 Richard Kelwick
Richard Kelwick James T. MacDonald
James T. MacDonald Alexander J. Webb
Alexander J. Webb Paul Freemont
Paul Freemont- 1Centre for Synthetic Biology and Innovation, Imperial College London, London, UK
- 2Department of Medicine, Imperial College London, London, UK
Synthetic biology is principally concerned with the rational design and engineering of biologically based parts, devices, or systems. However, biological systems are generally complex and unpredictable, and are therefore, intrinsically difficult to engineer. In order to address these fundamental challenges, synthetic biology is aiming to unify a “body of knowledge” from several foundational scientific fields, within the context of a set of engineering principles. This shift in perspective is enabling synthetic biologists to address complexity, such that robust biological systems can be designed, assembled, and tested as part of a biological design cycle. The design cycle takes a forward-design approach in which a biological system is specified, modeled, analyzed, assembled, and its functionality tested. At each stage of the design cycle, an expanding repertoire of tools is being developed. In this review, we highlight several of these tools in terms of their applications and benefits to the synthetic biology community.
Introduction
The synthetic biology toolkit has expanded greatly in recent years, which can be attributed to the efforts of a highly dynamic community of researchers, ambitious undergraduate students in the International Genetically Engineered Machine competition (iGEM), and the growing number of amateur scientists from the DIY BIO movement. Each of these groups has bold ambitions for the rapidly growing field of synthetic biology, which aims to rationally engineer biological systems for useful purposes (Purnick and Weiss, 2009; Anderson et al., 2012; Landrain et al., 2013; Jefferson et al., 2014). The merging of several foundational sciences, including molecular, cellular, and microbiology with a set of engineering principles, is a profound shift and is the key distinction between synthetic biology and genetic engineering (Andrianantoandro et al., 2006; Heinemann and Panke, 2006; Khalil and Collins, 2010; Kitney and Freemont, 2012). Indeed, many social scientists, who are themselves a part of the synthetic biology community, have extensively explored the ontological implications of this perspective (Schark, 2012; Preston, 2013). Although many of the social aspects of synthetic biology are beyond the scope of this review, they will continue to shape the synthetic biology toolkit. In particular, society is an important stakeholder that has some influence over chassis (host cell) choice, the design of biosafety measures, biosecurity considerations, and long-term research applications (Marris and Rose, 2010; Anderson et al., 2012; Agapakis, 2013; Moe-Behrens et al., 2013; Wright et al., 2013; Douglas and Stemerding, 2014).
From a biological perspective, there have been important developments in the field across several areas, some of which have been reviewed elsewhere (Arpino et al., 2013; Lienert et al., 2014; Way et al., 2014). For instance, the number, quality, and availability of biological parts (bioparts, e.g., promoters and ribosomal binding sites) have continued to increase. This is exemplified by the iGEM student registry of standard biological parts, which has increased its biopart collection to include over 12,000 parts, across 20 different categories (partsregistry.org). However, due to its open nature, the iGEM registry contains parts of variable quality that are mostly uncharacterized. There are also professional parts registries, such as those at BIOFAB, which include expansive libraries of characterized DNA-based regulatory elements (Mutalik et al., 2013a,b). Although libraries of bioparts are indeed useful, putting them together into predictable devices, pathways and systems are incredibly challenging as many biological design rules are not yet fully understood (Endy, 2005; Kitney and Freemont, 2012). Developing synthetic passive and active insulator sequences may help increase predictability and thus reduce context dependency (Davis et al., 2011; Lou et al., 2012; Qi et al., 2012; Mutalik et al., 2013a). Notwithstanding these challenges, the field is progressing across several areas. One such area is biopart characterization, which is critical to the field, primarily because it is fundamentally a realization of several of the core engineering principles adopted in synthetic biology, namely standardization, modularization, and abstraction. Discrete biological parts of known sequence and behavior can be abstracted based upon a descriptive function and thus, their true complexity can be masked behind a biological concept. For example, discrete DNA sequences (bioparts) that fit a standardized descriptive function, such as a promoter, can be functionally characterized and as a consequence bioparts become reusable (modularization) for use in other synthetic systems. Additionally, methods that provide standardized ways of assembling DNA parts such as the BioBrick standard can help establish platforms for the sharing and reuse of bioparts. At a higher level, abstraction and standardization are important because they permit the separation of design from assembly (Endy, 2005).
A desirable consequence of this perspective is that these engineering principles enable the separation of labor, expertise, and complexity at each level of the design hierarchy (Endy, 2005). In practical terms, this separation of biological design from DNA assembly enables innovation within these hierarchies to occur at different rates. For instance, it is generally true that with more recent DNA assembly methods it is currently easier to assemble multi-part genetic circuits consisting of several bioparts, or even entire genomes, than it is to reliably predict how these bioparts will interact in the final system (Purnick and Weiss, 2009; Ellis et al., 2011; Arpino et al., 2013; Ellefson et al., 2014). However, it is envisioned that this will change, with the increasing adoption of high-throughput characterization platforms that can test entire biopart libraries in parallel. These platforms typically use automated liquid-handling robots, coupled with plate readers although microfluidics approaches are also gaining traction (Lin and Levchenko, 2012; Boehm et al., 2013; Benedetto et al., 2014). In either case, when coupled with automated data analysis, modeling, and sophisticated forward-design strategies (Marchisio and Stelling, 2009; Wang et al., 2009; Esvelt et al., 2011; Ellefson et al., 2014; Marchisio, 2014; Stanton et al., 2014), these high-throughput platforms provide the basis for the rapid prototyping workflows required to realize a synthetic biology design cycle (Kitney and Freemont, 2012).
In this review, we focus on several significant tools, both classical and emerging, that the field of synthetic biology employs as part of a typical design cycle workflow. Building upon a design cycle template, the review is organized to explore prominent tools and research methodologies across three core areas: designing predictable biology (design), assembling DNA into bioparts, pathways, and genomes (build), and rapid prototyping (test) (Figure 1). We first describe several of the core challenges that are associated with designing predictable biology, including the complexities associated with chassis selection, biopart design, engineering, and characterization. In parallel, we highlight relevant tools and methodologies that are particularly aligned with the engineering principles of synthetic biology. We then discuss established and newly developed DNA assembly methodologies, and group them according to four broad assembly strategies: restriction enzyme-based, overlap-directed, recombination-based, and DNA synthesis. Finally, we highlight several emerging rapid prototyping technologies that are set to significantly improve the field’s capacity for testing synthetic parts, devices, and systems. We conclude with a summary of several of the core challenges that were described in each of the design, build, test sections of the review and discuss whether the synthetic biology toolbox is equipped to address them. In addition to this, we have also created an online community, the Synthetic Biology Index of Tools and Software (SynBITS) – http://www.synbits.co.uk, which has also been structured according to the design cycle (Figure 1).
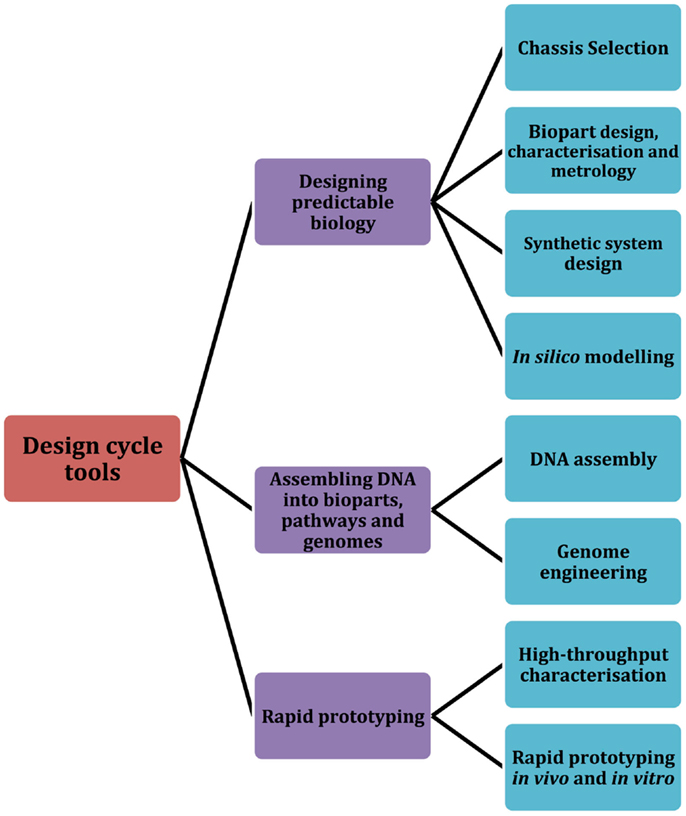
Figure 1. Synthetic Biology Index of Tools and Software (SynBITS). A schematic summary of the synthetic biology design cycle tools as depicted in SynBITS (www.synbits.co.uk), an online community-managed index of synthetic biology tools and software.
Designing Predictable Biology
From an engineering perspective, living systems can be perceived as overly complex, inefficient, and unpredictable (Csete and Doyle, 2002). It is this perception that has driven the concept of the biopart, in which a particular DNA sequence is defined by the function that it encodes (Endy, 2005). Thus, complex biological functions can be conceptually separated (abstracted) from the complexities of the sequence context from which they originated (Endy, 2005). As a consequence of this approach, biological pathways and circuits can potentially be redesigned into less complex and potentially more predictable designs. The defining examples of this perspective are the toggle switch (Gardner et al., 2000), a genetic circuit defined by two repressible promoters that were engineered to form a mutually inhibitory network, and the repressilator (Elowitz and Leibler, 2000), a type of oscillator (biological clock). What sets these examples apart from general genetic engineering is that modeling was used to predict and optimize the behavior of these genetic circuit designs prior to their construction.
While these forward-design approaches were hugely successful, the repressilator displayed noisy behavior as a result of stochastic fluctuations in components of the genetic circuit (Elowitz and Leibler, 2000). In other words, in silico modeling did not fully capture the true in vivo complexity of the synthetic circuit. Likewise, the toggle switch experienced natural fluctuations in gene expression that were sufficient to create variations in the level of inducer needed to switch the cells from one state to another. These variations were also not fully anticipated during in silico modeling (Gardner et al., 2000). While these genetic circuits have been improved, with novel oscillator (Stricker et al., 2008; Olson et al., 2014) and toggle switch designs, including those designed for mammalian cells (Muller et al., 2014b) and plants (Muller et al., 2014a), it is clear that the modeling of biological systems still requires a concerted and long-term effort. Critical to this effort is the availability of new synthetically designed bioparts and experimental data that accurately captures the behavior of the components or bioparts that constitute a synthetic system (Arkin, 2013) as well as the characteristics or influence that the chassis/host cell enacts upon them.
Chassis Selection
As an engineering concept, the chassis refers to a physical internal framework or structure that supports the addition of other components that combine to form a finalized engineered structure. From a synthetic biology perspective, the concept invokes an understanding that a biological chassis is a tool to provide the structures that accommodate (host) the execution of a synthetic system, including the provision of a metabolic environment, energy sources, transcription, and translation machinery, as well as other minimal cellular functions (Acevedo-Rocha et al., 2013; Danchin and Sekowska, 2014). Chassis selection is therefore a critical design decision that synthetic biologists are required to take, particularly since the chassis will directly influence the behavior and function of a synthetic system. Essentially, the chassis determines which bioparts can be used since they must be compatible with the biological machinery that is present. This can result in a difficult choice for the synthetic biologist: either to use an established chassis and design the circuit to be orthogonal with that host, or design a synthetic system that fits a requirement and then choose a host chassis that is compatible with the resultant bioparts or system. These constraints can to some degree be designed around, either by engineering the chassis to knockout genes that optimize its orthogonality and reduce burden, through codon optimization (Chung and Lee, 2012) or through the use of insulator sequences that negate context dependency effects (Guye et al., 2013; Torella et al., 2014a). Ultimately, however, chassis selection will dictate the downstream design considerations for any given synthetic system, and therefore, chassis selection must be coordinated with biopart design efforts.
In order to rationalize which chassis selection strategy is most appropriate for an intended application, it is important to consider the consequences and advantages of each strategy. Where a chassis is selected as a priority above that of the design considerations of the synthetic system, it is important to consider whether the chassis has been extensively characterized in the literature and/or if the chassis has known intrinsic capabilities that complement the intended application (Table 1). Additionally, access to detailed biological knowledge of a chassis will aid modeling-guided design efforts and the implementation of chassis optimization strategies for dealing with burden or metabolic flux effects. Likewise, the wealth of knowledge acquired about model organisms across several biological disciplines may encourage synthetic biologists to consider them as a potential chassis in preference to established favorites (Table 1). Indeed, there are already several emerging chassis that are gaining traction and are set to be utilized more frequently in the field (Table 1).
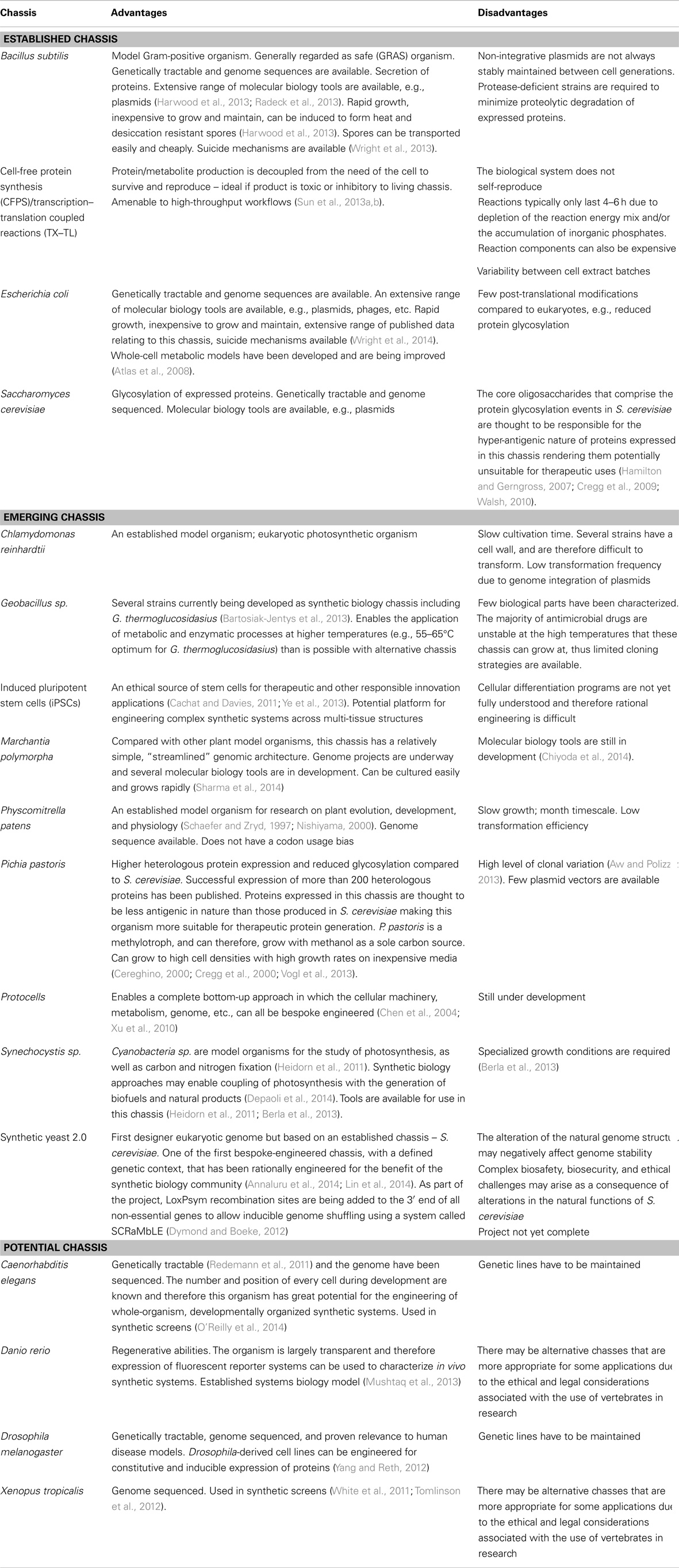
Table 1. Synthetic biology chassis.
Alternatively, a synthetic system could be specified and designed as a priority above that of chassis selection. As a consequence, there will be chassis, which are not compatible with the synthetic system and others that may require extensive engineering to accommodate its design. However, this approach is complementary to those chassis that are bespoke engineered. “Synthia,” the first organism to feature a fully synthetically manufactured genome, is indicative that the field of synthetic biology is shifting toward the development of rationally engineered chassis (Gibson et al., 2010a). Though it is important to recognize that the “Synthia” genome, while synthetic in origin, was not designed to significantly alter the characteristics of the chassis, and therefore, does not represent the first truly bespoke-engineered chassis. Yet, its successors, the synthetic yeast project (Annaluru et al., 2014), protocell developments (Xu et al., 2010), and even to some extent cell-free expression systems (Shin and Noireaux, 2012; Sun et al., 2013a) may all usher in an era in which the design of bespoke-engineered chassis is routine. Wholly rationally engineered chassis could conceivably be built around the specifications of a synthetic system, such that the chassis is both compatible with the synthetic system and the majority of its cellular resources are directed toward the execution of the synthetic system. In this sense, the function of the synthetic system would be free of chassis constraints; however, the full realization of this approach is still several decades away. Until then, chassis selection will remain a trade-off between which should be prioritized for each application, the chassis or the synthetic system? There are of course many other considerations to address, some of which we cover in the biopart design section of this review and others that have been previously discussed in the literature (Heinemann and Panke, 2006; Arpino et al., 2013; Danchin and Sekowska, 2014).
