 Yajuan Zhou1,2,3†
Yajuan Zhou1,2,3† Xinqiang Liu
Xinqiang Liu Jiacheng Wu
Jiacheng Wu Jin Wang
Jin Wang- 1CAS Key Laboratory of Synthetic Biology, Institute of Plant Physiology and Ecology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai, China
- 2University of Chinese Academy of Sciences, Beijing, China
- 3School of Life Sciences and Technology, Shanghai Tech University, Shanghai, China
- 4Department of Microbiology and Li Ka Shing Institute of Health Sciences, The Chinese University of Hong Kong, Prince of Wales Hospital, Shatin, Hong Kong
- 5College of Life Sciences, Shanghai Normal University, Shanghai, China
Amycolatopsis mediterranei U32 is an industrial producer of rifamycin SV, whose derivatives have long been the first-line antimycobacterial drugs. In order to perform genetic modification in this important industrial strain, a lot of efforts have been made in the past decades and a homologous recombination-based method was successfully developed in our laboratory, which, however, requires the employment of an antibiotic resistance gene for positive selection and did not support convenient markerless gene deletion. Here in this study, the clustered regularly interspaced short palindromic repeat (CRISPR) system was employed to establish a genome editing system in A. mediterranei U32. Specifically, the Francisella tularensis subsp. novicida Cas12a (FnCas12a) gene was first integrated into the U32 genome to generate target-specific double-stranded DNA (dsDNA) breaks (DSBs) under the guidance of CRISPR RNAs (crRNAs). Then, the DSBs could be repaired by either the non-homologous DNA end-joining (NHEJ) system or the homology-directed repair (HDR) pathway, generating inaccurate or accurate mutations in target genes, respectively. Besides of A. mediterranei, the present work may also shed light on the development of CRISPR-assisted genome editing systems in other species of the Amycolatopsis genus.
Introduction
Amycolatopsis mediterranei U32 is an industrial strain for production of rifamycin SV (Zhao et al., 2010), the first-line drug for anti-mycobacterial therapy till now (Rothstein, 2016). Due to the great importance of rifamycin, extensive efforts such as optimization of the fermentation conditions had been made to improve the yield of the antibiotics in the last century (Jiao et al., 1979; Lee et al., 1983; Mejia et al., 1998). Later, to facilitate the study of rifamycin biosynthesis as well as the molecular bioengineering of the producer, a genetic manipulation method based on native homologous recombination was developed for gene knockout in A. mediterranei (Ding et al., 2003). However, due to the relatively low efficiency of both DNA transformation and homologous recombination in A. mediterranei, an antibiotic cassette is usually employed to replace the target gene and the transformants are grown under antibiotic selection. To remove the antibiotic resistance cassette in the knockout mutant, the cassette should be flanked by site-specific recombination sequences such as the loxP sites or homologous arms, and a second cross-over recombination event is required. However, due to the relatively low genetic engineering efficiency, there are very few reports of successful construction of a markerless mutant in A. mediterranei. What is worse, since there are only a limited number of antibiotics applicable in A. mediterranei, it is difficult to perform continuous genetic engineering operations. Therefore, although the Amycolatopsis genus is well known to produce a huge diversity of secondary metabolites (Xu et al., 2014; Kumari et al., 2016; Adamek et al., 2018), the lack of efficient genome editing technology has severely impeded the research progress in this genus.
The clustered regularly interspaced short palindromic repeat (CRISPR) system is an adaptive immune system in bacteria and archaea (Horvath and Barrangou, 2010; Jinek et al., 2012; Mohanraju et al., 2016), where the CRISPR-associated (Cas) protein complex utilizes guide RNA for specific recognition, binding, and cutting of target nucleic acids with proper protospacer adjacent motifs (PAM) (Jinek et al., 2012). The CRISPR systems can be divided into class 1 and class 2 (Koonin et al., 2017), where the crRNA ribonucleoprotein (crRNP) effector of the class 1 system complexes are composed of multiple Cas proteins as subunits (Makarova et al., 2011, 2017a), whereas the class 2 system crRNP complexes contain single Cas protein such as the types II, V, and VI Cas proteins (Makarova et al., 2017b). With CRISPR-Cas-assisted accurate cleavage in target DNA sequences and thus introducing double-stranded DNA (dsDNA) breaks (DSBs), the genome engineering efficiency can be greatly improved. Up to date, both the type II Cas9 system and type V Cas12a system have been widely applied in genome editing in a large number of species (Cong et al., 2013; Jiang et al., 2014, 2015; Cobb et al., 2015; Huang et al., 2015; Matsu-Ura et al., 2015; Low et al., 2016; Jia et al., 2017; Harrison and Hart, 2018; Hu et al., 2019). Compared to Cas9, Cas12a has several distinct features, including the preference of T-rich PAM sequences and the staggered cleavage pattern against target dsDNA (Zetsche et al., 2015; Yamano et al., 2016). Besides, unlike Cas9, Cas12a only requires the CRISPR RNA (crRNA) but not the trans-activating RNA (tracrRNA), and is able to mature precursor crRNAs, thereby enabling Cas12a in multiple gene editing and regulation with much convenience (Fonfara et al., 2016; Zetsche et al., 2017).
Bacteria have evolved two mechanisms to efficiently repair DSB damage, including both homology-directed repairing (HDR) (Sung and Klein, 2006) and non-homologous DNA end-joining (NHEJ) (Lieber, 2010). Combined with the CRISPR system, HDR provides accurate and markerless target gene deletion, mutation, and insertion of foreign DNA sequences (Cobb et al., 2015). Alternatively, in some bacterial species such as Mycobacterium smegmatis, DSB can be repaired by the NHEJ system, which comprises an ATP-dependent DNA ligase and a Ku protein (Wright et al., 2017; Zheng et al., 2017). Unlike HDR, the NHEJ system does not require homologous DNA sequences for recombination, but directly joins the breaks, facilitating convenient gene deletion and insertion (Babynin, 2007). However, as the NHEJ repair may introduce errors at the joining site, it is inappropriate for accurate gene editing.
Here in this study, we successfully established the CRISPR-Cas12a-based genome editing system in A. mediterranei U32. We first demonstrated the existence of the NHEJ system in U32, and then combined NHEJ with Cas12a to construct site-specific markerless gene deletion mutants. Moreover, we also used the endogenous HDR to repair the Cas12a-introduced DSBs, facilitating efficient genome editing in U32.
Methodology
Strains, Media, and Growth Conditions
Strains and plasmids used in this study are listed in Supplementary Table S1. Escherichia coli DH10B was used for DNA cloning and was grown at 37°C in LB medium. A. mediterranei U32 was grown at 30°C in Bennet medium (yeast extract, 1 g/l; glycerol, 10 g/l; glucose, 10 g/l; beef powder, 1 g/l; tryptone, 2 g/l; and agar, 15 g/l; pH 7.0). To prepare the competent cells, A. mediterranei strains were cultured at 30°C in MYM medium (yeast extract, 3 g/l; malt extract, 3 g/l; peptone, 5 g/l; glucose, 10 g/l; sucrose, 170 g/l; KNO3, 6 g/l; glycine, 10 g/l; CaCl2⋅2H2O, 0.735 g/l; MgCl2⋅6H2O, 1.015 g/l; pH 7.0). For analyses of the growth phenotypes, A. mediterranei strains were grown in minimal medium (4% glucose, 0.05% NaCl, 0.2% K2HPO4, 0.1% MgSO4, 0.001% FeSO4⋅7H2O, 0.0001% MnCl2⋅4H2O, and 0.001% ZnSO4⋅7H2O) supplemented with either (NH4)2SO4 or KNO3 as the sole nitrogen sources. When necessary, appropriate antibiotics were added in the medium mentioned above at the following concentrations: 100 μg/ml ampicillin, 50 μg/ml kanamycin, 34 μg/ml chloramphenicol, 50 μg/ml apramycin, and 100 μg/ml hygromycin.
