 Samuel A. Bradley
Samuel A. Bradley Jie Zhang
Jie Zhang Michael K. Jensen
Michael K. Jensen- Novo Nordisk Foundation Center for Biosustainability, Technical University of Denmark, Lyngby, Denmark
Plants produce some of the most potent therapeutics and have been used for thousands of years to treat human diseases. Today, many medicinal natural products are still extracted from source plants at scale as their complexity precludes total synthesis from bulk chemicals. However, extraction from plants can be an unreliable and low-yielding source for human therapeutics, making the supply chain for some of these life-saving medicines expensive and unstable. There has therefore been significant interest in refactoring these plant pathways in genetically tractable microbes, which grow more reliably and where the plant pathways can be more easily engineered to improve the titer, rate and yield of medicinal natural products. In addition, refactoring plant biosynthetic pathways in microbes also offers the possibility to explore new-to-nature chemistry more systematically, and thereby help expand the chemical space that can be probed for drugs as well as enable the study of pharmacological properties of such new-to-nature chemistry. This perspective will review the recent progress toward heterologous production of plant medicinal alkaloids in microbial systems. In particular, we focus on the refactoring of halogenated alkaloids in yeast, which has created an unprecedented opportunity for biosynthesis of previously inaccessible new-to-nature variants of the natural alkaloid scaffolds.
Introduction
Plants express biosynthetic pathways capable of performing a fascinating plethora of complex chemistry (Wilson and Roberts, 2014; Kutchan et al., 2015; Wurtzel and Kutchan, 2016). Consequently, many of the biologically active compounds utilized commercially, particularly pharmaceuticals, agrochemicals, flavors and fragrances, are plant-derived natural products. Pharmaceutically important classes of plant natural products include the terpenes and terpenoids (Pichersky and Raguso, 2018; Zhou and Pichersky, 2020), polyketides (Ma et al., 2009), alkaloids (Springob and Kutchan, 2009), as well as other aromatic amino acids derivatives (Springob and Kutchan, 2009). Natural products display an impressive range and density of pharmaceutical activities, many of them are FDA-approved, and more than 50% of compounds recently introduced in drug discovery pipelines are natural products or derivatives thereof (Newman and Cragg, 2016). However, most bioactive compounds possess complex structures with multiple stereocenters and oxygenated functional groups which complicate, and even preclude, total synthesis as a means of production. For this reason extraction from natural plant resources remains indispensable for sourcing bioactive compounds. For example, vincristine and vinblastine are alkaloids found in the Madagascar periwinkle (Catharanthus roseus) and listed by the WHO as essential medicines (World Health Organisation Model Lists of Essential Medicines, 2019). They are commercially produced by semi-synthesis, in which the biological precursors vindoline and catharanthine are extracted from C. rosesus and subsequently chemically coupled in vitro (Courdavault et al., 2020). Yet, due to the vagaries inherent to agriculture and natural habitats, the low in planta accumulation, and the complex mixture of chemically similar compounds found in C. roseus, vincristine supply for clinical usage can be unstable (Groot et al., 2018). Consequently, there is considerable interest in producing plant medicinal alkaloids, and other bioactive plant natural products, by refactoring the biosynthetic pathways in microorganisms, so-called microbial synthesis (Chang et al., 2007; Ajikumar et al., 2010; Brown et al., 2015; Fossati et al., 2015; Galanie et al., 2015; Qu et al., 2015; Li et al., 2018).
Supplying plant-derived therapeutics for human illnesses using microbial synthesis could create cheaper, greener and more reliable sources of these compounds as microbes (i) grow faster (hours for yeast as compared to months/years for plants), (ii) can be engineered to produce less complex mixtures of plant natural product, and (iii) can be cultivated using more standardized and easily scalable fermentation and downstream processing methods (Junker, 2004; Jungbauer, 2013; Buyel et al., 2015; Liu et al., 2016; Zydney, 2016; Wang et al., 2019a). Budding yeast (Saccharomyces cerevisiae) presents an attractive chassis for refactoring complex biosynthetic pathways of bioactive natural products, thanks to its eukaryotic cell architecture capable of supporting biosynthetic pathways that require significant endomembrane systems (e.g., P450 enzymes) or compartmentalization (Avalos et al., 2013; Zhou et al., 2016). Indeed, the seminal demonstration of artemisinic acid production in yeast (Ro et al., 2006; Paddon and Keasling, 2014) has inspired research into microbial biosynthesis of many more bioactive plant natural products. This is exemplified by the recent engineering of the native yeast mevalonate pathway to enable high flux toward geranyl pyrophosphate (GPP), and introduction of a heterologous hexanoyl-CoA biosynthetic pathway for the complete biosynthesis of cannabinoids (Luo et al., 2019). From tyrosine derivatives, biosynthesis of the common benzylisoquinoline alkaloid (BIA) precursor (S)-reticuline was a landmark achievement toward de novo biosynthesis of medicinal alkaloids, including hydrocodone, thebaine, stylopine, and noscapine (Galanie and Smolke, 2015; Hori et al., 2016; Li et al., 2018). Likewise, morphinan BIAs codeine and morphine have also been synthesized in yeast based on feeding substrates (R)-reticuline, salutaridine, and codeine (Fossati et al., 2015). In addition to BIAs, another major class of alkaloids are the monoterpene indole alkaloids (MIAs) derived from GPP and tryptophan (De Luca et al., 2014). Brown et al. (2015) demonstrated the de novo synthesis of strictosidine, the common precursor of all MIAs, in yeast through the successful refactoring of 12 heterologous enzymatic steps. Ehrenworth et al. (2015) engineered yeast to produce tetrahydrobiopterin for a mono-oxidation of tryptophan to 5-hydroxytryptophan and further onto 5-hydroxytryptamine (serotonin), which when coupled to exogenously fed secologanin enabled production of 10-hydroxystrictosidine. Further downstream of the MIA building blocks, Qu et al. (2015) demonstrated the seven-step conversion of tabersonine to the marketed anticancer precursor vindoline. In addition to BIAs and MIAs, the ergot alkaloid precursor chanoclavine-1 and the complex ergot alkaloid cycloclavine derived from tryptophan and the C5 isoprenoid unit dimethylallyl diphosphate (DMAPP) also exemplifies successful hijacking of native yeast metabolites for microbial alkaloid synthesis (Nielsen et al., 2014; Jakubczyk et al., 2015). Milne et al. (2020) recently reported the refactoring of the hallucinogenic alkaloid psilocybin biosynthetic pathway extending from an engineered shikimate pathway and coupled via tryptophan decarboxylase to yield the starting block tryptamine for four-step psilocybin biosynthesis. Beyond the refactoring of complete alkaloid biosynthetic pathways derived from natural mevalonate pathway C10 and C15 precursor units, GPP and FPP, respectively, Ignea et al. (2018) also refactored biosynthesis of 40 different C11 non-canonical terpene scaffolds, based on 2-methyl GPP production and engineered C11-specific monoterpene synthases. Lastly, tropane alkaloids derived from the arginine and polyamine metabolism biosynthesis, also should be mentioned to emphasize the versatility of yeast metabolism and cell architecture for microbial synthesis of bioactive alkaloids (Ping et al., 2019; Srinivasan and Smolke, 2019). While this review will focus on two major classes of alkaloids, BIAs and MIAs, it deserves to be mentioned that other branches of yeast’s native metabolism have been harnessed for microbial biosynthesis of non-alkaloid bioactive natural products. This includes the production of methylxanthines from the S-adenosyl methionine (SAM) de novo purine synthesis pathways, and adenine nucleotide pools (McKeague et al., 2016), as well as phenylpropanoids resveratrol and breviscapin produced from the shikimate pathway (Becker et al., 2003; Liu et al., 2018). Together with an elaborate review of yeast metabolism for the production of broader classes of plant natural products, this has recently been excellently covered by Chen et al. (2020).
Beyond the rational refactoring of plant natural product pathways for microbial biosynthesis of natural alkaloids, synthetic biologists are taking inspiration from medicinal chemistry campaigns investigating small-molecule drug leads, with the objective of expanding the repertoire of medicinal plant alkaloids by including their unnatural derivatives. Bioactive natural products often proceed from lead to licensing without undergoing significant modification (Ganesan, 2008). However, there is evidence that the derivatives of plant alkaloids possess new or improved pharmaceutical activities (Leggans et al., 2013; Sears and Boger, 2015; Gautam et al., 2016; Li et al., 2018). This will be of interest for (i) finding drugs against therapeutic targets that are currently considered “undruggable,” such as B-class GPCRs (Barker et al., 2013), and ii) finding improved variants against current targets—for example, 10-fluorovinblastine, 10-fluorovincristine (Sears and Boger, 2015), and halogenated noscapine variants (DeBono et al., 2015) have demonstrated improved inhibition of tumor growth relative to the natural variants. Yet, the sparsity of derivatized natural products occurs because the same complexity that bedevils total synthesis of natural products also hinders systematic chemical modification. The high number of reactive oxygenic groups makes it difficult to chemically modify specific positions without also modifying others, creating a complex product mixture. Consequently, large collections of discrete natural product derivatives are not readily available for bioactivity screening (Dandapani and Marcaurelle, 2010). It has been often hypothesized that a biological approach could generate relatively pure lead compound derivatives by harnessing the intrinsically high substrate and product specificity of enzymes. Accordingly, a number of studies have investigated the natural promiscuity of the biosynthetic pathways for medicinal plants alkaloids by feeding building block analogs or expressing building-block modifying enzymes (McCoy et al., 2006; Yerkes et al., 2008; Runguphan et al., 2010; McDonald et al., 2019). However, the slow growth rate, relatively sparse molecular toolbox, and complex regulatory systems of plants have prevented systematic engineering of the pathway as a whole, and generally limited the biological exploration of chemical space to what is achievable with the natural promiscuity. Thus, while the refactoring of these pathways into microorganisms, as described above, holds great potential to yield valuable production workhorses for existing pharmaceuticals in the near future, microbial synthesis may furthermore facilitate the engineering of natural product pathways to open up entirely novel regions of chemical space that are currently inaccessible.
The recent acceleration of refactoring elements of plant metabolism in microorganisms, discussed above, is creating an unprecedented resource of plant alkaloid pathways in a context more amenable their engineering. Such engineering efforts can be used to improve the pathway turnover of complex intermediate derivatives, thereby allowing researchers to produce previously inaccessible classes of compounds that can be screened for therapeutic potential. It is therefore timely to bring together these refactoring efforts and the studies reporting parts that can be integrated into future cell factories optimized for producing alkaloid derivatives. While there is a near infinite number of possible natural product derivatives, this review will first focus on the introduction of halogens (fluorine, chlorine, bromine and iodine) into natural product scaffolds, using the MIA and BIA pathways as test cases (Figures 1A–C). Secondly, although no yeast cell factories optimized for producing halogenated MIAs and BIAs yet exist, a number of in vitro and in planta studies have characterized the turnover of halogenated intermediates and developed parts relevant to achieving this goal. These will be reviewed with respect to the pathway sections that have been refactored in yeast. Finally, how such strains would represent an unprecedented opportunity to develop semi-synthetic medicinal chemistry campaigns that probe entirely new regions of chemical space will be discussed (Figure 1D).
