 Lorenzo Maggi1*†
Lorenzo Maggi1*† Raffaella Brugnoni1†
Raffaella Brugnoni1† Eleonora Canioni1
Eleonora Canioni1 Paola Tonin2
Paola Tonin2 Veronica Saletti3Patrizia Sola4
Veronica Saletti3Patrizia Sola4 Stefano Cotti Piccinelli5
Stefano Cotti Piccinelli5 Lara Colleoni1Paola Ferrigno6
Lara Colleoni1Paola Ferrigno6 Antonella Pini7
Antonella Pini7 Riccardo Masson3
Riccardo Masson3 Fiore Manganelli8Daniele Lietti9Liliana Vercelli10
Fiore Manganelli8Daniele Lietti9Liliana Vercelli10 Giulia Ricci11Claudio Bruno12
Giulia Ricci11Claudio Bruno12 Giorgio Tasca13
Giorgio Tasca13 Antonio Pizzuti14,15
Antonio Pizzuti14,15 Alessandro Padovani5
Alessandro Padovani5 Carlo Fusco16Elena Pegoraro17
Carlo Fusco16Elena Pegoraro17 Lucia Ruggiero8Sabrina Ravaglia18Gabriele Siciliano11
Lucia Ruggiero8Sabrina Ravaglia18Gabriele Siciliano11 Lucia Morandi1
Lucia Morandi1 Raffaele Dubbioso8Tiziana Mongini10
Raffaele Dubbioso8Tiziana Mongini10 Massimiliano Filosto5
Massimiliano Filosto5 Irene Tramacere19
Irene Tramacere19 Renato Mantegazza1
Renato Mantegazza1 Pia Bernasconi1
Pia Bernasconi1- 1Neuroimmunology and Neuromuscular Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy
- 2Section of Clinical Neurology, Department of Neurosciences, Biomedicine and Movement Sciences, University of Verona, Verona, Italy
- 3Developmental Neurology Unit, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy
- 4Clinica Neurologica, Azienda Ospedaliero Universitaria di Modena, Modena, Italy
- 5Unit of Neurology, Center for Neuromuscular Diseases, ASST Spedali Civili and University of Brescia, Brescia, Italy
- 6SC Neurologia e Stroke Unit, Azienda Ospedaliera Brotzu, Cagliari, Italy
- 7Child Neurology and Psychiatry Unit, IRCCS Istituto delle Scienze Neurologiche di Bologna, Bologna, Italy
- 8Department of Neurosciences, Reproductive Sciences and Odontostomatology, University of Naples “Federico II,” Naples, Italy
- 9Pediatric Unit, Ospedale Valduce, Como, Italy
- 10Department of Neurosciences Rita Levi Montalcini, University of Turin, Turin, Italy
- 11Department of Clinical and Experimental Medicine, University of Pisa, Pisa, Italy
- 12Center of Translational and Experimental Myology, Istituto Giannina Gaslini, Genova, Italy
- 13Unità Operativa Complessa di Neurologia, Dipartimento di Scienze Dell'Invecchiamento, Neurologiche, Ortopediche e della Testa-Collo, Fondazione Policlinico Universitario A. Gemelli IRCCS, Rome, Italy
- 14Fondazione IRCCS Casa Sollievo della Sofferenza, Laboratory of Medical Genetics, San Giovanni Rotondo, Italy
- 15Department of Experimental Medicine, Sapienza University of Rome, Rome, Italy
- 16Dipartimento Materno-Infantile, S.C. Neuropsichiatria Infantile, Presidio Ospedaliero Provinciale Santa Maria Nuova, IRCCS di Reggio Emilia, Reggio Emilia, Italy
- 17Department of Neurosciences, University of Padova, Padova, Italy
- 18Emergency Neurology, IRCCS Mondino Foundation, Pavia, Italy
- 19Research and Clinical Development Department, Scientific Directorate, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy
Background: Four main clinical phenotypes have been traditionally described in patients mutated in SCN4A, including sodium-channel myotonia (SCM), paramyotonia congenita (PMC), Hypokaliemic type II (HypoPP2), and Hyperkaliemic/Normokaliemic periodic paralysis (HyperPP/NormoPP); in addition, rare phenotypes associated with mutations in SCN4A are congenital myasthenic syndrome and congenital myopathy. However, only scarce data have been reported in literature on large patient cohorts including phenotypes characterized by myotonia and episodes of paralysis.
Methods: We retrospectively investigated clinical and molecular features of 80 patients fulfilling the following criteria: (1) clinical and neurophysiological diagnosis of myotonia, or clinical diagnosis of PP, and (2) presence of a pathogenic SCN4A gene variant. Patients presenting at birth with episodic laryngospasm or congenital myopathy-like phenotype with later onset of myotonia were considered as neonatal SCN4A.
Results: PMC was observed in 36 (45%) patients, SCM in 30 (37.5%), Hyper/NormoPP in 7 (8.7%), HypoPP2 in 3 (3.7%), and neonatal SCN4A in 4 (5%). The median age at onset was significantly earlier in PMC than in SCM (p < 0.01) and in Hyper/NormoPP than in HypoPP2 (p = 0.02). Cold-induced myotonia was more frequently observed in PMC (n = 34) than in SCM (n = 23) (p = 0.04). No significant difference was found in age at onset of episodes of paralysis among PMC and PP or in frequency of permanent weakness between PP (n = 4), SCM (n = 5), and PMC (n = 10). PP was more frequently associated with mutations in the S4 region of the NaV1.4 channel protein compared to SCM and PMC (p < 0.01); mutations causing PMC were concentrated in the C-terminal region of the protein, while SCM-associated mutations were detected in all the protein domains.
Conclusions: Our data suggest that skeletal muscle channelopathies associated with mutations in SCN4A represent a continuum in the clinical spectrum.
