 Tayyabah Yousaf
Tayyabah Yousaf Gennaro Pagano
Gennaro Pagano Heather Wilson
Heather Wilson Marios Politis
Marios Politis- Neurodegeneration Imaging Group, Department of Basic and Clinical Neuroscience, Institute of Psychiatry, Psychology and Neuroscience, King's College London, London, United Kingdom
Sleep dysfunction is recognized as a distinct clinical manifestation in movement disorders, often reported early on in the disease course. Excessive daytime sleepiness, rapid eye movement sleep behavior disorder and restless leg syndrome, amidst several others, are common sleep disturbances that often result in significant morbidity. In this article, we review the spectrum of sleep abnormalities across atypical Parkinsonian disorders including multiple system atrophy (MSA), progressive supranuclear palsy (PSP) and corticobasal syndrome (CBS), as well as Parkinson's disease (PD) and Huntington's disease (HD). We also explore the current concepts on the neurobiological underpinnings of sleep disorders, including the role of dopaminergic and non-dopaminergic pathways, by evaluating the molecular, structural and functional neuroimaging evidence based on several novel techniques including magnetic resonance imaging (MRI), functional magnetic resonance imaging (fMRI), diffusion tensor imaging (DTI), single-photon emission computed tomography (SPECT) and positron emission tomography (PET). Based on the current state of research, we suggest that neuroimaging is an invaluable tool for assessing structural and functional correlates of sleep disturbances, harboring the ability to shed light on the sleep problems attached to the limited treatment options available today. As our understanding of the pathophysiology of sleep and wake disruption heightens, novel therapeutic approaches are certain to transpire.
Introduction
Sleep-wake disturbances are increasingly being recognized as common symptoms in neurodegenerative diseases, particularly movement disorders. Given that the quality of sleep is intrinsically associated to the quality of life, sleep disturbances inevitably induce significant morbidity. Therefore, early identification and effective treatment of sleep abnormalities is of considerable therapeutic interest, particularly for improving quality of life, delaying institutionalization and potentially reducing healthcare costs (1). Furthermore, emerging evidence has demonstrated that disturbed sleep-wake cycles may precede, influence and/or facilitate disease progression, thus possessing the potential to serve as early indicators of ongoing neurodegeneration and providing a window of opportunity to administer disease-modifying interventions early in the course of neurodegeneration.
Sleep disturbances can manifest as insomnia, sleep-disordered breathing, sleep fragmentation, restless leg syndrome (RLS), periodic limb movement disorder (PLMD), excessive daytime sleepiness (EDS) and parasomnias associated with rapid eye movements (REM) sleep. Whilst there is considerable overlap in the types of sleep disturbances reported across movement disorders, some sleep problems are either unique to, or substantially more prevalent in certain movement disorders (Figure 1). The origin of these sleep disorders is multifactorial, including neurodegeneration of central sleep regulatory structures, circadian dysfunction, impact of motor and non-motor symptoms, and adverse effects of medications (2–5).
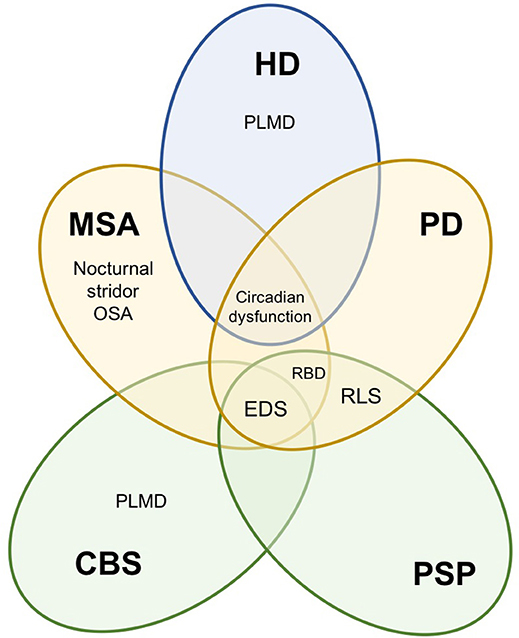
Figure 1. Summary of sleep disturbances found in movement disorders. Movement disorders exhibit some overlap in sleep disturbances. PSP and CBS are tauopathies (green), PD and MSA are synucleinopathies (yellow) and HD is a hereditary disorder (blue). PSP, progressive supranuclear palsy; CBS, corticobasal syndrome; PD, Parkinson's disease; MSA, multiple system atrophy; HD, Huntington's disease; EDS, excessive daytime sleepiness; RBD, REM behavior sleep disorder, RLS, restless leg syndrome; PLMD, periodic limb movement disorder; OSA, obstructive sleep apnoea.
Neuroimaging techniques, such as magnetic resonance imaging (MRI) and functional MRI (fMRI) have played a fundamental role in characterizing the structural and functional changes in patients suffering from sleep disturbances. Positron emission tomography (PET) is a powerful technique for investigating in vivo abnormalities in brain metabolism and receptor distribution, by enabling the measurement of radioligand distribution, which is introduced into the body on a biologically active molecule (6). By revealing the regional patterns of activation associated with specific sleep disorders, data from imaging methods, including single photon emission computed tomography (SPECT) (7–10), have complemented and extended previous findings predominantly based on electroencephalography (EEG) studies, thus improving and better characterizing the pathogenic mechanisms of major sleep disturbances.
In this article, we review the major sleep disturbances commonly experienced by patients with movement disorders, including Parkinson's disease (PD), Huntington's disease (HD) and atypical Parkinsonism, such as progressive supranuclear palsy (PSP), multiple system atrophy (MSA) and corticobasal syndrome (CBS). We discuss the role of neuroimaging in identifying neural correlates of various sleep disorders, with the aim of assessing how these studies have improved our knowledge via the insight gained into the underlying mechanisms of major sleep disturbances (Table 1).
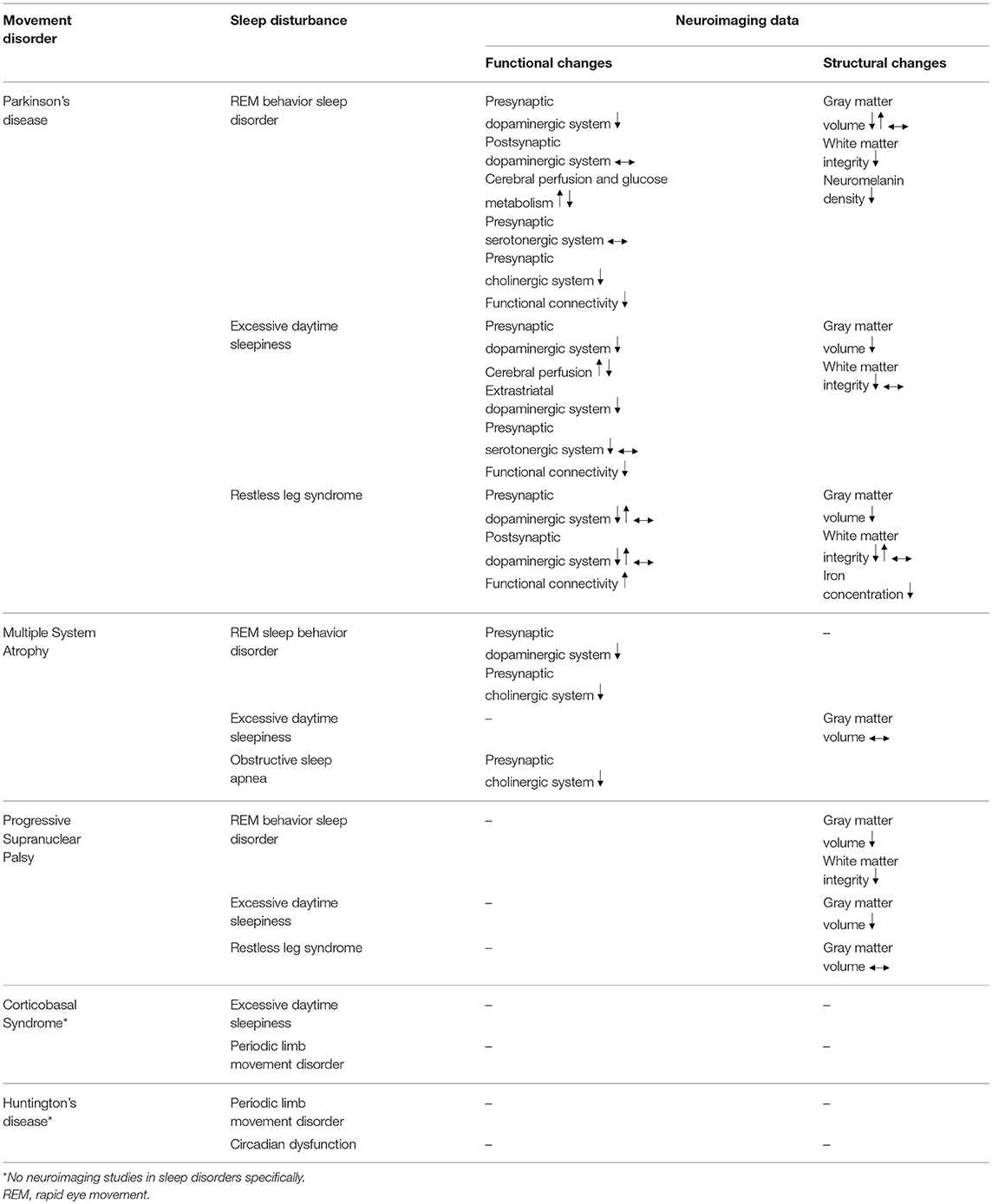
Table 1. Summary of functional and structural changes associated with common sleep disturbances in movement disorders.
Regulatory Circuits Involved in Sleep
Sleep is indispensable for promoting adequate health and bodily function, given that it is an active physiological process required for protection and normalization of all major organs and regulatory systems (11). Sleep is a global and dynamically regulated state, primarily by two intimately integrated internal biological mechanisms: circadian rhythm and homeostasis. These control mechanisms are expressed at all levels of biological organization, from genes and intracellular mechanisms to neural networks and central neuronal systems at the organismic level. Several brain structures, both subcortical and forebrain areas, have specific functions in sleep at different organizational levels, controlling sleep-wake transitions as well as alternating between rapid eye movement (REM) and non-REM sleep (Figure 2) (12).
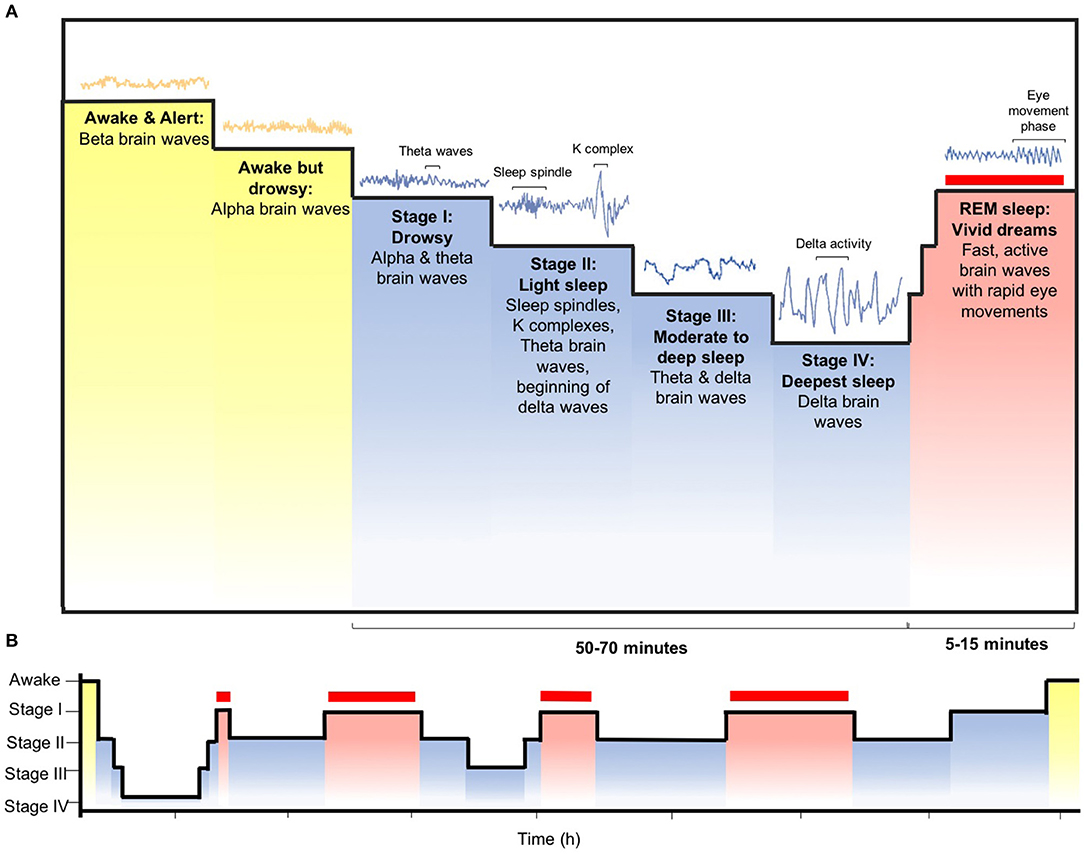
Figure 2. Sleep patterns and stages. (A) A complete sleep cycle typically takes, on average, between 90 and 110 min. There are two fundamental types of sleep: rapid eye movement (REM) sleep and non-REM sleep, which can be divided into stages I–IV. These non-REM stages correspond to the accumulating depth of sleep, as reflected by the progressive emergence of high-voltage, low-frequency brain wave activity, which dominate the deepest stages of non-REM sleep (stage III and IV, also identified as slow-wave sleep). Cardinal wave activity of stage II are sleep spindle and K-complex waveforms, who's timings are influenced by a slow oscillation (<1 Hz). REM sleep, also identified as active or paradoxical sleep, is characterized by wake-like, high-frequency, low-amplitude activity and atonia (i.e., low muscle tone). (B) During each of the four to five cycles that transpire each night of adult human sleep, non-REM (blue bars) and REM sleep (red bars) alternate. During the earlier proportion of the night, non-REM sleep is deeper, occupying a disproportionately large amount of time, particularly within the first cycle where the REM stage may be brief or terminated. As the night progresses, non-REM sleep becomes shallow, with more of each cycle being allocated to REM. A reliable oscillator times the sustained period of length of the non-REM and REM cycle, of which the amplitude varies according to extrinsic factors.
The Circadian System
Circadian rhythms, which can be defined as physiological and behavioral cycles with a periodicity of approximately 24 h, are orchestrated by sophisticated molecular loops. Given that the circadian system dictates the 24-h rhythmicity in biological processes such as rest-activity behavior, hormonal level, feeding and body temperature, disruption of this system can ultimately lead to negative affects imposed on alertness, sleep quality, cognitive performance, mental health, motor control and metabolism (4). These functions are often impaired in neurodegenerative diseases, with multiple brain areas, including nuclei involved in circadian and sleep regulation, being affected by neurodegenerative processes. Therefore, it is not surprising that neurodegenerative disorders often illustrate a progressive breakdown of the normal cycles of sleep and wakefulness, which not only contribute to poor quality of life and morbidity; but could also influence the disease process itself.
