 Marc Scherer
Marc Scherer Sarel J. Fleishman2
Sarel J. Fleishman2 Patrik R. Jones
Patrik R. Jones Thomas Dandekar
Thomas Dandekar Elena Bencurova
Elena Bencurova- 1Department of Bioinformatics, Julius-Maximilians University of Würzburg, Würzburg, Germany
- 2Department of Biomolecular Sciences, Weizmann Institute of Science, Rehovot, Israel
- 3Department of Life Sciences, Imperial College London, London, United Kingdom
To enable a sustainable supply of chemicals, novel biotechnological solutions are required that replace the reliance on fossil resources. One potential solution is to utilize tailored biosynthetic modules for the metabolic conversion of CO2 or organic waste to chemicals and fuel by microorganisms. Currently, it is challenging to commercialize biotechnological processes for renewable chemical biomanufacturing because of a lack of highly active and specific biocatalysts. As experimental methods to engineer biocatalysts are time- and cost-intensive, it is important to establish efficient and reliable computational tools that can speed up the identification or optimization of selective, highly active, and stable enzyme variants for utilization in the biotechnological industry. Here, we review and suggest combinations of effective state-of-the-art software and online tools available for computational enzyme engineering pipelines to optimize metabolic pathways for the biosynthesis of renewable chemicals. Using examples relevant for biotechnology, we explain the underlying principles of enzyme engineering and design and illuminate future directions for automated optimization of biocatalysts for the assembly of synthetic metabolic pathways.
Introduction
At the start of the third decade of the twenty-first century, humankind faces a multitude of challenges regarding climate change (Arnell et al., 2019), air pollution (Wang et al., 2019), and a shrinking number of intact ecosystems (Nolan et al., 2018) due to human activity. The demand for sustainable solutions addressing the basis of chemical production, transport, and agriculture to enable a net zero-carbon society is higher than ever before (Hoegh-Guldberg et al., 2019). Hence, the development of technologies substituting fossil resources is an important goal of current scientific research (Xu et al., 2018). Utilizing the synthetic power of microorganisms for the sustainable production of bulk chemicals and fuels to replace chemicals currently generated from fossil fuels and tropical plant agriculture is an important contributor toward the goal of achieving a net zero-carbon society (Rodionova et al., 2017). In this regard, biosynthesis of hydrocarbons in microorganisms can be a sustainable technological alternative to produce fuels for aviation (Schirmer et al., 2010; Kallio et al., 2014a), a model of transportation for which competitive electric solutions are still missing (Schäfer et al., 2019). The benefits of applying biocatalysts in the industrial production of commodity chemicals compared to inorganic catalysts lie mostly in their ability to facilitate enantioselective conversions at ambient conditions (temperature and pressure) (Woodley, 2020). Additionally, the usage of biocatalysts instead of metal catalysts, for example, can reduce the amount of waste of chemical production because biocatalysts can be recycled easily (Sheldon and Woodley, 2018).
Further optimization of biocatalysts can expand the solution space of an enzyme and enable the identification of novel synthetic pathways for biomanufacturing of chemicals (Erb et al., 2017). Hence, engineering enzymes for tailored substrate specificity (Amer et al., 2020; Eser et al., 2020), catalytic efficiency (Risso et al., 2020), and stability (Goldenzweig et al., 2016; Yu et al., 2017) are important for the implementation of novel biosynthetic systems.
Efforts for enzyme engineering are exponentially growing due to the demand for natural or biologically produced chemical compounds, such as alcohols, hormones, or essential oils, either by constructing de novo–designed pathways or by optimizing existing ones (Marcheschi et al., 2013). In addition, current progress in genome sequencing identified a number of new enzymes or strain-specific variants that may be an alternative for the application in biotechnology; however, in a lot of cases, they are not stable or suitable for standard expression strains. Currently, enzyme engineering efforts are mostly based on rational engineering with low- and medium-throughput screening of small libraries (Figure 1A) and directed evolution-based approaches and high- and ultrahigh-throughput screening (Figure 1B; Ma et al., 2021); nevertheless, also de novo approaches start to get more attention and had been already used in several works (DeLoache et al., 2015; Dou et al., 2018). Interestingly, including computational tools (Romero-Rivera et al., 2017) as evolutionary conservation analysis (Ashkenazy et al., 2016), mutant structure modeling (Khersonsky et al., 2018; Leman et al., 2020), and molecular dynamics (MD) simulations (Yu and Dalby, 2018; Surpeta et al., 2020) is becoming more abundant and has the potential to accelerate the identification of highly stable and productive biocatalysts for sustainable application (Figure 1C). The development of easy-to-use software and tools available as online servers makes it possible for researchers who are not experts in computational biology to apply state-of-the-art computational protein engineering methodology. On the other hand, the data from in silico engineering do not necessarily correlate with experimental data (Pucci and Rooman, 2016; Carlin et al., 2017), and thus more advanced pipelines using multiple computational tools are required for accurate mutant structure modeling and energy predictions. The application of engineered proteins is versatile and covering various technological branches from pharmaceutics to bioelectronic devices (Kalyoncu et al., 2017) and biosensors (Xiong et al., 2017; Kunjapur and Prather, 2019).
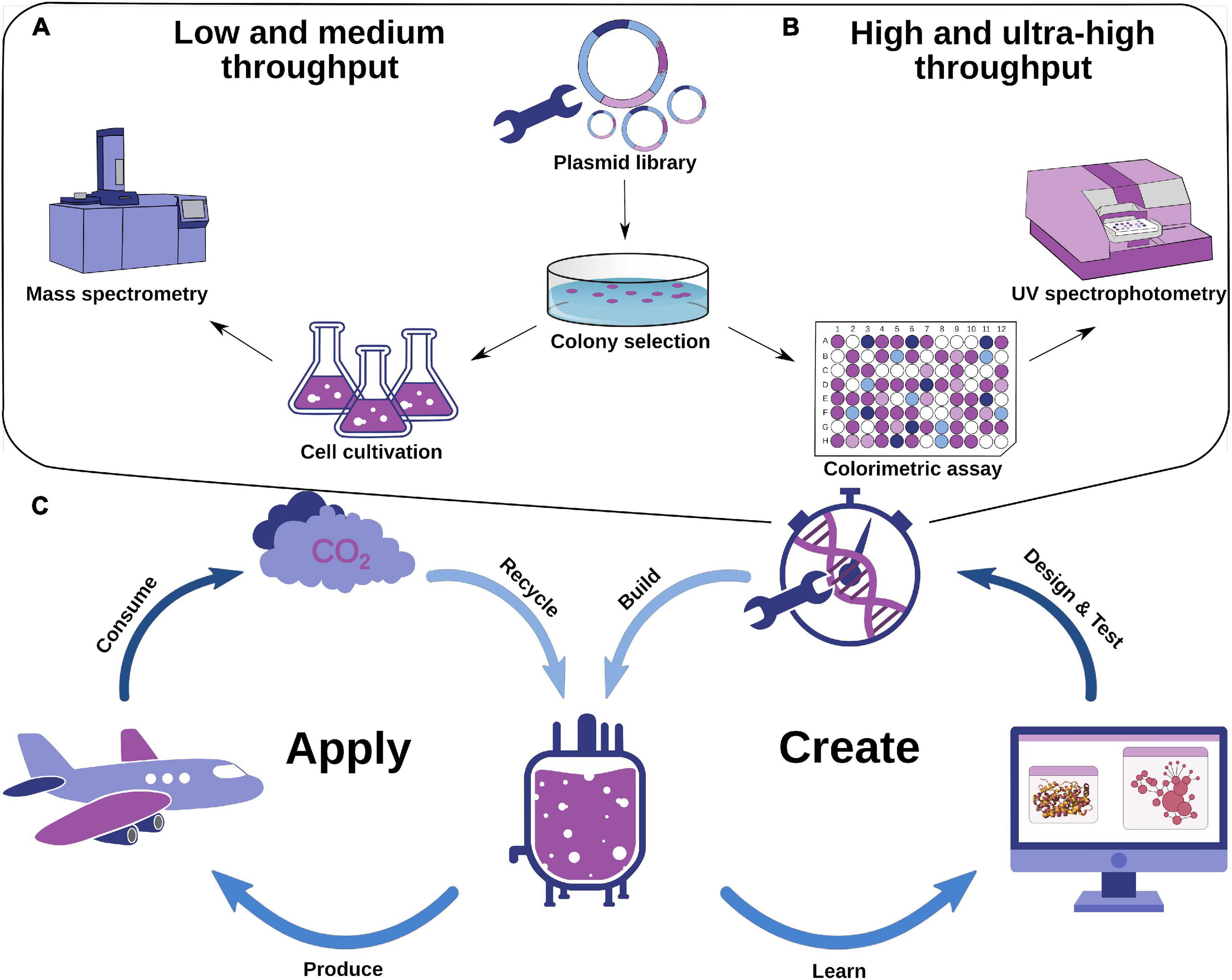
Figure 1. Experimental protein engineering strategies and an idealized scheme for a design–test–build–learn cycle of optimizing enzymes using computation. (A) Exemplified workflow for low- and medium-throughput enzyme engineering strategies. (B) Exemplified workflow for high- and ultrahigh-throughput enzyme engineering strategies. (C) Design–test–build–learn cycle for industrial chemical production including computational methodology and production–consumption–recycling cycle of chemical usage. Promising enzyme variants are identified computationally, which leads to targeted experimental testing. The metabolic systems are then applied in microorganisms for industrial-scale production. The experimental implementation provides additional information for computational optimization. Consumption of chemicals as biofuels results in the release of CO2, which can be recycled by microorganisms in bioreactors to close the cycle.
Our research in synthetic biology and metabolic engineering is directed toward developing methods for bioproduction of renewable chemicals with special emphasis on biofuel-producing pathways. We and others have found that conventional strategies such as directed evolution are not applicable to all enzymatic reactions for lack of high-throughput assays that are required for the effective use of laboratory-evolution strategies. This has turned our attention to computational enzyme engineering methodology that can guide the experimental efforts. It is important to note that the modules described here can be applied with necessary adjustments to all kinds of protein engineering tasks and are therefore not limited to the field of metabolic enzyme engineering. Still, the application of computational methodology will be discussed on the example of metabolic enzymes involved in biofuel production to highlight strengths and limitations of such approaches on a particular field of biotechnological research.
