 Ellen G. J. Ripmeester
Ellen G. J. Ripmeester Ufuk Tan Timur
Ufuk Tan Timur Marjolein M. J. Caron
Marjolein M. J. Caron Tim J. M. Welting
Tim J. M. Welting- Laboratory for Experimental Orthopedics, Department of Orthopedic Surgery, Maastricht University Medical Center, Maastricht, Netherlands
Osteoarthritis (OA) is an extremely prevalent age-related condition. The economic and societal burden due to the cost of symptomatic treatment, inability to work, joint replacement, and rehabilitation is huge and increasing. Currently, there are no effective medical therapies that delay or reverse the pathological manifestations of OA. Current treatment options are, without exception, focused on slowing down progression of the disease to postpone total joint replacement surgery for as long as possible and keeping the associated pain and joint immobility manageable. Alterations in the articular cartilage chondrocyte phenotype might be fundamental in the pathological mechanisms of OA development. In many ways, the changing chondrocyte phenotype in osteoarthritic cartilage resembles the process of endochondral ossification as seen, for instance, in developing growth plates. However, the relative contribution of endochondral ossification to the changing chondrocyte phenotype in the development and progression of OA remains poorly described. In this review, we will discuss the current knowledge regarding the cartilage endochondral phenotypic changes occurring during OA development and progression, as well as the molecular and environmental effectors driving these changes. Understanding how these molecular mechanisms determine the chondrocyte cell fate in OA will be essential in enabling cartilage regenerative approaches in future treatments of OA.
Introduction
Osteoarthritis (OA) is the most common degenerative joint disorder worldwide and its incidence rises with age (Loeser et al., 2012). The economic and societal burden due to costs of symptomatic treatment, inability to work, joint replacement surgery (and coinciding implant infections), rehabilitation, and social isolation is huge (Bijlsma et al., 2011; Le et al., 2012). Identifying the main molecular mechanisms by which OA is initiated and progresses is still one of the biggest challenges in this field. However, our current knowledge teaches us that the initial trigger for developing OA is multifactorial with risk factors, including obesity, diabetes, genetics, and trauma (Felson et al., 2000). Despite the diversity of initial triggers OA disease progression follows a predictable cell biological progression. While pain experience, joint immobility, and speed of disease progression are some of the few patient-variable parameters. Depending on the OA-stage, interventions are mainly based on alleviating chronic pain and preserving joint mobility by visco-supplementation, and physiotherapy to postpone joint replacement surgery (Lo et al., 2003; Zhang et al., 2010; Page et al., 2011). While there is a lack of treatment options that are disease-modifying and improve joint-tissue homeostasis. Substantial effort is required to identify targetable pathways or individual factors that alter the diseased cartilage phenotype in OA.
A prominent feature of OA is cartilage degradation. Other joint structures, such as the synovium, Hoffa’s fat pad (HFP), meniscus, and subchondral bone have been demonstrated to experience OA-specific pathologic changes. Changes in these joint structures include not only infiltration of active immune cells in the synovium and HFP, but also fibrillation of the meniscus and sclerosis of the subchondral bone. Together, these changes lead to a loss of joint mobility and function, accompanied by chronic pain (Loeser et al., 2012).
From a biochemical perspective, the cartilage degradation observed in OA has been attributed to an elevated production of proteolytic enzymes, such as matrix metalloprotease 13 (MMP13) and aggrecanases, such as a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS) 4 and 5. These degrade important cartilage matrix components, such as type II collagen (COL2A1) and aggrecan (Hunter, 2011). In addition to elevated production of proteolytic enzymes in OA cartilage, other observed features in OA cartilage include the expression of chondrocyte hypertrophic markers [such as type 10 collagen (COL10A1)] (Little et al., 2009), vascularization, and focal calcification. Since, these features resemble the endochondral ossification process that occurs in the hypertrophic zone of the growth plate, it has been hypothesized that OA is a disease characterized by ectopic recapitulation of the endochondral ossification process (Dreier, 2010; Pitsillides and Beier, 2011; van der Kraan and van den Berg, 2012).
Post-developmental healthy articular cartilage homeostasis is thought to be “protected” against hypertrophic or catabolic changes by several pathways employing soluble mediators, including BMP-, TGF-β-, and hedgehog signaling (Dreier, 2010; Pitsillides and Beier, 2011). These pathways transcriptionally control the chondrocyte phenotype by tuning the activity and levels of major chondrocyte phenotype-determining downstream transcription factors, such as SOX9, RUNX2, and SMADs (van der Kraan and van den Berg, 2012). Many risk factors for developing OA are thought to (in)directly influence the activity of these pathways, and thus ultimately resulting in a changing chondrocyte phenotype that becomes disposed to entering endochondral ossification. This may place, besides the local inflammatory condition (among others caused by synovitis), the chondrocyte/cartilage differentiation status central to the progression, or cause of OA.
The relation between the chondrocyte/cartilage differentiation status and OA development and -progression has been well-described and was acknowledged in the past by a number of excellent reviews (Dreier, 2010; Pitsillides and Beier, 2011; van der Kraan and van den Berg, 2012). In this review, we present an overview of the literature from the past 10 years, describing recent insights in the contribution of endochondral ossification-related processes in OA disease development and -progression. We conducted a PubMed literature search including papers from the past 10 years discussing endochondral ossification and its accompanying processes also occurring in OA disease progression. Molecular insight in the role of chondrocyte hypertrophic processes involved in OA initiation and progression is expected to provide valuable information for drug development targeting these processes for OA disease modification.
Review Procedure
To provide a current status on the role of hypertrophic changes in OA we searched for English manuscripts from the past 10 years on PubMed using the following search strategy (Figure 1): (OA OR osteoarthrosis OR osteoarthritis OR non-inflammatory arthritis OR degenerative arthritis OR osteoarthritic) AND (hypertrophic OR hypertrophy OR terminal differentiation OR hypertrophic differentiation OR end stage differentiation, OR endochondral ossification OR chondrocyte hypertrophy OR transdifferentiation OR mineralization OR mineralisation OR mineralized OR mineralised OR calcification) and chondrocyte. At May 3rd, 2017 a total of 461 papers were found and screened via title and abstract by two separate observers (ER and UT) using the following inclusion criteria: papers describing osteoarthritis, biomolecular data, and literature describing an OA process that resembles endochondral ossification in the title and abstract. Articles referring to apoptosis and autophagy were excluded as we wanted to focus on processes occurring during OA initiation and progression and we considered apoptosis and autophagy as end-stage processes. Besides apoptosis and autophagy, reviews were also excluded. When there was a discrepancy in paper selection, consensus was reached with all authors. This resulted in a short-list of 147 articles whose full-text was manually screened by four observers (ER, UT, MC, and TW) using the same inclusion criteria. Papers with missing full-texts were excluded. This resulted in a total of 76 papers being included in this review (Figure 1).
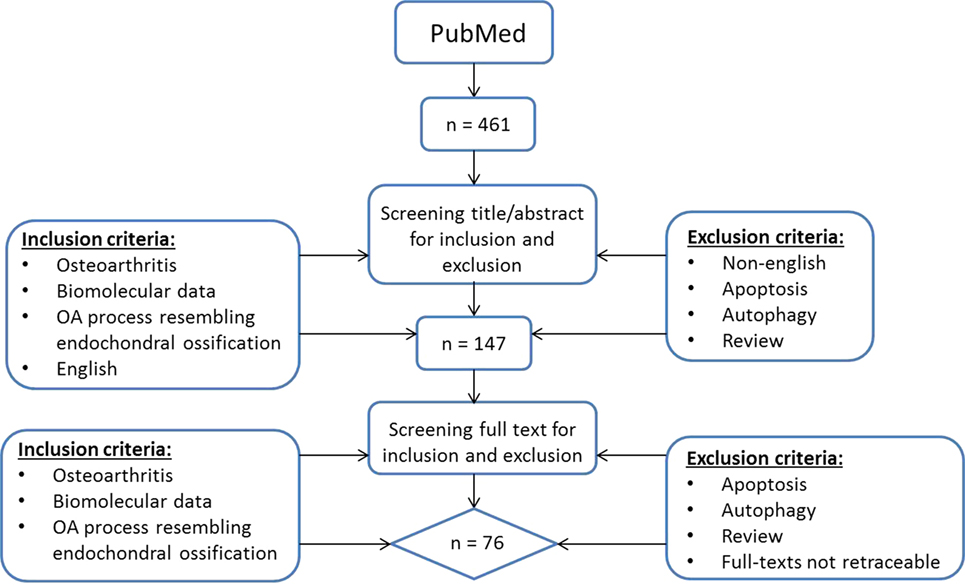
Figure 1. Literature search flowchart.
Results
Our search yielded a diverse range of publications from the past 10 years confirming earlier reports describing endochondral cellular phenotypic changes in OA cartilage and describing associations of these endochondral cellular phenotypic changes with the development and progression of OA. During our search, we noticed that the majority of the papers could be classified into signaling pathways known to be involved in endochondral ossification, such as Wnt-, Ihh/PTHrP-, TGF-β-, MAP-kinases, FGF-, Notch signaling, inflammatory signaling, and hypoxia-associated signaling pathways. Besides these pathways also papers describing processes, such as angiogenesis and matrix mineralization were found. These pathways and processes involved in endochondral ossification will be described now separately and new insights from the selected literature will be discussed with respect to these pathways.
Wnt Signaling
A thoroughly studied pathway involved in chondrocyte differentiation and hypertrophy is Wnt signaling. The canonical Wnt/β-catenin pathway (Wu et al., 2009; Borzi et al., 2010; Pitsillides and Beier, 2011; Castano Betancourt et al., 2012; Facchini et al., 2012; Papathanasiou et al., 2012; Leijten et al., 2013; van den Bosch et al., 2014; Guidotti et al., 2015; Chen et al., 2016; Staines et al., 2016) is activated by binding one of the canonical Wnt ligands to a Frizzled family receptor and an LRP5/6 co-receptor, which passes the signal via GSK-3β to inhibit the β-catenin destruction complex, causing an accumulation of β-catenin in the cytoplasm and eventual its translocation into the nucleus (MacDonald et al., 2009). This enables transcriptional coactivation with TCF/LEF transcription factors which are active in the transcription of Wnt-target genes, such as RUNX2 (MacDonald et al., 2009). In our review procedure, we did not find literature regarding non-canonical Wnt signaling that matched the search criteria. An overview of newly acquired insights into this pathway and its involvement in development of OA chondrocyte hypertrophy is provided in Table 1a and Figure 2.
Table 1. (A–K) Hypertrophy associated-factors.
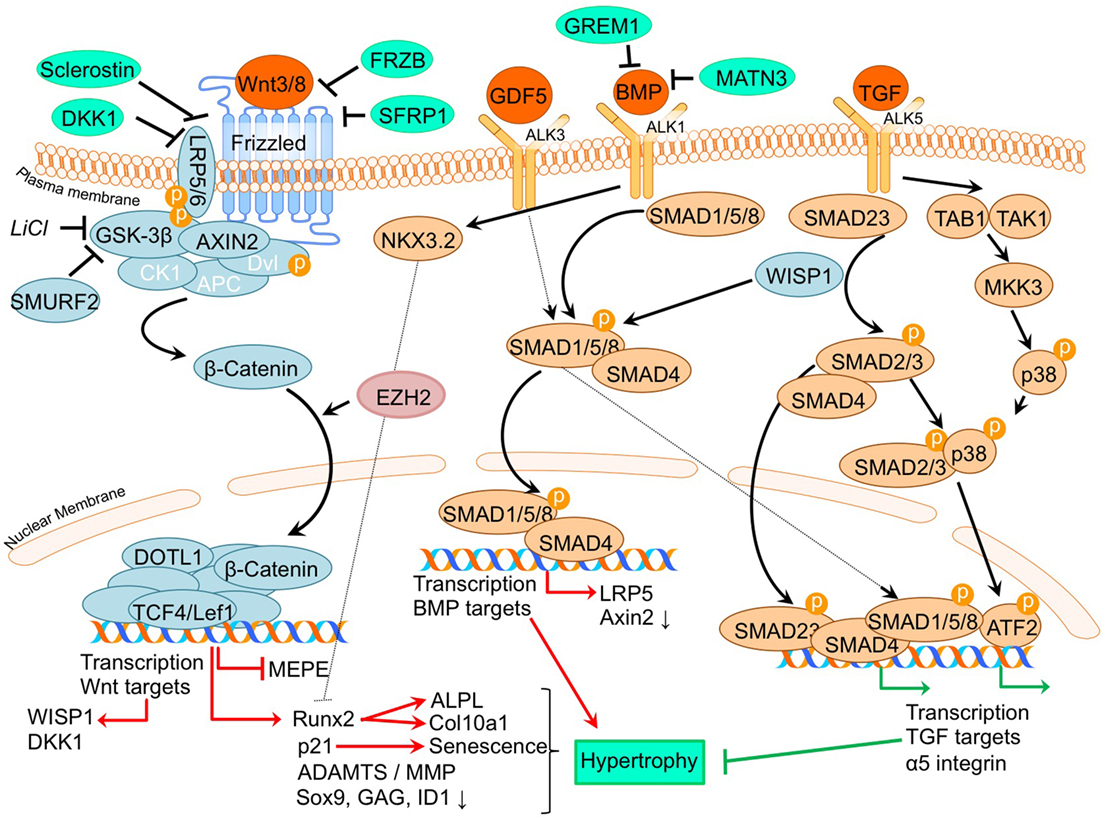
Figure 2. Overview of the canonical Wnt- and TGFβ superfamily signaling pathways and newly acquired insights into their relation to the development of the osteoarthritic (OA) chondrocyte hypertrophic phenotype. The canonical pathway was updated with recent insights in the role of Wnt- and TGFβ superfamily signaling in the hypertrophic switch occuring during OA development. In green pro-chondrogenic relations are shown, while pro-hypertrophic relations are depicted in red.
It is well established that β-catenin signaling is associated with chondrocyte hypertrophy (Wu et al., 2009; Borzi et al., 2010; Castano Betancourt et al., 2012; Facchini et al., 2012; Papathanasiou et al., 2012; Leijten et al., 2013; van den Bosch et al., 2014; Guidotti et al., 2015; Chen et al., 2016; Staines et al., 2016). Next to its role in chondrocyte hypertrophy, recent advancements in knowledge regarding OA and the involvement of Wnt signaling now demonstrate that expression levels of Wnt signaling- and β-catenin-inducing factors, as well as downstream Wnt effectors, such as LEF1 and AXIN2, are directly or indirectly associated with OA (Wu et al., 2009; Borzi et al., 2010; Castano Betancourt et al., 2012; Facchini et al., 2012; Papathanasiou et al., 2012; Leijten et al., 2013; van den Bosch et al., 2014; Guidotti et al., 2015; Chen et al., 2016; Staines et al., 2016).
