 Assia L. Angelova
Assia L. Angelova Karsten Geletneky
Karsten Geletneky Jürg P. F. Nüesch
Jürg P. F. Nüesch Jean Rommelaere
Jean Rommelaere- 1Infection and Cancer Program, Division of Tumor Virology, German Cancer Research Center (DKFZ), Heidelberg, Germany
- 2Department of Neurosurgery, University of Heidelberg, Heidelberg, Germany
Oncolytic virotherapy of cancer is among the innovative modalities being under development and especially promising for targeting tumors, which are resistant to conventional treatments. Presently, at least a dozen of viruses, belonging to nine different virus families, are being tested within the frames of various clinical studies in cancer patients. Continuously growing preclinical evidence showing that the autonomous rat parvovirus H-1 (H-1PV) is able to kill tumor cells that resist conventional treatments and to achieve a complete cure of various human tumors in animal models argues for its inclusion in the arsenal of oncolytic viruses with an especially promising bench to bedside translation potential. Oncolytic parvovirus safe administration to humans relies on the intrinsic preference of these agents for quickly proliferating, metabolically, and biochemically disturbed tumor versus normal cells (tumor selectivity or oncotropism). The present review summarizes and discusses (i) preclinical evidence of H-1PV innocuousness for normal cells and healthy tissues in vitro and in animals, respectively, (ii) toxicological assessments of H-1PV mono- or combined therapy in tumor-bearing virus-permissive animal models, as well as (iii) historical results of experimental infection of human cancer patients with H-1PV. Altogether, these data argue against a risk of H-1PV inducing significant toxic effects in human patients. This highly favorable safety profile allowed the translation of H-1PV preclinical research into a Phase I/IIa clinical trial being currently in progress.
The Concept of Oncolytic Virotherapy for Cancer Treatment
Oncolytic virotherapy is one of the innovative modalities under development to target tumors that are refractory to conventional surgical and radio/chemotherapeutic treatments. While the possibility of using some viruses to fight cancer was put forward at the beginning of the twentieth century, the field underwent striking revival in the last three decades, along with the development of molecular virology and genetic engineering. The term “oncolytic viruses” (OVs) designates non-pathogenic live viruses that can infect and kill malignant cells without causing any harm to normal tissues. Because of space constraints, the concept of oncolytic virotherapy will be only briefly outlined below, and citations will be limited to some recent review articles (Liu et al., 2007; Haseley et al., 2009; Meerani and Yao, 2010; Wong et al., 2010; Friedman et al., 2012; Russel et al., 2012; Singh et al., 2012; Ahmed, 2013; Bartlett et al., 2013; Goldufsky et al., 2013; Vacchelli et al., 2013; Lichty et al., 2014; Vähä-Koskela and Hinkkanen, 2014; Woller et al., 2014), to which readers are referred for further details.
Cancer virotherapy is based on four main properties of OVs, as depicted in Figure 1.
(i) Oncoselectivity is an obvious sine qua non-condition for oncolytic virotherapy. The selectivity of OV infection and replication for tumor cells is an inherent feature of certain virus species, or a result of virus adaptation or targeted genetic engineering. This oncotropism reflects the dependence of distinct step(s) of the OV life-cycle on tumor-specific molecular (epi)genetic alterations. It results in particular from the fact that, on the one hand, most tumors have evolved mechanisms to suppress responses used by normal cells to limit virus infection, and on the other hand, signaling pathways promoting the growth of tumor cells favor virus replication as well.
(ii) Oncolysis can be induced in a direct way by OVs as a result of virus replication and/or expression of viral cytotoxic gene products in infected tumor cells. A major asset of OVs lies in their multimechanistic mode of malignant cell killing, which differs from the cell death processes triggered by conventional anti-cancer agents. This peculiarity can be rationalized by the virus need to prevent premature cytopathic effects (CPE) from interfering with virus production, and is therapeutically exploited to overcome the resistance developed by many tumor cells to conventional therapies.
(iii) Indirect bystander oncolytic effects also contribute to a significant extent to the oncosuppressive activity of OVs, which can mediate in this way the killing of uninfected cancer cells. These indirect OV-induced oncolytic effects can result from tumor vasculature disruption and angiogenesis, release of toxic cytokines from infected tumor-resident or infiltrating immune cells, and even more importantly, from systemic anti-tumor immune responses. Indeed, the tumor cell death caused by OVs is often immunogenic, leading to the activation of innate immune cells and to efficient presentation of tumor-associated antigens eliciting adaptive anti-tumor immunity. Oncolytic virotherapy can be considered from either virocentric or immunocentric points of view, depending on whether emphasis is placed on direct virus-induced oncolysis or on virus-mediated stimulation of anti-tumor immune responses. In animal models, both mechanisms appear to act in combination, while having different relative weights depending on the individual target tumor.
(iv) Replication-competent OVs are used in the hope of achieving intratumoral amplification of the initial inoculum and production of progeny virions, which can spread to non-infected tumor cells, including distant metastases, until the entire tumor tissue is affected. While this multiplication has been observed in some animal tumor models, extensive OV spread at sites of tumor growth remains to be documented in clinical settings.
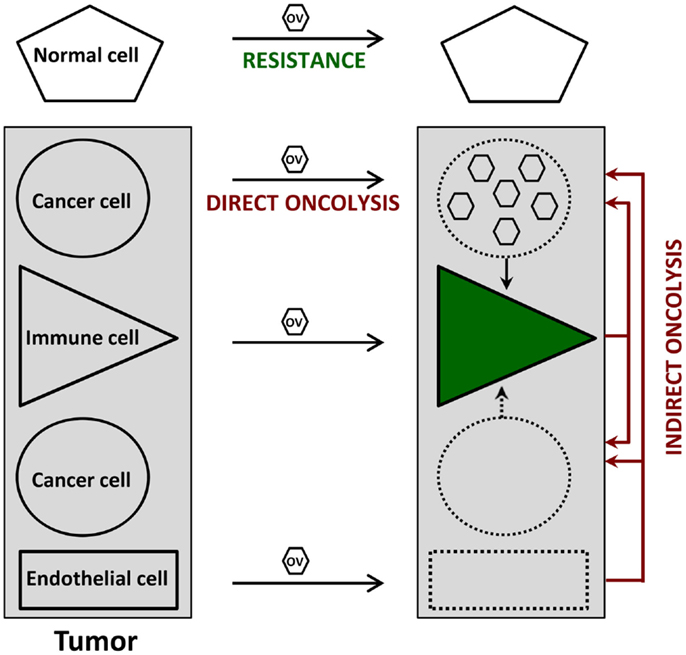
Figure 1. Direct and indirect anti-tumor effects of oncolytic viruses (OVs). Normal cells resist OV infection because of viral life-cycle blockage prior to the induction of cytopathic effects. In contrast, OVs can disturb at least three cell types within the tumor. Infected cancer cells undergo an immunogenic type of death (direct oncolysis), which is sometimes (e.g., figure) but not always accompanied by virus production; this oncolysis leads to the activation of various immune cells (green), irrespective of their direct virus infection, thereby priming anti-cancer immune responses. Furthermore, the (abortive) OV infection of immune and endothelial cells inside the tumor results in cytotoxic cytokine production and anti-vascular/angiogenic effects, respectively, which both contribute to virus-mediated oncolysis in an indirect way. Uninfected tumor cells can also serve as targets for these indirect oncolytic effects.
The potential of the oncolytic virotherapeutic approach is supported by substantial evidence of OV-induced oncosuppressive effects at both preclinical and clinical levels. More than a thousand patients have now been treated with OVs by intratumoral injection and/or intravenous infusion during Phase I–III clinical trials. Compelling evidence of OV-induced anti-cancer immunity was obtained for a recombinant oncolytic herpes simplex virus by showing that its intratumoral administration to patients with metastatic malignant melanoma led to the complete regression of injected and uninjected tumors in 8/50 treated patients (Kaufman et al., 2010). It has also been shown that an oncolytic vaccinia virus can reach tumor sites by extravasating from tumor blood vessels (Breitbach et al., 2013), lending credit to the use of systemic OV administration for metastases targeting. Indeed, in an earlier clinical trial, the authors have shown that a vaccinia vaccine-derived oncolytic poxvirus used as a vehicle for delivery and expression of transgenes, achieves cancer-selective replication after intravenous infusion in patients with advanced, treatment-refractory solid tumors (Breitbach et al., 2011). OV spread at tumor sites following systemic administration has been clinically demonstrated with yet another virus. An oncolytic reovirus was intravenously infused before surgery to resect colorectal cancer liver metastases. Reovirus immune cell carriage and delivery as well as recovery of replicating virus from the tumor was achieved, confirming intravenous reovirus targeting of metastatic colon carcinoma (Adair et al., 2012). The OVs, which are most advanced in clinical developments, include oncolytic herpes simplex, vaccinia, reo-, and adenoviruses, with the latter being licensed in China for use against head and neck cancer (Garber, 2006). The pace of clinical activities in the field has accelerated considerably, and OVs belonging to not less than nine different virus families are presently the subject of various clinical studies in cancer patients.
These clinical studies confirmed that OVs, as expected from their oncoselectivity, can be safely administered to humans. Indeed, the clinical tolerability of OVs has overall been excellent, and dose-limiting toxicities were only rarely observed. Interestingly, the most common adverse effects – transient flu-like symptoms – do not overlap with those caused by other anti-cancer agents, supporting the possibility of combining oncolytic virotherapy with current therapeutic modalities. Since future trials will likely use higher OV doses, it is not guaranteed that effective virotherapy will always be devoid of toxicity. However, this concern has to be balanced against the risk of current therapies, which approach the upper limit of tolerability. One OV-specific safety risk relates to viral spread from the treated patient to contacts. However, OV transmission has not been observed so far, although limited virus shedding in bodily fluids has occasionally been reported (Makower et al., 2003; Pecora and Lorence, 2007; Hughes et al., 2014).
Among OVs, the autonomous rat parvovirus H-1, the subject of this review, deserves special consideration as candidate anti-cancer agent. Continuously growing preclinical evidence demonstrates the ability of this virus to kill tumor cells that resist conventional anti-cancer treatments and to achieve a complete cure of various tumors in animal models, importantly, while being innocuous for non-transformed cells and normal tissues (Rommelaere et al., 2010). Currently, parvovirus H-1 is in a Phase I/IIa clinical trial designed to document its maximum tolerated dose and safety profile in patients with recurrent glioblastoma multiforme.
