Abstract
MiRNAs are small non-coding RNAs that interact with their target mRNAs for posttranscriptional gene regulation. Finely controlled miRNA biogenesis, target recognition and degradation indicate that maintaining miRNA homeostasis is essential for regulating cell proliferation, growth, differentiation and apoptosis. Increasingly, miRNAs have been recognized as a potential biomarker for disease diagnosis. MiRNAs can be found in blood, plasma, and tissues, and miRNA expression and activity differ in developmental stages, tissues and in response to external stimuli. MiRNA transcripts are matured from pri-miRNA over pre-miRNA to mature miRNA, a process that includes multiple steps and enzymes. Many tools are available to identify and quantify specific miRNAs, ranging from measuring total miRNA, specific miRNA activity, miRNA arrays and miRNA localization. The various miRNA assays differ in accuracy, cost, efficiency and convenience of monitoring miRNA dynamics. To acknowledge the significance and increasing research interest in miRNAs, we summarize the traditional as well as novel methods of miRNA quantification with strengths and limitations of various techniques in biochemical and medical research.
Introduction
Single-stranded microRNAs (miRNAs) are 21–24 nucleotides (nts) in length and are generated from non-coding RNA precursor transcripts. Mature miRNAs regulate gene expression by silencing messenger RNAs (mRNAs), or inducing mRNA cleavage and degradation (Bartel, 2004, 2009; Cai et al., 2009; Chekulaeva and Filipowicz, 2009). MiRNAs bind to partially complementary sequences of target mRNAs, thereby controlling mRNA stability and initiating translational repression of the target genes (Bartel, 2004; Valinezhad Orang et al., 2014). It became clear that miRNAs control almost every pathway and direct cellular proliferation, differentiation, morphogenesis, and apoptosis. Consequently, they are essential to maintain or determine cellular fate (Carthew, 2006; Cai et al., 2009). MiRNAs are encoded in introns or non-coding RNAs and are transcribed as primary transcripts (pri-miRNA) in the nucleus. Here, pri-miRNAs are cleaved into hairpin shaped precursor-miRNA (pre-miRNA) by the ribonuclease Drosha and the microprocessor complex subunit DGCR8. The resulting pre-miRNAs are exported into the cytoplasm via Exportin-5 and further processed by the endonuclease Dicer and other RNA binding proteins to produce mature miRNAs. Guided by Argonaute (AGO) proteins, mature miRNAs are loaded onto the RNA-induced silencing complex (RISC), to finally bind to the 3′-untranslated region (UTR) of target mRNA to suppress translation (Gebert and MacRae, 2019; Michlewski and Cáceres, 2019). Since the initial discovery of two miRNAs, lin-4 and let-7, over 38,000 miRNAs have been annotated in the miRNA reference repository miRbase [www.miRBase.org (Kozomara et al., 2019)].
While many miRNAs have been predicted computationally, development of miRNA reporters and experimental tools has only recently gained traction. To define the role of miRNAs in the cell, biogenesis, functionality and activity of the miRNA need to be experimentally determined, as well as their interaction with predicted targeting sites (Jin et al., 2013). As the consequence of gene regulation through miRNAs and transcription factors (TFs) are quite similar, identification of their cooperative actions has been researched in embryonic development, muscular development and macrophages differentiation. Yet, the individual contribution of mRNA regulation by canonical mRNA expression and degradation pathways vs. miRNA mediated silencing prove difficult to discern (Arora et al., 2013). There are many compelling reasons for monitoring and quantifying of miRNAs in tissues, cell cultures, and transgenic organisms. A conservative estimate is that more than 60% of mammalian genes are controlled by miRNAs, yet most of the target sites remain unexplored (Friedman et al., 2009). Many of these miRNA imperfectly base pair at the 3′-untranslated region (UTR) of their target mRNA, making predictions of miRNA targets even more difficult. It is, however, clear, that miRNAs negatively regulate protein output, and the dysregulated expression of miRNAs correlates with development of many diseases such as cardiovascular disease, retinal disorders, neurodegenerative diseases, diabetes and cancer (Lv et al., 2008; Gommans and Berezikov, 2012; Xin et al., 2012). Additionally, miRNAs may be cell-specific biomarkers for some human cancer as well as diagnostic and prognostic markers of old or novel therapeutic responses (Bartels and Tsongalis, 2009). Conserved miRNAs and changes in their expression can be used as a predictor for disease outcomes, sex chromosome evolution (Naqvi et al., 2018), T-cell development and neural cell specification (Smirnova et al., 2005; Neilson et al., 2007). MiRNAs are used as biomarkers in disease detection and treatment: changes in miRNAs concentrations in serum are utilized to study the effect of radiation treatments (Fendler et al., 2017), and the expression profile of certain miRNAs allows for a classification of cancer stage (Lu et al., 2005). Finally, miRNA levels are indicative of host cell inflammatory responses against certain viruses (O'Connell et al., 2007).
To investigate above mentioned miRNA concentrations and activity in cells, various miRNA assays have been developed to measure the level of miRNA expression, stability and abundance in cells, tissues and organs (Pritchard et al., 2012). It is challenging to detect and quantify miRNAs because of their short nt sequences, different forms of miRNA precursor transcripts, high copy numbers, variations in unique expression, and the difficulty in designing highly specific oligonucleotides for probe-based assays (Chen et al., 2011). Recent advances in the field have led to the development of a number of miRNA profiling methods. In this review we will discuss the different types of miRNA reporters and the approaches to measure miRNA activity with their advantages and disadvantages to give an overview of the current miRNA toolbox.
Quantification of Total MiRNA
While it has been shown that not all miRNAs in a cell are utilized for mRNA silencing at a given time, it has generally been assumed that the overall amount of miRNA – as regulated by expression, processing, and degradation - correlates with its biological availability to silence mRNA translation. Thus, many methods rely on the quantification of the overall miRNA amount. In general, we distinguish between hybridization, amplification, sequencing, and enzyme-based methods.
Hybridization Based Methods
Northern Blot
The most established method of miRNA detection is northern blotting, which is now used less frequently owing to the advancement of real-time PCR and microarrays. Nonetheless, Northern Blots are still considered the gold standard, as they detect mature and pre-miRNA without amplification bias and can simultaneously determine the size of the miRNA, including size changes caused by post-transcriptionally added nucleotides and accurate expression levels (Lagos-Quintana et al., 2001; Lee et al., 2002; Schmittgen et al., 2004). The general outline of miRNA northern blotting includes initial polyacrylamide gel electrophoresis of extracted RNAs to isolate fractionated small RNAs, followed by the transfer of the RNAs onto a nylon membrane. After crosslinking, radioactive, fluorescently or digoxygenin labeled probes are hybridized to the target miRNA (Lau et al., 2001; Li and Ruan, 2009). To improve the sensitivity for miRNA detection, chemically changed locked nucleic acid (LNA) induced oligonucleotide probes were developed and led to 10-times higher detection efficiency compared to traditional DNA probes, as well as increasing the thermal stability of probes (Várallyay et al., 2008). LNAs are RNA or DNA sequences with modified nucleotides that contain a methylene bridge in the ribose ring structure which is resistant to nucleases mediated degradation (Frieden et al., 2003; Válóczi et al., 2004; Várallyay et al., 2007). Using water-soluble 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide instead of UV radiation for RNA crosslinking to the membrane greatly reduces the movement of RNAs which is more suitable to complementary binding with nucleic acid probes and elevates the membranes' retention ability to bind larger amount of small RNAs (Pall et al., 2007; Pall and Hamilton, 2008). Finally, the combination of non-radioactive digoxigenin-labeled LNA probes and carbodiimide mediated cross-linking of RNAs enables high-resolution northern blotting as a distinctly useful tool for determining length variation of endogenous miRNA and its precursors, and showed higher reproducibility when comparing the blotting signals with deep-sequencing results (Kim et al., 2010; Koscianska et al., 2011). To overcome the problem of multiplexed blots of target RNAs, both radioactive and non-radioactive probes triggered hybridization chain reaction (HCR) can be operated independently for multiplex banding of target miRNA (Schwarzkopf and Pierce, 2016).
Strengths and Limitations
Northern blotting is a versatile tool to detect precursors and mature miRNAs with comparatively cheap and non-specialized equipment, but is nonetheless time consuming, and throughput and sensitivity is relatively low (Pritchard et al., 2012).
Microarrays
Microarrays are a universal analysis tool for profiling miRNA expression and are widely used to compare expression profiles. For example, the link between specific miRNAs to cancer cell proliferation and tumorigenesis has been investigated using arrays (Wang Y. W. et al., 2018; Wang Z. et al., 2018). Microarrays can assess the expression patterns of hundreds of miRNA genes in a single assay. They usually are solid surface slides with hundreds of probes spotted in a grid, each specific to a single miRNA gene. Generally, miRNA arrays encompass several major steps (Figure 1): purification of mature miRNA, reverse transcription and simultaneous labeling of the cDNA, and hybridization of the cDNA to high-affinity probes on a glass plate. Depending on the labeling method (biotin, radioactivity, fluorophores) the hybridized arrays are scanned for signals, allowing for quantification and normalization of the data to a control experiment (Zhao et al., 2006; Yin et al., 2008). Microarrays for miRNAs were utilized by Krichevsky et al., where a nylon membrane oligonucleotide array was incubated with radiolabeled cDNA to identify regulatory miRNAs in the mammalian brain (Krichevsky et al., 2003). Nowadays, radioactive labeling is largely avoided and oligonucleotide microchip based arrays on glass slides are coupled with 5′ biotin labeled cDNA probes, allowing for a robust miRnome analysis (Liu et al., 2004). Direct labeling of RNA through various types of fluorescence probes was successfully used to interrogate the role of non-coding miRNA in microarrays (Kampa et al., 2004; Liang et al., 2005). Importantly, miRNA array analysis must include post-data analysis steps, such as normalization and statistical analysis, and often requires data verification with alternate methods, such as reverse transcription quantitative PCR (RT-qPCR). Based on miRNA databases such as miRbase (Kozomara et al., 2019), several companies designed miRNA microarray platforms with specific probes, fluorescent dyes and hybridization steps (Callari et al., 2012), making microarrays a readily available tool and allow for the high throughput quantification of miRNAs. Several studies compared the available platform and analyzed inter-platform reproducibility, finding a poor overlap in miRNA determined to differentially expressed, but a general overlap was found in miRNAs considered to be enriched (Callari et al., 2012; Chatterjee et al., 2015; Franco et al., 2016). Therefore, the experimental approach (pooled biological samples vs. true triplicates, RNA extraction methods, etc.) and microarray platform need to be carefully chosen, and enrichment of miRNAs should be verified by an alternate method, such as RT-qPCR.
