 Sarah A. Wong1,2
Sarah A. Wong1,2 Kevin O. Rivera1,2
Kevin O. Rivera1,2 Theodore Miclau III1Eben Alsberg3
Theodore Miclau III1Eben Alsberg3 Ralph S. Marcucio1,2
Ralph S. Marcucio1,2 Chelsea S. Bahney1*
Chelsea S. Bahney1*- 1Department of Orthopaedic Surgery, Orthopaedic Trauma Institute, University of California, San Francisco, San Francisco, CA, United States
- 2School of Dentistry, University of California, San Francisco, San Francisco, CA, United States
- 3Department of Orthopaedic Surgery and Biomedical Engineering, Case Western Reserve University, Cleveland, OH, United States
The majority of fractures heal through the process of endochondral ossification, in which a cartilage intermediate forms between the fractured bone ends and is gradually replaced with bone. Recent studies have provided genetic evidence demonstrating that a significant portion of callus chondrocytes transform into osteoblasts that derive the new bone. This evidence has opened a new field of research aimed at identifying the regulatory mechanisms that govern chondrocyte transformation in the hope of developing improved fracture therapies. In this article, we review known and candidate molecular pathways that may stimulate chondrocyte-to-osteoblast transformation during endochondral fracture repair. We also examine additional extrinsic factors that may play a role in modulating chondrocyte and osteoblast fate during fracture healing such as angiogenesis and mineralization of the extracellular matrix. Taken together the mechanisms reviewed here demonstrate the promising potential of using developmental engineering to design therapeutic approaches that activate endogenous healing pathways to stimulate fracture repair.
Introduction
Fractures heal through two pathways: endochondral ossification and intramembranous ossification (Thompson et al., 2002; Bahney et al., 2015). Both processes begin with the differentiation of local osteochondral progenitor cells found within the periosteum and endosteum (Colnot, 2009; Duchamp de Lageneste et al., 2018). During endochondral ossification, or indirect bone healing, progenitor cells primarily derived from the periosteum differentiate into chondrocytes to form a cartilage callus between the fractured bone ends (Duchamp de Lageneste et al., 2018). This cartilage is gradually replaced with bone in a process that resembles embryonic bone development and post-natal growth. Intramembranous ossification, or direct bone healing, occurs when periosteal and endosteal progenitor cells differentiate directly into osteoblasts. Fate of the osteochondral progenitor is determined by the relative stability of the fracture site, with motion stimulating endochondral ossification and rigid microenvironments promoting intramembranous ossification (Thompson et al., 2002). In most cases, both healing pathways occur simultaneously such that a robust cartilage callus forms at the center of the fracture where the degree of motion is greatest, and intramembranous bone forms along the periosteal and endosteal surfaces (Thompson et al., 2002). Endochondral ossification is the predominant mechanism by which the majority of fractures heal and is the focus of this review (Silkstone et al., 2008; Bahney et al., 2015).
Formation of the cartilage callus functionally serves to stabilize the gap between the bone ends. To form the cartilage callus periosteal osteochondral progenitor cells migrate from the periosteum and undergo chondrogenic differentiation (Colnot, 2009). This occurs on top of the provisional fibrin matrix formed by the hematoma (Xing et al., 2010a). Growth factors produced by the hematoma promote cell migration and differentiation and also create a unique microenvironment with low pH and high lactate concentration (Wray, 1964). Formation of the hematoma and a strong pro-inflammatory response are essential to establishing a robust healing response (Park et al., 2002).
Following the initial hematoma, the subsequent steps of chondrogenesis and chondrocyte hypertrophy appear to parallel the molecular pathways involved in endochondral ossification in the growth plate during bone development (Kronenberg, 2003; Long and Ornitz, 2013). Chondrogenic programming is initiated by the expression of transcription factor Sox9, which is required for chondrogenesis (Bi et al., 1999; Akiyama et al., 2002). Sox9 regulates the expression of several chondrocyte-specific matrix components including collagen type II and aggrecan, the two predominant proteins within the cartilage matrix (Bell et al., 1997; Sekiya et al., 2000). This initial extracellular matrix is avascular and aneural until blood vessels and nerves penetrate the soft callus during later stages of healing (Gerber et al., 1999; Tatsuyama et al., 2000; Grässel, 2014; Hu et al., 2017). As chondrocytes mature, they produce collagen type X, mineralize their surrounding matrix, and undergo hypertrophy, increasing in volume and dry mass by ~20-fold (Cooper et al., 2013).
There has been a centuries-long debate regarding the subsequent fate of hypertrophic chondrocytes during endochondral bone development and repair. In the early 1800's, cartilage was believed to turn into bone (Beresford, 1981; Hall, 2015). However, in the mid-1800's, Muller and Sharpy changed this paradigm by claiming that chondrocytes are terminally-differentiated and ultimately undergo cell death, resulting in the replacement of cartilage with bone derived from a separate population of cells (Beresford, 1981; Hall, 2015). The latter model of chondrocyte fate, for the most part, dominated in textbooks and became the de facto model of endochondral ossification. In recent years, modern murine genetics has enabled lineage tracing studies that can more accurately follow the fate of cells. Using a combination of over five different genetic models, evidence now demonstrates that a significant portion of chondrocytes survive, proliferate, and transform into osteoblasts that derive the new bone (Bahney et al., 2014; Yang et al., 2014; Zhou et al., 2014; Jing et al., 2015; Park et al., 2015; Houben et al., 2016; Hu et al., 2017).
Pathways that regulate chondrocyte to bone conversion have practical implications on fracture healing. Importantly, since conversion of cartilage to bone is necessary for bone regeneration, it is critical to understand the molecular mechanisms regulating this process. Not only will these mechanistic data improve our understanding of impaired healing, especially in the context of hypertrophic non-unions where cartilage fails to convert to bone, but they will also enable new opportunities for therapeutic intervention through modulation of cartilage to bone transformation. Here, known and candidate molecular regulators of chondrocyte-to-osteoblast transformation, along with potential sources for these biological signals, are reviewed. Finally, we propose how tissue engineering can be used to translate the evidence reviewed here into new and improved fracture therapies.
Fracture Healing Standard of Care
Bone Grafting
Surgical intervention is currently the only effective treatment option for recalcitrant fractures (Bahney et al., 2015). Standard of care is to use bone autograft or allograft to stimulate healing (Hubble, 2002). Together this makes bone the second-most commonly transplanted tissue behind blood. While bone autografts stimulate strong bone repair, they come with the cost of significant donor site morbidity and limited supply. On the other hand, while bone allografts are readily available, they have significantly reduced bioactivity resulting in clinical failure associated with poor osteointegration and osteonecrosis of the graft (Brigman et al., 2004). Consequently, there is an unmet clinical need to develop pharmacologic agents, or “biologics,” which can be used either as a non-invasive alternative or in conjunction with surgical treatment to stimulate endogenous healing mechanisms and improve fracture outcomes.
Bone Morphogenetic Proteins
Bone morphogenetic proteins (BMPs) are currently the most common clinically-used biologics. BMP signal transduction occurs through the binding of BMP ligands to type I and type II serine/threonine kinase receptors (BMPR-I, BMPR-II). This induces phosphorylation of BMP receptors and subsequent phosphorylation of receptor SMADS (R-SMADs) 1, 5, and 8. R-SMADS then form a complex with SMAD4, enabling it to enter the nucleus where it regulates gene expression (Lin and Hankenson, 2011; Long and Ornitz, 2013; Katagiri and Watabe, 2016; Salazar et al., 2016) (Figure 1A).
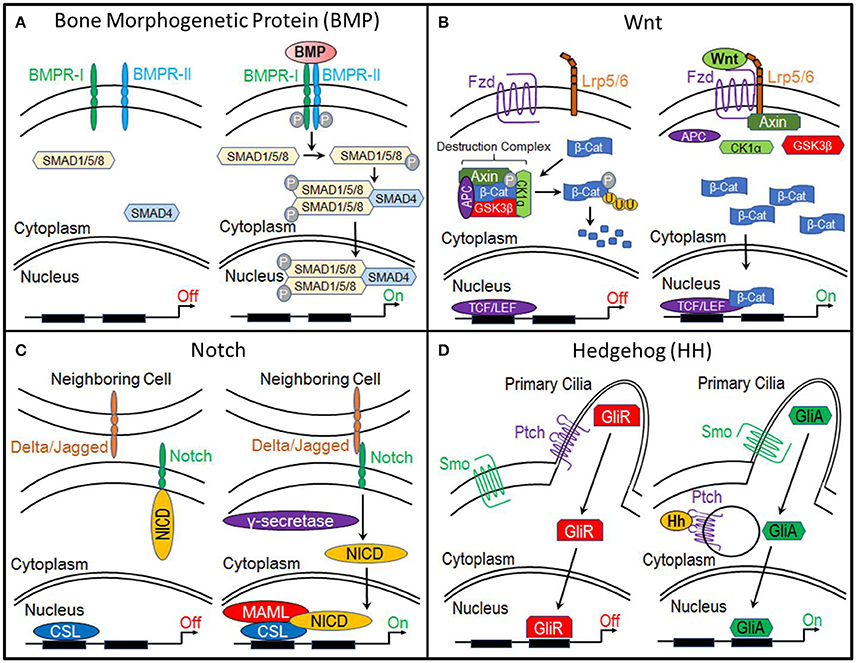
Figure 1. Molecular pathways. (A) Bone Morphogenetic Protein (BMP), (B) Canonical Wnt, (C) Notch, and (D) Hedgehog.
Pre-clinical studies indicated that the BMP pathway was an excellent target for therapeutic development due to its role in regulating osteoblastogenesis and the ability of several BMPs to strongly induce bone formation (Hoffmann and Gross, 2001; Karsenty and Wagner, 2002; Einhorn, 2010). This led to a series of clinical trials and FDA approval of two recombinant BMPs. Recombinant human BMP2 (INFUSE®) obtained pre-market approval for use in lumbar spinal fusion and for the treatment of compound tibial fractures (Einhorn, 2010; Chrastil et al., 2013). Recombinant human BMP7, also known as Osteogenic Protein 1 (OP-1), received a Humanitarian Device Exemption for the treatment of recalcitrant long bone non-unions and for revisions of lumbar spinal fusions (Einhorn, 2010; Chrastil et al., 2013). However, although rhBMP2 has exhibited clinical success in spinal fusion, both rhBMP2 and rhOP-1 have shown less impressive results in the treatment of fracture non-unions (Einhorn, 2010). rhOP-1 has now been taken off the market and use of rhBMP2 has been significantly diminished as a result of reports of serious side effects, including heterotopic ossification and tumorigenesis, and by the expense of treatment ($5,000–$15,000 per treatment) (Einhorn, 2010; DeVine et al., 2012; Chrastil et al., 2013; Almubarak et al., 2016).