The Oncolytic H-1 Parvovirus
The oncolytic H-1 parvovirus (H-1PV) belongs to the family Parvoviridae, subfamily Parvovirinae, genus Protoparvovirus, and species Rodent protoparvovirus 1 (Figure 2). Like all parvoviruses, H-1PV is a small (25 nm in diameter) non-enveloped virus characterized by an icosahedral capsid and a linear single-stranded DNA genome of approximately 5,000 nucleotides (Tattersall, 2006). Viral genomic DNA comprises two transcription units that are controlled by the P4 and P38 promoters and code for the non-structural (NS) and structural (VP) viral proteins, respectively (Cotmore and Tattersall, 2014; Cotmore et al., 2014). The NS protein NS1 plays a major role in the viral life-cycle, by being essential for viral DNA replication, gene expression, and virus-induced cytotoxic effects (Nüesch, 2006; Hristov et al., 2010; Nüesch et al., 2012; Li et al., 2013). H-1PV replicates autonomously in target cells, in close dependence on cellular proliferation and differentiation factors (Cornelis et al., 2006). The natural host of H-1PV is the rat, infection of whom can either be pathogenic and even lethal (in immunologically unprotected fetuses and neonates) or clinically inapparent (in adult animals). As documented below, under in vitro conditions, the virus preferentially replicates in and kills transformed or tumor-derived rat and human cell cultures, without inducing cytotoxicity in their corresponding non-transformed/non-malignant counterparts (Rommelaere et al., 2005, 2010). Moreover, the above observations illustrative of parvovirus H-1 oncoselectivity have been further extended to animal models, in which H-1PV was reported to efficiently suppress tumor formation and to cause striking regression of established tumors (oncosuppression, see below) (Rommelaere and Cornelis, 1991).
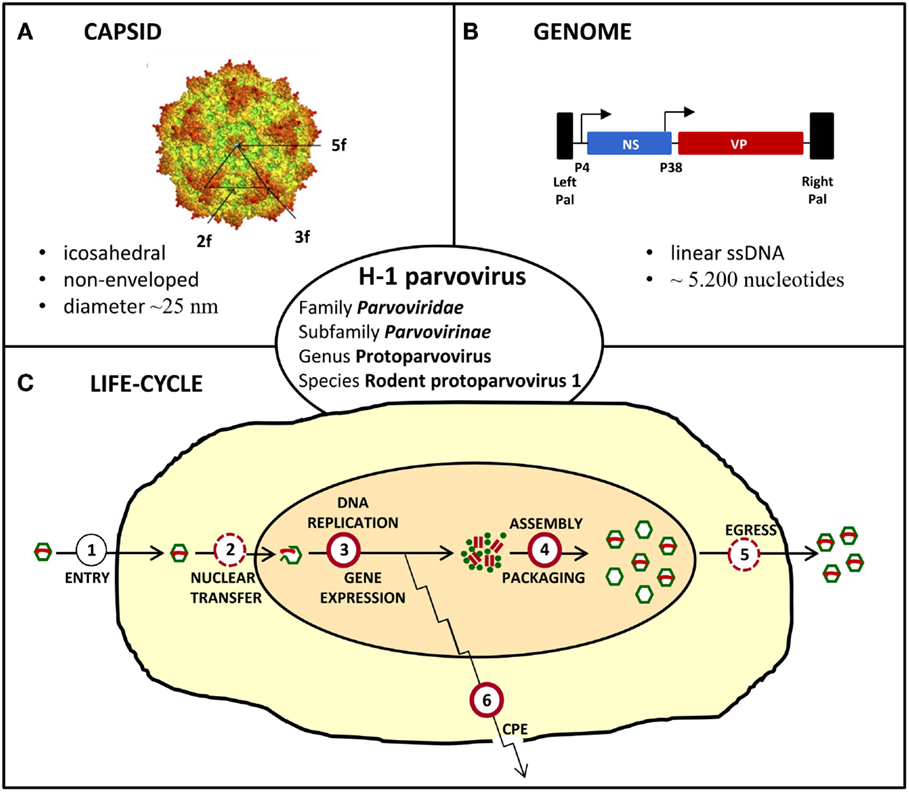
Figure 2. Characteristics of H-1 parvovirus. (A) In silico model of H-1PV capsid surface showing the two, three, and fivefold axes of symmetry (Allaume et al., 2012). (B) Simplified viral gene expression map. The viral single-stranded (ss) DNA genome ends in unique palindromic sequences (Pal), which serve as self-priming origins of replication for the synthesis of double-stranded replication forms/transcription templates. Transcription is controlled by the P4 and P38 promoters that are indicated by arrows and direct expression of non-structural (NS) and capsid (VP) proteins, respectively. (C) Simplified scheme of the viral life-cycle depicting the main steps leading to virus production and induction of cytopathic effects (CPE). Known and putative oncogenic transformation-stimulated steps are indicated by full and dashed red circles, respectively.
Mechanisms of Rodent Protoparvovirus Oncoselectivity
Parvovirus intrinsic oncoselectivity is a complex phenomenon based in part on multiple tumor cell-specific determinants, which are under-represented in non-malignant cells. The availability of cellular replication and transcription factors, the overexpression of cellular proteins known to interact with parvoviral ones (with NS1, in particular), the activation of metabolic pathways involved in the functional regulation of NS1, are all contributing to parvovirus preference for tumor, and not for normal cells. The molecular pathways involved in H-1PV tumor cell targeting are overviewed in a recent publication from our laboratory (Nüesch et al., 2012), while the most important in vitro and animal studies providing evidence of parvovirus oncoselectivity and innocuousness in a non-malignant environment are summarized in the present review (see following sections). Although it is feasible to attenuate PVs by genetic engineering (Daeffler et al., 2003), the use of these viruses as anti-cancer agents is not based on this strategy since the innocuousness of wild-type PVs to normal tissues avoids the need to attenuate undesirable toxicity.
To our knowledge, there is no evidence to indicate that PV preference for infecting tumor cells results from a higher competence of these cells for virus binding and internalization. Instead, neoplastic cells appear to provide an intracellular milieu that is especially permissive for at least part or the full course of the PV life-cycle. This higher permissiveness of tumor cells cannot be traced back to a single factor, but rather involves multiple cellular factors that control different steps of the PV life-cycle and each give infection a distinct boost (Figure 2). These factors are thus likely to cooperate in promoting virus infection, with the impact of later-acting factors depending on the completion of earlier steps in the viral cycle. A number of tumor cells provide all the factors that are necessary for full virus replication, resulting in progeny particle production and cell lysis. Some other tumor cells are semi-permissive (and get killed in the absence of progeny virions release), and a few remain resistant to infection. It should also be stated that man is not the natural host of rodent PVs, and normal human cells may pose different and/or additional restrictions to PV infection, compared to rodent cells. This can be exemplified by our recent work showing that the PDK1/PKC/PKB cascade – which regulates the functioning of the replicative and cytotoxic PV protein NS1 – is induced by the mouse parvovirus MVM in mouse but not in human cells (Lachmann et al., 2008; Bär et al., 2015). In consequence, the constitutive activation of this cascade through an unconventional mechanism in human tumor cells represents a human-specific determinant of the oncoselectivity of PVs.
Most of the cellular determinants of PV oncoselectivity, as identified so far, control the viral life-cycle in a positive way (Figure 3).
• Some of these factors are not strictly tumor-specific but characteristic of proliferating cells. Examples thereof are cyclin A/CDK2 (Bashir et al., 2000; Adeyemi and Pintel, 2012) and E2F (Deleu et al., 1999), which are cellular S-phase markers and control the conversion of the PV single-stranded DNA genome into double-stranded replicative forms/transcription templates, and the activation of the PV early promoter P4, respectively. Both factors are thus essential for the onset of PV replication, contributing to the S-phase dependency of these viruses. Although a fraction of tumor cells can be dormant and some normal tissues are rapidly self-renewing, the proliferating component of many tumors distinguishes them from essentially quiescent surrounding normal tissues, offering a target for PV infection and thereby contributing to the enhancement of virus replication and CPE in neoplastic versus normal tissues.
• Other determinants of PV oncoselectivity are more specific for malignantly transformed cells in which they are overexpressed or activated as a result of (epi)genetic alterations occurring in these cells. A few factors of this type have been shown to promote distinct steps of the PV life-cycle. These steps include:
(i) viral entry: PKCα- and CDK1-dependent nuclear envelope breakdown (Porwal et al., 2013).
(ii) viral gene expression: activation of the early PV promoter P4 by Ets and ATF transcription factors (Perros et al., 1995; Fuks et al., 1996).
(iii) viral DNA amplification: activation of the PV replicative protein NS1 through PDK1, PKC, and radixin (Rdx)-mediated phosphorylation (Dettwiler et al., 1999; Lachmann et al., 2003, 2008; Nüesch et al., 2009; Bär et al., 2015); HMGB1-dependent initiation of DNA amplification from the right-hand viral origin (Cotmore and Tattersall, 1998; Cotmore et al., 2000); facilitation of viral replication through an ATM kinase-mediated DNA damage response (Adeyemi et al., 2010).
(iv) viral progeny capsid assembly: Raf-1 phosphorylation-dependent nuclear transport of capsid intermediates (Riolobos et al., 2010).
(v) virions maturation: XPO1, PKB, PKC, and Rdx-dependent egress (Eichwald et al., 2002; Nüesch et al., 2009; Bär et al., 2013, 2015).
(vi) cytopathic effects: expression of potential PV targets, including distinct tropomyosin (TPM) isoforms (Nüesch and Rommelaere, 2007), or checkpoint factors, e.g., cyclin B1 (Adeyemi and Pintel, 2014); production of cytotoxic effectors, such as cathepsin B (CTSB) (Di Piazza et al., 2007) and CKII (Nüesch and Rommelaere, 2006); PDK1 and PKCι-dependent activation of NS1 cytotoxic function (Nüesch and Rommelaere, 2006; Bär et al., 2015).