Figure 1
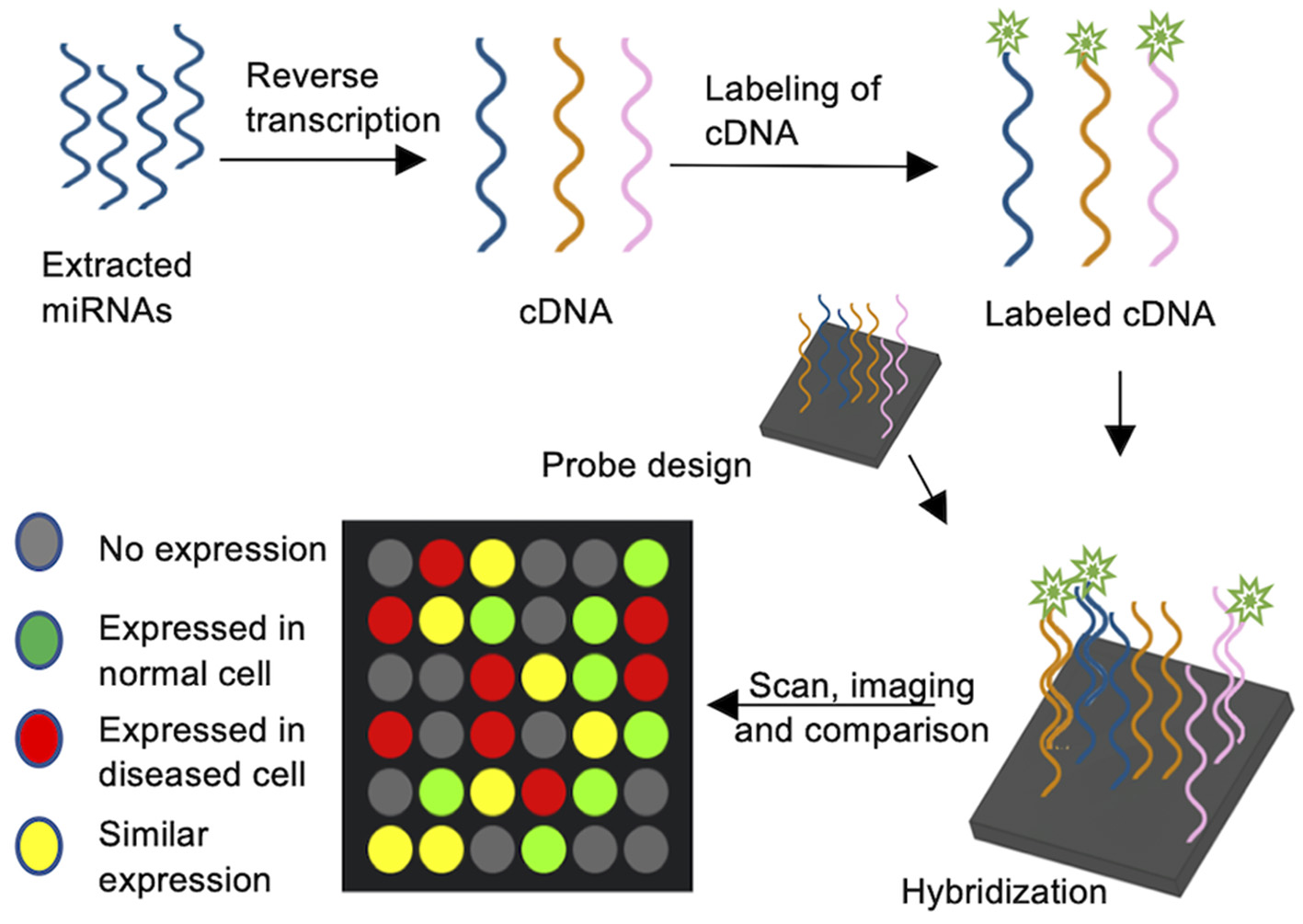
MiRNA Microarrays. cDNA is synthesized by reverse transcription of extracted miRNAs and the cDNAs are subsequently labeled with e.g., a fluorophore tag. Oligonucleotide probes complementary to target miRNAs are spotted on a carrier plate and the fluorescently labeled cDNA is hybridized. The signal intensity corresponds to miRNA abundance.
Strengths and Limitations
Microarrays can compare levels of miRNA expression in different organs, tissues and species, and are a major, high throughput tool to analyze hundreds of miRNAs simultaneously. Sensitivity of microarrays is relatively low, mostly due to the amount of miRNA applied to the array. Furthermore, specificity can be compromised in miRNAs with high sequence similarity due to the nature of miRNAs short hybridization sequences and low copy number. Finally, while many miRNAs can be analyzed simultaneously, microarrays do not allow for the discovery of new miRNAs and the cost is comparatively high.
Bead-Array Based Profiling
MiRNAs often only differ by a single nucleotide, posing significant challenges due to cross-hybridization of closely related miRNAs in microarrays. This led to the development of bead-based miRNA profiling. Bead-based profiling requires an initial adapter ligation step to the RNA 3′ and 5′ end. Oligonucleotide linkers are ligated at the 3′ and 5′ termini of miRNAs to maintain uniform lengths, avoiding amplification bias of to heterogeneously elongated miRNAs. A biotin tag is introduced during reverse transcription (Miska et al., 2004). Oligonucleotide probes complementary to the miRNAs are ligated to polystyrene beads. miRNAs are sequence specifically labeled with different dyes. The biotinylated cDNAs hybridize to the capture probes, unhybridized probes are washed away, and subsequently analyzed by flow cytometry to denote miRNA identity and abundance as determined by bead color and intensity (Lu et al., 2005).
Strengths and Limitations
Bead-based flow cytometry arrays are lower in cost compared to microarrays, involve fewer processing steps and are medium throughput, but require specialized equipment for flow cytometry.
Amplification Based Methods
RT-qPCR
To study miRNA and its functions, PCR- based methods generate quantitative data with efficiency, specificity and sensitivity, quickly revealing RNA expression variance between samples (Salone and Rederstorff, 2015). PCR based amplification of miRNA poses some challenges in specificity due to the short transcript lengths. Initially, conventional qPCR with a linear primer followed by poly (A) tailing was used to detect and quantify miRNA but was promptly replaced with RT-qPCR (Chen et al., 2005). A stem-loop RT-based assay (Figure 2) is the most commonly applied method for mature miRNA detection and can discriminate between miRNA species varying in as little as a single nucleotide. Here, the target miRNA is reverse transcribed by annealing a stem-loop primer to the 3′ end of the miRNA for reverse transcription, which is then used as template to amplify the miRNA with a specific forward primer and a universal reverse primer or TaqMan probe (Figure 2A) (Raymond et al., 2005; Hurley et al., 2012; Salone and Rederstorff, 2015). This specific preamplification is used by several RT-qPCR platforms, namely TaqMan array cards or open array cards (Liu et al., 2010). Stem-loop primer based RT-qPCR can be utilized to analyze the expression of one or a few miRNAs using gene specific primers, or be applied in high throughput analysis by utilizing universal stem-loop primers (Yang et al., 2014). The now widely used stem-loop method significantly enhanced specificity and reproducibility (Luo et al., 2012; Mohammadi-Yeganeh et al., 2013).
Figure 2
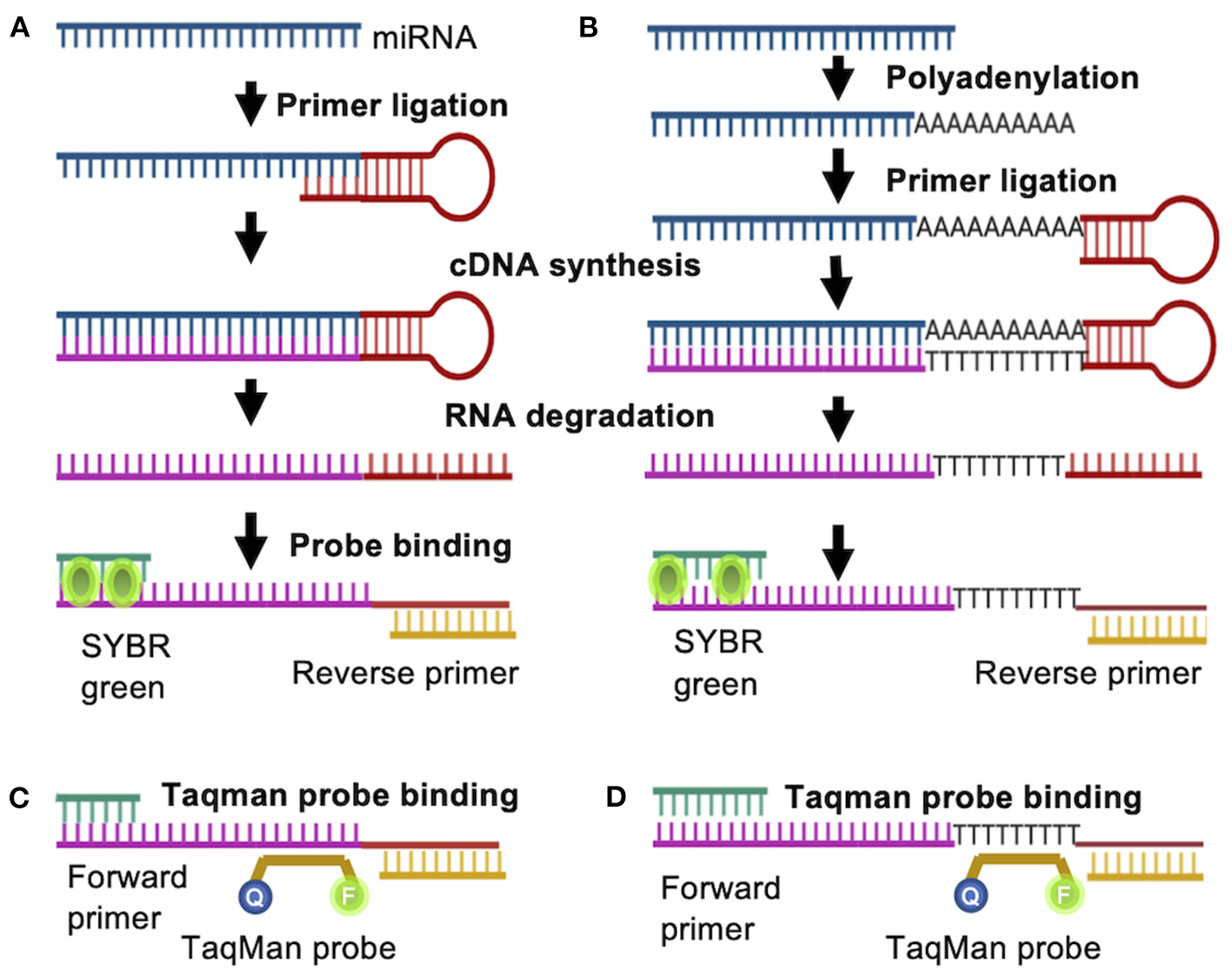
RT-PCR based quantification of miRNA. A Stem-loop RT primer binds to the target miRNA either (A) directly or (B) indirectly after addition of adenine nucleotides to the 3′- end, allowing for reverse transcription. The generated cDNA is amplified by PCR with a miRNA specific forward primer and a universal reverse primer. Instead of SYBR green, a gene specific (C) or universal (D) Taqman probe can be used.