Biopart Design and Engineering
The field of synthetic biology continues to benefit from decades of biological research that has built a knowledge base of biological systems that can be deconstructed and re-engineered as bioparts and synthetic systems. Here, we highlight prominent bioparts that are particularly aligned with the engineering principles of synthetic biology. In most cases, existing natural biological parts can be reused in synthetic devices or systems. However, there are situations where new bioparts need to be designed and synthesized by modifying existing bioparts or by creating entirely new parts de novo. These novel parts could be enzymes that catalyze unnatural reactions (Jiang et al., 2008; Rothlisberger et al., 2008), molecular biosensors (Penchovsky and Breaker, 2005), protein scaffold (Koga et al., 2012; Heider et al., 2014), DNA or RNA scaffolds (Rothemund, 2006; Delebecque et al., 2011), ribosome-binding sites with specifically designed transcription rates (Salis et al., 2009), promoters with novel regulatory features and/or specific translation rates (Marples et al., 2000; Kelly et al., 2009).
Transcriptional circuits use RNA polymerase operations per second (PoPS) as the common signal carrier but, until recently only a small set of DNA-binding proteins and associated operator sequences were used to regulate the flux of RNA polymerase (RNAP) and construct synthetic circuits. The lack of a large set of orthogonal regulatory proteins has limited the complexity of synthetic systems (Purnick and Weiss, 2009), but a new wave of engineered proteins has greatly increased the number of tools available to synthetic biology circuit designers. The clustered, regularly interspaced, short palindromic repeats (CRISPR)/Cas system consists of CRISPR and CRISPR associated genes (cas) coding for related proteins, which together constitute an adaptive prokaryotic immune system (Barrangou et al., 2007). The CRISPR loci consist of repeats interspaced with spacer sequences, which are transcribed and processed into crRNAs containing individual spacer sequences that are complementary to foreign DNA. The crRNAs bind Cas9 nuclease and the resulting complex recognizes and cleaves sequences complementary to the spacer sequences. This natural system has been repurposed as a transcriptional regulator by modifying the Cas proteins to deactivate nuclease activity and creating artificial guide RNA (gRNA) sequences to create the CRISPR interference (CRISPRi) system. Deactivated Cas9:gRNA complexes act as repressors by binding specific sites and inhibiting RNAP activity (Qi et al., 2013). Alternatively, Cas9 can be fused to domains that recruit RNAP in order to act as transcriptional activators (Bikard et al., 2013; Mali et al., 2013).
Transcription activator-like effectors (TALEs) are proteins secreted by Xanthomonas bacteria in order to activate expression of plant genes during the course of infection. They consist of tandem repeats of a small domain with two variable amino acid sites. The amino acid identities of the variable sites have a simple mapping to the DNA base recognized, enabling chains of domains to be stringed together in order to bind specific sequences (Boch et al., 2009; Moscou and Bogdanove, 2009). The simple modular nature of TALEs has enabled the engineering of synthetic proteins such as TAL effector nucleases (TALENs) (Mahfouz et al., 2011) and artificial orthogonal activators and repressors (Morbitzer et al., 2010; Blount et al., 2012).
Translation initiation regulators are relatively easy to de novo design as they rely on the reasonably well-characterized thermodynamics of RNA structure (Liang et al., 2011). However, unlike transcriptional circuits, there is no common signal carrier and thus they cannot be as easily composed into complex regulatory designs. By repurposing a regulatory element from the tnaCAB operon of E. coli, Liu et al. (2012), have created an adapter to convert translational regulators into transcriptional regulators (Liu et al., 2012). The 5′-end of the operon codes for a short leader peptide, TnaC that stalls the ribosome in the presence of free tryptophan. The stalled ribosome then blocks a Rho factor-binding site located adjacent to the stop codon of tnaC, allowing the transcription of the downstream genes tnaA and tnaB. The ribosome-binding site of tnaC in the native operon is constitutive but replacing this with translational regulator sequences, such as the RNA-IN/OUT system (Ross et al., 2013), enables the control of transcription of downstream genes.
In recent years, there has been rapid progress in developing software algorithms to enable the design of synthetic proteins that can be controlled at the atomic level of resolution (Leaver-Fay et al., 2011). Computational protein design is generally split into two components. Initially, a backbone scaffold is either artificially generated or taken from an existing known structure. Secondly, the amino acid sequence is optimized such that it minimizes the free energy of folding. It appears that minimizing a potential energy function by trialing different amino acid identities and rotamers is sufficient to achieve this. There have been a number of dramatic successes using this approach including the de novo design of enzymes. The design of a novel enzyme requires knowledge about the transition state structure of the reaction to be catalyzed and a predicted spatial arrangement of chemical groups that are likely to stabilize the transition state. The transition state structure and the stabilizing constellation of chemical groups around it can be designed theoretically (theozyme) (Tantillo et al., 1998), and using this knowledge, known protein structures can be searched for sites capable of accommodating side chain functional groups in the desired geometry (Zanghellini et al., 2006). These methods have resulted in a number of successful enzyme designs (Jiang et al., 2008; Rothlisberger et al., 2008).
In the cell, many biochemical processes are spatially organized in order to locally concentrate substrates or isolate toxic substances (e.g., the carboxysome or peroxisome) and reduce cross talk between components. Efforts to engineer high-level organization in synthetic biological systems is a major challenge with applications in encapsulating artificial organelles or protocells (Choi and Montemagno, 2005; Agapakis et al., 2012; Hammer and Kamat, 2012; Mali et al., 2013), the precise detection and delivery of payloads (Sukhorukov et al., 2005; Uchida et al., 2007). Methods based on the computational protein design methods described above have been applied to create new self-assembling biomaterials at the atomic level from protein subunits that do not naturally form into higher-order structures (King et al., 2012, 2014). Other work has focused on using hydrophobic patterning of peptides to produce higher-order structures based on coiled-coils (Rajagopal and Schneider, 2004; Woolfson and Mahmoud, 2010; Zaccai et al., 2011; Fletcher et al., 2013). However, these are not designed to atomic level accuracy, tend to be chemically synthesized and so have not yet been reported to assemble in vivo. An alternative approach is to reuse naturally occurring protein–protein interfaces and assemblies (Padilla et al., 2001; Howorka, 2011; Sinclair et al., 2011), although ultimately it may be more desirable to design completely artificial protein scaffolds that are more likely to be biologically neutral and avoid the Mullerian complexity of naturally evolved biological systems (Dutton and Moser, 2011).
Novel protein biomaterials have applications in metabolic engineering by co-locating enzymes in the same pathway on a structural scaffold. This has the advantage of increasing the local concentration of substrates improving reaction kinetics, helping to prevent the loss of intermediates to competing pathways and the accumulation of toxic intermediates (Dueber et al., 2009). Protein cages can be used to completely encapsulate metabolic pathways and create synthetic bacterial micro-compartments. In a recent example, genes from the propanediol utilization operon (pdu) encoding for an empty protein shell in Salmonella enterica were expressed in E. coli. Short peptide sequences known to bind to pdu shell proteins were used to target pyruvate decarboxylase and alcohol dehydrogenase to the micro compartment resulting in increased ethanol production (Lawrence et al., 2014). This approach promises to be particularly useful for biosynthetic pathways involving toxic metabolites.
Similarly, structural scaffolds can be constructed using nucleic acids. Base pairing in nucleic acids makes predicting and designing structures somewhat more tractable than for proteins. For example, 2D and 3D structures have been engineered in vitro using long single stranded DNA (ssDNA) and small ssDNA oligonucleotides called “staples,” that direct the folding of the long ssDNA into a pre-designed structure (Rothemund, 2006; Douglas et al., 2009; Han et al., 2011). It has also been shown to be possible to express simpler nanostructures in vivo using RNA transcribed by the cell (Delebecque et al., 2011). These structures were used together with specific protein-binding aptamers to efficiently channel substrates from one enzyme to another and substantially increase hydrogen production. At short distances, substrate channeling has been found to be more effective than expected by simple 3D Brownian diffusion models (Fu et al., 2012, 2014).
A number of tools for predicting and designing relative translation rates of ribosome-binding sites have been developed (Reeve et al., 2014) including the RBS Calculator (Salis et al., 2009; Salis, 2011), the RBS Designer (Na and Lee, 2010), and the UTR Designer (Seo et al., 2013), which can aid operon design (Arpino et al., 2013). These software tools are based on thermodynamic models of the pre-initiation complex of the 30S ribosomal subunit and the messenger RNA (mRNA) and include terms based on the free energy required to unfold the unbound mRNA, the free energy of hybridization of the mRNA and the 16S rRNA, and various other terms. If the pool of free 30S ribosomal subunits is assumed to be roughly constant then the translation initiation rate can be assumed to be proportional to exp(−βΔG). Mechanistic predictive models for promoters are somewhat more complicated as promoter strength is related to the binding of the sigma factor and RNAP, and also the efficiency of promoter escape. However, there has been some success in predicting the strength of promoters for the E. coli sigma factor σE using relatively simple position weight matrix models (Rhodius and Mutalik, 2010; Rhodius et al., 2012).
Most of the synthetic regulatory tools described above are used in the construction of transcriptional circuits. Nevertheless, post-transcriptional circuit design, particularly using RNA molecules, has attracted a great deal of interest in recent years (Liang et al., 2011; Wittmann and Suess, 2012). Unlike proteins, RNA molecules are somewhat easier to design due to their well-understood thermodynamics and the dominance of secondary structure formation on folding. One important application area is the use of RNA as switches (riboswitches) that respond to their environment. Riboswitches are RNA molecules that can regulate protein production in response to changes in the concentration of a small molecule and occur naturally as well as being synthetically designed. These molecules are composed of an RNA aptamer that binds a specific small-molecule ligand. On binding the small molecule, the RNA aptamer may then change conformation resulting in either the occlusion of the Shine–Dalgarno sequence or its increased accessibility. Expression of the downstream genes is then turned either on or off in response (Suess et al., 2004). Alternatively, aptamers may be coupled to a ribozyme that allosterically cleaves itself in response to ligand binding (Tang and Breaker, 1997; Penchovsky and Breaker, 2005). The de novo design of small-molecule binding RNA aptamers is a non-trivial task but novel aptamers can be evolved in vitro using methods such as SELEX (systematic evolution of ligands by exponential enrichment) (Ellington and Szostak, 1990; Tuerk and Gold, 1990; Jenison et al., 1994). Riboswitches may have uses in metabolic engineering such as down-regulating upstream genes if a metabolite reaches toxic levels (Zhang and Keasling, 2011). RNA aptamers have also found use as mimics of GFP by binding small-molecule fluorophores (Paige et al., 2011). As discussed in the metrology section, these aptamers can be used to monitor mRNA levels.
Biopart Characterization
Biopart characterization describes the functional and experimental metadata that is required to sufficiently capture the biological behavior of a biopart and the context in which it is being tested. The type and range of these characterization data have evolved over time as highlighted by refinements in biopart characterization data sheets (Arkin, 2008; Canton et al., 2008). Typically, these experimental metadata include information on the plasmid vector, the testing organism or strain, any relevant growth conditions, and the equipment or methodologies used to capture the bioparts functionality. The primary purpose of biopart characterization data is to provide the necessary experimental data for predictive in silico biological modeling. The determination of which biological data provide0 the greatest insight into the behavior of a given biological system is largely debatable; at least until more biological design rules are understood. The context dependency of bioparts in vivo provides significant challenges in predicting their function as modular components. Therefore, for biopart characterization, measurement standards should largely be defined by those biological data that can be measured (metrology), how relevant those data are for predicting the behavior of a biological process (modeling) and how widely these data can be adopted (standardization). This last point is particularly important since bioparts should ideally be reusable (modular) across multiple applications and contexts. To enable this, the formatting of these data should ideally be standardized to facilitate the measurement and use of biopart characterization data across different in silico design tools, forward-design strategies, and workflows.
The most concerted effort is the Synthetic Biology Open Language (SBOL) consortia, a group of life scientists, engineers, computer scientists and mathematicians that are actively building a set of standards that define a common data format for bioparts and their accompanying characterization data (Bower et al., 2010; Galdzicki et al., 2011; Quinn et al., 2013; Roehner and Myers, 2013). The concept is to create a file structure that can capture biopart sequence, characterization and experimental data in a format that is platform independent. Crucially, the format is designed to be extendable to include additional parameters as new characterization technologies and methodologies emerge. In combination with SBOL visual (SBOLv), which defines a standardized way to visually denote bioparts through symbols, the SBOL standard is set to enable the seamless sharing of genetic designs. Several bioinformatics and molecular cloning design tools have already adopted SBOL, and the intention for SBOL is to provide an interoperable standard between several in silico tools such that individuals can optimize their workflow as required, yet retain information between them. Several of these in silico tools have been extensively reviewed (MacDonald et al., 2011; Galdzicki et al., 2014); however, we include an updated list here, that combines in silico tools from these existing reviews, along with several new tools, in particular R2o designer and COOL (Table 2).
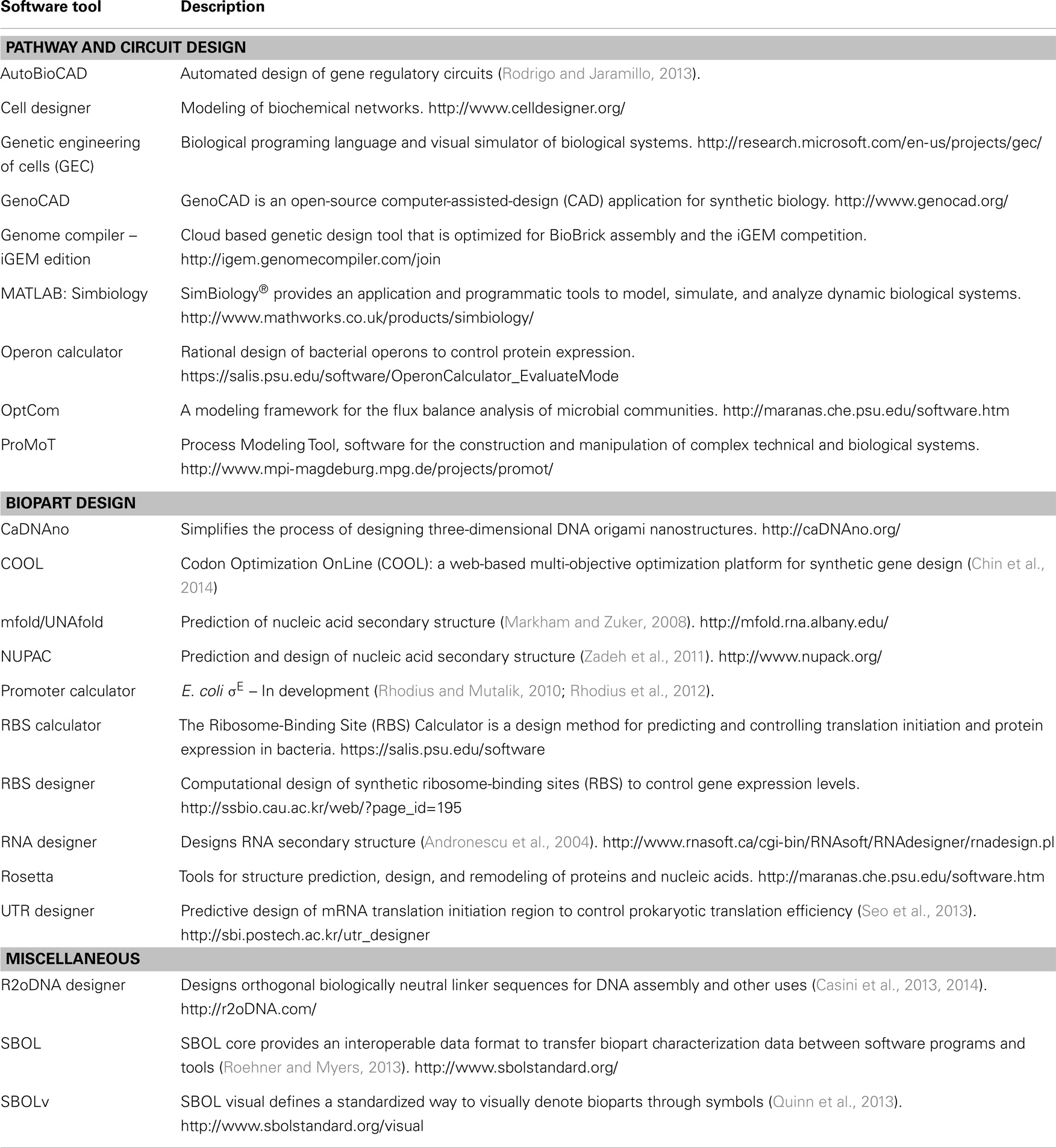
Table 2. Emerging tools for the forward-design of synthetic pathways and systems.
In contrast to SBOL, which is still under development, the iGEM registry of standard biological parts (http://parts.igem.org/) has provided a relatively large-scale and publically accessible repository of bioparts and some biopart characterization data for almost 10 years. Since its inception, the iGEM community has led, with mixed success, concerted efforts to improve the quality of its characterization data. The 2014 iGEM competition, for instance, has announced several specialist awards for teams that demonstrate advancements in metrology. This push for improvements in biopart characterization at the grassroots (undergraduate) level has permeated up to professional characterization efforts. For instance, early difficulties in the reproducibility of the behavior of DNA regulatory elements between iGEM teams and professional research groups provided the context for the emergence of the relative promoter unit (RPU) as a reference measurement standard (Kelly et al., 2009). The RPU standard compares the relative activity of a promoter against a reference standard, tested under the same experimental conditions, with an RPU arbitrarily set to 1. The rationale underpinning this standard is that while the absolute activity of a promoter may differ between experimental repeats, the relative activity should be less prone to such variability. Essentially, a promoter that is twice the strength of the standard should remain so, even between different experimental conditions and methodologies of different research groups. In agreement with this, Kelly et al. (2009) reported a 50% decrease in variability, when RPUs were independently reported for a set of Anderson constitutive promoters. Inter-experimental variability and reproducibility of data are a significant problem facing all scientific endeavors (Collins and Tabak, 2014), and for synthetic biologists the RPU measurement standard has highlighted these issues within the context of biopart characterization.