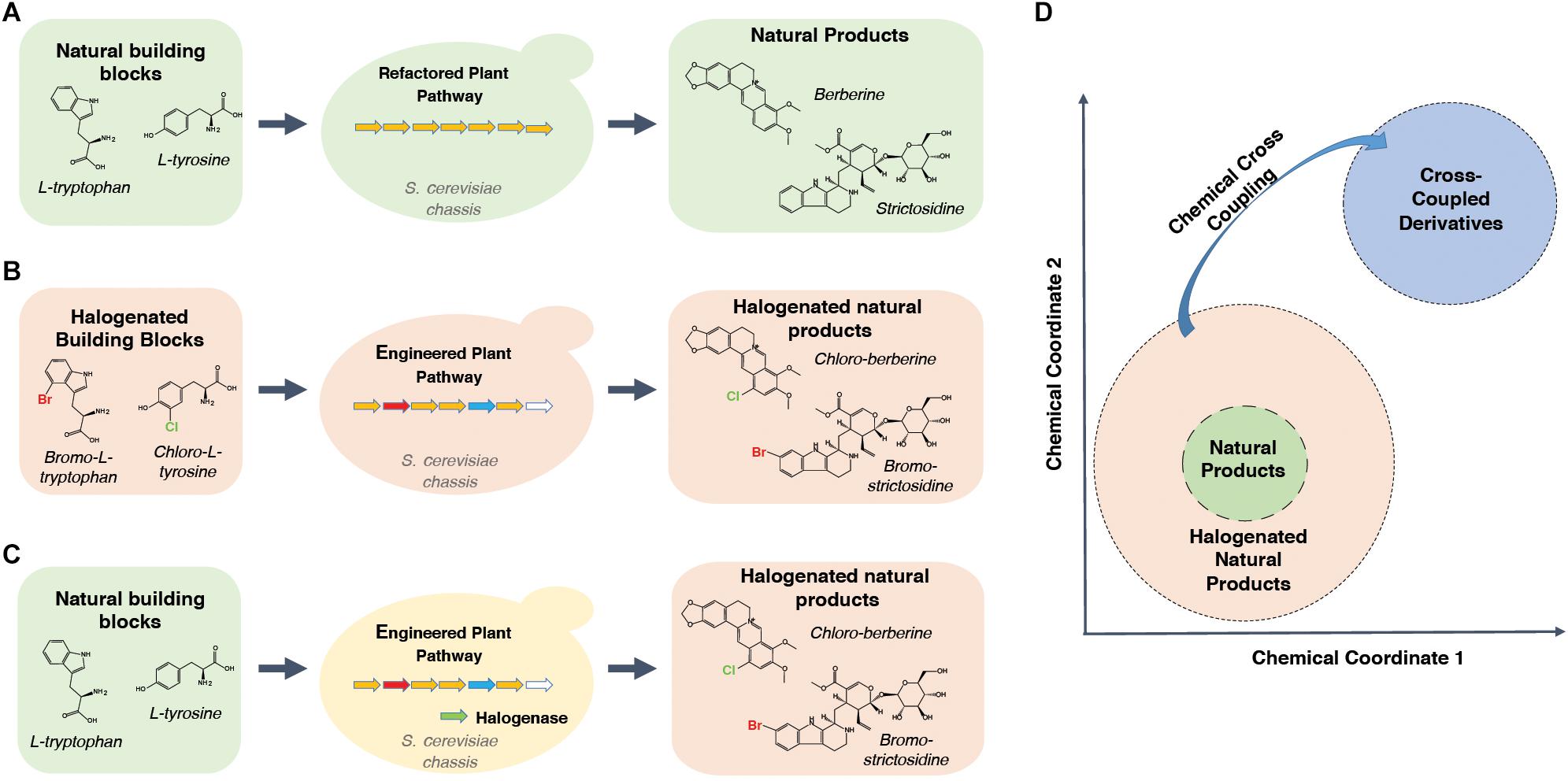
Figure 1. Deploying synthetic biology to access novel regions of chemical space. (A) Readily available bulk feedstocks can be fed to yeast strains hosting refactored plant biosynthetic pathways. Natural products can then be extracted from fermentation broth. (B) Engineering of the plant biosynthetic pathway within the yeast chassis can allow or improve turnover of halogenated substrate analogs. Halogenated natural product scaffolds can be extracted from the fermentation broth. Promiscuous or engineered enzyme variants in engineered plant pathways are colored by red, blue and white arrows in the middle panel. (C) Introduction of halogenases can yield halogenated products from natural substrates. Promiscuous or engineered enzyme variants in engineered plant pathways are colored by red, blue and white arrows in the middle panel. Halogenase is depicted by a green arrow. (D) Schematic depicting the halogen derivatives surrounding natural product scaffolds in a hypothetical chemical space in which more similar compounds appear closer together. Halogens can facilitate targeted cross-coupling reactions that allow “hopping” to entirely new and chemically distinct regions of chemical space.
Halogenation in Nature and Pharmacology
Halogens form group seven of the periodic table and the biologically relevant members are fluorine, chlorine, bromine and iodine. Although the prevalence of these elements in nature is becoming increasingly understood, they are not often found naturally in plant alkaloids (Runguphan et al., 2010). Conversely, halogens are highly prevalent in licensed pharmaceuticals, and often have beneficial effects on the ligand binding and their pharmacokinetic properties of human therapeutics (Fejzagić et al., 2019). This is due to a unique combination of chemical properties—bulkiness alters the sterics of ligand binding, the high electronegativity can alter the charge interactions of ligand binding, their specific orbital architectures support unique intermolecular interactions and their hydrophobicity can improve bioavailability (Figure 2). These properties and their effects on drugs have been recently reviewed (Fejzagić et al., 2019). As a consequence of these properties, organohalogens make up roughly 25% of licensed drugs (Xu et al., 2014) and 40% of all new drugs being tested (Fejzagić et al., 2019). Of these, 57% contain fluorine, 38% contain chlorine, whereas bromine and iodine make up just 5% between them (Xu et al., 2014; Fejzagić et al., 2019). In addition to directly altering pharmacokinetic properties of a compound, halogens can act as “chemical handles” for targeting further drug derivatization (Runguphan and O’Connor, 2013; Frese et al., 2016; Corr et al., 2017). They provide effective leaving groups that can allow synthetic chemists to more selectively alter the activated carbon without creating non-specific alterations at other points of the structure. This is significant when attempting to further modify complex natural products for drug use (Figure 1D) because their structural complexity makes specific substitution difficult to achieve, resulting in complex mixtures that can be costly to separate (Frese and Sewald, 2015; Schnepel and Sewald, 2017). Furthermore, FDA approval of novel compounds for therapeutic use is conditional on a pure compound being obtainable. Therefore, achieving regio-specific introduction of halogens into natural product scaffolds may also provide the key to building the natural product variant libraries that would allow the relatively unexplored derivative space surrounding natural products to be systematically probed for novel pharmaceutical activities.
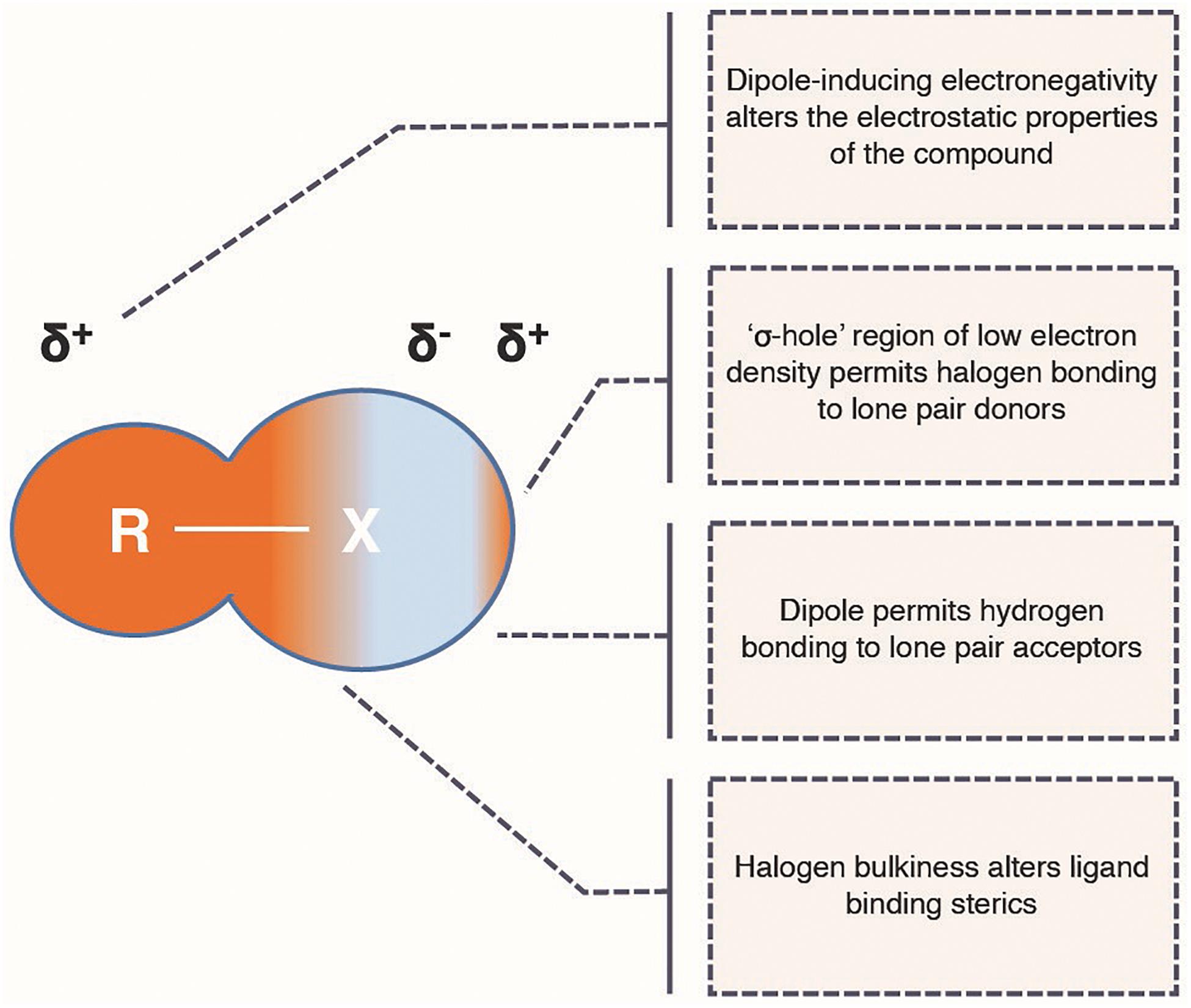
Figure 2. Ligand binding properties of halogen atoms. A schematic outlining the physiochemical properties of a halogen atom (X = F, Cl, Br, or I) and its potential effects on the ligand binding properties of a pharmaceutical (R = halogen-bound atom of natural product scaffold). δ± denote dipole partial charges.
However, despite considerable interest and a number of engineering studies targeting individual enzymes (Chen et al., 2006; McCoy et al., 2006; Yerkes et al., 2008; Glenn et al., 2011; Wang et al., 2019b), there has yet to be a systematic study aimed at optimizing pathway production of new-to-nature analogs of plant medicinal alkaloids. The complex chemistries of natural products that make halogenation so useful for targeting cross-coupling reactions (Figure 1D) also makes it extremely difficult to selectively halogenate the desired site in the first place (Chung and Vanderwal, 2016). Where it is possible chemically, this requires expensive catalysts (palladium) and elemental halogen, which is both toxic and energetically expensive to produce (Schnepel and Sewald, 2017; Fejzagić et al., 2019). The intrinsically high selectivity of enzymes therefore makes enzymatic halogenation a tempting alternative for achieving greener, cheaper, regio- and stereo-selective halogenation of natural products.
The potential for enzymatic halogenation emerged with the discovery of widespread halogenation in nature, which pointed toward a rich trove of halogenating enzymes operating under benign conditions (Gribble, 2003, 2018). Today, more than 6,000 naturally occurring organohalogens have been identified in a range of organisms across all three kingdoms of life, with functions including pheromones, hormones, antimicrobials, halogen recyclers, and structural proteins (Gribble, 2018). For the known organohalogens, chlorination, and bromination are the most common modifications although examples of iodination and fluorination are also reported (Gribble, 2003, 2018). Marine species and soil bacteria have provided the richest source of halogenated compounds due to the relative abundance of halogens in these habitats, particularly bromine in the ocean. A concerted effort to identify the enzymes responsible for incorporating inorganic halogen into metabolism has yielded a set of structurally and mechanistically diverse halogenases capable of catalyzing the formation of F-C, Cl-C, Br-C, and I-C bond formations (Schnepel and Sewald, 2017; Fejzagić et al., 2019).
