biochemical, molecular and medical aspects. Mol. Aspects Med. 25, 495–520. doi: 10.1016/j.mam.2004.06.004
Bouzamondo, A., Hulot, J. S., Sanchez, P., Cucherat, M., and Lechat, P. (2001). Beta-blocker treatment in heart failure. Fundam. Clin. Pharmacol. 15, 95–109. doi: 10.1046/j.1472-8206.2001.00019.x
Brahimi-Horn, M. C., Chiche, J., and Pouysségur, J. (2007). Hypoxia signalling controls metabolic demand. Curr. Opin. Cell Biol. 19, 223–229. doi: 10.1016/j.ceb.2007.02.003
Bricker, D. K., Taylor, E. B., Schell, J. C., Orsak, T., Boutron, A., Chen, Y.-C., et al. (2012). A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, drosophila, and humans. Science 337, 96–100. doi: 10.1126/science.1218099
Briston, S. J., Dibb, K. M., Solaro, R. J., Eisner, D. A., and Trafford, A. W. (2014). Balanced changes in Ca buffering by SERCA and troponin contribute to Ca handling during β-adrenergic stimulation in cardiac myocytes. Cardiovasc. Res. 104, 347–354. doi: 10.1093/cvr/cvu201
Budni, P., Pedrosa, R. C., Dalmarco, E. M., Dalmarco, J. B., Frode, T. S., and Wilhelm, D. (2013). Carvedilol enhances the antioxidant effect of vitamins e and c in chronic chagas heart disease. Arq. Bras. Cardiol. 101, 304–310. doi: 10.5935/abc.20130184
Burgoyne, J. R., Madhani, M., Cuello, F., Charles, R. L., Brennan, J. P., Schröder, E., et al. (2007). Cysteine redox sensor in PKGIα enables oxidant-induced activation. Science 317, 1393–1397. doi: 10.1126/science.1144318
Burkart, E. M., Sambandam, N., Han, X., Gross, R. W., Courtois, M., Gierasch, C. M., et al. (2007). Nuclear receptors PPARbeta/delta and PPARalpha direct distinct metabolic regulatory programs in the mouse heart. J. Clin. Invest. 117, 3930–3939. doi: 10.1172/JCI32578
Caminiti, G., Fossati, C., Battaglia, D., Massaro, R., Rosano, G., and Volterrani, M. (2016). Ranolazine improves insulin resistance in non-diabetic patients with coronary heart disease. A pilot study. Int. J. Cardiol. 219, 127–129. doi: 10.1016/j.ijcard.2016.06.003
Campbell, F. M., Kozak, R., Wagner, A., Altarejos, J. Y., Dyck, J. R. B., Belke, D. D., et al. (2002). A role for peroxisome proliferator-activated receptor alpha (PPARalpha) in the control of cardiac malonyl-CoA levels: reduced fatty acid oxidation rates and increased glucose oxidation rates in the hearts of mice lacking PPARalpha are associated with higher concentrations of malonyl-CoA and reduced expression of malonyl-CoA decarboxylase. J. Biol. Chem. 277, 4098–4103. doi: 10.1074/jbc.M106054200
Campbell, S. E., Tandon, N. N., Woldegiorgis, G., Luiken, J. J. F. P., Glatz, J. F. C., and Bonen, A. (2004). A novel function for fatty acid translocase (FAT)/CD36: involvement in long chain fatty acid transfer into the mitochondria. J. Biol. Chem. 279, 36235–36241. doi: 10.1074/jbc.M400566200
Cannavo, A., and Koch, W. J. (2017). Targeting β3-adrenergic receptors in the heart: selective agonism and β-blockade. J. Cardiovasc. Pharmacol. 69, 71–78. doi: 10.1097/FJC.0000000000000444
Cantu-Medellin, N., and Kelley, E. E. (2013). Xanthine oxidoreductase-catalyzed reactive species generation: a process in critical need of reevaluation. Redox Biol. 1, 353–358. doi: 10.1016/j.redox.2013.05.002
Castro, J. P., Jung, T., Grune, T., and Almeida, H. (2013). Actin carbonylation: from cell dysfunction to organism disorder. J. Proteomics 92, 171–180. doi: 10.1016/j.jprot.2013.05.006
Chang, G., Zhang, D., Liu, J., Zhang, P., Ye, L., Lu, K., et al. (2014). Exenatide protects against hypoxia/reoxygenation-induced apoptosis by improving mitochondrial function in H9c2 cells. Exp. Biol. Med. Maywood N.J. 239, 414–422. doi: 10.1177/1535370214522177
Chen, A. F., Chen, D.-D., Daiber, A., Faraci, F. M., Li, H., Rembold, C. M., et al. (2012). Free radical biology of the cardiovascular system. Clin. Sci. 123, 73–91. doi: 10.1042/CS20110562
Chen, A. P., Lau, J. Y. C., Alvares, R. D. A., and Cunningham, C. H. (2015). Using [1-(13) C]lactic acid for hyperpolarized (13) C MR cardiac studies. Magn. Reson. Med. 73, 2087–2093. doi: 10.1002/mrm.25354
Cheng, L., Ding, G., Qin, Q., Huang, Y., Lewis, W., He, N., et al. (2004). Cardiomyocyte-restricted peroxisome proliferator-activated receptor-δ deletion perturbs myocardial fatty acid oxidation and leads to cardiomyopathy. Nat. Med. 10, 1245–1250. doi: 10.1038/nm1116
Chinsomboon, J., Ruas, J., Gupta, R. K., Thom, R., Shoag, J., Rowe, G. C., et al. (2009). The transcriptional coactivator PGC-1α mediates exercise-induced angiogenesis in skeletal muscle. Proc. Natl. Acad. Sci. U.S.A. 106, 21401–21406. doi: 10.1073/pnas.0909131106
Cho, S. W., Park, J.-S., Heo, H. J., Park, S.-W., Song, S., Kim, I., et al. (2014). Dual modulation of the mitochondrial permeability transition pore and redox signaling synergistically promotes cardiomyocyte differentiation from pluripotent stem cells. J. Am. Heart Assoc. 3:e000693. doi: 10.1161/JAHA.113.000693
Chong, C.-R., Sallustio, B., and Horowitz, J. D. (2016). Drugs that affect cardiac metabolism: focus on perhexiline. Cardiovasc. Drugs Ther. 30, 399–405. doi: 10.1007/s10557-016-6664-3
Christe, M. E., and Rodgers, R. L. (1994). Altered glucose and fatty acid oxidation in hearts of the spontaneously hypertensive rat. J. Mol. Cell. Cardiol. 26, 1371–1375. doi: 10.1006/jmcc.1994.1155
Christian, B., El Alaoui-Talibi, Z., Moravec, M., and Moravec, J. (1998). Palmitate oxidation by the mitochondria from volume-overloaded rat hearts. Mol. Cell. Biochem. 180, 117–128. doi: 10.1023/A:1006851325669
Chu, W., Wan, L., Zhao, D., Qu, X., Cai, F., Huo, R., et al. (2012). Mild hypoxia-induced cardiomyocyte hypertrophy via up-regulation of HIF-1α-mediated TRPC signalling. J. Cell. Mol. Med. 16, 2022–2034. doi: 10.1111/j.1582-4934.2011.01497.x
Ciccarelli, M., Chuprun, J. K., Rengo, G., Gao, E., Wei, Z., Peroutka, R. J., et al. (2011). G protein-coupled receptor kinase 2 activity impairs cardiac glucose uptake and promotes insulin resistance after myocardial ischemia. Circulation 123, 1953–1962. doi: 10.1161/CIRCULATIONAHA.110.988642
Clark, A. L., Fonarow, G. C., and Horwich, T. B. (2011). Waist circumference, body mass index, and survival in systolic heart failure: the obesity paradox revisited. J. Card. Fail. 17, 374–380. doi: 10.1016/j.cardfail.2011.01.009
Clarke, B., Spedding, M., Patmore, L., and McCormack, J. G. (1993). Protective effects of ranolazine in guinea-pig hearts during low-flow ischaemia and their association with increases in active pyruvate dehydrogenase. Br. J. Pharmacol. 109, 748–750. doi: 10.1111/j.1476-5381.1993.tb13637.x
Collins-Nakai, R. L., Noseworthy, D., and Lopaschuk, G. D. (1994). Epinephrine increases ATP production in hearts by preferentially increasing glucose metabolism. Am. J. Physiol. 267, H1862–H1871.
Cook, S. A., Matsui, T., Li, L., and Rosenzweig, A. (2002). Transcriptional effects of chronic akt activation in the heart. J. Biol. Chem. 277, 22528–22533. doi: 10.1074/jbc.M201462200
Coppini, R., Mazzoni, L., Ferrantini, C., Gentile, F., Pioner, J. M., Laurino, A., et al. (2017). Ranolazine prevents phenotype development in a mouse model of hypertrophic cardiomyopathy. Circ. Heart Fail. 10:e003565. doi: 10.1161/CIRCHEARTFAILURE.116.003565.
