 David Wu
David Wu Konstantin Birukov
Konstantin Birukov- 1Section of Pulmonary and Critical Care, Department of Medicine, University of Chicago, Chicago, IL, United States
- 2Department of Anesthesia, University of Maryland, Baltimore, MD, United States
Lungs are the most vascular part of humans, accepting the totality of cardiac output in a volume much smaller than the body itself. Due to this cardiac output, the lung microvasculature is subject to mechanical forces including shear stress and cyclic stretch that vary with the cardiac and breathing cycle. Vessels are surrounded by extracellular matrix which dictates the stiffness which endothelial cells also sense and respond to. Shear stress, stiffness, and cyclic stretch are known to influence endothelial cell state. At high shear stress, endothelial cells exhibit cell quiescence marked by low inflammatory markers and high nitric oxide synthesis, whereas at low shear stress, endothelial cells are thought to “activate” into a pro-inflammatory state and have low nitric oxide. Shear stress' profound effect on vascular phenotype is most apparent in the arterial vasculature and in the pathophysiology of vascular inflammation. To conduct the flow of blood from the right heart, the lung microvasculature must be rigid yet compliant. It turns out that excessive substrate rigidity or stiffness is important in the development of pulmonary hypertension and chronic fibrosing lung diseases via excessive cell proliferation or the endothelial-mesenchymal transition. Recently, a new body of literature has evolved that couples mechanical sensing to endothelial phenotypic changes through metabolic signaling in clinically relevant contexts such as pulmonary hypertension, lung injury syndromes, as well as fibrosis, which is the focus of this review. Stretch, like flow, has profound effect on endothelial phenotype; metabolism studies due to stretch are in their infancy.
Introduction
In 2010, cardiovascular diseases accounted for 32% of all deaths (~780,000) in the U.S (NHLBI, 2013). Macrovascular arterial disease pre-disposes to microvascular dysfunction and is classically recognized as a complication in atherosclerosis (Shah et al., 2018), diabetes (Shi and Vanhoutte, 2017), and rheumatoid arthritis (Foster et al., 2010). Not surprisingly, microvascular dysfunction involves dysfunction of the organ proper. The lung has a prominent place in the microvasculature, as its estimated capillary surface area (as defined by diameter of a vessel 10 μm or less) is roughly 50–70 m2, which is one-fourth the size of a tennis court (Weibel, 1973). This is 20-times the surface area of all other vessels in the body combined (Albertine, 2016).
Endothelial cells (ECs) line the vasculature, including arteries, veins, and lymphatics. ECs are continuously subjected to shear stress (frictional force per unit area parallel to the cell) which can range from 10 to 50 dyne/cm2 in large arteries, 5 to 20 dyne/cm2 in the microvasculature, and about 10-fold less in veins (Paszkowiak and Dardik, 2003) [this is variable depending on tissue bed (Remuzzi et al., 1992)]. Shear forces play an especially important role in promoting cell quiescence or activation (Davies, 1995; Chiu and Chien, 2011; Davies et al., 2013). Lymph flow is slower even than microvascular flow, averaging 0.64 dyne/cm2 with peaks of 4–12 dyne/cm2 (Dixon et al., 2006). The specific aspects of flow are complex, vectorial, varying in both space and time (Noris et al., 1995; Blackman et al., 2000; Feaver et al., 2013; Wang et al., 2013). Furthermore, ECs are transcriptionally specialized according to their organ location (He et al., 2018) and accordingly have different responses to shear stress (Reinitz et al., 2015). ECs in the lungs are subject not only to shear stress due to the continuous pulsations from the cardiac cycle, but also to copious stretch due to the respiratory cycle. Stretch forces on the endothelium play a critical role in lung injury during mechanical ventilation (Acute Respiratory Distress Syndrome Network et al., 2000; Birukov et al., 2003; Slutsky and Ranieri, 2013).
Besides flow and stretch, endothelial cells are also responsive to stiffness (i.e., resistance to deformation) in the underlying extracellular matrix. Compared to hemodynamical forces, stiffness forces are much higher. Stresses and stiffness have the same units of measure, which allows for direct physical comparison. To put this into tactile perspective, 100 kPa is about the stiffness of muscle, whereas 1 kPa is about the stiffness of lung tissue (1 Pa = 0.1 dyne/cm2). Thus, stiffness forces are orders of magnitude larger than shear stress. Cellular traction forces or tensile stresses (also force per unit area, but this time a force that tends to lengthen or compress a material) are also much higher than hemodynamic shear stress—typically ~5 kPa at a focal adhesion (Balaban et al., 2001) and endothelial cells exert about 100–300 kPa (Shiu et al., 2004). Intuitively, cellular traction stress should match or be correlated substrate stiffness through force sensing at the focal adhesion (Califano and Reinhart-King, 2010); larger tensile stresses are necessary to overcome substrate stiffness (Balaban et al., 2001). Correspondingly, hemodynamic shear sensor sensors are thought to be activated with much less force (Fang et al., 2019). Substrate stiffness is important in chronic fibrosing lung diseases as well as promoting the endothelial to mesenchymal transformation.
Thus, ECs are mechanosensing machines (Davies, 1995), with the specific mechanical environment having a profound effect on EC phenotype in health and disease. There is now increasing evidence that mechanical forces causing endothelial activation—the conversion of quiescent phenotype to activated phenotype—and endothelial transformation—are associated with a change in cell metabolism (Sawada et al., 2014; Eelen et al., 2015). This review will focus on new developments in shear stress regulation of endothelial metabolism and signaling, and how substrate stiffness affects endothelial cell phenotype. Dysfunction in these pathways leads to the pathogenesis of major diseases of the pulmonary vasculature, including pulmonary hypertension, acute lung injury, and pulmonary fibrosis.
General Aspects of Endothelial Metabolism
Glycolysis is the pathway that converts glucose to pyruvate and then lactate. ECs are highly glycolytic. This was determined either by directly measuring glucose flux in cultured cells (De Bock et al., 2013b) or by inhibition of various energy pathways and measuring the resulting heat flux via calorimetry. In the latter experiment, it was discovered that inhibiting hexokinase (the first step of glycolysis which traps glucose inside the cell) with 2-deoxyglucose significantly reduced heat generated from ECs (Schrimpf et al., 1994). Furthermore, the glycolysis index (lactate/glucose ratio) in cultured human umbilical vein endothelial cells (HUVECs) is around 1.74 (Kim et al., 2017), suggesting that ECs metabolize glucose almost entirely into lactate. Thus, only a small fraction of glycolytic intermediate pyruvate is metabolized by mitochondria. ECs, in contrary to other cell types, are as glycolytic as tumor cells (De Bock et al., 2013b), and use very little oxygen to generate ATP (Schrimpf et al., 1994; Quintero et al., 2006; De Bock et al., 2013b). In support of this, older electron microscopy studies suggested that the total mitochondrial fraction of ECs is only 4–5% (slightly higher, up to 10% in blood brain barrier ECs) compared to 32% in cardiomyocytes (Oldendorf et al., 1977; Barth et al., 1992). This poses an interesting teleological conundrum as the vasculature holds the highest oxygen tension. One thought is that this evolutionary feature lets oxygen diffuse into the organ tissue instead of being consumed by the endothelium. Another hypothesis is that glycolysis powers the EC migratory or endocytotic machinery, whereas the tricarboxylic acid cycle/oxidative phosphorylation (TCA/OXPHOS) system is used for cell growth and division. In this scheme, glycolytic energy is necessary to continually modulate cell boundaries and perform transcytosis of macromolecules, whereas cell proliferation does not regularly occur in adult vasculature (Kim et al., 2017).
A caveat is that these are all in vitro studies and the true in vivo glycolytic rate of ECs is unknown. Furthermore, these studies were all done without hemodynamic shear stress and on cell culture plastic, which is 5 orders of magnitude stiffer than muscle. One in vitro study looked at blood brain barrier endothelial cells and lactate production and found that shear stress reduced lactate production, indicating that EC metabolism is dynamically modulated by hemodynamic forces and that measuring the metabolic properties of ECs without flow would introduce artifact (Cucullo et al., 2011). Another study found that shear stress increased the fraction of mitochondrial ATP production on ECs in vitro (Wu et al., 2017). Furthermore, the natural concentration of lactate in circulating human blood is < 2 mM, with glucose around 5 mM, suggesting a far lower glycolysis index in vivo than what is measured in vitro.
EC metabolism is a topic of intense, recent investigation (Theodorou and Boon, 2018). Briefly, glycolysis is important in mouse models of angiogenesis and lymphogenesis (De Bock et al., 2013b; Yu et al., 2017). Derivatives of the glycolytic pathway are involved in nucleic acid synthesis, NAD/NADH balance, and O-linked glycolsylation (Theodorou and Boon, 2018). Glycolysis plays a central role in energy and intermediate metabolite generation and is required for activation of a diverse set of cell types including immune cell activation, and tumor cell proliferation and survival (Ghesquière et al., 2014). Mitochondrial metabolism is, broadly speaking, important for cell growth. Amino acid metabolism through the TCA cycle (glutamine and its derivative, α-ketoglutarate) enhance anaplerosis (replenishing of TCA cycle intermediates that have been consumed for biosynthesis), critical for cell proliferation (Huang et al., 2017a; Kim et al., 2017). The electron transport chain (ETC) itself is also required for EC proliferation and angiogenesis as well as redox balance in vivo (Diebold et al., 2019). Interestingly, the ETC may be dispensable for EC migration (Diebold et al., 2019). Fatty acid metabolism is critical for de novo nucleotide synthesis, which is needed for EC proliferation in vivo (Schoors et al., 2015) as well as for generation of acetyl-CoA derivatives for histone acetylation in lymphatic ECs for differentiation and fate maintenance (Xiong et al., 2018).
Endothelial Cell Phenotype Is Dictated by Flow
In general, flow can be divided into unidirectional flow (high shear stress) and disturbed flow (low shear stress), or atheroprotective and atheroprone (against atherosclerosis), respectively, in the systemic arterial tree (Dai et al., 2004). Cardiovascular flow type has been shown to be critical for angiogenesis (Galie et al., 2014), cardiovascular development (Vermot et al., 2009; Chen et al., 2012; Franco et al., 2015) in zebrafish and mouse models, and plays a central role in the activation and subsequent inflammation of ECs in the pathogenesis of cardiovascular disease both in vitro and in vivo (Gimbrone and García-Cardeña, 2016). Besides macrovascular disease, flow patterns are also altered in the microvasculature in humans during sepsis (Vincent, 2001; De Backer et al., 2002) or in murine models of acute lung injury (Schmidt et al., 2012).
Tangential shear stress due to flowing blood is proportional to the change in velocity divided by the distance from the vessel wall. High shear stress promotes a healthy endothelium, characterized by quiescence and maintenance of vascular barrier integrity. In contrast, areas of atheroprone or disturbed flow (low shear stress) are sites where the endothelium undergoes “activation,” characterized by inflammation and a reduction in vascular barrier integrity (Dai et al., 2004; Davies et al., 2013). Endothelial cells have a “Goldilocks” set point of preferred shear stress: brain ECs lose barrier function under shear flow when it is twice as much as normal (40 dyne/cm2) (Garcia-Polite et al., 2017) and undergo apoptosis under low shear stress in developing zebrafish brains (Chen et al., 2012).
Shear-based endothelial dysfunction-activation (Liao, 2013) in the macrovasculature provides a useful model upon which to extrapolate mechanotransduction biology in microvascular endothelial cells. In the classical shear stress dependence of endothelial pathophysiology, atherosclerosis occurs at sites of disturbed flow: arterial curvature, branching, and bifurcation, characterized by flow separation, transient flow reversals, and low average shear forces (Davies, 1995). Microscopically, ECs align in the direction of flow, both in vivo and in vitro. In vitro, cells align in unidirectional flow producing a highly elongated shape, but not in disturbed flow, resulting in a cobblestone shape (Davies et al., 1986). Interestingly, in vitro flow that is perpendicular to the long axis of the cell and long axis of the cytoskeletal elements strongly activates inflammatory pathways (Wang et al., 2013). Failure to align with flow in vivo is a hallmark of an atherosclerosis-prone region (Davies, 2008). These atherosclerosis-prone regions have signs of chronic inflammation with increased expression of adhesion molecules and reduction of barrier integrity, all of which develop before any visible signs of disease—thus shear stress by itself could initiate clinical disease (Hajra et al., 2000; Won et al., 2007).
The lung microvasculature flow rate likely has a profound effect on pathophysiology as well. Microvascular endothelial cells are known to migrate against flow at low shear stress toward regions of high shear stress (Ostrowski et al., 2014). Microvascular ECs align with the flow field at lower shear stress levels and, interestingly, perpendicular to shear stress at higher values (>34 dyn/cm2) (Ostrowski et al., 2014). In one study, shear stress and microvascular barrier were fairly independent, but flow opposite of conditioned flow state resulted in a large increase in permeability (Adamson et al., 2013), which is similar to the arterial case described above. Cessation of flow in murine-derived microvascular ECs resulted in production of reactive oxygen species (ROS) and increased proliferation as well as cobblestone morphology (Milovanova et al., 2006). These data together highlight the sensitivity of lung microvascular ECs to shear stress.
Shear Stress Modulation of Endothelial Metabolism
The major regulator of unidirectional flow responses of microvascular ECs was discovered in the lung and is named Krüppel-like factor 2 (KLF2) (Dekker et al., 2002; Lee et al., 2006; Parmar et al., 2006). KLF2 or KLF4 [probably functionally redundant in mouse knockouts (Sangwung et al., 2017)] are thought of as master transcriptional regulators that mediate the vasodilatory, anti-inflammatory, and antithrombotic properties of quiescent endothelium (Atkins and Jain, 2007). Reduced expression of KLF2 or KLF4 has been mechanistically linked to increased expression of inflammatory gene markers in both cultured endothelial cells and in vivo in both the macro- and microvasculature (Lin et al., 2005, 2010; Parmar et al., 2006; Shen et al., 2009; Villarreal et al., 2010) (Figures 1A,D).
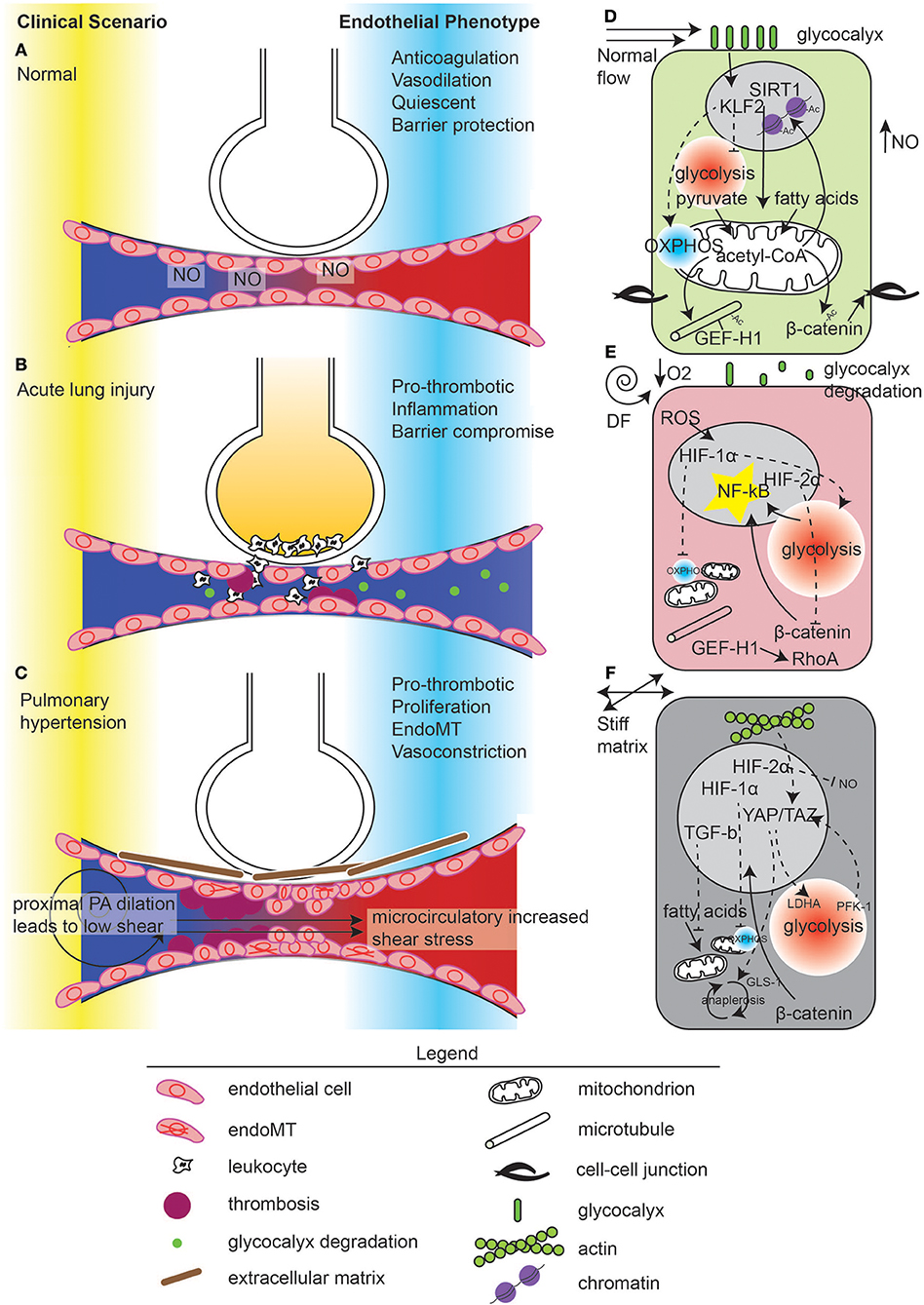
Figure 1. Mechanometabolomic coupling to disease states in the lung.Vascular disease states in the lung in (A) normal, (B) acute lung injury, and (C) pulmonary hypertension. In normal states, ventilation through the alveoli and perfusion occur through nitric oxide (NO) release by the endothelium. Normal microvascular perfusion (D) induces a KLF2 high state which suppresses glycolysis and promotes mitochondrial function, leading to pyruvate-derived acetyl-CoA and acetylation of microtubules, β-catenin, and maintenance of quiescence through chromatin modification. In contrast, during acute lung injury (B), flow becomes disturbed due to microvascular dysfunction, manifest by thrombosis and endothelial contraction leading to edema. Edema in the alveolus causes local hypoxia (E). Inflammation leads to glycocalyx degradation, ROS generation which, with hypoxia, activates HIF-1α. HIF-1α suppresses mitochondrial energy generation and induces glycolysis. De-acetylated microtubules release GEF-H1 for RhoA activation. Glycolysis induces NF-κB activation which can also be enhanced by β-catenin nuclear translocation, due to de-acetylation. However, hypoxia also induces HIF-2α, which inhibits β-catenin translocation. In pulmonary hypertension (C), the upstream pulmonary artery is dilated, reducing shear stress. In the microvasculature, flow is thought to be supra-normal due to thrombosis and vessel stiffness, causing endothelial damage. ECs undergo endoMT and synthesize connective tissue, which exacerbates these hemodynamical phenomena. (F) ECs in response to increased extracellular matrix stiffness stimulate YAP/TAZ activity which enhances glycolysis and anaplerosis, leading to cell proliferation and endoMT. Hypoxia pathways are also activated in pulmonary hypertension.
