 Lyuyin Wang
Lyuyin Wang Yue Sun1
Yue Sun1- 1National Institutes for Food and Drug Control, State Key Laboratory of Drug Regulatory Science, NHC Key Laboratory of Research on Quality and Standardization of Biotech Products, NMPA Key Laboratory for Quality Research and Evaluation of Biological Products, NMPA Key Laboratory for Quality Research and Evaluation of Chemical Drugs, Beijing, China
- 2Shanghai Yaxin Biotechnology Limited Company, Shanghai, China
- 3Sinovac Biotech Co., Ltd., Beijing, China
Introduction: Recombinant nuclease (RSN) is a critical raw material enzyme essential for removing host nucleic acids during the production of cell and gene therapy products. However, the lack of standardized quality evaluation methods and reference materials for RSN has resulted in poor reliability and comparability of testing results, hindering the standardized development of related industries. This study aimed to establish the first Chinese national standard for RSN activity, thereby enabling accurate and comparable quantification of enzymatic potency across laboratories and industries.
Methods: High-purity candidate RSN reference materials were prepared and structurally analyzed via mass spectrometry. A robust enzymatic activity assay using herring sperm DNA as the substrate was optimized and validated in accordance with ICH Q2 (R2) guidelines. The assay’s specificity, accuracy, and precision were evaluated, and a collaborative study involving three laboratories was subsequently conducted to determine the potency value.
Results: The assay exhibited excellent precision (intra-lab coefficient of variation (CV) < 5%, inter-lab CV 2.01%) and accuracy (recovery rate 90–110%). The candidate reference material showed high homogeneity and stability. Collaborative testing confirmed the statistical consistency of the study results (P > 0.05), and the potency of the candidate material was determined to be 518 U/μL.
Discussion: The establishment of this national standard will provide a unified reference for measuring RSN activity, enhance consistency in quality control, and support the safe development and production of biopharmaceuticals (including cell and gene therapies). This standard will serve as a critical tool for regulatory evaluation and industrial applications in China. The optimized content is consistent with the copyedited version of the article, featuring clear logic and comprehensive key information.
1 Introduction
The safety of biological products hinges on strict control over potentially hazardous impurities. Among these, residual host nucleic acids have emerged as a core indicator in global pharmaceutical regulation, given their potential to induce tumorigenicity, infectivity, and immune interference. Nucleic acid impurities may be generated during the production of vaccines, cell and gene therapy products, and recombinant protein drugs—spanning cell collection, viral vector construction, and final product purification. Therefore, the efficient removal of such impurities is pivotal to ensuring the clinical safety of these products. Nuclease (EC: 3.1.30.2), a non-specific endonuclease derived from Serratia marcescens (Eaves and Jeffries, 1963; Ball et al., 1987; Schwardmann et al., 2020), efficiently degrades DNA and RNA in all forms (e.g., double-stranded, single-stranded, linear, and circular), digesting them into oligonucleotides that are 2–5 bases in length (Biedermann et al., 1989; Lunin et al., 1997; Cheng et al., 2003). Recombinant nuclease (RSN) of non-animal origin, with its high purity, safety, stability, efficiency, broad spectrum, and ease of operation, has become the preferred tool for eliminating residual nucleic acids in biological products. In vaccine production, treating harvest fluids with RSN effectively reduces nucleic acid residues, enhances vaccine purity and stability, and ensures product quality and safety (Li et al., 2014; Kim et al., 2021; Moreira et al., 2021; Mayer et al., 2023; Ruan et al., 2023). In recombinant protein drug preparation, RSN lowers the nucleic acid content in fermentation broths, reduce solution viscosity, optimize downstream purification processes, and improve protein yield and quality (Biedermann et al., 1989; Yang et al., 2023). Within cell and gene therapy, RSN also plays an irreplaceable role in removing exogenous nucleic acids introduced during production, thereby safeguarding the safety and efficacy of therapeutic products (U.S. Department of Health and Human Services et al., 2020).
RSN is a recombinant endonuclease expressed in Escherichia coli via recombinant DNA technology. It is composed of 266 amino acids with two pairs of disulfide bonds and has a molecular weight of approximately 27 kDa. Despite its widespread application, RSN faces significant challenges in quality control and standardization. As it is not yet included in pharmacopoeias globally, manufacturers rely on in-house standards for production quality control, resulting in inconsistencies in enzymatic activity unit definitions, detection methods, and standard limits across commercial products. This not only complicates the uniform dosing of RSN—used as key process parameters during production (Liu et al., 2021; von Elling-Tammen et al., 2025)—but also increases the risk of residual host nucleic acids or nucleases in final products, creating hurdles for regulatory quality evaluation. In June 2025, the United States Pharmacopeia (USP) released the world’s first reference standard for RSN activity determination (The United States Pharmacopeial Convention, 2024), standardizing enzymatic activity assay methods and units. However, China has yet to establish a corresponding reference standard, leading to a lack of traceability for enzymatic activity values in production, quality control, application, and regulatory evaluation—severely impeding the standardized development of related industries. The demand for high-quality nucleases has surged with the rapid growth of emerging fields such as cell and gene therapy. Thus, establishing the first batch of national reference standards for RSN has become an urgent priority for China’s biopharmaceutical industry.
To this end, we prepared candidate RSN reference material samples. Structural characterization was performed using multiple analytical methods, while an enzymatic activity assay employing herring sperm DNA as a substrate was optimized and validated. We then comprehensively evaluated the candidate reference material in terms of activity, purity, protein content, homogeneity, and stability. Through a collaborative calibration process, the first Chinese national standard for RSN was established. This standard will provide a unified reference for quantifying RSN activity while ensuring measurement traceability, and it will also enhance quality control in biopharmaceutical production, particularly for cell and gene therapies. This work addresses a critical gap in China’s regulatory framework and promotes industrial standardization.
2 Materials and methods
2.1 Candidate reference material
The candidate RSN reference material (batch number: RSN240604) was provided by Shanghai Yaxin Biotechnology Co., Ltd. (Shanghai, China). It was prepared by expressing the endonuclease gene from Serratia marcescens in E. coli BL21 (DE3), followed by cultivation, bacterial harvesting, purification, sterilization, and the addition of 50% glycerol. The average filling volume was 100 μL per vial, with a coefficient of variation (CV) < 5%, indicating good filling uniformity.
2.2 Structural confirmation
2.2.1 Relative molecular weight
High-resolution mass spectrometry (Xevo G2-XS QTof, Waters, Shanghai, China) was used to analyze the relative molecular weight of the candidate reference material.
2.2.2 Peptide sequence coverage
A combined enzymatic hydrolysis strategy was employed, in which the candidate reference material was digested with trypsin (Promega), chymotrypsin (Sigma), and V8 protease Glu-C (Wako). The enzymatic hydrolysates were desalted and separated by high-performance liquid chromatography (Acquity I-Class, Waters), followed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis (Xevo G2-XS Q Tof, Waters). The raw data and theoretical sequence of the test sample were imported into the UNIFI software (Waters) for peptide coverage analysis.
2.2.3 Isoelectric point
Horizontal isoelectric focusing electrophoresis was performed. An appropriate amounts of the candidate reference material was placed in 10-kD ultrafiltration centrifugal tubes (Merck) and ultrafiltered with water; this was repeated three times, resulting in a protein concentration of approximately 1.0 mg/mL. An aliquot of this solution was mixed with 10% ampholyte and methyl red reagent to prepare the sample solution. For electrophoresis, 2 μL each of pI 3–10 isoelectric point markers and the sample solution were added. A standard curve of pI vs. migration distance (Rf) was plotted with the pI values of the markers as the X-axis and the Rf as the Y-axis. The isoelectric point of the candidate reference material was calculated by substituting its migration distance into the standard curve.
2.2.4 Maximum UV absorption wavelength
UV-visible spectrophotometry was used, with a 50% glycerol solution for baseline correction. An appropriate amounts of the candidate reference material was measured to determine their maximum absorption wavelength (λmax) in the range of 200–400 nm.
2.3 Purity
2.3.1 Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE)
An appropriate amount of the candidate reference material was diluted with water to prepare solutions of 1%, 0.5%, and 0.2%. Aliquots of the candidate reference material and its diluted solutions were mixed with 4× non-reducing loading buffer, heated in a boiling water bath at 100 °C for 5 min, cooled to room temperature, and centrifuged to obtain the sample solution and 1%, 0.5%, and 0.2% self-control solutions. A Novex WedgeWell 12% Tris-Glycine Gel was used, with 50 μL of each solution and 5 μL of Precision Plus Protein Standards (BIO-RAD) loaded into the wells. Electrophoresis was performed at a constant voltage of 100 V until bromophenol blue migrated to the bottom of the gel. The gel was fixed, stained with Coomassie brilliant blue, destained, photographed, and analyzed using a gel scanning system. The impurity limits were evaluated by comparing the staining intensity of any bands other than the main band in the sample electrophoresis profile with the main band of the 1% self-control solution.