Cortassa, S., Sollott, S. J., and Aon, M. A. (2017). Mitochondrial respiration and ROS emission during β-oxidation in the heart: an experimental-computational study. PLoS Comput. Biol. 13:e1005588. doi: 10.1371/journal.pcbi.1005588
Cox, K. B., Johnson, K. R., and Wood, P. A. (1998). Chromosomal locations of the mouse fatty acid oxidation genes Cpt1a, Cpt1b, Cpt2, Acadvl, and metabolically related Crat gene. Mamm. Genome Off. J. Int. Mamm. Genome Soc. 9, 608–610. doi: 10.1007/s003359900830
Csányi, G., and Miller, F. J. (2014). Oxidative Stress in Cardiovascular Disease. Int. J. Mol. Sci. 15, 6002–6008. doi: 10.3390/ijms15046002
Cunnane, S., Nugent, S., Roy, M., Courchesne-Loyer, A., Croteau, E., Tremblay, S., et al. (2011). Brain fuel metabolism, aging and alzheimer's disease. Nutr. Burbank Los Angel. Cty. Calif 27, 3–20. doi: 10.1016/j.nut.2010.07.021
Cunniff, B., Wozniak, A. N., Sweeney, P., DeCosta, K., and Heintz, N. H. (2014). Peroxiredoxin 3 levels regulate a mitochondrial redox setpoint in malignant mesothelioma cells. Redox Biol. 3, 79–87. doi: 10.1016/j.redox.2014.11.003
Dalle-Donne, I., Rossi, R., Giustarini, D., Gagliano, N., Lusini, L., Milzani, A., et al. (2001). Actin carbonylation: from a simple marker of protein oxidation to relevant signs of severe functional impairment. Free Radic. Biol. Med. 31, 1075–1083. doi: 10.1016/S0891-5849(01)00690-6
Dávila-Román, V. G., Vedala, G., Herrero, P., de las Fuentes, L., Rogers, J. G., Kelly, D. P., et al. (2002). Altered myocardial fatty acid and glucose metabolism in idiopathic dilated cardiomyopathy. J. Am. Coll. Cardiol. 40, 271–277. doi: 10.1016/S0735-1097(02)01967-8
Des Tombe, A. L., Van Beek-Harmsen, B. J., Lee-De Groot, M. B. E., and Van Der Laarse, W. J. (2002). Calibrated histochemistry applied to oxygen supply and demand in hypertrophied rat myocardium. Microsc. Res. Tech. 58, 412–420. doi: 10.1002/jemt.10153
D'hahan, N., Taouil, K., Dassouli, A., and Morel, J. E. (1997). Long-term therapy with trimetazidine in cardiomyopathic Syrian hamster BIO 14:6. Eur. J. Pharmacol. 328, 163–174.
DiNicolantonio, J. J., Lavie, C. J., Fares, H., Menezes, A. R., and O'Keefe, J. H. (2013). Meta-analysis of carvedilol versus beta 1 selective beta-blockers (atenolol, bisoprolol, metoprolol, and nebivolol). Am. J. Cardiol. 111, 765–769. doi: 10.1016/j.amjcard.2012.11.031
Djordjević, V. B., Zvezdanović, L., and Cosić, V. (2008). Oxidative stress in human diseases. Srp. Arh. Celok. Lek. 136(Suppl. 2), 158–165. doi: 10.2298/SARH08S2158D
Doenst, T., Nguyen, T. D., and Abel, E. D. (2013). Cardiac metabolism in heart failure - implications beyond ATP production. Circ. Res. 113, 709–724. doi: 10.1161/CIRCRESAHA.113.300376
Doenst, T., Pytel, G., Schrepper, A., Amorim, P., Färber, G., Shingu, Y., et al. (2010). Decreased rates of substrate oxidation ex vivo predict the onset of heart failure and contractile dysfunction in rats with pressure overload. Cardiovasc. Res. 86, 461–470. doi: 10.1093/cvr/cvp414
Donato, M., Evelson, P., and Gelpi, R. J. (2017). Protecting the heart from ischemia/reperfusion injury: an update on remote ischemic preconditioning and postconditioning. Curr. Opin. Cardiol. 32, 784–790. doi: 10.1097/HCO.0000000000000447
Douglas, W. R. (1963). Relationships of enzymology to cancer: a review. Br. J. Cancer 17, 415–445. doi: 10.1038/bjc.1963.58
Duncan, J. G., and Finck, B. N. (2008). The PPARα-PGC-1α axis controls cardiac energy metabolism in healthy and diseased myocardium. PPAR Res. 2008:253817. doi: 10.1155/2008/253817
Dyck, J. R. B., Cheng, J.-F., Stanley, W. C., Barr, R., Chandler, M. P., Brown, S., et al. (2004). Malonyl coenzyme a decarboxylase inhibition protects the ischemic heart by inhibiting fatty acid oxidation and stimulating glucose oxidation. Circ. Res. 94, e78–e84. doi: 10.1161/01.RES.0000129255.19569.8f
Eichhorn, E. J. (1998). Restoring function in failing hearts: the effects of beta blockers. Am. J. Med. 104, 163–169. doi: 10.1016/S0002-9343(97)00171-X
Eichhorn, E. J. (2000). Clinical use of beta-blockers in patients with heart failure. J. Card. Fail. 6, 40–46.
Eichhorn, E. J., Bedotto, J. B., Malloy, C. R., Hatfield, B. A., Deitchman, D., Brown, M., et al. (1990). Effect of beta-adrenergic blockade on myocardial function and energetics in congestive heart failure. Improvements in hemodynamic, contractile, and diastolic performance with bucindolol. Circulation 82, 473–483. doi: 10.1161/01.CIR.82.2.473
Eijkelenboom, A., and Burgering, B. M. T. (2013). FOXOs: signalling integrators for homeostasis maintenance. Nat. Rev. Mol. Cell Biol. 14:nrm3507. doi: 10.1038/nrm3507
Elezaby, A., Sverdlov, A. L., Tu, V. H., Soni, K., Luptak, I., Qin, F., et al. (2015). Mitochondrial remodeling in mice with cardiomyocyte-specific lipid overload. J. Mol. Cell. Cardiol. 79, 275–283. doi: 10.1016/j.yjmcc.2014.12.001
El-Shitany, N. A., and El-Desoky, K. (2016). Protective effects of carvedilol and vitamin C against azithromycin-induced cardiotoxicity in rats via decreasing ROS, IL1-β, and TNF-α production and inhibiting NF-κB and caspase-3 expression. Oxid. Med. Cell. Longev. 2016:1874762. doi: 10.1155/2016/1874762
Eltzschig, H. K., and Eckle, T. (2011). Ischemia and reperfusion–from mechanism to translation. Nat. Med. 17, 1391–1401. doi: 10.1038/nm.2507
Engberding, N., Spiekermann, S., Schaefer, A., Heineke, A., Wiencke, A., Müller, M., et al. (2004). Allopurinol attenuates left ventricular remodeling and dysfunction after experimental myocardial infarction: a new action for an old drug? Circulation 110, 2175–2179. doi: 10.1161/01.CIR.0000144303.24894.1C
Erickson, J. R., Joiner, M. A., Guan, X., Kutschke, W., Yang, J., Oddis, C. V., et al. (2008). A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 133, 462–474. doi: 10.1016/j.cell.2008.02.048
Farah, M. E., Sirotkin, V., Haarer, B., Kakhniashvili, D., and Amberg, D. C. (2011). Diverse protective roles of the actin cytoskeleton during oxidative stress. Cytoskelet. Hoboken N.J. 68, 340–354. doi: 10.1002/cm.20516
Ferrari, R., Guardigli, G., Mele, D., Percoco, G. F., Ceconi, C., and Curello, S. (2004). Oxidative stress during myocardial ischaemia and heart failure. Curr. Pharm. Des. 10, 1699–1711. doi: 10.2174/1381612043384718
Fillmore, N., and Lopaschuk, G. D. (2013). Targeting mitochondrial oxidative metabolism as an approach to treat heart failure. Biochim. Biophys. Acta BBA Mol. Cell Res. 1833, 857–865. doi: 10.1016/j.bbamcr.2012.08.014
Finck, B. N., Chinetti, G., and Staels, B. (2008). PPARs/RXRs in cardiovascular physiology and disease. PPAR Res. 2008:173780. doi: 10.1155/2008/173780
Fotino, A. D., Thompson-Paul, A. M., and Bazzano, L. A. (2013). Effect of coenzyme Q10 supplementation on heart failure: a meta-analysis. Am. J. Clin. Nutr. 97, 268–275. doi: 10.3945/ajcn.112.040741
Frank, A., Bonney, M., Bonney, S., Weitzel, L., Koeppen, M., and Eckle, T. (2012). Myocardial ischemia reperfusion injury - from basic science to clinical bedside. Semin. Cardiothorac. Vasc. Anesth. 16, 123–132. doi: 10.1177/1089253211436350
Fu, Y., Xiao, H., Ma, X., Jiang, S., Xu, M., and Zhang, Y. (2011). Metformin attenuates pressure overload-induced cardiac hypertrophy via AMPK activation. Acta Pharmacol. Sin. 32, 879–887. doi: 10.1038/aps.2010.229
Fukushima, A., Milner, K., Gupta, A., and Lopaschuk, G. D. (2015). Myocardial energy substrate metabolism in heart failure : from pathways to therapeutic targets. Curr. Pharm. Des. 21, 3654–3664. doi: 10.2174/1381612821666150710150445
Gao, W. D., Murray, C. I., Tian, Y., Zhong, X., DuMond, J. F., Shen, X., et al. (2012). Nitroxyl-mediated disulfide bond formation between cardiac myofilament cysteines enhances contractile function. Circ. Res. 111, 1002–1011. doi: 10.1161/CIRCRESAHA.112.270827
Gatto, G. J., Ao, Z., Kearse, M. G., Zhou, M., Morales, C. R., Daniels, E., et al. (2013). NADPH oxidase-dependent and -independent mechanisms of reported inhibitors of reactive oxygen generation. J. Enzyme Inhib. Med. Chem. 28, 95–104. doi: 10.3109/14756366.2011.636360
Gerstein, H. C. (2001). Diabetes and the HOPE study: implications for macrovascular and microvascular disease. Int. J. Clin. Pract (Suppl). 55, 8–12.
Giordano, F. J. (2005). Oxygen, oxidative stress, hypoxia, and heart failure. J. Clin. Invest. 115, 500–508. doi: 10.1172/JCI200524408
Goh, K. Y., Qu, J., Hong, H., Liu, T., Dell'Italia, L. J., Wu, Y., et al. (2016). Impaired mitochondrial network excitability in failing guinea-pig cardiomyocytes. Cardiovasc. Res. 109, 79–89. doi: 10.1093/cvr/cvv230
Goodwin, G. W., and Taegtmeyer, H. (2000). Improved energy homeostasis of the heart in the metabolic state of exercise. Am. J. Physiol. Heart Circ. Physiol. 279, H1490–H1501.
