 Maxime Fages-Lartaud
Maxime Fages-Lartaud Lisa Tietze
Lisa Tietze Florence Elie1
Florence Elie1 Rahmi Lale
Rahmi Lale- 1Department of Biotechnology, Norwegian University of Science and Technology, Trondheim, Norway
- 2United Scientists CORE (Limited), Dunedin, New Zealand
Fluorescent proteins are essential reporters in cell and molecular biology. Here, we found that red-fluorescent proteins possess an alternative translation initiation site that produces a short functional protein isoform in both prokaryotes and eukaryotes. The short isoform creates significant background fluorescence that biases the outcome of expression studies. In this study, we identified the short protein isoform, traced its origin, and determined the extent of the issue within the family of red fluorescent protein. Our analysis showed that the short isoform defect of the red fluorescent protein family may affect the interpretation of many published studies. We provided a re-engineered mCherry variant that lacks background expression as an improved tool for imaging and protein expression studies.
Introduction
The discovery of fluorescent proteins has played a major role in unraveling details of cellular functions (Day and Davidson, 2009; Chudakov et al., 2010). Fluorescent proteins display structural similarities, such as a fully amino acid-encoded chromophore present on an α-helix that is tightly packed into an 11-stranded ß-barrel (Ranganathan et al., 2000; Wachter et al., 2010). The chromophore is created by the autocatalytic cyclization of an amino acid triad (Gross et al., 2000; Shu et al., 2006; Wachter et al., 2010). The discovery of DsRed expanded the color range of fluorescent proteins to include red wavelengths (Matz et al., 1999). The natural versions of fluorescent proteins have been subject to a multitude of modifications to obtain different colors (Labas et al., 2002; Mishin et al., 2008; Wachter et al., 2010; Subach and Verkhusha, 2012) and improve their properties, such as solubility, maturation, stability, quantum yield, monomeric state, or the ability to uptake a fusion partner (Lauff and Hofer, 1984; Bevis and Glick, 2002; Campbell et al., 2002; Shaner et al., 2004; Rodriguez et al., 2017). This diversity of engineered fluorescent proteins have emerged as invaluable tools for molecular and cell biology, as they are excellent reporters for gene expression and subcellular protein localization in various biological systems (Day and Davidson, 2009; Chudakov et al., 2010; Rodriguez et al., 2017).
In a previous study, we used mCherry as one of the reporters for the development of a universal gene expression method that employs 200 random nucleotides as regulatory sequence to drive coding sequence expression (Lale et al., 1101). The efficiency of the method is gene- and context-dependent, but it usually yields between 30% and 40% of successful protein expression in Escherichia coli (Lale et al., 1101). However, when mCherry is used as a reporter, we observe fluorescence in about 65% of E. coli clones (Supplementary Figure S1). In addition, a large-scale analysis of the transcription start sites of these clones showed a nonnegligible fraction of leaderless mRNA sequences (Lale et al., 1101). Unsettled by these high proportions, our suspicion turned to the reporter sequence itself. We hypothesized that internal Shine–Dalgarno (SD) sequences within the reporter sequence followed by methionine codons just downstream the actual start codon could result in the expression of a shorter yet still functional version of mCherry. This constitutes an equivalent to Russell’s paradox in molecular biology, more specifically in relation with the red fluorescent protein mCherry (*see footer): mCherry does not contain itself; however, we suspected that it does (Supplementary Figure S2).
A fraction of eukaryotic and prokaryotic genes possess alternative translation initiation sites (ATIS) that lead to the production of different isoforms of a functional protein from a unique mRNA (Wegrzyn et al., 2008; Fritsch et al., 2012; Wan and Qian, 2014; Nakahigashi et al., 2016). The N-terminal sequence variation between protein isoforms can be the target of posttranslational regulation (Trulley et al., 2019), affect protein functionality (Ozin et al., 2001; Bernier et al., 2018), and even direct subcellular localization (Chabregas et al., 2003). The presence of an ATIS in mCherry greatly affects its function as a reporter and the outcome of experiments. A shorter version of mCherry contained in itself creates a nonnegligible background fluorescence, disturbing the results of gene expression and protein localization studies. For example, a genetic construct encoding a fusion protein composed of a C-terminal mCherry presents a risk of producing an independent, fusion-less mCherry protein, which interferes with protein localization. Likewise, studies using mCherry as reporter for gene expression would yield biased results because translation of the short mCherry isoform is included in the reporter gene sequence. As mCherry is widely used, the postulated interference may affect many studies, including our own work. However, like Frege, we believe that the advancement of knowledge deserves our full dedication. Therefore, we investigated mCherry expression in detail to uncover the source of the issue with the goal of providing solutions that eliminate the defectof translation initiation.
Results and discussion
Identification of the shorter protein isoform of mCherry
Our previous experiments with mCherry as a reporter gene (Lale et al., 1101) led us to suspect the presence of a shorter protein isoform of mCherry. We first analyzed the sequence of the codon-optimized version of mCherry and noticed three methionine residues in a relatively close proximity from the annotated start codon (Figure 1). In addition, we recognized an SD-like sequence ranging from −12 to −6 nucleotides upstream of the first internal methionine residue (Figure 1). This led us to hypothesize that a short functional isoform of mCherry is produced from one of the three downstream methionines and not from an alternative start codon.
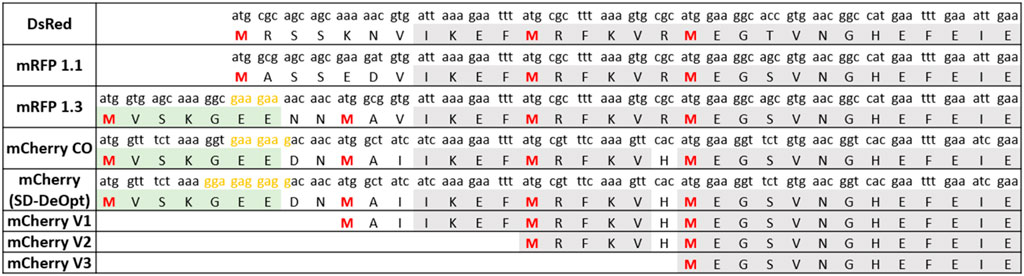
FIGURE 1. Amino acid sequence alignment of key engineering steps of red fluorescent proteins derived from DsRed. The isoform-causing N-terminal extension occurred in the engineering step from mRFP1.1 to mRFP1.3 due to of the addition of the eGFP fragment (in green) and the linker NNMA. The sequences of the short mCherry versions (V1, V2, and V3) used to determine the shortest functional isoform are also presented. Furthermore, the sequence of the “Shine–Dalgarno (SD)-deoptimized” version of mCherry is shown. Methionines potentially serving as start codons are shown in red (M1, M10, M17, and M23). SD-like sequences are presented in yellow. A codon usage variant of mCherry was created, named SD-deoptimized, which refers to the strongest SD sequence possible upstream M10 from codon usage variations that would increase background expression. The codon usage of each protein is indicated above each amino acid. The conserved amino acid sequence is presented in gray.