Construction of CRISPR-Cas12a-Based Genome Editing Plasmids
Primers for plasmid construction, mutant verification, and Sanger sequencing are listed in Supplementary Table S2. The Francisella tularensis subsp. novicida Cas12a gene (FnCas12a, previously known as FnCpf1) was PCR amplified from the plasmid pJV53-Cpf1 (Yan et al., 2017) using primer of FnCas12a-F and FnCas12a-R. Then, the linearized vector was obtained through PCR amplification of pDZL803 (Li et al., 2017b) with primers of pDZL803-apr-F and pDZL803-apr-R, which contained the promoter region of the apramycin resistance gene (Papr). Cas12a gene and the linearized pDZL803 vector were then assembled using the Ezmax seamless assembly kit (Tolo Biotech., Shanghai, China), and the obtained recombinant plasmid pDZLCas12a was further confirmed by Sanger sequencing (Supplementary Figure S1).
The crRNA guide sequences are listed in Supplementary Table S3. First, the BpmI and HindIII restriction sites in pCR-Hyg were replaced by the BbsI and AseI sites, obtaining the plasmid pCR1. In detail, the plasmid pCR-Hyg (Yan et al., 2017) was employed as the template for PCR amplification with primers of pCR-HYG-F and pCR-HYG-R, followed by template removal with DpnI and then self-assembly with the Ezmax seamless assembly kit (Tolo Biotech.). Then, the crRNA expressing cassette, containing the hsp60 promoter, two crRNA Direct Repeats (DR) sequences, the BbsI and AseI sites for insertion of crRNA guide sequences and the rrnB T1 terminator, was further amplified from pCR1 with paired primers of hsp60-rrnB-F and hsp60-rrnB-R, and the amplicon was then inserted into plasmid pULVK2A (Kumar et al., 1994), generating plasmid pULcrRNA (Supplementary Figure S1). Plasmid pULVK2A was generated from pRL1 (Lal et al., 1991) by spontaneous deletion of DNA sequences during passage, and can stably self-replicate in A. mediterranei. The Cas12a-expressing plasmid pDZLCas12a and the crRNA-expressing plasmid pULcrRNA were used to test the effectiveness of the CRISPR/Cas12a system in U32.
Deletion of rifZ and glnR Genes in U32
The CRISPR/Cas12a-assisted genome editing plasmids were constructed on the basis of plasmid pULcrRNA. First, 20-nt crRNA guide sequences for targeting rifZ and glnR were designed, synthesized, and individually annealed, and were then inserted into pULcrRNA that was digested by BbsI and AseI, generating pULrifZ and pULglnR, respectively. Then, 1.5-kb upstream and downstream sequences of the target genes (e.g., rifZ and glnR) were PCR amplified from U32 genome with paired primers (rifZL-F/rifZL-R, rifZR-F/rifZR-R, glnRL-F/glnRL-R, and glnRR-F/glnRR-R), the amplicons of which were used as homologous arms for HDR. The apramycin resistance gene was released from pBCAm plasmid by PstI digestion. Then, the upstream and downstream arms as well as the apramycin resistance cassette were assembled (designated as LAR donor fragment) by Ezmax seamless assembly kit (Tolo Biotech.) and introduced into the NdeI-treated plasmids of pULrifZ and pULglnR, obtaining the knout-out plasmids of pULrifZ-LAR and pULglnR-LAR, respectively (Supplementary Figures S1, S3). Alternatively, plasmids for markerless deletion of target genes were constructed through direct assembly of the upstream and downstream homologous arms and the crRNA expression cassette for guiding target-specific cleavage, and the obtained plasmids for rifZ and glnR markerless deletion were named pULrifZ-LR and pULglnR-LR, respectively (Supplementary Figure S1).
The U32 competent cells for electroporation were prepared as previously described (Ding et al., 2003). The Cas12a expression vector (pDZLCas12a) was electroporated into U32 competent cells and transformants were cultured on selective plates containing hygromycin. Specifically, about 500-ng pDZLCas12a was electroporated into 75-μl U32 competent cells with the following electroporation parameters: 1760 V, 1000 Ω, 25 μF, and 2 mm cuvette. Transformants were cultivated at 30°C for 7 days, and the clones were counted, analyzed, and verified by both PCR amplification and subsequent Sanger sequencing. The transformant expressing Cas12a was then employed for preparation of competent cells for subsequent gene editing. To test the NHEJ activities in U32, 300-ng crRNA-expressing plasmids of pULrifZ and pULglnR, targeting rifZ and glnR, respectively, were electroporated into the competent cells expressing Cas12a, and the transformants were then cultured and analyzed. Noticeably, there were no donor arms on plasmids pULrifZ and pULglnR for homologous recombination.
Similarly, to precisely delete target genes via HDR-mediated repair of DSBs, 300-ng plasmids of pULrifZ-LAR, pULrifZ-LR, pULglnR-LAR, and pULglnR-LR were individually electroporated into the Cas12a-expressing competent cells to delete the target gene rifZ and glnR, respectively. The transformants were cultivated on Bennet plate supplemented with apramycin at 30°C for 7 days, and the colonies were confirmed by colony PCR and Sanger sequencing.
Phenotypic Analyses of Gene Deletion Mutants
To analyze the growth phenotypes of the glnR deletion mutants, mutants were cultured with minimal medium with 20 mM KNO3 or 10 mM (NH4)2SO4 as the sole nitrogen sources. Specifically, mutants were first grown in Bennet medium and the cells were then washed with nitrogen-free medium. After that, a 10-fold serial dilution was made from the starting OD600 density of 1–1/400, and diluted cells were then spotted onto minimal medium plate (Dadura et al., 2017) before being further incubated at 30°C for 5 days. Three independent experiments were performed.
Results
As A. mediterranei is an important industrial strain for rifamycin production, many efforts have been made to study its genetic operation system, including the characterization of endogenous plasmids. However, up to now, there is only one stable replicon (namely, the pA-rep) characterized from the endogenous plasmid pA387 in Amycolatopsis sp. DSM 43387, and all self-replicable plasmids (e.g., pRL1 and pULVK2A) in A. mediterranei are generated from this plasmid (Lal et al., 1991; Kumar et al., 1994). Due to the plasmid incompatibility, it is hard to stably transform two plasmids with the same replication origin inside one cell. Therefore, to develop a CRISPR-based genetic engineering system in A. mediterranei U32, we decided to clone the Cas gene in an integrative plasmid and the crRNA expression cassette in a self-replicable plasmid. We ever tested the dead SpCas9 gene from Streptococcus pyogenes (Jinek et al., 2012), and cloned it into an integrative plasmid, which was then electroporated into U32 competent cells. However, no transformants were obtained (data not shown) after repeated electroporation experiments, which indicated that the expression of dCas9 alone was toxic to U32.
