 Laurice Beatrice Raphaelle O. dela Peña
Laurice Beatrice Raphaelle O. dela Peña Diana Rose D. Mamawal
Diana Rose D. Mamawal Mae Ashley G. Nacario
Mae Ashley G. Nacario Mark Raymond A. Vejano
Mark Raymond A. Vejano Windell L. Rivera*
Windell L. Rivera*- Pathogen–Host–Environment Interactions Research Laboratory, Institute of Biology, College of Science, University of the Philippines Diliman, Quezon City, Philippines
Fecal contamination of important water resources poses a significant public health concern. To protect the public’s health, the dominant sources and various factors that contribute to pathogenesis and fecal contamination must be assessed. This study aimed to assess the effectiveness of 16S rRNA gene amplicon sequencing in detecting pathogens and tracking their sources in Manila Bay, Philippines. We sequenced the 16S V3–V4 region from DNA extracts of fecal samples (n = 37) of chickens, ducks, pigs, cows, goats, dogs, and sewage; and environmental water sources (n = 55) from Manila Bay tributary rivers, coastal stations, and offshore sites, which represented the “source” and “sink” samples, respectively. We used SourceTracker2 to estimate the percent contribution of these sources to the microbial community in Manila Bay. Among the detected bacteria were human and animal pathogens, including Clostridiales, Alteromonadales, Campylobacterales, Pseudomonadales, and Aeromonadales. Phosphates, fecal coliform, and dissolved oxygen were the major drivers of the top bacterial groups. Microbial community signatures clustered according to their corresponding sample types based on the beta diversity distances, suggesting the potential application of source libraries for analyzing the sink samples. Validation of the fecal source library shows that SourceTracker2 correctly predicted the contribution of the six fecal sources, but had a lower distinction for bovine sources. Sewage accounted for 93% of the contamination in Manila Bay, followed by ducks (5.6%), indicating human waste as the primary source. This study demonstrates the utility of microbial source tracking in targeted water quality management strategies.
1 Introduction
Coastal recreational waters are continually at risk of fecal contamination, which poses a significant public health concern owing to the presence of pathogenic bacteria and viruses in feces. Manila Bay is one of the most important bodies of water in the Philippines as it supports the surrounding urban and rural areas and provides a primary source of livelihood for local residents (Jacinto et al., 2006). Once famous for its clean waters, it is now known as a polluted body of water because of increasing urbanization (Vallejo et al., 2019). Rehabilitation projects in Manila Bay aim to restore its waters to a safe level for swimming and recreational use. However, recent reports indicate high levels of fecal coliform in several areas of Manila Bay, which may pose health risks (Philippine News Agency, 2022; Manila Bay Office, 2015). Although the fecal coliform count is a standard parameter in measuring water quality and monitoring compliance, it does not account for the entire microbial pathogen community in the environment. Characterizing the pathogen community is important for addressing risks and developing prevention strategies to protect public health. Moreover, understanding the environmental parameters that influence pathogen loading into the system is important in predicting and preventing pathogen transmission. This has led to the development of microbial source tracking (MST) methods to trace contamination (Scott et al., 2002).
Pathogen detection techniques, such as culture-based assays, PCR, and microarrays, are limited by their labor-intensive nature, inability to detect most pathogens in the natural environment, sensitivity issues, and reliance on prior data, leading to bias toward models or well-studied organisms. The application of metagenomics to MST offers a powerful, culture-independent approach to comprehensively identify and characterize sources of fecal contamination. Metagenomics-based methods can characterize microbial communities through the use of rapid, high-throughput sequencing data extracted from environmental DNA samples, providing a comprehensive view of the microbial diversity (Pérez-Cobas et al., 2020). This approach, which has proven useful for MST, is crucial for understanding key ecological factors shaping microbial communities, developing bioindicators, and identifying toxin and antibiotic resistance genes, particularly in water quality studies (Tan et al., 2015; Aylagas et al., 2017). Unlike other library-independent methods that require several assays to measure different host source contributions to pollution, a community-based method can completely characterize microbial communities and determine the overlap of community compositions between the environment and suspected sources using computational methods (Unno et al., 2018; García-Aljaro et al., 2019). The Bayesian tool SourceTracker2 has been used to identify the causes of water quality degradation in watersheds (Kirs et al., 2017; Staley et al., 2018), coastal recreational beaches (Rothenheber and Jones, 2018; Henry et al., 2016; Neave et al., 2014), and other environmental sources (Nakatsu et al., 2019; Baral et al., 2018).
This study utilized next-generation sequencing to identify the microbial pathogen community from selected sites in Manila Bay and the contribution of fecal host sources. Information on microbial diversity was also correlated with various physico-chemical parameters to determine how these abiotic factors influence the microbial community. The high contribution of sewage sources can be used as a benchmark for the assessment of water treatment processes for the elimination of waterborne pathogens.
2 Materials and methods
2.1 Study area and sample collection
Manila Bay encompasses a watershed area of 17,000 km2 and a 190-km coastline surrounding Metro Manila and the provinces of Bulacan, Pampanga, and Bataan. The bay is influenced by a tropical monsoon climate, with the dry season lasting from November to April and the wet season occurring from May to October. On average, rainfall is highest in August and lowest in February (Szekielda, 2022).
The sampling design considered space and season. The sampling sites included (1) bathing beaches (Navotas Fish Port, MOA, PEATC, Dolomite Beach), (2) areas near the north and south piers (North and South Harbor), and (3) offshore sites coordinated with the Department of Environment and Natural Resources–Environmental Management Bureau (DENR-EMB). Two upstream and downstream sites in Pasig, Marikina, San Juan, and Las Piñas-Parañaque River were included to account for the freshwater input. Sampling stations for the Pasig River were coordinated with the Pasig River Coordinating Management Office (PRCMO) of the DENR. Sampling took place between December 2021 and May 2023 at least once per wet (May to October) and dry (November to April) season to account for the seasonality of microbial communities. Water samples (2 L) were collected at the surface (0–2 m depth) per sampling station and season. After collection, the water samples were immediately transported to the laboratory on ice for processing. Samples were collected concurrently with DENR-EMB and PRCMO. Physico-chemical parameters, such as pH, temperature (°C), phosphates (mg/L), nitrates (mg/L), total suspended solids (TSS, mg/L), dissolved oxygen (DO, mg/L), biochemical oxygen demand (BOD, mg/L), and fecal coliform (MPN/100 mL), were measured by PRCMO and DENR-EMB using standard protocols (Department of Environment and Natural Resources–Environmental Management Bureau, 2016). The corresponding data are available online and upon request from the respective agencies.
Fecal samples from agricultural and domestic animals including chickens, cows, dogs, ducks, goats, and pigs were collected from different farms in Malabon City, Valenzuela City, Bulacan, Cavite, and Laguna. Human-derived samples included 2-L sewage samples collected from three wastewater treatment plants (untreated sewage contribution) in Quezon City and Manila City during the dry and wet seasons. These treatment plants were selected as they reflect the broader sewage systems throughout Metro Manila. Untreated sewage, dominated by domestic wastewater, may be discharged into nearby water bodies, introducing potential pathogens into the water. Additionally, animal feces may enter adjacent water bodies via agricultural runoff. The collected fecal samples were immediately transported to the laboratory for sample processing. Samples were collected along various locations in Manila Bay, including river mouths, beaches, harbors, and offshore areas, as well as from fecal sources representing potential contributors to pollution (Figure 1).
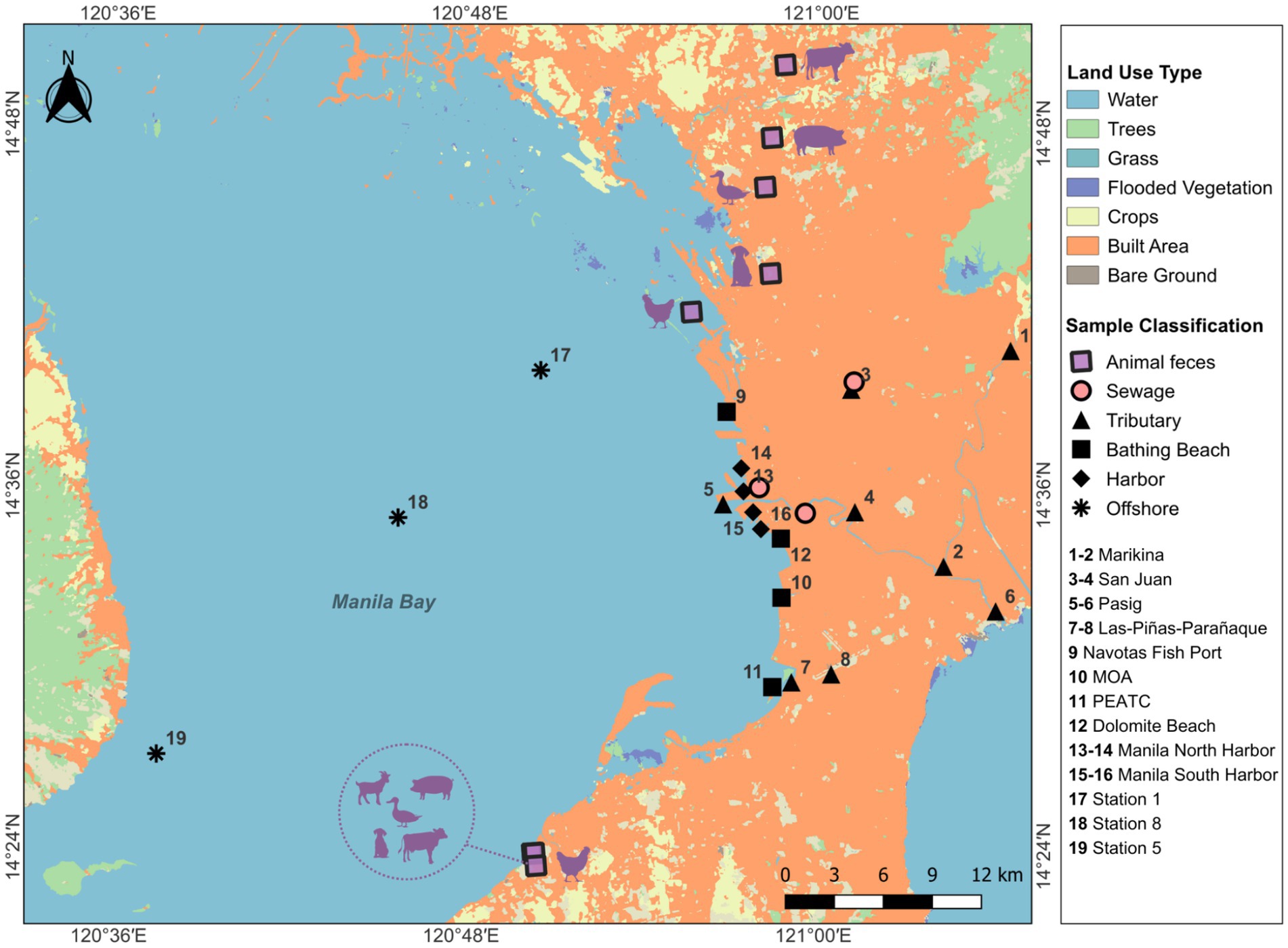
Figure 1. Sampling sites of environmental and fecal samples (QGIS Development Team, 2024). The environmental sites are represented by offshore, bathing beaches, and harbor areas in Manila Bay, and tributaries representing upstream and downstream points of Pasig River, San Juan River, Las Piñas-Parañaque River, and Marikina River. Animal fecal samples include excreta from chickens, cows, ducks, pigs, goats, and dogs. Sewage samples were collected from three wastewater treatment plants in Metro Manila.