It has been postulated that the lack of clinical success with BMPs is due to limited understanding of the molecular signals responsible for regulating fracture repair and that a combination of biologics applied during the appropriate phases of the repair process will be required to effectively stimulate healing (Simmons et al., 2004; Sukul et al., 2015; Dang et al., 2016a). Furthermore, supraphysiological dosing, burse release kinetics, and rapid diffusion of BMPs are key factors contributing to heterotopic ossification (Krishnan et al., 2017). As reviewed recently, engineering scaffolds and drug delivery systems to promote sustained and local delivery of BMPs is a significant and active area of research that can translate into improved clinical outcomes (Bessa et al., 2008; Bhattacharjee et al., 2015; Agrawal and Sinha, 2017).
Novel Molecular Targets for Fracture Healing
To study the molecular signals regulating chondrocyte-to-osteoblast transformation, we have defined the chondro-osseous border in the fracture callus as the “Transition Zone” (Hu et al., 2017). Here, mature hypertrophic chondrocytes have been shown to express classic osteogenic markers (i.e., runx2, osterix, collagen type I, osteocalcin, osteopontin) indicating that these cells adopt an osteogenic fate (Hu et al., 2017). Interestingly, a recent publication by Hu et al. demonstrated that hypertrophic chondrocytes at the Transition Zone also express pluripotency transcription factors Sox2, Oct4, and Nanog, suggesting that chondrocytes acquire a stem cell-like state during transformation (Hu et al., 2017). Sox2 was shown to play an important role during chondrocyte transformation since its deletion resulted in significantly reduced bone formation and increased cartilage retention within the fracture callus (Hu et al., 2017).
Despite advances in our understanding of chondrocyte gene expression during transformation, the signaling mechanisms that direct this process remain largely unknown. Evidence suggests numerous molecular pathways as regulatory candidates, including canonical Wnt, Notch, FGF, and Hedgehog signaling, each of which will be explored here (Figure 2).
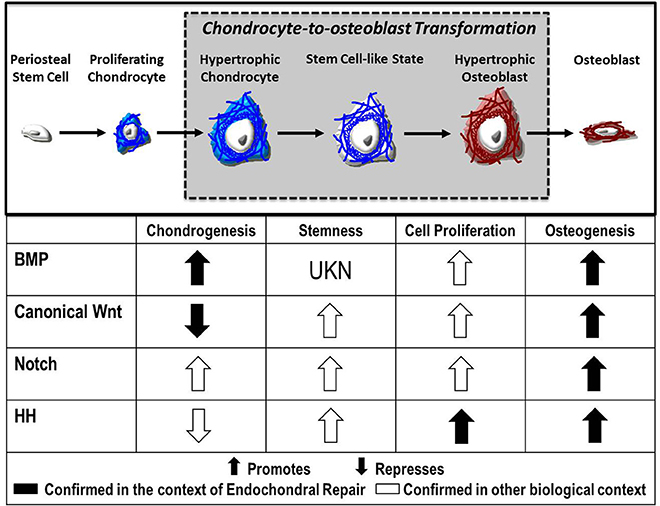
Figure 2. Fate of the chondrocyte. During endochondral ossification, the formation of the cartilage callus begins with the differentiation of periosteal stem cells into chondrocytes, which proliferate and mature to a hypertrophic state. These hypertrophic chondrocytes then re-enter the cell cycle, express stem cell markers, and finally transform into osteoblasts that contribute to the formation of new bone. Published evidence suggests the Bone Morphogenetic Protein (BMP), Canonical Wnt, Notch, and Hedgehog (HH) pathways as candidate regulators of chondrocyte-to-osteoblast transformation due to their effects on chondrogenesis, stemness, cell proliferation, and osteogenesis in the context of endochondral repair ( ) and in other biological contexts (
) and in other biological contexts ( ).
).
Canonical Wnt Signaling
Wnt signaling is traditionally categorized into the β-catenin-dependent canonical pathway and the β-catenin-independent non-canonical pathways (planar cell polarity and Ca2+-mediated pathways), as recently reviewed (Gammons and Bienz, 2018). While some evidence suggests that the non-canonical pathways may play a role in regulating osteogenesis (Chen et al., 2007), the canonical Wnt/β-catenin pathway is the most studied and has been shown to play a dominant role in bone development and fracture repair. Thus, this review focuses on the canonical Wnt pathway.
The primary function of canonical Wnt signaling is to regulate the transcription of genes involved in cellular processes such as proliferation, differentiation, self-renewal, and survival. When this pathway is inactive, β-catenin, a transcriptional co-activator and the primary effector of this pathway, is bound by a multiprotein “destruction” complex, which consists of Axin, adenomatous polyposis coli (APC), and serine/threonine kinases glycogen synthase kinase 3β (GSK3β) and casein kinase 1α (CK1α). This destruction complex phosphorylates β-catenin, targeting it for ubiquitination and ultimately proteosomal degradation. However, when the pathway is activated by the binding of Wnt ligands to Frizzled and LRP5/6 receptors, the destruction complex is disrupted, enabling β-catenin to accumulate within the cytoplasm and translocate to the nucleus, where it interacts with members of the T-cell factor/lymphocyte elongation factor (TCF/LEF) family to activate transcription of target genes (Gammons and Bienz, 2018) (Figure 1B).
The canonical Wnt pathway has an established role in osteogenesis and skeletal formation by functioning as a molecular switch regulating lineage commitment between osteogenesis and chondrogenesis (Hill et al., 2005; Topol et al., 2009). During development, inhibition of canonical Wnt signaling through conditional deletion of β-catenin from limb and head mesenchyme using Prx1-CreERT, or conditional deletion from skeletogenic mesenchyme using Dermo1-Cre, inhibits bone formation and results in early osteoblast differentiation arrest (Day et al., 2005; Hill et al., 2005). Osteoblastogenesis halts at the osteochondral progenitor stage and cells differentiate into chondrocytes, resulting in the formation of ectopic cartilage (Day et al., 2005; Hill et al., 2005). Although cells express Runx2, an early marker of the osteoblast lineage, they fail to express osterix, indicating that these cells are incapable of committing to an osteogenic fate (Day et al., 2005; Hill et al., 2005). In vitro experiments inhibiting canonical Wnt signaling in mesenchymal progenitor cells provide similar findings (Hill et al., 2005).
Canonical Wnt signaling also plays a key role in directing osteogenesis during intramembranous repair (Kim et al., 2007). Using a transcortical defect model, which heals through intramembranous ossification, inhibition of Wnt signaling through adenoviral expression of Dkk1 prevented the differentiation of osteoprogenitor cells into osteoblasts and significantly reduced bone regeneration compared to controls (Kim et al., 2007). Conversely, activating the canonical Wnt pathway through deletion of pathway inhibitors (sclerostin or Axin2) significantly improved intramembranous bone formation (McGee-Lawrence et al., 2013). Furthermore, treatment of bone grafts with Wnt3a protein restored the osteogenic potential of aged bone grafts and promoted intramembranous healing of critical-sized defects in mouse calvaria and rabbit ulna (Leucht et al., 2013).
Less work has been done to determine the role of canonical Wnt signaling during endochondral bone formation and repair since traditionally the Wnt pathway is thought to promote direct osteogenesis. However, the mounting data demonstrating chondrocytes can directly form bone in development and repair (Bahney et al., 2014; Yang et al., 2014; Zhou et al., 2014; Jing et al., 2015; Park et al., 2015; Houben et al., 2016; Hu et al., 2017) suggests that canonical Wnt signaling may have a functional role in chondrocyte-to-osteoblast transdifferentiation. This was directly tested recently by Houben et al. who showed conditional deletion of β-catenin in col10a1-expressing hypertrophic chondrocytes resulted in significantly reduced bone, whereas stabilized β-catenin produced osteopetrotic tissue during endochondral development (Houben et al., 2016).
Since fracture repair in many ways recapitulates bone development, canonical Wnt signaling may play a similar role in regulating chondrocyte-to-osteoblast transformation during endochondral repair. Indeed, during endochondral healing, nuclear localization of β-catenin was seen in hypertrophic chondrocytes at the fracture callus Transition Zone, indicating that these cells undergo active canonical Wnt signaling (Hu et al., 2017). RT-qPCR analysis of fracture calli revealed that numerous Wnt ligands, receptors, and transduction machinery are expressed during fracture repair (Chen et al., 2007; Leucht et al., 2008). Huang et al. demonstrated that inhibition of Wnt/β-catenin signaling in chondrocytes, using an 82-amino-acid peptide called Inhibitor of β-catenin/TCF (ICAT) driven by col2a1 expression, delayed cartilage formation and reduced bone formation (Huang et al., 2012b). Similarly, activation of canonical Wnt signaling through treatment with lithium chloride enhanced bone formation (Chen et al., 2007). Interestingly, enhanced bone regeneration was only observed when the Wnt pathway was activated at later time points, which corresponds biologically with chondrocyte-to-osteoblast transformation (Chen et al., 2007). Together, these data suggest that canonical Wnt signaling may play a role in regulating chondrocyte-to-osteoblast transformation during fracture healing.
The evidence outlined above are derived primarily from pre-clinical studies and in vitro systems. However, it is likely that the canonical Wnt pathway plays a similarly critical role in humans. Numerous human bone diseases are associated with mutations to components of the canonical Wnt pathway (Regard et al., 2012). Predisposition to osteoporosis has been associated with genomic polymorphisms in or close to Wnt/β-catenin signaling components (Regard et al., 2012). Loss-of-function mutations in the Wnt receptor LRP5 are associated with osteoporosis pseudoglioma (OPPG) syndrome and juvenile osteoporosis and gain-of-function mutations in the same receptor result in the opposite phenotype of high bone mass and enhanced bone strength (Einhorn, 2010; Regard et al., 2012). Sclerosteosis is a bone disease characterized by an overgrowth of bone and is caused by mutations in the gene and enhancer regions of the Wnt/β-catenin antagonist sclerostin (SOST) (Einhorn, 2010; Regard et al., 2012). Furthermore, the canonical Wnt pathway has been implicated in the context of human fracture repair since β-catenin and sclerostin levels have been shown to increase (Chen et al., 2007; Sarahrudi et al., 2012).
The canonical Wnt pathway is primed for translation. Numerous Wnt pathway regulators are being developed and several are already in clinical trials. The majority of these pathway modulators serve to activate the canonical Wnt pathway by neutralizing pathway inhibitors such as Dkk1 and sclerostin (Canalis, 2013). This indirect approach to pathway activation has been adopted primarily because direct pathway activation through treatment with Wnt ligands is clinically-irrelevant. Endogenous Wnts are hydrophobic due to palmitoylation, a form of lipidation required for the intracellular trafficking and full activation of Wnts (Willert et al., 2003; Takada et al., 2006; Janda et al., 2012). This makes Wnts challenging to extract and purify, requires that they be delivered using special liposome-based systems, and significantly increases the cost of treatment (Morrell et al., 2008). Fortunately, several of the Wnt pathway modulators acting to neutralize pathway inhibitors have shown promising osteogenic effects during clinical trials.
Of the Wnt pathway regulators currently in development, Romosozumab is closest to attaining FDA approval and is currently in Phase III clinical trials for treating osteoporosis (Regard et al., 2012; Canalis, 2013). It is a humanized monoclonal antibody that binds to and neutralizes the Wnt inhibitor sclerostin (Canalis, 2013). Studies show that treatment with Romosozumab significantly increases bone mineral density and reduces incidence of osteoporotic fractures (Canalis, 2013). Wnt pathway regulators, such as Romosozumab, could readily be repurposed for the context of fracture repair. However, the optimal dosage, timing, and the method of treatment still need to be determined.