Introduction
The SCN4A gene on chromosome 17q23 encodes the α-subunit of the voltage-gated sodium channel NaV1.4, responsible for the generation of action potentials and excitation of skeletal muscle fibers. The sodium channel is constituted by α subunits associated with β subunits (1). The α subunit is a single polypeptide chain that folds to four homologous but non-identical repeats (repeats I to IV). Each repeat contains six transmembrane segments (S1–S6). When inserted in the membrane, the four repeats form a central pore with segments five and six lining its wall, while the segment S4 is the channel voltage sensor due to positively charged amino acids. The loop (P loop) between S5 and S6 forming the extracellular domain is responsible for ion selectivity (1). The activation of the channel generates an action potential (AP) and the fast inactivation after AP can prevent repetitive discharge, which assures the physiological excitability changes of sarcolemma and the normal skeletal muscle contraction. Mutations in SCN4A lead to changes in skeletal muscle excitability, which is connected with the activation or inactivation speed of muscle ion channels (1). These mutations are responsible for a wide spectrum of clinical manifestations, ranging from myotonia to periodic paralysis (PP) and recently discovered phenotypes such as severe neonatal episodic laryngospasms, severe fetal hypokinesia or classical congenital myopathy, myalgia, and exercise intolerance, congenital myasthenic syndrome, and sudden infant death syndrome (2–4). However, four main clinical phenotypes have been traditionally described in patients mutated in SCN4A based on myotonia and paralysis features, including sodium-channel myotonia (SCM) and paramyotonia congenita (PMC) considered as non-dystrophic myotonias (NDM) and characterized by increased skeletal muscle excitability, and Hypokalemic type II (HypoPP2) and Hyperkalemic/Normokalemic PP (HyperPP/NormoPP), instead associated with reduced excitability. All three phenotypes represent a continuum in the clinical spectrum and their combined prevalence has been estimated in 0.5 per 100,000 in UK (5). However, only scarce data have been reported in literature on large cohorts of patients including all the aforementioned phenotypes, most of the studies being focused on single clinical subgroups (6, 7). Here, we describe the clinical, neurophysiological, and molecular features of a large cohort of Italian patients affected by skeletal muscle channelopathies associated with SCN4A mutations.
Materials and Methods
Patients
In this retrospective study, 80 patients genetically diagnosed in our laboratory at Fondazione IRCCS Istituto Neurologico Carlo Besta since 2004 were included fulfilling the following criteria: (1) clinical and neurophysiological diagnosis of myotonia, or clinical diagnosis of PP, and (2) presence of a pathogenic SCN4A gene variant. We also included two familial asymptomatic cases mutated in SCN4A. Clinical phenotypes were defined on the basis of predominant symptom (myotonia vs. periodic paralysis); in the myotonia subgroup, we identified PMC or SCM according to the presence of paradoxical myotonia or warm-up phenomenon, respectively. PP were classified as HyperPP/NormoPP and Hypo PP2 according to the potassium levels during paralytic attacks. Furthermore, patients presenting at birth with severe neonatal episodic laryngospasm (SNEL) or congenital myopathy-like phenotype and later onset of myotonia were considered as neonatal SCN4A. Pattern of muscle weakness was defined according to neurological examination at the end of the follow-up period.
The local ethics committees approved the study. All patients, parents/guardians provided written informed consent for genetic analysis and use of their anonymized clinical data at the time of their first visit at the individual centers.
Genetic Analyses
Direct Sanger sequencing of the SCN4A 24 exons was performed for 62 patients on genomic DNA extracted from peripheral blood as previously reported (8).
For 20 patients a targeted next generation sequencing panel covering the SCN4A gene was used. The panel was designed by Sure Design (https://earray.chem.agilent.com/suredesign/) (Agilent Technologies). DNA libraries were prepared using the HaloPlex Target Enrichment System (Agilent Technologies), following the manufacturer's instructions, and sequenced on the MiSeq Illumina (Illumina, San Diego, CA, USA). Alignment to human genome assembly hg19 (GRCh37) was carried out and Binary alignment/map (BAM file) and variant call format (VCF file) was generated. The variants were annotated with the free web server wANNOVAR (http://wannovar.usc.edu/) to generate the VCF files. To identify pathogenic variants and to exclude variants with allele frequency more than 1% public database (i.e., dbSNP, 1000 Genome project, ExAC, ClinVar, and HGMD) were used. To predict the functional effect of a novel variant Mutation Taster (http://www.mutationtaster.org/), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), Proven (http://provean.jcvi.org/genome_submit_2.php), and Human Splicing Finder (http://www.umd.be/HSF3/) were consulted.
All the NGS-discovered pathogenic variants were validated by Sanger sequencing on an ABI3500Dx DNA Analyzer (Thermo Fisher Scientific) in the DNA of the patient and, if any, the parents.
Statistics
Continuous variables were expressed as means with standard deviations and medians with value ranges, while categorical variables were expressed as numbers and percentages. Associations between variables were assessed by the Mann-Whitney test or Fisher exact test, as appropriate. P < 0.05 were considered statistically significant and all tests were two-sided. STATA statistical software, version 15 (StataCorp. 2017. Stata Statistical Software: Release 15. College Station, TX: StataCorp LLC) was used for the statistical analysis.