To determine which of these methionine residues function as an ATIS that still renders a functional mCherry protein, we designed three versions of mCherry (V1, V2, and V3), with increasing N-terminal truncations (Figure 1). Each version of mCherry was expressed in E. coli with a constitutive promoter/5′-UTR. The fluorescence measurements of the different mCherry versions are presented in Figure 2. The smallest truncation (V1) retains 73% of the fluorescence intensity of the original codon-optimized version (mCherry-CO); whereas, the other versions (V2 and V3) do not show any fluorescence. In addition, proteomics analysis of the red fluorescent protein mCherry V1 confirmed that the first 10 amino acids were absent (Supplementary Figure S3), although the fragment between M10 and M17 could not be detected due to the short length of the digested fragments. This is conclusive evidence that the mCherry gene produces a short functional protein isoform starting at the methionine in position 10 of the amino acid sequence.
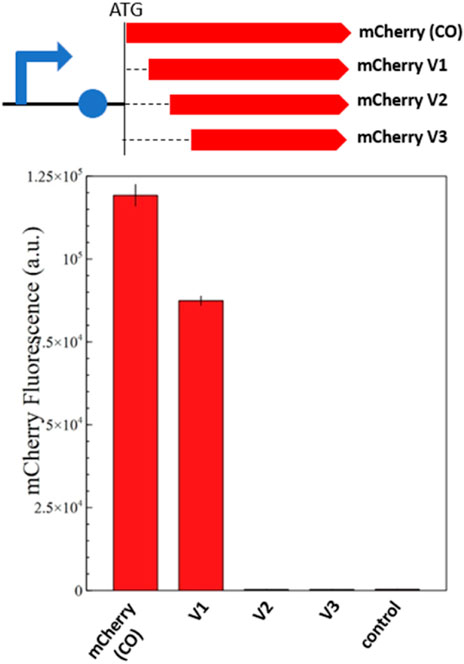
FIGURE 2. Fluorescence measurements of mCherry and its shorter isoforms (V1, V2, and V3). The genetic map of each constitutively expressed mCherry version is shown above the fluorescence quantification graph. The graph shows the in vivo fluorescence of the different mCherry versions in E. coli normalized by OD600. A plasmid without mCherry is used as a negative control. Four biological replicates were made for each mCherry version, and the respective standard deviation is presented with black error bars. Just mCherry V1 produces a functional protein isoform that retains 73% of the original fluorescence; whereas V2 and V3 are not fluorescent.
Phylogenic analysis reveals the origin and extent of the problem
Because most red fluorescent proteins originate from the modifications of DsRed (Matz et al., 1999), we imagined that the dual-isoform issue of mCherry could affect other members of the red fluorescent protein family. We performed protein sequence alignments to determine the extent of the issue across DsRed-derived fluorescent proteins (Figure 1). We were able to trace back the apparition of the second isoform to the engineering of mRFP1.3 (Figure 3).
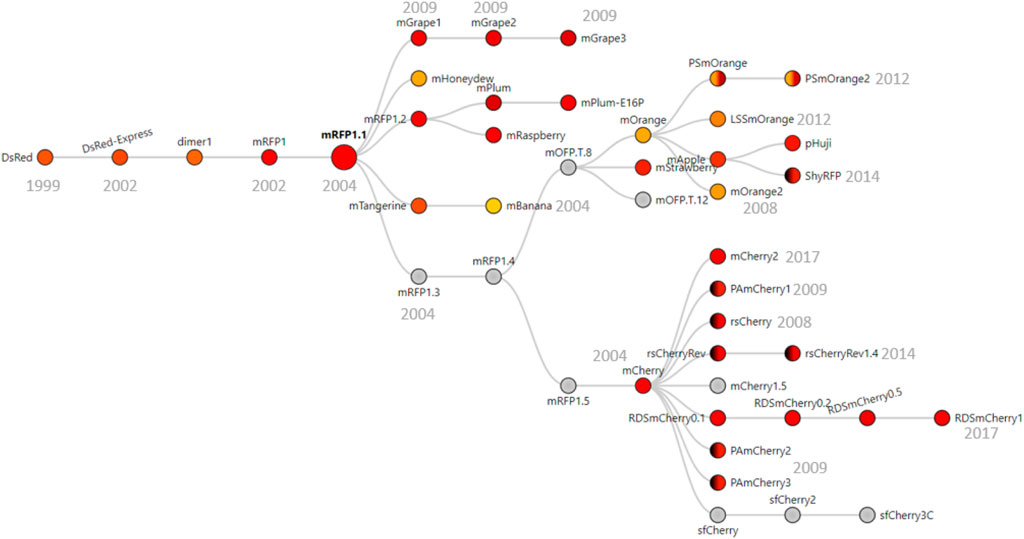
FIGURE 3. Phylogenic tree of DsRed-derived fluorescent proteins. Figure adapted from FPbase (Lambert, 2019) with their permission. The phylogenic tree shows all the fluorescent proteins derived from mRFP1.3 that contains the problem-causing N-terminal extension. Underlined proteins are derived from mRPF1.1 that also contain the extension. Color in circles corresponds to the emission wavelength(s) of the protein. Dark hemicycles represent an off-state of the protein. Gray indicates that the emission maximum was not entered in the database. The year in which the proteins were engineered is indicated in gray below/next to the corresponding protein.
DsRed was modified into mRFP1 to overcome obligate tetramerization, improve protein maturation, and modify the excitation/emission wavelength couple (Campbell et al., 2002). Then, mRFP1 was further modified into mRFP1.1 by the notable Q66M substitution in the chromophore to improve parameters such as photostability, quantum yield, and extinction coefficient (Shaner et al., 2004). During the same round of modifications, in an attempt to improve the poor N-terminal fusion properties of mRFP1.1, the initial residues of eGFP (MVSKGEE) followed by the linker (NNMA) were added to mRFP1.1 to yield mRFP1.312. This manipulation is responsible for the apparition of the short mCherry isoform. Indeed, the linker (NNMA) provides the alternative methionine start codon, and the eGFP fragment offers an SD-like sequence for ribosome entry (Figure 1).