SYBR green is commonly applied to visualize the formation of double stranded DNA and is often used in RT-qPCR applications. In solution, SYBR green exhibits low fluorescence, but once it binds to double stranded DNA, fluorescence is enhanced, making it an excellent reporter for double stranded DNA concentrations (Figures 2A,B). However, SYBR green dye can be somewhat inaccurate due to double stranded DNA contamination or primer dimers (Bustin, 2000). Alternatively, gene specific Taqman probes can be utilized to detect amplification rates. In this case, the probe is complementary to a DNA strand and is labeled with a fluorophore and a quencher. During amplification, the probe is displaced from the DNA strand, resulting in cleavage of the fluorophore and an increase in the fluorescence signal (Figures 2C,D). A modification of the stem loop primer (Figures 2A,C) approach includes an additional polyadenylation step prior to ligation of the stem loop primer (Figures 2B,D) (Luo et al., 2012; Mohammadi-Yeganeh et al., 2013). This allows for the utilization of universal poly(dT) probe, reducing reagent costs by more than 10-fold when a Taqman probe is utilized (Figure 2D) (Goldrick et al., 2013). This poly(T) containing primer mediated RT-qPCR method was successfully optimized for the analysis of circulating miRNAs as biomarkers for disease (Niu et al., 2015, 2019). RT-qPCR is a reliable, reproducible and highly quantitative method to detect even low levels of RNA and has been applied on the single cell level (Kang et al., 2012).
Strengths and Limitations
Depending on the target miRNAs, RT-qPCR is considered a low to medium throughput method, suitable for the targeted quantification of miRNAs. Variation in primer design, and inconsistent data analysis and normalization can negatively affect the reproducibility of RT-qPCR. Nonetheless, due to its high sensitivity and specificity, RT-qPCR is the current gold standard method to verify data obtained by microarrays or next generation sequencing approaches.
Amplification Assays
Different nucleic acid sequence-based amplification strategies, for example rolling circle amplification (RCA), hybridization chain reaction (HCR), strand displacement amplification (SDA), loop-mediated isothermal amplification (LAMP) and exponential isothermal amplification assay (EXPAR) have been utilized to analyze miRNAs (Xu et al., 2019). These strategies are constantly improved and modified to increase their analytical potential and we will only briefly introduce these methods, as they have recently been reviewed in detail (Reid et al., 2018). For example, a palindromic padlock probe was employed in RCA to detect let-7a in HeLa cells (Xu et al., 2019), or the application of an additional primer resulted in a branched RCA, that quantifies let-7 variants in single cells with high sensitivity (Cheng et al., 2009). EXPAR is a highly sensitive amplification method that detects miRNA at a constant temperature. The amplification template has two repeated flanking sequences at the 3′ and 5′ terminus, which are complementary to the desired miRNA and separated by a recognition site for nicking enzyme. Target miRNAs are hybridized with the complementary sequences to the template to add nucleotides using a DNA polymerase. Extended sequences containing a nicking endonuclease recognition site are cut by a nicking enzyme to release a sequence which triggers another amplification and is subsequently detected by SYBR green (Jia et al., 2010). SDA and modifications of the initiator primers in LAMP also lead to exponential amplification of the target miRNAs, generating fluorescence allowing quantification of miRNAs. In HCR, two hairpin DNA sequences carry out a series of multiple chain reactions using linear amplification initiated by the complementary binding of miRNA with a template (Dirks and Pierce, 2004).
Strengths and Limitations
Exponential amplification is gaining popularity due to the simplicity of procedures without the need of complex equipment, but these methods have proven less accurate in the identification of weakly expressed miRNA (Moody et al., 2017).
Next-Generation Sequencing
Next-generation sequencing (NGS) is not only able to profile known miRNAs expression but also suitable to identify various unknown miRNAs variants, which are not recuperated by the conventional targeted methods such as RT-qPCR and microarrays (Willenbrock et al., 2009; Liu et al., 2011). Applications of NGS in miRNA sequencing have evolved rapidly over the past few years. The basic procedure of miRNA sequencing is similar to DNA sequencing, with additional steps to account for RNA library generation. An initial step usually requires the enrichment of small RNAs, which are ligated to 3′ and 5′ adaptors simultaneously. Total small cDNA is generated by reverse transcription followed by amplification and subsequent sequencing (Bar et al., 2008; Hu et al., 2017). Next-generation RNA sequencing generates millions of reads and after raw data processing the expression of thousands of miRNAs can be quantified in one experiment, making NGS a powerful tool in identifying new and known miRNAs and quantifying miRNA expression levels (Metpally et al., 2013). The depth of NGS miRNA sequencing furthermore enabled the identification of new extracellular small RNAs from the blood and cerebrospinal fluid (Burgos et al., 2013), miRNAs associated with fibrosis progression in hepatitis C patients (Van Keuren-Jensen et al., 2016), circulating miRNAs as biomarkers of cervical cancers (Lv et al., 2021), and hippocampal miRNAs associated with memory formation (Tamming et al., 2020), demonstrating the power of NGS in discovery-driven research.
While acquisition of NGS data requires careful sample preparation and specialized machinery, it also requires computational infrastructure for data analysis. Bioinformatics tools are frequently optimized to analyze sequences and data is stored and analyzed in miRNA databases, including sequence reading platforms- Illumina (Hammond et al., 2012), Roche 454 (Johansen et al., 2011), SOLiD (Li et al., 2011), and Ion Torrent (Merriman et al., 2012). Alignment software includes SeqMap (Jiang and Wong, 2008), TopHat (Trapnell et al., 2009), Bowtie (Langmead et al., 2009), MAQ (Li et al., 2008), and megablast (Zhang et al., 2000). Processed sequences are analyzed by several web servers and programs notably including miRExpress, miRanda, miRDeep, DIANA microT, and PicTar (Motameny et al., 2010). While miRNA sequencing will surely gain even more attention as methods are being developed for single cell miRNA sequencing (Faridani et al., 2016), NGS of miRNAs already identified potential biomarkers for laryngeal cancer (Huang et al., 2018). Dysregulated expression of miRNA in inflammation and immune response (Zhang et al., 2019) and differentially expressed miRNAs in kidney disease act as new molecular targets, and NGS data is consistent to validated results of real-time PCR (Liu et al., 2019). NGS sequencing allows for massively parallel analysis and can capture the entire miRnome in a single experiment. NGS has resulted in the identification of previously unknown miRNAs and significantly expanded our knowledge on new miRNAs, that are now annotated in Database repositories [reviewed in Vlachos and Hatzigeorgiou, 2013; Aghaee-Bakhtiari, 2018].
Strengths and Limitations
Although NGS is more cost efficient these days, NGS remains expensive and requires extensive computational analysis, which may limit its applications. Nonetheless, the unbiased data acquisition, sequence coverage and depths of NGS is unparalleled by any other available method, and the only discovery-based approach allowing for the identification of novel miRNAs.
Enzyme Based Assays
Invader Assay
The Invader assay is structure-specific enzymatic reaction followed by fluorescence resonance energy transfer (FRET) mediated identification, and offers rapid and specific quantification of miRNA (Allawi et al., 2004). An oligonucleotide probe and an overlapping invasive oligonucleotide are annealed to the target miRNA, and subsequently recognized by an endonuclease that causes the release of 5′ oligonucleotide flaps (Figure 3). Each short flap initiates a second cleavage reaction on multiple matching FRET cassettes containing a fluorescence dye and a quencher. FRET cassettes generate thousands of fluorescence signals within a short period of time. A so-called arrester oligonucleotide is added to hybridize with un-cleaved probes, allowing for a quantification of miRNAs based on the 5′ flaps (Allawi et al., 2004; Olivier, 2005). Mass spectrometry or fluorescence polarization probes can be used for identifying cleaved oligonucleotides.
