 Qingqiu Yang
Qingqiu Yang Vanessa Marigo Rocha PintoWei DuanErica E. PaxtonJenna H. DessauerWilliam Ryan
Vanessa Marigo Rocha PintoWei DuanErica E. PaxtonJenna H. DessauerWilliam Ryan Mandi J. Lopez*
Mandi J. Lopez*- Laboratory for Equine and Comparative Orthopedic Research, Department of Veterinary Clinical Sciences, School of Veterinary Medicine, Louisiana State University, Baton Rouge, LA, United States
Damage to an ectodermal-mesodermal interface like that in the equine hoof and human finger nail bed can permanently alter tissue structure and associated function. The purpose of this study was to establish and validate in vitro culture of primary progenitor cell isolates from the ectodermal-mesodermal tissue junction in equine hooves, the stratum internum, with and without chronic inflammation known to contribute to lifelong tissue defects. The following were evaluated in hoof stratum internum cell isolates up to 5 cell passages (P): expansion capacity by cell doublings and doubling time; plasticity with multi-lineage differentiation and colony-forming unit (CFU) frequency percentage; immunophenotype with immunocytochemistry and flow cytometry; gene expression with RT-PCR; and ultrastructure with transmission electron microscopy. The presence of keratin (K)14, 15 and K19 as well as cluster of differentiation (CD)44 and CD29 was determined in situ with immunohistochemistry. To confirm in vivo extracellular matrix (ECM) formation, cell-scaffold (polyethylene glycol/poly-L-lactic acid and tricalcium phosphate/hydroxyapatite) constructs were evaluated with scanning electron microscopy 9 weeks after implantation in athymic mice. Cultured cells had characteristic progenitor cell morphology, expansion, CFU frequency percentage and adipocytic, osteoblastic, and neurocytic differentiation capacity. CD44, CD29, K14, K15 and K19 proteins were present in native hoof stratum internum. Cultured cells also expressed K15, K19 and desmogleins 1 and 3. Gene expression of CD105, CD44, K14, K15, sex determining region Y-box 2 (SOX2) and octamer-binding transcription factor 4 (OCT4) was confirmed in vitro. Cultured cells had large, eccentric nuclei, elongated mitochondria, and intracellular vacuoles. Scaffold implants with cells contained fibrous ECM 9 weeks after implantation compared to little or none on acellular scaffolds. In vitro expansion and plasticity and in vivo ECM deposition of heterogeneous, immature cell isolates from the ectodermal-mesodermal tissue interface of normal and chronically inflamed hooves are typical of primary cell isolates from other adult tissues, and they appear to have both mesodermal and ectodermal qualities in vitro. These results establish a unique cell culture model to target preventative and restorative therapies for ectodermal-mesodermal tissue junctions.
Introduction
The equine hoof is a complex anatomical structure that changed in size and shape as horses evolved from a multiple to a single toe ungulate (Pollitt, 1992; O'grady, 2002; Van Eps and Pollitt, 2009). Within a keratinized hoof wall, the last phalanx of the third digit is suspended by the stratum internum (lamellatum) (Pollitt, 1998), an attachment that can be permanently and fatally disrupted by inflammation within the hoof wall known as laminitis (Johnson et al., 1998; Pollitt and Daradka, 1998; Morgan et al., 1999; Longland and Byrd, 2006; Van Eps and Pollitt, 2009). Following damage, complex hoof architecture is replaced with abundant, poorly organized tissue (Hunt and Wharton, 2010; Barreto-Vianna et al., 2013), and current treatments to restore normal tissue after disruption are largely unsuccessful (Atala et al., 2010).
Tissue formation in the stratum medium, the cornified tissue layer directly above the stratum internum, results from basal cell proliferation in the coronary band (Daradka and Pollitt, 2004) located at the proximal aspect of the structure. The cells undergo maturation into partially, keratinocyte, or fully, corneocyte, keratinized cells as they move downward from the basal layer. Cells on the surface of the redundant, frond-like lamellae that constitute the ectodermal-mesodermal tissues in the stratum internum are connected to each other by tight cell junctions (French and Pollitt, 2004; Visser and Pollitt, 2011). As the ectodermal component of the tissue connected to the outer hoof wall grows downward relative to the mesodermal component fixed to the boney third phalanx via protein fibrils, cells are thought to detach and reattach via controlled enzymatic activity, largely within the basement membrane between them (Pollitt, 1994; Daradka and Pollitt, 2004). While cell growth and maturation is well-described for the cornified hoof tissue, the role of progenitor cells within the stratum internum to production and maintenance of the deeper hoof tissues, especially those at the ectodermal-mesodermal interface, are not well-elucidated (Anderson and Mcilwraith, 2004; Visser and Pollitt, 2010).
Isolation and characterization of progenitor cells from the ectodermal-mesodermal tissue nich of normal and inflamed, laminitic, hooves is necessary to advance understanding of cell behavior and reparative capacity under normal and inflamed conditions (Lian and Stein, 1992; Kisiday et al., 2002). Proliferating cells were identified in normal hoof lamellae with 5-bromo-2'-deoxyuridine (BRdU) (Daradka and Pollitt, 2004). Additionally, epithelial progenitor cells in the lamellae were localized by K14 and transcription factor p63 expression (Senoo et al., 2007; Bragulla and Homberger, 2009; Broeckx et al., 2014; Linardi et al., 2015). This information confirms that progenitor cells exist within the normal stratum internum. Altered progenitor cell behavior from disease and chronic inflammation disrupts tissue formation and healing in ectodermal-dermal tissue interfaces like the periodontal membrane and finger nail bed (Page and Schroeder, 1976; Ding et al., 2010; Bharathi and Bajantri, 2011). A mechanism to confidently isolate and culture hoof progenitor cells from the ectodermal-mesodermal tissue niche of normal and chronically inflamed hooves will provide a useful tool to evaluate mechanisms of cell damage and target therapies to prevent cell loss and restore normal growth following injury, local disease or systemic pathology. The purpose of this study was to establish and validate in vitro culture of progenitor cells from the stratum internum of equine hooves with and without chronic inflammation.
Materials and Methods
Study Design
Forelimbs from 22 horses belonging to the University research herd, 14 unaffected (U), and 8 with laminitis (L), were disarticulated at the metacarpophalangeal joint following humane euthanasia for reasons unrelated to this study. Cells were isolated from the stratum internum and progenitor cells selected by plastic affinity. Outcome measures included cell expansion rate for cell passages (P) 1-3 (n = 5 U; n = 6 L), P1 trilineage differentiation (n = 3 U; n = 3 L), P0, 2 and 5 colony forming unit frequency (CFU, n = 4 U; n = 3 L) and cell surface marker expression (n = 8 U; n = 6 L), hoof tissue immunohistochemistry (IHC) (n = 2 U; n = 1 L), immunocytochemistry (ICC) of P1 and 3 (n = 2 U; n = 2 L), P0, 2 and 5 gene expression of CD44, CD105, K14, K15, octamer-binding transcription factor 4 (OCT4), and sex determining region Y-box 2 (SOX2) (n = 4 U; n = 5 L) and transmission electron microscopy (TEM) of P1 cell ultrastructure (n = 2 U; n = 2 L). Scanning electron microscopy (SEM, n = 1 U) was used to assess extracellular matrix (ECM) deposition on polyethylene glycol/poly-L-lactic acid (GA) and tricalcium phosphate/hydroxyapatite (HT) scaffolds loaded with P3 cells 9 weeks after subcutaneous implantation in athymic mice (Table 1).
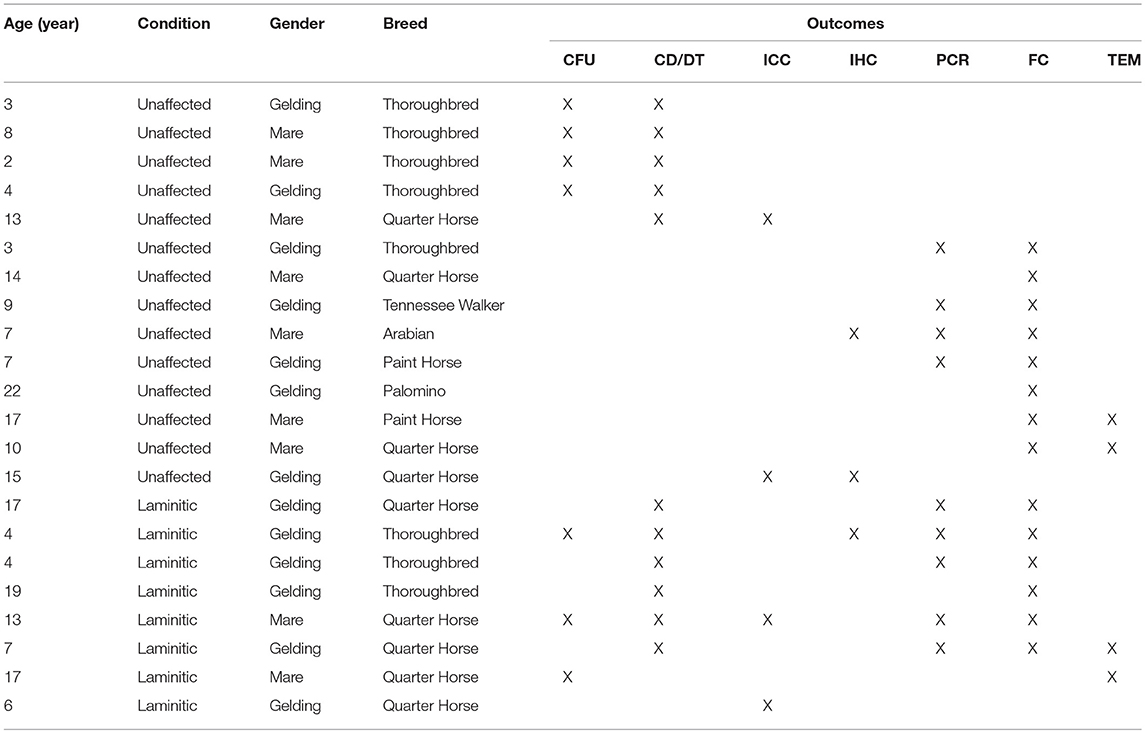
Table 1. Study samples and assays.
Radiographs
Hooves were cleaned with soap and water and any shoes removed. Lateral digital radiographs were performed immediately after limb collection with a portable radiography unit (70 kVp, 4l mAs, MiKasa X-ray Co, Japan) following placement of a radiopaque wire (length: 76.2 cm; diameter: 0.05 cm) on the dorsal midline of each hoof. A scale bar was included in radiographs to calibrate software tools used to make measures on digital images (Adobe Photoshop, version 5.5, Adobe Systems Inc., Seattle, WA). Dorsal rotation was quantified by subtracting the distance between the distal inner hoof wall and the third phalanx (P3) from the distance between the proximal hoof wall and P3 (Figure 1) (Kummer et al., 2004). Sinking was the distance between the proximal portion of the coronary band and the extensor process of P3.
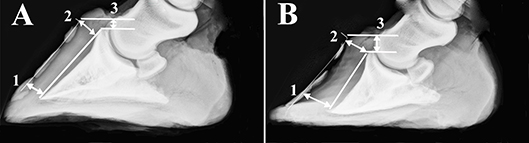
Figure 1. Radiographs of a representative unaffected (A) and laminitic hoof (B) demonstrating where distances were measured to determine capsular rotation (1-2) and distal descent (3). Specifically, dorsal rotation was quantified by subtracting the distance between the distal inner hoof wall and the third phalanx (1) from the distance between the proximal hoof wall and the third phalanx (2). Sinking was the distance between the proximal portion of the coronary band and the extensor process of the third phalanx (3).
Tissue Harvest
Within 2 h of harvest, hooves were cleansed with alternating washes of 10% sodium hypochlorite, sterile water and 0.1% chlorhexidine. With each lateral hoof surface gripped in a vice, two parallel cuts through hoof wall to bone were made 2 cm apart on the dorsal surface from coronary band to toe using a reciprocating saw and sterile blades (DC385, Dewalt®, Towson, MD). The cornified stratum externum and stratum medium between the cuts was elevated with a hoof nipper to expose the stratum internum. A square of approximately 2 × 2 cm was extracted from the midpoint of the dorsal surface with a #20 scalpel blade. The tissue was rinsed with 0.01% chlorhexidine then soaked in phosphate buffered saline (PBS, Hyclone, Logan, UT) containing 1% antibiotic solution (MP Biomedical, Irvine, CA) for 15–30 min at room temperature.