The phylogenic overview of red fluorescent proteins (Figure 3) shows all the proteins engineered from mRFP1.3 that are affected by the dual-isoform issue. In addition to the mRFP1.3-derived proteins, mBanana, mGrape2, and mGrape3 also received the N-terminal extension described above. The problem becomes even more concerning because it affects all commonly used variants in the red color spectrum. Furthermore, the timeline of engineering is important. As mRFP1.3 and mCherry were created in 2004, this entails that the results from a large number of publications using red fluorescent proteins since 2004 may have been affected by this issue.
Short mCherry isoform is also expressed both in prokaryotes and eukaryotes
In prokaryotes, translation initiation shares quasiuniversal principles (Nakagawa et al., 2010; Rodnina, 2018), with some variations in the SD sequences recruiting ribosomes. Because the short mCherry isoform is encoded in the mCherry gene sequence, it is highly likely that other prokaryotes also produce the short mCherry isoform. In addition, depending on the codon usage of the N-terminal extension, the short mCherry expression may be stronger or adapted to other organisms (as demonstrated by mCherry with SD-deoptimized) (Figure 1). We found that the short mCherry isoform (V1) was functional in Vibrio natriegens and Pseudomonas putida (Supplementary Figures S5 and S6), and another group had found the mCherry ATIS occurring in Mycoplasma (Carroll et al., 2014). This demonstrates that the mCherry defect occurs across a wide range of bacteria.
Translation initiation significantly differs between prokaryotes and eukaryotes. Bacteria mainly rely on the annealing of 16 S ribosomal RNA with the SD sequence and mRNA structures surrounding the start codon, whereas eukaryotes lack this type of interaction with the mRNA transcript. Instead, the small ribosomal subunit (40 S), charged with tRNAMet,i, recognizes an AUG start codon through a scanning mechanism of codons within the 5′UTR of mRNA strands (Sonenberg and Hinnebusch, 2009; Hinnebusch, 2014). The nucleotide context around the start codon also plays a role in initiating translation, favoring specific nucleotide motifs, sequence named after Kozak who first identified it (Kozak, 1986). The consensus yeast Kozak sequence was determined as (A/U)A (A/CA (A/C)A AUG UC(U/C) (Hamilton et al., 1986); however, this sequence varies a lot both within and across organisms (Nakagawa et al., 2008; Cuperus et al., 2017). To assess if the short mCherry isoform is also produced in eukaryotes, we replaced the AUG start codon with the UAG stop codon from a S. cerevisiae codon-optimized mCherry. In addition, we modified the codon usage surrounding M10 to GAAGAAGACAAC AUG GCC to resemble the optimal yeast Kozak sequence described by de Boer et al. (2020) and Hamilton et al. (1986). We demonstrated that this mCherry sequence provides in average a 19% background fluorescence compared with the original sequence (Figure 4; Supplementary Figure S7). It conclusively proves that the eukaryotic scanning mechanism of translation initiation allows the production of the short mCherry isoform. The nucleotide context surrounding the start codon may influence the level of translation in different eukaryotic cells. However, the ability of yeast to translate the short mCherry isoform raises concerns regarding studies using mCherry as a fluorescent marker across eukaryote species.
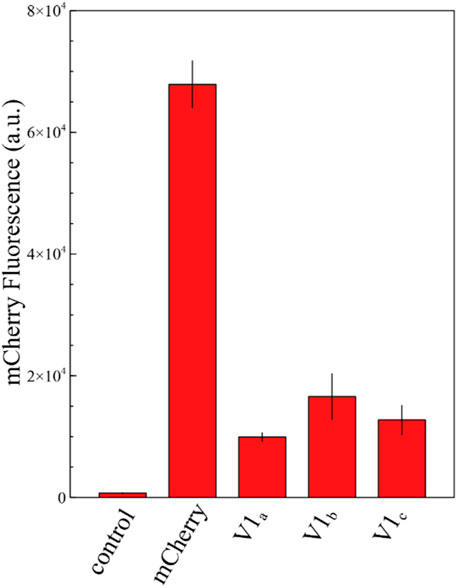
FIGURE 4. Fluorescence measurements of mCherry and its shorter isoforms V1 in the yeast Saccharomyces cerevisiae. The graph shows the fluorescence level of a S. cerevisiae strain carrying the yGPRA plasmid expressing the original mCherry sequence under the control of the TEF1 promoter and three independent yeast transformants carrying the mutated AUG to UAG original start codon of mCherry. The short mCherry isoform creates in average a 19% background fluorescence compared with the original sequence. The control shows the background fluorescence of the S. cerevisiae uracil-deficient strain. Each measurement was performed in biological triplicates.
Solutions deployed to circumvent the defect in mCherry
To provide a version of mCherry that is usable for gene expression and protein localization without conferring background expression, we tested different solutions in the context of fusion protein. The first solutions consisted in a substitution of the problem-causing M10 to glutamine or leucine, which aims to preserve the structural properties of the protein (Figure 5). As M10 stabilizes mCherry by interacting with the tyrosine residue Y43, the substitutions of M10 with glutamine or leucine allow similar stabilization through the formation of an H-bond with Y43 or with Van der Walls interactions, respectively, while occupying an equivalent steric space as methionine. The second solution was simply to use mCherry V1 as a reporter as it resembles mRFP1.1.
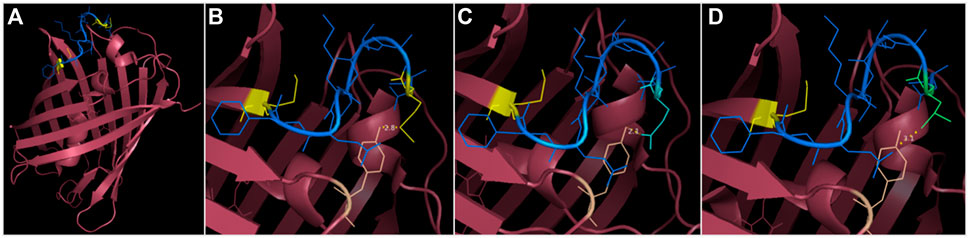
FIGURE 5. 3D structure visualization of mCherry and the substitutions provided to abolish the short isoform. (A) the segment between M10 and M17 (shown in blue, methionine in yellow) precedes the first ß-sheet but provides indispensable stabilization to the protein (as mCherry V2 is not functional). The eGFP segment preceding M10 is not present on any PDB files of mCherry, potentially because of a low resolution, due to mixed isoform population. (B) M10 (yellow) stabilizes mCherry by interacting with Y43 (wheat color). (C) M10Q substitution (in cyan) occupies the same volume as methionine and can produce an H-bond with Y43. (D) M10L substitution (in lime green) also mimics the steric occupancy of methionine and can weakly interact with Y43 through Van der Wall’s interaction.
