 Michael J. Greenberg
Michael J. Greenberg Neil J. Daily
Neil J. Daily Ann Wang2
Ann Wang2 Tetsuro Wakatsuki
Tetsuro Wakatsuki- 1Department of Biochemistry and Molecular Biophysics, Washington University School of Medicine, St. Louis, MO, United States
- 2InvivoSciences Inc., Madison, WI, United States
Heart failure is the leading cause of death in the western world and as such, there is a great need for new therapies. Heart failure has a variable presentation in patients and a complex etiology; however, it is fundamentally a condition that affects the mechanics of cardiac contraction, preventing the heart from generating sufficient cardiac output under normal operating pressures. One of the major issues hindering the development of new therapies has been difficulties in developing appropriate in vitro model systems of human heart failure that recapitulate the essential changes in cardiac mechanics seen in the disease. Recent advances in stem cell technologies, genetic engineering, and tissue engineering have the potential to revolutionize our ability to model and study heart failure in vitro. Here, we review how these technologies are being applied to develop personalized models of heart failure and discover novel therapeutics.
Heart failure (HF) is the leading cause of death in the United States, accounting for 1 in 9 deaths that occur each year and over $30 billion in annual health care costs (1). Chronic HF affects ~2% of the population <60 years old and >10% of adults >75 years old (2). HF is characterized by the inability of the heart to generate sufficient cardiac output to effectively pump blood to the body under normal physiological pressures. Clinically, HF patients are classified by their ejection fraction (i.e., the fraction of blood that is pumped out of the ventricles with each beat). HF with reduced ejection fraction (HFrEF) can be caused by several conditions, such as valvular disease, myocardial infarction, and some genetic cardiomyopathies (described in more detail below). Several treatment options are available for HFrEF, including ACE inhibitors, beta blockers, and implantable devices (3); however, many of these therapeutic options have significant side effects, including tachycardia and arrhythmia (4). HF with preserved ejection fraction (HFpEF) is characterized by diastolic dysfunction, such as impaired filling due to fibrotic stiffening of the ventricular wall, but a normal ejection fraction. HFpEF can be caused by several conditions including chronic hypertension, aging, metabolic syndrome, and several genetic cardiomyopathies. Despite the number of efforts in clinical trials to date, no efficacious therapies have been identified for HFpEF (5–8).
Even with the best treatments available, there are high rates of mortality and morbidity with both HFrEF and HFpEF (9). This is partly due to our lack of mechanistic understanding of the disease pathogenesis (10) and the lack of an appropriate in vitro model system that can recapitulate relevant aspects of cardiac mechanics with sufficient throughput for drug discovery. Here, we review several recent advances in the fields of genetic and tissue engineering that have made it possible to model aspects of these diseases in vitro, and we discuss the potential applications of these technologies to drug discovery and personalized medicine.
Current Challenges in Modeling Human Heart Diseases
Studying cardiovascular disease in vitro comes with several challenges. First, cardiac physiology is tightly regulated in whole organisms by complex neuronal and hormonal feedback systems (11–14). Perturbations affecting cardiac function can lead to both short-term adaptations of the heart (e.g., increased heart rate, length-dependent changes in contractility, increases in the phosphorylation of sarcomeric proteins such as troponin-I or titin, and force-induced changes on actomyosin contractility), as well as long-term adaptations (e.g., cellular reorganization, cardiac tissue remodeling, activation of fibroblasts, and changes in gene expression). Understanding the disease pathogenesis and the development of novel therapeutics requires tools for studying the disease phenotypes across multiple scales of organization, ranging from the level of single molecules to whole organisms.
Another major challenge to modeling HF is the heterogeneity in the prognosis and presentation of HF in patients (15). As described earlier, HF patients are typically characterized by ejection fraction (i.e., their symptoms), but there are multiple underlying conditions that cause HF. For example, non-genetic HF can be initiated by myocardial injury (e.g., myocardial infarction) (16), valvular disease (17), or as a side effect of some chemotherapies (18). There are also several forms of genetic heart disease that can lead to heart failure (19–22). Familial hypertrophic (HCM) and dilated (DCM) cardiomyopathies are primarily caused by mutations in proteins that regulate cardiac muscle power output. HCM is characterized by thickening of the ventricular wall, fibrosis, and myocyte disarray. It has an estimated prevalence of 1 in 500 people, and it is the leading cause of sudden cardiac death in people under 30 years old (23). Familial DCM is a closely related disease that is also strongly associated with sudden death, and it is a significant cause of HF (24). DCM is characterized by dilation of the myocardial wall, and it is often accompanied by necrosis and fibrosis. Even though these forms of genetic heart disease are relatively common, the clinical presentations and the prognoses of HCM and DCM are highly variable and depend on the exact pathogenic mutation. To date, hundreds of mutations have been associated with these diseases (19, 23). Point mutations within the same molecule can lead to either HCM or DCM, with the phenotype depending on the specific site of the mutation (25, 26). Therefore, when modeling these genetic diseases, it is perhaps more useful to think of these conditions as collections of rare diseases with a common presentation. As such, the design of therapeutics presents itself as an opportunity for personalized treatment (i.e., precision medicine) (27).
Several in vitro model systems have been developed to address the challenges associated with modeling HF, each with its own set of advantages and drawbacks. The choice of model system is dictated by the specific questions being asked. For example, in many patients with HCM or DCM, point mutations in sarcomeric proteins at the molecular level are the initial insults that lead to tissue remodeling in the disease. Understanding these diseases requires a molecular knowledge of the specific defects caused by the mutations (25), and excellent experiments using purified and/or expressed proteins have led to the development of several drugs that are currently in clinical or preclinical trials (28–31). While these experiments are needed to dissect the initial molecular insults that lead to the disease phenotype, they have several caveats. First, the majority of biochemical studies are conducted in the absence of load, and it has been shown that mechanical forces can change the kinetics (and thus functional properties) of proteins, including cardiac myosin (32–39). This is important since proteins in the heart experience both internally and externally generated forces during contraction, and aberrant forces are a primary driver of cardiomyopathies (40). In fact, for some HCM mutations, the molecular disease phenotype only becomes apparent under load (39), and thus one must consider the mechanobiology of the heart when studying these diseases. Second, changes in contractility at the molecular level in vitro are not necessarily predictive of how the disease affects contractility in cells or tissues. For example, the first mutation identified to cause HCM, R403Q in MYH7 (20), shows conflicting results at the molecular level (41–43) that do not necessarily correspond to the phenotype in mice (44–46). Moreover, some forms of genetic HF are due to haploinsufficiency rather than direct changes in protein function (47).
Another approach that has greatly furthered the understanding of both genetic and non-genetic HF is the use of transgenic mouse models for physiological and biochemical studies [e.g., (42, 44, 48–55)]. This system allows for control of the genetic environment and physiological studies. However, mouse hearts have very different physiology than human hearts. For example, mouse hearts can beat ~600 times per minute while human hearts beat ~60 times per min. To beat this quickly, mouse hearts have some different ion channels [e.g., different subunits for the K-ATP channel (56) and different IKr channels (57)] that define their action potentials, different machinery for handling calcium, and different myosin isoforms with disparate kinetics that drive contractility (58–60). Therefore, transgenic mouse models do not always recapitulate the human disease phenotype and pharmacological response (44, 46, 48, 61–64). Also, mouse hearts lack the hERG channel. Many drugs, both cardiac-specific and nonspecific, can bind to this protein, leading to cardiotoxicity and arrhythmias in humans, despite having no effects in mice. This missed cardiotoxicity is one of the reasons that drugs designed based on mouse studies fail in clinical trials (65, 66).
Tissue obtained from patients (67) gives unique insights into the disease pathogenesis that cannot be recapitulated in other systems. However, it is difficult to obtain human tissue, and the disease presentation is often complicated by the patient's genetic background and medical history. Moreover, human tissues are usually obtained from patients whose hearts have undergone major remodeling and changes in gene expression in response to the disease. As such, it is not necessarily a good model system for studying how the initial insult of the mutation affects cardiac functions including contractility. Also, in the case of genetic heart disease, it is difficult to obtain sufficient tissue with a given genotype for well controlled drug testing. Moreover, it is challenging to get appropriate control tissue, since differences in the genetic background and patient history can affect the observed phenotype (68).
Human Pluripotent Stem Cell Derived Cardiomyocytes as Models of Disease
Recent advances in stem cell and genome editing technologies have led to the development of human pluripotent stem cell (hPSC)-based models of genetic human cardiac diseases. A critical advance was the derivation of human embryonic stem cell lines (69, 70) and their subsequent differentiation to a cardiomyocyte lineage (71). These early studies, which relied on embryoid body formation, had a very low differentiation efficiency (<1%) (71). Several methods have been developed to increase the efficiency of differentiation of stem cells to hPSC-CMs in both embryoid bodies (72, 73) and adherent monolayers of cells (74, 75). One widely used method, where WNT signaling is initially stimulated to promote mesoderm formation and then repressed to induce a cardiomyocyte lineage (74), can produce >90% hPSC-CMs (75, 76).
One difficulty with hPSC-CMs is that the differentiation methods produce a mixture of atrial, ventricular, pacemaker, and non-myocyte cells; although techniques have been developed recently to promote differentiation toward a specific cardiac lineage (77–80) and to eliminate non-myocytes from the cell culture (81). An additional challenge with hPSC-CMs is that they are developmentally immature (82, 83). This immaturity can be seen in several aspects of the cell physiology, including the ratio of alpha (MYH6) to beta (MYH7) cardiac myosin, the shape of the action potential, the absence of t-tubules, and the orientation of sarcomeres within the cardiomyocyte (84, 85). Although hPSC-CMs are developmentally immature (86, 87), they are an ideal system for studying the early disease pathogenesis, before the heart undergoes many of the adaptations seen in older patients. Moreover, several approaches have been used to engineer more mature phenotypes in hPSC-CMs, including electrical pacing (88–91), addition of growth hormones or fatty acids (82, 92), providing mechanical or geometric cues that mimic the organization of the heart (93–96), and providing stretch/mechanical resistance (97–100). hPSC-CMs can also be matured through incorporation into 3D engineered tissues.