In this article, we review computational tools that can be used to create a platform for fast and customizable modeling and evaluation of promising enzyme variants in silico (Figure 2). We focus on methods that have been experimentally validated and shown to outperform conventional in vitro selection methods. We conclude that computational enzyme engineering can accelerate the development of synthetic metabolic pathways for industrial use.
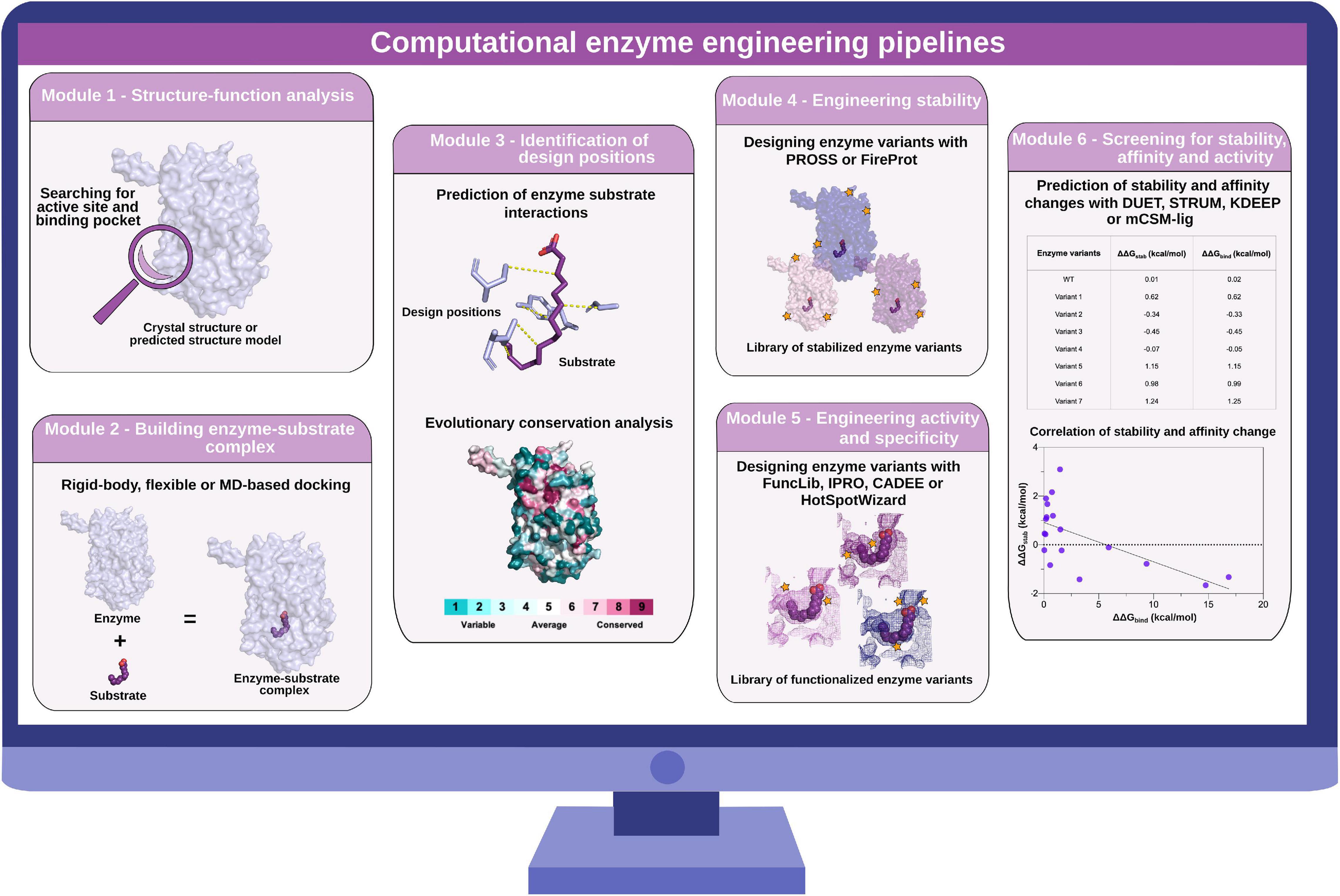
Figure 2. Computational enzyme engineering pipelines. Module 1: structure–function analysis to identify active site and substrate-binding pocket. Module 2: building enzyme–substrate complexes with molecular docking approaches. Module 3: identification of design positions for the subsequent sequence design. Module 4: engineering stability of enzymes with PROSS and FireProt. Module 5: engineering activity and specificity of enzymes with FuncLib, IPRO, CADEE, and HotSpotWizard. Module 6: screening for stability, affinity, and activity changes with DUET, STRUM, KDEEP, and mCSM-lig.
Metabolic Engineering of Fatty Acid Biosynthesis and Enzyme Engineering for Enhanced Production of Biofuels
Fatty acyl compounds are an important target for engineering microbial metabolism and chosen as example. By adding heterologous enzymatic modules, fatty acid metabolism can be redirected toward alkane/alkene biosynthesis (Liu and Li, 2020). Several metabolic pathways for the synthesis of alkanes of varying chain length have been reported (Schirmer et al., 2010; Bernard et al., 2012; Kallio et al., 2014b; Sorigué et al., 2017; Yunus et al., 2018; Amer et al., 2020). However, the production of the structurally similar class of alkenes, especially medium- and short-chain length alkenes, remains a challenge. Although first attempts at biosynthesis of medium- and short-chain alkenes have been made (Dennig et al., 2015; Zhang et al., 2019; Bauer et al., 2020), the substrate conversion efficiencies remain low. Further optimization of metabolic pathways will be required to facilitate future commercialization. The improvement of key enzymatic properties, such as stability and modified substrate specificity and activity, may be necessary, but that is traditionally a cost-intensive and time-consuming task. For example, in a study by Bao et al., single residues in the binding pocket of the Synechococcus elongatus cyanobacterial aldehyde-deformylating oxygenase (cADO) were targeted for site-directed mutagenesis experiments. Substitution of small residues by bulkier hydrophobic ones blocked parts of the binding pocket, which led to a shift in substrate specificity. Depending on the position of the substituted residue, specificity of the engineered cADO variants ranged from C4 to C12 substrates (Bao et al., 2016). With similar structure–function–based approaches, residues near the active site of Chlorella variabilis NC64A fatty acid photodecarboxylase (CvFAP) (Figure 3A) and Jeotgalicoccus sp. ATCC 8456 OleTJE were targeted recently to engineer substrate specificity of the enzymes for the short-chain-length substrate butyric acid, enabling increased production titers of propane (Amer et al., 2020) and propene (Bauer et al., 2020), respectively. All these examples have in common that a small library of rationally designed single-point or double mutants was synthesized and tested for elevated production levels and altered substrate specificities (Figure 1A). Despite these successes, however, in many cases multiple mutations are required to generate an enzyme variant with robust production titers (Grisewood et al., 2017; Khersonsky et al., 2018; Trudeau et al., 2018), and effects of enzyme destabilization upon mutations may interfere with a beneficial effect on substrate binding or catalysis (Trudeau and Tawfik, 2019). Therefore, adding computational prediction and engineering tools to the overall pipeline is likely to increase chances to identify an enzyme variant with enhanced properties. Until now, the most common method to engineer enzymes is by repetitive rounds of directed evolution-based sequence randomization and high-throughput screening (Figure 1B; Farinas et al., 2001). Such approaches are time-consuming and heavily rely on the availability of a suitable screening methodology, which has not been developed for terminal-alkene production yet (Sulzbach and Kunjapur, 2020). This emphasizes the importance of exploiting computational engineering solutions, at least in the special case of alkenes.
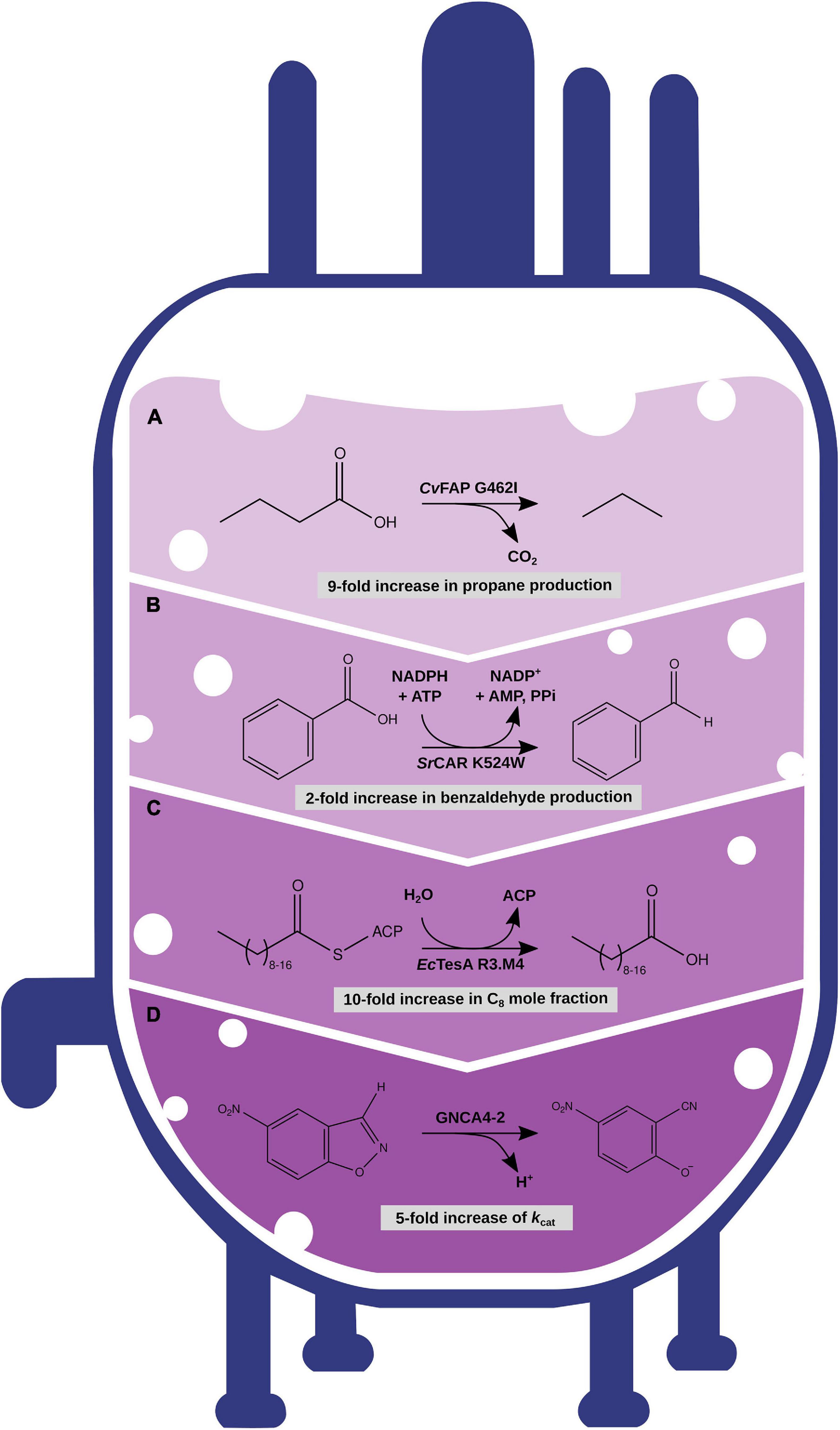
Figure 3. Optimization of enzymes with different engineering strategies with increasing amount of computational modeling and predictions. (A) Rational engineering of CvFAP enzyme for the increased production of propane. (B) Rational engineering based on MD simulation data of SrCAR increased production of benzaldehyde. (C) Semirational sequence design with IPRO of EcTesA for altered substrate specificity. (D) Semirational sequence design with FuncLib and subsequent screening with the EVB approach to increase the catalytic efficiency of a Kemp eliminase GNCA4.