Of the numerous halogenases, the flavin-dependent halogenases (FDHs) have been a focus for biotechnologists, due to their high substrate- and regio-specificity, and their ability to function independently of carrier proteins that bind the substrate. A subgroup of FDHs targeting the indole moiety of tryptophan are the most well studied group of FDHs, due to the prevalence of indole as a building block in both biology and synthetic chemistry (Frese and Sewald, 2015; Dhuguru and Skouta, 2020). Mechanistically, these tryptophan halogenases are similar to haem-dependent haloperoxidases and vanadium-dependent haloperoxidases in that they form a hypohalous acid (HOX) intermediate. However, unlike these other halogenases, the tryptophan FDHs do not release the HOX intermediate into solution. Instead, the HOX molecule moves through a 10 Å internal tunnel to the active site, where the halide is transferred, via a conserved lysine (Yeh et al., 2007; Flecks et al., 2008), to a specific carbon of the indole moiety of tryptophan (Karabencheva-Christova et al., 2017). Thus, this mechanism avoids the spurious halogenation associated with free HOX and achieves specific halogenation in a single step, compared to the 4–5 steps required for synthetic production of halogenated tryptophan. Indole moiety-targeting FDHs halogenating positions 5, 6 and 7 of the indole ring have been identified (Dong et al., 2005; Yeh et al., 2005; Zehner et al., 2005; Seibold et al., 2006; Fujimori et al., 2007; Heemstra and Walsh, 2008; Foulston and Bibb, 2010; Zeng and Zhan, 2011; Chang and Brady, 2013; Menon et al., 2016; Neubauer et al., 2018; Domergue et al., 2019; Ismail et al., 2019; Luhavaya et al., 2019; Lee et al., 2020; Lingkon and Bellizzi, 2020; Table 1) and, with the exception of a recently discovered brominase (Ismail et al., 2019), preferentially catalyze chlorination over bromination. Chlorination at position 4 has been observed but the responsible enzyme has not been identified (Payne et al., 2015). There are currently no known halogenases that will specifically fluorinate or iodinate tryptophan. Other common alkaloid building blocks also lack specific halogenases. However, more flavin-dependent halogenases continue to be discovered and characterized (Lingkon and Bellizzi, 2020).
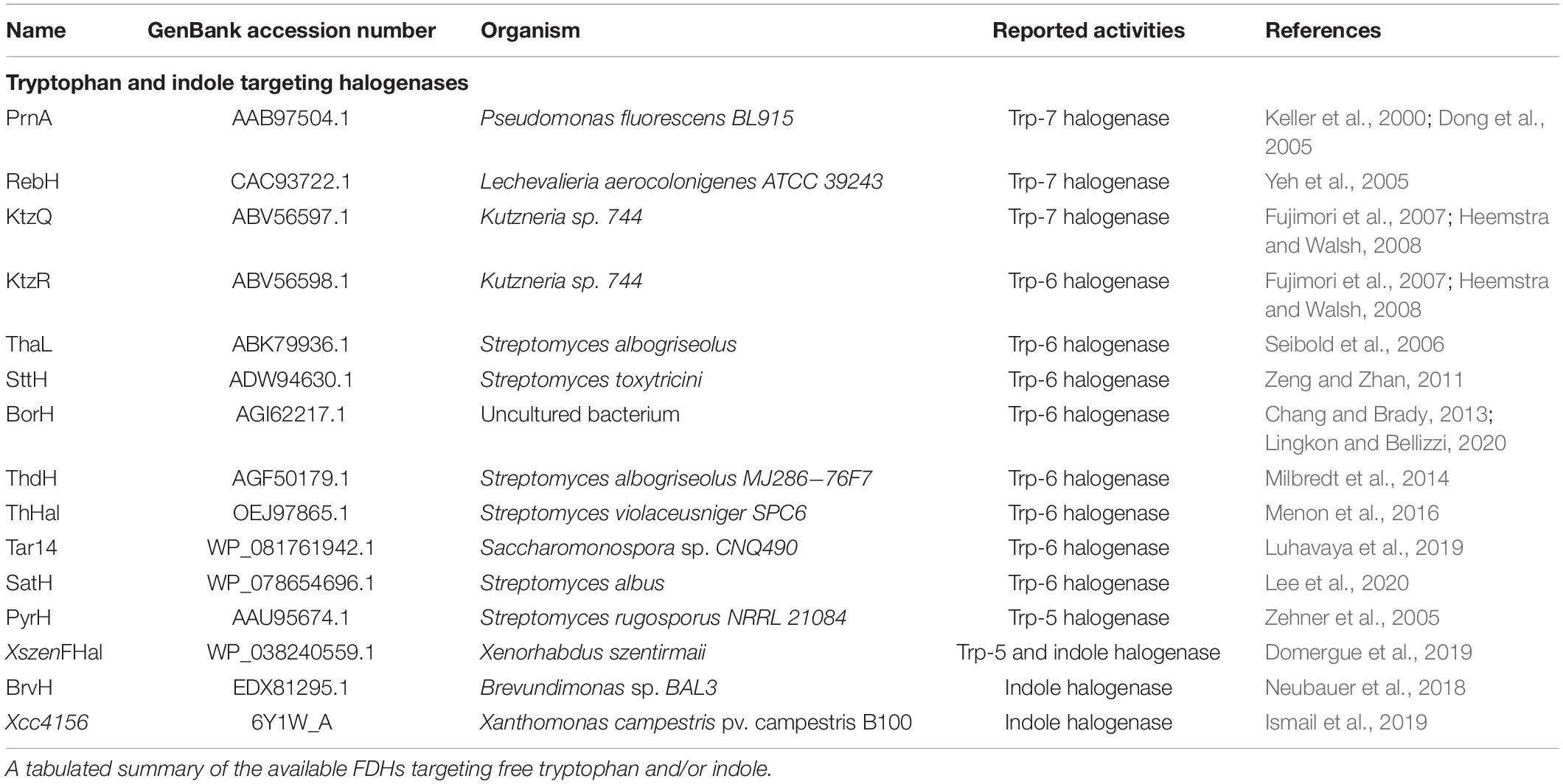
Table 1. Tryptophan and indole-targeting halogenases.
Overall, there is a convincing body of evidence to suggest that halogenated derivatives of natural product scaffolds could be a rich source of improved or entirely novel pharmaceuticals. Furthermore, the site-specific introduction of halogens can facilitate production of a near-infinite number of other derivatives via cross-coupling reactions (Figure 1D). While achieving the site selective halogenation required for this has been historically difficult, the integration of halogenases or derivatized feedstocks into natural product biosynthetic pathways is beginning to open these exciting regions of chemical space.
Expanding Natural Product Chemical Space Through Synthetic Biology
The well-documented effects of halogen atoms on the ligand binding and pharmacokinetic properties of pharmaceuticals present a tantalizing prospect of new or improved pharmaceuticals being created by introducing halogens into these classes of already bioactive compounds. This field remains in its infancy, but the number of studies reporting microbial synthesis of new-to-nature variants of medicinal plant alkaloids is growing. This section will review the progress made toward microbial synthesis of halogenated and other new-to-nature MIA and BIA variants, and describe protein engineering efforts predating microbial refactoring that can be integrated into emerging microbial strains. While this review will focus on MIAs and BIAs, the work performed to assess and engineer derivative turnover of these pathways provides an effective template that should be possible to emulate with other plant medicinal alkaloids (Table 2).
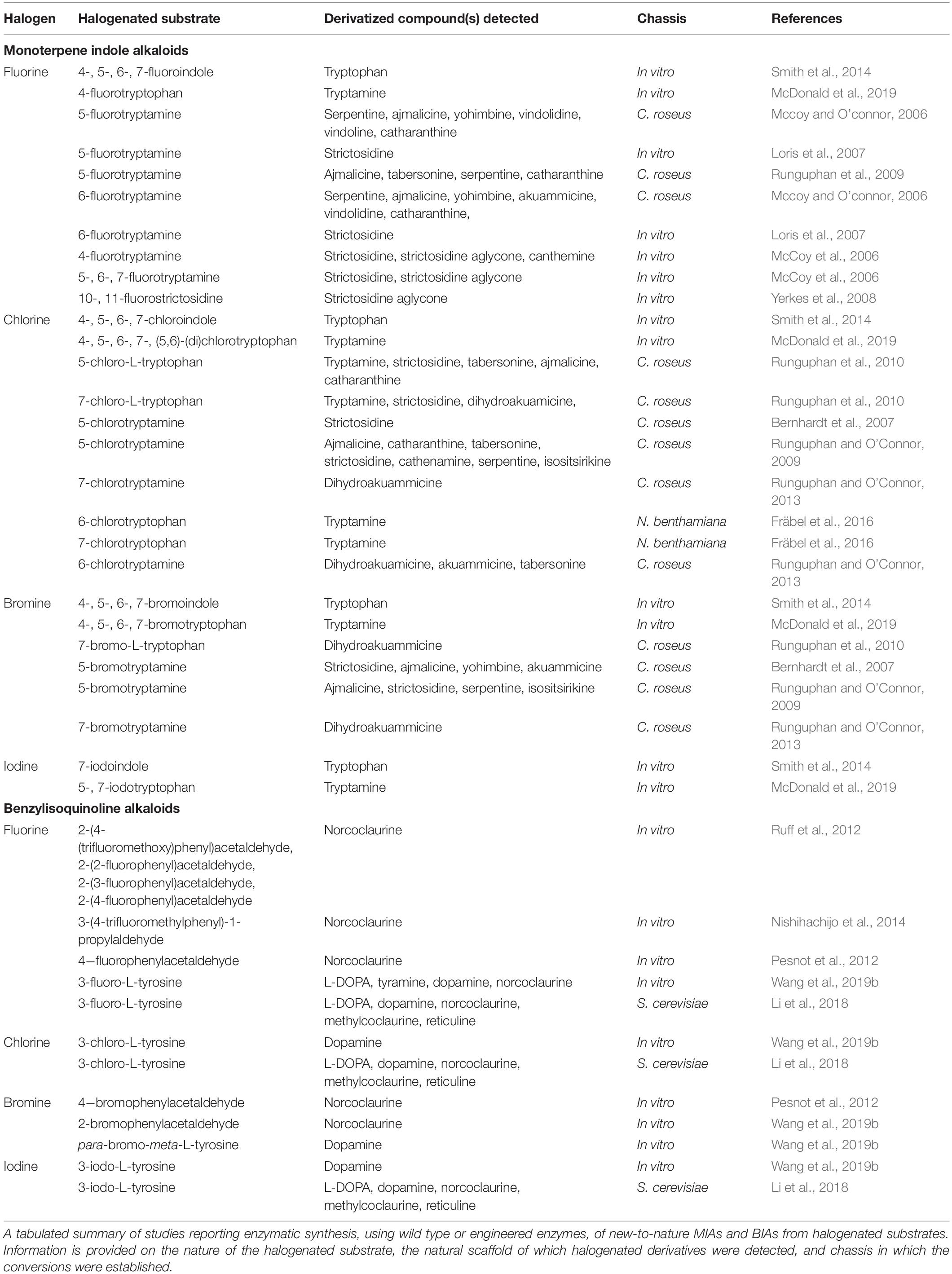
Table 2. Enzymatically produced alkaloids from halogenated substrates.
New-to-Nature Monoterpenoid Indole Alkaloids
MIA biosynthetic pathways have been popular testbeds, both in vivo and in vitro, for the biosynthesis of new-to-nature derivatives of plant medicinal alkaloids (Figure 3A). An early in vivo study, in which chemically synthesized tryptamines with substitutions on the indole ring were fed to C. roseus hairy root cultures or seedlings, observed production of fluorinated serpentine and ajmalicine, two “late-stage” MIA compounds with demonstrated pharmaceutical activities (Mccoy and O’connor, 2006). Furthermore, the authors speculated that fluorinated analogs could be widely incorporated into MIAs with minimal engineering effort due to the small size of fluorine. In another seminal study, two tryptophan halogenases (RebH and PyrH) were expressed in C. roseus and de novo production of 12-chloro-19,20-dihydroakuamicine, 10-chloroajmalicine, 15-chlorotabersonine, and 12-bromo-19,20-Dihydroakuammicine was observed (Runguphan et al., 2010). Detection of multiple “late-stage” MIA variants indicates an encouraging level of native promiscuity. However, it was observed that production of MIAs required expression of a previously identified promiscuous STR mutant (Bernhardt et al., 2007) and, interestingly, that the halogenated substrates shifted the major MIA products, most likely due to promiscuity differences forcing substrate flux into different branches (Bernhardt et al., 2007). It was further noted that accumulation of chlorotryptophan occurred, suggesting that the native tryptophan decarboxylase does not well tolerate substrate derivatives. The group were able to circumnavigate this in a follow up study in which one of the halogenases was engineered to target tryptamine instead of tryptophan (Glenn et al., 2011). This strategy may also greatly simplify separation of halogenated and unhalogenated MIAs, which would not be possible by targeting tryptophan as complete halogenation of this proteogenic amino acid would be toxic.