Several groups have derived stem cells from patient samples [e.g., (47, 101–103)] and then differentiated these cells to hPSC-CMs. These studies have shown that it is possible to recapitulate aspects of cardiac disease using these cells. Recent advances in genetic engineering, such as the application of the CRISPR/Cas9 system (104, 105), have opened the door to studying genetic forms of heart failure and the role of genetic modifiers in disease without the need for patient heart tissue. These tools have been harnessed to introduce disease-causing mutations into hPSCs and then study their phenotypes [e.g., (47, 103)]. The genome editing approach has the advantage that the mutant and WT lines are isogenic except for the pathogenic mutation. This is important since cardiomyopathies often show incomplete penetrance, and the disease presentation can vary depending on the genetic background (68, 106). A disadvantage to using genetic engineering of healthy cells instead of patient cells is the inability to directly correlate changes in vitro with relevant clinical data of cardiac function in vivo. Moreover, the disease presentation depends on the genetic background, and therefore, the presentation in a control cell line could differ from the presentation in a patient. However, it is possible to take cells from a patient with the disease and then fix the genetic mutation to generate genetically matched control cells (107). This later approach has the advantage that it enables the collection of in vivo clinical data from the patient and then the correlation of these parameters with properly controlled measurements in vitro.
Human Engineered Heart Tissues
The human heart has a complex three-dimensional structure composed of many cell types including cardiomyocytes, fibroblasts, macrophages, and endothelial cells. The cardiomyocytes interact with the other cell types, and these other cells can modulate the contractile and electrophysiological properties of cardiomyocytes (108–113). These cells are organized within the extracellular matrix to give rise to distinct regions within the heart with specific functions (e.g., sinoatrial node, ventricular wall, and papillary muscles). Moreover, these cells can be mechanically and electrically coupled, and the mechanical environment can affect the electrophysiological properties of these cells (114). The cells in the heart are thus subjected to an array of mechanical, chemical, and electrical signals that can affect their function. Generating in vitro models of heart disease that faithfully recapitulate cardiac dysfunction will require consideration of these complexities.
To recapitulate many of these aspects of cardiac functions in vitro, 3D engineered heart tissues (EHT) were first created more than two decades ago using cardiomyocytes isolated from chicken embryos (115). Since then, the successful fabrication of EHTs with hPSC-CMs has significantly advanced our ability to model human heart diseases in vitro, and these tissues faithfully recapitulate many features of the clinical disease phenotypes [e.g., (47, 116–118)]. In addition, miniaturization of the EHTs has enabled mass-production of EHTs for higher throughput assays (111–113, 119) The hPSC-CMs in EHTs exhibit more mature phenotypes than those grown in 2D culture, showing more normal sodium currents (120), organized sarcomeric arrangement (121), and improved mitochondrial function (88).
The 3D environment of EHTs allows researchers to control and recapitulate mechanical homeostasis unique to the heart (122, 123). Scaffold-free 3D spheroid tissue models have advantages for simple high-throughput assays, but they lack the mechanobiological cues necessary for tissue maturation and organization (124, 125). EHTs formed using parallel wires (89, 126–128), parallel posts (98, 129–131), or sheets (132) can provide an improved mechanical microenvironment for EHT development. Mechanically stretching EHTs improves the maturation of myocytes (98, 129, 133–135) and can increase cellular alignment (136–138). Combined electrical and mechanical conditioning of EHTs has shown promising results for cardiac tissue maturation (88, 137, 139).
While EHTs are powerful tools for studying heart disease, there are various limitations that must be considered. Since EHTs are fabricated in 3D, many cells are needed to fabricate a single sample. Therefore, the costs and times required to produce EHTs are generally higher than those of 2D cell culture. Additionally, the production of EHTs requires the quality control of many more parameters due to their complexity. For example, the differentiation efficiency of stem cells to hPSC-CMs, the number of stromal cells added to the tissue, and the formation of defined extracellular matrices are very important for reproducibility. Moreover, care must be taken when selecting an appropriate culture media, since supplements in the medium can affect certain cell types in the EHT and modulate the activity of enzymes that remodel the extracellular matrix (ECM). While cardiac tissues can be formed without adding any exogenous ECMs components using cell sheet technology (140), most EHTs use exogenously added ECMs. While collagen and fibrin are the most popular choices for the ECMs in EHTs, their hydrogel properties can be different depending on their methods of preparation (141, 142). Other ECM components such as basement membrane proteins can be doped into the base ECM to mimic the composition of ECMs in the heart. The field will benefit from continued examination of how different ECM compositions influence the physiological properties of EHTs, especially with regard to changes in the ECM associated with HF.
Measurement of Cardiac Mechanics Using Human Engineered Heart Tissues
To date, many different platforms for EHTs have been developed. These platforms have been tailored for specific applications, with systems that excel at modeling different aspects of the heart, including vascularization, microcirculation, cardiomyocyte maturity, structure, calcium handling, and contractility (47, 88, 119, 143–146). The selection of the appropriate EHT system will depend on the specific questions being asked.
In both HFpEF and HFrEF, the mechanics of the heart are altered; and therefore, when modeling HF in vitro, it is desirable to be able to examine the effects of the disease on cardiac contractility. In most EHT systems capable of modeling cardiac contractility, an EHT in a hydrogel is formed between two posts and the contractility of the tissue is measured using a transducer (Figure 1). Human hPSC-CMs and human cardiac fibroblasts in the EHT remodel the hydrogel to form cardiac tissue strips (or sheets), where the cells are aligned perpendicular to the parallel posts. The transducers used in most of these systems measure the force of contraction by monitoring the deflection of the posts. The deflection can be measured using electronic strain gauges or using optical detection of the post position.
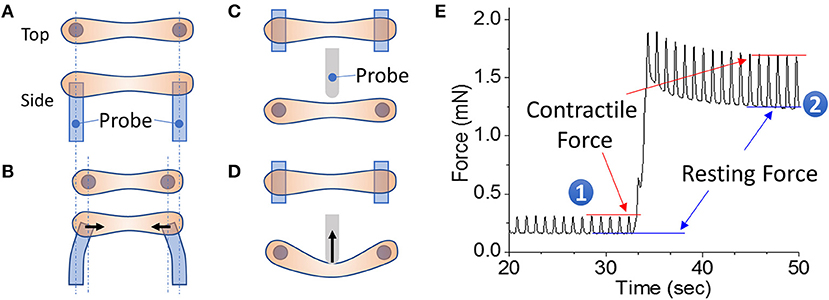
Figure 1. EHT strips formed in a 96-well format for phenotyping assays. (A,B) Example of a passive force system in which an EHT strip is formed between two parallel posts. The force generated during contraction can be monitored by the deflection of the posts. (C,D) Example of an active force system in which an EHT strip is formed between two parallel posts and then the tissue can be stretched using a probe that pushes on the side of the tissue. In this setup, the force is measured by a strain gauge in the probe. (E) Measurement of cardiac forces at two different muscle lengths (1 and 2). The peak and bottom of the cardiac twitch force profiles correspond to the contractile and resting forces, respectively. Note that stretching the EHT causes an increase in the contractile force, as would be expected from the Frank-Starling relationship.
EHT systems for measuring contractility can be broadly divided into passive and active force systems, depending on whether the tissue can be actively stretched in real time during an experiment or only passively monitored. Passive force systems are easier and cheaper to implement, but more limited in the parameters that they can measure. The choice of system will depend on the specific questions being asked. Passive force systems were first applied to examine skeletal muscle contractility (147), but now there are several passive force system for studying cardiac contractility (121, 148). In a passive force system, the tension in the EHT in between beats gives information about non-sarcomeric contractility and the peak tension in the EHT during contraction gives information about the force of cardiac myosin-driven contractility (Figures 1A,B).
In active force systems, the force on the tissue can be manipulated in real time during an experiment (98, 111, 115, 149–153). This can come from moving one of the posts or from using a probe to manipulate the tissue (Figures 1C,D). Using an active force system, it is possible to examine several important functional properties of the EHT that can be altered in HF (151). In a healthy heart, increasing the stretch of cardiac muscle during diastole causes an increase in cardiac output, an adaptation known as the Frank-Starling relationship. In HF, this relationship is altered, limiting the adaptive capability of the heart. To analyze this relationship, an active force system can stretch the EHT strips with preprogrammed wave forms (Figure 1D) (151). The forces generated during cardiac contraction (i.e., systolic force) and relaxation (i.e., diastolic force) at various tissue lengths are analyzed to generate a cardiac muscle-specific length-tension relationship, LTR (i.e., the Frank-Starling relationship) (Figures 1, 2).
