 Andreas Kremling
Andreas Kremling- Systems Biotechnology, School of Engineering and Design, Technical University of Munich, Munich, Germany
Mathematical models for cellular systems have become more and more important for understanding the complex interplay between metabolism, signalling, and gene expression.In this manuscript, starting from the well-known flux balance analysis, tools and methods are summarised and illustrated by various examples that describe the way to models with kinetics for individual reactions steps that are finally self-contained. While flux analysis requires known (measured) input fluxes, self-contained (or self-sustained) models only get information on concentrations of environmental components. Kinetic reaction laws, feedback structures, and protein allocation then determine the temporal output of all intracellular metabolites and macromolecules. Emphasis is placed on (i) mass conservation, a crucial system property frequently overlooked in models incorporating cellular structures like macromolecular structures like proteins, RNA, and DNA, and (ii) thermodynamic constraints which further limit the solution space. Matlab Live Scripts are provided for all simulation studies shown and additional reading material is given in the appendix.
1 Introduction
Understanding complex systems, both in technical and non-technical sciences, relies heavily on mathematical modeling. This holds true for life sciences, particularly in systems biology, where mathematical modeling serves as a formalization tool for comprehending the intricacies of a system. Systems theory provides a framework for constructing models with various dimensions. One dimension pertains to the level of detail in the model, spanning from simple qualitative interaction networks to extensive, mass-conservative quantitative models detailing processes within cells and their fluctuating environments. Another dimension involves whether the model represents an average cell or individual cells within an environment. Modeling individual cells demands sophisticated approaches, such as employing population balance equations developed by Ramkrishna and colleagues (Ramkrishna, 2000) or adopting an ensemble modeling approach. Both methods necessitate distinct numerical schemes for resolution. Additional dimensions encompass whether the system is static or dynamic, and if the model requires structural elements, like an objective function, to explore the potentially infinite solution space.
The text at hand endeavors to initiate the analysis of a biochemical reaction network using flux balance analysis (FBA), a well-established method in cellular systems modeling. Therefore, only a basic understanding of reaction engineering is needed, while in-depth knowledge of microbial physiology commonly applied in biotechnology is unnecessary, since the examples are based on a simplified (toy) network. It concludes by constructing self-contained models that consider cellular macromolecular units linked to metabolism. Emphasis is placed on mass conservation, a crucial system property frequently overlooked in models incorporating cellular structures, leading to an incorrect degree of freedom for selecting system unknowns. While standard FBA typically considers a single biomass reaction, the proposed framework accommodates the structured nature of cells, highlighting resource allocation importance. Simple, self-contained models derived from this framework serve as a foundation for more intricate models.
A systematic procedure is presented, starting with a basic biochemical network devoid of macromolecular units. It delves into thermodynamic properties and introduces methods for a proper description avoiding inconsistencies with physical basics. Conforming to the dimensions mentioned earlier, the manuscript confines itself to detailed quantitative models for an average cell, analyzing static properties.
Therefore, it is not only suitable for beginners with basic background in mathematics (algebraic knowledge and differential calculus is required) and modeling but also for advanced students who could deepen their knowledge, especially when the macromolecular unit structure of the cell is introduced (here two different approaches are used).
After studying the text, the reader gains an understanding of the fundamental mathematical framework underlying models of cellular systems, as introduced in Section 2.1. Although a simplified (toy) model is used for illustration, comprehensive stoichiometric models of real cellular systems are available in specialized databases and can serve as a foundation for further exploration. Section 2.2 addresses basic thermodynamic challenges, enabling the reader to formulate systems of equations that avoid thermodynamic inconsistencies. Section 2.3 expands the discussion to include cellular models that incorporate macromolecular structures. A key outcome here is the development of objective functions that account not only for intracellular fluxes but also for the enzyme requirements necessary to sustain those fluxes. Section 2.4 introduces an extended equation system that integrates a larger set of macromolecular components and explores their potential roles in feedback mechanisms—such as how proteome allocation can influence intracellular flux distributions. Finally, the text introduces the reader to kinetic modeling approaches (Section 2.5). Whereas fluxes were previously treated as unknown variables, this section presents kinetic equations for individual enzymatic reactions. This allows for the analysis of the dynamic relationship between metabolic fluxes and intracellular metabolite concentrations, illustrated through the ongoing example of a small metabolic network.
Last but not least, hopefully lecturers and also well-advanced scientist will find interesting examples for their courses. A quantitative description always aims for a better understanding of the system at hand. Although the examples used are small, the presented methodology is general and can be applied to larger stoichiometric networks to determine intracellular flux distributions given measured uptake and production rates. Moreover, problems of resource allocation can be considered and will allow researchers to compare simulation results with own data. Larger differences should lead to the formulation and testing of new hypotheses that again will lead to new sets of experimental experiments. Additional reading material (books and standard publications) are provided in the Supplementary Appendix.
The examples provided adhere strictly to quantitative principles, employing physiologically meaningful parameters and standard units such as
2 Case studies
2.1 Mass balance in biochemical reaction systems
A common feature of all deterministic models that were developed for applications in systems biology, medical systems biology, or metabolic engineering is mass conservation. This is valid on the level of single bio-chemical reactions but also for larger networks to whole-cell models. For the studies presented here, mass conservation is obtained for single biochemical reactions which also result in mass conservation on the level of biomass synthesis; closely related to this, it allows a clear calculation of the specific growth rate
A standard notation with stoichiometric coefficients
The numbers given in front of the metabolites are positive numbers (therefore they are written with value sign
with the vector
and as a results, a value of zero appears on the right side (scalar value since a row vector is multiplied with a column vector).