Shear induced metabolic changes to ECs have been studied in the systemic arterial tree, but these lessons may apply to the microvasculature. Generally speaking, metabolic changes in endothelial cells under disturbed flow in vitro and in vivo include reduced mitochondria mass and function (Chen et al., 2010; Kizhakekuttu et al., 2012), an upregulation of glycolysis via dependence on transcription factor hypoxia inducible factor-1α (HIF-1α) (Feng et al., 2017; Wu et al., 2017), an increase in ROS and advanced glycation end-products (such as in diabetes), and nitric oxide deficiency (Eelen et al., 2015) (such as in atherosclerosis). Additionally, changes in cell metabolism (specifically, upregulation of glycolysis by HIF-1α) are required for endothelial activation and inflammation (Feng et al., 2017; Wu et al., 2017), in addition to angiogenesis (De Bock et al., 2013a,b). Disturbed flow-upregulation of NA(P)DH oxidase 4 (NOX4) and consequent increase in ROS prevents HIF-1α degradation, leading to an increase in glycolytic gene expression. Furthermore, blocking glycolysis by inhibition of major glucose transporter SLC2A1 (GLUT1) can reduce Nuclear Factor-κB (NF-κB) activation in disturbed flow EC models in vitro (Wu et al., 2017). Endothelial specific heterozygous HIF-1α mouse demonstrated reduced inflammation and atherosclerosis (Feng et al., 2017). Additionally, HIF-1α, glycolytic gene expression, and inflammatory gene expression are all increased in the regions of disturbed flow in large arteries of swine (Wu et al., 2017).
On the contrary, unidirectional flow has been shown to reduce glycolysis in a KLF2-dependent manner (Doddaballapur et al., 2015) and increased mitochondria biogenesis in rodent endothelium (Chen et al., 2010; Kim et al., 2014). Suppression of glycolysis in activated ECs and ECs exposed to disturbed flow suppresses inflammatory and angiogenic phenotypes (Doddaballapur et al., 2015; Feng et al., 2017; Wu et al., 2017) whereas chemical suppression of ETC function under unidirectional flow increases EC inflammation in cultured flow models (Wu et al., 2017). These data suggest a role for mitochondrial function in modulating endothelial quiescence. However, no analogous genetic experiment has been performed. KLF2 also has a role in shear stress mediated changes in EC metabolism. KLF2 upregulation via disturbed flow represses 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3), an allosteric regulator of phosphofructokinase 1 (PFK1), which is a critical enzyme in glycolysis (Doddaballapur et al., 2015). PFKFB3 upregulation was shown to promote retinal angiogenesis (De Bock et al., 2013b). Furthermore, blocking PFKFB3 can improve barrier function and reduce cancer adhesion to ECs (Cantelmo et al., 2016). Interestingly, HIF-1α and KLF2 are counter-regulated: KLF2 has been shown to disrupt binding between HIF-1α and its chaperone, HSP90 (Kawanami et al., 2009; Wu et al., 2017).
Due to the relative paucity of mitochondria in ECs, it has been suggested that mitochondria play the role of signaling organelles (Quintero et al., 2006); accordingly, their role in EC biology is complex [angiogenesis, autophagy, ROS signaling, NO bioavailability (Kluge et al., 2013)]. Arterial segments exposed to high shear stress demonstrate increased mitochondrial activity in vivo (Kim et al., 2014). Unidirectional flow, in addition to increasing oxidative phosphorylation (OXPHOS), also upregulates mitochondrial biogenesis in a NAD-deacetylase sirtuin 1 (SIRT1)-dependent manner (Chen et al., 2010; Kim et al., 2015). SIRT1 is a key mediator of angiogenesis (Das et al., 2018) and stimulates endothelial NOS activity (Mattagajasingh et al., 2007). Unidirectional flow dynamically increases mitochondrial spare respiratory potential (Wu et al., 2017) probably in a KLF2/4-dependent manner, as KLFs are upregulated under unidirectional flow (Dekker et al., 2002) and increase mitochondrial biogenesis and metabolic function through transcriptional regulation of PPARγ/PGC1 (Liao et al., 2015). Unidirectional flow may also increase ETC activity through HIF-1α degradation, as HIF-1α is known to regulate the ETC by reducing complex I activity (Tello et al., 2011) and by shunting glucose substrates into glycolysis rather than mitochondria. HIF-1α induces pyruvate dehydrogenase kinase 1 (PDK1), which inhibits pyruvate dehydrogenase (PDH), the enzyme that traps pyruvate inside mitochondria as acetyl-CoA (Kim et al., 2006). Interestingly, the activation of SIRT1 is dependent on the cytoskeleton. In response to shear stress, AMPK phosphorylation of cortactin is followed by SIRT1 deacetylation. Due to deacetylation of cortactin, eNOS is able to translocate away from lipid raft domains, increasing NO bioavailability (Shentu et al., 2016).
Further changes in EC metabolism as a response to low shear stress or disturbed flow include an upregulation of transcription factors YAP/TAZ both in vitro and in vivo (Wang K. C et al., 2016; Wang L. et al., 2016), which also promotes glycolytic metabolism in turn. Yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ), are modulated by mechanotransduction signaling through the cytoskeleton (Dupont et al., 2011), and promotes a pro-inflammatory EC phenotype (Wang K. C et al., 2016; Wang L. et al., 2016). Nutrient and energy sensing also modulate YAP activity (more below) (Enzo et al., 2015; Wang et al., 2015; Bertero et al., 2016; Santinon et al., 2016). Cancer studies indicate that YAP and HIF-1α may interact to promote glycolysis as YAP may localize to the nucleus and prevent HIF-1α degradation (Zhang et al., 2018b). Interestingly, in the absence of disturbed flow, lymphatic endothelial cells undergo TAZ-dependent proliferation (Sabine et al., 2015).
EC quiescence seems to be associated with a mitochondrial phenotype whereas activation is associated with a glycolytic phenotype. Shear stress modulates the metabolic profile based on the endothelial cell bed. These macrovascular lessons are important for microvascular dysfunction, as macrovascular dysfunction causes disturbed flow in microvascular beds (Siasos et al., 2018). Furthermore, microvascular flow is probably highly altered in acute inflammatory disease processes (Vincent, 2001; De Backer et al., 2002; Schmidt et al., 2012). In animal models of sepsis or acute lung injury [sepsis is the top cause of acute lung injury and has a poor prognosis (Force et al., 2012; Bellani et al., 2016)], lung microvascular flow becomes disturbed or oscillatory (De Backer et al., 2002) and leads to microvascular mitochondrial dysfunction (Arulkumaran et al., 2016). In support of this, lung disease often involves pressure changes in the pulmonary vasculature due to either chronic hypoxic vasoconstriction, chronic thromboembolism, vascular obliteration due to plexiform lesions, or vascular apoptosis. The beneficial metabolic effects of constant high shear stress perfusion may actively repress ischemia sensing pathways and prevent reperfusion injury, perhaps accounting for the success of ex vivo lung perfusion technology (Cypel et al., 2011).
Though shear flow, mitochondrial function, and metabolism have been studied in atherosclerosis models of vascular dysfunction, microvascular flow, and metabolism in disease models such as diabetes and pulmonary hypertension or vascular injury are under investigated although glycolytic metabolism in particular is important in ECs in pulmonary hypertension (Xu et al., 2007; Michelakis et al., 2017; Plecitá-Hlavatá et al., 2017; Culley and Chan, 2018).
Shear Stress, Endothelial Metabolism, and Acute Lung Injury
Endothelial injury is critical for the promulgation of the acute respiratory distress syndrome (ARDS) during lung injury. One key mechanism is activation of cytoskeletal contractions which disassembles cell-cell junctions and causes ECs to shrink, leading to pulmonary edema (Kása et al., 2015) (Figures 1B,E).
In inflammatory states, the microvascular endothelium is exposed to disturbed flow and KLF2 is downregulated. In sepsis states, there is disseminated EC dysfunction manifested in part by oscillatory flow (Vincent, 2001; De Backer et al., 2002). In an experimental acute lung injury model [intratracheal lipopolysaccharide (LPS)], microvascular dysfunction due to downregulation of KLF2 leads to disruption of barrier function, and lung parenchymal edema formation. Rescue of depleted pulmonary microvascular KLF2, by intravascular delivery of KLF2 mRNA using nanoparticles was able to protect against pulmonary edema and neutrophil infiltration (Huang et al., 2017b). Mechanistically, lung microvascular ECs, when treated with TNFα, are unable to sense shear stress and lose their shear-imposed KLF2 upregulation, leading to loss of barrier function in vitro (Huang et al., 2017b).
One possible reason for ECs to becoming insensate to high shear stress is that under TNFα treatment in vivo, the pulmonary endothelial glycocalyx is lost (Schmidt et al., 2012). Glycocalyx is an essential part of the flow sensing apparatus in endothelial cells (Florian et al., 2003; Mochizuki et al., 2003; Baeyens et al., 2014; Yen et al., 2015). Endothelial glycocalyx is critical in maintaining capillary fluidity and perfusion homogeneity in the microvasculature (Mcclatchey et al., 2016). Glycocalyx is reduced in sepsis, diabetes, heart failure and sickle cell disease, suggesting a connection between mechanical sensing, NO production, and microvascular perfusion (Cabrales et al., 2007; Mcclatchey et al., 2016). Interestingly, normal shear stress remodeling of the actin cytoskeleton was changed after disruption of the glycocalyx (Thi et al., 2004) and orientation of the cells in response to flow was changed (Moon et al., 2005). Components of the glycocalyx interact with focal adheions (Zimmermann and David, 1999; Rapraeger, 2000) and also with G-protein coupled receptors (Davies et al., 1995). Pulmonary derived glycocalyx shedding may also be systemically toxic and damage other microvascular beds, causing acute kidney injury in clinical studies (Schmidt et al., 2016).
The connection between downregulation of KLF2 in endothelial dysfunction, an increase in HIF-1α-mediated glycolysis and worsened lung injury seems likely however has not been directly shown (Doddaballapur et al., 2015; Feng et al., 2017; Wu et al., 2017). Clinical data indicating that poor gas exchange occurs during acute lung injury suggests that the lung is locally hypoxic (Force et al., 2012). In ARDS, pulmonary lactate is increased, suggesting an upregulation of glycolysis has occurred (Brown et al., 1996; De Backer et al., 1997). Additionally, in a rat proteomic study of acute lung injury, glycolytic enzymes were discovered to be upregulated (Liu et al., 2014). Whereas resting humans have been shown to mount a lactate response to inspired O2 levels around 10% (Huckabee, 1958), HIF-1α stabilization in vitro occurs at <5% O2 in hypoxia chambers (Bracken et al., 2006); it is therefore reasonable to assume that tissue hypoxia occurs in vivo as tissue oxygenation must be below that of arterial oxygenation. In addition to frank hypoxia, HIF-1α can be stabilized in pseudo-hypoxic states in vivo which does not require such extreme levels of hypoxia. This accounts for the findings of focal HIF-1α expression in large arteries of pigs (Wu et al., 2017). Thus, persistent lung (real or pseudo-) hypoxia may allow for local HIF-1α expression (Mcclendon et al., 2017). A role for HIF-1α in the lung was also shown in a septic lymph model of sepsis, also showing that HIF-1α is activated in a pseudo-hypoxic manner (Sun et al., 2017). Compared with control, microvascular ECs treated with shRNA against HIF-1α incubated with septic lymph demonstrated lower levels of TNF-α, IL-6, and IL-1β cytokines, along with increased viability (Sun et al., 2017). Interestingly, HIF-1α in alveolar epithelial cells may be protective against lung injury (Mcclendon et al., 2017; Suresh et al., 2018).
In addition to HIF-1α, HIF-2α is also expressed in lung injury, and may act to protect against vascular barrier dysfunction (Gong et al., 2015). Notably, HIF-2α is also activated by disturbed flow (Wu et al., 2017). HIF-2α upregulation was shown to be protective in a lung injury mouse model as it promotes endothelial barrier maintenance through regulation of a vascular phosphotyrosine phosphatase (VE-PTP or PTPRB) which promotes endothelial cadherin junction maintenance (Gong et al., 2015). In a search for promoter sequences of genes that regulate adherens junction integrity, VE-PTP was found to be a transcriptional target of HIF-2α. Endothelial specific HIF-2α deletion had worse outcomes in the acute lung injury model due to decreased barrier function. The authors hypothesized that hypoxia induced stabilization of EC barrier is mediated by HIF-2α. Interestingly, VE-PTP also dephosphorylates TIE2 and ANG2 and regulates VEGFR2. This suggests that chronic ischemia, by inducing HIF-2α, may protect against hypoxic lung injury.
Metabolic Regulation of the Cytoskeleton in Pulmonary Microvasculature Through Acetyl-CoA
The role of the cytoskeleton in acute lung injury involves acetylation of microtubules. Acetylation of α-tubulin is known to promote microtubule stability (Szyk et al., 2014). Acetylation requires the key intermediate metabolite acetyl-CoA, which is the sole donor of acetyl groups for acetylation (Choudhary et al., 2014). Acetylation of proteins involves the mitochondria, as acetyl-CoA, the substrate of acetylation, is generated either by pyruvate dehydrogenase and subsequent export of acetyl-CoA into the cytoplasm via the malate-aspartate transporter, or from glutamine reductive decarboxylation or through acetate, the two main mitochondrial-independent ways in which acetyl-CoA is generated (Pietrocola et al., 2015).
Acetylation also affects epigenetic functions of chromatin. Indeed, fatty acid-derived acetyl-CoA (which is generated in the mitochondria) was found to be critical for maintaining lymphatic differentiation in vivo (Xiong et al., 2018). Thus, mitochondria function is critical for acetylation. As mitochondria activity of ECs is enhanced by unidirectional flow and suppressed by disturbed flow (Wu et al., 2017), it is critical to determine how mitochondria function, acetylation, and lung injury are linked.
In acute lung injury models, multiple studies invoke a final common pathway of RhoA-induced activation of myosin light chain, causing EC contractions and pulmonary vascular leak. Microtubules (MTs) can modulate RhoA activity because Rho-specific guanine exchange factors (GEFs) can bind MTs; disruption of MT network by oxidative stress causes release of GEF-H1 which activates Rho signaling in an LPS-dependent manner (Kratzer et al., 2012). Furthermore, GEF-H1 bound to MTs is dependent on microtubule acetylation. For instance, cigarette smoke exacerbates LPS induced ALI in vivo and EC barrier dysfunction in vitro via increased susceptibility to histone deacetylase 6 (HDAC6) phosphorylation, which deacetylates MTs and reduces barrier function, thus promulgating ALI (Kratzer et al., 2012; Borgas et al., 2016; Karki et al., 2019). In concordance, HDAC6 null mice were resistant to LPS-induced ALI (Zhang et al., 2008). Selective HDAC6 inhibition by tubastatin A blocked TNFα-induced lung endothelial cell hyperpermeability, which was associated with increased α-tubulin acetylation and microtubule stability (Yu et al., 2016). Mechanically, increased stiffness also can activate GEF-H1 expression and thereby exacerbate LPS-induced lung inflammation (Mambetsariev et al., 2014).
Thus, acetylation levels, and hence, acetyl-CoA metabolism and mitochondrial function, may be critical for vascular permeability in ALI, and is downstream of mechanical loading. Interestingly, HDAC6 may also regulate the canonical Wnt/β-catenin pathway. β-Catenin HDAC6-dependent deacetylation causes β-catenin nuclear translocation and disassembly of adherens junctions (Li et al., 2008); on the contrary, β-catenin acetylation promotes its membrane localization thus stabilizing adherens junctions (Valenta et al., 2012; Iaconelli et al., 2015).