Goodwin, G. W., Taylor, C. S., and Taegtmeyer, H. (1998). regulation of energy metabolism of the heart during acute increase in heart work. J. Biol. Chem. 273, 29530–29539. doi: 10.1074/jbc.273.45.29530
Gordan, R., Gwathmey, J. K., and Xie, L.-H. (2015). Autonomic and endocrine control of cardiovascular function. World J. Cardiol. 7, 204–214. doi: 10.4330/wjc.v7.i4.204
Goszcz, K., Deakin, S. J., Duthie, G. G., Stewart, D., Leslie, S. J., and Megson, I. L. (2015). Antioxidants in cardiovascular therapy: panacea or false hope? Front. Cardiovasc. Med. 2:29. doi: 10.3389/fcvm.2015.00029
Götz, F. M., Hertel, M., and Gröschel-Stewart, U. (1994). Fatty acid binding of myoglobin depends on its oxygenation. Biol. Chem. Hoppe. Seyler 375, 387–392. doi: 10.1515/bchm3.1994.375.6.387
Gralinski, M. R., Chi, L., Park, J. L., Friedrichs, G. S., Tanhehco, E. J., McCormack, J. G., et al. (1996). Protective effects of ranolazine on ventricular fibrillation induced by activation of the ATP-dependent potassium channel in the rabbit heart. J. Cardiovasc. Pharmacol. Ther. 1, 141–148. doi: 10.1177/107424849600100208
Grimm, M., Ling, H., Pereira, L., Willeford, A., Gray, C. B. B., Erickson, J. R., et al. (2015). CaMKIIδ mediates β-adrenergic effects on RyR2 phosphorylation and SR Ca2+ leak and the pathophysiological response to chronic β-adrenergic stimulation. J. Mol. Cell. Cardiol. 85, 282–291. doi: 10.1016/j.yjmcc.2015.06.007
Gray, L. R., Tompkins, S. C., and Taylor, E. B. (2014). Regulation of pyruvate metabolism and human disease. Cell. Mol. Life Sci. CMLS 71, 2577–2604. doi: 10.1007/s00018-013-1539-2
Gross, G. J., Hsu, A., Falck, J. R., and Nithipatikom, K. (2007). Mechanisms by which epoxyeicosatrienoic acids (EETs) elicit cardioprotection in rat hearts. J. Mol. Cell. Cardiol. 42, 687–691. doi: 10.1016/j.yjmcc.2006.11.020
Grynberg, A., and Demaison, L. (1996). Fatty acid oxidation in the heart. J. Cardiovasc. Pharmacol. 28(Suppl. 1), S11–S17.
Guo, Y., Fan, Y., Zhang, J., Lomberk, G. A., Zhou, Z., Sun, L., et al. (2015). Perhexiline activates KLF14 and reduces atherosclerosis by modulating ApoA-I production. J. Clin. Invest. 125, 3819–3830. doi: 10.1172/JCI79048
Gupta, A., Akki, A., Wang, Y., Leppo, M. K., Chacko, V. P., Foster, D. B., et al. (2012). Creatine kinase–mediated improvement of function in failing mouse hearts provides causal evidence the failing heart is energy starved. J. Clin. Invest. 122, 291–302. doi: 10.1172/JCI57426
Gutierrez, J. A., Karwatowska-Prokopczuk, E., Murphy, S. A., Belardinelli, L., Farzaneh-Far, R., Walker, G., et al. (2015). effects of ranolazine in patients with chronic angina in patients with and without percutaneous coronary intervention for acute coronary syndrome: observations From the MERLIN-TIMI 36 trial. Clin. Cardiol. 38, 469–475. doi: 10.1002/clc.22425
Hada, T., Kato, Y., Obana, E., Yamamoto, A., Yamazaki, N., Hashimoto, M., et al. (2012). Comparison of two expression systems using COS7 cells and yeast cells for expression of heart/muscle-type carnitine palmitoyltransferase 1. Protein Expr. Purif. 82, 192–196. doi: 10.1016/j.pep.2012.01.006
Hamilton, C., and Saggerson, E. D. (2000). Malonyl-CoA metabolism in cardiac myocytes. Biochem. J. 350(Pt 1), 61–67. doi: 10.1042/bj3500061
Hansford, R. G., and Cohen, L. (1978). Relative importance of pyruvate dehydrogenase interconversion and feed-back inhibition in the effect of fatty acids on pyruvate oxidation by rat heart mitochondria. Arch. Biochem. Biophys. 191, 65–81. doi: 10.1016/0003-9861(78)90068-1
Hardie, D. G. (2004). AMP-activated protein kinase: the guardian of cardiac energy status. J. Clin. Invest. 114, 465–468. doi: 10.1172/JCI200422683
He, L., Kim, T., Long, Q., Liu, J., Wang, P., Zhou, Y., et al. (2012). Carnitine Palmitoyltransferase-1b (CPT1b) deficiency aggravates pressure-overload-induced cardiac hypertrophy due to lipotoxicity. Circulation 126, 1705–1716. doi: 10.1161/CIRCULATIONAHA.111.075978
He, Y., Yu, S., Hu, J., Cui, Y., and Liu, P. (2016). Changes in the anatomic and microscopic structure and the expression of HIF-1α and VEGF of the yak heart with aging and hypoxia. PLoS ONE 11:e0149947. doi: 10.1371/journal.pone.0149947
Hemilä, H., and Kaprio, J. (2009). Modification of the effect of vitamin E supplementation on the mortality of male smokers by age and dietary vitamin C. Am. J. Epidemiol. 169, 946–953. doi: 10.1093/aje/kwn413
Hemilä, H., and Kaprio, J. (2011). Vitamin E may affect the life expectancy of men, depending on dietary vitamin C intake and smoking. Age Ageing 40, 215–220. doi: 10.1093/ageing/afq178
Hendgen-Cotta, U. B., Esfeld, S., Coman, C., Ahrends, R., Klein-Hitpass, L., Flögel, U., et al. (2017). A novel physiological role for cardiac myoglobin in lipid metabolism. Sci. Rep. 7:43219. doi: 10.1038/srep43219
Hesen, N. A., Riksen, N. P., Aalders, B., Ritskes-Hoitinga, M., El Messaoudi, S., and Wever, K. E. (2017). A systematic review and meta-analysis of the protective effects of metformin in experimental myocardial infarction. PLoS ONE 12:e0183664. doi: 10.1371/journal.pone.0183664
Hirsch, G. A., Bottomley, P. A., Gerstenblith, G., and Weiss, R. G. (2012). Allopurinol acutely increases adenosine triphospate energy delivery in failing human hearts. J. Am. Coll. Cardiol. 59, 802–808. doi: 10.1016/j.jacc.2011.10.895
Ho, H.-T., Belevych, A. E., Liu, B., Bonilla, I. M., Radwański, P. B., Kubasov, I. V., et al. (2016). Muscarinic stimulation facilitates sarcoplasmic reticulum Ca Release by modulating ryanodine receptor 2 phosphorylation through protein kinase G and Ca/calmodulin-dependent protein kinase II. Hypertension 68, 1171–1178. doi: 10.1161/HYPERTENSIONAHA.116.07666
Holubarsch, C. J. F., Rohrbach, M., Karrasch, M., Boehm, E., Polonski, L., Ponikowski, P., et al. (2007). A double-blind randomized multicentre clinical trial to evaluate the efficacy and safety of two doses of etomoxir in comparison with placebo in patients with moderate congestive heart failure: the ERGO (etomoxir for the recovery of glucose oxidation) study. Clin. Sci. Lond. Engl. 113, 205–212. doi: 10.1042/CS20060307
Hopkins, T. A., Sugden, M. C., Holness, M. J., Kozak, R., Dyck, J. R. B., and Lopaschuk, G. D. (2003). Control of cardiac pyruvate dehydrogenase activity in peroxisome proliferator-activated receptor-α transgenic mice. Am. J. Physiol. Heart Circ. Physiol. 285, H270–H276. doi: 10.1152/ajpheart.00852.2002
Hou, X., Song, J., Li, X.-N., Zhang, L., Wang, X., Chen, L., et al. (2010). Metformin reduces intracellular reactive oxygen species levels by upregulating expression of the antioxidant thioredoxin via the AMPK-FOXO3 pathway. Biochem. Biophys. Res. Commun. 396, 199–205. doi: 10.1016/j.bbrc.2010.04.017
Hsiao, Y.-S., Jogl, G., and Tong, L. (2006). Crystal structures of murine carnitine acetyltransferase in ternary complexes with its substrates. J. Biol. Chem. 281, 28480–28487. doi: 10.1074/jbc.M602622200
Huss, J. M., and Kelly, D. P. (2005). Mitochondrial energy metabolism in heart failure: a question of balance. J. Clin. Invest. 115, 547–555. doi: 10.1172/JCI24405
Imamura, F., Lemaitre, R. N., King, I. B., Song, X., Steffen, L. M., Folsom, A. R., et al. (2013). Long-chain monounsaturated Fatty acids and incidence of congestive heart failure in 2 prospective cohorts. Circulation 127, 1512–1521, 1521e1–18. doi: 10.1161/CIRCULATIONAHA.112.001197
Ingwall, J. S. (2009). Energy metabolism in heart failure and remodelling. Cardiovasc. Res. 81, 412–419. doi: 10.1093/cvr/cvn301
Ingwall, J. S., Kramer, M. F., Fifer, M. A., Lorell, B. H., Shemin, R., Grossman, W., et al. (1985). The creatine kinase system in normal and diseased human myocardium. N. Engl. J. Med. 313, 1050–1054. doi: 10.1056/NEJM198510243131704
Ingwall, J. S., and Weiss, R. G. (2004). Is the failing heart energy starved? Circ. Res. 95, 135–145. doi: 10.1161/01.RES.0000137170.41939.d9
Jäger, S., Handschin, C., St.-Pierre, J., and Spiegelman, B. M. (2007). AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1α. Proc. Natl. Acad. Sci. U.S.A. 104, 12017–12022. doi: 10.1073/pnas.0705070104