A causative relationship between canonical Wnt signaling and OA initiation/progression has been suggested by two recent studies. Cartilage-specific SMURF2-mediated ubiquitination and proteasomal degradation of GSK-3β resulted in increased β-catenin signaling (Wu et al., 2009). Tibial and femoral articular cartilage in this Col2a1-Smurf2 mouse model demonstrated that 2.5-week-old mice displayed an increased basal layer of the deep articular cartilage with higher Col10a1 expression. These early hypertrophic changes in the articular cartilage of these mice were subsequently followed by cartilage degeneration and osteophyte formation when the mice became older (Wu et al., 2009), suggesting a direct relationship between early hypertrophic changes followed by OA development. Another study by Chen et al. provided evidence for the activation of Wnt/β-catenin signaling in OA development. This study investigated the effects of EZH2 inhibition on OA development in a surgically induced OA mouse model (Chen et al., 2016). EZH2 is the catalytic unit of the polycomb repressive complex 2 (PRC2), responsible for transcriptional silencing of a multitude of genes involved in differentiation (Morey and Helin, 2010). EZH2 expression was higher in OA chondrocytes compared to healthy chondrocytes and overexpression of EZH2 in normal chondrocytes resulted in activation of β-catenin signaling, including higher mRNA expression of its downstream effectors, AXIN2 and LEF1. Confirming the association between increased β-catenin signaling and OA development, intra-articular injection with a pharmacological EZH2 inhibitor in a surgically induced mouse OA model resulted in reduced cartilage degradation compared to mice injected with a saline control. In the cartilage this was accompanied by reduced mRNA expression of Col10a1, Adamts5, Mmp13, Mmp3, and increased mRNA expression of the Wnt inhibitor Sfrp1 as well as lower mRNA expression of β-catenin.
Other studies provide additional links between the Wnt/β-catenin pathway and OA (Borzi et al., 2010; Castano Betancourt et al., 2012; Papathanasiou et al., 2012; Leijten et al., 2013; van den Bosch et al., 2014). The expression of Wnt and BMP antagonists dickkopf 1 homolog (DKK1), frizzled-related protein (FRZB), and Gremlin 1 (GREM1) were reduced in OA cartilage compared to post-mortem healthy controls (Leijten et al., 2013). These Wnt and BMP antagonists were able to inhibit hypertrophic chondrocyte differentiation when added to chondrogenically differentiated mesenchymal stem cells (MSCs). This study also described crosstalk between the Wnt pathway and BMP signaling pathway (Leijten et al., 2013). This was functionally revealed through the decreased expression of the Wnt target gene AXIN2 (axis inhibition protein 2) and the BMP target gene ID1 (DNA-binding inhibitor protein 1), following treatment with BMP-2 or WNT3A, respectively (Leijten et al., 2013). This feedback loop allows tight control and balance between BMP and Wnt signaling (Leijten et al., 2013). New insight into this crosstalk between Wnt and BMP signaling pathways was obtained through the observation that BMP-2-induced Wnt signaling through influencing the SMAD1/5/8-depending LRP5 promoter activity in human OA articular chondrocytes (Papathanasiou et al., 2012). LRP5 is a co-receptor of the Wnt/β-catenin signaling pathway and its expression is increased in OA chondrocytes (Papathanasiou et al., 2012). It was indicated that the increase in mRNA levels of genes, such as Col10a1, Mmp13, and Adamts5 after BMP stimulation could be abrogated by LRP5 siRNA-mediated knockdown, indicating that the hypertrophic effects of BMP signaling may promote cartilage destruction via increased Wnt/β-catenin signaling (Papathanasiou et al., 2012). Other intriguing crosstalk was identified between Wnt and TGF-β signaling through the finding that downstream TGF-β activity is altered after chondrocytes were exposed to WNT3A and the downstream canonical Wnt signaling protein WISP1 (van den Bosch et al., 2014). In vitro stimulation of chondrocytes or in vivo viral expression of WNT3, WNT8, and or WISP1 skews TGF-β signaling from ALK5 (resulting in SMAD 2/3 signaling), toward signaling via ALK1 (resulting in SMAD 1/5/8 phosphorylation), inducing a hypertrophic chondrocyte phenotype (van den Bosch et al., 2014).
Further support associating Wnt signaling to OA development comes from a study in which a single nucleotide polymorphism in the DOT1L gene results in a reduced risk for hip OA. The underlying mechanism was identified as decreased Wnt signaling activity, which was confirmed via reduced expression levels of the Wnt target genes, AXIN2 and TCF1. DOT1L was found to be involved in chondrogenic differentiation and is thought to co-transcriptionally regulate transcription of Wnt-target genes via direct interaction with TCF4 (Castano Betancourt et al., 2012). Activation of Wnt signaling via LiCl-mediated GSK3-β inactivation led to chondrocyte cellular senescence, as indicated by increased p21 expression, production of reactive oxygen species, SAβ galactosidase activity, and activation of the DNA damage response (Guidotti et al., 2015). Moreover it was concluded that the inhibition of GSK3-β activity promotes a chondrocyte hypertrophic phenotype, thereby supporting that Wnt signaling activity has an important balancing influence on major cell biological parameters with consequences for the chondrocyte phenotype (Guidotti et al., 2015). ShRNA-mediated MMP13 knockdown in primary human chondrocytes resulted in reduced expression of β-catenin. This was accompanied by reduced expression of RUNX2, and an increased nuclear presence of SOX9 as well as a higher glycosaminoglycan (GAG) content. This indicates that a loss of MMP13 may ameliorate chondrocyte homeostasis in a feedback loop via reduction of β-catenin levels (Borzi et al., 2010).
In another study, it was suggested that increased Wnt signaling may be associated with OA progression (Staines et al., 2016). STR/Ort mice develop OA spontaneously, and expression of the Wnt signaling inhibitor sclerostin (Sost) decreased during OA progression in these mice. Furthermore, sclerostin expression was reduced in regions with more cartilage degradation, again linking increased Wnt signaling to cartilage degradation. Intriguingly, this study also provided links between endochondral growth defects and OA progression. STR/Ort mice display an abnormal growth plate development, with greater expression levels of COL10A1 and MMP13. This supports an association between endochondral defects and cartilage degradation (Staines et al., 2016). In contrast to the study by Staines et al. Papathanasiou et al. found an increased expression of the Wnt inhibitor sclerostin in human OA chondrocytes compared to normal (Papathanasiou et al., 2015). This was accompanied with a decreased methylation of the SOST promotor, enhancing the binding affinity of SMAD1/5/8 to the CpG region of the SOST promotor. Whether upregulation of SOST expression in articular chondrocytes is a causal factor or a result in OA has to be further determined (Papathanasiou et al., 2015).
Overall these studies together confirm the involvement of canonical Wnt signaling in chondrocyte hypertrophy and revealed novel mechanisms that appear to tune canonical Wnt signaling responses and which relate to the development of OA.
Ihh/PTHrP Signaling
PTHrP- and Indian hedgehog signaling pathways generate a feedback loop which is involved in controlling the chondrocyte phenotype in the growth plate in skeletal development, as well as in determining the homeostasis that keeps articular cartilage healthy (Kronenberg, 2003). When IHH reaches its target cell, it binds to the Patched-1 (PTCH1) receptor. In the absence of ligand, PTCH1 inhibits Smoothened (SMO), the binding of IHH relieves SMO inhibition, leading to activation of the GLI transcription factors: the activators GLI1 and GLI2 and the repressor GLI3. Activated GLI accumulates in the nucleus and controls the transcription of hedgehog target genes (Yang et al., 2015). Insight into the mechanism of action of PTH and PTHrP has been provided by the discovery of the type I PTH/PTHrP receptor (PTHR). This G protein-coupled receptor (GPCR) is associated with, among others, the adenylyl cyclase/protein kinase A (PKA) pathway (Mannstadt et al., 1999). The PTHrP/Ihh feedback loop is a major determinant of the chondrocyte hypertrophic phenotype, and thus postulated to be pivotal in OA development. It is thought that part of the contra-hypertrophic action of PTHrP originates from inducing the expression of BAPX1/NKX3.2 (Provot et al., 2006), a potent hypertrophic switch (Caron et al., 2015). Some of the pertinent evidence that chondrocyte hypertrophy is an intrinsic part of OA development comes from papers regarding PTHrP/Ihh signaling. An overview of the newly acquired insights into this pathway and its involvement in development of OA chondrocyte hypertrophy is provided in Table 1b and Figure 3.
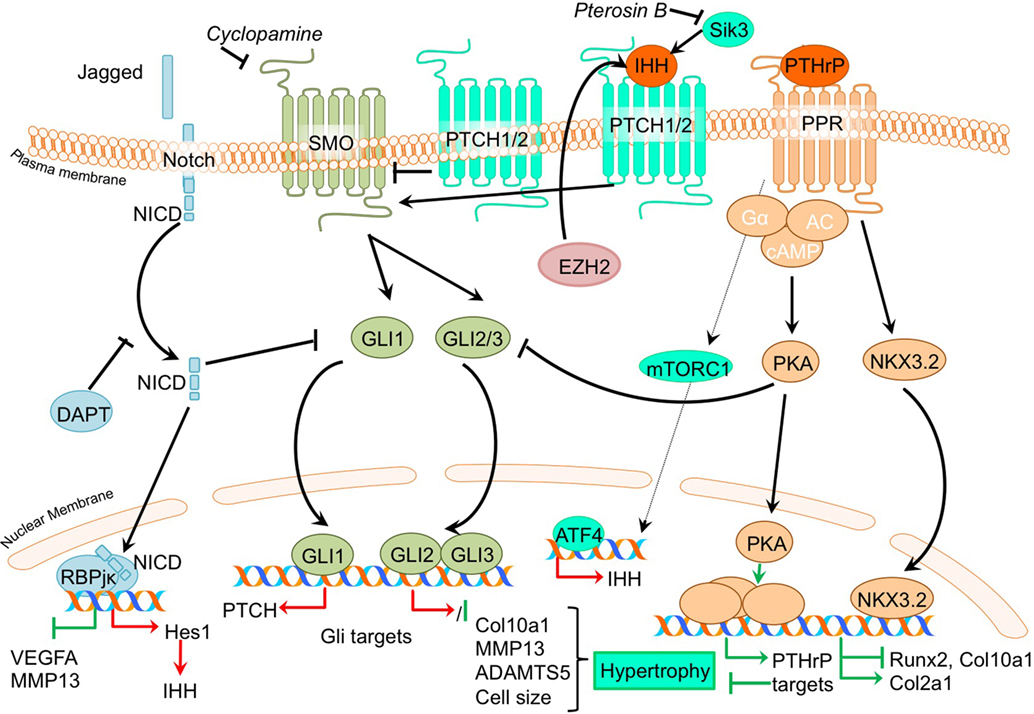
Figure 3. Overview of the canonical Ihh/PTHrP and Notch signaling pathways and newly acquired insights into their relation to the development of the osteoarthritic (OA) chondrocyte hypertrophic phenotype. The canonical pathway was updated with recent insights in the role of Ihh/PTHrP and Notch signaling in the hypertrophic switch occuring during OA development. In green pro-chondrogenic relations are shown, while pro-hypertrophic relations are depicted in red.
Given that PTHrP maintains the function of proliferating chondrocytes in the growth plate and inhibits chondrocyte differentiation toward hypertrophy, it has been suggested that PTHrP may be protective against OA (Kronenberg, 2003). Several studies demonstrate direct evidence that inhibiting hypertrophic processes results in protection against OA. Confirming an anti-hypertrophic and OA protective effect of PTHrP, it was shown that PTHrP can inhibit terminal differentiation of normal human articular chondrocytes and that intra-articular PTHrP administration in a chemically induced rat OA model reduces OA, as evidenced by reduced GAG loss (Chang et al., 2009). Along with reduced matrix loss, PTHrP treatment of chemically OA-induced knees also resulted in increased Col2a1 levels and reduced Col10a1 levels in the cartilage compared to OA-induced knees without PTHrP injection (Chang et al., 2009). Another study confirmed the chondroprotective and anti-hypertrophic role of PTHrP signaling in cartilage by revealing that incorporating PTHrP into a drug delivery system reduced OA severity in a chemically induced mouse OA model. This was evidenced through reduced GAG loss accompanied by decreased COL10A1 staining (Eswaramoorthy et al., 2012). The anti-hypertrophic role of PTHrP signaling was further confirmed in a study in which OA chondrocytes incorporated in alginate beads were treated with PTHrP, leading to reduced COL10A1 expression (Pesesse et al., 2014).
While PTHrP has been associated with anti-hypertrophic actions in the growth plate and in OA, Ihh expression is considered as a marker gene for hypertrophic chondrocytes in growth plates (Weisser et al., 2002). In concert with an important role for Ihh in chondrocyte hypertrophy, inhibition of the Ihh pathway protected against OA development (Zhou et al., 2014). In this study, a cartilage-specific Ihh knockout mouse was used to genetically confirm that Ihh drives the development of OA (Zhou et al., 2014). Cartilage-specific deletion of Ihh largely protected against the development of post-traumatic OA. Next to the decreased OA severity [as determined by the OOCHAS score (Osteoarthritis Cartilage Histopathology Assessment System)], Ihh-deleted mice revealed decreased expression of COL10A1 and MMP13, as well as decreased activity of cartilage proteolytic enzymes. A study by Yahara et al. illustrated a reduction of hypertrophic changes in Sik3 deficient mice including reduced Ihh levels (Yahara et al., 2016). The precise mechanism behind the anti-hypertrophic actions of Pterosin B via SIK3 is unknown, but KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway analysis indicated an inhibition of Ihh by Pterosin B, which is a SIK3 inhibitor.
It was demonstrated that the expression of IHH is increased in OA cartilage compared to cartilage obtained from non-OA patients (Wei et al., 2012). Furthermore, IHH levels were also higher in OA synovial fluid compared to non-OA synovial fluid (Wei et al., 2012). Since, expression of IHH was higher in more severely degenerated cartilage areas, as determined by the modified Mankin score, this also indicated an association of cartilage hypertrophic changes with OA progression (Wei et al., 2012). The authors in this study provided important evidence that chondrocyte hypertrophy is involved in the association between IHH and OA, showing that OA chondrocytes treated with Ihh displayed increased mRNA expression levels of markers of chondrocyte hypertrophy COL10A1 and MMP13. Furthermore, both IHH expression and chondrocyte size (the phenotypic hallmark of hypertrophy) were associated with OA severity. Modulation of Ihh signaling activity by the inhibitor cyclopamine caused a reduction of COL10A1 and MMP13 expression, suggesting that dampening the activity of the Ihh pathway may provide a target for OA treatment (Wei et al., 2012). Additional support connecting Ihh signaling to OA comes from an earlier mentioned study that described that the inhibition of EZH2 reduces cartilage degradation in vivo. It was found that IHH expression was increased after overexpression of EZH2 in chondrocytes, and was associated with an overall increased hypertrophic phenotype (Chen et al., 2016).
To isolate the effects of hedgehog pathway activation, the consequences of its activation for cartilage degradation have been investigated (Thompson et al., 2015). In contrast to the large body of evidence for an OA promoting role of Ihh, studies in bovine and human cartilage explants revealed that activation of hedgehog signaling by exposure to IHH did not increase ADAMTS5 or MMP13 expression. Chondrocyte hypertrophic phenotype development was not extensively addressed in this study, but it appears that the catabolic effects of hedgehog signaling in OA may be context dependent.