Cell Isolation and Culture
In a sterile petri dish (Nunc™ IVF petri dish, Thermo Fisher Scientific, Inc., Waltham, MA), tissue was minced into 1 × 1 mm squares using a #10 scalpel blade and then transferred to 50 ml sterile tubes containing collagenase digest composed of 1% bovine serum albumin (BSA) (Sigma-Aldrich, St. Louis, MO) and 0.1% type-1 collagenase (Worthington Biochemical Corporation, Lakewood, NJ) in Dulbecco's modified Eagle's medium with Ham's F12 nutrient mixture (DMEM-Ham's F12, Hyclone). Samples in digest at a 1:2 tissue to digest (v/v) ratio were incubated at 37°C for 2 h with 3-dimensional agitation (5.5 × g, GyroTwister™ GX-1000, LabNet, Inc., Edison, NJ). Subsequently, the digest was passed through a 100 μm followed by a 70 μm filter (Thermo Fisher Scientific) and then centrifuged at 260 × g for 5 min. The stromal vascular fraction (SVF) was resuspended in stromal medium [DMEM-Ham's F12, 1% antibiotic solution (MP Biomedical, Irvine, CA), 10% characterized fetal bovine serum (VWR Life Science, Radnor, PA)] and cultured on 10 cm culture plates (Corning Inc., Corning, NY). Medium was refreshed after 24 h and then every 3 days until 80% cell confluence. For study purposes, seeding density (5 × 103 cells/cm2), culture conditions (37°C, 5% CO2), and plate confluence (80%) for cell detachment (0.05% trypsin, Hyclone) and passage were conserved throughout unless otherwise indicated.
Colony Forming Unit Frequency (P0, 2, 5)
Limiting dilution assays were used to determine progenitor cell CFU frequency percentage by established methods (Zhang et al., 2014). Cells were added to a 48-well plate at 5 × 103, 2.5 × 103, 1.25 × 103, 6.25 × 102, 3.12 × 102, and 1.56 × 102 cells/well in individual columns (8 replicates per cell number). After 7 days of culture in stromal medium, wells were rinsed with PBS and fixed with 1% paraformaldehyde (Sigma-Aldrich) in PBS at room temperature for 20 min followed by staining with 0.1% toluidine blue (Sigma-Aldrich) for 1 h and a distilled water rinse. Digital images were obtained with an inverted phase contrast microscope (Olympus® CKX41SF, Japan) instrumented with a digital camera (Olympus DP21, Japan). Positive wells contained 10 or more stained colonies. The CFU frequency was calculated as F = e−x, where F = the ratio of negative to total wells, e = natural logarithm constant 2.71 and x = CFU number per well. The CFU frequency was expressed as a percentage (1/CFU frequency×100) (Zhang et al., 2013; Duan and Lopez, 2016).
Cell Doublings and Doubling Time (P1-3)
Cell expansion was quantified in 12 well plates (Nunclon®, Sigma-Aldrich) as previously described (Zhang et al., 2013). Cell numbers were evaluated after 2, 4, and 6 days of culture using trypan blue exclusion and a hemocytometer. Cell doublings (CD) and doubling time (DT) were calculated as CD = ln(Nf /Ni)/ln(2) and DT = CT/CD where CT = culture time, Nf = final cell number, and Ni = initial cell number.
Multi-Lineage Differentiation (P1)
Adipogenic, osteogenic and neurogenic differentiation was confirmed in cell isolates from U and L hooves cultured in 6 well plates (Thermo Fisher Scientific). Cells were cultured in stromal medium for 5 days to about 80% confluence followed by culture in induction medium. For adipogenesis, cells were cultured in adipogenic induction medium [DMEM-Ham's F12, 3% FBS, 1% antibiotic solution, biotin (33 mmol/L), pantothenate (17 mmol/L), insulin (1 mmol/L), dexamethasone (1 mmol/L), isobutylmethylxanthine (IBMX, 0.5 mmol/L), rosiglitazone (5 mmol/L) (TZD, AK Scientific, Union City, CA), 5% rabbit serum (Invitrogen Corporation, Carlsbad, CA)] for 3 days followed by adipogenic maintenance medium (adipogenic medium without IBMX or rosiglitazone) for 2 more days. To confirm lipid accumulation, cells were fixed for 2 h in 4% paraformaldehyde at room temperature and then stained with oil red O. To assess osteogenesis, cells were cultured in osteogenic induction medium [DMEM-Ham's F12, 10% FBS, 1% antibiotic solution, β-glycerophosphate (1 mol/L), dexamethasone (20 nmol/L), sodium 2-phosphate ascorbate (50 mg/mL)] for 14 days and then fixed with 70% cold ethanol for 2 h. Calcium in colonies was stained with 2% alizarin red in distilled water (pH 4.2) for 15 min at room temperature and then rinsed with distilled water (Vidal et al., 2007). Cells were also cultured in neurogenic pre-induction medium (DMEM, 10% FBS, 1 mM 2-mercaptoethanol) for 2 days followed by neurogenic induction medium (DMEM, 5.5 mM glucose, 120 μM indomethacin, 10% FBS, 3 μg/ml insulin, 300 μM IBMX) for 3 days. They were fixed with 1% paraformaldehyde overnight and then blocked with 10% goat serum for 1 h at 37°C. Cells were incubated with a mouse anti-microtubule-associated protein (MAP2) primary antibody (1:250) for 2 h at 37°C. They were then incubated with anti-mouse immunoglobulin G (IgG) secondary antibody labeled with Alexa Fluor 488 (1:1,000) (Table 2) for 1 h at room temperature. Controls included cells cultured in stromal medium and induced cells incubated with secondary antibody alone. Nuclei were stained with 4, 6-diamidino-2-phenylindole (DAPI, 1 μg/ml) for 10 min followed by a distilled water rinse. Photomicrographs were obtained with a camera (DFC480, Leica Microsystems, Germany) on a fluorescent microscope (DM 4500b, Leica Microsystems).
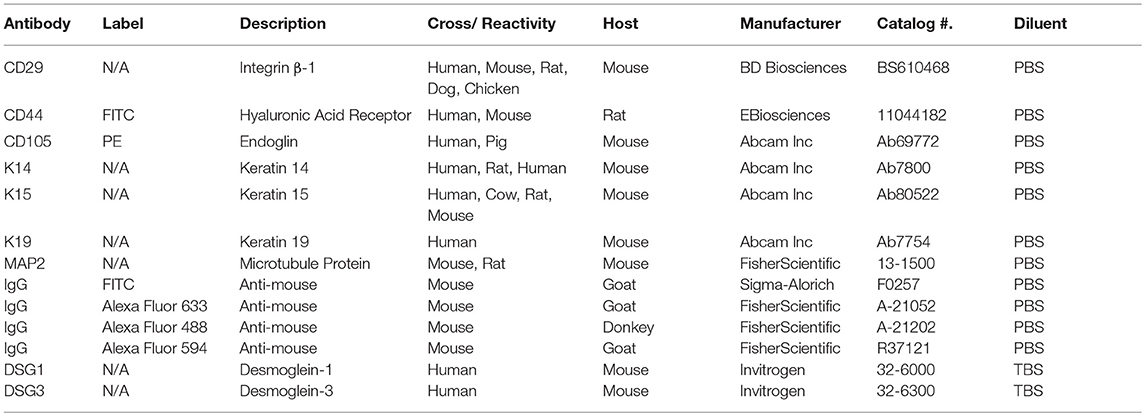
Table 2. Antibody information.
Immunohistochemistry (IHC), Immunocytochemistry (ICC) (P1, 3)
IHC (fluorescent)-Fresh tissue was embedded in optimal cutting temperature compound (OCT, Sakura Finetek Inc., Torrance, CA), solidified at −80°C, sectioned (5 μm) with a cryostat (Leica® CM1850, Sarasota, FL), and applied to slides (poly L-lysine coated, Sigma-Aldrich). Sections were blocked with 10% goat serum (Abcam Inc., Cambridge, MA) in PBS for 1 h at room temperature after rehydration in PBS for 10 min. Slides were incubated with individual primary antibodies (CD29, CD44, K14, K19) (Table 2) diluted in tris-buffered saline (TBS, 1:200) at 37°C for 2 h, rinsed with PBS, incubated with anti-mouse IgG-Alexa Fluor 594 at 37°C for 1 h in darkness, and then rinsed with PBS again. Nuclei were stained with Hoechst's dye (Biotium, Hayward, CA), for 10 min at room temperature in darkness. Digital images were obtained with a fluorescent microscope (DM 4500b, Leica) equipped with a digital camera (DFC 480, Leica). Negative controls for unlabeled antibodies included sections incubated with secondary antibody alone. Despite the fact that CD44 had a conjugated FITC label, sections labeled with CD44 were incubated with the same secondary antibody as unconjugated antibodies for consistency. The label does not interfere with the reaction between the primary and secondary antibodies.
IHC (chromogen)-Formalin fixed sections of laminae (1 × 0.5 × 0.5 cm) were paraffin embedded and sectioned (5 μm). Antigen retrieval was performed by incubating in citrate buffer (pH 6) for 30 min at 80°C. Sections were rinsed in PBS and endogenous peroxidase was blocked by incubation in 3% H2O2 for 30 min at room temperature. Non-specific binding of antibodies was blocked by incubation with 1% BSA (Sigma-Aldrich) and 1% pre-immune serum (Abcam) in PBS for 1 h at 37°C. Sections were then immunostained with murine anti-human antibodies against K14 or K15 (Table 2) overnight at room temperature. After rinsing in PBS, sections were incubated with horse radish peroxidase (HRP) conjugated anti-mouse IgG (DAKO EnVision, Dako, Carpinteria, CA) for 1 h at room temperature. Bound antibodies were then exposed with diaminobenzidine/H2O2 staining for 3 min at room temperature. Slides were counterstained with hematoxylin and digital images captured with a light microscope (DM5000B, Leica) instrumented with a camera (DFC 480, Leica). Sections treated with secondary antibody alone were included as negative controls.
ICC
Cells were seeded on eight well multi-chamber slides (7 × 103 cells/well, Thermo Fisher Scientific). At approximately 70% confluence, cells were fixed in 4% paraformaldehyde. They were then permeabilized with 1.0% Triton X-100 (Sigma-Aldrich) in PBS for 10 min at room temperature before blocking with 10% goat serum (Abcam) and 2% BSA (Sigma-Aldrich) in 1.0% Triton X-100. Cells were incubated with primary antibodies against desmoglein (DSG) 1, DSG3, K15, and K19 (Table 2) diluted 1:200 in the blocking solution above for 2 h at 37°C followed by goat anti-mouse IgG-Alexa Fluor 594 (1:1,000, Table 2) at room temperature for 2 h in darkness. Nuclei were stained with 4, 6-diamidino-2-phenylindole (DAPI). Photomicrographs were obtained with a fluorescent microscope (DM 4500b, Leica) equipped with a digital camera (DFC 480, Leica). Negative control samples consisted of cells incubated with secondary antibody alone.
Flow Cytometry (P0, 2, 5), Neurogenesis (P1)
Antibodies against K14, K15, K19, CD29, CD105, CD44 and MAP2 (Table 2) were used to label cells for flow cytometry. With the exception of MAP2, antibodies were conjugated with FITC (FluoroTag™ FITC conjugation kit, Sigma-Aldrich). Cell aliquots (105 cells) were suspended in 800 μl of 1% BSA and 0.2% sodium azide in PBS containing 2 μl (200 ug/ml) of labeled antibody, and the mixture was incubated at room temperature for 30 min in darkness. Incubation with keratin antibodies was performed after cell permeabilization (Cytofix/Cytoperm™ kit, BD Biosciences, San Jose, CA). Following incubation with antibodies, cells were rinsed with PBS and fixed with 4% neutral buffered formalin. For MAP2 detection, cells were additionally incubated with anti-mouse IgG-Alexa Fluor 633 (Thermo Fisher Scientific). Cell fluorescence was quantified using a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA) and CellQuest graphics software (BD Biosciences). Unlabeled cells and isotype controls were included for all assays.