Figure 3
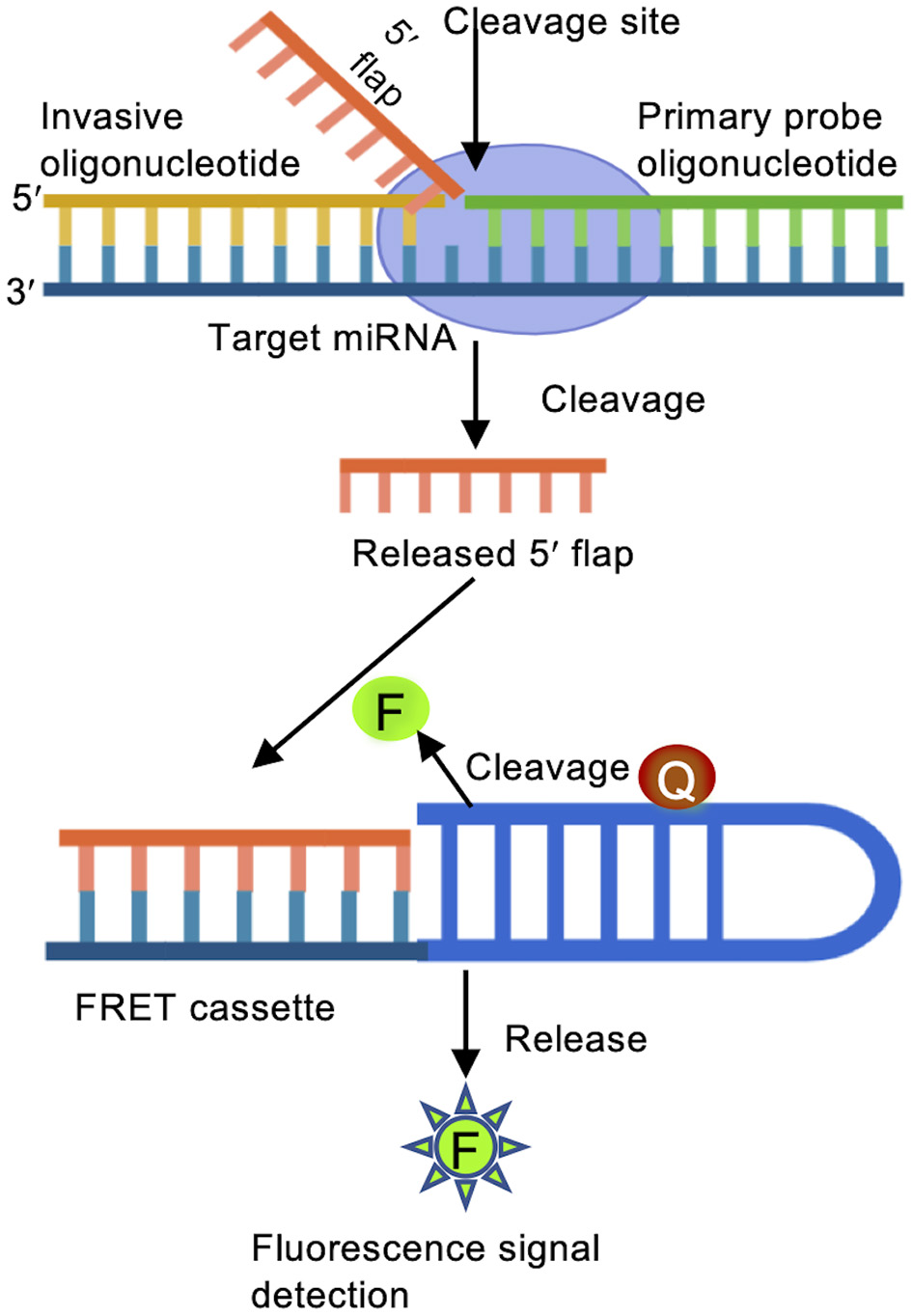
Invader assay. Binding of a primary probe and an over lapping invader oligonucleotide to the target miRNA initiates a cleavage reaction carried out by structure-specific 5′ flap nuclease to release the 5′ flap of the primary oligonucleotide probe. The free 5′ flap acts as an invader oligonucleotide for a second reaction to bind to a FRET cassette containing fluorescent (F) and quencher (Q) molecules. When the 2nd structure is recognized, the FRET cassette is cleaved, emitting detectable fluorescence signals.
Strengths and Limitations
The invader miRNA assay can identify single nucleotide variance as well as mature and pre-miRNA in parallel in multiple samples and provide medium-throughput results with generalized equipment (Olivier, 2005).
Quantification of Active MiRNA: MiRNA Activity Reporters
While total miRNA quantification is thought to provide an approximation of active miRNA, recent studies show that miRNAs can be silenced by mono nucleotide addition, or bound to proteins, making them unavailable for post-transcriptional suppression of mRNAs (Chung et al., 2017). These changes in miRNA sequence or availability are not captured in amplification, sequencing or hybridization-based assays. MiRNA activity reporters provide a more accurate snapshot of the amount of active miRNA, in some cases without lysis of cells, allowing for a time resolved or in-patient assessment of miRNA activity. Fluorescence or bioluminescence based optical imaging, magnetic resonance imaging (MRI) and positron emission tomography (PET) provide more or less long-time live cell monitoring of miRNA activity (Oh and Do Won Hwang, 2013).
Luciferase and GFP Based MiRNA Reporters
MiRNA Transcription and Target Site Reporters
The application of luciferase assays in miRNA research is 2-fold. On the one hand, miRNA expression can be investigated by fusing miRNA promoter to the reporter system. On the other hand, miRNA targets can be validated using the same system, by fusing the 3′UTR of a target gene to the reporter. In luciferase based reporter assays, the enzyme luciferase converts the substrate D-luciferin into oxyluciferin, thereby emitting light (Figure 4) (Haugwitz et al., 2008; Oh and Do Won Hwang, 2013). These reporters are used to monitor miRNA transcription and identify miRNA binding sites. Firefly (Fluc), Renilla (Rluc), and Gaussia luciferase (Gluc) are the most commonly used bioluminescent reporters due to their high sensitivity (Tannous et al., 2005; Haugwitz et al., 2008).
Figure 4
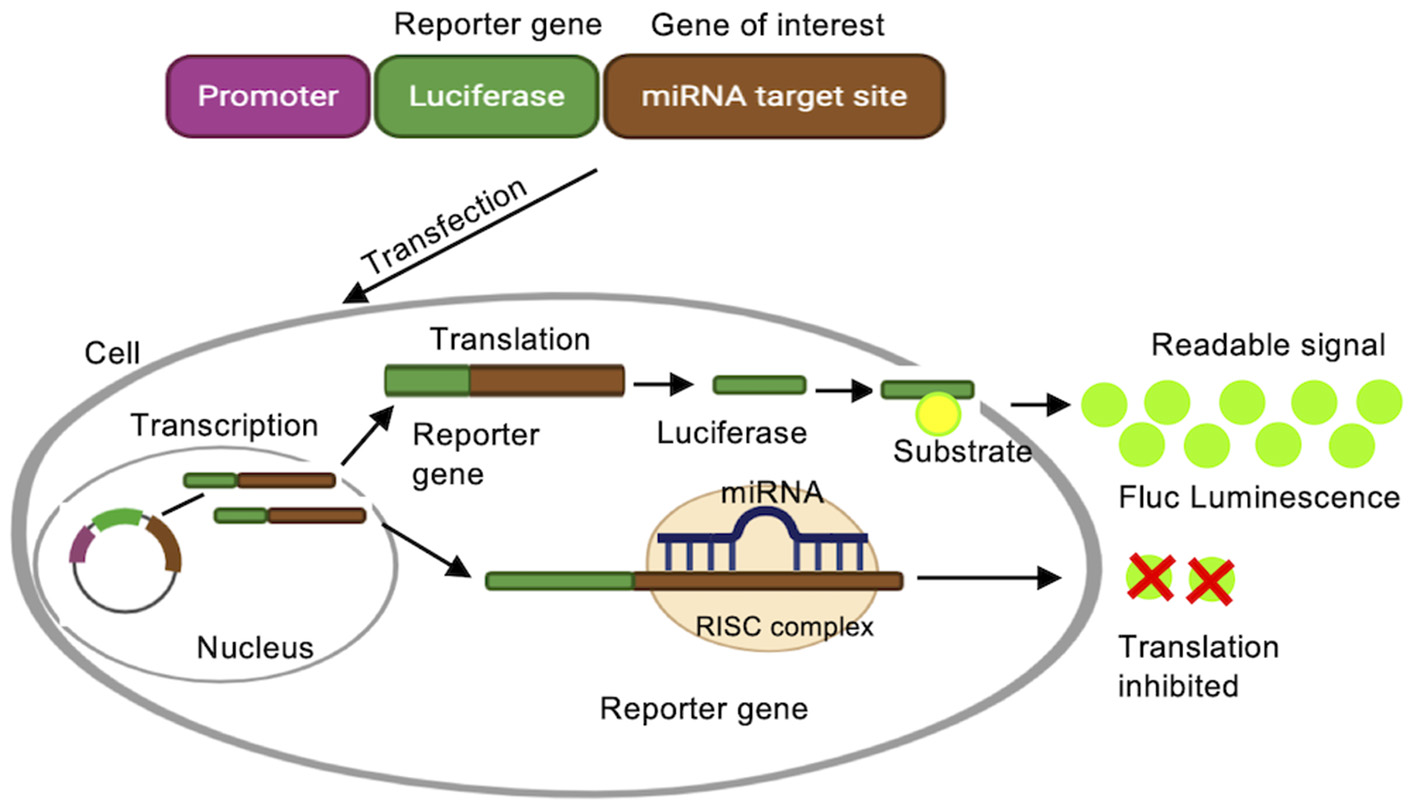
Luciferase reporter assay. A luciferase reporter gene is fused to the promoter sequence of a gene of interest, e.g., the promoter for a specific miRNA, or a target mRNA. Luciferase is transcribed when the native promoter is active, and activity of the reporter protein is detected by converting luciferin substrate to a detectable bioluminescence.
To monitor miRNA expression using a luciferase reporter, transcription of miRNAs and their precursors can be surveyed by fusing the promoter controlling specific miRNA expression to reporter genes. For example, both the 3′-end (miR-9) and the 5′-end (miR-9*) of the brain-specific miR-9 function biologically in brain development. Their expression was investigated by separately fusing the Fluc and Gluc genes to the upstream promoter regions of the 3′-end of miR-9 and 5′-end of miR-9* in P19 cells. Differential expression patterns of miR-9 and miR-9* were measured during neuronal cells differentiation (Ko et al., 2008).
