 Aneesh K. Ramaswamy
Aneesh K. Ramaswamy David A. Vorp1,2,3,4,5
David A. Vorp1,2,3,4,5 Justin S. Weinbaum
Justin S. Weinbaum- 1Department of Bioengineering, University of Pittsburgh, Pittsburgh, PA, United States
- 2McGowan Institute for Regenerative Medicine, University of Pittsburgh, Pittsburgh, PA, United States
- 3Department of Surgery, University of Pittsburgh, Pittsburgh, PA, United States
- 4Department of Cardiothoracic Surgery, University of Pittsburgh, Pittsburgh, PA, United States
- 5Department of Chemical and Petroleum Engineering, University of Pittsburgh, Pittsburgh, PA, United States
- 6Department of Pathology, University of Pittsburgh, Pittsburgh, PA, United States
Modern regenerative medicine, and tissue engineering specifically, has benefited from a greater appreciation of the native extracellular matrix (ECM). Fibronectin, collagen, and elastin have entered the tissue engineer's toolkit; however, as fully decellularized biomaterials have come to the forefront in vascular engineering it has become apparent that the ECM is comprised of more than just fibronectin, collagen, and elastin, and that cell-instructive molecules known as matricellular proteins are critical for desired outcomes. In brief, matricellular proteins are ECM constituents that contrast with the canonical structural proteins of the ECM in that their primary role is to interact with the cell. Of late, matricellular genes have been linked to diseases including connective tissue disorders, cardiovascular disease, and cancer. Despite the range of biological activities, this class of biomolecules has not been actively used in the field of regenerative medicine. The intent of this review is to bring matricellular proteins into wider use in the context of vascular tissue engineering. Matricellular proteins orchestrate the formation of new collagen and elastin fibers that have proper mechanical properties—these will be essential components for a fully biological small diameter tissue engineered vascular graft (TEVG). Matricellular proteins also regulate the initiation of thrombosis via fibrin deposition and platelet activation, and the clearance of thrombus when it is no longer needed—proper regulation of thrombosis will be critical for maintaining patency of a TEVG after implantation. Matricellular proteins regulate the adhesion, migration, and proliferation of endothelial cells—all are biological functions that will be critical for formation of a thrombus-resistant endothelium within a TEVG. Lastly, matricellular proteins regulate the adhesion, migration, proliferation, and activation of smooth muscle cells—proper control of these biological activities will be critical for a TEVG that recellularizes and resists neointimal formation/stenosis. We review all of these functions for matricellular proteins here, in addition to reviewing the few studies that have been performed at the intersection of matricellular protein biology and vascular tissue engineering.
Introduction
Modern regenerative medicine, and tissue engineering specifically, has benefited from a greater appreciation of the native extracellular matrix (ECM). Fibronectin, collagen, and elastin have entered the tissue engineer's toolkit: peptides derived from fibronectin are routinely added to synthetic scaffolds to promote adhesion of seeded cells, high collagen content has been desirable for mechanically strong engineered tissues, and elastin has been an agreed-upon target for tissues that rely on recoil such as engineered blood vessels, skin, and lungs. It has become apparent that ECM-based biomaterials obtained by decellularization of native intestine, bladder, and heart tissues have a strong positive influence on in vivo regeneration. This cell-instructive potential includes the recruitment of host stem cells, modulation of the immune system, and promotion of cell differentiation to a beneficial phenotype for repair and later homeostasis. Notably, the ECM molecules present in these decellularized biomaterials go beyond just fibronectin, collagen, and elastin and include cell-instructive molecules known as matricellular proteins.
The term “matricellular” was coined by Paul Bornstein and members of his group in 1995. Of note, a special issue of Matrix Biology was published in the summer of 2014 to celebrate Paul Bornstein's legacy, and is recommended reading for more details on the evolution of this field. In brief, matricellular proteins are ECM constituents that contrast with the canonical structural proteins of the ECM in that their primary role is to interact with the cell. General observations of matricellular activity have included a modulatory effect on cell adhesion (hence their alternate name, “modulatory adhesion proteins”) and mouse phenotypes characterized by an altered response to injury. Of late, matricellular genes have been linked to diseases including connective tissue disorders, cardiovascular disease, and cancer. Despite the range of biological activities, this class of biomolecules has not been actively used in the field of regenerative medicine. The intent of this review is to bring these proteins (and their constitutive active peptides) into wider use in the context of vascular tissue engineering. A list of the proteins reviewed here can be found in Table 1.
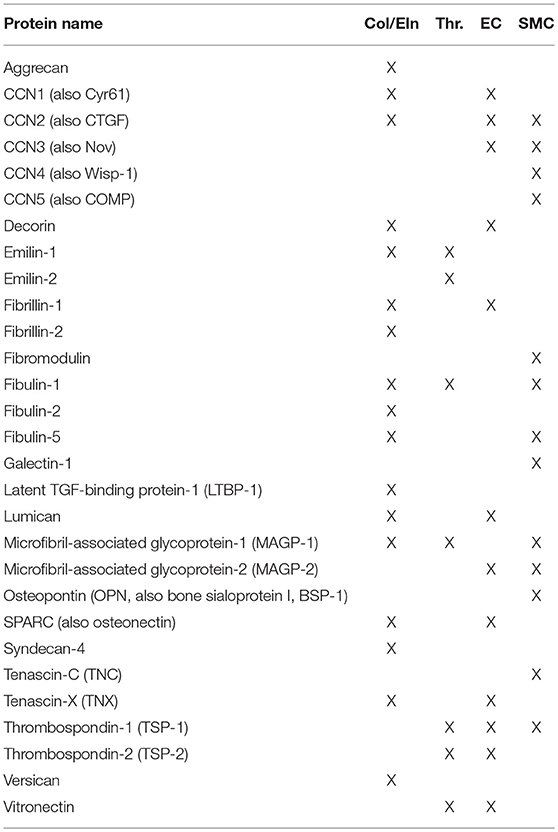
Table 1. Matricellular proteins discussed in this review.
In the realm of vascular engineering, there are four key design parameters that must be met for a successful tissue engineered vascular graft (TEVG). First, the graft must have mechanical strength compatible with hemodynamics and local tissue deformation. Second, the implanted graft must present a non-thrombogenic luminal surface; that is, it will not induce activation of the host extrinsic or intrinsic clotting pathways or promote platelet adhesion and activation. Third, the luminal surface of the graft must support the formation and maintenance of an endothelial lining, with the endothelial cell source either an implanted graft or the host. Finally, the interior of the graft wall should be, or become, populated by cells; the most important cell type to populate the new media will be vascular smooth muscle cells. Of note, the smooth muscle cells that repopulate the graft must be restricted from over-proliferation, which could lead to graft blood flow restrictions by neointimal formation and eventual stenosis. In this review, we will examine how the biological activity of matricellular proteins could help vascular engineers achieve each of these four parameters for graft design.
Matricellular Proteins Regulate the Production of Collagen and Elastin, the Key Molecular Determinants of Vascular Biomechanics
The compliance, elastic modulus, and burst strength of grafts have been primary criteria for success. In native vessels, the primary load-bearing proteins in the ECM are collagen and elastin, which combine to give the vessel a biphasic passive response to mechanical stretch (1). To mimic the native vessel, the presence of collagen, elastin, or synthetic substitutes for these molecules has been desirable in engineered grafts. In this section, we will focus on how matricellular proteins regulate the production of collagen and elastin in the context of biological grafts.
Collagen Overview
In the vasculature, the primary collagens which contribute to mechanical strength are the fibrillar collagens, Type I and Type III. Two human genes, COL1A1 and COL1A2, encode the protein components of Type I collagen. In contrast, Type III collagen is produced from a single gene, COL3A1. After transcription and translation, the collagen chains are then post-translationally modified (by proteins such as members of the prolyl hydroxylase and lysyl oxidase (LOX) families) and wound into a collagen triple-helix, comprised of three protein chains per collagen fiber. Collagen fibers are then combined with a staggered overlap into a larger bundle, resulting in fiber cooperativity and a high overall tensile strength of the higher order structure.
Collagen Transcription
Research into the transcriptional control of fibrillar collagens has benefited from the study of scarring in skin. Of note, one hallmark of scarring is the secretion of pro-inflammatory cytokines by immune cells during the wound healing response. The canonical signaling pathway for fibrosis involves transforming growth factor (TGF)-β1. After binding of TGF-β1 to its cellular receptor, the SMAD 2/3 signaling pathway is activated, leading to translocation of SMAD3/4 to the cell's nucleus. SMAD-binding enhancer sequences are found in the promoters for COL1A1 (2), COL1A2 (3), and COL3A1 (4). TGF-β1 functionality is primarily regulated in the vasculature by its binding to the matricellular proteins latent TGF-β binding protein (LTBP)-1 and−4 (5). A molecule with many overlapping functions to TGF-β1 in fibrosis, connective tissue growth factor (CTGF or CCN2) directly promotes collagen synthesis to support either new or healing vascular beds (6), and parallels collagen I expression in pericytes (7).
Collagen Post-translational Modification
In Ehlers-Danlos Syndrome, defects in the vascular wall, particularly in collagen fibers, lead to vascular fragility. One key player in this process is the matricellular protein tenascin-X (TNX). TNX absence results in the loss of structural integrity of the vascular collagenous matrix and a reduction of collagen-processing enzyme efficacy (8). Proteoglycan syndecan-4 promotes post-translational collagen LOX modification directly via its extracellular domain ECsyn4, and indirectly via osteopontin (9). Additionally, stimulation of cardiac fibroblasts with recombinant lumican, a collagen-binding proteoglycan, increased LOX and matrix metalloproteinase-9 (10), leading to hypotheses about the role of lumican in myocardial fibrosis (11) and as a biomarker for acute aortic dissection (12).
Collagen Fiber Assembly
Perhaps the greatest effect of matricellular proteins is on the assembly of collagen fibers, where they act as quality control regulators of collagen fiber size and stability. CCN family member 1 (CCN1), otherwise known as cysteine-rich angiogenic inducer 61 (CYR61), modulates collagen I stability through surface integrins in response to serum concentration levels of basic fibroblast growth factor (bFGF) and vascular endothelial growth factor (VEGF) (6). Secreted protein acidic and rich in cysteine (SPARC), or osteonectin, plays a vital role in binding and organizing collagen (13). SPARC primarily aggregates in vascular basement membranes, modulating procollagen processing and collagen interactions with cardiac fibroblasts (14) producing a series of smaller and more tightly organized collagen I fibrils (15).
Decorin is a member of the small leucine-rich proteoglycan family of matricellular proteins that sequesters TGF-(beta) activity, with glycoproteomic analysis showing decorin-derived peptide fragments present throughout the cardiac ECM (16). Decorin has been shown to influence collagen matrix organization and gel contraction, binding to collagens I-III, VI, and XIV (17) resulting in increased collagen fibril density and decreased diameter (18). The effects of decorin-derived peptides have been studied as a potential way of modulating collagen fibrillogenesis (19) and neointimal hyperplasia following balloon angioplasty (20, 21). Decorin in the context of aortic aneurysm has a complex role: decorin associates with a lowered risk of rupture in murine abdominal aortic aneurysm (22), but while decorin serves a protective role in early in murine aneurysm formation it also correlates strongly with macrophage matrix metalloproteinase (MMP)-9 expression and lesion location (23).
Elastin Overview
In the vasculature, the property of passive recoil in response to pulsatile blood flow is provided by the elastic fiber. While other proteins are responsible for the assembly and biological activity of the elastic fiber, the primary protein component is elastin. A single elastin gene is present in the mammalian genome, and mutation or heterozygosity of the gene is linked to several human diseases including Williams Syndrome (24), cutis laxa (25), and supravalvular aortic stenosis (26). After transcription and translation, elastin is trafficked to the surface of the cell, where it is chaperoned to fibrillin microfibrils and assembled. Intra and inter-molecular crosslinking of elastin is then performed by members of the lysyl oxidase family, producing a resilient, and elastic macrostructure.