There are, however, no universally agreed standards for advancing biopart characterization metrology, though in general the field is shifting away from relative measurements toward absolute measurements (Table 3). Many research groups are currently interested in measuring absolute numbers of cells, DNA molecules, proteins, or other components that constitute the synthetic system and its context. But this shift is largely incremental as certain types of biological data are very difficult to measure directly. These challenges are, however, worth addressing since it is assumed that such biological data are essential to improve the predictive capabilities of forward-design in silico models (Bower et al., 2010; Cooling et al., 2010). Yet, because of such data limitations, current-modeling approaches often depend upon inferred or assumed parameters that are derived from biological data that can be experimentally verified. One such modeling approach by Canton et al. (2008), proposed a set of standardized measurement units termed, PoPs and ribosomes per second (RIPS), even though the absolute biological data that underpin them has not been directly measured in vivo (Canton et al., 2008; Cooling et al., 2010; Marchisio, 2014). PoPs infers the flow of RNAP along a point of DNA per second and RIPS infers the flow of ribosomes across an mRNA molecule. As previously noted, PoPs and RIPS cannot be measured directly; instead they are calculated using fluorescence data from a reporter protein (e.g., GFP), growth data (OD), and largely assumed values for other parameters including protein or mRNA concentrations. These data are generally measured in vivo within a plate reader setup, though flow cytometry-based characterization efforts are increasingly being adopted and are set to progress metrology at the single cell level (Díaz et al., 2010; Tracy et al., 2010; Choi et al., 2013; Zuleta et al., 2014). In either case, if experimental setups are sufficiently standardized, it is possible to convert measurements between several widely adopted standards: RPU, PoPs/RIPS, and absolute measurements such as GFP cell−1 s−1 (Kelly et al., 2009).
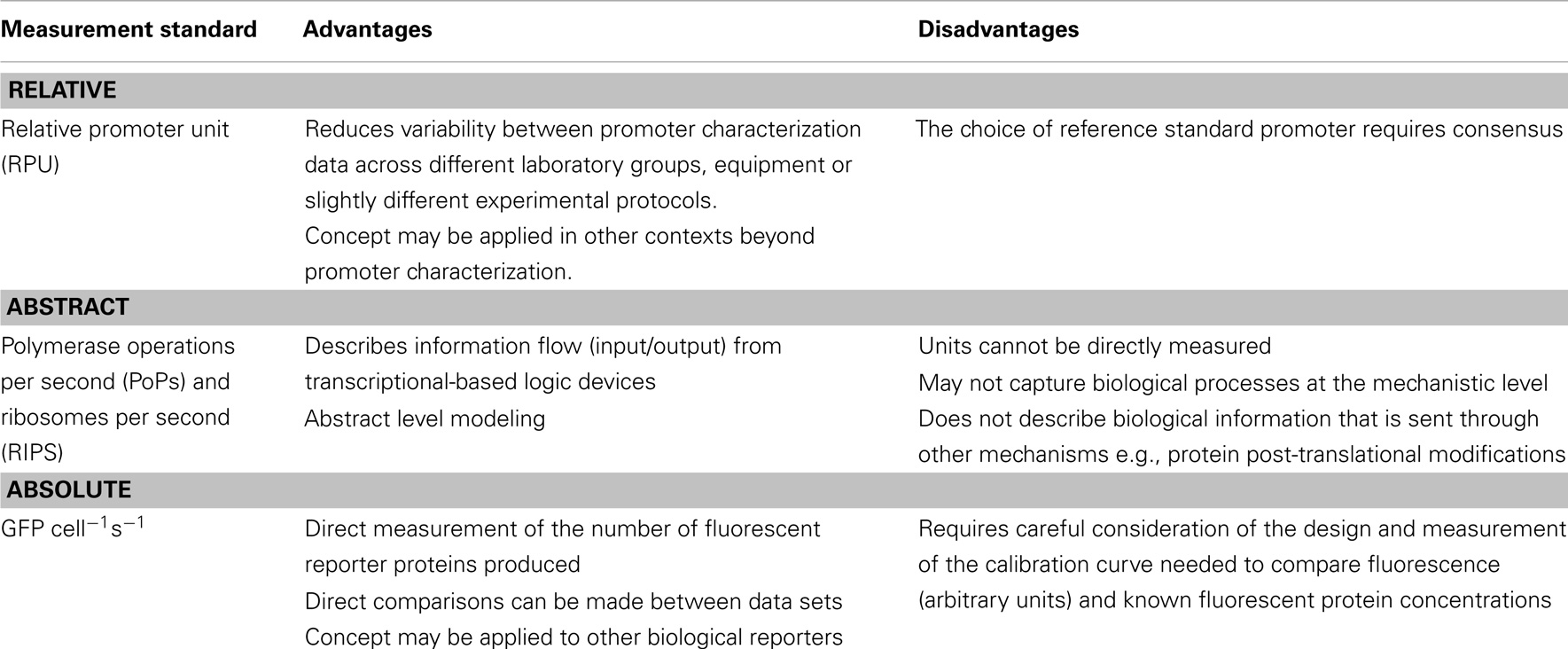
Table 3. Synthetic biology measurement standards.
Notwithstanding the above limitations of PoPs and RIPS, these units were primarily designed to reflect the behavior of genetic circuits at the level of information flow (inputs/outputs) rather than at the truly mechanistic level (Gardner et al., 2000; Canton et al., 2008; Stricker et al., 2008; Marchisio, 2014). For biologists, however, these terms represent an abstract merger of several elements of the transcriptional and translational machinery, which does not accurately reflect the mechanistic underpinning biology. However, abstract and mechanistic modeling approaches are not necessarily mutually exclusive since both approaches can provide insightful information for the forward-design of predictable biological pathways and systems.
Advances in metrology and novel measurement standards that are accessible, and hence, more widely adopted will clearly benefit the whole field of synthetic biology. Yet, it is challenging to achieve consensus for developing measurement standards, since standards intrinsically empower those that promote them above those that have not adopted them (Calvert, 2012; Frow and Calvert, 2013). Conversely, it should be noted that consensus in measurement standards and metrology does not preclude innovation if such standards are flexible enough to accommodate developments in the tools and methodologies that enable researchers to easily share, reuse, and build upon existing genetic designs. Likewise, standardized biological information can still be combined with expert knowledge, or novel forward-design strategies for the construction of complex, robust, and efficient biological systems.
Metrology in biology has been enabled in part to continual advancements in microscopy and in synthetic biology, technologies such as microfluidics coupled with quantitative microscopy are continuing to gain traction (Lin and Levchenko, 2012; Song et al., 2013; Walter and Bustamante, 2014). Microfluidic technologies enable the precise manipulation of fluids at small-scales through engineered channels, chambers, and valves. Microfluidic chip designs are sufficiently advanced to enable a high-degree of spatial-temporal control of liquid-flows to and between individual cells or cell populations seeded within the chambers of prefabricated microfluidics chips. With this level of control, small molecules that induce gene expression or influence other biological processes can be precisely delivered to elicit acute, basal, or morphogenic responses. Within a synthetic biology context, such systems have been used to characterize DNA regulatory elements, intercellular communication, and synthetic pathways at high spatial–temporal resolution. One notable example shown by Hansen and O’Shea (2013), in which the microfluidic control of the delivery of a small molecule (1-NM-PP1) was used to control the nuclear localization of a Yeast stress-inducible transcription factor, Msn2. Deliberate alterations in the oscillatory or acute dynamics of Msn2 trans-nuclear localization revealed the extent to which promoters respond differently to transcriptional-activation dynamics. From this, promoters could be modeled in silico, according to the extent that they could elicit differential gene expression patterns, as a consequence of their ability to distinguish a genuine nuclear-influx of Msn2 from background “noise” (Hansen and O’Shea, 2013). Manipulation of these dynamics could be used to reduce promoter leakiness; or conversely to exploit different classes of promoter transcriptional-signal processing to coordinate multiple genetic programs, through the modulation of a single transcription factor.
Another important technology for synthetic biology is flow cytometry, which relies upon hydrodynamic focusing to guide single cells through a fluidic channel where they are measured (Piyasena and Graves, 2014). Recent models of flow cytometers can simultaneously measure cell size, complexity, and up to 17 channels of fluorescence (Basiji et al., 2007; Piyasena and Graves, 2014), each of which could be used to capture data from different reporter outputs. Of the biological reporters available, RNA aptamers are particularly noteworthy, since they have the potential to increase the type and range of biological information that can be measured (Cho et al., 2013; Pothoulakis et al., 2014). For instance, several groups have reported the simultaneous measurement of both transcription (mRNA levels) and translation (protein levels) (Chizzolini et al., 2013; Pothoulakis et al., 2014). In both cases Spinach, an RNA aptamer that binds a fluorophore (Paige et al., 2011), was incorporated within the 3′ untranslated region (UTR) of a fluorescent reporter protein, either GFP or RFP (Chizzolini et al., 2013; Pothoulakis et al., 2014). Providing there is no spectral-overlap between fluorophores, this strategy could conceivably be up-scaled to measure entire synthetic pathways, and thus inform operon design strategies (Hiroe et al., 2012; Chizzolini et al., 2013). Metabolic engineering efforts may also benefit from engineered RNA aptamer-hybrids that simultaneously bind cellular metabolites and a fluorophore, effectively enabling the real-time reporting of intracellular metabolic flux (Barrick and Breaker, 2007; Roth and Breaker, 2009; Sefah et al., 2013; Szeto et al., 2014). These exponential increases in biological data could significantly impact whole-cell modeling (Atlas et al., 2008; Gama-Castro et al., 2011; Shuler et al., 2012; O’Brien et al., 2013) and pave the way for novel measurement standards or modeling approaches that are wholly based upon directly measured biological processes.
Assembling DNA into Bioparts, Pathways, and Genomes
Recombinant DNA technology, in which DNA sequences are “cut and pasted” together via restriction enzymes and DNA ligases respectively, form the foundations of the 1970s biotechnological revolution and have greatly expanded the possibilities of genetic engineering (Zimmerman et al., 1967; Cohen et al., 1973; Lobban and Kaiser, 1973). Synthetic biology continues to benefit from these foundational advancements in recombinant DNA-based biotechnology. For example, the BioBrick DNA assembly standard, uses a set of standardized restriction sites, termed the prefix (EcoRI XbaI) and suffix (SpeI PstI), that flank each biopart (BioBrick) (Rokke et al., 2014). Digestion and ligation using these sites allow several parts to be assembled together in a standard fashion. The BioBrick standard was originally developed by Tom Knight in 2003 and is still used within the synthetic biology community, particularly during the iGEM competition (Rokke et al., 2014). The BioBrick assembly standard is beneficial to the synthetic biology community for several reasons. Firstly, the flanking restriction site sequences set a physical border that defines individual bioparts. As a result, the BioBrick assembly standard realizes the idea that DNA sequences encode discrete functions and that these individual blocks (BioBricks) can be assembled together like “legotm bricks.” Additionally, the use of standardized restriction sites ensures that the cloning strategy for assembling BioBricks is standardized across the entire research community; thereby eliminating the requirement for some cloning-based tacit knowledge. Despite these advantages, a major limitation of the approach is that BioBrick sequences must not contain the prefix and suffix restriction sites, thus limiting the range of sequences that can be assembled. Additionally, when XbaI and SpeI sites are ligated together, the ligated sequence creates a “scar,” which does not contain either an XbaI or SpeI restriction site (Speer and Richard, 2011; Rokke et al., 2014). Scar sequences may alter the behavior of the flanking bioparts or prevent the generation of fusion proteins, and therefore, can be undesirable (Anderson et al., 2010; Ellis et al., 2011).
BioBrick assembly is also an inefficient way to create large multi-part constructs since it is limited to the assembly of two bioparts per reaction, as defined by the three antibiotic (3A) assembly method (Speer and Richard, 2011). ePathBrick potentially overcomes this limitation through the use of an expansive set of BioBrick-compatible isocaudomer pairs of restriction sites (Xu et al., 2012). The combinatorial assembly of multiple inserts is possible through the restriction digestion and ligation of different isocaudomer pairs into an ePathBrick vector. Backwards compatibility with the BioBrick standard is certainly advantageous from the perspective of modularity (re-useable bioparts); however, ePathBrick is still subject to the BioBrick limitations of forbidden sequences and post-assembly scar sequences. With these limitations in mind, several DNA assembly methods have been developed to address them (Figure 2) (Chao et al., 2014).
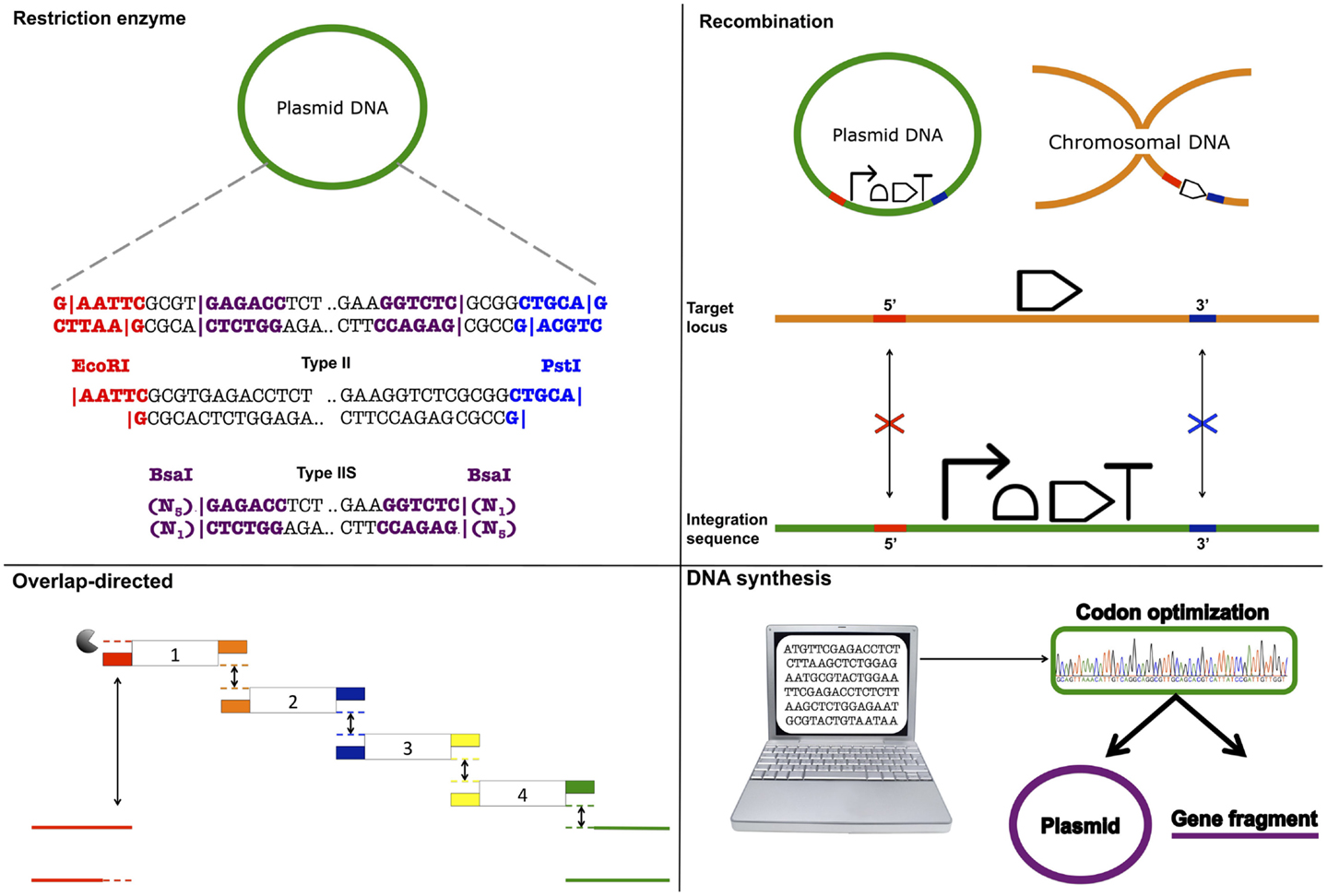
Figure 2. DNA assembly strategies. Restriction enzyme – restriction enzymes recognize specific DNA sequences and either cut within their recognition sequence (Type II) or adjacent to its recognition sequence (Type IIS) to create sticky or blunt-ended DNA fragments that can be ligated to other DNA fragments. Recombination – cellular DNA repair and recombination machinery can be utilized to integrate a DNA construct within a specific genomic locus. Integration is guided through 5′ and 3′ sequence complementarity of the integration sequence with the target locus. Overlap-directed – assembly order is guided by 20–40 bp overlaps at the ends of each DNA fragment that share sequence homology with adjacent DNA fragments. In the case of Gibson assembly, these homologous ends are processed (chew-back) and fused together (anneal) via the sequential activity of an exonuclease, a ligase, and a polymerase. DNA synthesis – DNA sequences are designed and optimized in silico for de novo synthesis. Commercial constructs are delivered as gene fragments or are pre-cloned within a plasmid vector.
Restriction-Directed Assembly: Bgl Bricks, Golden Gate, and SEVA
Golden gate assembly (Engler et al., 2008; Engler and Marillonnet, 2011, 2013), Bgl Bricks (Anderson et al., 2010), and the Standard European Vector Architecture (SEVA) (Silva-Rocha et al., 2013) use a set of restriction sites to standardize DNA assembly. However, in contrast to the BioBrick standard, these assembly methods use rare restriction site sequences, and therefore, support a greater range of sequences. The Bgl Brick standard uses BglII and BamHI restriction sites. Annealed BglII and BamHI restriction sites generate an inert, glycine-serine encoding scar sequence, which in contrast to the BioBrick standard scar allows the assembly of protein fusions. Golden Gate assembly supports scar-less assembly through the use of Type IIS restriction enzymes that act by cleaving outside of their recognition sequence leaving a variable overhang, which directs the assembly order and ligation reaction. If cleavage sites are designed appropriately, these overhangs can be designed so that the final assembled sequences are “scar-less.” More recently, combinatorial Golden Gate assembly methods have been described that allow multi-gene constructs, including synthetic pathways, to be assembled in parallel (Engler and Marillonnet, 2011, 2013). SEVA and to some extent ePathBrick, differ from the majority of assembly methods in that they are more correctly described as modular standards. SEVA describes a set of criteria for the physical assembly of plasmids according to a three-component architecture: an origin of replication segment, a selection marker segment, and a cargo segment (Silva-Rocha et al., 2013). These segments are flanked by insulator sequences and assembled together with a set of rare restriction sites. While the rationales for restriction site-based assembly methods support modularity, their limitations have led several research groups in the synthetic biology community to “trade-in” standardization and modularity, in favor of “bespoke” assembly methods that enable one-pot assembly of multiple DNA parts.