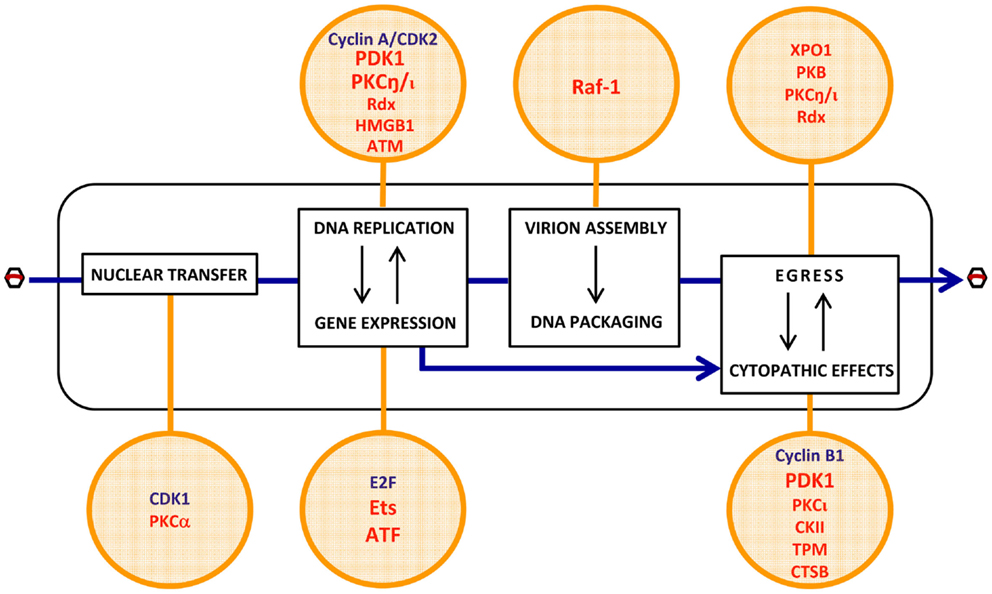
Figure 3. Putative cell determinants of PV oncoselectivity. Indicated steps of the PV life-cycle (rectangles) were shown to be controlled by cellular factors (circles) known to be regulated at gene amplification, expression, and functional levels by cell proliferation (blue) and oncogenic transformation (red). The list of factors is not exhaustive and exemplifies candidate mediators of the enhanced permissiveness of neoplastic cells for PV infection. Evidence of the contribution of these factors to PV oncotropism is experimental for a few of them (PDK1, PKCη, Ets, ATF, Raf-1) but circumstantial for the others. For more details, see main text (pp. 4–5). CDK2, cyclin-dependent kinase 2; PDK1, phosphoinositide-dependent kinase 1; PKC, protein kinase C; Rdx, radixin; HMGB1, high-mobility group box protein 1; ATM, ataxia-telangiectasia mutated protein; Raf-1, rapidly accelerated fibrosarcoma-1 protein; XPO1, exportin-1; PKB, protein kinase B; E2F, transcription factor E2F; Ets, E26 transformation-specific transcription factor; ATF, activating transcription factor; CKII, casein kinase II; TPM, tropomyosin; CTSB, cathepsin B.
The role played by some of these factors in the oncotropism of PVs was supported by the fact that their helper function was stimulated in transformed cells, rescuing at least to some extent the viral life-cycle defect in normal cells.
The oncoselectivity of a number of OVs can also be traced back to the frequent deficiency, in tumor cells, of mechanisms that allow normal cells to counteract virus infection, in particular, the type I interferon and stress responses. Along this line, the mouse Protoparvovirus MVM was shown to both induce the type I interferon response and be sensitive to it in normal mouse cells, while a still elusive evasion mechanism appeared to be triggered by the virus in their transformed counterparts, preventing interferons from being produced (Grekova et al., 2010a,b). It follows that in this system, PV oncoselectivity consisted of an additional component, namely, the absence of negative modulators of PV infection in transformed cells. It is unclear whether these data can be extrapolated to human neoplastic versus normal cells. Various human tumor cells were recently reported to fail to develop a type I interferon response upon PV (including H-1PV) infection. However, this failure was also observed in normal human cells (Paglino et al., 2014), questioning whether the interferon response plays any role in the oncoselectivity of rodent protoparvoviruses in human cells. It should be again recalled in this regard that man is not the natural host of rodent protoparvoviruses, and some of the factors limiting the viral life-cycle may therefore differ between cells from natural and heterologous hosts.
In vitro Evidence of H-1PV Oncoselectivity
The pioneering observations demonstrating that H-1PV fails to induce CPE in normal human (e.g., embryonic kidney and amnion) cultures were published by Toolan and Ledinko (1965). H-1PV innocuousness for non-malignant cells has been later confirmed in a number of in vitro studies, as summarized in Table 1.
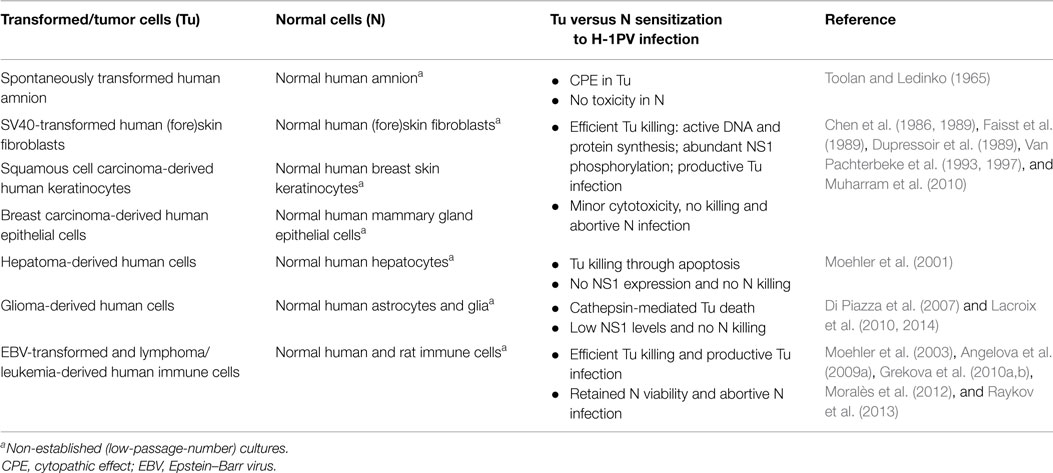
Table 1. In vitro evidence of H-1PV oncoselectivity.
For example, H-1PV infection was quantitatively compared in a series of non-permanent cultures of normal human (fore)skin fibroblasts and their respective SV40-transformed counterparts. Although the virus was adsorbed and taken up by both normal and transformed cells, only in the latter, H-1PV was able to induce killing and progressive culture degeneration. In addition, a striking difference in the capacity of normal versus transformed fibroblasts to support viral DNA and protein synthesis was found. Only the SV40-transformants, but not their non-transformed counterparts, could provide the intracellular environment required to support the completion of H-1PV lytic cycle and the release of infectious progeny virions (Chen et al., 1986; Faisst et al., 1989). Similar observations were made when established keratinocyte cell lines derived from human squamous cell carcinoma were used. Efficient virus-induced killing and productive infection were seen in human tongue, cheek, supraglottis, and face squamous cell carcinoma-derived keratinocytes. In contrast, normal human epidermal cells isolated from healthy adult breast and used after one passage in vitro (low-passage-number cultures) were resistant to H-1PV infection (Chen et al., 1989; Faisst et al., 1989). In another study, established lines or low-passage-number in vitro cultures derived from either human breast tumor specimens or normal peritumoral breast tissue from the same patient were used to evidence H-1PV oncoselectivity. The striking oncolytic virus-induced cytotoxic effects observed in tumor-derived cultures were absent in the respective normal tissue-derived controls (Dupressoir et al., 1989; Van Pachterbeke et al., 1993, 1997). Furthermore, in invasive breast carcinoma-derived cells, in contrast to normal breast epithelium, a distinct phosphorylation pattern of the major viral cytotoxic protein NS1 was documented (Muharram et al., 2010). Selective H-1PV-induced killing of human liver carcinoma cells versus normal hepatocytes was also reported. Low-passage number cultures established from healthy liver tissue failed to support NS1 expression and were therefore refractory to lytic infection (Moehler et al., 2001). In order to prepare a preclinical platform for H-1PV clinical application in brain tumor patients and patients with other nervous system-originating tumors, virus toxicity was evaluated in normal primary human infant astrocytes, glial cells, and cortical neurons as well. The data showed that the morphology, metabolic activity, and membrane integrity of the above cells remained unaltered even after high-dose H-1PV infection. Whereas in malignant neuroblastoma cells, abundant NS1 expression was observed and found to result in cellular G2 arrest, no or only low NS1 levels could be detected in normal astrocytes and mixed glial cultures. Indeed, although the latter could take a recombinant EGFP-transducing virus up, transduction efficiency was more than 10-fold lower than in the neuroblastoma cell line reported to be the least sensitive to H-1PV-induced cytotoxicity (Lacroix et al., 2010, 2014). In agreement with these data, virus innocuousness for normal human adult astrocytes was also described. It was shown that while triggering lysosomal membrane permeabilization and cathepsin-mediated death in human glioma-derived cells, H-1PV was non-toxic for normal astrocyte cultures (Di Piazza et al., 2007). Experimental data illustrating absence of H-1PV-induced toxicity in normal cells came also from studies using preclinical non-Hodgkin lymphoma models. In contrast to Burkitt lymphoma-derived cells, which were highly susceptible to H-1PV-induced killing and supported high levels of progeny virions production, non-malignant B lymphocytes, including normal memory B cells, were shown to resist infection, with minor toxicity being observed only when cultures were infected with high H-1PV doses. Since normal B lymphocytes were unable to express EGFP after transduction with a recombinant EGFP-transducing virus, this minimal toxic effect was unrelated to viral gene expression (Angelova et al., 2009a). Normal human immune cell resistance to PV infection was reported by other authors as well. Immature or mature dendritic cells and monocytes were shown to retain viability after H-1PV challenge (Moehler et al., 2003), while, in contrast, the human histiocytic lymphoma-derived monocyte cell line U937 is among the in vitro systems being the most sensitive to H-1PV lytic effects (Rayet et al., 1998). Proofs of PV innocuousness for normal peripheral blood mononuclear cells are of particular importance in view of current and future H-1PV clinical applications. It was shown that although basal levels of viral DNA replicative forms were detected in the latter cells, no productive infection, even after mitogen stimulation, could be observed (Grekova et al., 2010a,b). Similar results were reported by Moralès et al. (2012) and Raykov et al. (2013).
In vivo Evidence of H-1PV Oncoselectivity
After the in vitro experiments using different primary and low-passage-number cultures demonstrated that H-1PV infection of normal human cells is abortive, does not result in cell death and induces no or minor CPE (see above), various animal tumor models were next explored in our laboratory to test PV oncoselectivity under in vivo conditions. The most extensive animal studies were performed using two tumor models, namely glioma and pancreatic ductal adenocarcinoma (PDAC), which are distinguished by a dismal prognosis and represent potential targets for H-1PV-based oncolytic virotherapy clinical applications. Rats, i.e., the natural permissive host of the virus, were engrafted with the respective tumors, and a long-term follow-up of the animals was performed after H-1PV treatment, in order to assess the tumor-suppressing capacity of the virus, and to reveal any signs of virus-induced toxicity and/or other adverse effects.