2.3.2 Size-exclusion chromatography (SEC-HPLC)
A TSKgel G2000SWXL column (7.8 mm × 30 cm) was used with a mobile phase of isopropanol−0.075 mol/L phosphate buffer (15:85) at a flow rate of 0.5 mL/min, column temperature of 28 °C, and detection wavelength of 214 nm. An appropriate amounts of the candidate reference material was mixed with an equal volume of the mobile phase to prepare the sample solution. A blank solution was prepared using 50% glycerol solution in the same manner. Subsequently, 50 μL of each solution was injected into the HPLC system, and the chromatograms were recorded. Purity was calculated using the peak area normalization method. The specificity of the method was evaluated by checking for obvious interference peaks in the blank solution chromatogram. For precision assessment, two analysts each tested three vials of the candidate reference material, and the relative standard deviation (CV) of the main peak retention time and peak area percentage was calculated. For stability testing, the candidate reference material was incubated at 25 °C, 40 °C, and 60 °C for 1 h and at 60 °C for 1, 2, and 3 h. The purity was determined as described above to observe changes in the main component and high-molecular-weight polymers.
2.4 Protein content
2.4.1 Determination of experimental extinction coefficient using an amino acid analyzer
The RSN stock solution (batch number: NSY-RSN-400L-240601), internal standard amino acid (N-Val), and reference amino acids (Ser, Glu, Leu, Tyr, His, Arg, Pro) were hydrolyzed with 6N hydrochloric acid and 1% phenol solution. The amino acid composition of the stock solution was determined using an amino acid analyzer. Its molar concentration was calculated based on the theoretical number of amino acids, and the protein concentration was further calculated using the molecular weight determined in Section 2.2.1. The absorbance at 280 nm was measured as described in Section 2.4.2, and the experimental extinction coefficient of the RSN was calculated using the Lambert-Beer law (c = A/EL).
2.4.2 Determination of protein content by UV spectrophotometry
The protein content of the candidate reference material was measured using the theoretical molar extinction coefficient of RSN at 280 nm (1.68, defined as the absorption coefficient of 1 mg/mL RSN at 280 nm). Additionally, the protein content was determined by UV-visible spectrophotometry using the experimental extinction coefficient obtained from the amino acid analyzer. The average value from the two extinction coefficients was taken as the protein concentration of the candidate reference material.
2.5 Enzymatic activity assay
To establish enzymatic activity, 6 mL of pre-cooled (2 °C–8 °C) substrate solution (herring sperm DNA diluted with an assay buffer [4 mM MgCl2 in 0.05 M Tris buffer, pH 8.0] to a final concentration of 0.1 mg/mL) was mixed with 300 μL of the candidate reference material solution (0.2 mg/mL candidate reference material diluted 30,000-fold with assay buffer). Next, 500 μL aliquots of this mixture were transferred to 12 DNase-free EP tubes, with three replicate tubes for each of the four reaction time points. For blank solutions, 6 mL of pre-cooled substrate solution was mixed with 300 μL of assay buffer (containing 0.1 mg/mL herring sperm DNA). The EP tubes were incubated at 37 °C for 15, 30, 45, and 60 min, before adding 500 μL of stop solution (4% perchloric acid), mixing, and placing on ice for 30 min. After pre-cooling, the samples were centrifuged at 4 °C for 7 min at 14,000 rpm. The supernatant was collected, and the absorbance at 260 nm was measured for both sample and blank solutions, with water as the reference.
Enzymatic activity was defined as the amount of enzyme that causes a change in A260 of 1.0 per 30 min at 37 °C and pH 8.0, denoted as 1 unit (U). The formula for calculation is as follows:
where ΔA is the absorbance of the sample solution minus that of the blank solution at each time point; V1 is the total volume of the mixture, 6.3 mL; F is the dilution factor of the sample, 30,000; t is the incubation time of the mixture; V2 is the volume of the sample, 0.3 mL; 30 is the specified time for the defined enzymatic activity unit (30 min); 2 is the dilution factor from adding 500 μL stop solution to 500 μL reaction mixture; ΔA/t is the slope (a) of the linear regression equation y = ax + b, where y is ΔA and x is t; and 1,000 is the unit conversion from mL to μL.
2.6 Optimization of the enzymatic reaction conditions
2.6.1 Detection principle
Purified DNA from herring sperm was used as the substrate. RSN acts on phosphodiester bonds in DNA molecules, degrading long double-stranded DNA into oligonucleotides or even mononucleotides. This disrupts the double-helical structure, weakens base stacking, and exposes more bases, increasing UV absorption at 260 nm. After adding perchloric acid to precipitate DNA fragments longer than 100 bp (terminating the reaction), the increase in absorbance at 260 nm in the supernatant was measured to determine nuclease activity (Nestle and Roberts, 1969; Friedhoff et al., 1996; Benedik and Strych, 1998).
2.6.2 Quality release criteria for the substrate
Preliminary studies showed differences in activity when the candidate reference material was measured using herring sperm DNA substrates from three common brands (Invitrogen, Promega, and Solarbio), as described in Section 2.5. To investigate the cause, the molecular weights of the three substrates were analyzed by agarose gel electrophoresis. The ΔOD260 (OD260 at each time point minus blank OD260) was measured at 15, 30, 45, and 60 min using each substrate, and the slopes of the fitted linear equations (reaction rates) were compared by plotting ΔOD260 vs. time.
DNA substrates with larger and smaller molecular weights were used: after adding the candidate reference material, samples were taken at 5, 15, 30, 45, 60, and 90 min, and DNA fragments >100 bp were precipitated with perchloric acid. The precipitate was centrifuged, dissolved, and electrophoresed to observe changes in DNA molecular weight over time. The quality release criteria for the substrate were determined based on these results.
2.6.3 Optimization of experimental parameters
Enzymatic activity determination relies on the maximum reaction rate to characterize the enzyme quantity; thus, the key conditions in the enzymatic system were optimized to approach the maximum reaction rate. Herring sperm DNA substrates were diluted to 0.002, 0.004, 0.006, 0.020, 0.040, and 0.080 mg/mL, and the enzymatic activity was measured as described in Section 2.5. A Michaelis-Menten plot (substrate concentration vs. enzyme activity) was generated to calculate the kinetic parameters Km and Vmax; the optimal substrate concentration was selected as > 10 × Km based on enzymatic kinetics theory.
The candidate reference material was diluted 10,000-, 15,000-, 20,000-, 30,000-, 40,000-, 60,000-, and 80,000-fold with assay buffer, and the detection range was determined by measuring the activity. Samples were taken every 15 min for 15–180 min to determine the optimal reaction time.
Assay buffers with different Mg2+ concentrations (0, 1, 2, 3, 4, 5, 10, 20, and 50 mM) were prepared to measure the activity and determine the optimal Mg2+ concentration. Buffers with different pH values (4, 5, 6, 7, 8, 9, and 10) were used to measure the activity and determine the optimal pH range. The candidate reference material was diluted in buffers (pH 4, 5, 6, 7, 7.5, 8, 8.5, 9, and 10) with or without 0.1 mg/mL bovine serum albumin (BSA), incubated at 25 °C and 37 °C for 2 h, and then assayed at pH 8.0 to evaluate the effect of BSA on activity and stability. The activity was measured at 25 °C, 30 °C, 37 °C, 42 °C, and 50 °C to determine the optimal temperature range. Dilutions of the candidate reference material were incubated at 25 °C, 37 °C, and 42 °C for 2 h and then assayed to evaluate stability within 60 min, confirming the final reaction temperature.
2.7 Method validation
In-house method validation was performed according to the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) Q2 (R2), including evaluations of specificity, linearity, precision (intermediate precision), accuracy, and range using the RSN candidate reference material.
2.7.1 Specificity
The candidate reference material, 0.1 mg/mL BSA solution, and 0.2 mg/mL recombinant enterokinase (REK) solution were used to measure activity, as described in Section 2.5.
The candidate reference material was incubated at 25 °C, 40 °C, and 60 °C for 1 h and at 60 °C for 1, 2, and 3 h. The activity (Section 2.5) and purity (Section 2.3.2) were measured to evaluate interference from potential degradation products.
2.7.2 Intermediate precision
The candidate reference material was diluted 20,000-, 24,000-, 30,000-, and 40,000-fold with assay buffer to prepare four samples with different activity levels (150%, 125%, 100%, and 75%). Two analysts measured each sample 16 times over 2 days, and the mean and CV% were calculated.
2.7.3 Repeatability
One analyst prepared six replicate samples and assayed them in 1 day; the mean activity and CV% were calculated.
2.7.4 Accuracy
The activity result from multi-laboratory collaborative calibration (Section 2.8) was used as the reference value. Accuracy was evaluated using the recovery rate (recovery% = measured activity/reference activity × 100%).
2.7.5 Linearity and range
Linear regression analysis was performed using the reference reaction rates of samples at different levels as the X-axis and the corresponding measured values as the Y-axis.
2.8 Collaborative calibration
Collaborative calibration of the candidate reference material’s enzymatic activity was conducted by the National Institutes for Food and Drug Control, Shanghai Yaxin Biotechnology Co., Ltd., and Sinovac Life Sciences Co., Ltd. (randomly numbered Lab 1, Lab 2, and Lab 3). Each laboratory had two analysts performing two runs per day to obtain four valid results per day, totaling 20 results over 5 days. Data from the three laboratories were aggregated for precision and consistency analyses (Wang et al., 2023).
