 Ahmad S. Alotaibi1
Ahmad S. Alotaibi1 Musa Yilmaz1
Musa Yilmaz1 Sanam Loghavi2
Sanam Loghavi2 Courtney DiNardo1
Courtney DiNardo1 Gautam Borthakur1
Gautam Borthakur1 Tapan M. Kadia1
Tapan M. Kadia1 Beenu Thakral2
Beenu Thakral2 Naveen Pemmaraju1Ghayas C. Issa1
Naveen Pemmaraju1Ghayas C. Issa1 Marina Konopleva1
Marina Konopleva1 Nicholas J. Short1
Nicholas J. Short1 Keyur Patel2
Keyur Patel2 Guilin Tang2
Guilin Tang2 Farhad Ravandi1
Farhad Ravandi1 Naval Daver1*
Naval Daver1*- 1The Department of Leukemia, MD Anderson Cancer Center, Houston, TX, United States
- 2The Department of Hematopathology, MD Anderson Cancer Center, Houston, TX, United States
Despite the promising result with FLT3 inhibitors in AML, the emergence of resistance poses a significant challenge, leading to a shorter response duration and inferior survival. This is frequently driven by on-target or parallel prosurvival mutations. The emergence of BCR–ABL1 as a mechanism of possible clonal evolution in relapsed AML has rarely been reported. Here we report our experience with three patients who had emergent BCR–ABL1 fusion at relapse after FLT3 inhibitors–based therapies. The first patient was refractory to multiple lines of therapies, including FLT3 inhibitors–based therapy. Patients 2 and 3 showed some response to combined FLT3-inhibitor and BCR–ABL targeted therapy (gilteritinib and ponatinib). The availability of effective targeted therapies for BCR–ABL1 makes this an important aberration to proactively identify and possibly target at relapse post–FLT3-inhibitor therapies.
Introduction
The development of multiple small-molecule kinase inhibitors targeting FLT3 has improved the outcome of FLT3-mutated acute myeloid leukemia (AML) (1). Despite high response rates with FLT3 inhibitor–based therapies, the emergence of new mutations frequently drives resistance, and resulting in short durations of response and survival (2–5). These emergent mutations may involve the activating loop or gatekeeper residues of the FLT3 (on target resistance) (2, 3, 5) or genes regulating parallel prosurvival signaling pathways such as PI3K/AKT and RAS/MAPK (off-target resistance) (4, 5). Herein, we report the cases of three patients who relapsed following an FLT3 inhibitor–based therapy, with an emergent BCR–ABL1 fusion, rendering a potentially targetable mechanism of resistance.
Clinical Summary
Patient 1
A 33-year-old woman was diagnosed with AML with a normal karyotype (no molecular testing done locally at baseline). She received 7 + 3 induction without a response and was reinduced with fludarabine and cytarabine (FLAG) with complete remission (CR), followed by four cycles of high-dose cytarabine (HiDAC) consolidation. She relapsed 5 months after the last consolidation and was referred to our institution following an unsuccessful salvage attempt with FLAG.
Our initial bone marrow biopsy revealed 60% blasts, normal karyotype, FLT3–ITD (allelic ratio 0.33), with no BCR–ABL1 fusion or NPM1, RAS, IDH1&2, or KIT mutations. The patient received azacitidine with sorafenib with marrow remission after four cycles and underwent allogeneic stem cell transplant (ASCT), but relapsed 60 days post-ASCT with 58% bone marrow blasts, normal karyotype, and FLT3–ITD (allelic ratio 0.02). After achieving a short-term remission with idarubicin, cytarabine, and sorafenib combination, subsequent relapse bone marrow demonstrated t(9,22) in 7/20 metaphases with a positive BCR–ABL1 fusion transcript (55%) and FLT3–ITD (allelic ratio 0.12) (Figure 1A). The patient died after not responding to phase I agent BP-1001 (L-Grb-2 antisense oligonucleotide NCT01159028) (6).