Fusion proteins composed of sfGFP fused with C-terminal engineered versions of mCherry were constructed to assess the performance of each solution. First, sfGFP and mCherry versions were linked with an alanine/glycine linker to estimate the C-terminal fusion properties of the engineered mCherry versions (Figure 6). Second, the background expression due to the short isoform was assessed by placing two stop codons in the linker peptide right upstream the mCherry versions.
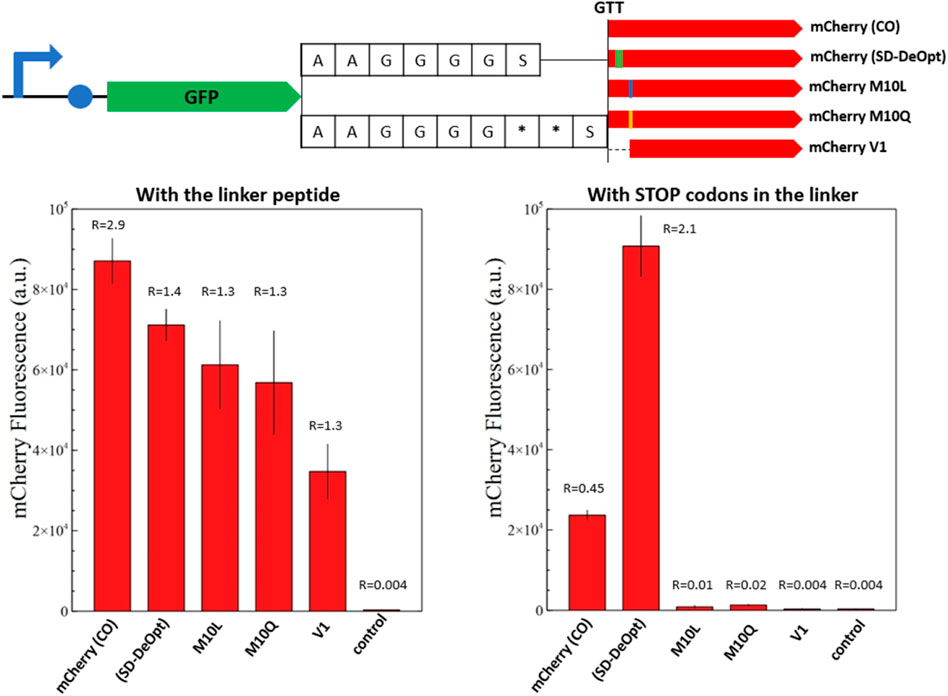
FIGURE 6. Fluorescence measurements of mCherry and its modified versions in the context of C-terminal fusion. The overview of the genetic constructs is presented (top). The versions of mCherry are fused to the C-terminal end of sfGFP with a standard alanine/glycine linker or with the linker containing two stop codons. The red fluorescence of the fusion proteins with the alanine/glycine linker is presented on the left-hand side and with stop codons on the right-hand side. The graphs show the in vivo fluorescence of the different mCherry versions in E. coli normalized by OD600. Four biological replicates were made for each mCherry version, and the respective standard deviation is represented with black error bars. A plasmid expressing only sfGFP is used as a negative control. The ratio between sfGFP and mCherry fluorescence is indicated above the corresponding histogram bar (from Supplementary Figure S4). Just mCherry (CO) and the “Shine–Dalgarno-deoptimized” version show background fluorescence with the linker containing stop codons. The M10 substitutions prove successful in abolishing the short mCherry isoform in the context of C-terminal fusion. Likewise, the short mCherry V1 does not show background fluorescence but appears to be less efficient in C-terminal fusions.
In both cases, GFP is used as an internal standard of gene expression to compare the mCherry production as fusion or discrete protein between samples. The fusion proteins composed of the mutated amino acids in position 10 or the V1 isoform display an mCherry/GFP ratio of 1.3, which constitutes a reference under the assumption of an equimolar production of mCherry and GFP because they are expressed as a fusion protein (Figure 6). The inclusion of stop codons in the protein linker confirms this assumption, as none of these constructs displays mCherry fluorescence when the full-length fusion protein is not produced.
In the context of fusion protein, the original mCherry-(CO) shows a nearly twofold increase in mCherry abundance that can be attributed to the short isoform; however, this was not clearly detected for the mCherry-(DeOpt) version that should present the same characteristics (Figure 6). This may suggest a higher proportion of V1 isoform and truncated translation of sfGFP-mCherry fusions because ribosomal entry at the internal SD sequence of mCherry-(DeOpt) may cause upstream-translating ribosomes to fall off at the protein junction with mCherry. The two M10-mutated versions of mCherry present the same expression intensities as the original protein sequences in the fusion context; whereas the fluorescence of the short mCherry V1 isoform is slightly lower but still reasonable (Figure 6). This demonstrates that all solutions provided to resolve the mCherry issue are functional in a fusion protein context.
For the fusion proteins containing stop codons, the original mCherry-(CO) sequence and the “SD-deoptimized” version present a background fluorescence due to the production of the short isoform, which is amplified in the case of “SD-deoptimized” due to higher ribosomal entry on the 5′end of the mCherry sequence as its internal SD sequence favors V1 isoform translation. On the contrary, the M10 substitutions and V1 isoform do not show any fluorescence, which proves that these solutions successfully abolish the production of the short mCherry isoform in the context of C-terminal fusion (Figure 6). Likewise, the short mCherry V1 did not show any background fluorescence but appeared to be less efficient in C-terminal fusions, as reported for mRFP1.1 (Shaner et al., 2004).
Subtle changes in the amino acid sequence of fluorescent proteins can cause changes in their spectral properties as well as their brightness and maturation (Arpino et al., 2014). The removal of the N-terminal sequence before M10, or the substitutions of M10 by leucine and glutamine, seems to affect the brightness and/or maturation of the protein and potentially its ability to be used as soluble fusion partner. However, we detected no changes in the excitation/emission spectra for the different variants (Supplementary figure S8). Further investigation and engineering are necessary to recover the properties of mCherry without generating the short protein isoform that creates background fluorescence.
Conclusion
The unexpected performance of mCherry as a reporter in our gene expression experiments led to the discovery of a short functional isoform of mCherry. The expression of this short isoform affects the outcome of gene expression and protein localization studies. The defect originates from a methionine residue in position 10 that is preceded by an SD-like sequence. We showed that the mutation of this residue to leucine or glutamine abolishes the production of the short isoform while conserving N-terminal fusion properties.