Endothelial Shear Stress in Pulmonary Hypertension
Shear stress in the pulmonary vasculature is altered in pulmonary arterial hypertension (PAH). Only through a combination of computational fluid dynamics and four-dimensional MRI has it recently become possible to measure wall shear stress in pulmonary arteries, as it was done to model carotid artery flow (Dai et al., 2004). PAH patients have more tortuous pulmonary artery branch vessels and larger proximal arteries (3.5 ± 0.4 cm compared to 2.7 ± 0.1 cm in healthy subjects) (Tang et al., 2012). This corresponds with radiology literature that increased main pulmonary artery diameter is highly specific for diagnosis of PAH (Kuriyama et al., 1984).
Additionally, the main pulmonary artery flow rate was significantly lower than in PAH patients by on average 1.1 L/m/m2. This combination of reduced flow rate and increased arterial diameter suggests that the shear stress is lower globally. Importantly, this may lead to a reduction in NO release from the endothelium (Hakim, 1994). In one patient during diastole, there was hardly flow at all (Tang et al., 2012). The calculated mean WSS of healthy subjects was almost 5-fold higher in healthy controls compared to PAH patients (20.5 ± 4.0 vs. 4.3 ± 2.8 dyne/cm2). Similarly, in distal pulmonary arteries, mean WSS was 1.4x higher in normal subjects than PAH subjects. Separate studies also found differences (2–3 fold) in shear stress between control and PAH patients (Barker et al., 2015; Odagiri et al., 2016; Schäfer et al., 2016; Zambrano et al., 2018). These values correlate with a reduction in NO bioavailability in PAH patients (Wolff et al., 2007; Gabrielli et al., 2011). This suggests that pruning of the distal vasculature in PAH may be a way for the lung to preserve microvascular perfusion—by increasing microvascular resistance, shear stress is elevated (Allen et al., 2014).
Counterintuitively, in PAH the microvasculature may also be subject to high shear states [such as in congenital heart disease (Gatzoulis et al., 2009)] or high oscillations in flow due to increased stiffness in the pulmonary arteries. Though the wall shear stress decreases in the larger pulmonary arteries, the pulsatility may increase in the microvasculature, which is how microvascular dysfunction is coupled to macrovascular dysfunction. Simply put, as the stiffness of arteries increases, cardiac pulsations are no longer dampened and instead are transmitted into the microvasculature. These high energy pulses can even be reflected back during, for instance, encounter with pulmonary thrombus (Castelain et al., 2001; Nakayama et al., 2001). Interestingly, in a rat model of PAH, reducing the flow to the lung via banding prevented development of plexiform lesions and inflammatory cell infiltrates, suggesting a causative role for force transmission in the development of PAH (Abe et al., 2016). However, the rats who had pulmonary artery banding had worse right ventricular function; moreover, hemodynamics were not measured. It is interesting that patients with a vasodilator challenge responsive PAH phenotype have increased survival (Rich and Brundage, 1987)—this suggests that reducing microvascular shear stress or pulsatility may improve PAH.
Mechanistically, microvascular cells isolated from patients with PAH demonstrated delayed shear adaptation and thus promoted shear induced endothelial injury and vascular remodeling (Szulcek et al., 2016). Additionally, pulmonary artery ECs subjected to high pulsatility but same mean shear stress exhibited increased NF-κB activation, inflammation, and extreme cell elongation, which could all be revered by microtubule stabilization with taxol (Li et al., 2009a, 2013). Decoupling the contributions from microvascular shear stress, pulsatility, oscillation index, and right ventricular function, not to mention cyclic stretch from breathing, would be necessary to fully work out the most important mechanistic and physiological parameters to survival in PAH.
ECs Are Metabolically Dysregulated in Pulmonary Hypertension
Morphologically, PAH is characterized by plexiform lesions, or disorganized endothelial cell growth, which is thought to be due to dysregulation of apoptosis and proliferation (Teichert-Kuliszewska et al., 2006; Abe et al., 2010). Indeed, isolated PAH patient cells have increased proliferation (Duong et al., 2011; Kim et al., 2013). Theoretically, the loss of BMPR2 (a hereditary mutation pre-disposing to PAH) pre-disposes cells to undergo apoptosis, and therefore causes the emergence of an apoptosis-resistant clonal cell population (Taraseviciene-Stewart et al., 2001). This suggests that a cycle of continual injury and healing happens in the pulmonary microvasculature is critical for the pathogenesis of PAH plexiform lesions. Correspondingly, patients with idiopathic pulmonary arterial hypertension (IPAH) showed increased uptake of fluorodeoxyglucose uptake in pulmonary arteries compared to controls. Furthermore, the glycolytic rate of endothelial cells derived from IPAH patients is 3-fold higher than controls. In IPAH cells, ATP content under hypoxia shows no decrease compared to normoxia, whereas ATP decreased by 35% in control (non-IPAH) cells, suggesting a greater dependence on respiration in control cells or reduced overall metabolic rate (Xu et al., 2007).
Mitochondria function in ECs is altered in PAH and can be reversed by promoting oxidative phosphorylation in rats (Rehman and Archer, 2010). Mechanisms include activation of HIF-1α, PDK-1, and downregulation of superoxide dismutase-2 (Rehman and Archer, 2010). The mitochondrial membrane potential, mitochondrial DNA, and regulators of mitochondrial biogenesis are reduced in a mouse BMPR2 ec−/− PAH model (Diebold et al., 2015). Notably, BMPR2 knockdown in normoxia also pre-disposes ECs to increased glycolysis (Diebold et al., 2015). Glycolysis is increased by activation of pyruvate kinase muscle (PKM) via a miR-124 induced splicing switch (from PKM isoform 1 to 2). Downregulation of miR-124 resulted in increased expression of PTBP1 splicing factor and enhanced glycolysis (Caruso et al., 2017). How the downregulation of OXPHOS, yet upregulation of anaplerosis interact (as suggested, through YAP/TAZ below) is unknown.
HIF-1α also decreases the expression of Fe-S cluster assembly ISCU1/2 by induction of microRNA-210 and therefore the electron transport chain components. This causes reduced mitochondrial respiration (decreased aconitase and Complex I activity) (Chan et al., 2009). NFU1 and BOLA3 (Fe-S scaffold genes) are critical for Fe-S dependent synthesis of lipoic acid, an important fatty acid which is a covalent moiety of mitochondrial enzymes including the E2 subunit of PDH, which regulates oxidative metabolism, and BOLA3 expression is downregulated by hypoxia in ECs in PH (Cameron et al., 2011; Yu et al., 2019). BOLA3 controls Fe-S integrity and mitochondria complex protein levels and regulates respiratory complex activity in pulmonary artery ECs (PAECs). Lipoate modified PDH is critical for PDH function. It is unclear how lipoation and phosphorylation interact with each other for PDH activity. BOLA3 knockdown expectantly increased glycolysis but also increased oxygen consumption. BOLA3 knockdown increased PAEC proliferation and angiogenesis in vitro and in vivo assays (Yu et al., 2019).
NOTCH1 as a Mechanotransducer of Metabolism in Pulmonary Hypertension (PH)
Endothelial cell metabolism-epigenetic regulation of cell proliferation/angiogenesis (Hautefort et al., 2017; Xu et al., 2017) and migration (Zheng et al., 2012) is also regulated by cell-cell contact. NOTCH1 is a known sensor of high shear stress in arteries and stabilizes cellular junctions through contact dependence (Mack et al., 2017). NOTCH1 signaling is required to coordinate metabolism and gene regulation that is critical for proliferation of stalk cells in contact with migrating tip cells (De Bock et al., 2013a). NOTCH1 is also downstream of BMPR2 which is implicated in PAH. BMPR2 is required for NOTCH1 activation; deletion of NOTCH1 in ECs worsens hypoxia-PH (Miyagawa et al., 2019). Bmpr2+/− mice demonstrate histological features of PAH such as impaired re-endothelialization, pulmonary neointimal thickening, and medial hypertrophy, suggestive of abnormal arterial remodeling. In endothelial specific NOTCH1 knockout mice, pulmonary hypertension was more pronounced under hypoxic conditions (Miyagawa et al., 2019).
It turns out that mutations or expression changes (Liu et al., 2017) in BMPR2 causes EC mitochondrial dysfunction (Diebold et al., 2015). NOTCH1 enhances glycolysis and mitochondria metabolism and promotes histone acetylation at enhancer binding sites for NOTCH1 and its target MYC, possibly responsible for wound healing after repeated injury (Miyagawa et al., 2019). Importantly, PFKFB3 is downstream of NOTCH1 signaling. NOTCH1 also increases mitochondrial replication and oxygen consumption rate due to a NOTCH1 intracellular domain-dependent mitochondrial transcription and replication. Interestingly, the authors postulated that PFKFB3 increased the production of acetyl-CoA through increased glucose-derived pyruvate flux through the mitochondria, which was also upregulated by NOTCH1 (Miyagawa et al., 2019).
Stiffness Accounts for Morbidity and Mortality in Vascular Diseases
Endothelial substrate stiffness regulates cardinal microvascular functions including angiogenesis (Zhao et al., 2018), monolayer integrity (Birukova et al., 2013; Andresen Eguiluz et al., 2017; Urbano et al., 2017), immune cell transmigration (Stroka and Aranda-Espinoza, 2011; Schaefer and Hordijk, 2015). In the systemic arterial circulation, increased vascular stiffness is associated with and precedes systemic hypertension (Beltran et al., 2001; Weisbrod et al., 2013). Furthermore, in a computational model of baroreceptors as strain receptors, arterial stiffening is by itself sufficient to explain primary hypertension (Pettersen et al., 2014). An increase of macrovascular stiffness is an independent predictor of cardiovascular morbidity (Benetos et al., 2012; Smulyan et al., 2016) and mortality (Laurent et al., 2001; Pannier et al., 2005). Increased stiffness (or reduced compliance) which equates with systolic hypertension and lower diastolic blood pressure (Safar et al., 2013) also impacts the shear stress experienced by vascular endothelia by increasing the pulse wave velocity, as described above. Impaired endothelial function was independently and inversely related to pulse wave velocity, a measure of vascular stiffness in a large-scale study among healthy participants (Mceniery et al., 2006), and is probably a measure of endogenous nitric oxide response (Kinlay et al., 2001). Chronic vascular smooth muscle changes also contribute to vascular stiffness (Lacolley et al., 2017). Indeed, there is now an emerging concept of modulating stiffness properties of the vasculature in order treat hypertension. Current standard of care antihypertensive treatment not only improves mortality (Brunström and Carlberg, 2018) but is now thought to have an anti-stiffness component which modulates this important outcome (Chen et al., 2017).
Macrovascular stiffness promotes microvascular damage (Mitchell, 2008; Cardoso and Salles, 2016; Cooper et al., 2018) and therefore end-organ damage and eventual end-organ failure. The mechanism is likely dysregulated transmission of large shear stresses, pulsatility, or dysregulation of autoregulation of blood pressure at the capillary bed level. These large pulse waves contribute to EC permeability and inflammatory response to vasoactive agonists through stiffness-dependent control of RhoA GTPase activity (Birukova et al., 2013; Mambetsariev et al., 2014; Meng et al., 2015). Increased flow pulsatility also increases cytokine expression and enhanced leukocyte adhesion and reduced NO/upregulated endothelin expression in a microvascular model of stiffened pulmonary microcirculation (Li et al., 2009a,b). Stiffness also increases endothelial inflammation via NF-κB in an in vitro model of PAECs and isolated human tissue (Tan et al., 2014).
Pulmonary vascular stiffness plays an important role in the progression of pulmonary hypertension and is highly associated with mortality (Mahapatra et al., 2006; Gan et al., 2007; Campo et al., 2010; Thenappan et al., 2016), and when added to pulmonary vascular resistance is better at predicting clinical outcomes (Hunter et al., 2008). Wall stiffness increased from 0.5 to 2.0 MPa; correspondingly, the compliance decreased from 2.9 to 0.3 ml/mmHg (Zambrano et al., 2018), and capacitance and elasticity were also reduced in a PAH cohort (Schäfer et al., 2016).
Stiffness and Metabolism in ECs in Pulmonary Hypertension
Extracellular matrix stiffness increases EC glycolysis and reduces mitochondrial oxygen consumption with concomitant increases in glutaminolysis in cultured PAECs. Glutaminolysis is the conversion of glutamine to glutamate which feeds into the TCA cycle as α-ketoglutarate. These PAECs also exhibited increased proliferation and collagen deposition (Bertero et al., 2016). Glutaminolysis is essential for replenishing the TCA cycle during anaplerosis. Further evidence for glutaminolysis was found in PAH patients with hereditary BMPR2 mutation whom exhibited enhanced blood levels of glutamine, glutaminolysis and exhibited hyperproliferative phenotype in their ECs. Interestingly, the requirement for glutamine was driven by normoxic stabilization of HIF-1α and loss of SIRT3 in microvascular cells deficient of BMPR2 (Egnatchik et al., 2017) (Figures 1C,F).
Stiffness originates in the extracellular matrix. In a monocrotaline rat model of PH, and humans with PAH, there was increased collagen deposition (Bertero et al., 2015). Isolation of pulmonary arteries from PAH patients, and in hypoxia rodent models of PAH, revealed increased expression of markers of extracellular matrix remodeling via lysyl oxidases (Nave et al., 2014). Microvascular ECs, as well as smooth muscle cells, are noted to have increased alpha smooth muscle actin (SMA) expression, suggesting a role for a fibroblastic phenotype ]or endothelial-mesenchymal transition (endoMT)] in matrix deposition and contractility (Li et al., 2009b; Scott et al., 2013). Furthermore, targeting vascular matrix production with lysyl oxidase inhibitors can improve PAH (Nave et al., 2014). Stiffness is also directly linked to metabolic signaling through HIF-1α in the pulmonary microvasculature. Hypoxic metabolic modeling of ECs has revealed that they deposit more collagen under HIF-1α expression (Stenmark et al., 2006; De Jong et al., 2016). As recent literature has shown that collagen production requires enhanced glycolytic flux, as well as glutamine metabolism (Nigdelioglu et al., 2016; Hamanaka et al., 2019), stiffness induced metabolic changes undoubtedly plays a role in the pathophysiology of PAH.
Stiffness, YAP/TAZ, and Metabolism
Surface stiffness by itself can cause mesenchymal stem cell differentiation (Engler et al., 2006). For instance, neuronal differentiation programs are activated on soft surfaces (0.1–1 kPa), whereas muscle or bone differentiation programs are activated by hard surfaces (10–100 kPa). For reference, tissue culture plastic is in the ~106 kPa range. The cytoskeleton plays a critical role in force transmission as these effects were mitigated by inhibitors of non-muscle-myosin II. Endothelial cells also demonstrate a strong cell shape dependence in their phenotype that is driven by surface stiffness and extracellular matrix contact (Chen et al., 1997). Investigations into factors which drive the transcriptional output of mammary epithelial cells based on low vs. high matrix stiffness led to the discovery of YAP and TAZ as being regulated by high stiffness (Dupont et al., 2011). YAP/TAZ have been shown to be activated in epithelial, fibroblast, endothelial, oncogenesis, neurons, and stem cells (Lian et al., 2010; Liu et al., 2015; Furukawa et al., 2017; Totaro et al., 2017; Chen et al., 2019). YAP/TAZ activity is regulated by interaction with the actin cytoskeleton—when perturbed by cytoskeleton remodeling, YAP is able to be de-phosphorylated and translocated into the nucleus. YAP/TAZ activate downstream pathways that are known to increase fibrotic pathways resulting in the synthesis of ECM (Totaro et al., 2018).
YAP/TAZ is controlled by and itself modulates cellular metabolism. In a rat pulmonary hypertension model, YAP promotes glutaminolysis through transcriptional activation of glutaminase (GLS1) in endothelia. Interestingly, in HUVECs glutamine was shown to be critical for cell growth but dispensable for cell migration (Kim et al., 2017). Stiffness-associated alterations in glycolysis through direct transactivation of lactate dehydrogenase are also in part due to YAP (Bertero et al., 2016). It has also been demonstrated recently that lipid accumulation is accelerated by YAP and TAZ in hepatocytes. Whether a similar mechanism exists in endothelial cells is unknown (Aylon et al., 2016; Jeong et al., 2018). Thus, it can be said that YAP/TAZ activation primes cellular metabolism for growth.
Cellular metabolism/energetics also drives YAP/TAZ: in the absence of favorable energy substrates or conditions, cellular growth or proliferation should be limited. Although the following studies were not performed in ECs, some of their findings may still be applicable, especially as it relates to energy/metabolic sensing. Depriving cells of glucose or inhibiting glycolysis reduces YAP/TAZ transcriptional activity through AMPK phosphorylation of AMOTL1, which inhibits YAP (Deran et al., 2014). AMPK also interacts with LATS in addition to directly phosphorylating YAP during energy starvation (Gailite et al., 2015; Mo et al., 2015). Conversely, energy abundance promotes YAP/TAZ transcriptional activity. PFK-1, the first enzyme that commits glucose to glycolysis, can bind YAP/TAZ TEADs to promote YAP/TAZ transcriptional programming (Enzo et al., 2015). In hyperglycemic conditions, the hexosamine biosynthesis pathway is induced and leads to O-GlcNAcylation of YAP, causing YAP-dependent transcription (Teo et al., 2010; Zhang et al., 2017). Unsaturated fatty acids may also stimulate β-catenin -dependent YAP/TAZ activity (Noto et al., 2017).