Results
We observed a PMC phenotype in 36/80 (45%) patients, SCM in 30 (37.5%), PP in 10 (12.5%), and neonatal SCN4A in 4 (5%), with a male/female ratio of 1.36. The median age at onset was 6 years (range 0–48) and the median disease duration was 22.5 years (range 2–61). Clinical features according to phenotypes are shown in Table 1. In addition, two familial asymptomatic patients were included, carrying the p.F1298C and p.A1156S mutations, respectively; the two mutations were associated with SCM in the probands. Despite not having symptoms or signs related to myotonia, both the subjects, aged 65 and 89 years at the end of the follow-up, showed myotonic discharges on electromyography. Fifty-six out of 82 (68.3%) patients were familial cases belonging to 20 different pedigrees. Phenotype concordance was observed in all families, except for the two aforementioned families including an asymptomatic subject, one family with the proband showing a neonatal SCN4A, and the mother displaying a mild SCM and two siblings carrying the p.M1592V mutation affected by Hyper/NormoPP and SCM, respectively. Electromyography response to short and long exercise test and cooling of muscle (9, 10) was performed in 26/80 (32.5%) patients, resulting in agreement with clinical phenotype in almost all the patients; in particular 15 PMC showed a pattern I, 8 SCM a pattern III, and 2 Hyper/NormoPP a pattern IV. The only exception was a patient displaying a neonatal SCN4A and showing a pattern III, suggestive of SCM, but with clear clinical paradoxical myotonia; a similar discrepancy has already been reported (11).
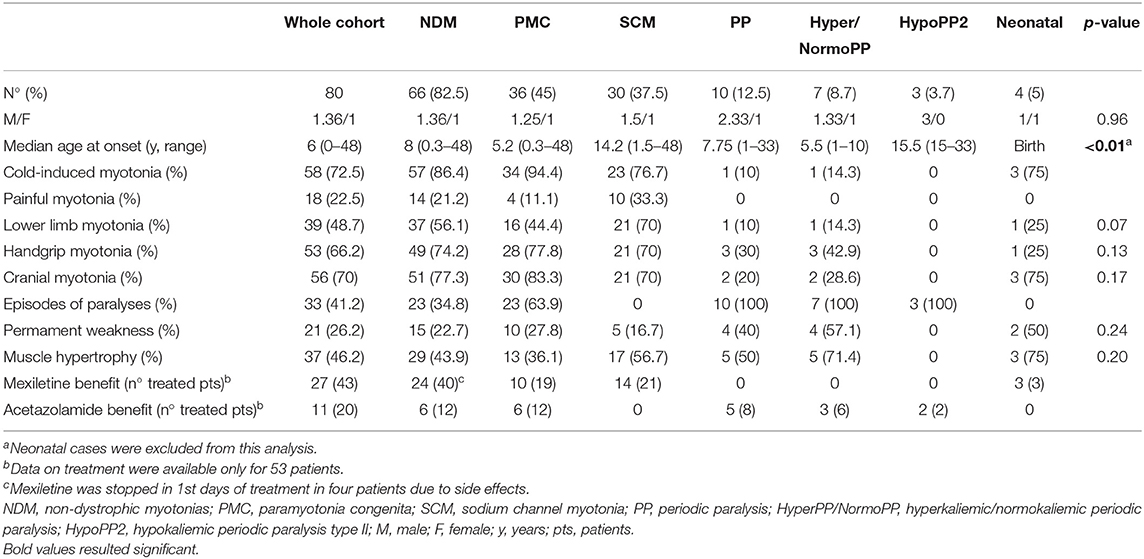
Table 1. Clinical features according to phenotype.
Non-dystrophic Myotonias
PMC (n = 36) was slightly more frequent than SCM (n = 30). Median age at onset was significantly (p < 0.01) earlier in PMC (5.2 years; range 0.3–48) than in SCM (14.2 years; range 1.5–48). Myotonia in NDM involved more frequently cranial muscles (n = 51), followed by hand (n = 49), and lower limb muscles (n = 37), especially at thigh level. Lower limb muscles were more frequently involved in SCM (n = 21) than in PMC (n = 16) (p = 0.05); although not significant, cranial muscle (p = 0.09) and handgrip myotonia (p = 0.59) were slightly more frequent in PMC than in SCM (see Table 1 for details). Cold-induced myotonia was more frequently observed in PMC (n = 34) than in SCM (n = 23) (p = 0.04). Painful myotonia tended to be more frequent in SCM (n = 10) than in PMC (n = 4), although not statistically significant. Permanent weakness was detected in 10/36 (27.8%) PMC and 5/30 (16.7%) SCM (p = 0.41), mainly in cranial (n = 8), thigh (n = 8), neck flexor (n = 6), and distal upper limb (n = 5) muscles. Of note, distal upper limb weakness was detected only in PMC. Muscle weakness was mild to moderate in most of the cases (Medical Research Council muscle power scale: MRC 3-4/5) in both NMD and PP. Muscle hypertrophy was evident in 13 (36.1%) PMC and 17 (56.7%) SCM (p = 0.15), mainly at calves (n = 18), and thigh (n = 12), while generalized hypertrophy with Hercules-like appearance was observed in 5 (16.7%) SCM and 3 (8.3%) PMC. Twenty-three out of 36 (63.9%) PMC patients had PP presenting at a median age of 5.5 years (range 1–20) and a median time from myotonia to PP onset of 0 years (range 0–15), without any case with PP preceding myotonia presentation. Of note, 1 SCM carrying the p.A699T mutation had stridor presenting in adult age. Triggering factors for episodes of paralysis in PMC were mainly cold temperature (n = 17), prolonged exercise (n = 14), and rest after exercise (n = 9). Data on treatment were available for 53 (66.2%) NDM patients; mexiletine was administered in 21/30 (70%) SCM and 19/36 (52.8%) PMC patients with benefit in 14 (66.7%) and 10 (71.4%), respectively; four out of 40 NDM patients stopped mexiletine due to side effects in 1st days of treatment. Acetazolamide was given to 12/36 (33.3%) PMC patients, with benefit in 6 (50%). Further seven patients had benefit from lamotrigine (n = 3), carbamazepine (n = 2), buprenorphine (n = 1), and propafenone (n = 1); all of them had PMC, except for the three patients taking lamotrigine, who displayed a SCM phenotype.