Then, instead of testing the wild-type Cas9, we tested the FnCas12a from F. tularensis (Zetsche et al., 2015), an alternative to Cas9 for CRISPR-mediated genome editing and has been successfully used in M. smegmatis (Yan et al., 2017). Similarly, the codon optimized FnCas12a gene was cloned in an integrative plasmid and was further electroporated into U32 competent cells to allow for integration into the attB site in the genome (Figure 1). Transformants were successfully obtained and the integrated FnCas12a gene was further confirmed by colony PCR verification and subsequent Sanger DNA sequencing; however, the transformation efficiency was much lower than that of the control plasmid with no Cas12a gene. Further phenotypic analysis showed that both bacterial growth and the rifamycin production of the transformant expressing FnCas12a were similar to those of the wild type U32 (Supplementary Figure S2), which implied that the constructed strain was a qualified system for genome editing analysis.
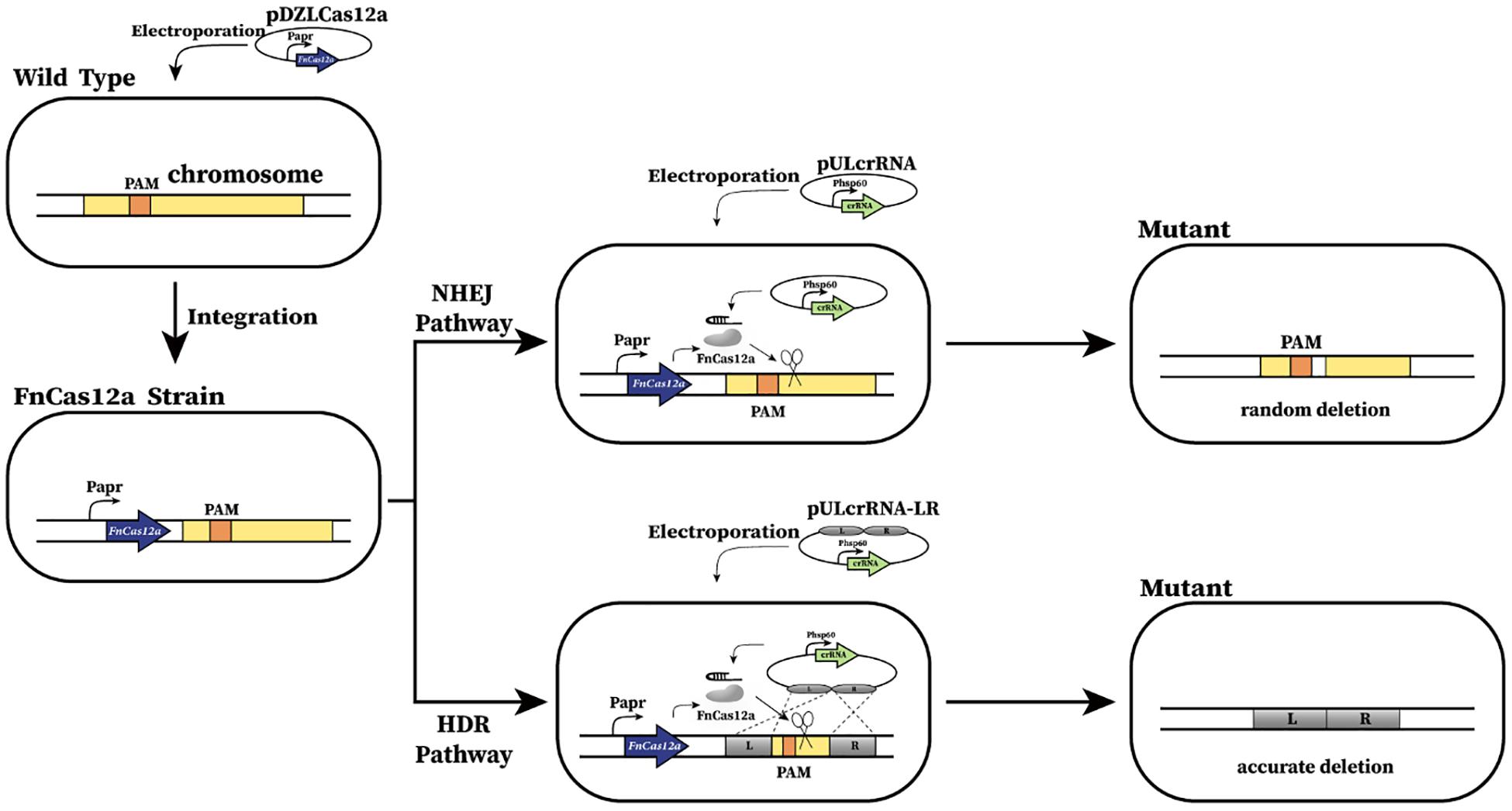
Figure 1. Schematic of CRISPR-Cas12a-assisted genome editing in A. mediterranei U32. First, FnCas12a was integrated into U32 chromosome and constitutively expressed under the promoter of Papr. Then, the crRNA transcribing plasmid was electroporated into the U32 competent cells that harbored FnCas12a. Guided by a target-specific crRNA, Cas12a specifically cleaved target dsDNA and generated DSBs on the chromosome, which could be repaired by either NHEJ or HDR, generating desired mutants. When DSBs were repaired by NHEJ, the breaks were directly jointed with no homologous donor DNA required; however, DNA sequences of ranging length around the Cas12a cleavage site will be deleted during the repair.
To test the whether Cas12a could introduce site-specific DSBs in U32, the glnR gene, which encodes the global regulator for nitrogen metabolisms, was chosen as the target gene. We designed three crRNAs targeting different coding regions in glnR, where crRNA1 and crRNA2 targeted the non-template strand (NT) and the crRNA-3 targeted the template strand (T) (Supplementary Table S3). The above three crRNAs were individually cloned into self-replicable pULVK2A, and the obtained plasmids were then electroporated into the U32 competent cells that constitutively expressed FnCas12a. In comparison with thousands of colonies obtained with the transformation of the control plasmid with no crRNA expression cassette, only 0, 9, and 1 transformants were obtained for plasmids pULglnR1, pULglnR2, and pULglnR3, respectively (Figure 2A), suggesting that the CRISPR-Cas12a system could efficiently cleave the genomic DNA.
