 Hao Zhang1†
Hao Zhang1† Bin Zhang
Bin Zhang- 1Department of Respiratory and Critical Care Medicine, Second Affiliated Hospital of Zhejiang University School of Medicine, Hangzhou, China
- 2Department of Respiration, Hospital of Traditional Chinese Medicine of Zhenhai, Ningbo, China
- 3Shanghai Engineering Research Center of Pharmaceutical Translation, Shanghai, China
Lung cancer is a highly prevalent type of cancer with a poor 5-year survival rate of about 4–17%. Eighty percent lung cancer belongs to non-small-cell lung cancer (NSCLC). For a long time, the treatment of NSCLC has been mostly guided by tumor stage, and there has been no significant difference between the therapy strategy of lung adenocarcinoma (LUAD) and squamous cell lung carcinoma (SCLC), the two major subtypes of NSCLC. In recent years, important molecular differences between LUAD and SCLC are increasingly identified, indicating that targeted therapy will be more and more histologically specific in the future. To investigate the LUAD and SCLC difference on multi-omics scale, we analyzed the methylation and gene expression data together. With the Boruta method to remove irrelevant features and the MCFS (Monte Carlo Feature Selection) method to identify the significantly important features, we identified 113 key methylation features and 23 key gene expression features. HNF1B and TP63 were found to be dysfunctional on both methylation and gene expression levels. The experimentally determined interaction network suggested that TP63 may play an important role in connecting methylation genes and expression genes. Many of the discovered signature genes have been supported by literature. Our results may provide directions of precision diagnosis and therapy of LUAD and SCLC.
Introduction
Lung cancer, considered to be a highly prevalent type of cancer, is a leading cause of cancer-related mortality worldwide, resulting in 1.6 million deaths each year with poor 5-year survival rate of about 4–17% (Hirsch et al., 2017; Altorki et al., 2019). Lung cancer is classified as follows: small-cell lung cancer (SCLC) and non-small-cell lung cancer (NSCLC), accounting for approximately 20 and 80% of all lung cancer cases, respectively (Oser et al., 2015). NSCLC is a complex systems disease with dysfunctions on multiple pathways and multiple molecular levels (Huang et al., 2012, 2015; Li et al., 2013; Zhou et al., 2015; Chen et al., 2016; Liu et al., 2017). It can also be typically divided into three main subtypes, lung adenocarcinoma (LUAD), squamous cell lung carcinoma (SCLC), and large cell cancer (LCC), according to standard pathology methods (Socinski et al., 2016; Swanton and Govindan, 2016; Herbst et al., 2018). Compared with squamous lung cancer, adenocarcinoma was associated with better prognosis. Despite the advances in diagnostic and therapeutic technology, lung cancer remains a serious global public health concern.
For a long time, the treatment of NSCLC has been mostly guided by tumor stage, and there has been no significant difference between the therapy strategy of LUAD and SCLC. Most lung cancers are usually diagnosed at an advanced stage and are treated primarily with systemic chemotherapy, typically with platinum-based regimens (Bishop et al., 2010). Recent progress in characterization of NSCLC by molecular typing, especially in adenocarcinomas of the lung, have brought new investigation of therapeutic agents that target dominant oncogenic mutations, such as epidermal growth factor receptor (EGFR)-targeted therapies, which have showed improved response rates in patients with NSCLC (Shigematsu et al., 2005).
Currently, progress in molecular biology of lung cancer has resulted in the identification of multiple potential biomarkers that may be related to the clinical management of NSCLC patients. In recent years, with the emergence of next-generation sequencing technologies, important molecular differences between LUAD and SCLC are increasingly identified, indicating that targeted therapy will be more and more histologically specific in the future (Kim et al., 2005; Sun et al., 2007; Li et al., 2014). Several studies have identified multiple gene expression subtypes that differ in prognosis, genomic alterations, clinical characteristics, including tumor differentiation, stage-specific survival, underlying drivers, and potential responses to treatment within LUAD and SCLC (Wilkerson et al., 2010; Thomas et al., 2014; Lu et al., 2016). For example, LUAD patients that harbor EGFR, ALK, ROS1, or BRAF mutations were discovered to benefit the most (Villalobos and Wistuba, 2017; Herbst et al., 2018). Targeted therapies for gene abnormalities of HER2, MET, RET, and NTRK1 appear to be an effective approach to treat LUAD (Dearden et al., 2013; Mazieres et al., 2013). SCLC shows different mutation spectrum from that of adenocarcinoma, and the mutation targeted therapy for SCLC has not been thoroughly studied to obtain approved treatment (Bunn et al., 2016; Soldera and Leighl, 2017).
A series of imaging studies suggested that NSCLC may progress rapidly between occurrence and primary treatment (Koh et al., 2017). Therefore, it is necessary for clinicians to identify between these two subtypes of NSCLC in a convenient and rapid way. With the improvement of the above clinical and molecular levels, growing evidences have shown that immunohistochemistry (IHC) is an effective tool for differentiating adenocarcinoma from squamous cell carcinoma (Bass et al., 2009; Weiss et al., 2010).
It is reported that the formation and development of lung cancer are related to the accumulation of permanent genetic changes and dynamic epigenetic changes. Therefore, enhancing our understanding of tumor biology and gene expression profiles will be critical for cancer treatment and diagnosis. In this study, an integrative analysis of lung cancer methylation data and gene expression data was performed, and mixed features were also screened out for analysis.
Materials and Methods
The Joint Methylation and Expression Profiles of Lung Cancer Patients
The methylation and gene expression profiles of lung cancer patients were obtained from GEO (Gene Expression Omnibus)1. The data were originally generated by Karlsson et al. (2014). They used the data to cluster the patients into five groups, and these groups showed different overall survival (Karlsson et al., 2014). We were more interested in how the methylation and expression differ from well-known subtypes, especially LUAD and SCLC. Therefore, we analyzed the 77 LUAD and 22 SCLC patients who had both methylation and expression data.
The methylation profiles were measured with Illumina HumanMethylation450 BeadChip while the gene expression profiles were measured with Illumina HumanHT-12 V4.0 expression BeadChip. The probe expression levels were averaged onto 20,178 genes. The 354,251 methylation sites within genes were analyzed. Therefore, each patient was represented with 20,178 genes and 354,251 methylation sites.
Screen for the Relevant Methylation and Expression Features
Since the number of methylation and expression features was very large, it was difficult to analyze directly. We applied the Boruta method (Kursa and Rudnicki, 2010) to screen the combined data and identify the relevant methylation and expression features. The Boruta method was based on random forest classification, and the relevance of features to sample classes was measured by the ensemble of the random forest classifier’s stochasticity.
Evaluate the Importance of Relevant Methylation and Expression Features
After the irrelevant features were removed, the relevant methylation and expression features were ranked based on their importance evaluated with MCFS (Monte Carlo Feature Selection) (Draminski et al., 2008). The MCFS was a widely used method to rank features based on classification trees (Chen et al., 2018, 2019; Pan et al., 2018, 2019a,b; Li et al., 2019). First, for the d features, we selected s subsets and each subset included m features (m was much smaller than d). Then, for each subset, t trees were constructed. Based on the s × t trees, we can estimate a feature’s importance by considering how many times it appeared in these trees and how well it performed in these trees as a node. By comparing the permutation results, the significance of features was evaluated.
