 Hisatake Ishida
Hisatake Ishida Cynthia Riginos
Cynthia Riginos Cheong Xin Chan
Cheong Xin Chan- 1Australian Centre for Ecogenomics, School of Chemistry and Molecular Biosciences, The University of Queensland, Brisbane, QLD, Australia
- 2School of the Environment, The University of Queensland, Brisbane, QLD, Australia
- 3Australian Institute of Marine Science, Townsville, QLD, Australia
Endosymbiotic dinoflagellates of the family Symbiodiniaceae are symbionts essential to corals and other marine organisms. A coral holobiont consists of the coral host, Symbiodiniaceae, and other microbes that together sustain the overall productivity and coral health. Coral hologenome data, generated from all interacting components of a coral holobiont, are key for elucidating the molecular mechanisms that underpin the resilience of the holobiont to changing environments. Although coral hologenome data are often dominated by host coral genomic sequences, they provide an avenue for recovering genomic sequences from Symbiodiniaceae in hospite. Here, we review recent advances in the approaches for assessing community diversity of in hospite Symbiodiniaceae from coral hologenome data. Using a case study based on existing hologenome datasets of the Acropora kenti coral, we highlight how hologenome datasets in large numbers can provide a useful analysis platform for assessing Symbiodiniaceae diversity and their function in coral holobionts.
1 Introduction
Coral reefs are marine biodiversity hotspots that are home to 25% of known marine species (Knowlton et al., 2010). This trophic foundation of coral reefs is critically supported by the mutualistic relationship between corals and dinoflagellates in the family Symbiodiniaceae (LaJeunesse et al., 2018). Through photosynthesis, Symbiodiniaceae provide fixed carbon and nutrients essential to the coral hosts. Under stress conditions, such as an episodic increase of water temperature or declining water quality, the coral-Symbiodiniaceae symbiosis can be disrupted (i.e. the phenomenon of coral bleaching), leading to coral death, and in the extreme, potential collapse of coral reef ecosystems (Hoegh-Guldberg, 1999). Corals are under threat from warming and acidifying oceans due in part to global climate change and anthropogenic activities (Suggett and Smith, 2020). Earlier research has revealed that coral tolerance to environmental changes is linked to the coral’s natural capacity to recover and adapt, and the coral’s association with symbionts that are robust against stressors (Voolstra et al., 2021b). A coral holobiont comprises the coral animal host and the associated microbial symbionts that together sustain a functional ecological unit. A sound understanding of how these biotic components respond to stressors enables an effective assessment of coral resilience to changing environments.
Genomic data provide an excellent analysis platform for deciphering the molecular mechanisms that underpin responses to stressors in coral holobionts (Fuller et al., 2020; Rose et al., 2021; Shinzato et al., 2021; Dougan et al., 2022a). Hologenome data, or genome data generated from all interacting biotic components of a holobiont, have been proven useful for taxonomic and functional assessment of the prokaryotic microbial community in the holobionts of humans, other animals, and plants (Alberdi et al., 2022). In recent studies of coral holobionts, hologenome data are generated by sequencing genomic DNA extracted from coral tissues, enabling recovery of whole-genome sequences from the coral host, and assessment of the host genotype (Figure 1); see recent reviews (Voolstra et al., 2021a; Riginos et al., 2024) for more detail. These samples are dominated by the host DNA (e.g. >90% sequenced reads derived from the coral genome) and are therefore often inadequate for the recovery of symbiont genomes at low data yield, particularly when Symbiodiniaceae genomes (1–3Gbp; (LaJeunesse et al., 2005; Saad et al., 2020) are larger than most coral genomes (<1Gbp; (Cowen and Putnam, 2022; Noel et al., 2023). For this reason, amplicon sequencing of the phylogenetic markers, e.g. the internally transcribed spacer 2 (ITS2) sequences, is commonly used for Symbiodiniaceae profiling among these samples (Hume et al., 2019; Quigley et al., 2022; Thomas et al., 2022). Although this approach is useful for characterising Symbiodiniaceae diversity, technical biases, such as primer misamplification and incomplete sampling of genetic variation, remain a challenge, calling for the use of multiple, carefully selected markers, enriched by genomic resources of Symbiodiniaceae (Davies et al., 2023).