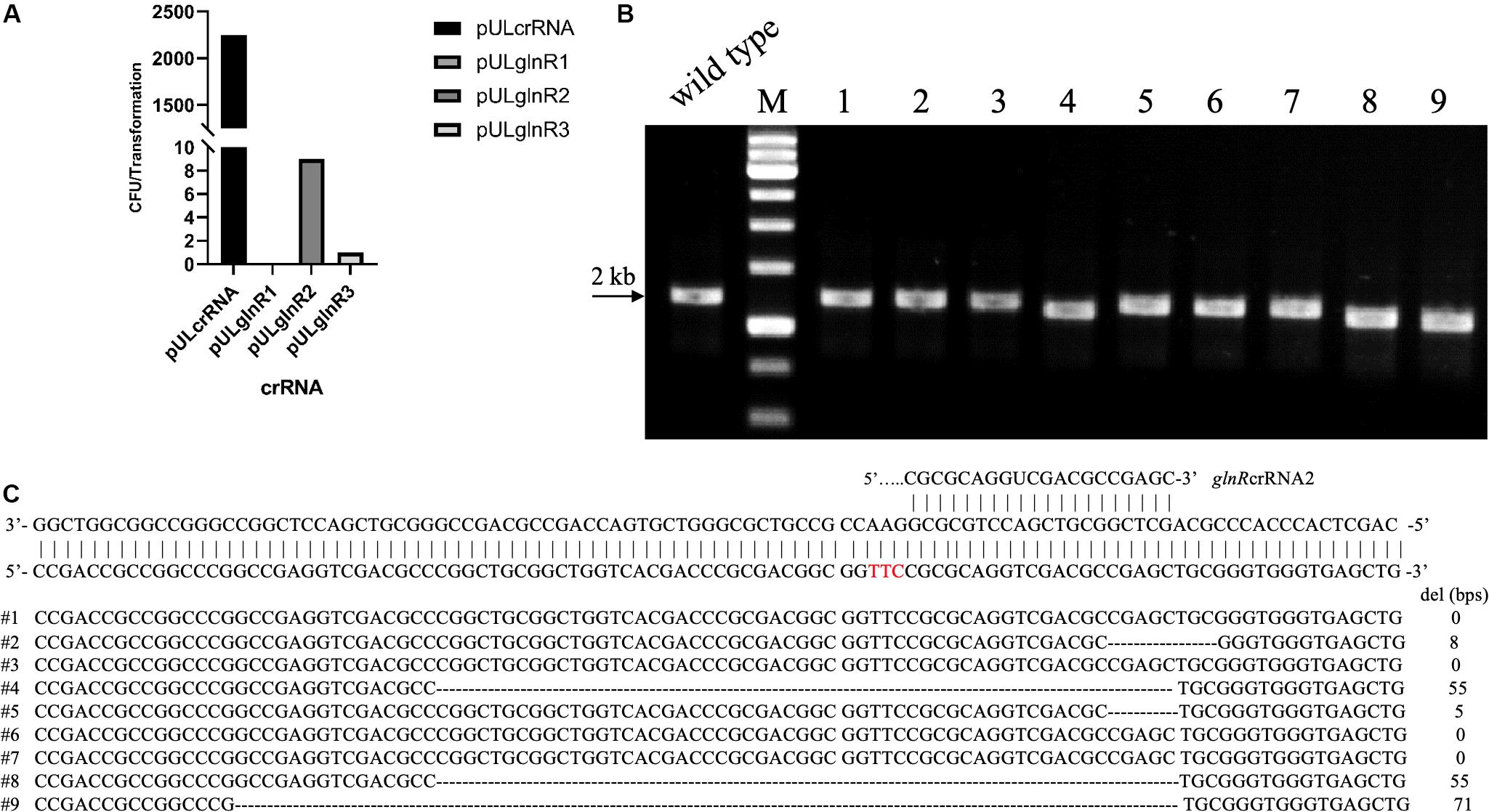
Figure 2. CRISPR-Cas12a-assisted glnR deletion in combination with the NHEJ repair in A. mediterranei U32. (A) The number of transformants obtained with different crRNAs transformed. Plasmid pULcrRNA, which contained no crRNA guide sequences, was employed as a control plasmid. The number stood for the total colonies obtained from three transformation experiments, where no transformants were obtained with crRNA1. The guide sequences are listed in Supplementary Table S3. (B) Characterization of pULglnR2 transformants by colony PCR amplification. M, GeneRuler 1 kb DNA Ladder (Thermo Scientific). Lanes 1–9, nine colonies transformed with CRISPR-Cas12a and pULglnR2 targeting glnR gene. (C) Sanger sequencing results of the PCR amplicons of the nine colonies in Figure 2B. Five colonies contained random deletion at the target site within glnR gene, which was repaired by the NHEJ pathway, and the deleted DNA sequences as well as the length were indicated. The PAM sequence was highlighted in red.
On the other hand, although no homologous DNA sequences were introduced for homologous recombination, we still obtained some transformants (Figure 2A), and the nine transformants with pULglnR2 were further verified by PCR amplification of the target regions. The PCR results showed that the amplicons were of different sizes (Figure 2B), which indicated that there might be random deletions inside the target gene. We further confirmed this hypothesis by Sanger sequencing of the PCR amplicons and found five of nine transformants contained deletions (ranging from 5 to 71 bps) at the glnRcrRNA2 targeting site (Figure 2C). With the identification of conserved homologues of ATP-dependent DNA ligase and Ku protein (Supplementary Tables S4, S5), one may conclude that the DSBs were probably repaired by NHEJ in U32, although the possibility of other template-independent repair such as alternative end-joining (A-EJ) (Chayot et al., 2010) and microhomology-mediated end-joining (MMEJ) (Sfeir and Symington, 2015) cannot be completely excluded. Meanwhile, there might also exist unknown mechanisms to inactivate the CRISPR-Cas12a system in U32 as no mutations were found in the target region among the rest four clones. Collectively, above findings not only demonstrated that Cas12a generated site-specific DSBs but also suggested that there was NHEJ in U32. As a consequence, with the combination of CRISPR-Cas12a-induced site-specific DSBs and NHEJ-mediated repair, markerless gene mutations can be easily acquired in this bacterium.
Besides, we also combined the CRISPR-Cas12a-assisted target cleavage with endogenous HDR activity to precisely delete target genes. To measure the efficiency of the HDR-mediated precise genome editing, we next knocked out the rifZ gene encoding the rifamycin pathway-specific activator by replacing it with the apramycin resistance cassette (Figure 3A). Three crRNAs were designed to target both the T strand (rifZcrRNA-1 and rifZcrRNA-2) and the NT strand (rifZcrRNA-3) of rifZ (Supplementary Table S3). The resistance cassette was in fusion assembled with both upstream and downstream homologous arms of rifZ, and the obtained donor fragment was then introduced into the plasmids expressing rifZ targeting crRNAs, generating plasmids pULrifZ1-LAR, pULrifZ2-LAR, and pULrifZ3-LAR, respectively. The obtained three plasmids as well as a control plasmid were then individually electroporated into the U32 competent cells harboring FnCas12a. In comparison with the more than 1000 colonies obtained from the transformation of the control plasmid, less than 10 colonies on average were obtained with the three plasmids with rifZ-specific crRNAs (Figure 3B). Subsequently, eight colonies from the transformation of pULrifZ1-LAR were verified by both PCR amplification and Sanger sequencing, and the results unambiguously showed that the rifZ gene was precisely replaced with the apramycin resistance cassette in all tested colonies (Figures 3C,D).