Glioma
Only about 50% of the patients with malignant brain tumors of glial origin survive 1 year after initial diagnosis. Furthermore, standard treatment options lead to only modest improvements in glioma outcome (Stummer et al., 2006). Oncolytic H-1PV, whose capacity to selectively kill glioma-derived cells through a cathepsin-mediated mechanism was demonstrated in vitro (Di Piazza et al., 2007), is believed to represent a promising therapeutic alternative. In this regard, experimental evidence obtained in our laboratory showing that intracerebral or systemic H-1PV injection leads to glioma regression in immunocompetent rats bearing orthotopic autologous RG-2 tumors and in immunodeficient animals implanted with human U87 gliomas without causing any toxic side effects, is of major preclinical importance (Di Piazza et al., 2007; Geletneky et al., 2010; Kiprianova et al., 2011). These effects could not only be achieved by direct local intratumoral therapy but also after intravenous and even intranasal virus inoculation, although significantly higher virus doses had to be used for systemic application. The effect of intracerebral H-1PV injection applied to healthy or glioma-bearing rats on CTSB activity was investigated by Di Piazza et al. (2007). In healthy brains, CTSB activity was found to be very low and not significantly affected by H-1PV treatment. In contrast, in H-1PV-treated gliomas a striking enhancement of CTSB activity was detected, together with an increase in the total amount of tumor-associated enzyme. Histological examination of H-1PV-treated gliomas raised in immunocompetent rats (Geletneky et al., 2010) demonstrated that, in addition to causing remission and improving animal survival, H-1PV treatment was not associated with normal brain tissue or other organs damage and was accompanied by only minor signs of inflammation. In agreement with previous in vitro and in vivo data (Di Piazza et al., 2007), CTSB activation was observed only in H-1PV-infected tumor cells but not in the surrounding peritumoral tissue. Parvoviral DNA could be detected in the tumor and in the peritumoral brain tissue 48 h post infection (p.i.). Virus spreading further increased with time and at 72 h p.i. viral DNA could additionally be detected in the contralateral brain hemisphere, in the cerebellum, and in distant organs (heart, lungs, liver, spleen, and kidneys), yet only transiently since no viral DNA could be revealed in any normal tissue 2 weeks p.i. Virus transcription and NS1 accumulation were strongly restricted to the tumor remnants, while undetectable in surrounding normal tissues, thus arguing for the selective H-1PV replication in tumor cells. To confirm this statement, virus replication was compared in two groups of animals, i.e., in glioma-bearing and in healthy control rats. The infectious virus yield in the brain of H-1PV-treated tumor-bearing rats was two orders of magnitude higher than in the brain of control animals, which were tumor-free but have been injected with the same amount of virus. This preferential replication of H-1PV in the neoplastic tissue, without inducing histopathological changes in normal brain tissue, provided convincing evidence that H-1PV preserves the oncotropism displayed in cell cultures under in vivo conditions as well (Geletneky et al., 2010).
In an immunodeficient rat model of human glioma, both intratumoral and multiple systemic (intravenous) H-1PV administrations were tested and shown to result in tumor suppression without being accompanied by any treatment-associated side effects. All animals subjected to H-1PV virotherapy remained active and gaining weight until the end of the observation period (Geletneky et al., 2010). Expression of the PV protein NS1 was detected in the necrotic tumor areas but not in the surrounding normal brain tissue, in agreement with the observations made in glioma-bearing immunocompetent animals (see above). The work of Geletneky et al. further raised the question whether an intranasal H-1PV application might also achieve therapeutic efficiency and suppress the growth of human glioma xenografts in immunodeficient rats. As reported by Kiprianova et al. (2011), a single intranasal virus instillation led to significant glioma regression and animal survival prolongation, without any toxicity for all but tumor tissues. Indeed, all virus replication markers were expressed exclusively in the tumor. These results suggest a safe and efficient alternative to H-1PV administration via the standard invasive intracranial route.
Pancreatic Ductal Adenocarcinoma
Pancreatic ductal adenocarcinoma is one of the most lethal gastrointestinal malignancies, causing every sixth cancer-related death in Europe (Jemal et al., 2007). The disease is highly resistant to current treatments: surgical resection, which achieves the best long-term survival so far, is feasible in only a minority of patients (Finlayson and Birkmeyer, 2003). Oncolytic H-1PV infection of PDAC-derived cells in vitro was shown to result in efficient virus-induced cell death, even when the cells were resistant to standard chemotherapeutics, e.g., gemcitabine (Angelova et al., 2009b). Furthermore, H-1PV capacity to suppress PDAC was also studied in vivo. In a syngeneic orthotopic rat model of PDAC, a single intratumoral H-1PV injection was applied 2 weeks after implantation of rat pancreatic carcinoma cells into the pancreas. Virus expression assessment demonstrated that H-1PV was expressed selectively in tumor as opposed to normal tissues. An initial burst of virus expression in tumor and surrounding pancreatic tissue was observed shortly after virus injection. H-1PV transcripts were also detected in lymphoid organs. From day 10 onwards, virus expression faded in normal pancreatic and other distant visceral tissues but remained persisting in the tumor (Angelova et al., 2009b). In an immunodeficient environment, a similar selective tumor targeting and absence of toxicity were observed in PDAC-bearing H-1PV-treated nude rats (Li et al., 2013).
In another study, human cervical carcinoma xenograft-bearing nude rats were used to demonstrate a virus dose-dependent tumor growth arrest and regression. NS1 expression was detected only in the kidneys and at very low levels. Remarkably, no weight loss or other adverse effects were documented in any of the treated animals (Li et al., 2013).
Recently, two large-scale animal experiments using virus-permissive immunocompetent rats were initiated in order to provide further preclinical proofs of H-1PV favorable safety profile. The virus was applied intravenously to healthy rats as either a single high dose or multiple injections. Virus doses were considered equivalent or higher than those resulting from viral amplification after infection of brain tumors. It is known that the rat is the natural host of H-1PV and the latter may persist in normal rat populations by a mechanism not yet known. This persistence indicates that also non-tumor-bearing rats are capable for parvovirus replication, making this animal model a suitable choice for the detection of possible side effects of H-1PV therapeutic application. Irrespective of the administration regimen used, animal mortality or macroscopic organ changes were not observed. Minimal diffuse bile tract hyperplasia and germinal center development in the spleen were detected after multiple H-1PV applications. However, liver changes were reversible within a 2-week recovery period. No virus-induced toxic effects could be revealed by measuring blood parameters (hematology, chemistry, coagulation). In agreement with in vitro data, blood mononuclear cells showed no functional alterations after virus injection, and measurable cytokine release could be detected. H-1PV treatment led to the development of IgG antibodies. The virus was shed mainly via feces (Geletneky et al., 2015a). Furthermore, the same authors demonstrated that H-1PV is non-pathogenic in adult rats and infection does not affect central or autonomous nervous system functions, even after a direct injection into the brain (Geletneky et al., 2015b).
Taken together, the above described data provide experimental evidence of the oncoselectivity of H-1PV in adult animals from its natural host, the rat, resulting in tumor suppression in absence of any pathological signs (Table 2). This innocuousness was also shown in young rats (Jacoby and Ball-Goodrich, 1995; Gaertner et al., 1996), in agreement with previous studies showing that pathological effects could only be observed if infection of the animals took place within the first few days after birth (Jacoby et al., 1979). Overall, this favorable safety profile supports the further translation of H-1PV applications into the clinic.
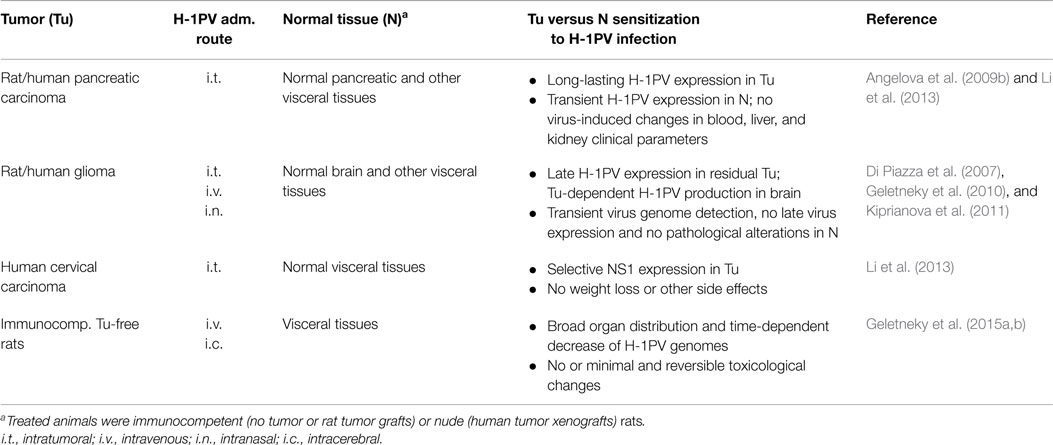
Table 2. In vivo evidence of H-1PV oncoselectivity.
H-1PV Anti-Tumor Efficacy Assessment Using Xenografts Models in Non-Permissive Mice
Additional studies (summarized in Table 3) were carried out to test the suppressive activity of H-1PV on human tumor xenografts in immunodeficient recipient mice. Since the mouse is a non-permissive host for H-1PV, these studies are not informative regarding the oncoselectivity of H-1PV, but are much relevant for the assessment of the suppressing capacity of the virus against human tumor targets.
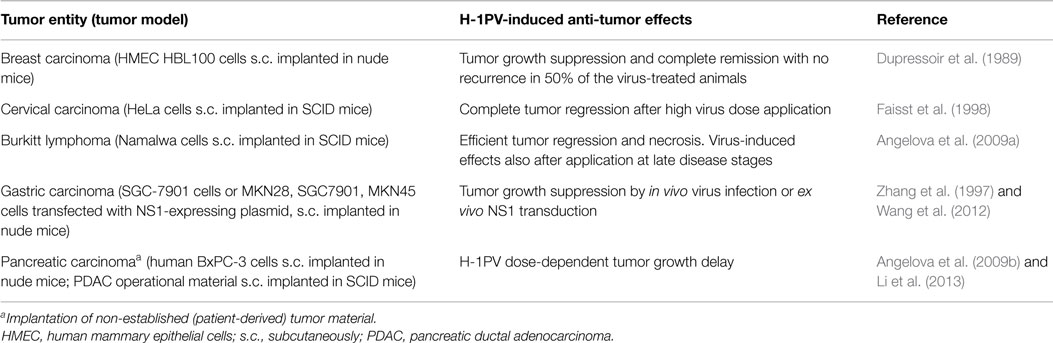
Table 3. H-1PV-induced suppression of human tumor xenografts in mouse models.
H-1PV capacity to inhibit the growth of human tumors xenotransplanted in recipient immunocompromised mice was first documented by Dupressoir et al. (1989) and further substantiated by Faisst et al. (1998). Dupressoir et al. reported that both local (intratumoral) and systemic (intravenous) virus application lead to significant human mammary carcinoma growth suppression and, in some cases, reversion, without any detectable infection-associated deleterious side effects. In the work of Faisst et al., quickly growing subcutaneous carcinomas were established by implantation of human cervical carcinoma cells into immunodeficient SCID mice. Tumor-bearing animals were subsequently intratumorally injected with different H-1PV doses. Intratumoral H-1PV gene expression and dose-dependent tumor regression were observed. This study provided the experimental evidence showing that a single local H-1PV injection is sufficient to induce regression, in an immunodeficient recipient environment, of certain human solid carcinomas.
Similar observations were made by Angelova et al. (2009a) in a human lymphoma model. A single intratumoral H-1PV injection in human Burkitt lymphoma-bearing SCID mice resulted in quick tumor regression. Moreover, it was shown that low H-1PV doses can achieve a strong therapeutic effect as well, even when the virus was applied at advanced disease stages. Histochemical analyses demonstrated H-1PV spreading to distant, non-virus-treated tumors, in addition to intratumoral replication and protein expression in virus-treated tumor tissues.