Periodic Paralyses
The median age at onset of PP was 7.75 years (range 1–33), not significantly different from age at onset of episodes of paralysis in PMC (p = 0.72). Hyper/NormoPP was observed in 7 (70%) patients and HypoPP2 in 3 (30%). Median age at onset was significantly (p = 0.02) earlier in Hyper/NormoPP (5.5 years; range 1–10) than in HypoPP2 (15.5 years; range 15–33). Five (50%) patients, all with Hyper/NormoPP, also had myotonia, with warm-up phenomenon, worsened by cold in only one case and never reported as painful. No significant difference was found in muscle distribution and age at onset of myotonia in PP and in NDM. Median time from PP and myotonia onset was 5 years (range 0–21); of note, PP onset always preceded myotonia presentation. Triggering factors for Hyper/Normo were mainly post-exercise period (n = 5) and cold temperature (n = 4), for HypoPP2 post-exercise period (n = 2). Episodes of paralysis were usually generalized in 3/7 (42.9%) Hyper/NormoPP and in all HypoPP2 or limited to lower limbs in 4/7 (57.1%) Hyper/NormoPP. Duration of PP was usually more than 24 h in all HypoPP2 patients and in 2/7 (28.6%) Hyper/NormoPP and <24 h in remaining 5 (71.4%) Hyper/NormoPP, in most of the cases lasting 1–4 h.
Permanent weakness was found in 4/10 (40%) patients, all affected by Hyper/NormoPP, and its occurrence was not statistically different from that in NDM; weakness was limited to thigh muscles in all patients, except for one with also involvement of distal upper limb muscles. Muscle hypertrophy was found in 5/7 (71.4%) Hyper/NormoPP patients and its frequency was not significantly different among PP and NDM.
Data on treatment were available for 8/10 (80%) PP patients; acetazolamide was administered in 6/7 (85.7%) Hyper/NormoPP and 2/3 (66.7%) HypoPP2, with benefit in 3 (50%) and 2 (100%), respectively.
Neonatal SCN4A
Three out of 4 (75%) patients with neonatal phenotype had typical SNEL and one had congenital myopathy-like presentation with bilateral clubfoot, hip dislocation, facial dysmorphism in association with myotonia characterized by warm-up phenomenon, and absence of stridor, previously reported in literature (12). This patient had an affected mother, carrying the same mutation p.N1180I and showing a SCM, with onset of hand and facial myotonia at the age of 12 years; mother's neurological examination revealed mild handgrip myotonia, diffuse muscle hypertrophy, pes cavus, hyporeflexia, and nasal speech. Notably, two SNEL patients were mother and son, respectively, carrying the p.I693T mutation and presenting with laryngeal stridor and hypotonia at birth, followed by paradoxical myotonia of facial and bulbar muscles at the age of 6 months followed by muscle hypertrophy. Stridor was triggered by cold temperature or cold food or beverages and spontaneously disappeared in adolescence in both patients. The sporadic patient carrying the p.G1306E mutation presented at birth with inguinal and hiatal hernia, hypotonia, myotonia with warm-up phenomenon, and laryngeal stridor, which disappeared after 1-year treatment with mexiletine. Permanent weakness was found in the patient with congenital myopathy-like presentation and limited to axial muscles and in the mother with paradoxical myotonia displaying weakness of neck flexor and tongue and facial muscles. Only the three patients with SNEL had cold-induced myotonia, and none of the neonatal SCN4A showed painful myotonia.
Molecular Genetics
Genetic analysis of SCN4A gene in our patient cohort revealed 28 potential disease-causing variants in the 80 patients and two asymptomatic familial cases. These variants involved nine different exons and the most mutated were exons 22, 24, 21, and 13 (Table 2). The most frequent mutations were p.T1313M, p.R1448C/G/H, and p.V1293I found in 18, 13, and 7 patients, respectively (Table 2).
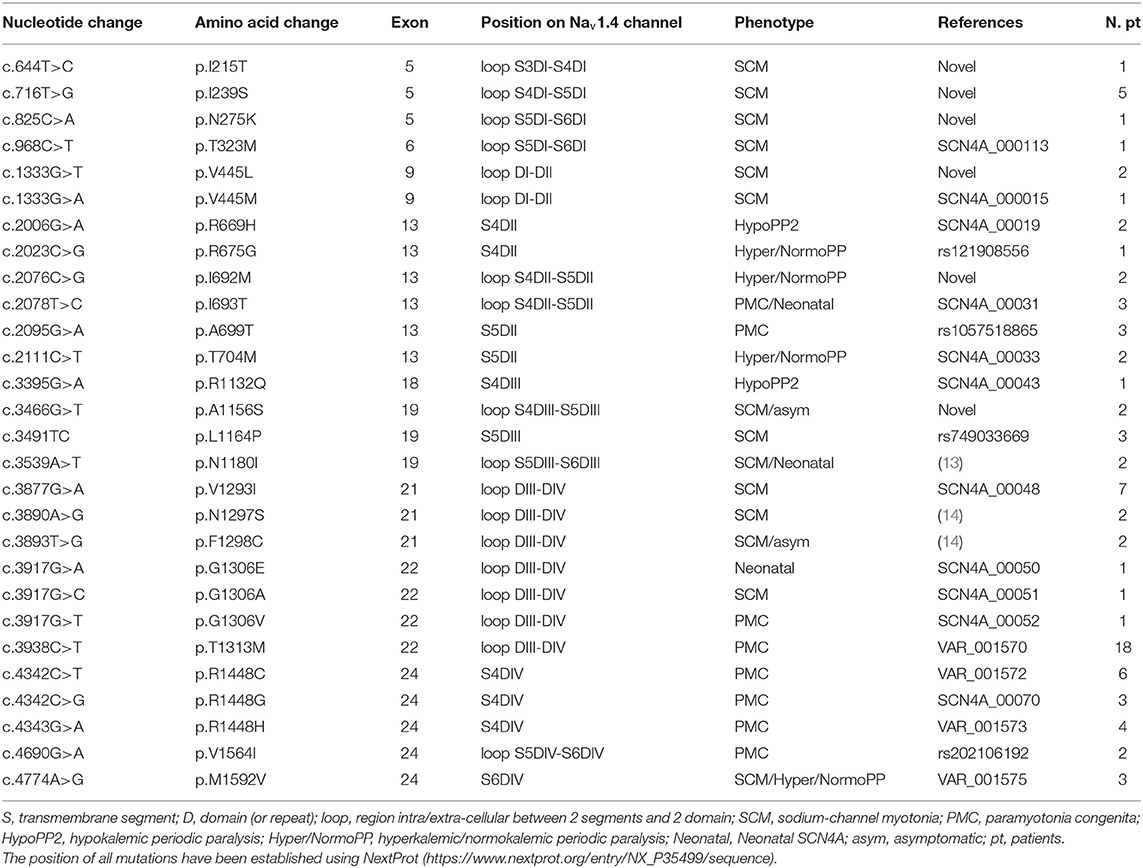
Table 2. Panel of the 28 mutations of the SCN4A gene found in our 80 patients.