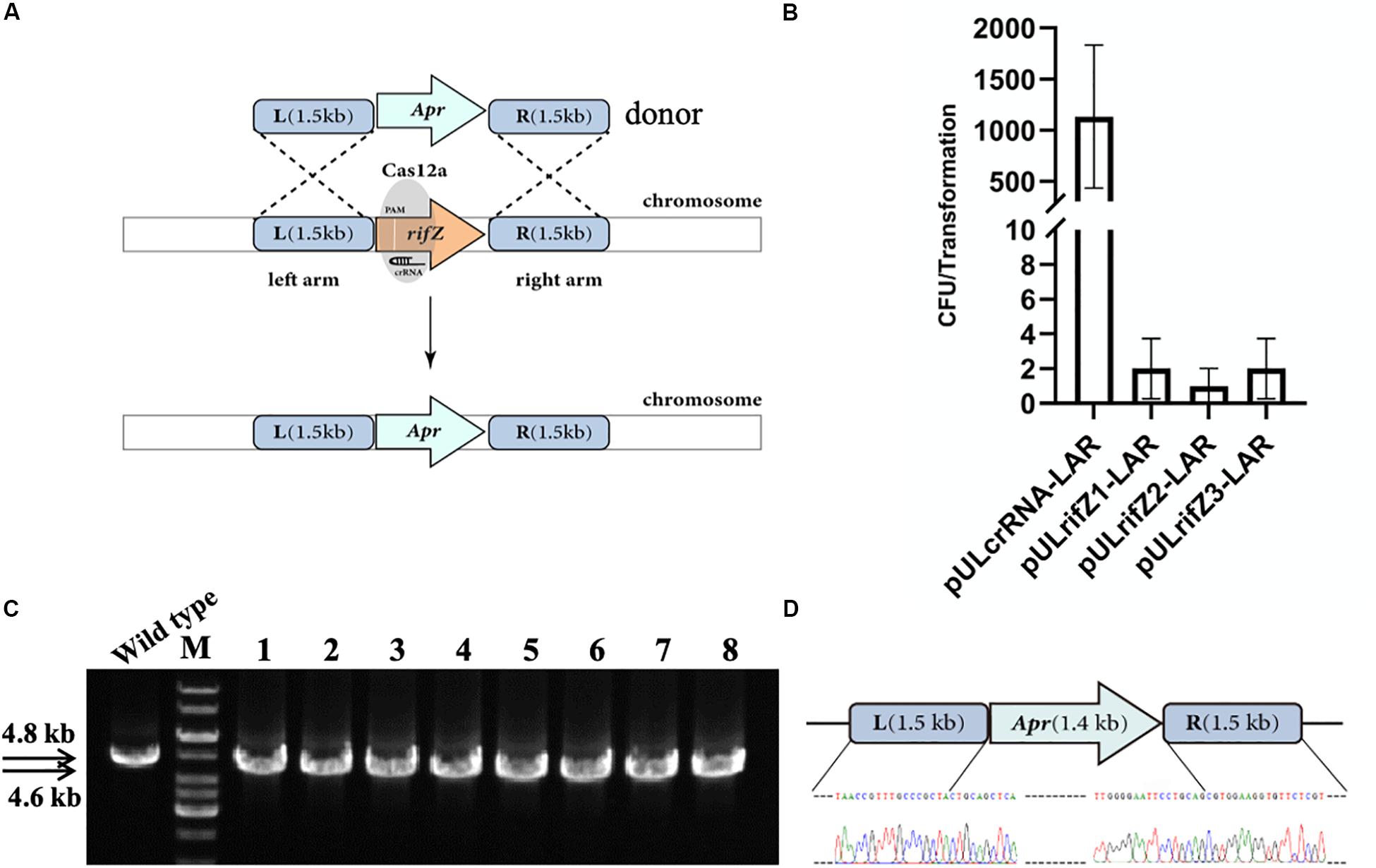
Figure 3. Replacement of rifZ by an antibiotic cassette with CRISPR-Cas12a-assisted precise cutting and HDR-mediated precise recombination in A. mediterranei U32. (A) Schematic of rifZ deletion. With the existence of a crRNA targeting rifZ, Cas12a introduced DSBs within rifZ gene, which could then be precisely repaired by a donor DNA fragment containing an apramycin resistance cassette and the flanking upstream and downstream homologous sequences. (B) The number of transformants obtained with three different crRNAs targeting different loci within rifZ. Plasmid pULcrRNA, which contained no crRNA guide sequences, was employed as a control plasmid. The data were obtained from three independent transformation experiments. (C) PCR amplification analysis of rifZ mutant with paired primers of rifZ-KO-F and rifZ-KO-R. The amplicon of the wild type was 4.8 kb in length, while was smaller in rifZ mutants. M, GeneRuler 1 kb DNA Ladder (Thermo Scientific); lanes 1 and 2, colonies obtained with pULrifZ1-LAR; lanes 3 and 4, colonies obtained with pULrifZ2-LAR; lanes 5–8, colonies obtained with pULrifZ3-LAR. (D) Sanger sequencing results of the amplicon from Figure 3C. All eight amplicons were sequenced and found to be correct, and only the results of the first colony were presented.
After confirmation of the effectiveness of HDR-mediated repair of CRISPR-Cas12a-generated DSBs, we then attempted to combine the CRISPR-Cas12a system and the endogenous HDR pathway to construct precise markerless mutants of both rifZ and glnR. The upstream and downstream homologous arms of the target genes were in fusion assembled and then inserted into the crRNA expressing plasmid, followed by electroporation into the U32 competent cells that constitutively expressed Cas12a protein (Figure 4A). For both target genes, i.e., glnR and rifZ, a dozen transformants were successfully obtained, which were further verified by both PCR amplification and Sanger sequencing. Among the four tested rifZ mutants, three had the HDR-assisted accurate rifZ gene deletion and one had inaccurate 692-bp deletion within rifZ gene, which was obviously repaired by NHEJ (Figures 4B–D). Phenotypic analysis showed that all four rifZ mutants produced no golden pigment (Figure 4E and Supplementary Figure S4a) and much reduced rifamycin SV yield (Supplementary Figure S4b) as indicated by the bactericidal test, which were consistent with the previous findings that RifZ functions as the pathway-specific activator for the whole rif cluster (Li et al., 2017a). Similarly, both the colony PCR and Sanger sequencing results demonstrated that the glnR gene, which encodes the central governor for nitrogen metabolisms, was precisely and markerlessly deleted (Supplementary Figures S5a,b) in all four tested transformants. Subsequent growth phenotypic analysis showed that all these glnR mutants grew poorly on minimal medium with nitrate as the sole nitrogen source (Supplementary Figure S5c). Collectively, above results clearly demonstrated that endogenous HDR pathway can be employed to efficiently repair the CRISPR-Cas12a-generated DSBs and engineer precise and markerless mutants.