Wang et al. (2012) reported that in an in vivo human gastric carcinoma model the ex vivo-induced NS1 expression in poorly differentiated gastric cancer cells prevents them from forming tumors in nude mice. This study was in agreement with an earlier report (Zhang et al., 1997) showing that intratumoral H-1PV application to human gastric carcinoma-bearing mice results in efficient tumor inhibition. Importantly, this oncosuppression was not accompanied by any toxic side reactions, even when the virus was applied as long as twice per week for 3 weeks. Similar lack of toxicity was observed after an intraperitoneal animal treatment.
Tumor regression and complete remission in human pancreatic carcinoma-bearing nude mice (Angelova et al., 2009b) or NOD/SCID mice (Li et al., 2013) were observed after the critical H-1PV dose was reached in a virus dose-escalation experiment. This regression was not associated with detectable toxicity. Importantly, pancreatic carcinoma patient-derived primary tumor material was used in the study by Li et al. (2013).
Preclinical Testing of H-1PV-Based Combinatorial Treatments
In order to evaluate the cooperative capacity of H-1PV and gemcitabine for suppressing pancreatic carcinoma growth in an orthotopic rat model, tumor-bearing animals were sequentially treated first with gemcitabine and 2 weeks later with H-1PV in a two-step protocol (Table 4). Since the two agents induce different death pathways (apoptosis and cathepsin-mediated cell death, respectively), H-1PV-triggered oncolysis is expected to circumvent the antiapoptotic features (i.e., gemcitabine resistance) acquired by many tumor cells during PDAC progression. The data have indeed demonstrated that the anti-PDAC potential of the drug was significantly improved when parvovirus was added to the treatment. The combined treatment was not accompanied by additive toxicity, as illustrated by the results of an extensive toxicological assessment carried out in this model (Angelova et al., 2009b). Rat bone marrow, liver, and kidney functions were monitored by measuring clinically relevant makers. The blood-borne markers of bone marrow activity were unaffected, except for the gemcitabine-induced drop in the reticulocyte and monocyte levels. Bilirubin, aspartate aminotransferase (ASAT), and alanine aminotransferase (ALAT) levels were elevated in both untreated and gemcitabine-treated groups of animals, reflecting the low-grade lytic processes typical of the livers of PDAC-bearing rats. H-1PV application as a second-line treatment restored the levels of these markers to their respective normal physiological ranges. Creatinine levels remained stable after the combinatorial treatment, showing an unaltered kidney clearance. It was concluded from the above toxicological assessments that the blood-borne marker variations detected were fully attributable to gemcitabine treatment and were not aggravated by subsequent H-1PV administration.

Table 4. Safety of H-1PV-based combinatorial treatments.
Similar observations were made when established PDAC or cervical carcinoma cells, as well as primary PDAC patient-derived tumor material, were used for implantation into immunodeficient rats or NOD/SCID mice, respectively (Li et al., 2013), in order to assess the anti-tumor efficacy and biosafety of H-1PV combined with valproic acid (VPA). VPA belongs to the class of histone deacetylase (HDAC) inhibitors and has been shown to reinforce the cytotoxicity of many OVs, including the vesicular stomatitis virus (Alvarez-Breckenridge et al., 2012), herpes simplex virus (Otsuki et al., 2008), and adenoviruses (Van Oosten et al., 2007), by inhibiting the expression of cellular genes involved in antiviral immune responses and/or by stimulating the expression of genes required for the viral life-cycle (Nguyen et al., 2010). In vitro, VPA has been shown to enhance H-1PV NS1-mediated cytotoxicity through increased protein acetylation and upregulated transcriptional activity (Li et al., 2013). In agreement with in vitro data, H-1PV treatment in vivo followed by VPA administration, was devoid of weight loss or other signs of toxicity, and resulted in synergistic tumor regression and survival prolongation. In addition, this combinatorial approach allowed reduction of H-1PV doses to a level, which is suboptimal in a monotherapeutical setting.
Altogether, preclinical data convincingly argue against a significant risk of oncolytic parvovirus H-1 inducing severe toxic effects when applied to the human patient. Moreover, H-1PV treatment is unlikely to pose a risk of insertional mutagenesis since autonomous parvoviruses are not known to integrate into the host cell genome (Richards and Armentrout, 1979; Ron and Tal, 1985). This highly favorable safety profile (Figure 4) together with prominent anti-cancer activities justify H-1PV consideration as mono-, combined, or second-line treatment alternative to current conventional toxic strategies.
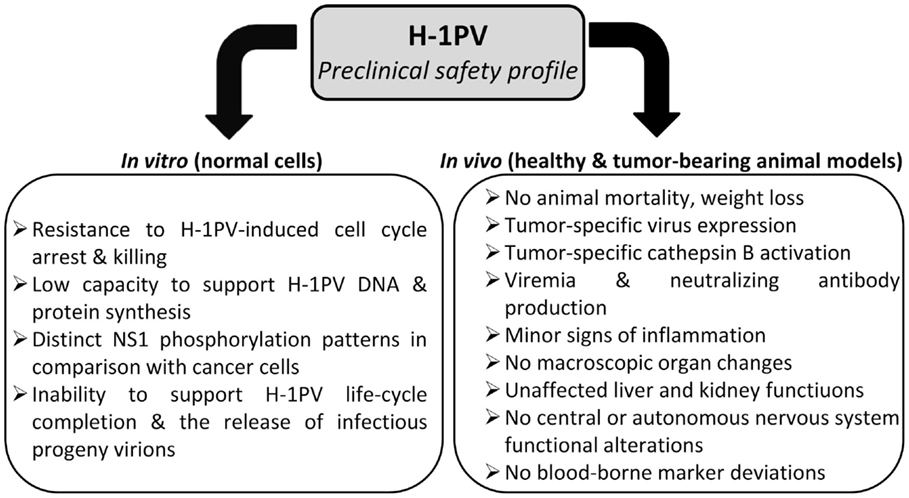
Figure 4. Preclinical evidence of H-1PV safety.
Clinical Evidence of H-1PV Tolerability
Despite frequent H-1PV isolation from cancer patients’ tissues and transplantable tumors or as a contaminant of human tumor-derived cell lines in 1970s (Toolan et al., 1960), all attempts to isolate the virus from normal non-malignant human tissues failed (Toolan et al., 1962; Rommelaere and Tattersall, 1989). No convincing experimental evidence of an association between any human disease and a previous H-1PV infection could be found either. Initial indications of a possible role of H-1PV in gestational problems were not confirmed by subsequent studies, which failed to reveal the presence of H-1PV-specific antibodies or H-1PV virions in sera or tissue specimens obtained after spontaneous human abortions (Monif et al., 1965; Neuman et al., 1970).
Important early clinical evidence showing that H-1PV application in humans is well tolerated and devoid of side toxicity was provided after experimental infection of human cancer patients under a compassionate use agreement. In the pioneering study by Toolan et al. (1965), two patients (a 12- and 13-year-old girl) with advanced disseminated osteosarcoma were intramuscularly inoculated with H-1PV at a dose of approximately 1 × 109 plaque-forming units (pfu) of a non-GMP-virus formulation. In one of the patients, a direct virus injection into the tumor of the right hip area was also applied. One of the patients developed fever of up to 38.5°C within the first 10 days after virus administration, yet it was not clearly attributable to the virus inoculation because of the presence of a concomitant urinary tract infection. Body temperature returned to normal after 5 days. The patient was discharged at day 15 without any additional clinical symptoms but she died of tumor progression shortly after hospital readmission at day 38 after H-1PV treatment. The second patient suffering from advanced metastatic disease did not experience any virus treatment-associated side effects. In both patients, H-1PV injection led to an extensive viremia and production of H-1PV-specific neutralizing antibodies. Overall, no virus-induced organ-specific side effects could be observed and H-1PV safety and tolerability was considered as good in both patients.
In a later study, which took place in France within the frame of a clinical trial entitled “Phase I clinical study on possible use of H-1 parvovirus in cancer treatment,” 12 patients with skin metastases originating from different types of solid tumors (breast adenocarcinoma, melanoma, lung large cell carcinoma, pancreatic carcinoma, and kidney leiomyosarcoma) were subjected to an intralesional dose-escalation (1 × 108, 1 × 109, 1 × 1010 pfu) H-1PV treatment (Le Cesne et al., 1993). The virus was administered repeatedly, with 10-day treatment-free intervals. Seroconversion was detected at days 10–15 after the first virus injection. Only a moderate fever up to approximately 38°C (in 3 out of the 12 patients), an isolated increase of creatinine and gamma-glutamyl transferase (GGT) but no other H-1PV-associated toxic side effects could be observed, arguing for an excellent safety profile of this oncolytic virus in humans. It is also noteworthy that in two out of seven breast carcinoma patients stable disease was documented throughout the observation period. The presence of virus DNA/proteins in tumor extracts was investigated in four subjects and in all of them viral genomes/proteins could be found after administration of H-1PV in both target lesions (metastases to which the virus was administered) and in control lesions (metastases distant from the site of injection). This confirmed a systemic exposure to the virus as already demonstrated by viremia.
With the support from this encouraging safety data in humans, a next clinical trial with H-1PV in patients with malignant brain tumors was planned and initiated (Geletneky et al., 2012, 2014a,b). This trial used three modes of virus application that were not tested in 1965 and 1993 reports, namely intratumoral injection, injection directly into the brain parenchyma bordering the tumor, and intravenous injections. From the aspect of safety and tolerability, these routes of parvovirus administration were potentially more challenging since active virus particles in the central nervous system (CNS) could lead to encephalitis or meningitis, and an intravenous infusion might result in a more rapid systemic exposure compared to the H-1PV release into the circulation after intramuscular, subcutaneous, or intracutaneous injection. The trial was planned as a dose-escalation trial at dose ranges of 1 × 106, 5 × 107, and 1 × 109 pfu. The H-1PV preparation was produced according to GMP standards thereby providing certified virus concentrations and purity.
The initial treatment was by intratumoral injection of half of the total dose per patient followed by a 9-day observation period in which the virus could interact with the tumor. Tumor resection was performed at day 10 and the second half of the dose was given by multiple injections into the tumor-surrounding brain. Three patients per dose group were treated and, for safety reasons, a time interval of 28 days (between the first and the second patient) or of 18 days (between the second and the third patient of one group) was allowed. All virus injections were well tolerated without any virus-associated side effects or pathology. Some adverse effects were detected but they were rated unrelated to virus treatment and no dose-limiting toxicity has been found. We did not detect any signs of fever or flu-like symptoms and in particular no signs of CNS pathology. Measurements of viral genomes in blood showed positive results in the intermediate-dose and high-dose group indicating penetration of H-1PV through the blood–brain barrier, again with no signs of systemic toxicity.