Six mutations in the SCN4A gene have not been previously reported in the molecular databases (Leiden Open Variation Database, NextProt, The Human Gene Mutation Database, Exome Variant Server and UniProtKB): p.I215T, p.I239S, p.N275K, p.V445L, p.I692M, p.A1156S, p.N1180I, p.N1297S, and p.F1298C (Table 2). Some of these variants were located in the same position or immediately nearby the amino acid of a codon already known to be mutated and associated with disease, or showing pathogenic score, or segregating with disease in familial genetic studies, strongly suggesting that our nine unknown variants are potential disease-causing mutations.
The p.I215T variant present in one SCM patient was placed in the extracellular loop between the transmembrane segments S3 and S4 of the first repeat (loop S3DI-S4DI) (Figure 1). In the same codon the known variant p.I215M (rs373289931) had been previously reported with a pathogenic score (score = 0.98).
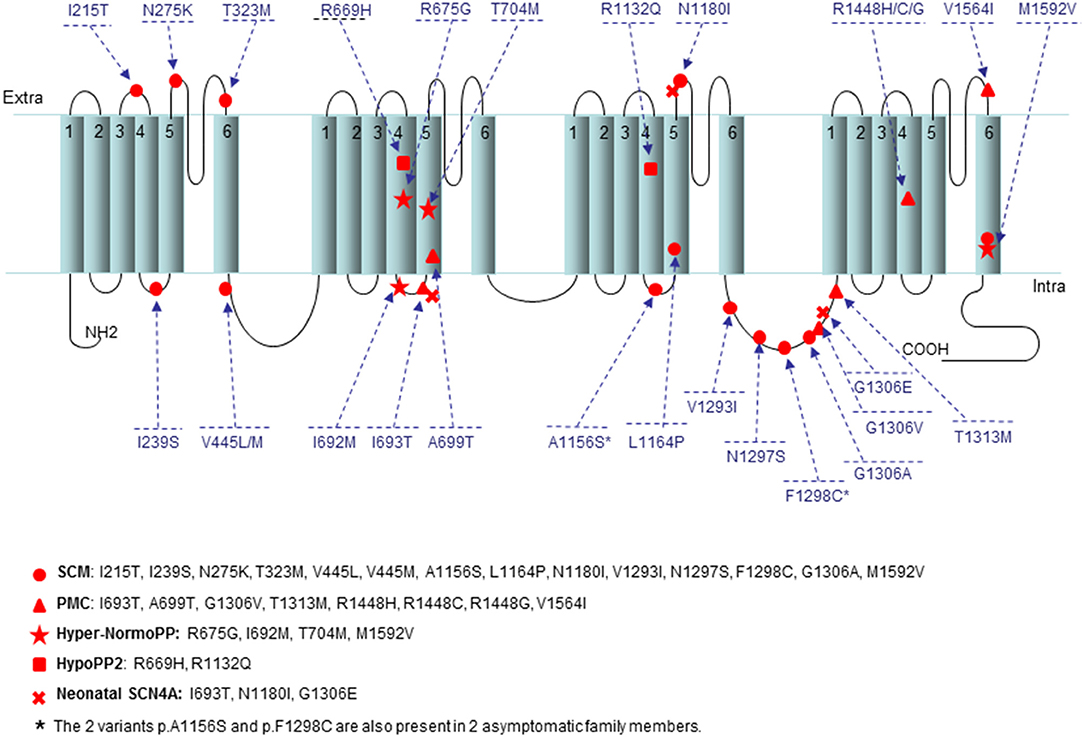
Figure 1. Location of the 28 mutations present in this study onto a secondary structure of Nav1.4 channel. The variants are indicated by small circle for SCM, triangle for PMC, star for Hyper/NormoPP, square for HypoPP2, and cross for SNEL. The two variants p.A1156S and p.F1298C are also present in two asymptomatic family members. The position of all variants has been established using NextProt (https://www.nextprot.org/entry/NX_P35499/sequence).
The p.I239S mutation detected in 5 SCM patients was located in the extracellular loop between the transmembrane segments S4 and S5 of the first repeat (loop S4DI-S5DI) (Figure 1); in the same loop the variants p.T238K/M and p.V240M (rs201661188 and rs746216167, respectively) had already been reported.
One SCM patient harbored the unreported p.N275K variant localized in the extracellular loop between the transmembrane segments S5 and S6 of the first repeat (loop S5DI-S6DI) (Figure 1); in the previous codon the variant p.G274E had been described (COSM3890178) with a pathogenic score (score 0.99).
The p.V445L mutation found in 2 SCM patients was located in the adjacent N-terminal area of the loop connecting repeats I and II (Figure 1). The novel p.V445L mutation could be pathogenic since the known mutation p.V445M at the same position has been reported in multiple families and individuals affected by myotonia congenita and was associated with marked phenotypic variability. According to the prediction sites the p.V445L also had a pathogenic score (score = 0.82).