Since in a cell, a large number of reactions run simultaneously, the stoichiometric coefficients are stored in a matrix
For
In this example, the notation for a chemical reaction is applied for a single reaction. A reaction equation reads (
and with the molecular weights
Variable
Mass conservation on the level of single reactions implies that also for each compound in the system, an equation can be written down that sums up all processes that either increase or decrease the mass (or number of molecules) of the compound. Unfortunately, we cannot write down directly an equation
The change of the mol number
That is, the reaction velocity
with the specific growth rate
Based on this fundamental equation, model analysis can be started. A standard tool here is the determination of all fluxes in a given metabolic network. Flux analysis focuses on balanced states, that is, it is assumed that all processes result in a quasi steady-state of the involved compounds. Furthermore, taking into account that in the upper equation the last summand has numeric values of different orders of magnitude, the standard equation for flux analysis simplifies:
Please note, that the dependencies of
Example 2
For the bacterium Escherichia coli, the uptake rate for glucose in a minimal medium is estimated to be
Equation 9 will be the starting point for the analysis of our first network. One can assume that not all of the rates
and the number of unknowns is reduced. In general the number of rows corresponds to the number
Formally, we are looking for a vector
with entries
2.2 Flux models without drain into biomass
On the first level, a simple network is considered, and the basic procedure to determine the flux maps is introduced. On a later stage, biomass synthesis will be considered. For the following examples, the stoichiometric matrix
and is illustrated in Figure 1.
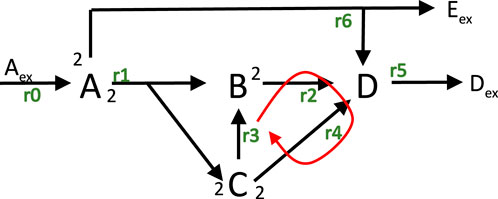
Figure 1. Network structure with input rate
Rates are numbered from
The calculation of flux distributions is shown for a number of different settings in Example 3. First, two different objective functions are applied and the different flux maps are compared Example 3a,b. For a selected objective function in Example 3c,it turns out, that some fluxes reach its maximal value. A closer inspection of the flux map reveals that thermodynamic laws are violated. After introducing the basic equations for a thermodynamic analysis for a single reaction and for biochemical reaction network, Example 3c is considered again and is now solved taking into account the presented equations.
Example 3
3a. Rate
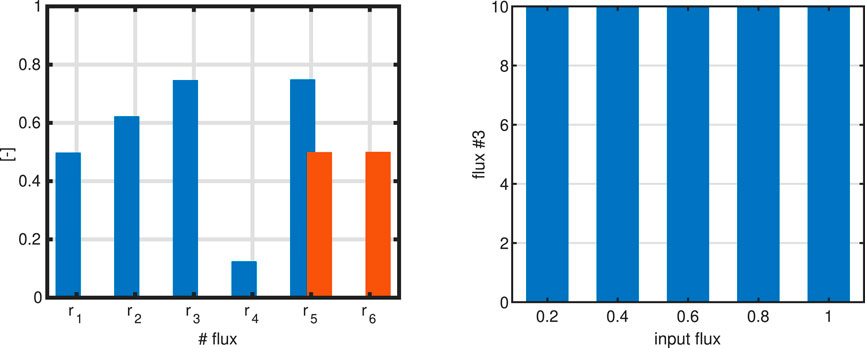
Figure 2. Left: Bar plot to compare a single flux objective
For 3c we note that–independent from the input flux–the objective always shows the same value. This value corresponds to the upper limit that was set by the boundaries. Boundaries limit the solution space and are based on (experts) knowledge on the system; they could be positive or negative, depending on the problem formulation. Since the direction of the fluxes are not known before in general, positive and negative values are used. How can we interpret this result? A closer and detailed look in this case reveals that not only flux
2.2.1 Thermodynamics
To solve the problem mentioned in the last example, thermodynamic considerations come into play. For reaction systems, the change of the free Gibbs energy
with index
•
•
This important equation relates the equilibrium constant of a reaction directly to the standard value of the Gibbs free energy.
•
Example 4
For a reaction with one substrate and two products (cf. Example 1):
The forward and the backward reaction are given by
In this case, Equation 13 reads:
and one obtains finally by combining the last two equations:
If
Taking advantage of the logarithmic function, the exponents can be written before the
with
From what we have seen before, a simple relationship between fluxs
That is, the product of the free Gibbs energy and the respective flux is always negative. For more than one reaction, the following notation is used:
2.2.2 Thermodynamics in networks
At this point, we have to note, that the values of
In the last section, we have seen that fluxes are coupled with metabolic concentrations via the Gibbs free energy. Considering large networks, this would require additional state variables (either the Gibbs free energy
As a starting point, an equilibrium is considered, that is, the net rate of all fluxes is zero as well as the three
and one can see by close inspection:
The equation can be re-written with the equilibrium constants of the reactions (Equation 18):
Can we obtain these equations also in a more systematic way? The cycle given by the three reactions can be obtained by the following equation; if only internal reactions are considered, that is, there is no exchange of mass with the environment, then, the null space
and one obtains for the null space:
Finally, we see that
is exactly Equation 23.