A total of 55 samples were collected for sinks (Table 1). For the tributary category, a total of 23 samples were collected in the upstream and downstream stations of Las Piñas-Parañaque River, Marikina River, Pasig River, and San Juan River, with representative samples from the wet and dry seasons. For the North and South Harbor samples, a total of 10 samples were collected, representing each season. Moreover, one representative was collected per season in the offshore stations I, V, and VIII. Lastly, stations from MOA, Navotas Fish Port, PEATC, and Dolomite Beach served as bathing beach representatives in Manila Bay, each having two representatives per season.
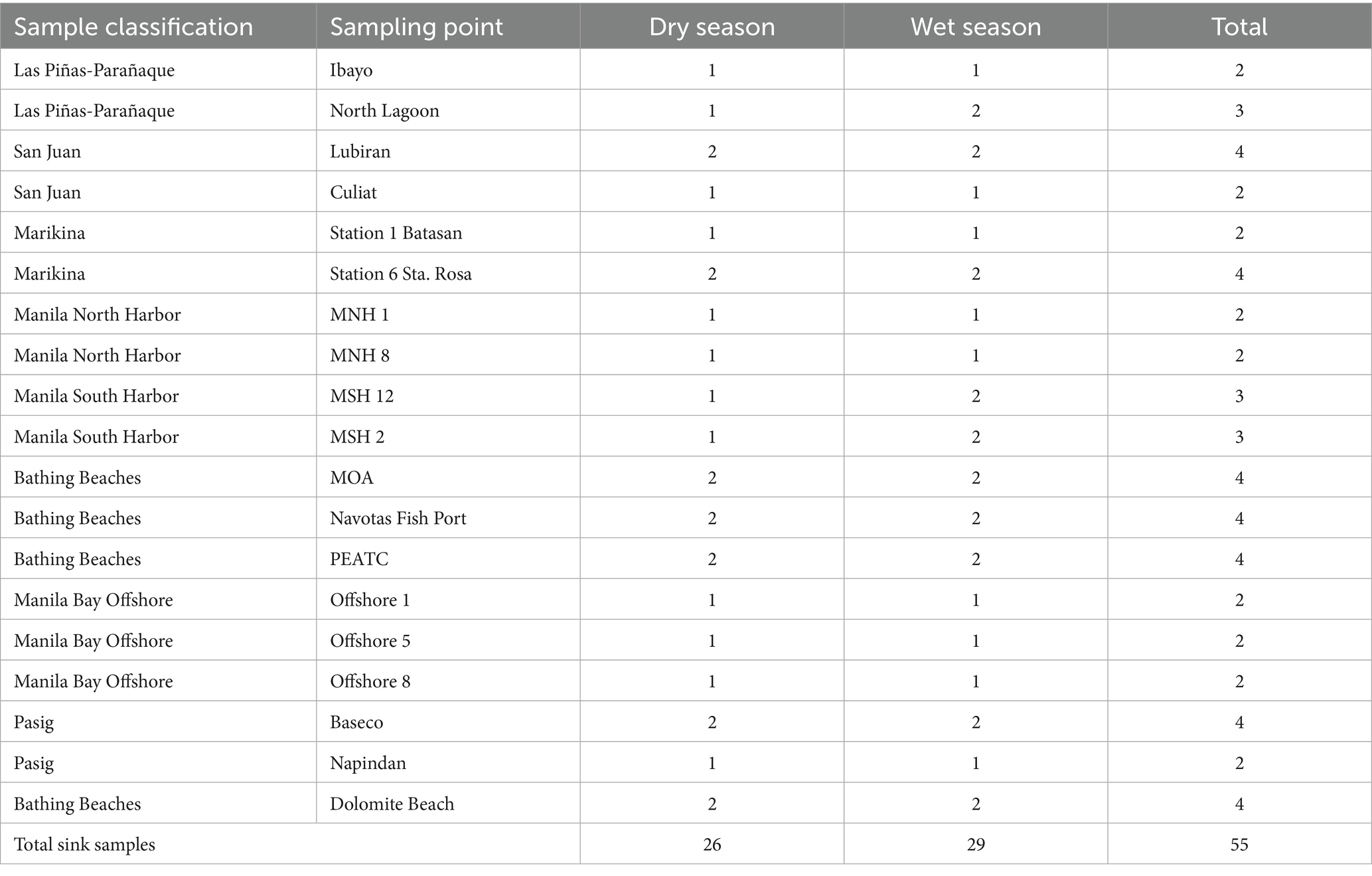
Table 1. Number of sink samples collected in the study.
Among the 37 source samples, individual fecal samples were collected for each of the following host sources: poultry (n = 6), bovine (n = 5), dog (n = 5), duck (n = 5), goat (n = 5), and pig (n = 5) (Table 2). Sewage samples collected during the wet and dry seasons served as representatives for human-derived samples.
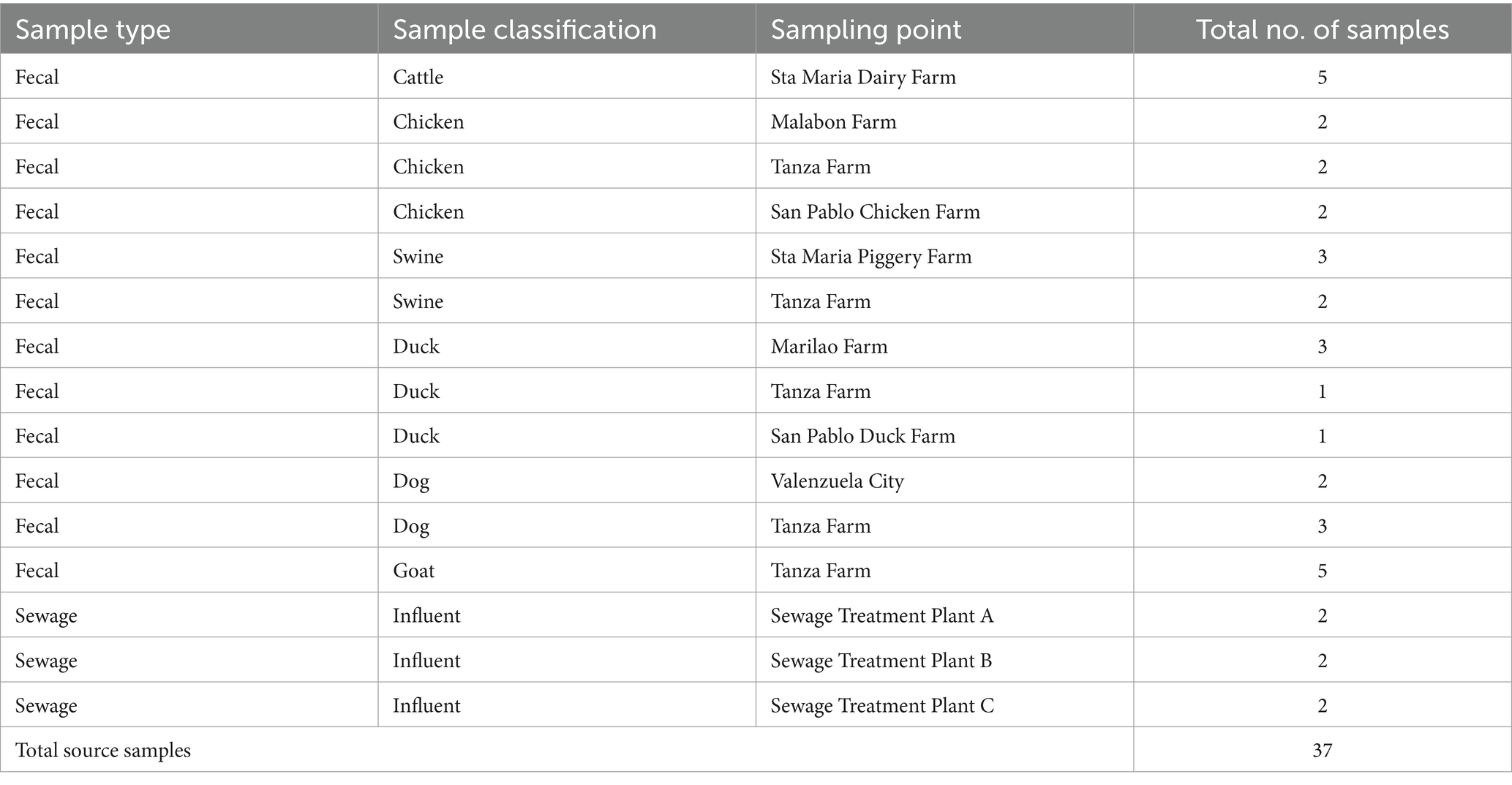
Table 2. Number of source samples (fecal and sewage) collected in the study.