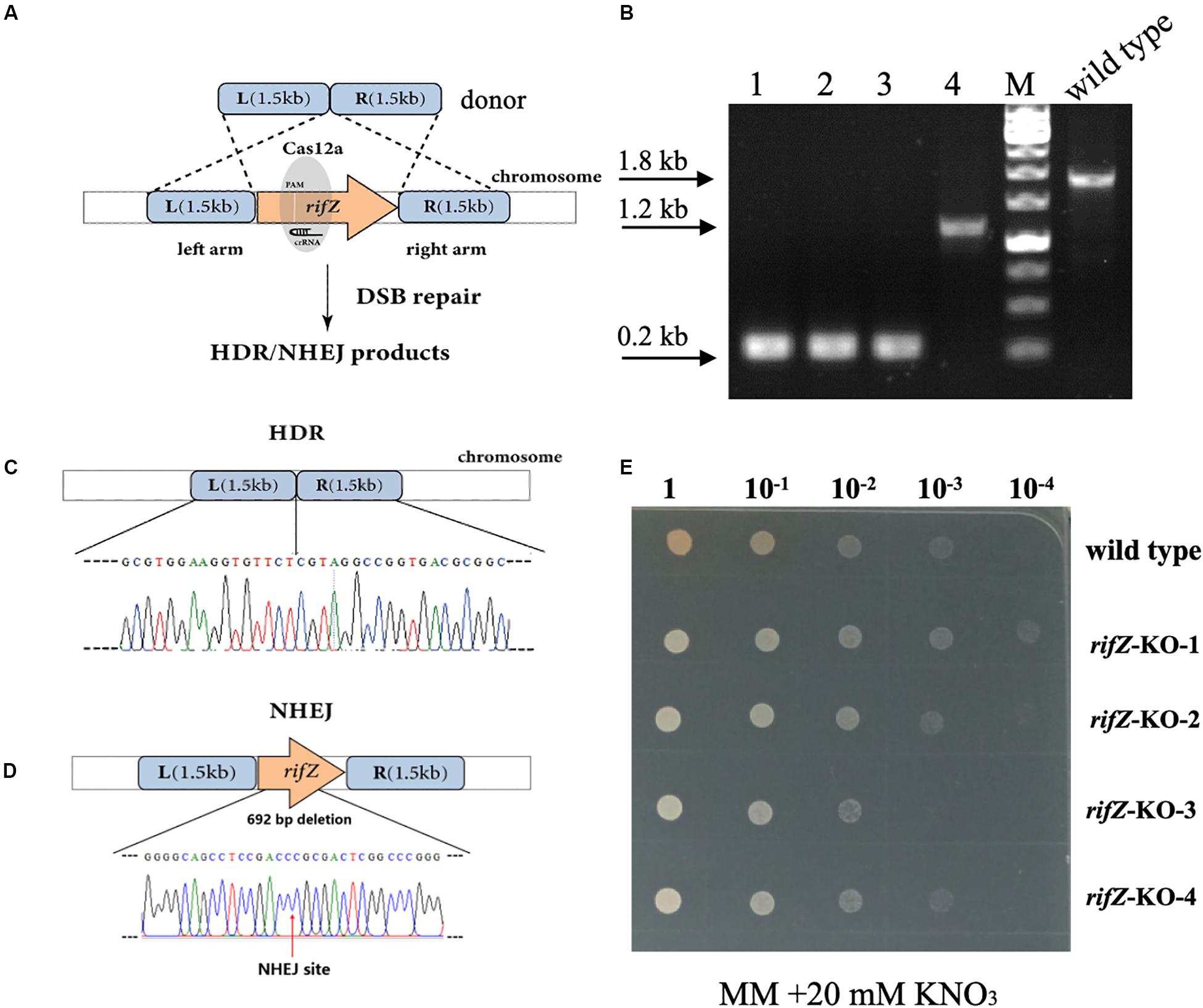
Figure 4. Markerless deletion of rifZ gene with the Cas12a and HDR systems in U32. (A) Schematic of markerless deletion of rifZ gene. Cas12a-generated DSBs in rifZ could be repaired by HDR. (B) Verification of rifZ deletion mutants by colony PCR with paired primers of rifZ+P-F and rifZ+P-R. M, GeneRuler 1 kb DNA Ladder (Thermo Scientific). Lanes 1–4 represented rifZ-KO-1 to rifZ-KO-4, respectively. (C) Sanger sequencing results from the amplicon of rifZ-KO-1 in Figure 4B. (D) Schematic of the rifZ-KO-4 mutant in Figure 4B. The mutant was obtained via NHEJ-mediated repair of the Cas12a-generated DSBs in rifZ gene. Confirmed by Sanger sequencing, 692-bp sequences were found to be deleted at the Cas12a cleavage site. (E) The growth phenotype of rifZ mutants on minimal medium. Serially diluted liquid culture was spotted on plates and cultured at 30°C for 5 days, and only the wild type produced pigmented rifamycin SV.
Discussion
In this study, we successfully employed CRISPR-Cas12a system to develop a genome editing system in A. mediterranei. To test the effectiveness of Cas12a-mediated site-specific DSBs, Cas12a and crRNAs were co-expressed in U32. And to our great surprise, even no homologous recombination arms were introduced, several transformants were obtained, leading to the identification of the endogenous NHEJ activities. Then, we showed that Cas12a-introduced DSBs could be efficiently repaired by either NHEJ or HDR, which therefore facilitates convenient genome editing in A. mediterranei.
We first tested dCas9 but found the protein was toxic to U32. As Cas9 has been demonstrated to be toxic in many other species (Jiang et al., 2017; Cho et al., 2018), we here directly used Cas12a to construct the genome editing system in U32 instead of testing the wild type Cas9. As the transformation efficiency of the plasmids containing Cas12a was much lower than that of the control plasmid, the Cas12a gene might also be harmful to U32 cells. However, once transformants were obtained, both the growth rate and the rifamycin yield of these transformants expressing Cas12a were similar to those of the wild type U32, and Cas12a was therefore employed to develop the genome editing system in U32.
Because of the relatively low transformation and recombination efficiencies in the genus Amycolatopsis, it is difficult to construct markerless mutants in this genus. Although an electroporation transformation system has been established in U32 years ago (Ding et al., 2002), the restricted condition for bacterial growth and the complex procedure for preparation of electro-competent cells make it difficult to prepare U32 competent cells of high transformation efficiency. Many factors have been known to affect bacterial transformation efficiency, including the restriction systems. With the CRISPR-Cas12a genome editing system established in this study, these factors can be efficiently modified to improve the U32 transformation efficiency. Furthermore, with the availability of the CRISPR-Cas12a system, precise DSBs can be introduced by the crRNA-guided Cas12a cleavage, then either NHEJ or HDR can be employed to repair the DNA damage, generating desired mutants with no markers left. Furthermore, CRISPR-Cas12a-assisted markerless mutagenesis makes it possible to perform continuous genome editing operations. As there is only one available self-replicating plasmid origin in U32, the original plasmid must be eliminated before a new plasmid can be transformed, expressing a new crRNA to target a new locus. Fortunately, plasmid curing experiments showed that nearly all original plasmids could be eliminated after one or two generations of passage in medium without selective pressure of antibiotics (Supplementary Figure S6). Alternatively, a new plasmid carrying a different antibiotic resistance cassette can be directly transformed and the transformants can be cultured on plates with the new antibiotic. Moreover, the new plasmid may also express a crRNA targeting the original antibiotic resistance cassette to help clear the original plasmid.
Cas12a is so far the most minimalistic of CRISPR systems and can process precursor crRNAs (Fonfara et al., 2016). Based on this characteristic, multiple gene editing can be easily achieved through simply constructing a crRNA array, expressing multiple precursor crRNAs driven under one promoter, which can be further processed by Cas12a to generate multiple mature crRNAs for multiple gene editing or gene regulation in vivo (Zetsche et al., 2017; Zhang et al., 2017b). After mutation of the RuvC domain, the DNase-dead Cas12a (namely, ddCas12a) can be employed for gene regulation. Similarly, multiple gene regulation can be achieved with the co-expression of both ddCas12a and a crRNA array (Zhang et al., 2017b). Moreover, with the mutagenesis of the crRNA DR, the ddCas12a binding affinities against mutant crRNAs can be precisely determined and hence the regulatory strength of target genes’ transcription (Wang et al., 2019). Although gene regulation was not tested in this study, one may easily perform transcriptional regulation through simply changing the wild type Cas12a to ddCas12a.
