 Faidra Parsopoulou1
Faidra Parsopoulou1 Gedeon Loules2Maria Zamanakou2
Gedeon Loules2Maria Zamanakou2 Dorottya Csuka3
Dorottya Csuka3 Agnes Szilagyi3Maria Kompoti1Grzegorz Porebski4
Agnes Szilagyi3Maria Kompoti1Grzegorz Porebski4 Fotis Psarros5
Fotis Psarros5 Markus Magerl6,7
Markus Magerl6,7 Anna Valerieva8Maria Staevska8Krystyna Obtulowicz4
Anna Valerieva8Maria Staevska8Krystyna Obtulowicz4 Marcus Maurer6,7
Marcus Maurer6,7 Matthaios Speletas1
Matthaios Speletas1 Henriette Farkas3
Henriette Farkas3 Anastasios E. Germenis1,2*
Anastasios E. Germenis1,2*- 1Department of Immunology and Histocompatibility, Faculty of Medicine, School of Health Sciences, University of Thessaly, Larissa, Greece
- 2CeMIA SA, Larissa, Greece
- 3Department of Internal Medicine and Haematology, Hungarian Angioedema Center of Reference and Excellence, Semmelweis University, Budapest, Hungary
- 4Department of Clinical and Environmental Allergology, Jagiellonian University Medical College, Krakow, Poland
- 5Department of Allergology, Navy Hospital, Athens, Greece
- 6Institute of Allergology, Charité – Universitätsmedizin Berlin, Freie Universität Berlin and Humboldt-Universität zu Berlin, Berlin, Germany
- 7Fraunhofer Institute for Translational Medicine and Pharmacology ITMP, Allergology and Immunology, Berlin, Germany
- 8Department of Allergology, Clinic of Allergology, University Hospital “Alexandrovska”, Medical University of Sofia, Sofia, Bulgaria
Existing evidence indicates that modifier genes could change the phenotypic outcome of the causal SERPING1 variant and thus explain the expression variability of hereditary angioedema due to C1-inhibitor deficiency (C1-INH-HAE). To further examine this hypothesis, we investigated the presence or absence of 18 functional variants of genes encoding proteins involved in the metabolism and function of bradykinin, the main mediator of C1-INH-HAE attacks, in relation to three distinct phenotypic traits of patients with C1-INH-HAE, i.e., the age at disease onset, the need for long-term prophylaxis (LTP), and the severity of the disease. Genetic analyses were performed by a validated next-generation sequencing platform. In total, 233 patients with C1-INH-HAE from 144 unrelated families from five European countries were enrolled in the study. Already described correlations between five common functional variants [F12-rs1801020, KLKB1-rs3733402, CPN1-rs61751507, and two in SERPING1 (rs4926 and rs28362944)] and C1-INH-HAE severity were confirmed. Furthermore, significant correlations were found between either the age at disease onset, the LTP, or the severity score of the disease and a series of other functional variants (F13B-rs6003, PLAU-rs2227564, SERPINA1-rs28929474, SERPINA1-rs17580, KLK1-rs5515, SERPINE1-rs6092, and F2-rs1799963). Interestingly, correlations uncovered in the entire cohort of patients were different from those discovered in the cohort of patients carrying missense causal SERPING1 variants. Our findings indicate that variants other than the SERPING1 causal variants act as independent modifiers of C1-INH-HAE severity and could be tested as possible prognostic biomarkers.
Introduction
Hereditary angioedema due to C1-inhibitor deficiency (C1-INH-HAE), an autosomal dominant disorder with recurrent attacks of edema spontaneously developing in any body location, is characterized by a large heterogeneity in its clinical expression, including the age at disease onset, the number and triggers of attacks, the severity and localization of edema, and prodromal signs and symptoms (1, 2). These features show variable expressivity, i.e., it may vary even among members of the same family carrying the same SERPING1 causal mutation, which, at present, is unpredictable and largely unexplained.
Accumulating evidence indicates that modifier genes could change the phenotypic outcome of the variant at the primary mutation in the target SERPING1 gene, and thus explain disease expression variability (3). Understanding genetic modification phenomena will obviously improve our ability to better manage the disabling and potentially fatal manifestations of C1-INH-HAE. To this aim, we investigated here the relationship between parameters associated with disease course severity and common functional variants in genes involved in the metabolism of bradykinin, the main mediator of angioedema attacks in patients with C1-INH-HAE.
Patients and Methods
This study included 233 patients (104 men and 129 women, mean age 40 years, range 2.5–85) with C1-INH-HAE (217 type I, 16 type II) from 144 unrelated families (16 Bulgarian, 23 German, 30 Greek, 43 Hungarian, and 32 Polish). All patients were previously genotyped for SERPING1 mutations. Demographic, clinical, and molecular data of the patients are presented in Table 1. Patients' medical records were reviewed and data regarding the age at disease onset and the long-term prophylaxis (LTP) were recorded. For patients not receiving long-term prophylactic treatment, the severity score Cutaneous Abdominal Laryngeal Score (CALS) was calculated according to the equation “CALS=1*Cutaneous+2*Abdominal+3*Laryngeal last year attacks”.
