 Erik D. Anderson
Erik D. Anderson Mohammadali E. Alishahedani
Mohammadali E. Alishahedani Ian A. Myles
Ian A. Myles- Epithelial Therapeutics Unit, National Institute of Allergy and Infectious Disease, National Institutes of Health, Bethesda, MD, United States
Atopic diseases, particularly atopic dermatitis (AD), asthma, and allergic rhinitis (AR) share a common pathogenesis of inflammation and barrier dysfunction. Epithelial to mesenchymal transition (EMT) is a process where epithelial cells take on a migratory mesenchymal phenotype and is essential for normal tissue repair and signal through multiple inflammatory pathways. However, while links between EMT and both asthma and AR have been demonstrated, as we outline in this mini-review, the literature investigating AD and EMT is far less well-elucidated. Furthermore, current studies on EMT and atopy are mostly animal models or ex vivo studies on cell cultures or tissue biopsies. The literature covered in this mini-review on EMT-related barrier dysfunction as a contributor to AD as well as the related (perhaps resultant) atopic diseases indicates a potential for therapeutic targeting and carry treatment implications for topical steroid use and environmental exposure assessments. Further research, particularly in vivo studies, may greatly advance the field and translate into benefit for patients and families.
Introduction
Atopic dermatitis (AD) is the most common inflammatory skin condition in industrialized societies. Symptoms present classically with xeroderma and pruritis. While 85% of cases diagnosed before the age of 5 (1), AD may persist into adulthood (2). The current pediatric prevalence is estimated to be between 11 and 15% in the U.S. (3, 4) and 5–25% globally (5) but, over the recent decades, has been increasing (6). Despite its increasing commonality the etiology of AD is still not fully understood, and standard treatments are not curative. Although most new treatments target the inflammatory component of the disease (7), epidermal barrier dysfunction is an important aspect of AD pathogenesis. Herein, we review the epidermal wound healing mechanisms' (specifically epithelial mesenchymal transition; EMT) established role in allergic rhinitis (AR) and asthma to contrast with the knowledge gaps present in AD. In doing this, we propose that the EMT pathway is a promising therapeutic target for AD and its associated atopic conditions.
Overview of Epithelial-to-Mesenchymal Transition
Epithelial-to-mesenchymal transition is a process where epithelial cells take on a migratory mesenchymal phenotype (8). Doctor Elizabeth Hay first described the process in the primitive streak of chick embryos (9, 10). Subsequently three main types of EMT have been described which, it should be noted, are defined by the context in which it occurs and not the molecular mediators. Type I occurs during embryo formation and organ development; type II occurs during wound healing, tissue regeneration, fibrosis, and inflammation; type III occurs when neoplastic cells that develop an invasive or metastatic phenotype (11). The initiation event is similar in all the types, with a transition from epithelial associated proteins like E-cadherin, cytokeratin, ZO-1, and Laminin-1 to express mesenchymal associated proteins such as N-cadherin, fibroblast-specific-protein 1 (FSP-1), α-smooth muscle actin, and vimentin (12, 13). The type of EMT relevant to atopic disease is type II (11) given the impact of epithelial turnover and inflammation in the lungs, nasal passages, gut, and skin. In vitro, epithelial cells undergoing EMT take on a fibroblastic appearance under light microscopy. The transitioned cells are referred to as fibroblasts or myofibroblasts because of their morphologic and molecular marker changes.
Chronic Inflammation and Epidermal Barrier Dysfunction in Atopic Dermatitis
While there is no consensus on the etiology of AD, it is generally agreed that it develops through a combination of inflammation and epidermal barrier dysfunction (14). The inflammatory component of disease progression has been stratified into an acute phase (<2 weeks) and a chronic phase (>2 weeks) (15, 16). The acute phase is understood to be primarily driven by TH2 cells (17), which release the cytokines interleukin (IL-) IL-4 (18, 19), IL-5, IL-13 (20), and IL-31 (21). During this phase TH1 cytokines such as INF-γ, and IL-1β are low, as well as downstream effectors like human β-defensin 2 and 3 and inducible NO synthetase (22–24). The immune profiles of the chronic phase is associated with disparate effects on TH2 cytokines such as an increase in IL-5 (25), increased IFN-γ, IL-12 (26), and IL-17A (27), with a decrease in IL-4 (25). An increase in IL-22 secreted by TH22 cells progresses through the acute into the chronic phase (24).
Epidermal barrier dysfunction is a major component known to play a role in AD initiation and progression. The epidermis is a stratified squamous epithelium and keratinocytes are the predominant cell type which form the barrier through progressive differentiation. It is histologically divided into four main layers based off of this differentiation pattern, beginning with the stratum basale, moving to the stratum spinosum, granulosum, and ending with the corneum (28). The stratum corneum is made up of terminally differentiated keratinocytes, also known as corneocytes, that have lost their nucleus and form a watertight layer through the formation of an extensive intra and extracellular protein-lipid matrix. Xeroderma is a hallmark of atopic dermatitis, and increased transepidermal water loss has been quantitatively recognized in lesional and non-lesional skin of patients with AD for many decades (29). Given this observation, it is not surprising that the first gene mutation identified to be associated with AD was in the protein filaggrin, an important component of the protein-lipid matrix in the stratum corneum (30). It is a part of the S100-fused type protein (SFTP) family and binds the intermediate filament keratin, which enables its cross linking via transglutaminases and ultimately corneum formation (31). The discovery that some mutations in filaggrin are associated with AD bolstered the theory that barrier dysfunction could be the precipitating event for the condition and cause the inflammatory component through increased antigen sensitization. Additionally, many other proteins and lipids associated with keratinocyte differentiation and epidermal integrity are aberrantly expressed in AD patients (32).
Epidermal Barrier Dysfunction and the Atopic March
Despite the known role for epidermal dysfunction in AD, EMT has not been directly studied in the disease. However, it has been investigated in other atopic conditions like asthma and allergic rhinitis (AR). Together with food allergy these four conditions make up a progression known as the atopic march. The term stems from the observation that these conditions co-occur with some temporal overlap in many individuals (33). Classically, the atopic march begins with AD in infancy and progresses to food allergy, asthma, and AR. Like AD, these other conditions involve a TH2 effector phase at some point in their progression and have barrier dysfunction as a hallmark. Despite the evidence for an epidermal component of AD pathogenesis, most of the new therapies for AD target only the inflammatory component of the disease (7, 14).
Inflammation Induced Epidermal Barrier Dysfunction
Chronic inflammation may both result from, and contribute to, barrier dysfunction (34). Microvascular permeability is essential for allowing immune cells to traffic to the site of inflammation but comes at the cost of reduced barrier function (35). Similarly, immune cells must traffic to the site of tissue damage in order to perform debridement functions (34). As inflammation persists, uninterrupted signals from inflammatory cytokines may create a positive feedback loop by inducting tissue destruction which releases additional pro-inflammatory danger- and pathogen-associated molecular patterns (36). Therefore, anti-inflammatory treatments may indirectly improve barrier function, even while directly inhibiting EMT. However, while these therapies in combination with hydration are effective, their use can be limited by side effects, lack of long term follow up, limited research in pediatric populations, and high costs (37–41).