Notch
Like, the canonical Wnt pathway, the functional roles of Notch signaling suggest it as a candidate regulator of chondrocyte-to-osteoblast transformation. Activation of this pathway begins when the Notch transmembrane receptor binds to membrane-bound ligands (Delta or Jagged) on the surface of neighboring cells. This triggers the proteolytic cleavage of the Notch intracellular domain (NICD) by y-secretase. NICD then translocates to the nucleus where it forms a complex with and activates the transcription factor CSL, which recruits its co-activator Mastermind-like (MAML) and initiates transcription of target genes (Lin and Hankenson, 2011) (Figure 1C).
Notch signaling has been shown to promote osteoblastogenesis. In vitro inhibition of Notch signaling in mouse MSCs impaired osteoblast differentiation as assessed by alizarin red staining for matrix mineralization (Dishowitz et al., 2013). In vivo, gain-of-function Notch signaling in osteoblasts through the overexpression of NICD resulted in abnormally dense or osteosclerotic bone attributed to increased cell proliferation of immature osteoblasts (Engin et al., 2008). Similarly, loss-of-function Notch signaling in osteoblasts, through mutations to y-secretase, led to late-onset osteoporosis (Engin et al., 2008).
Notch signaling also appears to play a role in promoting hypertrophic maturation of chondrocytes. During development, inhibition of Notch signaling in chondrocytes impaired terminal stages of endochondral ossification in the limb cartilage, resulting in shorter limbs with an increased hypertrophic zone and reduced bone (Hosaka et al., 2013). In the context of disease, Notch signaling may promote osteoarthritis (OA), which resembles pathological activation of endochondral ossification (Hosaka et al., 2013). Nuclear localization of the intracellular domains of Notch-1 and -2 was observed in chondrocytes in mouse and human OA articular cartilage, indicating active Notch signaling in these cells (Hosaka et al., 2013). Functionally, inhibition of Notch signaling in chondrocytes conferred resistance to OA development in the knee joint (Hosaka et al., 2013).
Notch signaling has also been shown to play an important role during fracture repair. Notch signaling is upregulated during both intramembranous and endochondral ossification, but data suggest it is more highly activated during endochondral ossification (Dishowitz et al., 2012). During endochondral ossification, Notch signaling decreases as progenitors differentiate into chondrocytes and as chondrocytes mature to hypertrophy. However, mature hypertrophic chondrocytes at the Transition Zone re-expressed Jag1 and NICD2, indicating that these cells have re-activated the Notch pathway (Dishowitz et al., 2012). Whether the Notch pathway plays a functional role in regulating chondrocyte-to-osteoblast transformation is unknown. However, systemic inhibition of Notch signaling using the Mx1-Cre;dnMAMLfl/− mouse impaired fracture healing primarily due to a prolonged inflammatory phase, decreased cartilage callus formation, and decreased osteoblast and osteoclast cell density (Dishowitz et al., 2013).
Hedgehog Signaling
The Hedgehog (Hh) pathway is essential to osteogenesis. When this pathway is inactive, cell surface receptor Patched (Ptch) prevents transmembrane protein Smoothened (Smo) from entering the primary cilia. This results in the proteolytic processing of Gli transcription factors into a repressor form (GliR). GliR then enters the nucleus and prevents Hedgehog target gene expression. Hedgehog signaling is activated by the binding of Hh ligands to Patched, thus relieving Patched-mediated suppression of Smoothened through Patched endocytosis. Smoothened enters the primary cilia where it prevents Gli transcription factors from being processed. Thus, Gli remains in its full-length, active form (GliA), which translocates to the nucleus and activates expression of Hedgehog target genes (Lin and Hankenson, 2011) (Figure 1D).
Of the three Hedgehog homologs, Sonic hedgehog (Shh) and Indian hedgehog (Ihh) have been implicated in osteoblastogenesis (Ehlen et al., 2006). Shh acts at early stages of development to direct patterning and growth (Zhu et al., 2008). Ihh is involved at later stages of endochondral ossification during limb development and consequently has been studied in greater depth in the context of bone formation and repair (Ehlen et al., 2006). Indian hedgehog is a central regulator of skeletogenesis and is required for osteoblastogenesis in endochondral, but not membranous bones (Kronenberg, 2003; Hill et al., 2005; Lin and Hankenson, 2011). Ihh is primarily expressed by pre- and early hypertrophic chondrocytes, where it controls proliferation and the onset of chondrocyte hypertrophy (St-Jacques et al., 1999; Long et al., 2001, 2004; Maeda et al., 2007). During development, chondrocyte expression of Ihh triggers Runx2 expression in the periosteum, thus coupling chondrocyte differentiation/maturation with osteoblastogenesis (Hill et al., 2005; Ehlen et al., 2006).
Like canonical Wnt signaling, evidence suggests that the Hedgehog pathway also serves as a molecular switch between osteogenesis and chondrogenesis. Chimeric embryos derived from Smoothened null and wild type embryonic cells exhibited abnormal bone collar formation (Long et al., 2004). Whereas, wild type cells underwent normal osteoblast differentiation, adjacent mutant cells failed to differentiate into osteoblasts and instead exhibited chondrocyte morphology, deposited cartilaginous matrix and expressed chondrocyte markers (collagen type II and X) (Long et al., 2004).
During development, Hedgehog signaling has also been shown to play an important role in trabecular bone formation. Inhibition of Hedgehog signaling through deletion of Smoothened in chondrocytes prevented formation of the primary spongiosa (Long et al., 2004). This loss in trabecular bone formation correlated with lost expression of the Hedgehog target gene, Patched1, at the chondro-osseous junction, suggesting that Hedgehog signaling promotes chondrocyte-to-osteoblast transformation (Long et al., 2004).
The Hedgehog pathway has also been implicated in regulating chondrocyte-to-osteoblast transformation during post-natal endochondral bone growth. Gli1-CreERT2 Hedgehog reporter mice demonstrated active Hedgehog signaling in hypertrophic chondrocytes and osteoprogenitors at the chondro-osseous junction of the growth plate (Haraguchi et al., 2018). Furthermore, deletion of Ihh from growth plate chondrocytes in post-natal mice resulted in continuous loss of trabecular bone with progression of age (Maeda et al., 2007).
Hedgehog signaling has been shown to promote osteogenesis during skeletal homeostasis. Systemic inhibition of Hedgehog signaling through treatment with cyclopamine decreased bone mass in adult mice (Ohba et al., 2008). In contrast, enhanced bone formation, was observed with forced activation of Hedgehog signaling in mature osteoblasts through global Patched1 haploinsufficiency or deletion (Ohba et al., 2008). Interestingly, enhanced Hedgehog activity also resulted in excessive bone resorption due to the role of Hedgehog signaling in promoting osteoclastogenesis (Mak et al., 2008).
Evidence suggests that the hedgehog pathway promotes endochondral repair as signaling is upregulated during fracture healing (Liu et al., 2017). Furthermore, Gli1 reporter mice demonstrated that cells actively signaling through the hedgehog pathway contribute to both chondrocytes and osteoblasts during fracture healing (Shi et al., 2017). Inhibition of the Hedgehog pathway through treatment with a systemic Hedgehog inhibitor GDC-0449, delayed fracture healing (Liu et al., 2017). Chondrogenesis was unaffected, suggesting that the effects were due to Hedgehog regulation of chondrocyte transformation (Liu et al., 2017). In contrast, activation of Hedgehog signaling through local administration of a Hedgehog agonist known as Smoothened Agonist (SAG) accelerated endochondral repair due to increased chondrocyte proliferation, an enlarged cartilaginous callus, and an increased number of cells expressing osteoblast markers within the bony callus (Kashiwagi et al., 2016).
Vasculature Regulation of Chondrocyte-to-Osteoblast Transformation
The vasculature plays a critical role during fracture repair. Whereas, the normal rate of impaired healing is 10–15%, this percentage increases to 46% when fractures occur in conjunction with severe vasculature injury (Bahney et al., 2015). The role of the vasculature begins at the outset of injury during hematoma formation where it helps to create the growth factor rich fibrin blood clot upon which periosteal stem cells differentiate to chondrocytes under a low pH, high lactate microenvironment (Wray, 1964; Xing et al., 2010a). After chondrogenic differentiation, the cartilage anlage is avascular and chondrogenic maturation happens in the absence of a regulatory role from the vasculature (Gerber et al., 1999; Tatsuyama et al., 2000; Hu et al., 2017).
In the later stages of repair, blood vessels are recruited into the cartilage fracture callus by hypertrophic chondrocytes expressing vascular endothelial growth factor (VEGF) (Gerber et al., 1999; Zelzer et al., 2002; Hu et al., 2017) and placental growth factor (PlGF) (Maes et al., 2006). Histologically, the cartilage to bone transition in the fracture callus occurs around this invading vasculature (Hu et al., 2017). Importantly, spatiotemporal expression of osteogenic genes and pluripotency transcription factors occurs in hypertrophic chondrocytes adjacent to the vasculature, suggesting that the vasculature plays a role in initiating chondrocyte-to-osteoblast transformation (Hu et al., 2017).
Growth Factor Secretion
Endothelial cells from the vasculature may functionally contribute to phenotypic modulation of the chondrocyte phenotype through secretion of pro-osteogenic growth factors. For example, it has been established that vascular tissues are a direct endogenous source of BMPs (Yu et al., 2010; Matsubara et al., 2012). Functionally it has been shown that secreted factors from vascular endothelial cell conditioned media were capable of inducing matrix mineralization and up-regulating the classic osteogenic gene osteocalcin (Bahney et al., 2014). It is likely that BMP expression contributed to this phenotype (Bahney et al., 2014). However, more recently it was also shown that the same vascular endothelial cell conditioned media induced expression of pluripotency transcription factors (Sox2, Oct4, Nanog) indicating that an additional factor may have a role in activating a stem-like state (Hu et al., 2017). While the complete secretome of vascular endothelial cells during fracture healing has not been detailed, it is known that this secretome is site specific (Nolan et al., 2013; Rafii et al., 2016). It is possible that fracture callus endothelial cells secrete factors other than BMP that may play a role in directing osteogenesis or chondrocyte plasticity.
Delivery of Macrophages
The vasculature is also responsible for delivering inflammatory cells to the fracture callus. These include circulatory macrophages, which are recruited by pro-inflammatory cytokines [Tumor necrosis factor (TNFα), Interleukin-1β (IL-1β), and IL-6] that activate a pro-inflammatory (M1) macrophage state (Wray, 1964). This pro-inflammatory phase has been shown to improve fracture repair by promoting cell proliferation and stem cell differentiation (Xing et al., 2010b; Wang et al., 2013).