In two patients affected by Hyper/NormoPP the p.I692M mutation was placed in the intracellular loop between the transmembrane segments S4 and S5 of the second repeat (loop S4DII-S5DII) (Figure 1), nearby the two known mutations p.I692T and p.I692F (rs757943588 and rs765779586).
The p.A1156S variant found in 1 SCM patient and the asymptomatic father (Table 2) affect the same codon of the p.A1156T mutation (LOVD: SCN4A_00044), which was reported to alter the function of Nav1.4 channel causing a channelopathy, interestingly associated with a mild phenotype without overt myotonia or periodic paralysis but with muscle pain (15).
Three mutations (p.N1180I, p.N1297S, and p.F1298C) found in six patients were already reported and associated to non-dystrophic myotonias (Table 2) (13, 14). Of note, p.N1297S was found in our cohort in two patients also carrying the p.F167L variant in CLCN1 gene, which probably mitigated the severe effect of the mutation in the Nav1.4 channel, as already reported (8). In our cohort of patients PP was more frequently associated with mutations distributed in the S4 region compared to NDM (p < 0.01). No significant association was found between specific domains of Nav1.4 channel and the presence of episodes of paralyses in PMC (p = 0.75).
Discussion
The present study includes a cohort of Italian patients mutated in SCN4A, displaying different phenotypes, spanning from SCM and PMC throughHyper/NormoPP and HypoPP2 to Neonatal SCN4A. To our knowledge, this is the first report on a large population of patients investigating clinical features of all these phenotypes together. Skeletal muscle channelopathies associated to SCN4A gene mutations indeed represent a continuum in the clinical spectrum, as also supported by the high frequency of episodes of paralysis in PMC and the relative high incidence (50%) of myotonia in Hyper/NormoPP. In addition, features of myotonia and episodes of paralysis did not differ between NDM and PP in terms of muscle distribution and age at onset. Phenotypes were consistent in the same family except for four out of 20 pedigrees, including two asymptomatic patients in late adult life, suggesting relative strong genotype-phenotype correlations in these diseases. On the other hand, clinical heterogeneity could also be a feature of sodium muscle channelopathies, as demonstrated by the p.G1306V/A/E mutations found in three patients displaying three different phenotypes (PMC, SCM, and SNEL), demonstrating how the replacement of glycine at the same codon of the NaV1.4 with three different amino acids (Valine/Alanine/Glutamic Acid) may lead to different phenotypes. Hence, other mechanisms, such as the coexistence of SCN4A and CLCN1 gene mutations and non-genetic factors (epigenetic, environmental, and hormonal), should be considered to explain the clinical variability in these disorders (8, 16).
In our cohort PMC was the most frequent phenotype, accounting for almost a half of the whole population, followed by SCM and then by PP and Neonatal SCN4A, which represent a minority of the cases. Our data showing greater frequency of NDM than PP caused by SCN4A gene mutations are in agreement with previous studies on large cohorts of British and Dutch patients (5, 17), although in our cohort PP seems to be underrepresented, being only 12.5% of the population.
In our study, PMC appeared to be significantly associated to earlier onset and cold-sensitivity than SCM and tended to affect more frequently cranial and hand muscles; conversely, SCM tended to show more frequently lower limb and painful myotonia and muscle hypertrophy than PMC, in agreement with previous reports (2, 6, 7). Hyper/NormoPP had earlier onset than HypoPP2 and tended to be associated more frequently with focal and shorter episodes of paralysis; myotonia and muscle hypertrophy were found only in Hyper/NormoPP patients (6, 18, 19). On the other hand, HypoPP2 displayed more frequently generalized PP than Hyper/NormoPP. Permanent weakness still represents an issue in skeletal muscle channelopathies, mainly due to the poor and somehow conflicting data on its frequency, presentation, and progression over time; in addition, its pathomechanisms are still not completely elucidated (20, 21). In our cohort, permanent weakness was detected in about one quarter of the patients, without any significant difference between NDM and PP or SCM and PMC. Of note, permanent muscle weakness was observed with neurological examination in about half of Hyper/NormoPP patients and in none of HypoPP2 patients, as previously reported (6). Muscle weakness was limited to thigh muscle in Hyper/NormoPP and more diffuse in NDM, including also cranial, axial, and distal upper limb muscles, with the latter appearing to be specific for PMC. To date, no predictive factors for permanent weakness are known and this phenomenon seems to occur regardless of the mutation.
Neonatal-SCN4A represents 5% of the whole cohort. There is a partial overlap with congenital myopathies due to the presentation at birth with hypotonia in three out four patients and bilateral clubfoot, hip dislocation, facial dysmorphism, or inguinal and hiatal hernia in two out of four patients. Autosomal recessive mutations in SCN4A gene have been reported in association with severe congenital myopathy without myotonia (22), while patients with simple heterozygous mutations may represent a milder phenotype associated with myotonia presenting at birth or in 1st years of age (23–26). Notably, myotonia in neonatal SCN4A may be associated to warm-up phenomenon or paradoxical, depending on the causing mutations, mainly the p.G1306E and p.I693T, respectively (23–25). Although the SNEL cases reported in literature are usually sporadic, we report here two familial cases (mother and son) with autosomal dominant inheritance displaying this phenotype.
The alpha-subunit of the voltage-gated sodium channel NaV1.4 is composed of four highly homologous domains (DI-DIV) each consisting of six transmembrane segments (S1–S6). When inserted in the membrane, the four repeats form a central pore with segments five and six lining its wall (Figure 1). The repeats are connected by intracellular loops; one of them is in the III–IV linker which contains the fast-inactivation particle and many mutations found in our population are located in this region, such as the frequent p.T1313M mutation found in 18 patients (Table 2). Notably, we found seven missense mutations (p.V1293I, p.N1297S, p.F1298C, p.G1306V/A/E, and p.T1313M) in this relevant portion of the NaV1.4 in 32 patients affected by different myotonic phenotype (SCM, PMC, and SNEL) and with various degrees of severity (Table 2, Figure 1). A further important region for the functionality of the Nav1.4 channel is the voltage sensor localized in the transmembrane segment S4. In this region we found six mutations: p.R669H and p.R675G in the second domain, p.R1132Q in the third domain, and p.R1448C/G/H in the fourth repeat (Figure 1).