Asthma and EMT
Asthma is one of the most common non-communicable respiratory diseases in the world, affecting ~7.5% of adults (42, 43). While heterogeneous in its pathology, asthma is characterized by intermittent cough, wheezing, and shortness of breath due to lower airway inflammation and/or spasms (42). Increased EMT in bronchial epithelial cells has been hypothesized to contribute to asthma barrier dysfunction (44). Hallmarks of asthma include airway hyperresponsiveness (to triggers such as allergens or exercise) as well as airway remodeling. The remodeling process involves an increase in fibroblasts present in the airway epithelium resulting in a type of fibrosis that may ultimately lead to the obstructive phenotype observed clinically (45). The question of where these fibroblasts originate became an important one for those studying asthma, and EMT served as a possible mechanism to explain this. The growth factor TGF-β, the quintessential stimulant of EMT (46, 47), is increased in the bronchoalveolar fluid of asthmatic patients (48), which suggests EMT could play a role in disease progression (49, 50).
Despite the deluge of subsequent studies on EMT and asthma (Table 1), the topic is not without controversy or limitations. Most of the studies are from in vitro/ex vivo cell culture models and therefore may not actually present in vivo (115, 116). Additionally, while there are a number of human studies that investigated the relevance of EMT in asthma treatment, vitamin D is the only therapy that has been studied by more than one group (98, 102, 103). However, several animal models have employed lineage tracing to bolster the EMT claim (56, 88). Furthermore, one of the central therapies for asthma, corticosteroids, are potent inhibitors of EMT in additional to being anti-inflammatory (94, 95).
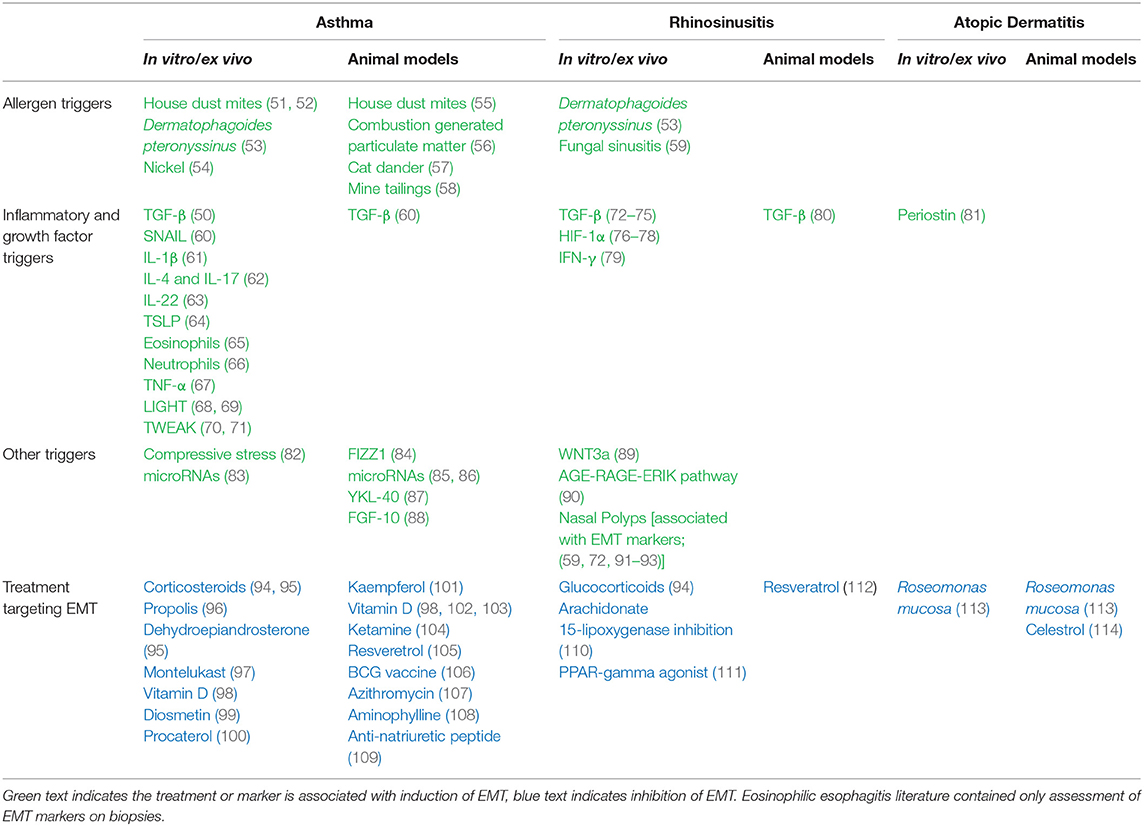
Table 1. Summary of publications directly assessing EMT and atopic diseases.
Allergic Rhinitis and EMT
Allergic rhinitis is marked by nasal congestion, runny nose, nasal itching, and sneezing (117). AR also has a high incidence, impacting 10–20% of adults in industrialized nations (118). Due to high incidence of co-morbid AR in asthmatics a relatively new model views the two conditions as a one airway disease (119). Despite this overlap between the two conditions, the role that EMT plays in AR is less well studied in asthma, and most of the studies that have been done primarily use chronic rhinosinusitis (CRS) tissue from patients without specifying the etiology (Table 1). This is important because the definition of AR is inflammation of the sinuses specifically due to allergy, whereas chronic rhinosinusitis is inflammation of the paranasal sinuses lasting more than 12 weeks. Although AR can lead to CRS, there are other causes like infection and granulomatous diseases (120). The difference in etiology of CRS could be why the condition can be subtyped into those that develop nasal polyps (CRSwNP) and those that do not (CRSsNP). The inflammatory response associated with nasal polyp development has classically been thought to be driven by TH2 polarization verses a TH1 response in those without polyps (121). However, some studies have found the picture to be more complex with CRSsNP patients showing TH2 polarization (122, 123).
Unfortunately, studies into the role of EMT in AR pathology most often only classify whether EMT markers can be observed in biopsies (59, 72, 91–93). Some studies have found that EMT markers and TGF-β are increased in CRSwNP vs. CRSsNP (72–74, 92), however others have found the opposite (93, 124). Furthermore, like the EMT asthma research, the AR studies are primarily in vitro and ex vivo models, leaving the need for lineage tracing experiments to determine whether the event is relevant in vivo. In addition to TGF-β, Hypoxia Inducible Factor-1α (HIF-1α) may play an important role in AR pathogenesis and is an important inducer of EMT (76–78). Over-expression of the histone acetylase SIRT1, an inhibitor of HIF-1α, may offer possible therapeutic benefit for suppressing NP formation (77). Another therapeutic avenue proposed is resveratrol (105, 112) either in native form or conjugated to a multimeric leucine and lysine rich peptide for enhanced permeability in the nasal epithelium (112).
Gaps in EMT and Atopic Dermatitis Research
Despite reported links between EMT and the other atopic diseases, few studies have been done on AD and EMT (Table 1). The most direct study that has been done was by Taniguchi et al. who used mouse keratinocytes in an organotypic model to study a role for periostin, a protein whose increased levels in AD correlate with disease severity (81). After finding that periostin could induce EMT, the authors hypothesized a mechanism for how AD epidermal differentiation becomes dysregulated and results in acanthosis. Prior to 2020, only two other publications had looked at the role of EMT in AD: one described triterpene celastrol as a Rac1-medicated inhibitor of EMT (114); the other described EMT markers in lens epithelial tissue from patients with AD associated subcapsular cataracts (125). More broadly, the underlying mechanism that could explain why asthma and AR are associated with enhanced EMT activation whereas AD is associated with reduced EMT activation remains to be elucidated.