2.2 Sample processing for prokaryotic microorganisms
Water and sewage samples were sequentially filtered using a 47 mm × 3.0-μm mixed cellulose ester membrane (Newstar, China) and 0.45-μm polycarbonate filters (Pall Corp., USA), with pre-filtration steps to remove larger-sized plankton. Filter membranes were then stored in 1.5-mL microcentrifuge tubes containing DNA/RNA Shield™ (Zymo Research, USA) reagent. For each fecal sample, 250 mg was measured and suspended in a 750-μL DNA/RNA Shield™ for DNA preservation. All samples were then stored at −20°C until further use.
The filtered particles were transferred into a sterile Petri dish for mechanical shredding using sterile forceps and scissors to increase DNA yield. Afterwards, the homogenized membranes were resuspended with DNA/RNA Shield™ up to the 1 mL mark. DNA from the homogenized filters was extracted using a ZymoBIOMICS™ DNA/RNA Extraction Kit (Zymo Research, USA) following the manufacturer’s protocols. The eluted DNA was stored at −20°C until further use. The extracted DNA samples represented the microbial community present in the water environment and were quantified using a Qubit 4 Fluorometer (Thermo Fisher Scientific, USA).
A similar kit was used for extracting the fecal samples, except that the suspended fecal samples were directly transferred to a ZR BashingBead Lysis Tube, vortexed, and secured in a high-speed bead-beater and processed at maximum speed for 15 min. This procedure was repeated twice. The fecal DNA samples were quantified using a Qubit 4 Fluorometer (Thermo Fisher Scientific, USA).
2.3 Next-generation sequencing and bioinformatics
Library preparation and sequencing were outsourced to Macrogen, Inc. (Korea). Briefly, the 16S rRNA V3–V4 region (prokaryotic fraction) was amplified from the environmental DNA using 341F (5′-CCTACGGGNGGCWGCAG-3′) and 805R (5′-GACTACHVGGTATCTAATCC-3′) primers, targeting a 440-bp region (Herlemann et al., 2011). Each sample had a unique barcode, and samples were sequenced using Illumina MiSeq.
The QIIME 2 bioinformatics pipeline was used to analyze the dataset (Bolyen et al., 2019). Quality control, demultiplexing, contig assembly, and operational taxonomic unit (OTU) picking were performed. Representative OTUs were shortlisted, and their taxonomic identities were assigned using public databases (SILVA ver. 138). The resulting OTU table was used to calculate the community composition as well as the alpha and beta diversity indices. Taxon abundance was correlated with physico-chemical parameters to assess the abiotic–biotic relationship. Regression models were developed to determine significant variables affecting pathogen abundance. All statistical analyses were performed using R v.4.2.1 (R Core Team, 2023). The data used in this study have been deposited in the National Center for Biotechnology Information (NCBI) BioProject database with BioProject accession number PRJNA1153485.1
2.4 Bacterial diversity and pathogen identification
Metagenomic sequences from representative source samples (chicken, cow, dog, duck, goat, pig, and sewage) and sink samples (beach, offshore, harbor, and river) were subjected to principal component analysis based on their calculated beta diversity values using unweighted UniFrac. This ordination analysis demonstrated differentiation in bacterial community composition across the various sample types.
Feature tables with taxonomies assigned by Greengenes were used to identify the presence of potential human pathogens. The Functional Annotation of Prokaryotic Taxa or FAPROTAX database (Louca et al., 2016) is a curated collection of prokaryotic clades that have been identified to have metabolic or ecological functions. This software was used to convert the microbial community profiles from the sink samples into functional profiles. FAPROTAX was run using the default parameters, and potential human pathogens associated with septicemia, pneumonia, nosocomial infections, gastroenteritis, meningitis, diarrhea, and other diseases were identified from the samples. The number of reads associated with human pathogens was divided by the total number of reads from each sample to calculate the percent abundance of each functional group in each sample.
2.5 SourceTracker2 validation and assessment of contribution
Samples from source types (tributaries, untreated sewage, agricultural discharges) were analyzed using SourceTracker2 (Knights et al., 2011) with default parameters. The output from the program indicates the predicted contribution of each attributed source to a specific sink. To validate the output of SourceTracker2, three spiked samples were prepared using equal proportions of DNA from different source samples. The same software was used to compare the percent contribution estimated with the existing library. Three independent SourceTracker2 runs were performed, and the mean percent contribution was calculated.
3 Results
3.1 Bacterial community profiles using metabarcoding sequence analysis
Bacterial communities were similar across the same host sources, indicating shared microbial profiles (Figure 2A). The orders Clostridiales (cow: 57%; goat: 49%; pig: 47%) and Bacteriodales (cow: 30%; goat: 33%; pig: 23%) were the dominant groups among cow, goat, and pig samples. The order Lactobacillales was dominant in chicken (35%) and dog (23%) samples. The orders Bacteroidales (8%) and Actinomycetales (12%) were the dominant groups for duck samples. In contrast, the orders Flavobacteriales (Phylum Bacteroidetes) (bay: 10%; river: 8%), Alteromonadales (bay: 8%; river: 2%), and Oceanospirillales (Phylum Proteobacteria) (bay: 12%; river: 2%) dominated the bay and river samples (Figure 2B). Synechococcales (Phylum Cyanobacteria) (23%) and Flavobacteriales (22%) were dominant in the offshore samples. Fecal and water samples share common groups of bacteria, most notably Campylobacterales (bay: 23%; rivers: 25%). Other bacterial groups detected included human and animal pathogens, such as Clostridiales, Alteromonadales, Pseudomonadales, and Aeromonadales.
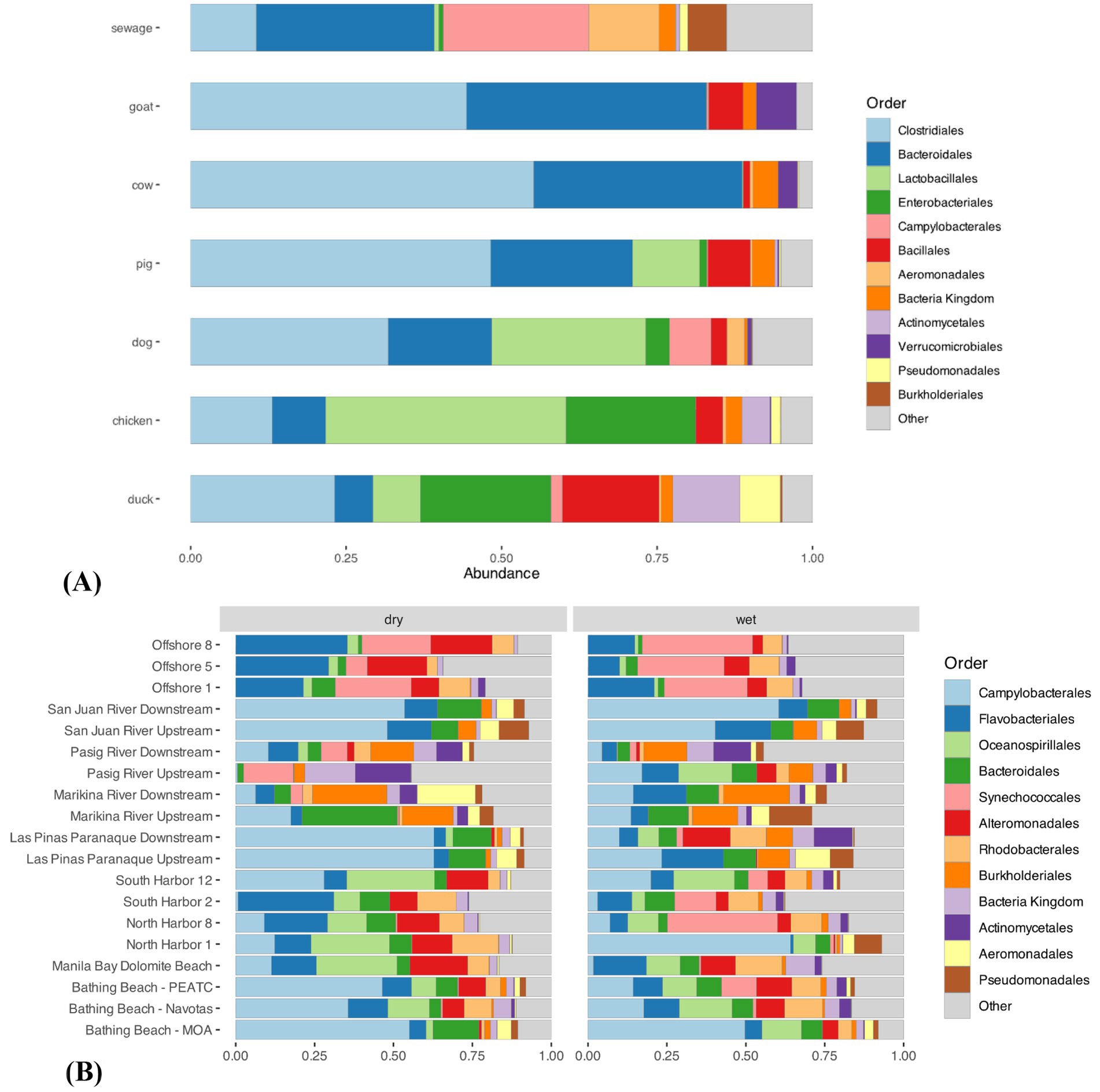
Figure 2. Average bacterial community profiles of (A) fecal and (B) water samples from different sites along Manila Bay.