While this inflammatory response is necessary for proper healing, it must be resolved in order for healing to progress (Wang et al., 2013). A prolonged pro-inflammatory state can delay fracture repair and is an underlying factor in impaired healing in elderly animals (Lu et al., 2008; Xing et al., 2010a,b; Abou-Khalil et al., 2014; Baht et al., 2015). Resolution of the pro-inflammatory state occurs when anti-inflammatory cytokines and growth factors [IL-10, arginase, TGFβ, EGF, PDGF, VEGF] push M1 macrophages toward the M2 phenotype (Laskin, 2009). Thus, it is possible that macrophages and their inflammatory resolution may help regulate chondrocyte-to-osteoblast transformation.
Matrix Mechanobiology
Recent studies have demonstrated that the extracellular matrix (ECM) plays an active role in regulating chondrogenic and osteogenic cell fate decisions. Changes in cell fate elicit changes to the surrounding matrix, thus producing a cycle of bi-directional interactions between cells and their surrounding matrix, a phenomenon known as “dynamic reciprocity” (Bissell et al., 1982). This cross-talk is modulated by the structural, mechanical, and biochemical cues provided by the ECM.
Remodeling of the ECM during endochondral ossification is a dynamic process that transforms the cartilaginous matrix into bone. This change in ECM contributes to the phenotypic adaptation that occurs during chondrocyte-to-osteoblast transformation. The major constituents of the cartilage ECM are collagens, hyaluronan, proteoglycans, and glycoproteins (Gentili and Cancedda, 2009). Collagens account for two-thirds of the tissue's dry weight, the most abundant of which is collagen type II (Eyre et al., 2006). Collagen type II is a fibril-forming collagen that creates nonparallel crosslinks with collagens type IX and XI. These crosslinks create a robust meshwork that gives cartilage its tensile strength. Cartilage is further characterized by its high aggrecan content (Martel-Pelletier et al., 2008). Aggrecan is anchored to hyaluronan within the matrix and is a negatively charged proteoglycan that attracts water (Roughley and Mort, 2014). This attraction of water to aggrecan creates osmotic pressure within the tissue, making cartilage shock-absorbent and resistant to high-load compression (Maldonado and Nam, 2013). Together, the collagen II and aggrecan ultrastructure allows for limited but necessary deformation under compressive forces that contributes to distribution of nutrients across the avascular tissue (Muir, 1995).
During endochondral ossification, there is a change in the amount and type of collagens present in the ECM. Chondrocyte hypertrophy is marked by the deposition of collagen type X and the up-regulation of matrix metalloproteinase-13 (MMP-13), which leads to the degradation of collagen II and aggrecan (Ortega et al., 2004; Maldonado and Nam, 2013). The loss of collagen II and aggrecan leads to a temporary reduction in tensile strength and stiffness of the tissue, which changes the mechanical microenvironment of chondrocytes and exposes the cells to greater strains that may induce phenotypic changes (Figure 3) (Stockwell, 1981; Ashman and Jae Young Rho, 1988; Rho et al., 1993; Chintala et al., 1994; Mente and Lewis, 1994; Liu et al., 2016). Proteolysis of collagen II likely contributes to chondrocyte hypertrophy and increased hydration experienced by the cartilage matrix as a consequence of a weakened fibril network losing the ability to resist the influx of proteoglycan-attracted water (Dejica et al., 2012; Akkiraju and Nohe, 2015). These changes in hydrostatic pressure could enhance mineralization of cartilage through the diffusion of ions (Tanck et al., 1999).
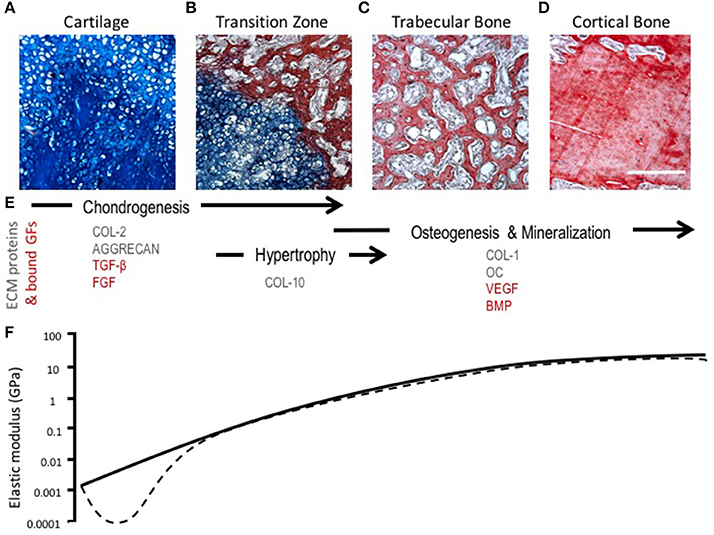
Figure 3. Morphological, compositional, and mechanical changes during endochondral ossification (EO). (A–D) HBQ histology (blue = cartilage, red = bone) of representative tissues from a murine fracture callus throughout stages of healing: (A) cartilage, (B) transition zone, (C) trabecular bone, and (D) cortical bone. Scale bar = 200 μm. (E) Tissue matrix components and matrix-bound growth factors corresponding to the location and phases of EO (Chintala et al., 1994; Shen, 2005; Eyre et al., 2006; Martel-Pelletier et al., 2008; Maldonado and Nam, 2013; Martino et al., 2013; Tampieri and Sprio, 2016; Tomlinson et al., 2016). (F) Log scale difference in elastic modulus of human samples corresponding to each tissue matrix listed above. Solid line represents normal ossification, dotted line accounts for the initial decline in elastic modulus (osteoarthritis model) (Ashman and Jae Young Rho, 1988; Rho et al., 1993; Mente and Lewis, 1994; Silver et al., 2002; Pal, 2014).
Numerous studies have demonstrated that chondrogenic and osteogenic gene expression can be directly modulated by compressive loading and microenvironmental stiffness, as recently reviewed (Park et al., 2011; Lv et al., 2015; Carrion et al., 2016). For example, MSCs subjected to cyclic equibiaxial strain up-reguated expression of markers specific to osteoblast differentiation and mineralization of the ECM (Thomas and el Haj, 1996; Simmons et al., 2003; Liu et al., 2016). Remarkably, when MSCs were subjected to both axial compression and sheer stress, these led to an increase in chondrogenic gene expression and elicited production and accumulation of collagen II and proteoglycan (Schätti et al., 2011; Huang et al., 2012a). Hadden et al. used adipose-derived stem cells (ASCs) cultured on hydrogels with a defined stiffness gradient to demonstrate a stiffness-dependent variation in cellular morphology, migration, and differentiation (Hadden et al., 2017). Furthermore, Engler et al confirmed stem cell fate plasticity by culturing MSCs on matrices with varying tissue-level elasticity. After several weeks of culture, MSCs committed to the lineage dictated by matrix stiffness such that softer, stiffer, and rigid matrices proved to be neurogenic, myogenic, and osteogenic, respectively (Engler et al., 2006). However, findings by Jha et al. suggested that high affinity adhesive ligands can serve as a substitute for a rigid matrix likely by signal transduction following focal adhesion assembly (Jha et al., 2014).
In the midst of an altering microenvironment, hypertrophic chondrocytes begin to predominantly express collagen type X. In contrast to the fibril-forming properties of collagen II, collagen X is a network-forming collagen that creates “basket weave-like” structures (Tampieri and Sprio, 2016). This collagen X ultrastructure is proposed to functionally compartmentalize matrix vesicles containing mineral and newly expressed alkaline phosphatase within the hypertrophic cartilage ECM (Kwan et al., 1997). Interactions between collagen X and matrix vesicles activate the influx of Ca2+ into matrix vesicles thus promoting mineralization and increasing stiffness of the matrix (Shen, 2005).
Tissue architecture, or the manner in which matrix components are structured and organized at the micro- and nanoscale, has been shown to be a factor in naïve cell differentiation. Thus, structural changes could be a driving factor for chondrocyte-to-osteoblast transformation (Healy, 2004). There have been numerous observations of matrix architecture influencing stem cell fate by controlling cell engagement with surrounding matrix and neighboring cells (Guilak et al., 2009; Ahmed and ffrench-Constant, 2016). Moreover, matrix architecture can alter cell surface receptor and cytoskeletal spatial arrangement subsequently altering ligand signaling (Ekerdt et al., 2013). For example, Lu et al. have shown that collagen type II enhances chondrogenesis in ASCs by affecting cell shape and size through the β1 integrin-mediated Rho A/Rock signaling pathway (Lu et al., 2010).
Likewise, research groups have also shown that tissue topography has the ability to guide mesenchymal stem cell fate to either chondrogenic or osteoblastic phenotypes. Shong et al. demonstrated the synergistic effect of microtopography and biochemical supplements to direct MSC fate toward an osteogenic phenotype (Guilak et al., 2009; Song et al., 2015). Additionally, work by Uskoković and Desai suggests that topography may potentially be more of a dominant factor in cell/material surface interaction than the surface chemistry or stiffness (Uskoković and Desai, 2014).
Matrix as a Growth Factor Reservoir
The bioavailability, local concentration, and stabilization of growth factors (GFs) within the ECM of cartilage are primarily modulated via electrostatic interactions between the negatively charged sulfate groups of proteoglycans and the positively charged surfaces of signaling molecules (Tampieri and Sprio, 2016). Moreover, GFs are immobilized by binding to heparan sulfate glycosaminoglycans, for example; Chintala et al. demonstrated that fibroblast growth factor (FGF) has a high affinity to heparan sulfate in the matrix of growth plate cartilage (Chintala et al., 1994). Similarly, Martino et al. identified various GFs from the PDGF, VEGF, TGF-β, and neurotrophin families that possess heparin-binding domains (Martino et al., 2013).
As chondrocytes mature into hypertrophic chondrocytes, they secrete VEGF to stimulate angiogenesis, alkaline phosphatase to induce mineralization, and BMPs to promote osteogenesis (Bahney et al., 2014). These growth factors are retained within the matrix due to the combination of collagen X in compartmentalizing matrix components during endochondral ossification and through interaction with the heparin and/or sulfated proteoglycans (Shen, 2005). Thus, the dynamic promiscuity of the ECM in hypertrophic cartilage likely has a role in cellular signaling affecting physiological functions of endochondral ossification.
For these reasons tissue engineers in recent years have begun to fabricate scaffolds and microparticles that are believed to mimic the release kinetics of GFs found in the cartilage ECM during endochondral ossification. Jeon et al. harnessed the high affinity GFs have to heparin by incorporating heparin into photocrosslinkable alginate gels, recapitulating matrix-growth factor interactions allowing for controlled and sustained release of therapeutic proteins (Jeon et al., 2011). Exploiting the well-documented affinity of proteins to hydroxyapatite (HAp), Dang et al. have fabricated HAp-based microparticles that exhibit sustained delivery of BMP alone as well as controlled dual delivery of BMP with TGF-β to enhance bone tissue engineering via endochondral ossification (Bernardi et al., 1972; Dang et al., 2016a,b). Likewise, glucosamine has also been incorporated into engineered scaffolds because of its effects on chondrocyte proliferation, matrix synthesis, and gene expression via modulation of TGF-β expression levels (Varghese et al., 2007; Murab et al., 2015).