Knowledge Gaps Beyond the Atopic March
Eosinophilic esophagitis (EoE) is an atopic disorder of the esophageal lining believed to be caused by aero- and food-allergen medicated inflammation (126, 127). Although often triggered by similar allergens as the other atopic disorders, EoE is not considered part of the atopic march. EoE is less common than the other atopic disorders, with a prevalence of ~60–120 per 100,000 children (128). Symptoms may range from food aversion and painful swallowing to food impaction and esophageal fibrosis (126). In one study of esophageal biopsies of children, treatment with topical steroids and elemental diets reversed the preexisting significant upregulation of EMT markers including N-cadherin, vimentin, and fibronectin (126). Similarly, biopsy levels of E-cadherin and vimentin were reduced with anti-IL-13 treatment in adults with EoE (127). While these studies add to the link between EMT and atopic disease, direct assessment of EMT in EoE has not been performed.
Discussion
Atopic dermatitis and its associated diseases asthma and allergic rhinitis represent a substantial burden to the world's health care systems. In this review we consolidated the studies that have tried to identify the role of EMT in AD, asthma, EoE, and AR, to highlight the growing, albeit incomplete, data suggesting a connection between the pathogenesis of these atopic diseases and EMT. They all involve chronic inflammation and epidermal dysfunction, but the underlying cause that sets their progression in motion is still unclear.
A recent publication from our group identified therapeutic benefit of topical microbiome transplantation of Roseomonas mucosa from healthy volunteers to the lesions of patients with AD (129). Subsequent research found that lipid mediators from R. mucosa stimulate EMT through potentiation of tumor necrosis factor receptor 2 and nicotinic acetylcholine activation; an additional role for flagella interactions with Toll-Like Receptor 5 was identified (113). This finding was consistent with our prior results in autosomal-dominant hyper IgE syndrome, a primary immune deficiency with an eczematous phenotype. In this report we showed dysregulation in EMT (130) downstream of a loss-of-function mutation in STAT3. Inhibiting TNF reversed this phenotype, as did treatment with PPAR-γ agonists. Overall, our work is consistent with the literature suggesting that both host and commensal derived lipid-mediators with influence over the ceramide-sphingolipid-arachidonic acid pathway play a role in EMT-related tissue maintenance of the skin (131, 132). However, our work also suggests that the pathologic increase in EMT seen in asthma and AR may be inversed in AD; thus, viable therapies may need to induce EMT in the skin but inhibit EMT in the airways.
The distinction between over-vs. under-active EMT as an underpinning of allergic disease may carry significant consequences. Although originally suggested in 2006, the possibility of “topical steroid addiction” or “topical steroid withdrawal” remains unelucidated (133–135); a PubMed search for (“topical steroid withdrawal” OR “topical steroid addiction”), (“atopic dermatitis” OR eczema) at the time of this manuscript yielded only nine total citations. The main feature of this purported syndrome is that withdrawal of topical corticosteroids leads to greater inflammation than was present prior to steroid application. Although the flare associated with the steroid withdrawal may be treated by resumption of topical steroids, the continued need for treatment may be perceived by the patient as “dependency” and thus “addiction.” While only speculative at this time, the possibility that steroid treatment may alleviate inflammation while worsening underlying defects in EMT is one that warrants further investigation. Such studies must contrast potential for steroid-induced barrier disruption stemming from EMT inhibition against countervailing enhancement of barrier function stemming from reduced inflammation. Furthermore, future work must look into other topical exposures (such as soaps, preservatives, etc.) that may alter EMT functions either directly or indirectly via impacts on commensal organisms. In conclusion, although sparse, the literature on EMT-related barrier dysfunction as a contributor to AD as well as the related (perhaps resultant) atopic diseases indicates a potential for therapeutic targeting. Further research, particularly in vivo studies, may greatly advance the field and translate into benefit for patients and families.
Author Contributions
All the authors meet all criteria for authorship in the ICMJE recommendations, since all authors made substantial contributions to the conception or design of the work, the acquisition and interpretation of data, critical review, and revision of the manuscript and approved the final version. All the authors agreed to be accountable for all aspects of the work.
Funding
This work was supported by the Intramural Research Program of the NIAID and the NIH.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/falgy.2020.628381/full#supplementary-material
References
1. Rudikoff D, and Lebwohl M. Atopic dermatitis. Lancet. (1998) 351:1715–21. doi: 10.1016/S0140-6736(97)12082-7
2. Margolis JS, Abuabara K, Bilker W, Hoffstad O, and Margolis DJ. Persistence of mild to moderate atopic dermatitis. JAMA Dermatol. (2014) 150:593–600. doi: 10.1001/jamadermatol.2013.10271
3. Shaw TE, Currie GP, Koudelka CW, and Simpson EL. Eczema prevalence in the United States: data from the 2003 national survey of children's health. J Invest Dermatol. (2011) 131:67–73. doi: 10.1038/jid.2010.251
4. McKenzie C, and Silverberg JI. The prevalence and persistence of atopic dermatitis in urban United States children. Ann Allergy Asthma Immunol. (2019) 123:173–8.e1. doi: 10.1016/j.anai.2019.05.014
5. Morales E, Strachan D, Asher I, Ellwood P, Pearce N, Garcia-Marcos L, et al. Combined impact of healthy lifestyle factors on risk of asthma, rhinoconjunctivitis and eczema in school children: ISAAC phase III. Thorax. (2019) 74:531–8. doi: 10.1136/thoraxjnl-2018-212668
6. Deckers IAG, McLean S, Linssen S, Mommers M, van Schayck CP, and Sheikh A. Investigating international time trends in the incidence and prevalence of atopic eczema 1990–2010: a systematic review of epidemiological studies. PLoS ONE. (2012) 7:e39803. doi: 10.1371/journal.pone.0039803
7. Kalamaha K, Reis E, Newton S, Roche C, Julson J, Fernandes H, et al. Atopic dermatitis: a review of evolving targeted therapies. Expert Rev Clin Immunol. (2019) 15:275–88. doi: 10.1080/1744666X.2019.1560267
8. Hay ED, and Zuk A. Transformations between epithelium and mesenchyme: normal, pathological, and experimentally induced. Am J Kidney Dis. (1995) 26:678–90. doi: 10.1016/0272-6386(95)90610-X
9. Hay ED. Organization and fine structure of epithelium and mesenchyme in the developing chick embryo. In: Epithelial-Mesenchymal Interactions; 18th Hahnemann Symposium Baltimore, MD (1968).