Recent advancements in research regarding crosstalk with other signaling pathways shows that hedgehog signaling crosstalks with the Notch signaling pathway (Lin et al., 2016) and with the Wnt signaling, BMP signaling, FGF (Zhou et al., 2016), and mTOR signaling pathways (Leijten et al., 2013). Inhibition of Notch1 resulted in an increased, HES1 dependent, hedgehog signaling activity (Lin et al., 2016). This increased activity led to an exacerbation of experimental OA, characterized by increased levels hedgehog target genes, osteocalcin, COL10A1, ADAMTS5, osteophyte formation, and reduced OARSI scores (Lin et al., 2016). It is still not completely understood as to how Notch signaling controls hedgehog signaling. It has, however, been suggested that Notch signaling activity may limit hypertrophy-provoking hedgehog signaling in articular chondrocytes (Lin et al., 2016). Interestingly, mTORC1 activation in mouse articular cartilage from 2-month-old mice resulted in upregulation of Ihh expression, along with other hypertrophic markers, such as RUNX2 and COL10A1, suggesting interplay between mTOR and Ihh signaling (Zhang et al., 2017). Along with an early increase in hypertrophic markers, mice in which mTORC1 was activated developed progressive OA, together with an increasing ratio of calcified cartilage relative to hyaline cartilage through time. In this study, it was also suggested that mTORC1 activation inhibits PTHrP signaling via downregulating the PPR receptor which ultimately results in impaired prevention of the initiation of chondrocyte hypertrophic differentiation (Zhang et al., 2017). Ihh signaling was also shown to crosstalk with the fibroblast growth factor (FGF) pathway, since the OA-like changes in the temporomandibular joints of FGFR3 knockout mice could be ameliorated using an Ihh signaling inhibitor (Zhou et al., 2016).
Together these studies underline the involvement of the PTHrP/Ihh feedback loop in determining the chondrocyte phenotype, and recent developments reveal that a disruption of this well-balanced system can initiate a hypertrophic switch, ultimately leading toward OA disease initiation and progression.
TGF-β Superfamily Signaling
The TGF-β superfamily is a group of multifunctional cytokines that play critical roles in cartilage homeostasis and have well established roles in endochondral ossification (Dangelo et al., 2001). In the classical TGF-β superfamily signaling pathway, superfamily ligands, such as TGF-β and BMP isoforms, bind to their respective cell surface receptors and, upon type I and -II receptor dimerization, activate a signaling cascade which includes the recruitment, phosphorylation, and interaction of different SMAD proteins. Upon activation, SMAD complexes translocate to the nucleus to drive transcription of genes regulating a variety of biological responses (van der Kraan et al., 2009). TGF-β has been strongly implicated in OA pathogenesis, since it has been well established that deregulation of TGF-β signaling is associated with OA (van der Kraan et al., 2009; van der Kraan, 2017). An overview of newly acquired insights into this pathway and its involvement in development of OA chondrocyte hypertrophy is provided in Table 1c and Figure 2.
Several studies have demonstrated an increased expression of TGF-β superfamily members or their receptors and down-stream signaling molecules in human OA chondrocytes compared to non-OA chondrocytes (Papathanasiou et al., 2012, 2015). TGF-β superfamily ligands can signal in chondrocytes via SMAD2/3 and also via SMAD1/5/8 (van der Kraan, 2017). The type I receptor ALK1 is associated with SMAD1/5/8 activity and hypertrophy and mineralization (Blaney Davidson et al., 2009). Additionally, a high correlation was found between ALK1 and the hypertrophic marker MMP13 (Blaney Davidson et al., 2009). In contrast, the type I receptor ALK5 is associated with SMAD2/3 activity and has anti-hypertrophic chondroprotective effects (van der Kraan et al., 2009). Together with the association of ALK1 with the hypertrophic marker MMP13, the study by Blaney Davidson et al. revealed that the ALK1/ALK5 ratio is increased in a post-traumatic model of OA using the destabilization of the medial meniscus (DMM) mouse OA model, and also increased with OA progression (Blaney Davidson et al., 2009), again highlighting the involvement of chondrocyte hypertrophic processes in OA development. The association of SMAD1/5/8 signaling with chondrocyte hypertrophy and OA is further confirmed in another study by Yang et al. Here, an association between the noncollagenous extracellular matrix protein Matrilin-3 (MATN3), and the SMAD1 pathway was confirmed (Yang et al., 2014). Authors of this study had earlier shown that Matn3 KO mice displayed premature articular cartilage hypertrophy and accelerated OA-like joint pathology (van der Weyden et al., 2006) and sought to determine molecular explanations for increased chondrocyte hypertrophy after Matn3 knockout. It was demonstrated that BMP-2 expression in embryonic chicken chondrocytes and a murine chondrocyte cell line lead to increased SMAD1 phosphorylation, resulting in increased Col10a1 promotor activity (Yang et al., 2014). MATN3 was able to directly interact with BMP-2 and acts as a BMP-2 antagonist inhibiting BMP-2-induced SMAD1 phosphorylation and reducing Col10a1 expression levels and chondrocyte hypertrophy.
TGF-β superfamily members can tune the activity and levels of chondrocyte phenotype-determining downstream transcriptional regulators. Examples of these downstream transcriptional regulators controlling the chondrocyte phenotype are BAPX1/NKX3.2 and ATF2. It was demonstrated that the anti-hypertrophic effect of BMP-7 on OA chondrocytes, evidenced by a reduction in mRNA expression levels of COL10A1, MMP13, and RUNX2, could be blocked with BAPX1/NKX3.2 knockdown, indicating that BMP-7 suppresses the hypertrophic phenotype in OA chondrocytes via BAPX1/NKX3.2 (Caron et al., 2015). ATF2 has been demonstrated to be expressed in the resting and proliferating zones of the growth plate, but not the hypertrophic zone (Reimold et al., 1996). It was established that ATF2 phosphorylation via TAK1 and p38 (which activates ATF2) was decreased as a consequence of SMAD3 ablation in DMM-induced OA in mice (Li et al., 2010). This study also revealed that ATF2 was able to reduce the increase in Col10a1 mRNA expression levels induced by BMP-2 stimulation, hinting at ATF2 as a protective factor to dampen chondrocyte hypertrophy.
TGF-β superfamily members have also been revealed to modulate the expression of integrins, which are ECM receptors involved in chondrocyte differentiation (Garciadiego-Cazares et al., 2015). Treatment of differentiating mesenchymal stem cells with TGF-β superfamily member GDF-5 induced α5 integrin expression and prevented chondrocyte hypertrophy (Garciadiego-Cazares et al., 2015). It was demonstrated that in articular cartilage of rats with surgically induced OA α5 integrin expression was reduced and associated with chondrocyte hypertrophy. It was concluded that α5 integrin expression is protective against hypertrophic changes.
In conclusion, these studies further establish that different TGF-β superfamily members are involved in regulating the chondrocyte phenotype via tuning of different downstream factors involved in gene transcription, which ultimately leads to either chondroprotective or pro-hypertrophic responses.
MAP Kinases
The MAP kinase (MAPK) pathway consists of a sequence of intracellular signaling proteins which transduce a signal from various cell receptors to the nucleus (Schaeffer and Weber, 1999). Activity of MAPKs is regulated in response to environmental stress and to cytokines and growth factors, such as members of the Wnt family (Bikkavilli and Malbon, 2009) or the TGF-β superfamily (Derynck and Zhang, 2003). After activation, a cascade of phosphorylating events takes place intracellularly, finally resulting in phosphorylation of the MAPKs themselves. The three major MAPK pathways, include p38, c-Jun N-terminal (JNK) kinase, and extra-cellular-regulated kinases (ERK) (Johnson and Lapadat, 2002). The currently held doctrine considers MAPKs as signaling mediators involved in the endochondral ossification process (Stanton et al., 2003), but they are also able to regulate the activity of multiple mediators of cartilage destruction (Loeser et al., 2008). Studies suggest that the ERK pathway is involved in destructive OA responses (Prasadam et al., 2013), while the p38 pathway is OA protective (Li et al., 2010; Prasadam et al., 2010). An overview of the newly acquired insights into this pathway and its involvement in development of OA chondrocyte hypertrophy is provided in Table 1d and Figure 4.
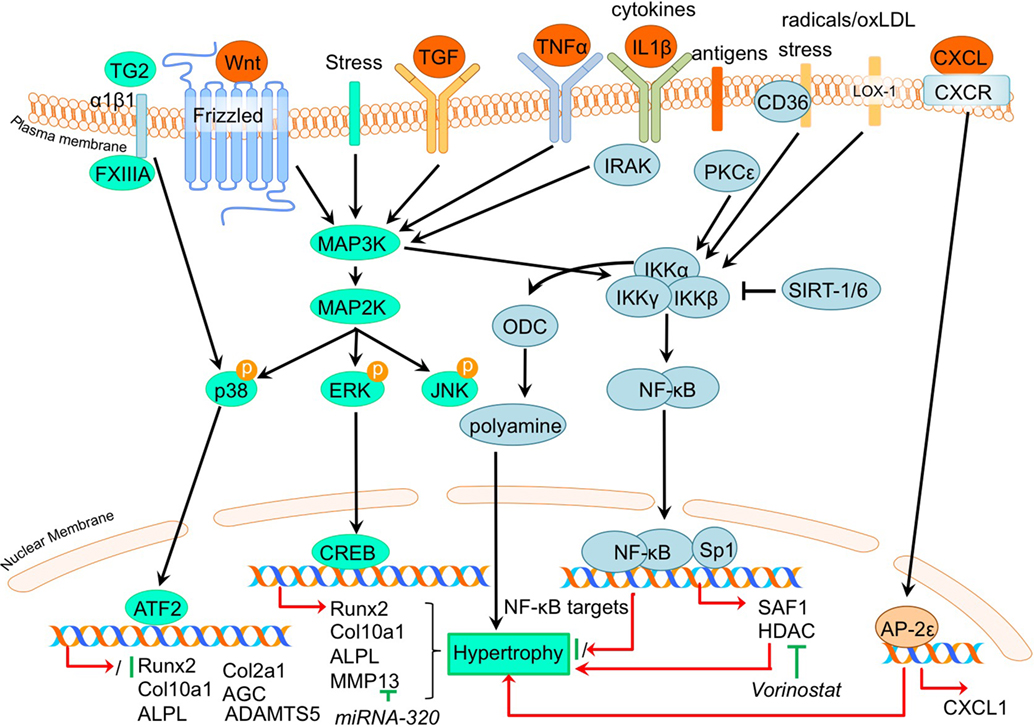
Figure 4. Overview of the canonical MAP kinase (MAPK) and inflammatory signaling pathways and newly acquired insights into their relation to the development of the osteoarthritic (OA) chondrocyte hypertrophic phenotype. The canonical pathway was updated with recent insights in the role of MAPK and inflammatory signaling in the hypertrophic switch occuring during OA development. In green pro-chondrogenic relations are shown, while pro-hypertrophic relations are depicted in red.
It was demonstrated that phosphorylation of ERK1/2 was increased in OA tibial cartilage together with an increased hypertrophic phenotype, characterized by increased COL10A1 and RUNX2 expression levels in a surgically induced rat OA model (Prasadam et al., 2013). On the other hand, expression levels of phosphorylated p38 were decreased in OA compared to non-OA cartilage (Prasadam et al., 2013). This study also revealed that pharmacological ERK inhibition together with hyaluronic acid treatment resulted in a synergistic chondroprotective effect compared to hyaluronic acid treatment only. This was characterized by significantly reduced Mankin scores, accompanied by reduced COL10A1 expression levels (Prasadam et al., 2013). Higher pERK levels, along with increased expression levels of hypertrophic markers, such as ALPL and RUNX2 were also found in OA articular chondrocytes compared to healthy articular chondrocytes (Prasadam et al., 2010). Furthermore, VEGF-induced hypertrophy of chondrocytes was accompanied by pERK activation (Zhang et al., 2016a,b).
In contrast to pERK, p38 has been negatively associated with OA. P38 expression levels were higher in wild-type murine articular cartilage compared to Smad3 knockout mice displaying cartilage damage (Li et al., 2010). P38 levels were also lower in human OA articular chondrocytes compared to healthy human articular chondrocytes (Prasadam et al., 2010).
The pro-hypertrophic effects of pERK and contra-hypertrophic effects of p38 were further substantiated in a co-culture system using subchondral osteoblasts (Prasadam et al., 2010). Co-culturing human articular chondrocytes with subchondral osteoblasts induced hypertrophic changes in the articular chondrocytes, evidenced by increased RUNX2, ALPL, and COL10A1 expression and decreased COL2A1 and AGC expression. Hypertrophic induction in these articular chondrocytes was accompanied by increased pERK phosphorylation and phosphorylation of p38 was decreased. Inhibition of pERK in cultures was able to reduce the hypertrophic induction of articular chondrocytes, in contrast to inhibition of p38 that resulted in hypertrophic induction (Prasadam et al., 2010). Furthermore, confirming a role for pERK in hypertrophic processes in OA chondrocytes, ERK1/2 inhibition abolished FGF23-induced MMP13 expression (Bianchi et al., 2016).
Although p38 is generally considered to act in a chondroprotective manner, it has been evidenced that under certain conditions the p38 activation may result in hypertrophic differentiation in cultured chondrocytes (Merz et al., 2003; Wang and Beier, 2005). In a study by Johnson et al. (2008) it was found that the transglutaminases (TG) TG2 and factor XIIIA (FXIIIA) were increased in hypertrophic chondrocytes from the growth plate and in OA articular chondrocytes (Johnson et al., 2008). Externalization of TG2 is mediated by FXIIIA via its interaction with α1β1 integrins and results in activation of the p38 MAPK signaling pathway, which ultimately led to increased COL10A1 expression in this study (Johnson et al., 2008). Additionally, TG2 expression was found to be associated with enhanced articular chondrocyte hypertrophy in a Hartley Guinea Pig Model of OA as determined by increased MMP13 and ADAMTS5 and an enhanced Col10a1:Col2a1 ratio (Huebner et al., 2009).
Taken together, specific MAP kinases have been demonstrated to regulate both hypertrophic and chondrogenic responses in the chondrocyte and inhibition of specific MAP kinases could potentially be a strategy to block OA progression via modulating the hypertrophic chondrocyte phenotype.
Inflammatory Signaling
Not surprisingly, regarding the inflammatory nature of OA, the involvement of the NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) pathway has been described as involved in the regulation of hypertrophic differentiation in OA (Marcu et al., 2010). Many stimuli (such as stress, cytokines, free radicals, heavy metals, ultraviolet irradiation, oxidized LDL, and bacterial or viral antigens), activate NF-κB, mostly through IκB kinase-dependent (IKK-dependent) phosphorylation and subsequent degradation of NF-κB inhibitory IκB proteins. The liberated NF-κB dimer enters the nucleus, where it regulates transcription of diverse target genes (Marcu et al., 2010). An overview of newly acquired insights into this pathway and its involvement in development of OA chondrocyte hypertrophy is provided in Table 1e and Figure 4.