YAP/TAZ activation in epithelial and mesenchymal stem cells mimics increased ECM stiffness as in ECs. YAP is induced after injury and promotes wound healing (proliferation, migration) (Wang et al., 2012; Kimura et al., 2016) in smooth muscle atherosclerosis models in vivo. Mechanical feedback through matrix sensing/remodeling is responsible for smooth muscle growth in pulmonary hypertension models (Bertero et al., 2015; Kudryashova et al., 2016; Dieffenbach et al., 2017). YAP/TAZ also promotes the development of pro-fibrotic phenotypes in fibroblasts in in vitro models of idiopathic pulmonary fibrosis (Liu et al., 2015).
Endothelial-Mesenchymal Transition (EndoMT) Is Related to Shear Stress, Stiffness, and Metabolic Changes
ECs display a significant amount of plasticity especially in pathological conditions. The endoMT, analogous to epithelial-mesenchymal transition, is a process by which endothelial cells have reduced expression of endothelial markers and increased expression of mesenchymal genes. EndoMT accounts for up to 30% of cancer associated fibroblasts in a murine melanoma model (Zeisberg et al., 2007a) and contributes to fibroblast stroma in a breast cancer model (Kidd et al., 2012). EndoMT is also thought to contribute to cardiac fibrosis (Zeisberg et al., 2007b). EndoMT has been shown to be involved in the pathogenesis of atherosclerosis and is thought to contribute to pulmonary fibrosis and pulmonary hypertension. Characteristic endoMT genes associated with fibroblast line include PDFGRα, SOX9, COL1A1, COL1A2, FSP1, vimentin, α-SMA, and periostin although the markers have different degrees of lineage specificity (Li et al., 2018).
EndoMT is dependent on mechanical forces including shear stress and stiffness. A time dependent shear stress/RNAseq study revealed that endoMT develops over time in cultured ECs exposed to oscillatory shear stress (disturbed flow) whereas pulsatile shear stress (unidirectional flow) did not induce endoMT genes. Metabolically, energy sensors AMPK and SIRT1 could inhibit oscillatory shear-induced endoMT. Mechanistically, compared to pulsatile flow, oscillatory flow increased DNA methylation of promoter regions of characteristic EC genes such as VWF, CD31, and Cadh5. However, there was decreased methylation of Cadh2, FSP1, and vimentin, markers of mesenchyme. The link between methylation and oscillatory flow is unknown (Lai et al., 2018). Correspondingly, statins and metformin are able to suppress endoMT (Osman et al., 2006).
EndoMT is also dependent on substrate stiffness. TGF-β-induced endoMT occurs preferentially on stiffer substrates and is inhibited by blockage of β-catenin/Wnt signaling pathway (Zhong et al., 2018). Furthermore, YAP is critical for developmental TGF-β-induced endoMT during heart valve mesenchyme formation (Zhang et al., 2014). Metabolically, TGF-β-induced endoMT is accompanied by inhibition of fatty acid oxidation, which is also required for endothelial cell proliferation through de novo nucleotide synthesis (Schoors et al., 2015). Inhibition of fatty acid oxidation alters intracellular acetyl-CoA levels, which maintains endothelial cell identity (Xiong et al., 2018).
Pulmonary Hypertension, Metabolism, and EndoMT
There is increasing evidence that endoMT contributes to EC dysfunction in the development in PAH and is regulated by HIFs (Good et al., 2015). Endothelial HIF-2α contributes to PAH through endoMT (Tang et al., 2018) leading to enhanced proliferation rate and down regulated prolyl hydroxylase 2 (and hence reduced ubiquitination-degradation of HIFs) (Dai et al., 2016). HIF-2α is thought to drive transcription of endoMT genes through SNAI1/2 (Snail) in a mouse hypoxia model whereas HIF-1α was dispensible; HIF-1α also did not regulate SNAI1/2 (Tang et al., 2018). However, in a separate rat hypoxia model, HIF-1α drove endoMT under Twist1 (Zhang et al., 2018a); additionally, SNAI1/2 was found to be a direct target of HIF-1α in endoMT of coronary endothelial cells (Xu et al., 2015). In an inducible HIF-1α endothelial-specific knock out mouse model, it was discovered that HIF-1α was necessary for formation of pulmonary hypertension in a chronic hypoxia model and fibrosis in a bleomycin model. Furthermore, HIF-1α expression was partially responsible for CTGF expression, an indicator of endoMT in PMVEC in pulmonary fibrosis induced pulmonary hypertension (Bryant et al., 2016).
Dysregulation of nitric oxide synthesis or reduction in NO bioavailability is also a hallmark of PAH. Nitric oxide generation requires arginine metabolism and is catalyzed by endothelial nitric oxide synthase. It turns out that in mice, HIF-1α regulates NOS2/iNOS and HIF-2α regulates Arg1 and Arg2. Arg2 is implicated in reducing airway NO and promotes remodeling and collagen deposition in PAH patients. Arg1 also decreases NO. Thus HIF-2α promotes PAH by upregulation of Arg1 and Arg2 (Cowburn et al., 2016). Interestingly, only the endothelial HIF-1α knockout model accrues severe right ventricular dysfunction (emblematic of PAH) in mouse models, whereas HIF-2α knockout does not; furthermore, patients with IPAH had higher HIF-2α levels than HIF-1α (Dai et al., 2016).
Pulmonary Fibrosis, Stiffness, and EndoMT
Bleomycin lung injury models are a classical mouse model of pulmonary fibrosis, in which the lung parenchyma becomes markedly stiff. Isolating ECs on day 7 and 21 of after treatment of bleomycin, one group found that ECs had increased expression of TGF-β, connective tissue growth factor and platelet-derived growth factor-C, compared with untreated lungs. Furthermore, those isolated ECs had decreased NO production, increased α-SMA, and collagen production compared to control (Kato et al., 2018). It is thus possible that lung ECs give rise to some portion of the mesenchymal population in pulmonary fibrosis in addition to the resident intrapulmonary mesenchymal cells, which constitute between 10 and 20% of the total cell population in the lung (Barron et al., 2016). In comparison, the total lung endothelial population constitutes around 30% of the total lung cells (Crapo et al., 1982). Using a pan-endothelial LacZ expression system, a bleomycin injury experiment showed coincident immunostaining of X-gal, COL1A1, and α-SMA, suggesting bleomycin-induced expression of mesenchymal lineage markers by lung endothelial cells. In this series of experiments, endoMT accounted for around 16% of bleomycin-induced fibroblasts. These results were recapitulated in vitro and established a role for ECs in promoting pulmonary fibrosis through endoMT in a Ras and TGF-β-dependent manner (Hashimoto et al., 2010).
The molecular mechanisms underpinning endoMT in pulmonary fibrosis are just starting to be elucidated but may be related to stiffness sensation. Endothelial HSP1 (HSPB1) suppresses markers of endoMT such as α-SMA after TGF-β stimulation. Furthermore, HSPB1 knockdown increased radiation induced endoMT in lung (Choi et al., 2016). A possible mechanism is through HSPB1 activation of Snail degradation (Wettstein et al., 2013). Snail is a transcription factor known to induce fibrosis in kidneys (Boutet et al., 2006) and epithelial-mesenchymal transition in pulmonary fibrosis (Jayachandran et al., 2009). HSPB1 also interacts with actin and is thought to regulate actin filament dynamics as it inhibits actin polymerization in vitro. HSPB1 colocalizes with cortical actin and is involved in EC adhesion and motility (Trott et al., 2009). Perhaps HSPB1 acts as a stress sensor, as cells depleted of HSPB1 are unable to close wounds or tissue planes (Doshi et al., 2009). This raises the possibility that disruption of tension homeostasis through the actin cytoskeleton might play a role in endoMT and may be a universal driver in development as well as fibrosis. Indeed, YAP/TAZ has been implicated in fibroblast activation during lung fibrosis (Liu et al., 2015; Noguchi et al., 2017) and thus likely plays a role in endoMT. On the flip side of protection against TGF-β-dependent endoMT, one study implicated TGF-β-dependent CXCR7 expression in ECs as a mechanism for negative feedback inhibition of TGF-β-induced endoMT by suppressing Notch signaling (Guan and Zhou, 2017).
Conclusion and Emerging Hypotheses
Altered mechanosensation with a reduction in shear stress mimics a reduction in cardiac output or reduced oxygen delivery. Similarly, lower mass flux is a perfect set up for accumulation of infectious material. Thus, it is conceivable that endothelial cells reduce oxygen consumption and increase glycolysis via HIF-1α and activate inflammatory signaling. Concomitant reduction in acetyl-CoA through a reduction in mitochondrial metabolism transmits this energetic condition into a different epigenetic state in addition to loss of barrier protection. Increased substrate stiffness leads to activation of YAP/TAZ which promotes cell proliferation through coordination of glycolysis with anaplerotic pathways. This results in a stiffening of the local circulatory system which may increase local blood flow to pathological levels in the microvasculature. Changes in stiffness also lead to activation of endoMT which again changes the metabolism of the cell with an increase in matrix deposition, causing localized fibrosis.
Not only are biosynthetic and energetic pathways in endothelium modulated by mechanical forces, but metabolic modifications of proteins also change their function. For example, endothelial barrier disruption clearly involve changes in microtubule acetylation states. It is possible that metabolic signaling through protein modifications play an essential role in regulating cellular phenotype in stretch-dependent lung injury; however, this is an under-investigated topic.
With technological advancements such as single cell RNA sequencing, single cell proteomics, and metabolomics, and new tissue clearing methodologies, many novel metabolic hypotheses will finally be able to be tested in animal disease models at the cellular level. With the lung as a privileged organ as a first pass filter for many injectable intravenous medications as well as inhaled medications, targeting pulmonary microvascular niches may be possible.
This review focused on mechanotransduction in the lung vasculature and its effects on endothelial metabolism in pulmonary hypertension, pulmonary fibrosis, and acute lung injury. While endothelial dysfunction is also a feature of chronic obstructive lung disease and asthma, the role of metabolism and mechanical factors is less clear and warrants further study. Furthermore, shear stress, traction forces, and stretch (though not discussed in this review) are often studied independently; it is unclear how combined loading affects cellular metabolism.
Author Contributions
DW conceived, wrote, and edited the manuscript. KB conceived and edited the manuscript.
Funding
DW was supported by NIH K99HL145113. KB was supported by NIH G01-M122940, R01-HL087823.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abe, K., Shinoda, M., Tanaka, M., Kuwabara, Y., Yoshida, K., Hirooka, Y., et al. (2016). Haemodynamic unloading reverses occlusive vascular lesions in severe pulmonary hypertension. Cardiovasc. Res. 111, 16–25. doi: 10.1093/cvr/cvw070
Abe, K., Toba, M., Alzoubi, A., Ito, M., Fagan, K. A., Cool, C. D., et al. (2010). Formation of plexiform lesions in experimental severe pulmonary arterial hypertension. Circulation 121, 2747–2754. doi: 10.1161/CIRCULATIONAHA.109.927681
Acute Respiratory Distress Syndrome Network, Brower, R. G., Matthay, M. A., Morris, A., Schoenfeld, D., Thompson, B. T., et al. (2000). Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N. Engl. J. Med. 342, 1301–1308. doi: 10.1056/NEJM200005043421801
Adamson, R. H., Sarai, R. K., Altangerel, A., Clark, J. F., Weinbaum, S., and Curry, F. E. (2013). Microvascular permeability to water is independent of shear stress, but dependent on flow direction. Am. J. Physiol. Heart Circ. Physiol. 304, H1077–H1084. doi: 10.1152/ajpheart.00956.2012
Albertine, K. H. (2016). “Anatomy of the lungs,” in Murray and Nadel's Textbook of Respiratory Medicine, eds V. C. Broaddus, J. D. Ernst, S. C. Lazarus, J. A. Nadel, M. B. Gotway, R. J. Mason, T. E. King, Jr., J. F. Murray, and A. S. Slutsky (Philadelphia, PA: Elsevier Saunders), 3–21.e25.
Allen, R. P., Schelegle, E. S., and Bennett, S. H. (2014). Diverse forms of pulmonary hypertension remodel the arterial tree to a high shear phenotype. Am. J. Physiol. Heart Circ. Physiol. 307, H405–417. doi: 10.1152/ajpheart.00144.2014
Andresen Eguiluz, R. C., Kaylan, K. B., Underhill, G. H., and Leckband, D. E. (2017). Substrate stiffness and VE-cadherin mechano-transduction coordinate to regulate endothelial monolayer integrity. Biomaterials 140, 45–57. doi: 10.1016/j.biomaterials.2017.06.010
Arulkumaran, N., Deutschman, C. S., Pinsky, M. R., Zuckerbraun, B., Schumacker, P. T., Gomez, H., et al. (2016). Mitochondrial function in sepsis. Shock 45, 271–281. doi: 10.1097/SHK.0000000000000463
Atkins, G. B., and Jain, M. K. (2007). Role of Kruppel-like transcription factors in endothelial biology. Circ. Res. 100, 1686–1695. doi: 10.1161/01.RES.0000267856.00713.0a
Aylon, Y., Gershoni, A., Rotkopf, R., Biton, I. E., Porat, Z., Koh, A. P., et al. (2016). The LATS2 tumor suppressor inhibits SREBP and suppresses hepatic cholesterol accumulation. Genes Dev. 30, 786–797. doi: 10.1101/gad.274167.115
Baeyens, N., Mulligan-Kehoe, M. J., Corti, F., Simon, D. D., Ross, T. D., Rhodes, J. M., et al. (2014). Syndecan 4 is required for endothelial alignment in flow and atheroprotective signaling. Proc. Natl. Acad. Sci. U.S.A. 111, 17308–17313. doi: 10.1073/pnas.1413725111
Balaban, N. Q., Schwarz, U. S., Riveline, D., Goichberg, P., Tzur, G., Sabanay, I., et al. (2001). Force and focal adhesion assembly: a close relationship studied using elastic micropatterned substrates. Nat. Cell Biol. 3, 466–472. doi: 10.1038/35074532
Barker, A. J., Roldán-Alzate, A., Entezari, P., Shah, S. J., Chesler, N. C., Wieben, O., et al. (2015). Four-dimensional flow assessment of pulmonary artery flow and wall shear stress in adult pulmonary arterial hypertension: results from two institutions. Magn. Reson. Med. 73, 1904–1913. doi: 10.1002/mrm.25326
Barron, L., Gharib, S. A., and Duffield, J. S. (2016). Lung pericytes and resident fibroblasts: busy multitaskers. Am. J. Pathol. 186, 2519–2531. doi: 10.1016/j.ajpath.2016.07.004
Barth, E., Stämmler, G., Speiser, B., and Schaper, J. (1992). Ultrastructural quantitation of mitochondria and myofilaments in cardiac muscle from 10 different animal species including man. J. Mol. Cell. Cardiol. 24, 669–681. doi: 10.1016/0022-2828(92)93381-S
Bellani, G., Laffey, J. G., Pham, T., Fan, E., Brochard, L., Esteban, A., et al. (2016). Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA 315, 788–800. doi: 10.1001/jama.2016.0291
Beltran, A., Mcveigh, G., Morgan, D., Glasser, S. P., Neutel, J. M., Weber, M., et al. (2001). Arterial compliance abnormalities in isolated systolic hypertension. Am. J. Hypertens. 14, 1007–1011. doi: 10.1016/S0895-7061(01)02160-4