After the safety data of the intratumoral patient arm had been reviewed by external experts and by the responsible federal agency (Paul-Ehrlich Institut, Langen, Germany), the permission was granted to move on to the intravenous trial arm (Geletneky et al., 2014a,b). Here, the patients were injected with a total dose of 5 × 107 or 1 × 109 pfu of H-1PV. All patients received daily virus infusions of 10% of the total dose at days 1 through 5, followed by a 4-day recovery period. At day 10, surgery and intraparenchymal injection of the second half of the planned dose of H-1PV were performed, as in the first trial arm. Also, after intravenous injection, we did not observe any side effects, in particular, no fever and no signs of typical virus infections. The pharmacokinetic measurements showed measurable concentrations of H-1PV over the first 5 days (and in some patients for another one), and virus DNA was constantly present in the 22 h period between the daily injections indicating continuous exposure.
Another important aspect of the safety of H-1PV treatment is biosafety and possible virus shedding by treated patients. Therefore, patients were hospitalized under quarantine conditions until they had generated an antibody response or were found to be negative for H-1PV in shedding samples (feces, urine, and saliva). Only small amounts of virus DNA could be detected in some feces probes while saliva and urine were constantly negative. It is still unknown whether the positive test results are indicative of active and infectious virus or of virus nucleic acids only.
In conclusion, based on three applications in humans, H-1PV can be considered safe and well tolerated at least to a cumulative dose of 1 × 109 pfu. Flu-like symptoms can occur under treatment but this could not be confirmed in the last trial with GMP-virus. Thus, it cannot be ruled out that these symptoms were related to impurities of the virus preparation and not caused by the virus itself. Another consequence of the current safety data is that the need for patients to be kept under isolation should be reconsidered for future trials.
Conflict of Interest Statement
Jean Rommelaere received research grants from ORYX GmbH & Co.KG. Assia L. Angelova, Karsten Geletneky, and Jean Rommelaere have an ownership interest (including patents) in the German Cancer Research Center. Jürg P. F. Nüesch has no conflicts of interest to declare.
Acknowledgments
AA was supported by a research grant from ORYX GmbH & Co.KG. The first Phase I/IIa clinical trial with parvovirus H-1 in glioblastoma multiforme patients (ParvOryx01) was funded by ORYX GmbH & Co.KG. The authors would like to acknowledge Dr. Jacek Hajda for ParvOryx01 safety data assessment and Dr. Michael Dahm for helpful discussions.
References
Adair, R. A., Roulstone, V., Scott, K. J., Morgan, R., Nuovo, G. J., Fuller, M., et al. (2012). Cell carriage, delivery, and selective replication of an oncolytic virus in tumor in patients. Sci. Transl. Med. 4, 138ra77. doi: 10.1126/scitranslmed.3003578
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Adeyemi, R., and Pintel, D. J. (2012). Replication of minute virus of mice in murine cells is facilitated by virally induced depletion of p21. J. Virol. 86, 8328–8332. doi:10.1128/JVI.00820-12
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Adeyemi, R. O., Landry, S., Davis, M. E., Weitzman, M. D., and Pintel, D. J. (2010). Parvovirus minute virus of mice induces a DNA damage response that facilitates viral replication. PLoS Pathog. 6:e1001141. doi:10.1371/journal.ppat.1001141
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Adeyemi, R. O., and Pintel, D. J. (2014). Parvovirus-induced depletion of cyclin B1 prevents mitotic entry of infected cells. PLoS Pathog. 10:e1003891. doi:10.1371/journal.ppat.1003891
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ahmed, M. (2013). Oncolytic viruses as therapeutic agents for prostate cancer. Adv. Tech. Biol. Med. 1, 107. doi:10.4172/atbm.1000107
Allaume, X., El-Andaloussi, N., Leuchs, B., Bonifati, S., Kulkarni, A., Marttila, T., et al. (2012). Retargeting of rat parvovirus H-1PV to cancer cells through genetic engineering of the viral capsid. J. Virol. 86, 3452–3465. doi:10.1128/JVI.06208-11
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Alvarez-Breckenridge, C. A., Yu, J., Price, R., Wei, M., Wang, Y., Nowicki, M. O., et al. (2012). The histone deacetylase inhibitor valproic acid lessens NK cell action against oncolytic virus-infected glioblastoma cells by inhibition of STAT5/T-BET signaling and generation of gamma interferon. J. Virol. 86, 4566–4577. doi:10.1128/JVI.05545-11
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Angelova, A. L., Aprahamian, M., Balboni, G., Delecluse, H. J., Feederle, R., Kiprianova, I., et al. (2009a). Oncolytic rat parvovirus H-1PV, a candidate for the treatment of human lymphoma: in vitro and in vivo studies. Mol. Ther. 17, 1164–1172. doi:10.1038/mt.2009.78
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Angelova, A. L., Aprahamian, M., Grekova, S. P., Hajri, A., Leuchs, B., Giese, N. A., et al. (2009b). Improvement of gemcitabine-based therapy of pancreatic carcinoma by means of oncolytic parvovirus H-1PV. Clin. Cancer Res. 15, 511–519. doi:10.1158/1078-0432.CCR-08-1088
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bär, S., Rommelaere, J., and Nüesch, J. P. F. (2013). Vesicular transport of progeny parvovirus particles through ER and Golgi regulates maturation and cytolysis. PLoS Pathog. 9:e1003605. doi:10.1371/journal.ppat.1003605
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bär, S., Rommelaere, J., and Nuesch, J. P. F. (2015). PKCη/Rdx-driven phosphorylation of PDK1: a novel mechanism promoting cancer cell survival and permissiveness for parvovirus-induced lysis. PLoS Pathog. 11:e1004703. doi:10.1371/journal.ppat.1004703
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bartlett, D. L., Liu, Z., Sathaiah, M., Ravindranathan, R., Guo, Z., He, Y., et al. (2013). Oncolytic viruses as therapeutic cancer vaccines. Mol. Cancer 12, 103. doi:10.1186/1476-4598-12-103
Bashir, T., Horlein, R., Rommelaere, J., and Willwand, K. (2000). Cyclin A activates the DNA polymerase delta-dependent elongation machinery in vitro: a parvovirus DNA replication model. Proc. Natl. Acad. Sci. U.S.A. 97, 5522–5527. doi:10.1073/pnas.090485297
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Breitbach, C. J., Arulanandam, R., De Silva, N., Thorne, S. H., Patt, R., Daneshmand, M., et al. (2013). Oncolytic vaccinia virus disrupts tumor-associated vasculature in humans. Cancer Res. 73, 1265–1275. doi:10.1158/0008-5472.CAN-12-2687
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Breitbach, C. J., Burke, J., Jonker, D., Stephenson, J., Haas, A. R., Chow, L. Q., et al. (2011). Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature 477, 99–104. doi:10.1038/nature10358
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, Y. Q., de Foresta, F., Hertoghs, J., Avalosse, B. L., Cornelis, J. J., and Rommelaere, J. (1986). Selective killing of simian virus 40-transformed human fibroblasts by parvovirus H-1. Cancer Res. 46, 3574–3579.
Chen, Y. Q., Tuynder, M. C., Cornelis, J. J., Boukamp, P., Fusenig, N. E., and Rommelaere, J. (1989). Sensitization of human keratinocytes to killing by parvovirus H-1 takes place during their malignant transformation but does not require them to be tumorigenic. Carcinogenesis 10, 163–167. doi:10.1093/carcin/10.1.163
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cornelis, J. J., Deleu, L., Koch, U., and Rommelaere, J. (2006). “Parvovirus oncosuppression,” in The Parvoviruses, eds J. R. Kerr, S. F. Cotmore, M. E. Bloom, R. M. Linden, and C. R. Parrish (London: Hodder Arnold), 365–384.
Cotmore, S. F., Agbandje-McKenna, M., Chiorini, J. A., Mukha, D. V., Pintel, D. J., Qiu, J., et al. (2014). The family Parvoviridae. Arch. Virol. 159, 1239–1247. doi:10.1007/s00705-013-1914-1
Cotmore, S. F., Christensen, J., and Tattersall, P. (2000). Two widely spaced initiator binding sites create an HMGB1-dependent parvovirus rolling-hairpin replication origin. J. Virol. 74, 1332–1341. doi:10.1128/JVI.74.3.1332-1341.2000
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cotmore, S. F., and Tattersall, P. (1998). High-mobility group 1/2 proteins are essential for initiating rolling-circle-type DNA replication at a parvovirus hairpin origin. J. Virol. 72, 8477–8484.
Cotmore, S. F., and Tattersall, P. (2014). Parvoviruses: small does not mean simple. Annu. Rev. Virol. 1, 517–537. doi:10.1146/annurev-virology-031413-085444
Daeffler, L., Hörlein, R., Rommelaere, J., and Nüesch, J. P. (2003). Modulation of minute virus of mice cytotoxic activities through site-directed mutagenesis within the NS coding region. J. Virol. 77, 12466–12478. doi:10.1128/JVI.77.23.12466-12478.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Deleu, L., Pujol, A., Faisst, S., and Rommelaere, J. (1999). Activation of promoter P4 of the autonomous parvovirus minute virus of mice at early S phase is required for productive infection. J. Virol. 73, 3877–3885.
Dettwiler, S., Rommelaere, J., and Nüesch, J. P. (1999). DNA unwinding functions of minute virus of mice NS1 protein are modulated specifically by the lambda isoform of protein kinase C. J. Virol. 73, 7410–7420.
Di Piazza, M., Mader, C., Geletneky, K., Herrero Y Calle, M., Weber, E., Schlehofer, J., et al. (2007). Cytosolic activation of cathepsins mediates parvovirus H-1-induced killing of cisplatin and TRAIL-resistant glioma cells. J. Virol. 81, 4186–4198. doi:10.1128/JVI.02601-06
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dupressoir, T., Vanacker, J. M., Cornelis, J. J., Duponchel, N., and Rommelaere, J. (1989). Inhibition by parvovirus H-1 of the formation of tumors in nude mice and colonies in vitro by transformed human mammary epithelial cells. Cancer Res. 49, 3203–3208.
Eichwald, V., Daeffler, L., Klein, M., Rommelaere, J., and Salomé, N. (2002). The NS2 proteins of parvovirus minute virus of mice are required for efficient nuclear egress of progeny virions in mouse cells. J. Virol. 76, 10307–10319. doi:10.1128/JVI.76.20.10307-10319.2002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Faisst, S., Guittard, D., Benner, A., Cesbron, J. Y., Schlehofer, J. R., Rommelaere, J., et al. (1998). Dose-dependent regression of HeLa cell-derived tumours in SCID mice after parvovirus H-1 infection. Int. J. Cancer 75, 584–589. doi:10.1002/(SICI)1097-0215(19980209)75:4<584::AID-IJC15>3.3.CO;2-G
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Faisst, S., Schlehoffer, J. R., and zur Hausen, H. (1989). Transformation of human cells by oncogenic viruses supports permissiveness for parvovirus H-1 propagation. J. Virol. 63, 2152–2158.