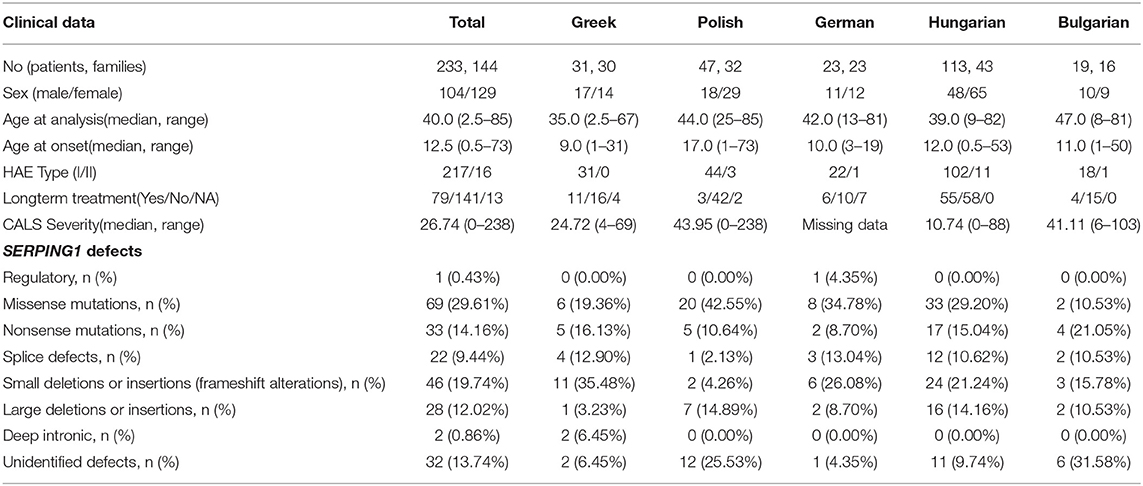
Table 1. Demographic, clinical, and molecular data of the C1-INH-HAE patients.
DNA samples were analyzed in a validated next-generation sequencing (NGS) platform (Ampliseq custom panel, Thermo Scientific, Waltham MA, USA), as previously described (4, 5). Briefly, DNA libraries were constructed for each sample using the Ion AmpliSeq Library Kit 2.0 (Thermo Scientific) and indexed with a unique adapter using the Ion Xpress barcode adapter kit (Thermo Scientific). Template preparation, enrichment, and chip loading were carried out on the Ion Chef System (Thermo Scientific). Sequencing was performed on S5XL on 520 and 530 chips, using the Ion 510, Ion 520, and Ion 530 Kit-Chef (Thermo Scientific). All procedures were performed according to the manufacturer's instructions. Base calling, demultiplexing, and alignment to the hg19 reference genome (GRCh37) of the raw sequencing data were performed in Torrent Suite 5.10 software (Thermo Scientific, Waltham, MA, USA) using the default parameters. Variant calling was performed by the VariantCaller v.5.8.0.19 plug-in and coverage analysis by the CoverageAnalysis v.5.8.0.8 plug-in in Torrent Suite 5.10. All variants were annotated on Ion Reporter Software (Thermo Scientific).
We investigated the presence or absence of 18 common functional variants (allele frequency ≥1%) in relation to three distinct phenotypic traits of patients, i.e., the age at disease onset, the need for LTP, and the severity of the disease based on the CALS score. The investigated variants were variants of genes encoding proteins involved in the metabolism and function of bradykinin, the main mediator of C1-INH-HAE attacks, which were chosen based on their effect on protein activity, their frequency, and the coverage that could be achieved by the platform (Table 2). The local institutional review boards approved this study, and written informed consent was obtained from each individual or an accompanying relative.
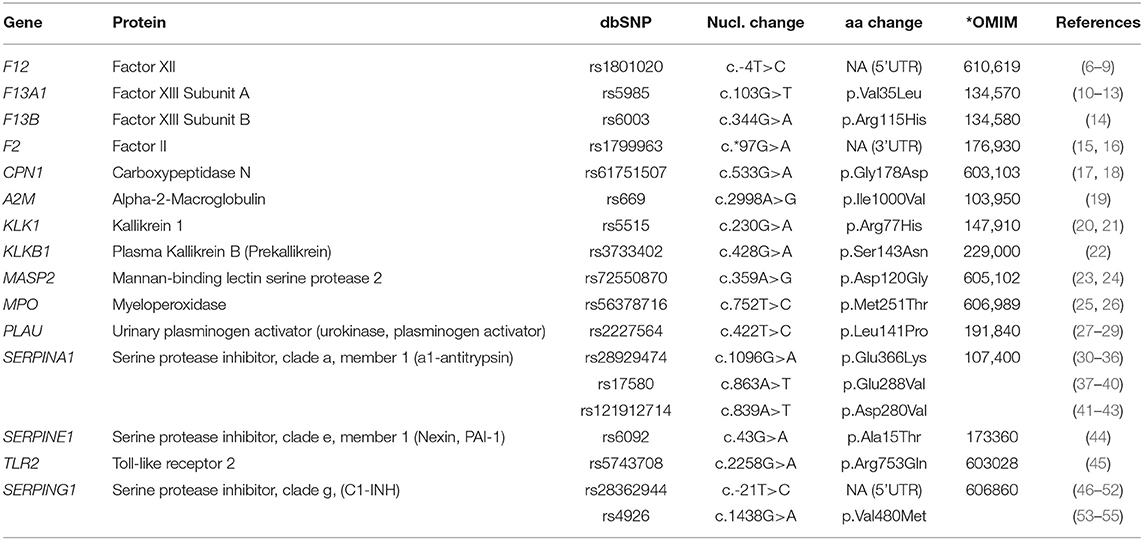
Table 2. Selected common functional variants.