Our findings indicate that a large proportion of the DsRed-derived proteins are affected by the production of the short isoform due to the presence of a problem-causing linker sequence (MVSKGEE-NNMA). In addition, we identified the presence of this linker in the bright-green fluorescent protein mNeonGreen (Shaner et al., 2013). We would like to highlight the distinct possibility that the production of short functional isoforms will also affect other fluorescent proteins. Indeed, the presence of methionine residues in the first 10–20 amino acids, which may act as an alternative translation start sites, is widespread among fluorescent proteins. For example, mTFP1 (Ai et al., 2006), KillerRed (Bulina et al., 2006), Dronpa (Ando et al., 2004), mEosEM (Fu et al., 2020), mKelly2 (Wannier et al., 2018), mGinger2 (Wannier et al., 2018), and their derivatives could be affected by the production of a short isoform as observed for mCherry.
In addition, we showed that the dual-isoform issue affects various prokaryotes as well as eukaryotes despite having distinct translation initiation mechanisms. On the one hand, prokaryotes produce the short isoform of mCherry due to the affinity of the 16 s ribosomal RNA and the internal SD sequence as well as the recognition of the downstream M10 methionine. On the other hand, the eukaryotic 40 S ribosome scans the 5′UTR region of the mRNA strand to find translation start sites. The nucleotide features of the 5′UTR sequence, such as the presence of an SD or a Kozak sequence, low mRNA structures, and presence of the alternative start codon, enable the translation of the short mCherry isoform both in prokaryotes and eukaryotes.
In this study, we showed that the presence of an SD-like sequence upstream M10 dramatically increases the expression of the short mCherry isoform. Hence, an unfortunate nucleotide combination upstream M10 due to a certain codon usage can promote translation of the short mCherry isoform and lead to a higher background fluorescence. We recommend using the mutated versions of mCherry, M10Q or M10L, to avoid any expression background related to alternative translation start sites.
We demonstrate that M10 substitution abolishes the expression of the short protein isoform. This solution should be investigated for all affected DsRed-derived proteins to ensure their accuracy and reliability as reporters. Moreover, we suggest that similar actions might be necessary for other fluorescent protein families. This work raises concerns on the outcome of studies that employed fluorescent proteins that possess an inherent background expression.
Materials and methods
Materials
Experiments were performed in E. coli DH5-α (NEB), grown in LB-Lennox (Oxoid) (10-g/L casein peptone, 5-g/L yeast extract, 5-g/L NaCl with additional 15-g/L agar for plates) supplemented with 100-µL/mL ampicillin (Sigma-Aldrich). PCR amplifications were performed using Q5 polymerase (NEB). All other necessary enzymes were also purchased from NEB. Primers were ordered from Eurofins Genomics – Sigma-Aldrich. Plasmids and PCR products were purified using the QIAprep Plasmid Miniprep Kit and QiaQuick PCR Purification Kit, respectively (Qiagen). Plasmid Sanger sequencing was performed by Eurofins Genomics. The E. coli codon-optimized sequence of mCherry was a gift from Yanina R. Sevastsyanovich (University of Birmingham).
Cloning and strain engineering
A pUC19 backbone containing a constitutive promoter/5′-UTR expressing sfGFP, generated in a previous study (Lale et al., 1101), was used as template for cloning the different versions of mCherry. The backbone and the different versions of mCherry were amplified by PCR using the respective primers presented in Supplementary Table S1. To test for short isoforms, sfGFP was replaced by the codon-optimized mCherry gene or its shorter versions (V1, V2, and V3) via Golden Gate assembly (Engler et al., 2008). To build fusion constructs, the different mCherry versions were fused downstream sfGFP using the Golden Gate assembly.
The Golden Gate assembly mixture was chemically transformed into competent E. coli by heat shock (45 s at 42°C). Cells were placed on LB plates containing 100-µg/mL ampicillin and grown overnight at 37°C. Positive clones were grown in 5-mL LB supplemented with 100-µg/mL ampicillin, and their respective plasmids were purified. DNA sequences were confirmed by Sanger sequencing.
For S. cerevisiae, we used the yGPRA plasmid kindly provided by Regev et al. (de Boer et al., 2020). In brief, it contains a yeast codon-optimized mCherry gene expressed by the TEF1 promoter. The AUG start codon was replaced by an UAG stop codon, and the codon usage surrounding M10 was modified to GAAGAAGACAAC AUG GCC to resemble yeast Kozak sequence via PCR using primers presented in Supplementary Table S1. The PCR products were subject to overnight DpnI digestion at 37°C, subsequently purified, assembled using the Gibson assembly, and transformed into E. coli Dh5α. Correct assembly was confirmed by restriction digest with the BbsI and speI enzymes of the plasmid as well as sequencing (Eurofins Genomics). The original yGPRA plasmid and the modified yGPRA_mCherry_V1 plasmid were transformed into the uracil-deficient S. cerevisiae strain using the previously described electroporation method (Benatuil et al., 2010). The transformants were selected onto SD–UT media (6.7-g/L yeast nitrogen base without amino acids, 1.6-g/L yeast synthetic dropout medium supplement without uracil, and 20-g/L agar).
Fluorescence measurements
Each E. coli strain bearing a given plasmid was inoculated into LB supplemented with 100-µg/mL ampicillin on 96-well plates and incubated overnight at 37°C with 800-rpm agitation in a Multitron Pro plate-shaking incubator (Infors HT). For yeast, positive clones for both yGPRA and yGPRA_mCherry_V1 plasmids were cultivated in triplicates in 5-mL SD–UT media. The uracil-deficient S. cerevisiae strain was cultivated in triplicates in 5-mL YPD media, collected via centrifugation at 5,000 rpm, and then washed with SD–UT media before fluorescence measurements. Each sample (100 µL) was used for fluorescence measurements. Fluorescence was measured using an Infinite M200 Pro TECAN fluorimeter (Noax Lab AS). The excitation/emission wavelengths were 488/525 nm for GFP (gain 67) and 576/610 nm for mCherry (gain 97 for E. coli and 163 for yeast). Fluorescence intensity was normalized by OD600 of the corresponding well.
Fluorescence microscopy
Cell cultures of E. coli and S. cerevisiae carrying the respective plasmids expressing mCherry and the V1 isoform were placed between glass slides for the microscopic analysis. An inverted fluorescence microscope (Zeiss Axio Observer. Z1, 2.3.64.0) with a ×20 air objective (NA 0.8) was used to detect mCherry and was localized within the cells using the bright field filter. Image processing was performed using the Zeiss image analysis software (2.3.64.0).
Bioinformatics analysis
The amino acid sequence of mCherry-(CO) was used as template to perform preliminary protein BLAST searches (blast.ncbi.nlm.nih.gov/Blast.cgi). Phylogeny of red fluorescent proteins was consulted on FPbase [27] (fpbase.org).