Elastin Transcription
Like collagen, elastin can be upregulated by TGF-β signaling. Uniquely, TGF-β regulation of elastin message level occurs post-transcriptionally through stabilization of the messenger RNA (27).
One component of the elastic fiber, the matricellular protein microfibril-associated glycoprotein-1 (MAGP-1), activates latent TGF-β signaling directly (28), potentially presenting an opportunity for molecular engineers looking to encourage elastogenesis. MAGP-1 inhibits binding of LTBP-1 to fibrillin-1 and could fine tune concentration and deposition of the large latent TGF-β (LLC) complex (29). MAGP-1 null mice showed a lack of competition for LLC-microfibril binding, resulting in a reduced concentration of active TGF-β (28).
An additional potential regulator of TGF-β activity upstream of elastin transcription is the matricellular protein emilin-1, which associates with elastic fiber components (30) and inhibits activation of all three pro-TGF-β molecules as a regulatory mechanism for blood pressure (31, 32).
Elastin Post-translational Modification
Elastic recoil is essential during aortic development and physiological blood flow. A murine vSMC lysyl oxidase (LOX) overexpression model resulted in more organized elastic fibers, stronger collagen assembly, and enhanced fibulin-5 production (33); however, an accompanying local H2O2 concentration increase was seen, suggesting LOX is a source of oxidative vascular wall stress. LOX knockout mice are prone to thoracic aortic aneurysm and dissection and die soon after birth, with a 60% reduction in elastin crosslinks observed in aortic tissue (34), underscoring LOX inactivation as a key prognostic indicator for aneurysm and dissection prognosis. MAGP-1 and fibronectin both repress endothelial cell LOX deposition (35), which is a secondary but significant LOX source within the vascular wall.
Elastic Fiber Assembly
Fibrillin-1 and fibrillin-2 are both important for elastic fiber development and maintenance of a healthy vasculature. Fibrillin-2 displays preferential accumulation in elastin rich areas (36) while fibrillin-1 is more widely expressed, making it unsurprising that Marfan Syndrome patients have a wide spectrum of pathologies (37). Fibrillin-1 (38) and fibrillin-2 (39) deficient mice both die soon after birth due to aortic rupture, although qualitative differences were observed in the medial wall between the two forms of fibrillin deficiency. One theory for the different modes of action of the fibrillin isoforms is that initial fibrillin-2 involvement aids aortic matrix stability, but continued fibrillin-1 expression is essential for maturation and post-neonatal vascular function (39). This suggests that vascular engineers concerned with repair modulation must give equal attention to the initial embryonic role of fibrillins as well as adult phenotypes in order to achieve a stable engineered vessel.
Fibulin-2, a protein that binds extracellular ligands, is thought to provide redundancy with fibulin-1 (which interacts with elastin precursor tropoelastin), since fibulin-1 knockout mice produce vessels with viable elastic fibers (40). Fibulin-2 interacts with virtually all elastin precursors [though it is not associated with tropoelastin deposition by fibroblasts (41)] and is expressed in basement membrane of heart, with an enhanced role during development (42). Fibulin-2 has the highest elastin-binding affinity of the fibulins and interacts with fibulin-5 to form the elastic lamina by directing elastic fiber microassembly during development and after injury (43). Fibulin-5 is itself an elastin binding protein involved in primary organization and assembly of elastic fibers (44). In a mouse fibrosis model, loss of fibulin-5 resulted in a reduction in aortic stiffness, possibly halting the stiffness-induced local inflammatory response and deposition of ECM (45). Based on the importance of the fibulins, a calcium-dependent elastin binding protein structure domain mimicking aspects of the fibulin family could be replicated to develop a therapeutic to aid in elastin assembly (46) or interrupt the pro-fibrotic feedback loop.
Fibulin-1 and aggrecan content within the aortic wall have recently shown key roles in age-related aortic stiffening (47), with significant deposits of aggrecan and versican accumulation found during proteomic analysis of human thoracic aortic aneurysm and dissection samples (48) due to either increased synthesis or decreased proteolytic turnover. Given high stiffening conditions and predisposition toward dissection and aneurysm, fibulin-1, aggrecan, and versican present as a potential biomarker combination for aneurysmal risk evaluation.
Matricellular Proteins That Limit Thrombosis
In large diameter vascular grafts, such as those used to repair the thoracic aorta, synthetic materials have shown great success. However, when the same synthetic materials (e.g., PTFE) have been used to make small diameter grafts (< 5 mm), thrombosis has been a major failure mechanism (49). Indeed, chemical incorporation of non-thrombogenic coatings (50) and seeding of cells that secrete pro-fibrinolytic factors (51) have been used by engineers to reduce thrombotic failure. The size of the vessel has a clear inverse relationship with the tendency to occlude, which results from the activation of thrombin and platelets (52). Upon activation of thrombin, fibrinogen in serum is converted to fibrin, which forms an insoluble fibrous mesh. Activated platelets adhere to the vascular wall and fibrin, and signal to circulating cells. The net effect of these two activation events is the formation of a thrombus loaded with fibrin and platelets that eventually occludes the small vessel. In this section we will examine how matricellular proteins regulate thrombin activation, ensuing fibrin homeostasis, as well as platelet activation in order to assess new anti-thrombotic engineering strategies.
Thrombin Activation
Thrombosis occurs when prothrombin in the blood is cleaved and thereby gains enzymatic activity. Thrombin activation can occur through one of two distinct pathways. The extrinsic pathway is initiated by trauma and is the primary initiator of the thrombosis cascade, while the intrinsic pathway is initiated through complex formation on collagen and functions as an amplification method for homeostasis. Both pathways result in downstream activation of Factor X and subsequent thrombin activation, but the intrinsic pathway is often associated with inflammatory responses following biomedical material implantation. In either case, activated thrombin cleaves its substrate, fibrinogen, which releases fibrinogen degradation products and initiates fibril formation.
The matricellular protein vitronectin binds to plasminogen activator inhibitor 1 (PAI-1), inhibiting downregulation of PAI-1 via its receptor, LRP (53). Vitronectin is co-released with PAI-1 as a counterbalance mechanism during platelet activation and incorporates into the clot where it can modulate PAI-1 activity (54).
Fibulin-1, a fibrinogen binding blood-borne matricellular protein, displays a characteristic overlapping pattern with fibrinogen within atheromatous regions displaying fresh thrombi. This suggests that fibulin-1 correlates with thrombotic aspects of atherosclerosis and on-site thrombus formation, which serve as the leading side-effect of vascular therapies (55).
Platelet Activation
Platelet activation is a key companion of thrombosis. Platelets interact with the vessel wall by first “rolling” along the surface, then attaching and spreading through integrin-mediated interactions. In order to activate, platelets must bind to exposed extracellular matrix, generally via the interaction of integrin on the cell surface with type I collagen.
Matricellular proteins in the thrombospondin (TSP) family (of which there are five known members) have key regulatory roles for both platelet recruitment and platelet aggregation. TSP-1 regulates platelet recruitment and anti-ADAMTS13 antibodies within stimulated endothelium (56). TSP-1 also stimulates platelet aggregation by blocking the antithrombotic activity of nitric oxide (NO)/cGMP signaling, resulting in vessel occlusion if it occurs in vascular engineered settings like balloon angioplasty, stent expansion, or therapeutic delivery (57). TSP-1 and CD36 have a co-dependent effect on platelet adhesion and collagen-dependent thrombus stabilization, with CD36 serving in an anchoring role in a TSP-1 knockout model (58). Emilin-2 regulates platelet activation/contractility and aggregation via adenosine diphosphate, collagen, and thrombin; emilin-1 also promotes platelet aggregation but in contrast reduces clot retraction (59). Both members of the emilin family therefore represent key molecules for platelet regulation.
Many alterations of vascular hemostasis have been identified in mouse models of matricellular protein deficiency. Notably, phenotypes of altered hemostasis have been difficult to identify because they are only revealed in response to injury. Mice deficient for either TSP-2 or MAGP-1 exhibit bleeding diathesis; in the case of the latter, this phenotype can be rescued with intravenous injection of recombinant protein (60). Lack of MAGP-1 alters the morphology of the thrombus, which is slow to permanently establish itself in the site of vessel damage (60, 61). The phenotype of TSP-2 deficient mice is inherent to the extracellular matrix. When fibroblasts from TSP-2 deficient mice are used as the source of a cell derived matrix, this matrix resists thrombosis compared to matrix synthesized by wild-type fibroblasts (62). Current vascular engineering strategies are exploiting this cell-derived matrix as an anti-thrombotic coating (63).
Matricellular Proteins That Support Endothelial Formation and Maintenance
Physiological control of thrombosis is primarily performed by the endothelium lining the luminal surface of a blood vessel. To mimic the native state, an implanted vascular graft should ideally be “pre-coated” with a permanent, antithrombotic lining; unfortunately, this is rarely a feasible strategy. In contrast, a vascular graft that encourages recellularization by endothelial cells (ECs) from the host, and treatment with anticoagulants during this recellularization period, is more often used. The design parameters responsible for proper formation of a new endothelial lining are endothelial cell adhesion, migration, proliferation, and survival. In this section, we will illustrate how these functions are modulated by matricellular proteins in vitro and in vivo.
Endothelial Cell Adhesion and Migration
CCN1 binding with αvβ3 and αIIβ3 integrins promotes both adhesion and migration of ECs (64). A dose-dependent adhesion enhancement of ECs was also shown using CCN2 within a 2D in vitro model (65). CCN1 also enhances VEGF signaling by promoting VEGF production (66) and enhancing VEGF-R2 phosphorylation (67). Together, enhanced EC tubule formation (68, 69) and nitric oxide production (70) are observed with CCN1 treatment. Additionally, CCN3 has been shown to stimulate EC adhesion and migration in an αvβ3 / α5β1 dependent manner (71). MAGP-2 antagonizes Notch signaling to promote cell sprouting and EC migration (72, 73). Initial formation of endothelial tubes is also promoted by SPARC, enabled primarily through TGF-β1 inhibition and preceded by pericyte migration (74).
Vitronectin, a serum glycoprotein, is found at high concentrations to promote differentiation of EC/SMC phenotypes (75). ECs bind to vitronectin via heparin sulfate proteoglycans (76), leading to EC spreading (77). αvβ3 integrin binding has shown to decrease cellular motility in vitro on HUVECs in vitronectin coated flasks (78), and can be a target for vitronectin-focused vascular engineering. Thrombogenic PAI-1, co-released with binding target vitronectin, induces HUVEC dysfunction and procoagulant states through mast cell exosome signaling (79), underscoring vitronectin and PAI-1 as EC-linked antithrombogenic vascular engineering targets.
The proteoglycan decorin regulates EC migration and adhesion indirectly, by binding to the receptors for EGF (80, 81) and IGF (82), thereby attenuating downstream Akt and MAPK signaling. An engineered decorin mimic has also been shown to directly promote both endothelial migration and proliferation (83). Lumican, another proteoglycan, inhibits EC migration by interfering with p38 MAPK signaling (84).