The last equation is not only true for the equilibrium. The Gibbs free energy can be written as a function of the stoichiometric matrix
Starting from the observation from above that the null space vector
A multiplication of the last equation from the left side with the vector
That is, the values for
Example 5
Considering the loop in the network above, one gets:
Now, assume that in the solution flux
In this case, a solution with a flux from
To incorporate these conditions in the linear framework used so far, we take advantage of a method described by Schellenberger et al. (2011). The methods uses integer variables
with
That is, for negative flux rates, the corresponding Gibbs energy is positive. In case (ii)
That is, for positive flux rates, the corresponding Gibbs energy is negative. This set of equation can now be written in matrix form in a very compact way. The full vector of variables that must be solved, now is as follows:
Note that matrix
A drawback of the procedure is, that additional variables including integer variables are needed that generate challenges while solving the equation system. An elegant way, described in (Beard et al., 2004), to avoid that fluxes are running in cycles is to formulate a non-linear equation based only on the sign of the fluxes (vector
Simply speaking, if a flux solution runs in a cycle, the left and the right hand side are equal; if this is not the case, then the left hand side is always smaller than the right hand side. Please note, however, that the strong constraint given in Equation 30 are not valid here; if there is potential between two components, a flux might be zero. In our case, for the sign vector we get
A further alternative, including the concentration of the metabolites and replacing the Gibbs energy by equations give above, allows us to find a further set of equations that could be used: Above, the condition
Example 6
The set of equation is applied for the cycle in the network, and let us assume that
From the first two lines, it follows:
with the condition for the equilibrium constants
That is a contradiction to the first two lines; the flux cannot run in the cycle. The presented equations are sufficient for a loopless network.
Example 3c (re-opened)
For the example, an extended optimization problem is formulated taking the concentrations of the compounds into consideration. The general form now reads:
with
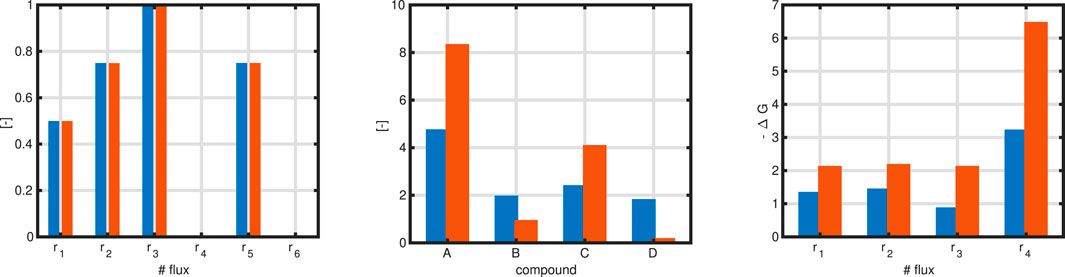
Figure 3. Left: Flux map with maximization of reaction
However, there is a broad range for the values for the concentrations fulfilling the given constraints. In analogy of selection one extraordinary flux for the optimization procedure, we proceed the same way and select from all possible values for a set of concentrations one which fulfills additional specifications. As proposed in literature, for a given flux map, all (absolute)
Example 3c (continued)
Figure 3 left shows the updated and thermodynamically valid flux distribution. In the middle, shown in red, are the values for the concentration of the compounds without and with the MDF design principle. Right: The minimal value for the negative
To complete the example, we perform a multi objective approach to maximize flux
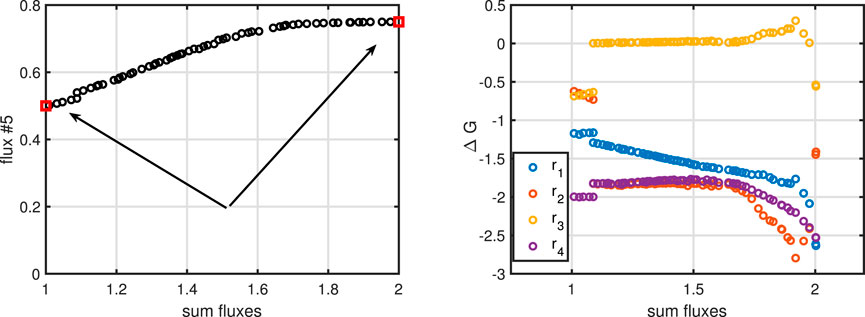
Figure 4. Left: Pareto front for maximizing flux
Lessons learned from Example 3: To determine valid flux distributions for cellular reaction networks, mass conservation as well as thermodynamic principles must be considered. Depending on the choice of the objective function, flux maps may differ. The selection of the objective function depend on the experimental set-up. For growth of bacteria in surplus carbohydrate the maximization of the growth rate is an appropriate choice. However, in case that substrates are limited or for applications in metabolic engineering, different objective functions are more suitable.
2.3 Flux models with drain into biomass
In this section, the network is extended to take into account that metabolites in the network often serve as precursors for biomass synthesis. Two different variants will be considered as shown in Figure 5; first, the standard notation used in FBA is introduced, then, a modeling approach with several macromolecular units such as protein, RNA and DNA is considered.
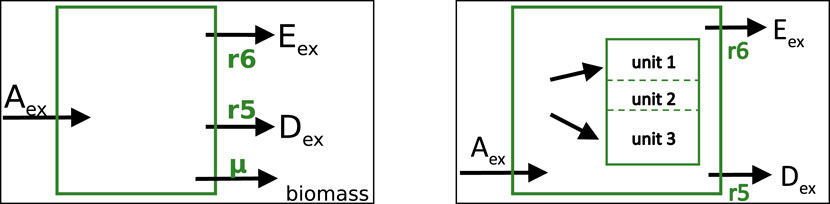
Figure 5. Left: Standard model representation in FBA with a single biomass flux as additional exchange reaction. Right: Model with several macromolecular units representing the major mass of a cell. Incoming and outgoing metabolic fluxes are the same as before and are the same in both approaches shown here.