10. Hay ED. An overview of epithelio-mesenchymal transformation. Acta Anat. (1995) 154:8–20. doi: 10.1159/000147748
11. Kalluri R, and Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. (2009) 119:1420–8. doi: 10.1172/JCI39104
12. Wheelock MJ, Shintani Y, Maeda M, Fukumoto Y, and Johnson KR. Cadherin switching. J Cell Sci. (2008) 121:727–35. doi: 10.1242/jcs.000455
13. Lamouille S, Xu J, and Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. (2014) 15:178–96. doi: 10.1038/nrm3758
14. Weidinger S, Beck LA, Bieber T, Kabashima K, and Irvine AD. Atopic dermatitis. Nat Rev Dis Primers. (2018) 4:1. doi: 10.1038/s41572-018-0001-z
16. Gittler JK, Shemer A, Suárez-Fariñas M, Fuentes-Duculan J, Gulewicz KJ, Wang CQF, et al. Progressive activation of T(H)2/T(H)22 cytokines and selective epidermal proteins characterizes acute and chronic atopic dermatitis. J Allergy Clin Immunol. (2012) 130:1344–54. doi: 10.1016/j.jaci.2012.07.012
17. Brown MA, and Hanifin JM. Atopic dermatitis. Curr Opin Immunol. (1989) 2:531–4. doi: 10.1016/0952-7915(90)90006-3
18. Miraglia del Giudice M, Decimo F, Leonardi S, Maioello N, Amelio R, Capasso A, et al. Immune dysregulation in atopic dermatitis. Allergy Asthma Proc. (2006) 27:451–5. doi: 10.2500/aap.2006.27.2887
19. van Reijsen FC, Bruijnzeel-Koomen CA, Kalthoff FS, Maggi E, Romagnani S, Westland JK, et al. Skin-derived aeroallergen-specific T-cell clones of Th2 phenotype in patients with atopic dermatitis. J Allergy Clin Immunol. (1992) 90:184–93. doi: 10.1016/0091-6749(92)90070-I
20. Akdis M, Akdis CA, Weigl L, Disch R, and Blaser K. Skin-homing, CLA+ memory T cells are activated in atopic dermatitis and regulate IgE by an IL-13-dominated cytokine pattern: IgG4 counter-regulation by CLA- memory T cells. J Immunol. (1997) 159:4611–9.
21. Oliva M, Renert-Yuval Y, and Guttman-Yassky E. The “omics” revolution: redefining the understanding and treatment of allergic skin diseases. Curr Opin Allergy Clin Immunol. (2016) 16:469–76. doi: 10.1097/ACI.0000000000000306
22. Nomura I, Goleva E, Howell MD, Hamid QA, Ong PY, Hall CF, et al. Cytokine milieu of atopic dermatitis, as compared to psoriasis, skin prevents induction of innate immune response genes. J Immunol. (2003) 171:3262–9. doi: 10.4049/jimmunol.171.6.3262
23. Raphael I, Nalawade S, Eagar TN, and Forsthuber TG. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine. (2015) 74:5–17. doi: 10.1016/j.cyto.2014.09.011
24. Nograles KE, Zaba LC, Shemer A, Fuentes-Duculan J, Cardinale I, Kikuchi T, et al. IL-22-producing “T22” T cells account for upregulated IL-22 in atopic dermatitis despite reduced IL-17-producing TH17 T cells. J Allergy Clin Immunol. (2009) 123:1244–52.e2. doi: 10.1016/j.jaci.2009.03.041
25. Hamid Q, Boguniewicz M, and Leung DY. Differential in situ cytokine gene expression in acute versus chronic atopic dermatitis. J Clin Invest. (1994) 94:870–6. doi: 10.1172/JCI117408
26. Hamid Q, Naseer T, Minshall EM, Song YL, Boguniewicz M, and Leung DY. In vivo expression of IL-12 and IL-13 in atopic dermatitis. J Allergy Clin Immunol. (1996) 98:225–31. doi: 10.1016/S0091-6749(96)70246-4
27. Noda S, Suárez-Fariñas M, Ungar B, Kim SJ, de Guzman Strong C, Xu H, et al. The Asian atopic dermatitis phenotype combines features of atopic dermatitis and psoriasis with increased TH17 polarization. J Allergy Clin Immunol. (2015) 136:1254–64. doi: 10.1016/j.jaci.2015.08.015
28. Kolarsick PAJ, Kolarsick MA, and Goodwin C. Anatomy and physiology of the skin. J Dermatol Nurses Assoc. (2011) 3:203–13. doi: 10.1097/JDN.0b013e3182274a98
29. Werner Y, and Lindberg M. Transepidermal water loss in dry and clinically normal skin in patients with atopic dermatitis. Acta Derm Venereol. (1985) 65:102–5.
30. Palmer CNA, Irvine AD, Terron-Kwiatkowski A, Zhao Y, Liao H, Lee SP, et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet. (2006) 38:441–6. doi: 10.1038/ng1767
31. Henry J, Toulza E, Hsu C-Y, Pellerin L, Balica S, Mazereeuw-Hautier J, et al. Update on the epidermal differentiation complex. Front Biosci. (2012) 17:1517–32. doi: 10.2741/4001
32. Kim BE, and Leung DYM. Significance of skin barrier dysfunction in atopic dermatitis. Allergy Asthma Immunol Res. (2018) 10:207–15. doi: 10.4168/aair.2018.10.3.207
33. Hill DA, and Spergel JM. The atopic march: critical evidence and clinical relevance. Ann Allergy Asthma Immunol. (2018) 120:131–7. doi: 10.1016/j.anai.2017.10.037
34. Thoo L, Noti M, and Krebs P. Keep calm: the intestinal barrier at the interface of peace and war. Cell Death Dis. (2019) 10:849. doi: 10.1038/s41419-019-2086-z
35. Aghajanian A, Wittchen ES, Allingham MJ, Garrett TA, and Burridge K. Endothelial cell junctions and the regulation of vascular permeability and leukocyte transmigration. J Thromb Haemost. (2008) 6:1453–60. doi: 10.1111/j.1538-7836.2008.03087.x
36. Chen GY, and Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. (2010) 10:826–37. doi: 10.1038/nri2873
37. Jury CS, McHenry P, Burden AD, Lever R, and Bilsland D. Narrowband ultraviolet B (UVB) phototherapy in children. Clin Exp Dermatol. (2006) 31:196–9. doi: 10.1111/j.1365-2230.2006.02061.x
38. Sidbury R, Davis DM, Cohen DE, Cordoro KM, Berger TG, Bergman JN, et al. Guidelines of care for the management of atopic dermatitis: section 3. Management and treatment with phototherapy and systemic agents. J Am Acad Dermatol. (2014) 71:327–49. doi: 10.1016/j.jaad.2014.03.030