RT-PCR (P0, 2, 5)
Equine specific primers were designed for CD44, CD105, K14, K15, OCT4 and SOX2 (Table 3) using publicly available software (Primer-BLAST) and according to standard criteria established by the National Center for Biotechnology. Cells were detached from culture ware with 0.05% trypsin (Hyclone), rinsed with PBS and centrifuged at 260 × g for 5 min. Total mRNA was isolated (RNAqueous Micro Kit Ambion, Inc., Austin, TX). Potential DNA contamination was removed by DNase I (Turbo DNA-free, Ambion, Inc., Austin, TX) digestion. Samples were reverse transcribed into complementary DNA (cDNA) using oligo (dT) primers and Moloney Murine Leukemia Virus (M-MLV) reverse transcriptase (Sigma-Aldrich). Target gene mRNA levels were quantified with qRT-PCR using SYBR Green technology (Thermo Fisher Scientific) and an MJ Research Chromo4 Detector (Bio-Rad Laboratories, Hercules, CA). Amplicons were sequenced to confirm target sequence amplification. The ΔCt values were determined relative to the reference gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH).
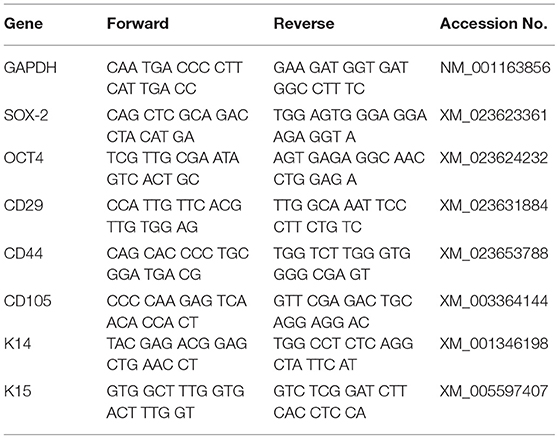
Table 3. Primer sequences.
In vivo Extracellular Matrix Production (P3)
Scaffold carriers composed of polyethylene glycol/poly-L-lactic acid (60:40, GA) and tricalcium phosphate/hydroxyapatite (40:60, HT) were fabricated by thermally induced phase separation as previously described (Smoak et al., 2015). Equine laminar progenitor cells (P1) from U hooves were cryopreserved in liquid nitrogen in custom medium (10% dimethyl sulfoxide, 10% DMEM/F-12, 80% FBS) after being gradually cooled to −80°C (−1°C/minute, CoolCell®, BioCision, Larkspur, CA). They were then culture expanded to P3 and loaded (6.3 × 103 cell/mm3) onto scaffolds (diameter: 10 mm; depth: 3 mm) for 2 h at 70 rpm stirring in a spinner flask (Bellco® Biotechnology, Newark, NJ) containing 120 ml of serum-free stromal medium. Cells that remained in the medium were quantified to estimate the number of cells on the scaffolds. Scaffolds without cells were treated identically to those with cells prior to implantation. One scaffold with or without cells divided into six equal pieces was surgically implanted in the dorsal subcutaneous tissues of each of four male athymic mice (nu/nu, Charles River Laboratories, Wilmington, MA). Specifically, six pieces of an HT cell-scaffold construct was implanted in one mouse, six pieces of a GA cell-scaffold construct in another, six pieces of an HT scaffold in another, and one mouse received six pieces of a GA scaffold.
For the implantation procedure, mice were premedicated with glycopyrrolate (0.02 mg/kg) and butorphanol (0.5 mg/kg) and subsequently anesthetized with isoflurane on oxygen delivered via a Baine circuit and mask. Following aseptic preparation, 6 skin incisions (5 mm) were created equidistantly along the dorsum extending from the scapula to the sacrum, approximately 1 cm lateral to each side of the spine, and scaffolds were placed after minimal blunt dissection. Subcutaneous tissues were closed with monocryl (Monocryl®, Cornelia, GA) and skin apposed with tissue glue (Sigma-Aldrich). Nine weeks after implantation, explants were fixed with 2.5% glutaradehyde in 0.1 M sodium carcodylate buffer, post-fixed with 0.1% osmium tetroxide, and dehydrated in a series of ethanol-distilled water solutions. After critical point drying, constructs were sputter coated with gold and imaged with a scanning electron microscope at 15 kVP (FEI Quanta 200, Netherlands) (Xie et al., 2013).
Transmission Electron Microscopy (P1)
Cells from U and L hooves were detached at 80% confluence and centrifuged at 260 × g for 5 min at room temperature. Pellets (3 × 106 cells/pellet) were fixed with 1.6% paraformaldehyde, 2.5% glutaraldehyde, 0.03% CaCl2 in 0.05 M cacodylate buffer (pH 7.4), washed in 0.1 M cacodylate buffer supplemented with 5% sucrose and post fixed in 2% osmium tetroxide for 1 h. They were washed with distilled water and incubated in a 1% water solution of tannic acid for 1 h. Following dehydration with propylene oxide in ascending ethanol concentrations, pellets were embedded in Epon-Araldite (EMS, Hatfield, PA). Blocks were sectioned (80–90 nm, Ultratome Leica EM UC7, Germany) and stained with 2% uranyl acetate in maleate buffer and lead citrate. Images were generated with a digital camera (Hamamatsu ORCA-HR, Japan) on an electron microscope (JEOL JEM 1011, Japan). All reagents for electron microscopy were from the same source (Electron Microscope Sciences, Hatfield, PA).
Results
Radiographs
Capsular rotation was 1.0 ± 1.4 mm (mean ± SEM) and 3.7 ± 1.7 mm, and distal descent 9.0 ± 0.8 mm and 10.6 ± 3.7 mm in U and L hooves, respectively (Figure 1).
Cell Isolation and Culture
Cell yield was 1.34 × 107 ± 1.25 × 106 and 1.70 × 107 ± 3.05 × 106 nucleated cells/g from U and L hoof tissue, respectively. Primary cell isolates from all hooves exhibited a rhomboid shape after 2 days of culture (Figures 2A,B). After 1 week's culture, most cells had a typical spindle shape consistent with progenitor cell morphology (Figures 2C,D).

Figure 2. Light photomicrographs of equine hoof progenitor cells from unaffected (A,C) and laminitic hooves (B,D) after 2 (A,B) and 7 days (C,D) of culture. Bar = 500 μm.
Colony Forming Unit Frequency (P0, 2, 5)
The CFU frequency percentages tended to increase with increasing passage (Figure 3). They were 0.05 ± 0.04%, 0.17 ± 0.07% and 0.32 ± 0.31% for passages P0, 2 and 5, respectively, for progenitor cells from U hooves. For the same cell passages from L hooves, CFU frequency percentages were 0.20 ± 0.09%, 0.10 ± 0.06% and 0.41 ± 0.21%.
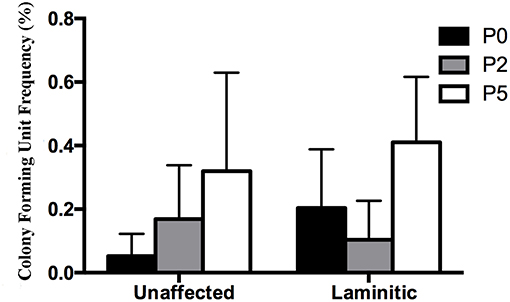
Figure 3. Colony forming unit frequency percentages (mean ± SEM) for P0, 2 and 5 progenitor cells from unaffected and laminitic hooves.
Cell Doublings and Doubling Time (P1-3)
The rate of in vitro expansion tended to increase with passage (Figures 4A–C). There was minimal variation in cell expansion between days for each sample, so data from all days for each sample were combined within passages for purposes of calculating mean and standard error. Cell doubling values for P1, P2 and P3 for cells from U hooves were 0.55 ± 0.65, 2.04 ± 1.20 and 1.58 ± 0.57 vs. 0.85 ± 0.50, 1.88 ± 1.10 and 1.99 ± 1.20 for the same passages from L hooves (Figure 4A). Doubling times for P1, P2 and P3 cells from U hooves were 0.26 ± 0.15, 0.12 ± 0.07 and 0.20 ± 0.11 days/CD vs. 0.32 ± 0.19, 0.13 ± 0.07 and 0.12 ± 0.07 days/CD from L hooves (Figure 4B).
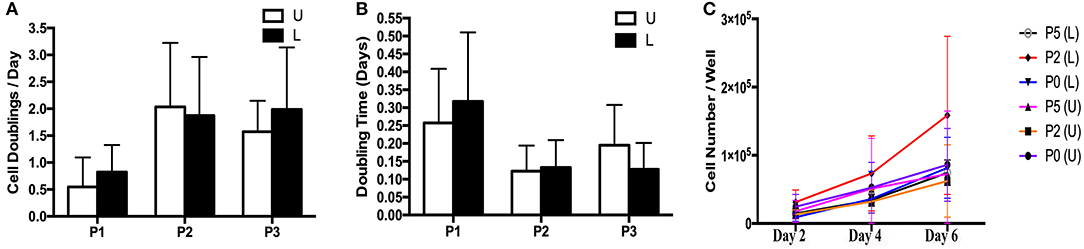
Figure 4. Cell doublings (A), doubling times (B) and growth curve (C) (mean ± SEM) for P1-3 progenitor cells from unaffected (U) and laminitic (L) hooves.
Multi-Lineage Differentiation (P1)
Passage 1 progenitor cells from U and L hooves all displayed characteristic adipocytic (Figure 5A), osteoblastic (Figure 5B) and neurocytic (Figure 5C) differentiation following culture in differentiation medium based on histochemical or immunocytochemical staining. Cells cultured in stromal medium for identical time periods did not differentiate (Figures 5D–F).
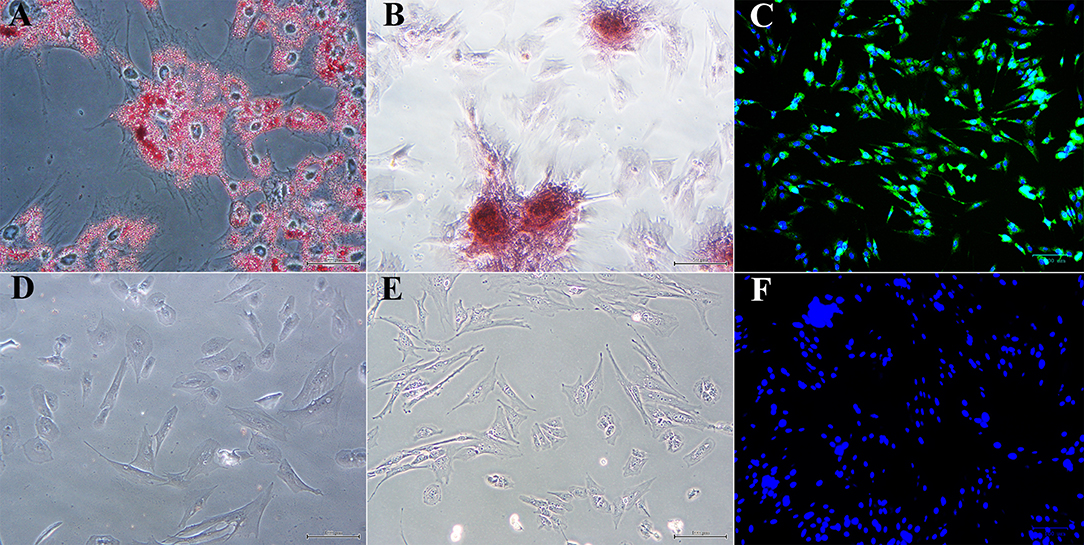
Figure 5. Photomicrographs of P1 laminar progenitor cells cultured in adipogenic (A), osteogenic (B), neurogenic (C) or stromal (D-F) medium and stained with oil red O (A,D), alizarin red (B,E) or anti-map2, anti-mouse IgG-488 (green, C,F). DAPI nuclear stain (blue, C,F). Magnification = 20X, Bar = 100 μm.