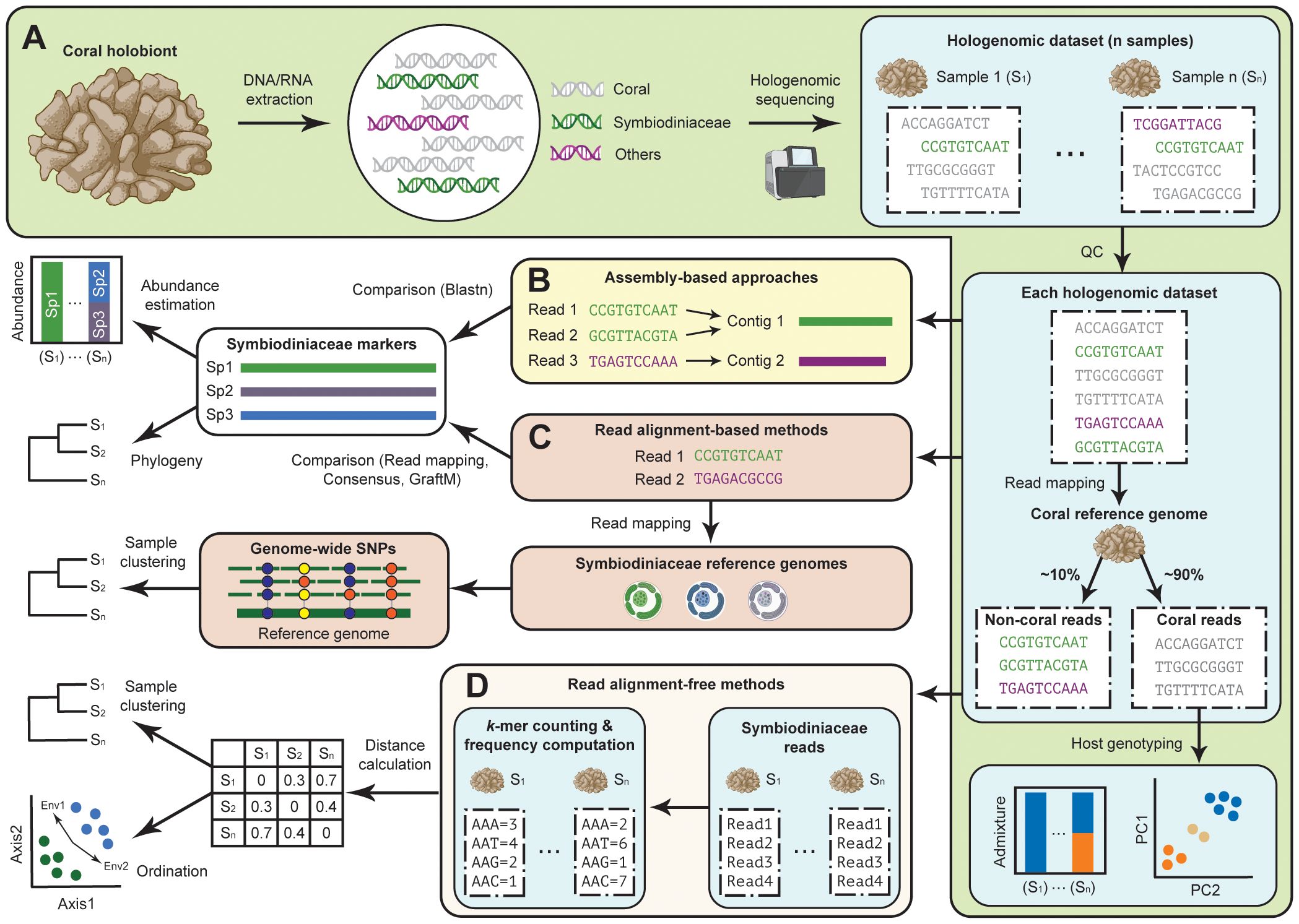
Figure 1 Assessing Symbiodiniaceae diversity using coral hologenome datasets. (A) Generation of high-throughput sequencing data directly from DNA of coral tissue enables the recovery of genome data from the coral animal host, Symbiodiniaceae, and other associated microbes, and the assessment of host genotype(s). Symbiodiniaceae diversity can then be assessed using (B) assembly-based approaches for which assembled genome sequences were used to recover specific marker genes, and read-based approaches for which sequence reads from the hologenome datasets are analysed using an (C) alignment-based or (D) alignment-free method based on k-mer.
Until recently, genomic resources of Symbiodiniaceae have been scarce, due in part to the technical challenges associated with the large genome sizes and genomic features atypical in eukaryotes (González-Pech et al., 2019; González-Pech et al., 2021; Lin et al., 2021). The advancement and increased affordability of sequencing technologies, in combination with the development of customised analytical workflows for dinoflagellate genomes (Chen et al., 2020, 2024), have empowered the development of genomic resources for Symbiodiniaceae and other dinoflagellate taxa (González-Pech et al., 2021; Dougan et al., 2023). These data have revealed remarkable sequence and structural divergence among Symbiodiniaceae genomes (Shoguchi et al., 2013; Lin et al., 2015; Aranda et al., 2016; González-Pech et al., 2021), phylogenetic diversity hidden behind subtly different morphologies (Dougan et al., 2022b; Shah et al., 2023a), and novel insights of genome evolution into how Symbiodiniaceae diversified to become symbionts that sustain coral reef health (Dougan et al., 2022a; Bhattacharya et al., 2023; Shah et al., 2023b).
Generation of these genome data thus far has been largely restricted to monoclonal cultures of Symbiodiniaceae cells isolated from (or nearby) hosts, for the ease of obtaining high-quality DNA and the need of generating high-quality genome assemblies as reference. As such, the available genomic resources represent only a small proportion of known Symbiodiniaceae diversity (Figure 2A). Technological challenges remain since many Symbiodiniaceae taxa, especially the host-specific taxa, are culture recalcitrant due to their narrow growth requirements (Krueger and Gates, 2012). In addition, genomes from ex hospite cell cultures may not necessarily be genetically representative of their in hospite counterparts (Maruyama et al., 2021). In this regard, coral hologenome data offer direct access to Symbiodiniaceae diversity that is more ecologically relevant, bypassing the need for cell cultures.
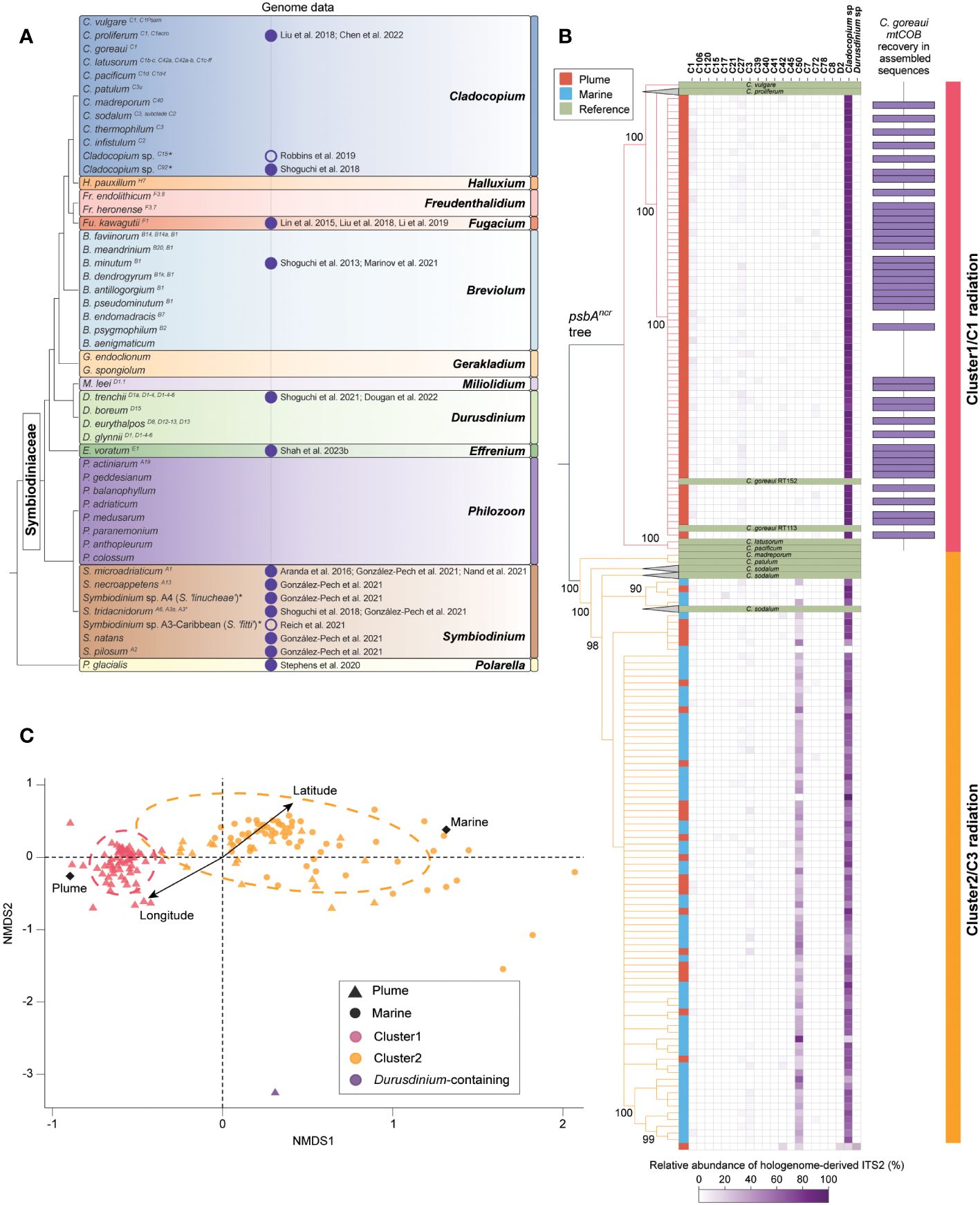
Figure 2 Symbiodiniaceae diversity based on current knowledge and an example of the rich information that can be mined from coral hologenome data, as illustrated by low-coverage whole-genome sequence datasets from holobionts of Acropora kenti. (A) Phylogeny of Symbiodiniaceae taxa for which formal description and/or genome data are available. The topology does not address the full extent of Symbiodiniaceae diversity. Position of genera follows LaJeunesse et al. (2022). Branching order among taxa within each genus is not shown. ITS2 “type” of each taxon based on (Davies et al., 2023), where applicable, are shown as superscript following the taxa name. Asterisks indicate taxa yet to be formally described. Vertical columns next to each taxon show the availability of genome data (fill circle: from culture, empty circle: from coral holobiont) as of 25 January 2024 with reference to publications that generated the assembly. (B) Bayesian phylogeny of the genus Cladocopium inferred based on alignment of psbAncr from formally described Cladocopium taxa and consensus psbAncr sequence reconstructed from the hologenome datasets of Acropora kenti. The vertical column next to the tree indicates whether the corresponding tree leaf is a