2.9 Evaluation of the homogeneity and stability of the candidate reference material
2.9.1 Homogeneity assessment
The purity of 12 vials of the candidate reference material was determined by SEC-HPLC. The homogeneity was evaluated by calculating the CV% of the main peak retention time and area percentage across the 12 vials.
2.9.2 Stability study
Two vials of the candidate reference material were stored at 4 °C for 5 and 10 days, and the SEC-HPLC purity and enzymatic activity were measured to evaluate accelerated stability. For long-term stability, enzymatic activity was measured at 0, 3, 6, 9, 12, 18, 24, and 36 months under −20 °C storage conditions.
3 Results
3.1 Structural confirmation of the candidate reference standard
3.1.1 Relative molecular weight
The molecular weight of the candidate reference standard was determined by high-resolution mass spectrometry. As shown in Figure 1A, the measured molecular weight was 26,836.50 Da, which is consistent with the theoretical molecular weight of 26,836.79 Da.
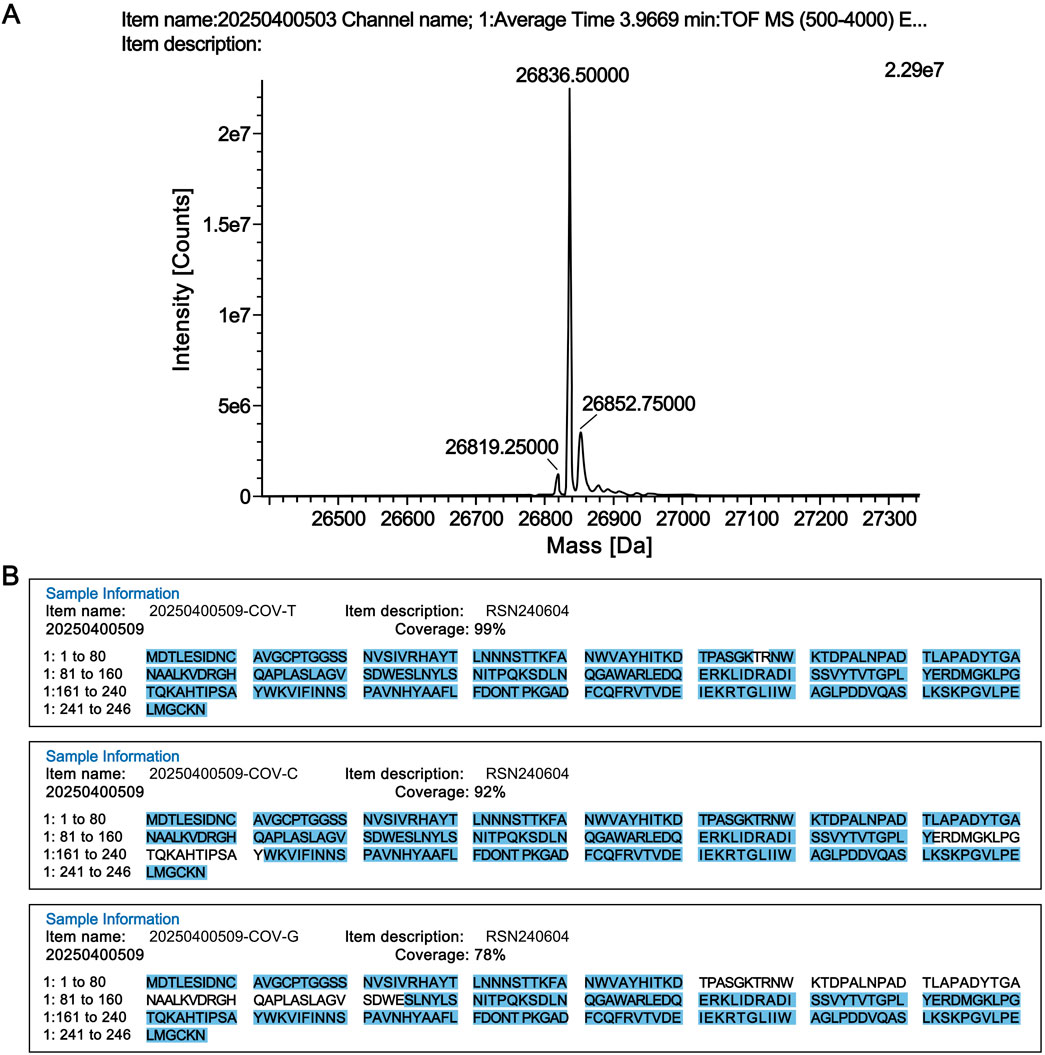
Figure 1. Structural confirmation of the candidate reference standard. (A) Molecular weight determination. (B) Peptide sequence coverage results of the candidate reference material digested by three proteases. COV-T: Trypsin; COV-C: Chymotrypsin; COV-G: Glu-C.
3.1.2 Peptide sequence coverage
The RSN candidate reference material was digested with proteases, and the digested peptide samples were analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS; Xevo G2-XS Q Tof, Waters). The LC-MS/MS data were further analyzed using UNIFI software (Waters), and the results confirmed that the product had 100% peptide sequence coverage (Figure 1B).
3.1.3 Isoelectric point
Using the method described in Section 2.2.3, the isoelectric point of this batch of RSN candidate reference material was determined to be 6.7.
3.1.4 Maximum UV absorption wavelength
Using the method described in Section 2.2.4, the maximum UV absorption wavelength (λmax) of this batch of the RSN candidate reference material was measured as 279.60 nm.
3.2 Purity
3.2.1 SDS-PAGE
The main component self-control method was used to prepare self-control solutions equivalent to 1.0%, 0.5%, and 0.2% of the candidate reference material concentration. Purity and impurity limit analyses were performed for these self-control solutions and the candidate reference material solution according to the method described in Section 2.3.1. The bands corresponding to 1.0%, 0.5%, and 0.2% of the candidate reference material concentration were all visible, confirming the method’s sufficient sensitivity (Figure 2A). The grayscale value of the impurity band with a molecular weight of approximately 19 kDa (10,056,624) in the candidate reference material lane (lane 2) was lower than that of the main component band in the 1.0% self-control solution lane (lane 3) (10,473,265), indicating that the purity of this batch of the candidate reference material was greater than 99.0%.
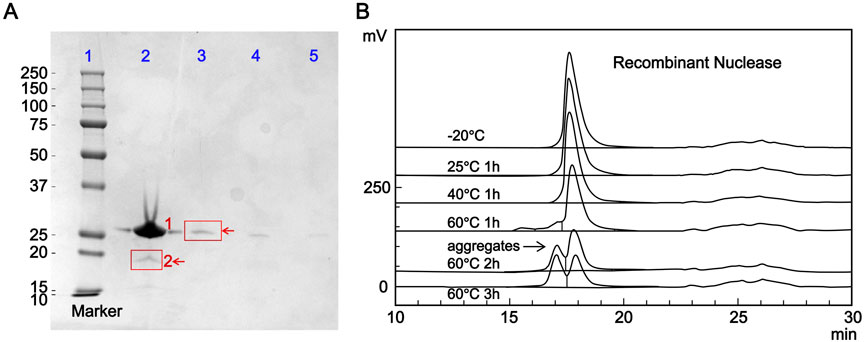
Figure 2. Purity analysis of the candidate reference standard. (A) Purity determination of the candidate reference material by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Lane 1: marker; Lane 2: candidate reference material (1: RSN, 2: impurity); Lane 3: 1.0% self-control solution; Lane 4: 0.5% self-control solution; Lane 5: 0.2% self-control solution. (B) Effect of different heating conditions on the purity of the candidate reference material evaluated by size-exclusion chromatography (SEC-HPLC).
3.2.2 Size-exclusion chromatography (SEC-HPLC)
Following the chromatographic conditions and method described in Section 2.3.2, 50 μL each of the blank solution and candidate reference material solution were injected into the HPLC system, and chromatograms were recorded. As shown in Figure 2B, no interference peaks were observed in the integration region of the main candidate reference material peak in the blank solution chromatogram, demonstrating the method’s good specificity. Two analysts separately determined the purity of three vials of the candidate reference material, and the results are shown in Table 1. The relative standard deviations (CV%) of the main peak retention time and peak area percentage were 0.03% and 0.12% (n = 6), respectively, confirming the accuracy of the method.
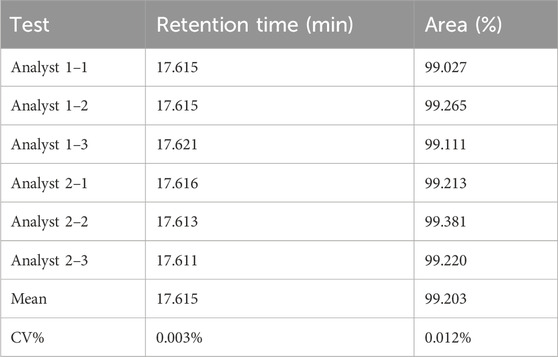
Table 1. Size-exclusion chromatography (SEC-HPLC) purity of RSN determined by two analysts (n = 6).