Benetos, A., Gautier, S., Labat, C., Salvi, P., Valbusa, F., Marino, F., et al. (2012). Mortality and cardiovascular events are best predicted by low central/peripheral pulse pressure amplification but not by high blood pressure levels in elderly nursing home subjects: the PARTAGE (Predictive Values of Blood Pressure and Arterial Stiffness in Institutionalized Very Aged Population) study. J. Am. Coll. Cardiol. 60, 1503–1511. doi: 10.1016/j.jacc.2012.04.055
Bertero, T., Cottrill, K. A., Lu, Y., Haeger, C. M., Dieffenbach, P., Annis, S., et al. (2015). Matrix remodeling promotes pulmonary hypertension through feedback mechanoactivation of the YAP/TAZ-miR-130/301 circuit. Cell Rep. 13, 1016–1032. doi: 10.1016/j.celrep.2015.09.049
Bertero, T., Oldham, W. M., Cottrill, K. A., Pisano, S., Vanderpool, R. R., Yu, Q., et al. (2016). Vascular stiffness mechanoactivates YAP/TAZ-dependent glutaminolysis to drive pulmonary hypertension. J. Clin. Invest. 126, 3313–3335. doi: 10.1172/JCI86387
Birukov, K. G., Jacobson, J. R., Flores, A. A., Ye, S. Q., Birukova, A. A., Verin, A. D., et al. (2003). Magnitude-dependent regulation of pulmonary endothelial cell barrier function by cyclic stretch. Am. J. Physiol. Lung Cell. Mol. Physiol. 285, L785–797. doi: 10.1152/ajplung.00336.2002
Birukova, A. A., Tian, X., Cokic, I., Beckham, Y., Gardel, M. L., and Birukov, K. G. (2013). Endothelial barrier disruption and recovery is controlled by substrate stiffness. Microvasc. Res. 87, 50–57. doi: 10.1016/j.mvr.2012.12.006
Blackman, B. R., Barbee, K. A., and Thibault, L. E. (2000). In vitro cell shearing device to investigate the dynamic response of cells in a controlled hydrodynamic environment. Ann. Biomed. Eng. 28, 363–372. doi: 10.1114/1.286
Borgas, D., Chambers, E., Newton, J., Ko, J., Rivera, S., Rounds, S., et al. (2016). Cigarette smoke disrupted lung endothelial barrier integrity and increased susceptibility to acute lung injury via histone deacetylase 6. Am. J. Respir. Cell Mol. Biol. 54, 683–696. doi: 10.1165/rcmb.2015-0149OC
Boutet, A., De Frutos, C. A., Maxwell, P. H., Mayol, M. J., Romero, J., and Nieto, M. A. (2006). Snail activation disrupts tissue homeostasis and induces fibrosis in the adult kidney. EMBO J. 25, 5603–5613. doi: 10.1038/sj.emboj.7601421
Bracken, C. P., Fedele, A. O., Linke, S., Balrak, W., Lisy, K., Whitelaw, M. L., et al. (2006). Cell-specific regulation of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha stabilization and transactivation in a graded oxygen environment. J. Biol. Chem. 281, 22575–22585. doi: 10.1074/jbc.M600288200
Brown, S. D., Clark, C., and Gutierrez, G. (1996). Pulmonary lactate release in patients with sepsis and the adult respiratory distress syndrome. J. Crit. Care 11, 2–8. doi: 10.1016/S0883-9441(96)90014-3
Brunström, M., and Carlberg, B. (2018). Association of blood pressure lowering with mortality and cardiovascular disease across blood pressure levels: a systematic review and meta-analysis. JAMA Intern. Med. 178, 28–36. doi: 10.1001/jamainternmed.2017.6015
Bryant, A. J., Carrick, R. P., Mcconaha, M. E., Jones, B. R., Shay, S. D., Moore, C. S., et al. (2016). Endothelial HIF signaling regulates pulmonary fibrosis-associated pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 310, L249–262. doi: 10.1152/ajplung.00258.2015
Cabrales, P., Vázquez, B. Y., Tsai, A. G., and Intaglietta, M. (2007). Microvascular and capillary perfusion following glycocalyx degradation. J. Appl. Physiol. (1985) 102, 2251–2259. doi: 10.1152/japplphysiol.01155.2006
Califano, J. P., and Reinhart-King, C. A. (2010). Substrate stiffness and cell area predict cellular traction stresses in single cells and cells in contact. Cell. Mol. Bioeng. 3, 68–75. doi: 10.1007/s12195-010-0102-6
Cameron, J. M., Janer, A., Levandovskiy, V., Mackay, N., Rouault, T. A., Tong, W. H., et al. (2011). Mutations in iron-sulfur cluster scaffold genes NFU1 and BOLA3 cause a fatal deficiency of multiple respiratory chain and 2-oxoacid dehydrogenase enzymes. Am. J. Hum. Genet. 89, 486–495. doi: 10.1016/j.ajhg.2011.08.011
Campo, A., Mathai, S. C., Le Pavec, J., Zaiman, A. L., Hummers, L. K., Boyce, D., et al. (2010). Hemodynamic predictors of survival in scleroderma-related pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 182, 252–260. doi: 10.1164/rccm.200912-1820OC
Cantelmo, A. R., Conradi, L. C., Brajic, A., Goveia, J., Kalucka, J., Pircher, A., et al. (2016). Inhibition of the glycolytic activator PFKFB3 in endothelium induces tumor vessel normalization, impairs metastasis, and improves chemotherapy. Cancer Cell 30, 968–985. doi: 10.1016/j.ccell.2016.10.006
Cardoso, C. R., and Salles, G. F. (2016). Aortic stiffness as a surrogate endpoint to micro- and macrovascular complications in patients with type 2 diabetes. Int. J. Mol. Sci. 17:2044. doi: 10.3390/ijms17122044
Caruso, P., Dunmore, B. J., Schlosser, K., Schoors, S., Dos Santos, C., Perez-Iratxeta, C., et al. (2017). Identification of MicroRNA-124 as a major regulator of enhanced endothelial cell glycolysis in pulmonary arterial hypertension via PTBP1 (polypyrimidine tract binding protein) and pyruvate kinase M2. Circulation 136, 2451–2467. doi: 10.1161/CIRCULATIONAHA.117.028034
Castelain, V., Hervé, P., Lecarpentier, Y., Duroux, P., Simonneau, G., and Chemla, D. (2001). Pulmonary artery pulse pressure and wave reflection in chronic pulmonary thromboembolism and primary pulmonary hypertension. J. Am. Coll. Cardiol. 37, 1085–1092. doi: 10.1016/S0735-1097(00)01212-2
Chan, S. Y., Zhang, Y. Y., Hemann, C., Mahoney, C. E., Zweier, J. L., and Loscalzo, J. (2009). MicroRNA-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins ISCU1/2. Cell Metab. 10, 273–284. doi: 10.1016/j.cmet.2009.08.015
Chen, C. S., Mrksich, M., Huang, S., Whitesides, G. M., and Ingber, D. E. (1997). Geometric control of cell life and death. Science 276, 1425–1428. doi: 10.1126/science.276.5317.1425
Chen, Q., Jiang, L., Li, C., Hu, D., Bu, J. W., Cai, D., et al. (2012). Haemodynamics-driven developmental pruning of brain vasculature in zebrafish. PLoS Biol. 10:e1001374. doi: 10.1371/journal.pbio.1001374
Chen, Y., Shen, F., Liu, J., and Yang, G. Y. (2017). Arterial stiffness and stroke: de-stiffening strategy, a therapeutic target for stroke. Stroke Vasc. Neurol. 2, 65–72. doi: 10.1136/svn-2016-000045
Chen, Y. A., Lu, C. Y., Cheng, T. Y., Pan, S. H., Chen, H. F., and Chang, N. S. (2019). WW domain-containing proteins YAP and TAZ in the hippo pathway as key regulators in stemness maintenance, tissue homeostasis, and tumorigenesis. Front. Oncol. 9:60. doi: 10.3389/fonc.2019.00060
Chen, Z., Peng, I. C., Cui, X., Li, Y. S., Chien, S., and Shyy, J. Y. (2010). Shear stress, SIRT1, and vascular homeostasis. Proc. Natl. Acad. Sci. U.S.A. 107, 10268–10273. doi: 10.1073/pnas.1003833107
Chiu, J. J., and Chien, S. (2011). Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiol. Rev. 91, 327–387. doi: 10.1152/physrev.00047.2009
Choi, S. H., Nam, J. K., Kim, B. Y., Jang, J., Jin, Y. B., Lee, H. J., et al. (2016). HSPB1 inhibits the endothelial-to-mesenchymal transition to suppress pulmonary fibrosis and lung tumorigenesis. Cancer Res. 76, 1019–1030. doi: 10.1158/0008-5472.CAN-15-0952
Choudhary, C., Weinert, B. T., Nishida, Y., Verdin, E., and Mann, M. (2014). The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 15, 536–550. doi: 10.1038/nrm3841
Cooper, L. L., Musani, S. K., Washington, F., Moore, J., Tripathi, A., Tsao, C. W., et al. (2018). Relations of microvascular function, cardiovascular disease risk factors, and aortic stiffness in blacks: the Jackson heart study. J. Am. Heart Assoc. 7:e004292. doi: 10.1161/JAHA.118.009515
Cowburn, A. S., Crosby, A., Macias, D., Branco, C., Colaco, R. D. D. R., Southwood, M., et al. (2016). HIF2 alpha-arginase axis is essential for the development of pulmonary hypertension. Proc. Natl. Acad. Sci. U.S.A. 113, 8801–8806. doi: 10.1073/pnas.1602978113
Crapo, J. D., Barry, B. E., Gehr, P., Bachofen, M., and Weibel, E. R. (1982). Cell number and cell characteristics of the normal human-lung. Am. Rev. Respir. Dis. 126, 332–337.
Cucullo, L., Hossain, M., Puvenna, V., Marchi, N., and Janigro, D. (2011). The role of shear stress in Blood-Brain Barrier endothelial physiology. BMC Neurosci. 12:40. doi: 10.1186/1471-2202-12-40
Culley, M. K., and Chan, S. Y. (2018). Mitochondrial metabolism in pulmonary hypertension: beyond mountains there are mountains. J. Clin. Investig. 128, 3704–3715. doi: 10.1172/JCI120847
Cypel, M., Yeung, J. C., Liu, M., Anraku, M., Chen, F., Karolak, W., et al. (2011). Normothermic ex vivo lung perfusion in clinical lung transplantation. N. Engl. J. Med. 364, 1431–1440. doi: 10.1056/NEJMoa1014597
Dai, G., Kaazempur-Mofrad, M. R., Natarajan, S., Zhang, Y. Z., Vaughn, S., Blackman, B. R., et al. (2004). Distinct endothelial phenotypes evoked by arterial waveforms derived from atherosclerosis-susceptible and -resistant regions of human vasculature. Proc. Natl. Acad. Sci. U.S.A. 101, 14871–14876. doi: 10.1073/pnas.0406073101
Dai, Z., Li, M., Wharton, J., Zhu, M. M., and Zhao, Y. Y. (2016). Prolyl-4 hydroxylase 2 (PHD2) deficiency in endothelial cells and hematopoietic cells induces obliterative vascular remodeling and severe pulmonary arterial hypertension in mice and humans through hypoxia-inducible factor-2alpha. Circulation 133, 2447–2458. doi: 10.1161/CIRCULATIONAHA.116.021494
Das, A., Huang, G. X., Bonkowski, M. S., Longchamp, A., Li, C., Schultz, M. B., et al. (2018). Impairment of an endothelial NAD(+)-H2S signaling network is a reversible cause of vascular aging. Cell 173, 74–89 e20. doi: 10.1016/j.cell.2018.02.008
Davies, P. F. (1995). Flow-mediated endothelial mechanotransduction. Physiol. Rev. 75, 519–560. doi: 10.1152/physrev.1995.75.3.519
Davies, P. F. (2008). Endothelial transcriptome profiles in vivo in complex arterial flow fields. Ann. Biomed. Eng. 36, 563–570. doi: 10.1007/s10439-007-9400-0
Davies, P. F., Civelek, M., Fang, Y., and Fleming, I. (2013). The atherosusceptible endothelium: endothelial phenotypes in complex haemodynamic shear stress regions in vivo. Cardiovasc. Res. 99, 315–327. doi: 10.1093/cvr/cvt101
Davies, P. F., Mundel, T., and Barbee, K. A. (1995). A mechanism for heterogeneous endothelial responses to flow in vivo and in vitro. J. Biomech. 28, 1553–1560. doi: 10.1016/0021-9290(95)00102-6
Davies, P. F., Remuzzi, A., Gordon, E. J., Dewey, C. F. Jr., and Gimbrone, M. A. Jr. (1986). Turbulent fluid shear stress induces vascular endothelial cell turnover in vitro. Proc. Natl. Acad. Sci. U.S.A. 83, 2114–2117. doi: 10.1073/pnas.83.7.2114
De Backer, D., Creteur, J., Preiser, J. C., Dubois, M. J., and Vincent, J. L. (2002). Microvascular blood flow is altered in patients with sepsis. Am. J. Respir. Crit. Care Med. 166, 98–104. doi: 10.1164/rccm.200109-016OC
De Backer, D., Creteur, J., Zhang, H., Norrenberg, M., and Vincent, J. L. (1997). Lactate production by the lungs in acute lung injury. Am. J. Respir. Crit. Care Med. 156, 1099–1104. doi: 10.1164/ajrccm.156.4.9701048
De Bock, K., Georgiadou, M., and Carmeliet, P. (2013a). Role of endothelial cell metabolism in vessel sprouting. Cell Metab. 18, 634–647. doi: 10.1016/j.cmet.2013.08.001
De Bock, K., Georgiadou, M., Schoors, S., Kuchnio, A., Wong, B. W., Cantelmo, A. R., et al. (2013b). Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 154, 651–663. doi: 10.1016/j.cell.2013.06.037
De Jong, O. G., Van Balkom, B. W., Gremmels, H., and Verhaar, M. C. (2016). Exosomes from hypoxic endothelial cells have increased collagen crosslinking activity through up-regulation of lysyl oxidase-like 2. J. Cell. Mol. Med. 20, 342–350. doi: 10.1111/jcmm.12730
Dekker, R. J., Van Soest, S., Fontijn, R. D., Salamanca, S., De Groot, P. G., Vanbavel, E., et al. (2002). Prolonged fluid shear stress induces a distinct set of endothelial cell genes, most specifically lung Kruppel-like factor (KLF2). Blood 100, 1689–1698. doi: 10.1182/blood-2002-01-0046
Deran, M., Yang, J., Shen, C. H., Peters, E. C., Fitamant, J., Chan, P., et al. (2014). Energy stress regulates hippo-YAP signaling involving AMPK-mediated regulation of angiomotin-like 1 protein. Cell Rep. 9, 495–503. doi: 10.1016/j.celrep.2014.09.036
Diebold, I., Hennigs, J. K., Miyagawa, K., Li, C. G., Nickel, N. P., Kaschwich, M., et al. (2015). BMPR2 preserves mitochondrial function and DNA during reoxygenation to promote endothelial cell survival and reverse pulmonary hypertension. Cell Metab. 21, 596–608. doi: 10.1016/j.cmet.2015.03.010
Diebold, L. P., Gil, H. J., Gao, P., Martinez, C. A., Weinberg, S. E., and Chandel, N. S. (2019). Mitochondrial complex III is necessary for endothelial cell proliferation during angiogenesis. Nat. Metab. 1, 158–171. doi: 10.1038/s42255-018-0011-x
Dieffenbach, P. B., Haeger, C. M., Coronata, A. M. F., Choi, K. M., Varelas, X., Tschumperlin, D. J., et al. (2017). Arterial stiffness induces remodeling phenotypes in pulmonary artery smooth muscle cells via YAP/TAZ-mediated repression of cyclooxygenase-2. Am. J. Physiol. Lung Cell. Mol. Physiol. 313, L628–L647. doi: 10.1152/ajplung.00173.2017
Dixon, J. B., Greiner, S. T., Gashev, A. A., Cote, G. L., Moore, J. E., and Zawieja, D. C. (2006). Lymph flow, shear stress, and lymphocyte velocity in rat mesenteric prenodal lymphatics. Microcirculation 13, 597–610. doi: 10.1080/10739680600893909
Doddaballapur, A., Michalik, K. M., Manavski, Y., Lucas, T., Houtkooper, R. H., You, X., et al. (2015). Laminar shear stress inhibits endothelial cell metabolism via KLF2-mediated repression of PFKFB3. Arterioscler. Thromb. Vasc. Biol. 35, 137–145. doi: 10.1161/ATVBAHA.114.304277
Doshi, B. M., Hightower, L. E., and Lee, J. (2009). The role of Hsp27 and actin in the regulation of movement in human cancer cells responding to heat shock. Cell Stress Chaperones 14, 445–457. doi: 10.1007/s12192-008-0098-1
Duong, H. T., Comhair, S. A., Aldred, M. A., Mavrakis, L., Savasky, B. M., Erzurum, S. C., et al. (2011). Pulmonary artery endothelium resident endothelial colony-forming cells in pulmonary arterial hypertension. Pulm. Circ. 1, 475–486. doi: 10.4103/2045-8932.93547
Dupont, S., Morsut, L., Aragona, M., Enzo, E., Giulitti, S., Cordenonsi, M., et al. (2011). Role of YAP/TAZ in mechanotransduction. Nature 474, 179–183. doi: 10.1038/nature10137
Eelen, G., De Zeeuw, P., Simons, M., and Carmeliet, P. (2015). Endothelial cell metabolism in normal and diseased vasculature. Circ. Res. 116, 1231–1244. doi: 10.1161/CIRCRESAHA.116.302855
Egnatchik, R. A., Brittain, E. L., Shah, A. T., Fares, W. H., Ford, H. J., Monahan, K., et al. (2017). Dysfunctional BMPR2 signaling drives an abnormal endothelial requirement for glutamine in pulmonary arterial hypertension. Pulm. Circ. 7, 186–199. doi: 10.1086/690236
Engler, A. J., Sen, S., Sweeney, H. L., and Discher, D. E. (2006). Matrix elasticity directs stem cell lineage specification. Cell 126, 677–689. doi: 10.1016/j.cell.2006.06.044
Enzo, E., Santinon, G., Pocaterra, A., Aragona, M., Bresolin, S., Forcato, M., et al. (2015). Aerobic glycolysis tunes YAP/TAZ transcriptional activity. EMBO J. 34, 1349–1370. doi: 10.15252/embj.201490379