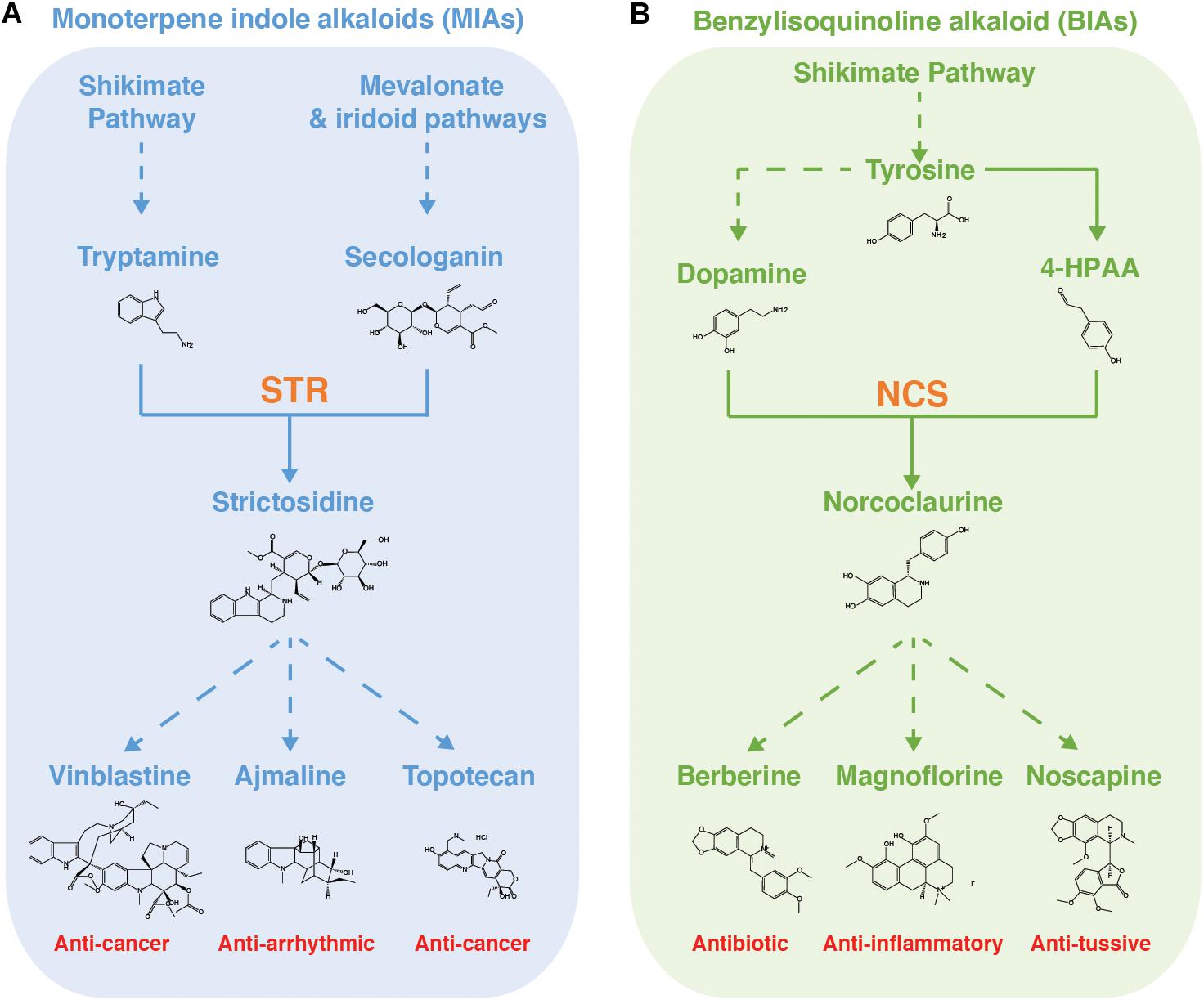
Figure 3. MIA and BIA biosynthetic pathways in yeast. Summaries of the (A) monoterpene indole alkaloid biosynthetic pathway, as refactored in yeast, and (B) benzylisoquinoline alkaloid biosynthetic pathway, with example compounds and, in red, their pharmaceutical applications. STR, strictosidine synthase. NCS, norcoclaurine synthase. Solid arrow denotes single reaction. Dashed arrow denotes multiple reactions.
Other studies have more systematically investigated individual enzymes in vitro. The promiscuity of strictosidine synthase for various analogs of tryptophan and secologanin has been studied (McCoy et al., 2006), and it has been found that substitutions on positions 4 and 7 of the indole ring of tryptamine and smaller substitutions in general are more well tolerated (McCoy et al., 2006). Another study found that strictosidine-β-glucosidase is promiscuous for a variety of indole ring substitutions and postulated that this is because the indole moiety faces outwards from the binding site (Yerkes et al., 2008).
In parallel to these studies, a number of engineering efforts to expand substrate and product promiscuity of MIA enzymes have been undertaken. This is significant because it means that many of individual parts are already defined and ready to be combinatorially tested in microbes. Engineering to improve MIA pathway turnover of substrate analogs was first reported more than 10 years ago with the rational engineering of strictosidine synthase, through introduction of point mutations, to accept analogs of secologanin with methyl ester and vinyl positions (Chen et al., 2006). Shortly following this, a V208A point mutation in the R. serpentina strictosidine synthase homolog was found to improve promiscuity for tryptamines with substitutions at positions 5 and 6 of the indole ring, which the wildtype enzyme does not readily accept (Loris et al., 2007). Following this, two further C. roseus strictosidine synthase homolog mutants (V214M and F232L) were designed to improve promiscuity for tryptamine derivatives, and the authors subsequently observed evidence of previously unobserved brominated MIAs (Bernhardt et al., 2007). Significantly, this study also noted that the expense of purchasing chemically synthesized brominated strictosidine analogs limited the scope of the study. This expense, contrasted with the utility of chlorine and bromine in targeting site-specific cross-coupling reactions and tryptophan’s ubiquity as a synthetic building block, has driven significant interest in the more cost effective semi-synthesis or de novo biosynthesis of halogenated tryptophan using tryptophan halogenases (Smith et al., 2014; Frese and Sewald, 2015; Latham et al., 2018; Fejzagić et al., 2019; Table 1). Both of these approaches are viable for the more efficient production of halogenated variants of MIAs and MIA precursors. For example, a recently identified promiscuous tryptophan synthase is able to combine halogenated indole with serine to yield enantiopure samples of fluorinated, chlorinated, brominated and iodinated tryptophan (Smith et al., 2014). This enzyme could facilitate the introduction of halogens into the MIA pathway by feeding halogenated indole. Due to its lack of chirality, halogenated indole is significantly cheaper to synthesize than halogenated tryptophan and is therefore a far more attractive feedstock. This consideration of different feedstocks is particularly relevant for the introduction of fluorine into alkaloid scaffolds because only one, relatively poor, enzyme has been identified as being able to catalyze fluorination (O’Hagan and Deng, 2015). Alternatively, the introduction of bacterial tryptophan halogenases that target the indole ring of tryptophan can facilitate de novo production of chlorinated and brominated MIAs.
To improve the utility of promiscuous enzymes, studies have implemented rational enzyme engineering, directed evolution and synthetic biology to assess and improve substrate scope (Glenn et al., 2011; Payne et al., 2015; Shepherd et al., 2015; Neubauer et al., 2020), regio-specificity (Lang et al., 2011; Andorfer et al., 2016; Shepherd et al., 2016; Moritzer et al., 2019), stability (Payne et al., 2013; Poor et al., 2014; Minges et al., 2020) and activity (Andorfer et al., 2016, 2017; Kong et al., 2020) of FDH halogenases (Table 3). Of particular interest are a study deploying directed evolution to yield RebH variants targeting positions 5, 6, and 7 of tryptamine (Andorfer et al., 2016), which could help to avoid the tryptophan decarboxylase bottleneck, and a study in which the substrate specificity of RebH was evolved to target, albeit with lower activities, “late-stage” MIAs such as the yohimbines (Payne et al., 2015), which may allow promiscuity requirements to be eschewed entirely. These studies have relied on HPLC as a detection method, often limiting the size of the library that can be screened (Schnepel and Sewald, 2017). The recent development of novel, transcription factor-based biosensors returning a fluorescent output in response to 5- or 6-bromotryptophan (Ellefson et al., 2018) may, in combination with multiplex screening (e.g., fluorescence-activated cell sorting), underpin the screening of larger mutant libraries for optimization of the aforementioned parameters in the intracellular environment of yeast strains.

Table 3. Engineered Tryptophan FDHs.
In addition to the ever-expanding repertoire of halogenase substrate specificities, a recent study elegantly demonstrated that these halogenases can be used in combination with a tryptophanase for biosynthesis of a halogenated indole (Fräbel et al., 2018), which is a common biosynthetic precursor. While engineering Nicotiana benthamiana to produce chlorinated precursors of indican dyes, the authors noted that the three tryptophan halogenases (RebH, SttH, and PyrH) did not accept indole as a substrate. They therefore allowed the halogenases to target tryptophan and expressed an E. coli-derived tryptophanase (TnaA) to convert this into a halogenated indole, which could then be converted, with varying efficiencies, to chloro-indican in planta. As indole is a common biosynthetic precursor, and a privileged heterocyclic scaffold (Dhuguru and Skouta, 2020), this system may help to unlock derivative space surrounding a number of diverse metabolites.
With the recent refactoring of the de novo strictosidine production in yeast (Figure 3A; Brown et al., 2015), it is expected that there will soon exist a microbial platform in which the combinatorial effects of the above-mentioned learnings can be more systematically tested. Such strains will be invaluable tools for further study because understanding of enzyme promiscuity remains patchy for the pathway leading up to strictosidine and extremely sparse for enzymes beyond this branch point. This is principally because it is only very recently that some of these modules have been fully elucidated and enzymes are still being identified (Caputi et al., 2018). The situation is further complicated by the fact that downstream substrates become increasingly complex, making derivatives difficult and expensive to chemically synthesis, leaving enzymatic synthesis as the only alternative (Figure 3A). This creates a catch-22 in which it is difficult to study in vitro enzyme promiscuity without the substrate analogs, yet we cannot effectively produce the substrate analogs without understanding the enzyme promiscuity. If the promiscuity of these downstream modules is to be systematically assessed and engineered in vitro or in vivo, biological platforms producing substrate analogs will have to be developed first. At first glance, source plants may look like attractive targets for these platforms. However, MIA-producing plants often possess multiple competing branches downstream of strictosidine, meaning that a small drop in turnover could cause another branch to outcompete and completely remove flux from the target branch, making it appear that there is no promiscuity. This effect was neatly demonstrated by Runguphan et al. (2010), who observed that substrate halogenation altered the major MIA products in C. roseus. The development of dependable, strictosidine analog-producing yeast strains, into which individual downstream MIA branches can be modularly expressed and systematically studied and engineered, will greatly accelerate our progress toward probing the derivative space surrounding MIA scaffolds.
Overall, the impressive array of pharmaceutical properties and the availability of a well-studied class of halogenases targeting direct precursors has provided the motivation and the means for more extensive new-to-nature integration of halogens into MIAs, relative to other plant medicinal alkaloids. The first production of an unnatural MIA in yeast has also already been reported—Ehrenworth et al. (2015) have reported semi-synthesis of 10-hydroxystrictosidine from yeast-derived 5-hydroxytryptamine and chemically synthesized secologanin. The principle challenge of achieving this was to engineer production of sufficient tetrahydrobiopterin cofactor for the mono-oxygenase activity yielding 5-hydroxytryptophan, which is not naturally produced in S. cerevisiae (Ehrenworth et al., 2015). This highlights the fact that a major challenge when microbially refactoring plant biosynthetic pathways is still often producing sufficient amounts of non-native cofactors or building blocks. A number of other significant challenges also remain, not least the enduringly low yields of microbially refactored plant biosynthetic pathways and the lack of structural information relating to enzymes downstream of strictosidine. It will therefore be some time before halogenated MIAs can be microbially produced at scale. A more attainable goal will be to produce sufficient quantities for bioactivity testing, and this would be a significant step toward unlocking the derivative space surrounding these fascinating compounds.
New-to-Nature Benzylisoquinoline Alkaloids
BIAs are an important class of specialized plant metabolites that include the antimicrobials berberine and sanguinarine as well as the opiate analgesics (Hagel and Facchini, 2013). Several semi-synthetic opiate analogs are widely used and are found on the WHO list of essential medicines (World Health Organisation Model Lists of Essential Medicines, 2019). In higher plants, all known BIAs share a common precursor, (S)-norcoclaurine, which is the product of the first committed step in the pathway. (S)-norcoclaurine is synthesized via the norcoclaurine synthase (NCS)-catalyzed Pictet–Spengler condensation of two tyrosine derivatives (4-hydroxyphenylacetaldehyde (4-HPAA and dopamine) (Figure 3B; Facchini, 2001). As with the MIA pathway, investigations into enzymatic production of BIA derivatives began with in vitro investigation of individual enzyme promiscuity. NCS has been a focus of these studies due to its central position catalyzing the committed step. This enzyme has been found to accept a variety of electron rich, electron deficient and polyfunctionalized analogs of the aldehyde 4-HPAA, including fluorine-containing (Ruff et al., 2012; Nishihachijo et al., 2014) and bromine-containing (Pesnot et al., 2012) derivatives, and ketones (Lichman et al., 2017) but excluding α-substituted aldehydes. This can be rationalized through analysis of the relatively shallow active site in which the R-group is partially solvent-exposed, while the α-carbon is more deeply buried (Ilari et al., 2009; Lichman et al., 2015). However, these studies found the substrate requirements for the dopamine to be stricter, with NCS failing to turn over phenethylamines or tryptamine.