The candidate reference material was subjected to heating at different temperatures and for different durations, with the untreated candidate reference material as a control, and its purity was evaluated. As shown in Figure 2B, the main peak area percentage remained above 99.0% (99.17%) after incubation at 25 °C for 1 h. After incubation at 40 °C for 1 h, the main peak area percentage dropped below 99.0% (98.97%). The purity decreased to 85.89% after incubation at 60 °C for 1 h, with a significant high-molecular-weight polymer peak (13.14%) appearing before the main peak. Further incubation at 60 °C for 2 and 3 h further decreased the purity to 63.49% and 50.75%, respectively, with the high-molecular-weight polymer content increasing to 35.08% and 47.82%, respectively. These results indicate that the proposed method can effectively detect heat-induced degradation products and reflect a decrease in purity.
3.3 Protein content
3.3.1 Determination of experimental extinction coefficient using an amino acid analyzer
The RSN stock solution was subjected to acid hydrolysis. Accurate protein quantification was performed using the molar amount determined by an amino acid analyzer (Figure 3) and the molecular weight measured by mass spectrometry. Based on the Lambert-Beer law (c = A/EL), the experimental extinction coefficient (E0.1%) of the RSN was calculated to be 1.81.
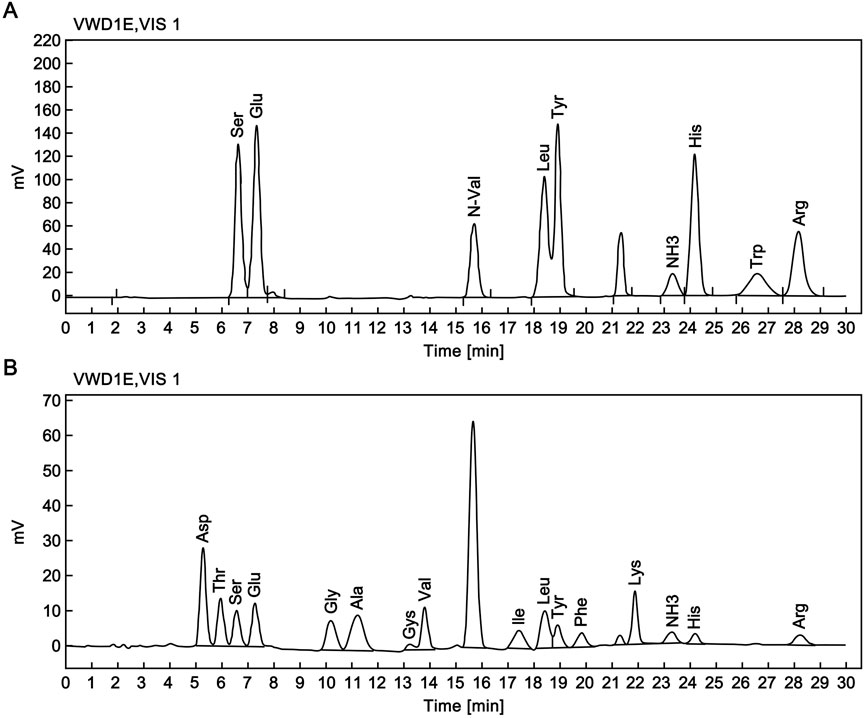
Figure 3. Amino acid composition analysis of the candidate reference standard. (A) Chromatogram of reference amino acid analysis. (B) Chromatogram of RSN amino acid analysis.
3.3.2 Determination of protein content by UV spectrophotometry
Using the theoretical extinction coefficient (E0.1% = 1.68, defined as the absorption coefficient of 1 mg/mL RSN at 280 nm) calculated from the amino acid sequence of the candidate reference material, the protein content of 32 vials of the candidate reference material was determined using the method described in Section 2.4.2, with an average value of 0.20 mg/mL. Using the experimentally measured extinction coefficient (E0.1% = 1.81), the protein content of the same 32 vials was calculated, with an average value of 0.19 mg/mL. The average of the two results (0.2 mg/mL) was taken as the protein content of the candidate reference material batch.
3.4 Optimization of enzymatic reaction conditions
3.4.1 Quality release criteria for the substrate
To investigate the cause of differences in the measured enzymatic activity of the candidate reference material when using different substrates, agarose gel electrophoresis was used to analyze the molecular weights of three herring sperm DNA substrates. As shown in Figure 4A, more than 75% of the DNA substrates from Invitrogen and Promega ranged from 100 to 1,500 bp, whereas the DNA substrate from Solarbio was concentrated in the 100–500 bp range. Using these three substrates, the ΔOD260 was measured at 15, 30, 45, and 60 min according to the method described in Section 2.5, and the ΔOD260 was plotted against time. As shown in Figure 4B, the slopes (k) of the linear plots (reaction rates) obtained with DNA substrates with higher molecular weights (from Invitrogen and Promega) were similar and more than twice that of the reaction rate measured with the low molecular weight substrate.
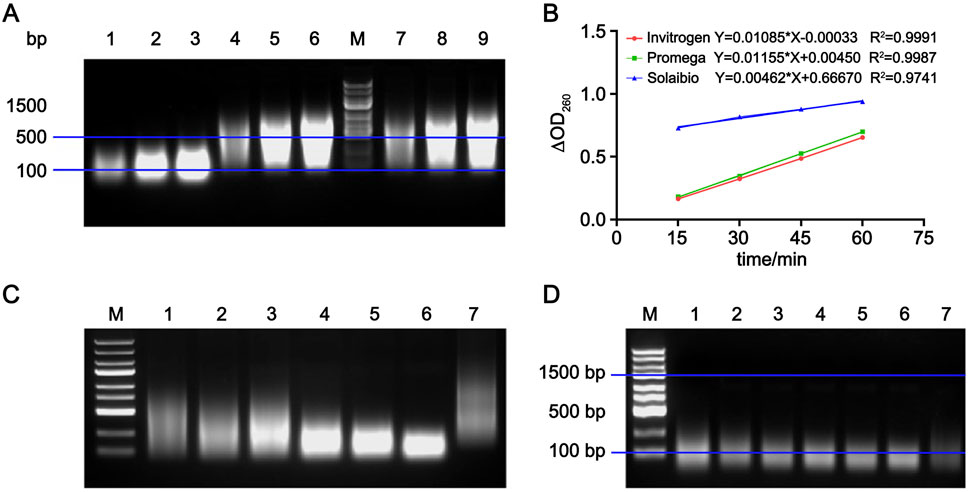
Figure 4. Establishment of substrate quality standards for the activity determination method. (A) Nucleic acid electrophoresis results of DNA substrates from three manufacturers. Lanes 1–3: DNA substrate from Solarbio; Lanes 4–6: DNA substrate from Invitrogen; Lanes 7–9: DNA substrate from Promega; M: marker. (B) Linear equations fitted by plotting ΔOD260 against time for reactions between RSN and DNA substrates from three manufacturers, n = 3. (C) Nucleic acid electrophoresis results of reactions between RSN and DNA substrate from Invitrogen at different times. Lanes 1–6: 5, 15, 30, 45, 60, and 90 min, respectively; Lane 7: unreacted DNA substrate control. (D) Nucleic acid electrophoresis results of reactions between RSN and DNA substrate from Solarbio at different times. Lanes 1–6: 5, 15, 30, 45, 60, and 90 min, respectively; Lane 7: unreacted control.
Diluted RSN candidate reference material was added to the DNA substrates from Invitrogen and Solarbio. Perchloric acid was added at 5, 15, 30, 45, 60, and 90 min, and the DNA precipitate was collected, dissolved, and electrophoresed. As shown in Figures 4C,D, the 100–1,500 bp DNA substrate from Invitrogen was gradually digested into 100–500 bp fragments with the extension of the enzymatic reaction time, while the low molecular weight (100–500 bp) DNA substrate from Solarbio showed no significant change in molecular weight. These results indicate that the greater the change in molecular weight of precipitated DNA over time, the larger the ΔOD260 in the supernatant, the higher the slope of the fitted line, and the faster the enzymatic reaction rate. Therefore, in this method, herring sperm DNA substrates with over 75% of fragments in the 100–1,500 bp range should be used.