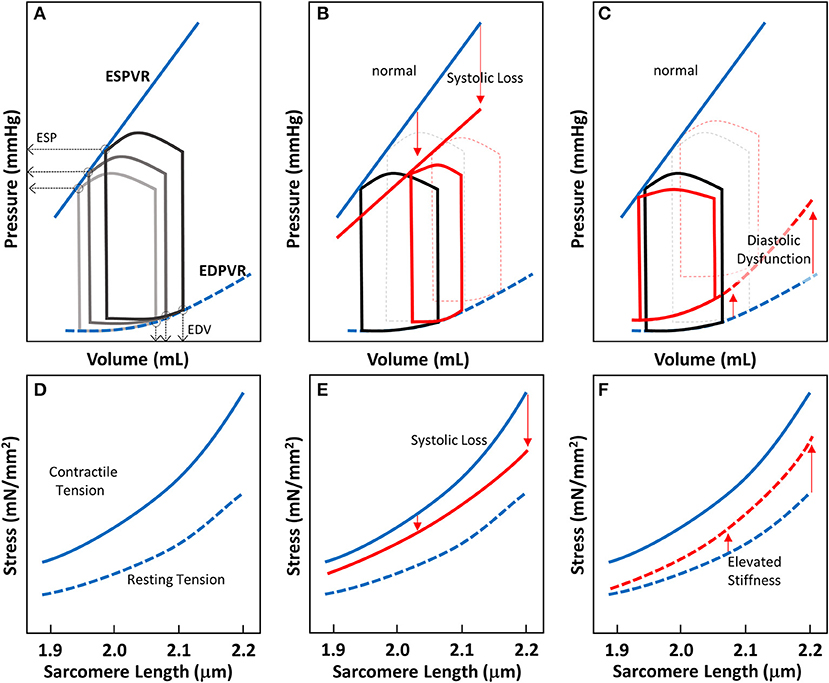
Figure 2. Cardiac pressure-volume (PV)-loop analysis and length-tension relationship of EHTs. (A) PV-loops are visualization tools to systematically analyze the contractile properties of heart chamber function. A series of PV-loops can be collected under various preloads (shown in gray). There is a linear relationship between the end systolic pressures (ESP) and their corresponding volume points, known as the end-systolic pressure volume relationship (ESPVR). Similarly, the line connecting the end diastolic volumes (EDVs) and their corresponding pressures is known as the end diastolic pressure volume relationship (EDPVR). (B) In systolic heart failure (HFrEF), both the slope of ESPVR and the ejection fraction decrease. (C) In diastolic heart failure, the ejection fraction may not change much, but EDPVR shifts upward, indicative of impaired myocardial relaxation during diastole. (D–F) Length-tension relationships (LTRs) of EHT strips are related to cardiac functions represented by PV-loops. The contractile tension (solid line) and resting tension (dotted line) are shown. (D) LTRs for healthy tissue, showing the contractile and resting stresses. (E) In systolic heart failure, there is a reduction in contractile tension that becomes more pronounced as the length is increased (red solid line). (F) In diastolic heart failure, the increase in tissue stiffness leads to an increase in resting tension that is more pronounced as the length is increased (red dotted line).
The LTR obtained for EHTs in vitro can be related to the work that the heart does in vivo during the cardiac cycle. The work done by the whole heart is calculated by measuring pressure-volume (PV) loops during the cardiac cycle (Figure 2A). The work equals the area enclosed within the loop. A family of PV loops can be collected with various preloads to assess cardiac function. As described in Figures 2A–C, one can analyze cardiac function by visualizing the end-systolic pressure-volume relationship (ESPVR) and end-diastolic pressure-volume relationship (EDPVR) at a given inotropic state. For a given stretch/preload, the peak and resting LTR values are related to the ESPVR and EDPVR respectively (Figures 2A,D).
For different types of heart failure, one would expect PV loops to exhibit different ESPVR and EDPVRs. In HFrEF, the loss of systolic function produces a reduced slope of the ESPVR (Figure 2B). To compensate for reduced efficiency of pump function in HFrEF, increasing preload on the heart forces the heart to operate at a higher diastolic volume. In EHTs, the reduction of systolic contractility should appear as a reduction in the contractile force (Figure 2E). The reduction of contractile force should be more pronounced at longer sarcomere lengths (Figure 2E). In HFpEF, there is no change in the ejection fraction, but impaired relaxation due to stiffening of the myocardium. Molecular analysis of the myocardium from patients with HFpEF shows that this elevated passive stiffness can be partly due to stiffening of titin and/or increases in collagen cross-linking (154). As a result of this stiffening, the EDPVR is elevated (Figure 2C). In the EHT strips, HFpEF would be expected to show elevated resting tensions due to this increase in stiffness, and this effect should be more pronounced when the tissue strips are stretched to higher levels of tension (Figure 2F). Taken together, this demonstrates the utility of EHTs for studying cardiac contractility and HF. While this assumption should be validated rigorously, the technology holds promise to be used in HF drug development.
Application of Engineered Heart Tissues to Drug Screening
One of the requirements for drug screening is the ability to rapidly screen through large libraries of compounds. The earliest studies of EHT contractility were performed on centimeter scale non-human cardiac tissues in an organ bath (115). In these experiments, over 1 million cells were used to generate a single tissue. The required organ bath was relatively large, requiring 20–50 mL of solution to test a single compound, and it would not be easy to analyze many samples simultaneously for drug screening at this scale. Moreover, the high cost of hPSC culture necessary to generate human tissues makes this system less amenable for drug screens.
To increase assay throughput for drug discovery, various excellent systems have been introduced over the last several years. For example, the Chen lab developed a passive force system where over 100 tissues are formed in microelectromechanical devices in each well of a tissue culture dish (119). Other approaches have focused on fabricating a single EHT in each well of a multi-well plate (e.g., 96/384 well plates) (150, 155). Both of these approaches can be tailored to enable high-throughput screens of libraries of compounds and to provide several physiological readouts of EHT function from a single sample.
As a proof of principle of how EHTs in an active force system can be used for drug screening, we present an example looking at the contractile effects of a drug that is currently in phase III clinical trials as a treatment for systolic heart failure, omecamtiv mecarbil (OM). OM was discovered through a high-throughput screen for compounds that increase cardiac myosin's actin-activated steady-state ATPase activity (156), and OM shows a high affinity for the cardiac myosin isoform (157). OM is a unique positive inotropic compound that was designed to directly activate myosin-based contractility without affecting calcium handling by the cell. This is significant because drugs that target calcium handling can be pro-arrhythmogenic (4). While the exact biophysical mechanism of OM's action on myosin is disputed (28, 158), it has clear positive inotropic effects over a range of dosages (159).
To demonstrate the effects of OM on EHT contractility, we used an active force system in which stem cell derived EHTs in hydrogels are formed between two parallel bars in each well of a 96-well plate (Figures 1C,D) (150–153, 160). A soft-tissue mechanical analyzer (Palpator, InvivoSciences) measures the mechanical properties of the EHT strips using micro-force transducers attached to its robotic head (150). We first analyzed OM's dose-dependent effects using human EHT strips (Figure 3A). As described previously in rat muscle fibers, active cardiac contractility was increased by concentrations of OM up to 1 μM and inhibited by high concentrations (113). Based on the integrated tension transient (Figure 3B), 1 μM was the most effective concentration tested to increase total contractility. To test the effects of OM on calcium transients, EHT strips were loaded with a biological calcium indicator (Fluo4, Thermo Fisher). As shown in Figure 3C, none of the OM doses tested changed the profiles of calcium transients, consistent with previous reports using non-human cardiomyocytes (28). To analyze OM's effects on metabolic activities, the mitochondrial membrane potential (MMP) was monitored. The mitochondrial activity showed no significant change upon the addition of OM, even with 1 h of incubation. As expected, 2,4-Dinitrophenol (DNP, 500 mM) uncouples the MMP activity. To analyze OM's effects on the LTR, OM (1 μM) was added to EHT tissue strips, and the tissue was stretched to various length using the soft tissue mechanical analyzer (Figure 4). As expected, in the absence of OM, the EHT strips produced increasing levels of cardiac contraction with increasing stretch. After the addition of OM, the tissues produced enhanced contractility with stretch. In general, the tissues generate more stress during a twitch after OM treatment compared to their stress before treatment; however, this difference becomes less pronounced with increasing stretch (Figure 4D). These results demonstrate the ability to use hPSC-CM EHT systems for drug testing. When combined with gene edited cells, this approach will open the door for targeted therapeutic design and precision medicine.
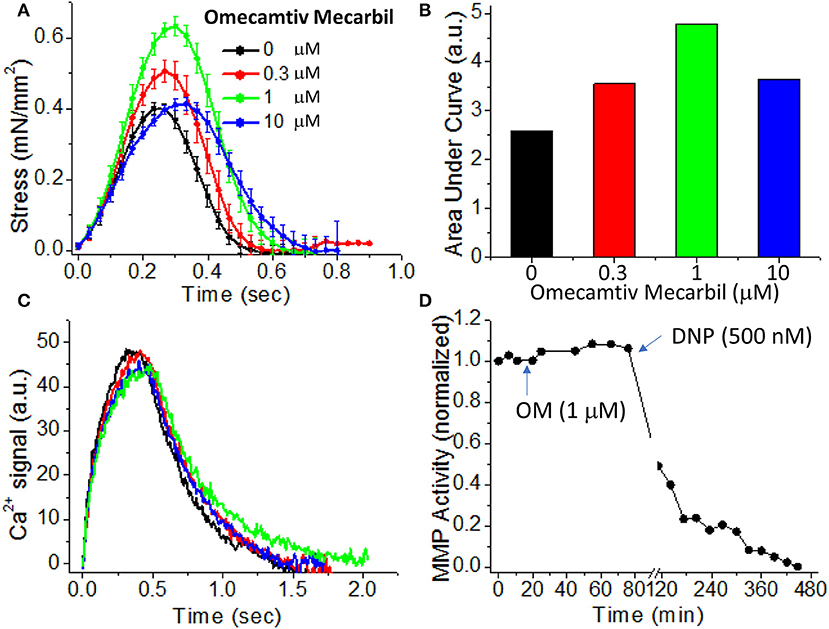
Figure 3. Effects of omecamtiv mecarbil (OM) on excitation-contraction-energy coupling in EHTs. (A) EHT strips were exposed to increasing concentrations (0, 0.3, 1, and 10 μM) of OM, and the average time courses of cardiac contraction were analyzed. For OM concentration up to 1 μM, the peak stress increases in a dose-dependent manner. (B) The area under each profile in (A) was calculated to compare the dose-dependent effects of OM on cardiac contraction profiles. An increase in total contractility was seen with OM treatment. (C) Average calcium-transient profiles in the presence of increasing concentrations of OM were measured using a fluorescent calcium dye (n = 4–6). OM has little effect on the calcium transient. (D) To analyze OM's effects on mitochondrial activity, mitochondrial membrane potential (MMP) activity was monitored for 1 h after compound addition. Treatment with 1 μM OM does not change MMP activity. DNP (2,4-Dinitrophenol), an agent for uncoupling oxidative phosphorylation, was added as a positive control to confirm that the tissue strip was energetically active.