Javadov, S., Chapa-Dubocq, X., and Makarov, V. (in press). Different approaches to modeling analysis of mitochondrial swelling. Mitochondrion. doi: 10.1016/j.mito.2017.08.004
Jeffrey, F. M., Diczku, V., Sherry, A. D., and Malloy, C. R. (1995). Substrate selection in the isolated working rat heart: effects of reperfusion, afterload, and concentration. Basic Res. Cardiol. 90, 388–396. doi: 10.1007/BF00788500
Jiang, Y.-Q., Chang, G.-L., Wang, Y., Zhang, D.-Y., Cao, L., and Liu, J. (2016). Geniposide prevents hypoxia/reoxygenation-induced apoptosis in H9c2 cells: improvement of mitochondrial dysfunction and activation of GLP-1R and the PI3K/AKT signaling pathway. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 39, 407–421. doi: 10.1159/000445634
Josan, S., Hurd, R., Park, J. M., Yen, Y.-F., Watkins, R., Pfefferbaum, A., et al. (2014). Dynamic metabolic imaging of hyperpolarized [2-13C]pyruvate using spiral CSI with alternating spectral band excitation. Magn. Reson. Med. Off. J. Soc. Magn. Reson. Med. Soc. Magn. Reson. Med. 71, 2051–2058. doi: 10.1002/mrm.24871
Jue, T., Simond, G., Wright, T. J., Shih, L., Chung, Y., Sriram, R., et al. (2016). Effect of fatty acid interaction on myoglobin oxygen affinity and triglyceride metabolism. J. Physiol. Biochem. 73, 359–370. doi: 10.1007/s13105-017-0559-z
Kaasik, A., Safiulina, D., Zharkovsky, A., and Veksler, V. (2007). Regulation of mitochondrial matrix volume. Am. J. Physiol. Cell Physiol. 292, C157–C163. doi: 10.1152/ajpcell.00272.2006
Kaijser, L., and Berglund, B. (1992). Myocardial lactate extraction and release at rest and during heavy exercise in healthy men. Acta Physiol. Scand. 144, 39–45. doi: 10.1111/j.1748-1716.1992.tb09265.x
Kaludercic, N., Mialet-Perez, J., Paolocci, N., Parini, A., and Di Lisa, F. (2014). Monoamine oxidases as sources of oxidants in the heart. J. Mol. Cell. Cardiol. 73, 34–42. doi: 10.1016/j.yjmcc.2013.12.032
Kanaan, G. N., and Harper, M.-E. (2017). Cellular redox dysfunction in the development of cardiovascular diseases. Biochim. Biophys. Acta BBA Gen. Subj. 1861, 2822–2829. doi: 10.1016/j.bbagen.2017.07.027
Kennedy, J. A., Unger, S. A., and Horowitz, J. D. (1996). Inhibition of carnitine palmitoyltransferase-1 in rat heart and liver by perhexiline and amiodarone. Biochem. Pharmacol. 52, 273–280. doi: 10.1016/0006-2952(96)00204-3
Khanna, G., Diwan, V., Singh, M., Singh, N., and Jaggi, A. S. (2008). Reduction of ischemic, pharmacological and remote preconditioning effects by an antioxidant N-acetyl cysteine pretreatment in isolated rat heart. Yakugaku Zasshi 128, 469–477. doi: 10.1248/yakushi.128.469
Khemtong, C., Carpenter, N. R., Lumata, L. L., Merritt, M. E., Moreno, K. X., Kovacs, Z., et al. (2015). Hyperpolarized 13C NMR detects rapid drug-induced changes in cardiac metabolism. Magn. Reson. Med. 74, 312–319. doi: 10.1002/mrm.25419
Kim, A. S., Miller, E. J., Wright, T. M., Li, J., Qi, D., Atsina, K., et al. (2011). A small molecule AMPK activator protects the heart against ischemia-reperfusion injury. J. Mol. Cell. Cardiol. 51, 24–32. doi: 10.1016/j.yjmcc.2011.03.003
King, K. L., Okere, I. C., Sharma, N., Dyck, J. R. B., Reszko, A. E., McElfresh, T. A., et al. (2005). Regulation of cardiac malonyl-CoA content and fatty acid oxidation during increased cardiac power. Am. J. Physiol. Heart Circ. Physiol. 289, H1033–H1037. doi: 10.1152/ajpheart.00210.2005
Klabunde, R. E. (2012). Cardiovascular Physiology Concepts, 2nd Edn. Philadelphia, PA: Lippincott Williams & Wilkins/Wolters Kluwer.
Kler, R. S., Jackson, S., Bartlett, K., Bindoff, L. A., Eaton, S., Pourfarzam, M., et al. (1991). Quantitation of acyl-CoA and acylcarnitine esters accumulated during abnormal mitochondrial fatty acid oxidation. J. Biol. Chem. 266, 22932–22938.
Kolwicz, S. C., Olson, D. P., Marney, L. C., Garcia-Menendez, L., Synovec, R. E., and Tian, R. (2012). Cardiac-specific deletion of acetyl CoA carboxylase 2 prevents metabolic remodeling during pressure-overload hypertrophy. Circ. Res. 111, 728–738. doi: 10.1161/CIRCRESAHA.112.268128
Kousteni, S. (2012). FoxO1, the transcriptional chief of staff of energy metabolism. Bone 50, 437–443. doi: 10.1016/j.bone.2011.06.034
Kuzmicic, J., Parra, V., Verdejo, H. E., López-Crisosto, C., Chiong, M., García, L., et al. (2014). Trimetazidine prevents palmitate-induced mitochondrial fission and dysfunction in cultured cardiomyocytes. Biochem. Pharmacol. 91, 323–336. doi: 10.1016/j.bcp.2014.07.022
Kveiborg, B., Major-Petersen, A., Christiansen, B., and Torp-Pedersen, C. (2007). Carvedilol in the treatment of chronic heart failure: lessons from the carvedilol or metoprolol european trial. Vasc. Health Risk Manag. 3, 31–37.
Labinskyy, V., Bellomo, M., Chandler, M. P., Young, M. E., Lionetti, V., Qanud, K., et al. (2007). Chronic activation of peroxisome proliferator-activated receptor-alpha with fenofibrate prevents alterations in cardiac metabolic phenotype without changing the onset of decompensation in pacing-induced heart failure. J. Pharmacol. Exp. Ther. 321, 165–171. doi: 10.1124/jpet.106.116871
Lassègue, B., San Martín, A., and Griendling, K. K. (2012). Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ. Res. 110, 1364–1390. doi: 10.1161/CIRCRESAHA.111.243972
Lechat, P. (2004). Clinical trials of beta-blockers in heart failure: history, overview and future prospects. Therapie 59, 517–526. doi: 10.2515/therapie:2004089
Lefort, B., Gouache, E., Acquaviva, C., Tardieu, M., Benoist, J. F., Dumas, J.-F., et al. (2017). Pharmacological inhibition of carnitine palmitoyltransferase 1 restores mitochondrial oxidative phosphorylation in human trifunctional protein deficient fibroblasts. Biochim. Biophys. Acta BBA Mol. Basis Dis. 1863, 1292–1299. doi: 10.1016/j.bbadis.2017.04.005
Lehman, J. J., and Kelly, D. P. (2002a). Gene regulatory mechanisms governing energy metabolism during cardiac hypertrophic growth. Heart Fail. Rev. 7, 175–185. doi: 10.1023/A:1015332726303
Lehman, J. J., and Kelly, D. P. (2002b). Transcriptional activation of energy metabolic switches in the developing and hypertrophied heart. Clin. Exp. Pharmacol. Physiol. 29, 339–345. doi: 10.1046/j.1440-1681.2002.03655.x
Leij, F. R., van der Cox, K. B., Jackson, V. N., Huijkman, N. C. A., Bartelds, B., Kuipers, J. R. G., et al. (2002). Structural and functional genomics of the CPT1B gene for muscle-type carnitine palmitoyltransferase I in mammals. J. Biol. Chem. 277, 26994–27005. doi: 10.1074/jbc.M203189200
Levy, D., Kenchaiah, S., Larson, M. G., Benjamin, E. J., Kupka, M. J., Ho, K. K. L., et al. (2002). Long-term trends in the incidence of and survival with heart failure. N. Engl. J. Med. 347, 1397–1402. doi: 10.1056/NEJMoa020265
Lexis, C. P. H., van der Horst, I. C. C., Lipsic, E., Wieringa, W. G., de Boer, R. A., van den Heuvel, A. F. M., et al. (2014). Effect of metformin on left ventricular function after acute myocardial infarction in patients without diabetes: the GIPS-III randomized clinical trial. JAMA 311, 1526–1535. doi: 10.1001/jama.2014.3315
Li, H., Xu, C., Li, Q., Gao, X., Sugano, E., Tomita, H., et al. (2017). Thioredoxin 2 offers protection against mitochondrial oxidative stress in H9c2 cells and against myocardial hypertrophy induced by hyperglycemia. Int. J. Mol. Sci. 18:E1958. doi: 10.3390/ijms18091958
Li, J., Chen, Z., Gao, X., Zhang, H., Xiong, W., Ju, J., et al. (2017). Meta-analysis comparing metoprolol and carvedilol on mortality benefits in patients with acute myocardial infarction. Am. J. Cardiol. 120, 1479–1486. doi: 10.1016/j.amjcard.2017.07.036
Liberts, E. A., Willoughby, S. R., Kennedy, J. A., and Horowitz, J. D. (2007). Effects of perhexiline and nitroglycerin on vascular, neutrophil and platelet function in patients with stable angina pectoris. Eur. J. Pharmacol. 560, 49–55. doi: 10.1016/j.ejphar.2007.01.001
Liu, R., Li, L., Chen, Y., Yang, M., Liu, H., and Yang, G. (2014). Effects of glucagon-like peptide-1 agents on left ventricular function: systematic review and meta-analysis. Ann. Med. 46, 664–671. doi: 10.3109/07853890.2014.949837
Lopaschuk, G. D., and Spafford, M. A. (1990). Energy substrate utilization by isolated working hearts from newborn rabbits. Am. J. Physiol. 258, H1274–H1280.