A standard approach in FBA adds a single reaction, also called a pseudo-reaction, representing 1 g of biomass. In this single reaction, the complete metabolism for biomass assembly is summarized. For the example network from above, we use on a gram basis:
The equation is converted with the molecular weight of the compounds and the stoichiometric matrix
In FBA, the right side in the last equation remains empty, that is, biomass is not explicitly taken into account as compound in the stoichiometric matrix and therefore, no additional row is necessary. This can also be seen in Figure 5 (right side); the reaction arrow points out of the system.
Aside note:
It is worth to have a closer look on the last equation since we changed the units from
Normally, as in the examples given in the beginning, the stoichiometric coefficients are dimensionless (essentially they are
but we do not consider compound biomass as an additional state variable. The o.d.e. for compound
is used and is now transformed by excluding the biomass concentration
The last equation is re-written:
We note a change of units in the stoichiometric coefficients;
In Example 7 the drain of metabolites to generate biomass is considered and as in the previous case, two different objective functions are compared. In addition, constraints by the available amount of enzyme for the network is studied, resulting in four different scenarios.
Example 7
The network considered so far is extended by a single biomass reaction (Equation 44). Simulations results are shown in Figure 6 with two cases: (i) maximization of growth rate, and (ii) minimization of the sum of all fluxes.
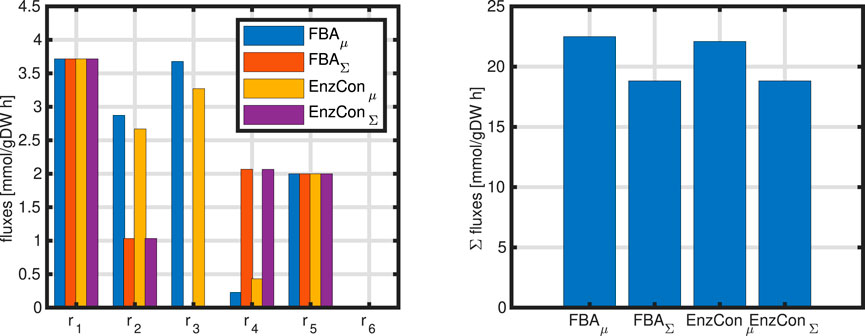
Figure 6. Left: Comparison of simulation results for four different cases; standard FBA (two objectives: maximizing growth rate and minimizing sum of fluxes) and enzyme constraint (two objectives: maximizing growth rate and minimizing sum of enzymes). The values for the incoming fluxes and for the growth rate are equal for all cases
For the next level, a further extension of the stoichiometric/thermodynamic approach is introduced. It becomes obvious during the last decade, that the standard approach has a strong disadvantage; the biomass constituents are not considered, and therefore, any feedback from the biomass parts back to the metabolic network is missing. This is true especially for proteins like enzymes that are involved in any metabolic processes. Measurement of the proteome now offers several possibilities to extend the flux analysis approach by taking into account not only different protein classes or sectors, but also information on individual enzymes. In a step-by-step approach we provide, and also combine, several methods from literature. The intention is to show that protein allocation has a strong impact on flux distributions, which has implications for the understanding of cellular physiology and but also is important for applications in Metabolic Engineering where one is interested to redirect fluxes to desired products.
Considering individual enzymes, from kinetics, it is convenient to split the mathematical term into two parts in a first step. Such rate laws depend on the concentration of all involved substrates
From here we see, that
Taking into account additional variable for the single enzyme
with
In case (ii) a different objective function is used; namely, the sum of all enzymes in the network is minimized. Here, the equation system reads with weighting factors, for example the molecular weight:
In case that only positive fluxes are considered (this can be achieved by splitting reversible fluxes into two positive ones), the two inequalities can be re-written in matrix notation with matrices/vectors with apparent size:
Example 8
For the given network, a rough estimation of the
and one obtains for the
Example 7 (continued)
Results of the different simulation studies introduced are shown in Figure 6. For a fair comparison, the growth rate was fixed in case of minimizing the sum of fluxes or minimizing the sum of enzymes to the same value that was obtained while maximizing the growth rate. The weights
Lessons learned from Example 7: The choice of the objective function and additional constraints show a strong impact on the flux distribution. However, while some fluxes does not change (for example
2.4 Flux models with several macromolecular units
The approaches so far still have a couple of limitations. The results are scalable since, typically, the main incoming fluxes must be given, and cannot get as a result from the optimization procedure; second, the biomass equation is an approximation, and does not account for different needs of the cell for growth and maintenance. In the next step, we omit the single biomass reaction and replace it with macromolecular units that are not subject to degradation (see Figure 5 right).
From the basic mass balance Equation 8, a steady state can only be reached, if the dilution term is considered. Also, to keep our mass conservative approach alive, the growth rate must be the result of incoming mass flow minus outgoing mass flow. Fortunately, from the basic equation, a simple, but powerful relationship could be derived for the specific growth rate (Doan et al., 2022):
The multiplication of
In comparison with standard FBA, an additional matrix
In Example 9, a simple model structure is used to analyze the influence of the biomass macromolecular structure. It is assumed that these elements are not actively degraded, and therefore only diluted by biomass growth. This allows model reduction, and, as shown in the extended case, also allows to study the influence of feedback structures, that is, the macromolecules influence their own synthesis.