39. Hernández-Martín A, Noguera-Morel L, Bernardino-Cuesta B, Torrelo A, Pérez-Martin MA, Aparicio-López C, et al. Cyclosporine A for severe atopic dermatitis in children. efficacy and safety in a retrospective study of 63 patients. J Eur Acad Dermatol Venereol. (2017) 31:837–42. doi: 10.1111/jdv.14066
40. Cork MJ, Thaçi D, Eichenfield LF, Arkwright PD, Hultsch T, Davis JD, et al. Dupilumab in adolescents with uncontrolled moderate-to-severe atopic dermatitis: results from a phase IIa open-label trial and subsequent phase III open-label extension. Br J Dermatol. (2020) 182:85–96. doi: 10.1111/bjd.18476
41. Sach TH, McManus E, and Levell NJ. Understanding economic evidence for the prevention and treatment of atopic eczema. Br J Dermatol. (2019) 181:707–16. doi: 10.1111/bjd.17696
42. McCracken JL, Veeranki SP, Ameredes BT, and Calhoun WJ. Diagnosis and management of asthma in adults: a review. JAMA. (2017) 318:279–90. doi: 10.1001/jama.2017.8372
43. Papi A, Brightling C, Pedersen SE, and Reddel HK. Asthma. Lancet. (2018) 391:783–800. doi: 10.1016/S0140-6736(17)33311-1
44. Faffe DS. Asthma: where is it going? Braz J Med Biol Res. (2008) 41:739–49. doi: 10.1590/S0100-879X2008005000031
45. Quirt J, Hildebrand KJ, Mazza J, Noya F, Kim H. Asthma. Allergy Asthma Clin Immunol. (2019) 14:50. doi: 10.1186/s13223-018-0279-0
46. Miettinen PJ, Ebner R, Lopez AR, and Derynck R. TGF-beta induced transdifferentiation of mammary epithelial cells to mesenchymal cells: involvement of type I receptors. J Cell Biol. (1994) 127:2021–36. doi: 10.1083/jcb.127.6.2021
47. Xu J, Lamouille S, and Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. (2009) 19:156–72. doi: 10.1038/cr.2009.5
48. Redington AE, Madden J, Frew AJ, Djukanovic R, Roche WR, Holgate ST, et al. Transforming growth factor-beta 1 in asthma. Measurement in bronchoalveolar lavage fluid. Am J Respir Crit Care Med. (1997) 156:642–7. doi: 10.1164/ajrccm.156.2.9605065
49. Holgate ST, Davies DE, Lackie PM, Wilson SJ, Puddicombe SM, and Lordan JL. Epithelial-mesenchymal interactions in the pathogenesis of asthma. J Allergy Clin Immunol. (2000) 105:193–204. doi: 10.1016/S0091-6749(00)90066-6
50. Hackett TL, Warner SM, Stefanowicz D, Shaheen F, Pechkovsky DV, Murray LA, et al. Induction of epithelial-mesenchymal transition in primary airway epithelial cells from patients with asthma by transforming growth factor-beta1. Am J Respir Crit Care Med. (2009) 180:122–33. doi: 10.1164/rccm.200811-1730OC
51. Heijink IH, Postma DS, Noordhoek JA, Broekema M, and Kapus A. House dust mite-promoted epithelial-to-mesenchymal transition in human bronchial epithelium. Am J Respir Cell Mol Biol. (2010) 42:69–79. doi: 10.1165/rcmb.2008-0449OC
52. Zou Y, Song W, Zhou L, Mao Y, and Hong W. House dust mite induces sonic hedgehog signaling that mediates epithelial-mesenchymal transition in human bronchial epithelial cells. Mol Med Rep. (2019) 20:4674–82. doi: 10.3892/mmr.2019.10707
53. Lin CH, Wang WC, and Kao SH. Der p 2 promotes motility of airway epithelial cell attributing to AKT/GSK3β-associated epithelial-to-mesenchymal transition. Mol Cell Biochem. (2014) 395:135–43. doi: 10.1007/s11010-014-2119-y
54. Jose CC, Jagannathan L, Tanwar VS, Zhang X, Zang C, and Cuddapah S. Nickel exposure induces persistent mesenchymal phenotype in human lung epithelial cells through epigenetic activation of ZEB1. Mol Carcinog. (2018) 57:794–806. doi: 10.1002/mc.22802
55. Johnson JR, Roos A, Berg T, Nord M, and Fuxe J. Chronic respiratory aeroallergen exposure in mice induces epithelial-mesenchymal transition in the large airways. PLoS ONE. 6:e16175. doi: 10.1371/journal.pone.0016175
56. Thevenot PT, Saravia J, Jin N, Giaimo JD, Chustz RE, Mahne S, et al. Radical-containing ultrafine particulate matter initiates epithelial-to-mesenchymal transitions in airway epithelial cells. Am J Respir Cell Mol Biol. (2013) 48:188–97. doi: 10.1165/rcmb.2012-0052OC
57. Tian B, Hosoki K, Liu Z, Yang J, Zhao Y, Sun H, et al. Mucosal bromodomain-containing protein 4 mediates aeroallergen-induced inflammation and remodeling. J Allergy Clin Immunol. (2019) 143:1380–1394.e9. doi: 10.1016/j.jaci.2018.09.029
58. Witten ML, Chau B, Sáez E, Boitano S, and Clark Lantz R. Early life inhalation exposure to mine tailings dust affects lung development. Toxicol Appl Pharmacol. (2019) 365:124–32. doi: 10.1016/j.taap.2019.01.009
59. Carroll WW, O'Connell BP, Schlosser RJ, Gudis DA, Karnezis TT, Lawrence LA, et al. Fibroblast levels are increased in chronic rhinosinusitis with nasal polyps and are associated with worse subjective disease severity. Int Forum Allergy Rhinol. (2016) 6:162–8. doi: 10.1002/alr.21636
60. Yang ZC, Yi MJ, Ran N, Wang C, Fu P, Feng XY, et al. Transforming growth factor-β1 induces bronchial epithelial cells to mesenchymal transition by activating the snail pathway and promotes airway remodeling in asthma. Mol Med Rep. (2013) 8:1663–8. doi: 10.3892/mmr.2013.1728
61. Doerner AM, and Zuraw BL. TGF-beta1 induced epithelial to mesenchymal transition (EMT) in human bronchial epithelial cells is enhanced by IL-1beta but not abrogated by corticosteroids. Respir Res. (2009) 10:100. doi: 10.1186/1465-9921-10-100
62. Ji X, Li J, Xu L, Wang W, Luo M, Luo S, et al. IL4 and IL-17A provide a Th2/Th17-polarized inflammatory milieu in favor of TGF-β1 to induce bronchial epithelial-mesenchymal transition (EMT). Int J Clin Exp Pathol. (2013) 6:1481–92.