3.4.2 Optimization of experimental parameters
The enzymatic kinetic parameters of the candidate reference material were determined using herring sperm DNA conforming to the aforementioned quality criteria as the substrate following the protocol described in Section 2.6.2. Based on the Michaelis-Menten equation, the calculated kinetic constants Km and Vmax were 0.0047 mg/mL and 417.6 U/μL, respectively. As illustrated in Figure 5A, at saturating substrate concentrations, the reaction rate approached Vmax and became independent of substrate concentration, exhibiting a linear correlation with the candidate reference material concentration. In accordance with enzymatic kinetic principles, the substrate concentration was selected to exceed 10×Km (i.e., 0.047 mg/mL), leading to the adoption of 0.1 mg/mL as the optimal substrate concentration. To define the detection range, the candidate reference material was serially diluted 10,000-, 15,000-, 20,000-, 30,000-, 40,000-, 60,000-, and 80,000-fold in assay buffer, with activity measured as detailed in Section 2.6.2. As shown in Figure 5B, the 10,000-fold dilution exhibited substantial variability across the 15-, 30-, 45-, and 60-min time points, whereas dilutions of 60,000-fold or higher resulted in diminished enzymatic activity. Consequently, the 20,000–40,000-fold dilution range was selected for subsequent analyses. For reaction time optimization, activity measurements were performed at 15-min intervals over 15–180 min. As shown in Figure 5C, the activity increased linearly within 15–90 min but declined after that. Within the 15–60-min window, linear regression of ΔOD260 against time yielded the equation Y = 0.01296X 0.04183 (R2 = 0.9979; Figure 5D), confirming robust linearity. Thus, this interval was designated as the optimal reaction period. Notably, the non-zero intercept of this regression precluded the use of the USP-recommended mean activity across time points; instead, the slope (ΔA/t) was substituted into the formula in Section 2.5 for activity calculation.
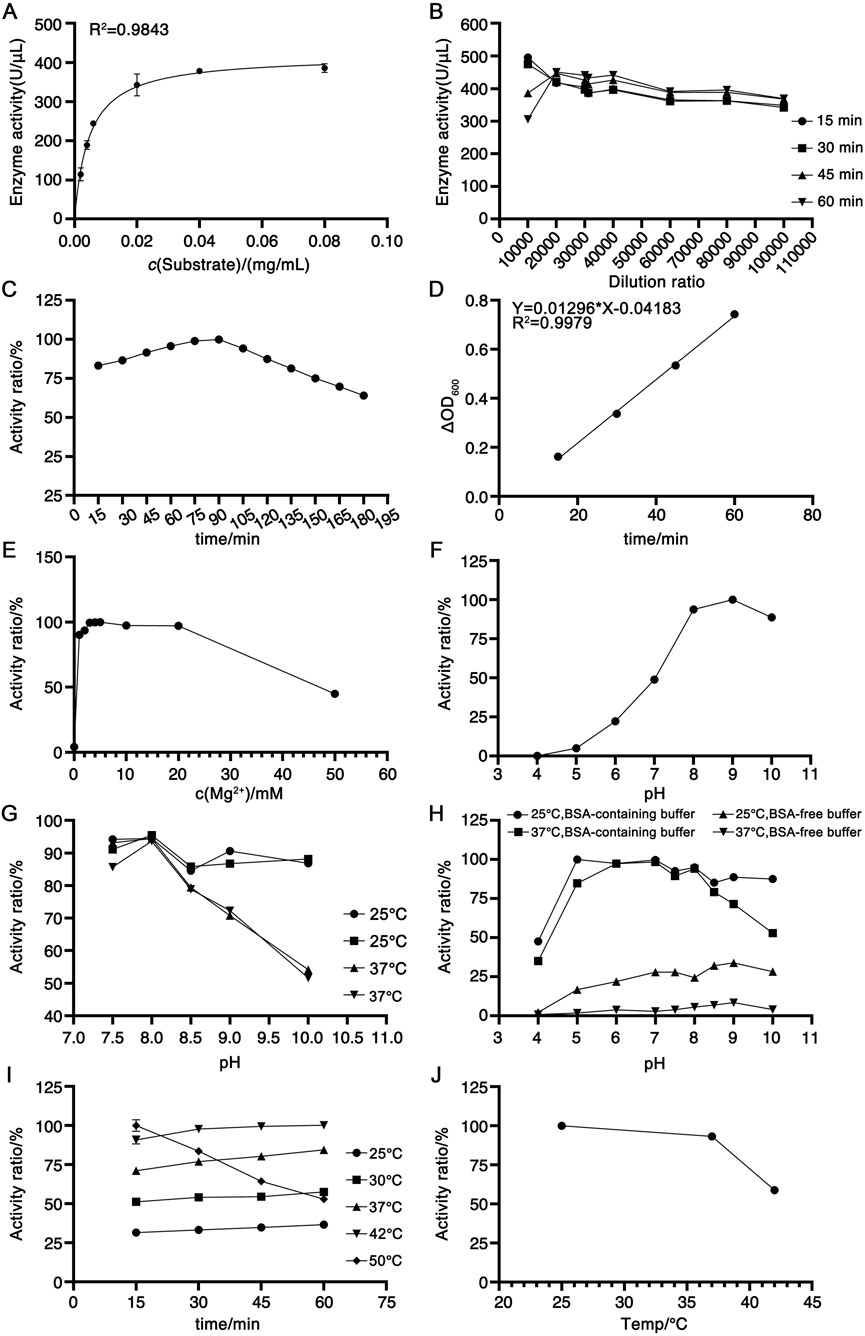
Figure 5. Optimization of experimental parameters for the newly established activity determination method. (A) Michaelis-Menten curve plotted with substrate concentration against RSN activity, n = 2. (B) Determination of candidate reference material activity at different dilution factors, n = 3. (C) Trend of candidate reference material activity changes with reaction time within 15–180 min, n = 3. (D) Linear equation fitted by plotting the ΔOD260 against time within 15–60 min, n = 3. (E) Effect of Mg2+ concentration on candidate reference material activity, n = 3. (F) Optimal pH of the assay buffer, n = 3. (G) Stability of the candidate reference material in assay buffers with different pH values, n = 3. (H) Effect of BSA on candidate reference material activity, n = 3. (I) Optimal enzymatic reaction temperature, n = 3. (J) Stability of the candidate reference material at different temperatures, n = 3.
Given that RSN requires divalent cations for activity, with Mg2+ as a critical cofactor (Miller et al., 1999; Shlyapnikov et al., 2001), assay buffers containing 0–50 mM Mg2+ were evaluated. As shown in Figure 5E, maximal activity was observed at 4 mM Mg2+, with a gradual decline at concentrations exceeding 10 mM, leading to the selection of 4 mM Mg2+ in the assay buffer. The pH dependence of activity was assessed using buffers ranging from pH 4 to 10, revealing optimal activity at pH 8–9 (Figure 5F). To evaluate pH stability under assay conditions (room temperature dilution, 60-min incubation), the candidate reference material was diluted in buffers of pH 7.5, 8, 8.5, 9, and 10, followed by an incubation at 25 °C or 37 °C for 2 h. As shown in Figure 5G, pH 8 consistently yielded the highest activity and stability at both temperatures, thus defining the optimal pH. The impact of BSA was investigated by diluting the candidate reference material in pH 4–10 buffers with or without 0.1 mg/mL BSA, followed by 2-h incubation at 25 °C or 37 °C. As depicted in Figure 5H, BSA-containing buffers significantly enhanced activity, with maximal values at pH 8, leading to the use of BSA-supplemented pH 8 buffer. Temperature optimization revealed increased activity from 25 °C to 42 °C, with inactivation at 50 °C within 60 min (Figure 5I). Stability testing (2-h incubation at 25 °C, 37 °C, or 42 °C) demonstrated robust stability at 25 °C and 37 °C but marked inactivation at 42 °C (Figure 5J), resulting in the selection of 37 °C as the reaction temperature.
3.5 Methodological validation
3.5.1 Specificity
No activity was detected for 0.1 mg/mL BSA solution or 0.2 mg/mL REK solution using this method, with relative activities of 0% and −0.9% compared to RSN, respectively.
Enzymatic activity was measured after incubating the candidate reference material at 25 °C, 40 °C, and 60 °C for 1 h, and the results are shown in Table 2. A paired t-test between the activity of the candidate reference material stored at −20 °C and that incubated at 25 °C for 1 h revealed no statistically significant difference (P = 0.1, >0.05). The RSN activity decreased with increasing temperature: incubation at 40 °C for 1 h resulted in approximately 10% activity loss, whereas incubation at 60 °C for 1 h led to ∼20% decrease. Extending the incubation time at 60 °C to 1–3 h further increased the activity loss from 20% to 60%, indicating that the method can reflect the reduction of RSN activity induced by heat. Size-exclusion chromatography (as described in Section 3.2.2) was used to analyze changes in the purity of the candidate reference material after incubation at 60 °C for 1, 2, and 3 h. As shown in Figure 6A, both activity and purity decreased gradually with prolonged heating, exhibiting consistent trends. This suggests that reduced purity is the primary cause of activity loss and confirms that the proposed method can effectively reflect heat-induced activity reduction.
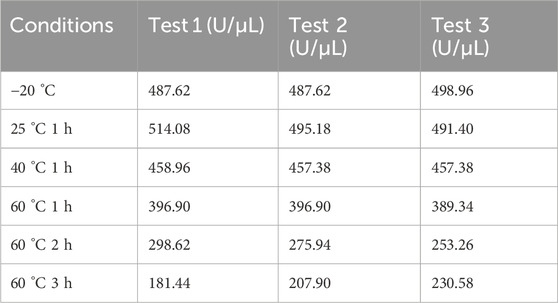
Table 2. Enzymatic activity of heat-treated RSN.