reference sequence or a psbAncr consensus sequence retrieved from each A. kenti hologenome (brown: Plume, blue: Marine). The heatmap shows the relative abundance of ITS2 sequences derived from the hologenome. ITS2 sequences that could not be further classified into distinct ITS2 types within a genus are represented by their genus, i.e. as Cladocopium sp. and Durusdinium sp. One hologenome of A. kenti, featuring ITS2 of Durusdinium taxa, is shown at the bottom. Bar plot at the right indicates samples that recovered mitochondrial cytochrome b (mtCOB) of Cladocopium goreaui RT152 (GenBank accession KF206028) in full-length from the assembly of non-coral reads. Tree branches and the most right vertical column are coloured according to the cluster (and the adaptive radiation of Cladocopium provisionally referred to) identified from the recovery of marker genes from the hologenome. (C) Non-metric multidimensional scaling (NMDS) plot showing relative pairwise D2S -derived distances among Symbiodiniaceae communities reflected in the Acropora kenti hologenome datasets. Each data point is coloured according to the cluster identified in (B). Ellipses were added by the cluster information. The arrow is the fitted vector for latitude and longitude of each reef location. Centroids of each local environment (Plume/Marine) are noted as diamonds.
In this article, we review the emerging computational methods for investigating Symbiodiniaceae community of coral holobionts using hologenome datasets, and discuss how these data are useful for assessing genomic diversity and function of in hospite and/or host-specific Symbiodiniaceae.
2 Assessing Symbiodiniaceae diversity using coral hologenome data
The microbial fraction of coral hologenome data provides a snapshot of the microbial symbionts from coral populations in the wild, but this fraction is usually small (5–10%) given that these data were derived directly from coral tissue (Kitchen et al., 2019; Baums et al., 2022). Although this microbial data fraction may be perceived as “contaminant” sequences in the analysis of host genome or genotype(s), it serves as a “gold mine” that provide direct access to genome data of in hospite microbial symbionts including host-specific Symbiodiniaceae. Importantly, the use of these data bypasses the amplification of marker genes in amplicon sequencing, and thus free from technical biases associated with DNA amplification. These genome-scale data also enable the recovery of genetic information regarding functional and metabolic capacity (e.g. via encoded gene functions) of microbial symbionts, which is inaccessible using the amplicon sequencing of phylogenetic marker genes. In this regard, a robust culture-independent approach leveraging coral hologenome data is highly desirable for assessing the diversity of the symbiont community and its function in the holobiont.
Increasingly, large-scale hologenome datasets are generated from coral holobiont research. For instance, datasets were generated from hundreds of DNA samples collected across environmental gradients in a region to investigate the adaptation and speciation processes of the target coral species (Cooke et al., 2020; Fuller et al., 2020; Bongaerts et al., 2021; Rose et al., 2021; Zhang et al., 2022). Although each dataset is commonly generated by a shallow sequencing approach (e.g. 3–10× coverage of the coral genome) to maximise cost effectiveness of such studies at scale, these large number of hologenome datasets in combination enable better recovery of the coral genome, and importantly, genome data from the microbial fraction.
Host-depleted sequences from the coral hologenome data (i.e. non-coral sequence reads) are generally assumed to be derived from the microbial symbionts, from which the Symbiodiniaceae genomic reads can be retrieved bioinformatically. Here, we discuss recently adopted bioinformatic approaches used to assess Symbiodiniaceae diversity using these host-depleted coral hologenome data, which we broadly categorised as assembly-based (Figure 1B) and read-based approaches (Figures 1C, D). We discuss how these approaches complement each other, and how these approaches can be used in combination to assess Symbiodiniaceae diversity among coral holobionts relative to conditions of water quality in a case study.