Perdition Performance of the Mixed Methylation and Expression Signature
The MCFS can find the significant top-ranking features by comparing with permutations. To objectively evaluate the significant top-ranking features’ prediction performance, we performed LOOCV (Leave One Out Cross Validation) using SVM (Support Vector Machine) classifier (Li et al., 2018; Sun et al., 2018; Pan et al., 2019a). Each time, one sample was chosen as test samples and all other samples were used to train the SVM predictor. After all samples were tested once, we compared the actual sample classes with predicted sample classes and calculated the sensitivity, specificity, accuracy, and Mathew’s correlation coefficient (MCC) based on the confusion matrix (Huang et al., 2011, 2013; Cai et al., 2012).
Results and Discussion
Rank the Methylation and Expression Features
The methylation and gene expression data were combined and, therefore, each lung cancer patient was represented with mixed methylation and gene expression features. The number of mixed features (20,178 gene expression features and 354,251 methylation features) was too large to conduct sophisticated statistical analysis. So, we removed irrelevant features using the Boruta method (Kursa and Rudnicki, 2010). At last, 711 relevant features were remained.
Then, these 711 Boruta selected features were further ranked with the MCFS method (Draminski et al., 2008). As a classification tree-based ensemble learning algorithm, MCFS can rank the features based on how many times and how much it contributed to the sample classification in s × t trees. By comparing with permutation results, it can evaluate the significance of features.
Identify the Methylation and Expression Signature
The 136 significant top-ranking features were identified using the latest dmLab version 2.3.0 software downloaded from2 with default parameters. These 136 methylation and expression signatures are given in Table 1.

Table 1. The 136 methylation and gene expression signature identified with the MCFS method.
It can be seen that within these 136 signature features, there were 113 methylation features and 23 gene expression features. The annotations of the 113 methylation features based on GPL135343 are provided in Supplementary Table S1. We plotted the heatmaps of LUAD and SCLC lung cancer patients with 113 methylation features and 23 gene expression features in Figures 1, 2, respectively. Both the 113 methylation features and 23 gene expression features can successfully group almost all samples with only three misclassified SCLC samples. They did not show difference on cluster results.
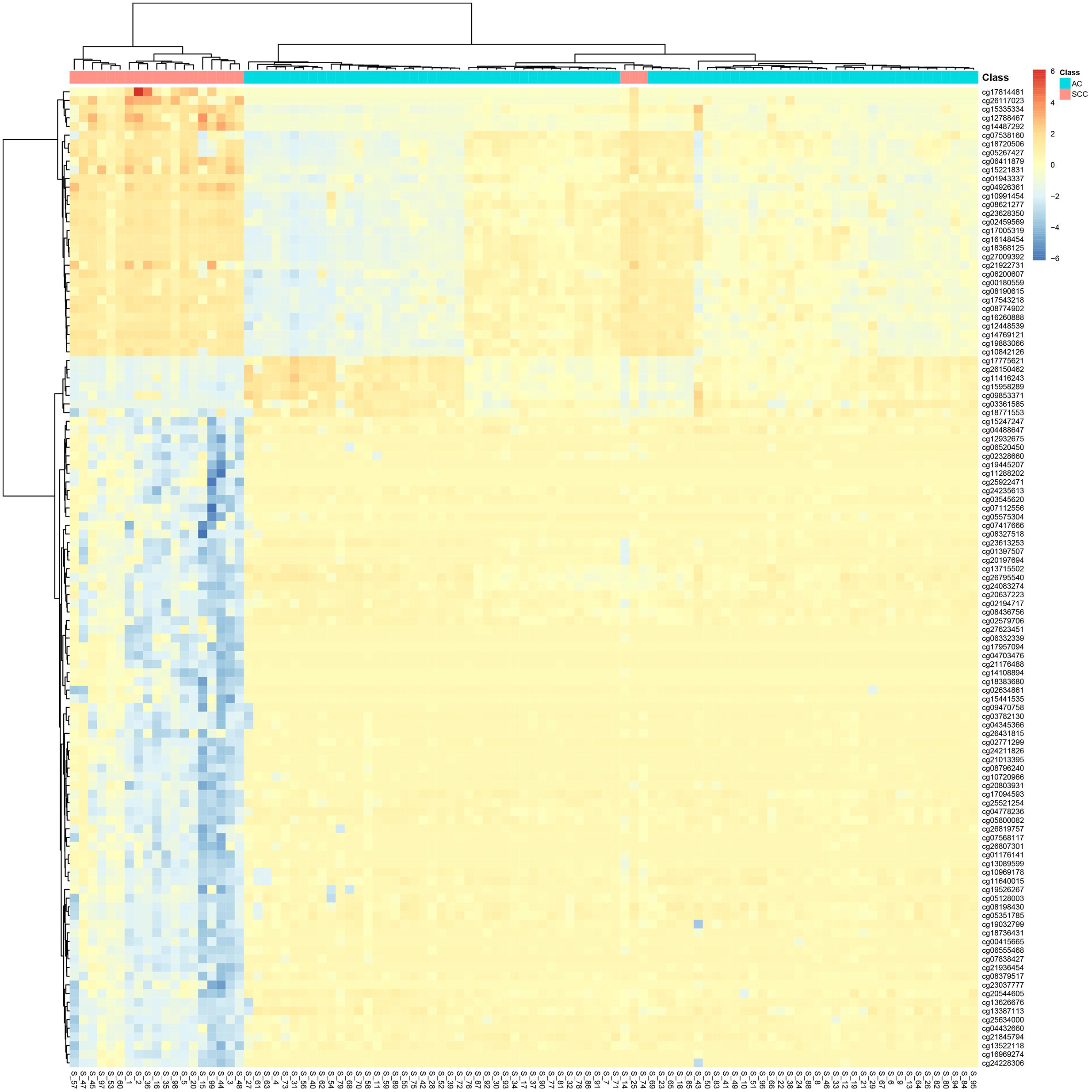
Figure 1. The heatmap of LUAD and SCLC lung cancer patients with 113 methylation features. Almost all samples were correctly clustered using the 113 methylation features and only three SCLC samples were misclassified.