Example 9
A simple scheme with one uptake reaction, one reaction that excreeds the metabolite directly back into the medium (this is based on experimental observations and is termed “overflow”5), and two cellular units
The stoichiometric matrix
with the first column represents the input flux as before. The molecular weights are in this case
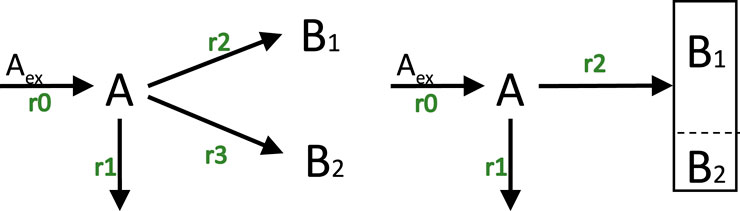
Figure 7. Left: Network with three unknown fluxes
While the masses of the compounds only fulfill the conservation condition (to sum up to
One gets for the steady state:
The nice thing with the last equations is, that scaling of the rate of synthesis with the molecular mass fractions always result in the specific growth rate, that is, the rates of synthesis are not independent (here:
The rates of synthesis for the macromolecular units can be obtained, and does not require the solving of an additional equation system. For this model, the null space
The first null space vector shows the way of the substrate through the network into the excreted overflow product (column 1) while the second vector represents the growth mode of the network (column 2), that is, the way from the substrate into the macromolecular units. A close inspection of the third element in the second vectors reveals that the element can be rewritten as
As indicated in Figure 7a reduced model can be obtained with only one single flux into the structured biomass (2 fractions). In the reduced model, the biomass is structured in two parts with only one rate.
Aside note:
If the stoichiometric
and the product
and the null space
Example 9 (continued)
Here, the reduced model is considered and a feedback structure is introduced as shown in Figure 8; both fractions of the biomass are needed to sustain the incoming flux
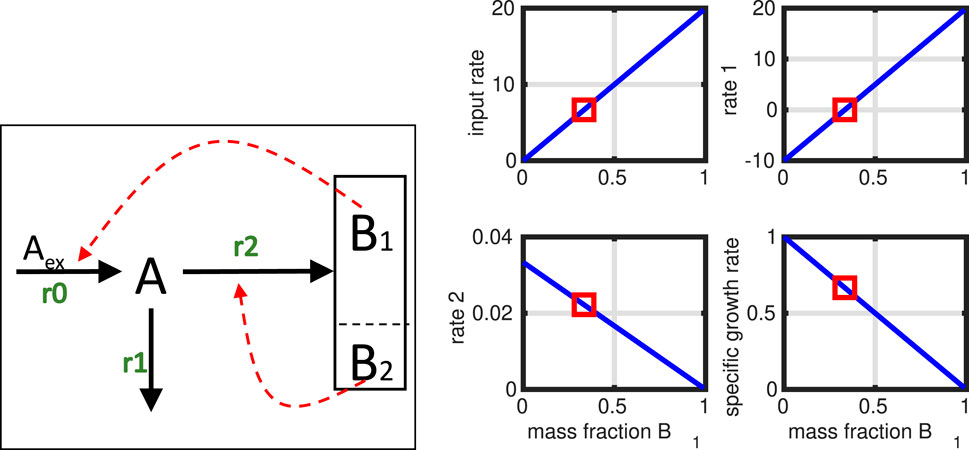
Figure 8. Controlled network; in a feedback loop, the macromolecular units directly influence the fluxes in network. Unit
The equation system starts from the standard equation system (Equation 58). Now, the rate vector (or parts of it) is replaced by a simple kinetic expression
However, we must add the conservation equation, saying that the sum of
and we optimize for the specific growth rate
Figure 8 shows the network structure and the simulation results. Since only only degree of freedom exists, the complete (linear) solution space could be determined (incoming flux
In the next Figure 9, we show two simulation studies with this simple model. First, we mimic a limitation of substrate in the medium (two left plots). This is realized by changing parameter
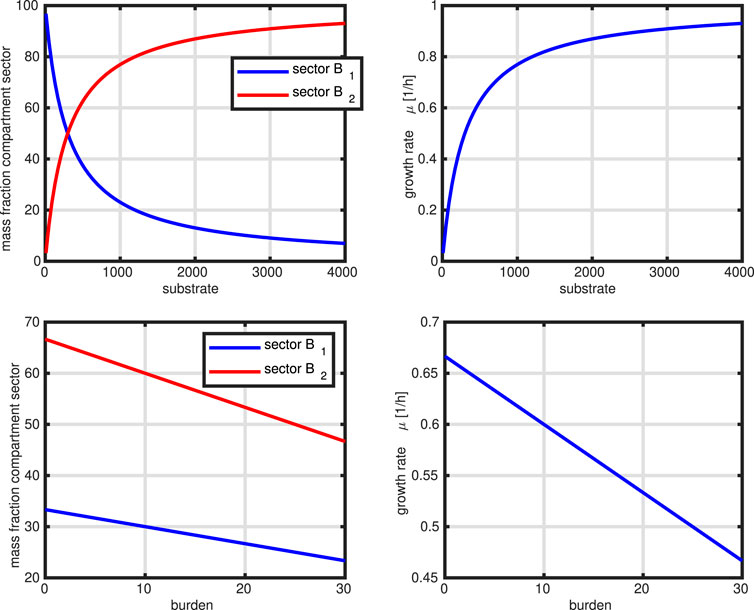
Figure 9. Controlled network. Upper row: mimic of substrate limitation (course of the two sectors, course of the growth rate) over the substrate concentration. Lower row: Mimic of heterologous protein production (course of the two sectors, course of the growth rate) over metabolic burden.