Lopaschuk, G. D., Ussher, J. R., Folmes, C. D. L., Jaswal, J. S., and Stanley, W. C. (2010). Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 90, 207–258. doi: 10.1152/physrev.00015.2009
Luiken, J. J. F. P., Coort, S. L. M., Koonen, D. P. Y., van der Horst, D. J., Bonen, A., Zorzano, A., et al. (2004). Regulation of cardiac long-chain fatty acid and glucose uptake by translocation of substrate transporters. Pflugers Arch. 448, 1–15. doi: 10.1007/s00424-003-1199-4
Ma, Y., and Li, J. (2015). Metabolic Shifts during Aging and Pathology. Compr. Physiol. 5, 667–686. doi: 10.1002/cphy.c140041
Mangmool, S., Denkaew, T., Phosri, S., Pinthong, D., Parichatikanond, W., Shimauchi, T., et al. (2016). Sustained βAR stimulation mediates cardiac insulin resistance in a PKA-dependent manner. Mol. Endocrinol. 30, 118–132. doi: 10.1210/me.2015-1201
Marazzi, G., Gebara, O., Vitale, C., Caminiti, G., Wajngarten, M., Volterrani, M., et al. (2009). Effect of trimetazidine on quality of life in elderly patients with ischemic dilated cardiomyopathy. Adv. Ther. 26, 455–461. doi: 10.1007/s12325-009-0024-7
Margulies, K. B., Anstrom, K. J., Hernandez, A. F., Redfield, M. M., Shah, M. R., Braunwald, E., et al. (2014). GLP-1 Agonist therapy for advanced heart failure with reduced ejection fraction. Circ. Heart Fail. 7, 673–679. doi: 10.1161/CIRCHEARTFAILURE.114.000346
Mariotti, E., Orton, M. R., Eerbeek, O., Ashruf, J. F., Zuurbier, C. J., Southworth, R., et al. (2016). Modeling non-linear kinetics of hyperpolarized [1-13C] pyruvate in the crystalloid-perfused rat heart. Nmr Biomed. 29, 377–386. doi: 10.1002/nbm.3464
Martin, W. H., Dalsky, G. P., Hurley, B. F., Matthews, D. E., Bier, D. M., Hagberg, J. M., et al. (1993). Effect of endurance training on plasma free fatty acid turnover and oxidation during exercise. Am. J. Physiol. 265, E708–E714.
Masoud, W. G. T., Ussher, J. R., Wang, W., Jaswal, J. S., Wagg, C. S., Dyck, J. R., et al. (2014). Failing mouse hearts utilize energy inefficiently and benefit from improved coupling of glycolysis and glucose oxidation. Cardiovasc. Res. 101, 30–38. doi: 10.1093/cvr/cvt216
Masson, S., Marchioli, R., Mozaffarian, D., Bernasconi, R., Milani, V., Dragani, L., et al. (2013). Plasma n-3 polyunsaturated fatty acids in chronic heart failure in the GISSI-Heart Failure Trial: relation with fish intake, circulating biomarkers, and mortality. Am. Heart J. 165, 208–215.e4. doi: 10.1016/j.ahj.2012.10.021
Matsushima, S., Kuroda, J., Ago, T., Zhai, P., Park, J. Y., Xie, L.-H., et al. (2013). Increased oxidative stress in the nucleus caused by Nox4 mediates oxidation of HDAC4 and cardiac hypertrophy. Circ. Res. 112, 651–663. doi: 10.1161/CIRCRESAHA.112.279760
Meng, D., Feng, L., Chen, X.-J., Yang, D., and Zhang, J.-N. (2006). Trimetazidine improved Ca2+ handling in isoprenaline-mediated myocardial injury of rats. Exp. Physiol. 91, 591–601. doi: 10.1113/expphysiol.2005.032615
Misra, M. K., Sarwat, M., Bhakuni, P., Tuteja, R., and Tuteja, N. (2009). Oxidative stress and ischemic myocardial syndromes. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 15, RA209–RA219.
Miura, S., Kai, Y., Kamei, Y., and Ezaki, O. (2008). Isoform-specific increases in murine skeletal muscle peroxisome proliferator-activated receptor-gamma coactivator-1alpha (PGC-1alpha) mRNA in response to beta2-adrenergic receptor activation and exercise. Endocrinology 149, 4527–4533. doi: 10.1210/en.2008-0466
Mjos, O. D. (1971). Effect of free fatty acids on myocardial function and oxygen consumption in intact dogs. J. Clin. Invest. 50, 1386–1389. doi: 10.1172/JCI106621
Morisco, C., Condorelli, G., Trimarco, V., Bellis, A., Marrone, C., Condorelli, G., et al. (2005). Akt mediates the cross-talk between beta-adrenergic and insulin receptors in neonatal cardiomyocytes. Circ. Res. 96, 180–188. doi: 10.1161/01.RES.0000152968.71868.c3
Mortensen, S. A., Rosenfeldt, F., Kumar, A., Dolliner, P., Filipiak, K. J., Pella, D., et al. (2014). The effect of coenzyme Q10 on morbidity and mortality in chronic heart failure: results from Q-SYMBIO: a randomized double-blind trial. JACC Heart Fail. 2, 641–649. doi: 10.1016/j.jchf.2014.06.008
Mozaffarian, D., Bryson, C. L., Lemaitre, R. N., Burke, G. L., and Siscovick, D. S. (2005). Fish intake and risk of incident heart failure. J. Am. Coll. Cardiol. 45, 2015–2021. doi: 10.1016/j.jacc.2005.03.038
Mueller-Hennessen, M., Sigl, J., Fuhrmann, J. C., Witt, H., Reszka, R., Schmitz, O., et al. (2017). Metabolic profiles in heart failure due to non-ischemic cardiomyopathy at rest and under exercise. Esc Heart Fail. 4:178. doi: 10.1002/ehf2.12133
Mutlak, M., and Kehat, I. (2015). Extracellular signal-regulated kinases 1/2 as regulators of cardiac hypertrophy. Front. Pharmacol. 6:149. doi: 10.3389/fphar.2015.00149
Nadkarni, P., Chepurny, O. G., and Holz, G. G. (2014). Regulation of glucose homeostasis by GLP-1. Prog. Mol. Biol. Transl. Sci. 121, 23–65. doi: 10.1016/B978-0-12-800101-1.00002-8
Nagarajan, V., Kohan, L., Holland, E., Keeley, E. C., and Mazimba, S. (2016). Obesity paradox in heart failure: a heavy matter. ESC Heart Fail. 3, 227–234. doi: 10.1002/ehf2.12120
Nagoshi, T., Yoshimura, M., Rosano, G. M. C., Lopaschuk, G. D., and Mochizuki, S. (2011). Optimization of cardiac metabolism in heart failure. Curr. Pharm. Des. 17, 3846–3853. doi: 10.2174/138161211798357773
Nakamura, K., Fushimi, K., Kouchi, H., Mihara, K., Miyazaki, M., Ohe, T., et al. (1998). Inhibitory effects of antioxidants on neonatal rat cardiac myocyte hypertrophy induced by tumor necrosis factor-alpha and angiotensin II. Circulation 98, 794–799. doi: 10.1161/01.CIR.98.8.794
Nakamura, T., Ranek, M. J., Lee, D. I., Shalkey Hahn, V., Kim, C., Eaton, P., et al. (2015). Prevention of PKG1α oxidation augments cardioprotection in the stressed heart. J. Clin. Invest. 125, 2468–2472. doi: 10.1172/JCI80275
Nediani, C., Raimondi, L., Borchi, E., and Cerbai, E. (2010). Nitric oxide/reactive oxygen species generation and nitroso/redox imbalance in heart failure: from molecular mechanisms to therapeutic implications. Antioxid. Redox Signal. 14, 289–331. doi: 10.1089/ars.2010.3198
Neely, J. R., Denton, R. M., England, P. J., and Randle, P. J. (1972). The effects of increased heart work on the tricarboxylate cycle and its interactions with glycolysis in the perfused rat heart. Biochem. J. 128, 147–159. doi: 10.1042/bj1280147
Neubauer, S. (2007). The failing heart — an engine out of fuel. N. Engl. J. Med. 356, 1140–1151. doi: 10.1056/NEJMra063052
Nikolaidis, L. A., Elahi, D., Hentosz, T., Doverspike, A., Huerbin, R., Zourelias, L., et al. (2004a). Recombinant glucagon-like peptide-1 increases myocardial glucose uptake and improves left ventricular performance in conscious dogs with pacing-induced dilated cardiomyopathy. Circulation 110, 955–961. doi: 10.1161/01.CIR.0000139339.85840.DD
Nikolaidis, L. A., Mankad, S., Sokos, G. G., Miske, G., Shah, A., Elahi, D., et al. (2004b). Effects of glucagon-like peptide-1 in patients with acute myocardial infarction and left ventricular dysfunction after successful reperfusion. Circulation 109, 962–965. doi: 10.1161/01.CIR.0000120505.91348.58
Nocito, L., Kleckner, A. S., Yoo, E. J., Jones, A. R. IV, Liesa, M., and Corkey, B. E. (2015). The extracellular redox state modulates mitochondrial function, gluconeogenesis, and glycogen synthesis in murine hepatocytes. PLoS ONE 10:e0122818. doi: 10.1371/journal.pone.0122818
O'Neill, B. T., Kim, J., Wende, A. R., Theobald, H. A., Tuinei, J., Buchanan, J., et al. (2007). A conserved role for phosphatidylinositol 3-kinase but not akt signaling in mitochondrial adaptations that accompany physiological cardiac hypertrophy. Cell Metab. 6, 294–306. doi: 10.1016/j.cmet.2007.09.001
Okere, I. C., Chess, D. J., McElfresh, T. A., Johnson, J., Rennison, J., Ernsberger, P., et al. (2005). High-fat diet prevents cardiac hypertrophy and improves contractile function in the hypertensive dahl salt-sensitive rat. Clin. Exp. Pharmacol. Physiol. 32, 825–831. doi: 10.1111/j.1440-1681.2005.04272.x
Ono, H., Ohara, N., and Hashimoto, K. (1982). Effect of an antianginal drug, perhexiline, on myocardial oxygen consumption in anesthetized open-chest dogs compared with verapamil and glyceryl trinitrate. Jpn. Circ. J. 46, 559–567. doi: 10.1253/jcj.46.559
Opie, L. H. (1968). Metabolism of the heart in health and disease. I. Am. Heart J. 76, 685–698. doi: 10.1016/0002-8703(68)90168-3
Opie, L. H. (1969). Metabolism of the heart in health and disease. Part II. Am. Heart J. 77, 100–122. doi: 10.1016/0002-8703(69)90135-5
Opie, L. H. (2004). Heart Physiology: From Cell to Circulation. Philadelphia, PA: Lippincott Williams & Wilkins.