Providing evidence for a role of NF-κB signaling in chondrocyte hypertrophy, it was demonstrated that IKKα and IKKβ knockdown in OA chondrocyte micromass cultures results in an increased GAG content, COL2A1 expression, and reduced calcium deposits. IKKα and IKKβ also appeared to have differential activities, since IKKα knockdown, but not IKKβ knockdown, resulted in smaller OA hypertrophic chondrocytes (Olivotto et al., 2008). Furthermore, only IKKα knockdown resulted in reduced RUNX2 levels, while IKKβ knockdown resulted in increased SOX9 levels. The pro-hypertrophic role of IKKα was later confirmed by the same group (Olivotto et al., 2013), as well as another group (Facchini et al., 2012). Facchini et al. revealed that expression of IKKα is related to chondrocyte hypertrophy and knockdown of IKKα results in reduced synthesis and activity of ornithine decarboxylase (ODC). ODC mediates the increase of polyamine levels that in turn increases RUNX2 expression, as well as RUNX2 nuclear translocation, leading to hypertrophic chondrocytes (Facchini et al., 2012). Additionally, a study by Meng et al. also revealed a role for NF-kB signaling in chondrocyte hypertrophy (Meng et al., 2016). In this study it was found that microRNA- (MiRNA)320 negatively regulates MMP13 expression by binding to the 3’UTR (3’ untranslated region) of the MMP13 mRNA. In turn, miRNA320 expression is reduced by IL-1β-induced NF-κB and MAPK signaling activity (Meng et al., 2016). This study thus revealed a link between a hypertrophy-dependent decrease of miRNA-320 and an increase of MMP13 via the NF-κB and MAPK signaling pathway. Oxidized low-density lipoprotein (ox-LDL) binding to lectin-like ox-LDL receptor-1 (LOX-1) in cultured bovine articular chondrocytes increased production of intracellular reactive oxygen species (ROS), resulting in the activation of the inflammatory signaling pathway via NF-κB (Nishimura et al., 2004). Hashimoto et al. demonstrated a role for LOX-1 in chondrocyte hypertrophy, as COL10A1 expression in a DMM mice OA model was induced by expression of the LOX-1. This resulted in decreased articular cartilage GAG content (Hashimoto et al., 2016). LOX-1−/− mice on the other hand, revealed a reduced OA score and a reduction in osteophyte formation. LOX-1 co-localized with RUNX2 and COL10A1 expression in articular chondrocytes as well as in osteophyte forming-cells, indicating a role in the pathogenesis of DMM-induced OA through endochondral ossification (Hashimoto et al., 2016).
In a study by Zhong et al. it was found that in IL-1β-treated OA chondrocytes, expression of the NF-κB subunit p65 was reduced in response to the HDAC-inhibitor vorinostat. Besides this inflammatory pathway, HDAC inhibition by vorinostat also resulted in decreased p38 and ERK1/2 activation in IL-1β-exposed human OA chondrocytes (Zhong et al., 2013). During OA progression in human articular chondrocytes, PKCε (protein kinase C epsilon) levels were reduced, which lead to an upregulation of HDAC2 and reduction of HDAC4 (Queirolo et al., 2016). This resulted in an increase of the HDAC2:HDAC4 ratio, which then induced RUNX2 and ultimately resulted in increased expression of MMP13, ADAMTS4, and ADAMTS5 (Queirolo et al., 2016). Interestingly, the decreased expression of HDAC4, related to the loss of PKCε in OA chondrocytes (Queirolo et al., 2016), was confirmed in another study by Lu et al. (2014a). However, in contradiction to the previous study, Lu et al. found that silencing of HDAC4 resulted in a decrease of chondrocyte hypertrophic marker expression, while additionally HDAC4 was decreased with increasing severity of OA, suggesting a role for HDAC4 in the onset of OA.
Besides NF-κB, ELR + CXC chemokines, which are characterized by their glutamic acid-leucine-arginine (ELR+) motif, provide new links connecting OA to hypertrophic changes (Wenke et al., 2011). AP-2ε is a transcription factor which acts in hypertrophic cartilage differentiation and its expression is increased in OA chondrocytes compared to primary chondrocytes (Wenke et al., 2009). This study described that CXCL1 expression accompanied the increase in AP-2ε expression levels during hypertrophic differentiation of MSCs (Wenke et al., 2011). Expression of CXCL1 appeared to be under transcriptional control of AP-2ε, since AP-2ε was demonstrated to bind and transactivate the CXCL1 promotor, indicating that the observed increase in AP-2ε in OA chondrocytes may lead to hypertrophic changes via regulating CXCL1 activity (Wenke et al., 2011). Another chemokine that was revealed to be involved in OA pathogenesis is CXCL6 (Sherwood et al., 2015). It was shown that the expression of this chemokine is reduced in human OA cartilage (Sherwood et al., 2015) and also in a mouse DMM OA model (Sherwood et al., 2015). Knockout of the CXCL6 receptor, CXCR2, resulted in more severe OA development after DMM surgery, as evidenced by higher OARSI scores (Sherwood et al., 2015). This was also accompanied by a more hypertrophic phenotype verified by increased COL10A1 protein expression. These data suggest a chondroprotective action of the CXCL6-CXCR2 axis.
In addition to chemokines, different inflammatory cytokines have been implicated in OA pathophysiology, one of them being TNFα (Lai et al., 2014). It was recently confirmed that TNFα induces Adamts7 expression in murine cartilage, and ADAMTS7 was able to induce the expression of Tnfα, creating a positive feedback loop (Lai et al., 2014). TNFα-mediated transactivation of an Adamts7 promotor reporter construct was dose-dependently inhibited by Bay 11-7082, a NF-κB-specific inhibitor, indicating NF-κB-mediated activation of Adamts7 after TNFα stimulation. Interestingly, mice overexpressing ADAMTS7 displayed OA-like phenotypes characterized by reduced cartilage GAG content, osteophyte formation, thinner cartilage, and an upregulation of hypertrophic marker expression such as Col10a1 and MMP13. Furthermore, ADAMTS7 overexpression resulted in an acceleration of OA development in a mouse DMM experiment which was also accompanied by increased expression of chondrocyte hypertrophic markers Col10a1 and Mmp13 (Lai et al., 2014). The ADAMTS7 overexpressing mice also displayed several skeletal developmental abnormalities, including a reduced hypertrophic zone and reduced COL10A1 levels in the growth plate and lower bone mineral density.
The inflammatory S100A11, a ligand for the receptor for advanced glycation end products (RAGE), is associated with chondrocyte hypertrophy (Cecil et al., 2009). However, the deletion of RAGE was not chondroprotective in an instability induced knee OA mouse model (Cecil et al., 2009). Cecil et al. (2009) demonstrated that the alternative patterning receptor CD36, a marker for growth plate chondrocyte hypertrophy, promotes OA via mediation of inflammatory and differentiation responses. Indeed, CD36 co-localized with COL10A1 expression in all zones of knee articular cartilage, as well as the aggrecan NITEGE aggrecanase neoepitope in the articular cartilage superficial zone. Surprisingly, overexpression of CD36 in CH-8 cells led to an inhibition of chondrocyte hypertrophic markers (Cecil et al., 2009), while S100A11 gained the capacity to induce proteoglycan synthesis in CH-8 chondrocytes (Cecil et al., 2009). These results indicate that CD36 is a hypertrophic chondrocyte-expressed patterning receptor that induces cartilage repair when exposed to inflammatory stimuli (Cecil et al., 2009). This demonstrates that besides pro-hypertrophic processes, inflammatory responses in articular chondrocytes can also result in a chondroprotective effect.
The semicarbazide-sensitive amine oxidase (SSAO) found in the hypertrophic chondrocytes of the growth plate is known to be involved in leukocyte extravasation from the blood to the inflammation site. Additionally, it may be associated with the differentiation of chondrocytes toward a hypertrophic phenotype (Filip et al., 2016). In line with this, inhibition of SSAO reduced Mmp13, Alpl, and Opn expression, potentially by modulation of glucose transport in rat hypertrophic articular chondrocytes (Filip et al., 2016). In humans, similar expression patterns were seen for SSAO in healthy and OA articular cartilage, supporting the idea that SSAO plays a role in the process of hypertrophic differentiation of the articular chondrocyte (Filip et al., 2016). OA is a disease associated with aging, which could be a result of tissue accumulation of p16INK4a positive cells. p16INK4a is suggested to support chronic inflammation, as p16INK4a positive cells exhibit a specific secretome called SASP (senescence-associated secretory phenotype) including pro-inflammatory cytokines. OA chondrocytes are characterized by an accumulation of p16INK4a as a result of reduced miRNA24 levels, which acts as a negative regulator for p16INK4a (Philipot et al., 2014). p16INK4a is also upregulated during chondrogenesis, indicating a recapitulation of a developmental process in OA.
Since inflammation and angiogenesis are closely correlated in the pathogenesis of OA, Ray et al. hypothesized the presence of common regulators controlling both processes simultaneously (Ray and Ray, 2008). In their study they found that overexpression of the inflammation responsive transcription factor SAF-1 in transgenic mice leads to the development of severe cartilage degradation and OA. SAF-1-overexpressing mice also showed neo-vasculature in the perichondrium and synovium, suggesting a link between angiogenesis and inflammation. Indeed, the VEGF promotor contains two tandem binding sites for SAF-1 (Ray and Ray, 2008), providing evidence for the link between inflammation and OA development and pathogenesis. Furthermore, SAF-1 has binding sites in the MMP1 and MMP9 promotor sequences (Ray and Ray, 2008), controlling their expression. In human OA articular chondrocytes, SAF-1 expression is increased and together with NF-κB and SP1 acts synergistically to induce MMP activation in OA chondrocytes.
Another factor involved in inflammatory signaling and OA disease progression is SIRT-1. SIRT-1 inhibits NF-κB by deacetylating the p65 NF-κB subunit, priming p65 for proteasome degradation, thus modulating the inflammatory signaling pathway (Liu-Bryan, 2015). The association of SIRT-1 with chondrocyte hypertrophy was demonstrated after inhibition of SIRT-1 by RNAi, which induced COL10A1 expression. Since OA chondrocytes display decreased SIRT-1 expression compared to healthy controls, this may provide a route for chondrocytes to acquire an endochondral cellular phenotype (Fujita et al., 2011). Besides SIRT-1, SIRT-6 also modulates inflammation. SIRT-6 haplo-insufficiency enhanced OA progression in high fat diet-induced obese mice by stimulating the inflammatory response as shown by increased expression of Tnfα and IL-6 in the HFP (Ailixiding et al., 2015). Additionally, SIRT-6 haplo-insufficient mice on a high fat diet revealed an increase in osteophytes and synovial tissue with increased infiltration of inflammatory cells, as well as increased MMP13 expression in the cartilage. These results indicate a higher inflammation status of the HFP and synovium in obese mice with SIRT-6 haplo-insufficiency accompanied with increased osteophyte formation, leading to OA.
To conclude cytokines and chemokines and their downstream intracellular pathways have been revealed to lead to hypertrophic as well as anti-hypertrophic changes in the chondrocyte. While inflammation-induced hypertrophic changes have been established as associated with processes involved in cartilage matrix degradation, inflammation-induced anti-hypertrophic changes have been described to lead to chondroprotective responses.
Hypoxic and Angiogenic Factors
The proliferative zone of the growth plate is an avascular hypoxic mesenchymal tissue (Maes et al., 2012). Intriguingly, chondrocytes are competent at surviving and differentiating in this challenging environment. It has been suggested that this survival and capability of differentiation is, at least in part, by virtue of the actions of hypoxia-inducible factors (HIFs), such as HIF1-α (Maes et al., 2012). Furthermore, the process of endochondral ossification in the growth plate is driven by vascularization and hypertrophic chondrocytes in the growth plate that secrete angiogenic stimuli which actively support vascularization (Maes et al., 2012). This hypertrophy associated angiogenic switch is a major driver of active growth plate cartilage remodeling toward bony tissue (Maes et al., 2012). The analogy between endochondral ossification processes and molecular processes observed in OA also led to the theory that OA pathophysiology may involve hypoxic/angiogenic mediators, such as HIFs and VEGF. An overview of the newly acquired insights into this pathway and its involvement in development of OA chondrocyte hypertrophy is provided in Table 1f and Figure 5.
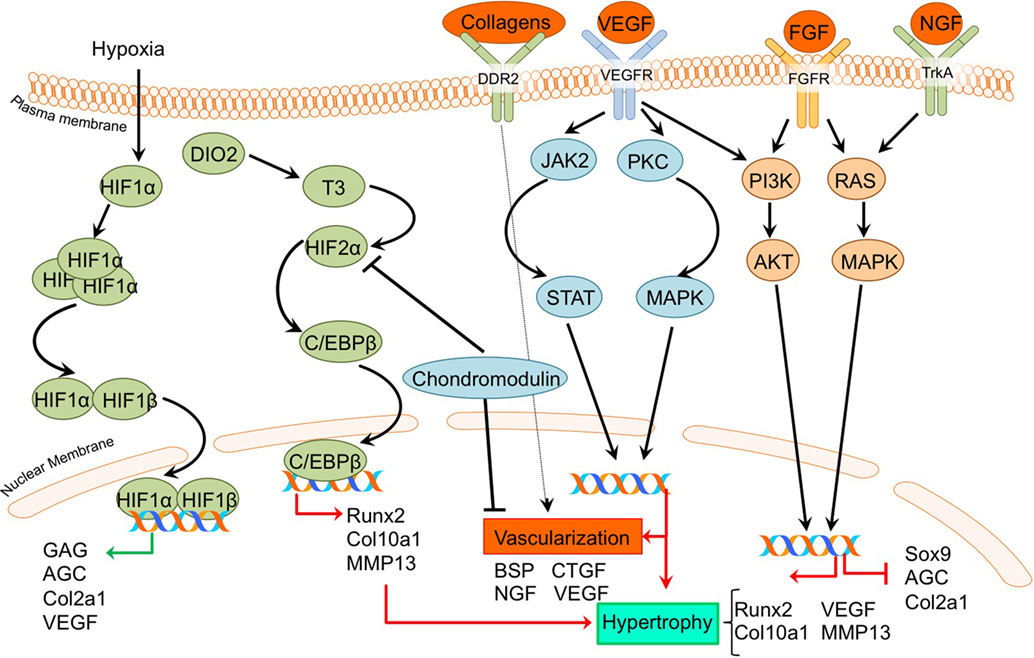
Figure 5. Overview of the processes of hypoxia and angiogenesis and the canonical fibroblast growth factor (FGF) signaling pathway and newly acquired insights into their relation to the development of the osteoarthritic (OA) chondrocyte hypertrophic phenotype. The processes and canonical FGF signaling pathway was updated with recent insights in the role of the processes of hypoxia and angiogenesis and the FGF signaling pathway in the hypertrohic switch occuring during OA development. In green pro-chondrogenic relations are shown, while pro-hypertrophic relations are depicted in red.
A direct link between HIF-2α and OA development became evident by a study reported by Saito et al (Saito et al., 2010). It revealed that HIF-2α is localized predominantly in the hypertrophic zone of mouse growth plates, and Hif-2α+/− mice display reduced proliferative and hypertrophic zone lengths, together with impaired bone length, indicating impaired endochondral ossification during limb development. Confirming its role in OA pathophysiology, HIF-2α expression was increased in articular chondrocytes of a surgically induced OA mouse model, while Hif-2α+/− caused significant resistance to cartilage degradation, osteophyte formation, and subchondral bone sclerosis in this model (Saito et al., 2010). Furthermore, in the same study HIF-2α expression was revealed to be higher in more severely OA-affected human cartilage samples than in mild OA affected regions. The crucial role of HIF-2α in OA pathophysiology was confirmed in another study, which described that Hif-2α+/− mice display less cartilage degradation in a surgically induced mouse OA model (Hirata et al., 2012). Furthermore, it was evident that in chondrocytes, HIF-2α is a potent inducer of C/EBPβ (Hirata et al., 2012). Confirming a role for C/EBPβ in OA pathophysiology, C/Ebpβ+/− mice developed less severe OA cartilage damage in a surgically induced mouse OA model (Hirata et al., 2012). Furthermore, C/EBPβ together with RUNX2 is higher expressed in human OA cartilage with higher Mankin scores compared to human OA cartilage with lower Mankin scores, and can activate MMP13 expression in chondrocytes (Hirata et al., 2012).