Statistical Analysis
Categorical variables were analyzed with Fisher's exact test. Normality of continuous variables was assessed with Kolmogorov–Smirnov test. Normally distributed data were analyzed with Student's t-test and one-way ANOVA as appropriate. Skewed data were analyzed with nonparametric methods (Mann–Whitney test or Kruskal–Wallis test as appropriate). Given the fact that our patient population consisted of correlated subjects (members of individual families), we implemented generalized estimating equations (GEE), an extension of the generalized linear model that accounts for the within-subject correlation. GEE was used to model the relationship between explanatory variables (polymorphisms, sex, etc.) and response variables (age at disease onset, need for long-term treatment, CALS severity). Age at disease onset and CALS severity score were modeled as continuous variables in linear GEE models and the need for long-term treatment was entered as a binary variable in logistic GEE models. In all GEE models, an unstructured correlation structure was used, and the Quasi-Likelihood Information Criterion (QIC) was used for model selection. Data analysis was performed with SPSS 17.0 (IBM Corporation, NY, 2008). For all analyses, alpha was set at 0.05 (two-tailed). In cases in which multivariable analysis was more appropriate, such analysis was performed with the dependent variable, including the age at disease onset, the LTP or the CALS, and with those of the variants presenting significant associations in univariable analysis fitted as independent variables. This type of analysis was performed for two groups of C1-INH-HAE patients (a) independently of the SERPING1 variant and (b) for patients carrying a missense variant in SERPING1 (n = 69). The common functional variants SERPINA1-rs121912714, SERPINA1-rs28929474, and MPO-rs56378716 were not detected in any patient of the missense group.
Results and Discussion
The allele frequency of the selected SNPs in our cohort did not differ significantly from the Global Allele Frequency (GMAF) and the European Non-Finnish Allele Frequency (ENFMAF) as recorded by GnomAD v2.1.1 (Supplementary Table 1). Similarly, the prevalence of the polymorphisms did not differ significantly between examined patient groups from different countries (Supplementary Table 2).
The correlations found between functional variants and the age at disease onset, the LTP, or the severity score of the disease are summarized in Table 3. Five common functional variants had been previously correlated with C1-INH-HAE severity – F12-rs1801020, KLKB1-rs3733402, CPN1-rs61751507, and two in SERPING1 (rs4926 and rs28362944).
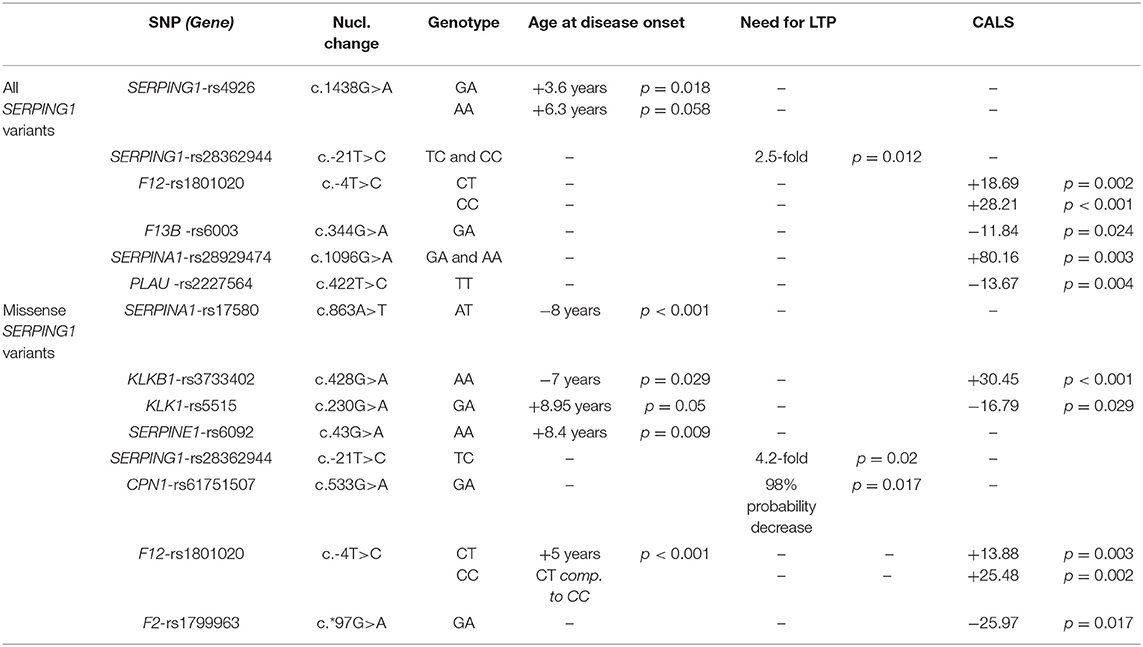
Table 3. Summary of the correlations of common functional variants with patients' phenotype – age at disease onset, need for LTP, CALS severity score.
The presence of the C allele of the F12-rs1801020 (c.-4T>C) was significantly correlated with an increase in disease severity. More precisely, independently of the type of SERPING1 mutations, homozygotes (CC) and heterozygotes of this variant present a mean disease severity score higher by 28.21 (p < 0.001) and 18.69 (p = 0.002) units of the CALS severity score, respectively. Similarly, homozygotes (CC) and heterozygotes (CT) of this variant carrying SERPING1 missense variants present a mean disease severity score higher by 25.48 (p = 0.002) and by 13.88 (p = 0.003) units of the CALS severity score, respectively, compared with CALS score in patients lacking the polymorphism. Our results agree with evidence provided by Bors et al. (6) who suggested that the carriage of the T allele of the F12-rs1801020 variant is independently associated with a less severe C1-INH-HAE clinical phenotype. Moreover, this result is in agreement with Rijavec et al. (7) who have shown that the C allele and the CC genotype were represented more in symptomatic patients, compared to asymptomatic. These authors suggested that carriers of the CC genotype have a 25-fold greater risk of developing the disease compared to those carrying the TT genotype. In our study, F12-rs1801020 displays a robust linear trend among the ordinal categories (homozygosity-heterozygosity-absence), indicating that the T allele provides a protective effect in regards to angioedema severity. Moreover, univariable analysis in patients with missense SERPING1 mutations uncovered a significant 5-year delay at disease onset in heterozygous (TC) compared to homozygous (CC) patients (p < 0.001), a result that agrees with our previous findings (8). These effects could be explained by the findings of Kanaji et al. (9). These authors observed different levels of FXII in plasma, depending on the genotype. Even though both alleles were equally transcribed in hepatocytes of heterozygotes, the cDNA containing the T allele was producing less FXII in vitro than the one containing the C allele. Therefore, the presence of the variant is affecting the efficiency of translation.