Fang, Y., Wu, D., and Birukov, K. G. (2019). Mechanosensing and mechanoregulation of endothelial cell functions. Compr. Physiol. 9, 873–904. doi: 10.1002/cphy.c180020
Feaver, R. E., Gelfand, B. D., and Blackman, B. R. (2013). Human haemodynamic frequency harmonics regulate the inflammatory phenotype of vascular endothelial cells. Nat. Commun. 4:1525. doi: 10.1038/ncomms2530
Feng, S., Bowden, N., Fragiadaki, M., Souilhol, C., Hsiao, S., Mahmoud, M., et al. (2017). Mechanical activation of hypoxia-inducible factor 1alpha drives endothelial dysfunction at atheroprone sites. Arterioscler. Thromb. Vasc. Biol. 37, 2087–2101. doi: 10.1161/ATVBAHA.117.309249
Florian, J. A., Kosky, J. R., Ainslie, K., Pang, Z. Y., Dull, R. O., and Tarbell, J. M. (2003). Heparan sulfate proteoglycan is a mechanosensor on endothelial cells. Circ. Res. 93, E136–E142. doi: 10.1161/01.RES.0000101744.47866.D5
Force, A. D. T., Ranieri, V. M., Rubenfeld, G. D., Thompson, B. T., Ferguson, N. D., Caldwell, E., et al. (2012). Acute respiratory distress syndrome: the Berlin definition. JAMA 307, 2526–2533. doi: 10.1001/jama.2012.5669
Foster, W., Carruthers, D., Lip, G. Y., and Blann, A. D. (2010). Inflammation and microvascular and macrovascular endothelial dysfunction in rheumatoid arthritis: effect of treatment. J. Rheumatol. 37, 711–716. doi: 10.3899/jrheum.090699
Franco, C. A., Jones, M. L., Bernabeu, M. O., Geudens, I., Mathivet, T., Rosa, A., et al. (2015). Dynamic endothelial cell rearrangements drive developmental vessel regression. PLoS Biol. 13:e1002125. doi: 10.1371/journal.pbio.1002125
Furukawa, K. T., Yamashita, K., Sakurai, N., and Ohno, S. (2017). The epithelial circumferential actin belt regulates YAP/TAZ through nucleocytoplasmic shuttling of merlin. Cell Rep. 20, 1435–1447. doi: 10.1016/j.celrep.2017.07.032
Gabrielli, L. A., Castro, P. F., Godoy, I., Mellado, R., Bourge, R. C., Alcaino, H., et al. (2011). Systemic oxidative stress and endothelial dysfunction is associated with an attenuated acute vascular response to inhaled prostanoid in pulmonary artery hypertension patients. J. Card. Fail. 17, 1012–1017. doi: 10.1016/j.cardfail.2011.08.008
Gailite, I., Aerne, B. L., and Tapon, N. (2015). Differential control of Yorkie activity by LKB1/AMPK and the Hippo/Warts cascade in the central nervous system. Proc. Natl. Acad. Sci. U.S.A. 112, E5169–5178. doi: 10.1073/pnas.1505512112
Galie, P. A., Nguyen, D. H., Choi, C. K., Cohen, D. M., Janmey, P. A., and Chen, C. S. (2014). Fluid shear stress threshold regulates angiogenic sprouting. Proc. Natl. Acad. Sci. U.S.A. 111, 7968–7973. doi: 10.1073/pnas.1310842111
Gan, C. T., Lankhaar, J. W., Westerhof, N., Marcus, J. T., Becker, A., Twisk, J. W., et al. (2007). Noninvasively assessed pulmonary artery stiffness predicts mortality in pulmonary arterial hypertension. Chest 132, 1906–1912. doi: 10.1378/chest.07-1246
Garcia-Polite, F., Martorell, J., Del Rey-Puech, P., Melgar-Lesmes, P., O'brien, C. C., Roquer, J., et al. (2017). Pulsatility and high shear stress deteriorate barrier phenotype in brain microvascular endothelium. J. Cereb. Blood Flow Metab. 37, 2614–2625. doi: 10.1177/0271678X16672482
Gatzoulis, M. A., Alonso-Gonzalez, R., and Beghetti, M. (2009). Pulmonary arterial hypertension in paediatric and adult patients with congenital heart disease. Eur. Respir. Rev. 18, 154–161. doi: 10.1183/09059180.00003309
Ghesquière, B., Wong, B. W., Kuchnio, A., and Carmeliet, P. (2014). Metabolism of stromal and immune cells in health and disease. Nature 511, 167–176. doi: 10.1038/nature13312
Gimbrone, M. A. Jr., and García-Cardeña, G. (2016). Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ. Res. 118, 620–636. doi: 10.1161/CIRCRESAHA.115.306301
Gong, H., Rehman, J., Tang, H., Wary, K., Mittal, M., Chaturvedi, P., et al. (2015). HIF2alpha signaling inhibits adherens junctional disruption in acute lung injury. J. Clin. Invest. 125, 652–664. doi: 10.1172/JCI77701
Good, R. B., Gilbane, A. J., Trinder, S. L., Denton, C. P., Coghlan, G., Abraham, D. J., et al. (2015). Endothelial to mesenchymal transition contributes to endothelial dysfunction in pulmonary arterial hypertension. Am. J. Pathol. 185, 1850–1858. doi: 10.1016/j.ajpath.2015.03.019
Guan, S., and Zhou, J. (2017). CXCR7 attenuates the TGF-beta-induced endothelial-to-mesenchymal transition and pulmonary fibrosis. Mol. Biosyst. 13, 2116–2124. doi: 10.1039/C7MB00247E
Hajra, L., Evans, A. I., Chen, M., Hyduk, S. J., Collins, T., and Cybulsky, M. I. (2000). The NF-kappa B signal transduction pathway in aortic endothelial cells is primed for activation in regions predisposed to atherosclerotic lesion formation. Proc. Natl. Acad. Sci. U.S.A. 97, 9052–9057. doi: 10.1073/pnas.97.16.9052
Hakim, T. S. (1994). Flow-induced release of EDRF in the pulmonary vasculature: site of release and action. Am. J. Physiol. 267, H363–369. doi: 10.1152/ajpheart.1994.267.1.H363
Hamanaka, R. B., O'leary, E. M., Witt, L. J., Tian, Y., Gokalp, G. A., Meliton, A. Y., et al. (2019). Glutamine metabolism is required for collagen protein synthesis in lung fibroblasts. Am. J. Respir. Cell Mol. Biol. doi: 10.1165/rcmb.2019-0008OC [Epub ahead of print].
Hashimoto, N., Phan, S. H., Imaizumi, K., Matsuo, M., Nakashima, H., Kawabe, T., et al. (2010). Endothelial-mesenchymal transition in bleomycin-induced pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 43, 161–172. doi: 10.1165/rcmb.2009-0031OC
Hautefort, A., Chesné, J., Preussner, J., Pullamsetti, S. S., Tost, J., Looso, M., et al. (2017). Pulmonary endothelial cell DNA methylation signature in pulmonary arterial hypertension. Oncotarget 8, 52995–53016. doi: 10.18632/oncotarget.18031
He, L., Vanlandewijck, M., Mäe, M. A., Andrae, J., Ando, K., Del Gaudio, F., et al. (2018). Single-cell RNA sequencing of mouse brain and lung vascular and vessel-associated cell types. Sci Data 5:180160. doi: 10.1038/sdata.2018.160
Huang, H., Vandekeere, S., Kalucka, J., Bierhansl, L., Zecchin, A., Brüning, U., et al. (2017a). Role of glutamine and interlinked asparagine metabolism in vessel formation. EMBO J. 36, 2334–2352. doi: 10.15252/embj.201695518
Huang, R. T., Wu, D., Meliton, A., Oh, M. J., Krause, M., Lloyd, J. A., et al. (2017b). Experimental lung injury reduces Kruppel-like factor 2 to increase endothelial permeability via regulation of RAPGEF3-Rac1 signaling. Am. J. Respir. Crit. Care Med. 195, 639–651. doi: 10.1164/rccm.201604-0668OC
Huckabee, W. E. (1958). Relationships of pyruvate and lactate during anaerobic metabolism. III. Effect of breathing low-oxygen gases. J. Clin. Invest. 37, 264–271. doi: 10.1172/JCI103605
Hunter, K. S., Lee, P. F., Lanning, C. J., Ivy, D. D., Kirby, K. S., Claussen, L. R., et al. (2008). Pulmonary vascular input impedance is a combined measure of pulmonary vascular resistance and stiffness and predicts clinical outcomes better than pulmonary vascular resistance alone in pediatric patients with pulmonary hypertension. Am. Heart J. 155, 166–174. doi: 10.1016/j.ahj.2007.08.014
Iaconelli, J., Huang, J. H., Berkovitch, S. S., Chattopadhyay, S., Mazitschek, R., Schreiber, S. L., et al. (2015). HDAC6 inhibitors modulate Lys49 acetylation and membrane localization of beta-catenin in human iPSC-derived neuronal cells. ACS Chem. Biol. 10, 883–890. doi: 10.1021/cb500838r
Jayachandran, A., Königshoff, M., Yu, H., Rupniewska, E., Hecker, M., Klepetko, W., et al. (2009). SNAI transcription factors mediate epithelial-mesenchymal transition in lung fibrosis. Thorax 64, 1053–1061. doi: 10.1136/thx.2009.121798
Jeong, S. H., Kim, H. B., Kim, M. C., Lee, J. M., Lee, J. H., Kim, J. H., et al. (2018). Hippo-mediated suppression of IRS2/AKT signaling prevents hepatic steatosis and liver cancer. J. Clin. Invest. 128, 1010–1025. doi: 10.1172/JCI95802
Karki, P., Ke, Y., Tian, Y., Ohmura, T., Sitikov, A., Sarich, N., et al. (2019). Staphylococcus aureus-induced endothelial permeability and inflammation are mediated by microtubule destabilization. J. Biol. Chem. 294, 3369–3384. doi: 10.1074/jbc.RA118.004030
Kása, A., Csortos, C., and Verin, A. D. (2015). Cytoskeletal mechanisms regulating vascular endothelial barrier function in response to acute lung injury. Tissue Barriers 3:e974448. doi: 10.4161/21688370.2014.974448
Kato, S., Inui, N., Hakamata, A., Suzuki, Y., Enomoto, N., Fujisawa, T., et al. (2018). Changes in pulmonary endothelial cell properties during bleomycin-induced pulmonary fibrosis. Respir. Res. 19:127. doi: 10.1186/s12931-018-0831-y
Kawanami, D., Mahabeleshwar, G. H., Lin, Z., Atkins, G. B., Hamik, A., Haldar, S. M., et al. (2009). Kruppel-like factor 2 inhibits hypoxia-inducible factor 1alpha expression and function in the endothelium. J. Biol. Chem. 284, 20522–20530. doi: 10.1074/jbc.M109.025346
Kidd, S., Spaeth, E., Watson, K., Burks, J., Lu, H., Klopp, A., et al. (2012). Origins of the tumor microenvironment: quantitative assessment of adipose-derived and bone marrow-derived stroma. PLoS ONE 7:e30563. doi: 10.1371/journal.pone.0030563
Kim, B., Lee, H., Kawata, K., and Park, J. Y. (2014). Exercise-mediated wall shear stress increases mitochondrial biogenesis in vascular endothelium. PLoS ONE 9:e111409. doi: 10.1371/journal.pone.0111409
Kim, B., Li, J., Jang, C., and Arany, Z. (2017). Glutamine fuels proliferation but not migration of endothelial cells. EMBO J. 36, 2321–2333. doi: 10.15252/embj.201796436
Kim, J., Kang, Y., Kojima, Y., Lighthouse, J. K., Hu, X., Aldred, M. A., et al. (2013). An endothelial apelin-FGF link mediated by miR-424 and miR-503 is disrupted in pulmonary arterial hypertension. Nat. Med. 19, 74–82. doi: 10.1038/nm.3040
Kim, J. S., Kim, B., Lee, H., Thakkar, S., Babbitt, D. M., Eguchi, S., et al. (2015). Shear stress-induced mitochondrial biogenesis decreases the release of microparticles from endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 309, H425–433. doi: 10.1152/ajpheart.00438.2014
Kim, J. W., Tchernyshyov, I., Semenza, G. L., and Dang, C. V. (2006). HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 3, 177–185. doi: 10.1016/j.cmet.2006.02.002
Kimura, T. E., Duggirala, A., Smith, M. C., White, S., Sala-Newby, G. B., Newby, A. C., et al. (2016). The Hippo pathway mediates inhibition of vascular smooth muscle cell proliferation by cAMP. J. Mol. Cell. Cardiol. 90, 1–10. doi: 10.1016/j.yjmcc.2015.11.024
Kinlay, S., Creager, M. A., Fukumoto, M., Hikita, H., Fang, J. C., Selwyn, A. P., et al. (2001). Endothelium-derived nitric oxide regulates arterial elasticity in human arteries in vivo. Hypertension 38, 1049–1053. doi: 10.1161/hy1101.095329
Kizhakekuttu, T. J., Wang, J., Dharmashankar, K., Ying, R., Gutterman, D. D., Vita, J. A., et al. (2012). Adverse alterations in mitochondrial function contribute to type 2 diabetes mellitus-related endothelial dysfunction in humans. Arterioscler. Thromb. Vasc. Biol. 32, 2531–2539. doi: 10.1161/ATVBAHA.112.256024
Kluge, M. A., Fetterman, J. L., and Vita, J. A. (2013). Mitochondria and endothelial function. Circ. Res. 112, 1171–1188. doi: 10.1161/CIRCRESAHA.111.300233
Kratzer, E., Tian, Y., Sarich, N., Wu, T., Meliton, A., Leff, A., et al. (2012). Oxidative stress contributes to lung injury and barrier dysfunction via microtubule destabilization. Am. J. Respir. Cell Mol. Biol. 47, 688–697. doi: 10.1165/rcmb.2012-0161OC
Kudryashova, T. V., Goncharov, D. A., Pena, A., Kelly, N., Vanderpool, R., Baust, J., et al. (2016). HIPPO-integrin-linked kinase cross-talk controls self-sustaining proliferation and survival in pulmonary hypertension. Am. J. Respir. Crit. Care Med. 194, 866–877. doi: 10.1164/rccm.201510-2003OC
Kuriyama, K., Gamsu, G., Stern, R. G., Cann, C. E., Herfkens, R. J., and Brundage, B. H. (1984). CT-determined pulmonary artery diameters in predicting pulmonary hypertension. Invest. Radiol. 19, 16–22. doi: 10.1097/00004424-198401000-00005
Lacolley, P., Regnault, V., Segers, P., and Laurent, S. (2017). Vascular smooth muscle cells and arterial stiffening: relevance in development, aging, and disease. Physiol. Rev. 97, 1555–1617. doi: 10.1152/physrev.00003.2017
Lai, B., Li, Z., He, M., Wang, Y., Chen, L., Zhang, J., et al. (2018). Atheroprone flow enhances the endothelial-to-mesenchymal transition. Am. J. Physiol. Heart Circ. Physiol. 315, H1293–H1303. doi: 10.1152/ajpheart.00213.2018
Laurent, S., Boutouyrie, P., Asmar, R., Gautier, I., Laloux, B., Guize, L., et al. (2001). Aortic stiffness is an independent predictor of all-cause and cardiovascular mortality in hypertensive patients. Hypertension 37, 1236–1241. doi: 10.1161/01.HYP.37.5.1236
Lee, J. S., Yu, Q., Shin, J. T., Sebzda, E., Bertozzi, C., Chen, M., et al. (2006). Klf2 is an essential regulator of vascular hemodynamic forces in vivo. Dev. Cell 11, 845–857. doi: 10.1016/j.devcel.2006.09.006
Li, M., Scott, D. E., Shandas, R., Stenmark, K. R., and Tan, W. (2009a). High pulsatility flow induces adhesion molecule and cytokine mRNA expression in distal pulmonary artery endothelial cells. Ann. Biomed. Eng. 37, 1082–1092. doi: 10.1007/s10439-009-9684-3
Li, M., Stenmark, K. R., Shandas, R., and Tan, W. (2009b). Effects of pathological flow on pulmonary artery endothelial production of vasoactive mediators and growth factors. J. Vasc. Res. 46, 561–571. doi: 10.1159/000226224
Li, M., Tan, Y., Stenmark, K. R., and Tan, W. (2013). High pulsatility flow induces acute endothelial inflammation through overpolarizing cells to activate NF-kappaB. Cardiovasc. Eng. Technol. 4, 26–38. doi: 10.1007/s13239-012-0115-5
Li, Y., Lui, K. O., and Zhou, B. (2018). Reassessing endothelial-to-mesenchymal transition in cardiovascular diseases. Nat. Rev. Cardiol. 15, 445–456. doi: 10.1038/s41569-018-0023-y
Li, Y., Zhang, X., Polakiewicz, R. D., Yao, T. P., and Comb, M. J. (2008). HDAC6 is required for epidermal growth factor-induced beta-catenin nuclear localization. J. Biol. Chem. 283, 12686–12690. doi: 10.1074/jbc.C700185200
Lian, I., Kim, J., Okazawa, H., Zhao, J., Zhao, B., Yu, J., et al. (2010). The role of YAP transcription coactivator in regulating stem cell self-renewal and differentiation. Genes Dev. 24, 1106–1118. doi: 10.1101/gad.1903310
Liao, J. K. (2013). Linking endothelial dysfunction with endothelial cell activation. J. Clin. Invest. 123, 540–541. doi: 10.1172/JCI66843
Liao, X., Zhang, R., Lu, Y., Prosdocimo, D. A., Sangwung, P., Zhang, L., et al. (2015). Kruppel-like factor 4 is critical for transcriptional control of cardiac mitochondrial homeostasis. J. Clin. Invest. 125, 3461–3476. doi: 10.1172/JCI79964
Lin, Z., Kumar, A., Senbanerjee, S., Staniszewski, K., Parmar, K., Vaughan, D. E., et al. (2005). Kruppel-like factor 2 (KLF2) regulates endothelial thrombotic function. Circ. Res. 96, e48–57. doi: 10.1161/01.RES.0000159707.05637.a1
Lin, Z., Natesan, V., Shi, H., Dong, F., Kawanami, D., Mahabeleshwar, G. H., et al. (2010). Kruppel-like factor 2 regulates endothelial barrier function. Arterioscler. Thromb. Vasc. Biol. 30, 1952–1959. doi: 10.1161/ATVBAHA.110.211474
Liu, D., Mao, P., Huang, Y., Liu, Y., Liu, X., Pang, X., et al. (2014). Proteomic analysis of lung tissue in a rat acute lung injury model: identification of PRDX1 as a promoter of inflammation. Mediat. Inflamm. 2014:469358. doi: 10.1155/2014/469358