As mentioned above, luciferase reporters are also utilized to verify predicted miRNA binding sites on miRNA target genes by monitoring both miRNA and target mRNA transcription. To validate miRNA targets, the 3′ UTR of the target mRNA is fused to the luciferase gene and can be probed in the presence (e.g., transfection of additional miRNAs or different growth conditions) and absence of a miRNA. In this case, miRNA binding to the UTR will decrease Luciferase production.
Dual reporter constructs allow for a simultaneous analysis of miRNAs and target mRNA. For example, expression of a Fluc and Rluc dual reporter construct was used to test the interaction between miR-138 with the 3′-UTR of its target rhoC mRNA, identifying the miRNA targeting sites in the coding region of the 3′-UTR (Jin et al., 2013). Fluc and Rluc mediated dual-reporter assays were also utilized to identify the multiple roles of miR-29b (Clément et al., 2015), and miR-529b (Moyle et al., 2017). A dual-fluorescence assay allowed for the detection of subtle changes in miRNAs and successfully identified mutations in targets sites of miR-212 in cardiac disorders (Goldoni et al., 2012). Dual reporter systems provide an efficient way to verify miRNA target sites, and gene regulation of miRNA expression. A large number of fluorescent reporters emitting at different wavelengths can be used to localize e.g., transcription sites and are useful molecular imaging tools (Wessels et al., 2010; Wang et al., 2011).
Strengths and Limitations
Luciferase-based reporters are exceedingly useful to investigate the expression of miRNAs or their target genes and generally do not require specialized equipment but are, in nature, low throughput and laborious assays. Furthermore, luciferase-based assays require the addition of luciferin, which makes them less suitable for a time resolved analysis of miRNA expression or activity.
In vivo MiRNA Activity Reporters
We recently developed a reporter gene construct that directly measures the activity on miRNAs over time and during different developmental stages. In these reporter constructs, the green fluorescence protein (GFP) gene is fused to a 3′UTR with specific miRNAs binding sites, making GFP expression responsive to changes in miRNA activity. While luciferase-based assays provide a similar reporter for miRNA activity, in contrast to luciferase expression, GFP expression can be monitored over time, allowing for a time and space resolved analysis of miRNA activity and fewer experimental steps than luciferase-based assays. This reporter was used to successfully measure cellular levels of miRNA let-7a and miR-122 in real-time (Turk et al., 2018). Here, GFP was fused to the 3′-UTR of the oncogene Ras, which encodes let-7a binding sites. GFP fluorescence showed an inverse relationship to let-7a levels (Figure 5). This system allows for the direct measurement of miRNA activity in living cells in a time resolved manner. The GFP reporter system could be expanded in its applications to match the dual reporter assays described above, fusing GFP to the 3′ UTR of a potential target mRNA, while fusing the miRNA promoter to a yellow fluorescent protein (YFP) or red fluorescent protein (RFP), further expanding the toolbox of time resolved miRNA assays.
Figure 5
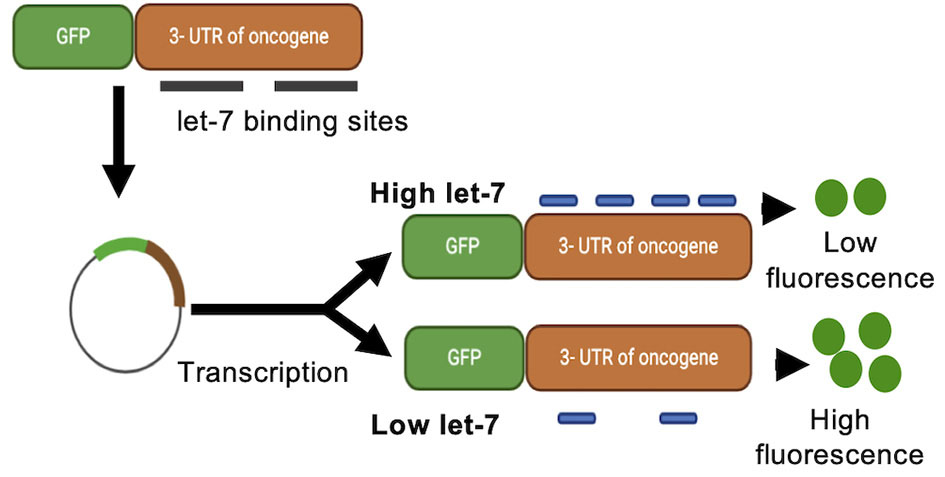
Green fluorescence protein reporter assay. A green fluorescence protein (GFP) reporter gene is fused to a 3′-UTR encoding miRNA binding sites. GFP mRNA translation is controlled by, and responsive to, changes in miRNA activity. Binding of the miRNA to the 3′-UTR of the reporter construct decreases the production of GFP and detectable fluorescence.
Strengths and Limitations
Similar to the luciferase-based assays, the GFP based assays are low throughput and time intensive, but do not require specialized equipment. Nonetheless, these assays provide a time resolved report on miRNA activity in cell lines, which is not easily accessible with alternate methods.
Molecular Beacon (MB) in vivo Imaging System
Optical reporter strategies, especially promoter studies, at times cannot differentiate whether the reduced fluorescence signals stem from deregulated gene expression or should be attributed to cellular loss. Plasmid based fluorescence reporters can also usually not be applied as a diagnostic in a living patient. To overcome these limitations, a molecular beacon (MB) in vivo imaging system was developed to trace miRNA biogenesis. The MB is a hairpin shaped oligonucleotide structure fused to a fluorescent dye at the 5′ end and a quencher at the 3′ end. In its hairpin form in the absence of miRNA, the fluorescent signal is quenched. Upon binding to the complementary sequence of a target miRNA, the quencher is displaced and a fluorescent signal emitted. MB-based emissions offer excellent tissue penetration and allow for molecular imaging to monitor miRNAs, as has been demonstrated for miR-26a and miR-206 dynamics in muscular cells (Kang et al., 2011). This general method was applied to image miR-124 expression by non-invasive magnetic resonance imaging (MRI). Magnetic nanoparticles conjugated to a fluorescent dye were attached to a double stranded oligonucleotide encoding a miR-124 binding site in one strand and a quencher on the complementary strand, effectively quenching fluorescence in the stem-loop, miRNA free form in the absence of the targets. Upon binding to the miR-124, quencher molecule is dispatched and results in enhancement of fluorescence signal which evaluates the level of miRNA expression. Moreover, magnetic nanoparticles offer in vivo cell tracking by imaging miRNA (Hwang et al., 2010; Hernandez et al., 2013). Recently, MRI has been applied to trace circulating miRNAs in serum and allowed for disease prognosis (Regev et al., 2017). Positron emission tomography (PET) imaging was used to localize specific miRNAs using radiolabeled tracer oligonucleotides complementary to the target miRNAs (Mäkilä et al., 2019).
Strengths and Limitations
Activity, localization and distribution of miRNA are disease biomarkers (Cheng et al., 2015; Cui et al., 2018), and the above mentioned methods effectively visualize miRNAs in patients as non-invasive imaging systems.
Conclusion
MiRNAs are differentially expressed, processed and silenced depending on the developmental and physiological context. MiRNAs can have multiple mRNA targets, differ in localization and tissue specific expression and show divergent effects in specific cell lines. The special characteristics of miRNAs, including their small size, high sequence similarity and sometimes low expression, required the development of new, highly sensitive, and specific protocols. MiRNAs in cells can be quantified by the available methods based on their biogenesis, miRNA types and research interest. Unlike intracellular miRNAs, circulating miRNAs are present in the extracellular environment or body fluids and often act as a biomarker for disease such as progression of cancer. Considering the low concentrations and high diversity of extracellular miRNAs, NGS is the preferred method due to its high sensitivity, but microarray and RT-qPCR have also been utilized to detect circulating miRNAs (Cui et al., 2019). In this review, we summarized many of the available methods to quantify miRNA expression, identify new miRNAs and miRNA targets, and measure miRNA activity. The approach of choice often depends on the research question: NGS is unparalleled in its ability to discover new miRNAs, while microarrays are highly useful when comparing the abundance of hundreds of known miRNAs in a more cost-efficient manner. Yet, these methods still depend on RT-qPCR for verification, and neither method illuminates the impact of miRNAs on target mRNAs similarly to luciferase of GFP-based reporter systems, or localize miRNAs in living systems as seen in molecular beacon systems. Thus, any one system is often not sufficient, and a combination of above approaches is usually employed to characterize miRNA functions in depth.
While new methods have been developed to address many of the challenges in miRNA research, the high throughput methods, such as microarrays and NGS remain cost prohibitive to be used on a day to day basis, challenging the science community to come up with novel, more cost-effective methods. Furthermore, the overlap in results from platforms is often lacking, as we outlined for the different microarray platforms. Here, data normalization plays a crucial role in the quantitative miRNA analysis tools. Usually, one or more endogenous control RNAs are used to normalize sample readout in RT-qPCR, microarrays and NGS, assuming that the control RNA does not vary in expression between samples to allow for an adjustment in data variation. While a similar invariance assumption is used in e.g., western blots, where GapdH is often used as a loading control and data is normalized to this protein, the invariance of a control RNA should be verified before designating a certain control RNA as reference. Alternatively, the global mean expression of all miRNAs has been used as a normalizer for microarray and NGS experiments, assuming that while individual miRNAs vary in expression, the overall miRNA content between samples would be similar, but again this may be based on a false assumption. The differences in normalization processes likely account for much or the inconsistency seen between different datasets (Gunaratne et al., 2010; Sales et al., 2010). Finally, RNA extraction and preparation methods, as well as the sample types can significantly impact the results of miRNA profiling, requiring rigorous quality controls. While cost efficiency, sensitivity and standardization remain challenging, the toolbox for miRNA detection, quantification, annotation and analysis are constantly expanding and evolve alongside with new discoveries in the field.
Statements
Author contributions
Both authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This research was funded by the Natural Sciences and Engineering Research Council of Canada to IUH (RGPIN 04776-2014), the Ontario Ministry of Research and Innovation (ER-18-14-183), and the Rare Disease Network (M20-00038).