Overlap-Directed Assembly: Gibson, SLiC, CPEC, SLiCE, and PaperClip
Daniel Gibson developed a widely adopted DNA assembly method that allows multiple DNA fragments to be assembled in a one-pot in vitro reaction (Gibson et al., 2009; Gibson, 2011). The Gibson assembly uses a linearized destination vector and PCR generated inserts as its starting material. Inserts are generated with PCR primers that include 20–40 bp overlaps that share sequence homology to adjacent DNA fragments. As a result, the correct arrangement of several inserts entering the same destination vector can be defined. During the reaction, a T5 exonuclease acts to chew-back at the 5′ ends of the linearized destination vector and inserts. The reaction occurs at 50°C and therefore the T5 exonuclease along with its activity is eventually inactivated. The destination vector and inserts anneal together, as defined by their exonuclease exposed homologous ends, and Phusion polymerase activity acts to fill in the gaps. Finally, Taq ligase seals nicks between the joined DNA fragments. Gibson assembly is simple, can assemble five or more parts in a single reaction, and the reaction itself only takes around 60 min, after which the final assembled product can be directly transformed into E. coli.
Sequence and Ligase-independent Cloning (SLiC) (Li and Elledge, 2007), Circular Polymerase Extension Cloning (CPEC) (Quan and Tian, 2009, 2011), Seamless Ligation Cloning Extract (SLiCE) (Zhang et al., 2012) are also overlap-directed DNA assembly methods that all result in the same final product. Therefore, inserts and destination vectors designed for Gibson assembly can also be used in SLiC, CPEC, and SLiCE assemblies. During SLiC reactions, the destination vector and inserts are independently treated in vitro with T4 DNA polymerase, which exhibits exonuclease activity in the absence of deoxynucleotide triphosphates (dNTPs). Exonuclease activity is subsequently inhibited with the addition of deoxycytidine triphosphate (dCTP) and the destination vector and inserts are then mixed together for annealing. However, because SLiC reactions do not include DNA ligase, gaps, or nicks in the DNA are repaired once the final product is transformed into E. coli. CPEC on the other hand, is a PCR-based approach in which the linearized destination vector and inserts are initially denatured to produce single DNA strands. These are then annealed together, as directed by the homologous DNA overlap regions. Once annealed, the destination vector and inserts act to prime each other for extension via the activity of Phusion DNA polymerase. A low number of PCR cycles act to prevent the propagation of PCR-based errors. SLiCE reactions markedly differ from the assembly methods just described in that they involve an ex vivo bacterial cell extract (PPY, E. coli DH10B λ–red) as the reaction mix. Since exogenous polymerases and DNA ligases are not required, this is a potentially cost-effective method and like Gibson, assembly reactions also typically take just 60 min, although at 37°C instead of 50°C as per Gibson assembly.
PaperClip DNA assembly is a relatively new overlap-directed assembly method that uses pairs of bridging oligonucleotides termed “Clips” to direct the assembly of multi-part constructs (Trubitsyna et al., 2014). Interestingly, PaperClip assembly protocols are derived from CPEC (PCR-based) and SLiCE (ex vivo-based) assembly methodologies. Yet, PaperClip assembly is advantageous over these assembly methods in that once the “Clips” have been prepared, the required assembly order of parts can be determined in a single reaction. While, “Clips” introduce an alanine encoding scar sequence between each part, the bridging oligos used to assemble multi-part constructs in ligase cycling reaction (LCR) assembly are scar-less (Rouillard et al., 2004; de Kok et al., 2014). Though as we describe below, PaperClip assembly differentiates itself from Gibson, CPEC, SLiCE, and LCR assembly methods in that de novo assembly fragments do not need to be generated each time the order assembly is changed (Trubitsyna et al., 2014).
Overlap-directed assembly methods use sequence homology to guide assembly and are therefore largely sequence independent. This is a clear advantage over restriction site-based DNA assembly methods and their forbidden sequences. It should be noted that repeat and short DNA sequences, particularly those that give rise to DNA secondary structures, can reduce the efficiency of overlap-directed methods and are best avoided. On the other hand, CPEC denaturation PCR cycles mitigate the effect of DNA secondary structures to some degree. Overlap-directed methods are also efficient at assembling multiple parts in a predefined order within a single one-pot reaction. Gibson assembly, for example, has been used to assemble genome-scale DNA fragments, including the complete assembly of the M. genitalium genome (583 kb) and more recently the entire mouse mitochondrial genome (16.3 kb) (Gibson et al., 2008, 2010b). It is clear therefore that overlap-directed assembly methods can be scaled toward the assembly of large genetic constructs, including synthetic genomes (Gibson et al., 2010a). Yet, despite their proven utility, they are inherently “bespoke” and are thus in conflict with the ideals of embedding standardization and modularity concepts within DNA assembly strategies. For instance, custom primers are needed to generate inserts de novo each time the assembly order is changed and while it is now possible to automate overlap-directed assembly primer design (Hillson et al., 2012), these assembly methods still require tacit knowledge. To this end, additional methodologies are being developed with the aim of making overlap-directed DNA assembly modular.
Overlap-Directed Assembly with Biologically Neutral Linker Sequences
Modular overlap-directed assembly with linkers (MODAL) makes use of standardized flanking sequences and biologically neutral (orthogonal) linkers as part of a modular overlap-directed DNA assembly strategy (Casini et al., 2013). MODAL assembly requires bioparts to be standardized with the addition of a common prefix and suffix sequence. The prefix and suffix sequences do not contain restriction sites and are not directly required for the assembly process. Instead, these sequences serve as a consistent set of PCR primer “landing pads” that enable all MODAL bioparts to be generated using the same primer set. Additionally, these sites serve as priming sites for the PCR-directed addition of biologically neutral linker sequences that serve as homologous sequences for overlap-directed assembly. These sequences can be designed with R2oDNA Designer (Casini et al., 2013, 2014), an in silico tool that was developed to automatically design orthogonal linker sequences for use in MODAL and other applications. Similar strategies have also been developed in parallel, in which biologically inactive unique nucleotide sequences (UNSes) were utilized to guide the Gibson assembly of insulated genetic circuits (Guye et al., 2013; Torella et al., 2014a,b). These neutral sequences are often standardized and may also incorporate BioBrick restriction sites, thus enabling modularity and standardization to be embedded within overlap-directed assembly strategies.
In vivo DNA Assembly and Genome Engineering
An array of chassis with a broad set of useful, extensively characterized genotypes and phenotypes are available to the synthetic biology community (Table 2). However, there are applications where it is appropriate to rationally engineer a chassis. For instance, an application may require a novel strain that is optimized, at the genome level, to fit a set of specific design requirements that may be difficult or otherwise impractical to bioprospect. Typically, genome-engineering efforts are geared toward maximizing compatibility between a chassis and a synthetic system, increasing the efficiency of the metabolic flux across a synthetic pathway or toward minimizing burden effects. The field is making progress in establishing rationally engineered genomes; of which the synthetic yeast 2.0 project (Dymond and Boeke, 2012; Annaluru et al., 2014; Lin et al., 2014) and minimal genome projects (Glass et al., 2006; Dewall and Cheng, 2011; Shuler et al., 2012), are currently the most prominent exemplars. These genome-engineering efforts are made possible due to the emergence and ongoing development of an expanding set of in vivo DNA assembly methods and genome-engineering tools.
Recombineering approaches, in which synthetic linear ds/ssDNA sequences are introduced into genomic regions through a process of homologous recombination, have proven utility as an efficient method to knockout or knock-in sequences of interest. Recombineering enables genomic engineering at all scales; from the introduction of single nucleotide polymorphisms, to the replacement of 40 kb+ DNA fragments or even toward the assembly of entire genomes (Narayanan and Chen, 2011; Zhao et al., 2011; Bonde et al., 2014; Song et al., 2014). S. cerevisiae transformation-associated recombination (TAR) cloning (Kouprina and Larionov, 2008), Bacillus Domino (Ohtani et al., 2012), and the E. coli Single-Selective-Marker Recombination Assembly System (SRAS) (Shi et al., 2013) uses the endogenous homologous recombination machinery of the indicated organisms to assemble DNA constructs in vivo. A variant of the yeast TAR method has successfully generated several genomes, including the first in vivo assembled synthetic genome of M. genitalium (Gibson et al., 2008). Bacillus domino has also shared similar successes in that this assembly method has also assembled DNA at the genomic scale, including the mouse mitochondrial genome and the rice chloroplast genome (Itaya et al., 2008; Ohtani et al., 2012; Iwata et al., 2013). While E. coli SRAS could potentially support the assembly of large DNA fragments, it is currently optimized for the assembly of multi-part constructs and their simultaneous integration into the E. coli genome (Shi et al., 2013).
The lambda-red (λ-red) recombinase system is another recombineering strategy, which is used for the integration of ssDNA or dsDNA constructs into the E. coli genome (Murphy, 1998; Murphy and Campellone, 2003). Optimized lambda-red recombination protocols can integrate linear DNA sequences into a specific genomic target, guided by only 35–50 bases of flanking homologous sequence (Murphy and Campellone, 2003). Interestingly, lambda-red-mediated recombination events do not require endogenous recombination proteins (e.g., RecA) and instead linear ssDNA or dsDNA constructs are integrated into the E. coli genome via the action of three λ-red proteins; Gam, Exo, and Beta. Gam protects linear dsDNA from the exonuclease activity of the endogenous proteins RecBCD, thus increasing the efficiency at which the introduced dsDNA will be recombined into the genome. λ-red-mediated recombination itself is primarily mediated by Exo, a 5′–3′ – dsDNA-specific exonuclease and Beta, a ssDNA annealing protein. It is interesting to note that Gam-associated protection of dsDNA is exploited in SLiCE ex vivo DNA assembly and as we discuss later, for in vitro transcription–translation (TX–TL) coupled reactions involving linear DNA as the input (Sitaraman et al., 2004).
The introduction of a large number of rationally engineered genomic changes is a potentially laborious process; however, multiplex automated genome engineering (MAGE) enables the automation of large-scale recombineering strategies. MAGE was originally characterized within EcNR2, a variant strain of E. coli MG1655. EcNR2 was modified to incorporate the λ-red recombination system and also to be deficient in DNA mismatch repair via the knockout of the mutS gene (Wang et al., 2009). MAGE relies upon the λ-red Beta protein-assisted incorporation of ssDNA oligonucleotides, typically 90mers, into the lagging strand during DNA replication (Wang et al., 2009). MAGE oligonucleotide pools can be designed to incorporate highly specific changes at a single genomic site, to introduce multiple changes across a single locus or to simultaneously target multiple genomic sites. These outcomes are largely defined through the diversity of the MAGE oligonucleotide pool, where mixtures of degenerate oligonucleotides can be designed to introduce divergent changes across a broad sequence and recombination efficiency space. Where a large number of simultaneous genomic changes are required, the process can be repeated through multiple MAGE cycles of cell growth, electroporation of oligonucleotides into the cell population, and phenotype/genotype characterization. MAGE cycles can be automated through a microfluidics-type setup and in combination with MODEST or optMAGE, which are in silico MAGE oligonucleotide design tools (Table 4), the directed evolution of a rationally designed chassis, can be accomplished within a timescale of several days. Indeed, MAGE has been used to optimize the DXP pathway in E. coli, such that isolated variants that are capable of a fivefold increase in lycopene production were engineered in just 3 days (Wang et al., 2009).
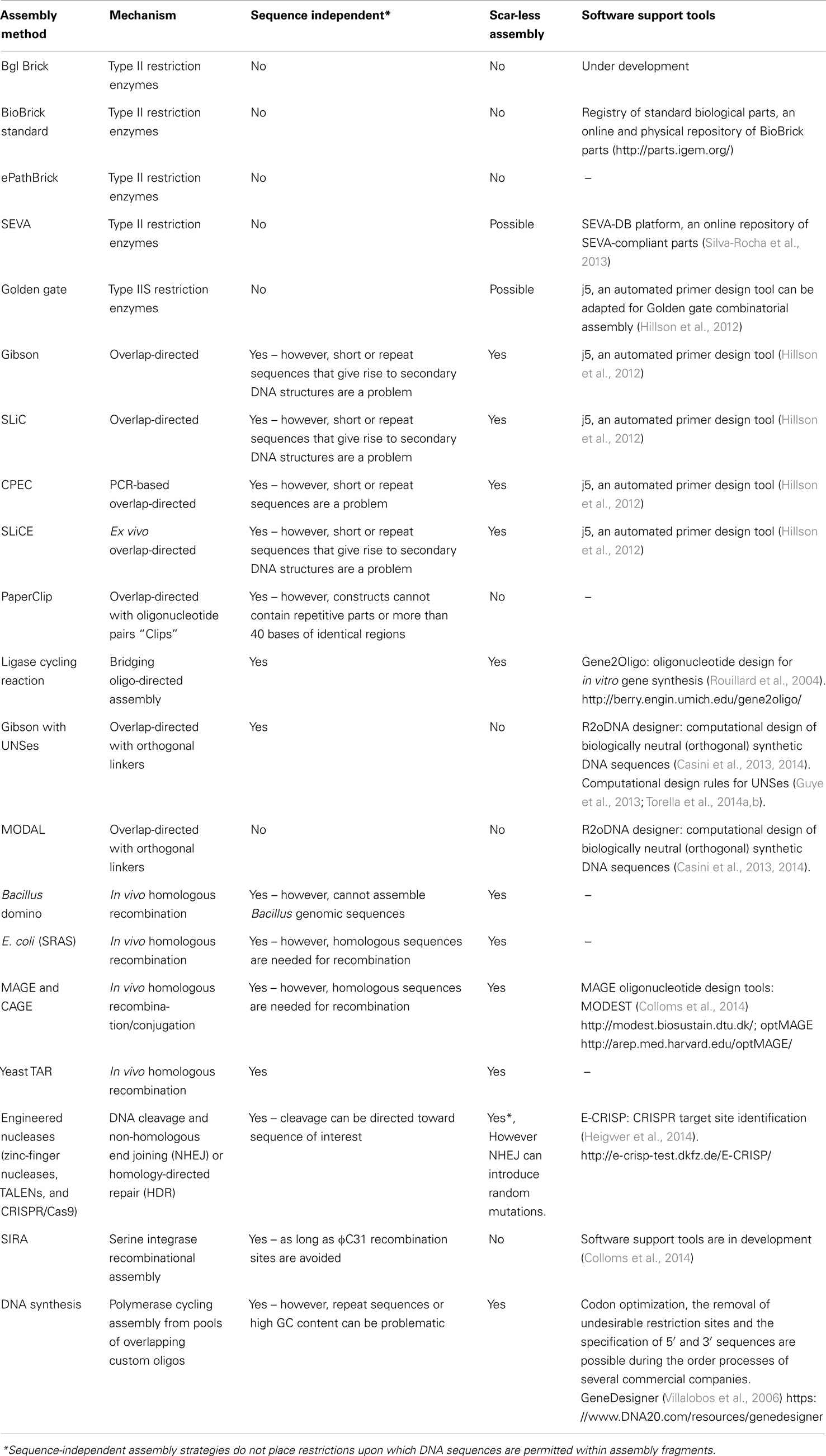
Table 4. DNA assembly and genome-engineering tools.
In parallel with MAGE, conjugative assembly genome engineering (CAGE) can be used to coordinate large-scale genomic engineering strategies across phases, such that subtle genetic combinations that are lethal can be screened out in a manner that does not impede overall progress toward the final strain. To achieve this, CAGE guides the conjugal transfer of MAGE genome modifications between hierarchical pairs of donor–recipient E. coli, such that a new strain emerges which incorporates all of the MAGE-optimized modifications from previous generations (Isaacs et al., 2011). Multiple MAGE–CAGE rounds enable a large set of genomic modifications to be generated and carefully integrated. As an example of such an approach, Isaacs et al. (2011) used a MAGE–CAGE strategy to replace 314 TAG stop codons with the synonymous TAA in E. coli across its entire genome (Isaacs et al., 2011).
Engineered nucleases, which cleave specific DNA sequences, creating double-stranded DNA breaks, can be used to introduce genomic changes. These strategies depend upon the random occurrence of perturbations in DNA repair mechanisms, where double-stranded breaks are inappropriately repaired, resulting in erroneous sequence insertions, deletions, or even significant chromosomal rearrangements. Screening strategies to identify cells that contain desirable genomic alterations can be subsequently isolated as an engineered population. Zinc-finger nucleases (Ellis et al., 2013), TALENS (Mahfouz et al., 2011), and the CRISPR/Cas system (Sander and Joung, 2014) have all been engineered for these types of genome editing applications. The CRISPR/Cas system is particularly interesting since as discussed above, a deactivated Cas9 nuclease:gRNA complex can also be fused with domains that act as transcriptional activators or repressors (Bikard et al., 2013; Mali et al., 2013; Qi et al., 2013). Nuclease-mediated genome editing strategies can also be combined with a recombineering-type approach, in which an engineered dsDNA can be introduced into the cell, which has sequence complementarity at the site of the nuclease breakage. Through the endogenous homologous recombination machinery (DNA repair mechanisms), it is possible to rationally integrate the engineered dsDNA into the genome (Cong et al., 2013; Sander and Joung, 2014). Thus in combination, MAGE, CAGE, and engineered, targeted nucleases (Zinc, TALENS and Cas9) represent a set of molecular tools that enable genome editing and the transcriptional control of natural and synthetic genomes.