There are vast majority of biosynthetic clusters in the genus Amycolatopsis, demonstrating the genus has great potential to produce diverse secondary metabolites (Adamek et al., 2018). Without efficient genome editing tools, heterologous expression is the main way to produce and characterize these metabolites, which could be of low efficiency. As CRISPR-based genome editing has been demonstrated as an efficient approach to discover unique metabolites in Streptomyces (Zhang et al., 2017a), the present work will certainly shed light on the development of CRISPR-assisted genome editing systems in other species in Amycolatopsis genus and further facilitates the genome mining in this genus.
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Author Contributions
YZ and XL performed most of the experiments. YZ prepared the draft. JWu drew the schematic maps. JWa and GZ designed the study. JWa revised the manuscript and supervised the whole project.
Funding
This work was supported by grants from the National Natural Science Foundation of China (31770057, 31670058, and 31430004). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledge the valuable assistance of Professor Yi-Cheng Sun for kindly providing plasmids of pJV53-Cpf1 and pCR-HYG. The authors thank Professor Hua Yuan for his careful revision of this manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbioe.2020.00698/full#supplementary-material
References
Adamek, M., Alanjary, M., Sales-Ortells, H., Goodfellow, M., Bull, A. T., Winkler, A., et al. (2018). Comparative genomics reveals phylogenetic distribution patterns of secondary metabolites in Amycolatopsis species. BMC Genomics 19:426. doi: 10.1186/s12864-018-4809-4
Babynin, E. V. (2007). [Molecular mechanism of homologous recombination in meiosis: origin and biological significance]. Tsitologiia 49, 182–193.
Chayot, R., Montagne, B., Mazel, D., and Ricchetti, M. (2010). An end-joining repair mechanism in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 107, 2141–2146. doi: 10.1073/pnas.0906355107
Cho, S., Choe, D., Lee, E., Kim, S. C., Palsson, B., and Cho, B. K. (2018). High-level dCas9 expression induces abnormal cell morphology in Escherichia coli. ACS Synth. Biol. 7, 1085–1094. doi: 10.1021/acssynbio.7b00462
Cobb, R. E., Wang, Y., and Zhao, H. (2015). High-efficiency multiplex genome editing of Streptomyces species using an engineered CRISPR/Cas system. ACS Synth. Biol. 4, 723–728. doi: 10.1021/sb500351f
Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto, R., Habib, N., et al. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823.
Dadura, K., Plocinska, R., Rumijowska-Galewicz, A., Plocinski, P., Zaczek, A., Dziadek, B., et al. (2017). PdtaS deficiency affects resistance of mycobacteria to ribosome targeting antibiotics. Front. Microbiol. 8:2145. doi: 10.3389/fmicb.2017.02145
Ding, X., Tian, Y., Chiao, J., Zhao, G., and Jiang, W. (2003). Stability of plasmid pA387 derivatives in Amycolatopsis mediterranei producing rifamycin. Biotechnol. Lett. 25, 1647–1652.
Ding, X. M., Zhang, N., Tian, Y. Q., Jiang, W. H., Zhao, G. P., and Jiao, R. S. (2002). [Establishment of gene replacement/disruption system through homologous recombination in Amycolatopsis mediterranei U32]. Sheng Wu Gong Cheng Xue Bao 18, 431–437.
Fonfara, I., Richter, H., Bratovic, M., Le Rhun, A., and Charpentier, E. (2016). The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature 532, 517–521. doi: 10.1038/nature17945
Harrison, P., and Hart, S. (2018). Gene editing and gene regulation with CRISPR. Exp. Physiol. 103, 437–438. doi: 10.1113/ep086864
Horvath, P., and Barrangou, R. (2010). CRISPR/Cas, the immune system of bacteria and archaea. Science 327, 167–170. doi: 10.1126/science.1179555
Hu, X. F., Zhang, B., Liao, C. H., and Zeng, Z. J. (2019). High-efficiency CRISPR/Cas9-mediated gene editing in honeybee (Apis mellifera) embryos. G3 9, 1759–1766. doi: 10.1534/g3.119.400130
Huang, H., Zheng, G., Jiang, W., Hu, H., and Lu, Y. (2015). One-step high-efficiency CRISPR/Cas9-mediated genome editing in Streptomyces. Acta Biochim. Biophys. Sin. 47, 231–243. doi: 10.1093/abbs/gmv007
Jia, H., Zhang, L., Wang, T., Han, J., Tang, H., and Zhang, L. (2017). Development of a CRISPR/Cas9-mediated gene-editing tool in Streptomyces rimosus. Microbiology 163, 1148–1155. doi: 10.1099/mic.0.000501
Jiang, W., Yang, B., and Weeks, D. P. (2014). Efficient CRISPR/Cas9-mediated gene editing in Arabidopsis thaliana and inheritance of modified genes in the T2 and T3 generations. PLoS One 9:e99225. doi: 10.1371/journal.pone.0099225
Jiang, Y., Chen, B., Duan, C., Sun, B., Yang, J., and Yang, S. (2015). Multigene editing in the Escherichia coli genome via the CRISPR-Cas9 system. Appl. Environ. Microbiol. 81, 2506–2514. doi: 10.1128/aem.04023-14
Jiang, Y., Qian, F., Yang, J., Liu, Y., Dong, F., Xu, C., et al. (2017). CRISPR-Cpf1 assisted genome editing of Corynebacterium glutamicum. Nat. Commun. 8:15179.
Jiao, R. S., Chen, Y. M., Wu, M. G., and Gu, W. L. (1979). Studies on the metabolic regulation of biosynthesis of rifamycin by Norcadia (Amycolatopsis) mediterranei I. The stimulative effect of nitrate on biosynthesis of rifamycin SV by Nocardia (Amycolatopsis) mediterranei. Acta Phytophysiol. Sin. 05, 395–402.
Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A., and Charpentier, E. (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. doi: 10.1126/science.1225829
Koonin, E. V., Makarova, K. S., and Zhang, F. (2017). Diversity, classification and evolution of CRISPR-Cas systems. Curr. Opin. Microbiol. 37, 67–78. doi: 10.1016/j.mib.2017.05.008
Kumar, C. V., Coque, J. J., and Martin, J. F. (1994). Efficient transformation of the cephamycin C producer nocardia lactamdurans and development of shuttle and promoter-probe cloning vectors. Appl. Environ. Microbiol. 60, 4086–4093. doi: 10.1128/aem.60.11.4086-4093.1994
Kumari, R., Singh, P., and Lal, R. (2016). Genetics and genomics of the Genus Amycolatopsis. Indian J Microbiol. 56, 233–246. doi: 10.1007/s12088-016-0590-8
Lal, R., Lal, S., Grund, E., and Eichenlaub, R. (1991). Construction of a hybrid plasmid capable of replication in Amycolatopsis mediterranei. Appl. Environ. Microbiol. 57, 665–671. doi: 10.1128/aem.57.3.665-671.1991
Lee, J. G., Choi, C. Y., Seong, B. L., and Han, M. H. (1983). Optimal Ph Profile in Rifamycin-B fermentation. J. Ferment. Technol. 61, 49–53.