Molecularly, an interlocking positive-negative feedback mechanism controlling gene transcription and their protein products in individual cells of the suprachiasmatic nucleus (SCN) of the hypothalamus, which can be entrained to ambient conditions by light, is the foundation of circadian rhythmicity in mammals (12). Intrinsic circadian rhythmicity is outputted by action potentials generated by SCN cells, impinging on nuclei adjacent to the anterior hypothalamus, such as the subparaventricular nucleus (SPZ), paraventricular nucleus, dorsomedial nucleus (DMH) and the medial preoptic area, which, sequentially, convey circadian rhythmicity to structures involved in regulating physiological processes, such as the GABA-containing ventrolateral preoptic area (VPLO) and the locus coeruleus. The SCN circadian oscillator can receive feedback via melatonin, which is a sleep-related hormone secreted reliably from the pineal gland in response to polysynaptically conveyed signals from the SCN. Additionally, other neuromodulatory systems, such as acetylecholine, modulate the SCN's responsiveness to photic input from the retinohypothalamic tract (RHT). Temporal specificity is also demonstrated, given that the circadian pacemaker is responsive to specific modulatory signals exclusively at certain periods of the circadian day (12).
Anatomy of Sleep and Neuroendocrine Mediators
Neural circuits involved in sleep regulation can be broadly categorized under four subsystems: (1) wake-promoting; (2) non-REM sleep promoting; (3) REM sleep promoting; (4) non-REM-REM switch (13, 14). The neurodegenerative nature of movement disorders ultimately disrupts sleep regulation by interrupting the normal feedback between neuronal subsystems following accumulation of local neuropathology and concomitant changes in neurotransmitters.
The VLPO serves as a key hypothalamic structure, responsible for promoting non-REM sleep. The VLPO may initiate the onset of sleep via its reciprocal inhibition of brainstem-residing serotonergic, cholinergic and noradrenergic systems, alongside histaminergic arousal systems of the posterior hypothalamus and cholinergic systems of the basal forebrain, of which all are modulated by orexinergic arousal system of the lateral hypothalamus. Activated brain states of waking are promoted by these arousal systems, whereas the activated state of REM sleep is promoted by the cholinergic system alone (12).
Sleep-wake homeostatic information from endogenous chemical signals, such as the accumulation of adenosine during wake hours, and circadian input from the anterior hypothalamus both trigger the VLPO to initiate sleep onset. Once sleep has commenced, the alternation between non-REM and REM sleep is controlled by an ultradian oscillator in the mesopontine junction, which, in turn, is executively controlled by the interaction between aminergic REM-off and cholinergic REM-on cell groups (Figure 2). Interposed autoregulatory, inhibitory and excitatory circuits that involve serotonin, noradrenaline, acetylcholine, glutamate and GABA mediate the interaction between the two cell groups.
Parkinson's Disease
Parkinson's disease (PD) is the second most common neurodegenerative disorder, pathologically characterized by dopaminergic neuronal atrophy in the substantia nigra and widespread intracellular inclusions containing aggregates of α-synuclein (15). PD may be perceived as an admirable illustration of where efforts to comprehend impaired sleep-wake homeostasis precipitated significant advances in understanding the disease biology and progression. Conversely, sleep disorders in HD and atypical Parkinsonism, including PSP, MSA and CBS, have not been systematically studied.
REM Sleep Behavior Disorder
Idiopathic REM sleep behavior disorder (iRBD) is a parasomnia characterized by the lack of normal skeletal muscle atonia during REM sleep, resulting in dream-enacting behaviors often associated with violent or aggressive dreams (16). Over recent years, it is now increasingly recognized that RBD is associated with neurodegenerative disease, particularly α-synucleinopathies such as PD, MSA and dementia with Lewy Bodies (DLB) (17–19). Presumed to be a preliminary symptom of progressive neurodegeneration, RBD can precede the clinical manifestation of these α-synucleinopathies by several years, with risk estimates of up to 33% at 5 years RBD diagnosis, 76% at 10 years and 91% at 14 years (20, 21), making RBD one of the strongest clinical predictors of synucleinopathy onset (22).
Diurnal and nocturnal sleep problems are highly prevalent in PD, affecting up to 88% of the PD population, with RBD, specifically, affecting up to 50% (23, 24). Studies have demonstrated that sleep difficulties are essential predictors of poor quality of life, with most reports proposing that disturbance of sleep, lack of independence and depression are the fundamental determinants of poor quality of life (25).
Though PD is currently diagnosed when patients present with key motor symptoms including bradykinesia, rigidity, tremor or postural instability (26), the term prodromal has surfaced to describe the period between the onset of motor symptoms and striatal neurodegeneration, with basal ganglia degeneration suggested to commence up to 7 years prior to diagnosis (27, 28). However, prodromal symptoms such as hyposmia and RBD can emerge decades earlier in some cases, with approximately 20% of PD patients reporting RBD onset prior to motor symptomology (29). This highlights that RBD has the potential to serve as the most promising prodromal marker (30, 31). Prominently, long-term cohort studies insinuate that up to 90% of individuals who suffer from isolated RBD will go onto develop PD or atypical Parkinsonism (21, 32). However, the pathophysiology of RBD in prodromal and established PD remains unclear.
Molecular Imaging
Early imaging studies using SPECT and PET to image presynaptic dopamine transporter (DAT) and presynaptic vesicular dopamine transporters in RBD confirmed that these patients exhibit subclinical striatal dopaminergic deficit (33, 34). Consequent larger cohorts comparing RBD patients with healthy controls illustrated that 20–40% of RBD subjects exhibit abnormal nigro-striatal functioning, particularly at the putamen level (35–37).
One of the earliest studies, however, was carried out by Eisensehret and colleagues who employed SPECT tracers [123I]IBZM and [123I]IPT-SPECT, as markers of post-synaptic dopamine type-2 receptors (D2R) density and presynaptic DAT, respectively, to gain an insight into striatal dopaminergic integrity in five patients with iRBD compared to 14 PD patients and seven controls (34). They found a reduction in striatal [123I]IPT uptake in iRBD compared to controls, though no differences were found in striatal [123I]IBZM. Despite the small sample sizes, this study was one of the first to report a reduction in striatal dopamine transporters in idiopathic clinically manifest RBD. Further, iRBD patients exhibit a decline in striatal [123I]FP-CIT uptake that reflects progressive nigrostriatal dopamine depletion (38). Whilst iRBD was reported to be associated with a loss of DAT within the entire basal ganglia compared to controls at baseline, the mean reduction in striatal [123I]FP-CIT uptake from baseline to 3 years was 19.36% in the right putamen, 15.57% in the left putamen, 7.14% in the right caudate and 10.81% in the left caudate (38). Given the extent of caudate denervation at baseline, these findings suggest that nigro-caudate deafferentation may reflect RBD pathophysiology and putaminal deafferentation may serve as a marker of increasing severity of PD. In this direction, caudate [123I]FP-CIT-SPECT uptake may serve as a marker of RBD specifically and putaminal [123I]FP-CIT-SPECT uptake may serve as a more sensitive marker of the progression of dopaminergic dysfunction, thus both useful for studying the potential disease-modifying compounds in iRBD.
Studies have also demonstrated that nigro-putaminal functionality is impaired to a mild extent in patients with subclinical RBD (REM sleep without muscle atonia, but not RBD on polysomography), with clinically manifest idiopathic RBD patients exhibiting moderate impairment and PD patients demonstrating the most severe dopaminergic impairment (39, 40). A [18F]DOPA PET study scanned PD patients twice with a 5-year interval to assess the rate of disease progression and found that the disease process initially affects the posterior putamen, followed by the anterior putamen and caudate nucleus (41), whereas PD patients with iRBD displayed a more sever nigro-caudate deafferentation compared to the PD patients without RBD, whilst preserving the nigro-putaminal functionality (40), which is in line with Eisensehr and colleagues (39). These findings corroborate with the notion that nigro-caudate deafferentation may be a hallmark of RBD itself, independent of PD diagnosis. Dopaminergic imaging may, therefore, hold the potential to monitor progression throughout the prodromal phase.
Although, taken together, these results implicate the nigrostriatal dopaminergic system in the pathophysiology of RBD, it remains unclear as to what extent this system is directly involved with the increase in motor activity during REM sleep. A [123I]FP-CIT-SPECT study by Eisensehr observed that muscle activity during REM sleep lasting persistently longer than 0.5 s was independently associated with reduced striatal [123I]IPT uptake in iRBD patients (39). Although these results were not reproduced by Kim and colleagues, who found that electromyography (EMG) activities during REM sleep were not associated with striatal DAT density (36), they were partially replicated by Zoetmulder and colleagues who illustrated that EMG-activity in the mentalis muscle correlated with putaminal [123I]FP-CIT uptake in iRBD patients, (42). Although these findings suggest that amplified muscle activity during REM sleep is associated to nigrostriatal dopaminergic system in RBD and dopaminergic function in PD, it may not be essential for the development of RBD, thus not directly associated with the severity of RBD symptomology in PD.
An interesting study applying [99mTc]-EDC SPECT with simultaneous polysomnographic (PSG) recordings demonstrated increased perfusion in the supplementary motor area during a REM sleep behavior episode (43), which was subsequently confirmed by Mayer and colleagues who reported bilateral activation of the premotor (supplementary motor) areas, the periaqueductal area, the interhemispheric cleft, the anterior lobe of the cerebellum and the dorsal and ventral pons (44). These studies implicate a common motor pathway in RBD, localizing the motor generators responsible for dream enactment behavior to include the supplementary motor area, bypassing the basal ganglia. These results corroborate with a previous study in PD patients with RBD, illustrating the “normalization” of movement during REM sleep, indicating that the motor cortex may generate movements during RBD, following the pyramidal tract bypassing the extrapyramidal system (45).
Prior observations of dopaminergic deficits in RBD have been extended by an imaging study with [18F]FDG PET and [99mTc]-EDC SPECT, which aimed to determine whether Parkinson disease-related covariance pattern (PDRP) of cerebral glucose metabolism and perfusion, respectively, is analogous in iRBD (46). PDRP topography is characterized by elevated activity in pontine, pallidothalamic and cerebellar metabolic activity and reduced activity in parietal and premotor association regions. iRBD patients exhibited increased expression of the PDRP, with long-term clinical follow-up data highlighting that iRBD patients with network-level functional abnormalities at baseline were more likely to phenoconvert to PD/DLB (46). These results indicate network abnormalities in iRBD patients, which hold the potential to predict those who are likely to develop PD.
How does dopaminergic denervation contribute to RBD? Rye hypothesized that GABAergic output from the basal ganglia targets the glutamatergic retrorubral field and/or midbrain extrapyramidal area, which subsequently activate the ventromedial medullary zone, promoting REM atonia (47). Nigral dopamine depletion occurs transiently or persistently in pathological states such as PD. Therefore, following dopaminergic neuronal loss in the substantia nigra, we can expect heightened phasic discharge of the internal segment of the globus pallidus to excessively inhibit neurons of the midbrain extrapyramidal area, thereby permitting the expression of movements that overcome REM atonia. This is supported by the reversal of excessive nocturnal movements following excessive inhibition of the midbrain extramyramidal area by pallidotomy (48).
The variability in RBD progression to Parkinsonism may be mediated by the spread of damage in the ventral mesopontine junction to the substantia nigra. In PD, neuronal degeneration has been suggested to originate at either the dorsal or ventral portion of the brainstem, extending rostrally or caudally (49). Therefore, PD would manifest first if the lesions originate in the rostroventral midbrain, nearby the caudoventral part of the substantia nigra pars compacta, where the putamen-labeled afferents reside (50). RBD, on the other hand, would manifest first if the lesions begin in the caudoventral mesopontine junction (49). The variability in paths of neurodegeneration could justify why RBD either precedes PD or develops at different stages of PD.
Whilst SPECT and PET imaging studies have demonstrated a contributory role for dopaminergic dysfunction in RBD pathophysiology, pharmacological evidence has indicated that serotonergic dysfunction may also play a fundamental role in RBD development and severity. Treatment with D2/D3 receptor agonists, including pramipexole, have proven to provide little benefit in alleviating RBD-specific symptomology, possibly because post-synaptic dopaminergic integrity remains in-tact, as demonstrated by Eisensehr and colleagues, who found no difference in striatal D2 receptor density between iRBD and controls (34). Nevertheless, therapies predominantly targeting serotonergic functionality, such as melatonin and clonazepam, effectively treat RBD symptomology, whilst serotonin reuptake inhibitors (SSRI) are typical causative agents of medication-induced RBD (51). Furthermore, nigrostriatal dopaminergic denervation is a primary feature of PD; whereas RBD manifests in a proportion of PD patients, indicating that additional factors are likely to contribute to the underlying pathogenesis of RBD.
To investigate this notion, a combined PET study enabled authors Kotagal et al. (52) to gain an insight into the role of the cholinergic, serotoninergic and dopaminergic systems in RBD pathophysiology. In this study, 80 non-demented PD patients were enrolled and divided into two groups: PD patients positive for RBD and PD patients negative for RBD, after being assessed by Mayo Sleep Questionnaire. All subjects were scanned with three selective radioligands: [11C]PMP, [11C]DASB and [11C]DTBZ, which served as markers for presynaptic acetylcholinersterase (AChE), presynaptic serotonin transporter (SERT) and presynaptic vesicular monoamine transporter type 2 (VMAT2), respectively. PD patients with RBD exhibited a reduction in AChE levels within the neocortex, limbic cortex and thalamus in comparison with PD patients without RBD (52). No differences were found in striatal VMAT2 levels or SERT availability within the striatum or raphe nucleus between the two groups. Furthermore, SERT availability within the brainstem and thalamus, as measured by [123I]FP-CIT-SPECT, was found to not significantly differ between iRBD patients and controls (53, 54). These results, taken together, indicate that serotonergic integrity does not have an impact on RBD pathogenesis, though cholinergic dysfunction appears to play a prominent role in the development of RBD symptoms in PD.
Molecular imaging has provided us with an insight into the mechanisms underlying RBD in PD, with most studies assessing dopaminergic function and highlighting that presynaptic dopaminergic deficit is strongly associated with RBD, thus likely to play a role in its pathogenesis. Molecular imaging investigating non-dopaminergic systems in RBD is incredibly limited, thus more studies are required to assess their roles in RBD pathophysiology, particularly the cholinergic and noradrenergic systems.
MR Imaging
Multi-modal MR imaging techniques have been utilized to investigate the mechanisms underlying RBD in patients with PD, often complimenting the findings reported from molecular imaging studies. A cross-sectional resting-state fMRI study investigating functional connectivity in nigrostriatal and nigrocortical pathways reported altered connectivity in both pathways (substantia nigra, putamen and occipital regions) in RBD patients compared to PD patients and controls (55), consistent with hypotheses of dopamimergic degeneration. This was further inenforced by a combined fMRI and [123I]FP-CIT-SPECT study, which aimed to explore whether connectivity dysfunction within basal ganglia networks is reflected in iRBD (56). Rolinski et al. (56) revealed that iRBD patients exhibit significant connectivity dysfunction of basal ganglia network, as well as frontal lobe regions, compared to healthy controls. No significant differences in basal ganglia connectivity was identified between iRBD and PD patients, suggesting that RBD and PD patients exhibit an analogous level of decline in basal ganglia functional connectivity. However, large scale longitudinal studies are compulsory to track the progression of functional connectivity in iRBD patients who convert to PD, in order to corroborate if basal ganglia connectivity could serve as a potential marker for identifying iRBD patients at risk of converting to PD.