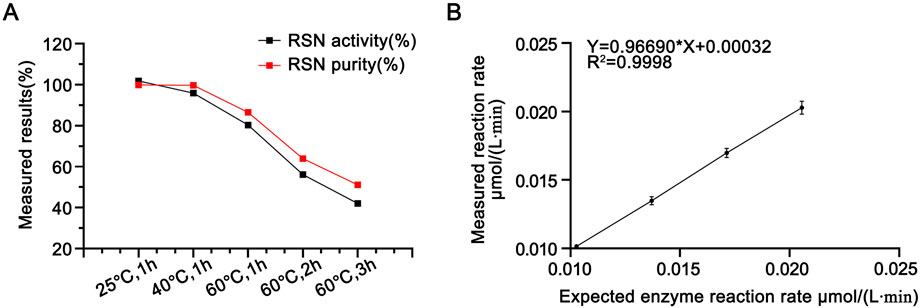
Figure 6. (A) Purity (determined by SEC-HPLC) and activity determination results of RSN with different heat treatments (with the enzymatic activity of untreated RSN set as 100%), n = 3. (B) Linearity of the newly established enzymatic activity determination method in this paper, n = 16.
3.5.2 Intermediate precision
The candidate reference material was prepared into test solutions at four dilution levels (150%, 125%, 100%, and 75%), with 16 replicate measurements performed for each level. The results are presented in Table 3. The mean activities of the test solutions at the four dilution levels were 511.06, 513.25, 509.66, and 511.40 U/μL, with corresponding coefficients of variation of 2.99%, 3.38%, 3.47%, and 4.42%, respectively. All CV% values were less than 5%, indicating that the method showed good in-house intermediate precision.
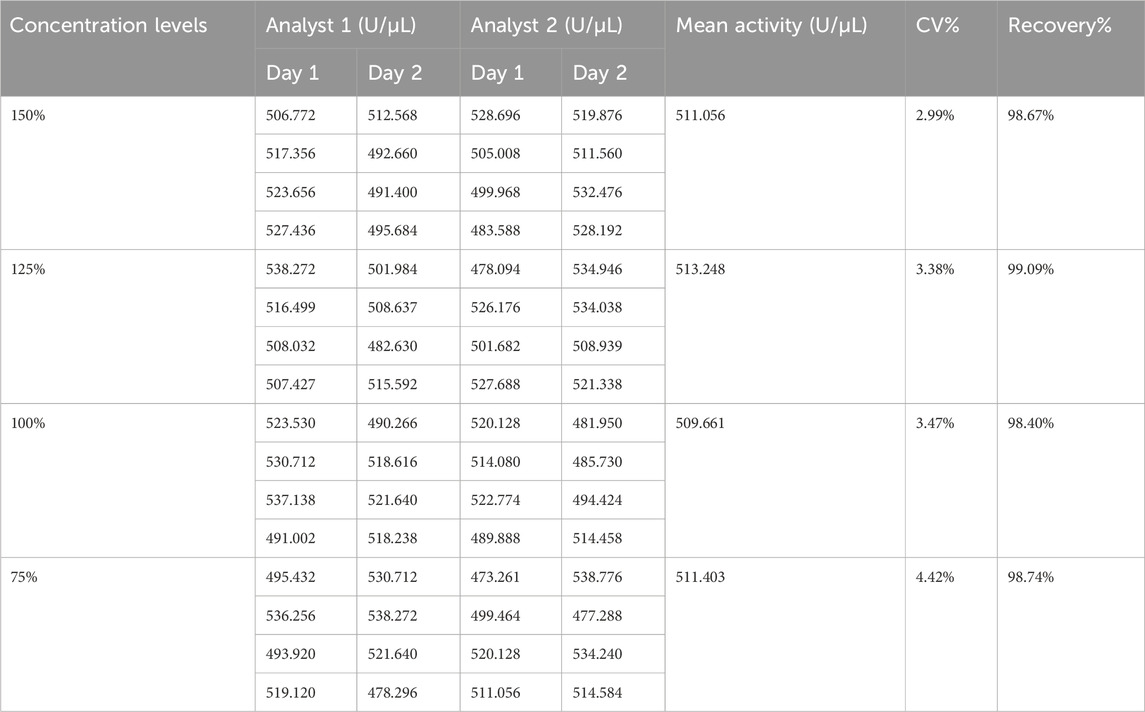
Table 3. Enzymatic activity of RSN at different dilution levels (n = 16).
3.5.3 Repeatability
One analyst determined the enzymatic activity of six samples within a day, with an average value of 514.269 U/μL and a CV% of 2.59%, indicating good repeatability of the method in the laboratory.
3.5.4 Accuracy
Taking the mean value of the collaborative calibration results under Section 3.6 as the reference value of the candidate reference material’s enzymatic activity (517.943 U/μL), the recoveries of the test solutions at the four dilution levels (150%, 125%, 100%, and 75%) in Table 2 were calculated to be 98.67%, 99.09%, 98.40%, and 98.74%, respectively, all within the range of 90%–110%, showing good accuracy of the method.
3.5.5 Linearity and range
The reference values of the enzymatic reaction rates measured for the test solutions at each level were plotted against the corresponding determined values, and the results are shown in Figure 6B. The linear regression equation was Y = 0.96690X + 0.00032, with R2 = 0.9998, and the slope was close to 1.0, indicating that the method had good linearity within the dilution level range of 75%–150%.
3.6 Collaborative calibration
Three laboratories conducted collaborative calibration according to the method mentioned in Section 2.8, and the results are shown in Table 4. Each laboratory obtained four valid determination results per day, totaling 20 determination results over 5 days.
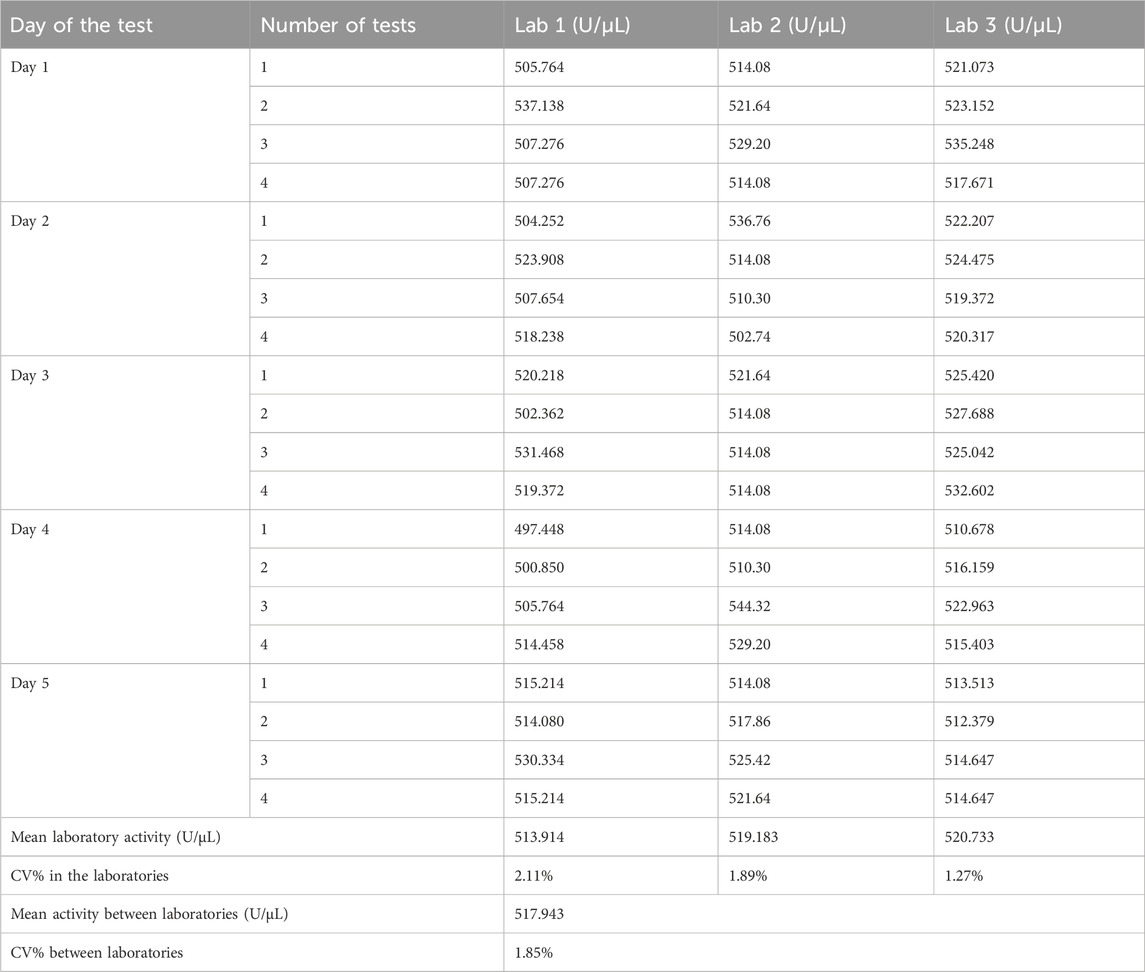
Table 4. Results of collaborative validation.