2.1 Assembly-based approaches
In an assembly-based approach (Figure 1B), the diversity of symbionts is assessed after the non-coral sequence reads have been assembled de novo as a metagenome and/or binned into metagenome-assembled genomes (MAGs). This approach was adopted in the hologenome study of Porites lutea coral from the Great Barrier Reef (Robbins et al., 2019), which provided an overview of microbial community composition and functional complementarity of distinct symbiotic partners including the dominant host-specific Symbiodiniaceae, Cladocopium sp. C15, and prokaryotic microbes based on 52 high-quality MAGs. This approach provides a genomic overview of microbial symbionts associated with the coral holobiont, including information about gene functions and metabolic capacity of the distinct members. A more targeted assembly approach for specific marker sequences has also been adopted. For instance, targeted assembly of a non-coding region of plastid genome (e.g. psbAncr of the psbA minicircle) directly from the hologenome data of Caribbean acroporid corals successfully elucidated the symbiont diversity of Symbiodinium taxa across samples (Reich et al., 2021). However, de novo assembly is computationally intensive (e.g. large memory usage). The recovery of microbial genomes and their assembly quality is also sensitive to technical (e.g. read coverage) and biological characteristics (e.g. richness and evenness of taxa represented) of the sequence data.
2.2 Read-based approaches
Another approach is to bypass the assembly process altogether, and use the non-coral sequence reads directly in an analysis. This approach can be broadly classified into two categories: alignment-based and alignment-free methods.
2.2.1 Alignment-based methods
Alignment of the sequence reads against a set of reference phylogenetic marker genes can elucidate the presence of microbial taxa in the hologenome data. This is commonly done by mapping the sequence reads against the reference sequences (Truong et al., 2017). For instance, the number of uniquely mapped reads can be interpreted as a proxy of abundance of Symbiodiniaceae taxa in the coral holobiont (Morikawa and Palumbi, 2019; Matias et al., 2023). This read mapping approach can be extended to reconstruct consensus sequences of target markers, e.g. the consensus psbAncr sequences were used to clarify symbiont diversity of Cladocopium in Pocillopora corals (Armstrong et al., 2023).
Furthermore, read mapping against the reference nuclear, mitochondrial, or plastid genomes can also be useful for identifying the population structure of Symbiodiniaceae based on analysis of single-nucleotide polymorphisms (SNPs). Such a method has been shown to uncover variations missed in the analysis of individual marker genes in earlier studies (Morikawa and Palumbi, 2019; Armstrong et al., 2023; Matias et al., 2023). Read-based taxonomic profiling tools, e.g. GraftM (Boyd et al., 2018), can also be used to identify microbial marker genes based on pairwise sequence alignment, and to estimate the abundance of microbial taxa based on read coverage.
2.2.2 Alignment-free methods
Microbial diversity among the non-coral sequence reads can also be assessed using methods not requiring sequence alignment, i.e. the so-called alignment-free (AF) methods. In general, AF methods aim to quantify sequence similarity (or rather, dissimilarity as a distance) among a set of biological sequences based on sequence features without the need for sequence alignment (Zielezinski et al., 2019). A common technique is to use short, sub-sequences of defined length k (i.e. k-mers) observed in the sequences. Many k-mer-based AF approaches work by transforming sequence information into numerical values through projection of each sequence into a feature space of k-mer counts.
By computing frequency of k-mers among non-coral reads for each hologenome dataset (i.e. the k-mer profile), the number of shared k-mers between two datasets can be calculated, e.g. using statistic (Reinert et al., 2009; Wan et al., 2010) that describes the number of shared k-mers observed in the two sequences, normalised by the overall occurrence of each k-mer observed in the sequences. The pairwise statistic is then converted into a measure of dissimilarity (i.e. distance), e.g. via logarithmic representation of the geometric mean (Chan et al., 2014), which can be used for downstream clustering analysis and/or phylogenetic inference. This AF method presents a good alternative to multiple sequence alignment in phylogenetic analysis, due in part to their high scalability to genome-scale data and capacity to capture phylogenetic signals that are robust against genetic rearrangements and/or transfer (Chan et al., 2014); see earlier reviews (Bernard et al., 2019; Zielezinski et al., 2019) for more detail. Importantly, the applicability of this method on genome data of Symbiodiniaceae has been demonstrated in several studies (González-Pech et al., 2021; Lo et al., 2022; Dougan et al., 2022b) to infer biologically meaningful relationships. The potential of this method in differentiating Symbiodiniaceae communities among coral hologenome datasets was also demonstrated recently (Zhang et al., 2022). High scalability of this method is advantageous for assessing Symbiodiniaceae composition among large number of datasets efficiently.
2.3 Case study: Symbiodiniaceae diversity in Acropora kenti holobionts of central Great Barrier Reef
To demonstrate the utility of these in silico approaches in assessing Symbiodiniaceae diversity, we used both assembly- and read-based approaches to analyse hologenome datasets of Acropora kenti (formerly Acropora tenuis (Bridge et al., 2023)), generated from an earlier study (Cooke et al., 2020). These Illumina short-read datasets (3× coverage of the coral genome per sample) were derived from 148 coral tissues collected from five inshore Great Barrier Reef locations across riverine plume influence gradients: Magnetic Island, Dunk Island, and Pandora Reef under high riverine influence (Plume), whereas Fitzroy and Pelorus Islands experience low riverine influence (Marine). These datasets were downloaded from NCBI GenBank via BioProject accession PRJEB37470. The workflow we adopted for this analysis is available at https://github.com/hisatakeishida/Symb-SHIN/.
From non-coral reads (mean 600,000 reads, ~5% from each hologenome dataset), ITS2 sequences of Symbiodiniaceae, extracted using GraftM (Figure 2B), revealed two distinct Symbiodiniaceae community clusters (i.e. Cluster1 and Cluster2) among the hologenome datasets; both featured only ITS2 of Cladocopium taxa, with a greater prevalence of C50 in Cluster2. External to these clusters, a sole dataset features ITS2 of both Cladocopium and Durusdinium taxa. For datasets in Cluster1 and Cluster2, consensus sequences of psbAncr were reconstructed using the read-mapping approach based on reference sequences of Cladocopium taxa. A Bayesian phylogeny inferred using alignment of these consensus sequences with reference Cladocopium psbAncr sequences separates Cluster1 and Cluster2 in distinct clades with robust support (Bayesian posterior probability > 0.9). Based on sequence similarity, the likely source of origin for psbAncr sequences recovered in Cluster1 is Cladocopium goreaui, representing the C1 radiation (LaJeunesse, 2005; Thornhill et al., 2014), whereas that for Cluster2 is Cladocopium sodalum, representing the C3 radiation (LaJeunesse, 2005; Thornhill et al., 2014; Butler et al., 2023). Interestingly, these two clusters of Cladocopium communities appear to be correlated with the gradients of local water quality, i.e. Plume and Marine (Figure 2B). Assembled data from samples in Cluster1 also contain mitochondrial cytochrome b (mtCOB) sequences that are identical to C. goreaui RT152 (GenBank accession KF206028) in full length (Figure 2B).