DNA Synthesis
Synthetic biology has greatly benefited from the rapid decline in the cost of commercial gene synthesis, a phenomenon popularized by the Carlson curve (Carlson, 2009), which is analogous to Moore’s law. Although the rate of decline has decreased in recent years, with DNA synthesis costs now relatively stable (Carlson, 2009, http://www.synthesis.cc/cgi-bin/mt/mt-search.cgi?blog_id=1&tag=CarlsonCurves&limit=20), it is likely that new disruptive technologies will decrease DNA synthesis costs in the near future. DNA synthesis costs are still sufficiently low that many research groups routinely order the synthesis of genes and gene fragments although still prohibitive for library generation or for the synthesis of large multi-part pathways. In these cases, gene synthesis can be combined with additional cloning techniques such as overlap-directed assembly or mutagenic PCR to generate large constructs or biopart libraries, respectively. It is likely that as DNA synthesis costs decline, there will be a continual shift away from DNA assembly toward de novo DNA synthesis, which will have a transformative effect on synthetic biology and the design-build-test cycle.
Rapid Prototyping
High-throughput platforms bring scalability to biopart characterization efforts, through the parallel characterization of function and context of entire biopart libraries (Arkin, 2013; Keren et al., 2013; Mutalik et al., 2013b). To ensure consistency at such scale, high-throughput workflows typically couple liquid-handling robots with plate readers (Keren et al., 2013), flow cytometry (Piyasena and Graves, 2014; Zuleta et al., 2014), or microfluidics (Lin and Levchenko, 2012; Benedetto et al., 2014) in order to automate the majority of the experimental workflow. Several high-throughput platforms have been described, the majority of which were used to characterize DNA regulatory elements (Keren et al., 2013; Mutalik et al., 2013a,b), however, this is expanding to include the characterization of enzymes (Choi et al., 2013), multi-gene operons (Chizzolini et al., 2013), and RNA aptamers (Cho et al., 2013; Szeto et al., 2014). When coupled with automated data analysis and modeling, these technologies and workflows could become rapid prototyping platforms, enabling a truly biological design cycle approach (Kitney and Freemont, 2012). At present, these high-throughput workflows are typically semi-rational design strategies in which thousands of biopart variants are tested and screened as part of a discovery workflow. Yet, at the same time, these approaches are simultaneously generating large data sets that provide useful insights into biological processes that may inform biological design rules. For example, characterization efforts have informed several systematic methodologies for the rational optimization of synthetic systems at the transcriptional, translational, and post-translational level (Table 2) (Arkin, 2008; Arpino et al., 2013; Reeve et al., 2014). In cases where synthetic systems could conceivably be rationally designed, it is still naïve to assume that the first iteration of a synthetic biological system will perfectly match the design specifications. Instead, multiple iterations of the design-build-test cycle will be needed until forward-design approaches are sufficiently advanced. Therefore, the requirements of interoperable standards in which researchers can apply the same protocols across different liquid-handling platforms are essential. To this end, Linshiz et al. (2013) have implemented a high-level robot programing language (PaR-PaR), which can translate biological protocols into instruction sets for an extendable range of liquid-handling robot platforms. As a consequence of this approach, the training requirements for end-users to implement the same biological protocol across different liquid-handlers are significantly reduced (Linshiz et al., 2013). If, as the authors propose, PaR–PaR is combined with SBOL, then the adoption of PaR–PaR scripts will enable researchers to share the same high-throughput DNA assembly or characterization protocols, but have them implemented across different experimental and equipment setups.
The majority of the rapid prototyping platforms that we have described so far have been optimized for testing biological parts, devices, and systems in vivo; however, in vitro systems are emerging as a useful testing platform. Cell-free protein synthesis (CFPS) systems based upon E. coli (Nirenberg and Matthaei, 1961; Sitaraman et al., 2004; Hong et al., 2014), B. subtilis (Zaghloul and Doi, 1987), S. cerevisiae (Hodgman and Jewett, 2013; Gan and Jewett, 2014), or other cell extracts have been reported in the scientific literature for several decades. Several CFPS systems are commercially available and are principally marketed as protein expression systems. Optimized E. coli CFPS systems can synthesize up to 2.3 mg/ml of the target protein (Caschera and Noireaux, 2014), including those that are toxic in vivo. In recent years, the synthetic biology community has repurposed CFPS systems as in vitro transcription–translation (TX–TL) coupled characterization platforms. A typical TX–TL reaction combines a synthetic system encoded into plasmid, linear or closed circular DNA, with cell-free extract, and a reaction buffer, the contents of which can be optimized (Sun et al., 2013a). For instance, the addition of maltodextrin (Wang and Zhang, 2009) and to a lesser degree maltose (Caschera and Noireaux, 2014) as an additional energy source can increase protein production, essentially prolonging the duration of in vitro reactions for up to 10 h.
Transcription–translation characterization systems provide characterization data within a timescale of hours (Chappell et al., 2013), and are therefore, amenable to a rapid prototyping workflow (Chappell et al., 2013; Sun et al., 2013b). For instance, Chappell et al. (2013) characterized a panel of Anderson constitutive promoters, using a commercially available TX–TL system, within a 5-h workflow. Interestingly, the in vitro characterization data of a set of Anderson promoters correlated with their performance in vivo (Chappell et al., 2013). Likewise, in the same study, a panel of LasR responsive, AHL-inducible promoters, also behaved similarly in vitro and in vivo, although meaningful comparisons could only be made where constructs were encoded into plasmid or closed circular DNA (Chappell et al., 2013). PCR-generated linear DNA templates did not produce sufficient transcription and translation of the reporter protein (Chappell et al., 2013). Based upon several reports, it is likely that linear DNA templates are unstable in vitro due to the presence of exonuclease activity in the cell-free extract (Sitaraman et al., 2004; Sun et al., 2013b). Expression of the phage lambda protein Gam, an inhibitor of RecBCD (ExoV), along with other modifications, can minimize linear DNA degradation, thus restoring protein expression to levels that are comparable to plasmid DNA constructs (Sitaraman et al., 2004; Sun et al., 2013b). Yet, in disagreement with several other studies (Chappell et al., 2013; Iyer et al., 2013; Lu and Ellington, 2014), Sun et al. (2013b) reported that in vitro characterization data were not comparable to in vivo data, though they did describe a methodology to calibrate between them. While the comparability between in vitro and in vivo characterization requires further investigation, several reports have demonstrated that cell-free TX–TL systems have proven utility in the rapid prototyping of logic-based genetic circuits (Karig et al., 2012; Shin and Noireaux, 2012; Iyer et al., 2013) or synthetic operons (Lu and Ellington, 2014). Within a systematic design context, in vitro characterization approaches have the potential to complement in vivo prototyping efforts by rapidly providing the characterization data required to rationally select a smaller number of designs for final testing (Figure 3).
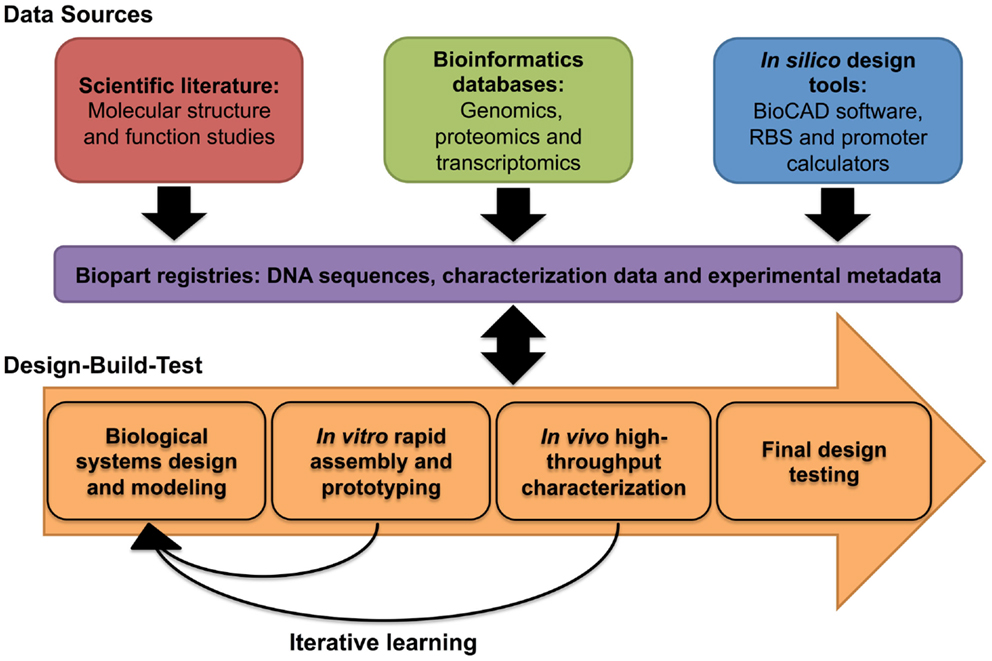
Figure 3. Systematic design of biological systems. The biological design cycle is one of several engineering principles that have been adopted in synthetic biology, and it describes the iterative process of designing a biological system through multiple rounds of design, build, and testing. To ensure that iterations of the design cycle are informative, the systematic capture, and integration of experimental and experiential data within a biological design workflow, such as the one shown here, is desirable.
Conclusion
Synthetic biology is generally described as the “engineering of biology” yet since its inception, the field has faced the well-understood reality that biological systems are complex, stochastic, and difficult to predict, and are therefore, intrinsically difficult to engineer. In order to address these fundamental challenges, synthetic biology must use and explore the existing large body of knowledge of biological systems at different scales from molecular to cellular to organismal. By establishing a systematic design framework in which existing biological knowledge can be adapted and utilized will ensure the rapid development of successful applications using synthetic biology. Furthermore, the accumulated measurements and acquired knowledge of many synthetic biology experiments will allow synthetic biologists to establish design rules that tackle biological complexity, such that robust biological systems can be designed, assembled, and prototyped as part of a biological design cycle. At each stage of the design cycle, an expanding repertoire of tools is being developed. In this review, we have highlighted several of these tools in terms of their applications and benefits to the synthetic biology community within the context of the synthetic biology design cycle namely, designing predictable biology (design), assembling DNA into bioparts, pathways, and genomes (build), and rapid prototyping (test).
Design encompasses the development of tools and methodologies that make it easier to forward-design predictable synthetic biological systems. While there are several areas that are critical to designing predictable biology including, chassis selection, biopart design, or engineering strategies, as well as, several accompanying in silico design tools, we would argue that measurement and characterization (metrology) of biological parts, devices, and systems is essential for the field of synthetic biology to fulfill its promise. It is only through improvements in our ability to measure and generate meaningful conclusions about the behavior of biological processes that the field can progress in terms of unlocking additional biological design rules. The RBS calculator is the current exemplar of this perspective, though further work is required to equip the synthetic biology toolbox with the tools to make it easier to engineer radically complex synthetic biological parts, devices, and systems.
Build encompasses DNA assembly and genome-engineering methods that enable synthetic systems to be assembled. The field has benefited immensely from the BioBrick assembly standard. BioBrick assembly, effectively making bioparts reusable (modular) at the physical DNA level, creates a standard that enables multiple research groups to use and share an expanding library of bioparts, without the need for bespoke cloning strategies. While limitations in the BioBrick assembly standard led to the emergence of powerful overlap-directed assembly methods, including Gibson, these methods also shifted away from several of the core principals of synthetic biology since these methods rely on bespoke cloning strategies. However, emerging DNA assembly methods including MODAL or Gibson with UNSes, aim to unify the advantages of overlap-directed assembly with the engineering principle of modularity. However, advances in DNA synthesis and resultant reduction of costs could radically transform the field, such that more time could be diverted away from DNA assembly toward the designing or testing of synthetic systems.
Test encompasses elements of biopart characterization, since even the testing of non-functional designs may provide insights into our understanding of the biological design rules. Liquid-handling robot high-throughput characterization platforms, along with plate readers are equipped to test prototypes of synthetic bioparts, devices, and systems. However, these systems benefit from the addition of flow cytometry and microfluidics, which bring single cell analysis to these platforms. Thus, individual cells could be analyzed and selected based upon preferred biological performance from a heterogeneous cell mix. Additionally, an array of emerging in vitro TX–TL cell-free characterization systems provide characterization data within a timescale of hours, and are therefore, amenable to a rapid prototyping workflow. Such systems are complementary to in vivo high-throughput approaches and may speed up iterations through the design cycle by reducing the number of final designs that need to be tested.
As the synthetic biology toolkit expands and more design rules are unlocked, the most successful forward-design strategies are likely to be those that encompass a diverse workflow that combines several interoperable tools at each stage of the design cycle.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We wish to acknowledge the support of the Engineering and Physical Science Research Council (EPSRC) and that of our colleagues in the Centre for Synthetic Biology and Innovation (CSynBI) at Imperial College. Funding: EPSRC [EP/K034359/1; EP/J02175X/1], Bill and Melinda Gates Foundation [OPP1046311], The Wellcome Trust [084369/Z/07/Z].