3.6.1 Precision analysis
The coefficient of variation (CV%) of the four daily detection results in each laboratory was calculated as the intra-day precision. A total of 15 intra-day precision values from three laboratories over 5 days were all less than 4%. The CV% of the mean detection results over 5 days in each laboratory was calculated as the inter-day precision, with three inter-day precision values from three laboratories being less than 2%. The CV% of detection results between two analysts in each laboratory was calculated as the inter-analyst precision, and all three inter-analyst precision values from three laboratories were less than 1%.
The CV% of 20 detection results in each laboratory was calculated as the intra-laboratory precision, which was 2.11%, 1.89%, and 1.27% for Laboratories 1–3, respectively, all being less than 5%. The CV% of 60 detection results across three laboratories was calculated as the inter-laboratory precision, which was 1.85%. These results indicate low variability within each laboratory and good reproducibility for the method between laboratories.
3.6.2 Consistency analysis
GraphPad Prism software was used to perform analysis of variance (ANOVA) on the enzymatic activity of the candidate reference material measured by the three laboratories. As shown in Figure 7, all three sets of data passed the D’Agostino & Pearson normality test (P > 0.05) and the homogeneity of variance test (P > 0.05). The results indicated no statistically significant difference in activity determination results among the three laboratories (P > 0.05), with statistical equivalence. Therefore, the mean value of the determination results from the three laboratories, 517.943 U/μL (rounded to 518 U/μL), was adopted as the activity of this batch of the candidate reference material.
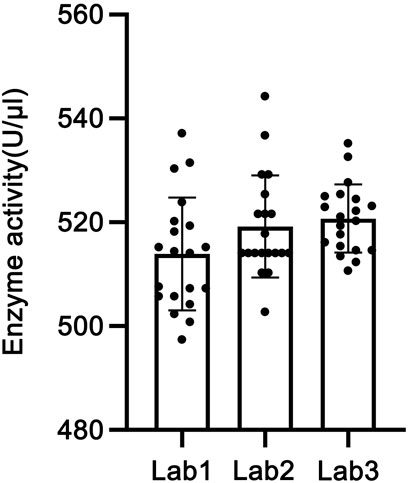
Figure 7. Results of the collaborative calibration of the candidate reference standard.
3.7 Homogeneity and stability evaluation of the candidate reference standard
3.7.1 Homogeneity assessment
Twelve vials of the candidate reference material were selected, and their purity was determined using the method described in Section 2.3.2. The CV% for the main peak area percentage of RSN was 0.14% (n = 12), and the CV% for the retention time was 0.03% (n = 12), indicating good homogeneity of the candidate reference material.
3.7.2 Stability study
As shown in Table 5, three vials of the candidate reference material were stored at 4 °C for 5 or 10 days, and their activities were measured. Paired t-tests were performed to compare the mean activities with that of the candidate reference material stored at −20 °C, yielding P-values of 0.1183 and 0.9775, respectively, both of which were >0.05, indicating no significant differences. Size-exclusion chromatography was used to analyze the purity of the candidate reference material after storage at 4 °C for 5 and 10 days, with the main peak area percentages of RSN being 99.140% and 99.452%, respectively, both exceeding 99.0%. These results demonstrate that the candidate reference material remains stable when stored at 4 °C for 5 and 10 days.
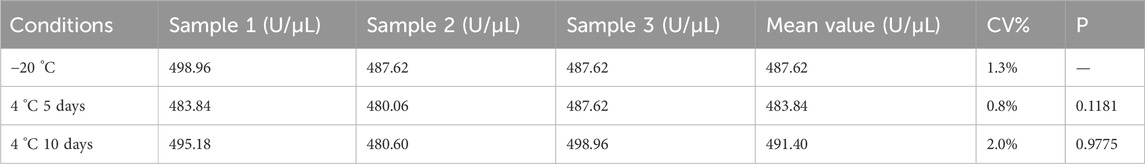
Table 5. Stability results.
For long-term stability, the enzymatic activity of the candidate reference material remained above 80% after 18 months of storage at −20 °C, indicating that it can be stably stored at −20 °C for up to 18 months. In summary, the transportation and long-term storage conditions for this product are designated as −20 °C.
4 Discussion
During the production of cell and gene therapy products, RSN is dosed based on activity units as a critical process parameter. However, changes in production process conditions, enzyme formulation, and transportation may alter enzymatic activity. This increases the risk of inadequate control over residual host cell DNA or RSN (National Medical Products Administration and National Health Commission, 2025). Therefore, the accurate evaluation of RSN activity is crucial for ensuring the stability of the host cell DNA removal process and controlling the cost of enzyme usage. To address the current lack of standard activity determination methods and national reference standards for RSN in China, we have developed and established the first national reference standard for RSN. First, necessary structural studies were conducted on the candidate reference standard. Its structure was confirmed to be consistent with the theoretical value through multiple analyses. These included isoelectric point identification, maximum ultraviolet absorption wavelength identification, mass spectrometric molecular weight determination, and peptide map sequence coverage analysis. The purity determined by size-exclusion chromatography was greater than 99.0%, and the protein content was confirmed to be 0.2 mg/mL by ultraviolet-visible spectrophotometry and amino acid analysis. Homogeneity assessment results showed a CV% of only 0.14% in the main peak area across 12 candidate reference standards. This low variation fully meets the basic requirements for reference standards. On this basis, to further improve the accuracy and reliability of RSN activity determination results, the key parameters of the enzymatic reaction were systematically studied. Substrate quality standards were established through comparative studies of different herring sperm DNA substrates. A complete methodological validation was conducted in accordance with ICH Q2 (R2), and the results showed that the method had good specificity, precision (intra-laboratory CV < 5%), and accuracy (recovery rate 90%–110%).
Activity unit definition methods include both the newly established method and that published by the USP in June 2025, along with its first batch of recombinant nuclease reference standards. They calculate the activity by measuring the ΔA260 after RSN acts on the herring sperm DNA substrate at 37 °C for 15, 30, 45, and 60 min. The optimized method uses an activity determination buffer with an Mg2+ concentration of 4 mM for activity determination. Compared with the USP RSN activity determination method, which uses an activity determination buffer containing 1 mM Mg2+, the enzymatic activity is increased by more than 10%. In addition,the USP RSN activity determination method as the mean of results measured at four time points: 15, 30, 45, and 60 min. This approach is based on the key assumption that a linear fit of the measured values at these four time points against time should pass through the origin. However, experimental data demonstrate that the fitted equation exhibits a significant intercept, indicating the presence of background absorption in the blank system. To address this issue, our method utilizes the slope of the linear equation (ΔA/t) to calculate RSN activity, effectively solve the problem of large variation in the results measured at the four time points in the USP method (CV% is approximately 15%). When applying the absolute definition method for activity assignment of national standards, high precision is essential. We recommend adopting the slope calculation approach described in our method to ensure the accuracy and reliability of RSN activity determination results. In the optimization of enzymatic reaction conditions, the stability of the enzyme during the determination is a crucial but easily overlooked factor. In this study, we investigated the effects of buffer pH and temperature on the stability of RSN during the enzyme–substrate reaction. Finally, we selected an activity determination buffer containing BSA with pH = 8 for RSN activity determination at 37 °C. As an activity unit definition method, this method is susceptible to factors such as changes in reaction temperature, ionic strength and pH of the activity determination solution, time control, and operation by experimental personnel. In recent years, an increasing number of enzyme products have begun to adopt relative activity determination methods based on activity reference standards, which can overcome the influence of the above-mentioned factors and improve the accuracy and stability of detection results.
Using the newly established activity determination method to assign activity to the RSN candidate reference standard, we formulated a detailed collaborative calibration plan and invited three laboratories to jointly calibrate the activity of the candidate reference standard. Each laboratory conducted 20 independent determinations, and the intra-laboratory precision (CV% 1.27%–2.11%) and inter-laboratory precision (CV% 1.85%) of the detection results were good. No significant difference was found in the results of each laboratory (P > 0.05), showing statistical equivalence. Therefore, the total mean of the collaborative calibration, 518 U/μL, was taken as the activity of this batch of candidate reference standards. During collaborative calibration, we found that not setting the centrifuge temperature led to significantly higher detection results. This occurred when centrifuging after terminating the enzymatic reaction. After setting the centrifugation temperature to 4 °C, the determination results of the candidate reference standard were closer to those of the other two laboratories. Therefore, we further specified the centrifugation conditions after terminating the reaction by centrifugation at 14,000 rpm for 7 min at 4 °C after pre-cooling the centrifuge.
Next, stability studies were conducted to clarify the suitable transportation conditions for the candidate reference standard and exclude factors that may affect the characteristic values. The results showed that the purity and activity of the candidate reference standard did not significantly change after being placed at 4 °C for 5 and 10 days. Therefore, this product should be stored and transported at −20 °C. Long-term stability studies indicated that the candidate reference standard remains stable over extended use. In addition to being an indispensable tool enzyme in the production process of biological products, nuclease is also an impurity in the final product. Regulatory authorities around the world have pointed out that nuclease must be removed and its residual amount in the final product should be controlled. Currently, the enzyme-linked immunosorbent assay remains the most common method for detecting residual nuclease. This method is based on antigen-antibody binding. However, the accuracy of this method depends on the quality of the antibody and batch consistency, and it cannot evaluate the residual enzymatic activity. The national reference standard for RSN established in this study shows a good specific activity relationship, which may be applied to the quantitative detection of residual enzymatic activity, realizing the dual quantification of residual enzymatic activity and protein content.