Oreopoulos, A., Padwal, R., Kalantar-Zadeh, K., Fonarow, G. C., Norris, C. M., and McAlister, F. A. (2008). Body mass index and mortality in heart failure: a meta-analysis. Am. Heart J. 156, 13–22. doi: 10.1016/j.ahj.2008.02.014
Park, K.-M., Teoh, J.-P., Wang, Y., Broskova, Z., Bayoumi, A. S., Tang, Y., et al. (2016). Carvedilol-responsive microRNAs, miR-199a-3p and -214 protect cardiomyocytes from simulated ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 311, H371–H383. doi: 10.1152/ajpheart.00807.2015
Penna, C., Perrelli, M.-G., and Pagliaro, P. (2013). Mitochondrial pathways, permeability transition pore, and redox signaling in cardioprotection: therapeutic implications. Antioxid. Redox Signal. 18, 556–599. doi: 10.1089/ars.2011.4459
Pereira, R. O., Wende, A. R., Crum, A., Hunter, D., Olsen, C. D., Rawlings, T., et al. (2014). Maintaining PGC-1α expression following pressure overload-induced cardiac hypertrophy preserves angiogenesis but not contractile or mitochondrial function. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 28, 3691–3702. doi: 10.1096/fj.14-253823
Pereira, R. O., Wende, A. R., Olsen, C., Soto, J., Rawlings, T., Zhu, Y., et al. (2013). Inducible overexpression of GLUT1 prevents mitochondrial dysfunction and attenuates structural remodeling in pressure overload but does not prevent left ventricular dysfunction. J. Am. Heart Assoc. 2:e000301. doi: 10.1161/JAHA.113.000301
Petersen, K. E., Rakipovski, G., Raun, K., and Lykkesfeldt, J. (2016). Does glucagon-like peptide-1 ameliorate oxidative stress in diabetes? evidence based on experimental and clinical studies. Curr. Diabetes Rev. 12, 331–358. doi: 10.2174/1573399812666150918150608
Phillips, D., Ten Hove, M., Schneider, J. E., Wu, C. O., Sebag-Montefiore, L., Aponte, A. M., et al. (2010). Mice over-expressing the myocardial creatine transporter develop progressive heart failure and show decreased glycolytic capacity. J. Mol. Cell. Cardiol. 48, 582–590. doi: 10.1016/j.yjmcc.2009.10.033
Poornima, I., Brown, S. B., Bhashyam, S., Parikh, P., Bolukoglu, H., and Shannon, R. P. (2008). Chronic glucagon-like peptide-1 infusion sustains left ventricular systolic function and prolongs survival in the spontaneously hypertensive, heart failure-prone rat. Circ. Heart Fail. 1, 153–160. doi: 10.1161/CIRCHEARTFAILURE.108.766402
Prosser, B. L., Khairallah, R. J., Ziman, A. P., Ward, C. W., and Lederer, W. J. (2013). X-ROS signaling in the heart and skeletal muscle: stretch-dependent local ROS regulates [Ca2+]i. J. Mol. Cell. Cardiol. 58, 172–181. doi: 10.1016/j.yjmcc.2012.11.011
Prosser, B. L., Ward, C. W., and Lederer, W. J. (2011). X-ROS signaling: rapid mechano- chemo transduction in heart. Science 333, 1440–1445. doi: 10.1126/science.1202768
Prysyazhna, O., Rudyk, O., and Eaton, P. (2012). Single atom substitution in mouse protein kinase G eliminates oxidant sensing to cause hypertension. Nat. Med. 18, 286–290. doi: 10.1038/nm.2603
Qiu, Y., Tang, X. L., Park, S. W., Sun, J. Z., Kalya, A., and Bolli, R. (1997). The early and late phases of ischemic preconditioning: a comparative analysis of their effects on infarct size, myocardial stunning, and arrhythmias in conscious pigs undergoing a 40-minute coronary occlusion. Circ. Res. 80, 730–742. doi: 10.1161/01.RES.80.5.730
Quyyumi, A. A., Dakak, N., Andrews, N. P., Gilligan, D. M., Panza, J. A., and Cannon, R. O. (1995). Contribution of nitric oxide to metabolic coronary vasodilation in the human heart. Circulation 92, 320–326. doi: 10.1161/01.CIR.92.3.320
Raedschelders, K., Ansley, D. M., and Chen, D. D. Y. (2012). The cellular and molecular origin of reactive oxygen species generation during myocardial ischemia and reperfusion. Pharmacol. Ther. 133, 230–255. doi: 10.1016/j.pharmthera.2011.11.004
Raher, M. J., Thibault, H. B., Buys, E. S., Kuruppu, D., Shimizu, N., Brownell, A.-L., et al. (2008). A short duration of high-fat diet induces insulin resistance and predisposes to adverse left ventricular remodeling after pressure overload. Am. J. Physiol. Heart Circ. Physiol. 295, H2495–H2502. doi: 10.1152/ajpheart.00139.2008
Randle, P. J. (1998). Regulatory interactions between lipids and carbohydrates: the glucose fatty acid cycle after 35 years. Diabetes. Metab. Rev. 14, 263–283.
Rassaf, T., Flögel, U., Drexhage, C., Hendgen-Cotta, U., Kelm, M., and Schrader, J. (2007). Nitrite reductase function of deoxymyoglobin: oxygen sensor and regulator of cardiac energetics and function. Circ. Res. 100, 1749–1754. doi: 10.1161/CIRCRESAHA.107.152488
Reszko, A. E., Kasumov, T., David, F., Thomas, K. R., Jobbins, K. A., Cheng, J.-F., et al. (2004). Regulation of malonyl-CoA concentration and turnover in the normal heart. J. Biol. Chem. 279, 34298–34301. doi: 10.1074/jbc.M405488200
Richters, L., Lange, N., Renner, R., Treiber, N., Ghanem, A., Tiemann, K., et al. (2011). Exercise-induced adaptations of cardiac redox homeostasis and remodeling in heterozygous SOD2-knockout mice. J. Appl. Physiol. 111, 1431–1440. doi: 10.1152/japplphysiol.01392.2010
Risérus, U., Sprecher, D., Johnson, T., Olson, E., Hirschberg, S., Liu, A., et al. (2008). Activation of peroxisome proliferator-activated receptor (PPAR)delta promotes reversal of multiple metabolic abnormalities, reduces oxidative stress, and increases fatty acid oxidation in moderately obese men. Diabetes 57, 332–339. doi: 10.2337/db07-1318
Ritchie, R. H., and Delbridge, L. M. D. (2006). Cardiac hypertrophy, substrate utilization and metabolic remodelling: cause or effect? Clin. Exp. Pharmacol. Physiol. 33, 159–166. doi: 10.1111/j.1440-1681.2006.04342.x
Roger, V. L., Go, A. S., Lloyd-Jones, D. M., Benjamin, E. J., Berry, J. D., Borden, W. B., et al. (2012). Heart disease and stroke statistics—2012 update. Circulation 125, e2–e220. doi: 10.1161/CIR.0b013e31823ac046
Rowe, G. C., Jiang, A., and Arany, Z. (2010). PGC-1 coactivators in cardiac development and disease. Circ. Res. 107, 825–838. doi: 10.1161/CIRCRESAHA.110.223818
Rupp, H., and Vetter, R. (2000). Sarcoplasmic reticulum function and carnitine palmitoyltransferase-1 inhibition during progression of heart failure. Br. J. Pharmacol. 131, 1748–1756. doi: 10.1038/sj.bjp.0703741
Sabri, A., Byron, K. L., Samarel, A. M., Bell, J., and Lucchesi, P. A. (1998). Hydrogen peroxide activates mitogen-activated protein kinases and Na+-H+ exchange in neonatal rat cardiac myocytes. Circ. Res. 82, 1053–1062. doi: 10.1161/01.RES.82.10.1053
Sack, M. N., and Kelly, D. P. (1998). The energy substrate switch during development of heart failure: gene regulatory mechanisms (review). Int. J. Mol. Med. 1, 17–24. doi: 10.3892/ijmm.1.1.17
Sack, M. N., Rader, T. A., Park, S., Bastin, J., McCune, S. A., and Kelly, D. P. (1996). Fatty acid oxidation enzyme gene expression is downregulated in the failing heart. Circulation 94, 2837–2842. doi: 10.1161/01.CIR.94.11.2837
Saeedi, R., Grist, M., Wambolt, R. B., Bescond-Jacquet, A., Lucien, A., and Allard, M. F. (2005). Trimetazidine normalizes postischemic function of hypertrophied rat hearts. J. Pharmacol. Exp. Ther. 314, 446–454. doi: 10.1124/jpet.104.082636
Saggerson, E. D. (1982). Carnitine acyltransferase activities in rat liver and heart measured with palmitoyl-CoA and octanoyl-CoA. Latency, effects of K+, bivalent metal ions and malonyl-CoA. Biochem. J. 202, 397–405. doi: 10.1042/bj2020397
Salzman, M. M., Cheng, Q., Deklotz, R. J., Dulai, G. K., Douglas, H. F., Dikalova, A. E., et al. (2017). Lipid emulsion enhances cardiac performance after ischemia–reperfusion in isolated hearts from summer-active arctic ground squirrels. J. Comp. Physiol. B 187, 715–724. doi: 10.1007/s00360-017-1071-z
Sanada, S., Komuro, I., and Kitakaze, M. (2011). Pathophysiology of myocardial reperfusion injury: preconditioning, postconditioning, and translational aspects of protective measures. Am. J. Physiol. Heart Circ. Physiol. 301, H1723–H1741. doi: 10.1152/ajpheart.00553.2011
Schaeffer, P. J., Wende, A. R., Magee, C. J., Neilson, J. R., Leone, T. C., Chen, F., et al. (2004). Calcineurin and calcium/calmodulin-dependent protein kinase activate distinct metabolic gene regulatory programs in cardiac muscle. J. Biol. Chem. 279, 39593–39603. doi: 10.1074/jbc.M403649200
Schlattner, U., Tokarska-Schlattner, M., and Wallimann, T. (2006). Mitochondrial creatine kinase in human health and disease. Biochim. Biophys. Acta 1762, 164–180. doi: 10.1016/j.bbadis.2005.09.004
Schmidt-Schweda, S., and Holubarsch, C. (2000). First clinical trial with etomoxir in patients with chronic congestive heart failure. Clin. Sci. Lond. Engl. 99, 27–35. doi: 10.1042/cs0990027
Schönekess, B. O. (1997). Competition between lactate and fatty acids as sources of ATP in the isolated working rat heart. J. Mol. Cell. Cardiol. 29, 2725–2733. doi: 10.1006/jmcc.1997.0504
Schuchardt, M., Herrmann, J., Tolle, M., and van der Giet, M. (2017). Xanthine oxidase and its role as target in cardiovascular disease: cardiovascular protection by enzyme inhibition? Curr. Pharm. Des. 23, 3391–3404. doi: 10.2174/1381612823666170417130115.