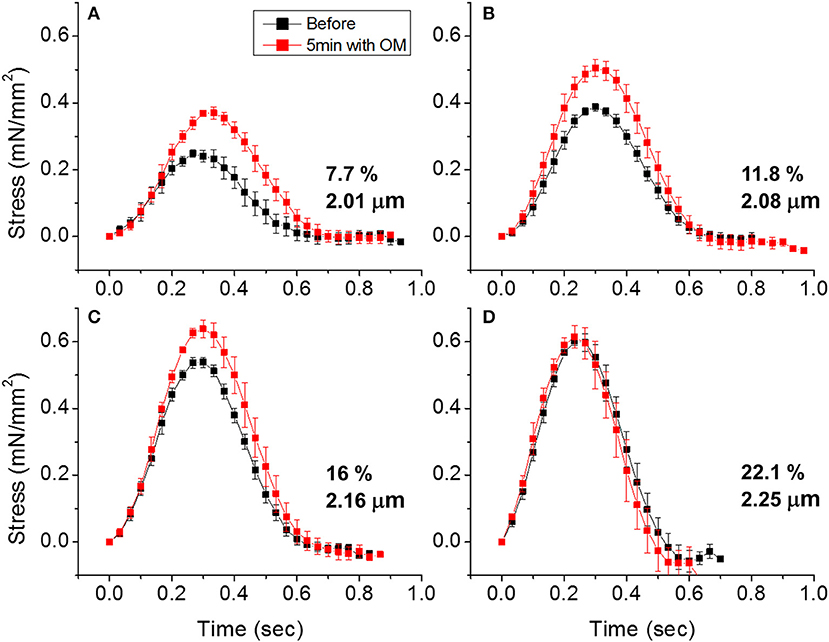
Figure 4. Effects of omecamtiv mecarbil (OM) on LTR in EHTs. (A) Cardiac contraction profiles were monitored before and after treating EHT strips with OM (1 μM). At ~2 μm sarcomere length, the OM increased both the peak and duration of cardiac contraction profiles. (B,C) As the EHT was stretched further, the effects of OM on contractility compared to the untreated tissue become less pronounced. (D) At ~2.25 μm sarcomere length, there is no difference in contractile profile before and after OM treatment.
Future Prospects for Drug Development and Precision Medicine
Even though deaths from cardiovascular disease accounted for >20% of all deaths in the US, cardiovascular drugs account for only 6.6% of compounds in Phase I clinical trials that were eventually approved for patient use (161). One of the difficulties with developing new cardiovascular treatments is the huge cost of clinical trials, which require large study cohorts to evaluate the efficacy of treatments for chronic diseases, such as age-associated HF. A recent analysis of 9,985 clinical and regulatory phase transition records between 2006 and 2015 indicates that the likelihood of approval increases three-fold when working with a targeted, well-defined patient population (161). The use of reliable biomarkers to select patients and monitor their responses has been shown to improve the performance of treatment candidates during trials. The use of genetically engineered cells in EHTs should allow for the development of preclinical disease models that mimic heart failure against a controlled genetic background.
The combined use of genetic engineering and tissue engineering can be used to model monogenic cardiac disease in vitro as well as the role of genetic modifiers in disease. Importantly, these tools can be harnessed for precision medicine. For example, one critical bottleneck in the treatment of genetic heart disease is identifying whether a given genetic variant identified in a patient is pathogenic or not. We envision that EHTs generated from genetically engineered cells will enable the direct testing of the consequences of specific genetic variants. Moreover, the use of reprogrammed cells taken from a patient cheek scraping or urine sample will open the door to the development of personalized medicine. EHTs generated from these cells can be used to evaluate the efficacy and side effects of precision therapies, enabling clinicians to optimize the treatment course for each patient. These applications will be aided by high-throughput EHT phenotyping. Taken together, these advances have the potential to revolutionize the treatment of cardiac disease.
Author Contributions
MG and TW wrote and edited the manuscript. ND, MC, AW, and TW designed and performed experiments, analyzed data, and created figures.
Funding
The authors acknowledge funding support partly from the National Institutes of Health (R01HL141086, R00HL123623 to MG, and R43GM109735, R43AG054270, R01HL109505 to TW) and the March of Dimes Foundation (FY18-BOC-430198 to MG).
Conflict of Interest Statement
MG's contributions were conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. ND, MC, AW, and TW are employees of a for-profit organization, InvivoSciences, Inc.
References
1. Writing Group M, Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, et al. Heart disease and stroke statistics-2016 update: a report from the American Heart Association. Circulation (2016) 133:e38–360. doi: 10.1161/CIR.0000000000000350
2. Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, et al. Heart disease and stroke statistics−2016 update. Circulation (2015)
3. Metra M, Teerlink JR. Heart failure. Lancet (2017) 390:1981–95. doi: 10.1016/S0140-6736(17)31071-1
4. Kass DA, Solaro RJ. Mechanisms and use of calcium-sensitizing agents in the failing heart. Circulation (2006) 113:305–15. doi: 10.1161/CIRCULATIONAHA.105.542407
5. Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE, Drazner MH, et al. 2013 ACCF/AHA Guideline for the Management of Heart Failure. J Am College Cardiol. (2013) 62:e147. doi: 10.1016/j.jacc.2013.05.019
6. Krittanawong C, Kukin ML. Current management and future directions of heart failure with preserved ejection fraction: a contemporary review. Curr Treat Options Cardiovasc Med. (2018) 20:28. doi: 10.1007/s11936-018-0623-1
7. Tschöpe C, Birner C, Böhm M, Bruder O, Frantz S, Luchner A, et al. Heart failure with preserved ejection fraction: current management and future strategies. Clin Res Cardiol. (2018) 107:1–19. doi: 10.1007/s00392-017-1170-6
8. Tschöpe C, Van Linthout S, Kherad B. Heart failure with preserved ejection fraction and future pharmacological strategies: a glance in the crystal ball. Curr Cardiol Rep. (2017) 19:70. doi: 10.1007/s11886-017-0874-6
9. Abebe TB, Gebreyohannes EA, Tefera YG, Abegaz TM. Patients with HFpEF and HFrEF have different clinical characteristics but similar prognosis: a retrospective cohort study. BMC Cardiovasc Disord. (2016) 16:232. doi: 10.1186/s12872-016-0418-9
10. Zakeri R, Cowie MR. Heart failure with preserved ejection fraction: controversies, challenges and future directions. Heart (2018) 104:377. doi: 10.1136/heartjnl-2016-310790
12. Morgan HE, Baker KM. Cardiac hypertrophy. Mechanical, neural, and endocrine dependence. Circulation (1991) 83:13–25.
13. Feng N, Huke S, Zhu G, Tocchetti CG, Shi S, Aiba T, et al. Constitutive BDNF/TrkB signaling is required for normal cardiac contraction and relaxation. Proc Natl Acad Sci USA. (2015) 112:1880–5. doi: 10.1073/pnas.1417949112
14. Gordan R, Gwathmey JK, Xie LH. Autonomic and endocrine control of cardiovascular function. World J Cardiol. (2015) 7:204–14. doi: 10.4330/wjc.v7.i4.204
15. Iorio A, Pozzi A, Senni M. Addressing the heterogeneity of heart failure in future randomized trials. Curr Heart Fail Rep. (2017) 14:197–202. doi: 10.1007/s11897-017-0332-1
16. Hendriks TR, Schurer AJ, Al Ali L, van den Heuvel AFM, van der Harst P. Left ventricular restoration devices post myocardial infarction. Heart Failure Rev. (2018) doi: 10.1007/s10741-018-9711-2. [Epub ahead of print].