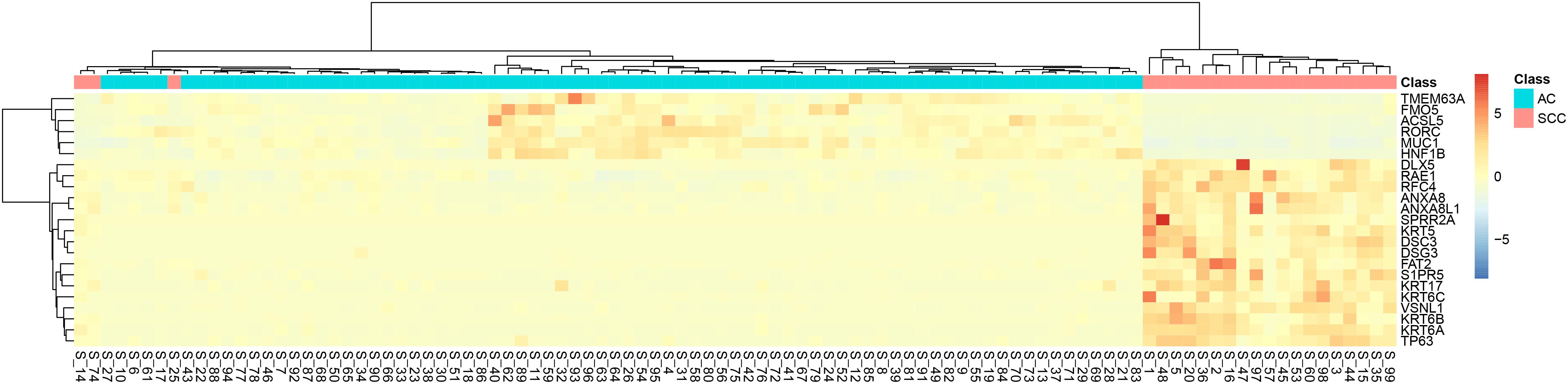
Figure 2. The heatmap of LUAD and SCLC lung cancer patients with 23 gene expression features. Almost all samples were correctly clustered using the 23 gene expression features and only three SCLC samples were misclassified.
To more objectively and carefully compare the performance of the 113 methylation features and 23 gene expression features, we conducted LOOCV with SVM classifier. The LOOCV prediction performances of the 136 mixed features, 113 methylation features and 23 gene expression features are listed in Tables 2–4. It can be seen that the prediction results of 113 methylation features were the same as the 136 mixed features and better than the 23 gene expression features. The 23 gene expression features had one more misclassified SCLC samples. It seemed that methylation had better performance.

Table 2. The confusion matrix using 136 mixed methylation and gene expression features.

Table 3. The confusion matrix using 113 methylation features.

Table 4. The confusion matrix using 23 gene expression features.
Comparison With CNV Signature
Comparing with the 136 LUAD and SQCLC CNV signatures identified by Li et al. (2014), we found that the methylated genes HORMAD2, KLHL3, LPP, and PTPN3 are also CNAs genes. HORMAD2 is expressed in nearly 10% of Chinese Han lung cancer tissues, which is a new target for lung cancer research (Liu et al., 2012). Lipoma preferred partner (LPP) may be an important candidate molecular marker for the classification of NSCLC tissue subtypes. PTPN3 can inhibit lung cancer by regulating EGFR signal (Li et al., 2015). However, there are no reports of KLHL3 in lung cancer, which also suggests a new idea of candidate molecular markers for the identification of lung cancer subtypes.
The Relationship Between Methylation and Expression Signature Genes
The 113 methylation features can be mapped onto 93 genes. We overlapped the selected methylation feature genes and expression feature genes and found that HNF1B and TP63 were dysfunctional on both methylation and gene expression levels. HNF1B was one of the DNA methylated markers of the same subtype (Matsuo et al., 2014; Shi et al., 2017). TP63, also known as P63, was considered to be the most common marker for SCLC (Bishop et al., 2012; Van de Laar et al., 2014).
We downloaded the 66 lung cancer genes from KEGG hsa05223 NSCLC4 and mapped them and the overlapped two genes: HNF1B and TP63, onto STRING network (Szklarczyk et al., 2018). TP63 interacted with 39 KEGG lung cancer genes: AKT1, AKT3, ALK, BAK1, BAX, CASP9, CCND1, CDK4, CDK6, CDKN1A, CDKN2A, DDB2, E2F1, E2F2, E2F3, EGF, EGFR, EML4, ERBB2, FHIT, FOXO3, GADD45A, GRB2, HRAS, KRAS, MAP2K1, MAPK1, MAPK3, NRAS, PIK3CA, PIK3CB, PIK3R1, RB1, STAT3, STAT5A, STAT5B, STK4, TGFA, and TP53. HNF1B interacted with 14 KEGG lung cancer genes: AKT1, AKT2, CCND1, CDKN1A, CDKN2A, EGF, HRAS, KRAS, MAPK1, MAPK3, PIK3CA, RXRA, STAT3, and TP53.
What’s more, we searched the methylation genes and expression genes in STRING database (Szklarczyk et al., 2018) and extracted the experimentally determined interaction and plotted the network in Figure 3. The light-yellow nodes were methylation genes, the light-blue nodes were expression genes. The overlapped methylation and expression genes were marked in red, the overlapped methylation and CNV genes from Li et al. (2014) were marked in pink. It can be seen that TP63 played an important role in connecting methylation genes and expression genes. The methylation genes and expression genes were closely connected to form a dense functional module on the network.
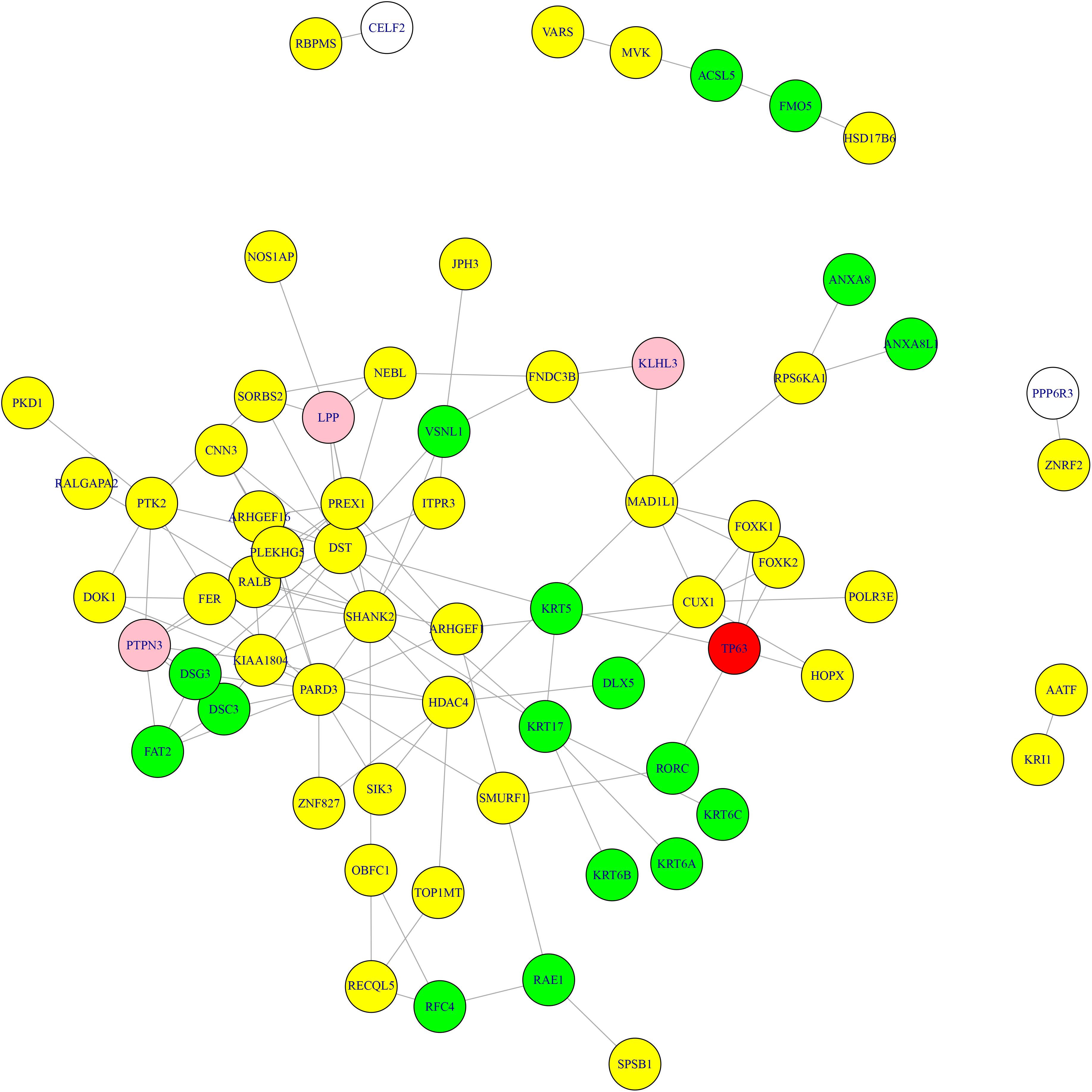
Figure 3. The methylation genes and expression genes with experimentally determined interactions on STRING network. The light-yellow nodes were methylation genes, and the light-blue nodes were expression genes. The overlapped methylation and expression genes were marked in red, and the overlapped methylation and CNV genes were marked in pink. TP63 played an important role in connecting methylation genes and expression genes.