Dio2 is upregulated in OA cartilage (Nagase et al., 2013) and is responsible for active thyroid hormone (T3) production. T3 in turn induces terminal chondrocyte differentiation with increased HIF-2α expression, as well as COL10A1, ALPL, osteocalcin, RUNX2, MMP13, and ADAMTS5 expression (Nagase et al., 2013; Bomer et al., 2015). The upregulation of HIF-2α expression after T3 treatment suggests a link between DIO2 levels and OA development via HIF-2α signaling and activating mutations in the DIO2 allele result in a predisposition for OA development in human patients (Bomer et al., 2015). Additionally, it was demonstrated in an experimental Dio2 transgenic rat OA model that overexpression of DIO2 results in increased OA-associated cartilage degradation with higher COL10A1 expression levels (Nagase et al., 2013). Confirming parallels between chondrocyte signaling events that take place in the growth plate and those of hypertrophic chondrocytes in OA-affected articular cartilage, DIO2 activity was also increased during chondrogenesis of BMSCs (Bomer et al., 2015).
Despite the general recognition that HIF expression in cartilage is associated with OA disease progression, a study by Markway et al. could not detect a differential expression of HIF-1α and HIF-2α in OA versus healthy chondrocytes under hypoxic conditions (Markway et al., 2013). In this study, hypoxia-induced HIF expression resulted in increased COL2A1, ACAN, and GAG levels (Markway et al., 2013). While HIF-2α is normally described as associated with hypertrophy, HIF-3α is not. HIF-3α expression is higher in the resting and proliferative zones of the growth plate compared to the hypertrophic zone (Markway et al., 2015). Furthermore, chondrogenically differentiated BMSCs showed MMP13 and COL10A1 induction, while HIF-3α expression was low.
In addition to HIF-2α, VEGF was demonstrated to be involved in OA development. In a surgically induced rat OA model, inhibition of Vegf with a shRNA resulted in less cartilage degradation compared to rats without Vegf inhibition at 5–9 weeks after OA induction. This chondroprotective effect was accompanied by reduced Col10a1 levels (Zhang et al., 2016a).
Another protein essential in vascularization during endochondral ossification is chondromodulin. Proliferative chondrocytes in the growth plate are resistant to vascular invasion because of the presence of angiogenic inhibitors, such as chondromodulin (ChM-1) (Hiraki and Shukunami, 2005). Chondromodulin overexpression protected against OA development in a surgically induced rat OA model, evidenced by lower Mankin scores, less COL10A1 expression and higher COL2A1 and AGC levels (Zhang et al., 2016a,b). Likewise, Chm-1 levels in OA cartilage were lower in a surgically induced rat OA model compared to cartilage from control joints (Zhang et al., 2016b). Furthermore, Chm-1 expression was decreased in more severely damaged human OA cartilage in comparison to mild OA cartilage (Zhang et al., 2016b). Chondromodulin also protected against TNFα-induced chondrocyte hypertrophy (Zhang et al., 2016b). Additional experiments in the C28/I2 chondrocyte cell line revealed that chondromodulin delays HIF-2α nuclear translocation, indicating that the anti-hypertrophic and chondroprotective effects of chondromodulin are caused by the repression of pro-hypertrophic HIF-2α activity (Zhang et al., 2016b).
Vascularization is essential for the endochondral ossification process and it has also been associated with OA. Indeed, OA cartilage is invaded by blood vessels into the non-calcified articular cartilage, likely due to an increased production of pro-angiogenic factors (Mapp and Walsh, 2012; Wang et al., 2012). This has been attributed to the increase in subchondral bone porosity (Botter et al., 2011), which has been hypothesized to result in disruption of the osteochondral junction. A disruption of the osteochondral junction with a subsequent invasion of blood vessels may lead to further structural damage, leading to progression of OA.
A role for chondrocyte hypertrophic processes in OA progression was further described by a study in which conditioned medium of hypertrophic OA chondrocytes induced wound healing in human umbilical vascular endothelial cells and increased endothelial cell adhesion and migration (Pesesse et al., 2013). Furthermore, the gene expression of pro-angiogenic factors, such as BSP and NGF was increased (Pesesse et al., 2013). Osteochondral angiogenesis was also assessed in a study by Wang et al. in which OA was evoked by disordered occlusion in rat mandibular joints (Wang et al., 2012). Experimental groups demonstrated OA-like changes, with a loss of cartilage surface integrity and osteophyte formation. Additionally, hypertrophic chondrocytes adjacent to the osteochondral interface showed increased expression of VEGF, CTGF, and MMP9 at 20 or 24 weeks post OA-induction surgery. Thus, hypertrophic chondrocytes may exacerbate the disruption of the osteochondral junction by stimulating angiogenesis, which can lead to progression of OA. Another study described the association between pro-angiogenic factors, hypertrophy, and OA severity. Discoidin domain receptor 2 (DDR2) was higher expressed in more severely damaged human OA cartilage compared to cartilage with lower Mankin scores (Zhang et al., 2014a). DDR2 is a receptor tyrosine kinase that can be activated by various collagens. Since DDR2 activity can induce COL10A1 expression in chicken chondrocytes (Zhang et al., 2014b), and given the fact that DDR2 has been associated with a pro-angiogenic function (Zhang et al., 2014a), this supports a link between chondrocyte hypertrophic pro-angiogenic factors and OA progression.
In conclusion, these results reveal the importance of angiogenesis in enabling OA disease progression. The differential activation of hypoxic and angiogenic pathways observed in OA appear to be key factors in OA development.
FGF Signaling
Fibroblast growth factors comprise a group of morphogens involved in wound healing, angiogenesis, and are involved in processes such as proliferation and differentiation in different cell types (Turner and Grose, 2010). They also have been described as being involved in endochondral ossification, since FGF23 and FGF receptor 1 are produced by growth plate hypertrophic chondrocytes (Raimann et al., 2013). Given that OA chondrocytes also produce FGF family members (Orfanidou et al., 2009), studies have investigated the involvement of FGF signaling in the chondrocyte phenotypic alterations observed in OA cartilage. An overview of newly acquired insights into this pathway and its involvement in development of OA chondrocyte hypertrophy is provided in Table 1g and Figure 5.
It has been demonstrated that the expression of FGFR1, FGF23, and its co-receptor KLOTHO is higher in OA chondrocytes compared to non-OA chondrocytes. Expression of these factors was also increased in cartilage samples with more severe macroscopic OA compared to less severe macroscopic OA within the same patient (Bianchi et al., 2016). Exogenous addition of FGF23 to human OA primary chondrocytes resulted in hypertrophic changes, as evidenced by COL10A1 and VEGFA induction via FGFR1 (Bianchi et al., 2016). Another study confirmed the increase in FGF23 expression levels in OA compared to non-OA chondrocytes (Orfanidou et al., 2009). This study confirmed the hypertrophic phenotype in OA chondrocytes with higher RUNX2 and lower SOX9 levels compared to non-OA chondrocytes, and also revealed that exogenous addition of FGF23 to non-OA chondrocytes induced a hypertrophic phenotype characterized by increased RUNX2 expression levels (Orfanidou et al., 2009). Involvement of FGF signaling as an OA-inducing pathway was demonstrated in a study in which G141, a pharmacological FGFR1 inhibitor was used. In a surgically induced mouse OA model, G141 delayed the progression of cartilage degradation, accompanied by a decrease of MMP13 and COL10A1 chondrocyte hypertrophic marker expression, suggesting that OA articular cartilage damage could be reduced via inhibition of hypertrophic processes (Xu et al., 2016).
On the other hand, FGFR3 seems to be crucial for cartilage homeostasis and inhibition of a hypertrophic phenotype, since FGFR3 knockout mice displayed OA-like defects in the temporomandibular joint together with an increased expression of hypertrophic markers COL10A1 and MMP13 (Zhou et al., 2016). Further support indicative of an association between chondrocyte hypertrophy and FGFR3 comes from a study in which it was revealed that FGFR3 levels were reduced in mtorc1 knockout mice which display cartilage degradation and chondrocyte hypertrophy (Zhang et al., 2017).
Taken together, recent studies have demonstrated that the FGF signaling pathway is active in OA and also provided new insights in the contribution of this signaling pathway to OA disease initiation or progression.
Notch Signaling
The Notch signaling pathway consists of five identified ligands (Jagged 1, Jagged 2, Dll1, Dll3, and Dll4) that can interact with four receptors (Notch 1–4) (Bray, 2016). Upon binding of Notch ligands, the Notch receptor cleavage site becomes accessible for cleavage by ADAM10. The residual transmembrane Notch fragment in turn is cleaved via proteolysis by the γ-secretase complex, releasing NICD. This intracellular domain of the receptor translocates to the nucleus, where it binds to the transcription factor recombination signal binding protein for immunoglobulin kappa J (RBPjκ), to participate in gene transcription of Notch target genes, such as Hes1. These target genes are involved in cellular processes, such as cell proliferation and differentiation (Nye and Kopan, 1995). Notch signaling is involved in endochondral ossification (Hosaka et al., 2013), and, therefore, studies have investigated its involvement in OA development as well. An overview of newly acquired insights into this pathway and its involvement in development of OA chondrocyte hypertrophy is provided in Table 1h and Figure 3.
Providing evidence for a role of Notch signaling in endochondral processes and OA development, it has been reported that RBPjκ is involved in endochondral ossification. Knockout of Rbpjκ resulted in reduced cartilage damage in a surgically induced OA model, together with reduced expression of MMP13, VEGFA, and HES1 in articular chondrocytes (Hosaka et al., 2013). Furthermore, in this study it was revealed that during OA development, the expression of Notch ligand Jagged1 is increased in OA cartilage, indicating that this ligand may regulate Notch signaling during OA progression (Hosaka et al., 2013). Confirming again a role for Notch signaling in OA development, it was demonstrated that the pharmacological Notch signaling inhibitor N-[N-(3,5-diflurophenylacetate)-l-alanyl]-(S)-phenylglycine t-butyl ester (DAPT), resulted in the same chondroprotective effect in a surgically induced mouse OA model (Hosaka et al., 2013), as observed with the Rbpjκ knockout mouse.
In concert with a role in chondrocyte hypertrophy, the intracellular domains of Notch ligands, NOTCH1 and NOTCH2 was shown to be located at the chondrocyte plasma membrane in resting and proliferative zone chondrocytes, while these intracellular domains were translocated (and thus potentially active) to the nucleus in hypertrophic zone chondrocytes (Hosaka et al., 2013). Similar as in the growth plate’s hypertrophic zone, the intracellular domains of NOTCH1 and NOTCH2 were also located to the chondrocyte’s nucleus in surgically induced mouse OA cartilage (Hosaka et al., 2013). An increase in Notch signaling activity was also observed in a further study. In this study, an increase in mRNA levels of Notch ligand Jagged 1 and its receptor Notch 1 in OA areas of human articular cartilage, together with induced mRNA levels of the Notch target gene HES1 was reported (Lin et al., 2016). Interestingly, inhibition of NOTCH1 resulted in an increased, HES1 dependent, hedgehog signaling activity, leading to an exacerbation of OA (Lin et al., 2016). These results are in contrast with the general theory that inhibition of Notch attenuates OA development. The increase in hypertrophy may be a result of a crosstalk between the Notch and hedgehog signaling pathway, previously described in the Ihh/PTHrP signaling paragraph.
In conclusion, these results provide evidence that the Notch signaling pathway is positively associated with hypertrophic changes in the chondrocyte. Inhibition of the Notch signaling pathway may be used as a therapeutic tool to block OA disease progression, based on reducing hypertrophic changes in the chondrocyte.
Mineralization
Mineralization is also an important consequence of chondrocyte hypertrophy in OA (Fuerst et al., 2009; Fukai et al., 2010; Wallin et al., 2010; Nguyen et al., 2013; Zhu et al., 2015; Cavaco et al., 2016; Nasi et al., 2016; Queirolo et al., 2016). The mineralization process is analogous to the last phase of endochondral ossification as it is observed in the growth plate (Kronenberg, 2003). Here, hypertrophic chondrocytes secrete matrix vesicles containing high concentrations of phosphatases, such as alkaline phosphatase (ALPL) and PHOSPHO1 (Anderson, 2003; Houston et al., 2004; Stewart et al., 2006; McKee et al., 2013). During matrix vesicle biogenesis, vesicles are formed by polarized budding and pinching-off of vesicles from the plasma membranes. Within these matrix vesicles, the first mineral crystals are formed by phosphatases hydrolyzing inorganic pyrophosphate (PPi) to create inorganic phosphate (Pi). Pi ions in turn crystallize with calcium, resulting in crystals which are released through the vesicles membranes. When these pre-formed hydroxyapatite crystals come in contact with the extracellular fluid containing Ca2+ and ions a process of continuous crystal formation takes place in the matrix (Anderson, 2003; Orimo, 2010). This mineralized matrix is then vascularized, further supporting mineralization and enabling the infiltration of osteoblasts and osteoclasts. Osteoblasts secrete osteoid, which forms the bone trabecula, while osteoclasts, formed from macrophages, breakdown spongy bone to form the medullary (bone marrow) cavity (Kronenberg, 2003). An overview of newly acquired insights into this process and its involvement in development of OA chondrocyte hypertrophy is provided in Table 1I and Figure 6.
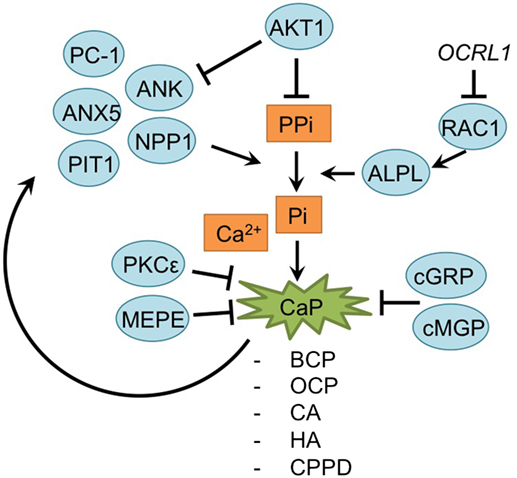
Figure 6. Overview of the mineralization process and newly acquired insights into its relation to the development of the osteoarthritic (OA) chondrocyte hypertrophic phenotype. The mineralization process was updated with recent insights in the role of mineralization in the hypertrophic switch occuring during OA development.
During endochondral ossification, AKT1 induces mineralization without affecting the hypertrophic and proliferative zone in the growth plate (Fukai et al., 2010). Surgically DMM OA-induced Akt1−/− mice specifically revealed a reduction of calcified osteophyte formation, while cartilage degradation was unaltered, supporting results observed in the growth plate. This may be the result of an increase of inorganic pyrophosphate (PPi) due to Akt1 inhibition, which antagonizes the ability of inorganic phosphate (Pi) ions to crystallize with calcium. Indeed, levels of PPi-regulators Ank and Npp1 were increased after Akt1 inhibition (Fukai et al., 2010).
A further relationship was found between RAC1 and hypertrophy (Zhu et al., 2015). Lenti-viral expression of the RAC1 inhibitor OCRL1 resulted in protection against cartilage degradation in a mouse OA model. Additionally, OCRL1 resulted in decreased ALPL activity in human primary chondrocytes. Pre-treatment of chondrocytes with IL-1β, resulted in an upregulation of ADAMTS5, Col10a1, RUNX2, MMP13, and ALPL activity, but overexpression of OCRL1 blocked these hypertrophic reactions, and reduced mineralization. Besides the mineralization modulators AKT1 and OCRL1, calcium deposition is also regulated by protein kinase C epsilon. In OA chondrocyte micromasses PKCε expression was reduced, resulting in an increase of calcium deposition and calcium crystals and thus increase in the matrix mineralization (Queirolo et al., 2016).