Proteomics analysis
The strain carrying the constitutively expressed mCherry V1 was grown overnight in 50 mL of LB media supplemented with 100-µg/mL ampicillin at 37°C, 225 rpm. Cells were harvested by centrifugation (5,000 rpm, 5 min) and resuspended in lysis buffer (50 mM Tris-HCl, 50 mM NaCl, 0.05% Triton X-100, pH 8.0) supplemented with 1 tablet EDTA-free cOmplete ULTRA protease inhibitor (Roche). Cell debris were eliminated via centrifugation (7,500 rpm, 15 min), and the soluble fraction was collected.
Sample preparation for LC-MS—protein digestion
Two samples of 200-µL cell lysate each were separated for LC-MS analysis, one to be digested by trypsin alone and the other by both trypsin and Lys-C. Soluble proteins were precipitated by methanol/chloroform/water precipitation. In brief, 800-µL methanol was added to the sample, followed by the addition of 200-µL chloroform and vortexing. After the addition of 600-µL ultrapure water (18 MΩ), samples were thoroughly mixed by vortexing and centrifuged at 16000 g for 2 min. The upper layer was discarded without disturbing the protein layer, and further 800-µL methanol was added, followed by vortexing and centrifugation as above. After removing the supernatant, the protein pellet was air-dried for 10 min and was then reconstituted in 150 µL of 50-mM ammonium bicarbonate (BioUltra; Sigma-Aldrich, Germany). Next, the proteins were reduced with 1.5 µL of DTT (Sigma-Aldrich, Canada) for 20 min at 70°C, brought back to room temperature, and then alkylated using 6 µm of iodoacetamide (BioUltra; Sigma-Aldrich, United States) in the dark at room temperature for 30 min. Excess iodoacetamide was quenched by adding 3.5 µm of DTT (Sigma-Aldrich, Canada) and incubating in the dark for 20 min at room temperature. At last, the proteins were digested with endoproteinase at 37°C overnight, one sample digested with 1.25-µg trypsin and the other by 1.25-µg trypsin and 1.25-µg Lys-C. After overnight digestion, 5 µL of formic acid was added to each sample, and then the peptides were dried in a vacuum concentrator at 60°C.
Sample preparation for LC-MS—peptide desalting
The samples were resuspended in 100-µL 0.1% formic acid and desalted in C18 stage tip columns, unless otherwise specified. The chemicals were of Optima grade obtained from Fisher Chemicals, and centrifugations were performed at 2000 g. In brief, stage tip columns consisting of three C18 plugs (Empore C18 47 mm SPE Disks, 3 M, United States) were made and activated using 50-µL methanol via centrifugation for 2 min; then, methanol activation was repeated. Then, the stage tip column was equilibrated with 60 µL of 0.1% formic acid in water via centrifugation for 2 min; then, the equilibration was repeated. The peptide samples were centrifuged (16000 g for 25 min), supernatants were loaded onto stage tip columns and centrifuged for 4 min, and flow-through solutions were reloaded to stage tip columns. The stage tip columns were washed with 60 µL 0.1% formic acid and centrifuged for 2 min; the wash was then repeated. The peptides were then eluted from the stage tip column using 40 µL of 70% acetonitrile, washed with 0.1% formic acid, and then centrifuged for 2 min; the elution step was then repeated. At last, desalted peptides were dried in a vacuum concentrator at 60°C and stored at −20°C until LC-MS analysis.
LC-MS analysis
Dried peptides were reconstituted in 50 µL of 0.1% formic acid in water and shaken at 6°C at 900 rpm for 1.5 h. The samples were centrifuged at 16000 g for 10 min, and 40-µL supernatants were transferred to MS vials for LC-MS analysis. LC-MS analysis was performed on an EASY-nLC 1200 UPLC system (Thermo Fisher Scientific) interfaced with a Q Exactive mass spectrometer (Thermo Fisher Scientific) via a Nanospray Flex ion source (Thermo Fisher Scientific). Peptides were injected onto an Acclaim PepMap100 C18 trap column (75 µm i. d., 2-cm long, 3 µm, 100 Å, Thermo Fisher Scientific) and further separated on an Acclaim PepMap100 C18 analytical column (75 µm i. d., 50-cm long, 2 µm, 100 Å, Thermo Fisher Scientific) using a 180-min multistep gradient (150 min 2%–40% B, 15 min 40%–100% B, 15 min at 100% B, where B is 0.1% formic acid and 80% CH3CN and A is 0.1% formic acid) at 250-nL/min flow. The peptides were analyzed in the positive ion mode under data-dependent acquisition using the following parameters: electrospray voltage, 1.9 kV; HCD fragmentation with normalized collision energy, 28. Each MS scan (200–2000 m/z, 2-m/z isolation width, profile) was acquired at a resolution of 70,000 FWHM in the Orbitrap analyzer, followed by MS/MS scans at a resolution of 17,500 (2 m/z isolation width, profile) triggered for the 12 most intense ions, with a 30-s dynamic exclusion, and analysis using the Orbitrap analyzer. Charge exclusion was set to unassigned, 1, and >4.
Processing of LC-MS data
The proteins were identified by processing the LC-MS data using Thermo Fisher Scientific Proteome Discoverer (Thermo Fisher Scientific) version 2.5. The following search parameters were used: enzyme specified as trypsin with maximum two missed cleavages allowed; acetylation of protein N-terminus with methionine loss, oxidation of methionine, and deamidation of asparagine/glutamine were considered as dynamic and carbamidomethylation of cysteine as static posttranslational modifications; precursor mass tolerance of 10 parts per million with a fragment mass tolerance of 0.02 Da. Sequest HT node was used to query the raw files against sequences for mCherry (original and short); E. coli (strain K-12) proteins were downloaded from UniProt (www.uniprot.org/proteomes/UP000000625) in September 2020 and a common LC-MS contaminants database. For downstream analysis of this peptide-spectrum matches (PSMs), for protein and peptide identifications the PSM FDR was set to 1% and as high and 5% as medium confidence; thus, only unique peptides with these confidence thresholds were used for the final protein group identification and for the labeling of the confidence level, respectively.
Protein 3D structure modeling
The 3D structure modeling of mCherry was performed using the software PyMOL and the PDB file 2H5Q. In all mCherry PDB files (2H5Q, 6YLM, 6IR1 and 2, 6MZ3, 4ZIN), the first eight amino acids were unmodeled, residues 9 and 10 were computationally added, but M10 was mismodeled. We corrected residue 10 to model methionine. Then, we changed M10 into glutamine and leucine on the 3D structure with PyMOL. Rotamers with 2–3 Å proximity with Y43 are shown to model their interactions.
Data availability statement
The proteomics data have been uploaded onto the ProteomeXchange database (project accession number: PXD032954).