Liu, D., Yan, Y., Chen, J. W., Yuan, P., Wang, X. J., Jiang, R., et al. (2017). Hypermethylation of BMPR2 promoter occurs in patients with heritable pulmonary arterial hypertension and inhibits BMPR2 expression. Am. J. Respir. Crit. Care Med. 196, 925–928. doi: 10.1164/rccm.201611-2273LE
Liu, F., Lagares, D., Choi, K. M., Stopfer, L., Marinković, A., Vrbanac, V., et al. (2015). Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 308, L344–L357. doi: 10.1152/ajplung.00300.2014
Mack, J. J., Mosqueiro, T. S., Archer, B. J., Jones, W. M., Sunshine, H., Faas, G. C., et al. (2017). NOTCH1 is a mechanosensor in adult arteries. Nat. Commun. 8:1620. doi: 10.1038/s41467-017-01741-8
Mahapatra, S., Nishimura, R. A., Sorajja, P., Cha, S., and Mcgoon, M. D. (2006). Relationship of pulmonary arterial capacitance and mortality in idiopathic pulmonary arterial hypertension. J. Am. Coll. Cardiol. 47, 799–803. doi: 10.1016/j.jacc.2005.09.054
Mambetsariev, I., Tian, Y., Wu, T., Lavoie, T., Solway, J., Birukov, K. G., et al. (2014). Stiffness-activated GEF-H1 expression exacerbates LPS-induced lung inflammation. PLoS ONE 9:e92670. doi: 10.1371/journal.pone.0092670
Mattagajasingh, I., Kim, C. S., Naqvi, A., Yamamori, T., Hoffman, T. A., Jung, S. B., et al. (2007). SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. U.S.A. 104, 14855–14860. doi: 10.1073/pnas.0704329104
Mcclatchey, P. M., Schafer, M., Hunter, K. S., and Reusch, J. E. B. (2016). The endothelial glycocalyx promotes homogenous blood flow distribution within the microvasculature. Am. J. Physiol. Heart Circ. Physiol. 311, H168–H176. doi: 10.1152/ajpheart.00132.2016
Mcclendon, J., Jansing, N. L., Redente, E. F., Gandjeva, A., Ito, Y., Colgan, S. P., et al. (2017). Hypoxia-inducible factor 1alpha signaling promotes repair of the alveolar epithelium after acute lung injury. Am. J. Pathol. 187, 1772–1786. doi: 10.1016/j.ajpath.2017.04.012
Mceniery, C. M., Wallace, S., Mackenzie, I. S., Mcdonnell, B., Yasmin, Newby, D. E., et al. (2006). Endothelial function is associated with pulse pressure, pulse wave velocity, and augmentation index in healthy humans. Hypertension 48, 602–608. doi: 10.1161/01.HYP.0000239206.64270.5f
Meng, F., Mambetsariev, I., Tian, Y., Beckham, Y., Meliton, A., Leff, A., et al. (2015). Attenuation of lipopolysaccharide-induced lung vascular stiffening by lipoxin reduces lung inflammation. Am. J. Respir. Cell Mol. Biol. 52, 152–161. doi: 10.1165/rcmb.2013-0468OC
Michelakis, E. D., Gurtu, V., Webster, L., Barnes, G., Watson, G., Howard, L., et al. (2017). Inhibition of pyruvate dehydrogenase kinase improves pulmonary arterial hypertension in genetically susceptible patients. Sci. Transl. Med. 9:eaao4583. doi: 10.1126/scitranslmed.aao4583
Milovanova, T., Chatterjee, S., Manevich, Y., Kotelnikova, I., Debolt, K., Madesh, M., et al. (2006). Lung endothelial cell proliferation with decreased shear stress is mediated by reactive oxygen species. Am. J. Physiol. Cell Physiol. 290, C66–C76. doi: 10.1152/ajpcell.00094.2005
Mitchell, G. F. (2008). Effects of central arterial aging on the structure and function of the peripheral vasculature: implications for end-organ damage. J. Appl. Physiol. (1985) 105, 1652–1660. doi: 10.1152/japplphysiol.90549.2008
Miyagawa, K., Shi, M., Chen, P. I., Hennigs, J. K., Zhao, Z., Wang, M., et al. (2019). Smooth muscle contact drives endothelial regeneration by BMPR2-Notch1-mediated metabolic and epigenetic changes. Circ. Res. 124, 211–224. doi: 10.1161/CIRCRESAHA.118.313374
Mo, J. S., Meng, Z., Kim, Y. C., Park, H. W., Hansen, C. G., Kim, S., et al. (2015). Cellular energy stress induces AMPK-mediated regulation of YAP and the Hippo pathway. Nat. Cell Biol. 17, 500–510. doi: 10.1038/ncb3111
Mochizuki, S., Vink, H., Hiramatsu, O., Kajita, T., Shigeto, F., Spaan, J. A., et al. (2003). Role of hyaluronic acid glycosaminoglycans in shear-induced endothelium-derived nitric oxide release. Am. J. Physiol. Heart Circ. Physiol. 285, H722–726. doi: 10.1152/ajpheart.00691.2002
Moon, J. J., Matsumoto, M., Patel, S., Lee, L., Guan, J. L., and Li, S. (2005). Role of cell surface heparan sulfate proteoglycans in endothelial cell migration and mechanotransduction. J. Cell. Physiol. 203, 166–176. doi: 10.1002/jcp.20220
Nakayama, Y., Nakanishi, N., Hayashi, T., Nagaya, N., Sakamaki, F., Satoh, N., et al. (2001). Pulmonary artery reflection for differentially diagnosing primary pulmonary hypertension and chronic pulmonary thromboembolism. J. Am. Coll. Cardiol. 38, 214–218. doi: 10.1016/S0735-1097(01)01365-1
Nave, A. H., MiŽíková, I., Niess, G., Steenbock, H., Reichenberger, F., Talavera, M. L., et al. (2014). Lysyl oxidases play a causal role in vascular remodeling in clinical and experimental pulmonary arterial hypertension. Arterioscler. Thromb. Vasc. Biol. 34, 1446–1458. doi: 10.1161/ATVBAHA.114.303534
Nigdelioglu, R., Hamanaka, R. B., Meliton, A. Y., O'leary, E., Witt, L. J., Cho, T., et al. (2016). Transforming growth factor (TGF)-beta promotes de novo serine synthesis for collagen production. J. Biol. Chem. 291, 27239–27251. doi: 10.1074/jbc.M116.756247
Noguchi, S., Saito, A., Mikami, Y., Urushiyama, H., Horie, M., Matsuzaki, H., et al. (2017). TAZ contributes to pulmonary fibrosis by activating profibrotic functions of lung fibroblasts. Sci. Rep. 7:42595. doi: 10.1038/srep42595
Noris, M., Morigi, M., Donadelli, R., Aiello, S., Foppolo, M., Todeschini, M., et al. (1995). Nitric-oxide synthesis by cultured endothelial-cells is modulated by flow conditions. Circ. Res. 76, 536–543. doi: 10.1161/01.RES.76.4.536
Noto, A., De Vitis, C., Pisanu, M. E., Roscilli, G., Ricci, G., Catizone, A., et al. (2017). Stearoyl-CoA-desaturase 1 regulates lung cancer stemness via stabilization and nuclear localization of YAP/TAZ. Oncogene 36, 4573–4584. doi: 10.1038/onc.2017.75
Odagiri, K., Inui, N., Hakamata, A., Inoue, Y., Suda, T., Takehara, Y., et al. (2016). Non-invasive evaluation of pulmonary arterial blood flow and wall shear stress in pulmonary arterial hypertension with 3D phase contrast magnetic resonance imaging. Springerplus 5:1071. doi: 10.1186/s40064-016-2755-7
Oldendorf, W. H., Cornford, M. E., and Brown, W. J. (1977). The large apparent work capability of the blood-brain barrier: a study of the mitochondrial content of capillary endothelial cells in brain and other tissues of the rat. Ann. Neurol. 1, 409–417. doi: 10.1002/ana.410010502
Osman, L., Yacoub, M. H., Latif, N., Amrani, M., and Chester, A. H. (2006). Role of human valve interstitial cells in valve calcification and their response to atorvastatin. Circulation 114, I547–I552. doi: 10.1161/CIRCULATIONAHA.105.001115
Ostrowski, M. A., Huang, N. F., Walker, T. W., Verwijlen, T., Poplawski, C., Khoo, A. S., et al. (2014). Microvascular endothelial cells migrate upstream and align against the shear stress field created by impinging flow. Biophys. J. 106, 366–374. doi: 10.1016/j.bpj.2013.11.4502
Pannier, B., Guérin, A. P., Marchais, S. J., Safar, M. E., and London, G. M. (2005). Stiffness of capacitive and conduit arteries: prognostic significance for end-stage renal disease patients. Hypertension 45, 592–596. doi: 10.1161/01.HYP.0000159190.71253.c3
Parmar, K. M., Larman, H. B., Dai, G., Zhang, Y., Wang, E. T., Moorthy, S. N., et al. (2006). Integration of flow-dependent endothelial phenotypes by Kruppel-like factor 2. J. Clin. Invest. 116, 49–58. doi: 10.1172/JCI24787
Paszkowiak, J. J., and Dardik, A. (2003). Arterial wall shear stress: observations from the bench to the bedside. Vasc. Endovasc. Surg. 37, 47–57. doi: 10.1177/153857440303700107
Pettersen, K. H., Bugenhagen, S. M., Nauman, J., Beard, D. A., and Omholt, S. W. (2014). Arterial stiffening provides sufficient explanation for primary hypertension. PLoS Comput. Biol. 10:e1003634. doi: 10.1371/journal.pcbi.1003634
Pietrocola, F., Galluzzi, L., Bravo-San Pedro, J. M., Madeo, F., and Kroemer, G. (2015). Acetyl coenzyme A: a central metabolite and second messenger. Cell Metab. 21, 805–821. doi: 10.1016/j.cmet.2015.05.014
Plecitá-Hlavatá, L., D'alessandro, A., El Kasmi, K., Li, M., Zhang, H., JeŽek, P., et al. (2017). Metabolic reprogramming and Redox signaling in pulmonary hypertension. Adv. Exp. Med. Biol. 967, 241–260. doi: 10.1007/978-3-319-63245-2_14
Quintero, M., Colombo, S. L., Godfrey, A., and Moncada, S. (2006). Mitochondria as signaling organelles in the vascular endothelium. Proc. Natl. Acad. Sci. U.S.A. 103, 5379–5384. doi: 10.1073/pnas.0601026103
Rapraeger, A. C. (2000). Syndecan-regulated receptor signaling. J. Cell Biol. 149, 995–998. doi: 10.1083/jcb.149.5.995
Rehman, J., and Archer, S. L. (2010). A proposed mitochondrial-metabolic mechanism for initiation and maintenance of pulmonary arterial hypertension in fawn-hooded rats: the Warburg model of pulmonary arterial hypertension. Adv. Exp. Med. Biol. 661, 171–185. doi: 10.1007/978-1-60761-500-2_11
Reinitz, A., Destefano, J., Ye, M., Wong, A. D., and Searson, P. C. (2015). Human brain microvascular endothelial cells resist elongation due to shear stress. Microvasc. Res. 99, 8–18. doi: 10.1016/j.mvr.2015.02.008
Remuzzi, A., Brenner, B. M., Pata, V., Tebaldi, G., Mariano, R., Belloro, A., et al. (1992). Three-dimensional reconstructed glomerular capillary network: blood flow distribution and local filtration. Am. J. Physiol. 263, F562–572. doi: 10.1152/ajprenal.1992.263.3.F562
Rich, S., and Brundage, B. H. (1987). High-dose calcium channel-blocking therapy for primary pulmonary hypertension: evidence for long-term reduction in pulmonary arterial pressure and regression of right ventricular hypertrophy. Circulation 76, 135–141. doi: 10.1161/01.CIR.76.1.135
Sabine, A., Bovay, E., Demir, C. S., Kimura, W., Jaquet, M., Agalarov, Y., et al. (2015). FOXC2 and fluid shear stress stabilize postnatal lymphatic vasculature. J. Clin. Invest. 125, 3861–3877. doi: 10.1172/JCI80454
Safar, M. E., Balkau, B., Lange, C., Protogerou, A. D., Czernichow, S., Blacher, J., et al. (2013). Hypertension and vascular dynamics in men and women with metabolic syndrome. J. Am. Coll. Cardiol. 61, 12–19. doi: 10.1016/j.jacc.2012.01.088
Sangwung, P., Zhou, G., Nayak, L., Chan, E. R., Kumar, S., Kang, D. W., et al. (2017). KLF2 and KLF4 control endothelial identity and vascular integrity. JCI Insight 2:e91700. doi: 10.1172/jci.insight.91700
Santinon, G., Pocaterra, A., and Dupont, S. (2016). Control of YAP/TAZ activity by metabolic and nutrient-sensing pathways. Trends Cell Biol. 26, 289–299. doi: 10.1016/j.tcb.2015.11.004
Sawada, N., Jiang, A., Takizawa, F., Safdar, A., Manika, A., Tesmenitsky, Y., et al. (2014). Endothelial PGC-1alpha mediates vascular dysfunction in diabetes. Cell Metab. 19, 246–258. doi: 10.1016/j.cmet.2013.12.014
Schaefer, A., and Hordijk, P. L. (2015). Cell-stiffness-induced mechanosignaling - a key driver of leukocyte transendothelial migration. J. Cell Sci. 128, 2221–2230. doi: 10.1242/jcs.163055
Schäfer, M., Kheyfets, V. O., Schroeder, J. D., Dunning, J., Shandas, R., Buckner, J. K., et al. (2016). Main pulmonary arterial wall shear stress correlates with invasive hemodynamics and stiffness in pulmonary hypertension. Pulm. Circ. 6, 37–45. doi: 10.1086/685024
Schmidt, E. P., Overdier, K. H., Sun, X., Lin, L., Liu, X., Yang, Y., et al. (2016). Urinary glycosaminoglycans predict outcomes in septic shock and acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 194, 439–449. doi: 10.1164/rccm.201511-2281OC
Schmidt, E. P., Yang, Y., Janssen, W. J., Gandjeva, A., Perez, M. J., Barthel, L., et al. (2012). The pulmonary endothelial glycocalyx regulates neutrophil adhesion and lung injury during experimental sepsis. Nat. Med. 18, 1217–1223. doi: 10.1038/nm.2843
Schoors, S., Bruning, U., Missiaen, R., Queiroz, K. C., Borgers, G., Elia, I., et al. (2015). Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature 520, 192–197. doi: 10.1038/nature14362
Schrimpf, G., Schroder, M., Weitnauer, E., and Friedl, P. (1994). Metabolic rates of vascular endothelial-cells in-vitro. Cytotechnology 16, 43–50. doi: 10.1007/BF00761778
Scott, D., Tan, Y., Shandas, R., Stenmark, K. R., and Tan, W. (2013). High pulsatility flow stimulates smooth muscle cell hypertrophy and contractile protein expression. Am. J. Physiol. Lung Cell. Mol. Physiol. 304, L70–81. doi: 10.1152/ajplung.00342.2012
Shah, S. J., Lam, C. S. P., Svedlund, S., Saraste, A., Hage, C., Tan, R. S., et al. (2018). Prevalence and correlates of coronary microvascular dysfunction in heart failure with preserved ejection fraction: PROMIS-HFpEF. Eur. Heart J. 39, 3439–3450. doi: 10.1093/eurheartj/ehy531
Shen, B., Smith, R. S. Jr., Hsu, Y. T., Chao, L., and Chao, J. (2009). Kruppel-like factor 4 is a novel mediator of Kallistatin in inhibiting endothelial inflammation via increased endothelial nitric-oxide synthase expression. J. Biol. Chem. 284, 35471–35478. doi: 10.1074/jbc.M109.046813
Shentu, T. P., He, M., Sun, X., Zhang, J., Zhang, F., Gongol, B., et al. (2016). AMP-activated protein Kinase and Sirtuin 1 coregulation of cortactin contributes to endothelial function. Arterioscler. Thromb. Vasc. Biol. 36, 2358–2368. doi: 10.1161/ATVBAHA.116.307871
Shi, Y., and Vanhoutte, P. M. (2017). Macro- and microvascular endothelial dysfunction in diabetes. J. Diabetes 9, 434–449. doi: 10.1111/1753-0407.12521
Shiu, Y. T., Li, S., Marganski, W. A., Usami, S., Schwartz, M. A., Wang, Y. L., et al. (2004). Rho mediates the shear-enhancement of endothelial cell migration and traction force generation. Biophys. J. 86, 2558–2565. doi: 10.1016/S0006-3495(04)74311-8
Siasos, G., Sara, J. D., Zaromytidou, M., Park, K. H., Coskun, A. U., Lerman, L. O., et al. (2018). Local low shear stress and endothelial dysfunction in patients with nonobstructive coronary atherosclerosis. J. Am. Coll. Cardiol. 71, 2092–2102. doi: 10.1016/j.jacc.2018.02.073
Slutsky, A. S., and Ranieri, V. M. (2013). Ventilator-induced lung injury. N. Engl. J. Med. 369, 2126–2136. doi: 10.1056/NEJMra1208707
Smulyan, H., Lieber, A., and Safar, M. E. (2016). Hypertension, diabetes type II, and their association: role of arterial stiffness. Am. J. Hypertens. 29, 5–13. doi: 10.1093/ajh/hpv107
Stenmark, K. R., Fagan, K. A., and Frid, M. G. (2006). Hypoxia-induced pulmonary vascular remodeling: cellular and molecular mechanisms. Circ. Res. 99, 675–691. doi: 10.1161/01.RES.0000243584.45145.3f
Stroka, K. M., and Aranda-Espinoza, H. (2011). Endothelial cell substrate stiffness influences neutrophil transmigration via myosin light chain kinase-dependent cell contraction. Blood 118, 1632–1640. doi: 10.1182/blood-2010-11-321125
Sun, H. D., Liu, Y. J., Chen, J., Chen, M. Y., Ouyang, B., and Guan, X. D. (2017). The pivotal role of HIF-1alpha in lung inflammatory injury induced by septic mesenteric lymph. Biomed. Pharmacother. 91, 476–484. doi: 10.1016/j.biopha.2017.04.103
Suresh, M. V., Balijepalli, S., Zhang, B., Singh, V. V., Swamy, S., Panicker, S., et al. (2018). Hypoxia-inducible factor (HIF)-1alpha promotes inflammation and injury following aspiration-induced lung injury in mice. Shock. doi: 10.1097/SHK.0000000000001312. [Epub ahead of print].