Mineralization is also modulated by MEPE, a matrix mineralization inhibitor (Staines et al., 2016). MEPE was detectable at the lateral OA-unaffected side of STR/Ort mice, while its expression was decreased in the medial affected OA-side of the knee joints. Interestingly, this endochondral phenotype in STR/Ort mice cartilage exists before OA-like changes appear, which suggest that these endochondral changes initiate later development of OA (Staines et al., 2016).
Articular cartilage of many OA patients hosts calcium-containing crystals, which are present in the superficial and deep layers of the cartilage, as well as in synovial fluid and the meniscus (Fuerst et al., 2009; Nguyen et al., 2013). The calcium deposits mainly consist of basic calcium phosphate (BCP), including octacalcium phosphate (OCP), carbonated-apatite (CA), and hydroxyapatite (HA) crystals or calcium pyrophosphate dihydrate (CPPD). Treatment of murine chondrocytes with BCP results in an increase of IL-6 secretion, which in turn induces the expression of pro-mineralizing genes, such as Ank, Anx5, Pc-1 [plasma-cell membrane glycoprotein 1/Enpp1 (ectonucleotide pyrophosphatase/ phosphodiesterase 1)] and Pit1 (Nasi et al., 2016). PC-1, ANK, and TNAPs control extracellular Pi and PPi levels, which are critical determinants of mineralization. Expression of these regulatory proteins is increased in chondrocytes with calcium-containing crystals, forming a positive feedback loop (Nguyen et al., 2013; Nasi et al., 2016).
Another factor involved in OA pathophysiology is Gla-rich protein (GRP). GRP was revealed to inhibit calcification and exerted anti-inflammatory effects in both chondrocytes and synoviocytes (Cavaco et al., 2016). Its expression was increased in OA chondrocytes and synoviocytes compared to non-OA controls. Furthermore, expression of GRP was also increased overtime when chondrocyte or synoviocyte mineralization was induced via addition of CaCl2, evidencing its association with mineralization. Interestingly, GRP was only able to inhibit calcification in its carboxylated form, and since OA chondrocytes and synoviocytes displayed reduced y-carboxylation capacity, it was hypothesized that the GRP increase in OA-derived cells is not able to inhibit calcification processes (Cavaco et al., 2016). Besides GRP, MGP, another mineralization inhibitor, has been described as becoming less carboxylated in OA chondrocytes. Lower levels of cMGP lead to a reduction of the fetuin-MGP complex in OA chondrocytes and in released vesicles. This reduction of fetuin-MGP containing vesicles, results in reduced extracellular transport of cMGP, lowering its extracellular concentration. Ultimately, the reduced extracellular cMGP levels lead to an impaired inhibition of matrix mineralization (Wallin et al., 2010).
Taken together, mineralization appears to be an important hallmark of osteoarthritic cartilage and is associated with OA chondrocyte hypertrophy.
Discussion
The analogs events which are observed during development of OA and endochondral ossification have been described in previous reviews (Dreier, 2010; Pitsillides and Beier, 2011; van der Kraan and van den Berg, 2012). On the contrary, Brew et al. found no evidence of a generalized chondrocyte hypertrophic change in OA (Brew et al., 2010). Despite the lack of evidence found by Brew et al., all other papers found in our literature search do describe the endochondral cellular phenotypic changes occurring in OA cartilage. Various signaling pathways and processes involved in endochondral processes, such as Wnt-, Ihh/PTHrP-, TGF-β-, MAP-kinases, FGF-, Notch signaling, inflammatory signaling, and hypoxia-associated signaling pathways, but also processes, such as angiogenesis and matrix mineralization have been described in OA disease progression. Interestingly, from some studies it became evident that crosstalk exists between the different signaling pathways involved in OA disease progression (Ray and Ray, 2008; Papathanasiou et al., 2012; Leijten et al., 2013; van den Bosch et al., 2014; Lin et al., 2016; Zhou et al., 2016). Next to cartilage degradation, which is the main hallmark of OA, it is well established that OA includes changes in several joint structures. Hence, OA should be considered as a total joint disease. Alterations in subchondral bone, meniscal degradation and inflammatory changes in intra-articular tissues, such as the synovium and Hoffa’s fat pad have been widely described. Unraveling mechanisms of feedback and crosstalk among these different joint tissues and their influence on cartilage hypertrophy will further provide important insights in OA pathophysiology.
In this review, we aimed to provide a recent overview of new insights into the contribution of the changing hypertrophic chondrocyte phenotype in the development and progression of OA. In order to include all papers from the past 10 years discussing the endochondral cellular phenotypic changes occurring during OA, we specifically used a search in PubMed which included the term “endochondral ossification,” but also includes terms such as “hypertrophic differentiation,” “hypertrophy,” “mineralization,” and “calcification,” which are all processes occurring during endochondral ossification. Interestingly, different studies use different molecular markers to evaluate the hypertrophic phenotype in the chondrocyte and an unambiguous definition of “hypertrophy” seems lacking (Table 1). This is most likely related to the complexity of the endochondral ossification process with various molecular pathways being involved. In future research, it will be important to reach a consensus and implement a standardized definition to describe “chondrocyte hypertrophy in OA.” We expect that this will support better data comparison between different studies.
A striking realization from our literature search is that it seems that in OA animal models the hypertrophic changes in OA cartilage pathology can be initiated or prevented by a single alteration in many different signaling pathways. While late OA disease progression follows a predictable cell biological progression, little is known about causal relations between the herein described pathways/processes and early OA disease initiation in man. Together this raises the question if OA can even be considered as a generalized single disease (Luyten et al., 2017). Indeed, it is becoming increasingly apparent that different OA phenotypes exist (van der Esch et al., 2015; Deveza et al., 2017; Dell’Isola and Steultjens, 2018). Different early OA phenotypes/disease initiation may result from alterations in molecular pathways that are specifically connected with clinical risk factors for developing OA, like obesity, aging, metabolic syndrome, joint shape/malalignment, or earlier cartilage trauma. We speculate that a phenotyping of early OA, in which dominant deregulation of a specific molecular pathway is taken into account, may provide additional resolution to the already described OA phenotypes. This will not only be important for diagnostic purposes, but equally important for developing new OA disease-modifying compounds and selecting the right patient populations for clinical studies testing novel OA disease-modifying drugs and -approaches.
It can be concluded that articular cartilage/chondrocyte homeostasis is fragile and disruption of chondrocyte homeostasis can be initiated by multiple factors leading to a cascade of intracellular changes in one or multiple pathways. This ultimately results in a chondrocyte hypertrophic/endochondral phenotype that is observed in OA. Understanding the relevance, mechanism-of-action, and crosstalk of processes and signaling pathways involved in OA initiation and progression is expected to further fuel the development of OA disease-modifying drugs and -approaches.
Author Contributions
All four authors ER, UT, MC, and TW have contributed to literature searches, reading, selecting, and interpretation of papers, and writing of the manuscript. ER and UT have contributed to layout of the tables and MC has contributed to the layout of the figures. The manuscript was approved by all four co-authors prior to submission. ER and UT have contributed equally as first author and MC and TW have contributed equally as last author.
Conflict of Interest Statement
MC and TW are listed as inventor on filed patents: WO2017178251 and WO2017178253. TW has shares in Chondropeptix BV.
The reviewer AL and handling editor declared their shared affiliation.
Funding
The work of the four authors is supported by grants from the Dutch Arthritis Foundation (grants LLP14, 15-3-403), the Annadal Foundation and Stichting de Weijerhorst.
References
Ailixiding, M., Aibibula, Z., Iwata, M., Piao, J., Hara, Y., Koga, D., et al. (2015). Pivotal role of Sirt6 in the crosstalk among ageing, metabolic syndrome and osteoarthritis. Biochem. Biophys. Res. Commun. 466, 319–326. doi: 10.1016/j.bbrc.2015.09.019
Aini, H., Ochi, H., Iwata, M., Okawa, A., Koga, D., Okazaki, M., et al. (2012). Procyanidin B3 prevents articular cartilage degeneration and heterotopic cartilage formation in a mouse surgical osteoarthritis model. PLoS One 7:e37728. doi:10.1371/journal.pone.0037728
Anderson, H. C. (2003). Matrix vesicles and calcification. Curr. Rheumatol. Rep. 5, 222–226. doi:10.1007/s11926-003-0071-z
Appleton, C. T., Pitelka, V., Henry, J., and Beier, F. (2007). Global analyses of gene expression in early experimental osteoarthritis. Arthritis Rheum 56, 1854–1868.
Bianchi, A., Guibert, M., Cailotto, F., Gasser, A., Presle, N., Mainard, D., et al. (2016). Fibroblast growth factor 23 drives MMP13 expression in human osteoarthritic chondrocytes in a Klotho-independent manner. Osteoarthr. Cartil. 24, 1961–1969. doi:10.1016/j.joca.2016.06.003
Bijlsma, J. W., Berenbaum, F., and Lafeber, F. P. (2011). Osteoarthritis: an update with relevance for clinical practice. Lancet 377, 2115–2126. doi:10.1016/s0140-6736(11)60243-2
Bikkavilli, R. K., and Malbon, C. C. (2009). Mitogen-activated protein kinases and Wnt/beta-catenin signaling: molecular conversations among signaling pathways. Commun. Integr. Biol. 2, 46–49. doi:10.4161/cib.2.1.7503
Blaney Davidson, E. N., Remst, D. F., Vitters, E. L., van Beuningen, H. M., Blom, A. B., Goumans, M. J., et al. (2009). Increase in ALK1/ALK5 ratio as a cause for elevated MMP-13 expression in osteoarthritis in humans and mice. J. Immunol. 182, 7937–7945. doi:10.4049/jimmunol.0803991
Bomer, N., den Hollander, W., Ramos, Y. F., Bos, S. D., van der Breggen, R., Lakenberg, N., et al. (2015). Underlying molecular mechanisms of DIO2 susceptibility in symptomatic osteoarthritis. Ann. Rheum. Dis. 74, 1571–1579. doi:10.1136/annrheumdis-2013-204739
Borzi, R. M., Olivotto, E., Pagani, S., Vitellozzi, R., Neri, S., Battistelli, M., et al. (2010). Matrix metalloproteinase 13 loss associated with impaired extracellular matrix remodeling disrupts chondrocyte differentiation by concerted effects on multiple regulatory factors. Arthritis Rheum. 62, 2370–2381. doi:10.1002/art.27512
Botter, S. M., van Osch, G. J., Clockaerts, S., Waarsing, J. H., Weinans, H., and van Leeuwen, J. P. (2011). Osteoarthritis induction leads to early and temporal subchondral plate porosity in the tibial plateau of mice: an in vivo microfocal computed tomography study. Arthritis Rheum. 63, 2690–2699. doi:10.1002/art.30307
Bray, S. J. (2016). Notch signalling in context. Nat. Rev. Mol. Cell Biol. 17, 722. doi:10.1038/nrm.2016.94
Brew, C. J., Clegg, P. D., Boot-Handford, R. P., Andrew, J. G., and Hardingham, T. (2010). Gene expression in human chondrocytes in late osteoarthritis is changed in both fibrillated and intact cartilage without evidence of generalised chondrocyte hypertrophy. Ann. Rheum. Dis. 69, 234–240. doi:10.1136/ard.2008.097139
Caron, M. M., Emans, P. J., Surtel, D. A., van der Kraan, P. M., van Rhijn, L. W., and Welting, T. J. (2015). BAPX-1/NKX-3.2 acts as a chondrocyte hypertrophy molecular switch in osteoarthritis. Arthritis Rheumatol. 67, 2944–2956. doi:10.1002/art.39293
Castano Betancourt, M. C., Cailotto, F., Kerkhof, H. J., Cornelis, F. M., Doherty, S. A., Hart, D. J., et al. (2012). Genome-wide association and functional studies identify the DOT1L gene to be involved in cartilage thickness and hip osteoarthritis. Proc. Natl. Acad. Sci. U.S.A. 109, 8218–8223. doi:10.1073/pnas.1119899109
Cavaco, S., Viegas, C. S., Rafael, M. S., Ramos, A., Magalhaes, J., Blanco, F. J., et al. (2016). Gla-rich protein is involved in the cross-talk between calcification and inflammation in osteoarthritis. Cell. Mol. Life Sci. 73, 1051–1065. doi:10.1007/s00018-015-2033-9
Cecil, D. L., Appleton, C. T., Polewski, M. D., Mort, J. S., Schmidt, A. M., Bendele, A., et al. (2009). The pattern recognition receptor CD36 is a chondrocyte hypertrophy marker associated with suppression of catabolic responses and promotion of repair responses to inflammatory stimuli. J. Immunol. 182, 5024–5031. doi:10.4049/jimmunol.0803603
Chang, J. K., Chang, L. H., Hung, S. H., Wu, S. C., Lee, H. Y., Lin, Y. S., et al. (2009). Parathyroid hormone 1-34 inhibits terminal differentiation of human articular chondrocytes and osteoarthritis progression in rats. Arthritis Rheum. 60, 3049–3060. doi:10.1002/art.24843
Chen, L., Wu, Y., Wu, Y., Wang, Y., Sun, L., and Li, F. (2016). The inhibition of EZH2 ameliorates osteoarthritis development through the Wnt/beta-catenin pathway. Sci. Rep. 6, 29176. doi:10.1038/srep29176