Author contributions
MF-L and LT are responsible for establishing the research perspective, protein alignments, and phylogenic investigations. MF-L designed the genetic experiments of shorter mCherry versions that were performed by FE, MF-L created and tested the solutions deployed for fusion proteins. LT performed the experiments related to P. putida and V. natriegens. MF-L wrote the manuscript with the help of LT, RL, and MFH-M, who also reviewed the manuscript.
Funding
This study was supported by PhD fellowships awarded to MF-L and LT by the Faculty of Natural Sciences of the Norwegian University of Science and Technology grant number (81771368) and (90809207).
Conflict of interest
MH-M was employed by the company United Scientists CORE (Limited).
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbioe.2022.892138/full#supplementary-material
Footnotes
*The mathematician Gottlob Frege dedicated his life to demonstrating that mathematics was reducible to logic. In June 1901, his ambitions were shattered by a letter from Bertrand Russell that pointed out a contradiction to his fundamental assumption that became famously known as “Russell’s paradox” (Chudakov et al., 2010). Russell’s paradox states the following: Consider R the set of all sets that do not contain themselves as members; if R is not a member of itself, then by definition, it is a member of itself, and reciprocally.
1R = {x | x ∉ x}, then R ∉ R ⬄ R ∈ R.
2This contradiction left Frege in a state of epistemic paralysis2, questioning the foundation of his lifelong hypothesis. However, Russell wrote that Frege responded to the challenge of his main hypothesis with intellectual pleasure and dedication to advance knowledge1.
References
Ai, H., Henderson, J. N., Remington, S. J., and Campbell, R. E. (2006). Directed evolution of a monomeric, bright and photostable version of clavularia cyan fluorescent protein: Structural characterization and applications in fluorescence imaging. Biochem. J. 400, 531–540. doi:10.1042/bj20060874
Ando, R., Mizuno, H., and Miyawaki, A. (2004). Regulated fast nucleocytoplasmic shuttling observed by reversible protein highlighting. Science 306, 1373. doi:10.1126/science.1102506
Arpino, J. A. J., Reddington, S. C., Halliwell, L. M., Rizkallah, P. J., and Jones, D. D. (2014). Random single amino acid deletion sampling unveils structural tolerance and the benefits of helical registry shift on GFP folding and structure. Structure 22, 889–898. doi:10.1016/j.str.2014.03.014
Benatuil, L., Perez, J. M., Belk, J., and Hsieh, C. M. (2010). An improved yeast transformation method for the generation of very large human antibody libraries. Protein Eng. Des. Sel. 23, 155–159. doi:10.1093/protein/gzq002
Bernier, S. C., Morency, L. P., Najmanovich, R., and Salesse, C. (2018). Identification of an alternative translation initiation site in the sequence of the commonly used Glutathione S-Transferase tag. J. Biotechnol. 286, 14–16. doi:10.1016/j.jbiotec.2018.09.003
Bevis, B. J., and Glick, B. S. (2002). Rapidly maturing variants of the Discosoma red fluorescent protein (DsRed). Nat. Biotechnol. 20 (1), 83–87. doi:10.1038/nbt0102-83
Bulina, M. E., Chudakov, D. M., Britanova, O. V., Yanushevich, Y. G., Staroverov, D. B., Chepurnykh, T. V., et al. (2006). A genetically encoded photosensitizer. Nat. Biotechnol. 24, 95–99. doi:10.1038/nbt1175
Campbell, R. E., Tour, O., Palmer, A. E., Steinbach, P. A., Baird, G. S., Zacharias, D. A., et al. (2002). A monomeric red fluorescent protein. Proc. Natl. Acad. Sci. U. S. A. 99, 7877–7882. doi:10.1073/pnas.082243699
Carroll, P., Muwanguzi-Karugaba, J., Melief, E., Files, M., and Parish, T. (2014). Identification of the translational start site of codon-optimized mCherry in Mycobacterium tuberculosis. BMC Res. Notes 7, 366. doi:10.1186/1756-0500-7-366
Chabregas, S. M., Luche, D. D., Van Sluys, M. A., Menck, C. F. M., and Silva-Filho, M. C. (2003). Differential usage of two in-frame translational start codons regulates subcellular localization of Arabidopsis thaliana THI1. J. Cell. Sci. 116, 285–291. doi:10.1242/jcs.00228
Chudakov, D. M., Matz, M. V., Lukyanov, S., and Lukyanov, K. A. (2010). Fluorescent proteins and their applications in imaging living cells and tissues. Physiol. Rev. 90, 1103–1163. doi:10.1152/physrev.00038.2009
Cuperus, J. T., Groves, B., Kuchina, A., Rosenberg, A. B., Jojic, N., Fields, S., et al. (2017). Deep learning of the regulatory grammar of yeast 5′ untranslated regions from 500, 000 random sequences. Genome Res. 27, 2015–2024. doi:10.1101/gr.224964.117
Day, R. N., and Davidson, M. W. (2009). The fluorescent protein palette: Tools for cellular imaging. Chem. Soc. Rev. 38, 2887. doi:10.1039/b901966a
de Boer, C. G., Vaishnav, E. D., Sadeh, R., Abeyta, E. L., Friedman, N., and Regev, A. (2020). Deciphering eukaryotic gene-regulatory logic with 100 million random promoters. Nat. Biotechnol. 38, 56–65. doi:10.1038/s41587-019-0315-8
Engler, C., Kandzia, R., and Marillonnet, S. (2008). A one pot, one step, precision cloning method with high throughput capability. PLoS One 3, e3647. doi:10.1371/journal.pone.0003647
Fritsch, C., Herrmann, A., Nothnagel, M., Szafranski, K., Huse, K., Schumann, F., et al. (2012). Genome-wide search for novel human uORFs and N-terminal protein extensions using ribosomal footprinting. Genome Res. 22, 2208–2218. doi:10.1101/gr.139568.112
Fu, Z., Peng, D., Zhang, M., Xue, F., Zhang, R., He, W., et al. (2020). mEosEM withstands osmium staining and Epon embedding for super-resolution CLEM. Nat. Methods 17, 55–58. doi:10.1038/s41592-019-0613-6
Gross, L. A., Baird, G. S., Hoffman, R. C., Baldridge, K. K., and Tsien, R. Y. (2000). The structure of the chromophore within DsRed, a red fluorescent protein from coral. Proc. Natl. Acad. Sci. U. S. A. 97, 11990–11995. doi:10.1073/pnas.97.22.11990