Immunohistochemistry (IHC), Immunocytochemistry (ICC) (P1, 3)
Fluorescent (Figure 6) and chromogen (Figure 7) immunohistochemical labeling confirmed the presence of CD44, CD29, K14, K15 and K19 in the stratum internum of both U and L hoof tissue. Based on immunocytochemical staining, cultured cells from U and L hooves expressed DSG1 and DSG3 (P3, Figure 8) as well as K15 and K19 (P1, Figure 9).
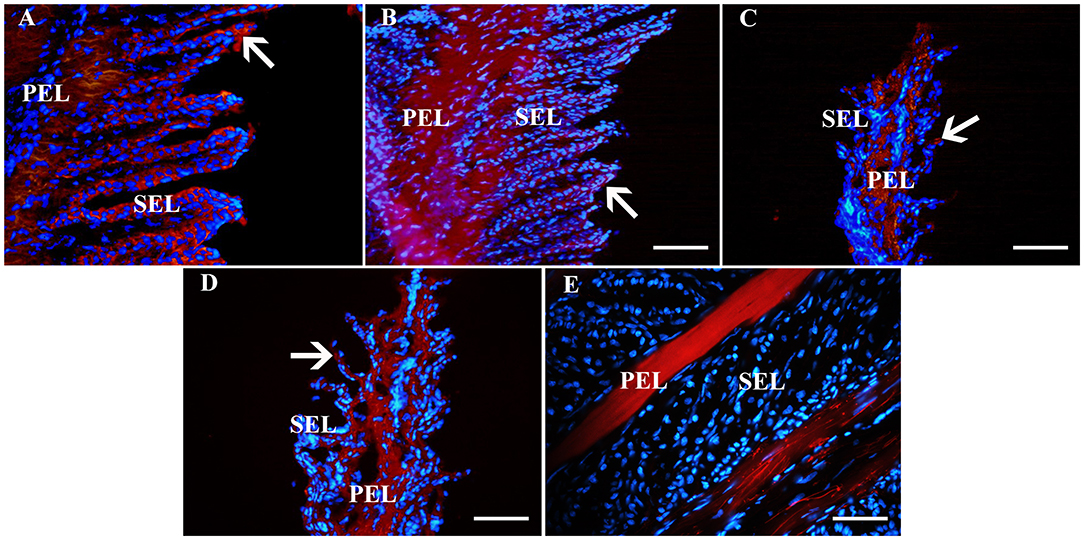
Figure 6. Fluorescent photomicrographs of primary and secondary epidermal lamellae from an unaffected hoof labeled with antibodies (red) against K14 (A), K19 (B), CD44 (C), CD29 (D) or no antibodies (E). Hoechst nuclear dye (blue) and Hoechst nuclear dye (blue). PEL, primary epidermal laminae; SEL, secondary epidermal laminae; Arrows = stained cells; Magnification = 40X, Bar = 50 μm.
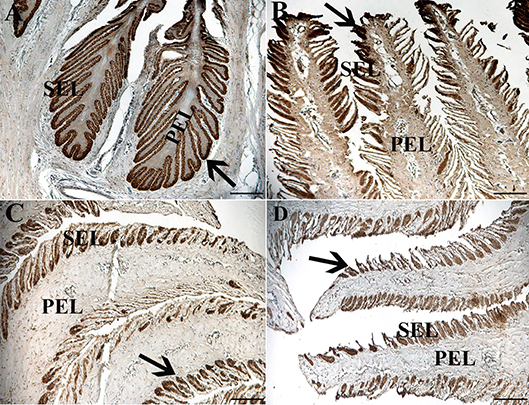
Figure 7. Light photomicrographs of lamellae from unaffected (A,B) and laminitic (C,D) hooves labeled (brown) with antibodies against K14 (A,C) and K15 (B,D). PEL, primary epidermal laminae; SEL, secondary epidermal laminae; Arrow = labeled cells; Magnification = 20X (A), Bar = 100 μm; Magnification = 40X (B–D), Bar = 50 μm.
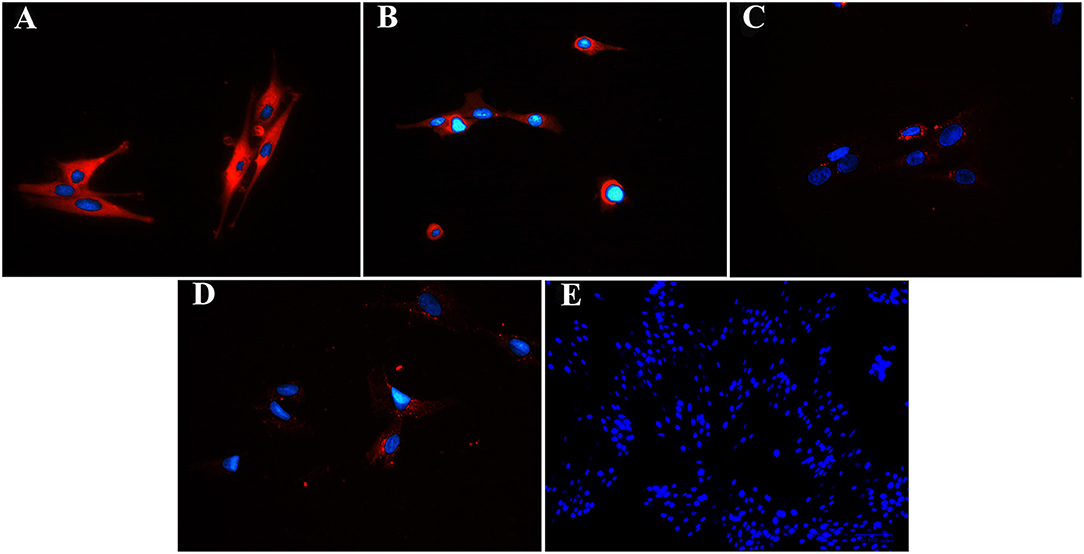
Figure 8. Fluorescent photomicrographs of cultured progenitor cells (P3) from unaffected (A,B) and laminitic (C,D) hooves labeled (red) with anti- DSG1 (A,C), DSG3 (B,D) antibodies or no antibodies (E). DAPI nuclear stain (blue); Magnification = 40X (A–D); 20X (E).
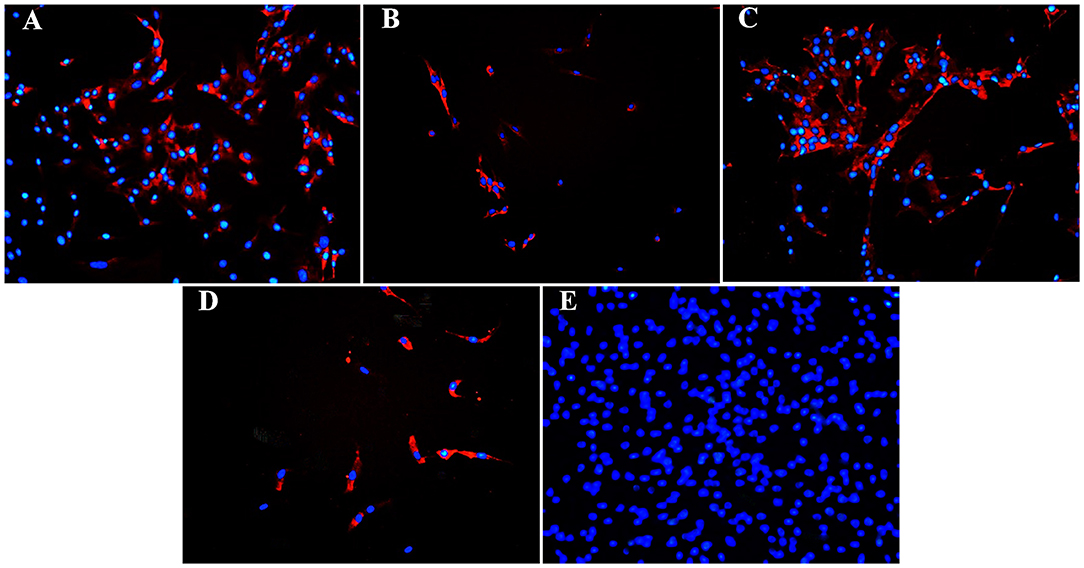
Figure 9. Fluorescent photomicrographs of cultured hoof progenitor cells (P1) from unaffected (A,B) and laminitic (C,D) hooves after labeling (red) with antibodies against K15 (A,C), K19 (B,D) or no antibodies (E). DAPI nuclear stain (blue); Magnification = 20X.
Flow Cytometry (P0, 2, 5), Neurogenesis (P1)
Passage P0, 2 and 5 cultured progenitor cells from U and L hooves expressed CD29, CD44, CD105, K14, K15, and K19 (Figure 10). The percentage of cells expressing CD105 or CD44 (Figures 10B,C) tended to decrease with passage while the percentage expressing CD29 (Figure 10A) remained fairly constant across passages. The majority of P2 and 5 from both hoof conditions expressed K14, K15 and K19 (>90%) (Figures 10D–F). Approximately 70% of P1 cells cultured in neurogenic induction medium expressed MAP2 compared to 18% of cells cultured in stromal medium (Figure 11).
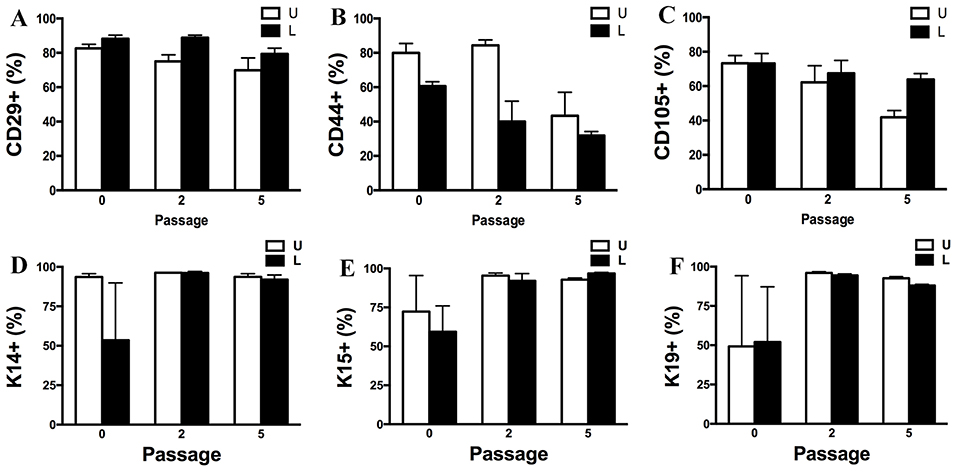
Figure 10. Percentage (mean ± SEM) of CD29+ (A), CD44+ (B), CD105+ (C), K14+ (D), K15+ (E) and K19+ (F) P0, 2 and 5 progenitor cells from unaffected (U) and laminitic (L) hooves based on flow cytometry.
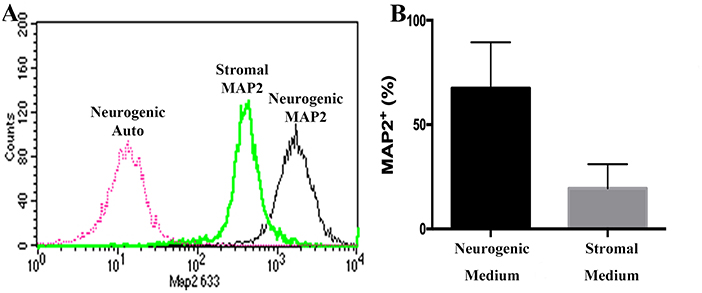
Figure 11. Flow cytometry histogram showing MAP2 labeling of P1 equine hoof progenitor cells from an unaffected hoof cultured in stromal (Stromal MAP2) or neurogenic medium (Neurogenic MAP2), and autofluorescence of unlabelled cells cultured in neurogenic medium (Neurogenic Auto, A). Percentage (mean ± SEM) of P1 hoof progenitor cells from unaffected hooves expressing MAP2 after culture in stromal (Stromal) or neurogenic (Neurogenic) induction medium (B).