Acknowledgments
We would like to acknowledge Rosan Kenana, Mallory Frederick, and Patrick O'Donoghue for critical discussions.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
Aghaee-BakhtiariS. H. (2018). Online databases and circular RNAs. Adv. Exp. Med. Biol. 1087, 35–38. 10.1007/978-981-13-1426-1_3
2
AllawiH. T.DahlbergJ.E.OlsonS.LundE.OlsonM.MaW.-P.et al. (2004). Quantitation of microRNAs using a modified Invader assay. RNA10, 1153–1161. 10.1261/rna.5250604
3
AroraS.RanaR.ChhabraA.JaiswalA.RaniV. (2013). miRNA–transcription factor interactions: a combinatorial regulation of gene expression. Mol. Genet. Genom.288, 77–87. 10.1007/s00438-013-0734-z
4
BarM.WymanS. K.FritzB. R.QiJ.GargK. S.ParkinR. K.et al. (2008). MicroRNA discovery and profiling in human embryonic stem cells by deep sequencing of small RNA libraries. Stem cells26, 2496–2505. 10.1634/stemcells.2008-0356
5
BartelD. P. (2004). MicroRNAs: genomics, biogenesis, mechanism, and function. Cell116, 281–297. 10.1016/S0092-8674(04)00045-5
6
BartelD. P. (2009). MicroRNAs: target recognition and regulatory functions. Cell136, 215–233. 10.1016/j.cell.2009.01.002
7
BartelsC. L.TsongalisG. J. (2009). MicroRNAs: novel biomarkers for human cancer. Clin. Chem. 55, 623–631. 10.1373/clinchem.2008.112805
8
BurgosK. L.JavaherianA.BomprezziR.GhaffariL.RhodesS.CourtrightA.et al. (2013). Identification of extracellular miRNA in human cerebrospinal fluid by next-generation sequencing. RNA19, 712–722. 10.1261/rna.036863.112
9
BustinS. A. (2000). Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J. Mol. Endocrinol. 25, 169–193. 10.1677/jme.0.0250169
10
CaiY.YuX.HuS.YuJ. (2009). A brief review on the mechanisms of miRNA regulation. Genom. Proteom. Bioinform.7, 147–154. 10.1016/S1672-0229(08)60044-3
11
CallariM.DugoM.MusellaV.MarchesiE.ChiorinoG.GrandM. M.et al. (2012). Comparison of microarray platforms for measuring differential microRNA expression in paired normal/cancer colon tissues. PLoS ONE7:e45105. 10.1371/journal.pone.0045105
12
CarthewR. W. (2006). Gene regulation by microRNAs. Curr. Opin. Genet. Dev. 16, 203–208. 10.1016/j.gde.2006.02.012
13
ChatterjeeA.LeichterA. L.FanV.TsaiP.PurcellR. V.SullivanM. J.et al. (2015). A cross comparison of technologies for the detection of microRNAs in clinical FFPE samples of hepatoblastoma patients. Sci. Rep. 5:10438. 10.1038/srep10438
14
ChekulaevaM.FilipowiczW. (2009). Mechanisms of miRNA-mediated post-transcriptional regulation in animal cells. Curr. Opin. Cell Biol. 21, 452–460. 10.1016/j.ceb.2009.04.009
15
ChenC.RidzonD. A.BroomerA. J.ZhouZ.LeeD. H.NguyenJ. T.et al. (2005). Real-time quantification of microRNAs by stem–loop RT–PCR. Nucleic Acids Res. 33:e179. 10.1093/nar/gni178
16
ChenC.TanR.WongL.FeketeR.HalseyJ. (2011). Quantitation of microRNAs by real-time RT-qPCR, in PCR Protocols, ed ParkD. (New York, NY: Springer), 113–134. 10.1007/978-1-60761-944-4_8
17
ChengL.DoeckeJ. D.SharplesR.VillemagneV. L.FowlerC. J.RembachA.et al. (2015). Prognostic serum miRNA biomarkers associated with Alzheimer's disease shows concordance with neuropsychological and neuroimaging assessment. Mol. Psychiatry20, 1188–1196. 10.1038/mp.2014.127
18
ChengY.ZhangX.LiZ.JiaoX.WangY.ZhangY. (2009). Highly sensitive determination of microRNA using target-primed and branched rolling-circle amplification. Angew. Chem. Int. Ed.48, 3268–3272. 10.1002/anie.200805665
19
ChungC. Z.SeidlL. E.MannM. R.HeinemannI. U. (2017). Tipping the balance of RNA stability by 3' editing of the transcriptome. Biochim Biophys Acta Gen Subj1861, 2971–2979. 10.1016/j.bbagen.2017.05.003
20
ClémentT.SaloneV.RederstorffM. (2015). Dual luciferase gene reporter assays to study miRNA function, in Small Non-Coding RNAs, ed RederstorffM. (New York, NY: Springer), 187–198. 10.1007/978-1-4939-2547-6_17
21
CuiM.WangH.YaoX.ZhangD.XieY.CuiR.et al. (2019). Circulating MicroRNAs in cancer: potential and challenge. Front. Genet. 10:626. 10.3389/fgene.2019.00626
22
CuiQ.VariF.CristinoA. S.SalomonC.RiceG. E.SabdiaM. B.et al. (2018). Circulating cell-free miR-494 and miR-21 are disease response biomarkers associated with interim-positron emission tomography response in patients with diffuse large B-cell lymphoma. Oncotarget9:34644. 10.18632/oncotarget.26141
23
DirksR. M.PierceN. A. (2004). Triggered amplification by hybridization chain reaction. Proc. Natl. Acad. Sci. U. S. A.101, 15275–15278. 10.1073/pnas.0407024101
24
FaridaniO. R.AbdullayevI.Hagemann-JensenM.SchellJ. P.LannerF.SandbergR. (2016). Single-cell sequencing of the small-RNA transcriptome. Nat. Biotechnol. 34, 1264–1266. 10.1038/nbt.3701
25
FendlerW.MalachowskaB.MeghaniK.KonstantinopoulosP. A.GuhaC.SinghV. K.et al. (2017). Evolutionarily conserved serum microRNAs predict radiation-induced fatality in nonhuman primates. Sci. Transl. Med.9:eaal2408. 10.1126/scitranslmed.aal2408
26
FrancoD.BonetF.Hernandez-TorresF.Lozano-VelascoE.EstebanF. J.AranegaA. E. (2016). Analysis of microRNA microarrays in cardiogenesis. Methods Mol. Biol. 1375, 207–221. 10.1007/7651_2015_247
27
FriedenM.HansenH. F.KochT. (2003). Nuclease stability of LNA oligonucleotides and LNA-DNA chimeras. Nucleosides Nucleotides Nucleic Acids22, 1041–1043. 10.1081/NCN-120022731
28
FriedmanR. C.FarhK. K.-H.BurgeC. B.BartelD. P. (2009). Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 19, 92–105. 10.1101/gr.082701.108
29
GebertL. F.MacRaeI. J. (2019). Regulation of microRNA function in animals. Nat. Rev. Mol. Cell Biol.20, 21–37. 10.1038/s41580-018-0045-7
30
GoldoniD.YarhamJ. M.McGahonM. K.O'ConnorA.Guduric-FuchsJ.EdgarK.et al. (2012). A novel dual-fluorescence strategy for functionally validating microRNA targets in 3′ untranslated regions: regulation of the inward rectifier potassium channel Kir2. 1 by miR-212. Biochem. J.448, 103–113. 10.1042/BJ20120578
31
GoldrickM.BuskP. K.LepovitzL. (2013). Reduce microRNA RT-qPCR Assay Costs by More Than 10-fold Without Compromising Results. Austin, TX: Bioo Scientific.