Computational Pipelines to Engineer Enzymes
Advances in the fields of structural bioinformatics, computational modeling, and the availability of huge amounts of DNA sequence data have led to the development of a variety of computational tools that can speed up enzyme engineering for biotechnological application. Computational enzyme engineering and design methodology has been reviewed recently for altering properties such as stability, substrate specificity, or activity of biocatalysts (Ebert and Pelletier, 2017; Chowdhury and Maranas, 2020; Sequeiros-Borja et al., 2020). Here, we describe which enzymatic features are important for enzyme engineering and design and how recently published computational tools (Table 1) can facilitate the required steps from preparing the input structure complexes to screening designed variants for best performance. Combining these engineering steps, we propose a pipeline for computational enzyme engineering and design (Figure 2) that includes workflows for engineering stability, activity, and specificity of enzymes. Depending on the research objective, different engineering efforts can be combined to first design a stable enzyme variant that can be functionalized afterward. Advantages and disadvantages of the most promising stand-alone software and web applications are summarized in Table 2. Initially, a structure–function analysis (Module 1) is performed. Then, Module 2 describes how to build enzyme–substrate complexes. Subsequent analysis of enzyme–substrate interaction and evolutionary conservation analysis leads to the identification of design positions (Module 3). Next, the sequence space of the enzyme–substrate complex can be designed for stability engineering (Module 4) and/or activity and specificity engineering (Module 5). The pipelines end with computational stability, affinity, and activity screening (Module 6) to identify the best variants for experimental testing. Additionally, we provide information on how computational modules have paved the way and will speed up the discovery of enzyme variants for increased chemical production in the future.
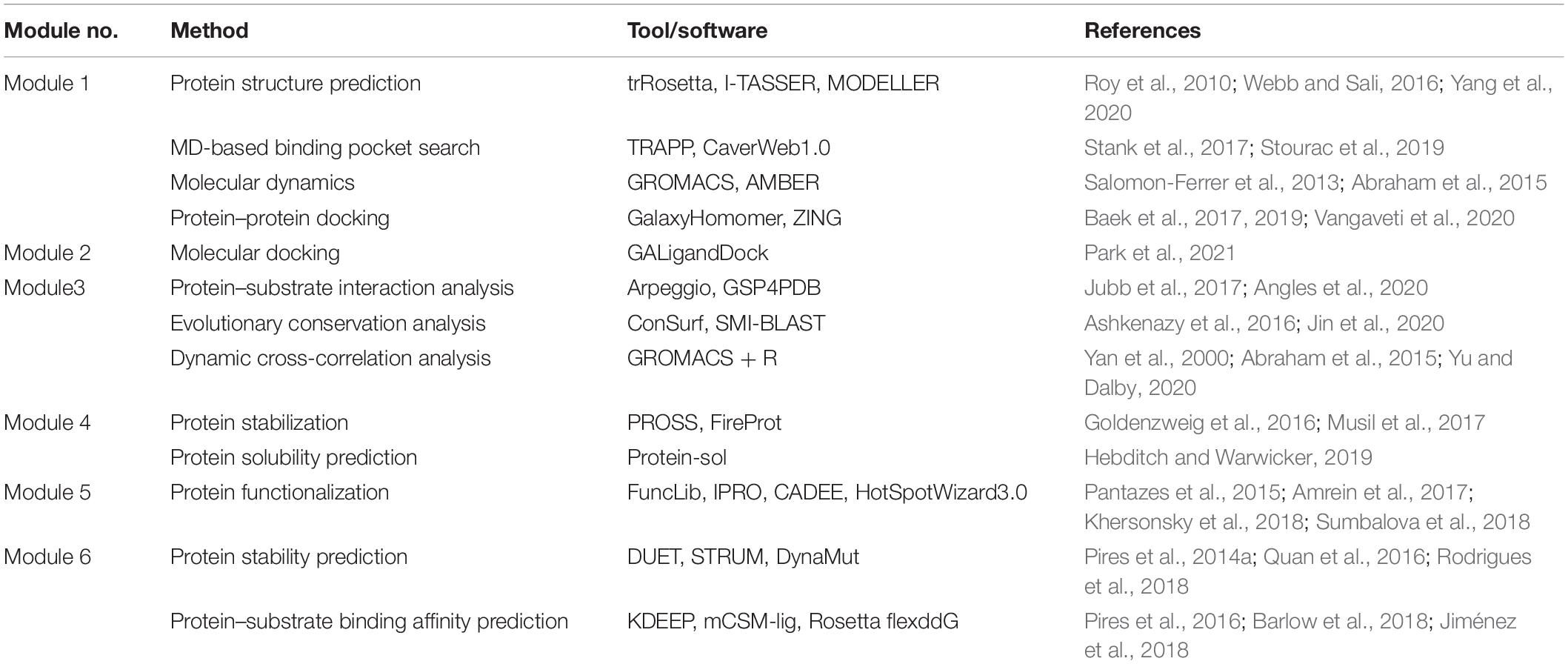
Table 1. State-of-the-art software and online tools for computational protein engineering pipelines.
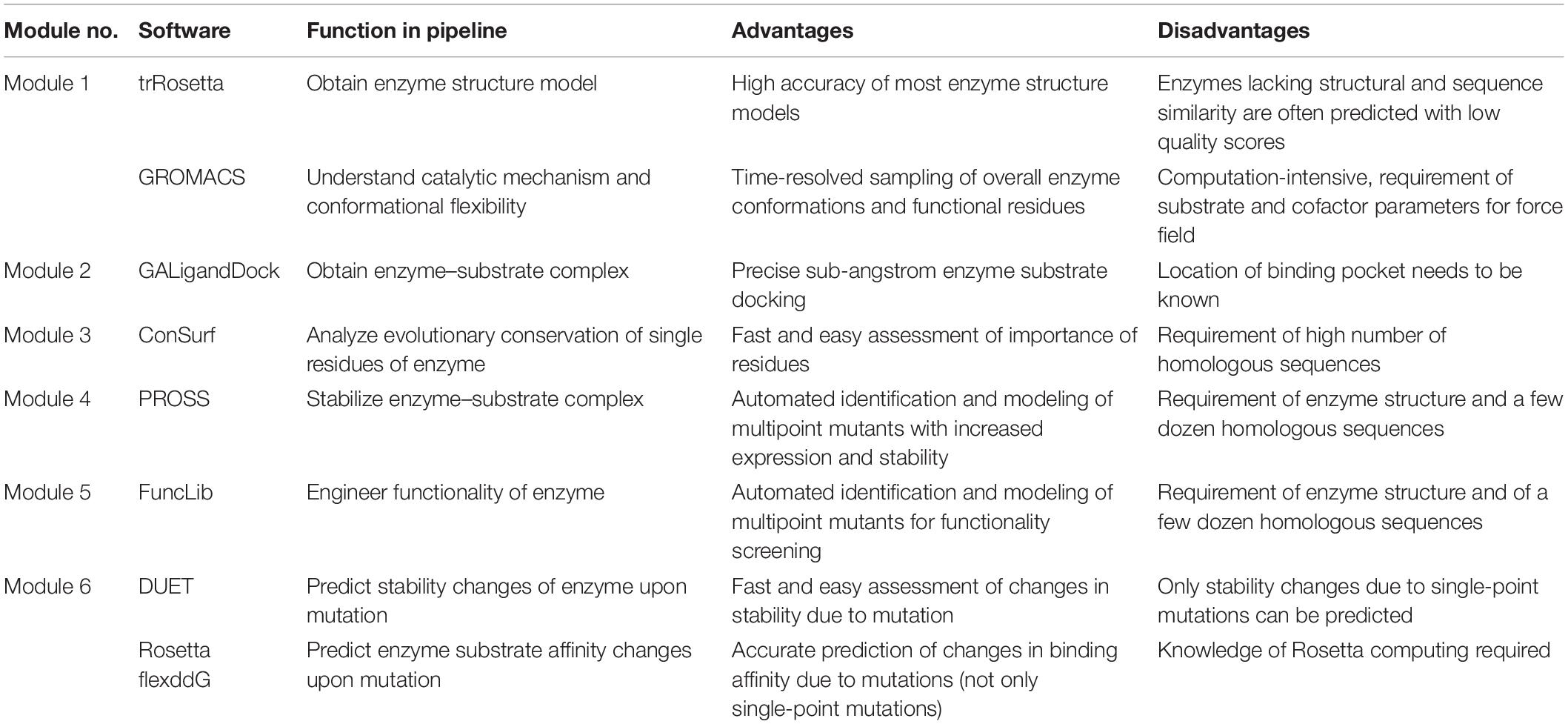
Table 2. Advantages and disadvantages of selected state-of-the-art software and online tools for computational protein engineering.