Finlayson, E., and Birkmeyer, J. D. (2003). Effects of hospital volume on life expectancy after selected cancer operations in older adults: a decision analysis. J. Am. Coll. Surg. 196, 410–417. doi:10.1016/S1072-7515(02)01753-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Friedman, G. K., Cassady, K. A., Beierle, E. A., Markert, J. M., and Gillespie, G. Y. (2012). Targeting pediatric cancer stem cells with oncolytic virotherapy. Pediatr. Res. 71(4 Pt 2), 500–510. doi:10.1038/pr.2011.58
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fuks, F., Deleu, L., Dinsart, C., Rommelaere, J., and Faisst, S. (1996). Ras oncogene-dependent activation of the P4 promoter of minute virus of mice through a proximal P4 element interacting with the Ets family of transcription factors. J. Virol. 70, 1331–1339.
Gaertner, D. G., Smith, A. L., and Jacoy, R. O. (1996). Efficient induction of persistent and prenatal parvovirus infection in rats. Virus Res. 44, 67–78. doi:10.1016/0168-1702(96)01351-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Garber, K. (2006). China approves world’s first oncolytic virus therapy for cancer treatment. J. Natl. Cancer Inst. 98, 298–300. doi:10.1093/jnci/djj111
Geletneky, K., Huesing, J., Rommelaere, J., Schlehofer, J. R., Leuchs, B., Dahm, M., et al. (2012). Phase I/IIa study of intratumoral/intracerebral or intravenous/intracerebral administration of parvovirus H-1 (ParvOryx) in patients with progressive primary or recurrent glioblastoma multiforme: ParvOryx01 protocol. BMC Cancer 12:99. doi:10.1186/1471-2407-12-99
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Geletneky, K., Angelova, A., Leuchs, B., Bhat, R., Just, A., Capper, D., et al. (2014a). Combination of intravenous and intracerebral injection of oncolytic parvovirus H-1 in a phase I/IIa clinical trial of patients with recurrent glioblastoma multiforme: penetration of H-1 virus across the blood-brain barrier. Neuro-oncology 16(Suppl. 5), v83–v84. doi:10.1093/neuonc/nou255.21
Geletneky, K., Huesing, J., Rommelaere, J., Schlehofer, J. R., Leuchs, B., Dahm, M., et al. (2014b). First combined intravenous and intracerebral application of an oncolytic virus, parvovirus H-1, in a phase I/Iia clinical trial in patients with recurrent glioblastoma multiforme (ParvOryx01). J. Clin. Oncol. 32, 5s. doi:10.1186/1471-2407-12-99
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Geletneky, K., Kiprianova, I., Ayache, A., Koch, R., Herrero Y Calle, M., Deleu, L., et al. (2010). Regression of advanced rat and human gliomas by local or systemic treatment with oncolytic parvovirus H-1 in rat models. Neuro-oncology 12, 804–814. doi:10.1093/neuonc/noq023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Geletneky, K., Leoni, A. L., Pohlmeyer-Esch, G., Loebhard, S., Baetz, A., Leuchs, B., et al. (2015a). Pathology, organ distribution, and immune responses after single and repeated intravenous injection of rats with clinical-grade parvovirus H1. Comp. Med. 65, 23–35.
Geletneky, K., Leoni, A. L., Pohlmeyer-Esch, G., Loebhard, S., Leuchs, B., Hoefer, C., et al. (2015b). Bioavailability, biodistribution, and CNS toxicity of clinical-grade parvovirus H1 after intravenous and intracerebral injection in rats. Comp. Med. 65, 36–45.
Goldufsky, J., Sivendran, S., Harcharik, S., Pan, M., Bernardo, S., Stern, R. H., et al. (2013). Oncolytic virus therapy for cancer. Oncolytic Virother. 2, 31–46. doi:10.2147/OV.S38901
Grekova, S., Aprahamian, M., Giese, N., Schmitt, S., Giese, T., Falk, C. S., et al. (2010a). Immune cells participate in the oncosuppressive activity of parvovirus H-1PV and are activated as a result of their abortive infection with this agent. Cancer Biol. Ther. 10, 1280–1289. doi:10.4161/cbt.10.12.13455
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Grekova, S., Zawatzky, R., Hörlein, R., Cziepluch, C., Mincberg, M., Davis, C., et al. (2010b). Activation of an antiviral response in normal but not transformed mouse cells: a new determinant of minute virus of mice oncotropism. J. Virol. 84, 516–531. doi:10.1128/JVI.01618-09
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Haseley, A., Alvarez-Breckenridge, C., Chaudhury, A. R., and Kaur, B. (2009). Advances in oncolytic virus therapy for glioma. Recent Pat. CNS Drug Discov. 4, 1–13. doi:10.2174/157488909787002573
Hristov, G., Krämer, M., Li, J., El-Andaloussi, N., Mora, R., Daeffler, L., et al. (2010). Through its non-structural protein NS1, parvovirus H-1 induces apoptosis via accumulation of reactive oxygen species. J. Virol. 84, 5909–5922. doi:10.1128/JVI.01797-09
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hughes, T., Coffin, R. S., Lilley, C. E., Ponce, R., and Kaufman, H. L. (2014). Critical analysis of an oncolytic herpesvirus encoding granulocyte-macrophage colony stimulating factor for the treatment of malignant melanoma. Oncolytic Virother. 3, 11–20. doi:10.2147/OV.S36701
Jacoby, R. O., and Ball-Goodrich, L. (1995). Parvovirus infections of mice and rats. Semin. Virol. 6, 329–337. doi:10.1006/smvy.1995.0039
Jacoby, R. O., Bhatt, P. N., and Jonas, A. M. (1979). “Viral diseases,” in The Laboratory Rat, Vol. 1: Biology and Diseases, eds H. J. Baker, J. R. Lindsey, and S. H. Weisbroth (New York, NY: Academic Press), 271–306.
Jemal, A., Siegel, R., Ward, E., Murray, T., Xu, J., and Thun, M. J. (2007). Cancer statistics, 2007. CA Cancer J. Clin. 57, 43–66. doi:10.3322/canjclin.57.1.43
Kaufman, H. L., Kim, D. W., DeRaffele, G., Mitcham, J., Coffin, R. S., and Kim-Schulze, S. (2010). Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Ann. Surg. Oncol. 17, 718–730. doi:10.1245/s10434-009-0809-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kiprianova, I., Thomas, N., Ayache, A., Fischer, M., Leuchs, B., Klein, M., et al. (2011). Regression of glioma in rat models by intranasal application of parvovirus H-1. Clin. Cancer Res. 17, 5333–5342. doi:10.1158/1078-0432.CCR-10-3124
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lachmann, S., Rommelaere, J., and Nüesch, J. P. F. (2003). Novel PKCeta is required to activate replicative functions of the major nonstructural protein NS1 of minute virus of mice. J. Virol. 77, 8048–8060. doi:10.1128/JVI.77.14.8048-8060.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lachmann, S., Bär, S., Rommelaere, J., and Nüesch, J. P. (2008). Parvovirus interference with intracellular signaling: mechanism of PKCeta activation in MVM-infected A9 fibroblasts. Cell. Microbiol. 10, 755–769. doi:10.1111/j.1462-5822.2007.01082.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lacroix, J., Leuchs, B., Li, J., Hristov, G., Deubzer, H. E., Kulozik, A. E., et al. (2010). Parvovirus H1 selectively induces cytotoxic effects on human neuroblastoma cells. Int. J. Cancer 127, 1230–1239. doi:10.1002/ijc.25168
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lacroix, J., Schlund, F., Leuchs, B., Adolph, K., Sturm, D., Bender, S., et al. (2014). Oncolytic effects of parvovirus H-1 in medulloblastomma are associated with repression of master regulators of early neurogenesis. Int. J. Cancer 134, 703–716. doi:10.1002/ijc.28386
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Le Cesne, A., Dupressoir, T., Janin, N., Spielmann, M., Le Chevalier, T., Sancho-Garnier, H., et al. (1993). Intra-lesional administration of a live virus, parvovirus H1 (PVH-1) in cancer patients: a feasibility study. Proc. Am. Soc. Clin. Oncol. 12, 297.
Li, J., Bonifati, S., Hristov, G., Marttila, T., Valmary-Degano, S., Stanzel, S., et al. (2013). Synergistic combination of valproic acid and oncolytic parvovirus H-1PV as a potential therapy against cervical and pancreatic carcinomas. EMBO Mol. Med. 5, 1537–1555. doi:10.1002/emmm.201302796
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lichty, B. D., Breitbach, C. J., Stojdl, D. F., and Bell, J. C. (2014). Going viral with cancer immunotherapy. Nat. Rev. 14, 559–567. doi:10.1038/nrc3770
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liu, T. C., Galanis, E., and Kirn, D. (2007). Clinical trial results with oncolytic virotherapy: a century of promise, a decade of progress. Nat. Clin. Pract. Oncol. 4, 101–117. doi:10.1038/ncponc0736
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Makower, D., Rozenblit, A., Kaufman, H., Edelman, M., Lane, M. E., Zwiebel, J., et al. (2003). Phase II clinical trial of intralesional administration of the oncolytic adenovirus ONYX-015 in patients with hepatobiliary tumors with correlative p53 studies. Clin. Cancer Res. 9, 693–702.
Meerani, S., and Yao, Y. (2010). Oncolytic viruses in cancer therapy. Eur. J. Sci. Res. 40, 156–171.
Moehler, M., Blechacz, B., Weiskopf, N., Zeidler, M., Stremmel, W., Rommelaere, J., et al. (2001). Effective infection, apoptotic cell killing and gene transfer of human hepatoma cells but not primary hepatocytes by parvovirus H1 and derived vectors. Cancer Gene Ther. 8, 158–167. doi:10.1038/sj.cgt.7700288
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moehler, M., Zeidler, M., Schede, J., Rommelaere, J., Galle, P. R., Cornelis, J. J., et al. (2003). Oncolytic parvovirus H1 induces release of heat-shock protein72 in susceptible human tumor cells but may not affect primary immune cells. Cancer Gene Ther. 10, 477–480. doi:10.1038/sj.cgt.7700591
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Monif, G. R., Sever, J. L., and Cochran, W. D. (1965). The H-1 and RV-viruses and pregnancy: serological studies of certain groups of pregnant women. J. Pediatr. 67, 253–256. doi:10.1016/S0022-3476(65)80248-7
Moralès, O., Richard, A., Martin, N., Mrizak, D., Sénéchal, M., Miroux, C., et al. (2012). Activation of a helper and not regulatory human CD4+ T cell response by oncolytic H-1 parvovirus. PLoS ONE 7:e32197. doi:10.1371/journal.pone.0032197
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Muharram, G., Le Rhun, E., Loison, I., Wizla, P., Richard, A., Martin, N., et al. (2010). Parvovirus H-1 induces cytopathic effects in breast carcinoma-derived cultures. Breast Cancer Res. Treat. 121, 23–33. doi:10.1007/s10549-009-0451-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Neuman, S. J., McCallin, P. F., and Sever, J. L. (1970). Attempts to isolate H-1 virus from spontaneous human abortions: a negative report. Teratology 3, 279–281. doi:10.1002/tera.1420030311
Nguyen, T. L., Wilson, M. G., and Hiscott, J. (2010). Oncolytic viruses and histone deacetylase inhibitors – a multi-pronged strategy to target tumor cells. Cytokine Growth Factor Rev. 21, 153–159. doi:10.1016/j.cytogfr.2010.03.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nüesch, J. P., Bär, S., Lachmann, S., and Rommelaere, J. (2009). Ezrin-radixin-moesin family proteins are involved in parvovirus replication and spreading. J. Virol. 83, 5854–5863. doi:10.1128/JVI.00039-09
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nüesch, J. P., Lacroix, J., Marchini, A., and Rommelaere, J. (2012). Molecular pathways: rodent parvoviruses – mechanisms of oncolysis and prospects for clinical cancer treatment. Clin. Cancer Res. 18, 3516–3523. doi:10.1158/1078-0432.CCR-11-2325
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nüesch, J. P. F. (2006). “Regulation of non-structural protein functions by differential synthesis, modification and trafficking,” in Parvoviruses, eds J. R. Kerr, S. F. Cotmore, M. E. Bloom, R. M. Linden, and C. R. Parrish (London: Hodder Arnold), 275–287.