The promiscuity of early-stage BIA pathway enzymes was also recently investigated in one pot, in vitro enzyme cascades converting tyrosine analogs to analogs of (S)-norcoclaurine (Wang et al., 2019b). This study identified a promiscuous tyrosine decarboxylase that accepted fluorinated, chlorinated, brominated and iodinated tyrosine analogs and observed production of six non-natural BIAs, including a fluorinated analog of (S)-norcoclaurine, when this enzyme was included in the cascade. The halogenated tyrosine used in this study was purchased commercially. However, stereoselective enzymatic synthesis of fluorinated tyrosine analogs has been reported from fluoro-phenols, which are non-chiral and significantly cheaper, using tyrosine-phenol lyases (VonTersch et al., 1996; Phillips et al., 1997). Although not the endogenous route for tyrosine biosynthesis in yeast, this opens up a potential for feeding simpler precursors as a way of introducing halogens into the BIA.
More recently, in vivo studies have emerged in the public domain. Pyne et al. (2020) have reported construction of a novel S. cerevisiae strain, containing several tens of modifications principally focusing of increasing precursor supply, capable of producing 4.6 g L–1 of (S)-reticuline. The authors took advantage of the improved yields of this strain to search for minor products indicative of NCS promiscuity, the enzymatic gateway to probing the derivative space surrounding the tetrahydroisoquinoline (THIQ) scaffold within norcoclaurine. By screening the liquid chromatography-mass spectrometry (LC-MS) spectra of supernatants for theoretical THIQ products, the authors identified peaks consistent with THIQs formed by the condensation of dopamine and endogenous yeast carbonyl species derived from L-phenylalanine, L-tryptophan and L-leucine (Pyne et al., 2020). By cultivating strains with single amino acids as the major nitrogen source, the authors were able to increase the yield of these derivatives but also force production of a more diverse suite of aromatic- and aliphatic-derived substituted THIQs. Interestingly, a peak corresponding to the condensation of dopamine and acetaldehyde that was independent of NCS activity was also observed. This is consistent with an earlier study reporting that inorganic phosphate can catalyze aqueous formation of 1-substituted-THIQs from dopamine and aldehydes (Pesnot et al., 2011) and is reminiscent of the pH-dependent chemical coupling of secologanin and serotonin observed by Ehrenworth et al. (2015) in their hydroxystrictosidine production study. Importantly, the chemically catalyzed reactions are not enantiospecific, thus highlighting the need for robust controls when assessing product yields of microbially refactored plant pathways. Equivalent substituted THIQs derived from L-isoleucine and L-valine were not observed due to NCS’s previously reported intolerance toward α-substituted aldehydes. In a promising result for future diversification, Pesnot et al. also demonstrated that the (S)-norcoclaurine methylating enzymes, OMT and CNMT, showed activity toward all substituted THIQs.
BIA pathway engineering efforts have been concomitant with exploration of NCS promiscuity (Figure 3B). For example, screening of NCS variants with active site point mutations yielded two variants, A79I and A79F, with increased turnover of methyl-ketone and cyclohexanone 4-HPAA analogs, respectively, was recently reported by Lichman et al. (2017). This is in keeping with previous work from the same group suggesting that the active site entrance loop is a key determinant of NCS promiscuity toward the aldehyde substrate (Lichman et al., 2015). Furthermore, Li et al. (2018) reported de novo biosynthesis of noscapine, a BIA with anti-tussive and anticancer properties, in S. cerevisiae, achieving a final titer of 2.2 mg/L. The authors note that halogenated noscapine variants have shown improved bioactivity against cancer cell lines (DeBono et al., 2015; Tomar et al., 2016) and hence attempted microbial semi-synthesis noscapine derivatives by supplementing the yeast media with 16 tyrosine analogs, including 3-fluoro-tyrosine, 3-chloro-tyrosine, and 3-iodo-tyrosine (Li et al., 2018). Although no noscapine derivatives were detected, peaks matching the exact masses of 8-fluoro-reticuline, 8-chloro-reticuline and 8-iodo substituted (S)-N-methylcoclaurine were observed. The authors present (i) limited promiscuities of the native enzymes, (ii) low reaction efficiencies, and/or (iii) low substrate abundances as possible explanations for missing downstream derivatives. It has been speculated that these short-comings could be addressed by either engineering tailoring enzymes to introduce “late-stage” derivatizations, or by engineering the promiscuity of BIA enzymes such that they turn over the derivatized substrates (Tomar et al., 2016; Li et al., 2018; Srinivasan and Smolke, 2019). Drawing parallels with the tryptophan halogenases supporting halogenated MIA production, enzymatic halogenation of hydroxyisoquinoline scaffolds is also an option that warrants earnest investigation. For example, direct halogenation of the THIQ scaffold by the fungal halogenase Rdc2 (Zeng et al., 2013) or of THIQ precursors by the Homo sapiens thyroperoxidase (Ruf and Carayon, 2006) are both options for de novo production of halogenated BIAs.
Discussion
Recent advances in the metabolic engineering toolbox have facilitated an explosion in the number of studies reporting microbial synthesis of plant medicinal alkaloids that cannot be chemically synthesized effectively. However, key challenges with both scale up of existing pathway sections and complete refactoring of both MIA and BIA extended pathways remain. Industrially competitive yeast strains able to produce even the naturally occurring plant medicinal alkaloids are still some distance into the future. Strains capable of producing industrial amounts of new-to-nature variants are yet further off. Despite this, these strains already represent an invaluable resource for the further engineering of these pathways for production of alkaloid derivatives. As described in this review, many relevant parts have already been developed, and we envision that the experimental advantages of microbial chasses will facilitate combinatorial testing of these parts to optimize derivative turnover as well as complete pathway refactoring.
Another prominent challenge will now be to progress from elegant proof-of-principles of microbial synthesis of alkaloid derivatives to more systematic explorations of the derivative space surrounding MIAs and BIAs and expanding these capabilities to include diversification of other natural product scaffolds. General approaches for achieving this will include (i) feeding of substrate analogs, (ii) pathway modification, and (iii) addition of enzymes, as recently reviewed by Cravens et al. (2019) For these non-mutually exclusive approaches to successfully produce sufficient yields for bioactivity testing of new drug leads, a number of challenges will have to be overcome. Chief among these, improving our ability to reliably engineer enzyme specificity and promiscuity would exponentially speed progress toward improved analog turnover. New computational methods designed to support protein design, such as the online webtool FuncLib (Khersonsky et al., 2018), may also aid this process. However, these approaches will also require new high-resolution structures to be published in many cases. Conversely, structure-agnostic methodologies, such as directed evolution, have already been highly successful at engineering enzyme specificities, but are limited by our ability to screen the variants (Payne et al., 2015; Andorfer et al., 2016). New biosensors targeting analogs of alkaloid precursors or intermediates may address this and help to improve analog turnover at pathway bottlenecks (Ellefson et al., 2018). Additionally, the large datasets generated by truly high throughput technologies open the door to the application of big data techniques such as machine learning, which has recently been used to successfully guide “semi-rational” protein engineering (Saito et al., 2018; Yang et al., 2019). Bioprospecting for promiscuous homologs may also prove fruitful. In the case of MIAs, the identification of two naturally occurring chlorinated alkaloids suggests that evolution may have already provided halo-tolerant enzymes and that screening alkaloid-producing plants native to halogen-rich habitats (e.g., coastal and volcanic soils) may save a significant amount of protein engineering (Al-Khdhairawi et al., 2017; Zeng et al., 2017).
Beyond improving the catalytic activity of enzymes directly catalyzing in alkaloid production, several systems level challenges will also have to be overcome. Substrate toxicity will likely be a major hurdle on the way to producing some variants in yeast. For example, it has been shown that some tryptophan analogs interfere with tryptophan synthesis in yeast and inhibit growth (Miozzari et al., 1977), and may also be erroneously incorporated into proteins. These issues could be overcome by reconsidering the halogen entry point into the pathway (Glenn et al., 2011) or engineering tryptophan tRNA synthetases to reject halogenated tryptophan. Alternatively, inspiration could be taken from a trans-acting aminoacyl-tRNA deacylase that selectively decouples fluoro-threonine from tRNA to prevent its integration into protein in Streptomyces cattleya (McMurry and Chang, 2017). Halogen salts, which are required for enzymatic halogenation, can also inhibit yeast growth. However, a number of non-conventional, halotolerant yeast strains are currently being investigated as chassis for biotechnological applications (Zaky et al., 2014; Musa et al., 2018). Although further characterization and toolbox development is still required, these strains could allow higher salt concentrations to be utilized. Functionality of the heterologously expressed plant enzymes at high salinity also remains an open question. However, we envision that both systems-level optimizations of existing chassis and further characterization of halo-tolerant yeasts could provide routes to improve the absolute yield of halogenated alkaloids, and the yield relative to the non-halogenated product, thus simplifying extraction.
Even as our understanding of how to engineer the promiscuity of these systems improves, the small number of successful studies on this topic have demonstrated the significant effort that is required to successfully produce new-to-nature variants. In addition to this, it is often difficult or expensive to source chemically synthesized intermediate analogs (Bernhardt et al., 2007) and this becomes exponentially more difficult downstream as the compounds become more complex. It will therefore be important to focus initial efforts on platform yeast strains that can produce usable quantities of multiple variants of branch point compounds, such as strictosidine in the MIA pathway and (S)-norcoclaurine in the BIA pathway. Downstream modules can then be expressed in a plug-and-play style that will allow them to be efficiently studied and engineered. Although a daunting task, several studies have reported microbial synthesis of unnatural strictosidine and (S)-norcoclaurine analogs (Chen et al., 2006; Bernhardt et al., 2007; Pesnot et al., 2012; Ehrenworth et al., 2015; Wang et al., 2019b). It should be noted that development of many of the parts required for this has already begun and these should be integrated into novel yeast strains with relative ease (Smith et al., 2014; Payne et al., 2015; Andorfer et al., 2016; McDonald et al., 2019; Moritzer et al., 2019). Thus, we argue that combinatorial effects of these optimizations are now ripe to be studied, hopefully yielding novel platform strains that produce sufficient amounts of specific derivatives to unlock new regions of chemical space.
In summary, although still relatively few, the growing number of studies reporting yeast-based microbial synthesis of plant medicinal alkaloids are providing experimentally amenable contexts in which these pathways can be further engineered. This has major implications for furthering our understanding of new-to-nature variants of natural products but will also allow the combinatorial effects of the many previous learnings from in vitro and in planta studies to be rapidly assessed. Although significant challenges still exist, if these challenges can be overcome, it is possible to envisage a new semi-synthetic approach to medicinal chemistry campaigns, specialized for diversifying natural product scaffolds. Such a system would involve microbial synthesis of medicinal alkaloid variants with halogen substitutions at specific points of the scaffold. These compounds can then be tested directly for pharmaceutical activities but also can be subjected to targeted, chemical cross-coupling reactions that can efficiently generate an almost infinite number of substituted variants. Thus, this system would combine natural mechanisms of generating diversity, i.e., using a small suite of organic building blocks combined through a large number of reactions, with synthetic mechanisms of generating diversity, i.e., using a wide range of building blocks combined with a small suite of reliable reactions (Ganesan, 2008). As these two approaches access very different regions of chemical space (Figure 1D), we foresee that this would create unprecedented opportunities for entirely new and promising regions of chemical space, possessing advantages of both the natural and synthetic spheres, to be systematically probed for pharmaceutical activities.