Module 1: Structure–Function Analysis of Enzymes
Before starting to engineer an enzyme, a deep understanding of the structural and dynamical underpinnings of enzymatic function has to be acquired. The availability of a crystal structure is still a prerequisite for successful rational engineering that can be exemplified by the discovery of mutations in the binding pocket changing the substrate specificity of the CvFAP (Figure 3A; Amer et al., 2020), the thioesterase TesA (Deng et al., 2020), and cADO (Bao et al., 2016). If the crystal structure has not been determined yet, computer-based protein structure prediction could generate models for structure–function analysis (Roy et al., 2010; Webb and Sali, 2016; Yang et al., 2020), but it is important to note that subsequent engineering heavily depends on the quality of the enzyme model (Kuhlman and Bradley, 2019). Recent advances in deep learning–based structure prediction are likely to change this situation completely, alleviating step-by-step the need for a crystal structure (Senior et al., 2020; Singh, 2020; Torrisi et al., 2020). The field of ab initio structure prediction has recently advanced tremendously through deep learning methods and that its accuracy even surpasses that of high-quality homology modeling and reaches the point of atomic accuracy. With this, it is quite likely that such models would be good starting points for protein design calculations, circumventing laborious structure determination by experimental methods.
In cases where the exact location of the binding pocket and active site of an enzyme is not known, MD simulation-based approaches can be applied to analyze substrate-binding trajectories and identify hidden binding pockets (Petřek et al., 2006; Stank et al., 2017; Stourac et al., 2019). The Caver 1.0 webserver, for example, was used to identify residues forming the access tunnel and substrate-binding cavity of the Serratia marcescens prodigiosin ligase PigC. Subsequent targeting of these residues in mutagenesis experiments revealed a double mutant with a shift in substrate preference by enhancing the catalytic efficiency (kcat) 3.4-fold for the pharmaceutically interesting short-chain prodiginines compared to the wild-type PigC (Brands et al., 2021).
Furthermore, understanding the catalytic mechanism of an enzyme can be helpful (Chen and Arnold, 2020). Simulating the MD of Segniliparus rugosus carboxylic acid reductase (SrCAR) (Qu et al., 2019a), which catalyzes the reduction of carboxylic acids to the corresponding aldehydes (Winkler, 2018; Figure 3B), sheds light on the structural underpinnings of catalytic mechanism and conformational flexibility. The information acquired by molecular dynamic simulation enabled the discovery of an SrCAR single-point mutant that showed an increase in enzymatic activity for benzoic acid by twofold (Qu et al., 2019b). This example highlights how studying the dynamic nature of catalysis in enzyme engineering can be used for optimization strategies.
Many enzymes form complexes by the assembly of protein subunits or monomers (Levy and Teichmann, 2013), as recently discovered for CvFAP (Lakavath et al., 2020), or interactions with additional molecules as cofactors to facilitate chemical conversions (Kara et al., 2014). Therefore, it is important to elucidate which residues are essential for such molecular interactions to restrain residue positions during the engineering process. Protein–protein interfaces and multimerization can be predicted by protein–protein docking (Baek et al., 2017, 2019; Vangaveti et al., 2020).
Module 2: Building the Enzyme–Substrate Complex
Modeling enzyme variants in the presence of the cognate substrate has shown to be helpful for both activity and specificity engineering (Jha et al., 2015; Grisewood et al., 2017; Risso et al., 2020). Hence, obtaining an enzyme–substrate complex with accurate substrate-binding poses or transition state complex is the first step in the proposed computational pipeline. Molecular docking is a widely used method in drug discovery for identification of substrate-binding poses by searching the conformational space for the best fit to the binding pocket of the protein (Pagadala et al., 2017) and can be applied similarly in enzyme engineering (Ebert and Pelletier, 2017). First molecular docking algorithms mainly focused on docking small molecules into a single static structure of a protein (rigid-body docking), hence immensely reducing the complexity of computation under the penalty of precision (Kuntz et al., 1982; Ebert and Pelletier, 2017). Over the years, molecular docking has advanced to take the dynamic nature of substrate binding and the conformational flexibility of enzymes into account (Pinzi and Rastelli, 2019). Such flexible docking (Carlson, 2002; Davis and Baker, 2009; Forli et al., 2016) or unbiased (Shan et al., 2011) and biased (Ebert et al., 2017; Kokh et al., 2020) MD-based docking approaches benefit from docking the substrate into an ensemble of structures rather than one single structure (static), therefore describing substrate-binding more accurately. Current methods in rigid-body, flexible, and MD-based substrate docking were compared elsewhere (Kotev et al., 2016). Still, engineering or design of an enzyme–substrate complex requires almost atomic-level accuracy, which may not be reached with the molecular docking methods mentioned previously. Recently, a novel approach called GALigandDock was reported that enabled small molecule docking with sub-angstrom accuracy in the Rosetta framework by force field optimization (Park et al., 2021). GALigandDock outperformed the top current docking approaches in a cross-docking benchmark set providing new directions for automated enzyme–substrate complex modeling.
Module 3: Identification of Design Positions
After the enzyme–substrate complex has been prepared, the next step is to identify residues (design positions or hotspots) that are allowed to mutate during the sequence design steps (see Modules 4 and 5). Computational tools that predict prominent interactions between enzyme and substrate can be used to analyze the basic molecular interaction network in the binding pocket (Jubb et al., 2017; Angles et al., 2020). A prediction of the importance of single residues or structural motifs can be achieved based on an evolutionary conservation analysis (Ashkenazy et al., 2016; Gil and Fiser, 2019; Jin et al., 2020), which can be helpful to assess suitability of residues as design positions. The ConSurf webserver for example was used to guide the engineering of PigC (see Module 1) to exclude those residues from mutagenesis experiments that showed high conservation scores as they are likely to be essential for enzymatic function (Brands et al., 2021). Depending on the engineering task, altering polarity and/or size of binding pocket residues has to be considered to increase activity or change specificity of the enzyme. It is important to keep in mind that changes in activity and substrate specificity are not always connected to mutations of the first shell. It has been shown that second shell mutations or even remote mutations can modulate functionality of enzymes (Trudeau et al., 2018; Wilding et al., 2019; Osuna, 2020). Interaction networks that may span the whole size of the enzyme facilitate conformational flexibility. With the aid of MD simulations and dynamic cross-correlation network analysis, such interaction networks can be identified and targeted for engineering (see also Module 4; Yu and Dalby, 2018, 2020). Additionally, biased MD simulations can be used to identify mutational hotspots by simulating substrate binding as shown for the cytochrome P450 enzyme CYP102A1 (BM3) binding palmitic acid (Ebert et al., 2017). In this study, simulating the steered movement of the substrate toward BM3 revealed all residues involved in substrate binding even a formally unknown Q73 of the second shell of the binding pocket. Its importance was proven by site-directed mutagenesis revealing a single-point mutant that showed a fivefold increase of the Michaelis constant (KM) compared to the wild-type BM3.
Module 4: Engineering Stability of Enzymes
For the engineering of protein stability, several strategies and computational tools have been reviewed (Liu et al., 2019). In general, engineering an enzyme to increase expression and thermostability represents a versatile starting point to diversify enzymatic function. The introduction of mutations into the wild-type structure is likely to interfere with configurational stability and hence functionality if a certain stability threshold is exceeded (Trudeau and Tawfik, 2019). From a biotechnological viewpoint, engineering the stability of a biocatalyst can improve its robustness against elevated temperature or organic solvents, which makes it possible to accelerate chemical conversion rates at industrial scale. Additionally, these enzymes may withstand the introduction of mutations that enhance enzymatic activity and specificity for a given substrate of interest.
To address this challenge, a combination of phylogenetic analysis and the outstanding capacity of the Rosetta software (Leaver-Fay et al., 2011) to screen and design conformational space of an enzyme scaffold enabled the identification of mutations that increased thermostability and expression levels. One application that was developed also as a webserver is called PROSS (Goldenzweig et al., 2016). PROSS uses sequence alignment to exclude mutations without occurrence in homologs sequences followed by modeling of mutant structures and Rosetta energy calculations of the native state to identify fewer than 10 candidates for experimental testing. Protein variants designed by PROSS showed dramatically increased heterologous protein expression yields underlining the ability of PROSS to optimize enzymes for heterologous expression in industrial microbial hosts as Escherichia coli (Goldenzweig et al., 2016). In a recently published study, PROSS was used to design three variants of the HIV-1 envelope glycoprotein gp140 with 17–45 mutations (Malladi et al., 2020). The gp140 variants maintained similar antigenicity profiles as the wild type and showed fourfold and twofold increases in protein yield, respectively. Additionally, PROSS stabilization was shown to be affective in designing enzyme variants of acetolactate synthase (AlsS) from Bacillus subtilis for increased solubility in 8% isobutanol while maintaining ∼80% of activity during a 5-day experiment (Sherkhanov et al., 2020). In this case, three mutant variants containing 20, 37, and 71 mutations, with and without three deletions (six mutant variants in total), respectively, were tested to identify one with increased solubility while maintaining activity in 8% isobutanol. These two examples highlight the applicability of PROSS-based protein design in biomedicine and biotechnology.
Alternatively, the FireProt webserver uses a similar approach that combines screening of stability changes (energy-based mutations) and back-to-consensus analysis (evolution-based mutations) that are summarized in a user-specific mutant library (Musil et al., 2017). FireProt was used, for example, to engineer a ketoreductase ChKRED12 for enhanced thermostability (Liu Y. et al., 2021). In this case, FireProt identified 12 (energy-based) and 17 (evolution-based) single-point mutations of ChKRED12, respectively. While the multipoint variants containing all identified mutations showed no activity, screening of 12 single-point mutants predicted with the evolution-based approach revealed four mutants with higher residual activity after 1.5 h of heat treatment at 50°C.
Both approaches, PROSS and FireProt, rely on Rosetta modeling and energy calculations but also show some differences as PROSS includes an option for specifying regions that will be excluded from the calculations as active sites or protein–protein interfaces. Additionally, PROSS designs a small library of multipoint mutants, whereas FireProt identifies a larger library of single-point mutations for experimental testing (Goldenzweig et al., 2016; Musil et al., 2017). To visualize changes in enzyme surface polarity due to the introduction of stabilizing mutations by PROSS and FireProt, the Protein-Sol web tool can be used (Hebditch and Warwicker, 2019).
An alternative strategy for stability engineering that requires a higher degree of computational skills is based on MD simulations and subsequent dynamic cross-correlation network analysis (Yu and Dalby, 2020). This approach was used to generate multipoint mutants of the E. coli transketolase (TK) based on four single mutations and a double-mutant variant (Yu and Dalby, 2018). The final quadruple mutant showed 10.2-fold increase in residual activity after 1-h incubation at 60°C.