The examples already demonstrate a self contained model system. In comparison to the previous models, substrate intake is limited by the available amount on sector
The last set of examples introduces a new aspect in modeling cellular systems, namely, the dependencies of the reaction rate from the concentration of substrates, products and parameters. The analysis start with the network from Example 3 and then extends it with kinetics. The final case takes into account that the resources to generate the biomass macromolecular structure is limited by the proteins. In this way, a self contained cellular model is obtained.
Example 10a
In the next step, our example with 4 metabolites is considered again and extended with three macromolecular units. In Figure 10 the structure of model variant is shown that is used for three different cases.
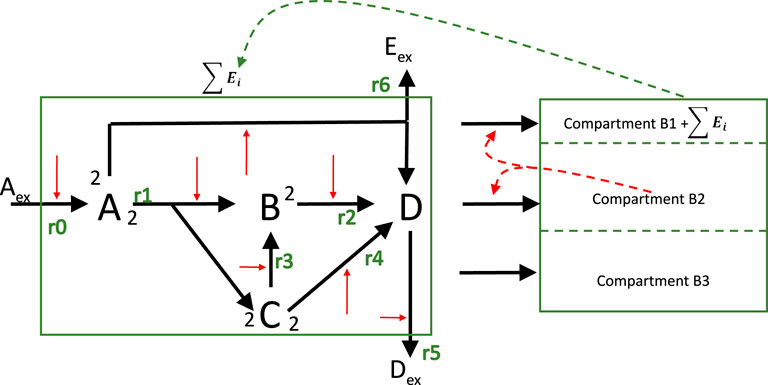
Figure 10. Overall structure of the model. The stoichiometric network is combined with the macromolecular unit modeling approach. For the model with resource allocation, feedback control is realized by using part of
In the first variant, the macromolecular units are fixed and the stoichiometric matrix contains the drain from the metabolites into the respective units. It is assumed that the sum of the mass fraction of the metabolites (
2.5 Kinetics–the step to dynamic models
Now we go back to Equation 51 and fill the kinetics with “life”, that is, we consider the dependencies of rates
with the substrate concentration
with the abbreviations
As seen above for a simple kinetic, a relationship in case of a reaction equilibrium could be obtained by setting
and the equilibrium constant is obtained like before:
Please note, that in this framework, the value of
Aside note:
In reversible enzyme kinetics, the equilibrium constant
We note that the kinetic parameters of substrate binding
Keeping
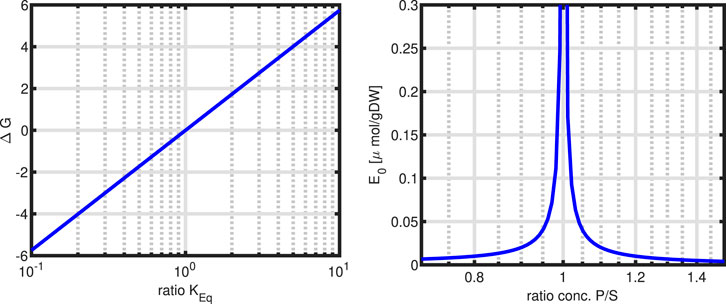
Figure 11. Left: Change of
However, taking Equation 75 to determine the amount of enzyme needed for varying product, a highly sensitive behaviour is observed (right plot in Figure 11). Near the equilibrium constant, the amount of enzyme needed to maintain a flux of
Furthermore, the above considerations on thermodynamic properties in cycles are taken into account, that is, the equilibrium constants are not independent, but must fulfill the condition given in Equation 24.
Example 10b (now with kinetics)
Now the same network as in Example 10a is considered, but kinetics for the metabolic network are taken into account. The system is now self-contained with a minimal number of constraints: mass fraction of metabolites is again fixed and the concentration of the unitss are given. For the models considered so far, the concentration of metabolites only had a modest role; they were only important for thermodynamic considerations. Figure 12 shows the results. Please note that for the equation for one macromolecular unit, the following equation holds in steady state:
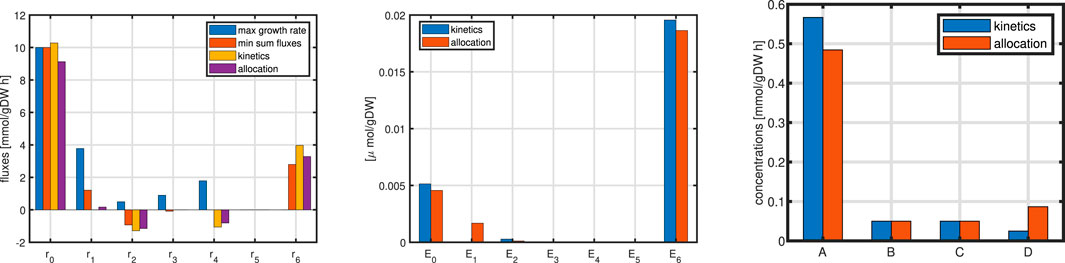
Figure 12. Left: Flux distribution for all rates for all cases. Middle: Comparison of enzyme concentrations for cases 10b and 10c. Right: metabolite concentrations for cases 10b and 10c.