3.2 Analysis of bacterial diversity
The first Principal Component (PC1 or Axis 1, Figure 3) accounted for 17% of the total community variation and mostly separated environmental samples from rivers, beaches, harbors, and offshore samples from the fecal sources. Samples mainly clustered according to sample type (fecal vs. environmental) and sample source (different animal hosts). Distinct microbial community signatures corresponded to the different fecal source types. The replicates of fecal source samples showed good agreement, demonstrated by their clustering using UPGMA based on Euclidean distances. Close clustering between goat and cow fecal samples suggests similar microbial community patterns. Furthermore, sewage samples clustered closer with sink samples than other sources, implying similarities in the microbial communities of sewage and sinks. This shows the potential application of source libraries for analyzing the sink samples.
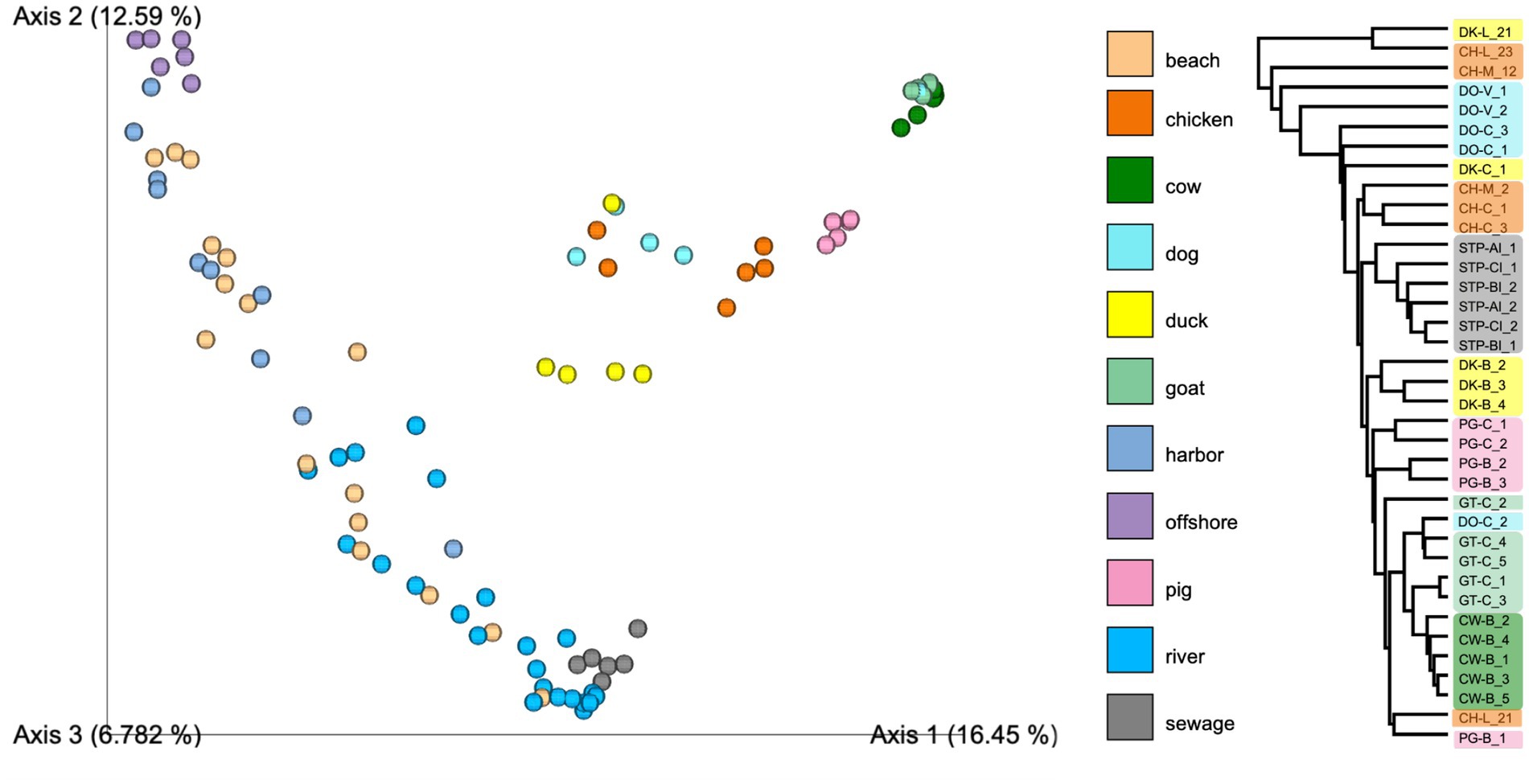
Figure 3. Bacterial diversity of fecal and water samples from different sites along Manila Bay using principal component analysis of unweighted Unifrac and UPGMA clustering using Euclidean distance.
3.3 Identification of pathogens
San Juan River showed the highest abundance of bacterial pathogens, followed by Las Piñas–Parañaque River, the upstream portion of Marikina River, and bathing beach sites (Figure 4). Specifically, septicemia-associated bacteria (Streptococcus, Campylobacter, and Providencia) dominantly contributed to the total abundance of pathogens observed. The same groups were also identified as potential causes of meningitis. This was followed by bacteria that can cause diarrhea and gastroenteritis, namely Clostridium spp., Vibrio, and Helicobacter. Lower abundances were observed for pathogens associated with nosocomial infections and pneumonia. Other potential human pathogens were also detected, notably Pseudomonas, Acinetobacter, Arcobacter, and Myroides spp.

Figure 4. Average percent of read abundance of human pathogenic taxa found in environmental samples.
3.4 Environmental parameters affecting the microbial communities
Overall, BOD, nitrate, phosphate, and TSS were higher in the dry season than in the wet season, whereas color, DO, fecal coliform, pH, and temperature were higher in the wet season (Table 3). A decreasing pattern of fecal coliform, phosphate, nitrate, TSS, and color levels was observed from tributaries to bathing beaches, harbors, and offshore stations during the wet season. Among the parameters, nitrate (p < 0.05) and pH (p < 0.05) were significantly different between the two seasons, indicating a high nitrate concentration in the dry season and alkalinity of the water samples during the wet season.

Table 3. Mean values of physico-chemical and microbiological parameters measured in sink samples.
Multiple linear correlations between physicochemical parameters and bacterial taxa showed a positive relationship between Campylobacterales, Enterobacteriales, Lactobacillales, and Clostridiales with phosphates and fecal coliform counts provided by the DENR, whereas the same taxa had a negative correlation with DO levels (Figure 5). This may indicate that the input of pollution in environmental waters, demonstrated by increased phosphate loading, may affect the abundance of these potentially pathogenic groups.
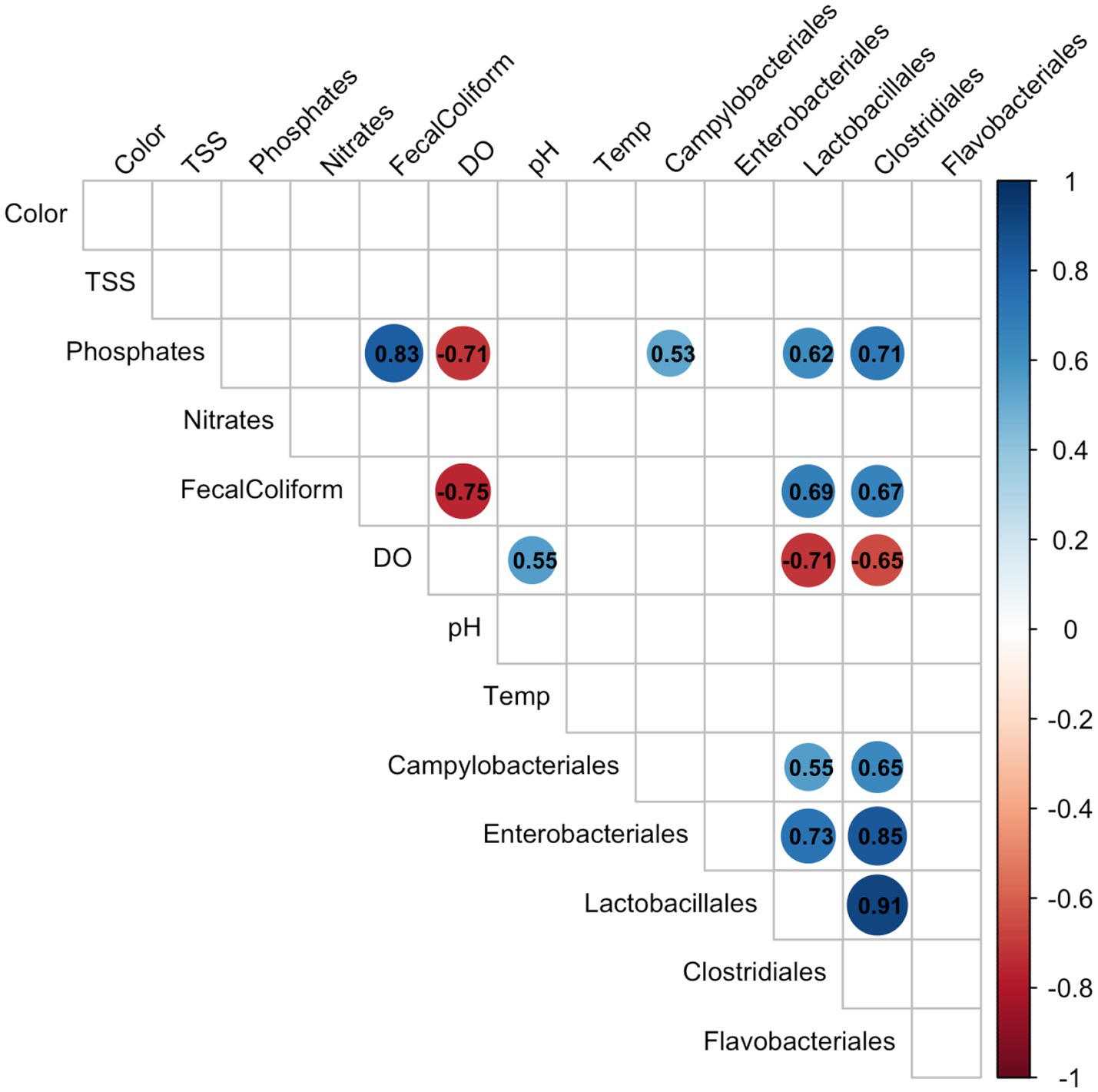
Figure 5. Spearman correlations between physicochemical parameters and top bacterial groups. Significant correlations (p < 0.05) are shown as colored circles with negative (red) or positive (blue) correlation. Insignificant correlations are not shown.