In regards to the carriage of the C allele of the SERPING1-rs28362944 (c.-21T>C) variant, the probability of the need for LTP was found increased by 4.2-fold (p = 0.02) and 2.5-fold (p = 0.012) among patients with C1-INH-HAE who were carrying missense SERPING1 mutations and independently of the SERPING1 variation, respectively. The variant had been previously characterized as likely pathogenic when in a homozygous state (49, 50), despite that no correlation between this variant and the biochemical values of C1-INH function or the clinical severity score has been reported by other investigators (51). Interestingly, however, Duponchel et al. (52) have proposed the variant as a modifier of disease severity as they had found that the variant yields low but significant levels of exon 2 skipping in transfected cells. Therefore, this allele may contribute, at the RNA level, to more severe forms of C1-INH-HAE. In accordance, Cumming et al. (54) reported an increased disease penetrance in carriers of c.-21T>C when the variant allele presented in trans with the SERPING1 mutation. Unfortunately, segregation analysis in our cohort could not be performed in many cases due to a lack of available family members.
The SERPING1-rs4926 (c.1438 G>A, p.Val480Met) had been predicted as deleterious and possibly damaging according to bioinformatic tools, because the highly conserved amino acid (Val) is important for the folding of the C1-INH protein into its native conformation (53). However, it is a well-documented common variant and characterized by different groups as benign in public databases. Independently of the SERPING1 causal mutation, a significant 3.6-year (p = 0.018) and a trend toward 6.3-year (p = 0.058) delay at the age of disease onset was found in heterozygous (GA) and homozygous (AA) carriers, respectively. Functional studies by Cumming et al. (54) found no detectable effect of this variant on C1-INH structure, function, stability, plasma levels, or disease expression. However, the authors did not exclude a consequence of the variant on other functions of C1-INH as a modulator of the coagulation and kinin release pathways.
Homozygosity (AA) for KLKB1-rs3733402 (c.428G>A, p.Ser143Asn) in carriers of a missense SERPING1 variant was significantly associated with 7-year earlier disease onset (p = 0.029) and increased disease severity by 30.45 units in CALS score (p < 0.001) compared to GG carriers. This result is in accordance with the results of a previous study by Gianni et al. (22). However, this is an unexpected finding. The KLKB1-rs3733402 variant locates in Apple domain 2 of the heavy chain where prekallikrein (PK) binds to high-molecular-weight kininogen (HMWK). The resulting reduced formation of the PK-HMWK complex interferes with optimal PK activation and reduces bradykinin formation and plasma kallikrein protection from inhibition by C1-INH.
Heterozygosity for CPN1-rs61751507 (c.533G>A, p.Gly178Asp) in carriers of missense SERPING1 variants were independently associated with a 98% decrease in the probability of LTP (p = 0.017). In the past, CPN1-rs61751507 has been once associated with HAE when found in compound heterozygosity with a rare frameshift mutation in CPN1-exon 1 (17, 18). The effect of this variant observed in our study might be related to the substitution of the Gly178 residue of CPN the significance of which is underlined by the fact that it has been conserved in diverse species and is also conserved among most members of the human carboxypeptidase family.
Apart from our above findings that were confirmatory of previously described correlations between functional variants and parameters of the C1-INH-HAE severity, a series of novel correlations were uncovered in this study. The variants F13B-rs6003, PLAU-rs2227564, and SERPINA1-rs28929474 were found significantly correlated with the severity of C1-INH-HAE, independently of the SERPING1 mutational status. Precisely, heterozygosity (GA) for F13B-rs6003 (c.344G>A, p.Arg115His) was correlated with decreased disease severity by 11.84 (p = 0.024) units of the CALS score. F13B-rs6003 has been considered benign concerning the FXIII subunit B deficiency. However, this variant has been characterized as a risk factor for venous thrombosis attributed to the substitution of Arg115 that prevents the dissociation between the A and B subunits of FXIII after activation by thrombin (14).
Carriage of T allele in homozygosity (TT) for PLAU-rs2227564 (c.422T>C, p.Leu141Pro) was found correlated with decreased disease severity by 13.67 units (p = 0.004) of the CALS score. Urokinase, encoded by PLAU, is an activator of plasminogen, which plays a significant role in the activation of the kinin-kallikrein system and the generation of bradykinin. The amino acid change p.Leu141Pro is located within the kringle domain of urokinase at the junction between two β-pleated sheets. The presence of the T allele does not appear to affect the activity of urokinase, but the zymogen containing Pro141 binds fibrin aggregates less efficiently than the one containing Leu141, suggesting a possibility of altered extracellular urokinase localization or stability (28).