Module 5: Engineering Activity and Specificity of Enzymes
While the search for stabilizing mutations takes the whole amino acid sequence of an enzyme into account (with exceptions), rational and semirational engineering of functionality focuses on residues that are directly or indirectly connected to the catalytic center or binding pocket of an enzyme. Assuming a simplistic correlation between binding pocket shape and the ligand structure based on polarity and non-covalent bonding, redesigning these properties allows the acceptance of novel substrates to bind to the enzyme and ideally being processed as the native substrate. This has been shown for substrate and activity engineering of several enzymes connected to biofuel production (see above). Of course, the reality is much more complex, and, in many cases, multiple mutations are required to substantially increase the activity toward a novel substrate (Grisewood et al., 2017; Khersonsky et al., 2018; Trudeau et al., 2018). Computational tools have been developed that target such a goal with sequence design algorithms and subsequent ranking of the energetics of enzyme variants (Pantazes et al., 2015; Amrein et al., 2017; Khersonsky et al., 2018; Lapidoth et al., 2018; Sumbalova et al., 2018). With the aid of the IPRO (iterative computational protein library redesign and optimization procedure) algorithm (Pantazes et al., 2015), for example, scientists were able to redesign the E. coli thioesterase TesA, which originally showed promiscuous activity to a wide range of substrates (Figure 3C). After four rounds of sequence design with IPRO, 54 TesA variants were screened experimentally, 3 and 27 of them showed an increased mole fraction of C12 and C8 fatty acids compared to the wild-type TesA, respectively (Grisewood et al., 2017).
Alternatively, the FuncLib webserver has been used recently to engineer the substrate specificity of the Salmonella enterica acetyl-CoA synthetase (ACS). Screening of 29 ACS designs revealed one multipoint mutant with five mutations in or adjacent to the binding pocket that showed increased specificity for the desired substrate glycolate by twofold and decreased specificity for the native substrate acetate by eightfold at the same time (Khersonsky et al., 2018; Trudeau et al., 2018). The FuncLib algorithm is based on a four-step workflow starting with a screening of the sequence space of the design positions of an enzyme input to allow only amino acid substitutions with at least a modest probability of occurrence in nature. Second, a mutant library in which all multipoint mutants of the allowed mutations is enumerated in Rosetta. The algorithm ranks the candidates after their stability and destabilizing mutations are excluded. To ensure the generation of a diverse set of multipoint mutants, the design candidates are compared based on the introduced mutations, and if similar designs occur, the less stable version(s) is (are) excluded to avoid clustering (Khersonsky et al., 2018), resulting in a condensed library (typically of a few dozen designs) for experimental screening or further pruning through additional computational steps. Forty-nine multipoint mutant variants of the phosphotriesterase (PTE) from Pseudomonas diminuta were tested, and 35 variants showed an increase in esterase efficiency up to 1,000-fold. As a more radical approach to engineer/design enzymes, the applicability of automated modular backbone assembly was recently reported to generate highly active enzymes with diverse substrate preferences (Lapidoth et al., 2018) and hence represent a platform for future directions of enzyme engineering and design. In both applications, FuncLib and modular assembly, no enzyme–substrate or transitions state complex was required.
An alternative web application, the HotSpot Wizard 3.0 webserver (Sumbalova et al., 2018), comprises a large number of computational tools for modeling and assessment of protein mutations for the generation of smart libraries. Again, Rosetta software is included to calculate energy changes upon mutations to select those mutations that have a stabilizing effect on the enzyme structure. Interestingly, with the third version of HotSpot Wizard, tools for enzyme structure prediction and model quality assessment were added to increase applicability for the vast amount of enzyme sequence data whose structure has not been determined yet. Success for the application of HotSpot Wizard 3.0 in enzyme engineering has been demonstrated recently on the examples of engineering the kinetic stability of a hyperthermostable β-mannanase (Liu Z. et al., 2021) and enzymatic efficiency of a lytic polysaccharide monooxygenase MtC1 (Guo et al., 2020).
Besides the computational methods described previously that can be classified as semirational engineering strategies, a tool for computer-aided directed evolution of enzymes (CADEE) has been reported (Amrein et al., 2017). The method connects automated high-throughput mutagenesis with computational screening based on the empirical valence bond (EVB) approach to identify substitutions that change the energetics of enzymatic activation barriers and therefore might increase activity for a given chemical reaction. On the downside, CADEE needs a calibrated reference state based on experimentally tested mutations to rigorously parameterize the EVB force field for high-quality prediction (Amrein et al., 2017). Interestingly, the EVB approach was successfully applied to identify variants of a de novo Kemp eliminase enzyme (Figure 3D) generated by FuncLib (Risso et al., 2020; see also Module 6).
Such pipelines in which fast bioinformatics and macromolecular modeling calculations are used to select designs for intensive but accurate transition-state modeling can be an important next step that increases the accuracy of enzyme-design methodology. The enzyme engineering applications described above represent a variety of approaches that can be used to engineer novel highly active and selective biocatalysts in silico for industrial fuel biomanufacturing.
Module 6: Screening for Stability, Affinity, and Activity
Depending on the approach chosen in Modules 4 and 5, the generated library of enzyme variants may exceed the size that can be screened experimentally. Furthermore, designing a diverse set of multimutation variants may require expensive gene synthesis when experimental mutagenesis will be too time-consuming. Therefore, computational screening of enzyme stability, substrate-binding affinity, or activity can speed up the process of identifying the best candidates for experimental testing and simultaneously decrease the research expenses. Nevertheless, prediction of the exact biophysical properties of an enzyme in terms of changes in stability (Pucci et al., 2018; Montanucci et al., 2019) and ligand-binding affinity (Wang et al., 2021) upon mutation are still a great challenge and therefore require intensive computation.
A variety of webservers were reported for fast prediction of stability changes upon single-point mutations mainly applying machine-learning approaches (Cheng et al., 2006; Pires et al., 2014a,b; Quan et al., 2016). A review comparing the most common machine learning–based approaches for protein stability prediction upon mutations was published recently (Fang, 2020). The impact of point mutations on dynamics of a protein and stability are provided by the DynaMut webserver (Rodrigues et al., 2018). These webservers can be useful for rational engineering of enzymatic function to screen single mutations for stabilizing effects. In contrast to the webservers mentioned previously, applications such as FireProt, PROSS, and FuncLib have an internal stability ranking with the Rosetta force field included. Thus, additional screening for stability changes is not necessary.
Similarly, the precision of applications for predicting the impact of mutations on binding affinity is growing steadily. A study by Aldeghi et al. (2018) compared different protein–ligand binding affinity prediction approaches based on a challenging benchmark set and showed that the Rosetta protocol (flex_ddG) (Barlow et al., 2018), although originally developed for prediction of protein–protein binding affinity changes upon mutations, produced comparable results to computation-intensive free energy calculations (Wang et al., 2021). A combination of both further increased the precision of the approach. Additionally, several webserver applications such as mCSM-lig (Pires et al., 2016) or KDEEP (Jiménez et al., 2018) were designed for the fast prediction of binding affinity changes upon single-point mutations based on machine learning algorithms.
Besides predictions of binding affinity changes, calculations of activation free energy changes of the transition state with an EVB approach proved to be useful for screening multipoint mutants of a Kemp eliminase GNCA4 designed by FuncLib (Risso et al., 2020). In this study, the 20 top-ranked GNCA4 variants were tested, and 4 variants showed enhanced kcat values of up to one magnitude compared to the GNCA4 WT (Figure 3D).
Still, accurate prediction on protein–ligand binding affinity changes upon mutations, especially if multiple mutations are involved, remains computationally demanding and requires a high amount of knowledge of the underlying methodology and biophysical parameters of ligand binding and enzymatic catalysis. Nevertheless, future improvements of computational enzyme engineering software led by a growing amount of experimental data and advanced machine learning–based predictions will further simplify and speed up the in silico analysis of mutant enzyme models, similarly, modern methods in drug design and drug delivery will speed up pharmacological applications.
Conclusion and Future Directions
Computational protein engineering promises a fast and efficient identification of enzyme variants with altered properties tailored for sustainable biomanufacturing of chemicals compared to experimental engineering techniques. The need for stability, specificity, and activity optimized biocatalysts drives the improvement of software along the engineering pipelines presented here. Still, proof of concept for the utilization of generalizable engineering pipelines including enzyme structure prediction, design, screening, and characterization of enzyme variants in silico is missing. The Rosetta modeling suite included in webserver applications and local software packages presents the option to perform and automate most tasks required in computational enzyme engineering by only one software. Implementation of automated molecular docking and screening platforms in webserver applications such as FuncLib could further increase the performance of Rosetta-based sequence design. The continuously increasing accuracy of protein structures prediction by machine learning–based approaches propels the development of fast and precise enzyme modeling and engineering pipelines to complement experimental engineering methodology or even open up the optimization of biocatalysts for which experimental techniques are missing. Complementing computational enzyme engineering with MD simulations provides powerful means to understand how mutations are affecting substrate binding and enzymatic activity. In the future, such computational approaches will accelerate the discovery of optimized protein-based solutions in general and of biocatalyst in particular for biotechnological and biomedical application. The computational pipelines described here can help to overcome the experimental limitations in metabolic pathway optimization on a protein level to enable, for example, industrial scale production of biofuels.
Author Contributions
MS wrote the article. SF critically revised the article. EB, PJ, and TD contributed to the design and writing of the manuscript. All authors discussed the results and contributed to the final manuscript.
Funding
Funded by the Land of Bavaria (contribution to DFG Project 324392634/TR221-INF; pipeline) and Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) Project 3 74031971/TRR 240-INF (TD).