As well as functional alterations, RBD-specific patterns of atrophy have also been explored, in order to better understand the contribution of subcortical and cortical atrophy to alterations in the sleep-wake cycle. Voxel-based morphometry (VBM) revealed early RBD-positive PD patients, with an average disease duration of 6.5 months, had reduced cortical gray matter volume in the parietal operculum, middle occipital gyrus, insular cortex, superior temporal gyrus and hippocampus compared to RBD-negative PD patients (57). Further, RBD-positive PD patients have been reported to exhibit prominent volume loss in the anterior cingulate cortex, pontomesencephalic tegmentum, thalamus, hypothalamus, amygdala, putamen and medullary reticular formation compared to those without RBD and controls (58). There has been a relative lack of consistency in the results dictated from structural imaging studies in iRBD, with only one demonstrating a smaller volume in the pontine tegmentum (59), two others revealing a subtle structural change in this region using MRI diffusion sequences (60, 61), and one repoting no volume change in this area (62). Furthermore, previous neuroimaging studies in PD-RBD reported no significant volume changes in the pontomesencephalic tegmentum (57, 63, 64). However, it is important to note that cholinergic, glutamatergic and GABAergic neurons reside in the pontomesencephalic tegmentum, thus postulating that dysregulation of non-dopaminergic systems may contribute to RBD, whilst the volume loss in the medullary reticular formation may be implicated in the loss of muscle atonia during REM sleep. A preclinical study using transgenic mice models with impaired glycine and GABA receptor function presented with an RBD phenotype, which was rescuable using melatonin and clonazepam (65). In this direction, dysfunctional neurotransmission of glycine and GABA could play a key role in the pathogenesis of RBD, though in vivo studies are required to confirm this in humans.
Diffusion tensor imaging, together with T1-weighted MR imaging has enabled Ford et al. (57) to study microstructural white matter changes and atrophy, simultaneously. PD patients with RBD exhibited reduced fractional anisotropy (FA) within the inferior and superior longitudinal fasciculus, inferior fronto-occipital, longitudinal fasciculi and corticospinal tract, as well as increased mean diffusivity (MD) within the inferior longitudinal fasciculi. These results highlight widespread pathological changes in white matter microstructure, as well as reduced gray matter volume constrained to parietal and temporal lobes (57). These findings are in line with previous DTI studies in subjects with iRBD (60, 61), suggesting that microstructural impairment may contribute to RBD symptomology.
Neuromelanin-sensitive MR imaging has demonstrated that PD patients with RBD exhibit a greater loss of neuromelanin within the locus coeruleus compared to PD patients without RBD (63). Specifically, loss of neuromelanin within the locus coeruleus was linked with abnormal muscle tone during REM sleep (63). This is particularly interesting as the locus coeruleus is acknowledged to play a critical role in regulating the sleep-wake cycle via its projections to the ventral tegmental area, hypothalamus, thalamus, hippocampus and amygdala, thus substantiating the contribution of the noradrenergic system in the pathophysiology of RBD.
Evidence from SPECT and PET studies have suggested a role for dopaminergic dysfunction in RBD pathogenesis, though dysfunction of non-dopaminergic systems, including the noradrenergic and cholinergic systems, are likely to also play a role in the development of RBD in PD. Additionally, MR imaging modalities have suggested that functional connectivity and microstructural changes contribute to the development of RBD in PD, giving an insight into the neural structures which may play a key role in RBD. However, largescale longitudinal studies are indispensable to fully comprehend the mechanisms underlying RBD, thus propelling the development of targeted therapies.
Excessive Daytime Sleepiness
Excessive daytime sleepiness (EDS) is one of the most common and burdensome non-motor symptoms in both early and advanced PD, substantively impacting on the patient's quality of life (66). EDS is described as an inappropriate and undesirable sleepiness during waking hours (67), which has been found to cause mild to severe cognitive impairment, with particular deficits in attention, memory and judgment (68). The prevalence of EDS has been reported to be higher in PD than in the general population, ranging from 16 to 74% (69, 70). This heterogeneity may reflect the fact that EDS has been found to rise with disease severity (71–73), as well as correlate with disease duration (74, 75), Hoehn and Yahr stage (76) and MDS Unified Parkinson's Disease Rating Scale (MDS-UPDRS) Part III (77, 76). However, the lack of correlation found between Hoehn and Yahr stage and EDS has led to the notion that sleepiness severity is not dependent on nocturnal sleep disruption, cognitive and motor impairment or anti-Parkinsonian therapy, but is actually associated with PD-specific pathology (75, 78–85).
Molecular Imaging
The pathophysiology of EDS may be a result of substantial degeneration of both the nigrostriatal dopaminergic system and extrastriatal neurons within the lower brainstem and midbrain, which pose as key regulators of the sleep-wake cycle (86). PET and SPECT molecular imaging studies have implicated both serotonergic and dopaminergic nigrostriatal dysfunction in EDS pathophysiology.
We recently carried out a [123I]FP-CIT SPECT study to investigate the potential implication of the presynaptic dopaminergic system in EDS. We found that PD patients with EDS generally had a worse clinical picture compared to PD without EDS, presenting with worse autonomic and cognitive function, depression and overall burden of non-motor symptom burden (87–90). Further, PD patients with EDS exhibited reduced DAT uptake within the caudate, with lower caudate DAT uptake correlating with worse EDS severity. More interestingly, abnormal caudate uptake and disease duration were found to predict the development of EDS, indicating that dopaminergic deficits in the caudate may contribute to EDS etiology (90). This is in corroboration with a study carried out by Happe and colleagues who reported a loss of [123I]FP-CIT uptake within the caudate, putamen and striatum, which inversely correlated with the severity of daytime sleepiness in moderate PD (H&Y stage 2) but not in early PD (H&Y stage 1), as measured by the Epworth sleepiness scale (91). Additionally, by investigating cerebral blood flow patterns in PD patients with EDS by employing [123I]iodoamphetamine SPECT, Matsui and colleagues reported hypoperfusion within the right caudate and the left parietal and temporal association cortex, as well as hyperperfusion within the right thalamus (92). These results suggest that cortical hypoperfusion relative to hyperperfusion within the brainstem may be related to EDS pathogenesis in PD. Interestingly, caudate functionality has been consistently demonstrated in these studies to be associated with EDS, with a reduction of caudate metabolism potentially reflecting dopamine depletion.
The caudate nucleus has recently surfaced as a common node in the regulator systems affiliated with sleep problems, given its complex role in modulating the impact of impulses between the thalamus and cortex. Neurotoxic lesion studies have revealed that bilateral striatal lesions cause a reduction in wakefulness, which is attenuated when the caudoputamen lesions include the nucleus accumbens (93). These findings emphasize that the caudate is implicated in promoting wakefulness, whereas the nucleus accumbens predominately enhances sleepiness. Therefore, hypometabolism and dopamine depletion within the caudate may cause sleepiness.
The extrastriatal dopaminergic system has also been explored in vivo (94, 95). A recent preliminary PET study employing [11C]PHNO, a radioligand selective for dopamine type-3 (D3) receptors, reported lower D3 receptor availability in the hypothalamus of 12 PD patients with EDS, which also correlated with daytime sleepiness severity (95). Although this study suggests that D3 receptor availability may be associated with EDS, it does not provide a principal cause for this decline. This led the authors to suggest that reduced monoamine transmission within important nuclei implicated in sleep control are accountable for adaptive changes in D3 receptor availability. A possible alternative explanation is that there is direct neuronal degeneration of dopamine receptor-expressing neurons within the hypothalamic dopaminergic networks (95).
Given that preclinical data implicates the brainstem raphe nuclei in the ascending arousal system, which promotes wakefulness and prevents EDS (96), the serotonergic system has also been investigated in vivo. A combined PET study utilizing [11C]DASB and [18F]DOPA, as markers of SERT and monoaminergic terminals function, respectively, revealed that PD subjects with EDS exhibited a reduction in SERT availability within the rostral raphe, locus coeruleus, thalamus and hypothalamus, as well as reduced [18F]DOPA uptake within the locus coeruleus, rostral raphe and ventral tegmental area (97). Since the authors controlled for depression and fatigue, it can be suggested that monoaminergic dysfunction may contribute to the mechanisms underlying EDS. However, these findings have not been replicated (54).
Molecular imaging has consistently demonstrated the caudate to be associated with EDS, as shown by a negative correlation between its metabolic activity and dopaminergic function and the severity of EDS. However, there is a real lack of non-dopaminergic exploration using molecular imaging techniques, which is critical to further expand our understading of daytime sleepiness, especially because wake-promoting neurotransmitters include norepinephrine, acetylcholine, hypocrein, histamine and hypocretin, which may be dysregulated in PD.
MR Imaging
At the structural level, whole-brain studies revealed widespread reductions of gray matter volume in the frontal, occipital, temporal and limbic lobes, as well as the right parahippocampus and nucleus basalis of Meynert in PD with EDS compared to PD without EDS (98, 99). Whole-brain white matter analysis of DTI data have revealed that PD patients with EDS exhibit elevated levels of white matter axonal damage, indexed by reduced fractional anisotropy within the fornix compared to those without EDS, which also correlated inversely with daytime sleepiness severity (100). Furthermore, Chondrogiorgi and colleagues revealed that PD patients with EDS exhibited a reduction of white matter integrity [as demonstrated by an increase in axial diffusivity (AD)] within the projection, association and brainstem fibers compared to PD patients without EDS (99), which corroborated with reports made by Gama et al. (101) and Kato et al. (98), who found widespread cortical and subcortical atrophy within frontal, temporal, limbic and insular regions, as well as the middle cerebellar preduncles. Although a larger cohort study is required to confirm these findings, these findings are suggestive of fornix fibers degeneration, which connects the hypothalamus and hippocampus, playing a potential role in EDS development. Hypothalamic dysfunction could lead to microstructural changes in the fornix, thus together contributing to the development of sleepiness in PD. Additionally, longitudinal studies are a necessity to gain an insight into the long-term role of structural and functional alterations in EDS pathophysiology in PD.
At the functional level, increased EDS was inversely associated with decreased functional connectivity within the thalamocortical and default mode networks, potentially reflecting the disengagement of sensory and motor processing from the stream of consciousness (102, 103). Additional rs-fMRI studies using whole-brain analysis identified regional homogeneity within the inferior gyrus and cerebellum, which inversely correlated with ESS score, and increased regional homogeneity in the paracentral lobule, which positively correlated with ESS scores (104). Subsequent functional connectivity analysis of these regions demonstrated a reduction in functional connectivity in the frontal, temporal, insula and limbic lobes, as well as the cerebellum, in EDS-positive PD patients compared to EDS-negative patients (104). The frontal, temporal and insula regions have been reported to play a role in the development of EDS, given the marked atrophy within the gray matter of these regions (92, 98).
Despite some negative findings, such as no significant EDS-related changes in FA (99), most studies observed a reduction in structural and/or functional features, including white matter integrity, white matter axonal damage, brain volume and functional connectivity in key neural structures involved in regulating alertness and sleepiness. However, further multimodal studies are required to gain a deeper understanding of the way in which structural changes are associated with EDS, particularly by carrying out longitudinal studies.
Restless Leg Syndrome
Idiopathic restless leg syndrome (iRLS), also known as Willis-Ekbom disease (WED), is a common neurological disorder characterized by strong feelings of restlessness and troublesome paraesthesia-like sensations in the lower legs, particularly at rest. Contrariwise, symptomology usually improves or disappears once the subject starts physical activity. This condition affects sleep and health (105–107), as well as quality of life, which has been reported to be analogous or worse than those suffering from congestive heart failure, osteoarthritis, stroke or depression (106, 108–116).
Most iRLS patients display some initial clinical benefit when administered dopaminergic drugs, including dopamine agonists and levodopa. Large-scale clinical trials encompassing diverse patient populations reported good clinical response to dopamine agonist in approximately 60–75% of participants (117). Therefore, in general clinical practice, a failure to respond to dopaminergic treatment should flag concern regarding the accuracy of diagnosis, but does not automatically exclude a diagnosis of iRLS.
Molecular Imaging
SPECT and PET molecular imaging techniques have been employed to investigate the role of dopaminergic dysfunction in the pathophysiology of iRLS, by imaging post-synaptic D2 receptor binding, and presynaptic DAT (118). However, few studies have aimed to characterize the underlying mechanisms of RLS in PD cohorts specifically. Generally, brain imaging studies evaluating dopaminergic function in iRLS patients have yielded inconclusive results though some have provided evidence for the pathogenic role of the dopaminergic dysfunction, which is in line with the effectiveness of dopaminergic therapies (119).
Striatal DAT density has been found to be compromised in iRLS patients, as demonstrated by two molecular studies employing [99MTc]TRODAT-1 SPECT (120) and [11C]methylphenidate PET (121). This presynaptic dopaminergic deficit is in line with studies employing [18F]DOPA, where iRLS patients exhibited reduced [18F]DOPA uptake in the putamen and caudate compared to controls, highlighting the potential role that dopaminergic dysfunction plays in RLS pathophysiology (122). However, a 4-year longitudinal [123I]-FP-CIT SPECT study in 88 de novo PD patients revealed that elevated DAT availability within the caudate and putamen was associated with the presence of RLS at baseline and follow-up, compared to those without RLS (123), suggesting that PD patients with RLS may have a comparatively preserved presynaptic dopaminergic pathways.
Striatal and extrastriatal D2 receptor availability in de novo RLS patients has been investigated using [11C]raclopride and [11C]FLB-457, respectively, with RLS participants illustrating increased availability of D2 receptor within the thalamus and cingulate cortex (124), but a reduction of D2 receptor binding within the caudate and putamen (125). The elevation of D2 receptor availability in RLS could be a result of the upregulation of D2 receptor in response to low levels of endogenous dopamine. Striatal and extrastriatal dopaminergic dysfunction and alterations in somatosensory processing may contribute to the mechanisms underlying RLS. Michaud et al. (126) employed [123I]-β-CIT and [123]IBZM to assess pre- and post-synaptic dopaminergic impairment, respectively. Reduced striatal D2 receptor binding was observed in RLS patients compared to healthy controls, though no differences were identified in presynaptic DAT binding (126). Given that presynaptic DAT uptake was within the normal range, reduced D2 receptor availability is likely to reflect downregulation of D2 receptor due to elevated synaptic dopamine levels, supporting the role of post-synaptic dopaminergic dysfunction in the pathophysiology of RLS.
Although there is good evidence supporting the notion that dopaminergic dysfunction contributes to RLS manifestation, some molecular imaging studies have revealed conflicting results. A [123I]-β-CIT and [123]IBZM SPECT study reported a reduction in putaminal and caudate DAT uptake, and no difference in striatal D2 receptor density, in de novo iRLS patients (n = 13) compared to controls (n = 12) (127). Conversely, other studies have demonstrated that no difference resides in dopaminergic activity between RLS patients and controls, with no striatal or frontal D2 receptor differences between RLS patients, early PD and healthy controls (128). These results, taken together, suggest that the underlying mechanisms of RLS are likely to involve non-dopaminergic systems, with the underlying mechanisms likely to be divergent from PD where presynaptic dopaminergic neuronal integrity is largely affected. In a study that examined real-time DAT binding potentials, RLS patients exhibited a reduction of striatal DAT binding in both day and night scans, suggesting that membrane-bound striatal DAT, but not total cellular DAT is depleted in RLS (121).
There are no studies, to date, exploring the non-dopaminergic basis of RLS in PD, which is required to enhance our understanding of this disorder, especially given the conflicting results in dopaminergic integrity between studies.