63. Johnson JR, Nishioka M, Chakir J, Risse PA, Almaghlouth I, Bazarbashi AN, et al. IL-22 contributes to TGF-β1-mediated epithelial-mesenchymal transition in asthmatic bronchial epithelial cells. Respir Res. (2013) 14:118. doi: 10.1186/1465-9921-14-118
64. Cai LM, Zhou YQ, Yang LF, Qu JX, Dai ZY, Li HT, et al. Thymic stromal lymphopoietin induced early stage of epithelial-mesenchymal transition in human bronchial epithelial cells through upregulation of transforming growth factor beta 1. Exp Lung Res. (2019) 45:221–35. doi: 10.1080/01902148.2019.1646841
65. Yasukawa A, Hosoki K, Toda M, Miyake Y, Matsushima Y, Matsumoto T, et al. Eosinophils promote epithelial to mesenchymal transition of bronchial epithelial cells. PLoS ONE. (2013) 8:e64281. doi: 10.1371/journal.pone.0064281
66. Haddad A, Gaudet M, Plesa M, Allakhverdi Z, Mogas AK, Audusseau S, et al. Neutrophils from severe asthmatic patients induce epithelial to mesenchymal transition in healthy bronchial epithelial cells. Respir Res. (2019) 20:234. doi: 10.1186/s12931-019-1186-8
67. Câmara J, Jarai G. Epithelial-mesenchymal transition in primary human bronchial epithelial cells is Smad-dependent and enhanced by fibronectin and TNF-alpha. Fibrogenesis Tissue Repair. (2010) 3:2. doi: 10.1186/1755-1536-3-2
68. Mikami Y, Yamauchi Y, Horie M, Kase M, Jo T, Takizawa H, et al. Tumor necrosis factor superfamily member LIGHT induces epithelial-mesenchymal transition in A549 human alveolar epithelial cells. Biochem Biophys Res Commun. (2012) 428:451–7. doi: 10.1016/j.bbrc.2012.10.097
69. Hung JY, Chiang SR, Tsai MJ, Tsai YM, Chong IW, Shieh JM, et al. LIGHT is a crucial mediator of airway remodeling. J Cell Physiol. (2015) 230:1042–53. doi: 10.1002/jcp.24832
70. Itoigawa Y, Harada N, Harada S, Katsura Y, Makino F, Ito J, et al. TWEAK enhances TGF-β-induced epithelial-mesenchymal transition in human bronchial epithelial cells. Respir Res. (2015) 16:48. doi: 10.1186/s12931-015-0207-5
71. Matsuno K, Harada N, Harada S, Takeshige T, Ishimori A, Itoigawa Y, et al. Combination of TWEAK and TGF-β1 induces the production of TSLP, RANTES, and TARC in BEAS-2B human bronchial epithelial cells during epithelial-mesenchymal transition. Exp Lung Res. (2018) 44:332–43. doi: 10.1080/01902148.2018.1522558
72. Könnecke M, Burmeister M, Pries R, Böscke R, Bruchhage KL, Ungefroren H, et al. Epithelial-mesenchymal transition in chronic rhinosinusitis: differences revealed between epithelial cells from nasal polyps and inferior turbinates. Arch Immunol Ther Exp. (2017) 65:157–73. doi: 10.1007/s00005-016-0409-7
73. Watelet JB, Claeys C, Perez-Novo C, Gevaert P, Van Cauwenberge P, and Bachert C. Transforming growth factor beta1 in nasal remodeling: differences between chronic rhinosinusitis and nasal polyposis. Am J Rhinol. (2004) 18:267–72. doi: 10.1177/194589240401800502
74. Li X, Li C, Zhu G, Yuan W, and Xiao ZA. TGF-β1 induces epithelial-mesenchymal transition of chronic sinusitis with nasal polyps through microRNA-21. Int Arch Allergy Immunol. (2019) 179:304–19. doi: 10.1159/000497829
75. Park I-H, Kang JH, Shin JM, and Lee HM. Trichostatin A inhibits epithelial mesenchymal transition induced by TGF-β1 in airway epithelium. PLoS ONE. (2016) 11:e0162058. doi: 10.1371/journal.pone.0162058
76. Shin HW, Cho K, Kim DW, Han DH, Khalmuratova R, Kim SW, et al. Hypoxia-inducible factor 1 mediates nasal polypogenesis by inducing epithelial-to-mesenchymal transition. Am J Respir Crit Care Med. (2012) 185:944–54. doi: 10.1164/rccm.201109-1706OC
77. Lee M, Kim DW, Yoon H, So D, Khalmuratova R, Rhee CS, et al. Sirtuin 1 attenuates nasal polypogenesis by suppressing epithelial-to-mesenchymal transition. J Allergy Clin Immunol. (2016) 137:87–98.e7. doi: 10.1016/j.jaci.2015.07.026
78. Do NY, Shin HJ, and Lee JE. Wheatgrass extract inhibits hypoxia-inducible factor-1-mediated epithelial-mesenchymal transition in A549 cells. Nutr Res Pract. (2017) 11:83–9. doi: 10.4162/nrp.2017.11.2.83
79. Lee M, Kim DW, Khalmuratova R, Shin SH, Kim YM, Han DH, et al. The IFN-γ-p38, ERK kinase axis exacerbates neutrophilic chronic rhinosinusitis by inducing the epithelial-to-mesenchymal transition. Mucosal Immunol. (2019) 12:601–11. doi: 10.1038/s41385-019-0149-1
80. Zhong Y, Li Y, and Zhang H. Silencing TBX1 exerts suppressive effects on epithelial-mesenchymal transition and inflammation of chronic rhinosinusitis through inhibition of the TGFβ-Smad2/3 signaling pathway in mice. Am J Rhinol Allergy. (2020) 34:16–25. doi: 10.1177/1945892419866543
81. Taniguchi K, Arima K, Masuoka M, Ohta S, Shiraishi H, Ontsuka K, et al. Periostin controls keratinocyte proliferation and differentiation by interacting with the paracrine IL-1α/IL-6 loop. J Invest Dermatol. (2014) 134:1295–304. doi: 10.1038/jid.2013.500
82. Park J-A, Mitchel JA, Qazvini NT, Kim JH, Park CY, Butler JP, et al. Compressive stress causes an unjamming transition and an epithelial-mesenchymal transition in the airway epithelium in asthma. Ann Am Thorac Soc. (2016) 13 (Suppl. 1):S102. doi: 10.1513/AnnalsATS.201506-382MG
83. Pacholewska A, Kraft MF, Gerber V, and Jagannathan V. Differential expression of serum micrornas supports CD4+ T Cell Differentiation into Th2/Th17 cells in severe equine asthma. Genes. (2017) 8:383. doi: 10.3390/genes8120383
84. Wang J, Li F, Yang M, Wu J, Zhao J, Gong W, et al. FIZZ1 promotes airway remodeling through the PI3K/Akt signaling pathway in asthma. Exp Ther Med. (2014) 7:1265–70. doi: 10.3892/etm.2014.1580
85. Yang ZC, Qu ZH, Yi MJ, Shan YC, Ran N, Xu L, et al. MiR-448-5p inhibits TGF-β1-induced epithelial-mesenchymal transition and pulmonary fibrosis by targeting Six1 in asthma. J Cell Physiol. (2019) 234:8804–14. doi: 10.1002/jcp.27540
86. Wang T, Zhou Q, and Shang Y. MiRNA-451a inhibits airway remodeling by targeting cadherin 11 in an allergic asthma model of neonatal mice. Int Immunopharmacol. (2020) 83:106440. doi: 10.1016/j.intimp.2020.106440