3.5 Source predictions using SourceTracker2
Spiked samples correctly predicted all source samples except for bovine sources, where low prediction was demonstrated (Table 4). The method was performed in triplicate, with the percent contribution estimated by SourceTracker2 compared to the known spiked DNA samples. Overall, SourceTracker2 exhibited good predictive accuracy for delineating the sources.
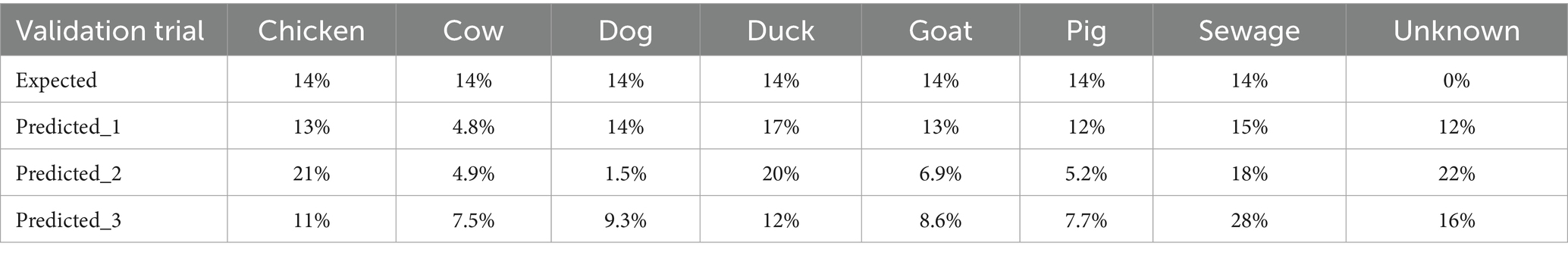
Table 4. Validation of SourceTracker2 predictive performance.
The contribution of each animal host to the microbial community found in water samples, including river, bay areas, and offshore sites, varied among sink types (Table 5). SourceTracker2 results revealed that sewage was the dominant known contributor, followed by avian species such as ducks (Figure 6). On average, sewage accounted for 93% of the known contamination, while ducks contributed 6%. However, a substantial proportion of unknown sources was also observed, ranging from 2 to 99% in offshore samples, with an average of 57%. These unknown contributions may indicate contamination from non-point sources or those not included in the study, such as human feces and other wildlife. A high percentage of unknown contributions may also indicate a low level of contamination from fecal sources, which was evident in the offshore samples.

Table 5. Average percentages (%) of SourceTracker2-predicted contributions of source samples to microbial communities.
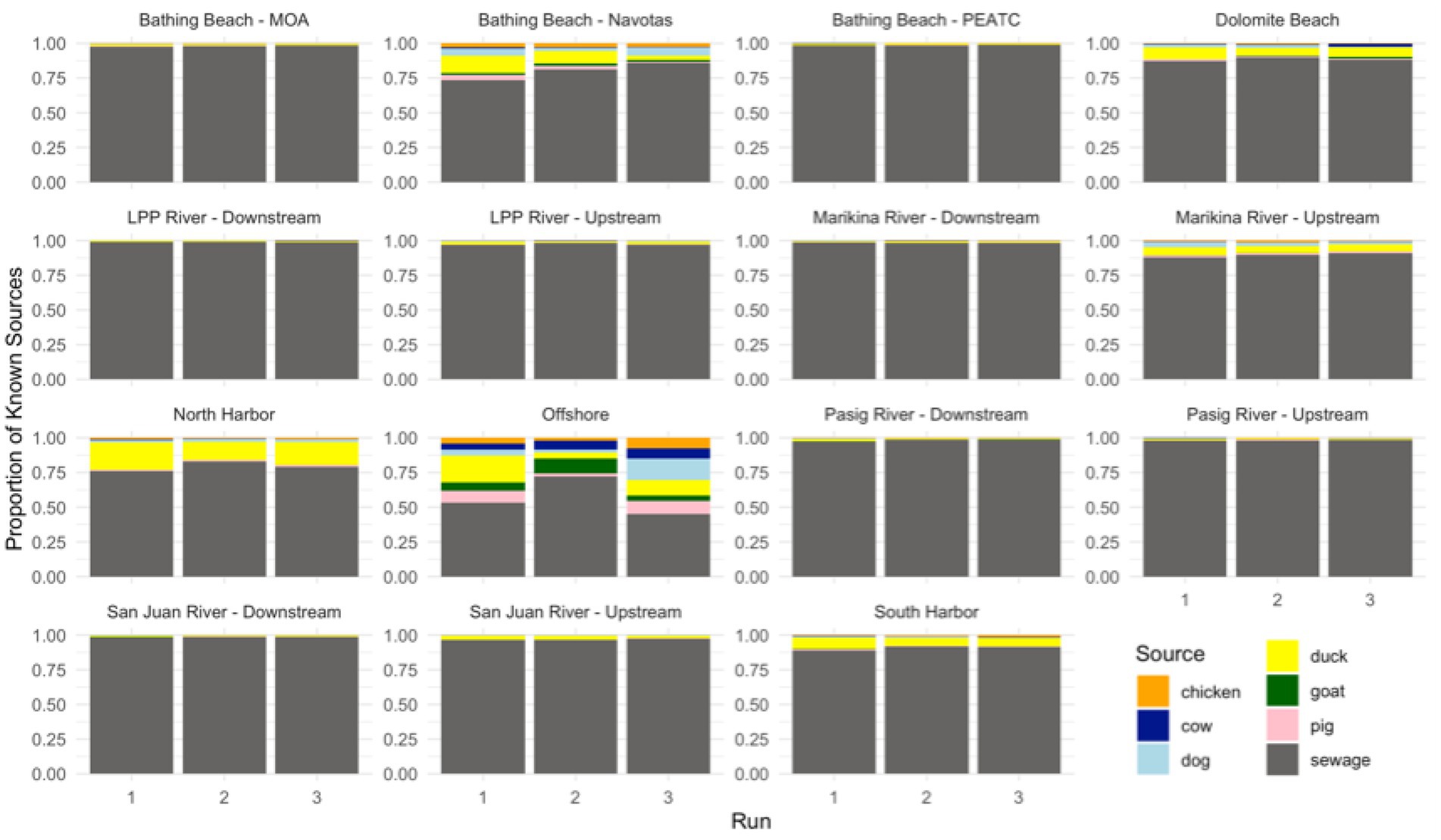
Figure 6. Preliminary source predictions of known fecal contamination in different sites along Manila Bay using SourceTracker2 (unknown sources not shown).
4 Discussion
Microbial source tracking is a valuable approach in identifying sources of fecal contamination in aquatic environments. Community-based MST can play a crucial role in informing strategies for the management of critical water bodies. However, the application of 16S rRNA gene sequencing in analyzing samples from Manila Bay and other major rivers in Metro Manila remains limited, leaving a gap in our knowledge of the source of fecal pollution in the bay. To address this, we applied microbial community analysis and showed that sewage is the major source of contamination in Manila Bay.
Analysis of the bacterial community profiles from the fecal samples showed a distinction between each source. Among the detected taxa, Bacteroidales was found to be the most dominant. Bacteroidales includes commensal bacteria that are commonly found in gut microbiota in warm-blooded animals, suggesting its dominance among the host sources (Kollarcikova et al., 2020). In contrast, the dominance of Lactobacillales in chicken fecal samples implies the common use of probiotics to improve production in chickens (Juricova et al., 2022). In addition, members of Lactobacillales reside as normal flora in the gastrointestinal tract of dogs, implying the abundance of the group in dog fecal samples (Kainulainen et al., 2015). For the sink samples, representatives under Bacteroidetes, Proteobacteria, and Cyanobacteria were the most abundant groups. Microorganisms under these taxa are known to be ubiquitously distributed in aquatic environments and thus are major communities in these ecosystems (Seo et al., 2017; Engloner et al., 2023). Various human and animal pathogens were observed across sample types. These include Clostridiales, Alteromonadales, Pseudomonadales, Aeromonadales, and Campylobacterales. Clostridiales and Aeromonadales consist of bacteria that cause diarrhea and other abdominal infections (Drancourt, 2010; Nagahama et al., 2019), while Alteromonadales and Pseudomonadales have been involved in bacteremia, sepsis, and other opportunistic infections (Berman, 2019). Moreover, Campylobacterales has been implicated in acute diarrhea and is present in the guts of several livestock, including poultry, cattle, and domestic animals, indicating its high abundance in several host sources (Shen et al., 2024). Cattle and goats also showed close composition clustering. The microbial community composition between cattle and goat guts depends heavily on their diet. These hosts are both ruminants and mostly consume roughage, thereby resulting in higher similarities in their gut microbiome composition compared to other host sources (Henderson et al., 2015). Overall, the distinction of microbial communities between sinks and sources is consistent with other studies (Henry et al., 2016; Rothenheber and Jones, 2018; Pantha et al., 2021), showing the potential use of source libraries for comparison against the sink samples.