The Biological Significance of the Identified Signature
To develop more specific and individualized targeted therapy, there is an urgent need to improve our knowledge on the molecular basis, in addition to different phenotypes. It is noteworthy that adenocarcinoma and squamous cell carcinoma show marked differences in expression profiles, DNA methylation, and lesion location. In this study, the features containing methylation and expression data were screened by Boruta and then further sorted by MCFS. After comparing the selected features with related literatures, a certain correlation was found between these features and lung cancer subtypes.
In this study, 113 methylation features were screened and mapped to 93 genes. We inquired about the functions of these genes and their relationship with lung cancer to discuss whether they have the potential as molecular markers to recognize LUAD and SQCLC. Many genes have been proved to promote or inhibit the progression of lung cancer. For instance, FOXK1 was expressed in many malignant tissues (Huang and Lee, 2004) and Ma et al. (2018) also found that FOXK1 plays a carcinogenic role in lung cancer. MAD1L1 is a checkpoint gene, with its mutation been proved to play a pathogenic role in lung cancer (Tsukasaki et al., 2001). Some genes have been reported to be related with the prognosis of NSCLC, such as HORMAD2 and ANO1. The overexpression of ANO1 is related to the high expression of EGFR, which can be used as a predictor of recurrence after NSCLC (He et al., 2017). In addition, according to Zhang et al. (2014) HORMAD2 gene polymorphism has great potential prognostic value in Chinese patients with NSCLC. Other genes are associated with NSCLC subtypes, such as another member of the FOX family, FOXK2, which was reported to be closely related to the overall survival of LUAD (Chen et al., 2017). DOK1 and HOPX were found to serve as lung tumor suppressors for LUAD (Berger et al., 2010; Chen et al., 2015). In the study of Zhou et al. (2017) the methylation locus of PARD3 gene was positively correlated with the expression of PARD3 and suppression of PARD3 intensified chemoresistance in LUAD cells. SFTA3 was found obviously overexpressed in LUAD, and its expression in LUAD and SQCLC was quite different. Therefore, the sensitivity and specificity of using SFTA3 to distinguish the two subtypes will be relatively high (Zhan et al., 2015). ARHGEF1 aliased p114RhoGEF and its expression might help to predict progression and survival of SQCLC patients (Song et al., 2013). Notably, LPP has multiple functions of actin binding protein and transcriptional coactivator (Kuriyama et al., 2016). Ngan et al. (2017) proved that the expression of LPP reduces the number of circulating tumor cells and inhibits lung cancer metastasis. Kang et al. (2009) used high-resolution array-CGH to find that the difference in genomic imbalance patterns between SQCLC and LUAD was most significant in 3q26.2-q29, while LPP (3q28) was significantly targeted in SQCLC, suggesting that LPP may be an attractive candidate molecular marker for histological subtype classification of NSCLC and may be involved in the pathogenesis of SQCLC.
We also investigated 23 expressed genes in lung cancer, and found that many studies clearly indicated that some genes were associated with LUAD or SQCLC. DSC3 (Han et al., 2014; Lv et al., 2015) and KRT5 (Xu et al., 2014; Travis et al., 2015) have been proved to be an effective marker of SQCLC. ANXA8 (Chao et al., 2006) and DSG3 (Savci-Heijink et al., 2009) were significantly over-expressed in SQCLC, and DSG3 could be an effective ancillary marker to identify SQCLC (Sanchez-Palencia et al., 2011; Gómez-Morales et al., 2013). VSNL1, also known as VILIP-1, was a tumor suppressor gene specific to SQCLC (Fu et al., 2008). KRT6A, KRT6B, and KRT6C, members of the keratin protein family, are specific to squamous cells and associated with epidermis of squamous epithelium (Fujii et al., 2002; Hawthorn et al., 2006; Chang et al., 2011). In addition, we also identified several genes primarily associated with LUAD. According to Balabko et al. (2014) RORC is a specific transcription factor in the tumor area of lung tissue in patients with LUAD. DLX5 (Kato et al., 2008; Balabko et al., 2014), MUC1 (Mashima et al., 2005; Molina-Pinelo et al., 2014), and KRT17 (Erdogan et al., 2009; Liu et al., 2018) were found to be overexpressed in LUAD.
The GO Enrichment Analysis of the Identified Signature
In order to further analyze the relationship between mixed characteristics and lung cancer, we carried out GO enrichment analysis. The results suggest that characteristic genes are mainly related to keratinization, epidermal cell differentiation, tissue development, and cytoplasm. The GO enriched results with FDR (False Discovery Rate) smaller than 0.05 are listed in Table 5. P63 appears to be useful in differentiating SQCLC from LUAD in small biopsies with no keratosis or glandular differentiation, helping to establish different treatments (Camilo et al., 2006). The expression of keratinocyte transglutaminase and cytokeratin 10 was measured as markers of squamous differentiation (Lokshin et al., 1999). Epidermal cell differentiation is related to EGFR signal pathway, which can inhibit the proliferation and metastasis of cancer cells, while EGFR mutation is largely limited to LUAD (Ladanyi and Pao, 2008). The expression of Promyelocytic leukemia zinc finger (PLZF) in SQCLC was weak or absent, which was significantly lower than that in LUAD (Xiao et al., 2015).