As permeability is typically very low in cartilage, this further accentuates the ECM's role in acting as a reservoir for latent growth factors (Pei et al., 2011). However, in the context of OA, a degenerative joint disease that exhibits endochondral ossification signaling, cartilage ECM degradation alters TGF-β signaling due to the displacement of TGF-β by fluid influx (Blaney Davidson et al., 2007). In native cartilaginous tissue, studies have shown that the loss of latent TGF-β induces chondrocyte hypertrophy and osteogenesis (Wu et al., 2016). Similarly, MSCs seeded onto tissue-engineered cartilage undergo hypertrophic differentiation in the presence of TGF-β, while in the absence of TGF-β MSCs undergo articular cartilage differentiation (Chawla et al., 2017). To that end, we can presume that changes in the properties of the matrix, whether directly or indirectly, have a significant role in the transformation of cartilage to bone during endochondral ossification.
Developmental Engineering to Recapitulate Endochondral Ossification
Bone injuries are extremely common with ~15 million fracture cases and over 2 million bone grafting procedures per year (Yelin et al., 2016). The current clinical gold standard for stimulating bone regeneration is to promote intramembranous bone formation through application of bone grafts, increased biomechanical stability of the fracture with additional orthopedic hardware, or less commonly, through implantation of BMP2-soaked scaffolds (INFUSE®). Given the clinical downsides of each, there is an unmet clinical need for regenerative techniques that could improve vascularized bone regeneration.
While the established clinical approaches to bone regeneration promote intramembranous bone formation, bones both develop and heal through the process of endochondral ossification during which the cartilage callus creates an angiogenic and osteoconductive scaffold for bone formation. Recent pre-clinical studies have capitalized on this, proposing therapeutic strategies that parallel the natural healing process by utilizing engineered hypertrophic cartilage grafts to stimulate bone regeneration (Scotti et al., 2010, 2013; Farrell et al., 2011; Sheehy et al., 2013, 2014; Bahney et al., 2014; Bourgine et al., 2014; Bhattacharjee et al., 2015; Dang et al., 2017). Translating these pre-clinical studies may be one strategy to improve clinical outcomes (Nishitani and Schwarz, 2014).
Further, new mechanistic understanding of endochondral ossification could have a significant impact on the design of novel therapeutic approaches to fracture healing and bone regeneration. Since we now understand chondrocytes can be a direct precursor of osteoblasts (Yang et al., 2014; Zhou et al., 2014; Jing et al., 2015; Park et al., 2015; Hu et al., 2017) stimulating transformation of chondrocytes into osteoblasts becomes a clinically-relevant therapeutic approach. Very little work has been done to understand how chondrocytes become osteoblasts during endochondral ossification. If we understood the extrinsic mediators of chondrocyte to osteoblast transformation, we would not only be able to engineer an ideal treatment for hypertrophic nonunions, but we could also accelerate fracture healing under normal conditions.
Author Contributions
SW, KR, and CB drafted the primary text. CB, TM, and RM financially supported this manuscript. All authors contributed to making the figures, editing the text, and approving the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This publication was supported financially by the National Institutes of Health (NIH) through the National Center for Advancing Translational Sciences Grant Numbers UCSF-UL1TR000004, the National Institute of Dental and Craniofacial Research F30 DE026359 (SW), National Institute of General Medical Sciences R25-GM056847, the AO Foundation Start-Up Grant (CB, #S-14-114B), the UCSF Core Center for Musculoskeletal Biology and Medicine Grant Number P30AR066262, and the Research Evaluation and Allocation Committee (REAC) UCSF School of Medicine. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH or other funding agencies. The authors acknowledge institutional support from the UCSF/ZSFG Orthopedic Trauma Institute, editorial support by Tiffany Shao, and administrative support from Gina Baldoza and Anna Lissa Wi.
References
Abou-Khalil, R., Yang, F., Mortreux, M., Lieu, S., Yu, Y. Y., Wurmser, M., et al. (2014). Delayed bone regeneration is linked to chronic inflammation in murine muscular dystrophy. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 29, 304–315. doi: 10.1002/jbmr.2038
Agrawal, V., and Sinha, M. (2017). A review on carrier systems for bone morphogenetic protein-2. J. Biomed. Mater. Res. B Appl. Biomater. 105, 904–925. doi: 10.1002/jbm.b.33599
Ahmed, M., and ffrench-Constant, C. (2016). Extracellular matrix regulation of stem cell behavior. Curr. Stem Cell Rep. 2, 197–206. doi: 10.1007/s40778-016-0056-2
Akiyama, H., Chaboissier, M. -C., Martin, J. F., Schedl, A., and de Crombrugghe, B. (2002). The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev. 16, 2813–2828. doi: 10.1101/gad.1017802
Akkiraju, H., and Nohe, A. (2015). Role of chondrocytes in cartilage formation, progression of osteoarthritis and cartilage regeneration. J. Dev. Biol. 3, 177–192. doi: 10.3390/jdb3040177
Almubarak, S., Nethercott, H., Freeberg, M., Beaudon, C., Jha, A., Jackson, W., et al. (2016). Tissue engineering strategies for promoting vascularized bone regeneration. Bone 83, 197–209. doi: 10.1016/j.bone.2015.11.011
Ashman, R. B., and Jae Young Rho (1988). Elastic modulus of trabecular bone material. J. Biomech. 21, 177–181. doi: 10.1016/0021-9290(88)90167-4
Bahney, C. S., Hu, D. P., Miclau, T., and Marcucio, R. S. (2015). The multifaceted role of the vasculature in endochondral fracture repair. Front. Endocrinol. (Lausanne). 6:4. doi: 10.3389/fendo.2015.00004
Bahney, C. S., Hu, D. P., Taylor, A. J., Ferro, F., Britz, H. M., Hallgrimsson, B., et al. (2014). Stem cell-derived endochondral cartilage stimulates bone healing by tissue transformation. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 29, 1269–1282. doi: 10.1002/jbmr.2148
Baht, G. S., Silkstone, D., Vi, L., Nadesan, P., Amani, Y., Whetstone, H., et al. (2015). Exposure to a youthful circulation rejuvenates bone repair through modulation of β-catenin. Nat. Commun. 6:7131. doi: 10.1038/ncomms8131
Bell, D. M., Leung, K. K. H., Wheatley, S. C., Ng, L. J., Zhou, S., Ling, K. W., et al. (1997). SOX9 directly regulates the type-ll collagen gene. Nat. Genet. 16, 174–178. doi: 10.1038/ng0697-174
Beresford, W. A. (1981). Chondroid bone, secondary cartilage and metaplasia. Q. Rev. Biol. 56, 471–471. doi: 10.1086/412472
Bernardi, G., Giro, M. -G., and Gaillard, C. (1972). Chromatography of polypeptides and proteins on hydroxyapatite columns: some new developments. Biochim. Biophys. Acta Protein Struct. 278, 409–420. doi: 10.1016/0005-2795(72)90001-3
Bessa, P. C., Casal, M., and Reis, R. L. (2008). Bone morphogenetic proteins in tissue engineering: the road from the laboratory to the clinic, part I (basic concepts). J. Tissue Eng. Regen. Med. 2, 1–13. doi: 10.1002/term.63
Bhattacharjee, M., Coburn, J., Centola, M., Murab, S., Barbero, A., Kaplan, D. L., et al. (2015). Tissue engineering strategies to study cartilage development, degeneration and regeneration. Adv. Drug Deliv. Rev. 84, 107–122. doi: 10.1016/j.addr.2014.08.010
Bi, W., Deng, J. M., Zhang, Z., Behringer, R. R., and de Crombrugghe, B. (1999). Sox9 is required for cartilage formation. Nat. Genet. 22, 85–89. doi: 10.1038/8792
Bissell, M. J., Hall, H. G., and Parry, G. (1982). How does the extracellular matrix direct gene expression? J. Theor. Biol. 99, 31–68. doi: 10.1016/0022-5193(82)90388-5
Blaney Davidson, E. N., van der Kraan, P. M., and van den Berg, W. B. (2007). TGF-beta and osteoarthritis. Osteoarthr. Cartil. 15, 597–604. doi: 10.1016/j.joca.2007.02.005
Bourgine, P. E., Scotti, C., Pigeot, S., Tchang, L. A., Todorov, A., and Martin, I. (2014). Osteoinductivity of engineered cartilaginous templates devitalized by inducible apoptosis. Proc. Natl. Acad. Sci. U.S.A. 111, 17426–17431. doi: 10.1073/pnas.1411975111
Brigman, B. E., Hornicek, F. J., Gebhardt, M. C., and Mankin, H. J. (2004). Allografts about the knee in young patients with high-grade sarcoma. Clin. Orthop. 232–239. doi: 10.1097/01.blo.0000127132.12576.05
Canalis, E. (2013). Wnt signalling in osteoporosis: mechanisms and novel therapeutic approaches. Nat. Rev. Endocrinol. 9, 575–583. doi: 10.1038/nrendo.2013.154
Carrion, B., Souzanchi, M. F., Wang, V. T., Tiruchinapally, G., Shikanov, A., Putnam, A. J., et al. (2016). The synergistic effects of matrix stiffness and composition on the response of chondroprogenitor cells in a 3D precondensation microenvironment. Adv. Healthc. Mater. 5, 1192–1202. doi: 10.1002/adhm.201501017
Chawla, S., Kumar, A., Admane, P., Bandyopadhyay, A., and Ghosh, S. (2017). Elucidating role of silk-gelatin bioink to recapitulate articular cartilage differentiation in 3D bioprinted constructs. Bioprinting 7, 1–13. doi: 10.1016/j.bprint.2017.05.001
Chen, Y., Whetstone, H. C., Lin, A. C., Nadesan, P., Wei, Q., Poon, R., et al. (2007). Beta-catenin signaling plays a disparate role in different phases of fracture repair: implications for therapy to improve bone healing. PLoS Med. 4:e249. doi: 10.1371/journal.pmed.0040249
Chintala, S. K., Miller, R. R., and McDevitt, C. A. (1994). Basic fibroblast growth factor binds to heparan sulfate in the extracellular matrix of rat growth plate chondrocytes. Arch. Biochem. Biophys. 310, 180–186. doi: 10.1006/abbi.1994.1155
Chrastil, J., Low, J. B., Whang, P. G., and Patel, A. A. (2013). Complications associated with the use of the recombinant human bone morphogenetic proteins for posterior interbody fusions of the lumbar spine. Spine 38, E1020–E1027. doi: 10.1097/BRS.0b013e3182982f8e
Colnot, C. (2009). Skeletal cell fate decisions within periosteum and bone marrow during bone regeneration. J. Bone Miner. Res. 24, 274–282. doi: 10.1359/jbmr.081003
Cooper, K. L., Oh, S., Sung, Y., Dasari, R. R., Kirschner, M. W., and Tabin, C. J. (2013). Multiple phases of chondrocyte enlargement underlie differences in skeletal proportions. Nature 495, 375–378. doi: 10.1038/nature11940
Dang, P. N., Dwivedi, N., Phillips, L. M., Yu, X., Herberg, S., Bowerman, C., et al. (2016a). Controlled dual growth factor delivery from microparticles incorporated within human bone marrow-derived mesenchymal stem cell aggregates for enhanced bone tissue engineering via endochondral ossification. Stem Cells Transl. Med. 5, 206–217. doi: 10.5966/sctm.2015-0115
Dang, P. N., Dwivedi, N., Yu, X., Phillips, L., Bowerman, C., Murphy, W. L., et al. (2016b). Guiding chondrogenesis and osteogenesis with mineral-coated hydroxyapatite and BMP-2 incorporated within high-density hMSC aggregates for bone regeneration. ACS Biomater. Sci. Eng. 2, 30–42. doi: 10.1021/acsbiomaterials.5b00277