RT- PCR (P0, 2, 5)
Gene expression of CD44, CD105, K14, K15, OCT4, and SOX2 by P0, 2 and 5 progenitor cells from U and L hooves was confirmed (Figure 12). Gene expression was relatively similar between conditions and stable across passages. Exceptions included an apparent decrease in K14 expression with increasing passage in cells from U hooves and an increase in CD44 expression with increasing passage in cells from L hooves. The stability of the reference gene, GAPDH, was confirmed by a CT value of 17.71 ± 0.20 for all samples.
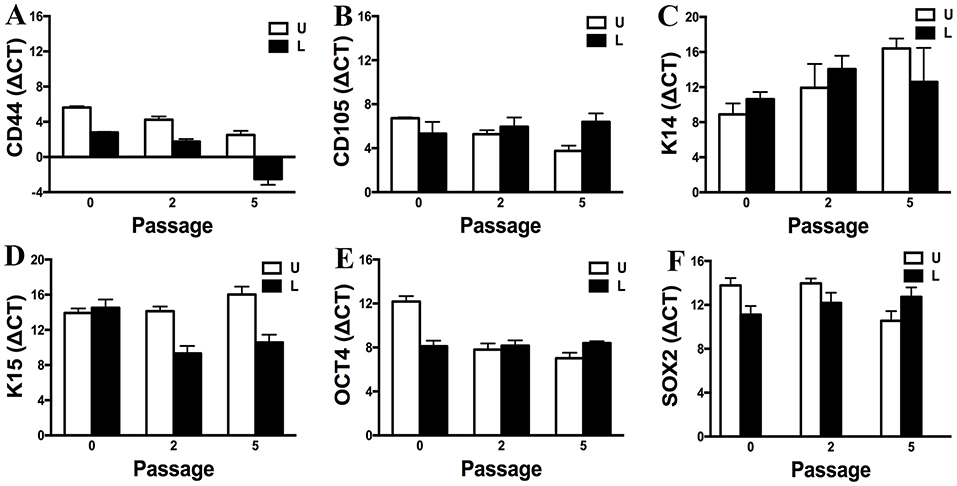
Figure 12. Expression levels (ΔCT, mean ± SEM) of CD44 (A), CD105 (B), K14 (C), K15 (D), OCT4 (E) and SOX2 (F) in progenitor cells from unaffected (U) and laminitic (L) hooves at the indicated passages. Reference gene: GAPDH; SOX2 = SRY (sex determining region Y)-box 2; OCT4 = octamer-binding transcription factor 4.
In vivo Extracellular Matrix Production (P3)
Approximately 5.60 × 103 cells/mm3 were loaded onto each scaffold. Abundant collagen fibrils and organized ECM was apparent on scaffolds containing cells (Figures 13A,B), while those without cells contained fewer collagen fibrils and little organized ECM (Figures 13C,D).
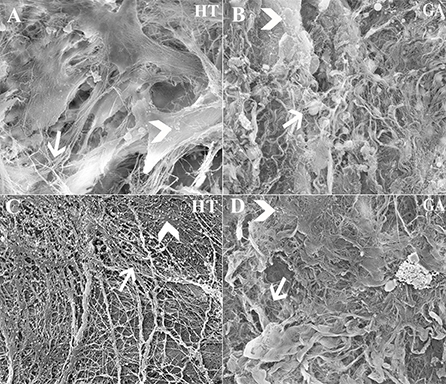
Figure 13. Scanning electrophotomicrographs of scaffold constructs with (A,B) and without (C,D) P3 equine hoof progenitor cells from an unaffected hoof 9 weeks after subcutaneous implantation in athymic mice. Arrow = collagen fibrils; Arrow head = organized extracellular matrix; Magnification = 6,000X (A,C), 3000X (B,D).
Transmission Electron Microscopy (P1)
Cell ultrastructure was similar between cells from U and L hooves (Figure 14). Large, irregular, eccentric nuclei, elongated mitochondria, rough endoplasmic reticulum surrounded by cisternae, and randomly distributed cytoplasmic vacuoles were present in all cells evaluated.
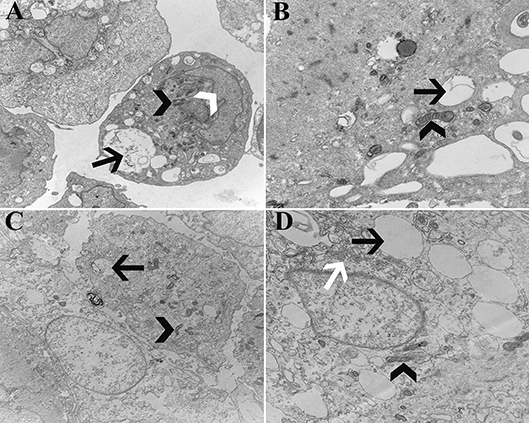
Figure 14. Transmission electromicrographs of progenitor cells (P1) from unaffected (A,B) and laminitic (C,D) hooves. Black arrow = vacuole; White arrow head = nucleus; Black arrow head = mitochondria; White arrow = rough endoplasmic reticulum; Magnification = 10,000X (A,C), 20,000X (B,D).
Discussion
The work in this study establishes a reproducible mechanism to isolate and culture progenitor cells from unaffected and chronically inflamed ectodermal-mesodermal tissue of the equine hoof, a tissue interface similar to the human and mouse periodontal membrane and the human fingernail (Leung et al., 2014; Maiolino et al., 2016). In vitro culture of adult progenitor cells provides a mechanism to evaluate cell behavior under distinct conditions (Gronthos et al., 2000; Caplan, 2007; Paschos et al., 2015; Khojasteh et al., 2017) to assess cell alterations mediated by chronic inflammation or disease. The methods will support cell and tissue culture for investigation of therapies to prevent or address pathogenic processes in ectodermal-mesodermal tissue. This work establishes a unique and important model to study progenitor cells in the distinct ectodermal-mesodermal niche, a site of important health challenges in both veterinary and human patients (Gross et al., 2005; Martins et al., 2016).
Plastic adherence of the cells in this study is consistent with cell immaturity and multi-lineage differentiation confirms plasticity (Dominici et al., 2006; Bourzac et al., 2010; Fulber et al., 2016). The immature status of the cell isolates is supported by relatively stable expression of genes associated with progenitor cell self-renewal and proliferation, OCT4 and SOX2 (Bourguignon et al., 2012). Progenitor cells from both inflamed and unaffected tissues showed similar in vitro behavior as equine progenitor cells from normal adult tissues (Koerner et al., 2006; Violini et al., 2009; Bourzac et al., 2010; Mccarthy et al., 2012), and characteristics appeared to be maintained through numerous passages (Jiang et al., 2017). Notably, mesodermal and ectodermal cell differentiation suggests the presence of cells from different lineages, cells capable of transdifferentiation, or, most likely, both given the heterogeneous composition of the isolates. Somewhat consistent expression of both mesodermal, CD44 and CD105, and ectodermal, K15 and K14, progenitor cell genes up to five passages further confirms the likely presence of multiple cell lineages and/or transitional cells and indicates that the unique cell identities are well maintained in culture (Ranera et al., 2011; Troy et al., 2011; Hayashi et al., 2016). In vitro expansion rates in this study were slightly higher than those reported for bone marrow, adipose and umbilical cord derived equine MSCs of about 1 day/CD (Vidal et al., 2012) and human epidermal keratinocytes of about 0.7 days/CD (Sun and Green, 1976). Distinct culture conditions and natural variation typically contribute to differences in expansion behavior among studies (Gerhard et al., 2004; Sotiropoulou et al., 2006). The CFU frequency percentages in this study are comparable to those from mesodermal and ectodermal equine progenitor cells of about 0.6 and 1.5%, respectively (Gargett et al., 2009; Corradetti et al., 2011). Taken together, these results confirm the immaturity and diversity of the heterogeneous primary cell isolates.
The presence of mesodermal and ectodermal proteins in hoof tissue and within heterogeneous cell isolates in vitro is important to translate information between in vitro and in vivo conditions (Michel et al., 1996; Ponta et al., 2003; Fonsatti and Maio, 2004; Schaffler and Buchler, 2007; De Mattos Carvalho et al., 2009; Abbas et al., 2011; Halfon et al., 2011; Bose et al., 2013; Liao et al., 2014). A prominent goal of this series of investigations was to validate parallel protein expression in cultured primary cell isolates and tissue. Despite a plethora of methods to culture progenitor cells from numerous tissues of most species, localization of progenitor cell antigens in situ and in vitro is rare (Galantino-Homer et al., 2014; Ghasemi, 2017). The fact that the cell isolates retained their characteristics in culture based on flow cytometry results is especially vital to the validity of the culture model. Results of this study are limited to cells that have been passaged beyond the initial stromal vascular fraction isolate. Culture of cells under ideal conditions can potentially mask natural attributes and behaviors, a recognized limitation of cell culture and impetus to maintain culture conditions that replicate distinct natural states (Duval et al., 2017).
The ability of cells within heterogeneous primary cell isolates to mature into distinct lineages in this study is consistent with current knowledge surrounding cell plasticity, especially in tissue interfaces (Pearton et al., 2005; Medvedev et al., 2010; Rohani et al., 2011). As indicated above, it is likely that the junction between epidermal and dermal tissues contains progenitor cells from ectodermal and mesodermal embryonic layers, and, potentially, transitional cells that have characteristics of both tissue types permitting ready transdifferentiation (Phinney and Prockop, 2007). Additional work is needed to isolate and characterize specific cell immunophenotypes from heterogeneous primary cell isolates.
Tight cell junctions that stabilize the ectodermal-mesodermal tissue interface are composed of a variety of proteins like desmogleins, cadherins, laminins and E-selectins, among others (Leise et al., 2011). Desmogleins are major proteins of desmosomes, cell to cell junctions (Pollitt, 2016), and hemidesmosomes, cell to tissue or ECM junctions. Loss of desmoglein integrity or diminished production is associated with sloughing of the epidermis (Miragliotta et al., 2006; Thomason et al., 2010; Amagai and Stanley, 2012; Saito et al., 2012) and abnormal tissue production (Walko et al., 2015; Najor and Green, 2018). There are fewer hemidesmosomes along the basement membrane between the ectodermal and dermal layers in inflamed vs. normal equine hoof tissue (Nourian et al., 2007). Future studies using cell culture may further confirm the potential for disruption of desmoglein function or production by inflammation.
The cells in this study shared some ultrastructural features with equine and human adult multipotent cells including polymorphic nuclei, well-developed dilated cisternae of rough endoplasmic reticulum, intracellular vacuoles and elongated mitochondria (Horstmann et al., 2002; Pascucci et al., 2010; Rodrigues et al., 2010; Alipour et al., 2015; Miko et al., 2015). High metabolic activity typical of immature cells is suggested by elongated mitochondria (Campello and Scorrano, 2010). Intracellular vacuoles are associated with protein recycling (Hwang et al., 2014; Zheng et al., 2014), and they are thought to be filled with proteinaceous material destined for secretion in human adult stem cells from bone marrow (Colter et al., 2001; Raimondo et al., 2006). There are also reports of small cytoplasmic vacuoles in Merkel cells within canine whisker pad basal epidermis (Hashimoto, 1972; Ramirez et al., 2015). The ultrastructure of the cells reported here is consistent with high metabolic activity of progenitor cells and a secretory function typically associated with keratinizing cells (Gallucci et al., 1979). The results are not consistent with any one cell type and likely reflect the heterogeneous nature of the cell isolates.