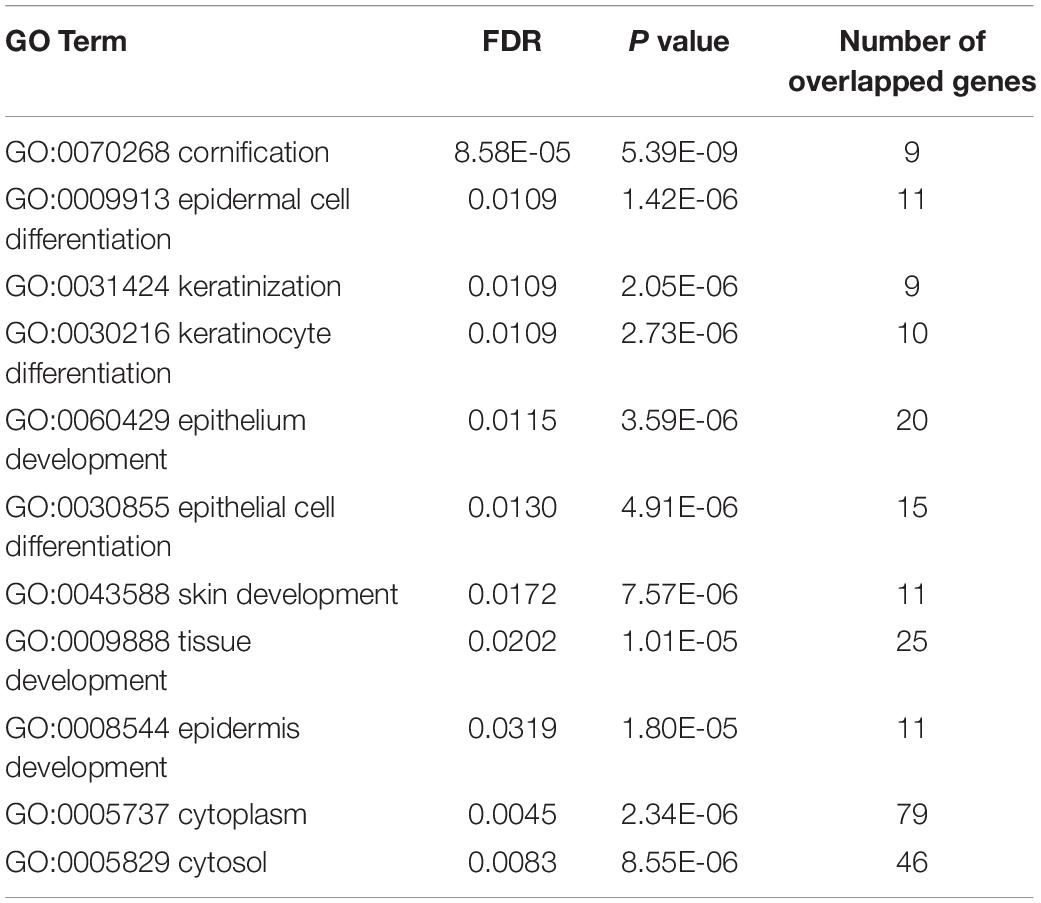
Table 5. The GO enrichment results of the identified signature.
To sum up, most of the 113 methylated genes and 23 expressed genes we found are closely related to lung cancer, and some of them have the possibility of distinguishing SQCLC from LUAD, which is helpful for the targeted selection of lung cancer treatment and provide more research support for lung cancer molecular markers.
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Author Contributions
HZ, ZJ, LC, and BZ contributed to the study design. HZ, ZJ, and LC conducted the literature search. HZ, ZJ, and BZ acquired the data. ZJ and LC wrote the manuscript. HZ and BZ performed the data analysis. All authors gave the final approval of the version to be submitted, read, and approved the final manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbioe.2020.00003/full#supplementary-material
TABLE S1 | The annotations of the 113 methylation features.
Footnotes
- ^ https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE60645
- ^ https://home.ipipan.waw.pl/m.draminski/mcfs.html
- ^ https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GPL13534
- ^ https://www.genome.jp/dbget-bin/www_bget?pathway+hsa05223
References
Altorki, N. K., Markowitz, G. J., Gao, D., Port, J. L., Saxena, A., Stiles, B., et al. (2019). The lung microenvironment: an important regulator of tumour growth and metastasis. Nat. Rev. Cancer 19, 9–31. doi: 10.1038/s41568-018-0081-9
Balabko, L., Andreev, K., Burmann, N., Schubert, M., Mathews, M., Trufa, D. I., et al. (2014). Increased expression of the Th17-IL-6R/pSTAT3/BATF/RorγT-axis in the tumoural region of adenocarcinoma as compared to squamous cell carcinoma of the lung. Sci. Rep. 4:7396. doi: 10.1038/srep07396
Bass, A. J., Watanabe, H., Mermel, C. H., Yu, S., Perner, S., Verhaak, R. G., et al. (2009). SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat. Genet. 41, 1238–1242. doi: 10.1038/ng.465
Berger, A. H., Niki, M., Morotti, A., Taylor, B. S., Socci, N. D., Viale, A., et al. (2010). Identification of DOK genes as lung tumor suppressors. Nat. Genet. 42, 216–223. doi: 10.1038/ng.527
Bishop, J. A., Benjamin, H., Cholakh, H., Chajut, A., Clark, D. P., and Westra, W. H. (2010). Accurate classification of non-small cell lung carcinoma using a novel microRNA-based approach. Clin. Cancer Res. 16, 610–619. doi: 10.1158/1078-0432.Ccr-09-2638
Bishop, J. A., Teruya-Feldstein, J., Westra, W. H., Pelosi, G., Travis, W. D., and Rekhtman, N. (2012). p40 (ΔNp63) is superior to p63 for the diagnosis of pulmonary squamous cell carcinoma. Mod. Pathol. 25, 405–415. doi: 10.1038/modpathol.2011.173
Bunn, P. A. Jr., Minna, J. D., Augustyn, A., Gazdar, A. F., Ouadah, Y., et al. (2016). Small cell lung cancer: can recent advances in biology and molecular biology be translated into improved outcomes? J. Thorac. Oncol. 11, 453–474. doi: 10.1016/j.jtho.2016.01.012
Cai, Y., Huang, T., Hu, L., Shi, X., Xie, L., and Li, Y. (2012). Prediction of lysine ubiquitination with mRMR feature selection and analysis. Amino Acids 42, 1387–1395. doi: 10.1007/s00726-011-0835-0
Camilo, R., Capelozzi, V. L., Siqueira, S. A., and Del Carlo Bernardi, F. (2006). Expression of p63, keratin 5/6, keratin 7, and surfactant-A in non-small cell lung carcinomas. Hum. Pathol. 37, 542–546. doi: 10.1016/j.humpath.2005.12.019
Chang, H. H., Dreyfuss, J. M., and Ramoni, M. F. (2011). A transcriptional network signature characterizes lung cancer subtypes. Cancer 117, 353–360. doi: 10.1002/cncr.25592
Chao, A., Wang, T. H., Lee, Y. S., Hsueh, S., Chao, A. S., Chang, T. C., et al. (2006). Molecular characterization of adenocarcinoma and squamous carcinoma of the uterine cervix using microarray analysis of gene expression. Int. J. Cancer 119, 91–98. doi: 10.1002/ijc.21813
Chen, L., Huang, T., Zhang, Y. H., Jiang, Y., Zheng, M., and Cai, Y. D. (2016). Identification of novel candidate drivers connecting different dysfunctional levels for lung adenocarcinoma using protein-protein interactions and a shortest path approach. Sci. Rep. 6:29849. doi: 10.1038/srep29849
Chen, L., Li, J., Zhang, Y. H., Feng, K., Wang, S., Zhang, Y., et al. (2018). Identification of gene expression signatures across different types of neural stem cells with the Monte-Carlo feature selection method. J. Cell. Biochem. 119, 3394–3403. doi: 10.1002/jcb.26507
Chen, L., Pan, X., Zhang, Y.-H., Kong, X., Huang, T., and Cai, Y.-D. (2019). Tissue differences revealed by gene expression profiles of various cell lines. J. Cell. Biochem. 120, 7068–7081. doi: 10.1002/jcb.27977
Chen, S., Jiang, S., Hu, F., Xu, Y., Wang, T., and Mei, Q. (2017). Foxk2 inhibits non-small cell lung cancer epithelial-mesenchymal transition and proliferation through the repression of different key target genes. Oncol. Rep. 37, 2335–2347. doi: 10.3892/or.2017.5461