MR Imaging
Studies exploring structural changes in RLS are very limited. The first study to report morphologic changes in RLS was published earlier this year, who found that RLS patients had a 7.5% decrease in cortical thickness in the bilateral post-central gyrus. Further, there was a decline in the corpus callosum posterior midbody, suggesting an alteration in white matter properties in the somatosensory pathway (129). Other studies exploring white matter microstructural changes have revealed altered fractional anisotropy in temporal regions, internal capsule cerebellum and pons in iRLS patients compared to controls, with no changes in gray matter (130). These results indicate that microstructural alterations in white matter, but not gray matter atrophy, may contribute to the development of iRLS. Nevertheless, Rizzo and colleagues reported no microstructural changes in iRLS subjects (131), thus arguing against structural or microstructural abnormalities having an association with iRLS. A functional neuroimaging study by Bucher and colleagues reported that iRLS patients exhibit activation in the cerebellum and thalamus (132), which is supported by structural cerebral and thalamic findings reported by Etgen et al. (133).
Iron deficiency has also been repeatedly shown to be associated with RLS, especially given that CSF ferritin levels are low and CSF transferrin levels are high in RLS patients (134). However, total iron and ferritin levels in the substantia nigra region are elevated in PD, resulting in oxidative stress and potentially leading to dopamine depletion (135). In idiopathic RLS, central nervous system iron storage may be impaired, though systemic iron may be normal (136). Utilizing MRI techniques, regional brain iron concentration has been assessed in iRLS patients, which reported decreased iron in the midbrain, which has been found to correlate with the severity of RLS (137). In a recent study with a 7-T MRI scanner, iRLS patients exhibited a reduction in iron levels, as measured by quantitative magnetic susceptibility mapping (SWI), within the thalamus and dentate nucleus, but not in the substantia nigra, when compared to controls (138). The mechanism by which iron deficiency leads to dopaminergic dysfunction remains uncertain. Iron has a complex effect on dopaminergic function, serving as a cofactor for tyrosine hydroxylase and is integral to D2 receptor function (139, 140).
Although PD and RLS are both related to central dopaminergic dysfunction, the evidence of an association between these disorders is limited to a few studies that report inconsistent results. Long-term prosective controlled studies are warranted to not only assess the pathophysiology of RLS in PD, but also the link between PD and RLS as clinical association studies differ widely. Currently, there are not functional MRI studies rhat have focused on patients with PD-RLS/Functional and molecular imaging studies of PD vs. PD-RLS are necessary to better understand the mechanisms involved in these disorders.
Multiple System Atrophy
MSA is an adult-onset, fatal neurodegenerative disorder characterized by a combination of autonomic dysfunction, Parkinsonian features and cerebellar and pyramidal features (141). The neuropathological hallmark of MSA is α-synuclein-immunoreactive inclusions that predominantly affect oligodendrocytes (142). The underlying neuropathology may contribute to the sleep disorders observed in this disease, with sleep dysfunction acknowledged to be an associated comorbidity. Some sleep disorders such as RBD and vocalization appear to have a higher prevalence in MSA than in PD, despite the same disease duration, and are typically associated with more severe motor symptoms, depression, longer disease duration and longer duration of levodopa treatment (143).
REM Sleep Behavior Disorder
MSA patients have reduced REM and slow-wave sleep, with a recent multicenter study revealing that polysomnography-confirmed RBD is present in up to 88% of patients with MSA. RBD has also been shown to precede the onset of MSA by more than 1 year in 44% of patients (144).
Given that both acetylcholine- and monoamine- containing neurons of the brainstem have been implicated in REM sleep (47), Gilman and colleagues employed [11C]DTBZ PET and [123I]IBVM SPECT, markers of striatal monoaminergic presynaptic terminals and cholinergic presynaptic terminals, respectively, to explore the neurochemical basis of RBD in 13 subjects with probable MSA (145). The MSA subjects exhibited a reduction in mean striatal [11C]DTBZ uptake, as well as a reduction in [123I]IBVM uptake within the thalamus (145). These results reflect the degeneration of dopaminergic neurons in the substantia nigra, as well as degeneration of cholinergic neurons in the pedunculopontine tegmental and laterodorsal tegmental nuclei. However, given the lack of correlation found between thalamic [123I]IBVM uptake and severity of REM atonia loss, it can be assumed that degenerative changes in the pedunculopontine tengmental and laterodorsal tegmental nuclei do not contribute to RBD. Striatal [11C]DTBZ binding, however, was inversely correlated with severity of REM atonia loss, which led the authors to conclude that loss of nigrostriatal dopaminergic projections may contribute to RBD in MSA.
Excessive Daytime Sleepiness
The prevalence and nature of EDS in MSA has not been systematically investigated. However, the few studies which have evaluated sleepiness have reported that 50% of MSA patients suffer from EDS (143), though Multiple Sleep Latency Tests in this subset of patients have revealed normal or mildly reduced sleep latencies (70, 146). In a multicenter survey, Epworth sleepiness scale scores from patients with MSA were comparable to PD, though higher compared to controls, with EDS present in 28% of MSA subjects (147). In contrast to PD, EDS did not correlate with dopaminergic treatment, and disease severity weakly correlated. However, sleep-disordered breathing and sleep efficiency predicted EDS in MSA (147).
One of the potential neuroanatomic substrates for EDS in MSA includes the hypocretin neuronal network within the lateral hypothalamus, which is acknowledged to play a fundamental role in promoting alertness. In an autopsy study of 7 MSA patients, the number of hypocretin neurons were significantly reduced compared to controls (148), though other studies have demonstrated that MSA patients have normal hypocretin levels in the cerebrospinal fluid (CSF) (146, 149). These findings suggest that substantial neurodegeneration is required in order for a measurable difference in CSF hypocretin levels to be detectable. Alternatively, EDS observed in MSA may be related to a loss of cholinergic alerting neurons from the brainstem, as Schmeichel and colleagues have reported that MSA subjects have a significant loss of cholinergic neurons within the laterodorsal tegmental and pedunculopontine tegmental nuclei within the pons (150). However, in vivo molecular imaging studies are required to confirm this notion in MSA subjects.
In an interesting cross-sectional MRI study including 16 PD cases, 13 MSA and 14 PSP, alongside 12 healthy controls, Gama and colleagues sought to evaluate and compare the three groups regarding EDS and its association to brain MRI morphometry. MSA subjects with EDS did not exhibit any EDS-specific brain atrophy, though PD subjects with EDS presented with more atrophy of the medial cerebellar peduncle compared to PD patients without EDS and PSP subjects exhibited atrophy within the midbrain compared to controls (101). The authors concluded that widespread neurodegeneration of brainstem sleep structures were related to sleep abnormalities in these subjects.
Sleep Disordered Breathing
Various types of sleep disordered breathing can arise in MSA. Nocturnal stridor and obstructive sleep apnea (OSA) occur more frequently than central sleep apnea, ranging from 40 to 42% for nocturnal stridor in MSA cases (151, 152) and 15–37% for OSA, with both carrying the risk off sudden death (144, 152). Laryngeal dysfunction and narrowing of the upper airway at the level of the vocal cords, laryngeal inlet, base of the tongue and soft palate has been reported in early descriptions of MSA (153, 154). Other factors, including rigidity, hypokinesia, dystonia, rigidity and paralysis of the upper airway muscles may predispose to OSA or nocturnal stridor in MSA (155).
Gilman and colleagues used [123I]IBVM SPECT to measure thalamic cholinergic terminal density and [11C]DTBZ to measure striatal monoaminergic terminal density to investigate the neuropshyiologic basis of OSA in MSA (156). The authors reported that thalamic [123I]IBVM uptake inversely correlated with OSA severity, with no correlations found between [11C]DTBZ and OSA severity. Given the limited number of cholinergic neuronal cell bodies in the thalamus (157), thalamic [123I]IBVM binding may serve as a surrogate marker for the density of cholinergic neurons in the pontine pedunculopontine tegmental and laterodorsal tegmental nuclei. Therefore, projections from pedunculopontine tegmental and laterodorsal tegmental nuclei to the medulla and pons may be more applicable in the pathophysiology of OSA, especially given that OSA mirrors collapse of the upper airway musculature, which receives cholinergic innervation from motor neurons of the lower brainstem.
To the best of our knowledge, there are no other PET or MR studies exploring the neurochemical or neuroanatomical correlates of sleep disorders in MSA. Further studies, both structural and functional neuroimaging, are required to enhance our understanding of these sleep disturbances in MSA.
Progressive Supranuclear Palsy
PSP belongs to the family of tauopathies, characterized by abnormalities in the hyperphosphorylation and aggregation of the microtubule-associated protein, tau, resulting in degeneration of cortical and subcortical brain structures, particularly within the midbrain (158). A PSP patient typically presents with postural instability, supranuclear ophthalmoplegia, Parkinsonism and pseudobulbar palsy (159).
Although more commonly reported in synucleinopathies, RBD has also been observed in PSP. In a relatively small study of 15 PSP patients, 15 PD patients and 15 age-matched controls, REM sleep without atonia and RBD were similarly present amongst the two cohorts (33 vs. 28%) (160), which was confirmed by Diederich et al. (161). In fact, a polysomnographically recorded study demonstrated that RBD and REM sleep without atonia are common also in PSP, and that sleep was more severely impaired in PSP than in PD (162).
Given the lack of neuroimaging studies assessing sleep disturbances in PSP, in this section, we discuss the findings from neuroimaging studies which may have relevance to sleep regulation.
PSP is pathologically characterized by abnormal accumulation of tau protein, with prominent atrophy in the midbrain (163). Most PSP patients also exhibit neurodegeneration of cholinergic neurons at the level of the pedunculopontine tegmentum, which is acknowledged to play a fundamental role in the regulation of REM sleep (164). Atrophy of the brainstem have been reflected in MR morphometry studies, which have described a reduction in the antero-posterior diameter of the midbrain and smaller ratio between midbrain area and pons (165–167).
Several subcortical structures degenerate over the course of PSP, as demonstrated by Saini et al. (168) and Whitwell et al. (169), who reported non-specific atrophy of the thalamus. However, after attempting to delineate the thalamus, Padovani and colleagues revealed that the pulvinar, dorsomedial and anterior nuclei of the thalamus was atrophic in PSP (170, 171). This was in corroboration with a previous study carried out by Price and colleagues who reported reduced gray matter in the thalamus and hypothalamus in PSP compared to controls (172). Studies have also reported prominent white matter atrophy within the midbrain and pons (169, 173, 174), with a loss of white matter volumes in the brainstem and subcortical structures of PSP patients. Whitwell and colleagues reported reduced fractional anisotropy within the pons and increased mean diffusivity within the thalamus of PSP patients (n = 18), with the midbrain exhibiting a decrease in fractional anisotropy and increase in mean diffusivity (175).
In an MRI study carried out by Lehericy et al. (176), 10 PSP subjects had marked gray matter atrophy in the medial thalamus and anterodorsal midbrain/hypothalamic area compared to controls. Reduced white matter volumes were evident in the basal forebrain/inferior globus pallidus/subthalamic area, midbrain and pons, amongst other cortical and subcortical regions. Further, DTI analysis revealed that PSP subjects exhibited lower fractional anisotropy in the white matter of the thalamus, midbrain, cerebellum and cortical regions (176).
The brainstem, thalamus and hypothalamus play essential roles in regulating sleep, with the brainstem and the hypothalamus centrally regulating the pace for sleep-related activity throughout the brain and the thalamus being targeted by both central and decentral regulators to induce global sleep-related oscillatory activity (177). The pons, in particular, is important for REM sleep, with several pontine structures contributing to the generation of each specific polygraphic event that characterizes REM sleep. The pontine tegmentum also encompasses the region where cholinergic activation can trigger behavioral and bioelectric signs of REM sleep (178), with the pedunculopontine tegmental nuclei, in particular, thought to be critical for generating REM sleep, and its connections to the reticular nuclei of the thalamus thought to be important for generating spindles (179). In this direction, degeneration and pathological changes within these regions could underlie RBD within PSP subjects, though neuroimaging studies are required to confirm whether an association is present in PSP.
Corticobasal Syndrome
CBS also belongs to the family of taupathies, though much less common than other atypical Parkinsonian disorders. CBS patients characteristically present with myoclonus, dystonia, progressive asymmetric levodopa-resistant Parkinsonism, and further cortical signs, such as alien limb phenomena, apraxia and cortical sensory loss (180). The neuropathological hallmark of CBS is unusual accumulation of hyperphosphorylated 4-repeat (4R) tau in the form of neurofibrillary tangles, coiled bodies and neuropil threads together with astrocytic plaques.
The literature on CBS is primarily limited to case reports, likely due to the rarity of CBS. Two case reports of RBD have been reported (181, 182), though in a larger case series of CBS patients, only 1 out of 11 patients had RBD (183). A descriptive study including five CBS patients reported that none had RBD, two had sleep disordered breathing, four had periodic limb movements during sleep and all five patients had insomnia (184).
CSF hypocretin levels have been evaluated in CBS and found to be significantly lower than in patients with PD. The sample size was quite small (n = 7) and the majority of the effect may have been primarily due to one outlier (185). Further, pathological studies have demonstrated that CBS patients have a significant loss of choline acetyltransferase (ChAT)-positive neurons within the nucleus basalis of Meynert compared to PSP (186).
There have been no studies, to-date, exploring the neuropathological basis of sleep disturbances in CBS using neuroimaging techniques. A data-driven meta-analysis identifying and comparing the neural correlates of CBS for atrophy measurements included 200 CBS patients and 318 controls (187). This study identified four brain regions with significant atrophy: (1) the bilateral anterior thalamus; (2) the posterior midcingulate cortex, bilateral posterior frontomedian cortex and the premotor area/supplementary motor area; (3) the posterior superior frontal sulcus and precentral gyrus/middle frontal gyrus; (4) the left posterior superior frontal sulcus and middle frontal gyrus. This is particularly interesting as thalamic neurons provide state-dependent gating of sensory information through their ability to create differential patterns of electrogenic activity during wakefulness and sleep. It has been argued that both central and decentral regulators target the thalamus, inducing global sleep-related oscillatory activity (177).
Striatal DAT uptake in patients with CBS is characterized by large variability, with [123I]FP-CIT found to be normal bilaterally in four CBS patients and only unilaterally reduced in the other four cases (188). These results highlight the hemispheric asymmetry in the putamen and caudate [123I]FP-CIT uptake, thus reflects the highly asymmetrical involvement and substantia nigra pars compacta neuronal loss implicating both ventral and dorsal tiers in CBS (189, 190), as opposed to PD, where cell loss is typically confined to the substantia nigra pars compacta ventral tiers. Given the association found between striatal dopamine depletion and sleep disturbances in PD, it could be assumed that a similar mechanism underlies sleep disorders in CBS. However, this needs to be investigated in MSA exhibiting specific sleep disorders in well-designed, controlled studies.
Huntington's Disease
Huntington's disease (HD) is a hereditary and fatal neurodegenerative disease, characterized by chorea, psychiatric symptoms and cognitive dysfunction, which manifest as a result of an expanded trinucleotide CAG sequence in the huntingtin gene (HTT) on chromosome 4 (191, 192). HD-specific pathology is defined by aggregations of intranuclear inclusions of mutated huntingtin in the brain. These aggregates have been reported to interact and impair the function of several transcription factors, ultimately inducing a loss of striatal GABAergic medium spiny neurons (MSNs), as well as cortical MSNs (193, 194). There is growing awareness that, alongside the psychiatric and cognitive syptoms, sleep and circadian abnormalities are also present. However, it remains unclear if they are directly caused by HD gene-related pathology or are merely a consequence of having a neurodegenerative disease.