Hamilton, R., Watanabe, C. K., and De Boer, H. A. (1986). Compilation and comparison of the sequence context around the AUG start codons in Saccharomyces cerevisiae mRNAs. Nucleic Acids Res. 15, 3581–3593. doi:10.1093/nar/15.8.3581
Hinnebusch, A. G. (2014). The scanning mechanism of eukaryotic translation initiation. Annu. Rev. Biochem. 83, 779–812. doi:10.1146/annurev-biochem-060713-035802
Kozak, M. (1986). Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell. 44, 283–292. doi:10.1016/0092-8674(86)90762-2
Labas, Y. A., Gurskaya, N. G., Yanushevich, Y. G., Fradkov, A. F., Lukyanov, K. A., Lukyanov, S. A., et al. (2002). Diversity and evolution of the green fluorescent protein family. Proc. Natl. Acad. Sci. U. S. A. 99, 4256–4261. doi:10.1073/pnas.062552299
Lale, R., Tietze, L., Nesje,, J., Onsager, I., Engelhardt, K., Rückert, C., et al. A universal method for gene expression engineering. NewYork: biorxiv. doi:10.1101/644989
Lambert, T. J. (2019). FPbase: A community-editable fluorescent protein database. Nat. Methods 16, 277–278. doi:10.1038/s41592-019-0352-8
Lauff, M., and Hofer, R. (1984). Proteolytic enzymes in fish development and the importance of dietary enzymes. Aquaculture 37, 335–346. doi:10.1016/0044-8486(84)90298-9
Matz, M. V., Fradkov, A. F., Labas, Y. A., Savitsky, A. P., Zaraisky, A. G., Markelov, M. L., et al. (1999). Fluorescent proteins from nonbioluminescent Anthozoa species. Nat. Biotechnol. 17, 969–973. doi:10.1038/13657
Mishin, A. S., Subach, F. V., Yampolsky, I. V., King, W., Lukyanov, K. A., and Verkhusha, V. V. (2008). The first mutant of the Aequorea victoria green fluorescent protein that forms a red chromophore. Biochemistry 47, 4666–4673. doi:10.1021/bi702130s
Nakagawa, S., Niimura, Y., Gojobori, T., Tanaka, H., and Miura, K. ichiro. (2008). Diversity of preferred nucleotide sequences around the translation initiation codon in eukaryote genomes. Nucleic Acids Res. 36, 861–871. doi:10.1093/nar/gkm1102
Nakagawa, S., Niimura, Y., Miura, K. I., and Gojobori, T. (2010). Dynamic evolution of translation initiation mechanisms in prokaryotes. Proc. Natl. Acad. Sci. U. S. A. 107, 6382–6387. doi:10.1073/pnas.1002036107
Nakahigashi, K., Takai, Y., Kimura, M., Abe, N., Nakayashiki, T., Shiwa, Y., et al. (2016). Comprehensive identification of translation start sites by tetracycline-inhibited ribosome profiling. DNA Res. 23, 193–201. doi:10.1093/dnares/dsw008
Ozin, A. J., Costa, T., Henriques, A. O., and Moran, J. (2001). Alternative translation initiation produces a short form of a spore coat protein in Bacillus subtilis. J. Bacteriol. 183, 2032–2040. doi:10.1128/jb.183.6.2032-2040.2001
Ranganathan, R., Wall, M. A., and Socolich, M. (2000). The structural basis for red fluorescence in the tetrameric GFP homolog DsRed. Nat. Struct. Biol. 7, 1133–1138. doi:10.1038/81992
Rodnina, M. V. (2018). Translation in prokaryotes. Cold Spring Harb. Perspect. Biol. 10, a032664. doi:10.1101/cshperspect.a032664
Rodriguez, E. A., Campbell, R. E., Lin, J. Y., Lin, M. Z., Miyawaki, A., Palmer, A. E., et al. (2017). The growing and glowing toolbox of fluorescent and photoactive proteins. Trends Biochem. Sci. 42, 111–129. doi:10.1016/j.tibs.2016.09.010
Shaner, N. C., Campbell, R. E., Steinbach, P. A., Giepmans, B. N. G., Palmer, A. E., and Tsien, R. Y. (2004). Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 22, 1567–1572. doi:10.1038/nbt1037
Shaner, N. C., Lambert, G. G., Chammas, A., Ni, Y., Cranfill, P. J., Baird, M. A., et al. (2013). A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nat. Methods 10, 407–409. doi:10.1038/nmeth.2413
Shu, X., Shaner, N. C., Yarbrough, C. A., Tsien, R. Y., and Remington, S. J. (2006). Novel chromophores and buried charges control color in mFruits. Biochemistry 45, 9639–9647. doi:10.1021/bi060773l
Sonenberg, N., and Hinnebusch, A. G. (2009). Regulation of translation initiation in eukaryotes: Mechanisms and biological targets. Cell. 136, 731–745. doi:10.1016/j.cell.2009.01.042
Subach, F. V., and Verkhusha, V. V. (2012). Chromophore transformations in red fluorescent proteins. Chem. Rev. 112, 4308–4327. doi:10.1021/cr2001965
Trulley, P., Snieckute, G., Bekker-Jensen, D., Menon, M. B., Freund, R., Kotlyarov, A., et al. (2019). Alternative translation initiation generates a functionally distinct isoform of the stress-activated protein kinase MK2. Cell. Rep. 27, 2859–2870.e6. doi:10.1016/j.celrep.2019.05.024
Wachter, R. M., Watkins, J. L., and Kim, H. (2010). Mechanistic diversity of red fluorescence acquisition by GFP-like proteins. Biochemistry 49, 7417–7427. doi:10.1021/bi100901h
Wan, J., and Qian, S.-B. (2014). TISdb: A database for alternative translation initiation in mammalian cells. Nucleic Acids Res. 42, D845–D850. doi:10.1093/nar/gkt1085
Wannier, T. M., Gillespie, S. K., Hutchins, N., McIsaac, R. S., Wu, S. Y., Shen, Y., et al. (2018). Monomerization of far-red fluorescent proteins. Proc. Natl. Acad. Sci. U. S. A. 115, E11294-E11301. doi:10.1073/pnas.1807449115
Keywords: mCherry, fluorescent protein, reporter gene, alternative translation initiation site, gene expression, fusion protein
Citation: Fages-Lartaud M, Tietze L, Elie F, Lale R and Hohmann-Marriott MF (2022) mCherry contains a fluorescent protein isoform that interferes with its reporter function. Front. Bioeng. Biotechnol. 10:892138. doi: 10.3389/fbioe.2022.892138
Received: 08 March 2022; Accepted: 30 June 2022;
Published: 09 August 2022.
Edited by:
Dafydd Jones, Cardiff University, United KingdomReviewed by:
Thomas -Gensch, Helmholtz Association of German Research Centres (HZ), GermanyHarley Worthy, University of Exeter, United Kingdom
Copyright © 2022 Fages-Lartaud, Tietze, Elie, Lale and Hohmann-Marriott. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Martin Frank Hohmann-Marriott, bXhpbWUuZmFnZXMtbGFydGF1ZEBudG51Lm5v