References
Acevedo-Rocha, C. G., Fang, G., Schmidt, M., Ussery, D. W., and Danchin, A. (2013). From essential to persistent genes: a functional approach to constructing synthetic life. Trends Genet. 29, 273–279. doi:10.1016/j.tig.2012.11.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Agapakis, C. M. (2013). Designing synthetic biology. ACS Synth. Biol. 3, 121–128. doi:10.1021/sb4001068
Agapakis, C. M., Boyle, P. M., and Silver, P. A. (2012). Natural strategies for the spatial optimization of metabolism in synthetic biology. Nat. Chem. Biol. 8, 527–535. doi:10.1038/nchembio.975
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Anderson, J., Strelkowa, N., Stan, G. B., Douglas, T., Savulescu, J., Barahona, M., et al. (2012). Engineering and ethical perspectives in synthetic biology. Rigorous, robust and predictable designs, public engagement and a modern ethical framework are vital to the continued success of synthetic biology. EMBO Rep. 13, 584–590. doi:10.1038/embor.2012.81
Anderson, J. C., Dueber, J. E., Leguia, M., Wu, G. C., Goler, J. A., Arkin, A. P., et al. (2010). BglBricks: a flexible standard for biological part assembly. J. Biol. Eng. 4, 1. doi:10.1186/1754-1611-4-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Andrianantoandro, E., Basu, S., Karig, D. K., and Weiss, R. (2006). Synthetic biology: new engineering rules for an emerging discipline. Mol. Syst. Biol. 2, 0028. doi:10.1038/msb4100073
Andronescu, M., Fejes, A. P., Hutter, F., Hoos, H. H., and Condon, A. (2004). A new algorithm for RNA secondary structure design. J. Mol. Biol. 336, 607–624. doi:10.1016/j.jmb.2003.12.041
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Annaluru, N., Muller, H., Mitchell, L. A., Ramalingam, S., Stracquadanio, G., Richardson, S. M., et al. (2014). Total synthesis of a functional designer eukaryotic chromosome. Science 344, 55–58. doi:10.1126/science.1249252
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Arkin, A. (2008). Setting the standard in synthetic biology. Nat. Biotechnol. 26, 771–774. doi:10.1038/nbt0708-771
Arkin, A. P. (2013). A wise consistency: engineering biology for conformity, reliability, predictability. Curr. Opin. Chem. Biol. 17, 893–901. doi:10.1016/j.cbpa.2013.09.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Arpino, J. A., Hancock, E. J., Anderson, J., Barahona, M., Stan, G. B., Papachristodoulou, A., et al. (2013). Tuning the dials of synthetic biology. Microbiology 159, 1236–1253. doi:10.1099/mic.0.067975-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Atlas, J. C., Nikolaev, E. V., Browning, S. T., and Shuler, M. L. (2008). Incorporating genome-wide DNA sequence information into a dynamic whole-cell model of Escherichia coli: application to DNA replication. IET Syst. Biol. 2, 369–382. doi:10.1049/iet-syb:20070079
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Aw, R., and Polizzi, K. M. (2013). Can too many copies spoil the broth? Microb. Cell Fact. 12, 128. doi:10.1186/1475-2859-12-128
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Barrangou, R., Fremaux, C., Deveau, H., Richards, M., Boyaval, P., Moineau, S., et al. (2007). CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712. doi:10.1126/science.1138140
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Barrick, J. E., and Breaker, R. R. (2007). The distributions, mechanisms, and structures of metabolite-binding riboswitches. Genome Biol. 8, R239. doi:10.1186/gb-2007-8-11-r239
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bartosiak-Jentys, J., Hussein, A. H., Lewis, C. J., and Leak, D. J. (2013). Modular system for assessment of glycosyl hydrolase secretion in Geobacillus thermoglucosidasius. Microbiology 159, 1267–1275. doi:10.1099/mic.0.066332-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Basiji, D. A., Ortyn, W. E., Liang, L., Venkatachalam, V., and Morrissey, P. (2007). Cellular image analysis and imaging by flow cytometry. Clin. Lab. Med. 27, 653–670. doi:10.1016/j.cll.2007.05.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Benedetto, A., Accetta, G., Fujita, Y., and Charras, G. (2014). Spatiotemporal control of gene expression using microfluidics. Lab. Chip 14, 1336–1347. doi:10.1039/c3lc51281a
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Berla, B. M., Saha, R., Immethun, C. M., Maranas, C. D., Moon, T. S., and Pakrasi, H. B. (2013). Synthetic biology of cyanobacteria: unique challenges and opportunities. Front. Microbiol. 4:246. doi:10.3389/fmicb.2013.00246
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bikard, D., Jiang, W., Samai, P., Hochschild, A., Zhang, F., and Marraffini, L. A. (2013). Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res. 41, 7429–7437. doi:10.1093/nar/gkt520
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Blount, B. A., Weenink, T., Vasylechko, S., and Ellis, T. (2012). Rational diversification of a promoter providing fine-tuned expression and orthogonal regulation for synthetic biology. PLoS ONE 7:e33279. doi:10.1371/journal.pone.0033279
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Boch, J., Scholze, H., Schornack, S., Landgraf, A., Hahn, S., Kay, S., et al. (2009). Breaking the code of DNA binding specificity of TAL-type III effectors. Science 326, 1509–1512. doi:10.1126/science.1178811
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Boehm, C. R., Freemont, P. S., and Ces, O. (2013). Design of a prototype flow microreactor for synthetic biology in vitro. Lab. Chip 13, 3426–3432. doi:10.1039/c3lc50231g
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bonde, M. T., Klausen, M. S., Anderson, M. V., Wallin, A. I., Wang, H. H., and Sommer, M. O. (2014). MODEST: a web-based design tool for oligonucleotide-mediated genome engineering and recombineering. Nucleic Acids Res. 42, W408–W415. doi:10.1093/nar/gku428
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bower, A. G., McClintock, M. K., and Fong, S. S. (2010). Synthetic biology: a foundation for multi-scale molecular biology. Bioeng. Bugs 1, 309–312. doi:10.4161/bbug.1.5.12391
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cachat, E., and Davies, J. A. (2011). Application of synthetic biology to regenerative medicine. J. Bioeng. Biomed. Sci. S2:003. doi:10.4172/2155-9538.S2-003
Calvert, J. (2012). Ownership and sharing in synthetic biology: a/‘diverse ecology/’ of the open and the proprietary[quest]. BioSocieties 7, 169–187. doi:10.1057/biosoc.2012.3
Canton, B., Labno, A., and Endy, D. (2008). Refinement and standardization of synthetic biological parts and devices. Nat. Biotechnol. 26, 787–793. doi:10.1038/nbt1413
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Carlson, R. (2009). The changing economics of DNA synthesis. Nat. Biotechnol. 27, 1091–1094. Available at: http://www.synthesis.cc/cgi-bin/mt/mt-search.cgi?blog_id=1&tag=CarlsonCurves&limit=20 doi:10.1038/nbt1209-1091
Caschera, F., and Noireaux, V. (2014). Synthesis of 2.3 mg/ml of protein with an all Escherichia coli cell-free transcription-translation system. Biochimie 99, 162–168. doi:10.1016/j.biochi.2013.11.025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Casini, A., Christodoulou, G., Freemont, P. S., Baldwin, G. S., Ellis, T., and MacDonald, J. T. (2014). R2oDNA designer: computational design of biologically neutral synthetic DNA Sequences. ACS Synth. Biol. 3, 525–528. doi:10.1021/sb4001323
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Casini, A., MacDonald, J. T., Jonghe, J. D., Christodoulou, G., Freemont, P. S., Baldwin, G. S., et al. (2013). One-pot DNA construction for synthetic biology: the modular overlap-directed assembly with linkers (MODAL) strategy. Nucleic Acids Res. 42, e7. doi:10.1093/nar/gkt915
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cereghino, J. (2000). Heterologous protein expression in the methylotrophic yeast Pichia pastoris. FEMS Microbiol. Rev. 24, 45–66. doi:10.1016/s0168-6445(99)00029-7
Chao, R., Yuan, Y., and Zhao, H. (2014). Recent advances in DNA assembly technologies. FEMS Yeast Res. doi:10.1111/1567-1364.12171
Chappell, J., Jensen, K., and Freemont, P. S. (2013). Validation of an entirely in vitro approach for rapid prototyping of DNA regulatory elements for synthetic biology. Nucleic Acids Res. 41, 3471–3481. doi:10.1093/nar/gkt052
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, I. A., Roberts, R. W., and Szostak, J. W. (2004). The emergence of competition between model protocells. Science 305, 1474–1476. doi:10.1126/science.1100757
Chin, J. X., Chung, B. K., and Lee, D. Y. (2014). Codon optimization online (COOL): a web-based multi-objective optimization platform for synthetic gene design. Bioinformatics 30, 2210–2212. doi:10.1093/bioinformatics/btu192
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chiyoda, S., Yamato, K. T., and Kohchi, T. (2014). Plastid transformation of sporelings and suspension-cultured cells from the liverwort Marchantia polymorpha L. Methods Mol. Biol. 1132, 439–447. doi:10.1007/978-1-62703-995-6_30
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chizzolini, F., Forlin, M., Cecchi, D., and Mansy, S. S. (2013). Gene position more strongly influences cell-free protein expression from operons than T7 transcriptional promoter strength. ACS Synth. Biol 3, 363–371. doi:10.1021/sb4000977
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cho, M., Soo Oh, S., Nie, J., Stewart, R., Eisenstein, M., Chambers, J., et al. (2013). Quantitative selection and parallel characterization of aptamers. Proc. Natl. Acad. Sci. U.S.A. 110, 18460–18465. doi:10.1073/pnas.1315866110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Choi, H. J., and Montemagno, C. D. (2005). Artificial organelle: ATP synthesis from cellular mimetic polymersomes. Nano Lett. 5, 2538–2542. doi:10.1021/nl051896e
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Choi, S. L., Rha, E., Lee, S. J., Kim, H., Kwon, K., Jeong, Y. S., et al. (2013). Toward a generalized and high-throughput enzyme screening system based on artificial genetic circuits. ACS Synth. Biol. 3, 163–171. doi:10.1021/sb400112u
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chung, B. K., and Lee, D. Y. (2012). Computational codon optimization of synthetic gene for protein expression. BMC Syst. Biol. 6:134. doi:10.1186/1752-0509-6-134
Cohen, S. N., Chang, A. C., Boyer, H. W., and Helling, R. B. (1973). Construction of biologically functional bacterial plasmids in vitro. Proc. Natl. Acad. Sci. U.S.A. 70, 3240–3244. doi:10.1073/pnas.70.11.3240
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Collins, F. S., and Tabak, L. A. (2014). Policy: NIH plans to enhance reproducibility. Nature 505, 612–613. doi:10.1038/505612a
Colloms, S. D., Merrick, C. A., Olorunniji, F. J., Stark, W. M., Smith, M. C., Osbourn, A., et al. (2014). Rapid metabolic pathway assembly and modification using serine integrase site-specific recombination. Nucleic Acids Res. 42, e23. doi:10.1093/nar/gkt1101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto, R., Habib, N., et al. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823. doi:10.1126/science.1231143
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cooling, M. T., Rouilly, V., Misirli, G., Lawson, J., Yu, T., Hallinan, J., et al. (2010). Standard virtual biological parts: a repository of modular modeling components for synthetic biology. Bioinformatics 26, 925–931. doi:10.1093/bioinformatics/btq063
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cregg, J. M., Cereghino, J. L., Shi, J., and Higgins, D. R. (2000). Recombinant protein expression in Pichia pastoris. Mol. Biotechnol. 16, 23–52. doi:10.1385/mb:16:1:23
Cregg, J. M., Tolstorukov, I., Kusari, A., Sunga, J., Madden, K., and Chappell, T. (2009). Chapter 13 expression in the yeast Pichia pastoris. Meth. Enzymol. 463, 169–189. doi:10.1016/s0076-6879(09)63013-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Csete, M. E., and Doyle, J. C. (2002). Reverse engineering of biological complexity. Science 295, 1664–1669. doi:10.1126/science.1069981
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Danchin, A., and Sekowska, A. (2014). The logic of metabolism and its fuzzy consequences. Environ. Microbiol. 16, 19–28. doi:10.1111/1462-2920.12270
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Davis, J. H., Rubin, A. J., and Sauer, R. T. (2011). Design, construction and characterization of a set of insulated bacterial promoters. Nucleic Acids Res. 39, 1131–1141. doi:10.1093/nar/gkq810
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
de Kok, S., Stanton, L. H., Slaby, T., Durot, M., Holmes, V. F., Patel, K. G., et al. (2014). Rapid and reliable DNA assembly via ligase cycling reaction. ACS Synth. Biol. 3, 97–106. doi:10.1021/sb4001992
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Delebecque, C. J., Lindner, A. B., Silver, P. A., and Aldaye, F. A. (2011). Organization of intracellular reactions with rationally designed RNA assemblies. Science 333, 470–474. doi:10.1126/science.1206938
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Depaoli, H. C., Borland, A. M., Tuskan, G. A., Cushman, J. C., and Yang, X. (2014). Synthetic biology as it relates to CAM photosynthesis: challenges and opportunities. J. Exp. Bot. 65, 3381–3393. doi:10.1093/jxb/eru038
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dewall, M. T., and Cheng, D. W. (2011). The minimal genome: a metabolic and environmental comparison. Brief. Funct. Genomics 10, 312–315. doi:10.1093/bfgp/elr030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Díaz, M., Herrero, M., García, L. A., and Quirós, C. (2010). Application of flow cytometry to industrial microbial bioprocesses. Biochem. Eng. J. 48, 385–407. doi:10.1016/j.bej.2009.07.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Douglas, C. M. W., and Stemerding, D. (2014). Challenges for the European governance of synthetic biology for human health. Life Sci. Soc. Policy 10, 6. doi:10.1186/s40504-014-0006-7
Douglas, S. M., Dietz, H., Liedl, T., Hogberg, B., Graf, F., and Shih, W. M. (2009). Self-assembly of DNA into nanoscale three-dimensional shapes. Nature 459, 414–418. doi:10.1038/nature08016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dueber, J. E., Wu, G. C., Malmirchegini, G. R., Moon, T. S., Petzold, C. J., Ullal, A. V., et al. (2009). Synthetic protein scaffolds provide modular control over metabolic flux. Nat. Biotechnol. 27, 753–759. doi:10.1038/nbt.1557
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dutton, P. L., and Moser, C. C. (2011). Engineering enzymes. Faraday Discuss. 148, 443. doi:10.1039/c005523a
Dymond, J., and Boeke, J. (2012). The Saccharomyces cerevisiae SCRaMbLE system and genome minimization. Bioeng. Bugs 3, 168–171. doi:10.4161/bbug.19543
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ellefson, J. W., Meyer, A. J., Hughes, R. A., Cannon, J. R., Brodbelt, J. S., and Ellington, A. D. (2014). Directed evolution of genetic parts and circuits by compartmentalized partnered replication. Nat. Biotechnol. 32, 97–101. doi:10.1038/nbt.2714
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ellington, A. D., and Szostak, J. W. (1990). In vitro selection of RNA molecules that bind specific ligands. Nature 346, 818–822. doi:10.1038/346818a0
Ellis, B. L., Hirsch, M. L., Porter, S. N., Samulski, R. J., and Porteus, M. H. (2013). Zinc-finger nuclease-mediated gene correction using single AAV vector transduction and enhancement by food and drug administration-approved drugs. Gene Ther. 20, 35–42. doi:10.1038/gt.2011.211
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ellis, T., Adie, T., and Baldwin, G. S. (2011). DNA assembly for synthetic biology: from parts to pathways and beyond. Integr. Biol. (Camb.) 3, 109–118. doi:10.1039/c0ib00070a
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Elowitz, M. B., and Leibler, S. (2000). A synthetic oscillatory network of transcriptional regulators. Nature 403, 335–338. doi:10.1038/35002125
Engler, C., Kandzia, R., and Marillonnet, S. (2008). A one pot, one step, precision cloning method with high throughput capability. PLoS ONE 3:e3647. doi:10.1371/journal.pone.0003647
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Engler, C., and Marillonnet, S. (2011). Generation of families of construct variants using golden gate shuffling. Methods Mol. Biol. 729, 167–181. doi:10.1007/978-1-61779-065-2_11
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Engler, C., and Marillonnet, S. (2013). Combinatorial DNA assembly using golden gate cloning. Methods Mol. Biol. 1073, 141–156. doi:10.1007/978-1-62703-625-2_12
Esvelt, K. M., Carlson, J. C., and Liu, D. R. (2011). A system for the continuous directed evolution of biomolecules. Nature 472, 499–503. doi:10.1038/nature09929
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fletcher, J. M., Harniman, R. L., Barnes, F. R., Boyle, A. L., Collins, A., Mantell, J., et al. (2013). Self-assembling cages from coiled-coil peptide modules. Science 340, 595–599. doi:10.1126/science.1233936
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Frow, E., and Calvert, J. (2013). Opening up the future(s) of synthetic biology. Futures 48, 32–43. doi:10.1016/j.futures.2013.03.001
Fu, J., Liu, M., Liu, Y., Woodbury, N. W., and Yan, H. (2012). Interenzyme substrate diffusion for an enzyme cascade organized on spatially addressable DNA nanostructures. J. Am. Chem. Soc. 134, 5516–5519. doi:10.1021/ja300897h
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fu, J., Yang, Y. R., Johnson-Buck, A., Liu, M., Liu, Y., Walter, N. G., et al. (2014). Multi-enzyme complexes on DNA scaffolds capable of substrate channelling with an artificial swinging arm. Nat. Nanotechnol. 9, 531–536. doi:10.1038/nnano.2014.100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Galdzicki, M., Clancy, K. P., Oberortner, E., Pocock, M., Quinn, J. Y., Rodriguez, C. A., et al. (2014). The synthetic biology open language (SBOL) provides a community standard for communicating designs in synthetic biology. Nat. Biotechnol. 32, 545–550. doi:10.1038/nbt.2891
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Galdzicki, M., Rodriguez, C., Chandran, D., Sauro, H. M., and Gennari, J. H. (2011). Standard biological parts knowledgebase. PLoS ONE 6:e17005. doi:10.1371/journal.pone.0017005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gama-Castro, S., Salgado, H., Peralta-Gil, M., Santos-Zavaleta, A., Muniz-Rascado, L., Solano-Lira, H., et al. (2011). RegulonDB version 7.0: transcriptional regulation of Escherichia coli K-12 integrated within genetic sensory response units (Gensor Units). Nucleic Acids Res. 39, D98–D105. doi:10.1093/nar/gkq1110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gan, R., and Jewett, M. C. (2014). A combined cell-free transcription-translation system from Saccharomyces cerevisiae for rapid and robust protein synthesis. Biotechnol. J. 9, 641–651. doi:10.1002/biot.201300545
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gardner, T. S., Cantor, C. R., and Collins, J. J. (2000). Construction of a genetic toggle switch in Escherichia coli. Nature 403, 339–342. doi:10.1038/35002131
Gibson, D. G. (2011). Enzymatic assembly of overlapping DNA fragments. Meth. Enzymol. 498, 349–361. doi:10.1016/B978-0-12-385120-8.00015-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gibson, D. G., Benders, G. A., Andrews-Pfannkoch, C., Denisova, E. A., Baden-Tillson, H., Zaveri, J., et al. (2008). Complete chemical synthesis, assembly, and cloning of a Mycoplasma genitalium genome. Science 319, 1215–1220. doi:10.1126/science.1151721