Human pathogens were detected in most sites along Manila Bay, except for Navotas Bathing Beach, Dolomite Beach, and offshore sites. Notably, human pathogens were most abundant in rivers that are more exposed to anthropogenic activities. Specifically, the San Juan River, followed by the Las Piñas-Parañaque River, and upstream of Marikina River all showed higher levels of bacterial pathogens. The presence of these potential pathogens poses a threat to public safety, especially as they were found in sites with human-water interface, such as in bathing beaches and rivers. The high levels of pathogens, especially in river samples, are linked to various sources of contamination such as untreated sewage, agricultural runoff, and nutrient pollution. Similarly, Cui et al. (2019) found several enteric pathogens, including Acinetobacter, in river samples in China, suggesting that domestic sewage is a major contributor to contamination. The same study also reported that environmental pathogens like Pseudomonas aeruginosa had more variable distributions, with some being more prevalent in nutrient-rich environments. In upstream sections of rivers, contaminants tend to be more concentrated and are more diluted downstream, which can result in higher levels of enteric and environmental pathogens in certain areas. However, we found that the downstream sections of the rivers generally exhibited the highest pathogen concentrations. The proximity of these rivers and the MOA and PEATC bathing beaches to residential and industrial zones likely contributed to the abundance of pathogens observed at these sites, a trend that has also been reported in studies examining similar urban-influenced environments (Abraham, 2011; Pandey et al., 2014).
The specific human pathogens detected are mostly septicemia-associated bacteria, including Streptococcus agalactiae, Campylobacter fetus, and Providencia stuartii. These bacteria can also cause meningitis. This was followed by bacteria that can cause diarrhea and gastroenteritis, namely Clostridium difficile, Clostridium spiriforme, Vibrio mimicus, and Helicobacter pullorum. Other potential human pathogens were detected, notably Pseudomonas, Acinetobacter, Arcobacter, and Myroides spp. Typically, these bacterial genera can be found in water systems near urbanized areas such as sewage-impacted rivers, estuaries, and lakes, especially those belonging to Pseudomonas, Acinetobacter, Arcobacter, Campylobacter, and Clostridium (Jin et al., 2018; Yang et al., 2020; Numberger et al., 2022). Nosocomial bacteria were also detected, although in lower abundance. This might be due to wastewater discharges from hospitals and other clinical settings, resulting in the transmission of pathogens into bays and rivers (Cabral, 2010).
Overall, the pathogenic bacterial taxa detected across the sites in Manila Bay are also commonly found in urban sources, such as treated wastewater effluent, stormwater runoff, and combined sewer overflow (McLellan et al., 2015; Jin et al., 2018). This suggests that urban sources, such as sewage, impact the sampling sites. Urbanization is associated with population growth and land use, which alter the natural microbial community and thereby promote the proliferation of pathogens in water bodies (Numberger et al., 2022).
Comparisons between dry and wet seasons across various Manila Bay sites revealed that only pH and nitrate were significantly different. The presence of alkaline waters during the wet season might be due to the untreated bicarbonate-rich wastewater discharges from various industries surrounding the bay. An example of such discharge is livestock wastewater, which can be a significant source of ammonium and bicarbonate ions, resulting in an increased pH in the water bodies (Han et al., 2022). The high nitrate concentration during the dry season suggests nitrate infiltration through the soil, which originates from domestic and industrial waste sources and enters nearby aquatic systems (Alsabti et al., 2023). During the wet season, a decreasing trend in fecal coliform, phosphate, nitrate, TSS, and color levels was observed from tributaries to bathing beaches, harbors, and offshore stations. This pattern suggests the dilution effects of rainfall runoff from upstream to downstream areas, resulting in lesser chemical and microbiological loads in downstream parts (Huang et al., 2020). Fecal coliform levels and microbial abundance were generally higher during the wet season due to increased runoff, nutrient enrichment, and sediment resuspension, all of which contribute to microbial loading in surface waters (Hong et al., 2010). In contrast, lower fecal coliform counts and microbial abundance during the dry season are likely due to limited surface runoff, reduced nutrient inputs, and higher doses of solar radiation, which together restrict the transport, survival, and growth of fecal-associated microbes in aquatic systems (Hughes, 2003). Moreover, the higher nutrient concentrations in the wet season were consistent with previous reports of high nitrate and phosphate levels in the rivers and Manila Bay (Sotto et al., 2015). A portion of these loads might originate from human excreta, sewer leakage, settlement of particles, and other anthropogenic-related activities. These nutrient loads contribute to eutrophication, which drives the observed drop in DO levels and creates hypoxic conditions that support the growth of certain bacterial groups (Rabalais and Turrner, 2001). In terms of microbial community dynamics, microbial groups like Flavobacteriales, Campylobacterales, Oceanospirillales, Alteromonadales, and Rhodobacterales were more abundant in the dry season, likely due to the stable, low-turbidity conditions, higher salinity, and reduced freshwater input. These groups thrive in nutrient-rich, less disturbed environments with higher salinity and lower oxygen levels (Ng and Chiu, 2020). In contrast, Synechococcales, a group of photosynthetic cyanobacteria, were more prominent in the wet season. This is likely driven by increased nutrient runoff, higher water turbidity, and sunlight availability, which supports their growth (Li et al., 2024).
A positive correlation of top bacterial groups (Campylobacterales, Enterobacteriales, Lactobacillales, and Clostridiales) with phosphates and fecal coliform counts and a negative correlation with DO suggests that the abundance of these potential pathogens is linked to fecal contamination. Douterelo et al. (2020) revealed that increased phosphate loads resulted in an increased abundance of various biofilm pathogenic groups, including Pseudomonas and Acinetobacter, because of the ability of these microorganisms to utilize phosphates. The direct relationship between fecal coliform count and certain bacterial groups, like coliforms, is expected because these microorganisms are commonly present in the intestines of humans and animals, leading to their excretion in feces (Patel et al., 2014; Some et al., 2021). Moreover, the decomposition of detritus by several bacterial species contributed to the decrease in DO of the water bodies. Energy transfer from aquatic fauna to microorganisms due to decomposition may have also contributed to the higher abundance of top pathogenic groups (Spietz et al., 2015).
SourceTracker2 analysis revealed that sewage is the dominant contaminant of Manila Bay. Sewage accounted for the 93% average known contribution and was predominant in the rivers (San Juan, Las Piñas-Parañaque, Marikina, and Pasig) and two bathing beaches sites (MOA and PEATC). These sites also had the most abundant presence of pathogens, which can be attributed to various anthropogenic activities in mostly urbanized areas. Similar findings were also observed in several source-tracking studies on urban rivers and beaches (Dickerson et al., 2007; Sidhu et al., 2013; Henry et al., 2016; Derx et al., 2023).
During periods of intense rainfall, the flow rate of the wastewater tends to increase, causing it to overspill and carry microbial loads, especially pathogens, into the environmental waters (McMahan, 2006). This can be linked to the heightened volume of water exceeding the capacity of sewage treatment plants, leading to the release of untreated wastewater. Similarly, the duck contribution in the sink samples indicates agricultural runoff from duck farm fields flowing into the ground and entering the water bodies (Baudišová, 2009; Jung et al., 2014). Agricultural runoff is a major contributor to microbial contamination in water bodies. This happens when rainwater transports pollutants from farmland into adjacent environmental waters (Devane et al., 2018). Thus, the detection of duck feces in sink samples suggests that runoff from these farms is likely adding to the microbial contamination. The analysis shows that ducks contribute approximately 6% of the total known source contribution, with smaller percentages from cows, pigs, and goats. Ducks may be the largest contributor among other animal sources because of factors such as higher farm density, their tendency to frequent water bodies, and the direct runoff from duck farms. The presence of pathogens in the water bodies poses a significant health risk to both humans and animals. Consequently, it is essential to adopt effective strategies to prevent and minimize microbial contamination, especially in urban waterways.
In our study, we considered the geographic proximity between sources and sink sites, with particular focus on farms located near Metro Manila. We sampled farms from multiple locations to ensure a broader representation of microbial community diversity within our source library. This combination of proximity and diversity increases the likelihood of observing microbial exchange, thus enhancing the potential to detect overlapping microbial taxa between the environment and the fecal source library. While we recognize that genus-level similarities do not necessarily imply a direct source-sink connection, the integration of spatial, environmental, and microbial evidence lends support to the hypothesis of microbial transfer from sources to sinks. In cases where the model cannot confidently assign a source category for the sink, SourceTracker2 includes an “unknown” category. This feature addresses the limitation in the source library, such as incomplete representation and missing sources, and recognizes that the sink microbiome may originate from uncharacterized environments. Samples from bathing beaches, harbors, and offshore stations showed a higher proportion of unknown contribution (Table 5), likely due to the ecological differences between freshwater and marine environments. This suggests that the metagenomic library is more representative of freshwater pollution sources, limiting its accuracy for marine pollution sources. Unidentified contributions suggest contamination from non-point sources not accounted for in the study, such as human feces and other wildlife. For example, droppings of migratory birds and other wildlife increases fecal coliform pollution in Metro Manila waterways (Raña et al., 2017). Several studies have also detected human fecal contamination in Pasig River, Laguna Lake, and other tributaries using other microbial source tracking methods, which might be due to direct fecal discharge into these water bodies (Abello et al., 2021; Nacario et al., 2022). To address the limitation of the fecal host source library, future work can incorporate the detection of human-associated markers, such as crAssphage, pBI143, and Bacteroides sequences, to confirm human fecal contamination more accurately (Malajacan et al., 2023). Another limitation of the library is the possible underestimation of bovine sources, as demonstrated in the validation test (Table 4). Several factors may contribute to this, including the high similarity of bovine gut microbiota to other ruminants, such as goats (Figure 3) or other environmental sources, which can result in reduced taxonomic resolution. Another possible factor is the dominance of shared bacterial taxa, which can make bovine contributions harder to distinguish through signature-based source-tracking (Hägglund et al., 2018). Despite these limitations in the metagenomic library, this method remains useful for classifying pollution sources in Metro Manila waterways draining into Manila Bay. It provides valuable insight into the dominant contributors and spatial patterns of pollution, supporting targeted management and mitigation efforts.
5 Conclusion
SourceTracker2 analysis revealed that sewage is the primary source of microbial contamination in Manila Bay, with water samples showing strong similarities to sewage-derived microorganisms. Distinct differences in microbial communities between source and sink samples highlight the effectiveness of using microbial libraries for source tracking. Several pathogenic bacteria linked to diseases such as septicemia, meningitis, gastroenteritis, and diarrhea were detected, particularly in rivers and bathing beaches, which are areas frequently used by the public. Additionally, strong correlations between bacterial groups and water quality indicators like phosphate, fecal coliform, and DO underscore the role of environmental conditions in shaping microbial communities. These findings reinforce the need for improved wastewater management and pollution control in Manila Bay to protect water quality and public health.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: NCBI, accession PRJNA1153485.