TSP-1 has a generally anti-angiogenic effect on ECs (85), mediated by CD36 (86). Similarly, TSP-2 is anti-angiogenic as TSP-2 knockout mice have shown increased angiogenesis (87) and impaired von Willebrand Factor accumulation on secreted ECM (62). In double TSP-1/TSP-2 knockout animals, neoangiogenesis is mediated by MMP9 activation (88). In contrast to the anti-angiogenic activities of TSP-1 and TSP-2, TSP-4 is required for proper increased adhesion, migration, and proliferation of ECs (89, 90). CD47, an essential TSP-1 receptor (91), can mediate TSP-1 function and is therefore another useful target for controlling angiogenesis. CD47 function is altered by a lipid environment (92), so angiogenesis can be manipulated by controlling the VLDL receptor in endothelial cells and thus decrease Akt and MAPK phosphorylation (93). From an engineering perspective, TSP-2 knockout mouse dermal fibroblast secreted ECM was used to coat decellularized rat aorta, resulting in an anti-thrombotic and pro-migratory vascular graft that improved EC recruitment and decreased failure rate following interpositional rat abdominal aortic implantation (63).
Endothelial Cell Proliferation and Survival
CCN1 enhances proliferation of endothelial cells by augmenting bFGF signaling (94), and also promotes endothelial cell survival (64). CCN2's effect is somewhat more complex, as it contains modules that can either enhance (95) or decrease (96) proliferative signaling. CCN3 overexpression suppressed VCAM-1 (which supports adhesion) and normal CCN3 expression reduced monocyte adhesion. TNX increases EC proliferation through interactions with VEGF (97). Fibrillin-1 RGD-containing sequences have the ability to stimulate EC proliferation in vitro (98). TSP-2 alone inhibits growth-factor induced microvascular EC proliferation in a caspase-independent manner (99). SPARC reduces VEGFR-1 after injury, resulting in anti-angiogenic activities early in the development cascade and opening the door for VEGF-A targeted therapeutics that route through VEGFR-1 pathway (100).
Matricellular Proteins That Recruit and Regulate Vascular Smooth Muscle Cells
As described above, a native vessel must possess the mechanical strength and resilience to survive the dynamic forces it will encounter over the course of a human life. In turn, an engineered vessel must be able to outlast the patient; in the case of pediatric patients, grafts must also grow with them. Vascular smooth muscle cells (vSMCs) are essential in the maintenance of vascular walls in vivo, and thus are prime targets for vascular engineers.
For the purposes of this review, we will highlight two main functions of vSMC in small vessels. First, vSMC are responsible for the synthesis, alignment, and maintenance of elastin during development to provide effective passive recoil in response to hemodynamics and local tissue deformation. vSMC also can potentially synthesize new elastin in response to damage; however, it appears that adult vSMC are limited in their elastin expression at the transcriptional level. Second, vSMC cooperate with EC to regulate vascular tone. EC receive signals from the circulation that stimulate production of the second messenger nitric oxide (NO), inducing vSMC relaxation and vessel dilation. In contrast, agents such as angiotensin II act directly on vSMC, causing the cell to contract and the vessel to constrict.
It is clear, then, that the presence of functional vSMC in a mature vascular graft would be ideal. As was the case for ECs, implanting a graft which already contains vSMC is rarely feasible, and more often recellularization by host cells is used. Therefore, in this section we will examine how matricellular proteins modulate vSMC adhesion, migration, proliferation, and survival, as well as vSMC function in the context of neointima formation.
Smooth Muscle Cell Adhesion and Migration
Angiotensin II promotes atherosclerosis by inducing the adhesion and migration of vSMCs, and is mediated by osteopontin using putative binding site miR181a in its inhibition (101). MAGP-1 is dependent on MAGP-2 in binding/activating Notch 1, which activates Notch signaling and modulates homeostasis during aortic SMC spreading (72).
Fibulin-5, in addition to its well-documented role on elastogenesis, is the primary mediator of uPA-driven migration of vSMCs (102). The mechanism for this appears to be that fibulin-5 binding facilitates activation of uPA, which then activates plasmin to cleave the portion of fibulin-5 that binds β1 integrin, driving cell migration. This action increases vSMC remodeling and vascularization after injury and antagonizes angiogenesis by inducing TSP-1 and antagonizing fibronectin receptors (102, 103). Galectin-1, a glycan-binding protein primarily involved within the anti-inflammatory cardiac tissue response to acute myocardial infarction, has been shown to restrict vSMC motility and modulates focal adhesion turnover on fibronectin, via in vitro assays using galectin-1 knockout mouse SMCs (104).
Smooth Muscle Cell Survival, Proliferation, and Neointimal Formation
In post-injury vasculature, galectin-1 is upregulated within proliferating SMCs, and in confirmatory studies an engineered galectin-1/GST fusion protein is able to drive SMC proliferation (105). Taken with its effects on increasing SMC proliferation and restricting SMC motility (105), galectin-1 presents as a potential supplement for cardiac therapeutics post-myocardial infarction and a serum biomarker for cardiac stress (106) with a well-characterized mouse model for in vivo evaluations.
The CCN family of matricellular proteins have significant, yet indirect, effects aiding vascularization and angiogenesis that might be considered in engineering microenvironment applications. Full length CCN2 utilizes integrin α6β1 to promote adhesion and spreading of vSMCs (65). CCN5 also inhibits vSMC proliferation and motility without affecting apoptosis and adhesion (107, 108), and has potential to be used as an delivered therapeutic CCN2 antagonist to control vSMC migration, adhesion, and phenotype (109). CCN5's effects are modulated in a dose-dependent fashion by PDGF and TGF-β, with some possible quiescence-related SMC properties (107). A reduction in CCN2 expression was observed in Notch1 haploinsufficiency murine abdominal aortic aneurysm models, leading to a maintenance of contractile vSMC phenotype and a reduction in aortic dilation (110). CCN2 presents with a key target for SMC phenotype maintenance in vasculopathy.
CCN4 (also known as Wisp-1), is upregulated in migrating vSMC in a Wnt2-dependent manner; loss of CCN4 leads to inhibited vSMC integrin mediated migration (111). Wnt5a, which induces β-catenin signaling in mouse via vSMCs, saw CCN4 rescue VSMCs from H2O2 induced apoptosis within atherosclerotic plaque (112, 113). CCN4 was expressed within advanced human coronary artery lesions (111) but absent in Wnt5a positive intimal SMCs, indicating that CCN4 deficiency may provoke vSMC apoptosis in coronary plaques, ultimately resulting in instability. Loss of CCN4 leads to reduced intimal thickening, related to CCN4's aforementioned positive effect on vSMC migration (111).
Tenascin-C (TNC) within the context of atherosclerotic plaques promotes SMC proliferation and migration via PDGF signaling (114). Areas of human abdominal aortic aneurysm with high TNC expression by medial SMCs correlate strongly with inflammation and tissue ECM destruction (115). TNC overexpression can also result in pulmonary hypertension, and it is expressed in adventitia and media of saphenous vein grafts under above average arterial stress pressure (116). Taken together, TNC presents with a SMC-linked biomarker target to assess atherosclerosis or aneurysm pathological state.
Neointimal hyperplasia is a common complication of vascular disease intervention (117). The main target for anti-neointimal formation is SMC phenotype, given the predisposition of unregulated hyperplastic vSMC proliferation to accelerate neointimal formation. Many matricellular proteins have shown efficacy in controlling vSMC phenotype and adhesion, and therapeutics often target at upregulation of these factors. TSP-1 targets NO-mediated vSMC relaxation to increase tissue survival and facilitate tissue perfusion to preserve tissues under ischemic stress (118). vSMC are also induced to proliferate by TSP-1 (119). Fibromodulin adenovirus-mediated gene transfer inhibits restenosis in an organ culture saphenous vein graft disease model of neointimal hyperplasia, at a much greater level than decorin or beta-galactosidase gene transfer (120). Fibromodulin could be a key SLRP for study within ECM-based vein grafts.
Another ECM-related inducer of neointimal formation is fibulin-1, where expression is closely overlapped with fibrinogen. Fibulin-1 and fibrinogen bind tightly within atherosclerotic legions, but this does not hold true in ECM associated with SMC surrounding the identified lesion. Fibulin-1 is associated with atherogenesis in diabetic patients, and at high levels fibulin-1 becomes an accurate indicator of cardiovascular risk in patients prior to acute cardiovascular events, obesity, or diabetes risk markers (121). vSMCs display increased SMC contractile differentiation markers with fibulin-5 overexpression (44), and vSMC phenotype has also been regulated through fibulin-4 expression in murine models (122). Overexpression of fibulin-5 has been shown post-injury to increase SMC motility in a step to aid in vascular remodeling (102).
CCN3 exhibited a protective role within the progression of murine abdominal aortic aneurysm model (123), and inhibited neointimal hyperplasia by inhibiting vSMC migration and proliferation (124). With this background, novel CCN3-based peptides have recently been developed, aiding in the modulation of pro-fibrotic factors CCN2, PAI-1, and post-translational fibrotic collagen modification (125).
Osteopontin (OPN, also known as bone sialoprotein I, abbr. BSP-1), a matricellular protein first identified within osteoblasts, is expressed by vSMC derived foam cells after immune response, and plays a large role in the calcification of atherosclerotic lesions (126). Increased OPN expression during neointimal hyperplasia formation was seen as a major microenvironment component of human atherosclerotic plaque progression. OPN downregulates vSMC differentiation markers αSMA and calponin, transitioning from quiescent status into an unregulated proliferative phenotype (127). Galectin-1 binds to lipoprotein(a), which binds tightly to LDL and are both co-implicated in atherogenesis. Lipoprotein(a) binds poorly to tissue with galectin-1 inhibition, suggesting that lipoprotein(a) accumulation changes have an effect on atherogenesis, possibly mediated through t-antigens on lipoprotein(a) and LDL (128). A novel engineering approach modulating OPN protein expression or galectin-1 binding within atherogenic arterial walls could present as a key mediating factor for downstream neointimal hyperplasia.
Matricellular Proteins in Vascular Tissue Engineering
To date, only a few of the matricellular proteins reviewed here have been pursued in the context of vascular tissue engineering, hence the need for future tissue engineering strategies harnessing these proteins. What follows is a brief summary of the findings from this small library of published work—note that by far the most predominant area of study has been how to encourage endothelial adhesion to biomaterials in the vascular context.
Production of New Vascular Matrix
Mature elastic fibers that are mechanically competent are a highly sought-after component of a TEVG. Several groups have investigated methods to improve elastin content, or production, within grafts and unsurprisingly a few of these studies have harnessed matricellular proteins to do so. Versican, as explored by the Wight group, has a complex relationship with elastogenesis. Full length glycosylated versican, known as V0, negatively impacts new elastin fiber formation. However, a splice variant of versican that cannot be glycosylated, known as V3, drives new elastin formation and cross linking by SMCs (129). With this background, the Wight group collaborated with a vascular tissue engineering company, Cytograft, to explore the potential for a TEVG with enhanced elastin content. Cell sheets of SMCs retrovirally expressing V3, or control cells, were cultured for 12 weeks to form a rich cell-derived matrix, rolled on a mandrel, and allowed to fuse for 18 more weeks. During the initial cell culture period (4 weeks), not only was expression of elastin elevated at the gene level, as expected, but also fibulin-5 (130). Mature crosslinking was also elevated by V3, as well as higher mechanical strength as judged by burst strength. It should be noted that in the cell-sheet TEVG strategy, the cells are generally removed before further TEVG processing, so the initial transduction of the cells should not serve as a hurdle to final FDA approval.