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gibson, D. G., Glass, J. I., Lartigue, C., Noskov, V. N., Chuang, R. Y., Algire, M. A., et al. (2010a). Creation of a bacterial cell controlled by a chemically synthesized genome. Science 329, 52–56. doi:10.1126/science.1190719
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gibson, D. G., Smith, H. O., Hutchison, C. A. III, Venter, J. C., and Merryman, C. (2010b). Chemical synthesis of the mouse mitochondrial genome. Nat. Methods 7, 901–903. doi:10.1038/nmeth.1515
Gibson, D. G., Young, L., Chuang, R. Y., Venter, J. C., Hutchison, C. A. III, and Smith, H. O. (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345. doi:10.1038/nmeth.1318
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Glass, J. I., Assad-Garcia, N., Alperovich, N., Yooseph, S., Lewis, M. R., Maruf, M., et al. (2006). Essential genes of a minimal bacterium. Proc. Natl. Acad. Sci. U.S.A. 103, 425–430. doi:10.1073/pnas.0510013103
Guye, P., Li, Y., Wroblewska, L., Duportet, X., and Weiss, R. (2013). Rapid, modular and reliable construction of complex mammalian gene circuits. Nucleic Acids Res. 41, e156. doi:10.1093/nar/gkt605
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hamilton, S. R., and Gerngross, T. U. (2007). Glycosylation engineering in yeast: the advent of fully humanized yeast. Curr. Opin. Biotechnol. 18, 387–392. doi:10.1016/j.copbio.2007.09.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hammer, D. A., and Kamat, N. P. (2012). Towards an artificial cell. FEBS Lett. 586, 2882–2890. doi:10.1016/j.febslet.2012.07.044
Han, D., Pal, S., Nangreave, J., Deng, Z., Liu, Y., and Yan, H. (2011). DNA origami with complex curvatures in three-dimensional space. Science 332, 342–346. doi:10.1126/science.1202998
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hansen, A. S., and O’Shea, E. K. (2013). Promoter decoding of transcription factor dynamics involves a trade-off between noise and control of gene expression. Mol. Syst. Biol. 9, 704. doi:10.1038/msb.2013.56
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Harwood, C. R., Pohl, S., Smith, W., and Wipat, A. (2013). Bacillus subtilis: model Gram-positive synthetic biology chassis. Comput. Sci. 40, 87–117. doi:10.1016/b978-0-12-417029-2.00004-2
Heider, S. A. E., Wolf, N., Hofemeier, A., Peters-Wendisch, P., and Wendisch, V. F. (2014). Optimization of the IPP precursor supply for the production of lycopene, decaprenoxanthin and astaxanthin by Corynebacterium glutamicum. Front. Bioeng. Biotechnol. 2:28. doi:10.3389/fbioe.2014.00028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Heidorn, T., Camsund, D., Huang, H. H., Lindberg, P., Oliveira, P., Stensjo, K., et al. (2011). Synthetic biology in cyanobacteria engineering and analyzing novel functions. Meth. Enzymol. 497, 539–579. doi:10.1016/B978-0-12-385075-1.00024-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Heigwer, F., Kerr, G., and Boutros, M. (2014). E-CRISP: fast CRISPR target site identification. Nat. Methods 11, 122–123. doi:10.1038/nmeth.2812
Heinemann, M., and Panke, S. (2006). Synthetic biology – putting engineering into biology. Bioinformatics 22, 2790–2799. doi:10.1093/bioinformatics/btl469
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hillson, N. J., Rosengarten, R. D., and Keasling, J. D. (2012). j5 DNA assembly design automation software. ACS Synth. Biol. 1, 14–21. doi:10.1021/sb2000116
Hiroe, A., Tsuge, K., Nomura, C. T., Itaya, M., and Tsuge, T. (2012). Rearrangement of gene order in the phaCAB operon leads to effective production of ultrahigh-molecular-weight poly[(R)-3-hydroxybutyrate] in genetically engineered Escherichia coli. Appl. Environ. Microbiol. 78, 3177–3184. doi:10.1128/AEM.07715-11
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hodgman, C. E., and Jewett, M. C. (2013). Optimized extract preparation methods and reaction conditions for improved yeast cell-free protein synthesis. Biotechnol. Bioeng. 110, 2643–2654. doi:10.1002/bit.24942
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hong, S. H., Kwon, Y.-C., and Jewett, M. C. (2014). Non-standard amino acid incorporation into proteins using Escherichia coli cell-free protein synthesis. Front. Chem. 2:34. doi:10.3389/fchem.2014.00034
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Howorka, S. (2011). Rationally engineering natural protein assemblies in nanobiotechnology. Curr. Opin. Biotechnol. 22, 485–491. doi:10.1016/j.copbio.2011.05.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Isaacs, F. J., Carr, P. A., Wang, H. H., Lajoie, M. J., Sterling, B., Kraal, L., et al. (2011). Precise manipulation of chromosomes in vivo enables genome-wide codon replacement. Science 333, 348–353. doi:10.1126/science.1205822
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Itaya, M., Fujita, K., Kuroki, A., and Tsuge, K. (2008). Bottom-up genome assembly using the Bacillus subtilis genome vector. Nat. Methods 5, 41–43. doi:10.1038/nmeth1143
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Iwata, T., Kaneko, S., Shiwa, Y., Enomoto, T., Yoshikawa, H., and Hirota, J. (2013). Bacillus subtilis genome vector-based complete manipulation and reconstruction of genomic DNA for mouse transgenesis. BMC Genomics 14:300. doi:10.1186/1471-2164-14-300
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Iyer, S., Karig, D. K., Norred, S. E., Simpson, M. L., and Doktycz, M. J. (2013). Multi-input regulation and logic with T7 promoters in cells and cell-free systems. PLoS ONE 8:e78442. doi:10.1371/journal.pone.0078442
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jefferson, C., Lentzos, F., and Marris, C. (2014). Synthetic biology and biosecurity: challenging the “myths”. Front. Public Health 2:115. doi:10.3389/fpubh.2014.00115
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jenison, R. D., Gill, S. C., Pardi, A., and Polisky, B. (1994). High-resolution molecular discrimination by RNA. Science 263, 1425–1429. doi:10.1126/science.7510417
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jiang, L., Althoff, E. A., Clemente, F. R., Doyle, L., Rothlisberger, D., Zanghellini, A., et al. (2008). De novo computational design of retro-aldol enzymes. Science 319, 1387–1391. doi:10.1126/science.1152692
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Karig, D. K., Iyer, S., Simpson, M. L., and Doktycz, M. J. (2012). Expression optimization and synthetic gene networks in cell-free systems. Nucleic Acids Res. 40, 3763–3774. doi:10.1093/nar/gkr1191
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kelly, J. R., Rubin, A. J., Davis, J. H., Ajo-Franklin, C. M., Cumbers, J., Czar, M. J., et al. (2009). Measuring the activity of BioBrick promoters using an in vivo reference standard. J. Biol. Eng. 3, 4. doi:10.1186/1754-1611-3-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keren, L., Zackay, O., Lotan-Pompan, M., Barenholz, U., Dekel, E., Sasson, V., et al. (2013). Promoters maintain their relative activity levels under different growth conditions. Mol. Syst. Biol. 9, 701. doi:10.1038/msb.2013.59
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Khalil, A. S., and Collins, J. J. (2010). Synthetic biology: applications come of age. Nat. Rev. Genet. 11, 367–379. doi:10.1038/nrg2775
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
King, N. P., Bale, J. B., Sheffler, W., McNamara, D. E., Gonen, S., Gonen, T., et al. (2014). Accurate design of co-assembling multi-component protein nanomaterials. Nature 510, 103–108. doi:10.1038/nature13404
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
King, N. P., Sheffler, W., Sawaya, M. R., Vollmar, B. S., Sumida, J. P., Andre, I., et al. (2012). Computational design of self-assembling protein nanomaterials with atomic level accuracy. Science 336, 1171–1174. doi:10.1126/science.1219364
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kitney, R., and Freemont, P. (2012). Synthetic biology – the state of play. FEBS Lett. 586, 2029–2036. doi:10.1016/j.febslet.2012.06.002
Koga, N., Tatsumi-Koga, R., Liu, G., Xiao, R., Acton, T. B., Montelione, G. T., et al. (2012). Principles for designing ideal protein structures. Nature 491, 222–227. doi:10.1038/nature11600
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kouprina, N., and Larionov, V. (2008). Selective isolation of genomic loci from complex genomes by transformation-associated recombination cloning in the yeast Saccharomyces cerevisiae. Nat. Protoc. 3, 371–377. doi:10.1038/nprot.2008.5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Landrain, T., Meyer, M., Perez, A. M., and Sussan, R. (2013). Do-it-yourself biology: challenges and promises for an open science and technology movement. Syst. Synth. Biol. 7, 115–126. doi:10.1007/s11693-013-9116-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lawrence, A. D., Frank, S., Newnham, S., Lee, M. J., Brown, I. R., Xue, W. F., et al. (2014). Solution structure of a bacterial microcompartment targeting peptide and its application in the construction of an ethanol bioreactor. ACS Synth. Biol. 3, 454–465. doi:10.1021/sb4001118
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Leaver-Fay, A., Tyka, M., Lewis, S. M., Lange, O. F., Thompson, J., Jacak, R., et al. (2011). ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Meth. Enzymol. 487, 545–574. doi:10.1016/B978-0-12-381270-4.00019-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, M. Z., and Elledge, S. J. (2007). Harnessing homologous recombination in vitro to generate recombinant DNA via SLIC. Nat. Methods 4, 251–256. doi:10.1038/nmeth1010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liang, J. C., Bloom, R. J., and Smolke, C. D. (2011). Engineering biological systems with synthetic RNA molecules. Mol. Cell 43, 915–926. doi:10.1016/j.molcel.2011.08.023
Lienert, F., Lohmueller, J. J., Garg, A., and Silver, P. A. (2014). Synthetic biology in mammalian cells: next generation research tools and therapeutics. Nat. Rev. Mol. Cell Biol. 15, 95–107. doi:10.1038/nrm3738
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lin, B., and Levchenko, A. (2012). Microfluidic technologies for studying synthetic circuits. Curr. Opin. Chem. Biol. 16, 307–317. doi:10.1016/j.cbpa.2012.04.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lin, Q., Jia, B., Mitchell, L. A., Luo, J., Yang, K., Zeller, K. I., et al. (2014). RADOM, an efficient in vivo method for assembling designed DNA fragments up to 10 kb long in Saccharomyces cerevisiae. ACS Synth. Biol. doi:10.1021/sb500241e
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Linshiz, G., Stawski, N., Poust, S., Bi, C., Keasling, J. D., and Hillson, N. J. (2013). PaR-PaR laboratory automation platform. ACS Synth. Biol. 2, 216–222. doi:10.1021/sb300075t
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liu, C. C., Qi, L., Lucks, J. B., Segall-Shapiro, T. H., Wang, D., Mutalik, V. K., et al. (2012). An adaptor from translational to transcriptional control enables predictable assembly of complex regulation. Nat. Methods 9, 1088–1094. doi:10.1038/nmeth.2184
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lobban, P. E., and Kaiser, A. D. (1973). Enzymatic end-to end joining of DNA molecules. J. Mol. Biol. 78, 453–471. doi:10.1016/0022-2836(73)90468-3
Lou, C., Stanton, B., Chen, Y. J., Munsky, B., and Voigt, C. A. (2012). Ribozyme-based insulator parts buffer synthetic circuits from genetic context. Nat. Biotechnol. 30, 1137–1142. doi:10.1038/nbt.2401
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lu, W.-C., and Ellington, A. D. (2014). Design and Selection of a Synthetic Operon. ACS Synth. Biol. 3, 410–415. doi:10.1021/sb400160m
MacDonald, J. T., Barnes, C., Kitney, R. I., Freemont, P. S., and Stan, G. B. (2011). Computational design approaches and tools for synthetic biology. Integr. Biol. (Camb.) 3, 97–108. doi:10.1039/c0ib00077a
Mahfouz, M. M., Li, L., Shamimuzzaman, M., Wibowo, A., Fang, X., and Zhu, J. K. (2011). De novo-engineered transcription activator-like effector (TALE) hybrid nuclease with novel DNA binding specificity creates double-strand breaks. Proc. Natl. Acad. Sci. U.S.A. 108, 2623–2628. doi:10.1073/pnas.1019533108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mali, P., Aach, J., Stranges, P. B., Esvelt, K. M., Moosburner, M., Kosuri, S., et al. (2013). CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat. Biotechnol. 31, 833–838. doi:10.1038/nbt.2675
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Marchisio, M. A. (2014). In silico design and in vivo implementation of yeast gene Boolean gates. J. Biol. Eng. 8, 6. doi:10.1186/1754-1611-8-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Marchisio, M. A., and Stelling, J. (2009). Computational design tools for synthetic biology. Curr. Opin. Biotechnol. 20, 479–485. doi:10.1016/j.copbio.2009.08.007
Markham, N. R., and Zuker, M. (2008). UNAFold: software for nucleic acid folding and hybridization. Methods Mol. Biol. 453, 3–31. doi:10.1007/978-1-60327-429-6_1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Marples, B., Scott, S. D., Hendry, J. H., Embleton, M. J., Lashford, L. S., and Margison, G. P. (2000). Development of synthetic promoters for radiation-mediated gene therapy. Gene Ther. 7, 511–517. doi:10.1038/sj.gt.3301116
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Marris, C., and Rose, N. (2010). Open engagement: exploring public participation in the biosciences. PLoS Biol. 8:e1000549. doi:10.1371/journal.pbio.1000549
Moe-Behrens, G. H., Davis, R., and Haynes, K. A. (2013). Preparing synthetic biology for the world. Front. Microbiol. 4:5. doi:10.3389/fmicb.2013.00005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Morbitzer, R., Romer, P., Boch, J., and Lahaye, T. (2010). Regulation of selected genome loci using de novo-engineered transcription activator-like effector (TALE)-type transcription factors. Proc. Natl. Acad. Sci. U.S.A. 107, 21617–21622. doi:10.1073/pnas.1013133107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moscou, M. J., and Bogdanove, A. J. (2009). A simple cipher governs DNA recognition by TAL effectors. Science 326, 1501. doi:10.1126/science.1178817
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Muller, K., Siegel, D., Rodriguez Jahnke, F., Gerrer, K., Wend, S., Decker, E. L., et al. (2014a). A red light-controlled synthetic gene expression switch for plant systems. Mol. Biosyst. 10, 1679–1688. doi:10.1039/c3mb70579j
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Muller, K., Zurbriggen, M. D., and Weber, W. (2014b). Control of gene expression using a red- and far-red light-responsive bi-stable toggle switch. Nat. Protoc. 9, 622–632. doi:10.1038/nprot.2014.038
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Murphy, K. C. (1998). Use of bacteriophage lambda recombination functions to promote gene replacement in Escherichia coli. J. Bacteriol. 180, 2063–2071.
Murphy, K. C., and Campellone, K. G. (2003). Lambda red-mediated recombinogenic engineering of enterohemorrhagic and enteropathogenic E. coli. BMC Mol. Biol. 4:11. doi:10.1186/1471-2199-4-11
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mushtaq, M. Y., Verpoorte, R., and Kim, H. K. (2013). Zebrafish as a model for systems biology. Biotechnol. Genet. Eng. Rev. 29, 187–205. doi:10.1080/02648725.2013.801238
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mutalik, V. K., Guimaraes, J. C., Cambray, G., Lam, C., Christoffersen, M. J., Mai, Q. A., et al. (2013a). Precise and reliable gene expression via standard transcription and translation initiation elements. Nat. Methods 10, 354–360. doi:10.1038/nmeth.2404
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mutalik, V. K., Guimaraes, J. C., Cambray, G., Mai, Q. A., Christoffersen, M. J., Martin, L., et al. (2013b). Quantitative estimation of activity and quality for collections of functional genetic elements. Nat. Methods 10, 347–353. doi:10.1038/nmeth.2403
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Na, D., and Lee, D. (2010). RBSDesigner: software for designing synthetic ribosome binding sites that yields a desired level of protein expression. Bioinformatics 26, 2633–2634. doi:10.1093/bioinformatics/btq458
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Narayanan, K., and Chen, Q. (2011). Bacterial artificial chromosome mutagenesis using recombineering. J. Biomed. Biotechnol. 2011, 971296. doi:10.1155/2011/971296
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nirenberg, M. W., and Matthaei, J. H. (1961). The dependence of cell-free protein synthesis in E. coli upon naturally occurring or synthetic polyribonucleotides. Proc. Natl. Acad. Sci. U.S.A. 47, 1588–1602. doi:10.1073/pnas.47.10.1588
Nishiyama, T. (2000). Tagged mutagenesis and gene-trap in the moss, Physcomitrella patens by shuttle mutagenesis. DNA Res. 7, 9–17. doi:10.1093/dnares/7.1.9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
O’Brien, E. J., Lerman, J. A., Chang, R. L., Hyduke, D. R., and Palsson, B. O. (2013). Genome-scale models of metabolism and gene expression extend and refine growth phenotype prediction. Mol. Syst. Biol. 9, 693. doi:10.1038/msb.2013.52
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ohtani, N., Hasegawa, M., Sato, M., Tomita, M., Kaneko, S., and Itaya, M. (2012). Serial assembly of Thermus megaplasmid DNA in the genome of Bacillus subtilis 168: a BAC-based domino method applied to DNA with a high GC content. Biotechnol. J. 7, 867–876. doi:10.1002/biot.201100396
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Olson, E. J., Hartsough, L. A., Landry, B. P., Shroff, R., and Tabor, J. J. (2014). Characterizing bacterial gene circuit dynamics with optically programmed gene expression signals. Nat. Methods 11, 449–455. doi:10.1038/nmeth.2884
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
O’Reilly, L. P., Luke, C. J., Perlmutter, D. H., Silverman, G. A., and Pak, S. C. (2014). C. elegans in high-throughput drug discovery. Adv. Drug Deliv. Rev. 6, 247–253. doi:10.1016/j.addr.2013.12.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Padilla, J. E., Colovos, C., and Yeates, T. O. (2001). Nanohedra: using symmetry to design self assembling protein cages, layers, crystals, and filaments. Proc. Natl. Acad. Sci. U.S.A. 98, 2217–2221. doi:10.1073/pnas.041614998
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Paige, J. S., Wu, K. Y., and Jaffrey, S. R. (2011). RNA mimics of green fluorescent protein. Science 333, 642–646. doi:10.1126/science.1207339
Penchovsky, R., and Breaker, R. R. (2005). Computational design and experimental validation of oligonucleotide-sensing allosteric ribozymes. Nat. Biotechnol. 23, 1424–1433. doi:10.1038/nbt1155
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Piyasena, M. E., and Graves, S. W. (2014). The intersection of flow cytometry with microfluidics and microfabrication. Lab. Chip 14, 1044–1059. doi:10.1039/c3lc51152a
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pothoulakis, G., Ceroni, F., Reeve, B., and Ellis, T. (2014). The spinach RNA aptamer as a characterization tool for synthetic biology. ACS Synth. Biol. 3, 182–187. doi:10.1021/sb400089c
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Preston, B. (2013). Synthetic biology as red herring. Stud. Hist. Philos. Biol. Biomed. Sci. 44, 649–659. doi:10.1016/j.shpsc.2013.05.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Purnick, P. E., and Weiss, R. (2009). The second wave of synthetic biology: from modules to systems. Nat. Rev. Mol. Cell Biol. 10, 410–422. doi:10.1038/nrm2698