Abnormal tissue formation in inflamed ectodermal-mesodermal interface tissues has been reported to be the result of altered maturation (Eastham et al., 2007; Rucker, 2010; Galantino-Homer et al., 2014; Lamouille et al., 2014; Bailey, 2015; Tomasello et al., 2017). As such, appropriate cell direction is essential for stem cell transplantation therapies to drive restorative tissue formation (Shokrgozar et al., 2012; Takahashi et al., 2013). The present study was not designed to assess treatment outcomes in contrast to clinical reports of stem cell therapies (Peroni, 2013; Morrison et al., 2014) or to compare characteristics of cells from normal and inflamed tissue, but to establish a model of cell culture that accommodates cells from both tissue conditions. Robust, organized ECM formation by hoof progenitor cells on scaffold carriers confirms the translational potential of the cell isolates from this study (Rustad et al., 2012; Duan et al., 2018). However, future work is necessary to formulate scaffold carriers to direct hoof progenitor cell differentiation in vivo or support hoof tissue formation in vitro to create implantable graft tissue that guides native progenitor cells (Sundelacruz and Kaplan, 2009; Qi et al., 2014). Evaluation of the broad cell signaling pathways represented by OCT4 and SOX2 in primary cell isolates cultured under conditions that replicate native tissue may be natural starting points for study of mechanisms to modulate cell behaviors in tissue constructs (Zhou et al., 2013; Amini et al., 2014; Matic et al., 2016; Sardarabadi et al., 2016). In the future, de novo tissue formation with progenitor cells may provide a new therapeutic option for chronic conditions of the ectodermal-mesodermal interface.
The authors recognize several limitations of this preliminary study to establish isolation and culture of progenitor cell isolates from an ectodermal-mesodermal interface. The characteristics of a cell population reflect but are not identical to those of individual cells (Altschuler and Wu, 2010). As such, work is necessary to isolate and evaluate differences among distinct immunophenotypes within the mixed (heterogeneous) populations included in this study. Also, as pointed out above, the initial 2D cell culture in mesodermal basal medium as well as the isolation process very likely selected for distinct progenitor cell populations that may have stronger mesodermal than ectodermal predilections (Arsenian et al., 1998; Alipour et al., 2015; Lombana et al., 2015). Selection of cell immunophenotypes and subsequent culture in mesodermal and ectodermal basal medium will provide important information about the novel progenitor cells residing in the ectodermal-mesodermal interface. Nonetheless, the presence of mesodermal and ectodermal antigens and differentiation potential over multiple cell passages confirm that cells of both embryonic origins survived under the culture conditions employed in this study.
Conclusions
Progenitor cell isolates from the ectodermal-mesodermal tissue interface of the equine hoof display characteristics of both tissue types and maintain progenitor qualities through multiple in vitro cell passages. The cells are present in both normal and inflamed tissue and key protein antigens, including those in cell to cell and cell to tissue junctions, are retained in culture. Hoof progenitor cells promote collagenous extracellular matrix formation on distinct scaffold carriers in vivo, confirming future opportunities for cell directed tissue formation. Results from this study set the stage for work to ascertain mechanisms by which inflammation impacts progenitor cells in the ectodermal-mesodermal niche. Methods to direct appropriate cell differentiation and tissue formation will provide new therapeutic approaches to inflammatory conditions of tissue interfaces.
Ethics Statement
This study was approved by the Louisiana State University Animal Care and Use Committee prior to study initiation (protocol # 16-036).
Author Contributions
QY and WD contributed to the study design, data collection and reduction, manuscript writing, and final approval of manuscript. VP and ML contributed to the study design, data collection and reduction, data analysis and interpretation, manuscript writing, and final approval of manuscript. EP, JD, and WR were responsible for data collection, manuscript writing, and final approval of manuscript.
Funding
This work was funded in part by the Louisiana State University Equine Health Studies Program (EHSP) and the United States Department of Agriculture (USDA 1443 award).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Special thanks to Mr. Michael Keowen, Ms. Marilyn Dietrich, Ms. Ying Xiao and Dr. Yuliya Sokolova for their technical support.
References
Abbas, O., Richards, J. E., Yaar, R., and Mahalingam, M. (2011). Stem cell markers (cytokeratin 15, cytokeratin 19 and p63) in in situ and invasive cutaneous epithelial lesions. Mod. Pathol. 24, 90–97. doi: 10.1038/modpathol.2010.180
Alipour, F., Parham, A., Kazemi Mehrjerdi, H., and Dehghani, H. (2015). Equine adipose-derived mesenchymal stem cells: phenotype and growth characteristics, gene expression profile and differentiation potentials. Cell J. 16, 456–465. doi: 10.22074/cellj.2015.491
Altschuler, S. J., and Wu, L. F. (2010). Cellular heterogeneity: do differences make a difference? Cell 141, 559–563. doi: 10.1016/j.cell.2010.04.033
Amagai, M., and Stanley, J. R. (2012). Desmoglein as a target in skin disease and beyond. J. Invest. Dermatol. 132, 776–784. doi: 10.1038/jid.2011.390
Amini, S., Fathi, F., Mobalegi, J., Sofimajidpour, H., and Ghadimi, T. (2014). The expressions of stem cell markers: Oct4, Nanog, Sox2, nucleostemin, Bmi, Zfx, Tcl1, Tbx3, Dppa4, and Esrrb in bladder, colon, and prostate cancer, and certain cancer cell lines. Anat. Cell Biol. 47, 1–11. doi: 10.5115/acb.2014.47.1.1
Anderson, T. M., and Mcilwraith, C. W. (2004). Longitudinal development of equine conformation from weanling to age 3 years in the Thoroughbred. Equine Vet. J. 36, 563–570. doi: 10.2746/0425164044864507
Arsenian, S., Weinhold, B., Oelgeschlager, M., Ruther, U., and Nordheim, A. (1998). Serum response factor is essential for mesoderm formation during mouse embryogenesis. EMBO J. 17, 6289–6299. doi: 10.1093/emboj/17.21.6289
Atala, A., Irvine, D. J., Moses, M., and Shaunak, S. (2010). Wound healing versus regeneration: role of the tissue environment in regenerative medicine. MRS Bull. 35, 597–606. doi: 10.1557/mrs2010.528
Bailey, S. R. (2015). Acute equine laminitis: exciting prospects afoot. Vet. J. 206, 121–122. doi: 10.1016/j.tvjl.2015.07.034
Barreto-Vianna, A. R., Oliveira, L. S., Leonardo, A. S., Santana, M. I., Godoy, R. F., and De Lima, E. M. (2013). Density of primary and secondary epidermal laminae of equine hoof. Pesqui. Vet. Bras. 33, 543–548. doi: 10.1590/S0100-736X2013000400020
Bharathi, R. R., and Bajantri, B. (2011). Nail bed injuries and deformities of nail. Indian J. Plast. Surg. 44, 197–202. doi: 10.4103/0970-0358.85340
Bose, A., Teh, M. T., Mackenzie, I. C., and Waseem, A. (2013). Keratin k15 as a biomarker of epidermal stem cells. Int. J. Mol. Sci. 14, 19385–19398. doi: 10.3390/ijms141019385
Bourguignon, L. Y., Wong, G., Earle, C., and Chen, L. (2012). Hyaluronan-CD44v3 interaction with Oct4-Sox2-Nanog promotes miR-302 expression leading to self-renewal, clonal formation, and cisplatin resistance in cancer stem cells from head and neck squamous cell carcinoma. J. Biol. Chem. 287, 32800–32824. doi: 10.1074/jbc.M111.308528
Bourzac, C., Smith, L. C., Vincent, P., Beauchamp, G., Lavoie, J. P., and Laverty, S. (2010). Isolation of equine bone marrow-derived mesenchymal stem cells: a comparison between three protocols. Equine Vet. J. 42, 519–527. doi: 10.1111/j.2042-3306.2010.00098.x
Bragulla, H. H., and Homberger, D. G. (2009). Structure and functions of keratin proteins in simple, stratified, keratinized and cornified epithelia. J. Anat. 214, 516–559. doi: 10.1111/j.1469-7580.2009.01066.x
Broeckx, S. Y., Maes, S., Martinello, T., Aerts, D., Chiers, K., Marien, T., et al. (2014). Equine epidermis: a source of epithelial-like stem/progenitor cells with in vitro and in vivo regenerative capacities. Stem Cells Dev. 23, 1134–1148. doi: 10.1089/scd.2013.0203
Campello, S., and Scorrano, L. (2010). Mitochondrial shape changes: orchestrating cell pathophysiology. EMBO Rep. 11, 678–684. doi: 10.1038/embor.2010.115
Caplan, A. I. (2007). Adult mesenchymal stem cells for tissue engineering versus regenerative medicine. J. Cell. Physiol. 213, 341–347. doi: 10.1002/jcp.21200
Colter, D. C., Sekiya, I., and Prockop, D. J. (2001). Identification of a subpopulation of rapidly self-renewing and multipotential adult stem cells in colonies of human marrow stromal cells. Proc. Natl. Acad. Sci. U.S.A 98, 7841–7845. doi: 10.1073/pnas.141221698
Corradetti, B., Lange-Consiglio, A., Barucca, M., Cremonesi, F., and Bizzaro, D. (2011). Size-sieved subpopulations of mesenchymal stem cells from intervascular and perivascular equine umbilical cord matrix. Cell Prolif. 44, 330–342. doi: 10.1111/j.1365-2184.2011.00759.x
Daradka, M., and Pollitt, C. C. (2004). Epidermal cell proliferation in the equine hoof wall. Equine Vet. J. 36, 236–241. doi: 10.2746/0425164044877198
De Mattos Carvalho, A., Alves, A. L., Golim, M. A., Moroz, A., Hussni, C. A., De Oliveira, P. G., et al. (2009). Isolation and immunophenotypic characterization of mesenchymal stem cells derived from equine species adipose tissue. Vet. Immunol. Immunopathol. 132, 303–306. doi: 10.1016/j.vetimm.2009.06.014
Ding, G., Liu, Y., Wang, W., Wei, F., Liu, D., Fan, Z., et al. (2010). Allogeneic periodontal ligament stem cell therapy for periodontitis in swine. Stem Cells 28, 1829–1838. doi: 10.1002/stem.512
Dominici, M., Le Blanc, K., Mueller, I., Slaper-Cortenbach, I., Marini, F., Krause, D., et al. (2006). Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 8, 315–317. doi: 10.1080/14653240600855905
Duan, W., Chen, C., Haque, M., Hayes, D., and Lopez, M. J. (2018). Polymer-mineral scaffold augments in vivo equine multipotent stromal cell osteogenesis. Stem Cell Res. Ther. 9:60. doi: 10.1186/s13287-018-0790-8
Duan, W., and Lopez, M. J. (2016). Effects of cryopreservation on canine multipotent stromal cells from subcutaneous and infrapatellar adipose tissue. Stem Cell Rev. 12, 257–268. doi: 10.1007/s12015-015-9634-4
Duval, K., Grover, H., Han, L. H., Mou, Y., Pegoraro, A. F., Fredberg, J., et al. (2017). Modeling physiological events in 2D vs. 3D cell culture. Physiol. (Bethesda) 32, 266–277. doi: 10.1152/physiol.00036.2016
Eastham, A. M., Spencer, H., Soncin, F., Ritson, S., Merry, C. L., Stern, P. L., et al. (2007). Epithelial-mesenchymal transition events during human embryonic stem cell differentiation. Cancer Res. 67, 11254–11262. doi: 10.1158/0008-5472.CAN-07-2253
Fonsatti, E., and Maio, M. (2004). Highlights on endoglin (CD105): from basic findings towards clinical applications in human cancer. J. Transl. Med. 2:18. doi: 10.1186/1479-5876-2-18
French, K. R., and Pollitt, C. C. (2004). Equine laminitis: loss of hemidesmosomes in hoof secondary epidermal lamellae correlates to dose in an oligofructose induction model: an ultrastructural study. Equine Vet. J. 36, 230–235. doi: 10.2746/0425164044877125
Fulber, J., Maria, D. A., Da Silva, L. C., Massoco, C. O., Agreste, F., and Baccarin, R. Y. (2016). Comparative study of equine mesenchymal stem cells from healthy and injured synovial tissues: an in vitro assessment. Stem Cell Res. Ther. 7:35. doi: 10.1186/s13287-016-0294-3
Galantino-Homer, H. L., Clark, R. K., and Linardi, R. L. (2014). “Characterization of equine hoof lamellar tissue microanatomy with fluorescent markers,” in Proceedings of the 60th Annual Convention of the American Association of Equine Practitioners (Salt Lake City, UT: American Association of Equine Practitioners (AAEP)), 73–78.