Scirica, B. M., and Morrow, D. A. (2007). Ranolazine in patients with angina and coronary artery disease. Curr. Cardiol. Rep. 9, 272–278. doi: 10.1007/BF02938375
Scirica, B. M., Morrow, D. A., Hod, H., Murphy, S. A., Belardinelli, L., Hedgepeth, C. M., et al. (2007). Effect of ranolazine, an antianginal agent with novel electrophysiological properties, on the incidence of arrhythmias in patients with non ST-segment elevation acute coronary syndrome: results from the Metabolic Efficiency With Ranolazine for Less Ischemia in Non ST-Elevation Acute Coronary Syndrome Thrombolysis in Myocardial Infarction 36 (MERLIN-TIMI 36) randomized controlled trial. Circulation 116, 1647–1652. doi: 10.1161/CIRCULATIONAHA.107.724880
Scolletta, S., and Biagioli, B. (2010). Energetic myocardial metabolism and oxidative stress: let's make them our friends in the fight against heart failure. Biomed. Pharmacother. Biomed. Pharmacother. 64, 203–207. doi: 10.1016/j.biopha.2009.10.002
Seiler, S. E., Martin, O. J., Noland, R. C., Slentz, D. H., DeBalsi, K. L., Ilkayeva, O. R., et al. (2014). Obesity and lipid stress inhibit carnitine acetyltransferase activity. J. Lipid Res. 55, 635–644. doi: 10.1194/jlr.M043448
Sepa-Kishi, D. M., Wu, M. V., Uthayakumar, A., Mohasses, A., and Ceddia, R. B. (2016). Anti-lipolytic and anti-lipogenic effects of the CPT-1b inhibitor oxfenicine in the white adipose tissue of rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 311, R779–R787. doi: 10.1152/ajpregu.00243.2016
Shah, D. D., Fonarow, G. C., and Horwich, T. B. (2010). Metformin therapy and outcomes in patients with advanced systolic heart failure and diabetes. J. Card. Fail. 16, 200–206. doi: 10.1016/j.cardfail.2009.10.022
Shah, R. R., Oates, N. S., Idle, J. R., Smith, R. L., and Lockhart, J. D. (1982). Impaired oxidation of debrisoquine in patients with perhexiline neuropathy. Br. Med. J. Clin. Res. Ed 284, 295–299. doi: 10.1136/bmj.284.6312.295
Sharma, N., Okere, I. C., Brunengraber, D. Z., McElfresh, T. A., King, K. L., Sterk, J. P., et al. (2005). Regulation of pyruvate dehydrogenase activity and citric acid cycle intermediates during high cardiac power generation. J. Physiol. 562, 593–603. doi: 10.1113/jphysiol.2004.075713
Sharov, V. S., Dremina, E. S., Galeva, N. A., Williams, T. D., and Schöneich, C. (2006). Quantitative mapping of oxidation-sensitive cysteine residues in SERCA in vivo and in vitro by HPLC–electrospray-tandem MS: selective protein oxidation during biological aging. Biochem. J. 394, 605–615. doi: 10.1042/BJ20051214
Shih, L., Chung, Y., Sriram, R., and Jue, T. (2014). Palmitate Interaction with Physiological States of Myoglobin. Biochim. Biophys. Acta 1840, 656–666. doi: 10.1016/j.bbagen.2013.10.028
Shih, L., Chung, Y., Sriram, R., and Jue, T. (2015). Interaction of myoglobin with oleic acid. Chem. Phys. Lipids 191, 115–122. doi: 10.1016/j.chemphyslip.2015.07.010
Shirwany, N. A., and Zou, M.-H. (2014). AMPK: a cellular metabolic and redox sensor. A minireview. Front. Biosci. (Landmark Ed.) 19, 447–474. doi: 10.2741/4218
Simon, J. N., Duglan, D., Casadei, B., and Carnicer, R. (2014). Nitric oxide synthase regulation of cardiac excitation-contraction coupling in health and disease. J. Mol. Cell. Cardiol. 73, 80–91. doi: 10.1016/j.yjmcc.2014.03.004
Skyschally, A., Schulz, R., Gres, P., Korth, H.-G., and Heusch, G. (2003). Attenuation of ischemic preconditioning in pigs by scavenging of free oxyradicals with ascorbic acid. Am. J. Physiol. Heart Circ. Physiol. 284, H698–H703. doi: 10.1152/ajpheart.00693.2002
Slatton, M. L., and Eichhorn, E. J. (1996). Beta-blocker therapy for heart failure. Curr. Opin. Cardiol. 11, 263–268. doi: 10.1097/00001573-199605000-00006
Sleight, P. (2000). The HOPE study (heart outcomes prevention evaluation). J. Renin-Angiotensin Aldosterone Syst. JRAAS 1, 18–20. doi: 10.3317/jraas.2000.002
Snijder, J., Rose, R. J., Raijmakers, R., and Heck, A. J. R. (2011). Site-specific methionine oxidation in calmodulin affects structural integrity and interaction with Ca2+/calmodulin-dependent protein kinase II. J. Struct. Biol. 174, 187–195. doi: 10.1016/j.jsb.2010.12.002
Sokos, G. G., Nikolaidis, L. A., Mankad, S., Elahi, D., and Shannon, R. P. (2006). Glucagon-like peptide-1 infusion improves left ventricular ejection fraction and functional status in patients with chronic heart failure. J. Card. Fail. 12, 694–699. doi: 10.1016/j.cardfail.2006.08.211
Stanley, B. A., Sivakumaran, V., Shi, S., McDonald, I., Lloyd, D., Watson, W. H., et al. (2011). Thioredoxin reductase-2 is essential for keeping low levels of H2O2 emission from isolated heart mitochondria. J. Biol. Chem. 286, 33669–33677. doi: 10.1074/jbc.M111.284612
Stanley, W. C., Hernandez, L. A., Spires, D., Bringas, J., Wallace, S., and McCormack, J. G. (1996). Pyruvate dehydrogenase activity and malonyl CoA levels in normal and ischemic swine myocardium: effects of dichloroacetate. J. Mol. Cell. Cardiol. 28, 905–914. doi: 10.1006/jmcc.1996.0085
Stanley, W. C., Lopaschuk, G. D., Hall, J. L., and McCormack, J. G. (1997). Regulation of myocardial carbohydrate metabolism under normal and ischaemic conditions. Cardiovasc. Res. 33, 243–257. doi: 10.1016/S0008-6363(96)00245-3
Sugden, M. C., and Holness, M. J. (2006). Mechanisms underlying regulation of the expression and activities of the mammalian pyruvate dehydrogenase kinases. Arch. Physiol. Biochem. 112, 139–149. doi: 10.1080/13813450600935263
Sugden, P. H. (2003). Ras, akt, and mechanotransduction in the cardiac myocyte. Circ. Res. 93, 1179–1192. doi: 10.1161/01.RES.0000106132.04301.F5
Sun, W., Liu, Q., Leng, J., Zheng, Y., and Li, J. (2015). The role of pyruvate dehydrogenase complex in cardiovascular diseases. Life Sci. 121, 97–103. doi: 10.1016/j.lfs.2014.11.030
Sverdlov, A. L., Elezaby, A., Qin, F., Behring, J. B., Luptak, I., Calamaras, T. D., et al. (2016). Mitochondrial reactive oxygen species mediate cardiac structural, functional, and mitochondrial consequences of diet-induced metabolic heart disease. J. Am. Heart Assoc. 5:e002555. doi: 10.1161/JAHA.115.002555
Taegtmeyer, H., and Overturf, M. L. (1988). Effects of moderate hypertension on cardiac function and metabolism in the rabbit. Hypertens. Dallas Tex 1979 11, 416–426. doi: 10.1161/01.HYP.11.5.416
Taegtmeyer, H., Young, M. E., Lopaschuk, G. D., Abel, E. D., Brunengraber, H., Darley-Usmar, V., et al. (2016). Assessing cardiac metabolism: a scientific statement from the american heart association. Circ. Res. 118. 1659–1701. doi: 10.1161/RES.0000000000000097
Tang, W. H., Kravtsov, G. M., Sauert, M., Tong, X. Y., Hou, X. Y., Wong, T. M., et al. (2010). Polyol pathway impairs the function of SERCA and RyR in ischemic-reperfused rat hearts by increasing oxidative modifications of these proteins. J. Mol. Cell. Cardiol. 49, 58–69. doi: 10.1016/j.yjmcc.2009.12.003
Totzeck, M., Hendgen-Cotta, U. B., Kelm, M., and Rassaf, T. (2014). Crosstalk between nitrite, myoglobin and reactive oxygen species to regulate vasodilation under hypoxia. PLoS ONE 9:e105951. doi: 10.1371/journal.pone.0105951
Trujillo, J. M., Nuffer, W., and Ellis, S. L. (2015). GLP-1 receptor agonists: a review of head-to-head clinical studies. Ther. Adv. Endocrinol. Metab. 6, 19–28. doi: 10.1177/2042018814559725
Tsutsui, H., Kinugawa, S., and Matsushima, S. (2011). Oxidative stress and heart failure. Am. J. Physiol. Heart Circ. Physiol. 301, H2181–H2190. doi: 10.1152/ajpheart.00554.2011
Turer, A. T., Malloy, C. R., Newgard, C. B., and Podgoreanu, M. V. (2010). Energetics and metabolism in the failing heart: important but poorly understood. Curr. Opin. Clin. Nutr. Metab. Care 13, 458–465. doi: 10.1097/MCO.0b013e32833a55a5
Turer, A. T., Stevens, R. D., Bain, J. R., Muehlbauer, M. J., van der Westhuizen, J., Mathew, J. P., et al. (2009). Metabolomic profiling reveals distinct patterns of myocardial substrate utilization in humans with coronary artery disease or left ventricular dysfunction during surgical ischemia-reperfusion. Circulation 119, 1736–1746. doi: 10.1161/CIRCULATIONAHA.108.816116
Turrell, H. E., Rodrigo, G. C., Norman, R. I., Dickens, M., and Standen, N. B. (2011). Phenylephrine preconditioning involves modulation of cardiac sarcolemmal K(ATP) current by PKC delta, AMPK and p38 MAPK. J. Mol. Cell. Cardiol. 51, 370–380. doi: 10.1016/j.yjmcc.2011.06.015
Ussher, J. R. (2014). Sweet as sugar: excessive glucose metabolism in the failing heart. Future Cardiol. 10, 465–468. doi: 10.2217/fca.14.28
van der Leij, F. R., Huijkman, N. C., Boomsma, C., Kuipers, J. R., and Bartelds, B. (2000). Genomics of the human carnitine acyltransferase genes. Mol. Genet. Metab. 71, 139–153. doi: 10.1006/mgme.2000.3055
van der Vusse, G. J., van Bilsen, M., and Glatz, J. F. (2000). Cardiac fatty acid uptake and transport in health and disease. Cardiovasc. Res. 45, 279–293. doi: 10.1016/S0008-6363(99)00263-1
Vendrov, A. E., Vendrov, K. C., Smith, A., Yuan, J., Sumida, A., Robidoux, J., et al. (2015). NOX4 NADPH oxidase-dependent mitochondrial oxidative stress in aging-associated cardiovascular disease. Antioxid. Redox Signal. 23, 1389–1409. doi: 10.1089/ars.2014.6221
Viel, E. C., Benkirane, K., Javeshghani, D., Touyz, R. M., and Schiffrin, E. L. (2008). Xanthine oxidase and mitochondria contribute to vascular superoxide anion generation in DOCA-salt hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 295, H281–H288. doi: 10.1152/ajpheart.00304.2008
Wallimann, T., Dolder, M., Schlattner, U., Eder, M., Hornemann, T., O'Gorman, E., et al. (1998). Some new aspects of creatine kinase (CK): compartmentation, structure, function and regulation for cellular and mitochondrial bioenergetics and physiology. BioFactors Oxf. Engl. 8, 229–234.