The changes seen in HD appear to have very little in common with sleep disturbances in the neurological disorders mentioned above. For example, the incidence of RBD in the early stages of HD are reported to be fairly low, which is consistent with the fact that although dopaminergic pathology is evident in HD, it does not dominate at early stages (195). At subsequent stages of HD when dopamine-mediated function has deteriorated, the degeneration of this pathway is only part of a much wider picture. Other parasomnias that are associated with neurodegenerative disease, such as RLS, is not significantly increased in the HD population, though periodic limb movement disorder (PLMD) appears to be more frequent in this population compared to controls. It is important to note, however, that it is difficult to distinguish between PLMD and chorea—which is part of their movement disorder. Although sleep disorder studies in HD are rare or non-existent, we could gain critical insight into the potential sleep comorbidity in HD by investigating the molecular, structural and functional basis of sleep-related circuitry in HD. Here, we discuss the neuronal circuitry known to be implicated in sleep and affected in HD, as demonstrated by neuroimaging techniques.
HD pathology is characterized by atrophy and degeneration of the neostriatum and cortex that eventually comprises of the whole brain including subcortical structures (85, 196). Although sleep disturbances were originally reported to be associated with the degree of atrophy within the caudate (197), more recent findings revealed this correlation to be weak (198).
Molecular Imaging
The hypothalamus is critical for the regulation of sleep and metabolism, as well as playing a key role in the regulation of automatic functions such as heartbeat and breathing. A [11C]PK11195 and [11C]raclopride PET study with the aim of assessing in vivo D2 receptor integrity and microglial activation in premanifest (n = 10) and manifest (n = 9) HD gene carriers reported a significant decrease in hypothalamic [11C]raclopride uptake in both premanifest and manifest HD gene carriers, as well as a significant increases in mean hypothalamic [11C]PK11195 uptake (199). This study was the first to demonstrate a significant loss of D2 receptors, as well as increased microglial activation within the hypothalamus of HD (199). This is in corroboration with a multimodal imaging study which used MRI, [11C]PK11195 and [11C]raclopride PET to investigate volumetric differences, as well as microglial activation and D2/D3 receptor binding in premanifest and symptomatic HD gene carriers (200). The authors found increased microglial activation within the hypothalamus, amongst other regions, as well as a reduction in hypothalamic D2/D3 receptor binding in premanifest HD gene carriers. This is particularly interesting as, even though sleep was not specifically investigated in this cohort, the hypothalamus is known to play a critical role in regulation sleep, emotion and appetite (201–203).
MR Imaging
Studies employing voxel-based morphometry analyses have reported a reduction in voxel signal intensity within the hypothalamus in early symptomatic stages of the disease (204, 205). Soneson and colleagues have revealed, using both VBM and logistic regression analyses, that hypothalamic changes are visible from up to 15 years before the predicted onset of motor symptoms (206). Hypothalamic dysfunction in HD patients would not only have an impact on circadian rhythm and daytime sleepiness, but would affect additional relevant functions that have an influence on sleep. Although evidence from MRI studies have suggested that neurodegeneration in the hypothalamus presents very early in HD, the function or structure of the suprachiasmatic nuclei, which resides in the anterior, ventral region of the hypothalamus, has not been rigorously examined in post-mortem brain. However, significant loss and atrophy of orexin/hypocrein neurons have been identified in HD (207), alongside the degeneration of the thalamus (208–210), which is crucial for the generation of sleep and wakefulness. However, it has been poorly studied, with little known about if or how thalamic pathology impacts on sleep in HD.
Given the lack of neuroimaging studies specifically exploring the pathology underlying changes in sleep and circadian activity in HD, the pathophysiology of sleep comorbidities in HD remains unclear. There is, therefore a dire need for studies investigating HD-specific sleep issues to enhance our understanding the cause of these symptoms in HD, thus help us understand how they can be treated.
Conclusion and Future Directions
Sleep is a multifaceted phenomenon, regulated by an intricate interplay between several neurotransmitter systems. Although efforts have been made to delineate structural and functional correlates of sleep disturbances, there is a need for standardization across studies, especially given the discord between various studies. Furthermore, multimodal neuroimaging studies hold the key to further understand the simultaneous disruption of various systems, which may all, in some way, play a role in sleep disturbances.
Overall, neuroimaging techniques are unparalleled tools for investigating the mechanisms underlying sleep impairments in movement disorders. These invaluable techniques have provided some insights into the pathophysiology of sleep disorders, unraveling the involvement of both dopaminergic and non-dopaminergic systems. Nonetheless, the visualization of functional and neuroanatomical hallmarks of these sleep disorders remains an active and challenging area. There is a great need of imaging studies specifically seeking to understand neuropathological substrates of sleep disorders in atypical Parkinsonism and HD, which could potentially serve as novel targets for pharmacotherapy.
Author Contributions
TY gave input into the article design; wrote the first draft; prepared and revised the manuscript and generated the figures. GP, HW revised the manuscript for intellectual content. MP revised the manuscript for intellectual content and gave final approval of the manuscript.
Funding
MP research is supported by Parkinson's UK, Lily and Edmond J. Safra Foundation, Michael J. Fox Foundation (MJFF) for Parkinson's research, KCL's NIHR Biomedical Research Unit, Medical Research Council (grant reference: MR/N013700/1) and CHDI Foundation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Shulman LM, Taback RL, Rabinstein AA, Weiner WJ. Non-recognition of depression and other non-motor symptoms in Parkinson's disease. Parkinsonism Relat Disord. (2002) 8:193–7. doi: 10.1016/S1353-8020(01)00015-3
2. Kaynak D, Kiziltan G, Kaynak H, Benbir G, Uysal O. Sleep and sleepiness in patients with Parkinson's disease before and after dopaminergic treatment. Eur J Neurol. (2005) 12:199–207. doi: 10.1111/j.1468-1331.2004.00971.x
3. Fronczek R, Overeem S, Lee SY, Hegeman IM, Van Pelt J, Van Duinen SG, et al. Hypocretin (orexin) loss in Parkinson's disease. Brain (2007) 130:1577–85. doi: 10.1093/brain/awm090
4. Videnovic A, Lazar AS, Barker RA, Overeem S. ‘The clocks that time us’–circadian rhythms in neurodegenerative disorders. Nat Rev Neurol. (2014) 10:683–93. doi: 10.1038/nrneurol.2014.206
5. Wilson H, Giordano B, Turkheimer FE, Chaudhuri KR, Politis M. Serotonergic dysregulation is linked to sleep problems in Parkinson's disease. Neuroimage Clin. (2018) 18:630–7. doi: 10.1016/j.nicl.2018.03.001
6. Politis M, Piccini P. Positron emission tomography imaging in neurological disorders. J Neurol. (2012) 259:1769–80. doi: 10.1007/s00415-012-6428-3
7. Pagano G, Ferrara N, Brooks DJ, Pavese N. Age at onset and Parkinson disease phenotype. Neurology (2016) 86:1400–7. doi: 10.1212/WNL.0000000000002461
8. Pagano G, Niccolini F, Politis M. The serotonergic system in Parkinson's patients with dyskinesia: evidence from imaging studies. J Neural Transm. (2017) 125:1217–23. doi: 10.1007/s00702-017-1823-7
9. Pagano G, Yousaf T, Politis M. PET molecular imaging research of levodopa-induced dyskinesias in Parkinson's disease. Curr Neurol Neurosci Rep. (2017) 17:90. doi: 10.1007/s11910-017-0794-2
10. Pagano G, Polychronis S, Wilson H, Giordano B, Ferrara N, Niccolini F, et al. Diabetes mellitus and Parkinson disease. Neurology (2018) 90:e1654–62. doi: 10.1212/WNL.0000000000005475
11. Colten HR, Altevogt BM. In: Colten HR, Altevogt BM, editors. Sleep disorders and sleep deprivation: An unmet public health problem. Sleep Disorders and Sleep Deprivation: An Unmet Public Health Problem. Washington DC: National Academy of Sciences (2006).
12. Pace-Schott EF, Hobson JA. The neurobiology of sleep: genetics, cellular physiology and subcortical networks. Nat Rev Neurosci. (2002) 3:591–605. doi: 10.1038/nrn895
13. Saper CB, Fuller PM, Pedersen NP, Lu J, Scammell TE. Sleep state switching. Neuron (2010) 68:1023–42. doi: 10.1016/j.neuron.2010.11.032
14. Brown RE, Basheer R, Mckenna JT, Strecker RE, Mccarley RW. Control of sleep and wakefulness. Physiol Rev. (2012) 92:1087–187. doi: 10.1152/physrev.00032.2011
15. Poewe W, Seppi K, Tanner CM, Halliday GM, Brundin P, Volkmann J, et al. Parkinson disease. Nat Rev Dis Primers (2017) 3:17013. doi: 10.1038/nrdp.2017.13
16. Iranzo A, Santamaria J, Tolosa E. Idiopathic rapid eye movement sleep behaviour disorder: diagnosis, management, and the need for neuroprotective interventions. Lancet Neurol. (2016) 15:405–19. doi: 10.1016/S1474-4422(16)00057-0
17. Iranzo A, Molinuevo JL, Santamaria J, Serradell M, Marti MJ, Valldeoriola F, et al. Rapid-eye-movement sleep behaviour disorder as an early marker for a neurodegenerative disorder: a descriptive study. Lancet Neurol. (2006) 5:572–7. doi: 10.1016/S1474-4422(06)70476-8
18. Boeve BF. REM sleep behavior disorder: updated review of the core features, the REM sleep behavior disorder-neurodegenerative disease association, evolving concepts, controversies, and future directions. Ann N Y Acad Sci. (2010) 1184:15–54. doi: 10.1111/j.1749-6632.2009.05115.x
19. Oertel WH, Depboylu C, Krenzer M, Vadasz D, Ries V, Sixel-Doring F, et al. REM sleep behavior disorder as a prodromal stage of alpha-synucleinopathies: symptoms, epidemiology, pathophysiology, diagnosis and therapy. Nervenarzt (2014) 85:19–25. doi: 10.1007/s00115-013-3891-8
20. Postuma RB, Gagnon JF, Vendette M, Fantini ML, Massicotte-Marquez J, Montplaisir J. Quantifying the risk of neurodegenerative disease in idiopathic REM sleep behavior disorder. Neurology (2009) 72:1296–300. doi: 10.1212/01.wnl.0000340980.19702.6e
21. Iranzo A, Fernandez-Arcos A, Tolosa E, Serradell M, Molinuevo JL, Valldeoriola F, et al. Neurodegenerative disorder risk in idiopathic REM sleep behavior disorder: study in 174 patients. PLoS ONE (2014) 9:e89741. doi: 10.1371/journal.pone.0089741
22. Pagano G, De Micco R, Yousaf T, Wilson H, Chandra A, Politis M. REM behaviour disorder predicts motor progression and cognitive decline in Parkinson's disease. Neurology (2018). doi: 10.1212/WNL.0000000000006134
23. Factor SA, Mcalarney T, Sanchez-Ramos JR, Weiner WJ. Sleep disorders and sleep effect in Parkinson's disease. Mov Disord. (1990) 5:280–5. doi: 10.1002/mds.870050404
24. Rolinski M, Szewczyk-Krolikowski K, Tomlinson PR, Nithi K, Talbot K, Ben-Shlomo Y, et al. REM sleep behaviour disorder is associated with worse quality of life and other non-motor features in early Parkinson's disease. J Neurol Neurosurg Psychiatry (2014) 85:560–6. doi: 10.1136/jnnp-2013-306104
25. Karlsen KH, Tandberg E, Arsland D, Larsen JP. Health related quality of life in Parkinson's disease: a prospective longitudinal study. J Neurol Neurosurg Psychiatry (2000) 69:584–9. doi: 10.1136/jnnp.69.5.584
26. Reichmann H. Clinical criteria for the diagnosis of Parkinson's disease. Neurodegener Dis. (2010) 7:284–90. doi: 10.1159/000314478
27. Hawkes CH. The prodromal phase of sporadic Parkinson's disease: does it exist and if so how long is it? Mov Disord. (2008) 23:1799–807. doi: 10.1002/mds.22242
28. Stern MB, Lang A, Poewe W. Toward a redefinition of Parkinson's disease. Mov Disord. (2012) 27:54–60. doi: 10.1002/mds.24051
29. Postuma RB, Berg D. Advances in markers of prodromal Parkinson disease. Nat Rev Neurol. (2016) 12:622–34. doi: 10.1038/nrneurol.2016.152
30. Iranzo A, Tolosa E, Gelpi E, Molinuevo JL, Valldeoriola F, Serradell M, et al. Neurodegenerative disease status and post-mortem pathology in idiopathic rapid-eye-movement sleep behaviour disorder: an observational cohort study. Lancet Neurol. (2013) 12:443–53. doi: 10.1016/S1474-4422(13)70056-5
31. Schenck CH, Boeve BF, Mahowald MW. Delayed emergence of a Parkinsonian disorder or dementia in 81% of older men initially diagnosed with idiopathic rapid eye movement sleep behavior disorder: a 16-year update on a previously reported series. Sleep Med. (2013) 14:744–8. doi: 10.1016/j.sleep.2012.10.009
32. Howell MJ, Schenck CH. Rapid eye movement sleep behavior disorder and neurodegenerative disease. JAMA Neurol. (2015) 72:707–12. doi: 10.1001/jamaneurol.2014.4563
33. Albin RL, Koeppe RA, Chervin RD, Consens FB, Wernette K, Frey KA, et al. Decreased striatal dopaminergic innervation in REM sleep behavior disorder. Neurology (2000) 55:1410–2. doi: 10.1212/WNL.55.9.1410
34. Eisensehr I, Linke R, Noachtar S, Schwarz J, Gildehaus FJ, Tatsch K. Reduced striatal dopamine transporters in idiopathic rapid eye movement sleep behaviour disorder. Comparison with Parkinson's disease and controls. Brain (2000) 123:1155–60. doi: 10.1093/brain/123.6.1155
35. Iranzo A, Lomena F, Stockner H, Valldeoriola F, Vilaseca I, Salamero M, et al. Decreased striatal dopamine transporter uptake and substantia nigra hyperechogenicity as risk markers of synucleinopathy in patients with idiopathic rapid-eye-movement sleep behaviour disorder: a prospective study [corrected]. Lancet Neurol. (2010) 9:1070–7. doi: 10.1016/S1474-4422(10)70216-7
36. Kim YK, Yoon IY, Kim JM, Jeong SH, Kim KW, Shin YK, et al. The implication of nigrostriatal dopaminergic degeneration in the pathogenesis of REM sleep behavior disorder. Eur J Neurol. (2010) 17:487–92. doi: 10.1111/j.1468-1331.2009.02854.x
37. Heller J, Brcina N, Dogan I, Holtbernd F, Romanzetti S, Schulz JB, et al. Brain imaging findings in idiopathic REM sleep behavior disorder (RBD)–a systematic review on potential biomarkers for neurodegeneration. Sleep Med Rev. (2017) 34:23–33. doi: 10.1016/j.smrv.2016.06.006
38. Iranzo A, Valldeoriola F, Lomena F, Molinuevo JL, Serradell M, Salamero M, et al. Serial dopamine transporter imaging of nigrostriatal function in patients with idiopathic rapid-eye-movement sleep behaviour disorder: a prospective study. Lancet Neurol. (2011) 10:797–805. doi: 10.1016/S1474-4422(11)70152-1
39. Eisensehr I, Linke R, Tatsch K, Kharraz B, Gildehaus JF, Wetter CT, et al. Increased muscle activity during rapid eye movement sleep correlates with decrease of striatal presynaptic dopamine transporters. IPT and IBZM SPECT imaging in subclinical and clinically manifest idiopathic REM sleep behavior disorder, Parkinson's disease, and controls. Sleep (2003) 26:507–12. doi: 10.1093/sleep/26.5.507
40. Arnaldi D, De Carli F, Picco A, Ferrara M, Accardo J, Bossert I, et al. Nigro-caudate dopaminergic deafferentation: a marker of REM sleep behavior disorder? Neurobiol Aging (2015) 36:3300–5. doi: 10.1016/j.neurobiolaging.2015.08.025
41. Nurmi E, Ruottinen HM, Bergman J, Haaparanta M, Solin O, Sonninen P, et al. Rate of progression in Parkinson's disease: a 6-[18F]fluoro-L-dopa PET study. Mov Disord. (2001) 16:608–15. doi: 10.1002/mds.1139
42. Zoetmulder M, Nikolic M, Biernat H, Korbo L, Friberg L, Jennum P. Increased motor activity during REM sleep is linked with dopamine function in idiopathic REM sleep behavior disorder and Parkinson disease. J Clin Sleep Med. (2016) 12:895–903. doi: 10.5664/jcsm.5896
43. Dauvilliers Y, Boudousq V, Lopez R, Gabelle A, De Cock VC, Bayard S, et al. Increased perfusion in supplementary motor area during a REM sleep behaviour episode. Sleep Med. (2011) 12:531–2. doi: 10.1016/j.sleep.2011.10.001
44. Mayer G, Bitterlich M, Kuwert T, Ritt P, Stefan H. Ictal SPECT in patients with rapid eye movement sleep behaviour disorder. Brain (2015) 138:1263–70. doi: 10.1093/brain/awv042
45. De Cock VC, Vidailhet M, Leu S, Texeira A, Apartis E, Elbaz A, et al. Restoration of normal motor control in Parkinson's disease during REM sleep. Brain (2007) 130:450–6. doi: 10.1093/brain/awl363
46. Holtbernd F, Gagnon JF, Postuma RB, Ma Y, Tang CC, Feigin A, et al. Abnormal metabolic network activity in REM sleep behavior disorder. Neurology (2014) 82:620–7. doi: 10.1212/WNL.0000000000000130
47. Rye DB. Contributions of the pedunculopontine region to normal and altered REM sleep. Sleep (1997) 20:757–88. doi: 10.1093/sleep/20.9.757
48. Vitek J, Kaneoke Y, Turner R, Baron M, Bakay R, De Long M. Neuronal activity in the internal (GPi) and external (GPe) segments of the globus pallidus (GP) of Parkinsonian patients is similar to that in the MPTP-treated primate model of Parkinsonism [abstract]. Soc Neurosci Abstr. (1993) 19:1584.