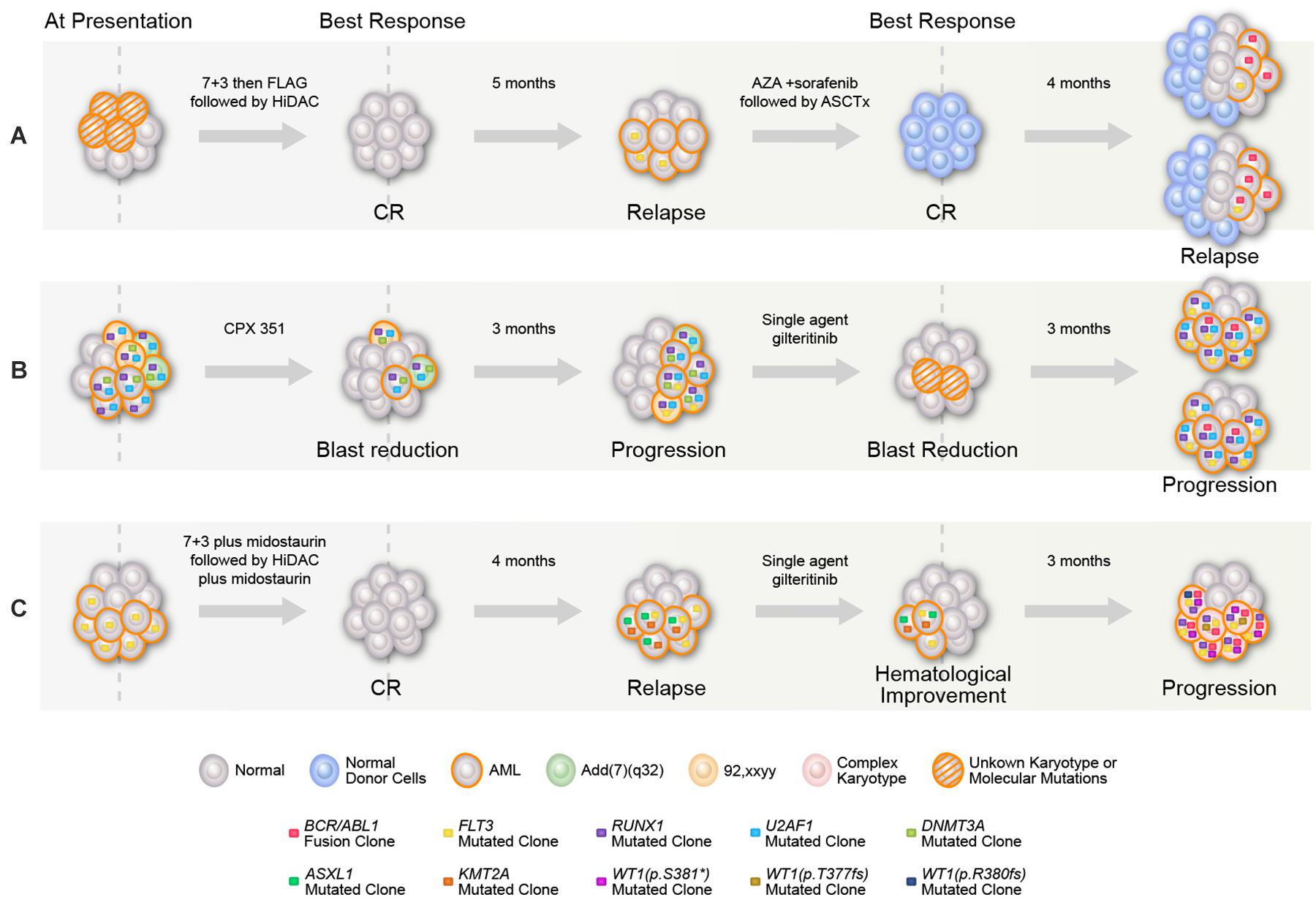
Figure 1. Graphical representation of mutational acquisition/expansion. (A) Patient 1, (B) patient 2, and (C) patient 3. Clonal size was estimated based on variant allelic frequency (VAF) for mutations detected by NGS, allelic ratio for FLT3, percentage of BCR–ABL to ABL transcripts, and number of aberrant metaphases in karyotype. Clones could have coexisted or be mutually exclusive, mutations with high clonal size at the same given point of time were considered to be coexisted, for other mutations, the two possibilities were represented. At last relapse/progression in (A,B), the two branch graphs represent the possibility of coexistence or mutually exclusivity of FLT3 mutation and BCR/ABL.
Patient 2
A 47-year-old man was diagnosed with AML with [add (7) (q32)], and DNMT3A, RUNX1, and U2AF1 mutations on a myeloid NGS panel [no FLT3 ITD/tyrosine kinase domain (TKD) mutation] at diagnosis at a local institution. He received consecutive therapies with CPX-351, decitabine, and FLAG-IDA, without a response. Repeat bone marrow analysis revealed an emergent FLT3–ITD mutation (allelic ratio not known). After receiving a single-agent gilteritinib salvage therapy for 2 months, he was referred to our institution with progressive disease. The initial bone marrow revealed 70% blasts, normal karyotype [no t(9,22) detected], positive for BCR–ABL1 rearrangement (2.8% by FISH) and BCR–ABL1 fusion (1.92%), FLT-3 ITD (allelic ratio 0.49), RUNX1, U2AF1, and KRAS (Figure 1B) (7). The patient did not respond to cladribine, idarubicin, cytarabine, and sorafenib combination, with increased BCR–ABL1 fusion transcript (7.05%) posttreatment. After failing the next salvage therapy (decitabine, venetoclax, and ponatinib), the patient received CPX-351 plus venetoclax, ponatinib, and gilteritinib combination with a marrow remission (MLFS) (BCR–ABL1 fusion transcript 0.05%). ASCT was performed with a CR on day 30 post-ASCT (no detectable measurable residual disease by multiparametric flow cytometry, undetectable BCR–ABL1 fusion). The patient died 8 months post-ASCT in CR because of infectious complications.