Dangelo, M., Sarment, D. P., Billings, P. C., and Pacifici, M. (2001). Activation of transforming growth factor beta in chondrocytes undergoing endochondral ossification. J. Bone Miner. Res. 16, 2339–2347. doi:10.1359/jbmr.2001.16.12.2339
Dell’Isola, A., and Steultjens, M. (2018). Classification of patients with knee osteoarthritis in clinical phenotypes: data from the osteoarthritis initiative. PLoS ONE 13:e0191045. doi:10.1371/journal.pone.0191045
Derynck, R., and Zhang, Y. E. (2003). Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 425, 577–584. doi:10.1038/nature02006
Deveza, L. A., Melo, L., Yamato, T. P., Mills, K., Ravi, V., and Hunter, D. J. (2017). Knee osteoarthritis phenotypes and their relevance for outcomes: a systematic review. Osteoarthr. Cartil. 25, 1926–1941. doi:10.1016/j.joca.2017.08.009
Dreier, R. (2010). Hypertrophic differentiation of chondrocytes in osteoarthritis: the developmental aspect of degenerative joint disorders. Arthritis Res. Ther. 12, 216. doi:10.1186/ar3117
Eswaramoorthy, R., Chang, C. C., Wu, S. C., Wang, G. J., Chang, J. K., and Ho, M. L. (2012). Sustained release of PTH(1-34) from PLGA microspheres suppresses osteoarthritis progression in rats. Acta Biomater. 8, 2254–2262. doi:10.1016/j.actbio.2012.03.015
Facchini, A., Borzi, R. M., Olivotto, E., Platano, D., Pagani, S., Cetrullo, S., et al. (2012). Role of polyamines in hypertrophy and terminal differentiation of osteoarthritic chondrocytes. Amino Acids 42, 667–678. doi:10.1007/s00726-011-1041-9
Felson, D. T., Lawrence, R. C., Dieppe, P. A., Hirsch, R., Helmick, C. G., Jordan, J. M., et al. (2000). Osteoarthritis: new insights. Part 1: the disease and its risk factors. Ann. Intern. Med. 133, 635–646. doi:10.7326/0003-4819-133-8-200010170-00016
Filip, A., Pinzano, A., Bianchi, A., Feve, B., Jalkanen, S., Gillet, P., et al. (2016). Expression of the semicarbazide-sensitive amine oxidase in articular cartilage: its role in terminal differentiation of chondrocytes in rat and human. Osteoarthr. Cartil. 24, 1223–1234. doi:10.1016/j.joca.2016.01.340
Fuerst, M., Bertrand, J., Lammers, L., Dreier, R., Echtermeyer, F., Nitschke, Y., et al. (2009). Calcification of articular cartilage in human osteoarthritis. Arthritis Rheum. 60, 2694–2703. doi:10.1002/art.24774
Fujita, N., Matsushita, T., Ishida, K., Kubo, S., Matsumoto, T., Takayama, K., et al. (2011). Potential involvement of SIRT1 in the pathogenesis of osteoarthritis through the modulation of chondrocyte gene expressions. J. Orthop. Res. 29, 511–515. doi:10.1002/jor.21284
Fukai, A., Kawamura, N., Saito, T., Oshima, Y., Ikeda, T., Kugimiya, F., et al. (2010). Akt1 in murine chondrocytes controls cartilage calcification during endochondral ossification under physiologic and pathologic conditions. Arthritis Rheum. 62, 826–836. doi:10.1002/art.27296
Gao, Z. Q., Guo, X., Duan, C., Ma, W., Xu, P., Wang, W., et al. (2012). Altered aggrecan synthesis and collagen expression profiles in chondrocytes from patients with Kashin-Beck disease and osteoarthritis. J. Int. Med. Res. 40, 1325–1334. doi:10.1177/147323001204000411
Garciadiego-Cazares, D., Aguirre-Sanchez, H. I., Abarca-Buis, R. F., Kouri, J. B., Velasquillo, C., and Ibarra, C. (2015). Regulation of alpha5 and alphaV integrin expression by GDF-5 and BMP-7 in chondrocyte differentiation and osteoarthritis. PLoS ONE 10:e0127166. doi:10.1371/journal.pone.0127166
Guidotti, S., Minguzzi, M., Platano, D., Cattini, L., Trisolino, G., Mariani, E., et al. (2015). Lithium chloride dependent glycogen synthase kinase 3 inactivation links oxidative DNA damage, hypertrophy and senescence in human articular chondrocytes and reproduces chondrocyte phenotype of obese osteoarthritis patients. PLoS ONE 10:e0143865. doi:10.1371/journal.pone.0143865
Hashimoto, K., Mori, S., Oda, Y., Nakano, A., Sawamura, T., and Akagi, M. (2016). Lectin-like oxidized low density lipoprotein receptor 1-deficient mice show resistance to instability-induced osteoarthritis. Scand. J. Rheumatol. 45, 412–422. doi:10.3109/03009742.2015.1135979
Hiraki, Y., and Shukunami, C. (2005). Angiogenesis inhibitors localized in hypovascular mesenchymal tissues: chondromodulin-I and tenomodulin. Connect. Tissue Res. 46, 3–11. doi:10.1080/03008200590935547
Hirata, M., Kugimiya, F., Fukai, A., Saito, T., Yano, F., Ikeda, T., et al. (2012). C/EBPbeta and RUNX2 cooperate to degrade cartilage with MMP-13 as the target and HIF-2alpha as the inducer in chondrocytes. Hum. Mol. Genet. 21, 1111–1123. doi:10.1093/hmg/ddr540
Hosaka, Y., Saito, T., Sugita, S., Hikata, T., Kobayashi, H., Fukai, A., et al. (2013). Notch signaling in chondrocytes modulates endochondral ossification and osteoarthritis development. Proc. Natl. Acad. Sci. U.S.A. 110, 1875–1880. doi:10.1073/pnas.1207458110
Houston, B., Stewart, A. J., and Farquharson, C. (2004). PHOSPHO1 – a novel phosphatase specifically expressed at sites of mineralisation in bone and cartilage. Bone 34, 629–637. doi:10.1016/j.bone.2003.12.023
Huebner, J. L., Johnson, K. A., Kraus, V. B., and Terkeltaub, R. A. (2009). Transglutaminase 2 is a marker of chondrocyte hypertrophy and osteoarthritis severity in the Hartley guinea pig model of knee OA. Osteoarthr. Cartil. 17, 1056–1064. doi:10.1016/j.joca.2009.02.015
Hunter, D. J. (2011). Pharmacologic therapy for osteoarthritis – the era of disease modification. Nat. Rev. Rheumatol. 7, 13–22. doi:10.1038/nrrheum.2010.178
Ijiri, K., Zerbini, L. F., Peng, H., Otu, H. H., Tsuchimochi, K., Otero, M., et al. (2008). Differential expression of GADD45beta in normal and osteoarthritic cartilage: potential role in homeostasis of articular chondrocytes. Arthritis. Rheum. 58, 2075–2087. doi:10.1002/art.23504
Johnson, G. L., and Lapadat, R. (2002). Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 298, 1911–1912. doi:10.1126/science.1072682
Johnson, K. A., Rose, D. M., and Terkeltaub, R. A. (2008). Factor XIIIA mobilizes transglutaminase 2 to induce chondrocyte hypertrophic differentiation. J. Cell. Sci. 121(Pt 13), 2256–2264. doi:10.1242/jcs.011262
Kronenberg, H. M. (2003). Developmental regulation of the growth plate. Nature 423, 332–336. doi:10.1038/nature01657
Lai, Y., Bai, X., Zhao, Y., Tian, Q., Liu, B., Lin, E. A., et al. (2014). ADAMTS-7 forms a positive feedback loop with TNF-alpha in the pathogenesis of osteoarthritis. Ann. Rheum. Dis. 73, 1575–1584. doi:10.1136/annrheumdis-2013-203561
Le, T. K., Montejano, L. B., Cao, Z., Zhao, Y., and Ang, D. (2012). Health care costs in US patients with and without a diagnosis of osteoarthritis. J. Pain Res. 5, 23–30. doi:10.2147/jpr.s27275
Leijten, J. C., Bos, S. D., Landman, E. B., Georgi, N., Jahr, H., Meulenbelt, I., et al. (2013). GREM1, FRZB and DKK1 mRNA levels correlate with osteoarthritis and are regulated by osteoarthritis-associated factors. Arthritis Res. Ther. 15, R126. doi:10.1186/ar4306
Li, T. F., Gao, L., Sheu, T. J., Sampson, E. R., Flick, L. M., Konttinen, Y. T., et al. (2010). Aberrant hypertrophy in Smad3-deficient murine chondrocytes is rescued by restoring transforming growth factor beta-activated kinase 1/activating transcription factor 2 signaling: a potential clinical implication for osteoarthritis. Arthritis Rheum. 62, 2359–2369. doi:10.1002/art.27537
Lin, N. Y., Distler, A., Beyer, C., Philipi-Schobinger, A., Breda, S., Dees, C., et al. (2016). Inhibition of Notch1 promotes hedgehog signalling in a HES1-dependent manner in chondrocytes and exacerbates experimental osteoarthritis. Ann. Rheum. Dis. 75, 2037–2044. doi:10.1136/annrheumdis-2015-208420
Little, C. B., Barai, A., Burkhardt, D., Smith, S. M., Fosang, A. J., Werb, Z., et al. (2009). Matrix metalloproteinase 13-deficient mice are resistant to osteoarthritic cartilage erosion but not chondrocyte hypertrophy or osteophyte development. Arthritis Rheum. 60, 3723–3733. doi:10.1002/art.25002
Liu-Bryan, R. (2015). Inflammation and intracellular metabolism: new targets in OA. Osteoarthr. Cartil. 23, 1835–1842. doi:10.1016/j.joca.2014.12.016
Lo, G. H., LaValley, M., McAlindon, T., and Felson, D. T. (2003). Intra-articular hyaluronic acid in treatment of knee osteoarthritis: a meta-analysis. JAMA 290, 3115–3121. doi:10.1001/jama.290.23.3115
Loeser, R. F., Erickson, E. A., and Long, D. L. (2008). Mitogen-activated protein kinases as therapeutic targets in osteoarthritis. Curr. Opin. Rheumatol. 20, 581–586. doi:10.1097/BOR.0b013e3283090463
Loeser, R. F., Goldring, S. R., Scanzello, C. R., and Goldring, M. B. (2012). Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum. 64, 1697–1707. doi:10.1002/art.34453
Lu, J., Sun, Y., Ge, Q., Teng, H., and Jiang, Q. (2014a). Histone deacetylase 4 alters cartilage homeostasis in human osteoarthritis. BMC Musculoskelet. Disord. 15:438. doi:10.1186/1471-2474-15-438
Lu, Y., Ding, M., Li, N., Wang, Q., Li, J., Li, X., et al. (2014b). Col10a1-Runx2 transgenic mice with delayed chondrocyte maturation are less susceptible to developing osteoarthritis. Am. J. Transl. Res. 6, 736–745.
Luyten, F. P., Bierma-Zeinstra, S., Dell’Accio, F., Kraus, V. B., Nakata, K., Sekiya, I., et al. (2017). Toward classification criteria for early osteoarthritis of the knee. Semin. Arthritis Rheum. 4, 457–463. doi:10.1016/j.semarthrit.2017.08.006
MacDonald, B. T., Tamai, K., and He, X. (2009). Wnt/β-catenin signaling: components, mechanisms, and diseases. Dev. Cell 17, 9–26. doi:10.1016/j.devcel.2009.06.016
Maes, C., Carmeliet, G., and Schipani, E. (2012). Hypoxia-driven pathways in bone development, regeneration and disease. Nat. Rev. Rheumatol. 8, 358–366. doi:10.1038/nrrheum.2012.36
Mannstadt, M., Juppner, H., and Gardella, T. J. (1999). Receptors for PTH and PTHrP: their biological importance and functional properties. Am. J. Physiol. 277(5 Pt 2), F665–F675.
Mapp, P. I., and Walsh, D. A. (2012). Mechanisms and targets of angiogenesis and nerve growth in osteoarthritis. Nat. Rev. Rheumatol. 8, 390–398. doi:10.1038/nrrheum.2012.80
Marcu, K. B., Otero, M., Olivotto, E., Borzi, R. M., and Goldring, M. B. (2010). NF-κB signaling: multiple angles to target OA. Curr. Drug Targets 11, 599–613. doi:10.2174/138945010791011938
Markway, B. D., Cho, H., and Johnstone, B. (2013). Hypoxia promotes redifferentiation and suppresses markers of hypertrophy and degeneration in both healthy and osteoarthritic chondrocytes. Arthritis Res. Ther. 15, R92. doi:10.1186/ar4272
Markway, B. D., Cho, H., Zilberman-Rudenko, J., Holden, P., McAlinden, A., and Johnstone, B. (2015). Hypoxia-inducible factor 3-alpha expression is associated with the stable chondrocyte phenotype. J. Orthop. Res. 33, 1561–1570. doi:10.1002/jor.22930
McKee, M. D., Yadav, M. C., Foster, B. L., Somerman, M. J., Farquharson, C., and Millan, J. L. (2013). Compounded PHOSPHO1/ALPL deficiencies reduce dentin mineralization. J. Dent. Res. 92, 721–727. doi:10.1177/0022034513490958
Meng, F., Zhang, Z., Chen, W., Huang, G., He, A., Hou, C., et al. (2016). MicroRNA-320 regulates matrix metalloproteinase-13 expression in chondrogenesis and interleukin-1beta-induced chondrocyte responses. Osteoarthr. Cartil. 24, 932–941. doi:10.1016/j.joca.2015.12.012
Merz, D., Liu, R., Johnson, K., and Terkeltaub, R. (2003). IL-8/CXCL8 and growth-related oncogene α/CXCL1 induce chondrocyte hypertrophic differentiation. J. Immunol. 171, 4406–4415. doi:10.4049/jimmunol.171.8.4406
Morey, L., and Helin, K. (2010). Polycomb group protein-mediated repression of transcription. Trends Biochem. Sci. 35, 323–332. doi:10.1016/j.tibs.2010.02.009
Nagase, H., Nagasawa, Y., Tachida, Y., Sakakibara, S., Okutsu, J., Suematsu, N., et al. (2013). Deiodinase 2 upregulation demonstrated in osteoarthritis patients cartilage causes cartilage destruction in tissue-specific transgenic rats. Osteoarthr. Cartil. 21, 514–523. doi:10.1016/j.joca.2012.12.013
Nasi, S., So, A., Combes, C., Daudon, M., and Busso, N. (2016). Interleukin-6 and chondrocyte mineralisation act in tandem to promote experimental osteoarthritis. Ann. Rheum. Dis. 75, 1372–1379. doi:10.1136/annrheumdis-2015-207487
Nguyen, C., Bazin, D., Daudon, M., Chatron-Colliet, A., Hannouche, D., Bianchi, A., et al. (2013). Revisiting spatial distribution and biochemical composition of calcium-containing crystals in human osteoarthritic articular cartilage. Arthritis Res. Ther. 15, R103. doi:10.1186/ar4283
Nishimura, S., Akagi, M., Yoshida, K., Hayakawa, S., Sawamura, T., Munakata, H., et al. (2004). Oxidized low-density lipoprotein (ox-LDL) binding to lectin-like ox-LDL receptor-1 (LOX-1) in cultured bovine articular chondrocytes increases production of intracellular reactive oxygen species (ROS) resulting in the activation of NF-κB. Osteoarthr. Cartil. 12, 568–576. doi:10.1016/j.joca.2004.04.005
Nye, J. S., and Kopan, R. (1995). Developmental signaling. Vertebrate ligands for Notch. Curr. Biol. 5, 966–969. doi:10.1016/S0960-9822(95)00189-8
Olivotto, E., Borzi, R. M., Vitellozzi, R., Pagani, S., Facchini, A., Battistelli, M., et al. (2008). Differential requirements for IKKalpha and IKKbeta in the differentiation of primary human osteoarthritic chondrocytes. Arthritis Rheum. 58, 227–239. doi:10.1002/art.23211
Olivotto, E., Otero, M., Astolfi, A., Platano, D., Facchini, A., Pagani, S., et al. (2013). IKKalpha/CHUK regulates extracellular matrix remodeling independent of its kinase activity to facilitate articular chondrocyte differentiation. PLoS ONE 8:e73024. doi:10.1371/journal.pone.0073024