The presence of the A allele of SERPINA1-rs28929474 (c.1096G>A, p.Glu366Lys) was correlated with increased disease severity by 80.16 units (p = 0.003) of CALS score. SERPINA1-rs28929474, commonly known as the Z allele of a1-antitrypsin (A1AT), is five times less effective than the normal M allele as an inhibitor of neutrophil elastase. It forms polymers in the lung that can be chemoattractants for neutrophils, thereby increasing inflammation (31–33), while it alters the global structural dynamics of A1AT (34). When found in a homozygous state, the Z allele is responsible for 95% of all clinical cases of A1AT deficiency, and in compound heterozygosity with the S allele, it is associated with 20–50% risk for emphysema (35, 36).
In addition to the above correlations detected between functional variants and disease severity, independently of SERPING1 variation, further correlations were uncovered only in carriers of missense SERPING1 variants. Heterozygosity (AT) for SERPINA1-rs17580 (c.863A>T, p.Glu288Val) was correlated with an 8-year earlier age at disease onset (p < 0.001) compared to AA carriers. SERPINA1-rs17580, commonly known as the S allele of A1AT, causes reduced cellular secretion of A1AT because the newly synthesized protein is degraded intracellularly before secretion (37). The S allele is not disease causing. Even homozygous carriers do not present the common expressions of A1AT deficiency. However, it represents a risk factor when in compound heterozygosity with the Z allele. Such compound heterozygotes are relatively frequent due to the high frequency of this allele. However, this compound heterozygosity was not detected among our patients.
Another functional variant related to disease severity in SERPING1 missense carriers is SERPINE1-rs6092 (c.43G>A, p.Ala15Thr). Homozygous carriers (AA) of SERPINE1-rs6092 displayed a significantly higher mean age at disease onset by 8.4 years (p = 0.009). The variant has been previously characterized as likely benign for plasminogen activator inhibitor-1 (PAI-1) deficiency, but functional studies in a heterozygous patient showed activity at about 70%. Zhang et al. (44) suggested that the change from a hydrophobic non-polar amino acid (Ala) to a hydrophilic polar amino acid (Thr) in the hydrophobic core region (h-region) of the signal peptide of the protein may disturb its function.
Moreover, in the same group of C1-INH-HAE patients, heterozygous carriers (GA) of the gain-of-function mutation F2-rs1799963 (c*97G>A) had a significant decrease in the disease severity by 25.97 units in CALS score (p = 0.017) compared to GG carriers. According to Gehring et al. (15), this variant probably affects the generation of prothrombin. Finally, the KLK1-rs5515 (c.230G>A, p.Arg77His) variant was correlated with both the mean age at disease onset and the disease severity. Heterozygous carriers of this variant were presented with an 8.95-year later age at disease onset (p = 0.05) and with decreased disease severity by 16.79 units of the CALS score (p = 0.029). This variant had been characterized as a loss-of-function polymorphism resulting in reduced kallikrein activity. Slim et al. (20) detected 50–60% lower urinary kallikrein activity to carriers, while in studies concerning branchial artery function (21), which exhibited arterial dysfunction. Interestingly, this is an inverse correlation to what has been detected for the KLKB1-rs3733402 variant, as it is described above.
Conclusion
Our study provides clear evidence that variants other than the SERPING1 causal variants act as independent modifiers of C1-INH-HAE severity and could serve as possible prognostic biomarkers. The next step is the validation of detected correlations in a large cohort of patients. Such a study would best be performed by a global consortium of angioedema centers such as the ACARE network (56) to allow for the inclusion of a diverse and sizable population of clinically well-characterized patients. Enrolling large cohorts of patients could also allow examining the different effects possibly exerted by variants of modifier genes on carriers of different kinds of causal SERPING1 mutations (nonsense, frameshift, large defects, etc.) with different impacts on C1-INH production, structure, and function. Furthermore, functional studies on the effect of these variants could shed light on missing parts of the pathogenesis of the disease. Finally, our results indicate that functional variants of genes involved in pathways other than contact activation system/kallikrein kinin system but recently recognized as participating in C1-INH-HAE pathogenesis (e.g., endothelial cells) should be investigated.
Data Availability Statement
The data have been submitted to European Variation Archive (EVA) EMBL-EBI (https://www.ebi.ac.uk). The accessions associated with submission are: Project: PRJEB51008, Analyses: ERZ5253849.
Ethics Statement
The studies involving human participants were reviewed and approved by the local institutional review boards, and written informed consent was obtained from each individual or an accompanying relative. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author Contributions
FP performed genetic analyses, analyzed the results, and wrote the manuscript. GL performed genetic analyses and critically read the manuscript. MK analyzed the results. DC designed the study and collected clinical data. GP, FP, MMag, AV, MS, and KO collected clinical data. MZ, AS, MMau, MS, and HF critically read and commented on the manuscript. AG designed and supervised the study, critically read and revised the manuscript. All authors approved the submitted version of the article.