17. Steiner J, Rodés-Cabau J, Holmes DR, LeWinter MM, Dauerman HL. Mechanical intervention for aortic valve stenosis in patients with heart failure and reduced ejection fraction. J Am Coll Cardiol. (2017) 70:3026–41. doi: 10.1016/j.jacc.2017.10.040
18. Lenneman CG, Sawyer DB. Cardio-Oncology. Circ Res. (2016) 118:1008. doi: 10.1161/CIRCRESAHA.115.303633
19. Burke MA, Cook SA, Seidman JG, Seidman CE. Clinical and Mechanistic Insights Into the Genetics of Cardiomyopathy. J Am Coll Cardiol. (2016) 68:2871–86. doi: 10.1016/j.jacc.2016.08.079
20. Geisterfer-Lowrance AA, Kass S, Tanigawa G, Vosberg HP, McKenna W, Seidman CE, et al. A molecular basis for familial hypertrophic cardiomyopathy: a beta cardiac myosin heavy chain gene missense mutation. Cell (1990) 62:999–1006. doi: 10.1016/0092-8674(90)90274-I
21. Jarcho JA, McKenna W, Pare JA, Solomon SD, Holcombe RF, Dickie S, et al. Mapping a gene for familial hypertrophic cardiomyopathy to chromosome 14q1. N Eng J Med. (1989) 321:1372–8. doi: 10.1056/NEJM198911163212005
22. Schonberger J, Seidman CE. Many roads lead to a broken heart: the genetics of dilated cardiomyopathy. Am J Hum Genet. (2001) 69:249–60. doi: 10.1086/321978
23. Alcalai R, Seidman JG, Seidman CE. Genetic basis of hypertrophic cardiomyopathy: from bench to the clinics. J Cardiovasc Electrophysiol. (2008) 19:104–10. doi: 10.1111/j.1540-8167.2007.00965.x
24. Lu QW, Wu XY, Morimoto S. Inherited cardiomyopathies caused by troponin mutations. J Geriatr Cardiol. (2013) 10:91–101. doi: 10.3969/j.issn.1671-5411.2013.01.014
25. Spudich JA. Hypertrophic and dilated cardiomyopathy: four decades of basic research on muscle lead to potential therapeutic approaches to these devastating genetic diseases. Biophys J. (2014) 106:1236–49. doi: 10.1016/j.bpj.2014.02.011
26. Lynn ML, Lehman SJ, Tardiff JC. Biophysical derangements in genetic cardiomyopathies. Heart Fail Clin. (2018) 14:147–59. doi: 10.1016/j.hfc.2017.12.002
27. Jameson JL, Longo DL. Precision medicine — personalized, problematic, and promising. N Eng J Med. (2015) 372:2229–34. doi: 10.1056/NEJMsb1503104
28. Malik FI, Hartman JJ, Elias KA, Morgan BP, Rodriguez H, Brejc K, et al. Cardiac myosin activation: a potential therapeutic approach for systolic heart failure. Science (2011) 331:1439–43. doi: 10.1126/science.1200113
29. Green EM, Wakimoto H, Anderson RL, Evanchik MJ, Gorham JM, Harrison BC, et al. A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science (2016) 351:617–21. doi: 10.1126/science.aad3456
30. Regnier M, Rivera AJ, Chen Y, Chase PB. 2-deoxy-ATP enhances contractility of rat cardiac muscle. Circ Res. (2000) 86:1211–7. doi: 10.1161/01.RES.86.12.1211
31. Cheng Y, Hogarth KA, O'Sullivan ML, Regnier M, Pyle WG. 2-Deoxyadenosine triphosphate restores the contractile function of cardiac myofibril from adult dogs with naturally occurring dilated cardiomyopathy. Am J Physiol. (2016) 310:H80–91. doi: 10.1152/ajpheart.00530.2015
32. Greenberg MJ, Arpag G, Tuzel E, Ostap EM. A Perspective on the role of myosins as mechanosensors. Biophys J. (2016) 110:2568–76. doi: 10.1016/j.bpj.2016.05.021
33. Jansen KA, Donato DM, Balcioglu HE, Schmidt T, Danen EH, Koenderink GH. A guide to mechanobiology: Where biology and physics meet. Biochim Biophys Acta (2015) 1853:3043–52. doi: 10.1016/j.bbamcr.2015.05.007
34. Hill AV. The heat of shortening and the dynamic constants of muscle. Proc R Soc Lond Ser B Biol Sci. (1938) 126:136–95.
35. Hill AV. The maximum work and mechanical efficiency of human muscles, and their most economical speed. J Physiol. (1922) 56:19–41.
36. Fenn WO. A quantitative comparison between the energy liberated and the work performed by the isolated sartorius muscle of the frog. J Physiol. (1923) 58:175–203.
37. Sung J, Nag S, Mortensen KI, Vestergaard CL, Sutton S, Ruppel K, et al. Harmonic force spectroscopy measures load-dependent kinetics of individual human beta-cardiac myosin molecules. Nat Commun. (2015) 6:7931. doi: 10.1038/ncomms8931
38. Greenberg MJ, Shuman H, Ostap EM. Inherent force-dependent properties of beta-cardiac myosin contribute to the force-velocity relationship of cardiac muscle. Biophys J. (2014) 107:L41–4. doi: 10.1016/j.bpj.2014.11.005
39. Greenberg MJ, Kazmierczak K, Szczesna-Cordary D, Moore JR. Cardiomyopathy-linked myosin regulatory light chain mutations disrupt myosin strain-dependent biochemistry. Proc Natl Acad Sci USA. (2010) 107:17403–8. doi: 10.1073/pnas.1009619107
40. Davis J, Davis LC, Correll RN, Makarewich CA, Schwanekamp JA, Moussavi-Harami F, et al. A tension-based model distinguishes hypertrophic versus dilated cardiomyopathy. Cell (2016) 165:1147–59. doi: 10.1016/j.cell.2016.04.002
41. Palmiter KA, Tyska MJ, Haeberle JR, Alpert NR, Fananapazir L, Warshaw DM. R403Q and L908V mutant beta-cardiac myosin from patients with familial hypertrophic cardiomyopathy exhibit enhanced mechanical performance at the single molecule level. J Muscle Res Cell Motil. (2000) 21:609–20. doi: 10.1023/A:1005678905119
42. Tyska MJ, Hayes E, Giewat M, Seidman CE, Seidman JG, et al. Single-molecule mechanics of R403Q cardiac myosin isolated from the mouse model of familial hypertrophic cardiomyopathy. Circ Res. (2000) 86:737–44. doi: 10.1161/01.RES.86.7.737
43. Nag S, Sommese RF, Ujfalusi Z, Combs A, Langer S, Sutton S, et al. Contractility parameters of human beta-cardiac myosin with the hypertrophic cardiomyopathy mutation R403Q show loss of motor function. Sci Adv. (2015) 1:e1500511. doi: 10.1126/sciadv.1500511
44. Lowey S, Bretton V, Gulick J, Robbins J, Trybus KM. Transgenic mouse alpha- and beta-cardiac myosins containing the R403Q mutation show isoform-dependent transient kinetic differences. J Biol Chem. (2013) 288:14780–7. doi: 10.1074/jbc.M113.450668
45. Chuan P, Sivaramakrishnan S, Ashley EA, Spudich JA. Cell-intrinsic functional effects of the alpha-cardiac myosin Arg-403-Gln mutation in familial hypertrophic cardiomyopathy. Biophys J. (2012) 102:2782–90. doi: 10.1016/j.bpj.2012.04.049
46. Lowey S, Lesko LM, Rovner AS, Hodges AR, White SL, Low RB, et al. Functional effects of the hypertrophic cardiomyopathy R403Q mutation are different in an alpha- or beta-myosin heavy chain backbone. J Biol Chem. (2008) 283:20579–89. doi: 10.1074/jbc.M800554200
47. Hinson JT, Chopra A, Nafissi N, Polacheck WJ, Benson CC, Swist S, et al. Titin mutations in iPS cells define sarcomere 575 insufficiency as a cause of dilated cardiomyopathy. Science (2015) 349:982–6. doi: 10.1126/science.aaa5458
48. Ford SJ, Mamidi R, Jimenez J, Tardiff JC, Chandra M. Effects of R92 mutations in mouse cardiac troponin T are influenced by changes in myosin heavy chain isoform. J Mol Cell Cardiol. (2012) 53:542–51. doi: 10.1016/j.yjmcc.2012.07.018
49. Miller T, Szczesna D, Housmans PR, Zhao J, de Freitas F, Gomes AV, et al. Abnormal contractile function in transgenic mice expressing a familial hypertrophic cardiomyopathy-linked troponin T (I79N) mutation. J Biol Chem. (2001) 276:3743–55. doi: 10.1074/jbc.M006746200
50. van Dijk SJ, Witt CC, Harris SP. Normal cardiac contraction in mice lacking the proline-alanine rich region and C1 domain of cardiac myosin binding protein C. J Mol Cell Cardiol. (2015) 88:124–32. doi: 10.1016/j.yjmcc.2015.09.006
51. Greenberg MJ, Watt JD, Jones M, Kazmierczak K, Szczesna-Cordary D, Moore JR. Regulatory light chain mutations associated with cardiomyopathy affect myosin mechanics and kinetics. J Mol Cell Cardiol. (2009) 46:108–15. doi: 10.1016/j.yjmcc.2008.09.126
52. Dorn GW II, Molkentin JD. Manipulating cardiac contractility in heart failure: data from mice and men. Circulation (2004) 109:150–8. doi: 10.1161/01.CIR.0000111581.15521.F5
53. Molkentin JD, Robbins J. With great power comes great responsibility: using mouse genetics to study cardiac hypertrophy and failure. J Mol Cell Cardiol. (2009) 46:130–6. doi: 10.1016/j.yjmcc.2008.09.002
54. Cook SA, Clerk A, Sugden PH. Are transgenic mice the 'alkahest' to understanding myocardial hypertrophy and failure? J Mol Cell Cardiol. (2009) 46:118–29. doi: 10.1016/j.yjmcc.2008.11.005
55. Bulfield G, Siller WG, Wight PA, Moore KJ. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc Natl Acad Sci USA. (1984) 81:1189–92. doi: 10.1073/pnas.81.4.1189