Patient 3
A 53-year-old woman with AML, diploid karyotype, and FLT3–ITD mutation achieved CR with 7 + 3 plus midostaurin. She relapsed 2 months later with t(6,11) (p24,q14), FLT3–ITD, ASXL1, and KMT2A (ASXL1, KMT2A were not detectable at baseline). After failing mitoxantrone, etoposide, and cytarabine combination and then salvage with single-agent gilteritinib, the bone marrow revealed 50% blasts with BCR–ABL1 rearrangement (91% by FISH). Upon referral to our institution, repeat bone marrow biopsy showed 38% blasts, complex cytogenetics with t(9,22), BCR–ABL1 fusion transcript (63.9%), FLT3–ITD (allelic ratio 0.48), RUNX1, WT1 (three distinct mutations) (Figure 1C). On day 21 of the salvage therapy (decitabine, gilteritinib, and ponatinib), the patient had MLFS (persistent BCR–ABL1 at a level of 76.19%) and later died because of infectious complications.
Discussion
Targeting FLT3 mutations has improved outcomes in AML. Acquisition/expansion of mutations drives secondary resistance to FLT3-inhibitor therapy. Activation of parallel signaling pathways, such as PI3K/AKT and RAS/MAPK, is an increasingly recognized mechanism of FLT3-inhibitor resistance (2, 4). Targeted NGS at baseline and relapse in 41 relapsed FLT3-mutated patients before and after single-agent gilteritinib demonstrated emergent RAS/MAPK pathway mutations (NRAS, KRAS, PTPN11, and NF1) in 15 (36.6%) and newly detectable BCR/ABL1 fusions in 2 (5%) patients (5).
The Philadelphia chromosome, t(9,22) (q34,q11.2), results in a BCR–ABL1 fusion gene, encoding a constitutively active oncogenic tyrosine kinase. The incidence of the Ph chromosome in de novo AML ranges from 0.5 to 3% (8). Acquisition of BCR–ABL1 as a secondary abnormality and a mechanism of possible clonal evolution in relapsed AML has rarely been reported postchemotherapy treatment (9) and now post–FLT3 inhibitor–based therapy (5, 10).
Although rare (3–5%), identification of BCR–ABL1 fusion at relapse has clinical significance as it is a targetable mutation. In this report, patients 2 and 3 were refractory to FLT3 inhibitor–based therapies, but eventually responded to combined FLT3-inhibitor and BCR–ABL targeted therapy (gilteritinib and ponatinib). Patient 1 remained refractory to multiple conventional salvage chemotherapy plus FLT3-inhibitor regimens, possibly in some part due to BCR–ABL–mediated resistance to FLT3 inhibitor–based therapies. Ponatinib is a potent kinase inhibitor with pan-BCR–ABL1 inhibitor activity and strong FLT3-inhibitor activity. Smith et al. (11) demonstrated in vitro activity of ponatinib against FLT3–ITD and F691 gatekeeper mutation. Gilteritinib is a selective FLT3 inhibitor with potent activity against FLT3–ITD, as well as TKD mutations, although 5 (12.2%) of 42 patients acquired F691 gatekeeper mutations at relapse post–gilteritinib therapy (5). Combinatorial or sequential use of gilteritinib and ponatinib to overcome each individual drug resistance could be of interest for prospective evaluation, especially in FLT3-mutated patients with detectable subclonal acquisition of BCR–ABL1 fusion. Combining these two potent FLT 3 inhibitors should ideally be performed under clinical investigation/trial setting, with close monitoring and caution for myelosuppression, ideally in large leukemia centers with significant expertise. An increased understanding of the mechanisms of FLT3-inhibitor resistance may help identify and target known druggable pathways of resistance to overcome primary and secondary resistance in clinical practice.
Data Availability Statement
All datasets generated in this study are included in the article/supplementary material.
Ethics Statement
The studies involving human participants were reviewed and approved by the Institutional Review Board (IRB), MD Anderson Cancer Center (MDACC) IRB protocol DR09-0223 and PA12-0395. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
ND, AA, and MY collected the data, conceived, designed, and wrote the manuscript. SL, KP, BT, and GT analyzed and reported the molecular and cytogenetics data. CD, GB, TK, NP, GI, MK, NS, and FR treated the patients, read, revised, and approved the final manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported in part by the MD Anderson Cancer Centre Support Grant (CCSG) CA016672, the MD Anderson Cancer Center Leukemia SPORE CA100632, and the Charif Souki Cancer Research Fund.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Special thanks to Jordan Pietz, MA, CMI for his assistance in creating the visual artwork. All copyrights to the visual artwork are retained by MD Anderson Cancer Center.