Funding
This work was partly funded by the Hellenic Society of Angioedema. The funder had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/falgy.2022.868185/full#supplementary-material
References
1. Longhurst HJ, Bork K. Hereditary angioedema: an update on causes, manifestations and treatment. Br J Hosp Med. (2019) 80:391–8. doi: 10.12968/hmed.2019.80.7.391
2. Busse PJ, Christiansen SC. Hereditary angioedema. N Engl J Med. (2020) 382:1136–48. doi: 10.1056/NEJMra1808012
3. Margaglione M, D'Apolito M, Santocroce R, Maffione AB. Hereditary angioedema: Looking for bradykinin production and triggers of vascular permeability. Clin Exp Allergy. (2019) 49:1395–402. doi: 10.1111/cea.13506
4. Loules G, Zamanakou M, Parsopoulou F, Vatsiou S, Psarros F, Csuka D, et al. Targeted next-generation sequencing for the molecular diagnosis of hereditary angioedema due to C1-inhibitor deficiency. Gene. (2018) 667:76–82. doi: 10.1016/j.gene.2018.05.029
5. Loules G, Parsopoulou F, Zamanakou M, Csuka D, Bova M, González-Quevedo T, et al. Deciphering the genetics of primary angioedema with normal levels of C1 inhibitor. J Clin Med. (2020) 9:3402. doi: 10.3390/jcm9113402
6. Bors A, Csuka D, Varga L, Farkas H, Tordai A, Füst G, et al. Less severe clinical manifestations in patients with hereditary angioedema with missense C1INH gene mutations. J Allergy Clin Immunol. (2013) 131:1708–11. doi: 10.1016/j.jaci.2012.11.015
7. Rijavec M, Košnik M, Andrejević S, KaradŽa-Lapić L, Grivčeva-Panovska V, Korošec P. The functional promoter F12-46C/T variant predicts the asymptomatic phenotype of C1-INH-HAE. Clin Exp Allergy. (2019) 49:1520–2. doi: 10.1111/cea.13470
8. Speletas M, Szilágyi Á, Csuka D, Koutsostathis N, Psarros F, Moldovan D, et al. F12-46C/T polymorphism as modifier of the clinical phenotype of hereditary angioedema. Allergy. (2015) 70:1661–4. doi: 10.1111/all.12714
9. Kanaji T, Okamura T, Osaki K, Kuroiwa M, Shimoda K, Hamasaki N, et al. common genetic polymorphism (46 C to T substitution) in the 5'-untranslated region of the coagulation factor XII gene is associated with low translation efficiency and decrease in plasma factor XII level. Blood. (1998) 91:2010–4. doi: 10.1182/blood.V91.6.2010
10. Ariëns RA, Philippou H, Nagaswami C, Weisel JW, Lane DA, Grant PJ. The factor XIII V34L polymorphism accelerates thrombin activation of factor XIII and affects cross-linked fibrin structure. Blood. (2000) 96:988–95. doi: 10.1182/blood.V96.3.988
11. Göpel W, Kattner E, Seidenberg J, Kohlmann T, Segerer H, Möller J; Genetic factors in Neonatology Study Group. The effect of the Val34Leu polymorphism in the factor XIII gene in infants with a birth weight below 1500 g. J Pediatr. (2002) 140:688–92. doi: 10.1067/mpd.2002.123666
12. Reiner AP, Heckbert SR, Vos HL, Ariëns RA, Lemaitre RN, Smith NL, et al. Genetic variants of coagulation factor XIII, postmenopausal estrogen therapy, and risk of nonfatal myocardial infarction. Blood. (2003) 102:25–30. doi: 10.1182/blood-2002-07-2308
13. Shafey M, Anderson JL, Scarvelis D, Doucette SP, Gagnon F, Wells PS. Factor XIII Val34Leu variant and the risk of myocardial infarction: a meta-analysis. Thromb Haemost. (2007) 97:635–41. doi: 10.1160/TH06-09-0517
14. Komanasin N, Catto AJ, Futers TS, van Hylckama Vlieg A, Rosendaal FR, Ariëns RA, et al. novel polymorphism in the factor XIII B-subunit (His95Arg): relationship to subunit dissociation and venous thrombosis. J Thromb Haemost. (2005) 3:2487–96. doi: 10.1111/j.1538-7836.2005.01624.x
15. Gehring NH, Frede U, Neu-Yilik G, Hundsdoerfer P, Vetter B, Hentze MW, et al. Increased efficiency of mRNA 3' end formation: a new genetic mechanism contributing to hereditary thrombophilia. Nat Genet. (2001) 28:389–92. doi: 10.1038/ng578
16. Segal JB, Brotman DJ, Necochea AJ, Emadi A, Samal L, Wilson LM, et al. Predictive value of factor V Leiden and prothrombin G20210A in adults with venous thromboembolism and in family members of those with a mutation: a systematic review. JAMA. (2009) 301:2472–85. doi: 10.1001/jama.2009.853
17. Mathews KP, Pan PM, Gardner NJ, Hugli TE. Familial carboxypeptidase N deficiency. Ann Intern Med. (1980) 93:443–5. doi: 10.7326/0003-4819-93-3-443
18. Cao H, Hegele RA, DNA. polymorphism and mutations in CPN1, including the genomic basis of carboxypeptidase N deficiency. J Hum Genet. (2003) 48:20–2. doi: 10.1007/s100380300003
19. Poller W, Faber JP, Klobeck G, Olek K. Cloning of the human alpha 2-macroglobulin gene and detection of mutations in two functional domains: the bait region and the thiolester site. Hum Genet. (1992) 88:313–9. doi: 10.1007/BF00197266
20. Slim R, Torremocha F, Moreau T, Pizard A, Hunt SC, Vuagnat A, et al. Loss-of-function polymorphism of the human kallikrein gene with reduced urinary kallikrein activity. J Am Soc Nephrol. (2002) 13:968–76. doi: 10.1681/ASN.V134968
21. Azizi M, Boutouyrie P, Bissery A, Agharazii M, Verbeke F, Stern N, et al. Arterial and renal consequences of partial genetic deficiency in tissue kallikrein activity in humans. J Clin Invest. (2005) 115:780–7. doi: 10.1172/JCI200523669
22. Gianni P, Loules G, Zamanakou M, Kompoti M, Csuka D, Psarros F, et al. Genetic determinants of C1 inhibitor deficiency angioedema age of onset. Int Arch Allergy Immunol. (2017) 174:200–4. doi: 10.1159/000481987
23. Stengaard-Pedersen K, Thiel S, Gadjeva M, Møller-Kristensen M, Sørensen R, Jensen LT, et al. Inherited deficiency of mannan- binding lectin-associated serine protease 2. N Engl J Med. (2003) 349:554–60. doi: 10.1056/NEJMoa022836