56. Fedorov VV, Glukhov AV, Ambrosi CM, Kostecki G, Chang R, Janks D, et al. Effects of KATP channel openers diazoxide and pinacidil in coronary-perfused atria and ventricles from failing and non-failing human hearts. J Mol Cell Cardiol. (2011) 51:215–25. doi: 10.1016/j.yjmcc.2011.04.016
57. Pond AL, Scheve BK, Benedict AT, Petrecca K, Van Wagoner DR, Shrier A, et al. Expression of distinct ERG proteins in rat, mouse, and human heart. Relation to functional I(Kr) channels. J Biol Chem. (2000) 275:5997–6006. doi: 10.1074/jbc.275.8.5997
58. Alpert NR, Brosseau C, Federico A, Krenz M, Robbins J, Warshaw DM. Molecular mechanics of mouse cardiac myosin isoforms. Am J Physiol Heart Circ Physiol. (2002) 283:H1446–54. doi: 10.1152/ajpheart.00274.2002
59. Krenz M, Sanbe A, Bouyer-Dalloz F, Gulick J, Klevitsky R, Hewett TE, et al. Analysis of myosin heavy chain functionality in the heart. J Biol Chem. (2003) 278:17466–74. doi: 10.1074/jbc.M210804200
60. Deacon JC, Bloemink MJ, Rezavandi H, Geeves MA, Leinwand LA. Identification of functional differences between recombinant human alpha and beta cardiac myosin motors. Cell Mol Life Sci. (2012) 69:2261–77. doi: 10.1007/s00018-012-0927-3
61. He H, Javadpour MM, Latif F, Tardiff JC, Ingwall JS. R-92L and R-92W mutations in cardiac troponin T lead to distinct energetic phenotypes in intact mouse hearts. Biophys J. (2007) 93:1834–44. doi: 10.1529/biophysj.107.107557
62. Roh J, Houstis N, Rosenzweig A. Why don't we have proven treatments for HFpEF? Circ Res. (2017) 120:1243. doi: 10.1161/CIRCRESAHA.116.310119
63. Conceição G, Heinonen I, Lourenço AP, Duncker DJ, Falcão-Pires I. Animal models of heart failure with preserved ejection fraction. Netherlands Heart J. (2016) 24:275–86. doi: 10.1007/s12471-016-0815-9
64. Sacco A, Mourkioti F, Tran R, Choi J, Llewellyn M, Kraft P, et al. Short telomeres and stem cell exhaustion model Duchenne muscular dystrophy in mdx/mTR mice. Cell (2010) 143:1059–71. doi: 10.1016/j.cell.2010.11.039
65. Liang P, Lan F, Lee AS, Gong T, Sanchez-Freire V, Wang Y, et al. Drug screening using a library of human induced pluripotent stem cell-derived cardiomyocytes reveals disease-specific patterns of cardiotoxicity. Circulation (2013) 127:1677–91. doi: 10.1161/CIRCULATIONAHA.113.001883
66. Sharma A, Burridge PW, McKeithan WL, Serrano R, Shukla P, Sayed N, et al. High-throughput screening of tyrosine kinase inhibitor cardiotoxicity with human induced pluripotent stem cells. Sci Transl Med. (2017) 9:eaaf2584. doi: 10.1126/scitranslmed.aaf2584
67. Fananapazir L, Dalakas MC, Cyran F, Cohn G, Epstein ND. Missense mutations in the beta-myosin heavy-chain gene cause central core disease in hypertrophic cardiomyopathy. Proc Natl Acad Sci USA. (1993) 90:3993–7. doi: 10.1073/pnas.90.9.3993
68. Wilcox JE, Hershberger RE. Genetic cardiomyopathies. Curr Opin Cardiol. (2018) 33:354–62. doi: 10.1097/HCO.0000000000000512
69. Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, et al. Embryonic stem cell lines derived from human blastocysts. Science (1998) 282:1145–7. doi: 10.1126/science.282.5391.1145
70. Reubinoff BE, Pera MF, Fong CY, Trounson A, Bongso A. Embryonic stem cell lines from human blastocysts: somatic differentiation in vitro. Nat Biotechnol. (2000) 18:399–404. doi: 10.1038/74447
71. Kehat I, Kenyagin-Karsenti D, Snir M, Segev H, Amit M, Gepstein A, et al. Human embryonic stem cells can differentiate into myocytes with structural and functional properties of cardiomyocytes. J Clin Invest. (2001) 108:407–14. doi: 10.1172/JCI200112131
72. Yang L, Soonpaa MH, Adler ED, Roepke TK, Kattman SJ, Kennedy M, et al. Human cardiovascular progenitor cells develop from a KDR+ embryonic-stem-cell-derived population. Nature (2008) 453:524–8. doi: 10.1038/nature06894
73. Laflamme MA, Chen KY, Naumova AV, Muskheli V, Fugate JA, Dupras SK, e al. Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nat Biotechnol. (2007) 25:1015–24. doi: 10.1038/nbt1327
74. Lian X, Hsiao C, Wilson G, Zhu K, Hazeltine LB, Azarin SM, et al. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc Natl Acad Sci USA. (2012) 109:E1848–57. doi: 10.1073/pnas.1200250109
75. Lian X, Zhang J, Azarin SM, Zhu K, Hazeltine LB, Bao X, et al. Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/beta-catenin signaling under fully defined conditions. Nat Protoc. (2013) 8:162–75. doi: 10.1038/nprot.2012.150
76. Burridge PW, Matsa E, Shukla P, Lin ZC, Churko JM, Ebert AD, et al. Chemically defined generation of human cardiomyocytes. Nat Methods (2014) 11:855–60. doi: 10.1038/nmeth.2999
77. Weng Z, Kong CW, Ren L, Karakikes I, Geng L, He J, et al. A simple, cost-effective but highly efficient system for deriving ventricular cardiomyocytes from human pluripotent stem cells. Stem Cells Dev. (2014) 23:1704–16. doi: 10.1089/scd.2013.0509
78. Zhang Q, Jiang J, Han P, Yuan Q, Zhang J, Zhang X, et al. Direct differentiation of atrial and ventricular myocytes from human embryonic stem cells by alternating retinoid signals. Cell Res. (2011) 21:579–87. doi: 10.1038/cr.2010.163
79. Lee JH, Protze SI, Laksman Z, Backx PH, Keller GM. Human pluripotent stem cell-derived atrial and ventricular cardiomyocytes develop from distinct mesoderm populations. Cell Stem Cell (2017) 21:179–94 e4. doi: 10.1016/j.stem.2017.07.003
80. Devalla HD, Schwach V, Ford JW, Milnes JT, El-Haou S, Jackson C, et al. Atrial-like cardiomyocytes from human pluripotent stem cells are a robust preclinical model for assessing atrial-selective pharmacology. EMBO Mol Med. (2015) 7:394–410. doi: 10.15252/emmm.201404757
81. Sharma A, Li G, Rajarajan K, Hamaguchi R, Burridge PW, Wu SM. Derivation of highly purified cardiomyocytes from human induced pluripotent stem cells using small molecule-modulated differentiation and subsequent glucose starvation. J Vis Exp. (2015) e52628. doi: 10.3791/52628
82. DeLaughter DM, Bick AG, Wakimoto H, McKean D, Gorham JM, Kathiriya IS, et al. Single-cell resolution of temporal gene expression during heart development. Dev Cell (2016) 39:480–90. doi: 10.1016/j.devcel.2016.10.001
83. Matsa E, Ahrens JH, Wu JC. Human induced pluripotent stem cells as a platform for personalized and precision cardiovascular medicine. Physiol Rev. (2016) 96:1093–126. doi: 10.1152/physrev.00036.2015
84. Addis RC, Epstein JA. Induced regeneration–the progress and promise of direct reprogramming for heart repair. Nat Med. (2013) 19:829–36. doi: 10.1038/nm.3225
85. Iorga B, Schwanke K, Weber N, Wendland M, Greten S, Piep B, et al. Differences in contractile function of myofibrils within human embryonic stem cell-derived cardiomyocytes vs. adult ventricular myofibrils are related to distinct sarcomeric protein isoforms. Front Physiol. (2017) 8:1111. doi: 10.3389/fphys.2017.01111
86. Sallam K, Kodo K, Wu JC. Modeling inherited cardiac disorders. Circ J. (2014) 78:784–94. doi: 10.1253/circj.CJ-14-0182
87. Addis RC, Ifkovits JL, Pinto F, Kellam LD, Esteso P, Rentschler S, et al. Optimization of direct fibroblast reprogramming to cardiomyocytes using calcium activity as a functional measure of success. J Mol Cell Cardiol. (2013) 60:97–106. doi: 10.1016/j.yjmcc.2013.04.004
88. Ronaldson-Bouchard K, Ma SP, Yeager K, Chen T, Song L, Sirabella D, Morikawa K, et al. Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nature (2018) 556:239–43. doi: 10.1038/s41586-018-0016-3
89. Nunes SS, Miklas JW, Liu J, Aschar-Sobbi R, Xiao Y, Zhang B, et al. Biowire: a platform for maturation of human pluripotent stem cell-derived cardiomyocytes. Nat Methods (2013) 10:781–7. doi: 10.1038/nmeth.2524
90. Radisic M, Park H, Shing H, Consi T, Schoen FJ, Langer R, et al. Functional assembly of engineered myocardium by electrical stimulation of cardiac myocytes cultured on scaffolds. Proc Natl Acad Sci USA. (2004) 101:18129–34. doi: 10.1073/pnas.0407817101
91. Hirt MN, Boeddinghaus J, Mitchell A, Schaaf S, Bornchen C, Muller C, et al. Functional improvement and maturation of rat and human engineered heart tissue by chronic electrical stimulation. J Mol Cell Cardiol. (2014) 74:151–61. doi: 10.1016/j.yjmcc.2014.05.009
92. Birket MJ, Casini S, Kosmidis G, Elliott DA, Gerencser AA, Baartscheer A, et al. PGC-1alpha and reactive oxygen species regulate human embryonic stem cell-derived cardiomyocyte function. Stem Cell Rep. (2013) 1:560–74. doi: 10.1016/j.stemcr.2013.11.008