Both the p.R669H and p.R1132Q mutations found in 3 HypoPP2 patients cause a change of an arginine in S4 segments of the second and third repeats, respectively; conversely, as also reported by Cannon (1). In addition, the p.R675G variant found in 1 patient affected by Hyper/NormoPP, and the mutations p.R1448C/G/H in 13 patients affected by PMC, further confirm that the S4 is an essential region for the function of the sodium channel and that changes in this transmembrane segment are associated with a large phenotypic variability. These mutations may increase the continuous current of the sodium channel, change the voltage-dependent activation process, lead to abnormal depolarization of resting potential, or slow down the inactivation process, affecting the normal function of sodium channel and leading to disease. However, mutations in the S4 segments appeared to be significantly more frequent in PP than NDM patients.
Based on the distribution of mutations along the SCN4A gene, we may conclude that the hot-spot regions for sodium channelopathies in our cohort patients involve the exons 13, 21, 22, 24, in agreement with the literature (27, 28). These exons encoding the S4–S5 segments of the second domain, the loop between the third and the fourth domain and the segments S4 and S6 of the fourth domain, as shown in Figure 1 and Table 2.
Genetic analysis of SCN4A gene in our cohort patients revealed a greater prevalence of PMC-associated mutations in the C-terminal region of the NaV1.4 channel protein than the more diffuse distribution of SCM-associated mutations, detected in all 4 domains of the protein (Table 2, Figure 1).
In conclusion, our data provide further insight in the field of skeletal muscle channelopathies due to mutations in SCN4A; however, prospective studies on large cohort of patients are needed to better clarify the natural history of these diseases and investigate possible genetic and non-genetic modifiers of the phenotype.
Data Availability Statement
The datasets generated for this study can be found in the https://databases.lovd.nl/shared/individuals/SCN4A.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics Committee at Fondazione IRCCS Istituto Neurologico Carlo Besta. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author Contributions
LM, RB, and PB conceived and designed the study. LM, RB, PB, RM, and IT drafted manuscript, figure, and tables. All the authors acquired and analyzed the data and gave the final approval to the current version of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Cannon SC. Sodium channelopathies of skeletal muscle. Handb Exp Pharmacol. (2018) 246:309–30. doi: 10.1007/164_2017_52
2. Matthews E, Fialho D, Tan SV, Venance SL, Cannon SC, Sternberg D, et al. The non-dystrophic myotonias: molecular pathogenesis, diagnosis and treatment. Brain. (2010) 133:9–22. doi: 10.1093/brain/awp294
3. Arnold WD, Feldman DH, Ramirez S, He L, Kassar D, Quick A, et al. Defective fast inactivation recovery of Nav 1.4 in congenital myasthenic syndrome. Ann Neurol. (2015) 77:840–50. doi: 10.1002/ana.24389
4. Männikkö R, Wong L, Tester DJ, Thor MG, Sud R, Kullmann DM, et al. Dysfunction of NaV1.4, a skeletal muscle voltage-gated sodium channel, in sudden infant death syndrome: a case-control study. Lancet. (2018) 391:1483–92. doi: 10.1016/S0140-6736(18)30021-7
5. Horga A, Raja Rayan DL, Matthews E, Sud R, Fialho D, Durran SC, et al. Prevalence study of genetically defined skeletal muscle channelopathies in England. Neurology. (2013) 80:1472–5. doi: 10.1212/WNL.0b013e31828cf8d0
6. Miller TM, Dias da Silva MR, Miller HA, Kwiecinski H, Mendell JR, Tawil R, et al. Correlating phenotype and genotype in the periodic paralyses. Neurology. (2004) 63:1647–55. doi: 10.1212/01.WNL.0000143383.91137.00
7. Trip J, Drost G, Ginjaar HB, Nieman FH, van der Kooi AJ, de Visser M, et al. Redefining the clinical phenotypes of non-dystrophic myotonic syndromes. J Neurol Neurosurg Psychiatry. (2009) 80:647–52. doi: 10.1136/jnnp.2008.162396
8. Maggi L, Ravaglia S, Farinato A, Brugnoni R, Altamura C, Imbrici P, et al. Coexistence of CLCN1 and SCN4A mutations in one family suffering from myotonia. Neurogenetics. (2017) 18:219–25. doi: 10.1007/s10048-017-0525-5
9. Fournier E, Arzel M, Sternberg D, Vicart S, Laforet P, Eymard B, et al. Electromyography guides toward subgroups of mutations in muscle channelopathies. Ann Neurol. (2004) 56:650–61. doi: 10.1002/ana.20241
10. Fournier E, Viala K, Gervais H, Sternberg D, Arzel-Hézode M, Laforet P, et al. Cold extends electromyography distinction between ion channel mutations causing myotonia. Ann Neurol. (2006) 60:356–65. doi: 10.1002/ana.20905
11. Trivedi JR, Bundy B, Statland J, Salajegheh M, Rayan DR, Venance SL, et al. Non-dystrophic myotonia: prospective study of objective and patient reported outcomes. Brain. (2013) 136:2189–200. doi: 10.1093/brain/awt133