49. Lai YY, Hsieh KC, Nguyen D, Peever J, Siegel JM. Neurotoxic lesions at the ventral mesopontine junction change sleep time and muscle activity during sleep: an animal model of motor disorders in sleep. Neuroscience (2008) 154:431–43. doi: 10.1016/j.neuroscience.2008.03.085
50. Parent A, Mackey A, De Bellefeuille L. The subcortical afferents to caudate nucleus and putamen in primate: a fluorescence retrograde double labeling study. Neuroscience (1983) 10:1137–50. doi: 10.1016/0306-4522(83)90104-5
51. Mccarter SJ, Boswell CL, St Louis EK, Dueffert LG, Slocumb N, Boeve BF, et al. Treatment outcomes in REM sleep behavior disorder. Sleep Med. (2013) 14:237–42. doi: 10.1016/j.sleep.2012.09.018
52. Kotagal V, Albin RL, Muller ML, Koeppe RA, Chervin RD, Frey KA, et al. Symptoms of rapid eye movement sleep behavior disorder are associated with cholinergic denervation in Parkinson disease. Ann Neurol. (2012) 71:560–8. doi: 10.1002/ana.22691
53. Arnaldi D, Fama F, De Carli F, Morbelli S, Ferrara M, Picco A, et al. The role of the serotonergic system in REM sleep behavior disorder. Sleep (2015) 38:1505–9. doi: 10.5665/sleep.5000
54. Qamhawi Z, Towey D, Shah B, Pagano G, Seibyl J, Marek K, et al. Clinical correlates of raphe serotonergic dysfunction in early Parkinson's disease. Brain (2015) 138:2964–73. doi: 10.1093/brain/awv215
55. Ellmore TM, Castriotta RJ, Hendley KL, Aalbers BM, Furr-Stimming E, Hood AJ, et al. Altered nigrostriatal and nigrocortical functional connectivity in rapid eye movement sleep behavior disorder. Sleep (2013) 36:1885–92. doi: 10.5665/sleep.3222
56. Rolinski M, Griffanti L, Piccini P, Roussakis AA, Szewczyk-Krolikowski K, Menke RA, et al. Basal ganglia dysfunction in idiopathic REM sleep behaviour disorder parallels that in early Parkinson's disease. Brain (2016) 139:2224–34. doi: 10.1093/brain/aww124
57. Ford AH, Duncan GW, Firbank MJ, Yarnall AJ, Khoo TK, Burn DJ, et al. Rapid eye movement sleep behavior disorder in Parkinson's disease: magnetic resonance imaging study. Mov Disord. (2013) 28:832–6. doi: 10.1002/mds.25367
58. Boucetta S, Salimi A, Dadar M, Jones BE, Collins DL, Dang-Vu TT. Structural brain alterations associated with rapid eye movement sleep behavior disorder in Parkinson's disease. Sci Rep. (2016) 6:26782. doi: 10.1038/srep26782
59. Hanyu H, Inoue Y, Sakurai H, Kanetaka H, Nakamura M, Miyamoto T, et al. Voxel-based magnetic resonance imaging study of structural brain changes in patients with idiopathic REM sleep behavior disorder. Parkinsonism Relat Disord. (2012) 18:136–9. doi: 10.1016/j.parkreldis.2011.08.023
60. Unger MM, Belke M, Menzler K, Heverhagen JT, Keil B, Stiasny-Kolster K, et al. Diffusion tensor imaging in idiopathic REM sleep behavior disorder reveals microstructural changes in the brainstem, substantia nigra, olfactory region, and other brain regions. Sleep (2010) 33:767–73. doi: 10.1093/sleep/33.6.767
61. Scherfler C, Frauscher B, Schocke M, Iranzo A, Gschliesser V, Seppi K, et al. White and gray matter abnormalities in idiopathic rapid eye movement sleep behavior disorder: a diffusion-tensor imaging and voxel-based morphometry study. Ann Neurol. (2011) 69:400–7. doi: 10.1002/ana.22245
62. Rahayel S, Montplaisir J, Monchi O, Bedetti C, Postuma RB, Brambati S, et al. Patterns of cortical thinning in idiopathic rapid eye movement sleep behavior disorder. Mov Disord. (2015) 30:680–7. doi: 10.1002/mds.25820
63. Garcia-Lorenzo D, Longo-Dos Santos C, Ewenczyk C, Leu-Semenescu S, Gallea C, Quattrocchi G, et al. The coeruleus/subcoeruleus complex in rapid eye movement sleep behaviour disorders in Parkinson's disease. Brain (2013) 136:2120–9. doi: 10.1093/brain/awt152
64. Salsone M, Cerasa A, Arabia G, Morelli M, Gambardella A, Mumoli L, et al. Reduced thalamic volume in Parkinson disease with REM sleep behavior disorder: volumetric study. Parkinsonism Relat Disord. (2014) 20:1004–8. doi: 10.1016/j.parkreldis.2014.06.012
65. Brooks PL, Peever JH. Impaired GABA and glycine transmission triggers cardinal features of rapid eye movement sleep behavior disorder in mice. J Neurosci. (2011) 31:7111–21. doi: 10.1523/JNEUROSCI.0347-11.2011
66. Politis M, Wu K, Molloy S, G Bain P, Chaudhuri KR, Piccini P. Parkinson's disease symptoms: the patient's perspective. Mov Disord. (2010) 25:1646–51. doi: 10.1002/mds.23135
67. Verbaan D, Van Rooden SM, Visser M, Marinus J, Van Hilten JJ. Nighttime sleep problems and daytime sleepiness in Parkinson's disease. Mov Disord. (2008) 23:35–41. doi: 10.1002/mds.21727
68. Adler CH, Thorpy MJ. Sleep issues in Parkinson's disease. Neurology (2005) 64:S12–20. doi: 10.1212/WNL.64.12_suppl_3.S12
69. Hobson DE, Lang AE, Martin WR, Razmy A, Rivest J, Fleming J. Excessive daytime sleepiness and sudden-onset sleep in Parkinson disease: a survey by the Canadian Movement Disorders Group. JAMA (2002) 287:455–63. doi: 10.1001/jama.287.4.455
70. Arnulf I. Excessive daytime sleepiness in Parkinsonism. Sleep Med Rev. (2005) 9:185–200. doi: 10.1016/j.smrv.2005.01.001
71. O'suilleabhain PE, Dewey RB Jr. Contributions of dopaminergic drugs and disease severity to daytime sleepiness in Parkinson disease. Arch Neurol. (2002) 59:986–9. doi: 10.1001/archneur.59.6.986
72. Diederich NJ, Vaillant M, Mancuso G, Lyen P, Tiete J. Progressive sleep ‘destructuring’ in Parkinson's disease. A polysomnographic study in 46 patients. Sleep Med. (2005) 6:313–8. doi: 10.1016/j.sleep.2005.03.011
73. Thannickal TC, Lai YY, Siegel JM. Hypocretin (orexin) cell loss in Parkinson's disease. Brain (2007) 130:1586–95. doi: 10.1093/brain/awm097
74. Ondo WG, Dat Vuong K, Khan H, Atassi F, Kwak C, Jankovic J. Daytime sleepiness and other sleep disorders in Parkinson's disease. Neurology (2001) 57:1392–6. doi: 10.1212/WNL.57.8.1392
75. Tan EK, Lum SY, Fook-Chong SM, Teoh ML, Yih Y, Tan L, et al. Evaluation of somnolence in Parkinson's disease: comparison with age- and sex-matched controls. Neurology (2002) 58:465–8. doi: 10.1212/WNL.58.3.465
76. Kumar S, Bhatia M, Behari M. Excessive daytime sleepiness in Parkinson's disease as assessed by Epworth Sleepiness Scale (ESS). Sleep Med. (2003) 4:339–42. doi: 10.1016/S1389-9457(03)00105-9
77. Pal S, Bhattacharya KF, Agapito C, Chaudhuri KR. A study of excessive daytime sleepiness and its clinical significance in three groups of Parkinson's disease patients taking pramipexole, cabergoline and levodopa mono and combination therapy. J Neural Transm. (2001) 108:71–7. doi: 10.1007/s007020170098
78. Rye DB, Bliwise DL, Dihenia B, Gurecki P. FAST TRACK: daytime sleepiness in Parkinson's disease. J Sleep Res. (2000) 9:63–9. doi: 10.1046/j.1365-2869.2000.00201.x
79. Arnulf I, Konofal E, Merino-Andreu M, Houeto JL, Mesnage V, Welter ML, et al. Parkinson's disease and sleepiness: an integral part of PD. Neurology (2002) 58:1019–24. doi: 10.1212/WNL.58.7.1019
80. Brodsky MA, Godbold J, Roth T, Olanow CW. Sleepiness in Parkinson's disease: a controlled study. Mov Disord. (2003) 18:668–72. doi: 10.1002/mds.10429
81. Pagano G, Rengo G, Pasqualetti G, Femminella GD, Monzani F, Ferrara N, et al. Cholinesterase inhibitors for Parkinson's disease: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry (2015) 86:767–73. doi: 10.1136/jnnp-2014-308764
82. Pagano G, Niccolini F, Politis M. Imaging in Parkinson's disease. Clin Med. (2016) 16:371–5. doi: 10.7861/clinmedicine.16-4-371
83. Roy R, Niccolini F, Pagano G, Politis M. Cholinergic imaging in dementia spectrum disorders. Eur J Nucl Med Mol Imaging (2016) 43:1376–86. doi: 10.1007/s00259-016-3349-x
84. Schulz J, Pagano G, Fernandez Bonfante JA, Wilson H, Politis M. Nucleus basalis of Meynert degeneration precedes and predicts cognitive impairment in Parkinson's disease. Brain (2018) 141:1501–16. doi: 10.1093/brain/awy072
85. Pagano G, Niccolini F, Politis M. Current status of PET imaging in Huntington's disease. Eur J Nucl Med Mol Imaging (2016) 43:1171–82. doi: 10.1007/s00259-016-3324-6
86. Poryazova R, Benninger D, Waldvogel D, Bassetti CL. Excessive daytime sleepiness in Parkinson's disease: characteristics and determinants. Eur Neurol. (2010) 63:129–35. doi: 10.1159/000276402
87. Pagano G, Tan EE, Haider JM, Bautista A, Tagliati M. Constipation is reduced by beta-blockers and increased by dopaminergic medications in Parkinson's disease. Parkinsonism Relat Disord. (2015) 21:120–5. doi: 10.1016/j.parkreldis.2014.11.015
88. Pagano G, Niccolini F, Yousaf T, Wilson H, Polychronis S, Chaudhuri KR, et al. Urinary dysfunction in early de novo patients with Parkinson's disease. Mov Disord. (2017) 32:939–40. doi: 10.1002/mds.26967
89. Pagano G, Yousaf T, Wilson H, Niccolini F, Polychronis S, Chaudhuri KR, et al. Constipation is not associated with dopamine transporter pathology in early drug-naive patients with Parkinson's disease. Eur J Neurol. (2018) 25:307–12. doi: 10.1111/ene.13503
90. Yousaf T, Pagano G, Niccolini F, Politis M. Excessive daytime sleepiness may be associated with caudate denervation in Parkinson disease. J Neurol Sci. (2018) 387:220–7. doi: 10.1016/j.jns.2018.02.032
91. Happe S, Baier PC, Helmschmied K, Meller J, Tatsch K, Paulus W. Association of daytime sleepiness with nigrostriatal dopaminergic degeneration in early Parkinson's disease. J Neurol. (2007) 254:1037–43. doi: 10.1007/s00415-006-0483-6
92. Matsui H, Nishinaka K, Oda M, Hara N, Komatsu K, Kubori T, et al. Excessive daytime sleepiness in Parkinson disease: a SPECT study. Sleep (2006) 29:917–20. doi: 10.1093/sleep/29.7.917
93. Qiu MH, Vetrivelan R, Fuller PM, Lu J. Basal ganglia control of sleep-wake behavior and cortical activation. Eur J Neurosci. (2010) 31:499–507. doi: 10.1111/j.1460-9568.2009.07062.x
94. Politis M, Piccini P, Pavese N, Koh SB, Brooks DJ. Evidence of dopamine dysfunction in the hypothalamus of patients with Parkinson's disease: an In vivo 11C-raclopride PET study. Exp Neurol. (2008) 214:112–6. doi: 10.1016/j.expneurol.2008.07.021
95. Pagano G, Molloy S, Bain PG, Rabiner EA, Chaudhuri KR, Brooks DJ, et al. Sleep problems and hypothalamic dopamine D3 receptor availability in Parkinson disease. Neurology (2016) 87:2451–6. doi: 10.1212/WNL.0000000000003396
96. Schwartz JR, Roth T. Neurophysiology of sleep and wakefulness: basic science and clinical implications. Curr Neuropharmacol. (2008) 6:367–78. doi: 10.2174/157015908787386050
97. Pavese N, Metta V, Simpson BS, Marohy TA, Ramlackhansingh A, Chaudhuri KR, et al. Sleep regulatory centres dysfunction in Parkinson's disease patients with excessive daytime sleepiness. An In vivo pet study.Parkinsonism Relat Disord. (2012) 18:S24–5. doi: 10.1016/S1353-8020(11)70174-2
98. Kato S, Watanabe H, Senda J, Hirayama M, Ito M, Atsuta N, et al. Widespread cortical and subcortical brain atrophy in Parkinson's disease with excessive daytime sleepiness. J Neurol. (2012) 259:318–26. doi: 10.1007/s00415-011-6187-6
99. Chondrogiorgi M, Tzarouchi LC, Zikou AK, Astrakas LG, Kosta P, Argyropoulou MI, et al. Multimodal imaging evaluation of excessive daytime sleepiness in Parkinson's disease. Int J Neurosci. (2016) 126:422–8. doi: 10.3109/00207454.2015.1023437