Conflict of Interest
SF is a named inventor on patent filings regarding the PROSS and FuncLib methods and several proteins designed using these tools.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abraham, M. J., Murtola, T., Schulz, R., Páll, S., Smith, J. C., Hess, B., et al. (2015). GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25. doi: 10.1016/j.softx.2015.06.001
Aldeghi, M., Gapsys, V., and de Groot, B. L. (2018). Accurate estimation of ligand binding affinity changes upon protein mutation. ACS Cent. Sci. 4, 1708–1718. doi: 10.1021/acscentsci.8b00717
Amer, M., Wojcik, E. Z., Sun, C., Hoeven, R., Hughes, J. M. X., Faulkner, M., et al. (2020). Low carbon strategies for sustainable bio-alkane gas production and renewable energy. Energy Environ. Sci. 13, 1818–1831. doi: 10.1039/D0EE00095G
Amrein, B. A., Steffen-Munsberg, F., Szeler, I., Purg, M., Kulkarni, Y., and Kamerlin, S. C. L. (2017). CADEE: computer-aided directed evolution of enzymes. IUCrJ 4, 50–64. doi: 10.1107/S2052252516018017
Angles, R., Arenas-Salinas, M., García, R., Reyes-Suarez, J. A., and Pohl, E. (2020). GSP4PDB: a web tool to visualize, search and explore protein-ligand structural patterns. BMC Bioinformatics 21:85. doi: 10.1186/s12859-020-3352-x
Arnell, N. W., Lowe, J. A., Challinor, A. J., and Osborn, T. J. (2019). Global and regional impacts of climate change at different levels of global temperature increase. Clim. Change 155, 377–391. doi: 10.1007/s10584-019-02464-z
Ashkenazy, H., Abadi, S., Martz, E., Chay, O., Mayrose, I., Pupko, T., et al. (2016). ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 44, W344–W350. doi: 10.1093/nar/gkw408
Baek, M., Park, T., Heo, L., Park, C., and Seok, C. (2017). GalaxyHomomer: a web server for protein homo-oligomer structure prediction from a monomer sequence or structure. Nucleic Acids Res. 45, W320–W324. doi: 10.1093/nar/gkx246
Baek, M., Park, T., Woo, H., and Seok, C. (2019). Prediction of protein oligomer structures using GALAXY in CASP13. Proteins Struct. Funct. Bioinform. 87, 1233–1240. doi: 10.1002/prot.25814
Bao, L., Li, J. J., Jia, C., Li, M., and Lu, X. (2016). Structure-oriented substrate specificity engineering of aldehyde-deformylating oxygenase towards aldehydes carbon chain length. Biotechnol. Biofuels 9:185. doi: 10.1186/s13068-016-0596-9
Barlow, K. A., Ó Conchúir, S., Thompson, S., Suresh, P., Lucas, J. E., Heinonen, M., et al. (2018). Flex ddG: rosetta ensemble-based estimation of changes in protein–protein binding affinity upon mutation. J. Phys. Chem. B 122, 5389–5399. doi: 10.1021/acs.jpcb.7b11367
Bauer, D., Zachos, I., and Sieber, V. (2020). Production of propene from n-butanol: a three-step cascade utilizing the cytochrome P450 fatty acid decarboxylase OleTJE. ChemBioChem 21, 3273–3281. doi: 10.1002/cbic.202000378
Bernard, A., Domergue, F., Pascal, S., Jetter, R., Renne, C., Faure, J.-D., et al. (2012). Reconstitution of plant alkane biosynthesis in yeast demonstrates that Arabidopsis ECERIFERUM1 and ECERIFERUM3 are core components of a very-long-chain alkane synthesis complex. Plant Cell 24, 3106–3118. doi: 10.1105/tpc.112.099796
Brands, S., Sikkens, J. G., Davari, M. D., Brass, H. U. C., Klein, A. S., Pietruszka, J., et al. (2021). Understanding substrate binding and the role of gatekeeping residues in PigC access tunnels. Chem. Commun. 57, 2681–2684. doi: 10.1039/D0CC08226K
Carlin, D. A., Hapig-Ward, S., Chan, B. W., Damrau, N., Riley, M., Caster, R. W., et al. (2017). Thermal stability and kinetic constants for 129 variants of a family 1 glycoside hydrolase reveal that enzyme activity and stability can be separately designed. PLoS One 12:e0176255. doi: 10.1371/journal.pone.0176255
Carlson, H. A. (2002). Protein flexibility and drug design: how to hit a moving target. Curr. Opin. Chem. Biol. 6, 447–452. doi: 10.1016/S1367-5931(02)00341-1
Chen, K., and Arnold, F. H. (2020). Engineering new catalytic activities in enzymes. Nat. Catal. 3, 203–213. doi: 10.1038/s41929-019-0385-5
Cheng, J., Randall, A., and Baldi, P. (2006). Prediction of protein stability changes for single-site mutations using support vector machines. Proteins Struct. Funct. Bioinform. 62, 1125–1132. doi: 10.1002/prot.20810
Chowdhury, R., and Maranas, C. D. (2020). From directed evolution to computational enzyme engineering—a review. AIChE J. 66:e16847. doi: 10.1002/aic.16847
Davis, I. W., and Baker, D. (2009). RosettaLigand docking with full ligand and receptor flexibility. J. Mol. Biol. 385, 381–392. doi: 10.1016/j.jmb.2008.11.010
DeLoache, W. C., Russ, Z. N., Narcross, L., Gonzales, A. M., Martin, V. J. J., and Dueber, J. E. (2015). An enzyme-coupled biosensor enables (S)-reticuline production in yeast from glucose. Nat. Chem. Biol. 11, 465–471. doi: 10.1038/nchembio.1816
Deng, X., Chen, L., Hei, M., Liu, T., Feng, Y., and Yang, G.-Y. (2020). Structure-guided reshaping of the acyl binding pocket of ‘TesA thioesterase enhances octanoic acid production in E. coli. Metab. Eng. 61, 24–32. doi: 10.1016/j.ymben.2020.04.010
Dennig, A., Kuhn, M., Tassoti, S., Thiessenhusen, A., Gilch, S., Bülter, T., et al. (2015). Oxidative decarboxylierung von kurzkettigen fettsäuren zu 1-alkenen. Angew. Chem. 127, 8943–8946. doi: 10.1002/ange.201502925
Dou, J., Vorobieva, A. A., Sheffler, W., Doyle, L. A., Park, H., Bick, M. J., et al. (2018). De novo design of a fluorescence-activating β-barrel. Nature 561, 485–491. doi: 10.1038/s41586-018-0509-0
Ebert, M. C., and Pelletier, J. N. (2017). Computational tools for enzyme improvement: why everyone can – and should – use them. Curr. Opin. Chem. Biol. 37, 89–96. doi: 10.1016/j.cbpa.2017.01.021
Ebert, M. C. C. J. C., Guzman Espinola, J., Lamoureux, G., and Pelletier, J. N. (2017). Substrate-specific screening for mutational hotspots using biased molecular dynamics simulations. ACS Catal. 7, 6786–6797. doi: 10.1021/acscatal.7b02634
Erb, T. J., Jones, P. R., and Bar-Even, A. (2017). Synthetic metabolism: metabolic engineering meets enzyme design. Curr. Opin. Chem. Biol. 37, 56–62. doi: 10.1016/j.cbpa.2016.12.023
Eser, B. E., Poborsky, M., Dai, R., Kishino, S., Ljubic, A., Takeuchi, M., et al. (2020). Rational engineering of hydratase from Lactobacillus acidophilus reveals critical residues directing substrate specificity and regioselectivity. ChemBioChem 21, 550–563. doi: 10.1002/cbic.201900389
Fang, J. (2020). A critical review of five machine learning-based algorithms for predicting protein stability changes upon mutation. Brief. Bioinform. 21, 1285–1292. doi: 10.1093/bib/bbz071
Farinas, E. T., Bulter, T., and Arnold, F. H. (2001). Directed enzyme evolution. Curr. Opin. Biotechnol. 12, 545–551. doi: 10.1016/S0958-1669(01)00261-0
Forli, S., Huey, R., Pique, M. E., Sanner, M. F., Goodsell, D. S., and Olson, A. J. (2016). Computational protein–ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 11, 905–919. doi: 10.1038/nprot.2016.051
Gil, N., and Fiser, A. (2019). The choice of sequence homologs included in multiple sequence alignments has a dramatic impact on evolutionary conservation analysis. Bioinformatics 35, 12–19. doi: 10.1093/bioinformatics/bty523
Goldenzweig, A., Goldsmith, M., Hill, S. E., Gertman, O., Laurino, P., Ashani, Y., et al. (2016). Automated structure- and sequence-based design of proteins for high bacterial expression and stability. Mol. Cell 63, 337–346. doi: 10.1016/J.MOLCEL.2016.06.012
Grisewood, M. J., Hernández-Lozada, N. J., Thoden, J. B., Gifford, N. P., Mendez-Perez, D., Schoenberger, H. A., et al. (2017). Computational redesign of Acyl-ACP thioesterase with improved selectivity toward medium-chain-length fatty acids. ACS Catal. 7, 3837–3849. doi: 10.1021/acscatal.7b00408
Guo, X., An, Y., Chai, C., Sang, J., Jiang, L., Lu, F., et al. (2020). Construction of the R17L mutant of MtC1LPMO for improved lignocellulosic biomass conversion by rational point mutation and investigation of the mechanism by molecular dynamics simulations. Bioresour. Technol. 317, 124024. doi: 10.1016/j.biortech.2020.124024
Hebditch, M., and Warwicker, J. (2019). Web-based display of protein surface and pH-dependent properties for assessing the developability of biotherapeutics. Sci. Rep. 9:1969. doi: 10.1038/s41598-018-36950-8
Hoegh-Guldberg, O., Jacob, D., Taylor, M., Guillén Bolaños, T., Bindi, M., Brown, S., et al. (2019). The human imperative of stabilizing global climate change at 1.5∘C. Science 365:eaaw6974. doi: 10.1126/science.aaw6974
Jha, R. K., Chakraborti, S., Kern, T. L., Fox, D. T., and Strauss, C. E. M. (2015). Rosetta comparative modeling for library design: engineering alternative inducer specificity in a transcription factor. Proteins Struct. Funct. Bioinform. 83, 1327–1340. doi: 10.1002/prot.24828