To examine the genomic variation of Symbiodiniaceae communities among the datasets, we adopted the k-mer-based AF method to calculate pairwise -derived distances, from which non-metric multidimensional scaling (NMDS) was applied for dimensionality reduction. The NMDS plot (Figure 2C) shows the two distinct Cladocopium-dominant clusters, whereby the holobionts were separated along the NMDS1 axis. The plot also revealed significant variation of holobionts within Cluster2, suggesting more-extensive genomic diversity that is not evident in our earlier analysis based on hologenome-derived markers alone. This observation may also reflect the fact that psbAncr of the C3 lineages are more conserved relative to those of the C1 lineages (Butler et al., 2023). Overall, these analyses revealed that Symbiodiniaceae community is structured along environmental gradients among conspecifics of A. kenti relative to the levels of exposure to riverine influences, lending support to results of the earlier study (Cooke et al., 2020).
3 Finding “gold” in the “contaminant” heap of coral hologenomes
The access to genome data of in hospite and/or host-specific Symbiodiniaceae remains challenging. Coral hologenome datasets present a “gold mine” for recovering genome data from the symbionts as well as the host. Although the microbial fraction remains small in each coral hologenome dataset, these datasets in large numbers may provide sufficient data yield for recovering this fraction. Importantly, the analysis of these data enables direct and more-comprehensive diversity assessment of in hospite Symbiodiniaceae. Scalable approaches, such as alignment-free read-based approaches, are key for analysing large number (e.g. >100) of datasets, thereby enabling genomic investigations of in hospite Symbiodiniaceae relative to their coral host. Integration of these genomic-scale data will provide novel insights into the biology and ecology of coral-Symbiodiniaceae partnerships (e.g. symbiosis specificity and flexibility).
The use of these approaches relies on the availability of high-quality marker genes and genomes as reference. Curated databases for symbiodiniacean marker genes thus far have been largely restricted to ITS2 (Hume et al., 2019) that is known to vary even within a genome (Thornhill et al., 2007). As more genome data from Symbiodiniaceae become available, other suitable (phylogenetic) marker genes can be identified and functionally validated. Such resources, proven useful for assessing prokaryote diversity (Chaumeil et al., 2019), will enhance our capacity in assessing Symbiodiniaceae diversity (including characterising unknown taxa), more accurately at finer resolution (Davies et al., 2023).
The development of these resources for Symbiodiniaceae will benefit from having genome-scale data from more broadly sampled taxa, both taxonomically and ecologically (Ishida et al., 2023). The integration (i.e. co-assembly) of microbial fraction of coral hologenome datasets en masse presents an innovative strategy for recovering genomes of Symbiodiniaceae taxa in hospite, which have been largely inaccessible due to the technical challenges associated with maintaining these taxa in lab cultures. Leveraging already existing coral hologenome datasets, this strategy complements other approaches that adopt more-advanced sequencing technologies and more-elaborate experimental designs yet to be optimised for Symbiodiniaceae, such as adaptive sampling sequencing (Martin et al., 2022) to enrich for microbial fraction in a metagenome/hologenome sample, and single-cell sequencing (Delmont et al., 2022) to recover genome data from DNA of a single cell.
Comparative genomic analyses of broadly sampled Symbiodiniaceae taxa will clarify the structural and functional features related to their diversification and niche specialisation. These genomic resources will facilitate other omics studies, e.g. integrating transcriptomics, proteomics, and/or metabolomics (Camp et al., 2022), to elucidate their function in a coral holobiont (Williams et al., 2023), and in deciphering the molecular regulatory mechanisms that underpin gene expression (Liew et al., 2017; Roy et al., 2018; Dougan et al., 2022a).
Due to the high cost and intricacy of coral sampling strategies associated with generating large number of coral hologenome datasets, concerted efforts through large international research consortia, e.g. the Aquatic Symbiosis Genomics Project (McKenna et al., 2021) funded by the Moore Foundation, and the Reef Adaptation and Restoration Program (McLeod et al., 2022) funded by the Australian Commonwealth Government, are essential for such an endeavour. The “gold mine” of microbial fraction from these datasets is highly valuable for research of coral symbiosis, particularly in our quest to understand how Symbiodiniaceae have diversified to sustain symbiosis with corals and other marine organisms.
Author contributions
HI: Conceptualization, Formal analysis, Visualization, Writing – original draft, Writing – review & editing. CR: Conceptualization, Supervision, Writing – review & editing. CXC: Conceptualization, Supervision, Visualization, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Australian Research Council grant DP190102474 awarded to CXC, and the Australian Academy of Science Thomas Davies Research Grant for Marine, Soil, and Plant Biology awarded to CXC. HI was supported by the University of Queensland Research Training Program.
Acknowledgments
We thank Dr Ira Cooke and Dr. Jia Zhang from James Cook University for sharing the coral hologenome dataset of Acropora kenti.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Alberdi A., Andersen S. B., Limborg M. T., Dunn R. R., Gilbert M. T. P. (2022). Disentangling host–microbiota complexity through hologenomics. Nat. Rev. Genet. 23, 281–297. doi: 10.1038/s41576-021-00421-0
Aranda M., Li Y., Liew Y. J., Baumgarten S., Simakov O., Wilson M. C., et al. (2016). Genomes of coral dinoflagellate symbionts highlight evolutionary adaptations conducive to a symbiotic lifestyle. Sci. Rep. 6, 39734. doi: 10.1038/srep39734
Armstrong E. J., Lê-Hoang J., Carradec Q., Aury J. M., Noel B., Hume B. C. C., et al. (2023). Host transcriptomic plasticity and photosymbiotic fidelity underpin Pocillopora acclimatization across thermal regimes in the Pacific Ocean. Nat. Commun. 14, 3056. doi: 10.1038/s41467-023-38610-6
Baums I. B., Chamberland V. F., Locatelli N. S., Conn T. (2022). ““Maximizing genetic diversity in coral restoration projects,”,” in Coral Reef Conservation and Restoration in the Omics Age. Eds. Aranda Lastra M., van Oppen. M. J. H. (Cham, Switzerland: Springer International Publishing), 33–53.