Szulcek, R., Happé, C. M., Rol, N., Fontijn, R. D., Dickhoff, C., Hartemink, K. J., et al. (2016). Delayed microvascular shear adaptation in pulmonary arterial hypertension. role of platelet endothelial cell adhesion molecule-1 cleavage. Am. J. Respir. Crit. Care Med. 193, 1410–1420. doi: 10.1164/rccm.201506-1231OC
Szyk, A., Deaconescu, A. M., Spector, J., Goodman, B., Valenstein, M. L., Ziolkowska, N. E., et al. (2014). Molecular basis for age-dependent microtubule acetylation by tubulin acetyltransferase. Cell 157, 1405–1415. doi: 10.1016/j.cell.2014.03.061
Tan, Y., Tseng, P. O., Wang, D., Zhang, H., Hunter, K., Hertzberg, J., et al. (2014). Stiffening-induced high pulsatility flow activates endothelial inflammation via a TLR2/NF-kappaB pathway. PLoS ONE 9:e102195. doi: 10.1371/journal.pone.0102195
Tang, B. T., Pickard, S. S., Chan, F. P., Tsao, P. S., Taylor, C. A., and Feinstein, J. A. (2012). Wall shear stress is decreased in the pulmonary arteries of patients with pulmonary arterial hypertension: an image-based, computational fluid dynamics study. Pulm. Circ. 2, 470–476. doi: 10.4103/2045-8932.105035
Tang, H. Y., Babicheva, A., Mcdermott, K. M., Gu, Y. L., Ayon, R. J., Song, S. S., et al. (2018). Endothelial HIF-2 alpha contributes to severe pulmonary hypertension due to endothelial-to-mesenchymal transition. Am. J. Physiol. Lung Cell. Mol. Physiol. 314, L256–L275. doi: 10.1152/ajplung.00096.2017
Taraseviciene-Stewart, L., Kasahara, Y., Alger, L., Hirth, P., Mc Mahon, G., Waltenberger, J., et al. (2001). Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J. 15, 427–438. doi: 10.1096/fj.00-0343com
Teichert-Kuliszewska, K., Kutryk, M. J., Kuliszewski, M. A., Karoubi, G., Courtman, D. W., Zucco, L., et al. (2006). Bone morphogenetic protein receptor-2 signaling promotes pulmonary arterial endothelial cell survival: implications for loss-of-function mutations in the pathogenesis of pulmonary hypertension. Circ. Res. 98, 209–217. doi: 10.1161/01.RES.0000200180.01710.e6
Tello, D., Balsa, E., Acosta-Iborra, B., Fuertes-Yebra, E., Elorza, A., Ordóñez, A., et al. (2011). Induction of the mitochondrial NDUFA4L2 protein by HIF-1alpha decreases oxygen consumption by inhibiting complex I activity. Cell Metab. 14, 768–779. doi: 10.1016/j.cmet.2011.10.008
Teo, C. F., Wollaston-Hayden, E. E., and Wells, L. (2010). Hexosamine flux, the O-GlcNAc modification, and the development of insulin resistance in adipocytes. Mol. Cell. Endocrinol. 318, 44–53. doi: 10.1016/j.mce.2009.09.022
Thenappan, T., Prins, K. W., Pritzker, M. R., Scandurra, J., Volmers, K., and Weir, E. K. (2016). The critical role of pulmonary arterial compliance in pulmonary hypertension. Ann. Am. Thorac. Soc. 13, 276–284. doi: 10.1513/AnnalsATS.201509-599FR
Theodorou, K., and Boon, R. A. (2018). Endothelial cell metabolism in atherosclerosis. Front. Cell. Dev. Biol. 6:82. doi: 10.3389/fcell.2018.00082
Thi, M. M., Tarbell, J. M., Weinbaum, S., and Spray, D. C. (2004). The role of the glycocalyx in reorganization of the actin cytoskeleton under fluid shear stress: a “bumper-car” model. Proc. Natl. Acad. Sci. U.S.A. 101, 16483–16488. doi: 10.1073/pnas.0407474101
Totaro, A., Castellan, M., Battilana, G., Zanconato, F., Azzolin, L., Giulitti, S., et al. (2017). YAP/TAZ link cell mechanics to Notch signalling to control epidermal stem cell fate. Nat. Commun. 8:15206. doi: 10.1038/ncomms15206
Totaro, A., Panciera, T., and Piccolo, S. (2018). YAP/TAZ upstream signals and downstream responses. Nat. Cell Biol. 20, 888–899. doi: 10.1038/s41556-018-0142-z
Trott, D., Mcmanus, C. A., Martin, J. L., Brennan, B., Dunn, M. J., and Rose, M. L. (2009). Effect of phosphorylated hsp27 on proliferation of human endothelial and smooth muscle cells. Proteomics 9, 3383–3394. doi: 10.1002/pmic.200800961
Urbano, R. L., Furia, C., Basehore, S., and Clyne, A. M. (2017). Stiff substrates increase inflammation-induced endothelial monolayer tension and permeability. Biophys. J. 113, 645–655. doi: 10.1016/j.bpj.2017.06.033
Valenta, T., Hausmann, G., and Basler, K. (2012). The many faces and functions of beta-catenin. EMBO J. 31, 2714–2736. doi: 10.1038/emboj.2012.150
Vermot, J., Forouhar, A. S., Liebling, M., Wu, D., Plummer, D., Gharib, M., et al. (2009). Reversing blood flows act through klf2a to ensure normal valvulogenesis in the developing heart. PLoS Biol. 7:e1000246. doi: 10.1371/journal.pbio.1000246
Villarreal, G. Jr., Zhang, Y., Larman, H. B., Graca-Sancho, J., Koo, A., and García-Cardeña, G. (2010). Defining the regulation of KLF4 expression and its downstream transcriptional targets in vascular endothelial cells. Biochem. Biophys. Res. Commun. 391, 984–989. doi: 10.1016/j.bbrc.2009.12.002
Vincent, J. L. (2001). Microvascular endothelial dysfunction: a renewed appreciation of sepsis pathophysiology. Crit Care 5, S1–5. doi: 10.1186/cc1332
Wang, C., Baker, B. M., Chen, C. S., and Schwartz, M. A. (2013). Endothelial cell sensing of flow direction. Arterioscler. Thromb. Vasc. Biol. 33, 2130–2136. doi: 10.1161/ATVBAHA.113.301826
Wang, K. C., Yeh, Y. T., Nguyen, P., Limqueco, E., Lopez, J., Thorossian, S., et al. (2016). Flow-dependent YAP/TAZ activities regulate endothelial phenotypes and atherosclerosis. Proc. Natl. Acad. Sci. U.S.A. 113, 11525–11530. doi: 10.1073/pnas.1613121113
Wang, L., Luo, J. Y., Li, B., Tian, X. Y., Chen, L. J., Huang, Y., et al. (2016). Integrin-YAP/TAZ-JNK cascade mediates atheroprotective effect of unidirectional shear flow. Nature 540, 579–582. doi: 10.1038/nature20602
Wang, W., Xiao, Z. D., Li, X., Aziz, K. E., Gan, B., Johnson, R. L., et al. (2015). AMPK modulates Hippo pathway activity to regulate energy homeostasis. Nat. Cell Biol. 17, 490–499. doi: 10.1038/ncb3113
Wang, X., Hu, G., Gao, X., Wang, Y., Zhang, W., Harmon, E. Y., et al. (2012). The induction of Yesz-associated protein expression after arterial injury is crucial for smooth muscle phenotypic modulation and neointima formation. Arterioscler. Thromb. Vasc. Biol. 32, 2662–2669. doi: 10.1161/ATVBAHA.112.254730
Weibel, E. R. (1973). Morphological basis of alveolar-capillary gas exchange. Physiol. Rev. 53, 419–495. doi: 10.1152/physrev.1973.53.2.419
Weisbrod, R. M., Shiang, T., Al Sayah, L., Fry, J. L., Bajpai, S., Reinhart-King, C. A., et al. (2013). Arterial stiffening precedes systolic hypertension in diet-induced obesity. Hypertension 62, 1105–1110. doi: 10.1161/HYPERTENSIONAHA.113.01744
Wettstein, G., Bellaye, P. S., Kolb, M., Hammann, A., Crestani, B., Soler, P., et al. (2013). Inhibition of HSP27 blocks fibrosis development and EMT features by promoting Snail degradation. FASEB J. 27, 1549–1560. doi: 10.1096/fj.12-220053
Wolff, B., Lodziewski, S., Bollmann, T., Opitz, C. F., and Ewert, R. (2007). Impaired peripheral endothelial function in severe idiopathic pulmonary hypertension correlates with the pulmonary vascular response to inhaled iloprost. Am. Heart J. 153, 1088 e1081–1087. doi: 10.1016/j.ahj.2007.03.005
Won, D., Zhu, S. N., Chen, M., Teichert, A. M., Fish, J. E., Matouk, C. C., et al. (2007). Relative reduction of endothelial nitric-oxide synthase expression and transcription in atherosclerosis-prone regions of the mouse aorta and in an in vitro model of disturbed flow. Am. J. Pathol. 171, 1691–1704. doi: 10.2353/ajpath.2007.060860
Wu, D., Huang, R. T., Hamanaka, R. B., Krause, M., Oh, M. J., Kuo, C. H., et al. (2017). HIF-1alpha is required for disturbed flow-induced metabolic reprogramming in human and porcine vascular endothelium. Elife 6:e25217. doi: 10.7554/eLife.25217
Xiong, J., Kawagishi, H., Yan, Y., Liu, J., Wells, Q. S., Edmunds, L. R., et al. (2018). A metabolic basis for endothelial-to-mesenchymal transition. Mol. Cell 69, 689–698 e687. doi: 10.1016/j.molcel.2018.01.010
Xu, W., Koeck, T., Lara, A. R., Neumann, D., Difilippo, F. P., Koo, M., et al. (2007). Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc. Natl. Acad. Sci. U.S.A. 104, 1342–1347. doi: 10.1073/pnas.0605080104
Xu, X., Tan, X., Tampe, B., Sanchez, E., Zeisberg, M., and Zeisberg, E. M. (2015). Snail is a direct target of hypoxia-inducible factor 1alpha (HIF1alpha) in hypoxia-induced endothelial to mesenchymal transition of human coronary endothelial cells. J. Biol. Chem. 290, 16653–16664. doi: 10.1074/jbc.M115.636944
Xu, Y., Wang, Y., Yan, S., Zhou, Y., Yang, Q., Pan, Y., et al. (2017). Intracellular adenosine regulates epigenetic programming in endothelial cells to promote angiogenesis. EMBO Mol. Med. 9, 1263–1278. doi: 10.15252/emmm.201607066
Yen, W., Cai, B., Yang, J., Zhang, L., Zeng, M., Tarbell, J. M., et al. (2015). Endothelial surface glycocalyx can regulate flow-induced nitric oxide production in microvessels in vivo. PLoS ONE 10:e0117133. doi: 10.1371/journal.pone.0117133
Yu, J., Ma, Z., Shetty, S., Ma, M., and Fu, J. (2016). Selective HDAC6 inhibition prevents TNF-alpha-induced lung endothelial cell barrier disruption and endotoxin-induced pulmonary edema. Am. J. Physiol. Lung Cell. Mol. Physiol. 311, L39–47. doi: 10.1152/ajplung.00051.2016
Yu, P., Wilhelm, K., Dubrac, A., Tung, J. K., Alves, T. C., Fang, J. S., et al. (2017). FGF-dependent metabolic control of vascular development. Nature 545, 224–228. doi: 10.1038/nature22322
Yu, Q., Tai, Y. Y., Tang, Y., Zhao, J., Negi, V., Culley, M. K., et al. (2019). BOLA3 deficiency controls endothelial metabolism and glycine homeostasis in pulmonary hypertension. Circulation. 139, 2238–2255. doi: 10.1161/CIRCULATIONAHA.118.035889
Zambrano, B. A., Mclean, N. A., Zhao, X., Tan, J. L., Zhong, L., Figueroa, C. A., et al. (2018). Image-based computational assessment of vascular wall mechanics and hemodynamics in pulmonary arterial hypertension patients. J. Biomech. 68, 84–92. doi: 10.1016/j.jbiomech.2017.12.022
Zeisberg, E. M., Potenta, S., Xie, L., Zeisberg, M., and Kalluri, R. (2007a). Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 67, 10123–10128. doi: 10.1158/0008-5472.CAN-07-3127
Zeisberg, E. M., Tarnavski, O., Zeisberg, M., Dorfman, A. L., Mcmullen, J. R., Gustafsson, E., et al. (2007b). Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 13, 952–961. doi: 10.1038/nm1613
Zhang, B., Niu, W., Dong, H. Y., Liu, M. L., Luo, Y., and Li, Z. C. (2018a). Hypoxia induces endothelialmesenchymal transition in pulmonary vascular remodeling. Int. J. Mol. Med. 42, 270–278. doi: 10.3892/ijmm.2018.3584
Zhang, H., Von Gise, A., Liu, Q., Hu, T., Tian, X., He, L., et al. (2014). Yap1 is required for endothelial to mesenchymal transition of the atrioventricular cushion. J. Biol. Chem. 289, 18681–18692. doi: 10.1074/jbc.M114.554584
Zhang, X., Li, Y., Ma, Y., Yang, L., Wang, T., Meng, X., et al. (2018b). Yes-associated protein (YAP) binds to HIF-1alpha and sustains HIF-1alpha protein stability to promote hepatocellular carcinoma cell glycolysis under hypoxic stress. J. Exp. Clin. Cancer Res. 37:216. doi: 10.1186/s13046-018-0892-2
Zhang, X., Qiao, Y., Wu, Q., Chen, Y., Zou, S., Liu, X., et al. (2017). The essential role of YAP O-GlcNAcylation in high-glucose-stimulated liver tumorigenesis. Nat. Commun. 8:15280. doi: 10.1038/ncomms15280
Zhang, Y., Kwon, S., Yamaguchi, T., Cubizolles, F., Rousseaux, S., Kneissel, M., et al. (2008). Mice lacking histone deacetylase 6 have hyperacetylated tubulin but are viable and develop normally. Mol. Cell. Biol. 28, 1688–1701. doi: 10.1128/MCB.01154-06
Zhao, D., Xue, C., Li, Q., Liu, M., Ma, W., Zhou, T., et al. (2018). Substrate stiffness regulated migration and angiogenesis potential of A549 cells and HUVECs. J. Cell. Physiol. 233, 3407–3417. doi: 10.1002/jcp.26189
Zheng, C., Yu, Z., Zhou, Y., Tao, L., Pang, Y., Chen, T., et al. (2012). Live cell imaging analysis of the epigenetic regulation of the human endothelial cell migration at single-cell resolution. Lab Chip 12, 3063–3072. doi: 10.1039/c2lc40192d
Zhong, A., Mirzaei, Z., and Simmons, C. A. (2018). The roles of matrix stiffness and ss-catenin signaling in endothelial-to-mesenchymal transition of aortic valve endothelial cells. Cardiovasc. Eng. Technol. 9, 158–167. doi: 10.1007/s13239-018-0363-0
Keywords: metabolism, pulmonary, endothelial (dys)function, microvasculature, endothelial mesenchymal transition
Citation: Wu D and Birukov K (2019) Endothelial Cell Mechano-Metabolomic Coupling to Disease States in the Lung Microvasculature. Front. Bioeng. Biotechnol. 7:172. doi: 10.3389/fbioe.2019.00172
Received: 30 April 2019; Accepted: 03 July 2019;
Published: 19 July 2019.
Edited by:
Martijn van Griensven, Technical University of Munich, GermanyReviewed by:
Duncan J. Stewart, University of Ottawa, CanadaRebecca Heise, Virginia Commonwealth University, United States
Paul Brian Dieffenbach, Brigham and Women's Hospital and Harvard Medical School, United States
Copyright © 2019 Wu and Birukov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David Wu, ZHd1MUBtZWRpY2luZS5ic2QudWNoaWNhZ28uZWR1