Jiménez, J., Škalič, M., Martínez-Rosell, G., and De Fabritiis, G. (2018). KDEEP: protein–ligand absolute binding affinity prediction via 3D-convolutional neural networks. J. Chem. Inf. Model. 58, 287–296. doi: 10.1021/acs.jcim.7b00650
Jin, X., Liao, Q., Wei, H., Zhang, J., and Liu, B. (2020). SMI-BLAST: a novel supervised search framework based on PSI-BLAST for protein remote homology detection. Bioinformatics 37, 913–920. doi: 10.1093/bioinformatics/btaa772
Jubb, H. C., Higueruelo, A. P., Ochoa-Montaño, B., Pitt, W. R., Ascher, D. B., and Blundell, T. L. (2017). Arpeggio: a web server for calculating and visualising interatomic interactions in protein structures. J. Mol. Biol. 429, 365–371. doi: 10.1016/j.jmb.2016.12.004
Kallio, P., Pásztor, A., Akhtar, M. K., and Jones, P. R. (2014a). Renewable jet fuel. Curr. Opin. Biotechnol. 26, 50–55. doi: 10.1016/j.copbio.2013.09.006
Kallio, P., Pásztor, A., Thiel, K., Akhtar, M. K., and Jones, P. R. (2014b). An engineered pathway for the biosynthesis of renewable propane. Nat. Commun. 5:4731. doi: 10.1038/ncomms5731
Kalyoncu, E., Ahan, R. E., Olmez, T. T., and Safak Seker, U. O. (2017). Genetically encoded conductive protein nanofibers secreted by engineered cells. RSC Adv. 7, 32543–32551. doi: 10.1039/C7RA06289C
Kara, S., Schrittwieser, J. H., Hollmann, F., and Ansorge-Schumacher, M. B. (2014). Recent trends and novel concepts in cofactor-dependent biotransformations. Appl. Microbiol. Biotechnol. 98, 1517–1529. doi: 10.1007/s00253-013-5441-5
Khersonsky, O., Lipsh, R., Avizemer, Z., Ashani, Y., Goldsmith, M., Leader, H., et al. (2018). Automated design of efficient and functionally diverse enzyme repertoires. Mol. Cell 72, 178–186.e5. doi: 10.1016/j.molcel.2018.08.033
Kokh, D. B., Doser, B., Richter, S., Ormersbach, F., Cheng, X., and Wade, R. C. (2020). A workflow for exploring ligand dissociation from a macromolecule: efficient random acceleration molecular dynamics simulation and interaction fingerprint analysis of ligand trajectories. J. Chem. Phys. 153:125102. doi: 10.1063/5.0019088
Kotev, M., Soliva, R., and Orozco, M. (2016). Challenges of docking in large, flexible and promiscuous binding sites. Bioorg. Med. Chem. 24, 4961–4969. doi: 10.1016/j.bmc.2016.08.010
Kuhlman, B., and Bradley, P. (2019). Advances in protein structure prediction and design. Nat. Rev. Mol. Cell Biol. 20, 681–697. doi: 10.1038/s41580-019-0163-x
Kunjapur, A. M., and Prather, K. L. J. (2019). Development of a vanillate biosensor for the vanillin biosynthesis pathway in E. coli. ACS Synth. Biol. 8, 1958–1967. doi: 10.1021/acssynbio.9b00071
Kuntz, I. D., Blaney, J. M., Oatley, S. J., Langridge, R., and Ferrin, T. E. (1982). A geometric approach to macromolecule-ligand interactions. J. Mol. Biol. 161, 269–288. doi: 10.1016/0022-2836(82)90153-X
Lakavath, B., Hedison, T. M., Heyes, D. J., Shanmugam, M., Sakuma, M., Hoeven, R., et al. (2020). Radical-based photoinactivation of fatty acid photodecarboxylases. Anal. Biochem. 600:113749. doi: 10.1016/j.ab.2020.113749
Lapidoth, G., Khersonsky, O., Lipsh, R., Dym, O., Albeck, S., Rogotner, S., et al. (2018). Highly active enzymes by automated combinatorial backbone assembly and sequence design. Nat. Commun. 9:2780. doi: 10.1038/s41467-018-05205-5
Leaver-Fay, A., Tyka, M., Lewis, S. M., Lange, O. F., Thompson, J., Jacak, R., et al. (2011). ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 487, 545–574. doi: 10.1016/B978-0-12-381270-4.00019-6
Leman, J. K., Weitzner, B. D., Lewis, S. M., Adolf-Bryfogle, J., Alam, N., Alford, R. F., et al. (2020). Macromolecular modeling and design in Rosetta: recent methods and frameworks. Nat. Methods 17, 665–680. doi: 10.1038/s41592-020-0848-2
Levy, E. D., and Teichmann, S. A. (2013). “Chapter Two - structural, evolutionary, and assembly principles of protein oligomerization,” in Oligomerization in Health and Disease, eds J. Giraldo and T. S. Ciruela (Cambridge, MA: Academic Press), 25–51. doi: 10.1016/B978-0-12-386931-9.00002-7
Liu, K., and Li, S. (2020). Biosynthesis of fatty acid-derived hydrocarbons: perspectives on enzymology and enzyme engineering. Curr. Opin. Biotechnol. 62, 7–14. doi: 10.1016/j.copbio.2019.07.005
Liu, Q., Xun, G., and Feng, Y. (2019). The state-of-the-art strategies of protein engineering for enzyme stabilization. Biotechnol. Adv. 37, 530–537. doi: 10.1016/j.biotechadv.2018.10.011
Liu, Y., Li, Z.-Y., Guo, C., Cui, C., Lin, H., and Wu, Z.-L. (2021). Enhancing the thermal stability of ketoreductase ChKRED12 using the FireProt web server. Process Biochem. 101, 207–212. doi: 10.1016/j.procbio.2020.11.018
Liu, Z., Liang, Q., Wang, P., Kong, Q., Fu, X., and Mou, H. (2021). Improving the kinetic stability of a hyperthermostable β-mannanase by a rationally combined strategy. Int. J. Biol. Macromol. 167, 405–414. doi: 10.1016/j.ijbiomac.2020.11.202
Ma, F., Guo, T., Zhang, Y., Bai, X., Li, C., Lu, Z., et al. (2021). An ultrahigh-throughput screening platform based on flow cytometric droplet sorting for mining novel enzymes from metagenomic libraries. Environ. Microbiol. 23, 996–1008.
Malladi, S. K., Schreiber, D., Pramanick, I., Sridevi, M. A., Goldenzweig, A., Dutta, S., et al. (2020). One-step sequence and structure-guided optimization of HIV-1 envelope gp140. Curr. Res. Struct. Biol. 2, 45–55. doi: 10.1016/j.crstbi.2020.04.001
Marcheschi, R. J., Gronenberg, L. S., and Liao, J. C. (2013). Protein engineering for metabolic engineering: current and next-generation tools. Biotechnol. J. 8, 545–555. doi: 10.1002/biot.201200371
Montanucci, L., Martelli, P. L., Ben-Tal, N., and Fariselli, P. (2019). A natural upper bound to the accuracy of predicting protein stability changes upon mutations. Bioinformatics 35, 1513–1517. doi: 10.1093/bioinformatics/bty880
Musil, M., Stourac, J., Bendl, J., Brezovsky, J., Prokop, Z., Zendulka, J., et al. (2017). FireProt: web server for automated design of thermostable proteins. Nucleic Acids Res. 45, W393–W399. doi: 10.1093/nar/gkx285
Nolan, C., Overpeck, J. T., Allen, J. R. M., Anderson, P. M., Betancourt, J. L., Binney, H. A., et al. (2018). Past and future global transformation of terrestrial ecosystems under climate change. Science 361, 920L–923. doi: 10.1126/science.aan5360
Osuna, S. (2020). The challenge of predicting distal active site mutations in computational enzyme design. WIREs Comput. Mol. Sci. 11:e1502. doi: 10.1002/wcms.1502
Pagadala, N. S., Syed, K., and Tuszynski, J. (2017). Software for molecular docking: a review. Biophys. Rev. 9, 91–102. doi: 10.1007/s12551-016-0247-1
Pantazes, R. J., Grisewood, M. J., Li, T., Gifford, N. P., and Maranas, C. D. (2015). The iterative protein redesign and optimization (IPRO) suite of programs. J. Comput. Chem. 36, 251–263. doi: 10.1002/jcc.23796
Park, H., Zhou, G., Baek, M., Baker, D., and DiMaio, F. (2021). Force field optimization guided by small molecule crystal lattice data enables consistent sub-angstrom protein–ligand docking. J. Chem. Theory Comput. 17, 2000–2010. doi: 10.1021/acs.jctc.0c01184
Petřek, M., Otyepka, M., Banáš, P., Košinová, P., Koča, J., and Damborský, J. (2006). CAVER: a new tool to explore routes from protein clefts, pockets and cavities. BMC Bioinformatics 7:316. doi: 10.1186/1471-2105-7-316
Pinzi, L., and Rastelli, G. (2019). Molecular docking: shifting paradigms in drug discovery. Int. J. Mol. Sci. 20:4331. doi: 10.3390/ijms20184331
Pires, D. E. V., Ascher, D. B., and Blundell, T. L. (2014a). DUET: a server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res. 42, W314–W319. doi: 10.1093/nar/gku411
Pires, D. E. V., Ascher, D. B., and Blundell, T. L. (2014b). mCSM: predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics 30, 335–342. doi: 10.1093/bioinformatics/btt691
Pires, D. E. V., Blundell, T. L., and Ascher, D. B. (2016). mCSM-lig: quantifying the effects of mutations on protein-small molecule affinity in genetic disease and emergence of drug resistance. Sci. Rep. 6:29575. doi: 10.1038/srep29575
Pucci, F., Bernaerts, K. V., Kwasigroch, J. M., and Rooman, M. (2018). Quantification of biases in predictions of protein stability changes upon mutations. Bioinformatics 34, 3659–3665. doi: 10.1093/bioinformatics/bty348
Pucci, F., and Rooman, M. (2016). Towards an accurate prediction of the thermal stability of homologous proteins. J. Biomol. Struct. Dyn. 34, 1132–1142. doi: 10.1080/07391102.2015.1073631
Qu, G., Fu, M., Zhao, L., Liu, B., Liu, P., Fan, W., et al. (2019a). Computational insights into the catalytic mechanism of bacterial carboxylic acid reductase. J. Chem. Inf. Model. 59, 832–841. doi: 10.1021/acs.jcim.8b00763
Qu, G., Liu, B., Zhang, K., Jiang, Y., Guo, J., Wang, R., et al. (2019b). Computer-assisted engineering of the catalytic activity of a carboxylic acid reductase. J. Biotechnol. 306, 97–104. doi: 10.1016/j.jbiotec.2019.09.006