Chen, Y., Yang, L., Cui, T., Pacyna-Gengelbach, M., and Petersen, I. (2015). HOPX is methylated and exerts tumour-suppressive function through Ras-induced senescence in human lung cancer. J. Pathol. 235, 397–407. doi: 10.1002/path.4469
Dearden, S., Stevens, J., Wu, Y. L., and Blowers, D. (2013). Mutation incidence and coincidence in non small-cell lung cancer: meta-analyses by ethnicity and histology (mutMap). Ann. Oncol. 24, 2371–2376. doi: 10.1093/annonc/mdt205
Draminski, M., Rada-Iglesias, A., Enroth, S., Wadelius, C., Koronacki, J., and Komorowski, J. (2008). Monte Carlo feature selection for supervised classification. Bioinformatics 24, 110–117. doi: 10.1093/bioinformatics/btm486
Erdogan, E., Klee, E. W., Thompson, E. A., and Fields, A. P. (2009). Meta-analysis of oncogenic protein kinase Ciota signaling in lung adenocarcinoma. Clin. Cancer Res. 15, 1527–1533. doi: 10.1158/1078-0432.Ccr-08-2459
Fu, J., Fong, K., Bellacosa, A., Ross, E., Apostolou, S., Bassi, D. E., et al. (2008). VILIP-1 downregulation in non-small cell lung carcinomas: mechanisms and prediction of survival. PLoS One 3:e1698. doi: 10.1371/journal.pone.0001698
Fujii, T., Dracheva, T., Player, A., Chacko, S., Clifford, R., Strausberg, R. L., et al. (2002). A preliminary transcriptome map of non-small cell lung cancer. Cancer Res. 62, 3340–3346.
Gómez-Morales, M., Cámara-Pulido, M., Miranda-León, M. T., Sánchez-Palencia, A., Boyero, L., Gómez-Capilla, J. A., et al. (2013). Differential immunohistochemical localization of desmosomal plaque-related proteins in non-small-cell lung cancer. Histopathology 63, 103–113. doi: 10.1111/his.12126
Han, F., Dong, Y., Liu, W., Ma, X., Shi, R., Chen, H., et al. (2014). Epigenetic regulation of sox30 is associated with testis development in mice. PLoS One 9:e97203. doi: 10.1371/journal.pone.0097203
Hawthorn, L., Stein, L., Panzarella, J., Loewen, G. M., and Baumann, H. (2006). Characterization of cell-type specific profiles in tissues and isolated cells from squamous cell carcinomas of the lung. Lung Cancer 53, 129–142. doi: 10.1016/j.lungcan.2006.04.015
He, Y., Li, H., Chen, Y., Li, P., Gao, L., Zheng, Y., et al. (2017). Expression of anoctamin 1 is associated with advanced tumor stage in patients with non-small cell lung cancer and predicts recurrence after surgery. Clin. Transl. Oncol. 19, 1091–1098. doi: 10.1007/s12094-017-1643-0
Herbst, R. S., Morgensztern, D., and Boshoff, C. (2018). The biology and management of non-small cell lung cancer. Nature 553, 446–454. doi: 10.1038/nature25183
Hirsch, F. R., Scagliotti, G. V., Mulshine, J. L., Kwon, R., Curran, W. J., Wu, Y. L., et al. (2017). Lung cancer: current therapies and new targeted treatments. Lancet 389, 299–311. doi: 10.1016/S0140-6736(16)30958-8
Huang, J. T., and Lee, V. (2004). Identification and characterization of a novel human FOXK1 gene in silico. Int. J. Oncol. 25, 751–757. doi: 10.3892/ijo.25.3.751
Huang, T., He, Z. S., Cui, W. R., Cai, Y. D., Shi, X. H., Hu, L. L., et al. (2013). A sequence-based approach for predicting protein disordered regions. Protein Pept. Lett. 20, 243–248. doi: 10.2174/0929866511320030002
Huang, T., Jiang, M., Kong, X., and Cai, Y. D. (2012). Dysfunctions associated with methylation, MicroRNA expression and gene expression in lung cancer. PLoS One 7:e43441. doi: 10.1371/journal.pone.0043441
Huang, T., Niu, S., Xu, Z., Huang, Y., Kong, X., Cai, Y. D., et al. (2011). Predicting transcriptional activity of multiple site p53 mutants based on hybrid properties. PLoS One 6:e22940. doi: 10.1371/journal.pone.0022940
Huang, T., Yang, J., and Cai, Y.-D. (2015). Novel candidate key drivers in the integrative network of genes, MicroRNAs, methylations, and copy number variations in squamous cell lung carcinoma. BioMed Res. Int. 2015:358125. doi: 10.1155/2015/358125
Kang, J. U., Koo, S. H., Kwon, K. C., Park, J. W., and Kim, J. M. (2009). Identification of novel candidate target genes, including EPHB3, MASP1 and SST at 3q26.2-q29 in squamous cell carcinoma of the lung. BMC Cancer 9:237. doi: 10.1186/1471-2407-9-237
Karlsson, A., Jonsson, M., Lauss, M., Brunnstrom, H., Jonsson, P., Borg, A., et al. (2014). Genome-wide DNA methylation analysis of lung carcinoma reveals one neuroendocrine and four adenocarcinoma epitypes associated with patient outcome. Clin. Cancer Res. 20, 6127–6140. doi: 10.1158/1078-0432.Ccr-14-1087
Kato, T., Sato, N., Takano, A., Miyamoto, M., Nishimura, H., Tsuchiya, E., et al. (2008). Activation of placenta-specific transcription factor distal-less homeobox 5 predicts clinical outcome in primary lung cancer patients. Clin. Cancer Res. 14, 2363–2370. doi: 10.1158/1078-0432.Ccr-07-1523
Kim, C. F., Jackson, E. L., Woolfenden, A. E., Lawrence, S., Babar, I., Vogel, S., et al. (2005). Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell 121, 823–835. doi: 10.1016/j.cell.2005.03.032
Koh, W. J., Greer, B. E., Abu-Rustum, N. R., Campos, S. M., Cho, K. R., Chon, H. S., et al. (2017). Vulvar cancer, version 1.2017, NCCN clinical practice guidelines in oncology. J. Natl. Compr. Canc. Netw. 15, 92–120.