Bernard G., Chan C. X., Chan Y. B., Chua X. Y., Cong Y., Hogan J. M., et al. (2019). Alignment-free inference of hierarchical and reticulate phylogenomic relationships. Brief. Bioinform. 20, 426–435. doi: 10.1093/bib/bbx067
Bhattacharya D., Stephens T. G., Chille E. E., Benites L. F., Chan C. X. (2023). Facultative lifestyle drives diversity of coral algal symbionts. Trends Ecol. Evol. 39, 426–435. doi: 10.1016/j.tree.2023.10.005
Bongaerts P., Cooke I. R., Ying H., Wels D., den Haan S., Hernandez-Agreda A., et al. (2021). Morphological stasis masks ecologically divergent coral species on tropical reefs. Curr. Biol. 31, 2286–2298. doi: 10.1016/j.cub.2021.03.028
Boyd J. A., Woodcroft B. J., Tyson G. W. (2018). GraftM: a tool for scalable, phylogenetically informed classification of genes within metagenomes. Nucleic Acids Res. 46, e59. doi: 10.1093/nar/gky174
Bridge T. C. L., Cowman P. F., Quattrini A. M., Bonito V. E., Sinniger F., Harii S., et al. (2023). A tenuis relationship: traditional taxonomy obscures systematics and biogeography of the ‘Acropora tenuis’ (Scleractinia: Acroporidae) species complex. Zool. J. Linn. Soc., zlad062. doi: 10.1093/zoolinnean/zlad062
Butler C. C., Turnham K. E., Lewis A. M., Nitschke M. R., Warner M. E., Kemp D. W., et al. (2023). Formal recognition of host-generalist species of dinoflagellate (Cladocopium, Symbiodiniaceae) mutualistic with Indo-Pacific reef corals. J. Phycol. 59, 698–711. doi: 10.1111/jpy.13340
Camp E. F., Kahlke T., Signal B., Oakley C. A., Lutz A., Davy S. K., et al. (2022). Proteome metabolome and transcriptome data for three Symbiodiniaceae under ambient and heat stress conditions. Sci. Data 9, 153. doi: 10.1038/s41597-022-01258-w
Chan C. X., Bernard G., Poirion O., Hogan J. M., Ragan M. A. (2014). Inferring phylogenies of evolving sequences without multiple sequence alignment. Sci. Rep. 4, 6504. doi: 10.1038/srep06504
Chaumeil P.-A., Mussig A. J., Hugenholtz P., Parks D. H. (2019). GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 36, 1925–1927. doi: 10.1093/bioinformatics/btz848
Chen Y., Dougan K. E., Nguyen Q., Bhattacharya D., Chan C. X. (2024). Genome-wide transcriptome analysis reveals the diversity and function of long non-coding RNAs in dinoflagellates. NAR Genom. Bioinform. 6, lqae016. doi: 10.1093/nargab/lqae016
Chen Y., González-Pech R. A., Stephens T. G., Bhattacharya D., Chan C. X. (2020). Evidence that inconsistent gene prediction can mislead analysis of dinoflagellate genomes. J. Phycol. 56, 6–10. doi: 10.1111/jpy.12947
Cooke I., Ying H., Forêt S., Bongaerts P., Strugnell J. M., Simakov O., et al. (2020). Genomic signatures in the coral holobiont reveal host adaptations driven by Holocene climate change and reef specific symbionts. Sci. Adv. 6, eabc6318. doi: 10.1126/sciadv.abc6318
Cowen L. J., Putnam H. M. (2022). Bioinformatics of corals: Investigating heterogeneous omics data from coral holobionts for insight into reef health and resilience. Annu. Rev. Biomed. Data Sci. 5, 205–231. doi: 10.1146/annurev-biodatasci-122120-030732
Davies S. W., Gamache M. H., Howe-Kerr L. I., Kriefall N. G., Baker A. C., Banaszak A. T., et al. (2023). Building consensus around the assessment and interpretation of Symbiodiniaceae diversity. PeerJ 11, e15023. doi: 10.7717/peerj.15023
Delmont T. O., Gaia M., Hinsinger D. D., Frémont P., Vanni C., Fernandez-Guerra A., et al. (2022). Functional repertoire convergence of distantly related eukaryotic plankton lineages abundant in the sunlit ocean. Cell Genom. 2, 100123. doi: 10.1016/j.xgen.2022.100123
Dougan K. E., Bellantuono A. J., Kahlke T., Abbriano R. M., Chen Y., Shah S., et al. (2022a). Whole-genome duplication in an algal symbiont bolsters coral heat tolerance. bioRxiv 2022.04.10.487810. doi: 10.1101/2022.04.10.487810
Dougan K. E., Deng Z. L., Wöhlbrand L., Reuse C., Bunk B., Chen Y., et al. (2023). Multi-omics analysis reveals the molecular response to heat stress in a “red tide” dinoflagellate. Genome Biol. 24, 265. doi: 10.1186/s13059-023-03107-4
Dougan K. E., González-Pech R. A., Stephens T. G., Shah S., Chen Y., Ragan M. A., et al. (2022b). Genome-powered classification of microbial eukaryotes: focus on coral algal symbionts. Trends Microbiol. 30, 831–840. doi: 10.1016/j.tim.2022.02.001
Fuller Z. L., Mocellin V. J. L., Morris L. A., Cantin N., Shepherd J., Sarre L., et al. (2020). Population genetics of the coral Acropora millepora: toward genomic prediction of bleaching. Science 369, 268. doi: 10.1126/science.aba4674
González-Pech R. A., Bhattacharya D., Ragan M. A., Chan C. X. (2019). Genome evolution of coral reef symbionts as intracellular residents. Trends Ecol. Evol. 34, 799–806. doi: 10.1016/j.tree.2019.04.010
González-Pech R. A., Stephens T. G., Chen Y., Mohamed A. R., Cheng Y., Shah S., et al. (2021). Comparison of 15 dinoflagellate genomes reveals extensive sequence and structural divergence in family Symbiodiniaceae and genus Symbiodinium. BMC Biol. 19, 73. doi: 10.1186/s12915-021-00994-6
Hoegh-Guldberg O. (1999). Climate change, coral bleaching and the future of the world’s coral reefs. Mar. Freshw. Res. 50, 839–866. doi: 10.1071/MF99078
Hume B. C. C., Smith E. G., Ziegler M., Warrington H. J. M., Burt J. A., LaJeunesse T. C., et al. (2019). SymPortal: a novel analytical framework and platform for coral algal symbiont next-generation sequencing ITS2 profiling. Mol. Ecol. Resour. 19, 1063–1080. doi: 10.1111/1755-0998.13004
Ishida H., John U., Murray S. A., Bhattacharya D., Chan C. X. (2023). Developing model systems for dinoflagellates in the post-genomic era. J. Phycol. 59, 799–808. doi: 10.1111/jpy.13386
Kitchen S. A., Ratan A., Bedoya-Reina O. C., Burhans R., Fogarty N. D., Miller W., et al. (2019). Genomic variants among threatened Acropora corals. G3 9, 1633–1646. doi: 10.1534/g3.119.400125
Knowlton N., Brainard R. E., Fisher R., Moews M., Plaisance L., Caley M. J. (2010). “Coral Reef Biodiversity,” in Life in the World’s Oceans: Diversity, Distribution, and Abundance. (Wiley-Blackwell, Oxford, UK), 65–78.