Quan, L., Lv, Q., and Zhang, Y. (2016). STRUM: structure-based prediction of protein stability changes upon single-point mutation. Bioinformatics 32, 2936–2946. doi: 10.1093/bioinformatics/btw361
Risso, V. A., Romero-Rivera, A., Gutierrez-Rus, L. I., Ortega-Muñoz, M., Santoyo-Gonzalez, F., Gavira, J. A., et al. (2020). Enhancing a de novo enzyme activity by computationally-focused ultra-low-throughput screening. Chem. Sci. 11, 6134–6148. doi: 10.1039/D0SC01935F
Rodionova, M. V., Poudyal, R. S., Tiwari, I., Voloshin, R. A., Zharmukhamedov, S. K., Nam, H. G., et al. (2017). Biofuel production: challenges and opportunities. Int. J. Hydrogen Energy 42, 8450–8461. doi: 10.1016/j.ijhydene.2016.11.125
Rodrigues, C. H. M., Pires, D. E. V., and Ascher, D. B. (2018). DynaMut: predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res. 46, W350–W355. doi: 10.1093/nar/gky300
Romero-Rivera, A., Garcia-Borràs, M., and Osuna, S. (2017). Computational tools for the evaluation of laboratory-engineered biocatalysts. Chem. Commun. 53, 284–297. doi: 10.1039/C6CC06055B
Roy, A., Kucukural, A., and Zhang, Y. (2010). I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 5, 725–738. doi: 10.1038/nprot.2010.5
Salomon-Ferrer, R., Case, D. A., and Walker, R. C. (2013). An overview of the Amber biomolecular simulation package. WIREs Comput. Mol. Sci. 3, 198–210. doi: 10.1002/wcms.1121
Schäfer, A. W., Barrett, S. R. H., Doyme, K., Dray, L. M., Gnadt, A. R., Self, R., et al. (2019). Technological, economic and environmental prospects of all-electric aircraft. Nat. Energy 4, 160–166. doi: 10.1038/s41560-018-0294-x
Schirmer, A., Rude, M. A., Li, X., Popova, E., and del Cardayre, S. B. (2010). Microbial biosynthesis of alkanes. Science 329, 559–562. doi: 10.1126/science.1187936
Senior, A. W., Evans, R., Jumper, J., Kirkpatrick, J., Sifre, L., Green, T., et al. (2020). Improved protein structure prediction using potentials from deep learning. Nature 577, 706–710. doi: 10.1038/s41586-019-1923-7
Sequeiros-Borja, C. E., Surpeta, B., and Brezovsky, J. (2020). Recent advances in user-friendly computational tools to engineer protein function. Brief. Bioinform. 22:bbaa150. doi: 10.1093/bib/bbaa150
Shan, Y., Kim, E. T., Eastwood, M. P., Dror, R. O., Seeliger, M. A., and Shaw, D. E. (2011). How does a drug molecule find its target binding site? J. Am. Chem. Soc. 133, 9181–9183. doi: 10.1021/ja202726y
Sheldon, R. A., and Woodley, J. M. (2018). Role of biocatalysis in sustainable chemistry. Chem. Rev. 118, 801–838. doi: 10.1021/acs.chemrev.7b00203
Sherkhanov, S., Korman, T. P., Chan, S., Faham, S., Liu, H., Sawaya, M. R., et al. (2020). Isobutanol production freed from biological limits using synthetic biochemistry. Nat. Commun. 11:4292. doi: 10.1038/s41467-020-18124-1
Sorigué, D., Légeret, B., Cuiné, S., Blangy, S., Moulin, S., Billon, E., et al. (2017). An algal photoenzyme converts fatty acids to hydrocarbons. Science 357, 903–907. doi: 10.1126/science.aan6349
Stank, A., Kokh, D. B., Horn, M., Sizikova, E., Neil, R., Panecka, J., et al. (2017). TRAPP webserver: predicting protein binding site flexibility and detecting transient binding pockets. Nucleic Acids Res. 45, W325–W330. doi: 10.1093/nar/gkx277
Stourac, J., Vavra, O., Kokkonen, P., Filipovic, J., Pinto, G., Brezovsky, J., et al. (2019). Caver Web 1.0: identification of tunnels and channels in proteins and analysis of ligand transport. Nucleic Acids Res. 47, W414–W422. doi: 10.1093/nar/gkz378
Sulzbach, M., and Kunjapur, A. M. (2020). The pathway less traveled: engineering biosynthesis of nonstandard functional groups. Trends Biotechnol. 38, 532–545. doi: 10.1016/j.tibtech.2019.12.014
Sumbalova, L., Stourac, J., Martinek, T., Bednar, D., and Damborsky, J. (2018). HotSpot Wizard 3.0: web server for automated design of mutations and smart libraries based on sequence input information. Nucleic Acids Res. 46, W356–W362. doi: 10.1093/nar/gky417
Surpeta, B., Sequeiros-Borja, C. E., and Brezovsky, J. (2020). Dynamics, a powerful component of current and future in silico approaches for protein design and engineering. Int. J. Mol. Sci. 21:2713. doi: 10.3390/ijms21082713
Torrisi, M., Pollastri, G., and Le, Q. (2020). Deep learning methods in protein structure prediction. Comput. Struct. Biotechnol. J. 18, 1301–1310. doi: 10.1016/j.csbj.2019.12.011
Trudeau, D. L., Edlich-Muth, C., Zarzycki, J., Scheffen, M., Goldsmith, M., Khersonsky, O., et al. (2018). Design and in vitro realization of carbon-conserving photorespiration. Proc. Natl. Acad. Sci. U.S.A. 115, E11455–E11464. doi: 10.1073/pnas.1812605115
Trudeau, D. L., and Tawfik, D. S. (2019). Protein engineers turned evolutionists—the quest for the optimal starting point. Curr. Opin. Biotechnol. 60, 46–52. doi: 10.1016/j.copbio.2018.12.002
Vangaveti, S., Vreven, T., Zhang, Y., and Weng, Z. (2020). Integrating ab initio and template-based algorithms for protein–protein complex structure prediction. Bioinformatics 36, 751–757. doi: 10.1093/bioinformatics/btz623
Wang, D. D., Zhu, M., and Yan, H. (2021). Computationally predicting binding affinity in protein–ligand complexes: free energy-based simulations and machine learning-based scoring functions. Brief. Bioinform. 22:bbaa107. doi: 10.1093/bib/bbaa107
Wang, X.-C., Klemeš, J. J., Dong, X., Fan, W., Xu, Z., Wang, Y., et al. (2019). Air pollution terrain nexus: a review considering energy generation and consumption. Renew. Sustain. Energy Rev. 105, 71–85. doi: 10.1016/j.rser.2019.01.049
Webb, B., and Sali, A. (2016). Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinformatics 54, 5.6.1–5.6.37. doi: 10.1002/cpbi.3
Wilding, M., Hong, N., Spence, M., Buckle, A. M., and Jackson, C. J. (2019). Protein engineering: the potential of remote mutations. Biochem. Soc. Trans. 47, 701–711. doi: 10.1042/BST20180614
Winkler, M. (2018). Carboxylic acid reductase enzymes (CARs). Curr. Opin. Chem. Biol. 43, 23–29. doi: 10.1016/j.cbpa.2017.10.006
Woodley, J. M. (2020). New frontiers in biocatalysis for sustainable synthesis. Curr. Opin. Green Sustain. Chem. 21, 22–26. doi: 10.1016/j.cogsc.2019.08.006
Xiong, D., Lu, S., Wu, J., Liang, C., Wang, W., Wang, W., et al. (2017). Improving key enzyme activity in phenylpropanoid pathway with a designed biosensor. Metab. Eng. 40, 115–123. doi: 10.1016/j.ymben.2017.01.006
Xu, K., Lv, B., Huo, Y. X., and Li, C. (2018). Toward the lowest energy consumption and emission in biofuel production: combination of ideal reactors and robust hosts. Curr. Opin. Biotechnol. 50, 19–24. doi: 10.1016/j.copbio.2017.08.011
Yan, W., Hunt, L., Sheng, Q., and Szlavnies, Z. (2000). R: Development Core Team (2005): R: A Language and Environment Interaction for Statistical Computing. Vienna: R Found. Stat. Comput.
Yang, J., Anishchenko, I., Park, H., Peng, Z., Ovchinnikov, S., and Baker, D. (2020). Improved protein structure prediction using predicted interresidue orientations. Proc. Natl. Acad. Sci. U.S.A. 117, 1496–1503. doi: 10.1073/pnas.1914677117
Yu, H., and Dalby, P. A. (2018). Exploiting correlated molecular-dynamics networks to counteract enzyme activity–stability trade-off. Proc. Natl. Acad. Sci. U.S.A. 115, E12192–E12200. doi: 10.1073/pnas.1812204115
Yu, H., and Dalby, P. A. (2020). “Chapter Two - a beginner’s guide to molecular dynamics simulations and the identification of cross-correlation networks for enzyme engineering,” in Enzyme Engineering and Evolution: General Methods, ed. D. S. Tawfik (Cambridge, MA: Academic Press), 15–49. doi: 10.1016/bs.mie.2020.04.020
Yu, H., Yan, Y., Zhang, C., and Dalby, P. A. (2017). Two strategies to engineer flexible loops for improved enzyme thermostability. Sci. Rep. 7:41212. doi: 10.1038/srep41212
Yunus, I. S., Wichmann, J., Wördenweber, R., Lauersen, K. J., Kruse, O., and Jones, P. R. (2018). Synthetic metabolic pathways for photobiological conversion of CO 2 into hydrocarbon fuel. Metab. Eng. 49, 201–211. doi: 10.1016/j.ymben.2018.08.008
Keywords: computational, enzyme, engineering, design, biomanufacturing, biofuel, microbes, metabolism
Citation: Scherer M, Fleishman SJ, Jones PR, Dandekar T and Bencurova E (2021) Computational Enzyme Engineering Pipelines for Optimized Production of Renewable Chemicals. Front. Bioeng. Biotechnol. 9:673005. doi: 10.3389/fbioe.2021.673005
Received: 26 February 2021; Accepted: 06 May 2021;
Published: 15 June 2021.
Edited by:
Stephan Klähn, Helmholtz Centre for Environmental Research (UFZ), GermanyReviewed by:
John Michael Woodley, Technical University of Denmark, DenmarkMartin Weissenborn, Leibniz Institute of Plant Biochemistry, Germany
Copyright © 2021 Scherer, Fleishman, Jones, Dandekar and Bencurova. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Patrik R. Jones, cC5qb25lc0BpbXBlcmlhbC5hYy51aw==; Thomas Dandekar, ZGFuZGVrYXJAYmlvemVudHJ1bS51bmktd3VlcnpidXJnLmRl; Elena Bencurova, ZWxlbmEuYmVuY3Vyb3ZhQHVuaS13dWVyemJ1cmcuZGU=