24. Sokolowska A, Szala A, St Swierzko A, Kozinska M, Niemiec T, Blachnio M, et al. Mannan-binding lectin-associated serine protease-2 (MASP-2) deficiency in two patients with pulmonary tuberculosis and one healthy control. Cell Mol Immunol. (2015) 12:119–21. doi: 10.1038/cmi.2014.19
25. Romano M, Dri P, Da Dalt L, Patriarca P, Baralle FE. Biochemical and molecular characterization of hereditary myeloperoxidase deficiency. Blood. (1997) 90:4126–34. doi: 10.1182/blood.V90.10.4126
26. Marchetti C, Patriarca P, Solero GP, Baralle FE, Romano M. Genetic characterization of myeloperoxidase deficiency in Italy. Hum Mutat. (2004) 23:496–505. doi: 10.1002/humu.20027
27. Finckh U, van Hadeln K, Müller-Thomsen T, Alberici A, Binetti G, Hock C. et al. Association of late-onset Alzheimer disease with a genotype of PLAU, the gene encoding urokinase-type plasminogen activator on chromosome 10q222. Neurogenetics. (2003) 4:213–7. doi: 10.1007/s10048-003-0157-9
28. Yoshimoto M, Ushiyama Y, Sakai M, Tamaki S, Hara H, Takahashi K, et al. Characterization of single chain urokinase-type plasminogen activator with a novel amino-acid substitution in the kringle structure. Biochim Biophys Acta. (1996) 1293:83–9. doi: 10.1016/0167-4838(95)00228-6
29. Wu W, Jiang H, Wang M, Zhang D. Meta-analysis of the association between urokinase-plasminogen activator gene rs2227564 polymorphism and Alzheimer's disease. Am J Alzheimers Dis Other Demen. (2013) 28:517–23. doi: 10.1177/1533317513494450
30. Crystal RG. The alpha 1-antitrypsin gene and its deficiency states. Trends Genet. (1989) 5:411–7. doi: 10.1016/0168-9525(89)90200-X
31. Ogushi F, Fells GA, Hubbard RC, Straus SD, Crystal RG. Z-type alpha 1-antitrypsin is less competent than M1-type alpha 1-antitrypsin as an inhibitor of neutrophil elastase. J Clin Invest. (1987) 80:1366–74. doi: 10.1172/JCI113214
32. Elliott PR, Bilton D, Lomas DA. Lung polymers in Z alpha1-antitrypsin deficiency-related emphysema. Am J Respir Cell Mol Biol. (1998) 18:670–4. doi: 10.1165/ajrcmb.18.5.3065
33. Parmar JS, Mahadeva R, Reed BJ, Farahi N, Cadwallader KA, Keogan MT, et al. Polymers of alpha(1)-antitrypsin are chemotactic for human neutrophils: a new paradigm for the pathogenesis of emphysema. Am J Respir Cell Mol Biol. (2002) 26:723–30. doi: 10.1165/ajrcmb.26.6.4739
34. Hughes VA, Meklemburg R, Bottomley SP, Wintrode PL. The Z mutation alters the global structural dynamics of α1-antitrypsin. PLoS ONE. (2014) 9:e102617. doi: 10.1371/journal.pone.0102617
35. Stoller JK, Aboussouan LS. Alpha1-antitrypsin deficiency. Lancet. (2005) 365:2225–36. doi: 10.1016/S0140-6736(05)66781-5
36. de Serres FJ, Blanco I. Prevalence of α1-antitrypsin deficiency alleles PI*S and PI*Z worldwide and effective screening for each of the five phenotypic classes PI*MS, PI*MZ, PI*SS, PI*SZ, and PI*ZZ: a comprehensive review. Ther Adv Respir Dis. (2012) 6:277–95. doi: 10.1177/1753465812457113
37. Curiel DT, Chytil A, Courtney M, Crystal RG. Serum alpha 1-antitrypsin deficiency associated with the common S-type (Glu264—Val) mutation results from intracellular degradation of alpha 1-antitrypsin prior to secretion. J Biol Chem. (1989) 264:10477–86. doi: 10.1016/S0021-9258(18)81646-5
38. Brantly ML, Wittes JT, Vogelmeier CF, Hubbard RC, Fells GA, Crystal RG. Use of a highly purified alpha 1-antitrypsin standard to establish ranges for the common normal and deficient alpha 1-antitrypsin phenotypes. Chest. (1991) 100:703–8. doi: 10.1378/chest.100.3.703
39. Bornhorst JA, Greene DN, Ashwood ER, Grenache DG. α1-Antitrypsin phenotypes and associated serum protein concentrations in a large clinical population. Chest. (2013) 143:1000–8. doi: 10.1378/chest.12-0564
40. Dahl M, Hersh CP, Ly NP, Berkey CS, Silverman EK, Nordestgaard BG. The protease inhibitor PI*S allele and COPD: a meta-analysis. Eur Respir J. (2005) 26:67–76. doi: 10.1183/09031936.05.00135704
41. Holmes MD, Brantly ML, Crystal RG. Molecular analysis of the heterogeneity among the P-family of alpha-1-antitrypsin alleles. Am Rev Respir Dis. (1990) 142:1185–92. doi: 10.1164/ajrccm/142.5.1185
42. Jung CH, Na YR, Im H. Retarded protein folding of deficient human alpha 1-antitrypsin D256V and L41P variants. Protein Sci. (2004) 13:694–702. doi: 10.1110/ps.03356604
43. Ray S, Mickleborough TD, Brown JL. Comparison of the properties of rare variants of alpha1-proteinase inhibitor expressed in COS-1 cells and assessment of their potential as risk factors in human disease. Biochim Biophys Acta. (2005) 1740:390–402. doi: 10.1016/j.bbadis.2005.03.010
44. Zhang ZY, Wang ZY, Dong NZ, Bai X, Zhang W, Ruan CG, et al. case of deficiency of plasma plasminogen activator inhibitor-1 related to Ala15Thr mutation in its signal peptide. Blood Coagul Fibrinolysis. (2005) 16:79–84. doi: 10.1097/00001721-200501000-00013
45. Xiong Y, Song C, Snyder GA, Sundberg EJ, Medvedev AE. R753Q polymorphism inhibits Toll-like receptor (TLR) 2 tyrosine phosphorylation, dimerization with TLR6, and recruitment of myeloid differentiation primary response protein 88. J Biol Chem. (2012) 287:38327–37. doi: 10.1074/jbc.M112.375493
46. Verpy E, Biasotto M, Brai M, Misiano G, Meo T, Tosi M. Exhaustive mutation scanning by fluorescence-assisted mismatch analysis discloses new genotype-phenotype correlations in angiodema. Am J Hum Genet. (1996) 59:308–19.