Krueger T., Gates R. D. (2012). Cultivating endosymbionts — Host environmental mimics support the survival of Symbiodinium C15 ex hospite. J. Exp. Mar. Biol. Ecol. 413, 169–176. doi: 10.1016/j.jembe.2011.12.002
LaJeunesse T. C. (2005). “Species” radiations of symbiotic dinoflagellates in the Atlantic and Indo-Pacific since the Miocene-Pliocene transition. Mol. Biol. Evol. 22, 570–581. doi: 10.1093/molbev/msi042
LaJeunesse T. C., Lambert G., Andersen R. A., Coffroth M. A., Galbraith D. W. (2005). Symbiodinium (Pyrrhophyta) genome sizes (DNA content) are smallest among dinoflagellates. J. Phycol. 41, 880–886. doi: 10.1111/j.0022-3646.2005.04231.x
LaJeunesse T. C., Parkinson J. E., Gabrielson P. W., Jeong H. J., Reimer J. D., Voolstra C. R., et al. (2018). Systematic revision of Symbiodiniaceae highlights the antiquity and diversity of coral endosymbionts. Curr. Biol. 28, 2570–2580. doi: 10.1016/j.cub.2018.07.008
LaJeunesse T. C., Wiedenmann J., Casado-Amezúa P., D’Ambra I., Turnham K. E., Nitschke M. R., et al. (2022). Revival of Philozoon Geddes for host-specialized dinoflagellates, ‘zooxanthellae’, in animals from coastal temperate zones of northern and southern hemispheres. Eur. J. Phycol. 57, 166–180. doi: 10.1080/09670262.2021.1914863
Liew Y. J., Li Y., Baumgarten S., Voolstra C. R., Aranda M. (2017). Condition-specific RNA editing in the coral symbiont Symbiodinium microadriaticum. PLoS Genet. 13, e1006619. doi: 10.1371/journal.pgen.1006619
Lin S., Cheng S., Song B., Zhong X., Lin X., Li W., et al. (2015). The Symbiodinium kawagutii genome illuminates dinoflagellate gene expression and coral symbiosis. Science 350, 691. doi: 10.1126/science.aac0408
Lin S., Song B., Morse D., Palenik B. (2021). Spatial organization of dinoflagellate genomes: novel insights and remaining critical questions. J. Phycol. 57, 1674–1678. doi: 10.1111/jpy.13206
Lo R., Dougan K. E., Chen Y., Shah S., Bhattacharya D., Chan C. X. (2022). Alignment-free analysis of whole-genome sequences from Symbiodiniaceae reveals different phylogenetic signals in distinct regions. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.815714
Martin S., Heavens D., Lan Y., Horsfield S., Clark M. D., Leggett R. M. (2022). Nanopore adaptive sampling: a tool for enrichment of low abundance species in metagenomic samples. Genome Biol. 23, 11. doi: 10.1186/s13059-021-02582-x
Maruyama S., Weis V. M., Smith J. (2021). Limitations of using cultured algae to study cnidarian-algal symbioses and suggestions for future studies. J. Phycol. 57, 30–38. doi: 10.1111/jpy.13102
Matias A. M. A., Popovic I., Thia J. A., Cooke I. R., Torda G., Lukoschek V., et al. (2023). Cryptic diversity and spatial genetic variation in the coral Acropora tenuis and its endosymbionts across the Great Barrier Reef. Evol. Appl. 16, 293–310. doi: 10.1111/eva.13435
McKenna V., Archibald J. M., Beinart R., Dawson M. N., Hentschel U., Keeling P. J., et al. (2021). The Aquatic Symbiosis Genomics Project: probing the evolution of symbiosis across the tree of life. Wellcome Open Res. 6, 254. doi: 10.12688/wellcomeopenres.17222.1
McLeod I. M., Hein M. Y., Babcock R., Bay L., Bourne D. G., Cook N., et al. (2022). Coral restoration and adaptation in Australia: the first five years. PLoS One 17, e0273325. doi: 10.1371/journal.pone.0273325
Morikawa M. K., Palumbi S. R. (2019). Using naturally occurring climate resilient corals to construct bleaching-resistant nurseries. Proc. Natl. Acad. Sci. U. S. A. 116, 10586–10591. doi: 10.1073/pnas.1721415116
Noel B., Denoeud F., Rouan A., Buitrago-López C., Capasso L., Poulain J., et al. (2023). Pervasive tandem duplications and convergent evolution shape coral genomes. Genome Biol. 24, 123. doi: 10.1186/s13059-023-02960-7
Quigley K. M., Ramsby B., Laffy P., Harris J., Mocellin V. J. L., Bay L. K. (2022). Symbioses are restructured by repeated mass coral bleaching. Sci. Adv. 8, eabq8349. doi: 10.1126/sciadv.abq8349
Reich H. G., Kitchen S. A., Stankiewicz K. H., Devlin-Durante M., Fogarty N. D., Baums I. B. (2021). Genomic variation of an endosymbiotic dinoflagellate (Symbiodinium ‘fitti’) among closely related coral hosts. Mol. Ecol. 30, 3500–3514. doi: 10.1111/mec.15952
Reinert G., Chew D., Sun F., Waterman M. S. (2009). Alignment-free sequence comparison (I): statistics and power. J. Comput. Biol. 16, 1615–1634. doi: 10.1089/cmb.2009.0198
Riginos C., Popovic I., Meziere Z., Garcia V. O., Byrne I., Howitt S. M., et al. (2024). Cryptic species and hybridisation in corals: challenges and opportunities for conservation and restoration. EcoEvoRxiv. doi: 10.32942/X2502X