Since in this case, the concentration of the unit is fixed and
Example 10c (resource limitation)
In the last case, the first unit
A simple first order dependency in the form
The optimization problem is now as follows (for all cases in Example 10):
The substrate uptake rates in all cases are comparable (left plot) while the flux maps differ: rate
The last model variant finally is used to mimic the behavior for a decreasing substrate concentration. In the previous example, the enzyme for substrate uptake is working in saturation, that is, the concentration of the substrate
Although the input flux is linear, the resulting growth rate tends to saturate for high values. This is based on (i) the saturation kinetics of the individual enzymes in the network, and (ii) on the limited capacity of the enzyme fraction that can be allocated for substrate uptake; please note, that for the synthesis of enzymes (and unit
We finally compare and illustrate the results of the simulation studies with biomass using elementary flux modes (see Supplementary Appendix for a definition and an example). For both networks, elementary flux modes with substrate uptake (from
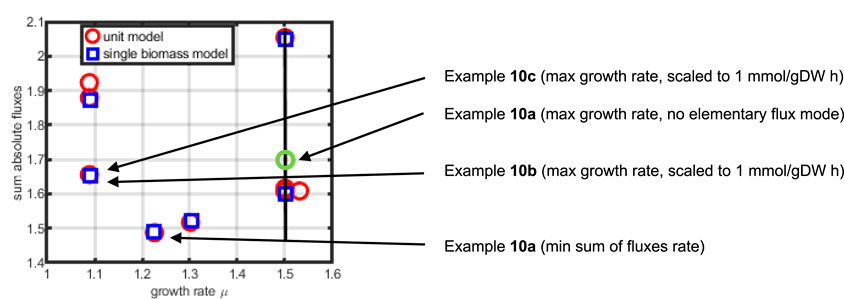
Figure 13. Sum of fluxes as a function of the growth rate for elementary flux modes (red circles indicate the elementary flux modes for the three unit model, blue squares for the single biomass equation model). Shown are the results for example 10.
As can be seen, the macromolecular unit model has more elementary flux modes than the single biomass model, showing a higher flexibility in providing precursors for biomass. However, the elementary flux mode with a higher growth rate than
A further analysis can be done by focusing on one node in the network and characterizing the incoming fluxes, that are named supply fluxes, and the outgoing fluxes, named demand fluxes. In steady-state, supply and demand are exactly balanced, that is, the sum of supply fluxes must match the demand fluxes. Moreover, since kinetic expressions are used for the last examples, the influence of metabolic concentrations can be studied. In the last example shown in Figure 14, the distribution of enzyme over the substrate input abruptly changes. In the following figure, two values of substrate input, corresponding to growth rates
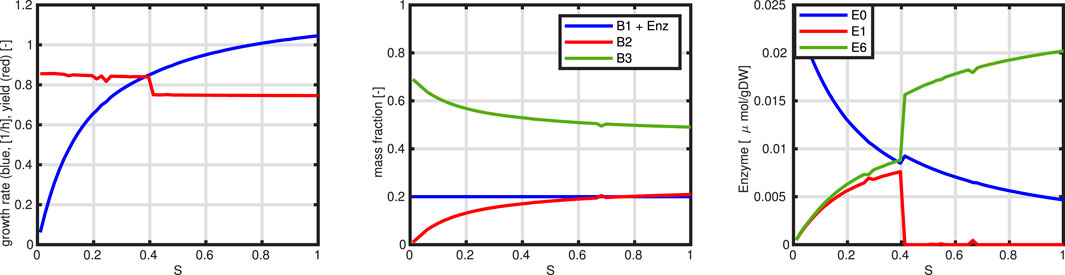
Figure 14. Left: Growth rate
In the network, the demand is split in several parts. Metabolite
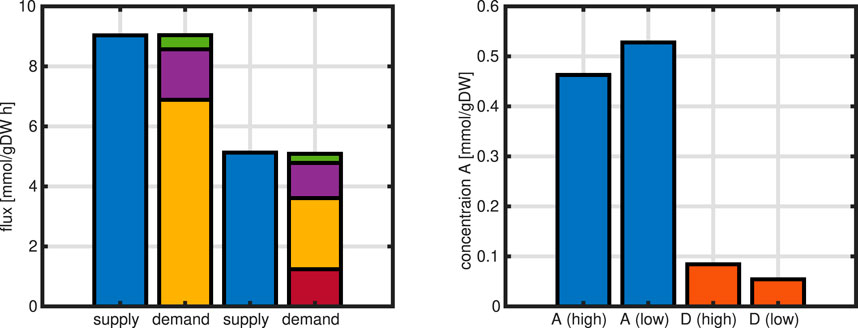
Figure 15. Left: first 2 bars are supply (left) and demand (right) for the high growth rate for metabolite
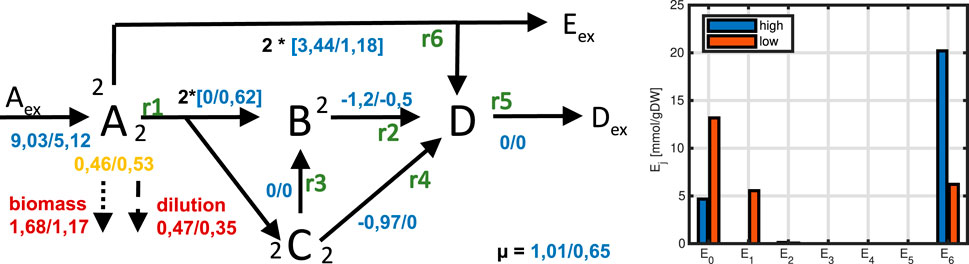
Figure 16. Left: Flux maps for high and low substrate input. For metabolite
3 Summary
The provided examples demonstrate an increasing complexity starting from a detached network with a couple of reactions and ending with a prototype of a cellular system with metabolism and macromolecular synthesis as well as kinetics for the metabolic network. While the simple network structure without biomass reactions must be scaled by the incoming flux, the last examples provide self-contained models. Thermodynamic considerations play an important role for the outcome of the simulations, but could be considered in the kinetic reaction expressions directly, bridging pure fluxes with metabolite concentrations.