Gallucci, B. B., Shulman, H. M., Sale, G. E., Lerner, K. G., Caldwell, L. E., and Thomas, E. D. (1979). The ultrastructure of the human epidermis in chronic graft-versus-host disease. Am. J. Pathol. 95, 643–662.
Gargett, C. E., Schwab, K. E., Zillwood, R. M., Nguyen, H. P., and Wu, D. (2009). Isolation and culture of epithelial progenitors and mesenchymal stem cells from human endometrium. Biol. Reprod. 80, 1136–1145. doi: 10.1095/biolreprod.108.075226
Gerhard, D. S., Wagner, L., Feingold, E. A., Shenmen, C. M., Grouse, L. H., Schuler, G., et al. (2004). The status, quality, and expansion of the NIH full-length cDNA project: the mammalian gene collection (MGC). Genome Res. 14, 2121–2127. doi: 10.1101/gr.2596504
Ghasemi, N. (2017). Comparison of two protocols for induction of differentiation of human adipose derived stem cells into oligodendrocyte progenitor cells. Sci. J. Kurdistan Univ. Med. Sci. 22, 93–102. doi: 10.22102/22.5.93
Gronthos, S., Mankani, M., Brahim, J., Robey, P. G., and Shi, S. (2000). Postnatal human dental pulp stem cells (DPSCs) in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A 97, 13625–13630. doi: 10.1073/pnas.240309797
Gross, T. L., Ihrke, P. J., Walder, E. J., and Affolter, V. K. (2005). Skin Diseases of the Dog and Cat: Clinical and Histopathologic Diagnosis, 2nd Edn. Wiley.
Halfon, S., Abramov, N., Grinblat, B., and Ginis, I. (2011). Markers distinguishing mesenchymal stem cells from fibroblasts are downregulated with passaging. Stem Cells Dev. 20, 53–66. doi: 10.1089/scd.2010.0040
Hashimoto, K. (1972). The ultrastructure of the skin of human embryos. X. Merkel tactile cells in the finger and nail. J. Anat. 111(Pt 1), 99–120.
Hayashi, R., Ishikawa, Y., Sasamoto, Y., Katori, R., Nomura, N., Ichikawa, T., et al. (2016). Co-ordinated ocular development from human iPS cells and recovery of corneal function. Nature 531, 376–380. doi: 10.1038/nature17000
Horstmann, H., Ng, C. P., Tang, B. L., and Hong, W. (2002). Ultrastructural characterization of endoplasmic reticulum–Golgi transport containers (EGTC). J. Cell Sci. 115, 4263–4273. doi: 10.1242/jcs.00115
Hunt, R. J., and Wharton, R. E. (2010). Clinical presentation, diagnosis, and prognosis of chronic laminitis in North America. Vet. Clin. North Am. Equine Pract. 26, 141–153. doi: 10.1016/j.cveq.2009.12.006
Hwang, G. W., Murai, Y., Takahashi, T., and Naganuma, A. (2014). The protein transportation pathway from Golgi to vacuoles via endosomes plays a role in enhancement of methylmercury toxicity. Sci. Rep. 4:5888. doi: 10.1038/srep05888
Jiang, T., Xu, G., Wang, Q., Yang, L., Zheng, L., Zhao, J., et al. (2017). In vitro expansion impaired the stemness of early passage mesenchymal stem cells for treatment of cartilage defects. Cell Death Dis. 8:e2851. doi: 10.1038/cddis.2017.215
Johnson, P. J., Tyagi, S. C., Katwa, L. C., Ganjam, V. K., Moore, L. A., Kreeger, J. M., et al. (1998). Activation of extracellular matrix metalloproteinases in equine laminitis. Vet. Rec. 142, 392–396. doi: 10.1136/vr.142.15.392
Khojasteh, A., Fahimipour, F., Jafarian, M., Sharifi, D., Jahangir, S., Khayyatan, F., et al. (2017). Bone engineering in dog mandible: coculturing mesenchymal stem cells with endothelial progenitor cells in a composite scaffold containing vascular endothelial growth factor. J. Biomed. Mater. Res. B Appl. Biomater. 105, 1767–1777. doi: 10.1002/jbm.b.33707
Kisiday, J., Jin, M., Kurz, B., Hung, H., Semino, C., Zhang, S., et al. (2002). Self-assembling peptide hydrogel fosters chondrocyte extracellular matrix production and cell division: implications for cartilage tissue repair. Proc. Natl. Acad. Sci. U.S.A 99, 9996–10001. doi: 10.1073/pnas.142309999
Koerner, J., Nesic, D., Romero, J. D., Brehm, W., Mainil-Varlet, P., and Grogan, S. P. (2006). Equine peripheral blood-derived progenitors in comparison to bone marrow-derived mesenchymal stem cells. Stem Cells 24, 1613–1619. doi: 10.1634/stemcells.2005-0264
Kummer, M., Lischer, C., Ohlerth, S., Vargas, J., and Auer, J. (2004). Evaluation of a standardised radiographic technique of the equine hoof. Schweiz Arch. Tierheilkd. 146, 507–514. doi: 10.1024/0036-7281.146.11.507
Lamouille, S., Xu, J., and Derynck, R. (2014). Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell. Biol. 15, 178–196. doi: 10.1038/nrm3758
Leise, B. S., Faleiros, R. R., Watts, M., Johnson, P. J., Black, S. J., and Belknap, J. K. (2011). Laminar inflammatory gene expression in the carbohydrate overload model of equine laminitis. Equine Vet. J. 43, 54–61. doi: 10.1111/j.2042-3306.2010.00122.x
Leung, Y., Kandyba, E., Chen, Y. B., Ruffins, S., Chuong, C. M., and Kobielak, K. (2014). Bifunctional ectodermal stem cells around the nail display dual fate homeostasis and adaptive wounding response toward nail regeneration. Proc. Natl. Acad. Sci. U.S.A 111, 15114–15119. doi: 10.1073/pnas.1318848111
Lian, J. B., and Stein, G. S. (1992). Concepts of osteoblast growth and differentiation: basis for modulation of bone cell development and tissue formation. Crit. Rev. Oral Biol. Med. 3, 269–305. doi: 10.1177/10454411920030030501
Liao, Y., Itoh, M., Yang, A., Zhu, H., Roberts, S., Highet, A. M., et al. (2014). Human cord blood-derived unrestricted somatic stem cells promote wound healing and have therapeutic potential for patients with recessive dystrophic epidermolysis bullosa. Cell Transplant. 23, 303–317. doi: 10.3727/096368913X663569
Linardi, R. L., Megee, S. O., Mainardi, S. R., Senoo, M., and Galantino-Homer, H. L. (2015). Expression and localization of epithelial stem cell and differentiation markers in equine skin, eye and hoof. Vet. Dermatol. 26, 213–e247. doi: 10.1111/vde.12214
Lombana, K. G., Goodrich, L. R., Phillips, J. N., Kisiday, J. D., Ruple-Czerniak, A., and Mcilwraith, C. W. (2015). An investigation of equine mesenchymal stem cell characteristics from different harvest sites: more similar than not. Front. Vet. Sci. 2:67. doi: 10.3389/fvets.2015.00067
Longland, A. C., and Byrd, B. M. (2006). Pasture nonstructural carbohydrates and equine laminitis. J. Nutr. 136, 2099S−2102S. doi: 10.1093/jn/136.7.2099S
Maiolino, S. A., Kingston, A. K., and Lemelin, P. (2016). “Comparative and functional morphology of the primate hand integument,” in The Evolution of the Primate Hand. Developments in Primatology: Progress and Prospects, eds T. Kivell, P. Lemelin, B. Richmond, and D. Schmitt (New York, NY: Springer).
Martins, L., Valente, F., Reis, E., Sepúlveda, R., Perdigão, A., and Borges, A. (2016). Treatment of periodontal disease with guided tissue regeneration technique using a hydroxyapatite and polycaprolactone membrane. Arq. Bras. Med. Vet. Zootec. 68, 1413–1421. doi: 10.1590/1678-4162-8322
Matic, I., Antunovic, M., Brkic, S., Josipovic, P., Mihalic, K. C., Karlak, I., et al. (2016). Expression of OCT-4 and SOX-2 in bone marrow-derived human mesenchymal stem cells during osteogenic differentiation. Open Access Maced. J. Med. Sci. 4, 9–16. doi: 10.3889/oamjms.2016.008
Mccarthy, H. E., Bara, J. J., Brakspear, K., Singhrao, S. K., and Archer, C. W. (2012). The comparison of equine articular cartilage progenitor cells and bone marrow-derived stromal cells as potential cell sources for cartilage repair in the horse. Vet. J. 192, 345–351. doi: 10.1016/j.tvjl.2011.08.036
Medvedev, S. P., Malakhova, A. A., Grigor'eva, E. V., Shevchenko, A. I., Dementyeva, E. V., Sobolev, I. A., et al. (2010). Derivation of induced pluripotent stem cells from fetal human skin fibroblasts. Acta Nat. 2, 102–106.
Michel, M., Torok, N., Godbout, M. J., Lussier, M., Gaudreau, P., Royal, A., et al. (1996). Keratin 19 as a biochemical marker of skin stem cells in vivo and in vitro: keratin 19 expressing cells are differentially localized in function of anatomic sites, and their number varies with donor age and culture stage. J. Cell Sci. 109, 1017–1028.
Miko, M., Danisovic, L., Majidi, A., and Varga, I. (2015). Ultrastructural analysis of different human mesenchymal stem cells after in vitro expansion: a technical review. Eur. J. Histochem. 59:2528. doi: 10.4081/ejh.2015.2528
Miragliotta, V., Donadio, E., Felicioli, A., Podesta, A., Ricciardi, M. P., Ceccardi, S., et al. (2006). Immunolocalisation of desmoglein-1 in equine muzzle skin. Equine Vet. J. 38, 485–487. doi: 10.2746/042516406778400592
Morgan, S. J., Grosenbaugh, D. A., and Hood, D. M. (1999). The pathophysiology of chronic laminitis. pain and anatomic pathology. Vet. Clin. North Am. Equine Pract. 15, 395–417. doi: 10.1016/S0749-0739(17)30152-9
Morrison, S., Dryden, V. C., Bras, R., and Morrell, S. (2014). “How to use stem cells in clinical laminitis cases,” in Proceedings of the 60th Annual Convention of the American Association of Equine Practitioners, (Salt Lake City, UT: American Association of Equine Practitioners (AAEP)), 499–503.
Najor, N. A., and Green, K. J. (2018). Desmosomes in Human Disease. Ann. Rev. Pathol. Mechanisms of Disease 13. doi: 10.1146/annurev-pathol-020117-044030
Nourian, A. R., Baldwin, G. I., Van Eps, A. W., and Pollitt, C. C. (2007). Equine laminitis: ultrastructural lesions detected 24-30 hours after induction with oligofructose. Equine Vet. J. 39, 360–364. doi: 10.2746/042516407X177448
O'grady, S. E. (2002). White line disease - an update equine veterinary education. Equine Vet. Educ. 14, 51–55. doi: 10.1111/j.2042-3292.2002.tb00138.x
Page, R. C., and Schroeder, H. E. (1976). Pathogenesis of inflammatory periodontal disease. A summary of current work. Lab. Invest. 34, 235–249.