47. Kalmár L, Bors A, Farkas H, Vas S, Fandl B, Varga L, et al. Mutation screening of the C1 inhibitor gene among Hungarian patients with hereditary angioedema. Hum Mutat. (2003) 22:498. doi: 10.1002/humu.9202
48. Pappalardo E, Caccia S, Suffritti C, Tordai A, Zingale LC, Cicardi M. Mutation screening of C1 inhibitor gene in 108 unrelated families with hereditary angioedema: functional and structural correlates. Mol Immunol. (2008) 45:3536–44. doi: 10.1016/j.molimm.2008.05.007
49. Varga L, Bors A, Tordai A, Csuka D, Farkas H. Diagnostic pitfalls in hereditary angioedema. 7th C1 Inhibitor Deficiency Workshop in Budapest, Hungary, May 20–22, 2011. J Angioedema, special preview issue. doi: 10.1016/j.molimm.2011.06.396
50. Rijavec M, Korošec P, Šilar M, Zidarn M, Miljković J, Košnik M. Hereditary angioedema nationwide study in Slovenia reveals four novel mutations in SERPING1 gene. PLoS ONE. (2013) 8:e56712. doi: 10.1371/journal.pone.0056712
51. Bygum A, Fagerberg CR, Ponard D, Monnier N, Lunardi J, Drouet C. Mutational spectrum and phenotypes in Danish families with hereditary angioedema because of C1 inhibitor deficiency. Allergy. (2011) 66:76–84. doi: 10.1111/j.1398-9995.2010.02456.x
52. Duponchel C, Djenouhat K, Frémeaux-Bacchi V, Monnier N, Drouet C, Tosi M. Functional analysis of splicing mutations and of an exon 2 polymorphic variant of SERPING1/C1NH. Hum Mutat. (2006) 27:295–6. doi: 10.1002/humu.9414
53. Gösswein T, Kocot A, Emmert G, Kreuz W, Martinez-Saguer I, Aygören-Pürsün E, et al. Mutational spectrum of the C1INH (SERPING1) gene in patients with hereditary angioedema. Cytogenet Genome Res. (2008) 121:181–8. doi: 10.1159/000138883
54. Cumming SA, Halsall DJ, Ewan PW, Lomas DA. The effect of sequence variations within the coding region of the C1 inhibitor gene on disease expression and protein function in families with hereditary angio-oedema. J Med Genet. (2003) 40:e114. doi: 10.1136/jmg.40.10.e114
55. Blanch A, Roche O, López-Granados E, Fontán G, López-Trascasa M. Detection of C1 inhibitor (SERPING1/C1NH) mutations in exon 8 in patients with hereditary angioedema: evidence for 10 novel mutations. Hum Mutat. (2002) 20:405–6. doi: 10.1002/humu.9073
Keywords: C1-inhibitor deficiency, genetic biomarkers, functional variants, hereditary angioedema, long-term prophylaxis, next-generation sequencing, severity score
Citation: Parsopoulou F, Loules G, Zamanakou M, Csuka D, Szilagyi A, Kompoti M, Porebski G, Psarros F, Magerl M, Valerieva A, Staevska M, Obtulowicz K, Maurer M, Speletas M, Farkas H and Germenis AE (2022) Searching for Genetic Biomarkers for Hereditary Angioedema Due to C1-Inhibitor Deficiency (C1-INH-HAE). Front. Allergy 3:868185. doi: 10.3389/falgy.2022.868185
Received: 02 February 2022; Accepted: 15 March 2022;
Published: 07 July 2022.
Edited by:
Riccardo Castagnoli, National Institute of Allergy and Infectious Diseases (NIH), United StatesReviewed by:
Sonia Caccia, Università degli studi di Milano, ItalyAlvin H. Schmaier, Case Western Reserve University, United States
Copyright © 2022 Parsopoulou, Loules, Zamanakou, Csuka, Szilagyi, Kompoti, Porebski, Psarros, Magerl, Valerieva, Staevska, Obtulowicz, Maurer, Speletas, Farkas and Germenis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anastasios E. Germenis, YWdlcm1lbkBtZWQudXRoLmdy