For the first time in China, the RSN reference standard established in this study has achieved the unification of the characteristic value of the enzyme, namely, enzymatic activity. This effectively solves the problem of incompatibility of RSN activity units from different sources and provides a reliable basis for the precise feeding of RSNs in the production process. Moreover, it provides a standardized activity determination method and traceable standard for RSN manufacturers’ release inspection. This significantly improves the accuracy and comparability of detection results. More importantly, this reference standard has accumulated key data for the future inclusion of RSN general rules in the Chinese Pharmacopoeia, promoting the standardization process of recombinant nuclease and providing quality assurance for the industrial development of emerging fields such as cell and gene therapy.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Author contributions
LW: Methodology, Investigation, Formal Analysis, Data curation, Writing – original draft, Writing – review and editing. YS: Investigation, Data curation, Formal Analysis, Writing – original draft. KX: Data curation, Investigation, Writing – review and editing. XH: Investigation, Writing – review and editing. YL: Resources, Investigation, Writing – review and editing. PL: Investigation, Data curation, Writing – review and editing. XL: Methodology, Investigation, Writing – review and editing. JL: Supervision, Conceptualization, Writing – review and editing. JL: Supervision, Conceptualization, Project administration, Validation, Funding acquisition, Writing – review and editing.
Funding
The authors declare that financial support was received for the research and/or publication of this article. This study was funded by the State Key Laboratory of Drug Regulatory Science (Grant No. 2024SKLDRS0203).
Acknowledgements
We thank LetPub (www.letpub.com.cn) for its linguistic assistance during the preparation of this manuscript.
Conflict of interest
Author XL was employed by the company Shanghai Yaxin Biotechnology Limited Company. Author JL was employed by the company Sinovac Biotech Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ball, T. K., Saurugger, P. N., and Benedik, M. J. (1987). The extracellular nuclease gene of Serratia marcescens and its secretion from Escherichia coli. Gene 57, 183–192. doi:10.1016/0378-1119(87)90121-1
Benedik, M. J., and Strych, U. (1998). Serratia marcescens and its extracellular nuclease. FEMS Microbiol. Lett. 165, 1–13. doi:10.1111/j.1574-6968.1998.tb13120.x
Biedermann, K., Jepsen, P. K., Riise, E., and Svendsen, I. (1989). Purification and characterization of a Serratia marcescens nuclease produced by Escherichia coli. Carlsb. Res. Commun. 54, 17–27. doi:10.1007/bf02910469
Cheng, L.-C., Hor, L.-I., Wu, J.-Y., and Chen, T.-L. (2003). Effect of specific growth rate on the production of a recombinant nuclease by Escherichia coli. Biochem. Eng. J. 14, 101–107. doi:10.1016/s1369-703x(02)00156-0
Eaves, G. N., and Jeffries, C. D. (1963). Isolation and properties of an exocellular nuclease of Serratia marcescens. J. Bacteriol. 85, 273–278. doi:10.1128/jb.85.2.273-278.1963
Friedhoff, P., Meiss, G., Kolmes, B., Pieper, U., Gimadutdinow, O., Urbanke, C., et al. (1996). Kinetic analysis of the cleavage of natural and synthetic substrates by the serratia nuclease. Eur. J. Biochem. 241, 572–580. doi:10.1111/j.1432-1033.1996.00572.x
Kim, A.-Y., Park, S. Y., Park, S. H., Jin, J. S., Kim, E.-S., Kim, J. Y., et al. (2021). Validation of pretreatment methods for the in-process quantification of foot-and-mouth disease vaccine antigens. Vaccines 9, 1361. doi:10.3390/vaccines9111361
Li, S.-M., Bai, F.-L., Xu, W.-J., Yang, Y.-B., An, Y., Li, T.-H., et al. (2014). Removing residual DNA from Vero-cell culture-derived human rabies vaccine by using nuclease. Biologicals 42, 271–276. doi:10.1016/j.biologicals.2014.06.005
Liu, L., Guo, Q., Hao, J., Guo, D., Xuemei, Z., and Wu, Y. (2021). Preliminary development of purification process for influenza vaccine cultured in serum-free suspended MDCK cells. Chin. J. Biol. 34, 1100–1104.
Lunin, V. Y., Levdikov, V. M., Shlyapnikov, S. V., Blagova, E. V., Lunin, V. V., Wilson, K. S., et al. (1997). Three-dimensional structure of Serratia marcescens nuclease at 1.7 Å resolution and mechanism of its action. FEBS Lett. 412, 217–222. doi:10.1016/s0014-5793(97)00512-7
Mayer, V., Frank, A. C., Preinsperger, S., Csar, P., Steppert, P., Jungbauer, A., et al. (2023). Removal of chromatin by salt-tolerant endonucleases for production of recombinant measles virus. Biotechnol. Prog. 39, e3342. doi:10.1002/btpr.3342
Miller, M. D., Cai, J., and Krause, K. L. (1999). The active site of Serratia endonuclease contains a conserved magnesium-water cluster. J. Mol. Biol. 288, 975–987. doi:10.1006/jmbi.1999.2729
Moreira, A. S., Cavaco, D. G., Faria, T. Q., Alves, P. M., Carrondo, M. J. T., and Peixoto, C. (2021). Advances in lentivirus purification. Biotechnol. J. 16, 2000019. doi:10.1002/biot.202000019
National Medical Products Administration, and National Health Commission (2025). Pharmacopoeia of the People's Republic of China, 3. Beijing: China Medical Science Press. Available online at: https://www.nmpa.gov.cn/xxgk/fgwj/gzwj/gzwjyp/20250325183810122.html.
Nestle, M., and Roberts, W. K. (1969). An extracellular nuclease from Serratia marcescens. J. Biol. Chem. 244, 5213–5218. doi:10.1016/s0021-9258(18)63648-8
Ruan, C., Shi, J., Li, W., Liu, B., Bao, X., Liao, G., et al. (2023). Application of benzonase nuclease in manufacture of vero cell inactivated influenza virus vaccine. Chin. J. Virol. 39, 980–990. doi:10.13242/j.cnki.bingduxuebao.004341
Schwardmann, L. S., Nölle, V., and Elleuche, S. (2020). Bacterial non-specific nucleases of the phospholipase D superfamily and their biotechnological potential. Appl. Microbiol. Biotechnol. 104, 3293–3304. doi:10.1007/s00253-020-10459-5
Shlyapnikov, S. V., Lunin, V. V., Blagova, E. V., Pembrandt, M., Betzel, C., and Mikhailov, A. M. (2001). X-ray analysis of the magnesium-containing endonuclease from Serratia marcescens. Russ. J. Bioorg. Chem. 27, 370–377. doi:10.1023/a:1012932601549
The United States Pharmacopeial Convention (2024). Endonuclease (Serratia marcescens) 35 μL. Volume USP reference standard certificate. Available online at: https://static.usp.org/pdf/EN/referenceStandards/certificates/1235470-F233D0.pdf.
U.S. Department of Health and Human Services, Food and Drug Administration, and Center for Biologics Evaluation and Research (2020). Chemistry, manufacturing, and control (CMC) information for human gene therapy investigational new drug applications (INDs), guidance for industry. Available online at: https://www.fda.gov/media/113760/download.
von Elling-Tammen, M. S., Taft, F., Thom, V., Stitz, J., Barbe, S., and Krause, A. (2025). Optimizing nuclease treatment to enhance anion exchange chromatography of HIV-derived virus-like particles. J. Chromatogr. B 1256, 124539. doi:10.1016/j.jchromb.2025.124539
Wang, L., Lu, P., Zhang, H., Li, J., and Liang, C. (2023). Collaborative study to evaluate a reporter gene assay for recombinant human follicle-stimulating hormone bioactivity. Acta Pharm. Sin. 58, 760–766.
Keywords: recombinant nuclease, national standard, enzymatic activity assay, herring sperm DNA substrate, methodological validation, collaborative calibration
Citation: Wang L, Sun Y, Xu K, Hu X, Li Y, Lyu P, Liu X, Li J and Li J (2025) Establishment of the first Chinese national standard for recombinant nuclease used as a process material in pharmaceutical manufacturing. Front. Bioeng. Biotechnol. 13:1695176. doi: 10.3389/fbioe.2025.1695176
Received: 29 August 2025; Accepted: 03 November 2025;
Published: 02 December 2025.
Edited by:
Aravind Madhavan, Amrita Vishwa Vidyapeetham University, IndiaReviewed by:
Ayswarya Ravi, Corteva Agriscience, United StatesGanesha Antarnusa, Sultan Ageng Tirtayasa University, Indonesia
Copyright © 2025 Wang, Sun, Xu, Hu, Li, Lyu, Liu, Li and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jing Li, bGlfamluZ0BuaWZkYy5vcmcuY24=