Nüesch, J. P. F., and Rommelaere, J. (2006). NS1 interaction with CKII alpha: novel protein complex mediating parvovirus-induced cytotoxicity. J. Virol. 80, 4729–4739. doi:10.1128/JVI.80.10.4729-4739.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nüesch, J. P. F., and Rommelaere, J. (2007). A viral adaptor protein modulating casein kinase II activity induces cytopathic effects in permissive cells. Proc. Natl. Acad. Sci. U.S.A. 104, 12482–12487. doi:10.1073/pnas.0705533104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Otsuki, A., Patel, A., Kasai, K., Suzuki, M., Kurozumi, K., Chiocca, E. A., et al. (2008). Histone deacetylase inhibitors augment antitumor efficacy of herpes-based oncolytic viruses. Mol. Ther. 16, 1546–1555. doi:10.1038/mt.2008.155
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Paglino, J. C., Andres, W., and van den Pol, A. N. (2014). Autonomous parvoviruses neither stimulate nor are inhibited by the type I interferon response in human normal or cancer cells. J. Virol. 88, 4932–4942. doi:10.1128/JVI.03508-13
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pecora, A. L., and Lorence, R. M. (2007). “Phase I clinical experience with intravenous administration of PV701, an oncolytic virus,” in Cancer Drug Discovery and Development: Gene Therapy for Cancer, eds K. K. Hunt, S. A. Vorburger, and S. G. Swisher (Totowa, NJ: Humana Press Inc), 399–411.
Perros, M., Deleu, L., Vanacker, J. M., Kherrouche, Z., Spruyt, N., Faisst, S., et al. (1995). Upstream CREs participate in the basal activity of minute virus of mice promoter P4 and in its stimulation in ras-transformed cells. J. Virol. 69, 5506–5515.
Porwal, M., Cohen, S., Snoussi, K., Popa-Wagner, R., Anderson, F., Dugot-Senant, N., et al. (2013). Parvoviruses cause nuclear envelope breakdown by activating key enzymes of mitosis. PLoS Pathog. 9:e1003671. doi:10.1371/journal.ppat.1003671
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rayet, B., Lopez-Guerrero, J. A., Rommelaere, J., and Dinsart, C. (1998). Induction of programmed cell death by parvovirus H-1 in U937 cells: connection with the TNFα signaling pathway. J. Virol. 72, 8893–8903.
Raykov, Z., Grekova, S. P., Hörlein, R., Leuchs, B., Giese, T., Giese, N. A., et al. (2013). TLR-9 contributes to the antiviral innate immune sensing of rodent parvoviruses MVMp and H-1PV by normal human immune cells. PLoS ONE 8:e55086. doi:10.1371/journal.pone.0055086
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Richards, R. G., and Armentrout, R. W. (1979). Early events in parvovirus replication: lack of integration by minute virus of mice into host cell DNA. J. Virol. 30, 397–399.
Riolobos, L., Valle, N., Hernando, E., Maroto, B., Kann, M., and Almendral, J. M. (2010). Viral oncolysis that targets Raf-1 signaling control of nuclear transport. J. Virol. 84, 2090–2099. doi:10.1128/JVI.01550-09
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rommelaere, J., and Cornelis, J. J. (1991). Antineoplastic activity of parvoviruses. J. Virol. Methods 33, 233–251. doi:10.1016/0166-0934(91)90024-T
Rommelaere, J., Giese, N., Cziepluch, C., and Cornelis, J. J. (2005). “Parvoviruses as anticancer agents,” in Viral Therapy of Human Cancers, eds J. G. Sinkovics and J. C. Horvath (New York, NY: Marcel Dekker), 627–675.
Rommelaere, J., Geletneky, K., Angelova, A. L., Daeffler, L., Dinsart, C., Kiprianova, I., et al. (2010). Oncolytic parvoviruses as cancer therapeutics. Cytokine Growth Factor Rev. 21, 185–195. doi:10.1016/j.cytogfr.2010.02.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rommelaere, J., and Tattersall, P. (1989). “Oncosuppression by parvoviruses,” in CRC Handbook of Parvoviruses, ed. P. Tijssen (Boca Raton, FL: CRC Press), 41.
Ron, D., and Tal, J. (1985). Coevolution of cells and virus as a mechanism for the persistence of lymphotropic minute virus of mice in L-cells. J. Virol. 55, 424–430.
Russel, S. J., Peng, K. W., and Bell, J. C. (2012). Oncolytic virotherapy. Nat. Biotechnol. 30, 658–670. doi:10.1038/nbt.2287
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Singh, P. K., Doley, J., Ravi Kumar, G., Saho, A. P., and Tiwari, A. K. (2012). Oncolytic viruses & their specific targeting to tumour cells. Indian J. Med. Res. 136, 571–584.
Stummer, W., Pichlmeier, U., Meinel, T., Wiestler, O. D., Zanella, F., Reulen, H. J., et al. (2006). Fluorescence-guided surgery with 5-aminolevulinic acid for resection of malignant glioma: a randomised controlled multicentre phase III trial. Lancet Oncol. 7, 392–401. doi:10.1016/S1470-2045(06)70665-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tattersall, P. (2006). “The evolution of parvoviral taxonomy,” in The Parvoviruses, eds J. R. Kerr, S. F. Cotmore, M. E. Bloom, R. M. Linden, and C. R. Parrish (London: Hodder Arnold), 5–14.
Toolan, H., and Ledinko, N. (1965). Growth and cytopathogenicity of H-viruses in human and simian cell cultures. Nature 208, 812–813. doi:10.1038/208812a0
Toolan, H. W., Buttle, G. A. H., and Kay, H. E. M. (1962). Isolation of the H-1 and H-3 viruses directly from human embryos. Proc. Am. Assoc. Cancer Res. 3, 368.
Toolan, H. W., Dalldorf, G., Barclay, M., Chandra, S., and Moore, A. E. (1960). An unidentified, filterable agent isolated from transplanted human tumors. Proc. Natl. Acad. Sci. U.S.A. 46, 1256–1258. doi:10.1073/pnas.46.9.1256
Toolan, H. W., Saunders, E. L., Southam, C. M., Moore, A. E., and Levin, A. G. (1965). H-1 virus viremia in the human. Proc. Soc. Exp. Biol. Med. 119, 711–715. doi:10.3181/00379727-119-30278
Vacchelli, E., Eggermont, A., Sautès-Fridman, C., Galon, J., Zitvogel, L., Kroemer, G., et al. (2013). Oncolytic viruses for cancer therapy. Oncoimmunology 2, e24612. doi:10.4161/onci.24612
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vähä-Koskela, M., and Hinkkanen, A. (2014). Tumor restrictions to oncolytic virus. Biomedicines 2, 163–194. doi:10.3390/biomedicines2020163
Van Pachterbeke, C., Tuynder, M., Cosyn, J. P., Lespagnard, L., Larsimont, D., and Rommelaere, J. (1993). Parvovirus H-1 inhibits growth of short-term tumor-derived but not normal mammary tissue cultures. Int. J. Cancer 55, 672–677. doi:10.1002/ijc.2910550427
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Van Pachterbeke, C., Tuynder, M., Brandenburger, A., Leclercq, G., Borras, M., and Rommelaere, J. (1997). Varying sensitivity of human mammary carcinoma cells to the toxic effect of parvovirus H-1. Eur. J. Cancer 33, 1648–1653. doi:10.1016/S0959-8049(97)00153-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Van Oosten, R. L., Earel, J. K. Jr., and Griffith, T. S. (2007). Histone deacetylase inhibitors enhance Ad5-TRAIL killing of TRAIL-resistant prostate tumor cells through increased caspase-2 activity. Apoptosis 12, 561–571. doi:10.1007/s10495-006-0009-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, Y. Y., Liu, J., Zheng, Q., Ran, Z. H., Salomé, N., Vogel, M., et al. (2012). Effect of the parvovirus H-1 non-structural protein NS1 on the tumorigenicity of human gastric cancer cells. J. Dig. Dis. 13, 366–373. doi:10.1111/j.1751-2980.2012.00601.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Woller, N., Gürlevik, E., Ureche, C. I., Schumacher, A., and Kühnel, F. (2014). Oncolytic viruses as anticancer vaccines. Front. Oncol. 4:188. doi:10.3389/fonc.2014.00188
Wong, H. H., Lemoine, N. R., and Wang, Y. (2010). Oncolytic viruses for cancer therapy: overcoming the obstacles. Viruses 2, 78–106. doi:10.3390/v2010078
Keywords: oncolytic virotherapy of cancer, oncolytic viruses, oncotropism, oncoselectivity, parvovirus H-1, preclinical assessment, toxicological studies, safety profile
Citation: Angelova AL, Geletneky K, Nüesch JPF and Rommelaere J (2015) Tumor selectivity of oncolytic parvoviruses: from in vitro and animal models to cancer patients. Front. Bioeng. Biotechnol. 3:55. doi: 10.3389/fbioe.2015.00055
Received: 16 December 2014; Paper pending published: 25 February 2015;
Accepted: 05 April 2015; Published: 22 April 2015
Edited by:
John M. Hardham, Zoetis Inc., USAReviewed by:
Isabel Cristina Pires, University of Trás-os-Monts e Alto Douro, PortugalCaroline Breitbach, Jennerex Inc., USA
Copyright: © 2015 Angelova, Geletneky, Nüesch and Rommelaere. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jean Rommelaere, Infection and Cancer Program, Division of Tumor Virology, German Cancer Research Center (DKFZ), Im Neuenheimerfeld 280, Heidelberg 69120, Germanyai5yb21tZWxhZXJlQGRrZnotaGVpZGVsYmVyZy5kZQ==