Kuriyama, S., Yoshida, M., Yano, S., Aiba, N., Kohno, T., Minamiya, Y., et al. (2016). LPP inhibits collective cell migration during lung cancer dissemination. Oncogene 35, 952–964. doi: 10.1038/onc.2015.155
Kursa, M., and Rudnicki, W. (2010). Feature selection with the Boruta Package. J. Stat. Softw. Artic. 36, 1–13. doi: 10.18637/jss.v036.i11
Ladanyi, M., and Pao, W. (2008). Lung adenocarcinoma: guiding EGFR-targeted therapy and beyond. Mod. Pathol. 21(Suppl. 2) S16–S22. doi: 10.1038/modpathol.3801018
Li, B. Q., You, J., Chen, L., Zhang, J., Zhang, N., Li, H. P., et al. (2013). Identification of lung-cancer-related genes with the shortest path approach in a protein-protein interaction network. BioMed Res. Int. 2013:267375. doi: 10.1155/2013/267375
Li, B. Q., You, J., Huang, T., and Cai, Y. D. (2014). Classification of non-small cell lung cancer based on copy number alterations. PLoS One 9:e88300. doi: 10.1371/journal.pone.0088300
Li, J., Lan, C.-N., Kong, Y., Feng, S.-S., and Huang, T. (2018). Identification and analysis of blood gene expression signature for osteoarthritis with Advanced feature selection methods. Front. Genet. 9:246. doi: 10.3389/fgene.2018.00246
Li, J., Lu, L., Zhang, Y. H., Xu, Y., Liu, M., Feng, K., et al. (2019). Identification of leukemia stem cell expression signatures through Monte Carlo feature selection strategy and support vector machine. Cancer Gene Ther. doi: 10.1038/s41417-019-0105-y
Li, M. Y., Lai, P. L., Chou, Y. T., Chi, A. P., Mi, Y. Z., Khoo, K. H., et al. (2015). Protein tyrosine phosphatase PTPN3 inhibits lung cancer cell proliferation and migration by promoting EGFR endocytic degradation. Oncogene 34, 3791–3803. doi: 10.1038/onc.2014.312
Liu, C., Zhang, Y. H., Huang, T., and Cai, Y. (2017). Identification of transcription factors that may reprogram lung adenocarcinoma. Artif. Intell. Med. 83, 52–57. doi: 10.1016/j.artmed.2017.03.010
Liu, J., Liu, L., Cao, L., and Wen, Q. (2018). Keratin 17 promotes lung adenocarcinoma progression by enhancing cell proliferation and invasion. Med. Sci. Monit. 24, 4782–4790. doi: 10.12659/msm.909350
Liu, M., Chen, J., Hu, L., Shi, X., Zhou, Z., Hu, Z., et al. (2012). HORMAD2/CT46.2, a novel cancer/testis gene, is ectopically expressed in lung cancer tissues. Mol. Hum. Reprod. 18, 599–604. doi: 10.1093/molehr/gas033
Lokshin, A., Zhang, H., Mayotte, J., Lokshin, M., and Levitt, M. L. (1999). Early effects of retinoic acid on proliferation, differentiation and apoptosis in non-small cell lung cancer cell lines. Anticancer Res. 19, 5251–5254.
Lu, C., Chen, H., Shan, Z., and Yang, L. (2016). Identification of differentially expressed genes between lung adenocarcinoma and lung squamous cell carcinoma by gene expression profiling. Mol. Med. Rep. 14, 1483–1490. doi: 10.3892/mmr.2016.5420
Lv, J., Zhu, P., Yang, Z., Li, M., Zhang, X., Cheng, J., et al. (2015). PCDH20 functions as a tumour-suppressor gene through antagonizing the Wnt/β-catenin signalling pathway in hepatocellular carcinoma. J. Viral Hepat. 22, 201–211. doi: 10.1111/jvh.12265
Ma, X., Yang, X., Bao, W., Li, S., Liang, S., and Sun, Y. (2018). Circular RNA circMAN2B2 facilitates lung cancer cell proliferation and invasion via miR-1275/FOXK1 axis. Biochem. Biophys. Res. Commun. 498, 1009–1015. doi: 10.1016/j.bbrc.2018.03.105
Mashima, T., Oh-hara, T., Sato, S., Mochizuki, M., Sugimoto, Y., Yamazaki, K., et al. (2005). p53-defective tumors with a functional apoptosome-mediated pathway: a new therapeutic target. J. Natl. Cancer Inst. 97, 765–777. doi: 10.1093/jnci/dji133
Matsuo, T., Dat le, T., Komatsu, M., Yoshimaru, T., Daizumoto, K., and Sone, S. (2014). Early growth response 4 is involved in cell proliferation of small cell lung cancer through transcriptional activation of its downstream genes. PLoS One 9:e113606. doi: 10.1371/journal.pone.0113606
Mazieres, J., Peters, S., Lepage, B., Cortot, A. B., Barlesi, F., Beau-Faller, M., et al. (2013). Lung cancer that harbors an HER2 mutation: epidemiologic characteristics and therapeutic perspectives. J. Clin. Oncol. 31, 1997–2003. doi: 10.1200/jco.2012.45.6095
Molina-Pinelo, S., Gutiérrez, G., Pastor, M. D., Hergueta, M., Moreno-Bueno, G., García-Carbonero, R., et al. (2014). MicroRNA-dependent regulation of transcription in non-small cell lung cancer. PLoS One 9:e90524. doi: 10.1371/journal.pone.0090524
Ngan, E., Stoletov, K., Smith, H. W., Common, J., Muller, W. J., Lewis, J. D., et al. (2017). LPP is a Src substrate required for invadopodia formation and efficient breast cancer lung metastasis. Nat. Commun. 8:15059. doi: 10.1038/ncomms15059
Oser, M. G., Niederst, M. J., Sequist, L. V., and Engelman, J. A. (2015). Transformation from non-small-cell lung cancer to small-cell lung cancer: molecular drivers and cells of origin. Lancet Oncol. 16, e165–e172. doi: 10.1016/s1470-2045(14)71180-5
Pan, X., Chen, L., Feng, K. Y., Hu, X. H., Zhang, Y. H., and Kong, X. Y. (2019a). Analysis of expression pattern of snoRNAs in different cancer types with machine learning algorithms. Int. J. Mol. Sci. 20:2185. doi: 10.3390/ijms20092185
Pan, X., Hu, X., Zhang, Y.-H., Chen, L., Zhu, L., Wan, S., et al. (2019b). Identification of the copy number variant biomarkers for breast cancer subtypes. Mol. Genet. Genomics 294, 95–110. doi: 10.1007/s00438-018-1488-4
Pan, X., Hu, X., Zhang, Y. H., Feng, K., Wang, S. P., and Chen, L. (2018). Identifying patients with atrioventricular septal defect in down syndrome populations by using self-normalizing neural networks and feature selection. Genes (Basel) 9:208. doi: 10.3390/genes9040208
Sanchez-Palencia, A., Gomez-Morales, M., Gomez-Capilla, J. A., Pedraza, V., Boyero, L., Rosell, R., et al. (2011). Gene expression profiling reveals novel biomarkers in nonsmall cell lung cancer. Int. J. Cancer 129, 355–364. doi: 10.1002/ijc.25704