Dang, P. N., Herberg, S., Varghai, D., Riazi, H., Varghai, D., McMillan, A., et al. (2017). Endochondral ossification in critical-sized bone defects via readily implantable scaffold-free stem cell constructs. Stem Cells Transl. Med. 6, 1644–1659. doi: 10.1002/sctm.16-0222
Day, T. F., Guo, X., Garrett-Beal, L., and Yang, Y. (2005). Wnt/β-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev. Cell 8, 739–750. doi: 10.1016/j.devcel.2005.03.016
Dejica, V. M., Mort, J. S., Laverty, S., Antoniou, J., Zukor, D. J., Tanzer, M., et al. (2012). Increased type II collagen cleavage by cathepsin K and collagenase activities with aging and osteoarthritis in human articular cartilage. Arthritis Res. Ther. 14:R113. doi: 10.1186/ar3839
DeVine, J. G., Dettori, J. R., France, J. C., Brodt, E., and McGuire, R. A. (2012). The use of rhBMP in spine surgery: is there a cancer risk? Evid. Based Spine Care J. 3, 35–41. doi: 10.1055/s-0031-1298616
Dishowitz, M. I., Mutyaba, P. L., Takacs, J. D., Barr, A. M., Engiles, J. B., Ahn, J., et al. (2013). Systemic inhibition of canonical notch signaling results in sustained callus inflammation and alters multiple phases of fracture healing. PLoS ONE 8:e68726. doi: 10.1371/journal.pone.0068726
Dishowitz, M. I., Terkhorn, S. P., Bostic, S. A., and Hankenson, K. D. (2012). Notch signaling components are upregulated during both endochondral and intramembranous bone regeneration. J. Orthop. Res. Off. Publ. Orthop. Res. Soc. 30, 296–303. doi: 10.1002/jor.21518
Duchamp de Lageneste, O., Julien, A., Abou-Khalil, R., Frangi, G., Carvalho, C., Cagnard, N., et al. (2018). Periosteum contains skeletal stem cells with high bone regenerative potential controlled by Periostin. Nat. Commun. 9:773. doi: 10.1038/s41467-018-03124-z
Ehlen, H. W. A., Buelens, L. A., and Vortkamp, A. (2006). Hedgehog signaling in skeletal development. Birth Defects Res. C Embryo Today Rev. 78, 267–279. doi: 10.1002/bdrc.20076
Einhorn, T. A. (2010). The Wnt signaling pathway as a potential target for therapies to enhance bone repair. Sci. Transl. Med. 2:42ps36. doi: 10.1126/scitranslmed.3001149
Ekerdt, B. L., Segalman, R. A., and Schaffer, D. V. (2013). Spatial organization of cell-adhesive ligands for advanced cell culture. Biotechnol. J. 8, 1411–1423. doi: 10.1002/biot.201300302
Engin, F., Yao, Z., Yang, T., Zhou, G., Bertin, T., Jiang, M. M., et al. (2008). Dimorphic effects of Notch signaling in bone homeostasis. Nat. Med. 14, 299–305. doi: 10.1038/nm1712
Engler, A. J., Sen, S., Sweeney, H. L., and Discher, D. E. (2006). Matrix elasticity directs stem cell lineage specification. Cell 126, 677–689. doi: 10.1016/j.cell.2006.06.044
Eyre, D. R., Weis, M. A., and Wu, J. -J. (2006). Articular cartilage collagen: an irreplaceable framework? Eur. Cell. Mater. 12, 57–63. doi: 10.22203/eCM.v012a07
Farrell, E., Both, S. K., Odörfer, K. I., Koevoet, W., Kops, N., O'Brien, F. J., et al. (2011). In-vivo generation of bone via endochondral ossification by in-vitro chondrogenic priming of adult human and rat mesenchymal stem cells. BMC Musculoskelet. Disord. 12:31. doi: 10.1186/1471-2474-12-31
Gammons, M., and Bienz, M. (2018). Multiprotein complexes governing Wnt signal transduction. Curr. Opin. Cell Biol. 51, 42–49. doi: 10.1016/j.ceb.2017.10.008
Gentili, C., and Cancedda, R. (2009). Cartilage and bone extracellular matrix. Curr. Pharm. Des. 15, 1334–1348. doi: 10.2174/138161209787846739
Gerber, H. -P., Vu, T. H., Ryan, A. M., Kowalski, J., Werb, Z., and Ferrara, N. (1999). VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat. Med. 5:623. doi: 10.1038/9467
Grässel, S. (2014). The role of peripheral nerve fibers and their neurotransmitters in cartilage and bone physiology and pathophysiology. Arthritis Res. Ther. 16:485. doi: 10.1186/s13075-014-0485-1
Guilak, F., Cohen, D. M., Estes, B. T., Gimble, J. M., Liedtke, W., and Chen, C. S. (2009). Control of stem cell fate by physical interactions with the extracellular matrix. Cell Stem Cell 5, 17–26. doi: 10.1016/j.stem.2009.06.016
Hadden, W. J., Young, J. L., Holle, A. W., McFetridge, M. L., Kim, D. Y., Wijesinghe, P., et al. (2017). Stem cell migration and mechanotransduction on linear stiffness gradient hydrogels. Proc. Natl. Acad. Sci. U.S.A. 114, 5647–5652. doi: 10.1073/pnas.1618239114
Hall, B. K. (2015). Bones and Cartilage: Developmental and Evolutionary Skeletal Biology. Academic Press. Available online at: https://www.sciencedirect.com/science/article/pii/B9780124166783000495
Haraguchi, R., Kitazawa, R., Imai, Y., and Kitazawa, S. (2018). Growth plate-derived hedgehog-signal-responsive cells provide skeletal tissue components in growing bone. Histochem. Cell Biol. 149, 365–373. doi: 10.1007/s00418-018-1641-5
Healy, K. E. (2004). Control of cell function with tunable hydrogel networks. Conf. Proc. Annu. Int. Conf. IEEE Eng. Med. Biol. Soc. 7:5035. doi: 10.1109/IEMBS.2004.1404391
Hill, T. P., Später, D., Taketo, M. M., Birchmeier, W., and Hartmann, C. (2005). Canonical Wnt/β-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev. Cell 8, 727–738. doi: 10.1016/j.devcel.2005.02.013
Hoffmann, A., and Gross, G. (2001). BMP signaling pathways in cartilage and bone formation. Crit. Rev. Eukaryot. Gene Expr. 11, 23–45. doi: 10.1615/CritRevEukarGeneExpr.v11.i1-3.20
Hosaka, Y., Saito, T., Sugita, S., Hikata, T., Kobayashi, H., Fukai, A., et al. (2013). Notch signaling in chondrocytes modulates endochondral ossification and osteoarthritis development. Proc. Natl. Acad. Sci. U.S.A. 110, 1875–1880. doi: 10.1073/pnas.1207458110
Houben, A., Kostanova-Poliakova, D., Weissenböck, M., Graf, J., Teufel, S., Mark, K., et al. (2016). β-catenin activity in late hypertrophic chondrocytes locally orchestrates osteoblastogenesis and osteoclastogenesis. Development 143, 3826–3838. doi: 10.1242/dev.137489
Hu, D. P., Ferro, F., Yang, F., Taylor, A. J., Chang, W., Miclau, T., et al. (2017). Cartilage to bone transformation during fracture healing is coordinated by the invading vasculature and induction of the core pluripotency genes. Development 144, 221–234. doi: 10.1242/dev.130807
Huang, A. H., Baker, B. M., Ateshian, G. A., and Mauck, R. L. (2012a). Sliding contact loading enhances the tensile properties of mesenchymal stem cell-seeded hydrogels. Eur. Cell. Mater. 24, 29–45. doi: 10.22203/eCM.v024a03
Huang, Y., Zhang, X., Du, K., Yang, F., Shi, Y., Huang, J., et al. (2012b). Inhibition of β-catenin signaling in chondrocytes induces delayed fracture healing in mice. J. Orthop. Res. 30, 304–310. doi: 10.1002/jor.21505
Janda, C. Y., Waghray, D., Levin, A. M., Thomas, C., and Garcia, K. C. (2012). Structural basis of Wnt recognition by frizzled. Science 337, 59–64. doi: 10.1126/science.1222879
Jeon, O., Powell, C., Solorio, L. D., Krebs, M. D., and Alsberg, E. (2011). Affinity-based growth factor delivery using biodegradable, photocrosslinked heparin-alginate hydrogels. J. Control. Release Off. J. Control. Release Soc. 154, 258–266. doi: 10.1016/j.jconrel.2011.06.027
Jha, A. K., Jackson, W. M., and Healy, K. E. (2014). Controlling osteogenic stem cell differentiation via soft bioinspired hydrogels. PLoS ONE 9:e98640. doi: 10.1371/journal.pone.0098640
Jing, Y., Zhou, X., Han, X., Jing, J., von der Mark, K., Wang, J., et al. (2015). Chondrocytes directly transform into bone cells in mandibular condyle growth. J. Dent. Res. 94, 1668–1675. doi: 10.1177/0022034515598135
Karsenty, G., and Wagner, E. F. (2002). Reaching a genetic and molecular understanding of skeletal development. Dev. Cell 2, 389–406. doi: 10.1016/S1534-5807(02)00157-0
Kashiwagi, M., Hojo, H., Kitaura, Y., Maeda, Y., Aini, H., Takato, T., et al. (2016). Local administration of a hedgehog agonist accelerates fracture healing in a mouse model. Biochem. Biophys. Res. Commun. 479, 772–778. doi: 10.1016/j.bbrc.2016.09.134
Katagiri, T., and Watabe, T. (2016). Bone morphogenetic proteins. Cold Spring Harb. Perspect. Biol. 8:a021899. doi: 10.1101/cshperspect.a021899
Kim, J. -B., Leucht, P., Lam, K., Luppen, C., Ten Berge, D., Nusse, R., et al. (2007). Bone regeneration is regulated by Wnt signaling. J. Bone Miner. Res. 22, 1913–1923. doi: 10.1359/jbmr.070802
Krishnan, L., Priddy, L. B., Esancy, C., Klosterhoff, B. S., Stevens, H. Y., Tran, L., et al. (2017). Delivery vehicle effects on bone regeneration and heterotopic ossification induced by high dose BMP-2. Acta Biomater. 49, 101–112. doi: 10.1016/j.actbio.2016.12.012
Kronenberg, H. M. (2003). Developmental regulation of the growth plate. Nature 423, 332–336. doi: 10.1038/nature01657
Kwan, K. M., Pang, M. K. M., Zhou, S., Cowan, S. K., Kong, R. Y. C., Pfordte, T., et al. (1997). Abnormal compartmentalization of cartilage matrix components in mice lacking collagen X: implications for function. J. Cell Biol. 136, 459–471. doi: 10.1083/jcb.136.2.459
Laskin, D. L. (2009). Macrophages and inflammatory mediators in chemical toxicity: a battle of forces. Chem. Res. Toxicol. 22, 1376–1385. doi: 10.1021/tx900086v
Leucht, P., Jiang, J., Cheng, D., Liu, B., Dhamdhere, G., Fang, M. Y., et al. (2013). Wnt3a reestablishes osteogenic capacity to bone grafts from aged animals. J. Bone Joint Surg. Am. 95, 1278–1288. doi: 10.2106/JBJS.L.01502
Leucht, P., Minear, S., Ten Berge, D., Nusse, R., and Helms, J. A. (2008). Translating insights from development into regenerative medicine: the function of Wnts in bone biology. Semin. Cell Dev. Biol. 19, 434–443. doi: 10.1016/j.semcdb.2008.09.002
Lin, G. L., and Hankenson, K. D. (2011). Integration of BMP, Wnt, and notch signaling pathways in osteoblast differentiation. J. Cell. Biochem. 112, 3491–3501. doi: 10.1002/jcb.23287
Liu, Q., Hu, X., Zhang, X., Duan, X., Yang, P., Zhao, F., et al. (2016). Effects of mechanical stress on chondrocyte phenotype and chondrocyte extracellular matrix expression. Sci. Rep. 6:37268. doi: 10.1038/srep37268
Liu, X., McKenzie, J. A., Maschhoff, C. W., Gardner, M. J., and Silva, M. J. (2017). Exogenous hedgehog antagonist delays but does not prevent fracture healing in young mice. Bone 103, 241–251. doi: 10.1016/j.bone.2017.07.017
Long, F., Chung, U., Ohba, S., McMahon, J., Kronenberg, H. M., and McMahon, A. P. (2004). Ihh signaling is directly required for the osteoblast lineage in the endochondral skeleton. Dev. Camb. Engl. 131, 1309–1318. doi: 10.1242/dev.01006
Long, F., and Ornitz, D. M. (2013). Development of the endochondral skeleton. Cold Spring Harb. Perspect. Biol. 5:a008334. doi: 10.1101/cshperspect.a008334
Long, F., Zhang, X. M., Karp, S., Yang, Y., and McMahon, A. P. (2001). Genetic manipulation of hedgehog signaling in the endochondral skeleton reveals a direct role in the regulation of chondrocyte proliferation. Development 128, 5099–5108.