Ethics statement
The manuscript presents research on animals that do not require ethical approval for their study.
Author contributions
LP: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Software, Validation, Visualization, Writing – original draft, Writing – review & editing. DM: Data curation, Formal analysis, Investigation, Methodology, Validation, Visualization, Writing – original draft, Writing – review & editing. MN: Data curation, Formal analysis, Investigation, Methodology, Validation, Visualization, Writing – original draft, Writing – review & editing. MV: Data curation, Formal analysis, Investigation, Methodology, Validation, Visualization, Writing – original draft, Writing – review & editing. WR: Conceptualization, Funding acquisition, Project administration, Resources, Software, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. Financial support was received from the National Research Council of the Philippines (NRCP; Project No. E-249 “Manila Bay Monitoring Using Metagenomics for Pathogen Detection and Source-Tracking”) of the Department of Science and Technology.
Acknowledgments
We thank the Environmental Management Bureau and the Pasig River Coordination and Monitoring Office of the Department of Environment and Natural Resources for their valuable contributions and fruitful collaboration during the collection of water samples. Moreover, we extend this appreciation to farm owners who granted our request to collect fecal samples from their farm animals.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
References
Abello, J. J. M., Malajacan, G. T., Labrador, K. L., Nacario, M. A. G., Galarion, L. H., Obusan, M. C. M., et al. (2021). Library-independent source tracking of fecal contamination in selected stations and tributaries of Laguna Lake, Philippines. J. Water Health 19, 846–854. doi: 10.2166/wh.2021.058
Abraham, W. R. (2011). Megacities as sources for pathogenic bacteria in rivers and their fate downstream. Int. J. Microbiol. 2011:798292. doi: 10.1155/2011/798292
Alsabti, B., Sabarathinam, C., and Svv, D. R. (2023). Identification of high nitrate concentration in shallow groundwater of an arid region: a case study of South Kuwait’s bay. Environ. Monit. Assess. 195:143. doi: 10.1007/s10661-022-10698-1
Aylagas, E., Borja, Á., Tangherlini, M., Dell’Anno, A., Corinaldesi, C., Michell, C. T., et al. (2017). A bacterial community-based index to assess the ecological status of estuarine and coastal environments. Mar. Pollut. Bull. 114, 679–688. doi: 10.1016/j.marpolbul.2016.10.050
Baral, D., Speicher, A., Dvorak, B., Admiraal, D., and Li, X. (2018). Quantifying the relative contributions of environmental sources to the microbial community in an urban stream under dry and wet weather conditions. Appl. Environ. Microbiol. 84, e00896–e00818. doi: 10.1128/AEM.00896-18
Baudišová, D. (2009). Microbial pollution of water from agriculture. Plant Soil Environ. 55, 429–435. doi: 10.17221/131/2009-PSE
Berman, J. J. (2019). “Bacteria” in Taxonomic guide to infectious diseases: understanding the biologic classes of pathogenic organisms (USA: Academic Press), 39–119.
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C. C., Al-Ghalith, G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. doi: 10.1038/s41587-019-0209-9
Cabral, J. P. S. (2010). Water microbiology. Bacterial pathogens and water. Int. J. Environ. Res. Public Health 7, 3657–3703. doi: 10.3390/ijerph7103657
Cui, Q., Huang, Y., Wang, H., and Fang, T. (2019). Diversity and abundance of bacterial pathogens in urban rivers impacted by domestic sewage. Environ. Pollut. 249, 24–35. doi: 10.1016/j.envpol.2019.02.094
Department of Environment and Natural Resources–Environmental Management Bureau (2016) EMB Approved Methods of Analysis for Water and Wastewater. Available online at: https://emb.gov.ph/wp-content/uploads/2019/04/EMB-MC_2016-012_APPROVED-METHODS-OF-ANALYSIS-FOR-WATER-AND-WASTEWATER.pdf
Derx, J., Kılıç, H. S., Linke, R., Cervero-Aragó, S., Frick, C., Schijven, J., et al. (2023). Probabilistic fecal pollution source profiling and microbial source tracking for an urban river catchment. Sci. Total Environ. 857:159533. doi: 10.1016/j.scitotenv.2022.159533
Devane, M. L., Weaver, L., Singh, S. K., and Gilpin, B. J. (2018). Fecal source tracking methods to elucidate critical sources of pathogens and contaminant microbial transport through New Zealand agricultural watersheds – a review. J. Environ. Manag. 222, 293–303. doi: 10.1016/j.jenvman.2018.05.033
Dickerson, J. W. Jr., Hagedorn, C., and Hassall, A. (2007). Detection and remediation of human-origin pollution at two public beaches in Virginia using multiple source tracking methods. Water Res. 41, 3758–3770. doi: 10.1016/j.watres.2007.02.055
Douterelo, I., Dutilh, B. E., Calero, C., Rosales, E., Martin, K., and Husband, S. (2020). Impact of phosphate dosing on the microbial ecology of drinking water distribution systems: fieldwork studies in chlorinated networks. Water Res. 187:116416. doi: 10.1016/j.watres.2020.116416
Drancourt, M. (2010). Acute Diarrhea. Infect. Dis., 381–388. doi: 10.1016/B978-0-323-04579-7.00035-6
Engloner, A. I., Vargha, M., Kós, P., and Borsodi, A. K. (2023). Planktonic and epilithic Prokaryota community compositions in a large temperate river reflect climate change related seasonal shifts. PLoS One 18:e0292057. doi: 10.1371/journal.pone.0292057
García-Aljaro, C., Blanch, A. R., Campos, C., Jofre, J., and Lucena, F. (2019). Pathogens, faecal indicators and human-specific microbial source-tracking markers in sewage. J. Appl. Microbiol. 126, 701–717. doi: 10.1111/jam.14112
Hägglund, M., Bäckman, S., Macellaro, A., Lindgren, P., Borgmästars, E., Jacobsson, K., et al. (2018). Accounting for bacterial overlap between raw water communities and contaminating sources improves the accuracy of signature-based microbial source tracking. Front. Microbiol. 9:2364. doi: 10.3389/fmicb.2018.02364
Han, S., Jeon, S., Lee, J., Ahn, J., Lee, C., Lee, J., et al. (2022). Efficient bicarbonate removal and recovery of ammonium bicarbonate as CO2 utilization using flow-electrode capacitive deionization. Chem. Eng. J. 431:134233. doi: 10.1016/j.cej.2021.134233
Henderson, G., Cox, F., Ganesh, S., Jonker, A., and Young, W. Global Rumen Census Collaborators, et al. (2015). Rumen microbial community composition varies with diet and host, but a core microbiome is found across a wide geographical range. Sci. Rep. 5:14567. doi: 10.1038/srep14567
Henry, R., Schang, C., Coutts, S., Kolotelo, P., Prosser, T., Crosbie, N., et al. (2016). Into the deep: evaluation of SourceTracker for assessment of faecal contamination of coastal waters. Water Res. 93, 242–253. doi: 10.1016/j.watres.2016.02.029
Herlemann, D. P. R., Labrenz, M., Jürgens, K., Bertilsson, S., Waniek, J. J., and Andersson, A. F. (2011). Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J. 5 1571–1579. doi: 10.1038/ismej.2011.41
Hong, H., Qiu, J., and Liang, Y. (2010). Environmental factors influencing the distribution of total and fecal coliform bacteria in six water storage reservoirs in the Pearl River Delta region, China. J. Environ. Sci. 22, 663–668. doi: 10.1016/S1001-0742(09)60160-1
Huang, G. Z., Hsu, T. C., Yu, C. K., Huang, J. C., and Lin, T. C. (2020). Dilution and precipitation dominated regulation of stream water chemistry of a volcanic watershed. J. Hydrol. 583:124564. doi: 10.1016/j.jhydrol.2020.124564
Hughes, K. A. (2003). Influence of seasonal environmental variables on the distribution of presumptive fecal coliforms around an Antarctic research station. Appl. Environ. Microbiol. 69. doi: 10.1128/AEM.69.8.4884-4891.2003
Jacinto, G. S., Azanza, R. V., Velasquez, I. B., and Siringan, F. P. (2006). “Manila Bay: environmental challenges and opportunities” in The environment in Asia Pacific Harbours. ed. E. Wolanski (Dordrecht, NL: Springer), 309–328.