12. Matthews E, Manzur AY, Sud R, Muntoni F, Hanna MG. Stridor as a neonatal presentation of skeletal muscle sodium channelopathy. Arch Neurol. (2011) 68:127–9. doi: 10.1001/archneurol.2010.347
13. Fusco C, Frattini D, Salerno GG, Canali E, Bernasconi P, Maggi L. New phenotype and neonatal onset of sodium channel myotonia in a child with a novel mutation of SCN4A gene. Brain Dev. (2015) 37:891–3. doi: 10.1016/j.braindev.2015.02.004
14. Farinato A, Altamura C, Imbrici P, Maggi L, Bernasconi P, Mantegazza R, et al. Pharmacogenetics of myotonic hNav1.4 sodium channel variants situated near the fast inactivation gate. Pharmacol Res. (2019) 141:224–35. doi: 10.1016/j.phrs.2019.01.004
15. Suokas K, Palmio J, Sandell S, Udd B, Hietaharju A. Pain in SCN4A mutated P.A1156T muscle sodium channelopathy-a postal survey. Muscle Nerve. (2018) 57:1014–1017. doi: 10.1002/mus.26050
16. Suetterlin K, Männikkö R, Hanna MG. Muscle channelopathies: recent advances in genetics, pathophysiology and therapy. Curr Opin Neurol. (2014) 27:583–90. doi: 10.1097/WCO.0000000000000127
17. Stunnenberg BC, Raaphorst J, Deenen JCW, Links TP, Wilde AA, Verbove DJ, et al. Prevalence and mutation spectrum of skeletal muscle channelopathies in the Netherlands. Neuromuscul Disord. (2018) 28:402–7. doi: 10.1016/j.nmd.2018.03.006
18. Venance SL, Cannon SC, Fialho D, Fontaine B, Hanna MG, Ptacek LJ, et al. The primary periodic paralyses: diagnosis, pathogenesis and treatment. Brain. (2006) 129:8–17. doi: 10.1093/brain/awh639
19. Statland JM, Fontaine B, Hanna MG, Johnson NE, Kissel JT, Sansone VA, et al. Review of the diagnosis and treatment of periodic paralysis. Muscle Nerve. (2018) 57:522–30. doi: 10.1002/mus.26009
20. Amarteifio E, Nagel AM, Weber MA, Jurkat-Rott K, Lehmann-Horn F. Hyperkalemic periodic paralysis and permanent weakness: 3-T MR imaging depicts intracellular 23Na overload–initial results. Radiology. (2012) 264:154–63. doi: 10.1148/radiol.12110980
21. Charles G, Zheng C, Lehmann-Horn F, Jurkat-Rott K, Levitt J. Characterization of hyperkalemic periodic paralysis: a survey of genetically diagnosed individuals. J Neurol. (2013) 260:2606–13. doi: 10.1007/s00415-013-7025-9
22. Zaharieva IT, Thor MG, Oates EC, van Karnebeek C, Hendson G, Blom E, et al. Loss-of-function mutations in SCN4A cause severe foetal hypokinesia or ‘classical’ congenital myopathy. Brain. (2016) 139:674–91.
23. Gay S, Dupuis D, Faivre L, Masurel-Paulet A, Labenne M, Colombani M, et al. Severe neonatal non-dystrophic myotonia secondary to a novel mutation of the voltage-gated sodium channel (SCN4A) gene. Am J Med Genet. (2008) 146A:380–3. doi: 10.1002/ajmg.a.32141
24. Lion-Francois L, Mignot C, Vicart S, Manel V, Sternberg D, Landrieu P, et al. Severe neonatal episodic laryngospasm due to de novo SCN4A mutations: a new treatable disorder. Neurology. (2010) 75:641–5. doi: 10.1212/WNL.0b013e3181ed9e96
25. Caietta E, Milh M, Sternberg D, Lépine A, Boulay C, McGonigal A, et al. Diagnosis and outcome of SCN4A-related severe neonatal episodic laryngospasm (SNEL):2 new cases. Pediatrics. (2013) 132:784–7. doi: 10.1542/peds.2012-3065
26. Waldrop M, Amornvit J, Pierson CR, Boue DR, Sahenk Z. A novel de novo heterozygous SCN4A mutation causing congenital myopathy, myotonia and multiple congenital anomalies. J Neuromuscul Dis. (2019) 6:467–73. doi: 10.3233/JND-190425
27. Matthews E, Tan SV, Fialho D, Sweeney MG, Sud R, Haworth A, Stanley E, et al. What causes paramyotonia in the United Kingdom? Common and new SCN4A mutations revealed. Neurology. (2008) 70:50–3. doi: 10.1212/01.wnl.0000287069.21162.94
Keywords: myotonia, periodic paralysis, SNEL, channelopathies, voltage-gated sodium channel NaV1.4, SCN4A gene mutation
Citation: Maggi L, Brugnoni R, Canioni E, Tonin P, Saletti V, Sola P, Piccinelli SC, Colleoni L, Ferrigno P, Pini A, Masson R, Manganelli F, Lietti D, Vercelli L, Ricci G, Bruno C, Tasca G, Pizzuti A, Padovani A, Fusco C, Pegoraro E, Ruggiero L, Ravaglia S, Siciliano G, Morandi L, Dubbioso R, Mongini T, Filosto M, Tramacere I, Mantegazza R and Bernasconi P (2020) Clinical and Molecular Spectrum of Myotonia and Periodic Paralyses Associated With Mutations in SCN4A in a Large Cohort of Italian Patients. Front. Neurol. 11:646. doi: 10.3389/fneur.2020.00646
Received: 23 December 2019; Accepted: 29 May 2020;
Published: 29 July 2020.
Edited by:
Irene Litvan, University of California, San Diego, United StatesCopyright © 2020 Maggi, Brugnoni, Canioni, Tonin, Saletti, Sola, Piccinelli, Colleoni, Ferrigno, Pini, Masson, Manganelli, Lietti, Vercelli, Ricci, Bruno, Tasca, Pizzuti, Padovani, Fusco, Pegoraro, Ruggiero, Ravaglia, Siciliano, Morandi, Dubbioso, Mongini, Filosto, Tramacere, Mantegazza and Bernasconi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lorenzo Maggi, bG9yZW56by5tYWdnaUBpc3RpdHV0by1iZXN0YS5pdA==
†These authors have contributed equally to this work and share first authorship