Wang, L., Ma, X., Xiang, L., Lu, M., Yan, C., Zhao, S., et al. (2016). The characterization and prognostic significance of right ventricular glucose metabolism in non-ischemic dilated cardiomyopathy. J. Nucl. Cardiol. 23, 758–767. doi: 10.1007/s12350-015-0165-9
Wang, X.-F., Zhang, J.-Y., Li, L., Zhao, X.-Y., Tao, H.-L., and Zhang, L. (2011). Metformin improves cardiac function in rats via activation of AMP-activated protein kinase. Clin. Exp. Pharmacol. Physiol. 38, 94–101. doi: 10.1111/j.1440-1681.2010.05470.x
Wang, Z., Ding, J., Luo, X., Zhang, S., Yang, G., Zhu, Q., et al. (2016). Effect of allopurinol on myocardial energy metabolism in chronic heart failure rats after myocardial infarct. Int. Heart. J. 57, 753–759. doi: 10.1536/ihj.16-149
Wang, Z. V., Li, D. L., and Hill, J. A. (2014). heart failure and loss of metabolic control. J. Cardiovasc. Pharmacol. 63, 302–313. doi: 10.1097/FJC.0000000000000054
Warburton, D. E. R., Haykowsky, M. J., Quinney, H. A., Blackmore, D., Teo, K. K., and Humen, D. P. (2002). myocardial response to incremental exercise in endurance-trained athletes: influence of heart rate, contractility and the frank-starling effect. Exp. Physiol. 87, 613–622. doi: 10.1113/eph8702372
Ward, C. W., Prosser, B. L., and Lederer, W. J. (2014). Mechanical stretch-induced activation of ROS/RNS signaling in striated muscle. Antioxid. Redox Signal. 20, 929–936. doi: 10.1089/ars.2013.5517
Watson, R. T., and Pessin, J. E. (2001). Intracellular organization of insulin signaling and GLUT4 translocation. Recent Prog. Horm. Res. 56, 175–193. doi: 10.1210/rp.56.1.175
Wei, S., Rothstein, E. C., Fliegel, L., Dell'Italia, L. J., and Lucchesi, P. A. (2001). Differential MAP kinase activation and Na+/H+exchanger phosphorylation by H2O2 in rat cardiac myocytes. Am. J. Physiol. Cell Physiol. 281, C1542–C1550.
Wilson, C., Terman, J. R., González-Billault, C., and Ahmed, G. (2016). Actin filaments-A target for redox regulation. Cytoskelet. Hoboken N.J. 73, 577–595. doi: 10.1002/cm.21315
Winder, W. W. (1998). Malonyl-CoA–regulator of fatty acid oxidation in muscle during exercise. Exerc. Sport Sci. Rev. 26, 117–132. doi: 10.1249/00003677-199800260-00007
Wisneski, J. A., Gertz, E. W., Neese, R. A., Gruenke, L. D., Morris, D. L., and Craig, J. C. (1985). Metabolic fate of extracted glucose in normal human myocardium. J. Clin. Invest. 76, 1819–1827. doi: 10.1172/JCI112174
Wong, A. K. F., Howie, J., Petrie, J. R., and Lang, C. C. (2009). AMP-activated protein kinase pathway: a potential therapeutic target in cardiometabolic disease. Clin. Sci. Lond. Engl. 116, 607–620. doi: 10.1042/CS20080066
Wu, H., Zhu, Q., Cai, M., Tong, X., Liu, D., Huang, J., et al. (2014). Effect of inhibiting malonyl-CoA decarboxylase on cardiac remodeling after myocardial infarction in rats. Cardiology 127, 236–244. doi: 10.1159/000356471
Xin, W., Wei, W., and Li, X. (2012). Effects of fish oil supplementation on cardiac function in chronic heart failure: a meta-analysis of randomised controlled trials. Heart Br. Card. Soc. 98, 1620–1625. doi: 10.1136/heartjnl-2012-302119
Yang, X., Cohen, M. V., and Downey, J. M. (2010). mechanism of cardioprotection by early ischemic preconditioning. Cardiovasc. Drugs Ther. Spons. Int. Soc. Cardiovasc. Pharmacother. 24, 225–234. doi: 10.1007/s10557-010-6236-x
Ye, Y., Gong, G., Ochiai, K., Liu, J., and Zhang, J. (2001). high-energy phosphate metabolism and creatine kinase in failing hearts. Circulation 103, 1570–1576. doi: 10.1161/01.CIR.103.11.1570
Yin, M., van der Horst, I. C. C., van Melle, J. P., Qian, C., van Gilst, W. H., Silljé, H. H. W., et al. (2011). Metformin improves cardiac function in a nondiabetic rat model of post-MI heart failure. Am. J. Physiol. Heart Circ. Physiol. 301, H459–H468. doi: 10.1152/ajpheart.00054.2011
Yin, X., Dwyer, J., Langley, S. R., Mayr, U., Xing, Q., Drozdov, I., et al. (2013). Effects of perhexiline-induced fuel switch on the cardiac proteome and metabolome. J. Mol. Cell. Cardiol. 55, 27–30. doi: 10.1016/j.yjmcc.2012.12.014
Zarain-Herzberg, A., Afzal, N., Elimban, V., and Dhalla, N. S. (1996). Decreased expression of cardiac sarcoplasmic reticulum Ca2+-pump ATPase in congestive heart failure due to myocardial infarction. Mol. Cell. Biochem. 163-164, 285–290. doi: 10.1007/BF00408669
Zebrowski, D. C., and Engel, F. B. (2013). The cardiomyocyte cell cycle in hypertrophy, tissue homeostasis, and regeneration. Rev. Physiol. Biochem. Pharmacol. 165, 67–96. doi: 10.1007/112_2013_12
Zervou, S., Whittington, H. J., Russell, A. J., and Lygate, C. A. (2016). Augmentation of creatine in the heart. Mini Rev. Med. Chem. 16, 19–28. doi: 10.2174/1389557515666150722102151
Zhang, X., Shen, C., Zhai, S., Liu, Y., Yue, W.-W., and Han, L. (2016). A meta-analysis of the effects of β-adrenergic blockers in chronic heart failure. Exp. Ther. Med. 12, 2489–2496. doi: 10.3892/etm.2016.3657
Zhao, Y., Hu, X., Liu, Y., Dong, S., Wen, Z., He, W., et al. (2017). ROS signaling under metabolic stress: cross-talk between AMPK and AKT pathway. Mol. Cancer 16:79. doi: 10.1186/s12943-017-0648-1
Zhong, M., Alonso, C. E., Taegtmeyer, H., and Kundu, B. K. (2013). Quantitative PET imaging detects early metabolic remodeling in a mouse model of pressure-overload left ventricular hypertrophy in vivo. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 54, 609–615. doi: 10.2967/jnumed.112.108092
Zhou, L., Huang, H., Yuan, C. L., Keung, W., Lopaschuk, G. D., and Stanley, W. C. (2008). Metabolic response to an acute jump in cardiac workload: effects on malonyl-CoA, mechanical efficiency, and fatty acid oxidation. Am. J. Physiol. Heart Circ. Physiol. 294, H954–H960. doi: 10.1152/ajpheart.00557.2007
Zhou, X., and Chen, J. (2014). Is treatment with trimetazidine beneficial in patients with chronic heart failure? PLoS ONE 9:e94660. doi: 10.1371/journal.pone.0094660
Zhu, L., Wang, Q., Zhang, L., Fang, Z., Zhao, F., Lv, Z., et al. (2010). Hypoxia induces PGC-1α expression and mitochondrial biogenesis in the myocardium of TOF patients. Cell Res. 20, 676–687. doi: 10.1038/cr.2010.46
Zhu, X., and Zuo, L. (2013). Characterization of oxygen radical formation mechanism at early cardiac ischemia. Cell Death Dis. 4:e787. doi: 10.1038/cddis.2013.313
Keywords: cardiac metabolism, cardiac function, heart failure, oxidative stress, hypoxia, fatty acids β-oxidation, glycolysis
Citation: Angelini A, Pi X and Xie L (2017) Dioxygen and Metabolism; Dangerous Liaisons in Cardiac Function and Disease. Front. Physiol. 8:1044. doi: 10.3389/fphys.2017.01044
Received: 18 September 2017; Accepted: 29 November 2017;
Published: 12 December 2017.
Edited by:
Li Zuo, The Ohio State University, United StatesReviewed by:
Lewis J. Watson, University of Pikeville, United StatesNazareno Paolocci, Johns Hopkins University, United States
Zewen Liu, Wuhan University, China
Copyright © 2017 Angelini, Pi and Xie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liang Xie, bGlhbmcueGllQGJjbS5lZHU=