100. Matsui H, Nishinaka K, Oda M, Niikawa H, Komatsu K, Kubori T, et al. Disruptions of the fornix fiber in Parkinsonian patients with excessive daytime sleepiness. Parkinsonism Relat Disord. (2006) 12:319–22. doi: 10.1016/j.parkreldis.2006.01.007
101. Gama RL, Tavora DG, Bomfim RC, Silva CE, De Bruin VM, De Bruin PF. Sleep disturbances and brain MRI morphometry in Parkinson's disease, multiple system atrophy and progressive supranuclear palsy–a comparative study. Parkinsonism Relat Disord. (2010) 16:275–9. doi: 10.1016/j.parkreldis.2010.01.002
102. Ward AM, Mclaren DG, Schultz AP, Chhatwal J, Boot BP, Hedden T, et al. Daytime sleepiness is associated with decreased default mode network connectivity in both young and cognitively intact elderly subjects. Sleep (2013) 36:1609–15. doi: 10.5665/sleep.3108
103. Killgore WD, Vanuk JR, Knight SA, Markowski SM, Pisner D, Shane B, et al. Daytime sleepiness is associated with altered resting thalamocortical connectivity. Neuroreport (2015) 26:779–84. doi: 10.1097/WNR.0000000000000418
104. Wen MC, Ng SY, Heng HS, Chao YX, Chan LL, Tan EK, et al. Neural substrates of excessive daytime sleepiness in early drug naive Parkinson's disease: a resting state functional MRI study. Parkinsonism Relat Disord. (2016) 24:63–8. doi: 10.1016/j.parkreldis.2016.01.012
105. Earley CJ, Heckler D, Allen RP. Repeated IV doses of iron provides effective supplemental treatment of restless legs syndrome. Sleep Med. (2005) 6:301–5. doi: 10.1016/j.sleep.2005.01.008
106. Winkelman JW, Redline S, Baldwin CM, Resnick HE, Newman AB, Gottlieb DJ. Polysomnographic and health-related quality of life correlates of restless legs syndrome in the Sleep Heart Health Study. Sleep (2009) 32:772–8. doi: 10.1093/sleep/32.6.772
107. Earley CJ, Silber MH. Restless legs syndrome: understanding its consequences and the need for better treatment. Sleep Med. (2010) 11:807–15. doi: 10.1016/j.sleep.2010.07.007
108. Abetz L, Allen R, Follet A, Washburn T, Earley C, Kirsch J, et al. Evaluating the quality of life of patients with restless legs syndrome. Clin Ther. (2004) 26:925–35. doi: 10.1016/S0149-2918(04)90136-1
109. Allen RP, Walters AS, Montplaisir J, Hening W, Myers A, Bell TJ, et al. Restless legs syndrome prevalence and impact: REST general population study. Arch Intern Med. (2005) 165:1286–92. doi: 10.1001/archinte.165.11.1286
110. Kushida C, Martin M, Nikam P, Blaisdell B, Wallenstein G, Ferini-Strambi L, et al. Burden of restless legs syndrome on health-related quality of life. Qual Life Res. (2007) 16:617–24. doi: 10.1007/s11136-006-9142-8
111. Happe S, Reese JP, Stiasny-Kolster K, Peglau I, Mayer G, Klotsche J, et al. Assessing health-related quality of life in patients with restless legs syndrome. Sleep Med. (2009) 10:295–305. doi: 10.1016/j.sleep.2008.01.002
112. Allen RP, Bharmal M, Calloway M. Prevalence and disease burden of primary restless legs syndrome: results of a general population survey in the United States. Mov Disord. (2011) 26:114–20. doi: 10.1002/mds.23430
113. Salas RE, Kwan AB. The real burden of restless legs syndrome: clinical and economic outcomes. Am J Manag Care (2012) 18:S207–212.
114. Innes KE, Flack KL, Selfe TK, Kandati S, Agarwal P. Restless legs syndrome in an appalachian primary care population: prevalence, demographic and lifestyle correlates, and burden. J Clin Sleep Med. (2013) 9:1065–75. doi: 10.5664/jcsm.3084
115. Durgin T, Witt EA, Fishman J. The humanistic and economic burden of restless legs syndrome. PLoS ONE (2015) 10:e0140632. doi: 10.1371/journal.pone.0140632
116. Stevens MS. Restless legs syndrome/willis-ekbom disease morbidity: burden, quality of life, cardiovascular aspects, and sleep. Sleep Med Clin. (2015) 10:369–73, xv–xvi. doi: 10.1016/j.jsmc.2015.05.017
117. Fulda S, Wetter TC. Where dopamine meets opioids: a meta-analysis of the placebo effect in restless legs syndrome treatment studies. Brain (2008) 131:902–17. doi: 10.1093/brain/awm244
118. Taylor S, Gafton J, Shah B, Pagano G, Chaudhuri KR, Brooks DJ, et al. Progression of nonmotor symptoms in subgroups of patients with non-dopamine-deficient Parkinsonism. Mov Disord. (2016) 31:344–51. doi: 10.1002/mds.26456
119. Ferini-Strambi L, Marelli S, Galbiati A. Clinical pharmacology and efficacy of rotigotine (Neupro(R) patch) in the treatment of restless leg syndrome. Expert Opin Drug Metab Toxicol. (2016) 12:967–75. doi: 10.1080/17425255.2016.1194393
120. Lin CC, Fan YM, Lin GY, Yang FC, Cheng CA, Lu KC, et al. 99mTc-TRODAT-1 SPECT as a potential neuroimaging biomarker in patients with restless legs syndrome. Clin Nucl Med. (2016) 41:e14–17. doi: 10.1097/RLU.0000000000000916
121. Earley CJ, Kuwabara H, Wong DF, Gamaldo C, Salas R, Brasic J, et al. The dopamine transporter is decreased in the striatum of subjects with restless legs syndrome. Sleep (2011) 34:341–7. doi: 10.1093/sleep/34.3.341
122. Ruottinen HM, Partinen M, Hublin C, Bergman J, Haaparanta M, Solin O, et al. An FDOPA PET study in patients with periodic limb movement disorder and restless legs syndrome. Neurology (2000) 54:502–4. doi: 10.1212/WNL.54.2.502
123. Moccia M, Erro R, Picillo M, Santangelo G, Spina E, Allocca R, et al. A four-year longitudinal study on restless legs syndrome in Parkinson disease. Sleep (2016) 39:405–12. doi: 10.5665/sleep.5452
124. Cervenka S, Palhagen SE, Comley RA, Panagiotidis G, Cselenyi Z, Matthews JC, et al. Support for dopaminergic hypoactivity in restless legs syndrome: a PET study on D2-receptor binding. Brain (2006) 129:2017–28. doi: 10.1093/brain/awl163
125. Turjanski N, Lees AJ, Brooks DJ. Striatal dopaminergic function in restless legs syndrome: 18F-dopa and 11C-raclopride PET studies. Neurology (1999) 52:932–7. doi: 10.1212/WNL.52.5.932
126. Michaud M, Soucy JP, Chabli A, Lavigne G, Montplaisir J. SPECT imaging of striatal pre- and postsynaptic dopaminergic status in restless legs syndrome with periodic leg movements in sleep. J Neurol. (2002) 249:164–70. doi: 10.1007/PL00007859
127. Kim KW, Jhoo JH, Lee SB, Lee SD, Kim TH, Kim SE, et al. Increased striatal dopamine transporter density in moderately severe old restless legs syndrome patients. Eur J Neurol. (2012) 19:1213–8. doi: 10.1111/j.1468-1331.2012.03705.x
128. Tribl GG, Asenbaum S, Happe S, Bonelli RM, Zeitlhofer J, Auff E. Normal striatal D2 receptor binding in idiopathic restless legs syndrome with periodic leg movements in sleep. Nucl Med Commun. (2004) 25:55–60. doi: 10.1097/00006231-200401000-00008
129. Lee BY, Kim J, Connor JR, Podskalny GD, Ryu Y, Yang QX. Involvement of the central somatosensory system in restless legs syndrome: a neuroimaging study. Neurology (2018) 90:e1834–41. doi: 10.1212/WNL.0000000000005562
130. Belke M, Heverhagen JT, Keil B, Rosenow F, Oertel WH, Stiasny-Kolster K, et al. DTI and VBM reveal white matter changes without associated gray matter changes in patients with idiopathic restless legs syndrome. Brain Behav. (2015) 5:e00327. doi: 10.1002/brb3.327
131. Rizzo G, Manners D, Vetrugno R, Tonon C, Malucelli E, Plazzi G, et al. Combined brain voxel-based morphometry and diffusion tensor imaging study in idiopathic restless legs syndrome patients. Eur J Neurol. (2012) 19:1045–9. doi: 10.1111/j.1468-1331.2011.03604.x
132. Bucher SF, Seelos KC, Oertel WH, Reiser M, Trenkwalder C. Cerebral generators involved in the pathogenesis of the restless legs syndrome. Ann Neurol. (1997) 41:639–45. doi: 10.1002/ana.410410513
133. Etgen T, Draganski B, Ilg C, Schroder M, Geisler P, Hajak G, et al. Bilateral thalamic gray matter changes in patients with restless legs syndrome. Neuroimage (2005) 24:1242–7. doi: 10.1016/j.neuroimage.2004.10.021
134. Earley CJ, Allen RP, Beard JL, Connor JR. Insight into the pathophysiology of restless legs syndrome. J Neurosci Res. (2015) 62:623–8. doi: 10.1002/1097-4547(20001201)62:5<623::AID-JNR1>3.0.CO;2-H
135. Oshiro S, Morioka MS, Kikuchi M. Dysregulation of iron metabolism in Alzheimer's disease, Parkinson's disease, and amyotrophic lateral sclerosis. Adv Pharmacol Sci. (2011) 2011:378278. doi: 10.1155/2011/378278
136. Allen RP, Earley CJ. The role of iron in restless legs syndrome. Mov Disord. (2007) 22:S440–8. doi: 10.1002/mds.21607
137. Allen RP, Barker PB, Wehrl FW, Song HK, Earley CJ. MRI measurement of brain iron in patients with restless legs syndrome. Neurology (2001) 56:263–5. doi: 10.1212/WNL.56.2.263
138. Li X, Allen RP, Earley CJ, Liu H, Cruz TE, Edden R, et al. Brain iron deficiency in idiopathic restless legs syndrome measured by quantitative magnetic susceptibility at 7 tesla. Sleep Med. (2016) 22:75–82. doi: 10.1016/j.sleep.2016.05.001
139. Allen RP, Earley CJ. Restless legs syndrome: a review of clinical and pathophysiologic features. J Clin Neurophysiol. (2001) 18:128–47. doi: 10.1097/00004691-200103000-00004
140. Connor JR, Boyer PJ, Menzies SL, Dellinger B, Allen RP, Ondo WG, et al. Neuropathological examination suggests impaired brain iron acquisition in restless legs syndrome. Neurology (2003) 61:304–9. doi: 10.1212/01.WNL.0000078887.16593.12
141. Abbott SM, Videnovic A. sleep disorders in atypical Parkinsonism. Mov Disord Clin Pract. (2014) 1:89–96. doi: 10.1002/mdc3.12025
142. Trojanowski JQ, Revesz T, Neuropathology Working Group On MSA. Proposed neuropathological criteria for the post mortem diagnosis of multiple system atrophy. Neuropathol Appl Neurobiol. (2007) 33:615–20. doi: 10.1111/j.1365-2990.2007.00907.x
143. Ghorayeb I, Yekhlef F, Chrysostome V, Balestre E, Bioulac B, Tison F. Sleep disorders and their determinants in multiple system atrophy. J Neurol Neurosurg Psychiatry (2002) 72:798–800. doi: 10.1136/jnnp.72.6.798
144. Plazzi G, Corsini R, Provini F, Pierangeli G, Martinelli P, Montagna P, et al. REM sleep behavior disorders in multiple system atrophy. Neurology (1997) 48:1094–7. doi: 10.1212/WNL.48.4.1094
145. Gilman S, Koeppe RA, Chervin RD, Consens FB, Little R, An H, et al. REM sleep behavior disorder is related to striatal monoaminergic deficit in MSA. Neurology (2003) 61:29–34. doi: 10.1212/01.WNL.0000073745.68744.94
146. Martinez-Rodriguez JE, Seppi K, Cardozo A, Iranzo A, Stampfer-Kountchev M, Wenning G, et al. Cerebrospinal fluid hypocretin-1 levels in multiple system atrophy. Mov Disord. (2007) 22:1822–4. doi: 10.1002/mds.21668
147. Moreno-Lopez C, Santamaria J, Salamero M, Del Sorbo F, Albanese A, Pellecchia MT, et al. Excessive daytime sleepiness in multiple system atrophy (SLEEMSA study). Arch Neurol. (2011) 68:223–30. doi: 10.1001/archneurol.2010.359
148. Benarroch EE, Schmeichel AM, Low PA, Parisi JE. Depletion of putative chemosensitive respiratory neurons in the ventral medullary surface in multiple system atrophy. Brain (2007) 130:469–75. doi: 10.1093/brain/awl357
149. Abdo WF, Bloem BR, Kremer HP, Lammers GJ, Verbeek MM, Overeem S. CSF hypocretin-1 levels are normal in multiple-system atrophy. Parkinsonism Relat Disord. (2008) 14:342–4. doi: 10.1016/j.parkreldis.2007.08.012
150. Schmeichel AM, Buchhalter LC, Low PA, Parisi JE, Boeve BW, Sandroni P, et al. Mesopontine cholinergic neuron involvement in Lewy body dementia and multiple system atrophy. Neurology (2008) 70:368–73. doi: 10.1212/01.wnl.0000298691.71637.96