An alternate method for exploring improved elastin production was published by the Suzuki group. As reported by others (131), collagen is generally a poor 3D matrix for cellular elastic fiber production. The Suzuki group had previously discovered a beneficial effect of LTBP-4 on elastic fiber assembly (132) and reasoned that it could also then encourage elastic fiber formation in collagen gels. Here, human skin fibroblasts were seeded on top of porous collagen sponges, allowing the cells to infiltrate the sponge during culture. Recombinant LTBP-4 was added to experimental cultures at a concentration of 5 μg/ml, and replaced along with media changes every week for 3 weeks. Increased LTBP-4 immunostaining was observed in the LTBP-4 treated cultures (both recombinant and native human protein), as well as improved fibrillin-1 and elastin (133). Interestingly, the benefit on elastin was also observed with a single dose of LTBP-4 at 1 week, as opposed to the 3 week treatment described above—although immunostaining for fibrillin-1 was not explored. This work was limited by lacking more rigorous analysis of the elastic fibers (such as desmosine assay) but the simple addition of recombinant LTBP-4 to culture media is more amenable to the typical tissue engineer than retroviral work, and therefore of note.
Limiting Thrombosis
Because thrombosis can be initiated upon contact of blood with ECM such as the vascular basement membrane and internal elastic lamella, most groups have focused on endothelialization (see next section). However, in work by the Kyriakides lab analyzing a mouse model for TSP-2 deficiency, it became clear that TSP-2 deficient arteries are resistant to thrombosis for reasons linked to the ECM of the vessel wall (not just inherent to thrombus formation) (62). In a similar situation to the Wight group collaboration with Cytograft explored in the previous section, Kyriakides teamed up with Laura Niklason from the vascular tissue engineering company Humacyte to explore if a TSP-2 deficient matrix could serve as a thrombosis-resistant lumenal coating for the Humacyte graft. Modification with the TSP-2 deficient matrix had no effect on the mechanical properties of the graft, but both inhibited platelet activation in vitro and improved endothelialization of interpositional abdominal aortic implants after 4 weeks in vivo (63). They do not report an effect on patency as seen by our group when evaluating TEVGs in a similar model (51), but it could be expected that as they move to a larger animal model a patency effect could be revealed.
Endothelial Formation and Maintenance
As discussed above, CCN1 has a pro-adhesive and pro-migratory effect on ECs. Hilfiker, Haverich, Wilhelmi, and Boer have performed collaborative work for nearly a decade using recombinant CCN1 to promote re-endothelialization of decellularized small intestine with preserved artery and vein pedicles (134), heart valves (135), and decellularized arteries (136, 137). Introduction of recombinant CCN1 (100 ng/ml overnight for the intestine and arteries, 400 μg/ml 6 h for the heart valves) was performed either by perfusion of cannulated tissue (intestines) or end-over-end incubation of the tissue and CCN1 solution (heart valves and arteries). In either case, introduction of matricellular protein to engineered tissues appears to be both simple and effective using these methods, and could be extended to other molecules.
The Chaikof group has investigated the use of elastin-like peptides (ELPs) as the basis of a new biocompatible and tunable hydrogel scaffold for vascular tissue engineering. Since the ELPs self-assemble, coacervating at a tunable transition temperature, it is relatively simple to incorporate other cargo into the gelation mixture. Two matricellular proteins have been investigated in this context—fibronectin (138) and CCN1 (139). Two methods were used for incorporation of fibronectin into ELPs. In one method, ELPs were coacervated into hydrogels, which were then incubated in a 1 mg/ml solution of fibronectin for 4 h of adsorption before genipin crosslinking of the ELPs. In the second method, ELPs and fibronectin were blended at 4°C before coacervation into hydrogels at 37°C and subsequent genipin cross-linking. While improved pro-adhesive activity toward ECs was observed with the adsorption method, both fibronectin-loaded hydrogel strategies outperformed ELPs alone. Other beneficial outcomes included improved spreading and migration of ECs (138). A distinct strategy was used to incorporate beneficial CCN1 activities into ELP hydrogels—instead of adding recombinant protein to the gelation mixture the peptides themselves were engineered to contain a biologically active sequence derived from CCN1. Specifically, this 20-residue sequence, known as the V2 domain, is critical for integrin αvβ3-dependent binding of ECs to CCN1 (140) and therefore represents a new cell-interaction site for engineered biomaterials. Ten repeats of the V2 sequence were introduced into the standard triblock structure of the ELP; hydrogels containing V2 had improved EC adhesion that could be blocked by soluble V2 peptide or an antibody that interferes with αvβ3 function (139). As was the case with fibronectin-functionalized ELPs, beneficial outcomes included improved migration and spreading of ECs.
In a similar vein to the Chaikof CCN1 experiments, the Heath lab has recently published work exploring the synergistic EC-adhesive effect of traditional integrin-binding RGD peptide along with peptides used for binding to syndecans −1, −2, and −4 (141). In this case, the hydrogel system used was polyethylene glycol (PEG), a common biomaterial explored in tissue engineering; within the PEG network, RGD peptide and/or one of two different syndecan-binding peptides were conjugated. While RGD and syndecan-binding peptides did promote EC adhesion beyond their respective controls (RGE and a scrambled syndecan-binding peptide) the combination of RGD and syndecan peptide at ratios of 1:1 or 3:1 promoted superior adhesion. Interestingly, this worked whether the two peptides were in separate PEG chains (nanoclusters) or in the same PEG chain (heteroclusters). Beyond adhesion, proliferation was also accelerated with the peptide mixture, vs. single peptides. Perhaps the most astonishing finding of this work was revealed with flow. ECs adherent to single peptides stayed adherent under 10 h of flow (3.1 dyn/cm2) but did not form proper focal adhesions or align to flow—in contrast ECs adherent to the peptide mixture formed prominent focal adhesions, stress fibers, and re-aligned with flow (141). A recent review on syndecans has explored their role in the context of regeneration and their potential to regulate stem cell function, though not specifically in the context of vascular tissue engineering (142).
As discussed earlier, small diameter PTFE grafts typically undergo thrombotic failure. While seeding of ECs has been explored, they are generally unable to remain within the graft long term and ultimately the graft continues to fail. The Flugelman group has explored the use of fibulin-5, in addition to VEGF, as a method to retain ECs (143). Specifically, ECs were retrovirally transduced to express both fibulin-5 and VEGF, and then dually transduced, GFP transduced, or naïve ECs were seeded into PTFE scaffolds for in vivo testing. Both short term (2 weeks, interpositional) and middle term (3 or 6 months, end-to-side) carotid artery implantations were performed in sheep. A substantial benefit was observed using the fibulin-5/VEGF ECs with respect to patency, vs. GFP transduced and naïve EC controls. While there are some limitations to the study concerning a lack of evaluation of fibulin-5/elastin deposition in explants or a fibulin-5 only control, it is an exciting foray in the context of matricellular proteins in vascular tissue engineering.
An alternate method for inducing endothelialization of vascular scaffolds is to recruit circulating endothelial progenitor cells (EPCs). Work by the Schenke-Layland has explored the use of decorin as a ligand to attract EPCs from the circulation and thereby quickly coat an implanted scaffold with a “pre-endothelium.” An electrospun PEGdma-PLA scaffold, meant to serve as a heart valve equivalent, was coated with recombinantly expressed decorin, stromal-derived factor 1, or were left uncoated. Using a dynamic in vitro cell capture system, human and mouse EPCs were exposed to the scaffolds for 24 or 48 h, respectively. Decorin and stromal-derived factor 1 both led to higher capture of EPCs compared to bare scaffolds, in addition the captured cells proceeded to form focal adhesions at the conclusion of the incubation period (144). While it should be noted that this has not yet been tested with whole blood, or under flow, the system described here could be a novel method for endothelializing an a cellular implanted scaffold.
Smooth Muscle Cell Recruitment
Aside from the above studies regarding new elastin production by SMCs, little has been published regarding the role of matricellular proteins in SMC recruitment for vascular tissue engineering. One study of note, however, investigated the chemical conjugation of whole fibronectin, not just the RGD peptide, to porous poly(carbonate) urethane scaffolds (145). Considering that fibronectin is a 500 kD protein, this conjugation was not trivial. In this work, scaffolds functionalized with full-length fibronectin encouraged SMC recruitment to a depth of up to 300 μm within the material; non-functionalized scaffolds only supported SMC growth on the surface. Therefore, fibronectin incorporation into porous scaffolds for vascular tissue engineering may promote host cell SMC recruitment.
Summary
Much of the groundwork for matricellular protein-based protein engineering has been laid by the two decades of biological studies reviewed here. The goal of this review is to get the results of these studies into the hands of bioengineers developing new cardiovascular technologies, and to spark interdisciplinary discussions that can lead to novel solutions for cardiovascular disease. A summary of the biological effects of each individual matricellular protein is shown in Table 2. Admittedly, the knowledge base reviewed here is constantly evolving, but the intent is that established collaborations will then stay informed about their protein(s) of interest. Another caveat is that regeneration of the vasculature likely involves the cooperation of several of these factors in concert, which may not have a simple summative effect compared to the individual activities.
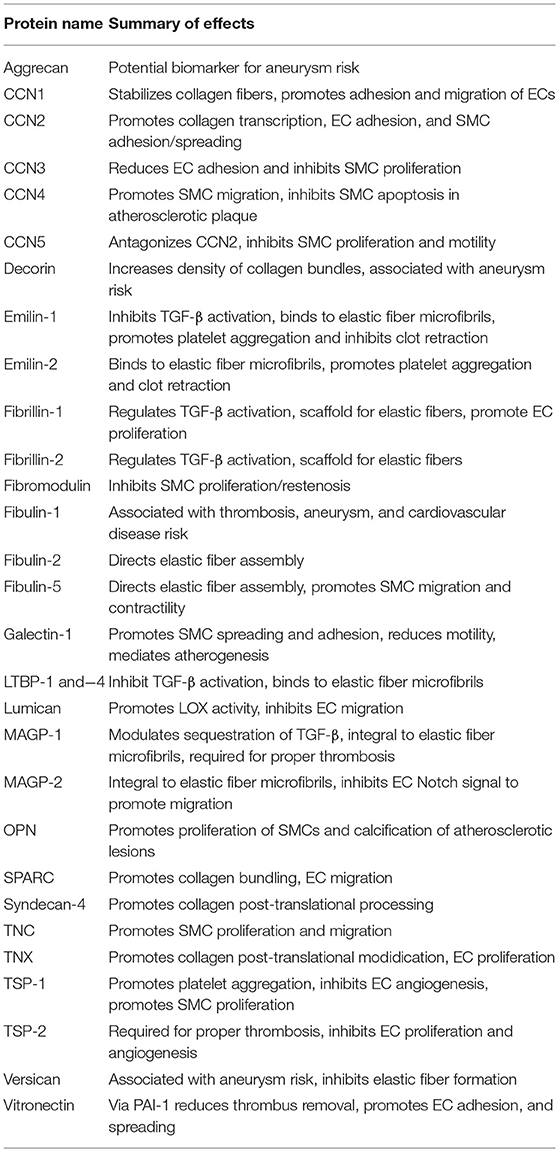
Table 2. Summary of biological effects for each matricellular protein discussed in this review (see respective sections for details and references).
Matricellular proteins can be used to enhance production of new collagen and elastin, the primary contributors to vascular strength and resilience. Much of the literature on collagen transcriptional regulation can be informative for future engineering of collagen-rich tissues, and reciprocally informative for strategies to reduce graft fibrosis or the foreign body response to cardiovascular devices. Understanding how matricellular proteins guide collagen bundling can be used to inform future collagen-based biomaterials. Notably, new elastin production by adult cells has been something of a holy grail in the field of vascular regenerative medicine, with several recent studies in the field of aneurysm therapeutics focused on production of this important molecule (146, 147). Including matricellular proteins such as members of the fibulin family in future cardiovascular tissue engineering strategies will be critical for achieving fully mature, mechanically sound, elastic fibers. Understanding how these elastin chaperones work will also be important for designing devices to help patients with deficiency in these important proteins.