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Qi, L., Haurwitz, R. E., Shao, W., Doudna, J. A., and Arkin, A. P. (2012). RNA processing enables predictable programming of gene expression. Nat. Biotechnol. 30, 1002–1006. doi:10.1038/nbt.2355
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Qi, L. S., Larson, M. H., Gilbert, L. A., Doudna, J. A., Weissman, J. S., Arkin, A. P., et al. (2013). Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152, 1173–1183. doi:10.1016/j.cell.2013.02.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Quan, J., and Tian, J. (2009). Circular polymerase extension cloning of complex gene libraries and pathways. PLoS ONE 4:e6441. doi:10.1371/journal.pone.0006441
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Quan, J., and Tian, J. (2011). Circular polymerase extension cloning for high-throughput cloning of complex and combinatorial DNA libraries. Nat. Protoc. 6, 242–251. doi:10.1038/nprot.2010.181
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Quinn, J., Beal, J., Bhatia, S., Cai, P., Chen, J., Clancy, K., et al. (2013). Synthetic biology open language visual (SBOL visual), version 1.0.0, BBF RFC #93. doi:1721.1/78249
Radeck, J., Kraft, K., Bartels, J., Cikovic, T., Durr, F., Emenegger, J., et al. (2013). The Bacillus BioBrick Box: generation and evaluation of essential genetic building blocks for standardized work with Bacillus subtilis. J. Biol. Eng. 7, 29. doi:10.1186/1754-1611-7-29
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rajagopal, K., and Schneider, J. P. (2004). Self-assembling peptides and proteins for nanotechnological applications. Curr. Opin. Struct. Biol. 14, 480–486. doi:10.1016/j.sbi.2004.06.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Redemann, S., Schloissnig, S., Ernst, S., Pozniakowsky, A., Ayloo, S., Hyman, A. A., et al. (2011). Codon adaptation-based control of protein expression in C. elegans. Nat. Methods 8, 250–252. doi:10.1038/nmeth.1565
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Reeve, B., Hargest, T., Gilbert, C., and Ellis, T. (2014). Predicting translation initiation rates for designing synthetic biology. Front. Bioeng. Biotechnol. 2:1. doi:10.3389/fbioe.2014.00001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rhodius, V. A., and Mutalik, V. K. (2010). Predicting strength and function for promoters of the Escherichia coli alternative sigma factor, sigmaE. Proc. Natl. Acad. Sci. U.S.A. 107, 2854–2859. doi:10.1073/pnas.0915066107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rhodius, V. A., Mutalik, V. K., and Gross, C. A. (2012). Predicting the strength of UP-elements and full-length E. coli sigmaE promoters. Nucleic Acids Res. 40, 2907–2924. doi:10.1093/nar/gkr1190
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rodrigo, G., and Jaramillo, A. (2013). AutoBioCAD: full biodesign automation of genetic circuits. ACS Synth. Biol. 2, 230–236. doi:10.1021/sb300084h
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Roehner, N., and Myers, C. J. (2013). A methodology to annotate systems biology markup language models with the synthetic biology open language. ACS Synth. Biol. 3, 57–66. doi:10.1021/sb400066m
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rokke, G., Korvald, E., Pahr, J., Oyas, O., and Lale, R. (2014). BioBrick assembly standards and techniques and associated software tools. Methods Mol. Biol. 1116, 1–24. doi:10.1007/978-1-62703-764-8_1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ross, J. A., Ellis, M. J., Hossain, S., and Haniford, D. B. (2013). Hfq restructures RNA-IN and RNA-OUT and facilitates antisense pairing in the Tn10/IS10 system. RNA 19, 670–684. doi:10.1261/rna.037747.112
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Roth, A., and Breaker, R. R. (2009). The structural and functional diversity of metabolite-binding riboswitches. Annu. Rev. Biochem. 78, 305–334. doi:10.1146/annurev.biochem.78.070507.135656
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rothemund, P. W. (2006). Folding DNA to create nanoscale shapes and patterns. Nature 440, 297–302. doi:10.1038/nature04586
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rothlisberger, D., Khersonsky, O., Wollacott, A. M., Jiang, L., Dechancie, J., Betker, J., et al. (2008). Kemp elimination catalysts by computational enzyme design. Nature 453, 190–195. doi:10.1038/nature06879
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rouillard, J. M., Lee, W., Truan, G., Gao, X., Zhou, X., and Gulari, E. (2004). Gene2Oligo: oligonucleotide design for in vitro gene synthesis. Nucleic Acids Res. 32, W176–W180. doi:10.1093/nar/gkh401
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Salis, H. M. (2011). The ribosome binding site calculator. Meth. Enzymol. 498, 19–42. doi:10.1016/B978-0-12-385120-8.00002-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Salis, H. M., Mirsky, E. A., and Voigt, C. A. (2009). Automated design of synthetic ribosome binding sites to control protein expression. Nat. Biotechnol. 27, 946–950. doi:10.1038/nbt.1568
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sander, J. D., and Joung, J. K. (2014). CRISPR-Cas systems for editing, regulating and targeting genomes. Nat. Biotechnol. 32, 347–355. doi:10.1038/nbt.2842
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schaefer, D. G., and Zryd, J.-P. (1997). Efficient gene targeting in the moss Physcomitrella patens. Plant J. 11, 1195–1206. doi:10.1046/j.1365-313X.1997.11061195.x
Schark, M. (2012). Synthetic biology and the distinction between organisms and machines. Environ. Values 21, 19–41. doi:10.3197/096327112x13225063227943
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sefah, K., Yang, Z., Bradley, K. M., Hoshika, S., Jimenez, E., Zhang, L., et al. (2013). In vitro selection with artificial expanded genetic information systems. Proc. Natl. Acad. Sci. U.S.A. 111, 1449–1454. doi:10.1073/pnas.1311778111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Seo, S. W., Yang, J. S., Kim, I., Yang, J., Min, B. E., Kim, S., et al. (2013). Predictive design of mRNA translation initiation region to control prokaryotic translation efficiency. Metab. Eng. 15, 67–74. doi:10.1016/j.ymben.2012.10.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sharma, N., Jung, C. H., Bhalla, P. L., and Singh, M. B. (2014). RNA sequencing analysis of the gametophyte transcriptome from the liverwort, Marchantia polymorpha. PLoS One 9:e97497. doi:10.1371/journal.pone.0097497
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shi, Z., Wedd, A. G., and Gras, S. L. (2013). Parallel in vivo DNA assembly by recombination: experimental demonstration and theoretical approaches. PLoS ONE 8:e56854. doi:10.1371/journal.pone.0056854
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shin, J., and Noireaux, V. (2012). An E. coli cell-free expression toolbox: application to synthetic gene circuits and artificial cells. ACS Synth. Biol. 1, 29–41. doi:10.1021/sb200016s
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shuler, M. L., Foley, P., and Atlas, J. (2012). Modeling a minimal cell. Methods Mol. Biol. 881, 573–610. doi:10.1007/978-1-61779-827-6_20
Silva-Rocha, R., Martinez-Garcia, E., Calles, B., Chavarria, M., Arce-Rodriguez, A., De Las Heras, A., et al. (2013). The standard European vector architecture (SEVA): a coherent platform for the analysis and deployment of complex prokaryotic phenotypes. Nucleic Acids Res. 41, D666–D675. doi:10.1093/nar/gks1119
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sinclair, J. C., Davies, K. M., Venien-Bryan, C., and Noble, M. E. (2011). Generation of protein lattices by fusing proteins with matching rotational symmetry. Nat. Nanotechnol. 6, 558–562. doi:10.1038/nnano.2011.122
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sitaraman, K., Esposito, D., Klarmann, G., Le Grice, S. F., Hartley, J. L., and Chatterjee, D. K. (2004). A novel cell-free protein synthesis system. J. Biotechnol. 110, 257–263. doi:10.1016/j.jbiotec.2004.02.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Song, C. W., Lee, J., and Lee, S. Y. (2014). Genome engineering and gene expression control for bacterial strain development. Biotechnol. J. doi:10.1002/biot.201400057
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Song, Y., Sauret, A., and Cheung Shum, H. (2013). All-aqueous multiphase microfluidics. Biomicrofluidics 7, 61301. doi:10.1063/1.4827916
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Speer, M. A., and Richard, T. L. (2011). Amplified insert assembly: an optimized approach to standard assembly of BioBrickTM genetic circuits. J. Biol. Eng. 5, 17. doi:10.1186/1754-1611-5-17
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stanton, B. C., Nielsen, A. A., Tamsir, A., Clancy, K., Peterson, T., and Voigt, C. A. (2014). Genomic mining of prokaryotic repressors for orthogonal logic gates. Nat. Chem. Biol. 10, 99–105. doi:10.1038/nchembio.1411
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stricker, J., Cookson, S., Bennett, M. R., Mather, W. H., Tsimring, L. S., and Hasty, J. (2008). A fast, robust and tunable synthetic gene oscillator. Nature 456, 516–519. doi:10.1038/nature07389
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Suess, B., Fink, B., Berens, C., Stentz, R., and Hillen, W. (2004). A theophylline responsive riboswitch based on helix slipping controls gene expression in vivo. Nucleic Acids Res. 32, 1610–1614. doi:10.1093/nar/gkh321
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sukhorukov, G. B., Rogach, A. L., Zebli, B., Liedl, T., Skirtach, A. G., Kohler, K., et al. (2005). Nanoengineered polymer capsules: tools for detection, controlled delivery, and site-specific manipulation. Small 1, 194–200. doi:10.1002/smll.200400075
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sun, Z. Z., Hayes, C. A., Shin, J., Caschera, F., Murray, R. M., and Noireaux, V. (2013a). Protocols for implementing an Escherichia coli based TX-TL cell-free expression system for synthetic biology. J. Vis. Exp. 79:e50762. doi:10.3791/50762
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sun, Z. Z., Yeung, E., Hayes, C. A., Noireaux, V., and Murray, R. M. (2013b). Linear DNA for rapid prototyping of synthetic biological circuits in an Escherichia coli based TX-TL cell-free system. ACS Synth. Biol 3, 387–397. doi:10.1021/sb400131a
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Szeto, K., Reinholt, S. J., Duarte, F. M., Pagano, J. M., Ozer, A., Yao, L., et al. (2014). High-throughput binding characterization of RNA aptamer selections using a microplate-based multiplex microcolumn device. Anal. Bioanal. Chem. 406, 2727–2732. doi:10.1007/s00216-014-7661-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tang, J., and Breaker, R. R. (1997). Rational design of allosteric ribozymes. Chem. Biol. 4, 453–459. doi:10.1016/S1074-5521(97)90197-6
Tantillo, D. J., Chen, J., and Houk, K. N. (1998). Theozymes and compuzymes: theoretical models for biological catalysis. Curr. Opin. Chem. Biol. 2, 743–750. doi:10.1016/S1367-5931(98)80112-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tomlinson, M. L., Hendry, A. E., and Wheeler, G. N. (2012). Chemical genetics and drug discovery in Xenopus. Methods Mol. Biol. 917, 155–166. doi:10.1007/978-1-61779-992-1_9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Torella, J. P., Boehm, C. R., Lienert, F., Chen, J. H., Way, J. C., and Silver, P. A. (2014a). Rapid construction of insulated genetic circuits via synthetic sequence-guided isothermal assembly. Nucleic Acids Res. 42, 681–689. doi:10.1093/nar/gkt860
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Torella, J. P., Lienert, F., Boehm, C. R., Chen, J. H., Way, J. C., and Silver, P. A. (2014b). Unique nucleotide sequence-guided assembly of repetitive DNA parts for synthetic biology applications. Nat. Protoc. 9, 2075–2089. doi:10.1038/nprot.2014.145
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tracy, B. P., Gaida, S. M., and Papoutsakis, E. T. (2010). Flow cytometry for bacteria: enabling metabolic engineering, synthetic biology and the elucidation of complex phenotypes. Curr. Opin. Biotechnol. 21, 85–99. doi:10.1016/j.copbio.2010.02.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Trubitsyna, M., Michlewski, G., Cai, Y., Elfick, A., and French, C. E. (2014). PaperClip: rapid multi-part DNA assembly from existing libraries. Nucleic Acids Res. 42, e154. doi:10.1093/nar/gku829
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tuerk, C., and Gold, L. (1990). Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 249, 505–510. doi:10.1126/science.2200121
Uchida, M., Klem, M. T., Allen, M., Suci, P., Flenniken, M., Gillitzer, E., et al. (2007). Biological containers: protein cages as multifunctional nanoplatforms. Adv. Mater. Weinheim 19, 1025–1042. doi:10.1002/adma.200601168
Villalobos, A., Ness, J. E., Gustafsson, C., Minshull, J., and Govindarajan, S. (2006). Gene designer: a synthetic biology tool for constructing artificial DNA segments. BMC Bioinformatics 7:285. doi:10.1186/1471-2105-7-285
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vogl, T., Hartner, F. S., and Glieder, A. (2013). New opportunities by synthetic biology for biopharmaceutical production in Pichia pastoris. Curr. Opin. Biotechnol. 24, 1094–1101. doi:10.1016/j.copbio.2013.02.024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Walsh, G. (2010). Post-translational modifications of protein biopharmaceuticals. Drug Discov. Today 15, 773–780. doi:10.1016/j.drudis.2010.06.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Walter, N. G., and Bustamante, C. (2014). Introduction to single molecule imaging and mechanics: seeing and touching molecules one at a time. Chem. Rev. 114, 3069–3071. doi:10.1021/cr500059w
Wang, H. H., Isaacs, F. J., Carr, P. A., Sun, Z. Z., Xu, G., Forest, C. R., et al. (2009). Programming cells by multiplex genome engineering and accelerated evolution. Nature 460, 894–898. doi:10.1038/nature08187
Wang, Y., and Zhang, Y. H. (2009). Cell-free protein synthesis energized by slowly-metabolized maltodextrin. BMC Biotechnol. 9:58. doi:10.1186/1472-6750-9-58
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Way, J. C., Collins, J. J., Keasling, J. D., and Silver, P. A. (2014). Integrating biological redesign: where synthetic biology came from and where it needs to go. Cell 157, 151–161. doi:10.1016/j.cell.2014.02.039
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
White, R. M., Cech, J., Ratanasirintrawoot, S., Lin, C. Y., Rahl, P. B., Burke, C. J., et al. (2011). DHODH modulates transcriptional elongation in the neural crest and melanoma. Nature 471, 518–522. doi:10.1038/nature09882
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wittmann, A., and Suess, B. (2012). Engineered riboswitches: expanding researchers’ toolbox with synthetic RNA regulators. FEBS Lett. 586, 2076–2083. doi:10.1016/j.febslet.2012.02.038
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Woolfson, D. N., and Mahmoud, Z. N. (2010). More than just bare scaffolds: towards multi-component and decorated fibrous biomaterials. Chem. Soc. Rev. 39, 3464–3479. doi:10.1039/c0cs00032a
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wright, O., Delmans, M., Stan, G. B., and Ellis, T. (2014). GeneGuard: a modular plasmid system designed for biosafety. ACS Synth. Biol. doi:10.1021/sb500234s
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wright, O., Stan, G. B., and Ellis, T. (2013). Building-in biosafety for synthetic biology. Microbiology 159, 1221–1235. doi:10.1099/mic.0.066308-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xu, J., Sigworth, F. J., and Lavan, D. A. (2010). Synthetic protocells to mimic and test cell function. Adv. Mater. 22, 120–127. doi:10.1002/adma.200901945
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xu, P., Vansiri, A., Bhan, N., and Koffas, M. A. (2012). ePathBrick: a synthetic biology platform for engineering metabolic pathways in E. coli. ACS Synth. Biol. 1, 256–266. doi:10.1021/sb300016b
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yang, J., and Reth, M. (2012). Drosophila S2 Schneider cells: a useful tool for rebuilding and redesigning approaches in synthetic biology. Methods Mol. Biol. 813, 331–341. doi:10.1007/978-1-61779-412-4_20
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ye, H., Aubel, D., and Fussenegger, M. (2013). Synthetic mammalian gene circuits for biomedical applications. Curr. Opin. Chem. Biol. 17, 910–917. doi:10.1016/j.cbpa.2013.10.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zaccai, N. R., Chi, B., Thomson, A. R., Boyle, A. L., Bartlett, G. J., Bruning, M., et al. (2011). A de novo peptide hexamer with a mutable channel. Nat. Chem. Biol. 7, 935–941. doi:10.1038/nchembio.692
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zadeh, J. N., Wolfe, B. R., and Pierce, N. A. (2011). Nucleic acid sequence design via efficient ensemble defect optimization. J. Comput. Chem. 32, 439–452. doi:10.1002/jcc.21633
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zaghloul, T., and Doi, R. (1987). In vitro expression of a Tn9-derived chloramphenicol acetyltransferase gene fusion by using a Bacillus subtilis system. J. Bacteriol. 169, 1212–1216.
Zanghellini, A., Jiang, L., Wollacott, A. M., Cheng, G., Meiler, J., Althoff, E. A., et al. (2006). New algorithms and an in silico benchmark for computational enzyme design. Protein Sci. 15, 2785–2794. doi:10.1110/ps.062353106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, F., and Keasling, J. (2011). Biosensors and their applications in microbial metabolic engineering. Trends Microbiol. 19, 323–329. doi:10.1016/j.tim.2011.05.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, Y., Werling, U., and Edelmann, W. (2012). SLiCE: a novel bacterial cell extract-based DNA cloning method. Nucleic Acids Res. 40, e55. doi:10.1093/nar/gkr1288
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhao, Y., Wang, S., and Zhu, J. (2011). A multi-step strategy for BAC recombineering of large DNA fragments. Int J Biochem Mol Biol 2, 199–206. Available from http://www.ijbmb.org/files/IJBMB1104005.pdf
Zimmerman, S. B., Little, J. W., Oshinsky, C. K., and Gellert, M. (1967). Enzymatic joining of DNA strands: a novel reaction of diphosphopyridine nucleotide. Proc. Natl. Acad. Sci. U.S.A. 57, 1841–1848. doi:10.1073/pnas.57.6.1841
Zuleta, I. A., Aranda-Diaz, A., Li, H., and El-Samad, H. (2014). Dynamic characterization of growth and gene expression using high-throughput automated flow cytometry. Nat. Methods 11, 443–448. doi:10.1038/nmeth.2879
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: synthetic biology, engineering biology, design cycle, tools, standardization
Citation: Kelwick R, MacDonald JT, Webb AJ and Freemont P (2014) Developments in the tools and methodologies of synthetic biology. Front. Bioeng. Biotechnol. 2:60. doi: 10.3389/fbioe.2014.00060
Received: 09 September 2014; Accepted: 12 November 2014;
Published online: 26 November 2014.
Edited by:
Karmella Ann Haynes, Arizona State University, USAReviewed by:
M. Kalim Akhtar, University College London, UKDong-Yup Lee, National University of Singapore, Singapore
Copyright: © 2014 Kelwick, MacDonald, Webb and Freemont. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Richard Kelwick and Paul Freemont, Department of Medicine, Centre for Synthetic Biology and Innovation, Sir Ernst Chain Building, South Kensington Campus, Exhibition Road, London SW7 2AZ, UK e-mail:ci5rZWx3aWNrQGltcGVyaWFsLmFjLnVr;cC5mcmVlbW9udEBpbXBlcmlhbC5hYy51aw==