93. Ribeiro AJ, Ang YS, Fu JD, Rivas RN, Mohamed TM, Higgs GC, et al. Contractility of single cardiomyocytes differentiated from pluripotent stem cells depends on physiological shape and substrate stiffness. Proc Natl Acad Sci USA. (2015) 112:12705–10. doi: 10.1073/pnas.1508073112
94. Feaster TK, Cadar AG, Wang L, Williams CH, Chun YW, Hempel JE, et al. Matrigel mattress: a method for the generation of single contracting human-induced pluripotent stem cell-derived cardiomyocytes. Circ Res. (2015) 117:995–1000. doi: 10.1161/CIRCRESAHA.115.307580
95. Motlagh D, Senyo SE, Desai TA, Russell B. Microtextured substrata alter gene expression, protein localization and the shape of cardiac myocytes. Biomaterials (2003) 24:2463–76. doi: 10.1016/S0142-9612(02)00644-0
96. Engler AJ, Carag-Krieger C, Johnson CP, Raab M, Tang HY, Speicher DW, et al. Embryonic cardiomyocytes beat best on a matrix with heart-like elasticity: scar-like rigidity inhibits beating. J Cell Sci. (2008) 121:3794–802. doi: 10.1242/jcs.029678
97. Fink C, Ergun S, Kralisch D, Remmers U, Weil J, Eschenhagen T. Chronic stretch of engineered heart tissue induces hypertrophy and functional improvement. FASEB J. (2000) 14:669–79. doi: 10.1096/fasebj.14.5.669
98. Leonard A, Bertero A, Powers JD, Beussman KM, Bhandari S, Regnier M, et al. Afterload promotes maturation of human induced pluripotent stem cell derived cardiomyocytes in engineered heart tissues. J Mol Cell Cardiol. (2018) 118:147–58. doi: 10.1016/j.yjmcc.2018.03.016
99. Shimko VF, Claycomb WC. Effect of mechanical loading on three-dimensional cultures of embryonic stem cell-derived cardiomyocytes. Tissue Eng. (2008) 14:49–58. doi: 10.1089/ten.a.2007.0092
100. Mihic A, Li J, Miyagi Y, Gagliardi M, Li SH, Zu J, Weisel RD, et al. The effect of cyclic stretch on maturation and 3D tissue formation of human embryonic stem cell-derived cardiomyocytes. Biomaterials (2014) 35:2798–808. doi: 10.1016/j.biomaterials.2013.12.052
101. Sun N, Yazawa M, Liu J, Han L, Sanchez-Freire V, Abilez OJ, et al. Patient-specific induced pluripotent stem cells as a model for familial dilated cardiomyopathy. Sci Transl Med. (2012) 4:130ra47. doi: 10.1126/scitranslmed.3003552
102. Broughton KM, Li J, Sarmah E, Warren CM, Lin YH, Henze MP, et al. A myosin activator improves actin assembly and sarcomere function of human-induced pluripotent stem cell-derived cardiomyocytes with a troponin T point mutation. Am J Physiol. (2016) 311:H107–17. doi: 10.1152/ajpheart.00162.2016
103. Han L, Li Y, Tchao J, Kaplan AD, Lin B, Li Y, et al. Study familial hypertrophic cardiomyopathy using patient-specific induced pluripotent stem cells. Cardiovasc Res. (2014) 104:258–69. doi: 10.1093/cvr/cvu205
104. Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. (2013) 31:827–32. doi: 10.1038/nbt.2647
105. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science (2012) 337:816–21. doi: 10.1126/science.1225829
106. Mathew J, Zahavich L, Lafreniere-Roula M, Wilson J, George K, Benson L, Bowdin S, et al. Utility of genetics for risk stratification in pediatric hypertrophic cardiomyopathy. Clin Genet. (2018) 93:310–9. doi: 10.1111/cge.13157
107. Ma N, Zhang J, Itzhaki I, Zhang SL, Chen H, Haddad F, et al. Determining the pathogenicity of a genomic variant of uncertain significance using CRISPR/Cas9 and human-induced pluripotent stem cells. Circulation (2018) doi: 10.1161/CIRCULATIONAHA.117.032273
108. Hulsmans M, Clauss S, Xiao L, Aguirre AD, King KR, Hanley A, et al. Macrophages facilitate electrical conduction in the heart. Cell (2017) 169:510–22 e20. doi: 10.1016/j.cell.2017.03.050
109. McArthur L, Chilton L, Smith GL, Nicklin SA. Electrical consequences of cardiac myocyte: fibroblast coupling. Biochem Soc Trans. (2015) 43:513–8. doi: 10.1042/BST20150035
110. Miragoli M, Gaudesius G, Rohr S. Electrotonic modulation of cardiac impulse conduction by myofibroblasts. Circ Res. (2006) 98:801–10. doi: 10.1161/01.RES.0000214537.44195.a3
111. Daily NJ, Du Z-W, Wakatsuki T. High-throughput phenotyping of human induced pluripotent stem cell-derived cardiomyocytes and neurons using electric field stimulation and high-speed fluorescence imaging. ASSAY Drug Dev Tech. (2017) 15:178–88. doi: 10.1089/adt.2017.781
112. Daily NJ, Santos R, Vecchi J, Kemanli P, Wakatsuki T. Calcium transient assays for compound screening with human iPSC-derived cardiomyocytes: evaluating new tools. J Evol Stem Cell Res. (2017) 1:1–11. doi: 10.14302/issn.2574-4372.jesr-16-1395
113. Daily NJ, Yin Y, Kemanli P, Wakatsuki T. Improving cardiac action potential measurements: 2d and 3d cell culture. J Bioeng Biomed Sci. (2015) 5:168. doi: 10.4172/2155-9538.1000168
114. Timmermann V, Dejgaard LA, Haugaa KH, Edwards AG, Sundnes J, McCulloch AD, et al. An integrative appraisal of mechano-electric feedback mechanisms in the heart. Prog Biophys Mol Biol. (2017) 130:404–17. doi: 10.1016/j.pbiomolbio.2017.08.008
115. Eschenhagen T, Fink C, Remmers U, Scholz H, Wattchow J, Weil J, et al. Three-dimensional reconstitution of embryonic cardiomyocytes in a collagen matrix: a new heart muscle model system. FASEB J. (1997) 11:683–94. doi: 10.1096/fasebj.11.8.9240969
116. Wijnker PJ, Friedrich FW, Dutsch A, Reischmann S, Eder A, Mannhardt I, et al. Comparison of the effects of a truncating and a missense MYBPC3 mutation on contractile parameters of engineered heart tissue. J Mol Cell Cardiol. (2016) 97:82–92. doi: 10.1016/j.yjmcc.2016.03.003
117. Mosqueira D, Mannhardt I, Bhagwan JR, Lis-Slimak K, Katili P, Scott E, et al. CRISPR/Cas9 editing in human pluripotent stem cell-cardiomyocytes highlights arrhythmias, hypocontractility, and energy depletion as potential therapeutic targets for hypertrophic cardiomyopathy. Eur Heart J. (2018). doi: 10.1093/eurheartj/ehy249. [Epub ahead of print].
118. Stillitano F, Turnbull IC, Karakikes I, Nonnenmacher M, Backeris P, Hulot JS, et al. Genomic correction of familial cardiomyopathy in human engineered cardiac tissues. Eur Heart J. (2016) 37:3282–4. doi: 10.1093/eurheartj/ehw307
119. Legant WR, Pathak A, Yang MT, Deshpande VS, McMeeking RM, et al. Microfabricated tissue gauges to measure and manipulate forces from 3D microtissues. Proc Natl Acad Sci USA. (2009) 106:10097–102. doi: 10.1073/pnas.0900174106
120. Lemoine MD, Mannhardt I, Breckwoldt K, Prondzynski M, Flenner F, Ulmer B, et al. Human iPSC-derived cardiomyocytes cultured in 3D engineered heart tissue show physiological upstroke velocity and sodium current density. Sci Rep. (2017) 7:5464. doi: 10.1038/s41598-017-05600-w
121. Boudou T, Legant WR, Mu A, Borochin MA, Thavandiran N, Radisic M, et al. A microfabricated platform to measure and manipulate the mechanics of engineered cardiac microtissues. Tissue Eng Part A (2012) 18:910–9. doi: 10.1089/ten.tea.2011.0341
122. Kural MH, Billiar KL. Regulating tension in three-dimensional culture environments. Exp Cell Res. (2013) 319:2447–59. doi: 10.1016/j.yexcr.2013.06.019
123. Eyckmans J, Chen CS. 3D culture models of tissues under tension. J Cell Sci. (2017) 130:63–70. doi: 10.1242/jcs.198630
124. Beauchamp P, Moritz W, Kelm JM, Ullrich ND, Agarkova I, Anson BD, et al. Development and characterization of a scaffold-free 3D spheroid model of induced pluripotent stem cell-derived human cardiomyocytes. Tissue Eng Part C Methods (2015) 21:852–61. doi: 10.1089/ten.tec.2014.0376
125. Kim TY, Kofron CM, King ME, Markes AR, Okundaye AO, Qu Z, et al. Directed fusion of cardiac spheroids into larger heterocellular microtissues enables investigation of cardiac action potential propagation via cardiac fibroblasts. PLoS One (2018) 13:e0196714. doi: 10.1371/journal.pone.0196714
126. Sun X, Nunes SS. Biowire platform for maturation of human pluripotent stem cell-derived cardiomyocytes. Methods (2016) 101:21–6. doi: 10.1016/j.ymeth.2015.11.005
127. Schroer AK, Shotwell MS, Sidorov VY, Wikswo JP, Merryman WD. I-Wire heart-on-a-chip II: biomechanical analysis of contractile, three-dimensional cardiomyocyte tissue constructs. Acta Biomater. (2017) 48:79–87. doi: 10.1016/j.actbio.2016.11.010