Li, C., Liu, X., Lei, C., Yan, H., Shao, Z., Wang, Y., et al. (2017a). RifZ (AMED_0655) is a pathway-specific regulator for rifamycin biosynthesis in Amycolatopsis mediterranei. Appl. Environ. Microbiol. 83, e3201–e3216.
Li, C., Zhou, L., Wang, Y., Zhao, G., and Ding, X. (2017b). Conjugation of varphiBT1-derived integrative plasmid pDZL802 in Amycolatopsis mediterranei U32. Bioengineered 8, 549–554. doi: 10.1080/21655979.2016.1270808
Lieber, M. R. (2010). The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 79, 181–211. doi: 10.1146/annurev.biochem.052308.093131
Low, B. E., Kutny, P. M., and Wiles, M. V. (2016). Simple, efficient CRISPR-Cas9-mediated gene editing in mice: strategies and methods. Methods Mol. Biol. 1438, 19–53. doi: 10.1007/978-1-4939-3661-8_2
Makarova, K. S., Haft, D. H., Barrangou, R., Brouns, S. J., Charpentier, E., Horvath, P., et al. (2011). Evolution and classification of the CRISPR-Cas systems. Nat. Rev. Microbiol. 9, 467–477.
Makarova, K. S., Zhang, F., and Koonin, E. V. (2017a). SnapShot: class 1 CRISPR-Cas systems. Cell 168, 946–946.
Makarova, K. S., Zhang, F., and Koonin, E. V. (2017b). SnapShot: class 2 CRISPR-Cas systems. Cell 168, 328–328.
Matsu-Ura, T., Baek, M., Kwon, J., and Hong, C. (2015). Efficient gene editing in Neurospora crassa with CRISPR technology. Fungal Biol. Biotechnol. 2:4.
Mejia, A., Barrios-Gonzalez, J., and Viniegra-Gonzalez, G. (1998). Overproduction of rifamycin B by Amycolatopsis mediterranei and its relationship with the toxic effect of barbital on growth. J. Antibiot. 51, 58–63. doi: 10.7164/antibiotics.51.58
Mohanraju, P., Makarova, K. S., Zetsche, B., Zhang, F., Koonin, E. V., and van der Oost, J. (2016). Diverse evolutionary roots and mechanistic variations of the CRISPR-Cas systems. Science 353:aad5147. doi: 10.1126/science.aad5147
Rothstein, D. M. (2016). Rifamycins, alone and in combination. Cold Spring Harbor Perspect. Med. 6:a027011. doi: 10.1101/cshperspect.a027011
Sfeir, A., and Symington, L. S. (2015). Microhomology-mediated end joining: a back-up survival mechanism or dedicated pathway? Trends Biochem Sci. 40, 701–714. doi: 10.1016/j.tibs.2015.08.006
Sung, P., and Klein, H. (2006). Mechanism of homologous recombination: mediators and helicases take on regulatory functions. Nat. Rev. Mol. Cell Biol. 7, 739–750. doi: 10.1038/nrm2008
Wang, J., Lu, A., Bei, J., Zhao, G., and Wang, J. (2019). CRISPR/ddCas12a-based programmable and accurate gene regulation. Cell Discovery 5:15.
Wright, D. G., Castore, R., Shi, R., Mallick, A., Ennis, D. G., and Harrison, L. (2017). Mycobacterium tuberculosis and Mycobacterium marinum non-homologous end-joining proteins can function together to join DNA ends in Escherichia coli. Mutagenesis 32, 245–256.
Xu, L., Huang, H., Wei, W., Zhong, Y., Tang, B., Yuan, H., et al. (2014). Complete genome sequence and comparative genomic analyses of the vancomycin-producing Amycolatopsis orientalis. BMC Genomics 15:363. doi: 10.1186/1471-2164-15-363
Yamano, T., Nishimasu, H., Zetsche, B., Hirano, H., Slaymaker, I. M., Li, Y., et al. (2016). Crystal structure of cpf1 in complex with guide RNA and target DNA. Cell 165, 949–962.
Yan, M. Y., Yan, H. Q., Ren, G. X., Zhao, J. P., Guo, X. P., and Sun, Y. C. (2017). CRISPR-Cas12a-assisted recombineering in bacteria. Appl. Environ. Microbiol. 83, e00947-17.
Zetsche, B., Gootenberg, J. S., Abudayyeh, O. O., Slaymaker, I. M., Makarova, K. S., Essletzbichler, P., et al. (2015). Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163, 759–771. doi: 10.1016/j.cell.2015.09.038
Zetsche, B., Heidenreich, M., Mohanraju, P., Fedorova, I., Kneppers, J., DeGennaro, E. M., et al. (2017). Multiplex gene editing by CRISPR-Cpf1 using a single crRNA array. Nat. Biotechnol. 35, 31–34. doi: 10.1038/nbt.3737
Zhang, M. M., Wong, F. T., Wang, Y., Luo, S., Lim, Y. H., Heng, E., et al. (2017a). CRISPR-Cas9 strategy for activation of silent Streptomyces biosynthetic gene clusters. Nat. Chem. Biol. 13, 607–609. doi: 10.1038/nchembio.2341
Zhang, X., Wang, J., Cheng, Q., Zheng, X., Zhao, G., and Wang, J. (2017b). Multiplex gene regulation by CRISPR-ddCpf1. Cell Discovery 3:17018.
Zhao, W., Zhong, Y., Yuan, H., Wang, J., Zheng, H., Wang, Y., et al. (2010). Complete genome sequence of the rifamycin SV-producing Amycolatopsis mediterranei U32 revealed its genetic characteristics in phylogeny and metabolism. Cell Res. 20, 1096–1108. doi: 10.1038/cr.2010.87
Zheng, X., Li, S. Y., Zhao, G. P., and Wang, J. (2017). An efficient system for deletion of large DNA fragments in Escherichia coli via introduction of both Cas9 and the non-homologous end joining system from Mycobacterium smegmatis. Biochem. Biophys. Res. Commun. 485, 768–774. doi: 10.1016/j.bbrc.2017.02.129
Keywords: Amycolatopsis mediterranei, CRISPR-Cas12a, genome editing, NHEJ, HDR
Citation: Zhou Y, Liu X, Wu J, Zhao G and Wang J (2020) CRISPR-Cas12a-Assisted Genome Editing in Amycolatopsis mediterranei. Front. Bioeng. Biotechnol. 8:698. doi: 10.3389/fbioe.2020.00698
Received: 13 March 2020; Accepted: 03 June 2020;
Published: 26 June 2020.
Edited by:
Jiazhang Lian, Zhejiang University, ChinaReviewed by:
Feng-Qing Wang, East China University of Science and Technology, ChinaZehua Bao, Boston University, United States
John Van Der Oost, Wageningen University and Research, Netherlands
Copyright © 2020 Zhou, Liu, Wu, Zhao and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jin Wang, d2FuZ2owMUBob3RtYWlsLmNvbQ==
†These authors have contributed equally to this work