32
GommansW. M.BerezikovE. (2012). Controlling miRNA regulation in disease, in Next-Generation MicroRNA Expression Profiling Technology, ed FanJ.-B. (New York, NY: Springer), 1–18. 10.1007/978-1-61779-427-8_1
33
GunaratneP. H.CreightonC. J.WatsonM.TennakoonJ. B. (2010). Large-scale integration of MicroRNA and gene expression data for identification of enriched microRNA-mRNA associations in biological systems. Methods Mol. Biol. 667, 297–315. 10.1007/978-1-60761-811-9_20
34
HammondS.SwanbergJ. C.PolsonS. W.LeeK. H. (2012). Profiling conserved microRNA expression in recombinant CHO cell lines using Illumina sequencing. Biotechnol. Bioeng. 109, 1371–1375. 10.1002/bit.24415
35
HaugwitzM.NourzaieO.GarachtchenkoT.HuL.GandlurS.OlsenC.et al. (2008). Multiplexing bioluminescent and fluorescent reporters to monitor live cells. Curr. Chem. Genomics1:11. 10.2174/1875397300801010011
36
HernandezR.OrbayH.CaiW. (2013). Molecular imaging strategies for in vivo tracking of microRNAs: a comprehensive review. Curr. Med. Chem. 20, 3594–3603. 10.2174/0929867311320290005
37
HuY.LanW.MillerD. (2017). Next-generation sequencing for MicroRNA expression profile, in Bioinformatics in MicroRNA Research, eds HuangJ.BorchertG. M.DouD.HuanJ.LanW.TanM.et al. (New York, NY: Springer), 169–177. 10.1007/978-1-4939-7046-9_12
38
HuangQ.YangJ.ZhengJ.HsuehC.GuoY.ZhouL. (2018). Characterization of selective exosomal microRNA expression profile derived from laryngeal squamous cell carcinoma detected by next generation sequencing. Oncol. Rep. 40, 2584–2594. 10.3892/or.2018.6672
39
HurleyJ.RobertsD.BondA.KeysD.ChenC. (2012). Stem-loop RT-qPCR for microRNA expression profiling, in Next-generation MicroRNA Expression Profiling Technology, ed FanJ.-B. (New York, NY: Springer), 33–52. 10.1007/978-1-61779-427-8_3
40
HwangD. W.SongI. C.LeeD. S.KimS. (2010). Smart magnetic fluorescent nanoparticle imaging probes to monitor microRNAs. Small6, 81–88. 10.1002/smll.200901262
41
JiaH.LiZ.LiuC.ChengY. (2010). Ultrasensitive detection of microRNAs by exponential isothermal amplification. Angew. Chem. Int. Ed.49, 5498–5501. 10.1002/anie.201001375
42
JiangH.WongW. H. (2008). SeqMap: mapping massive amount of oligonucleotides to the genome. Bioinformatics24, 2395–2396. 10.1093/bioinformatics/btn429
43
JinY.ChenZ.LiuX.ZhouX. (2013). Evaluating the microRNA targeting sites by luciferase reporter gene assay, in MicroRNA Protocols, ed YingS.-Y. (Totowa, NJ: Springer), 117–127. 10.1007/978-1-62703-083-0_10
44
JohansenS. D.KarlsenB. O.FurmanekT.AndreassenM.JørgensenT. E.BizuayehuT. T.et al. (2011). RNA deep sequencing of the Atlantic cod transcriptome. Comp. Biochem. Physiol. Part D Genomics Proteomics6, 18–22. 10.1016/j.cbd.2010.04.005
45
KampaD.ChengJ.KapranovP.YamanakaM.BrubakerS.CawleyS.et al. (2004). Novel RNAs identified from an in-depth analysis of the transcriptome of human chromosomes 21 and 22. Genome Res.14, 331–342. 10.1101/gr.2094104
46
KangK.PengX.LuoJ.GouD. (2012). Identification of circulating miRNA biomarkers based on global quantitative real-time PCR profiling. J. Anim. Sci. Biotechnol.3:4. 10.1186/2049-1891-3-4
47
KangW. J.ChoY. L.ChaeJ. R.LeeJ. D.ChoiK.-J.KimS. (2011). Molecular beacon-based bioimaging of multiple microRNAs during myogenesis. Biomaterials32, 1915–1922. 10.1016/j.biomaterials.2010.11.007
48
KimS. W.LiZ.MooreP. S.MonaghanA. P.ChangY.NicholsM.et al. (2010). A sensitive non-radioactive northern blot method to detect small RNAs. Nucleic Acids Res. 38:e98. 10.1093/nar/gkp1235
49
KoM. H.KimS.HwangD. W.KoH. Y.KimY. H.LeeD. S. (2008). Bioimaging of the unbalanced expression of microRNA9 and microRNA9* during the neuronal differentiation of P19 cells. FEBS J.275, 2605–2616. 10.1111/j.1742-4658.2008.06408.x
50
KoscianskaE.Starega-RoslanJ.SznajderL. J.OlejniczakM.Galka-MarciniakP.KrzyzosiakW. J. (2011). Northern blotting analysis of microRNAs, their precursors and RNA interference triggers. BMC Mol. Biol. 12:14. 10.1186/1471-2199-12-14
51
KozomaraA.BirgaoanuM.Griffiths-JonesS. (2019). miRBase: from microRNA sequences to function. Nucleic Acids Res. 47, D155–D162. 10.1093/nar/gky1141
52
KrichevskyA. M.KingK. S.DonahueC. P.KhrapkoK.KosikK. S. (2003). A microRNA array reveals extensive regulation of microRNAs during brain development. RNA9, 1274–1281. 10.1261/rna.5980303
53
Lagos-QuintanaM.RauhutR.LendeckelW.TuschlT. (2001). Identification of novel genes coding for small expressed RNAs. Science294, 853–858. 10.1126/science.1064921
54
LangmeadB.TrapnellC.PopM.SalzbergS. L. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10:R25. 10.1186/gb-2009-10-3-r25
55
LauN. C.LimL. P.WeinsteinE. G.BartelD. P. (2001). An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science294, 858–862. 10.1126/science.1065062
56
LeeY.JeonK.LeeJ. T.KimS.KimV. N. (2002). MicroRNA maturation: stepwise processing and subcellular localization. EMBO J. 21, 4663–4670. 10.1093/emboj/cdf476
57
LiH.RuanJ.DurbinR. (2008). Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 18, 1851–1858. 10.1101/gr.078212.108
58
LiS.WangH.QiY.TuJ.BaiY.TianT.et al. (2011). Assessment of nanomaterial cytotoxicity with SOLiD sequencing-based microRNA expression profiling. Biomaterials32, 9021–9030. 10.1016/j.biomaterials.2011.08.033
59
LiW.RuanK. (2009). MicroRNA detection by microarray. Anal. Bioanal. Chem. 394, 1117–1124. 10.1007/s00216-008-2570-2
60
LiangR.-Q.LiW.LiY.TanC.-y.LiJ.-X.JinY.-X.et al. (2005). An oligonucleotide microarray for microRNA expression analysis based on labeling RNA with quantum dot and nanogold probe. Nucleic Acids Res. 33:e17. 10.1093/nar/gni019
61
LiuC.-G.CalinG. A.MeloonB.GamlielN.SevignaniC.FerracinM.et al. (2004). An oligonucleotide microchip for genome-wide microRNA profiling in human and mouse tissues. Proc. Natl. Acad. Sci. U. S. A.101, 9740–9744. 10.1073/pnas.0403293101
62
LiuD.-Z.TianY.AnderB. P.XuH.StamovaB. S.ZhanX.et al. (2010). Brain and blood microRNA expression profiling of ischemic stroke, intracerebral hemorrhage, and kainate seizures. J. Cereb. Blood Flow Metab. 30, 92–101. 10.1038/jcbfm.2009.186
63
LiuJ.JenningsS. F.TongW.HongH. (2011). Next generation sequencing for profiling expression of miRNAs: technical progress and applications in drug development. J. Biomed. Sci. Eng. 4:666. 10.4236/jbise.2011.410083
64
LiuY.LiuB.LiuY.ChenS.YangJ.LiuJ.et al. (2019). MicroRNA expression profile by next-generation sequencing in a novel rat model of contrast-induced acute kidney injury. Ann. Transl. Med.7:44. 10.21037/atm.2019.04.44
65
LuJ.GetzG.MiskaE. A.Alvarez-SaavedraE.LambJ.PeckD.et al. (2005). MicroRNA expression profiles classify human cancers. Nature435, 834–838. 10.1038/nature03702
66
LuoX.ZhangJ.WangH.DuY.YangL.ZhengF.et al. (2012). PolyA RT-PCR-based quantification of microRNA by using universal TaqMan probe. Biotechnol. Lett. 34, 627–633. 10.1007/s10529-011-0813-3
67
LvA.TuZ.HuangY.LuW.XieB. (2021). Circulating exosomal miR-125a-5p as a novel biomarker for cervical cancer. Oncol. Lett. 21:54. 10.3892/ol.2020.12316
68
LvK.GuoY.ZhangY.WangK.JiaY.SunS. (2008). Allele-specific targeting of hsa-miR-657 to human IGF2R creates a potential mechanism underlying the association of ACAA-insertion/deletion polymorphism with type 2 diabetes. Biochem. Biophys. Res. Commun. 374, 101–105. 10.1016/j.bbrc.2008.06.102
69
MäkiläJ.KiviniemiA.SaanijokiT.LiljenbäckH.KäkeläM.JadhavS.et al. (2019). Noninvasive and quantitative monitoring of the distributions and kinetics of microRNA-targeting molecules in vivo by positron emission tomography. Mol. Pharm. 16, 1507–1515. 10.1021/acs.molpharmaceut.8b01169
70
MerrimanB. D.TeamI. T.RothbergJ. M. (2012). Progress in ion torrent semiconductor chip based sequencing. Electrophoresis33, 3397–3417. 10.1002/elps.201200424
71
MetpallyR. P.NasserS.MalenicaI.CourtrightA.CarlsonE.GhaffariL.et al. (2013). Comparison of analysis tools for miRNA high throughput sequencing using nerve crush as a model. Front. Genet. 4:20. 10.3389/fgene.2013.00020
72
MichlewskiG.CáceresJ. F. (2019). Post-transcriptional control of miRNA biogenesis. RNA25, 1–16. 10.1261/rna.068692.118
73
MiskaE. A.Alvarez-SaavedraE.TownsendM.YoshiiA.ŠestanN.RakicP.et al. (2004). Microarray analysis of microRNA expression in the developing mammalian brain. Genome Biol. 5:R68. 10.1186/gb-2004-5-9-r68
74
Mohammadi-YeganehS.ParyanM.SamieeS. M.SoleimaniM.ArefianE.AzadmaneshK.et al. (2013). Development of a robust, low cost stem-loop real-time quantification PCR technique for miRNA expression analysis. Mol. Biol. Rep. 40, 3665–3674. 10.1007/s11033-012-2442-x
75
MoodyL.HeH.PanY.-X.ChenH. (2017). Methods and novel technology for microRNA quantification in colorectal cancer screening. Clin. Epigenetics9:119. 10.1186/s13148-017-0420-9
76
MotamenyS.WoltersS.NürnbergP.SchumacherB. (2010). Next generation sequencing of miRNAs–strategies, resources and methods. Genes1, 70–84. 10.3390/genes1010070