A key limitation of synthetic small diameter grafts is the occurrence of thrombotic occlusion; as explored here it has become clear that matricellular proteins such as members of the TSP family can have a significant effect on how thrombosis occurs. The work by Kyriakides et al. demonstrating thrombosis resistance in the absence of TSP-2 is particularly striking in this regard (62, 63). Regulation of thrombosis by matricellular proteins can be due to inhibition of thrombin activation, platelet activation, or both. While the focus in this review has been on thrombotic failure in vascular grafts, these findings also have implications for the design of cardiovascular devices with lower risk of embolism and stroke.
In a tissue-engineered graft, formation of an intact, thromboresistant endothelium is critical to enable prolonged function. In development and maintenance of the endothelium, matricellular proteins found in both the circulation and the underlying basement membrane have critical roles. Not only are these proteins important to support endothelial adhesion, they also are important for promoting survival of adherent ECs, which will be critical for a small-diameter engineered graft. Maintenance of ECs in a non-pathologic phenotype is also an area of interest in vascular engineering, so understanding the role of matricellular proteins on EC differentiation state will also be critical for future device design.
Finally, an understanding of the effects of matricellular proteins on vascular SMCs will be beneficial for future vascular engineering, since SMCs not only are critical for production (and turnover) of new vascular matrix in the graft context but also perform multiple roles in normal vascular physiology. Implanted cell-free grafts must support survival and ingrowth of host SMCs to better mimic the native artery. Matricellular proteins have the ability to promote both of these outcomes. In addition, a common failure method for the most used graft in coronary bypass operations—an autologous saphenous vein graft—is neointimal formation and occlusion due to excessive proliferation of SMCs. Control of SMC overproliferation by peptides derived from matricellular proteins could replace conventional drug-eluting stent technologies, and also be used to augment current vascular graft strategies.
In summary, we would propose that the use of matricellular proteins and matricellular-derived peptides could serve as a paradigm shift in the field of vascular tissue engineering.
Author Contributions
Conception of this article was primarily by JW. Literature research was performed by AR and JW. Writing and critical revision were performed by AR, DV, and JW.
Funding
This work was supported by the Leonard H. Berenfield Graduate Fellowship in Cardiovascular Bioengineering and NIH T32 HL094295 (AR), the NIH grants HL129066, HL130784, and HL13077 as well as McCune Foundation Pediatric Device Initiative (DV), and the NIH grant HL104768 and University of Pittsburgh Competitive Medical Research Fund (JW).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Wagenseil JE, Mecham RP. Vascular extracellular matrix and arterial mechanics. Physiol Rev. (2009) 89:957–89. doi: 10.1152/physrev.00041.2008
2. Lok CN, Ehrlich HP, White SL, Buttolph TR, Cutroneo KR, Chiu JF. Oligodeoxynucleotide decoy therapy blocks type 1 procollagen transcription and the prolyl hydroxylase beta subunit translation. J Cell Biochem. (2008) 103:1066–75. doi: 10.1002/jcb.21477
3. Bagchi RA, Czubryt MP. Synergistic roles of scleraxis and Smads in the regulation of collagen 1alpha2 gene expression. Biochim Biophys Acta. (2012) 1823:1936–44. doi: 10.1016/j.bbamcr.2012.07.002
4. Kim HJ, Kim MY, Jin H, Kim HJ, Kang SS, Kim HJ, et al. Peroxisome proliferator-activated receptor {delta} regulates extracellular matrix and apoptosis of vascular smooth muscle cells through the activation of transforming growth factor-β1/Smad3. Circ Res. (2009) 105:16–24. doi: 10.1161/CIRCRESAHA.108.189159
5. Robertson IB, Horiguchi M, Zilberberg L, Dabovic B, Hadjiolova K, Rifkin DB. Latent TGF-beta-binding proteins. Matrix Biol. (2015) 47:44–53. doi: 10.1016/j.matbio.2015.05.005
6. Brigstock DR. Regulation of angiogenesis and endothelial cell function by connective tissue growth factor (CTGF) and cysteine-rich 61 (CYR61). Angiogenesis. (2002) 5:153–65. doi: 10.1023/A:1023823803510
7. Shiwen X, Rajkumar V, Denton CP, Leask A, Abraham DJ. Pericytes display increased CCN2 expression upon culturing. J Cell Commun Signal. (2009) 3:61–4. doi: 10.1007/s12079-009-0053-7
8. Schalkwijk J, Zweers MC, Steijlen PM, Dean WB, Taylor G, van Vlijmen IM, et al. A recessive form of the Ehlers-Danlos syndrome caused by tenascin-X deficiency. N Engl J Med. (2001) 345:1167–75. doi: 10.1056/NEJMoa002939
9. Herum KM, Lunde IG, Skrbic B, Louch WE, Hasic A, Boye S, et al. Syndecan-4 is a key determinant of collagen cross-linking and passive myocardial stiffness in the pressure-overloaded heart. Cardiovasc Res. (2015) 106:217–26. doi: 10.1093/cvr/cvv002
10. Engebretsen KVT, Lunde IG, Strand ME, Waehre A, Sjaastad I, Marstein HS, et al. Lumican is increased in experimental and clinical heart failure, and its production by cardiac fibroblasts is induced by mechanical and proinflammatory stimuli. Febs J. (2013) 280:2382–98. doi: 10.1111/febs.12235
11. Christensen G, Herum KM, Lunde IG. Sweet, yet underappreciated: proteoglycans and extracellular matrix remodeling in heart disease. Matrix Biol. (2018) 75–6:286–99. doi: 10.1016/j.matbio.2018.01.001
12. Gu G, Wan F, Xue Y, Cheng W, Zheng H, Zhao Y, et al. Lumican as a novel potential clinical indicator for acute aortic dissection: a comparative study, based on multi-slice computed tomography angiography. Experi Therap Med. (2016) 11:923–8. doi: 10.3892/etm.2016.3020
13. Bradshaw AD, Sage EH. SPARC, a matricellular protein that functions in cellular differentiation and tissue response to injury. J Clin Invest. (2001) 107:1049–54. doi: 10.1172/JCI12939
14. Harris BS, Zhang Y, Card L, Rivera LB, Brekken RA, Bradshaw AD. SPARC regulates collagen interaction with cardiac fibroblast cell surfaces. Am J Physiol Heart Circ Physiol. (2011) 301:H841–847. doi: 10.1152/ajpheart.01247.2010
15. Puolakkainen P, Bradshaw AD, Kyriakides TR, Reed M, Brekken R, Wight T, et al. Compromised production of extracellular matrix in mice lacking secreted protein, acidic and rich in cysteine (SPARC) leads to a reduced foreign body reaction to implanted biomaterials. Am J Pathol. (2003) 162:627–35. doi: 10.1016/S0002-9440(10)63856-4
16. Barallobre-Barreiro J, Gupta SK, Zoccarato A, Kitazume-Taneike R, Fava M, Yin X, et al. Glycoproteomics reveals decorin peptides with anti-myostatin activity in human atrial fibrillation. Circulation. (2016) 134:817–32. doi: 10.1161/CIRCULATIONAHA.115.016423
17. Ferdous Z, Wei VM, Iozzo R, Hook M, Grande-Allen KJ. Decorin-transforming growth factor- interaction regulates matrix organization and mechanical characteristics of three-dimensional collagen matrices. J Biol Chem. (2007) 282:35887–98. doi: 10.1074/jbc.M705180200
18. Ferdous Z, Lazaro LD, Iozzo RV, Hook M, Grande-Allen KJ. Influence of cyclic strain and decorin deficiency on 3D cellularized collagen matrices. Biomaterials. (2008) 29:2740–8. doi: 10.1016/j.biomaterials.2008.03.018
19. Paderi JE, Panitch A. Design of a synthetic collagen-binding peptidoglycan that modulates collagen fibrillogenesis. Biomacromolecules. (2008) 9:2562–6. doi: 10.1021/bm8006852
20. Paderi JE, Stuart K, Sturek M, Park K, Panitch A. The inhibition of platelet adhesion and activation on collagen during balloon angioplasty by collagen-binding peptidoglycans. Biomaterials. (2011) 32:2516–23. doi: 10.1016/j.biomaterials.2010.12.025
21. Scott RA, Paderi JE, Sturek M, Panitch A. Decorin mimic inhibits vascular smooth muscle proliferation and migration. PLoS ONE. (2013) 8:e82456. doi: 10.1371/journal.pone.0082456
22. Ang LS, Boivin WA, Williams SJ, Zhao H, Abraham T, Carmine-Simmen K, et al. Serpina3n attenuates granzyme B-mediated decorin cleavage and rupture in a murine model of aortic aneurysm. Cell Death Dis. (2011) 2:e209. doi: 10.1038/cddis.2011.88
23. Ueda K, Yoshimura K, Yamashita O, Harada T, Morikage N, Hamano K. Possible dual role of decorin in abdominal aortic aneurysm. PLoS ONE. (2015) 10:e0120689. doi: 10.1371/journal.pone.0120689
24. Kozel BA, Danback JR, Waxler JL, Knutsen RH, de Las Fuentes L, Reusz GS, et al. Williams syndrome predisposes to vascular stiffness modified by antihypertensive use and copy number changes in NCF1. Hypertension. (2014) 63:74–9. doi: 10.1161/HYPERTENSIONAHA.113.02087
25. Callewaert B, Renard M, Hucthagowder V, Albrecht B, Hausser I, Blair E, et al. New insights into the pathogenesis of autosomal-dominant cutis laxa with report of five ELN mutations. Hum Mutat. (2011) 32:445–55. doi: 10.1002/humu.21462
26. Li DY, Faury G, Taylor DG, Davis EC, Boyle WA, Mecham RP, et al. Novel arterial pathology in mice and humans hemizygous for elastin. J Clin Invest. (1998) 102:1783–7. doi: 10.1172/JCI4487
27. Zhang M, Pierce RA, Wachi H, Mecham RP, Parks WC. An open reading frame element mediates posttranscriptional regulation of tropoelastin and responsiveness to transforming growth factor beta1. Mol Cell Biol. (1999) 19:7314–26. doi: 10.1128/MCB.19.11.7314
28. Weinbaum JS, Broekelmann TJ, Pierce RA, Werneck CC, Segade F, Craft CS, et al. Deficiency in microfibril-associated glycoprotein-1 leads to complex phenotypes in multiple organ systems. J Biol Chem. (2008) 283:25533–43. doi: 10.1074/jbc.M709962200