Starting from flux balance analysis, using different objective functions to finally a three macromolecular unit model, increasing levels of complexity are developed and demonstrated by numerical examples. A final summary of basic equations and examples are given in Figure 17.

Figure 17. Overview equation systems and examples.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://sourceforge.net/projects/fba-to-self-contained-models/.
Author contributions
AK: Conceptualization, investigation, Writing – review and editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
This work was inspired by discussion on several book chapters with colleagues of the “Economic Principles in Cell Physiology” community, Meike Wortel, Wolf Liebermeister, and Elad Noor.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fsysb.2025.1546072/full#supplementary-material
Footnotes
1Please note, that symbol
2Conversion between both number can be done with the cellular density
3For basic properties of equation systems, we refer to the Supplementary Appendix.
4In the cited publication, 11 objective functions are analysed for E. coli under different environmental conditions. However, in completely different settings or when considering growth of different species in mixed cultures, it might be challenging to find a suitable objective function.
5The concept of overflow addresses the excretion of metabolites from central metabolism for example during high growth rates. It is observed for E. coli that the uptake of carbohydrates and oxygen consumption are not perfectly balanced, that is, for high rates of glucose uptake, the cell cannot use the carbon for growth due to the low availability of enzymes in the respiration chain. The surplus carbohydrates are converted for example into acetate that is excreted.
References
Beard, D. A., Babson, E., Curtis, E., and Qian, H. (2004). Thermodynamic constraints for biochemical networks. J. Theor. Biol. 228 (3), 327–333. ISSN 0022-5193. doi:10.1016/j.jtbi.2004.01.008
Doan, D. T., Hoang, M. D., Heins, A.-L., and Kremling, A. (2022). Applications of coarse-grained models in metabolic engineering. Front. Mol. Biosci. 9, 806213. doi:10.3389/fmolb.2022.806213
Ebrahim, A., Lerman, J. A., Palsson, B. O., and Hyduke, D. R. (2013). COBRApy: constraints-based reconstruction and analysis for python. BMC Syst. Biol. 7, 74. doi:10.1186/1752-0509-7-74
Heirendt, L., Arreckx, S., Pfau, T., Mendoza, S. N., Richelle, A., Heinken, A., et al. (2019). Creation and analysis of biochemical constraint-based models using the COBRA Toolbox v.3.0. Nat. Protoc. 14, 639–702. doi:10.1038/s41596-018-0098-2
Kremling, A. (2021). A counting-strategy together with a spatial structured model describes RNA polymerase and ribosome availability in Escherichia coli. Metab. Eng. 67, 145–152. doi:10.1016/j.ymben.2021.06.006
Noor, E., Bar-Even, A., Flamholz, A., Reznik, E., Liebermeister, W., and Milo, R. (2014). Pathway thermodynamics highlights kinetic obstacles in central metabolism. PLOS Comput. Biol. 10 (2), e1003483–12. doi:10.1371/journal.pcbi.1003483
Noor, E., Flamholz, A., Bar-Even, A., Davidi, D., Milo, R., and Liebermeister, W. (2016). The protein cost of metabolic fluxes: prediction from enzymatic rate laws and cost minimization. Plos Comp. Biol. 12, e1005167. doi:10.1371/journal.pcbi.1005167
Noor, E., Flamholz, A., Liebermeister, W., Bar-Even, A., and Milo, R. (2013). A note on the kinetics of enzyme action: a decomposition that highlights thermodynamic effects. FEBS Lett. 587 (17), 2772–2777. doi:10.1016/j.febslet.2013.07.028
Sanchez, B., Zhang, C., Nilsson, A., Lahtvee, P.-J., Kerkhoven, E. J., and Nielsen, J. (2017). Improving the phenotype predictions of a yeast genome-scale metabolic model by incorporating enzymatic constraints. Mol. Sys.Biol. 13 (8), 935. doi:10.15252/msb.20167411
Schellenberger, J., Lewis, N. E., and Palsson, B. O. (2011). Elimination of thermodynamically infeasible loops in steady-state metabolic models. Biophysical J. 100 (3), 544–553. doi:10.1016/j.bpj.2010.12.3707
Schmidt, A., Kochanowski, K., Vedelaar, S., Ahrne, E., Volkmer, B., Callipo, L., et al. (2016). The quantitative and condition-dependent Escherichia coli proteome. Nat. Biotechnol. 34 (1), 104–110. doi:10.1038/nbt.3418
Schuetz, R., Kuepfer, L., and Sauer, U. (2007). Systematic evaluation of objective functions for predicting intracellular fluxes in Escherichia coli. Mol. Syst. Biol. 3, 119. doi:10.1038/msb4100162
Wortel, M. T., Peters, H., Hulshof, J., Teusink, B., and Bruggeman, F. J. (2014). Metabolic states with maximal specific rate carry flux through an elementary flux mode. FEBS J. 281 (6), 1547–1555. doi:10.1111/febs.12722
Keywords: flux balance analysis, coarse-grained models, thermodynamics, resource allocation, enzyme kinetics
Citation: Kremling A (2025) From flux analysis to self contained cellular models. Front. Syst. Biol. 5:1546072. doi: 10.3389/fsysb.2025.1546072
Received: 16 December 2024; Accepted: 18 June 2025;
Published: 22 August 2025.
Edited by:
John Hancock, University of Ljubljana, SloveniaReviewed by:
Maria Suarez-Diez, Wageningen University and Research, NetherlandsYin Hoon Chew, University of Birmingham, United Kingdom
Copyright © 2025 Kremling. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andreas Kremling, YS5rcmVtbGluZ0B0dW0uZGU=