Author Contributions
SB, JZ, and MJ conceived the scope of the review content and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Novo Nordisk Foundation, Novo Nordisk Foundation Bioscience Ph.D. Programme grant No. NNF19SA0035438, and the European Commission Horizon 2020 programme (MIAMi; No. 722287).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ajikumar, P. K., Xiao, W. H., Tyo, K. E. J., Wang, Y., Simeon, F., Leonard, E., et al. (2010). Isoprenoid pathway optimization for Taxol precursor overproduction in Escherichia coli. Science 330, 70–74. doi: 10.1126/science.1191652
Al-Khdhairawi, A. A. Q., Krishnan, P., Mai, C. W., Chung, F. F. L., Leong, C. O., Yong, K. T., et al. (2017). A bis-benzopyrroloisoquinoline alkaloid incorporating a cyclobutane core and a chlorophenanthroindolizidine alkaloid with cytotoxic activity from Ficus fistulosa var. tengerensis. J. Nat. Prod. 80, 2734–2740. doi: 10.1021/acs.jnatprod.7b00500
Andorfer, M. C., Belsare, K. D., Girlich, A. M., and Lewis, J. C. (2017). Aromatic halogenation by using bifunctional flavin reductase–halogenase fusion enzymes. ChemBioChem 18, 2099–2103. doi: 10.1002/cbic.201700391
Andorfer, M. C., Park, H. J., Vergara-Coll, J., and Lewis, J. C. (2016). Directed evolution of RebH for catalyst-controlled halogenation of indole C-H bonds. Chem. Sci. 7, 3720–3729. doi: 10.1039/c5sc04680g
Avalos, L., Fink, G. R., Stephanopoulos, G., Avalos, J. L., Fink, G. R., and Stephanopoulos, G. (2013). Compartmentalization of metabolic pathways in yeast mitochondria improves the production of branched-chain alcohols. Nat. Biotechnol. 31, 335–341. doi: 10.1038/nbt.2509
Barker, A., Kettle, J. G., Nowak, T., and Pease, J. E. (2013). Expanding medicinal chemistry space. Drug Discov. Today 18, 298–304. doi: 10.1016/j.drudis.2012.10.008
Becker, J. V. W., Armstrong, G. O., Van Der Merwe, M. J., Lambrechts, M. G., Vivier, M. A., and Pretorius, I. S. (2003). Metabolic engineering of Saccharomyces cerevisiae for the synthesis of the wine-related antioxidant resveratrol. FEMS Yeast Res. 4, 79–85. doi: 10.1016/S1567-1356(03)00157-0
Bernhardt, P., McCoy, E., and O’Connor, S. E. (2007). Rapid identification of enzyme variants for reengineered alkaloid biosynthesis in periwinkle. Chem. Biol. 14, 888–897. doi: 10.1016/j.chembiol.2007.07.008
Brown, S., Clastre, M., Courdavault, V., and O’Connor, S. E. (2015). De novo production of the plant-derived alkaloid strictosidine in yeast. Proc. Natl. Acad. Sci. U. S. A. 112, 3205–3210. doi: 10.1073/pnas.1423555112
Buyel, J. F., Twyman, R. M., and Fischer, R. (2015). Extraction and downstream processing of plant-derived recombinant proteins. Biotechnol. Adv. 33, 902–913. doi: 10.1016/j.biotechadv.2015.04.010
Caputi, L., Franke, J., Farrow, S. C., Chung, K., Payne, R. M. E., Nguyen, T. D., et al. (2018). Missing enzymes in the biosynthesis of the anticancer drug vinblastine in Madagascar periwinkle. Science 360, 1235–1239. doi: 10.1126/science.aat4100
Chang, F. Y., and Brady, S. F. (2013). Discovery of indolotryptoline antiproliferative agents by homology-guided metagenomic screening. Proc. Natl. Acad. Sci. U. S. A. 110, 2478–2483. doi: 10.1073/pnas.1218073110
Chang, M. C. Y., Eachus, R. A., Trieu, W., Ro, D. K., and Keasling, J. D. (2007). Engineering Escherichia coli for production of functionalized terpenoids using plant P450s. Nat. Chem. Biol. 3, 274–277. doi: 10.1038/nchembio875
Chen, R., Yang, S., Zhang, L., and Zhou, Y. J. (2020). Advanced strategies for production of natural products in yeast. iScience 23:100879. doi: 10.1016/j.isci.2020.100879
Chen, S., Galan, M. C., Coltharp, C., and O’Connor, S. E. (2006). Redesign of a central enzyme in alkaloid biosynthesis. Chem. Biol. 13, 1137–1141. doi: 10.1016/j.chembiol.2006.10.009
Chung, W. J., and Vanderwal, C. D. (2016). Stereoselective halogenation in natural product synthesis. Angew. Chemie Int. Ed. 55, 4396–4434. doi: 10.1002/anie.201506388
Corr, M. J., Sharma, S. V., Pubill-Ulldemolins, C., Bown, R. T., Poirot, P., Smith, D. R. M., et al. (2017). Sonogashira diversification of unprotected halotryptophans, halotryptophan containing tripeptides; and generation of a new to nature bromo-natural product and its diversification in water. Chem. Sci. 8, 2039–2046. doi: 10.1039/c6sc04423a
Courdavault, V., O’Connor, S. E., Oudin, A., Besseau, S., and Papon, N. (2020). Towards the microbial production of plant-derived anticancer drugs. Trends Cancer 6, 444–448. doi: 10.1016/j.trecan.2020.02.004
Cravens, A., Payne, J., and Smolke, C. D. (2019). Synthetic biology strategies for microbial biosynthesis of plant natural products. Nat. Commun. 10, 1–12. doi: 10.1038/s41467-019-09848-w
Dandapani, S., and Marcaurelle, L. A. (2010). Grand challenge commentary: accessing new chemical space for “undruggable” targets. Nat. Chem. Biol. 6, 861–863. doi: 10.1038/nchembio.479
De Luca, V., Salim, V., Thamm, A., Masada, S. A., and Yu, F. (2014). Making iridoids/secoiridoids and monoterpenoid indole alkaloids: progress on pathway elucidation. Curr. Opin. Plant Biol. 19, 35–42. doi: 10.1016/j.pbi.2014.03.006
DeBono, A., Capuano, B., and Scammells, P. J. (2015). Progress toward the development of noscapine and derivatives as anticancer agents. J. Med. Chem. 58, 5699–5727. doi: 10.1021/jm501180v
Dhuguru, J., and Skouta, R. (2020). Role of indole scaffolds as pharmacophores in the development of anti-lung cancer agents. Molecules 25:1615. doi: 10.3390/molecules25071615
Domergue, J., Erdmann, D., Fossey-Jouenne, A., Petit, J. L., Debard, A., de Berardinis, V., et al. (2019). XszenFHal, a novel tryptophan 5-halogenase from Xenorhabdus szentirmaii. AMB Express 9:175. doi: 10.1186/s13568-019-0898-y
Dong, C., Flecks, S., Unversucht, S., Haupt, C., Van Pée, K. H., and Naismith, J. H. (2005). Structural biology: tryptophan 7-halogenase (PrnA) structure suggests a mechanism for regioselective chlorination. Science 309, 2216–2219. doi: 10.1126/science.1116510
Ehrenworth, A. M., Sarria, S., and Peralta-Yahya, P. (2015). Pterin-dependent mono-oxidation for the microbial synthesis of a modified monoterpene indole alkaloid. ACS Synth. Biol. 4, 1295–1307. doi: 10.1021/acssynbio.5b00025
Ellefson, J. W., Ledbetter, M. P., and Ellington, A. D. (2018). Directed evolution of a synthetic phylogeny of programmable Trp repressors article. Nat. Chem. Biol. 14, 361–367. doi: 10.1038/s41589-018-0006-7
Facchini, P. J. (2001). Alkaloid biosynthesis in plants: biochemistry, cell biology, molecular regulation, and metabolic engineering applications. Annu. Rev. Plant Physiol. Plant Mol. Biol. 52, 1–26.
Fejzagić, A. V., Gebauer, J., Huwa, N., and Classen, T. (2019). Halogenating enzymes for active agent synthesis: first steps are done and many have to follow. Molecules 24:4008. doi: 10.3390/molecules24214008
Flecks, S., Patallo, E. P., Zhu, X., Ernyei, A. J., Seifert, G., Schneider, A., et al. (2008). New insights into the mechanism of enzymatic chlorination of tryptophan. Angew. Chemie Int. Ed. 47, 9533–9536. doi: 10.1002/anie.200802466
Fossati, E., Narcross, L., Ekins, A., Falgueyret, J.-P., and Martin, V. J. J. (2015). Synthesis of morphinan alkaloids in Saccharomyces cerevisiae. PLoS One 10:e0124459. doi: 10.1371/journal.pone.0124459
Foulston, L. C., and Bibb, M. J. (2010). Microbisporicin gene cluster reveals unusual features of lantibiotic biosynthesis in actinomycetes. Proc. Natl. Acad. Sci. U. S. A. 107, 13461–13466. doi: 10.1073/pnas.1008285107
Fräbel, S., Krischke, M., Staniek, A., and Warzecha, H. (2016). Recombinant flavin-dependent halogenases are functional in tobacco chloroplasts without co-expression of flavin reductase genes. Biotechnol. J. 11, 1586–1594. doi: 10.1002/biot.201600337
Fräbel, S., Wagner, B., Krischke, M., Schmidts, V., Thiele, C. M., Staniek, A., et al. (2018). Engineering of new-to-nature halogenated indigo precursors in plants. Metab. Eng. 46, 20–27. doi: 10.1016/j.ymben.2018.02.003
Frese, M., Schnepel, C., Minges, H., Voß, H., Feiner, R., and Sewald, N. (2016). Modular combination of enzymatic halogenation of tryptophan with suzuki-miyaura cross-coupling reactions. ChemCatChem 8, 1799–1803. doi: 10.1002/cctc.201600317
Frese, M., and Sewald, N. (2015). Enzymatic halogenation of tryptophan on a gram scale. Angew. Chemie Int. Ed. 54, 298–301. doi: 10.1002/anie.201408561
Fujimori, D. G., Hrvatin, S., Neumann, C. S., Strieker, M., Marahiel, M. A., and Walsh, C. T. (2007). Cloning and characterization of the biosynthetic gene cluster for kutznerides. Proc. Natl. Acad Sci. U. S. A. 104, 16498–16503.
Galanie, S., and Smolke, C. D. (2015). Optimization of yeast-based production of medicinal protoberberine alkaloids. Microb. Cell Fact. 14:144. doi: 10.1186/s12934-015-0332-3
Galanie, S., Thodey, K., Trenchard, I. J., Interrante, M. F., and Smolke, C. D. (2015). Complete biosynthesis of opioids in yeast. Science 349, 1095–1100. doi: 10.1126/science.aac9373
Ganesan, A. (2008). The impact of natural products upon modern drug discovery. Curr. Opin. Chem. Biol. 12, 306–317. doi: 10.1016/j.cbpa.2008.03.016
Gautam, L. N., Ling, T., Lang, W., and Rivas, F. (2016). Anti-proliferative evaluation of monoterpene derivatives against leukemia. Eur. J. Med. Chem. 113, 75–80. doi: 10.1016/j.ejmech.2016.02.034
Glenn, W. S., Nims, E., and O’Connor, S. E. (2011). Reengineering a tryptophan halogenase to preferentially chlorinate a direct alkaloid precursor. J. Am. Chem. Soc. 133, 19346–19349. doi: 10.1021/ja2089348
Gribble, G. W. (2003). The diversity of naturally produced organohalogens. Chemosphere 56, 289–297. doi: 10.1016/S0045-6535(03)00207-8
Gribble, G. W. (2018). Newly discovered naturally occurring organohalogens. Arkivoc 2018, 372–410. doi: 10.24820/ark.5550190.p010.610
Groot, H. J., Lubberts, S., de Wit, R., Witjes, J. A., Martijn Kerst, J., de Jong, I. J., et al. (2018). Journal of clinical oncology risk of solid cancer after treatment of testicular germ cell cancer in the platinum era. J. Clin. Oncol. 36, 319–325. doi: 10.1200/JCO
Hagel, J. M., and Facchini, P. J. (2013). Benzylisoquinoline alkaloid metabolism: a century of discovery and a brave new world. Plant Cell Physiol. 54, 647–672. doi: 10.1093/pcp/pct020
Heemstra, J. R., and Walsh, C. T. (2008). Tandem action of the O2- and FADH2-dependent halogenases KtzQ and KtzR produce 6,7-dichlorotryptophan for kutzneride assembly. J. Am. Chem. Soc. 130, 14024–14025. doi: 10.1021/ja806467a
Hori, K., Okano, S., and Sato, F. (2016). Efficient microbial production of stylopine using a Pichia pastoris expression system. Sci. Rep. 6:22201. doi: 10.1038/srep22201
Ignea, C., Pontini, M., Motawia, M. S., Maffei, M. E., Makris, A. M., and Kampranis, S. C. (2018). Synthesis of 11-carbon terpenoids in yeast using protein and metabolic engineering. Nat. Chem. Biol. 14, 1090–1098. doi: 10.1038/s41589-018-0166-5
Ilari, A., Franceschini, S., Bonamore, A., Arenghi, F., Botta, B., Macone, A., et al. (2009). Structural basis of enzymatic (S)-norcoclaurine biosynthesis. J. Biol. Chem. 284, 897–904. doi: 10.1074/jbc.M803738200
Ismail, M., Frese, M., Patschkowski, T., Ortseifen, V., Niehaus, K., and Sewald, N. (2019). Flavin-dependent halogenases from Xanthomonas campestris pv. campestris B100 Prefer bromination over chlorination. Adv. Synth. Catal. 361, 2475–2486. doi: 10.1002/adsc.201801591
Jakubczyk, D., Caputi, L., Hatsch, A., Nielsen, C. A. F., Diefenbacher, M., Klein, J., et al. (2015). Discovery and reconstitution of the cycloclavine biosynthetic pathway - enzymatic formation of a cyclopropyl group. Angew. Chemie Int. Ed. 54, 5117–5121. doi: 10.1002/anie.201410002
Jungbauer, A. (2013). Continuous downstream processing of biopharmaceuticals. Trends Biotechnol. 31, 479–492. doi: 10.1016/j.tibtech.2013.05.011
Junker, B. H. (2004). Scale-up methodologies for Escherichia coli and yeast fermentation processes. J. Biosci. Bioeng. 97, 347–364. doi: 10.1016/S1389-1723(04)70218-2
Karabencheva-Christova, T. G., Torras, J., Mulholland, A. J., Lodola, A., and Christov, C. Z. (2017). Mechanistic insights into the reaction of chlorination of tryptophan catalyzed by tryptophan 7-halogenase. Sci. Rep. 7:17395. doi: 10.1038/s41598-017-17789-x
Keller, S., Wage, T., Hohaus, K., Hölzer, M., Eichhorn, E., and Van Pée, K. H. (2000). Purification and partial characterization of tryptophan 7- halogenase (PrnA) from Pseudomonas fluorescens. Angew. Chemie Int. Ed. 39, 2300–2302.