29. Massam-Wu T, Chiu M, Choudhury R, Chaudhry SS, Baldwin AK, McGovern A, et al. Assembly of fibrillin microfibrils governs extracellular deposition of latent TGF beta. J Cell Sci. (2010) 123:3006–18. doi: 10.1242/jcs.073437
30. Schiavinato A, Keene DR, Wohl AP, Corallo D, Colombatti A, Wagener R, et al. Targeting of EMILIN-1 and EMILIN-2 to fibrillin microfibrils facilitates their incorporation into the extracellular matrix. J Invest Dermatol. (2016) 136:1150–60. doi: 10.1016/j.jid.2016.02.021
31. Zacchigna L, Vecchione C, Notte A, Cordenonsi M, Dupont S, Maretto S, et al. Emilin1 links TGF-beta maturation to blood pressure homeostasis. Cell. (2006) 124:929–42. doi: 10.1016/j.cell.2005.12.035
32. Litteri G, Carnevale D, D'Urso A, Cifelli G, Braghetta P, Damato A, et al. Vascular smooth muscle Emilin-1 is a regulator of arteriolar myogenic response and blood pressure. Arterioscl Thromb Vasc Biol. (2012) 32:2178–84. doi: 10.1161/ATVBAHA.112.254664
33. Varona S, García-Redondo AB, Martínez-González J, Salaices M, Briones AM, Rodríguez C. La sobreexpresión vascular de la lisil oxidasa altera la estructura de la matriz extracelular e induce estrés oxidativo. Clín Invest Arteriosc. (2017) 29:157–65. doi: 10.1016/j.arteri.2017.01.004
34. Staiculescu MC, Kim J, Mecham RP, Wagenseil JE. Mechanical behavior and matrisome gene expression in the aneurysm-prone thoracic aorta of newborn lysyl oxidase knockout mice. Am J Physiol-Heart Circul Physiol. (2017) 313:H446–56. doi: 10.1152/ajpheart.00712.2016
35. Villain G, Lelievre E, Broekelmann T, Gayet O, Havet C, Werkmeister E, et al. MAGP-1 and fibronectin control EGFL7 functions by driving its deposition into distinct endothelial extracellular matrix locations. FEBS J. (2018) 285:4394–412. doi: 10.1111/febs.14680
36. Zhang H, Apfelroth SD, Hu W, Davis EC, Sanguineti C, Bonadio J, et al. Structure and expression of fibrillin-2, a novel microfibrillar component preferentially located in elastic matrices. J Cell Biol. (1994) 124:855–63. doi: 10.1083/jcb.124.5.855
37. Carta L, Smaldone S, Zilberberg L, Loch D, Dietz HC, Rifkin DB, et al. p38 MAPK is an early determinant of promiscuous Smad2/3 signaling in the aortas of fibrillin-1 (Fbn1)-null mice. J Biol Chem. (2009) 284:5630–6. doi: 10.1074/jbc.M806962200
38. Pereira L, Andrikopoulos K, Tian J, Lee SY, Keene DR, Ono R, et al. Targetting of the gene encoding fibrillin-1 recapitulates the vascular aspect of Marfan syndrome. Nat Genet. (1997) 17:218–22. doi: 10.1038/ng1097-218
39. Carta L, Pereira L, Arteaga-Solis E, Lee-Arteaga SY, Lenart B, Starcher B, et al. Fibrillins 1 and 2 perform partially overlapping functions during aortic development. J Biol Chem. (2006) 281:8016–23. doi: 10.1074/jbc.M511599200
40. Sicot FX, Tsuda T, Markova D, Klement JF, Arita M, Zhang RZ, et al. Fibulin-2 is dispensable for mouse development and elastic fiber formation. Mol Cell Biol. (2008) 28:1061–7. doi: 10.1128/MCB.01876-07
41. Yamauchi Y, Tsuruga E, Nakashima K, Sawa Y, Ishikawa H. Fibulin-4 and−5, but not Fibulin-2, are associated with tropoelastin deposition in elastin-producing cell culture. Acta Histochem Cytochem. (2010) 43:131–8. doi: 10.1267/ahc.10026
42. de Vega S, Iwamoto T, Yamada Y. Fibulins: multiple roles in matrix structures and tissue functions. Cell Mol Life Sci. (2009) 66:1890–902. doi: 10.1007/s00018-009-8632-6
43. Chapman SL, Sicot FX, Davis EC, Huang J, Sasaki T, Chu ML, et al. Fibulin-2 and fibulin-5 cooperatively function to form the internal elastic lamina and protect from vascular injury. Arterioscler Thromb Vasc Biol. (2010) 30:68–74. doi: 10.1161/ATVBAHA.109.196725
44. Spencer JA, Hacker SL, Davis EC, Mecham RP, Knutsen RH, Li DY, et al. Altered vascular remodeling in fibulin-5-deficient mice reveals a role of fibulin-5 in smooth muscle cell proliferation and migration. Proc Natl Acad Sci USA. (2005) 102:2946–51. doi: 10.1073/pnas.0500058102
45. Nakasaki M, Hwang Y, Xie Y, Kataria S, Gund R, Hajam EY, et al. The matrix protein Fibulin-5 is at the interface of tissue stiffness and inflammation in fibrosis. Nat Commun. (2015) 6:8574. doi: 10.1038/ncomms9574
46. Yanagisawa H, Davis EC, Starcher BC, Ouchi T, Yanagisawa M, Richardson JA, et al. Fibulin-5 is an elastin-binding protein essential for elastic fibre development in vivo. Nature. (2002) 415:168–71. doi: 10.1038/415168a
47. Yasmin MRA, McEniery CM, Cleary SE, Li Y, Siew K, Figg NL, et al. The matrix proteins aggrecan and fibulin-1 play a key role in determining aortic stiffness. Sci Rep. (2018) 8:8550. doi: 10.1038/s41598-018-25851-5
48. Cikach FS, Koch CD, Mead TJ, Galatioto J, Willard BB, Emerton KB, et al. Massive aggrecan and versican accumulation in thoracic aortic aneurysm and dissection. JCI Insight. (2018) 3:e97167. doi: 10.1172/jci.insight.97167
49. Kaufman JS, O'Connor TZ, Zhang JH, Cronin RE, Fiore LD, Ganz MB, et al. Randomized controlled trial of clopidogrel plus aspirin to prevent hemodialysis access graft thrombosis. J Am Soc Nephrol. (2003) 14:2313–21. doi: 10.1097/01.ASN.0000081661.10246.33
50. Soletti L, Nieponice A, Hong Y, Ye SH, Stankus JJ, Wagner WR, et al. In vivo performance of a phospholipid-coated bioerodable elastomeric graft for small-diameter vascular applications. J Biomed Mater Res A. (2011) 96:436–48. doi: 10.1002/jbm.a.32997
51. Krawiec JT, Weinbaum JS, Liao HT, Ramaswamy AK, Pezzone DJ, Josowitz AD, et al. In vivo functional evaluation of tissue-engineered vascular grafts fabricated using human adipose-derived stem cells from high cardiovascular risk populations. Tissue Eng Part A. (2016) 22:765–75. doi: 10.1089/ten.tea.2015.0379
52. Wootton DM, Ku DN. Fluid mechanics of vascular systems, diseases, and thrombosis. Annu Rev Biomed Eng. (1999) 1:299–329. doi: 10.1146/annurev.bioeng.1.1.299
53. Kamikubo Y, Neels JG, Degryse B. Vitronectin inhibits plasminogen activator inhibitor-1-induced signalling and chemotaxis by blocking plasminogen activator inhibitor-1 binding to the low-density lipoprotein receptor-related protein. Int J Biochem Cell Biol. (2009) 41:578–85. doi: 10.1016/j.biocel.2008.07.006
54. Podor TJ, Campbell S, Chindemi P, Foulon DM, Farrell DH, Walton PD, et al. Incorporation of vitronectin into fibrin clots. Evidence for a binding interaction between vitronectin and gamma A/gamma' fibrinogen. J Biol Chem. (2002) 277:7520–8. doi: 10.1074/jbc.M109677200
55. Argraves WS, Tanaka A, Smith EP, Twal WO, Argraves KM, Fan D, et al. Fibulin-1 and fibrinogen in human atherosclerotic lesions. Histochem Cell Biol. (2009) 132:559–65. doi: 10.1007/s00418-009-0628-7
56. Bonnefoy A, Daenens K, Feys HB, De Vos R, Vandervoort P, Vermylen J, et al. Thrombospondin-1 controls vascular platelet recruitment and thrombus adherence in mice by protecting (sub)endothelial VWF from cleavage by ADAMTS13. Blood. (2006) 107:955–64. doi: 10.1182/blood-2004-12-4856
57. Isenberg JS, Hyodo F, Ridnour LA, Shannon CS, Wink DA, Krishna MC, et al. Thrombospondin 1 and vasoactive agents indirectly alter tumor blood flow. Neoplasia. (2008) 10:886–96. doi: 10.1593/neo.08264
58. Kuijpers MJ, de Witt S, Nergiz-Unal R, van Kruchten R, Korporaal SJ, Verhamme P, et al. Supporting roles of platelet thrombospondin-1 and CD36 in thrombus formation on collagen. Arteriosc Thromb Vasc Biol. (2014) 34:1187–92. doi: 10.1161/ATVBAHA.113.302917
59. Huang M, Sannaningaiah D, Zhao N, Gong Y, Grondolsky J, Hoover-Plow J. EMILIN2 regulates platelet activation, thrombus formation, and clot retraction. PLoS ONE. (2015) 10:e0115284. doi: 10.1371/journal.pone.0115284
60. Werneck CC, Vicente CP, Weinberg JS, Shifren A, Pierce RA, Broekelmann TJ, et al. Mice lacking the extracellular matrix protein MAGP1 display delayed thrombotic occlusion following vessel injury. Blood. (2008) 111:4137–44. doi: 10.1182/blood-2007-07-101733
61. Vassequi-Silva T, Pereira DS, Nery Diez ACC, Braga GG, Godoy JA, Mendes CB, et al. Losartan and captopril treatment rescue normal thrombus formation in microfibril associated glycoprotein-1 (MAGP1) deficient mice. Thromb Res. (2016) 138:7–15. doi: 10.1016/j.thromres.2015.12.004
62. Kristofik N, Calabro NE, Tian W, Meng A, MacLauchlan S, Wang Y, et al. Impaired von Willebrand factor adhesion and platelet response in thrombospondin-2 knockout mice. Blood. (2016) 128:1642–50. doi: 10.1182/blood-2016-03-702845
63. Kristofik NJ, Qin L, Calabro NE, Dimitrievska S, Li G, Tellides G, et al. Improving In vivo outcomes of decellularized vascular grafts via incorporation of a novel extracellular matrix. Biomaterials. (2017) 141:63–73. doi: 10.1016/j.biomaterials.2017.06.025
64. Leu SJ, Lam SC, Lau LF. Pro-angiogenic activities of CYR61 (CCN1) mediated through integrins alphavbeta3 and alpha6beta1 in human umbilical vein endothelial cells. J Biol Chem. (2002) 277:46248–55. doi: 10.1074/jbc.M209288200
65. Ball DK, Rachfal AW, Kemper SA, Brigstock DR. The heparin-binding 10 kDa fragment of connective tissue growth factor (CTGF) containing module 4 alone stimulates cell adhesion. J Endocrinol. (2003) 176:R1–7. doi: 10.1677/joe.0.176r001
66. Chen CY, Su CM, Hsu CJ, Huang CC, Wang SW, Liu SC, et al. CCN1 Promotes VEGF production in osteoblasts and induces endothelial progenitor cell angiogenesis by inhibiting miR-126 expression in rheumatoid arthritis. J Bone Miner Res. (2017) 32:34–45. doi: 10.1002/jbmr.2926
67. Chintala H, Krupska I, Yan L, Lau L, Grant M, Chaqour B. The matricellular protein CCN1 controls retinal angiogenesis by targeting VEGF, Src homology 2 domain phosphatase-1 and Notch signaling. Development. (2015) 142:2364–74. doi: 10.1242/dev.121913
68. Di Y, Zhang Y, Nie Q, Chen X. CCN1/Cyr61-PI3K/AKT signaling promotes retinal neovascularization in oxygen-induced retinopathy. Int J Mol Med. (2015) 36:1507–18. doi: 10.3892/ijmm.2015.2371