References
1. Daver N, Schlenk RF, Russell NH, Levis MJ. Targeting FLT3 mutations in AML: review of current knowledge and evidence. Leukemia. (2019) 33:299–312. doi: 10.1038/s41375-018-0357-9
2. Daver N, Cortes J, Ravandi F, Patel KP, Burger JA, Konopleva M, et al. Secondary mutations as mediators of resistance to targeted therapy in leukemia. Blood. (2015) 125:3236–45. doi: 10.1182/blood-2014-10-605808
3. Cools J, Mentens N, Furet P, Fabbro D, Clark JJ, Griffin JD, et al. Prediction of resistance to small molecule FLT3 inhibitors: implications for molecularly targeted therapy of acute leukemia. Cancer Res. (2004) 64:6385–9. doi: 10.1158/0008-5472.CAN-04-2148
4. Piloto O, Wright M, Brown P, Kim KT, Levis M, Small D. Prolonged exposure to FLT3 inhibitors leads to resistance via activation of parallel signaling pathways. Blood. (2007) 109:1643–52. doi: 10.1182/blood-2006-05-023804
5. McMahon CM, Ferng T, Canaani J, Wang ES, Morrissette JJD, Eastburn DJ, et al. Clonal selection with RAS pathway activation mediates secondary clinical resistance to selective FLT3 inhibition in acute myeloid leukemia. Cancer Discov. (2019) 9:1050–63. doi: 10.1158/2159-8290.CD-18-1453
6. Ohanian M, Kantarjian HM, Ravandi F, Borthakur G, Garcia-Manero G, Andreeff M, et al. Safety, pharmacokinetics, and efficacy of BP-100-1.01 (Liposomal Grb-2 antisense oligonucleotide) in patients with refractory or relapsed acute myeloid leukemia (AML), philadelphia chromosome positive chronic myelogenous leukemia (CML), acute lymphoblasti. Blood. (2015) 126:3801. doi: 10.1182/blood.v126.23.3801.3801
7. Luthra R, Patel KP, Reddy NG, Haghshenas V, Routbort MJ, Harmon MA, et al. Next-generation sequencing-based multigene mutational screening for acute myeloid leukemia using MiSeq: applicability for diagnostics and disease monitoring. Haematologica. (2014) 99:465–73. doi: 10.3324/haematol.2013.093765
8. Konoplev S, Yin CC, Kornblau SM, Kantarjian HM, Konopleva M, Andreeff M, et al. Molecular characterization of de novo Philadelphia chromosome-positive acute myeloid leukemia. Leuk Lymphoma. (2013) 54:138–44. doi: 10.3109/10428194.2012.701739
9. Kurt H, Zheng L, Kantarjian HM, Tang G, Ravandi F, Garcia-Manero G, et al. Secondary philadelphia chromosome acquired during therapy of acute leukemia and myelodysplastic syndrome. Mod Pathol. (2018) 31:1141–54. doi: 10.1038/s41379-018-0014-x
10. Kasi PM, Litzow MR, Patnaik MM, Hashmi SK, Gangat N. Clonal evolution of AML on novel FMS-like tyrosine kinase-3 (FLT3) inhibitor therapy with evolving actionable targets. Leuk Res Rep. (2016) 5:7–10. doi: 10.1016/j.lrr.2016.01.002
Keywords: FLT3, BCR-ABL, FLT3 inhibitors, secondary mutations, AML
Citation: Alotaibi AS, Yilmaz M, Loghavi S, DiNardo C, Borthakur G, Kadia TM, Thakral B, Pemmaraju N, Issa GC, Konopleva M, Short NJ, Patel K, Tang G, Ravandi F and Daver N (2020) Emergence of BCR–ABL1 Fusion in AML Post–FLT3 Inhibitor-Based Therapy: A Potentially Targetable Mechanism of Resistance – A Case Series. Front. Oncol. 10:588876. doi: 10.3389/fonc.2020.588876
Received: 29 July 2020; Accepted: 11 September 2020;
Published: 20 October 2020.
Edited by:
Alessandro Isidori, AORMN Hospital, ItalyReviewed by:
Amir Fathi, Massachusetts General Hospital and Harvard Medical School, United StatesPrasanth Ganesan, Jawaharlal Institute of Postgraduate Medical Education and Research (JIPMER), India
Copyright © 2020 Alotaibi, Yilmaz, Loghavi, DiNardo, Borthakur, Kadia, Thakral, Pemmaraju, Issa, Konopleva, Short, Patel, Tang, Ravandi and Daver. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Naval Daver, ndaver@mdanderson.org