77
MoyleR. L.CarvalhaisL. C.PretoriusL.-S.NowakE.SubramaniamG.Dalton-MorganJ.et al. (2017). An optimized transient dual luciferase assay for quantifying MicroRNA directed repression of targeted sequences. Front. Plant Sci. 8:1631. 10.3389/fpls.2017.01631
78
NaqviS.BellottD. W.LinK. S.PageD. C. (2018). Conserved microRNA targeting reveals preexisting gene dosage sensitivities that shaped amniote sex chromosome evolution. Genome Res. 28, 474–483. 10.1101/gr.230433.117
79
NeilsonJ. R.ZhengG. X.BurgeC. B.SharpP. A. (2007). Dynamic regulation of miRNA expression in ordered stages of cellular development. Genes Develop.21, 578–589. 10.1101/gad.1522907
80
NiuY.XiaS.SuM.DangQ.KangK.LiL.et al. (2019). Direct S-Poly (T) Plus assay in quantification of microRNAs without RNA extraction and its implications in colorectal cancer biomarker studies. J. Transl. Med. 17, 1–12. 10.1186/s12967-019-2061-6
81
NiuY.ZhangL.QiuH.WuY.WangZ.ZaiY.et al. (2015). An improved method for detecting circulating microRNAs with S-Poly (T) Plus real-time PCR. Sci. Rep. 5:15100. 10.1038/srep15100
82
O'ConnellR. M.TaganovK. D.BoldinM. P.ChengG.BaltimoreD. (2007). MicroRNA-155 is induced during the macrophage inflammatory response. Proc. Natl. Acad. Sci. U. S. A.104, 1604–1609. 10.1073/pnas.0610731104
83
OhS. W.Do Won HwangD. S. L. (2013). In vivo monitoring of microRNA biogenesis using reporter gene imaging. Theranostics3:1004. 10.7150/thno.4580
84
OlivierM. (2005). The Invader® assay for SNP genotyping. Mutation Res. Fundamental Mol. Mech. Mutagen.573, 103–110. 10.1016/j.mrfmmm.2004.08.016
85
PallG. S.Codony-ServatC.ByrneJ.RitchieL.HamiltonA. (2007). Carbodiimide-mediated cross-linking of RNA to nylon membranes improves the detection of siRNA, miRNA and piRNA by northern blot. Nucleic Acids Res. 35:e60. 10.1093/nar/gkm112
86
PallG. S.HamiltonA. J. (2008). Improved northern blot method for enhanced detection of small RNA. Nat. Protoc. 3:1077. 10.1038/nprot.2008.67
87
PritchardC. C.ChengH. H.TewariM. (2012). MicroRNA profiling: approaches and considerations. Nat. Rev. Genet. 13, 358–369. 10.1038/nrg3198
88
RaymondC. K.RobertsB. S.Garrett-EngeleP.LimL. P.JohnsonJ. M. (2005). Simple, quantitative primer-extension PCR assay for direct monitoring of microRNAs and short-interfering RNAs. RNA11, 1737–1744. 10.1261/rna.2148705
89
RegevK.HealyB. C.KhalidF.PaulA.ChuR.TauhidS.et al. (2017). Association between serum MicroRNAs and magnetic resonance imaging measures of multiple sclerosis severity. JAMA Neurol. 74, 275–285. 10.1001/jamaneurol.2016.5197
90
ReidM. S.LeX. C.ZhangH. (2018). Exponential isothermal amplification of nucleic acids and assays for proteins, cells, small molecules, and enzyme activities: an EXPAR example. Angew. Chem. Int. Ed. Engl. 57, 11856–11866. 10.1002/anie.201712217
91
SalesG.CoppeA.BisogninA.BiasioloM.BortoluzziS.RomualdiC. (2010). MAGIA, a web-based tool for miRNA and Genes Integrated Analysis. Nucleic Acids Res. 38, W352–359. 10.1093/nar/gkq423
92
SaloneV.RederstorffM. (2015). Stem-loop RT-PCR based quantification of small non-coding RNAs, in Small Non-Coding RNAs, ed RederstorffM. (New York, NY: Springer), 103–108. 10.1007/978-1-4939-2547-6_10
93
SchmittgenT. D.JiangJ.LiuQ.YangL. (2004). A high-throughput method to monitor the expression of microRNA precursors. Nucleic Acids Res. 32,:e43. 10.1093/nar/gnh040
94
SchwarzkopfM.PierceN. A. (2016). Multiplexed miRNA northern blots via hybridization chain reaction. Nucleic Acids Res. 44:e129. 10.1093/nar/gkw503
95
SmirnovaL.GräfeA.SeilerA.SchumacherS.NitschR.WulczynF. G. (2005). Regulation of miRNA expression during neural cell specification. Eur. J. Neurosci.21, 1469–1477. 10.1111/j.1460-9568.2005.03978.x
96
TammingR. J.DumeauxV.JiangY.ShafiqS.LangloisL.EllegoodJ.et al. (2020). Atrx deletion in neurons leads to sexually dimorphic dysregulation of miR-137 and spatial learning and memory deficits. Cell Rep. 31:107838. 10.1016/j.celrep.2020.107838
97
TannousB. A.KimD.-E.FernandezJ. L.WeisslederR.BreakefieldX. O. (2005). Codon-optimized Gaussia luciferase cDNA for mammalian gene expression in culture and in vivo. Mol. Therapy11, 435–443. 10.1016/j.ymthe.2004.10.016
98
TrapnellC.PachterL.SalzbergS. L. (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics25, 1105–1111. 10.1093/bioinformatics/btp120
99
TurkM. A.ChungC. Z.ManniE.ZukowskiS. A.EngineerA.BadakhshiY.et al. (2018). MiRAR—miRNA activity reporter for living cells. Genes (Basel)9:305. 10.3390/genes9060305
100
Valinezhad OrangA.SafaralizadehR.Kazemzadeh-BaviliM. (2014). Mechanisms of miRNA-mediated gene regulation from common downregulation to mRNA-specific upregulation. Int. J. Genom.2014:970607. 10.1155/2014/970607
101
VálócziA.HornyikC.VargaN.BurgyanJ.KauppinenS.HaveldaZ. (2004). Sensitive and specific detection of microRNAs by northern blot analysis using LNA-modified oligonucleotide probes. Nucleic Acids Res. 32:e175. 10.1093/nar/gnh171
102
Van Keuren-JensenK. R.MalenicaI.CourtrightA. L.GhaffariL. T.StarrA. P.et al. (2016). microRNA changes in liver tissue associated with fibrosis progression in patients with hepatitis C. Liver Int. 36, 334–343. 10.1111/liv.12919
103
VárallyayE.BurgyánJ.HaveldaZ. (2007). Detection of microRNAs by Northern blot analyses using LNA probes. Methods43, 140–145. 10.1016/j.ymeth.2007.04.004
104
VárallyayE.BurgyánJ.HaveldaZ. (2008). MicroRNA detection by northern blotting using locked nucleic acid probes. Nat. Protoc. 3, 190–196. 10.1038/nprot.2007.528
105
VlachosI. S.HatzigeorgiouA. G. (2013). Online resources for miRNA analysis. Clin. Biochem.46, 879–900. 10.1016/j.clinbiochem.2013.03.006
106
WangF.NiuG.ChenX.CaoF. (2011). Molecular imaging of microRNAs. Eur. J. Nucl. Med. Mol. Imaging38, 1572–1579. 10.1007/s00259-011-1786-0
107
WangY. W.ZhangW.MaR. (2018). Bioinformatic identification of chemoresistance-associated microRNAs in breast cancer based on microarray data. Oncol. Rep. 39, 1003–1010. 10.3892/or.2018.6205
108
WangZ.JiG.WuQ.FengS.ZhaoY.CaoZ.et al. (2018). Integrated microarray meta-analysis identifies miRNA-27a as an oncogene in ovarian cancer by inhibiting FOXO1. Life Sci. 210, 263–270. 10.1016/j.lfs.2018.08.043
109
WesselsJ. T.YamauchiK.HoffmanR. M.WoutersF. S. (2010). Advances in cellular, subcellular, and nanoscale imaging in vitro and in vivo. Cytometry Part A77, 667–676. 10.1002/cyto.a.20931
110
WillenbrockH.SalomonJ.SøkildeR.BarkenK. B.HansenT. N.NielsenF. C.et al. (2009). Quantitative miRNA expression analysis: comparing microarrays with next-generation sequencing. RNA15, 2028–2034. 10.1261/rna.1699809
111
XinH.LiY.BullerB.KatakowskiM.ZhangY.WangX.et al. (2012). Exosome-mediated transfer of miR-133b from multipotent mesenchymal stromal cells to neural cells contributes to neurite outgrowth. Stem Cells.30, 1556–1564. 10.1002/stem.1129
112
XuH.ZhangS.OuyangC.WangZ.WuD.LiuY.et al. (2019). DNA nanostructures from palindromic rolling circle amplification for the fluorescent detection of cancer-related microRNAs. Talanta192, 175–181. 10.1016/j.talanta.2018.07.090
113
YangL. H.WangS. L.TangL. L.LiuB.YeW. L.WangL. L.et al. (2014). Universal stem-loop primer method for screening and quantification of microRNA. PLoS ONE9:e115293. 10.1371/journal.pone.0115293
114
YinJ. Q.ZhaoR. C.MorrisK. V. (2008). Profiling microRNA expression with microarrays. Trends Biotechnol. 26, 70–76. 10.1016/j.tibtech.2007.11.007
115
ZhangL.LiuY.ChenX.-G.ZhangY.ChenJ.HaoZ.-Y.et al. (2019). MicroRNA expression profile in chronic nonbacterial prostatitis revealed by next-generation small RNA sequencing. Asian J. Androl. 21:351. 10.4103/aja.aja_97_18
116
ZhangZ.SchwartzS.WagnerL.MillerW. (2000). A greedy algorithm for aligning DNA sequences. J. Comput. Biol.7, 203–214. 10.1089/10665270050081478
117
ZhaoJ.-J.HuaY.-J.SunD.-G.MengX.-X.XiaoH.-S.MaX. (2006). Genome-wide microRNA profiling in human fetal nervous tissues by oligonucleotide microarray. Child's Nervous Syst.22, 1419–1425. 10.1007/s00381-006-0173-9
Summary
Keywords
microRNA, reporter activity, fluorescence, RT-qPCR, next generation sequencing
Citation
Siddika T and Heinemann IU (2021) Bringing MicroRNAs to Light: Methods for MicroRNA Quantification and Visualization in Live Cells. Front. Bioeng. Biotechnol. 8:619583. doi: 10.3389/fbioe.2020.619583
Received
20 October 2020
Accepted
15 December 2020
Published
18 January 2021
Volume
8 - 2020
Edited by
Michele Galluccio, University of Calabria, Italy
Reviewed by
Gracjan Michlewski, University of Edinburgh, United Kingdom; Simona Panni, University of Calabria, Italy
Updates
Copyright
© 2021 Siddika and Heinemann.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ilka U. Heinemann ilka.heinemann@uwo.ca
This article was submitted to Synthetic Biology, a section of the journal Frontiers in Bioengineering and Biotechnology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.