Lu, C., Hansen, E., Sapozhnikova, A., Hu, D., Miclau, T., and Marcucio, R. S. (2008). Effect of age on vascularization during fracture repair. J. Orthop. Res. Off. Publ. Orthop. Res. Soc. 26, 1384–1389. doi: 10.1002/jor.20667
Lu, Z., Doulabi, B. Z., Huang, C., Bank, R. A., and Helder, M. N. (2010). Collagen type II enhances chondrogenesis in adipose tissue-derived stem cells by affecting cell shape. Tissue Eng. A 16, 81–90. doi: 10.1089/ten.tea.2009.0222
Lv, H., Li, L., Sun, M., Zhang, Y., Chen, L., Rong, Y., et al. (2015). Mechanism of regulation of stem cell differentiation by matrix stiffness. Stem Cell Res. Ther. 6:103. doi: 10.1186/s13287-015-0083-4
Maeda, Y., Nakamura, E., Nguyen, M. -T., Suva, L. J., Swain, F. L., Razzaque, M. S., et al. (2007). Indian Hedgehog produced by postnatal chondrocytes is essential for maintaining a growth plate and trabecular bone. Proc. Natl. Acad. Sci. U.S.A. 104, 6382–6387. doi: 10.1073/pnas.0608449104
Maes, C., Coenegrachts, L., Stockmans, I., Daci, E., Luttun, A., Petryk, A., et al. (2006). Placental growth factor mediates mesenchymal cell development, cartilage turnover, and bone remodeling during fracture repair. J. Clin. Invest. 116, 1230–1242. doi: 10.1172/JCI26772
Mak, K. K., Bi, Y., Wan, C., Chuang, P. -T., Clemens, T., Young, M., et al. (2008). Hedgehog signaling in mature osteoblasts regulates bone formation and resorption by controlling PTHrP and RANKL expression. Dev. Cell 14, 674–688. doi: 10.1016/j.devcel.2008.02.003
Maldonado, M., and Nam, J. (2013). The role of changes in extracellular matrix of cartilage in the presence of inflammation on the pathology of osteoarthritis. Biomed. Res. Int. 2013:284873. doi: 10.1155/2013/284873
Martel-Pelletier, J., Boileau, C., Pelletier, J. -P., and Roughley, P. J. (2008). Cartilage in normal and osteoarthritis conditions. Best Pract. Res. Clin. Rheumatol. 22, 351–384. doi: 10.1016/j.berh.2008.02.001
Martino, M. M., Briquez, P. S., Ranga, A., Lutolf, M. P., and Hubbell, J. A. (2013). Heparin-binding domain of fibrin(ogen) binds growth factors and promotes tissue repair when incorporated within a synthetic matrix. Proc. Natl. Acad. Sci. U.S.A. 110, 4563–4568. doi: 10.1073/pnas.1221602110
Matsubara, H., Hogan, D. E., Morgan, E. F., Mortlock, D. P., Einhorn, T. A., and Gerstenfeld, L. C. (2012). Vascular tissues are a primary source of BMP2 expression during bone formation induced by distraction osteogenesis. Bone 51, 168–180. doi: 10.1016/j.bone.2012.02.017
McGee-Lawrence, M. E., Ryan, Z. C., Carpio, L. R., Kakar, S., Westendorf, J. J., and Kumar, R. (2013). Sclerostin deficient mice rapidly heal bone defects by activating β-catenin and increasing intramembranous ossification. Biochem. Biophys. Res. Commun. 441, 886–890. doi: 10.1016/j.bbrc.2013.10.155
Mente, P. L., and Lewis, J. L. (1994). Elastic modulus of calcified cartilage is an order of magnitude less than that of subchondral bone. J. Orthop. Res. Off. Publ. Orthop. Res. Soc. 12, 637–647. doi: 10.1002/jor.1100120506
Morrell, N. T., Leucht, P., Zhao, L., Kim, J. -B., ten Berge, D., Ponnusamy, K., et al. (2008). Liposomal packaging generates wnt protein with in vivo biological activity. PLoS ONE 3:e2930. doi: 10.1371/journal.pone.0002930
Muir, H. (1995). The chondrocyte, architect of cartilage. Biomechanics, structure, function and molecular biology of cartilage matrix macromolecules. BioEssays News Rev. Mol. Cell. Dev. Biol. 17, 1039–1048.
Murab, S., Samal, J., Shrivastava, A., Ray, A. R., Pandit, A., and Ghosh, S. (2015). Glucosamine loaded injectable silk-in-silk integrated system modulate mechanical properties in bovine ex-vivo degenerated intervertebral disc model. Biomaterials 55, 64–83. doi: 10.1016/j.biomaterials.2015.03.032
Nishitani, K., and Schwarz, E. M. (2014). Cartilage transplants hold promise for challenging bone defects. Nat. Rev. Rheumatol. 10, 129–130. doi: 10.1038/nrrheum.2013.216
Nolan, D. J., Ginsberg, M., Israely, E., Palikuqi, B., Poulos, M. G., James, D., et al. (2013). Molecular signatures of tissue-specific microvascular endothelial cell heterogeneity in organ maintenance and regeneration. Dev. Cell 26, 204–219. doi: 10.1016/j.devcel.2013.06.017
Ohba, S., Kawaguchi, H., Kugimiya, F., Ogasawara, T., Kawamura, N., Saito, T., et al. (2008). Patched1 haploinsufficiency increases adult bone mass and modulates Gli3 repressor activity. Dev. Cell 14, 689–699. doi: 10.1016/j.devcel.2008.03.007
Ortega, N., Behonick, D. J., and Werb, Z. (2004). Matrix remodeling during endochondral ossification. Trends Cell Biol. 14, 86–93. doi: 10.1016/j.tcb.2003.12.003
Pal, S. (2014). Design of Artificial Human Joints & Organs. New York, NY; Heidelberg; Dordrecht; London: Springer US.
Park, J., Gebhardt, M., Golovchenko, S., Perez-Branguli, F., Hattori, T., Hartmann, C., et al. (2015). Dual pathways to endochondral osteoblasts: a novel chondrocyte-derived osteoprogenitor cell identified in hypertrophic cartilage. Biol. Open 4, 608–621. doi: 10.1242/bio.201411031
Park, J. S., Chu, J. S., Tsou, A. D., Diop, R., Tang, Z., Wang, A., et al. (2011). The effect of matrix stiffness on the differentiation of mesenchymal stem cells in response to TGF-β. Biomaterials 32, 3921–3930. doi: 10.1016/j.biomaterials.2011.02.019
Park, S. -H., Silva, M., Bahk, W. -J., McKellop, H., and Lieberman, J. R. (2002). Effect of repeated irrigation and debridement on fracture healing in an animal model. J. Orthop. Res. 20, 1197–1204. doi: 10.1016/S0736-0266(02)00072-4
Pei, M., Li, J. T., Shoukry, M., and Zhang, Y. (2011). A review of decellularized stem cell matrix: a novel cell expansion system for cartilage tissue engineering. Eur. Cell. Mater. 22, 333–343; discussion 343. doi: 10.22203/eCM.v022a25
Rafii, S., Butler, J. M., and Ding, B. -S. (2016). Angiocrine functions of organ-specific endothelial cells. Nature 529, 316–325. doi: 10.1038/nature17040
Regard, J. B., Zhong, Z., Williams, B. O., and Yang, Y. (2012). Wnt signaling in bone development and disease: making stronger bone with Wnts. Cold Spring Harb. Perspect. Biol. 4:a007997. doi: 10.1101/cshperspect.a007997
Rho, J. Y., Ashman, R. B., and Turner, C. H. (1993). Young's modulus of trabecular and cortical bone material: ultrasonic and microtensile measurements. J. Biomech. 26, 111–119.