Savci-Heijink, C. D., Kosari, F., Aubry, M. C., Caron, B. L., Sun, Z., Yang, P., et al. (2009). The role of desmoglein-3 in the diagnosis of squamous cell carcinoma of the lung. Am. J. Pathol. 174, 1629–1637. doi: 10.2353/ajpath.2009.080778
Shi, Y. X., Wang, Y., Li, X., Zhang, W., Zhou, H. H., Yin, J. Y., et al. (2017). Genome-wide DNA methylation profiling reveals novel epigenetic signatures in squamous cell lung cancer. BMC Genomics 18:901. doi: 10.1186/s12864-017-4223-3
Shigematsu, H., Lin, L., Takahashi, T., Nomura, M., Suzuki, M., Wistuba, I. I., et al. (2005). Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J. Natl. Cancer Inst. 97, 339–346. doi: 10.1093/jnci/dji055
Socinski, M. A., Obasaju, C., Gandara, D., Hirsch, F. R., Bonomi, P., Bunn, P., et al. (2016). Clinicopathologic features of advanced squamous NSCLC. J. Thorac. Oncol. 11, 1411–1422. doi: 10.1016/j.jtho.2016.05.024
Soldera, S. V., and Leighl, N. B. (2017). Update on the treatment of metastatic squamous non-small cell lung cancer in new era of personalized medicine. Front. Oncol. 7:50. doi: 10.3389/fonc.2017.00050
Song, C., Gao, Y., Tian, Y., Han, X., Chen, Y., and Tian, D. L. (2013). Expression of p114RhoGEF predicts lymph node metastasis and poor survival of squamous-cell lung carcinoma patients. Tumour. Biol. 34, 1925–1933. doi: 10.1007/s13277-013-0737-8
Sun, S., Schiller, J. H., and Gazdar, A. F. (2007). Lung cancer in never smokers–a different disease. Nat. Rev. Cancer 7, 778–790. doi: 10.1038/nrc2190
Sun, X., Li, J., Gu, L., Wang, S., Zhang, Y., Huang, T., et al. (2018). Identifying the characteristics of the hypusination sites using SMOTE and SVM algorithm with feature selection. Curr. Proteom. 15, 111–118. doi: 10.2174/1570164614666171109120615
Swanton, C., and Govindan, R. (2016). Clinical implications of genomic discoveries in lung cancer. N. Engl. J. Med. 374, 1864–1873. doi: 10.1056/NEJMra1504688
Szklarczyk, D., Gable, A. L., Lyon, D., Junge, A., Wyder, S., and Huerta-Cepas, J. (2018). STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613. doi: 10.1093/nar/gky1131
Thomas, J. K., Kim, M. S., Balakrishnan, L., Nanjappa, V., Raju, R., Marimuthu, A., et al. (2014). Pancreatic cancer database: an integrative resource for pancreatic cancer. Cancer Biol. Ther. 15, 963–967. doi: 10.4161/cbt.29188
Travis, W. D., Brambilla, E., Nicholson, A. G., Yatabe, Y., Austin, J. H. M., Beasley, M. B., et al. (2015). The 2015 world health organization classification of lung tumors: impact of genetic. clinical and radiologic advances since the 2004 classification. J. Thorac. Oncol. 10, 1243–1260.
Tsukasaki, K., Miller, C. W., Greenspun, E., Eshaghian, S., Kawabata, H., and Fujimoto, T. (2001). Mutations in the mitotic check point gene, MAD1L1, in human cancers. Oncogene 20, 3301–3305. doi: 10.1038/sj.onc.1204421
Van de Laar, E., Clifford, M., Hasenoeder, S., Kim, B. R., Wang, D., Lee, S., et al. (2014). Cell surface marker profiling of human tracheal basal cells reveals distinct subpopulations, identifies MST1/MSP as a mitogenic signal, and identifies new biomarkers for lung squamous cell carcinomas. Respir. Res. 15:160. doi: 10.1186/s12931-014-0160-8
Villalobos, P., and Wistuba, I. I. (2017). Lung cancer biomarkers. Hematol. Oncol. Clin. North Am. 31, 13–29. doi: 10.1016/j.hoc.2016.08.006
Weiss, J., Sos, M. L., Seidel, D., Peifer, M., Zander, T., and Heuckmann, J. M. (2010). Frequent and focal FGFR1 amplification associates with therapeutically tractable FGFR1 dependency in squamous cell lung cancer. Sci. Transl. Med. 2:62ra93. doi: 10.1126/scitranslmed.3001451
Wilkerson, M. D., Yin, X., Hoadley, K. A., Liu, Y., Hayward, M. C., Cabanski, C. R., et al. (2010). Lung squamous cell carcinoma mRNA expression subtypes are reproducible, clinically important, and correspond to normal cell types. Clin. Cancer Res. 16, 4864–4875. doi: 10.1158/1078-0432.ccr-10-0199
Xiao, G. Q., Li, F., Findeis-Hosey, J., Hyrien, O., Unger, P. D., and Xiao, L. (2015). Down-regulation of cytoplasmic PLZF correlates with high tumor grade and tumor aggression in non-small cell lung carcinoma. Hum. Pathol. 46, 1607–1615. doi: 10.1016/j.humpath.2015.06.021
Xu, C., Fillmore, C. M., Koyama, S., Wu, H., Zhao, Y., Chen, Z., et al. (2014). Loss of Lkb1 and Pten leads to lung squamous cell carcinoma with elevated PD-L1 expression. Cancer Cell 25, 590–604. doi: 10.1016/j.ccr.2014.03.033
Zhan, C., Yan, L., Wang, L., Sun, Y., Wang, X., Lin, Z., et al. (2015). Identification of immunohistochemical markers for distinguishing lung adenocarcinoma from squamous cell carcinoma. J. Thorac. Dis. 7, 1398–1405. doi: 10.3978/j.issn.2072-1439.2015.07.25
Zhang, K., Tang, S., Cao, S., Hu, L., Pan, Y., and Ma, H. (2014). Association of polymorphisms at HORMAD2 and prognosis in advanced non-small-cell lung cancer patients. Cancer Epidemiol. 38, 414–418. doi: 10.1016/j.canep.2014.03.013
Zhou, Q., Dai, J., Chen, T., Dada, L. A., Zhang, X., Zhang, W., et al. (2017). Downregulation of PKCζ/Pard3/Pard6b is responsible for lung adenocarcinoma cell EMT and invasion. Cell. Signal. 38, 49–59. doi: 10.1016/j.cellsig.2017.06.016
Keywords: lung adenocarcinoma, squamous cell lung carcinoma, methylation, gene expression, Boruta, Monte Carlo Feature Selection
Citation: Zhang H, Jin Z, Cheng L and Zhang B (2020) Integrative Analysis of Methylation and Gene Expression in Lung Adenocarcinoma and Squamous Cell Lung Carcinoma. Front. Bioeng. Biotechnol. 8:3. doi: 10.3389/fbioe.2020.00003
Received: 31 October 2019; Accepted: 03 January 2020;
Published: 07 February 2020.
Edited by:
Tao Huang, Shanghai Institutes for Biological Sciences (CAS), ChinaReviewed by:
Yunguang Tong, University of California, Los Angeles, United StatesHong Liu, Temple University, United States
Copyright © 2020 Zhang, Jin, Cheng and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bin Zhang, YmVhbnpoYW5nMDgyMUB6anUuZWR1LmNu; YmluemhhbmcwODIxQHpqdS5lZHUuY24=
†These authors have contributed equally to this work