87. Sun Y, Shi Z, Liu B, Li X, Li G, Yang F, et al. YKL-40 mediates airway remodeling in asthma via activating FAK and MAPK signaling pathway. Cell Cycle. (2020) 19:1378–90. doi: 10.1080/15384101.2020.1750811
88. Volckaert T, Dill E, Campbell A, Tiozzo C, Majka S, Bellusci S, et al. Parabronchial smooth muscle constitutes an airway epithelial stem cell niche in the mouse lung after injury. J Clin Invest. (2011) 121:4409–19. doi: 10.1172/JCI58097
89. Dobzanski A, Khalil SM, and Lane AP. Nasal polyp fibroblasts modulate epithelial characteristics via Wnt signaling. Int Forum Allergy Rhinol. (2018) 8:1412–20. doi: 10.1002/alr.22199
90. Vetuschi A, Pompili S, Di Marco GP, Calvaruso F, Iacomino E, Angelosante L, et al. Can the AGE/RAGE/ERK signalling pathway and the epithelial-to-mesenchymal transition interact in the pathogenesis of chronic rhinosinusitis with nasal polyps? Eur J Histochem. (2020) 64:3079. doi: 10.4081/ejh.2020.3079
91. Hupin C, Gohy S, Bouzin C, Lecocq M, Polette M, and Pilette C. Features of mesenchymal transition in the airway epithelium from chronic rhinosinusitis. Allergy. (2014) 69:1540–9. doi: 10.1111/all.12503
92. Meng J, Zhou P, Liu Y, Liu F, Yi X, Liu S, et al. The development of nasal polyp disease involves early nasal mucosal inflammation and remodelling. PLoS ONE. (2013) 8:e82373. doi: 10.1371/journal.pone.0082373
93. Li H, Liu Q, Wang H, Sun XC, Yu HP, Hu L, et al. Epithelial-mesenchymal transition in chronic rhinosinusitis (CRS) and the prognostic value of α-SMA in postoperative outcomes of patients with CRS. Mol Med Rep. (2019) 20:2441–9. doi: 10.3892/mmr.2019.10461
94. Yang HW, Lee SA, Shin JM, Park IH, and Lee HM. Glucocorticoids ameliorate TGF-β1-mediated epithelial-to-mesenchymal transition of airway epithelium through MAPK and snail/slug signaling pathways. Sci Rep. (2017) 7:3486. doi: 10.1038/s41598-017-02358-z
95. Xu L, Xiang X, Ji X, Wang W, Luo M, Luo S, et al. Effects and mechanism of dehydroepiandrosterone on epithelial-mesenchymal transition in bronchial epithelial cells. Exp Lung Res. (2014) 40:211–21. doi: 10.3109/01902148.2013.879966
96. Kao HF, Chang-Chien PW, Chang WT, Yeh TM, and Wang JY. Propolis inhibits TGF-β1-induced epithelial-mesenchymal transition in human alveolar epithelial cells via PPARγ activation. Int Immunopharmacol. (2013) 15:565–74. doi: 10.1016/j.intimp.2012.12.018
97. Hosoki K, Kainuma K, Toda M, Harada E, Chelakkot-Govindalayathila AL, Roeen Z, et al. Montelukast suppresses epithelial to mesenchymal transition of bronchial epithelial cells induced by eosinophils. Biochem Biophys Res Commun. (2014) 449:351–6. doi: 10.1016/j.bbrc.2014.05.033
98. Fischer KD, and Agrawal DK. Vitamin D regulating TGF-β induced epithelial-mesenchymal transition. Respir Res. (2014) 15:146. doi: 10.1186/s12931-014-0146-6
99. Ge A, Ma Y, Liu YN, Li YS, Gu H, Zhang JX, et al. Diosmetin prevents TGF-β1-induced epithelial-mesenchymal transition via ROS/MAPK signaling pathways. Life Sci. (2016) 153:1–8. doi: 10.1016/j.lfs.2016.04.023
100. Kainuma K, Kobayashi T, D'Alessandro-Gabazza CN, Toda M, Yasuma T, Nishihama K, et al. β2 adrenergic agonist suppresses eosinophil-induced epithelial-to-mesenchymal transition of bronchial epithelial cells. Respir Res. (2017) 18:79. doi: 10.1186/s12931-017-0563-4
101. Gong JH, Cho IH, Shin D, Han SY, Park SH, and Kang YH. Inhibition of airway epithelial-to-mesenchymal transition and fibrosis by kaempferol in endotoxin-induced epithelial cells and ovalbumin-sensitized mice. Lab Invest. (2014) 94:297–308. doi: 10.1038/labinvest.2013.137
102. Yurt M, Liu J, Sakurai R, Gong M, Husain SM, Siddiqui MA, et al. Vitamin D supplementation blocks pulmonary structural and functional changes in a rat model of perinatal vitamin D deficiency. Am J Physiol Lung Cell Mol Physiol. (2014) 307:L859–67. doi: 10.1152/ajplung.00032.2014
103. Fischer KD, Hall SC, and Agrawal DK. Vitamin D supplementation reduces induction of epithelial-mesenchymal transition in allergen sensitized and challenged mice. PLoS ONE. (2016) 11:e0149180. doi: 10.1371/journal.pone.0149180
104. Song L, Sen S, Sun Y, Zhou J, Mo L, and He Y. Ketamine inhalation ameliorates ovalbumin-induced murine asthma by suppressing the epithelial-mesenchymal transition. Med Sci Monit. (2016) 22:2471–83. doi: 10.12659/MSM.899955
105. Lee HY, Kim IK, Yoon HK, Kwon SS, Rhee CK, and Lee SY. Inhibitory effects of resveratrol on airway remodeling by transforming growth factor-β/Smad signaling pathway in chronic asthma model. Allergy Asthma Immunol Res. (2017) 9:25–34. doi: 10.4168/aair.2017.9.1.25
106. Tian X, Tian X, Huo R, Chang Q, Zheng G, Du Y, et al. Bacillus calmette-guerin alleviates airway inflammation and remodeling by preventing TGF-β1 induced epithelial-mesenchymal transition. Hum Vaccin Immunother. (2017) 13:1758–64. doi: 10.1080/21645515.2017.1313366
107. Pu Y, Liu Y, Liao S, Miao S, Zhou L, and Wan L. Azithromycin ameliorates OVA-induced airway remodeling in Balb/c mice via suppression of epithelial-to-mesenchymal transition. Int Immunopharmacol. (2018) 58:87–93. doi: 10.1016/j.intimp.2018.03.016
108. Lin C-C, Chen WJ, Liaw SF, Lin MW, and Lin SC. Effects of aminophylline on airway epithelial-mesenchymal transition in brown Norway rats after repeated allergen challenge. Exp Lung Res. (2019) 45:42–53. doi: 10.1080/01902148.2019.1610116
109. Chu S, Zhang X, Sun Y, Liang Y, Sun J, Lu M, et al. Atrial natriuretic peptide inhibits epithelial-mesenchymal transition (EMT) of bronchial epithelial cells through cGMP/PKG signaling by targeting Smad3 in a murine model of allergic asthma. Exp. Lung. Res. (2019) 45:245–54. doi: 10.1080/01902148.2019.1660734
110. Yan B, Wang Y, Li Y, Wang C, and Zhang L. Inhibition of arachidonate 15-lipoxygenase reduces the epithelial-mesenchymal transition in eosinophilic chronic rhinosinusitis with nasal polyps. Int Forum Allergy Rhinol. (2019) 9:270–80. doi: 10.1002/alr.22243
111. Yang P, Chen S, Zhong G, Kong W, and Wang Y. Agonist of PPAR-γ reduced epithelial-mesenchymal transition in eosinophilic chronic rhinosinusitis with nasal polyps via inhibition of high mobility group box1. Int J Med Sci. (2019) 16:1631–41. doi: 10.7150/ijms.35936