Paschos, N. K., Brown, W. E., Eswaramoorthy, R., Hu, J. C., and Athanasiou, K. A. (2015). Advances in tissue engineering through stem cell-based co-culture. J. Tissue Eng. Regen. Med. 9, 488–503. doi: 10.1002/term.1870
Pascucci, L., Mercati, F., Marini, C., Ceccarelli, P., Dall'aglio, C., Pedini, V., et al. (2010). Ultrastructural morphology of equine adipose-derived mesenchymal stem cells. Histol. Histopathol. 25, 1277–1285. doi: 10.14670/HH-25.1277
Pearton, D. J., Yang, Y., and Dhouailly, D. (2005). Transdifferentiation of corneal epithelium into epidermis occurs by means of a multistep process triggered by dermal developmental signals. Proc. Natl. Acad. Sci. U.S.A 102, 3714–3719. doi: 10.1073/pnas.0500344102
Peroni, J. (2013). Regenerative medicine and laminitis: where are we? J. Equine Vet. Sci. 33, 865–866. doi: 10.1016/j.jevs.2013.08.045
Phinney, D. G., and Prockop, D. J. (2007). Concise review: mesenchymal stem/multipotent stromal cells: the state of transdifferentiation and modes of tissue repair—current views. Stem cells 25, 2896–2902. doi: 10.1634/stemcells.2007-0637
Pollitt, C. C. (1992). Clinical anatomy and physiology of the normal equine foot equine veterinary education. Equine Vet. Educ. 4, 219–224. doi: 10.1111/j.2042-3292.1992.tb01623.x
Pollitt, C. C. (1994). The basement membrane at the equine hoof dermal epidermal junction. Equine Vet. J. 26, 399–407. doi: 10.1111/j.2042-3306.1994.tb04410.x
Pollitt, C. C. (1998). The anatomy and physiology of the hoof wall. Equine Vet. Educ. 10, 318–325. doi: 10.1111/j.2042-3292.1998.tb00902.x
Pollitt, C. C. (2016). Lamellar function at the cellular level. Equ. Lamin. 22–38. doi: 10.1002/9781119169239.ch4
Pollitt, C. C., and Daradka, M. (1998). Equine laminitis basement membrane pathology: loss of type IV collagen, type VII collagen and laminin immunostaining. Equine Vet. J. (Suppl.) 26, 139–144. doi: 10.1111/j.2042-3306.1998.tb05133.x
Ponta, H., Sherman, L., and Herrlich, P. A. (2003). CD44: from adhesion molecules to signalling regulators. Nat. Rev. Mol. Cell Biol. 4, 33–45. doi: 10.1038/nrm1004
Qi, Y., Du, Y., Li, W., Dai, X., Zhao, T., and Yan, W. (2014). Cartilage repair using mesenchymal stem cell (MSC) sheet and MSCs-loaded bilayer PLGA scaffold in a rabbit model. Knee Surg. Sports Traumatol. Arthrosc. 22, 1424–1433. doi: 10.1007/s00167-012-2256-3
Raimondo, S., Penna, C., Pagliaro, P., and Geuna, S. (2006). Morphological characterization of GFP stably transfected adult mesenchymal bone marrow stem cells. J. Anat. 208, 3–12. doi: 10.1111/j.1469-7580.2006.00511.x
Ramirez, G. A., Rodriguez, F., Herraez, P., Castro-Alonso, A., Andrada, M., and Espinosa-De-Los-Monteros, A. (2015). Ultrastructural characterization of normal merkel cells in the dog. Vet. Dermatol. 26, 328–333, e368–e329. doi: 10.1111/vde.12230
Ranera, B., Lyahyai, J., Romero, A., Vazquez, F. J., Remacha, A. R., Bernal, M. L., et al. (2011). Immunophenotype and gene expression profiles of cell surface markers of mesenchymal stem cells derived from equine bone marrow and adipose tissue. Vet. Immunol. Immunopathol. 144, 147–154. doi: 10.1016/j.vetimm.2011.06.033
Rodrigues, G. C., Oliveira, L. J., Monteiro, J. M., De Lima, A. R., Gonçalez, P. O., Hernandez-Blazquez, F. J., et al. (2010). Ultrastructural characterization of bovine umbilical cord blood cells. Pesqui. Vet. Bras. 30, 897–902. doi: 10.1590/S0100-736X2010001000015
Rohani, N., Canty, L., Luu, O., Fagotto, F., and Winklbauer, R. (2011). EphrinB/EphB signaling controls embryonic germ layer separation by contact-induced cell detachment. PLoS Biol. 9:e1000597. doi: 10.1371/journal.pbio.1000597
Rucker, A. (2010). Chronic laminitis: strategic hoof wall resection. Vet. Clin. North Am. Equine Pract. 26, 197–205. doi: 10.1016/j.cveq.2009.12.009
Rustad, K. C., Wong, V. W., Sorkin, M., Glotzbach, J. P., Major, M. R., Rajadas, J., et al. (2012). Enhancement of mesenchymal stem cell angiogenic capacity and stemness by a biomimetic hydrogel scaffold. Biomaterials 33, 80–90. doi: 10.1016/j.biomaterials.2011.09.041
Saito, M., Tucker, D. K., Kohlhorst, D., Niessen, C. M., and Kowalczyk, A. P. (2012). Classical and desmosomal cadherins at a glance. J. Cell Sci. 125, 2547–2552. doi: 10.1242/jcs.066654
Sardarabadi, P., Soleimani, M., Atashi, A., Beiranvand, S. P., Rahnama, M. A., and Anbarlou, A. (2016). Gene expression analysis of SOX2, NANOG, KLF4, OCT4, and REX1 genes in cord blood mononuclear cells treated with external stresses. Int. J. Health Stud. 2, 10–13. doi: 10.22100/ijhs.v2i3.150
Schaffler, A., and Buchler, C. (2007). Concise review: adipose tissue-derived stromal cells–basic and clinical implications for novel cell-based therapies. Stem Cells 25, 818–827. doi: 10.1634/stemcells.2006-0589
Senoo, M., Pinto, F., Crum, C. P., and Mckeon, F. (2007). p63 Is essential for the proliferative potential of stem cells in stratified epithelia. Cell 129, 523–536. doi: 10.1016/j.cell.2007.02.045
Shokrgozar, M. A., Fattahi, M., Bonakdar, S., Ragerdi Kashani, I., Majidi, M., Haghighipour, N., et al. (2012). Healing potential of mesenchymal stem cells cultured on a collagen-based scaffold for skin regeneration. Iran. Biomed. J. 16, 68–76. doi: 10.6091/ibj.1053.2012
Smoak, M., Hogan, K., Kriegh, L., Chen, C., Terrell, L. B., Qureshi, A. T., et al. (2015). Modulation of mesenchymal stem cell behavior by nano-and micro-sized β-tricalcium phosphate particles in suspension and composite structures. J. Nanopart. Res. 17:182. doi: 10.1007/s11051-015-2985-6
Sotiropoulou, P. A., Perez, S. A., Salagianni, M., Baxevanis, C. N., and Papamichail, M. (2006). Characterization of the optimal culture conditions for clinical scale production of human mesenchymal stem cells. Stem Cells 24, 462–471. doi: 10.1634/stemcells.2004-0331
Sun, T. T., and Green, H. (1976). Differentiation of the epidermal keratinocyte in cell culture: formation of the cornified envelope. Cell 9, 511–521. doi: 10.1016/0092-8674(76)90033-7
Sundelacruz, S., and Kaplan, D. L. (2009). Stem cell- and scaffold-based tissue engineering approaches to osteochondral regenerative medicine. Semin. Cell Dev. Biol. 20, 646–655. doi: 10.1016/j.semcdb.2009.03.017
Takahashi, H., Shimizu, T., Nakayama, M., Yamato, M., and Okano, T. (2013). The use of anisotropic cell sheets to control orientation during the self-organization of 3D muscle tissue. Biomaterials 34, 7372–7380. doi: 10.1016/j.biomaterials.2013.06.033
Thomason, H. A., Scothern, A., Mcharg, S., and Garrod, D. R. (2010). Desmosomes: adhesive strength and signalling in health and disease. Biochem. J. 429, 419–433. doi: 10.1042/BJ20100567
Tomasello, L., Mauceri, R., Coppola, A., Pitrone, M., Pizzo, G., Campisi, G., et al. (2017). Mesenchymal stem cells derived from inflamed dental pulpal and gingival tissue: a potential application for bone formation. Stem Cell Res. Ther. 8:179. doi: 10.1186/s13287-017-0633-z
Troy, T. C., Arabzadeh, A., and Turksen, K. (2011). Re-assessing K15 as an epidermal stem cell marker. Stem Cell Rev. 7, 927–934. doi: 10.1007/s12015-011-9243-9
Van Eps, A. W., and Pollitt, C. C. (2009). Equine laminitis model: lamellar histopathology seven days after inductionwith oligofructose. Equine Vet. J. 41, 735–740. doi: 10.2746/042516409X444953
Vidal, M. A., Kilroy, G. E., Lopez, M. J., Johnson, J. R., Moore, R. M., and Gimble, J. M. (2007). Characterization of equine adipose tissue-derived stromal cells: adipogenic and osteogenic capacity and comparison with bone marrow-derived mesenchymal stromal cells. Vet. Surg. 36, 613–622. doi: 10.1111/j.1532-950X.2007.00313.x
Vidal, M. A., Walker, N. J., Napoli, E., and Borjesson, D. L. (2012). Evaluation of senescence in mesenchymal stem cells isolated from equine bone marrow, adipose tissue, and umbilical cord tissue. Stem Cells Dev. 21, 273–283. doi: 10.1089/scd.2010.0589
Violini, S., Ramelli, P., Pisani, L. F., Gorni, C., and Mariani, P. (2009). Horse bone marrow mesenchymal stem cells express embryo stem cell markers and show the ability for tenogenic differentiation by in vitro exposure to BMP-12. BMC Cell Biol. 10:29. doi: 10.1186/1471-2121-10-29
Visser, M. B., and Pollitt, C. C. (2010). Characterization of extracellular matrix macromolecules in primary cultures of equine keratinocytes. BMC Vet. Res. 6:16. doi: 10.1186/1746-6148-6-16
Visser, M. B., and Pollitt, C. C. (2011). Immunohistochemical distribution of laminin-332 and collagen type IV in the basement membrane of normal horses and horses with induced laminitis. J. Comp. Pathol. 145, 80–87. doi: 10.1016/j.jcpa.2010.11.009
Walko, G., Castanon, M. J., and Wiche, G. (2015). Molecular architecture and function of the hemidesmosome. Cell Tissue Res. 360, 529–544. doi: 10.1007/s00441-015-2216-6
Xie, L., Zhang, N., Marsano, A., Vunjak-Novakovic, G., Zhang, Y., and Lopez, M. J. (2013). In vitro mesenchymal trilineage differentiation and extracellular matrix production by adipose and bone marrow derived adult equine multipotent stromal cells on a collagen scaffold. Stem Cell Rev. 9, 858–872. doi: 10.1007/s12015-013-9456-1
Zhang, N., Dietrich, M. A., and Lopez, M. J. (2013). Canine intra-articular multipotent stromal cells (MSC) from adipose tissue have the highest in vitro expansion rates, multipotentiality, and MSC immunophenotypes. Vet. Surg. 42, 137–146. doi: 10.1111/j.1532-950X.2013.01091.x
Zhang, N., Dietrich, M. A., and Lopez, M. J. (2014). Therapeutic doses of multipotent stromal cells from minimal adipose tissue. Stem Cell Rev. 10, 600–611. doi: 10.1007/s12015-014-9508-1
Zheng, Y., Hu, C. J., Zhuo, R. H., Lei, Y. S., Han, N. N., and He, L. (2014). Inhibition of autophagy alleviates the senescent state of rat mesenchymal stem cells during long-term culture. Mol. Med. Rep. 10, 3003–3008. doi: 10.3892/mmr.2014.2624
Keywords: mesoderm, ectoderm, ultrastructure, scaffold, horse, stem, keratin
Citation: Yang Q, Pinto VMR, Duan W, Paxton EE, Dessauer JH, Ryan W and Lopez MJ (2019) In vitro Characteristics of Heterogeneous Equine Hoof Progenitor Cell Isolates. Front. Bioeng. Biotechnol. 7:155. doi: 10.3389/fbioe.2019.00155
Received: 19 December 2018; Accepted: 10 June 2019;
Published: 11 July 2019.
Edited by:
Cláudia Lobato da Silva, University of Lisbon, PortugalReviewed by:
Fausto Cremonesi, University of Milan, ItalyRenata Linardi, School of Veterinary Medicine, University of Pennsylvania, United States
Copyright © 2019 Yang, Pinto, Duan, Paxton, Dessauer, Ryan and Lopez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mandi J. Lopez, bWxvcGV6QGxzdS5lZHU=