69. Di Y, Zhang Y, Yang H, Wang A, Chen X. The mechanism of CCN1-enhanced retinal neovascularization in oxygen-induced retinopathy through PI3K/Akt-VEGF signaling pathway. Drug Des Dev Ther. (2015) 9:2463–73. doi: 10.2147/DDDT.S79782
70. Hwang S, Lee HJ, Kim G, Won KJ, Park YS, Jo I. CCN1 acutely increases nitric oxide production via integrin alphavbeta3-Akt-S6K-phosphorylation of endothelial nitric oxide synthase at the serine 1177 signaling axis. Free Radic Biol Med. (2015) 89:229–40. doi: 10.1016/j.freeradbiomed.2015.08.005
71. Lin CG, Leu SJ, Chen N, Tebeau CM, Lin SX, Yeung CY, et al. CCN3 (NOV) is a novel angiogenic regulator of the CCN protein family. J Biol Chem. (2003) 278:24200–8. doi: 10.1074/jbc.M302028200
72. Miyamoto A, Lau R, Hein PW, Shipley JM, Weinmaster G. Microfibrillar proteins MAGP-1 and MAGP-2 induce Notch1 extracellular domain dissociation and receptor activation. J Biol Chem. (2006) 281:10089–97. doi: 10.1074/jbc.M600298200
73. Albig AR, Becenti DJ, Roy TG, Schiemann WP. Microfibril-associate glycoprotein-2 (MAGP-2) promotes angiogenic cell sprouting by blocking notch signaling in endothelial cells. Microvasc Res. (2008) 76:7–14. doi: 10.1016/j.mvr.2008.01.001
74. Rivera LB, Brekken RA. SPARC promotes pericyte recruitment via inhibition of endoglin-dependent TGF-beta1 activity. J Cell Biol. (2011) 193:1305–19. doi: 10.1083/jcb.201011143
75. Heydarkhan-Hagvall S, Gluck JM, Delman C, Jung M, Ehsani N, Full S, et al. The effect of vitronectin on the differentiation of embryonic stem cells in a 3D culture system. Biomaterials. (2012) 33:2032–40. doi: 10.1016/j.biomaterials.2011.11.065
76. Preissner KT, Seiffert D. Role of vitronectin and its receptors in haemostasis and vascular remodeling. Thromb Res. (1998) 89:1–21. doi: 10.1016/S0049-3848(97)00298-3
77. Re F, Zanetti A, Sironi M, Polentarutti N, Lanfrancone L, Dejana E, et al. Inhibition of anchorage-dependent cell spreading triggers apoptosis in cultured human endothelial cells. J Cell Biol. (1994) 127:537–46. doi: 10.1083/jcb.127.2.537
78. Reinmuth N, Liu W, Ahmad SA, Fan F, Stoeltzing O, Parikh AA, et al. αvβ3 integrin antagonist S247 decreases colon cancer metastasis and angiogenesis and improves survival in mice. Cancer Res. (2003) 63:2079–87. Available online at: http://cancerres.aacrjournals.org/content/63/9/2079.long
79. Al-Nedawi K, Szemraj J, Cierniewski CS. Mast Cell-Derived derived exosomes activate endothelial cells to secrete plasminogen activator inhibitor type 1. Arterioscler Thromb Vasc Biol. (2005) 25:1744–9. doi: 10.1161/01.ATV.0000172007.86541.76
80. Iozzo RV, Moscatello DK, McQuillan DJ, Eichstetter I. Decorin is a biological ligand for the epidermal growth factor receptor. J Biol Chem. (1999) 274:4489–92. doi: 10.1074/jbc.274.8.4489
81. Seidler DG, Goldoni S, Agnew C, Cardi C, Thakur ML, Owens RT, et al. Decorin protein core inhibits In vivo cancer growth and metabolism by hindering epidermal growth factor receptor function and triggering apoptosis via caspase-3 activation. J Biol Chem. (2006) 281:26408–18. doi: 10.1074/jbc.M602853200
82. Iozzo RV, Buraschi S, Genua M, Xu SQ, Solomides CC, Peiper SC, et al. Decorin antagonizes IGF receptor I (IGF-IR) function by interfering with IGF-IR activity and attenuating downstream signaling. J Biol Chem. (2011) 286:34712–21. doi: 10.1074/jbc.M111.262766
83. Scott RA, Ramaswamy AK, Park K, Panitch A. Decorin mimic promotes endothelial cell health in endothelial monolayers and endothelial-smooth muscle co-cultures. J Tissue Eng Regen Med. (2017) 11:1365–76. doi: 10.1002/term.2035
84. Albig AR, Roy TG, Becenti DJ, Schiemann WP. Transcriptome analysis of endothelial cell gene expression induced by growth on matrigel matrices: identification and characterization of MAGP-2 and lumican as novel regulators of angiogenesis. Angiogenesis. (2007) 10:197–216. doi: 10.1007/s10456-007-9075-z
85. Iruela-Arispe ML, Bornstein P, Sage H. Thrombospondin exerts an antiangiogenic effect on cord formation by endothelial cells in vitro. Proc Natl Acad Sci USA. (1991) 88:5026–30. doi: 10.1073/pnas.88.11.5026
86. Dawson DW, Pearce SF, Zhong R, Silverstein RL, Frazier WA, Bouck NP. CD36 mediates the in vitro inhibitory effects of thrombospondin-1 on endothelial cells. J Cell Biol. (1997) 138:707–17. doi: 10.1083/jcb.138.3.707
87. Yang Z, Kyriakides TR, Bornstein P. Matricellular proteins as modulators of cell-matrix interactions: adhesive defect in thrombospondin 2-null fibroblasts is a consequence of increased levels of matrix metalloproteinase-2. Mol Biol Cell. (2000) 11:3353–64. doi: 10.1091/mbc.11.10.3353
88. Kopp HG, Hooper AT, Broekman MJ, Avecilla ST, Petit I, Luo M, et al. Thrombospondins deployed by thrombopoietic cells determine angiogenic switch and extent of revascularization. J Clin Invest. (2006) 116:3277–91. doi: 10.1172/JCI29314
89. Muppala S, Frolova E, Xiao R, Krukovets I, Yoon S, Hoppe G, et al. Proangiogenic properties of thrombospondin-4. Arterioscler Thromb Vasc Biol. (2015) 35:1975–86. doi: 10.1161/ATVBAHA.115.305912
90. Muppala S, Xiao R, Krukovets I, Verbovetsky D, Yendamuri R, Habib N, et al. Thrombospondin-4 mediates TGF-beta-induced angiogenesis. Oncogene. (2017) 36:5189–98. doi: 10.1038/onc.2017.140
91. Isenberg JS, Ridnour LA, Dimitry J, Frazier WA, Wink DA, Roberts DD. CD47 is necessary for inhibition of nitric oxide-stimulated vascular cell responses by thrombospondin-1. J Biol Chem. (2006) 281:26069–80. doi: 10.1074/jbc.M605040200
92. McDonald JF, Zheleznyak A, Frazier WA. Cholesterol-independent interactions with CD47 enhance alphavbeta3 avidity. J Biol Chem. (2004) 279:17301–11. doi: 10.1074/jbc.M312782200
93. Oganesian A, Armstrong LC, Migliorini MM, Strickland DK, Bornstein P. Thrombospondins use the VLDL receptor and a nonapoptotic pathway to inhibit cell division in microvascular endothelial cells. Mol Biol Cell. (2008) 19:563–71. doi: 10.1091/mbc.e07-07-0649
94. Kolesnikova TV, Lau LF. Human CYR61-mediated enhancement of bFGF-induced DNA synthesis in human umbilical vein endothelial cells. Oncogene. (1998) 16:747–54. doi: 10.1038/sj.onc.1201572
95. Kubota S, Kawaki H, Kondo S, Yosimichi G, Minato M, Nishida T, et al. Multiple activation of mitogen-activated protein kinases by purified independent CCN2 modules in vascular endothelial cells and chondrocytes in culture. Biochimie. (2006) 88:1973–81. doi: 10.1016/j.biochi.2006.07.007
96. Karagiannis ED, Popel AS. Peptides derived from type I thrombospondin repeat-containing proteins of the CCN family inhibit proliferation and migration of endothelial cells. Int J Biochem Cell Biol. (2007) 39:2314–23. doi: 10.1016/j.biocel.2007.06.018
97. Ishitsuka T, Ikuta T, Ariga H, Matsumoto K. Serum tenascin-X strongly binds to vascular endothelial growth factor. Biol Pharm Bull. (2009) 32:1004–11. doi: 10.1248/bpb.32.1004
98. Mariko B, Ghandour Z, Raveaud S, Quentin M, Usson Y, Verdetti J, et al. Microfibrils and fibrillin-1 induce integrin-mediated signaling, proliferation and migration in human endothelial cells. Am J Physiol Cell Physiol. (2010) 299:C977–87. doi: 10.1152/ajpcell.00377.2009
99. Armstrong LC, Bjorkblom B, Hankenson KD, Siadak AW, Stiles CE, Bornstein P. Thrombospondin 2 inhibits microvascular endothelial cell proliferation by a caspase-independent mechanism. Mol Biol Cell. (2002) 13:1893–905. doi: 10.1091/mbc.e01-09-0066
100. Nozaki M, Sakurai E, Raisler BJ, Baffi JZ, Witta J, Ogura Y, et al. Loss of SPARC-mediated VEGFR-1 suppression after injury reveals a novel antiangiogenic activity of VEGF-A. J Clin Invest. (2006) 116:422–9. doi: 10.1172/JCI26316
101. Remus EW, Lyle AN, Weiss D, Landazuri N, Weber M, Searles C, et al. miR181a protects against angiotensin II-induced osteopontin expression in vascular smooth muscle cells. Atherosclerosis. (2013) 228:168–74. doi: 10.1016/j.atherosclerosis.2013.01.037
102. Kapustin A, Stepanova V, Aniol N, Cines DB, Poliakov A, Yarovoi S, et al. Fibulin-5 binds urokinase-type plasminogen activator and mediates urokinase-stimulated beta1-integrin-dependent cell migration. Biochem J. (2012) 443:491–503. doi: 10.1042/BJ20110348
103. Albig AR, Schiemann WP. Fibulin-5 Antagonizes Vascular Endothelial Growth Factor (VEGF) signaling and angiogenic sprouting by endothelial cells. DNA Cell Biol. (2004) 23:367–79. doi: 10.1089/104454904323145254
104. Tsai M-S, Chiang M-T, Tsai D-L, Yang C-W, Hou H-S, Li Y-R, et al. Galectin-1 restricts vascular smooth muscle cell motility via modulating adhesion force and focal adhesion dynamics. Scient Rep. (2018) 8:11497. doi: 10.1038/s41598-018-29843-3
105. Moiseeva EP, Javed Q, Spring EL, de Bono DP. Galectin 1 is involved in vascular smooth muscle cell proliferation. Cardiovasc Res. (2000) 45:493–502. doi: 10.1016/S0008-6363(99)00276-X
106. Hashmi S, Al-Salam S. Galectin-1: a biomarker of surgical stress in murine model of cardiac surgery. Int J Clini Exp Pathol. (2015) 8:7157–64. Available online at: http://www.ijcep.com/files/ijcep0008555.pdf
107. Lake AC, Bialik A, Walsh K, Castellot JJ Jr. CCN5 is a growth arrest-specific gene that regulates smooth muscle cell proliferation and motility. Am J Pathol. (2003) 162:219–31. doi: 10.1016/S0002-9440(10)63813-8
108. Lake AC, Castellot JJ Jr. CCN5 modulates the antiproliferative effect of heparin and regulates cell motility in vascular smooth muscle cells. Cell Commun Signal. (2003) 1:5. doi: 10.1186/1478-811X-1-5
109. Castellot J, Gray MR, Simon AR. Use of CCN5 for Treatment Of Smooth Muscle Proliferation Disorders. USA patent application US20080207489A1 (2007).