128. Sidorov VY, Samson PC, Sidorova TN, Davidson JM, Lim CC, Wikswo JP. I-wire heart-on-a-chip I: three-dimensional cardiac tissue constructs for physiology and pharmacology. Acta Biomater. (2017) 48:68–78. doi: 10.1016/j.actbio.2016.11.009
129. Mannhardt I, Breckwoldt K, Letuffe-Breniere D, Schaaf S, Schulz H, Neuber C, et al. Human engineered heart tissue: analysis of contractile force. Stem Cell Rep. (2016) 7:29–42. doi: 10.1016/j.stemcr.2016.04.011
130. Mannhardt I, Saleem U, Benzin A, Schulze T, Klampe B, Eschenhagen T, et al. Automated contraction analysis of human engineered heart tissue for cardiac drug safety screening. J Vis Exp. (2017) e55461. doi: 10.3791/55461
131. Bielawski KS, Leonard A, Bhandari S, Murry CE, Sniadecki NJ. Real-time force and frequency analysis of engineered human heart tissue derived from induced pluripotent stem cells using magnetic sensing. Tissue Eng Part C Methods (2016) 22:932–40. doi: 10.1089/ten.tec.2016.0257
132. Shadrin IY, Allen BW, Qian Y, Jackman CP, Carlson AL, Juhas ME, et al. Cardiopatch platform enables maturation and scale-up of human pluripotent stem cell-derived engineered heart tissues. Nat Commun. (2017) 8:1825. doi: 10.1038/s41467-017-01946-x
133. Abilez OJ, Tzatzalos E, Yang H, Zhao MT, Jung G, Zollner AM, et al. Passive stretch induces structural and functional maturation of engineered heart muscle as predicted by computational modeling. Stem Cells (2018) 36:265–77. doi: 10.1002/stem.2732
134. Liaw NY, Zimmermann WH. Mechanical stimulation in the engineering of heart muscle. Adv Drug Deliv Rev. (2016) 96:156–60. doi: 10.1016/j.addr.2015.09.001
135. Ruan JL, Tulloch NL, Razumova MV, Saiget M, Muskheli V, Pabon L, et al. Mechanical stress conditioning and electrical stimulation promote contractility and force maturation of induced pluripotent stem cell-derived human cardiac tissue. Circulation (2016) 134:1557–67. doi: 10.1161/CIRCULATIONAHA.114.014998
136. Chen K, Vigliotti A, Bacca M, McMeeking RM, Deshpande VS, Holmes JW. Role of boundary conditions in determining cell alignment in response to stretch. Proc Natl Acad Sci USA. (2018) 115:986–91. doi: 10.1073/pnas.1715059115
137. Zhu R, Blazeski A, Poon E, Costa KD, Tung L, Boheler KR. Physical developmental cues for the maturation of human pluripotent stem cell-derived cardiomyocytes. Stem Cell Res Ther. (2014) 5:117. doi: 10.1186/scrt507
138. Ulmer BM, Stoehr A, Schulze ML, Patel S, Gucek M, Mannhardt I, Funcke S, Murphy E, Eschenhagen T, et al. Contractile work contributes to maturation of energy metabolism in hiPSC-derived cardiomyocytes. Stem Cell Rep. (2018) 10:834–47. doi: 10.1016/j.stemcr.2018.01.039
139. Sun X, Nunes SS. Bioengineering approaches to mature human pluripotent stem cell-derived cardiomyocytes. Front Cell Dev Biol. (2017) 5:19. doi: 10.3389/fcell.2017.00019
140. Yamada N, Okano T, Sakai H, Karikusa F, Sawasaki Y, Sakurai Y. Thermo-responsive polymeric surfaces; control of attachment and detachment of cultured cells. Die Makromolekulare Chemie (1990) 11:571–6. doi: 10.1002/marc.1990.030111109
141. Bailey JL, Critser PJ, Whittington C, Kuske JL, Yoder MC, Voytik-Harbin SL. Collagen oligomers modulate physical and biological properties of three-dimensional self-assembled matrices. Biopolymers (2011) 95:77–93. doi: 10.1002/bip.21537
142. Dempfle CE, Heene DL. Isolation and purification of fibrinogen/fibrin degradation products by chromatography on protamine-agarose. Thromb Res. (1987) 48:223–32. doi: 10.1016/0049-3848(87)90419-1
143. Weinberger F, Mannhardt I, Eschenhagen T. Engineering cardiac muscle tissue: a maturating field of research. Circ Res. (2017) 120:1487–500. doi: 10.1161/CIRCRESAHA.117.310738
144. Lesman A, Gepstein L, Levenberg S. Cell tri-culture for cardiac vascularization. Methods Mol Biol. (2014) 1181:131–7. doi: 10.1007/978-1-4939-1047-2_12
145. Kurokawa YK, George SC. Tissue engineering the cardiac microenvironment: multicellular microphysiological systems for drug screening. Adv Drug Deliv Rev. (2016) 96:225–33. doi: 10.1016/j.addr.2015.07.004
146. Huebsch N, Loskill P, Deveshwar N, Spencer CI, Judge LM, Mandegar MA, et al. Miniaturized iPS-cell-derived cardiac muscles for physiologically relevant drug response analyses. Sci Rep. (2016) 6:24726. doi: 10.1038/srep24726
147. Vandenburgh H, Shansky J, Benesch-Lee F, Barbata V, Reid J, Thorrez L, et al. Drug-screening platform based on the contractility of tissue-engineered muscle. Muscle Nerve (2008) 37:438–47. doi: 10.1002/mus.20931
148. Pasqualini FS, Agarwal A, O'Connor BB, Liu Q, Sheehy SP, Parker KK. Traction force microscopy of engineered cardiac tissues. PLoS ONE (2018) 13:e0194706. doi: 10.1371/journal.pone.0194706
149. Zhao R, Boudou T, Wang WG, Chen CS, Reich DH. Decoupling cell and matrix mechanics in engineered microtissues using magnetically actuated microcantilevers. Adv Mater. (2013) 25:1699–705. doi: 10.1002/adma.201203585
150. Marquez JP, Legant W, Lam V, Cayemberg A, Elson E, Wakatsuki T. High-throughput measurements of hydrogel tissue construct mechanics. Tissue Eng Part C Methods (2009) 15:181–90. doi: 10.1089/ten.tec.2008.0347
151. Asnes CF, Marquez JP, Elson EL, Wakatsuki T. Reconstitution of the Frank-Starling mechanism in engineered heart tissues. Biophys J. (2006) 91:1800–10. doi: 10.1529/biophysj.105.065961
152. Lam V, Bigley T, Terhune SS, Wakatsuki T. A method for quantifying mechanical properties of tissue following viral infection. PLoS ONE (2012) 7:e42197. doi: 10.1371/journal.pone.0042197
153. Lam V, Wakatsuki T. Hydrogel tissue construct-based high-content compound screening. J Biomol Screen (2011) 16:120–8. doi: 10.1177/1087057110388269
154. Wu Y, Cazorla O, Labeit D, Labeit S, Granzier H. Changes in titin and collagen underlie diastolic stiffness diversity of cardiac muscle. J Mol Cell Cardiol. (2000) 32:2151–62. doi: 10.1006/jmcc.2000.1281
155. Tiburcy M, Hudson JE, Balfanz P, Schlick S, Meyer T, Chang Liao ML, et al. Defined engineered human myocardium with advanced maturation for applications in heart failure modeling and repair. Circulation (2017) 135:1832–47. doi: 10.1161/CIRCULATIONAHA.116.024145
156. Morgan BP, Muci AP, Lu P, Qian X, Tochimoto T, Smith WW, et al. Discovery of omecamtiv mecarbil the first, selective, small molecule activator of cardiac myosin. ACS Med Chem Lett. (2010) 1:472–7. doi: 10.1021/ml100138q
157. Liu Y, White HD, Belknap B, Winkelmann DA, Forgacs E. Omecamtiv mecarbil modulates the kinetic and motile properties of porcine beta-cardiac myosin. Biochemistry (2015) 54:1963–75. doi: 10.1021/bi5015166
158. Woody SG, Barua B, Winkelmann DA, Goldman YE, Ostap EM. Positive cardiac inotrope, omecamtiv mecarbil, activates muscle despite suppressing the myosin working stroke. Biorxiv [Preprint] (2018). doi: 10.1101/298141
159. Nagy L, Kovacs A, Bodi B, Pasztor ET, Fulop GA, Toth A, et al. The novel cardiac myosin activator omecamtiv mecarbil increases the calcium sensitivity of force production in isolated cardiomyocytes and skeletal muscle fibres of the rat. Br J Pharmacol. (2015) 172:4506–18. doi: 10.1111/bph.13235
160. Sonin DL, Wakatsuki T, Routhu KV, Harmann LM, Petersen M, Meyer J, et al. Protease-activated receptor 1 inhibition by SCH79797 attenuates left ventricular remodeling and profibrotic activities of cardiac fibroblasts. J Cardiovasc Pharmacol Ther. (2013) 18:460–75. doi: 10.1177/1074248413485434
Keywords: heart failure, tissue engineering, length-tension relationship, gene editing, human induced pluripotent stem cells, high-throughput screening, rare heart disease, drug discovery
Citation: Greenberg MJ, Daily NJ, Wang A, Conway MK and Wakatsuki T (2018) Genetic and Tissue Engineering Approaches to Modeling the Mechanics of Human Heart Failure for Drug Discovery. Front. Cardiovasc. Med. 5:120. doi: 10.3389/fcvm.2018.00120
Received: 30 May 2018; Accepted: 13 August 2018;
Published: 19 September 2018.
Edited by:
David F. Stowe, Medical College of Wisconsin, United StatesReviewed by:
Nazareno Paolocci, Johns Hopkins University, United StatesRaffaele Altara, Oslo University Hospital, Norway
Justus Stenzig, Universität Hamburg, Germany
Copyright © 2018 Greenberg, Daily, Wang, Conway and Wakatsuki. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael J. Greenberg, Z3JlZW5iZXJnQHd1c3RsLmVkdQ==
Tetsuro Wakatsuki, dGV0c3Vyb0BpbnZpdm9zY2llbmNlcy5jb20=