Orfanidou, T., Iliopoulos, D., Malizos, K. N., and Tsezou, A. (2009). Involvement of SOX-9 and FGF-23 in RUNX-2 regulation in osteoarthritic chondrocytes. J. Cell. Mol. Med. 13, 3186–3194. doi:10.1111/j.1582-4934.2009.00678.x
Orimo, H. (2010). The mechanism of mineralization and the role of alkaline phosphatase in health and disease. J. Nippon Med. Sch. 77, 4–12. doi:10.1272/jnms.77.4
Page, C. J., Hinman, R. S., and Bennell, K. L. (2011). Physiotherapy management of knee osteoarthritis. Int. J. Rheum. Dis. 14, 145–151. doi:10.1111/j.1756-185X.2011.01612.x
Papathanasiou, I., Kostopoulou, F., Malizos, K. N., and Tsezou, A. (2015). DNA methylation regulates sclerostin (SOST) expression in osteoarthritic chondrocytes by bone morphogenetic protein 2 (BMP-2) induced changes in Smads binding affinity to the CpG region of SOST promoter. Arthritis Res. Ther. 17, 160. doi:10.1186/s13075-015-0674-6
Papathanasiou, I., Malizos, K. N., and Tsezou, A. (2012). Bone morphogenetic protein-2-induced Wnt/beta-catenin signaling pathway activation through enhanced low-density-lipoprotein receptor-related protein 5 catabolic activity contributes to hypertrophy in osteoarthritic chondrocytes. Arthritis Res. Ther. 14, R82. doi:10.1186/ar3805
Pesesse, L., Sanchez, C., Delcour, J. P., Bellahcene, A., Baudouin, C., Msika, P., et al. (2013). Consequences of chondrocyte hypertrophy on osteoarthritic cartilage: potential effect on angiogenesis. Osteoarthr. Cartil. 21, 1913–1923. doi:10.1016/j.joca.2013.08.018
Pesesse, L., Sanchez, C., Walsh, D. A., Delcour, J. P., Baudouin, C., Msika, P., et al. (2014). Bone sialoprotein as a potential key factor implicated in the pathophysiology of osteoarthritis. Osteoarthr. Cartil. 22, 547–556. doi:10.1016/j.joca.2014.01.010
Philipot, D., Guerit, D., Platano, D., Chuchana, P., Olivotto, E., Espinoza, F., et al. (2014). p16INK4a and its regulator miR-24 link senescence and chondrocyte terminal differentiation-associated matrix remodeling in osteoarthritis. Arthritis Res. Ther. 16, R58. doi:10.1186/ar4494
Pitsillides, A. A., and Beier, F. (2011). Cartilage biology in osteoarthritis – lessons from developmental biology. Nat. Rev. Rheumatol. 7, 654–663. doi:10.1038/nrrheum.2011.129
Prasadam, I., Mao, X., Shi, W., Crawford, R., and Xiao, Y. (2013). Combination of MEK-ERK inhibitor and hyaluronic acid has a synergistic effect on anti-hypertrophic and pro-chondrogenic activities in osteoarthritis treatment. J. Mol. Med. (Berl) 91, 369–380. doi:10.1007/s00109-012-0953-5
Prasadam, I., van Gennip, S., Friis, T., Shi, W., Crawford, R., and Xiao, Y. (2010). ERK-1/2 and p38 in the regulation of hypertrophic changes of normal articular cartilage chondrocytes induced by osteoarthritic subchondral osteoblasts. Arthritis Rheum. 62, 1349–1360. doi:10.1002/art.27397
Provot, S., Kempf, H., Murtaugh, L. C., Chung, U. I., Kim, D. W., Chyung, J., et al. (2006). Nkx3.2/Bapx1 acts as a negative regulator of chondrocyte maturation. Development 133, 651–662. doi:10.1242/dev.02258
Queirolo, V., Galli, D., Masselli, E., Borzi, R. M., Martini, S., Vitale, F., et al. (2016). PKCepsilon is a regulator of hypertrophic differentiation of chondrocytes in osteoarthritis. Osteoarthr. Cartil. 24, 1451–1460. doi:10.1016/j.joca.2016.04.003
Raimann, A., Ertl, D. A., Helmreich, M., Sagmeister, S., Egerbacher, M., and Haeusler, G. (2013). Fibroblast growth factor 23 and Klotho are present in the growth plate. Connect. Tissue Res. 54, 108–117. doi:10.3109/03008207.2012.753879
Ray, A., and Ray, B. K. (2008). An inflammation-responsive transcription factor in the pathophysiology of osteoarthritis. Biorheology 45, 399–409. doi:10.3233/BIR-2008-0500
Reimold, A. M., Grusby, M. J., Kosaras, B., Fries, J. W., Mori, R., Maniwa, S., et al. (1996). Chondrodysplasia and neurological abnormalities in ATF-2-deficient mice. Nature 379, 262–265. doi:10.1038/379262a0
Saito, T., Fukai, A., Mabuchi, A., Ikeda, T., Yano, F., Ohba, S., et al. (2010). Transcriptional regulation of endochondral ossification by HIF-2alpha during skeletal growth and osteoarthritis development. Nat. Med. 16, 678–686. doi:10.1038/nm.2146
Schaeffer, H. J., and Weber, M. J. (1999). Mitogen-activated protein kinases: specific messages from ubiquitous messengers. Mol. Cell. Biol. 19, 2435–2444. doi:10.1128/MCB.19.4.2435
Sherwood, J., Bertrand, J., Nalesso, G., Poulet, B., Pitsillides, A., Brandolini, L., et al. (2015). A homeostatic function of CXCR2 signalling in articular cartilage. Ann. Rheum. Dis. 74, 2207–2215. doi:10.1136/annrheumdis-2014-205546
Staines, K. A., Madi, K., Mirczuk, S. M., Parker, S., Burleigh, A., Poulet, B., et al. (2016). Endochondral growth defect and deployment of transient chondrocyte behaviors underlie osteoarthritis onset in a natural murine model. Arthritis Rheumatol. 68, 880–891. doi:10.1002/art.39508
Stanton, L. A., Underhill, T. M., and Beier, F. (2003). MAP kinases in chondrocyte differentiation. Dev. Biol. 263, 165–175. doi:10.1016/S0012-1606(03)00321-X
Stewart, A. J., Roberts, S. J., Seawright, E., Davey, M. G., Fleming, R. H., and Farquharson, C. (2006). The presence of PHOSPHO1 in matrix vesicles and its developmental expression prior to skeletal mineralization. Bone 39, 1000–1007. doi:10.1016/j.bone.2006.05.014
Thompson, C. L., Patel, R., Kelly, T. A., Wann, A. K., Hung, C. T., Chapple, J. P., et al. (2015). Hedgehog signalling does not stimulate cartilage catabolism and is inhibited by Interleukin-1beta. Arthritis Res. Ther. 17, 373. doi:10.1186/s13075-015-0891-z
Turner, N., and Grose, R. (2010). Fibroblast growth factor signalling: from development to cancer. Nat. Rev. Cancer 10, 116–129. doi:10.1038/nrc2780
van den Bosch, M. H., Blom, A. B., van Lent, P. L., van Beuningen, H. M., Blaney Davidson, E. N., van der Kraan, P. M., et al. (2014). Canonical Wnt signaling skews TGF-beta signaling in chondrocytes towards signaling via ALK1 and Smad 1/5/8. Cell. Signal. 26, 951–958. doi:10.1016/j.cellsig.2014.01.021
van der Esch, M., Knoop, J., van der Leeden, M., Roorda, L. D., Lems, W. F., Knol, D. L., et al. (2015). Clinical phenotypes in patients with knee osteoarthritis: a study in the Amsterdam osteoarthritis cohort. Osteoarthr. Cartil. 23, 544–549. doi:10.1016/j.joca.2015.01.006
van der Kraan, P. M. (2017). The changing role of TGFβ in healthy, ageing and osteoarthritic joints. Nat. Rev. Rheumatol. 13, 155. doi:10.1038/nrrheum.2016.219
van der Kraan, P. M., Blaney Davidson, E. N., Blom, A., and van den Berg, W. B. (2009). TGF-beta signaling in chondrocyte terminal differentiation and osteoarthritis: modulation and integration of signaling pathways through receptor-Smads. Osteoarthr. Cartil. 17, 1539–1545. doi:10.1016/j.joca.2009.06.008
van der Kraan, P. M., and van den Berg, W. B. (2012). Chondrocyte hypertrophy and osteoarthritis: role in initiation and progression of cartilage degeneration? Osteoarthr. Cartil. 20, 223–232. doi:10.1016/j.joca.2011.12.003
van der Weyden, L., Wei, L., Luo, J., Yang, X., Birk, D. E., Adams, D. J., et al. (2006). Functional knockout of the matrilin-3 gene causes premature chondrocyte maturation to hypertrophy and increases bone mineral density and osteoarthritis. Am. J. Pathol. 169, 515–527. doi:10.2353/ajpath.2006.050981
Wallin, R., Schurgers, L. J., and Loeser, R. F. (2010). Biosynthesis of the vitamin K-dependent matrix Gla protein (MGP) in chondrocytes: a fetuin-MGP protein complex is assembled in vesicles shed from normal but not from osteoarthritic chondrocytes. Osteoarthr. Cartil. 18, 1096–1103. doi:10.1016/j.joca.2010.05.013
Wang, G., and Beier, F. (2005). Rac1/Cdc42 and RhoA GTPases antagonistically regulate chondrocyte proliferation, hypertrophy, and apoptosis. J. Bone Miner. Res. 20, 1022–1031. doi:10.1359/jbmr.050113
Wang, Q. Y., Dai, J., Kuang, B., Zhang, J., Yu, S. B., Duan, Y. Z., et al. (2012). Osteochondral angiogenesis in rat mandibular condyles with osteoarthritis-like changes. Arch. Oral Biol. 57, 620–629. doi:10.1016/j.archoralbio.2011.12.006
Wei, F., Zhou, J., Wei, X., Zhang, J., Fleming, B. C., Terek, R., et al. (2012). Activation of Indian hedgehog promotes chondrocyte hypertrophy and upregulation of MMP-13 in human osteoarthritic cartilage. Osteoarthr. Cartil. 20, 755–763. doi:10.1016/j.joca.2012.03.010
Weisser, J., Riemer, S., Schmidl, M., Suva, L. J., Poschl, E., Brauer, R., et al. (2002). Four distinct chondrocyte populations in the fetal bovine growth plate: highest expression levels of PTH/PTHrP receptor, Indian hedgehog, and MMP-13 in hypertrophic chondrocytes and their suppression by PTH (1-34) and PTHrP (1-40). Exp. Cell Res. 279, 1–13. doi:10.1006/excr.2002.5580
Wenke, A. K., Grassel, S., Moser, M., and Bosserhoff, A. K. (2009). The cartilage-specific transcription factor Sox9 regulates AP-2epsilon expression in chondrocytes. FEBS J. 276, 2494–2504. doi:10.1111/j.1742-4658.2009.06973.x
Wenke, A. K., Niebler, S., Grassel, S., and Bosserhoff, A. K. (2011). The transcription factor AP-2 epsilon regulates CXCL1 during cartilage development and in osteoarthritis. Osteoarthr. Cartil. 19, 206–212. doi:10.1016/j.joca.2010.11.011
Wu, Q., Huang, J. H., Sampson, E. R., Kim, K. O., Zuscik, M. J., O’Keefe, R. J., et al. (2009). Smurf2 induces degradation of GSK-3beta and upregulates beta-catenin in chondrocytes: a potential mechanism for Smurf2-induced degeneration of articular cartilage. Exp. Cell Res. 315, 2386–2398. doi:10.1016/j.yexcr.2009.05.019
Xu, W., Xie, Y., Wang, Q., Wang, X., Luo, F., Zhou, S., et al. (2016). A novel fibroblast growth factor receptor 1 inhibitor protects against cartilage degradation in a murine model of osteoarthritis. Sci. Rep. 6, 24042. doi:10.1038/srep24042
Yahara, Y., Takemori, H., Okada, M., Kosai, A., Yamashita, A., Kobayashi, T., et al. (2016). Corrigendum: pterosin B prevents chondrocyte hypertrophy and osteoarthritis in mice by inhibiting Sik3. Nat. Commun. 7, 12117. doi:10.1038/ncomms12117
Yang, J., Andre, P., Ye, L., and Yang, Y.-Z. (2015). The Hedgehog signalling pathway in bone formation. Int. J. Oral Sci. 7, 73. doi:10.1038/ijos.2015.14
Yang, X., Trehan, S. K., Guan, Y., Sun, C., Moore, D. C., Jayasuriya, C. T., et al. (2014). Matrilin-3 inhibits chondrocyte hypertrophy as a bone morphogenetic protein-2 antagonist. J. Biol. Chem. 289, 34768–34779. doi:10.1074/jbc.M114.583104
Zhang, H., Wang, H., Zeng, C., Yan, B., Ouyang, J., Liu, X., et al. (2017). mTORC1 activation downregulates FGFR3 and PTH/PTHrP receptor in articular chondrocytes to initiate osteoarthritis. Osteoarthr. Cartil. 25, 952–963. doi:10.1016/j.joca.2016.12.024
Zhang, S., Bu, X., Zhao, H., Yu, J., Wang, Y., Li, D., et al. (2014a). A host deficiency of discoidin domain receptor 2 (DDR2) inhibits both tumour angiogenesis and metastasis. J. Pathol. 232, 436–448. doi:10.1002/path.4311
Zhang, S., Zhong, Y., Li, R., Wang, W., Zeng, L., Wang, Z., et al. (2014b). Experimental chondrocyte hypertrophy is promoted by the activation of discoidin domain receptor 2. Mol. Med. Rep. 10, 1543–1548. doi:10.3892/mmr.2014.2340
Zhang, W., Nuki, G., Moskowitz, R. W., Abramson, S., Altman, R. D., Arden, N. K., et al. (2010). OARSI recommendations for the management of hip and knee osteoarthritis: part III: changes in evidence following systematic cumulative update of research published through January 2009. Osteoarthr. Cartil. 18, 476–499. doi:10.1016/j.joca.2010.01.013
Zhang, X., Crawford, R., and Xiao, Y. (2016a). Inhibition of vascular endothelial growth factor with shRNA in chondrocytes ameliorates osteoarthritis. J. Mol. Med. (Berl) 94, 787–798. doi:10.1007/s00109-016-1425-0
Zhang, X., Prasadam, I., Fang, W., Crawford, R., and Xiao, Y. (2016b). Chondromodulin-1 ameliorates osteoarthritis progression by inhibiting HIF-2alpha activity. Osteoarthr. Cartil. 24, 1970–1980. doi:10.1016/j.joca.2016.06.005
Zhong, H.-M., Ding, Q.-H., Chen, W.-P., and Luo, R.-B. (2013). Vorinostat, a HDAC inhibitor, showed anti-osteoarthritic activities through inhibition of iNOS and MMP expression, p38 and ERK phosphorylation and blocking NF-κB nuclear translocation. Int. Immunopharmacol. 17, 329–335. doi:10.1016/j.intimp.2013.06.027
Zhou, J., Chen, Q., Lanske, B., Fleming, B. C., Terek, R., Wei, X., et al. (2014). Disrupting the Indian hedgehog signaling pathway in vivo attenuates surgically induced osteoarthritis progression in Col2a1-CreERT2; Ihhfl/fl mice. Arthritis Res. Ther. 16, R11. doi:10.1186/ar4437
Zhou, S., Xie, Y., Li, W., Huang, J., Wang, Z., Tang, J., et al. (2016). Conditional deletion of Fgfr3 in chondrocytes leads to osteoarthritis-like defects in temporomandibular joint of adult mice. Sci. Rep. 6, 24039. doi:10.1038/srep24039
Keywords: chondrocytes, osteoarthritis, hypertrophy, cartilage, phenotype, endochondral ossification
Citation: Ripmeester EGJ, Timur UT, Caron MMJ and Welting TJM (2018) Recent Insights into the Contribution of the Changing Hypertrophic Chondrocyte Phenotype in the Development and Progression of Osteoarthritis. Front. Bioeng. Biotechnol. 6:18. doi: 10.3389/fbioe.2018.00018
Received: 18 December 2017; Accepted: 08 February 2018;
Published: 19 March 2018
Edited by:
Eric Farrell, Erasmus University Rotterdam, NetherlandsReviewed by:
Katherine A. Staines, Edinburgh Napier University, United KingdomAndrea Lolli, Erasmus Medical Center, Erasmus University Rotterdam, Netherlands
Copyright: © 2018 Ripmeester, Timur, Caron and Welting. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tim J. M. Welting, dC53ZWx0aW5nQG1hYXN0cmljaHR1bml2ZXJzaXR5Lm5s
†Co-first authors.
‡Co-last authors.