112. Kim Y, Hwang S, Khalmuratova R, Kang S, Lee M, Song Y, et al. α-Helical cell-penetrating peptide-mediated nasal delivery of resveratrol for inhibition of epithelial-to-mesenchymal transition. J Control Release. (2020) 317:181–94. doi: 10.1016/j.jconrel.2019.11.034
113. Myles IA, Castillo CR, Barbian KD, Kanakabandi K, Virtaneva K, Fitzmeyer E, et al. Therapeutic responses to roseomonas mucosa for atopic dermatitis involve lipid mediated TNF-related epithelial repair. Sci Transl Med. (2020) 12:eaaz8631. doi: 10.1126/scitranslmed.aaz8631
114. Kim Y, Kim K, Lee H, Han S, Lee Y-S, Choe J, et al. Celastrol binds to ERK and inhibits FcepsilonRI signaling to exert an anti-allergic effect. Eur J Pharmacol. (2009) 612:131–42. doi: 10.1016/j.ejphar.2009.03.071
115. Knight DA, Grainge CL, Stick SM, Kicic A, and Schuliga M. Epithelial mesenchymal transition in respiratory disease: fact or fiction. Chest. (2020) 157:1591–6. doi: 10.1016/j.chest.2019.12.014
116. Bartis D, Mise N, Mahida RY, Eickelberg O, and Thickett DR. Epithelial-mesenchymal transition in lung development and disease: does it exist and is it important? Thorax. (2014) 69:760–5. doi: 10.1136/thoraxjnl-2013-204608
117. Small P, Keith PK, and Kim H. Allergic rhinitis. Allergy Asthma Clin Immunol. (2018) 14 (Suppl. 2):51. doi: 10.1186/s13223-018-0280-7
118. Dykewicz MS, and Hamilos DL. Rhinitis and sinusitis. J Allergy Clin Immunol. (2010) 125 (2 Suppl 2):S103–15. doi: 10.1016/j.jaci.2009.12.989
119. Cingi C, Gevaert P, Mösges R, Rondon C, Hox V, Rudenko M, et al. Multi-morbidities of allergic rhinitis in adults: European academy of allergy and clinical immunology task force report. Clin Transl Allergy. (2017) 7:17. doi: 10.1186/s13601-017-0153-z
120. Marcus S, DelGaudio JM, Roland LT, and Wise SK. Chronic rhinosinusitis: does allergy play a role? Med Sci. (2019) 7:30. doi: 10.3390/medsci7020030
121. Van Zele T, Claeys S, Gevaert P, Van Maele G, Holtappels G, Van Cauwenberge P, et al. Differentiation of chronic sinus diseases by measurement of inflammatory mediators. Allergy. (2006) 61:1280–9. doi: 10.1111/j.1398-9995.2006.01225.x
122. Tan BK, Klingler AI, Poposki JA, Stevens WW, Peters AT, Suh LA, et al. Heterogeneous inflammatory patterns in chronic rhinosinusitis without nasal polyps in Chicago, Illinois. J Allergy Clin Immunol. (2017) 139:699–703.e7. doi: 10.1016/j.jaci.2016.06.063
123. Nagarkar DR, Poposki JA, Tan BK, Comeau MR, Peters AT, Hulse KE, et al. Thymic stromal lymphopoietin activity is increased in nasal polyps of patients with chronic rhinosinusitis. J Allergy Clin Immunol. (2013) 132:593–600.e12. doi: 10.1016/j.jaci.2013.04.005
124. Van Bruaene N, Pérez-Novo CA, Basinski TM, Van Zele T, Holtappels G, De Ruyck N, et al. T-cell regulation in chronic paranasal sinus disease. J Allergy Clin Immunol. (2008) 121:1435–41.e1. doi: 10.1016/j.jaci.2008.02.018
125. Shu DY, Ong K, and Lovicu FJ. Histopathology of subcapsular cataract in a patient with atopic dermatitis. Optom Vis Sci. (2017) 94:270–6. doi: 10.1097/OPX.0000000000001011
126. Kagalwalla AF, Akhtar N, Woodruff SA, Rea BA, Masterson JC, Mukkada V, et al. Eosinophilic esophagitis: epithelial mesenchymal transition contributes to esophageal remodeling and reverses with treatment. J Allergy Clin Immunol. (2012) 129:1387–96.e7. doi: 10.1016/j.jaci.2012.03.005
127. Gann PH, Deaton RJ, McMahon N, Collins MH, Dellon ES, Hirano I, et al. An anti-IL-13 antibody reverses epithelial-mesenchymal transition biomarkers in eosinophilic esophagitis: phase 2 trial results. J Allergy Clin Immunol. (2020) 146:367–76.e3. doi: 10.1016/j.jaci.2020.03.045
128. Robson J, O'Gorman M, McClain A, Mutyala K, Davis C, Barbagelata C, et al. Incidence and prevalence of pediatric eosinophilic esophagitis in utah based on a 5-year population-based study. Clin Gastroenterol Hepatol. (2019) 17:107–14.e1. doi: 10.1016/j.cgh.2018.06.028
129. Myles IA, Earland NJ, Anderson ED, Moore IN, Kieh MD, Williams KW, et al. First-in-human topical microbiome transplantation with Roseomonas mucosa for atopic dermatitis. JCI Insight. (2018) 3:e120608. doi: 10.1172/jci.insight.120608
130. Myles IA, Anderson ED, Earland NJ, Zarember KA, Sastalla I, Williams KW, et al. TNF overproduction impairs epithelial staphylococcal response in hyper IgE syndrome. J Clin Invest. (2018) 128:3595–604. doi: 10.1172/JCI121486
131. Chen PC, Lee WY, Ling HH, Cheng CH, Chen KC, and Lin CW. Activation of fibroblasts by nicotine promotes the epithelial-mesenchymal transition and motility of breast cancer cells. J Cell Physiol. (2018) 233:4972–80. doi: 10.1002/jcp.26334
132. Sampaio JL, Gerl MJ, Klose C, Ejsing CS, Beug H, Simons K, et al. Membrane lipidome of an epithelial cell line. Proc Natl Acad Sci USA. (2011) 108:1903–7. doi: 10.1073/pnas.1019267108
133. Fukaya M, Sato K, Sato M, Kimata H, Fukisawa S, Dozono H, et al. Topical steroid addiction in atopic dermatitis. Drug Healthc Patient Saf. (2014) 6:131–8. doi: 10.2147/DHPS.S69201
134. Llyod-Lavery A, Rodgers NK, Davies E, Grindlay DJC, and Thomas KS. What's new in atopic eczema? An analysis of systemic reviews published in 2015. Part 2: prevention and treatment. Clin Exp Dermatol. (2018) 43:653–8. doi: 10.1111/ced.13554
Keywords: epithelial-mesechymal transition, atopy, atopic dermatitis, asthma, allergic rhinitis, tissue repair
Citation: Anderson ED, Alishahedani ME and Myles IA (2020) Epithelial-Mesenchymal Transition in Atopy: A Mini-Review. Front. Allergy 1:628381. doi: 10.3389/falgy.2020.628381
Received: 11 November 2020; Accepted: 01 December 2020;
Published: 18 December 2020.
Edited by:
Oscar Palomares, Complutense University of Madrid, SpainReviewed by:
Helene Salvator, Hôpital Foch, FranceCristina Benito-Villalvilla, Complutense University of Madrid, Spain
Copyright © 2020 Anderson, Alishahedani and Myles. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ian A. Myles, bXlsZXNpQG5pYWlkLm5paC5nb3Y=