Khersonsky, O., Lipsh, R., Avizemer, Z., Ashani, Y., Goldsmith, M., Leader, H., et al. (2018). Automated design of efficient and functionally diverse enzyme repertoires. Mol. Cell 72, 178.e5–186.e5. doi: 10.1016/j.molcel.2018.08.033
Kong, L., Wang, Q., Deng, Z., and You, D. (2020). Flavin adenine dinucleotide-dependent halogenase xanh and engineering of multifunctional fusion halogenases. Appl. Environ. Microbiol. 86:e01225-20. doi: 10.1128/AEM.01225-0
Kutchan, T. M., Gershenzon, J., Moller, B. L., and Gang, D. R. (2015). “Natural products,” in Biochemistry and Molecular Biology Of Plants, eds B. Buchanan, W. Gruissem, and R. Jones (Hoboken, NY: Blackwell Publishing Ltd).
Lang, A., Polnick, S., Nicke, T., William, P., Patallo, E. P., Naismith, J. H., et al. (2011). Changing the regioselectivity of the tryptophan 7-halogenase PrnA by site-directed mutagenesis. Angew. Chemie Int. Ed. 50, 2951–2953. doi: 10.1002/anie.201007896
Latham, J., Brandenburger, E., Shepherd, S. A., Menon, B. R. K., and Micklefield, J. (2018). Development of halogenase enzymes for use in synthesis. Chem. Rev. 118, 232–269. doi: 10.1021/acs.chemrev.7b00032
Lee, J., Kim, J., Kim, H., Kim, E. J., Jeong, H. J., Choi, K. Y., et al. (2020). Characterization of a tryptophan 6-halogenase from Streptomyces albus and its regioselectivity determinants. ChemBioChem 21, 1446–1452. doi: 10.1002/cbic.201900723
Leggans, E. K., Duncan, K. K., Barker, T. J., Schleicher, K. D., and Boger, D. L. (2013). A remarkable series of vinblastine analogues displaying enhanced activity and an unprecedented tubulin binding steric tolerance: c20’ urea derivatives. J. Med. Chem. 56, 628–639. doi: 10.1021/jm3015684
Li, Y., Li, S., Thodey, K., Trenchard, I., Cravens, A., and Smolke, C. D. (2018). Complete biosynthesis of noscapine and halogenated alkaloids in yeast. Proc. Natl. Acad. Sci. U. S. A. 115, E3922–E3931. doi: 10.1073/pnas.1721469115
Lichman, B. R., Gershater, M. C., Lamming, E. D., Pesnot, T., Sula, A., Keep, N. H., et al. (2015). Dopamine-first mechanism enables the rational engineering of the norcoclaurine synthase aldehyde activity profile. FEBS J. 282, 1137–1151. doi: 10.1111/febs.13208
Lichman, B. R., Zhao, J., Hailes, H. C., and Ward, J. M. (2017). Enzyme catalysed Pictet-Spengler formation of chiral 1,1’-disubstituted- A nd spiro-tetrahydroisoquinolines. Nat. Commun. 8:14883. doi: 10.1038/ncomms14883
Lingkon, K., and Bellizzi, J. J. (2020). Structure and activity of the thermophilic tryptophan-6 halogenase BorH. ChemBioChem 21, 1121–1128. doi: 10.1002/cbic.201900667
Liu, D., Ding, L., Sun, J., Boussetta, N., and Vorobiev, E. (2016). Yeast cell disruption strategies for recovery of intracellular bio-active compounds — A review. Innov. Food Sci. Emerg. Technol. 36, 181–192. doi: 10.1016/j.ifset.2016.06.017
Liu, X., Cheng, J., Zhang, G., Ding, W., Duan, L., Yang, J., et al. (2018). Engineering yeast for the production of breviscapine by genomic analysis and synthetic biology approaches. Nat. Commun. 9:448. doi: 10.1038/s41467-018-02883-z
Loris, E. A., Panjikar, S., Ruppert, M., Barleben, L., Unger, M., Schübel, H., et al. (2007). Structure-based engineering of strictosidine synthase: auxiliary for alkaloid libraries. Chem. Biol. 14, 979–985. doi: 10.1016/j.chembiol.2007.08.009
Luhavaya, H., Sigrist, R., Chekan, J. R., McKinnie, S. M. K., and Moore, B. S. (2019). Biosynthesis of l-4-chlorokynurenine, an antidepressant prodrug and a non-proteinogenic amino acid found in lipopeptide antibiotics. Angew. Chemie Int. Ed. 58, 8394–8399. doi: 10.1002/anie.201901571
Luo, X., Reiter, M. A., D’Espaux, L., Wong, J., Denby, C. M., Lechner, A., et al. (2019). Complete biosynthesis of cannabinoids and their unnatural analogues in yeast. Nature 567, 123–126. doi: 10.1038/s41586-019-0978-9
Ma, S. M., Li, J. W. H., Choi, J. W., Zhou, H., Lee, K. K. M., Moorthie, V. A., et al. (2009). Complete reconstitution of a highly reducing iterative polyketide synthase. Science 326, 589–592. doi: 10.1126/science.1175602
McCoy, E., Galan, M. C., and O’Connor, S. E. (2006). Substrate specificity of strictosidine synthase. Bioorganic Med. Chem. Lett. 16, 2475–2478. doi: 10.1016/j.bmcl.2006.01.098
Mccoy, E., and O’connor, S. E. (2006). Directed biosynthesis of alkaloid analogs in the medicinal plant Catharanthus roseus. J. Am. Chem. Soc. 128, 14276–14277. doi: 10.1021/ja066787w
McDonald, A. D., Perkins, L. J., and Buller, A. R. (2019). Facile in vitro biocatalytic production of diverse tryptamines. ChemBioChem 20, 1939–1944. doi: 10.1002/cbic.201900069
McKeague, M., Wang, Y. H., Cravens, A., Win, M. N., and Smolke, C. D. (2016). Engineering a microbial platform for de novo biosynthesis of diverse methylxanthines. Metab. Eng. 38, 191–203. doi: 10.1016/j.ymben.2016.08.003
McMurry, J. L., and Chang, M. C. Y. (2017). Fluorothreonyl-tRNA deacylase prevents mistranslation in the organofluorine producer Streptomyces cattleya. Proc. Natl. Acad. Sci. U. S. A. 114, 11920–11925. doi: 10.1073/pnas.1711482114
Menon, B. R. K., Latham, J., Dunstan, M. S., Brandenburger, E., Klemstein, U., Leys, D., et al. (2016). Structure and biocatalytic scope of thermophilic flavin-dependent halogenase and flavin reductase enzymes. Org. Biomol. Chem. 14, 9354–9361. doi: 10.1039/c6ob01861k
Milbredt, D., Patallo, E. P., and Van Pée, K. H. (2014). A tryptophan 6-halogenase and an amidotransferase are involved in thienodolin biosynthesis. ChemBioChem 15, 1011–1020. doi: 10.1002/cbic.201400016
Milne, N., Thomsen, P., Mølgaard Knudsen, N., Rubaszka, P., Kristensen, M., and Borodina, I. (2020). Metabolic engineering of Saccharomyces cerevisiae for the de novo production of psilocybin and related tryptamine derivatives. Metab. Eng. 60, 25–36. doi: 10.1016/j.ymben.2019.12.007
Minges, H., Schnepel, C., Böttcher, D., Weiß, M. S., Sproß, J., Bornscheuer, U. T., et al. (2020). Targeted enzyme engineering unveiled unexpected patterns of halogenase stabilization. ChemCatChem 12, 818–831. doi: 10.1002/cctc.201901827
Miozzari, G., Niederberger, P., and Hütter, R. (1977). Action of tryptophan analogues in Saccharomyces cerevisiae. Arch. Microbiol. 115, 307–316. doi: 10.1007/BF00446457
Moritzer, A. C., Minges, H., Prior, T., Frese, M., Sewald, N., and Niemann, H. H. (2019). Structure-based switch of regioselectivity in the flavin-dependent tryptophan 6-halogenase Thal. J. Biol. Chem. 294, 2529–2542. doi: 10.1074/jbc.RA118.005393
Musa, H., Kasim, F. H., Nagoor Gunny, A. A., and Gopinath, S. C. B. (2018). Salt-adapted moulds and yeasts: potentials in industrial and environmental biotechnology. Process Biochem. 69, 33–44. doi: 10.1016/j.procbio.2018.03.026
Neubauer, P. R., Pienkny, S., Wessjohann, L., Brandt, W., and Sewald, N. (2020). Predicting the substrate scope of the flavin-dependent halogenase BrvH. ChemBioChem doi: 10.1002/cbic.202000444 Online ahead of print.