Roughley, P. J., and Mort, J. S. (2014). The role of aggrecan in normal and osteoarthritic cartilage. J. Exp. Orthop. 1:8. doi: 10.1186/s40634-014-0008-7
Salazar, V. S., Gamer, L. W., and Rosen, V. (2016). BMP signalling in skeletal development, disease and repair. Nat. Rev. Endocrinol. 12, 203–221. doi: 10.1038/nrendo.2016.12
Sarahrudi, K., Thomas, A., Albrecht, C., and Aharinejad, S. (2012). Strongly enhanced levels of sclerostin during human fracture healing. J. Orthop. Res. 30, 1549–1555. doi: 10.1002/jor.22129
Schätti, O., Grad, S., Goldhahn, J., Salzmann, G., Li, Z., Alini, M., et al. (2011). A combination of shear and dynamic compression leads to mechanically induced chondrogenesis of human mesenchymal stem cells. Eur. Cell. Mater. 22, 214–225. doi: 10.22203/eCM.v022a17
Scotti, C., Piccinini, E., Takizawa, H., Todorov, A., Bourgine, P., Papadimitropoulos, A., et al. (2013). Engineering of a functional bone organ through endochondral ossification. Proc. Natl. Acad. Sci. U.S.A. 110, 3997–4002. doi: 10.1073/pnas.1220108110
Scotti, C., Tonnarelli, B., Papadimitropoulos, A., Scherberich, A., Schaeren, S., Schauerte, A., et al. (2010). Recapitulation of endochondral bone formation using human adult mesenchymal stem cells as a paradigm for developmental engineering. Proc. Natl. Acad. Sci. U.S.A. 107, 7251–7256. doi: 10.1073/pnas.1000302107
Sekiya, I., Tsuji, K., Koopman, P., Watanabe, H., Yamada, Y., Shinomiya, K., et al. (2000). SOX9 enhances aggrecan gene promoter/enhancer activity and is up-regulated by retinoic acid in a cartilage-derived cell line, TC6. J. Biol. Chem. 275, 10738–10744. doi: 10.1074/jbc.275.15.10738
Sheehy, E. J., Vinardell, T., Buckley, C. T., and Kelly, D. J. (2013). Engineering osteochondral constructs through spatial regulation of endochondral ossification. Acta Biomater. 9, 5484–5492. doi: 10.1016/j.actbio.2012.11.008
Sheehy, E. J., Vinardell, T., Toner, M. E., Buckley, C. T., and Kelly, D. J. (2014). Altering the architecture of tissue engineered hypertrophic cartilaginous grafts facilitates vascularisation and accelerates mineralisation. PLoS ONE 9:e90716. doi: 10.1371/journal.pone.0090716
Shen, G. (2005). The role of type X collagen in facilitating and regulating endochondral ossification of articular cartilage. Orthod. Craniofac. Res. 8, 11–17. doi: 10.1111/j.1601-6343.2004.00308.x
Shi, Y., He, G., Lee, W. -C., McKenzie, J. A., Silva, M. J., and Long, F. (2017). Gli1 identifies osteogenic progenitors for bone formation and fracture repair. Nat. Commun. 8:2043. doi: 10.1038/s41467-017-02171-2
Silkstone, D., Hong, H., and Alman, B. A. (2008). Beta-catenin in the race to fracture repair: in it to Wnt. Nat. Clin. Pract. Rheumatol. 4, 413–419. doi: 10.1038/ncprheum0838
Silver, F. H., Bradica, G., and Tria, A. (2002). Elastic energy storage in human articular cartilage: estimation of the elastic modulus for type II collagen and changes associated with osteoarthritis. Matrix Biol. J. Int. Soc. Matrix Biol. 21, 129–137. doi: 10.1016/S0945-053X(01)00195-0
Simmons, C. A., Alsberg, E., Hsiong, S., Kim, W. J., and Mooney, D. J. (2004). Dual growth factor delivery and controlled scaffold degradation enhance in vivo bone formation by transplanted bone marrow stromal cells. Bone 35, 562–569. doi: 10.1016/j.bone.2004.02.027
Simmons, C. A., Matlis, S., Thornton, A. J., Chen, S., Wang, C. Y., and Mooney, D. J. (2003). Cyclic strain enhances matrix mineralization by adult human mesenchymal stem cells via the extracellular signal-regulated kinase (ERK1/2) signaling pathway. J. Biomech. 36, 1087–1096. doi: 10.1016/S0021-9290(03)00110-6
Song, S., Kim, E. J., Bahney, C. S., Miclau, T., Marcucio, R., and Roy, S. (2015). The synergistic effect of micro-topography and biochemical culture environment to promote angiogenesis and osteogenic differentiation of human mesenchymal stem cells. Acta Biomater. 18, 100–111. doi: 10.1016/j.actbio.2015.02.021
St-Jacques, B., Hammerschmidt, M., and McMahon, A. P. (1999). Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 13, 2072–2086. doi: 10.1101/gad.13.16.2072
Sukul, M., Nguyen, T. B. L., Min, Y. -K., Lee, S. -Y., and Lee, B. -T. (2015). Effect of local sustainable release of BMP2-VEGF from nano-cellulose loaded in sponge biphasic calcium phosphate on bone regeneration. Tissue Eng. A 21, 1822–1836. doi: 10.1089/ten.tea.2014.0497
Takada, R., Satomi, Y., Kurata, T., Ueno, N., Norioka, S., Kondoh, H., et al. (2006). Monounsaturated fatty acid modification of Wnt protein: its role in Wnt secretion. Dev. Cell 11, 791–801. doi: 10.1016/j.devcel.2006.10.003
Tampieri, A., and Sprio, S. (2016). Bio-Inspired Regenerative Medicine: Materials, Processes, and Clinical Applications. Boca Raton, FL: CRC Press.
Tanck, E., van Driel, W. D., Hagen, J. W., Burger, E. H., Blankevoort, L., and Huiskes, R. (1999). Why does intermittent hydrostatic pressure enhance the mineralization process in fetal cartilage? J. Biomech. 32, 153–161. doi: 10.1016/S0021-9290(98)00165-1
Tatsuyama, K., Maezawa, Y., Baba, H., Imamura, Y., and Fukuda, M. (2000). Expression of various growth factors for cell proliferation and cytodifferentiation during fracture repair of bone. Eur. J. Histochem. 44, 269–278.
Thomas, G. P., and el Haj, A. J. (1996). Bone marrow stromal cells are load responsive in vitro. Calcif. Tissue Int. 58, 101–108. doi: 10.1007/BF02529731
Thompson, Z., Miclau, T., Hu, D., and Helms, J. A. (2002). A model for intramembranous ossification during fracture healing. J. Orthop. Res. 20, 1091–1098. doi: 10.1016/S0736-0266(02)00017-7
Tomlinson, R. E., Li, Z., Zhang, Q., Goh, B. C., Li, Z., Thorek, D. L. J., et al. (2016). NGF-TrkA signaling by sensory nerves coordinates the vascularization and ossification of developing endochondral bone. Cell Rep. 16, 2723–2735. doi: 10.1016/j.celrep.2016.08.002
Topol, L., Chen, W., Song, H., Day, T. F., and Yang, Y. (2009). Sox9 inhibits Wnt signaling by promoting beta-catenin phosphorylation in the nucleus. J. Biol. Chem. 284, 3323–3333. doi: 10.1074/jbc.M808048200
Uskoković, V., and Desai, T. A. (2014). Does translational symmetry matter on the micro scale? Fibroblastic and osteoblastic interactions with the topographically distinct poly(ε-caprolactone)/hydroxyapatite thin films. ACS Appl. Mater. Interfaces 6, 13209–13220. doi: 10.1021/am503043t
Varghese, S., Theprungsirikul, P., Sahani, S., Hwang, N., Yarema, K. J., and Elisseeff, J. H. (2007). Glucosamine modulates chondrocyte proliferation, matrix synthesis, and gene expression. Osteoarthr. Cartil. 15, 59–68. doi: 10.1016/j.joca.2006.06.008
Wang, X., Yu, Y. Y., Lieu, S., Yang, F., Lang, J., Lu, C., et al. (2013). MMP9 regulates the cellular response to inflammation after skeletal injury. Bone 52, 111–119. doi: 10.1016/j.bone.2012.09.018
Willert, K., Brown, J. D., Danenberg, E., Duncan, A. W., Weissman, I. L., Reya, T., et al. (2003). Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature 423, 448–452. doi: 10.1038/nature01611
Wray, J. B. (1964). Acute changes in femoral arterial blood flow after closed tibial fracture in dogs. J. Bone Joint Surg. Am. 46:1262. doi: 10.2106/00004623-196446060-00012
Wu, M., Chen, G., and Li, Y. -P. (2016). TGF-β and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone Res. 4:16009. doi: 10.1038/boneres.2016.9
Xing, Z., Lu, C., Hu, D., Miclau, T., and Marcucio, R. S. (2010a). Rejuvenation of the inflammatory system stimulates fracture repair in aged mice. J. Orthop. Res. 28, 1000–1006. doi: 10.1002/jor.21087
Xing, Z., Lu, C., Hu, D., Yu, Y., Wang, X., Colnot, C., et al. (2010b). Multiple roles for CCR2 during fracture healing. Dis. Model. Mech. 3, 451–458. doi: 10.1242/dmm.003186
Yang, L., Tsang, K. Y., Tang, H. C., Chan, D., and Cheah, K. S. E. (2014). Hypertrophic chondrocytes can become osteoblasts and osteocytes in endochondral bone formation. Proc. Natl. Acad. Sci. U.S.A. 111, 12097–12102. doi: 10.1073/pnas.1302703111
Yelin, E., Weinstein, S., and King, T. (2016). The burden of musculoskeletal diseases in the United States. Semin. Arthritis Rheum. 46, 259–260. doi: 10.1016/j.semarthrit.2016.07.013
Yu, Y. Y., Lieu, S., Lu, C., Miclau, T., Marcucio, R. S., and Colnot, C. (2010). Immunolocalization of BMPs, BMP antagonists, receptors, and effectors during fracture repair. Bone 46, 841–851. doi: 10.1016/j.bone.2009.11.005
Zelzer, E., McLean, W., Ng, Y. -S., Fukai, N., Reginato, A. M., Lovejoy, S., et al. (2002). Skeletal defects in VEGF(120/120) mice reveal multiple roles for VEGF in skeletogenesis. Development 129, 1893–1904.
Zhou, X., von der Mark, K., Henry, S., Norton, W., Adams, H., and de Crombrugghe, B. (2014). Chondrocytes transdifferentiate into osteoblasts in endochondral bone during development, postnatal growth and fracture healing in mice. PLoS Genet. 10:e1004820. doi: 10.1371/journal.pgen.1004820
Keywords: fracture, endochondral ossification, chondrocyte fate, developmental engineering, transdifferentiation
Citation: Wong SA, Rivera KO, Miclau T III, Alsberg E, Marcucio RS and Bahney CS (2018) Microenvironmental Regulation of Chondrocyte Plasticity in Endochondral Repair—A New Frontier for Developmental Engineering. Front. Bioeng. Biotechnol. 6:58. doi: 10.3389/fbioe.2018.00058
Received: 01 February 2018; Accepted: 23 April 2018;
Published: 15 May 2018.
Edited by:
Eric Farrell, Erasmus University Rotterdam, NetherlandsReviewed by:
Natalina Quarto, University of Naples Federico II, ItalySourabh Ghosh, Indian Institute of Technology Delhi, India
Copyright © 2018 Wong, Rivera, Miclau, Alsberg, Marcucio and Bahney. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chelsea S. Bahney, Q2hlbHNlYS5CYWhuZXlAdWNzZi5lZHU=