151. Silber MH, Levine S. Stridor and death in multiple system atrophy. Mov Disord. (2000) 15:699–704.
152. Vetrugno R, Provini F, Cortelli P, Plazzi G, Lotti EM, Pierangeli G, et al. Sleep disorders in multiple system atrophy: a correlative video-polysomnographic study. Sleep Med. (2004) 5:21–30. doi: 10.1016/j.sleep.2003.07.002
153. Shy GM, Drager GA. A neurological syndrome associated with orthostatic hypotension: a clinical-pathologic study. Arch Neurol. (1960) 2:511–27. doi: 10.1001/archneur.1960.03840110025004
154. Shimohata T, Shinoda H, Nakayama H, Ozawa T, Terajima K, Yoshizawa H, et al. Daytime hypoxemia, sleep-disordered breathing, and laryngopharyngeal findings in multiple system atrophy. Arch Neurol. (2007) 64:856–61. doi: 10.1001/archneur.64.6.856
155. Vincken WG, Gauthier SG, Dollfuss RE, Hanson RE, Darauay CM, Cosio MG. Involvement of upper-airway muscles in extrapyramidal disorders. A cause of airflow limitation. N Engl J Med. (1984) 311:438–42. doi: 10.1056/NEJM198408163110704
156. Gilman S, Chervin RD, Koeppe RA, Consens FB, Little R, An H, et al. Obstructive sleep apnea is related to a thalamic cholinergic deficit in MSA. Neurology (2003) 61:35–9. doi: 10.1212/01.WNL.0000073624.13436.32
157. Rico B, Cavada C. A population of cholinergic neurons is present in the macaque monkey thalamus. Eur J Neurosci. (1998) 10:2346–52. doi: 10.1046/j.1460-9568.1998.00246.x
158. Rampello L, Butta V, Raffaele R, Vecchio I, Battaglia G, Cormaci G, et al. Progressive supranuclear palsy: a systematic review. Neurobiol Dis. (2005) 20:179–86. doi: 10.1016/j.nbd.2005.03.013
159. Steele JC, Richardson JC, Olszewski J. Progressive supranuclear palsy. A heterogeneous degeneration involving the brain stem, basal ganglia and cerebellum with vertical gaze and pseudobulbar palsy, nuchal dystonia and dementia Arch Neurol. (1964) 10:333–59. doi: 10.1001/archneur.1964.00460160003001
160. Arnulf I, Merino-Andreu M, Bloch F, Konofal E, Vidailhet M, Cochen V, et al. REM sleep behavior disorder and REM sleep without atonia in patients with progressive supranuclear palsy. Sleep (2005) 28:349–54. doi: 10.1093/sleep/28.3.349
161. Diederich NJ, Leurgans S, Fan W, Chmura TA, Goetz CG. Visual hallucinations and symptoms of REM sleep behavior disorder in Parkinsonian tauopathies. Int J Geriatr Psychiatry (2008) 23:598–603. doi: 10.1002/gps.1945
162. Sixel-Doring F, Schweitzer M, Mollenhauer B, Trenkwalder C. Polysomnographic findings, video-based sleep analysis and sleep perception in progressive supranuclear palsy. Sleep Med. (2009) 10:407–15. doi: 10.1016/j.sleep.2008.05.004
163. Dickson DW, Ahmed Z, Algom AA, Tsuboi Y, Josephs KA. Neuropathology of variants of progressive supranuclear palsy. Curr Opin Neurol. (2010) 23:394–400. doi: 10.1097/WCO.0b013e32833be924
164. Warren NM, Piggott MA, Perry EK, Burn DJ. Cholinergic systems in progressive supranuclear palsy. Brain (2005) 128:239–49. doi: 10.1093/brain/awh391
165. Oba H, Yagishita A, Terada H, Barkovich AJ, Kutomi K, Yamauchi T, et al. New and reliable MRI diagnosis for progressive supranuclear palsy. Neurology (2005) 64:2050–5. doi: 10.1212/01.WNL.0000165960.04422.D0
166. Longoni G, Agosta F, Kostic VS, Stojkovic T, Pagani E, Stosic-Opincal T, et al. MRI measurements of brainstem structures in patients with Richardson's syndrome, progressive supranuclear Palsy-Parkinsonism, and Parkinson's disease. Mov Disord. (2011) 26:247–55. doi: 10.1002/mds.23293
167. Massey LA, Jager HR, Paviour DC, O'sullivan SS, Ling H, Williams DR, et al. The midbrain to pons ratio: a simple and specific MRI sign of progressive supranuclear palsy. Neurology (2013) 80:1856–861. doi: 10.1212/WNL.0b013e318292a2d2
168. Saini J, Bagepally BS, Sandhya M, Pasha SA, Yadav R, Thennarasu K, et al. Subcortical structures in progressive supranuclear palsy: vertex-based analysis. Eur J Neurol. (2013) 20:493–501. doi: 10.1111/j.1468-1331.2012.03884.x
169. Whitwell JL, Duffy JR, Strand EA, Machulda MM, Senjem ML, Gunter JL, et al. Neuroimaging comparison of primary progressive apraxia of speech and progressive supranuclear palsy. Eur J Neurol. (2013) 20:629–37. doi: 10.1111/ene.12004
170. Padovani A, Borroni B, Brambati SM, Agosti C, Broli M, Alonso R, et al. Diffusion tensor imaging and voxel based morphometry study in early progressive supranuclear palsy. J Neurol Neurosurg Psychiatry (2006) 77:457–63. doi: 10.1136/jnnp.2005.075713
171. Cordato NJ, Duggins AJ, Halliday GM, Morris JG, Pantelis C. Clinical deficits correlate with regional cerebral atrophy in progressive supranuclear palsy. Brain (2005) 128:1259–66. doi: 10.1093/brain/awh508
172. Price S, Paviour D, Scahill R, Stevens J, Rossor M, Lees A, et al. Voxel-based morphometry detects patterns of atrophy that help differentiate progressive supranuclear palsy and Parkinson's disease. Neuroimage (2004) 23:663–9. doi: 10.1016/j.neuroimage.2004.06.013
173. Canu E, Agosta F, Baglio F, Galantucci S, Nemni R, Filippi M. Diffusion tensor magnetic resonance imaging tractography in progressive supranuclear palsy. Mov Disord. (2011) 26:1752–5. doi: 10.1002/mds.23739
174. Saini J, Bagepally BS, Sandhya M, Pasha SA, Yadav R, Pal PK. In vivo evaluation of white matter pathology in patients of progressive supranuclear palsy using TBSS. Neuroradiology (2012) 54:771–80. doi: 10.1007/s00234-011-0983-7
175. Whitwell JL, Schwarz CG, Reid RI, Kantarci K, Jack CRJr, Josephs KA. Diffusion tensor imaging comparison of progressive supranuclear palsy and corticobasal syndromes. Parkinsonism Relat Disord. (2014) 20:493–8. doi: 10.1016/j.parkreldis.2014.01.023
176. Lehericy S, Hartmann A, Lannuzel A, Galanaud D, Delmaire C, Bienaimee MJ, et al. Magnetic resonance imaging lesion pattern in Guadeloupean Parkinsonism is distinct from progressive supranuclear palsy. Brain (2010) 133:2410–25. doi: 10.1093/brain/awq162
177. Coulon P, Budde T, Pape HC. The sleep relay–the role of the thalamus in central and decentral sleep regulation. Pflugers Arch. (2012) 463:53–71. doi: 10.1007/s00424-011-1014-6
178. Reinoso-Suarez F, De Andres I, Rodrigo-Angulo ML, Garzon M. Brain structures and mechanisms involved in the generation of REM sleep. Sleep Med Rev. (2001) 5:63–77. doi: 10.1053/smrv.2000.0136
179. Benarroch EE. Pedunculopontine nucleus: functional organization and clinical implications. Neurology (2013) 80:1148–55. doi: 10.1212/WNL.0b013e3182886a76
180. Gibb WR, Luthert PJ, Marsden CD. Corticobasal degeneration. Brain (1989) 112:1171–92. doi: 10.1093/brain/112.5.1171
181. Kimura K, Tachibana N, Aso T, Kimura J, Shibasaki H. Subclinical REM sleep behavior disorder in a patient with corticobasal degeneration. Sleep (1997) 20:891–4. doi: 10.1093/sleep/20.10.891
182. Wetter TC, Brunner H, Collado-Seidel V, Trenkwalder C, Winkelmann J. Sleep and periodic limb movements in corticobasal degeneration. Sleep Med. (2002) 3:33–6. doi: 10.1016/S1389-9457(01)00097-1
183. Cooper AD, Josephs KA. Photophobia, visual hallucinations, and REM sleep behavior disorder in progressive supranuclear palsy and corticobasal degeneration: a prospective study. Parkinsonism Relat Disord. (2009) 15:59–61. doi: 10.1016/j.parkreldis.2008.01.011
184. Roche S, Jacquesson JM, Destee A, Defebvre L, Derambure P, Monaca C. Sleep and vigilance in corticobasal degeneration: a descriptive study. Neurophysiol Clin. (2007) 37:261–4. doi: 10.1016/j.neucli.2007.05.002
185. Yasui K, Inoue Y, Kanbayashi T, Nomura T, Kusumi M, Nakashima K. CSF orexin levels of Parkinson's disease, dementia with Lewy bodies, progressive supranuclear palsy and corticobasal degeneration. J Neurol Sci. (2006) 250:120–3. doi: 10.1016/j.jns.2006.08.004
186. Kasashima S, Oda Y. Cholinergic neuronal loss in the basal forebrain and mesopontine tegmentum of progressive supranuclear palsy and corticobasal degeneration. Acta Neuropathol. (2003) 105:117–24. doi: 10.1007/s00401-002-0621-x
187. Albrecht F, Bisenius S, Morales Schaack R, Neumann J, Schroeter ML. Disentangling the neural correlates of corticobasal syndrome and corticobasal degeneration with systematic and quantitative ALE meta-analyses. NPJ Parkinsons Dis. (2017) 3:12. doi: 10.1038/s41531-017-0012-6
188. Cilia R, Rossi C, Frosini D, Volterrani D, Siri C, Pagni C, et al. Dopamine transporter SPECT imaging in corticobasal syndrome. PLoS ONE (2011) 6:e18301. doi: 10.1371/journal.pone.0018301
189. Oyanagi K, Tsuchiya K, Yamazaki M, Ikeda K. Substantia nigra in progressive supranuclear palsy, corticobasal degeneration, and Parkinsonism-dementia complex of Guam: specific pathological features. J Neuropathol Exp Neurol. (2001) 60:393–402. doi: 10.1093/jnen/60.4.393
190. Dickson DW, Bergeron C, Chin SS, Duyckaerts C, Horoupian D, Ikeda K, et al. Office of Rare Diseases neuropathologic criteria for corticobasal degeneration. J Neuropathol Exp Neurol. (2002) 61:935–46. doi: 10.1093/jnen/61.11.935
191. Niccolini F, Haider S, Reis Marques T, Muhlert N, Tziortzi AC, Searle GE, et al. Altered PDE10A expression detectable early before symptomatic onset in Huntington's disease. Brain (2015) 138:3016–29. doi: 10.1093/brain/awv214
192. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell (1993) 72:971–83.
193. Davies SW, Turmaine M, Cozens BA, Difiglia M, Sharp AH, Ross CA, et al. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell (1997) 90:537–48. doi: 10.1016/S0092-8674(00)80513-9
194. Li SH, Cheng AL, Zhou H, Lam S, Rao M, Li H, et al. Interaction of Huntington disease protein with transcriptional activator Sp1. Mol Cell Biol. (2002) 22:1277–87. doi: 10.1128/MCB.22.5.1277-1287.2002
195. Andre VM, Cepeda C, Levine MS. Dopamine and glutamate in Huntington's disease: a balancing act. CNS Neurosci Ther. (2010) 16:163–78. doi: 10.1111/j.1755-5949.2010.00134.x
196. Niccolini F, Politis M. Neuroimaging in Huntington's disease. World J Radiol. (2014) 6:301–12. doi: 10.4329/wjr.v6.i6.301
197. Hansotia P, Wall R, Berendes J. Sleep disturbances and severity of Huntington's disease. Neurology (1985) 35:1672–4. doi: 10.1212/WNL.35.11.1672
198. Arnulf I, Nielsen J, Lohmann E, Schiefer J, Wild E, Jennum P, et al. Rapid eye movement sleep disturbances in Huntington disease. Arch Neurol. (2008) 65:482–8. doi: 10.1001/archneur.65.4.482
199. Politis M, Pavese N, Tai YF, Tabrizi SJ, Barker RA, Piccini P. Hypothalamic involvement in Huntington's disease: an In vivo PET study. Brain (2008) 131:2860–9. doi: 10.1093/brain/awn244
200. Politis M, Pavese N, Tai YF, Kiferle L, Mason SL, Brooks DJ, et al. Microglial activation in regions related to cognitive function predicts disease onset in Huntington's disease: a multimodal imaging study. Hum Brain Mapp. (2011) 32:258–70. doi: 10.1002/hbm.21008
201. Swaab DF. Neuropeptides in hypothalamic neuronal disorders. Int Rev Cytol. (2004) 240:305–75. doi: 10.1016/S0074-7696(04)40003-5
202. Petersen A, Bjorkqvist M. Hypothalamic-endocrine aspects in Huntington's disease. Eur J Neurosci. (2006) 24:961–7. doi: 10.1111/j.1460-9568.2006.04985.x
203. Petersen A, Gabery S. Hypothalamic and limbic system changes in Huntington's Disease. J Huntingtons Dis. (2012) 1:5–16. doi: 10.3233/JHD-2012-120006
204. Kassubek J, Juengling FD, Kioschies T, Henkel K, Karitzky J, Kramer B, et al. Topography of cerebral atrophy in early Huntington's disease: a voxel based morphometric MRI study. J Neurol Neurosurg Psychiatry (2004) 75:213–20.
205. Douaud G, Gaura V, Ribeiro MJ, Lethimonnier F, Maroy R, Verny C, et al. Distribution of grey matter atrophy in Huntington's disease patients: a combined ROI-based and voxel-based morphometric study. Neuroimage (2006) 32:1562–75. doi: 10.1016/j.neuroimage.2006.05.057
206. Soneson C, Fontes M, Zhou Y, Denisov V, Paulsen JS, Kirik D, et al. Early changes in the hypothalamic region in prodromal Huntington disease revealed by MRI analysis. Neurobiol Dis. (2010) 40:531–43. doi: 10.1016/j.nbd.2010.07.013
207. Roos RA, Aziz NA. Hypocretin-1 and secondary signs in Huntington's disease. Parkinsonism Relat Disord. (2007) 13:S387–390. doi: 10.1016/S1353-8020(08)70035-X
208. Aron AR, Schlaghecken F, Fletcher PC, Bullmore ET, Eimer M, Barker R, et al. Inhibition of subliminally primed responses is mediated by the caudate and thalamus: evidence from functional MRI and Huntington's disease. Brain (2003) 126:713–23. doi: 10.1093/brain/awg067
209. Kassubek J, Juengling FD, Ecker D, Landwehrmeyer GB. Thalamic atrophy in Huntington's disease co-varies with cognitive performance: a morphometric MRI analysis. Cereb Cortex (2005) 15:846–53. doi: 10.1093/cercor/bhh185
Keywords: neuroimaging, magnetic resonance imaging, positron emission tomography, sleep, Parkinson's disease, atypical Parkinsonism, REM behavior sleep disorder, excessive daytime sleepiness
Citation: Yousaf T, Pagano G, Wilson H and Politis M (2018) Neuroimaging of Sleep Disturbances in Movement Disorders. Front. Neurol. 9:767. doi: 10.3389/fneur.2018.00767
Received: 29 November 2017; Accepted: 23 August 2018;
Published: 11 September 2018.
Edited by:
Cristian Falup-Pecurariu, Transilvania University of Braşov, RomaniaReviewed by:
Antonella Conte, Università degli Studi di Roma La Sapienza, ItalyOvidiu Lungu, Université de Montréal, Canada
Copyright © 2018 Yousaf, Pagano, Wilson and Politis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marios Politis, bWFyaW9zLnBvbGl0aXNAa2NsLmFjLnVr