Neubauer, P. R., Widmann, C., Wibberg, D., Schröder, L., Frese, M., Kottke, T., et al. (2018). A flavin-dependent halogenase from metagenomic analysis prefers bromination over chlorination. PLoS One 13:e0196797. doi: 10.1371/journal.pone.0196797
Newman, D. J., and Cragg, G. M. (2016). Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 79, 629–661. doi: 10.1021/acs.jnatprod.5b01055
Nielsen, C. A. F., Folly, C., Hatsch, A., Molt, A., Schröder, H., O’Connor, S. E., et al. (2014). The important ergot alkaloid intermediate chanoclavine-I produced in the yeast Saccharomyces cerevisiae by the combined action of EasC and EasE from aspergillus japonicus. Microb. Cell Fact. 13:95. doi: 10.1186/s12934-014-0095-2
Nishihachijo, M., Hirai, Y., Kawano, S., Nishiyama, A., Minami, H., Katayama, T., et al. (2014). Asymmetric synthesis of tetrahydroisoquinolines by enzymatic Pictet-Spengler reaction. Biosci. Biotechnol. Biochem. 78, 701–707. doi: 10.1080/09168451.2014.890039
O’Hagan, D., and Deng, H. (2015). Enzymatic fluorination and biotechnological developments of the fluorinase. Chem. Rev. 115, 634–649. doi: 10.1021/cr500209t
Paddon, C. J., and Keasling, J. D. (2014). Semi-synthetic artemisinin: a model for the use of synthetic biology in pharmaceutical development. Nat. Rev. Microbiol. 12, 355–367. doi: 10.1038/nrmicro3240
Payne, J. T., Andorfer, M. C., and Lewis, J. C. (2013). Regioselective arene halogenation using the FAD-dependent halogenase RebH. Angew. Chemie Int. Ed. 52, 5271–5274. doi: 10.1002/anie.201300762
Payne, J. T., Poor, C. B., and Lewis, J. C. (2015). Directed evolution of rebh for site-selective halogenation of large biologically active molecules. Angew. Chemie Int. Ed. 54, 4226–4230. doi: 10.1002/anie.201411901
Pesnot, T., Gershater, M. C., Ward, J. M., and Hailes, H. C. (2011). Phosphate mediated biomimetic synthesis of tetrahydroisoquinoline alkaloids. Chem. Commun. 47, 3242–3244. doi: 10.1039/c0cc05282e
Pesnot, T., Gershater, M. C., Ward, J. M., and Hailes, H. C. (2012). The catalytic potential of Coptis japonica NCS2 revealed - Development and utilisation of a fluorescamine-based assay ETI. Adv. Synth. Catal. 354, 2997–3008. doi: 10.1002/adsc.201200641
Phillips, R. S., Von Tersch, R. L., and Secundo, F. (1997). Effects of tyrosine ring fluorination on rates and equilibria of formation of intermediates in the reactions of carbon-carbon lyases. Eur. J. Biochem. 244, 658–663. doi: 10.1111/j.1432-1033.1997.00658.x
Pichersky, E., and Raguso, R. A. (2018). Why do plants produce so many terpenoid compounds? New Phytol. 220, 692–702. doi: 10.1111/nph.14178
Ping, Y., Li, X., You, W., Li, G., Yang, M., Wei, W., et al. (2019). De Novo production of the plant-derived tropine and pseudotropine in yeast. ACS Synth. Biol. 8, 1257–1262. doi: 10.1021/acssynbio.9b00152
Poor, C. B., Andorfer, M. C., and Lewis, J. C. (2014). Improving the stability and catalyst lifetime of the halogenase RebH by directed evolution. ChemBioChem 15, 1286–1289. doi: 10.1002/cbic.201300780
Pyne, M. E., Kevvai, K., Grewal, P. S., Narcross, L., Choi, B., Bourgeois, L., et al. (2020). A yeast platform for high-level synthesis of tetrahydroisoquinoline alkaloids. Nat. Commun. 11:3337. doi: 10.1038/s41467-020-17172-x
Qu, Y., Easson, M. L. A. E., Froese, J., Simionescu, R., Hudlicky, T., and DeLuca, V. (2015). Completion of the seven-step pathway from tabersonine to the anticancer drug precursor vindoline and its assembly in yeast. Proc. Natl. Acad. Sci. U. S. A. 112, 6224–6229. doi: 10.1073/pnas.1501821112
Ro, D. K., Paradise, E. M., Quellet, M., Fisher, K. J., Newman, K. L., Ndungu, J. M., et al. (2006). Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature 440, 940–943. doi: 10.1038/nature04640
Ruf, J., and Carayon, P. (2006). Structural and functional aspects of thyroid peroxidase. Arch. Biochem. Biophys. 445, 269–277. doi: 10.1016/j.abb.2005.06.023
Ruff, B. M., Bräse, S., and O’Connor, S. E. (2012). Biocatalytic production of tetrahydroisoquinolines. Tetrahedron Lett. 53, 1071–1074. doi: 10.1016/j.tetlet.2011.12.089
Runguphan, W., Maresh, J. J., and O’Connor, S. E. (2009). Silencing of tryptamine biosynthesis for production of nonnatural alkaloids in plant culture. Proc. Natl. Acad. Sci. U. S. A. 106, 13673–13678. doi: 10.1073/pnas.0903393106
Runguphan, W., and O’Connor, S. E. (2009). Metabolic reprogramming of periwinkle plant culture. Nat. Chem. Biol. 5, 151–153. doi: 10.1038/nchembio.141
Runguphan, W., and O’Connor, S. E. (2013). Diversification of monoterpene indole alkaloid analogs through cross-coupling. Org. Lett. 15, 2850–2853. doi: 10.1021/ol401179k
Runguphan, W., Qu, X., and O’Connor, S. E. (2010). Integrating carbon-halogen bond formation into medicinal plant metabolism. Nature 468, 461–467. doi: 10.1038/nature09524
Saito, Y., Oikawa, M., Nakazawa, H., Niide, T., Kameda, T., Tsuda, K., et al. (2018). Machine-learning-guided mutagenesis for directed evolution of fluorescent proteins. ACS Synth. Biol. 7, 2014–2022. doi: 10.1021/acssynbio.8b00155
Schnepel, C., and Sewald, N. (2017). Enzymatic halogenation: a timely strategy for regioselective C-H activation. Chem. A Eur. J. 23, 12064–12086. doi: 10.1002/chem.201701209
Sears, J. E., and Boger, D. L. (2015). Total synthesis of vinblastine, related natural products, and key analogues and development of inspired methodology suitable for the systematic study of their structure-function properties. Acc. Chem. Res. 48, 653–662. doi: 10.1021/ar500400w
Seibold, C., Schnerr, H., Rumpf, J., Kunzendorf, A., Hatscher, C., Wage, T., et al. (2006). A flavin-dependent tryptophan 6-halogenase and its use in modification of pyrrolnitrin biosynthesis. Biocatal. Biotransformation 24, 401–408. doi: 10.1080/10242420601033738
Shepherd, S. A., Karthikeyan, C., Latham, J., Struck, A. W., Thompson, M. L., Menon, B. R. K., et al. (2015). Extending the biocatalytic scope of regiocomplementary flavin-dependent halogenase enzymes. Chem. Sci. 6, 3454–3460. doi: 10.1039/c5sc00913h
Shepherd, S. A., Menon, B. R. K., Fisk, H., Struck, A. W., Levy, C., Leys, D., et al. (2016). a structure-guided switch in the regioselectivity of a tryptophan halogenase. ChemBioChem 17, 821–824. doi: 10.1002/cbic.201600051
Smith, D. R. M. M., Willemse, T., Gkotsi, D. S., Schepens, W., Maes, B. U. W. W., Ballet, S., et al. (2014). The first one-pot synthesis of L-7-iodotryptophan from 7-iodoindole and serine, and an improved synthesis of other L-7-halotryptophans. Org. Lett. 16, 2622–2625. doi: 10.1021/ol5007746
Springob, K., and Kutchan, T. M. (2009). “Introduction to the different classes of natural products,” in Plant-Derived Natural Products: Synthesis, Function, and Application, eds A. E. Osbourn and V. Lanzotti (New York, NY: Springer), 3–50. doi: 10.1007/978-0-387-85498-4_1
Srinivasan, P., and Smolke, C. D. (2019). Engineering a microbial biosynthesis platform for de novo production of tropane alkaloids. Nat. Commun. 10, 1–15. doi: 10.1038/s41467-019-11588-w
Tomar, V., Kukreti, S., Prakash, S., Madan, J., and Chandra, R. (2016). Noscapine and its analogs as chemotherapeutic agent: current updates. Curr. Top. Med. Chem. 17, 174–188. doi: 10.2174/1568026616666160530153518
VonTersch, R. L., Secundo, F., Phillips, R. S., and Newton, M. G. (1996). Preparation of fluorinated amino acids with tyrosine phenol lyase. Biomed. Front. Fluor. Chem. 95–104. doi: 10.1021/bk-1996-0639.ch007
Wang, Y., Ling, C., Chen, Y., Jiang, X., and Chen, G. Q. (2019a). Microbial engineering for easy downstream processing. Biotechnol. Adv. 37:107365. doi: 10.1016/j.biotechadv.2019.03.004
Wang, Y., Tappertzhofen, N., Méndez-Sánchez, D., Bawn, M., Lyu, B., Ward, J. M., et al. (2019b). Design and use of de novo cascades for the biosynthesis of new benzylisoquinoline alkaloids. Angew. Chemie 131, 10226–10231. doi: 10.1002/ange.201902761
Wilson, S. A., and Roberts, S. C. (2014). Metabolic engineering approaches for production of biochemicals in food and medicinal plants. Curr. Opin. Biotechnol. 26, 174–182. doi: 10.1016/j.copbio.2014.01.006
World Health Organisation Model Lists of Essential Medicines (2019). World Health Organisation Model Lists of Essential Medicines. Geneva: World Health Organisation. Available online at: https://apps.who.int/iris/bitstream/handle/10665/325771/WHO-MVP-EMP-IAU-2019.06-eng.pdf?ua=1
Wurtzel, E. T., and Kutchan, T. M. (2016). Plant metabolism, the diverse chemistry set of the future. Science 353, 1232–1236. doi: 10.1126/science.aad2062
Xu, Z., Yang, Z., Liu, Y., Lu, Y., Chen, K., and Zhu, W. (2014). Halogen bond: its role beyond drug–target binding affinity for drug discovery and development. J. Chem. Inf. Model. 54, 69–78. doi: 10.1021/ci400539q
Yang, K. K., Wu, Z., and Arnold, F. H. (2019). Machine-learning-guided directed evolution for protein engineering. Nat. Methods 16, 687–694. doi: 10.1038/s41592-019-0496-6
Yeh, E., Blasiak, L. C., Koglin, A., Drennan, C. L., and Walsh, C. T. (2007). Chlorination by a long-lived intermediate in the mechanism of flavin-dependent halogenases. Biochemistry 46, 1284–1292. doi: 10.1021/bi0621213
Yeh, E., Garneau, S., and Walsh, C. T. (2005). Robust in vitro activity of RebF and RebH, a two-component reductase/halogenase, generating 7-chlorotryptophan during rebeccamycin biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 102, 3960–3965. doi: 10.1073/pnas.0500755102
Yerkes, N., Wu, J. X., McCoy, E., Galan, M. C., Chen, S., and O’Connor, S. E. (2008). Substrate specificity and diastereoselectivity of strictosidine glucosidase, a key enzyme in monoterpene indole alkaloid biosynthesis. Bioorganic Med. Chem. Lett. 18, 3095–3098. doi: 10.1016/j.bmcl.2007.11.063
Zaky, A. S., Tucker, G. A., Daw, Z. Y., and Du, C. (2014). Marine yeast isolation and industrial application. FEMS Yeast Res. 14, 813–825. doi: 10.1111/1567-1364.12158
Zehner, S., Kotzsch, A., Bister, B., Süssmuth, R. D., Méndez, C., Salas, J. A., et al. (2005). A regioselective tryptophan 5-halogenase is involved in pyrroindomycin biosynthesis in Streptomyces rugosporus LL-42D005. Chem. Biol. 12, 445–452. doi: 10.1016/j.chembiol.2005.02.005
Zeng, J., Lytle, A. K., Gage, D., Johnson, S. J., and Zhan, J. (2013). Specific chlorination of isoquinolines by a fungal flavin-dependent halogenase. Bioorganic Med. Chem. Lett. 23, 1001–1003. doi: 10.1016/j.bmcl.2012.12.038
Zeng, J., and Zhan, J. (2011). Characterization of a tryptophan 6-halogenase from Streptomyces toxytricini. Biotechnol. Lett. 33, 1607–1613. doi: 10.1007/s10529-011-0595-7
Zeng, J., Zhang, D. B., Zhou, P. P., Zhang, Q. L., Zhao, L., Chen, J. J., et al. (2017). Rauvomines A and B. Two monoterpenoid indole alkaloids from Rauvolfia vomitoria. Org. Lett. 19, 3998–4001. doi: 10.1021/acs.orglett.7b01723
Zhou, F., and Pichersky, E. (2020). More is better: the diversity of terpene metabolism in plants. Curr. Opin. Plant Biol. 55, 1–10. doi: 10.1016/j.pbi.2020.01.005
Zhou, Y. J., Buijs, N. A., Zhu, Z., Gómez, D. O., Boonsombuti, A., Siewers, V., et al. (2016). Harnessing yeast peroxisomes for biosynthesis of fatty-acid-derived biofuels and chemicals with relieved side-pathway competition. J. Am. Chem. Soc. 138, 15368–15377. doi: 10.1021/jacs.6b07394
Keywords: alkaloids, yeast, halogenation, plants, new-to-nature
Citation: Bradley SA, Zhang J and Jensen MK (2020) Deploying Microbial Synthesis for Halogenating and Diversifying Medicinal Alkaloid Scaffolds. Front. Bioeng. Biotechnol. 8:594126. doi: 10.3389/fbioe.2020.594126
Received: 12 August 2020; Accepted: 02 October 2020;
Published: 23 October 2020.
Edited by:
Farshad Darvishi, Alzahra University, IranCopyright © 2020 Bradley, Zhang and Jensen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael K. Jensen, bWlqZUBiaW9zdXN0YWluLmR0dS5kaw==