Robbins S. J., Singleton C. M., Chan C. X., Messer L. F., Geers A. U., Ying H., et al. (2019). A genomic view of the reef-building coral Porites lutea and its microbial symbionts. Nat. Microbiol. 4, 2090–2100. doi: 10.1038/s41564-019-0532-4
Rose N. H., Bay R. A., Morikawa M. K., Thomas L., Sheets E. A., Palumbi S. R. (2021). Genomic analysis of distinct bleaching tolerances among cryptic coral species. Proc. Biol. Sci. 288, 20210678. doi: 10.1098/rspb.2021.0678
Roy S., Jagus R., Morse D. (2018). Translation and translational control in dinoflagellates. Microorganisms 6, 30. doi: 10.3390/microorganisms6020030
Saad O. S., Lin X., Ng T. Y., Li L., Ang P., Lin S. (2020). Genome size, rDNA Copy, and qPCR assays for Symbiodiniaceae. Front. Microbiol. 11. doi: 10.3389/fmicb.2020.00847
Shah S., Dougan K. E., Chen Y., Bhattacharya D., Chan C. X. (2023a). Gene duplication is the primary driver of intraspecific genomic divergence in coral algal symbionts. Open Biol. 13, 230182. doi: 10.1098/rsob.230182
Shah S., Dougan K. E., Chen Y., Lo R., Laird G., Fortuin M. D. A., et al. (2023b). Massive genome reduction occurred prior to the origin of coral algal symbionts. bioRxiv 2023, 3.24.534093. doi: 10.1101/2023.03.24.534093
Shinzato C., Khalturin K., Inoue J., Zayasu Y., Kanda M., Kawamitsu M., et al. (2021). Eighteen coral genomes reveal the evolutionary origin of Acropora strategies to accommodate environmental changes. Mol. Biol. Evol. 38, 16–30. doi: 10.1093/molbev/msaa216
Shoguchi E., Shinzato C., Kawashima T., Gyoja F., Mungpakdee S., Koyanagi R., et al. (2013). Draft assembly of the Symbiodinium minutum nuclear genome reveals dinoflagellate gene structure. Curr. Biol. 23, 1399–1408. doi: 10.1016/j.cub.2013.05.062
Suggett D. J., Smith D. J. (2020). Coral bleaching patterns are the outcome of complex biological and environmental networking. Glob. Change Biol. 26, 68–79. doi: 10.1111/gcb.14871
Thomas L., Underwood J. N., Rose N. H., Fuller Z. L., Richards Z. T., Dugal L., et al. (2022). Spatially varying selection between habitats drives physiological shifts and local adaptation in a broadcast spawning coral on a remote atoll in Western Australia. Sci. Adv. 8, eabl9185. doi: 10.1126/sciadv.abl9185
Thornhill D. J., LaJeunesse T. C., Santos S. R. (2007). Measuring rDNA diversity in eukaryotic microbial systems: how intragenomic variation, pseudogenes, and PCR artifacts confound biodiversity estimates. Mol. Ecol. 16, 5326–5340. doi: 10.1111/j.1365-294X.2007.03576.x
Thornhill D. J., Lewis A. M., Wham D. C., LaJeunesse T. C. (2014). Host-specialist lineages dominate the adaptive radiation of reef coral endosymbionts: adaptive radiation of symbiotic dinoflagellates. Evolution 68, 352–367. doi: 10.1111/evo.12270
Truong D. T., Tett A., Pasolli E., Huttenhower C., Segata N. (2017). Microbial strain-level population structure and genetic diversity from metagenomes. Genome Res. 27, 626–638. doi: 10.1101/gr.216242.116
Voolstra C. R., Quigley K. M., Davies S. W., Parkinson J. E., Peixoto R. S., Aranda M., et al. (2021a). Consensus guidelines for advancing coral holobiont genome and specimen voucher deposition. Front. Mar. Sci. 8. doi: 10.3389/fmars.2021.701784
Voolstra C. R., Suggett D. J., Peixoto R. S., Parkinson J. E., Quigley K. M., Silveira C. B., et al. (2021b). Extending the natural adaptive capacity of coral holobionts. Nat. Rev. Earth Environ. 2, 747–762. doi: 10.1038/s43017-021-00214-3
Wan L., Reinert G., Sun F., Waterman M. S. (2010). Alignment-free sequence comparison (II): theoretical power of comparison statistics. J. Comput. Biol. 17, 1467–1490. doi: 10.1089/cmb.2010.0056
Williams A., Stephens T. G., Shumaker A., Bhattacharya D. (2023). Peeling back the layers of coral holobiont multi-omics data. iScience 26, 107623. doi: 10.1016/j.isci.2023.107623
Zhang J., Richards Z. T., Adam A. A. S., Chan C. X., Shinzato C., Gilmour J., et al. (2022). Evolutionary responses of a reef-building coral to climate change at the end of the last glacial maximum. Mol. Biol. Evol. 39, msac201. doi: 10.1093/molbev/msac201
Keywords: Symbiodiniaceae, coral, holobiont, hologenome, bioinformatics, genomics, symbiosis
Citation: Ishida H, Riginos C and Chan CX (2024) Contaminant or goldmine? In silico assessment of Symbiodiniaceae community using coral hologenomes. Front. Protistol. 2:1376877. doi: 10.3389/frpro.2024.1376877
Received: 26 January 2024; Accepted: 21 March 2024;
Published: 02 April 2024.
Edited by:
Alexey Potekhin, University of Innsbruck, AustriaReviewed by:
Patricia Elena Thome, National Autonomous University of Mexico, MexicoEiichi Shoguchi, Okinawa Institute of Science and Technology Graduate University, Japan
Copyright © 2024 Ishida, Riginos and Chan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cheong Xin Chan, Yy5jaGFuMUB1cS5lZHUuYXU=