Jin, D., Kong, X., Cui, B., Jin, S., Xie, Y., Wang, X., et al. (2018). Bacterial communities and potential waterborne pathogens within the typical urban surface waters. Sci. Rep. 8:13368. doi: 10.1038/s41598-018-31706-w
Jung, A. V., Le Cann, P., Roig, B., Thomas, O., Baurès, E., and Thomas, M. F. (2014). Microbial contamination detection in water resources: interest of current optical methods, trends and needs in the context of climate change. Int. J. Environ. Res. Public Health 11, 4292–4310. doi: 10.3390/ijerph110404292
Juricova, H., Matiasovicova, J., Faldynova, M., Sebkova, A., Kubasova, T., Prikrylova, H., et al. (2022). Probiotic lactobacilli do not protect chickens against Salmonella enteritidis infection by competitive exclusion in the intestinal tract but in feed, outside the chicken host. Microorganisms 10:219. doi: 10.3390/microorganisms10020219
Kainulainen, V., Tang, Y., Spillmann, T., Kilpinen, S., Reunanen, J., Saris, P. E. J., et al. (2015). The canine isolate Lactobacillus acidophilus LAB20 adheres to intestinal epithelium and attenuates LPS-induced IL-8 secretion of enterocytes in vitro. BMC Microbiol. 15:4. doi: 10.1186/s12866-014-0337-9
Kirs, M., Kisand, V., Wong, M., Caffaro-Filho, R. A., Moravcik, P., Harwood, V. J., et al. (2017). Multiple lines of evidence to identify sewage as the cause of water quality impairment in an urbanized tropical watershed. Water Res. 116, 23–33. doi: 10.1016/j.watres.2017.03.024
Knights, D., Kuczynski, J., Charlson, E. S., Zaneveld, J., Mozer, M. C., Collman, R. G., et al. (2011). Bayesian community-wide culture-independent microbial source tracking. Nat. Methods 8, 761–763. doi: 10.1038/nmeth.1650
Kollarcikova, M., Faldynova, M., Matiasovicova, J., Jahodarova, E., Kubasova, T., Seidlerova, Z., et al. (2020). Different Bacteroides species colonise human and chicken intestinal tract. Microorganisms 8:1483. doi: 10.3390/microorganisms8101483
Li, H., Jiang, M., Li, P., Xu, Z., Jiang, P., and Chen, L. (2024). Picocyanobacterial-bacterial interactions sustain cyanobacterial blooms in nutrient-limited aquatic environments. Envi. Res. 260:119508. doi: 10.1016/j.envres.2024.119508
Louca, S., Parfrey, L. W., and Doebeli, M. (2016). Decoupling function and taxonomy in the global ocean microbiome. Science 353, 1272–1277. doi: 10.1126/science.aaf4507
Malajacan, G. T., Nacario, M. A. G., Obusan, M. C. M., and Rivera, W. L. (2023). Host-associated Bacteroides 16S rDNA-based markers for source tracking of fecal pollution in Laguna Lake, Philippines. Microorganisms 11:1142. doi: 10.3390/microorganisms11051142
Manila Bay Office (2015). Water Quality Monitoring. Available online at: https://mbo.emb.gov.ph (Accessed July 15, 2021).
McLellan, S. L., Fisher, J. C., and Newton, R. J. (2015). The microbiome of urban waters. Int. Microbiol. 18, 141–149. doi: 10.2436/20.1501.01.244
McMahan, E. (2006). Impacts of rainfall events on wastewater treatment processes. [Dissertation/Master’s Thesis] [Tampa (FL)]: University of South Florida. Available online at: https://digitalcommons.usf.edu/etd/3846
Nacario, M. A. G., dela Pena, L. B. R. O., Labrador, K. L., and Rivera, W. L. (2022). DNA fingerprinting using BOX-A1R and (GTG)5 primers identify spatial variations of fecal contamination along Pasig River, Philippines. Environ. Monit. Assess. 194:868. doi: 10.1007/s10661-022-10504-y
Nagahama, M., Takehara, M., and Rood, J. I. (2019). Histotoxic clostridial infections. Microbiol. Spectr. 7. doi: 10.1128/microbiolspec.GPP3-0024-2018
Nakatsu, C. H., Byappanahalli, M. N., and Nevers, M. B. (2019). Bacterial community 16S rRNA gene sequencing characterizes riverine microbial impact on Lake Michigan. Front. Microbiol. 10:996. doi: 10.3389/fmicb.2019.00996
Neave, M., Luter, H., Padovan, A., Townsend, S., Schobben, X., and Gibb, K. (2014). Multiple approaches to microbial source tracking in tropical northern Australia. Microbiologyopen 3, 860–874. doi: 10.1002/mbo3.209
Ng, J. C., and Chiu, J. M. (2020). Changes in biofilm bacterial communities in response to combined effects of hypoxia, ocean acidification and nutrients from aquaculture activity in three fathoms cove. Mar. Pollut. Bull. 156:111256. doi: 10.1016/j.marpolbul.2020.111256
Numberger, D., Zoccarato, L., Woodhouse, J., Ganzert, L., Sauer, S., Márquez, J. R. G., et al. (2022). Urbanization promotes specific bacteria in freshwater microbiomes including potential pathogens. Sci. Total Environ. 845:157321. doi: 10.1016/j.scitotenv.2022.157321
Pandey, P. K., Kass, P. H., Soupir, M. L., Biswas, S., and Singh, V. P. (2014). Contamination of water resources by pathogenic bacteria. AMB Express 4:51. doi: 10.1186/s13568-014-0051-x
Pantha, K., Acharya, K., Mohapatra, S., Khanal, S., Amatya, N., Ospina-Betancourth, C., et al. (2021). Faecal pollution source tracking in the holy Bagmati River by portable 16S rRNA gene sequencing. NPJ Clean Water 4:12. doi: 10.1038/s41545-021-00099-1
Patel, A. K., Singhania, R. R., Pandey, A., Joshi, V. K., Nigam, P. S., and Soccol, C. R. (2014). “Enterobacteriaceae, coliforms and E. coli: introduction” in Encyclopedia of food microbiology (United Kingdom: Elsevier), 659–666.
Pérez-Cobas, A. E., Gomez-Valero, L., and Buchrieser, C. (2020). Metagenomic approaches in microbial ecology: an update on whole-genome and marker gene sequencing analyses. Microb. Genom. 6:mgen000409. doi: 10.1099/mgen.0.000409
Philippine News Agency. (2022). DENR cites significant improvement in Manila Bay water quality. Available online at: https://www.pna.gov.ph/articles/1179088 (Accessed April 11, 2025).
QGIS Development Team. (2024). QGIS geographic information system (open-source geospatial foundation project). Available online at: http://qgis.osgeo.org
R Core Team (2023). R: A language and environment for statistical computing (R Foundation for Statistical Computing, Vienna, Austria). Available online at: https://www.R-project.org/
Rabalais, N., and Turrner, R. (2001). Coastal hypoxia: consequences for living resources and ecosystems. Washington, DC: American Geophysical Union.
Raña, J. A., Domingo, J. E., Opinion, A. G. R., and Cambia, F. D. (2017). Contamination of coliform bacteria in water and fishery resources in Manila Bay aquaculture farms. Philipp. J. Fish. 24, 98–126. doi: 10.31398/tpjf/24.2.2016A0015
Rothenheber, D., and Jones, S. (2018). Enterococcal concentrations in a coastal ecosystem are a function of fecal source input, environmental conditions, and environmental sources. Appl. Environ. Microbiol. 84, e01038–e01018. doi: 10.1128/AEM.01038-18
Scott, T. M., Rose, J. B., Jenkins, T. M., Farrah, S. R., and Lukasik, J. (2002). Microbial source tracking: current methodology and future directions. Appl. Environ. Microbiol. 68, 5796–5803. doi: 10.1128/AEM.68.12.5796-5803.2002
Seo, J. H., Kang, I., Yang, S. J., and Cho, J. C. (2017). Characterization of spatial distribution of the bacterial community in the South Sea of Korea. PLoS One 12:e0174159. doi: 10.1371/journal.pone.0174159
Shen, Z., Wang, Y., and Shen, J. (2024). “Campylobacter” in Molecular Medical Microbiology (United States: Academic Press), 1097–1132.
Sidhu, J. P. S., Ahmed, W., Gernjak, W., Aryal, R., McCarthy, D., Palmer, A., et al. (2013). Sewage pollution in urban stormwater runoff as evident from the widespread presence of multiple microbial and chemical source tracking markers. Sci. Total Environ. 463–464, 488–496. doi: 10.1016/j.scitotenv.2013.06.020
Some, S., Mondal, R., Mitra, D., Jain, D., Verma, D., and Das, S. (2021). Microbial pollution of water with special reference to coliform bacteria and their nexus with environment. Energy Nexus 1:100008. doi: 10.1016/j.nexus.2021.100008
Sotto, L. P. A., Beusen, A. H. W., Villanoy, C. L., Bouwman, L. F., and Jacinto, G. S. (2015). Nutrient load estimates for Manila Bay, Philippines using population data. Ocean Sci. J. 50, 467–474. doi: 10.1007/s12601-015-0042-0
Spietz, R. L., Williams, C. M., Rocap, G., and Horner-Devine, M. C. (2015). A dissolved oxygen threshold for shifts in bacterial community structure in a seasonally hypoxic estuary. PLoS One 10:e0135731. doi: 10.1371/journal.pone.0135731
Staley, C., Kaiser, T., Lobos, A., Ahmed, W., Harwood, V. J., Brown, C. M., et al. (2018). Application of SourceTracker for accurate identification of fecal pollution in recreational freshwater: a double-blinded study. Environ. Sci. Technol. 52, 4207–4217. doi: 10.1021/acs.est.7b05401
Tan, B., Ng, C. M., Nshimyimana, J. P., Loh, L. L., Gin, K. Y. H., and Thompson, J. R. (2015). Next-generation sequencing (NGS) for assessment of microbial water quality: current progress, challenges, and future opportunities. Front. Microbiol. 6:1027. doi: 10.3389/fmicb.2015.01027
Unno, T., Staley, C., Brown, C. M., Han, D., Sadowsky, M. J., and Hur, H. G. (2018). Fecal pollution: new trends and challenges in microbial source tracking using next-generation sequencing. Environ. Microbiol. 20, 3132–3140. doi: 10.1111/1462-2920.14281
Vallejo, B. M., Aloy, A. B., Ocampo, M., Conejar-Espedido, J., and Manubag, L. M. (2019). “Manila Bay ecology and associated invasive species” in Impacts of invasive species on coastal environments. eds. C. Makowski and C. W. Finkl (Cham: Springer), 145–169.
Keywords: Manila Bay, metagenomics, pathogen, 16S rDNA, SourceTracker2
Citation: dela Peña LBRO, Mamawal DRD, Nacario MAG, Vejano MRA and Rivera WL (2025) Application of 16S rDNA metagenomic library for source-tracking of fecal pollution in selected stations and tributaries of Manila Bay, Philippines. Front. Water. 7:1589330. doi: 10.3389/frwa.2025.1589330
Edited by:
Pamela Monaco, University of Molise, ItalyReviewed by:
Michael G. LaMontagne, University of Houston–Clear Lake, United StatesAtif Khurshid Wani, Lovely Professional University, India
Hai Pham, VNU University of Science, Vietnam
Copyright © 2025 dela Peña, Mamawal, Nacario, Vejano and Rivera. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Windell L. Rivera, d2xyaXZlcmFAc2NpZW5jZS51cGQuZWR1LnBo