110. Sachdeva J, Mahajan A, Cheng J, Baeten JT, Lilly B, Kuivaniemi H, et al. Smooth muscle cell-specific Notch1 haploinsufficiency restricts the progression of abdominal aortic aneurysm by modulating CTGF expression. PLoS ONE. (2017) 12:e0178538. doi: 10.1371/journal.pone.0178538
111. Williams H, Mill CA, Monk BA, Hulin-Curtis S, Johnson JL, George SJ. Wnt2 and WISP-1/CCN4 induce intimal thickening via promotion of smooth muscle cell migration. Arterioscler Thromb Vasc Biol. (2016) 36:1417–24. doi: 10.1161/ATVBAHA.116.307626
112. Mill C, Jeremy JY, George SJ. WNT5A signalling promotes vsmc survival via WISP-1: consequences for vsmc viability in atherosclerotic plaques. Heart. (2011) 97:1–2. doi: 10.1136/heartjnl-2011-300920a.4
113. Mill C, Monk BA, Williams H, Simmonds SJ, Jeremy JY, Johnson JL, et al. Wnt5a-induced Wnt1-inducible secreted protein-1 suppresses vascular smooth muscle cell apoptosis induced by oxidative stress. Arterioscler Thromb Vasc Biol. (2014) 34:2449–56. doi: 10.1161/ATVBAHA.114.303922
114. Ishigaki T, Imanaka-Yoshida K, Shimojo N, Matsushima S, Taki W, Yoshida T. Tenascin-C enhances crosstalk signaling of integrin αvβ3/PDGFR-β complex by SRC recruitment promoting PDGF-induced proliferation and migration in smooth muscle cells. J Cell Physiol. (2011) 226:2617–24. doi: 10.1002/jcp.22614
115. Kimura T, Yoshimura K, Aoki H, Imanaka-Yoshida K, Yoshida T, Ikeda Y, et al. Tenascin-C is expressed in abdominal aortic aneurysm tissue with an active degradation process. Pathol Int. (2011) 61:559–64. doi: 10.1111/j.1440-1827.2011.02699.x
116. Wallner K, Li C, Fishbein MC, Shah PK, Sharifi BG. Arterialization of human vein grafts is associated with tenascin-C expression. J Am Coll Cardiol. (1999) 34:871–5. doi: 10.1016/S0735-1097(99)00272-7
117. Isaji T, Hashimoto T, Yamamoto K, Santana JM, Yatsula B, Hu H, et al. Improving the outcome of vein grafts: should vascular surgeons turn veins into arteries? Ann Vasc Dis. (2017) 10:8–16. doi: 10.3400/avd.ra.17-00008
118. Isenberg JS, Hyodo F, Matsumoto K, Romeo MJ, Abu-Asab M, Tsokos M, et al. Thrombospondin-1 limits ischemic tissue survival by inhibiting nitric oxide-mediated vascular smooth muscle relaxation. Blood. (2007) 109:1945–52. doi: 10.1182/blood-2006-08-041368
119. Majack RA, Cook SC, Bornstein P. Control of smooth muscle cell growth by components of the extracellular matrix: autocrine role for thrombospondin. Proc Natl Acad Sci USA. (1986) 83:9050–4. doi: 10.1073/pnas.83.23.9050
120. Ranjzad P, Salem HK, Kingston PA. Adenovirus-mediated gene transfer of fibromodulin inhibits neointimal hyperplasia in an organ culture model of human saphenous vein graft disease. Gene Ther. (2009) 16:1154–62. doi: 10.1038/gt.2009.63
121. Bergmann K, Mankowska-Cyl A, Kretowicz M, Manitius J, Sypniewska G. Fibulin-1 is associated with cardiovascular risk in non-obese, non-diabetic individuals. Folia Medica Copernicana. (2013) 1:18–22. Available online at: https://journals.viamedica.pl/medical_research_journal/article/view/35429
122. Huang J, Davis EC, Chapman SL, Budatha M, Marmorstein LY, Word RA, et al. Fibulin-4 deficiency results in ascending aortic aneurysms: a potential link between abnormal smooth muscle cell phenotype and aneurysm progression. Circul Res. (2010) 106:583–92. doi: 10.1161/CIRCRESAHA.109.207852
123. Zhang C, van der Voort D, Shi H, Zhang R, Qing Y, Hiraoka S, et al. Matricellular protein CCN3 mitigates abdominal aortic aneurysm. J Clini Invest. (2016) 126:1282–99. doi: 10.1172/JCI82337
124. Shimoyama T, Hiraoka S, Takemoto M, Koshizaka M, Tokuyama H, Tokuyama T, et al. CCN3 inhibits neointimal hyperplasia through modulation of smooth muscle cell growth and migration. Arterioscler Thromb Vasc Biol. (2010) 30:675–U680. doi: 10.1161/ATVBAHA.110.203356
125. Riser BL, Barnes JL, Varani J. Balanced regulation of the CCN family of matricellular proteins: a novel approach to the prevention and treatment of fibrosis and cancer. J Cell Comm Signal. (2015) 9:327–39. doi: 10.1007/s12079-015-0309-3
126. Ikeda T, Shirasawa T, Esaki Y, Yoshiki S, Hirokawa K. Osteopontin messenger-rna is expressed by smooth muscle-derived foam cells in human atherosclerotic lesions of the aorta. J Clini Invest. (1993) 92:2814–20. doi: 10.1172/JCI116901
127. Gao H, Steffen MC, Ramos KS. Osteopontin regulates alpha-smooth muscle actin and calponin in vascular smooth muscle cells. Cell Biol Int. (2012) 36:155–61. doi: 10.1042/CBI20100240
128. Chellan B, Narayani J, Appukuttan PS. Galectin-1, an endogenous lectin produced by arterial cells, binds lipoprotein(a) [Lp(a)] in situ: relevance to atherogenesis. Exp Mol Pathol. (2007) 83:399–404. doi: 10.1016/j.yexmp.2007.04.004
129. Merrilees MJ, Lemire JM, Fischer JW, Kinsella MG, Braun KR, Clowes AW, et al. Retrovirally mediated overexpression of versican v3 by arterial smooth muscle cells induces tropoelastin synthesis and elastic fiber formation in vitro and in neointima after vascular injury. Circ Res. (2002) 90:481–7. doi: 10.1161/hh0402.105791
130. Keire PA, L'Heureux N, Vernon RB, Merrilees MJ, Starcher B, Okon E, et al. Expression of versican isoform V3 in the absence of ascorbate improves elastogenesis in engineered vascular constructs. Tissue Eng Part A. (2010) 16:501–12. doi: 10.1089/ten.tea.2009.0129
131. Long JL, Tranquillo RT. Elastic fiber production in cardiovascular tissue-equivalents. Matrix Biol. (2003) 22:339–50. doi: 10.1016/S0945-053X(03)00052-0
132. Noda K, Dabovic B, Takagi K, Inoue T, Horiguchi M, Hirai M, et al. Latent TGF-beta binding protein 4 promotes elastic fiber assembly by interacting with fibulin-5. Proc Natl Acad Sci USA. (2013) 110:2852–7. doi: 10.1073/pnas.1215779110
133. Aya R, Ishiko T, Noda K, Yamawaki S, Sakamoto Y, Tomihata K, et al. Regeneration of elastic fibers by three-dimensional culture on a collagen scaffold and the addition of latent TGF-beta binding protein 4 to improve elastic matrix deposition. Biomaterials. (2015) 72:29–37. doi: 10.1016/j.biomaterials.2015.08.036
134. Bar A, Dorfman SE, Fischer P, Hilfiker-Kleiner D, Cebotari S, Tudorache I, et al. The pro-angiogenic factor CCN1 enhances the re-endothelialization of biological vascularized matrices in vitro. Cardiovasc Res. (2010) 85:806–13. doi: 10.1093/cvr/cvp370
135. Theodoridis K, Tudorache I, Calistru A, Cebotari S, Meyer T, Sarikouch S, et al. Successful matrix guided tissue regeneration of decellularized pulmonary heart valve allografts in elderly sheep. Biomaterials. (2015) 52:221–8. doi: 10.1016/j.biomaterials.2015.02.023
136. Boer U, Spengler C, Jonigk D, Klingenberg M, Schrimpf C, Lutzner S, et al. Coating decellularized equine carotid arteries with CCN1 improves cellular repopulation, local biocompatibility, and immune response in sheep. Tissue Eng Part A. (2013) 19:1829–42. doi: 10.1089/ten.tea.2012.0558
137. Jeinsen N, Magel L, Jonigk D, Klingenberg M, Haverich A, Wilhelmi M, et al. Biocompatibility of intensified decellularized equine carotid arteries in a rat subcutaneous implantation model and in a human in vitro model. Tissue Eng Part A. (2018) 24:310–21. doi: 10.1089/ten.tea.2016.0542
138. Ravi S, Caves JM, Martinez AW, Haller CA, Chaikof EL. Incorporation of fibronectin to enhance cytocompatibility in multilayer elastin-like protein scaffolds for tissue engineering. J Biomed Mater Res A. (2013) 101:1915–25. doi: 10.1002/jbm.a.34484
139. Ravi S, Haller CA, Sallach RE, Chaikof EL. Cell behavior on a CCN1 functionalized elastin-mimetic protein polymer. Biomaterials. (2012) 33:2431–8. doi: 10.1016/j.biomaterials.2011.11.055
140. Chen N, Leu SJ, Todorovic V, Lam SC, Lau LF. Identification of a novel integrin alphavbeta3 binding site in CCN1 (CYR61) critical for pro-angiogenic activities in vascular endothelial cells. J Biol Chem. (2004) 279:44166–76. doi: 10.1074/jbc.M406813200
141. Karimi F, Thombare VJ, Hutton CA, O'Connor AJ, Qiao GG, Heath DE. Beyond RGD; nanoclusters of syndecan- and integrin-binding ligands synergistically enhance cell/material interactions. Biomaterials. (2018) 187:81–92. doi: 10.1016/j.biomaterials.2018.10.002
142. Chung H, Multhaupt HA, Oh ES, Couchman JR. Minireview: Syndecans and their crucial roles during tissue regeneration. FEBS Lett. (2016) 590:2408–17. doi: 10.1002/1873-3468.12280
143. Preis M, Schneiderman J, Koren B, Ben-Yosef Y, Levin-Ashkenazi D, Shapiro S, et al. Co-expression of fibulin-5 and VEGF165 increases long-term patency of synthetic vascular grafts seeded with autologous endothelial cells. Gene Ther. (2016) 23:237–46. doi: 10.1038/gt.2015.104
144. Hinderer S, Sudrow K, Schneider M, Holeiter M, Layland SL, Seifert M, et al. Surface functionalization of electrospun scaffolds using recombinant human decorin attracts circulating endothelial progenitor cells. Sci Rep. (2018) 8:110. doi: 10.1038/s41598-017-18382-y
145. Dubey G, Mequanint K. Conjugation of fibronectin onto three-dimensional porous scaffolds for vascular tissue engineering applications. Acta Biomater. (2011) 7:1114–25. doi: 10.1016/j.actbio.2010.11.010
146. Blose KJ, Ennis TL, Arif B, Weinbaum JS, Curci JA, Vorp DA. Periadventitial adipose-derived stem cell treatment halts elastase-induced abdominal aortic aneurysm progression. Regen Med. (2014) 9:733–41. doi: 10.2217/rme.14.61
Keywords: collagen, elastin, extracellular matrix, endothelial cells, smooth muscle cells, thrombosis
Citation: Ramaswamy AK, Vorp DA and Weinbaum JS (2019) Functional Vascular Tissue Engineering Inspired by Matricellular Proteins. Front. Cardiovasc. Med. 6:74. doi: 10.3389/fcvm.2019.00074
Received: 28 February 2019; Accepted: 15 May 2019;
Published: 31 May 2019.
Edited by:
Steve Bibevski, Joe DiMaggio Children's Hospital, United StatesReviewed by:
Axel Haverich, Hannover Medical School, GermanyXuechong Hong, Boston Children's Hospital, Harvard Medical School, United States
Copyright © 2019 Ramaswamy, Vorp and Weinbaum. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Justin S. Weinbaum, anV3NTFAcGl0dC5lZHU=