 Yuan Wang
Yuan Wang Xu Qian
Xu Qian Guokui Zheng4
Guokui Zheng4 Ziqi Tian
Ziqi Tian- 1Ningbo Institute of Materials Technology and Engineering, Chinese Academy of Sciences, Ningbo, China
- 2Department of Materials Science and Opto-Electronic Technology, University of Chinese Academy of Sciences, Beijing, China
- 3Nano Science and Technology Institute, University of Science and Technology of China, Suzhou, China
- 4Key Laboratory of Biomass Chemical Engineering of Ministry of Education, College of Chemical and Biological Engineering, Zhejiang University, Hangzhou, China
Electrocatalytic nitrogen reduction reaction (NRR) is a promising and sustainable approach for ammonia production. Since boron as an active center possesses electronic structure similar to that of transition metals with d-orbitals (J. Am. Chem. Soc., 2019, 141 (7), 2884), it is supposed to be able to effectively activate the triple bond in N2. MXenes can be applied as substrates due to the large specific surface area, high conductivity, and tunable surface composition. In this work, the catalytic performance of a series of MXenes-supported single boron atom systems (labeled as B@MXenes) has been systematically studied by using density functional theory (DFT). B@Nb4C3O2, B@Ti4N3O2, and B@Ti3N2O2 were screened out owing to outstanding catalytic activity with limiting potentials of −0.26, −0.15, and −0.10 V, respectively. Further analysis shows that the unique property of boron that can intensely accept lone pair and back-donate the anti-bond of nitrogen contributes to the activation of inert triple bond. This work provides a new idea for the rational design of NRR catalyst and is of great significance for the future development of nitrogen reduction catalysts.
Introduction
NH3 is one of the most important basic chemicals used in the downstream manufacturing of various products in the agricultural industry, synthetic fiber, and fine chemicals (Giddey et al., 2013; Zhan and Zhang, 2021). Moreover, it is supposed to be promising energy storage intermediate. Although the atmosphere consists of more than 70% of nitrogen and the reduction of nitrogen to produce NH3 is an exothermic reaction under standard conditions, the nitrogen fixation from N2 to NH3 is still tough because of the inherent nature of the N≡N bond with extremely high bond energy (Gambarotta and Scott, 2004). Currently, industrial production of NH3 relies on the high energy-intensive Haber–Bosch process, which results in a huge amount of energy consumption and carbon emissions around the world (Cui et al., 2018).
In contrast, inspired by the biological N2 fixation in bacteria (Singh et al., 2021), the electrochemical nitrogen reduction reaction (NRR) provides a green and sustainable alternative to produce NH3 at relatively mild conditions (Liu et al., 2021a). The development of highly active and stable catalysts for the electrochemical NRR is urgently demanded and challenging. In many cases (Kugler et al., 2015; Manjunatha and Schechter, 2018; Liu et al., 2020a; Wang et al., 2020a; Wang et al., 2020b; Cai et al., 2020; Yang et al., 2020; Zheng et al., 2020; Dai et al., 2021), electrocatalysts contain transition metals as the catalytic center. The empty d-orbitals accept the σ-electron pair and the filled d-orbitals back-donate the anti-bonding π orbitals, activating the inert bond (Rao and Rao, 1991; Légaré et al., 2018), while there is much room for improvement of Faraday efficiency and reaction selectivity. In recent years, nonmetallic boron-based catalysts, such as boron-doped graphene and dicoordinate borylene, have attracted great interest in N2 reduction (Légaré et al., 2018; Yu et al., 2018). The boron sites can provide enhanced affinity to N2 molecules, leading to high NH3 productivity (Hering-Junghans, 2018).
Introducing boron dopant into an appropriate substrate is a strategy to construct boron-centered catalytic system (Ji et al., 2019; Liu et al., 2019; Zheng et al., 2019). The transition metal carbides/nitrides/carbonitrides, known as MXenes, is a new class of two-dimensional materials (Sun et al., 2020). MXenes materials are generally expressed in chemical formula as Mn + 1XnTx. M represents early transition metals (Hf, Zr, Nb, Ti, and Ta), X represents the nonmetal element C or N, and T refers to the surface terminal groups, like hydroxyl group, −F, or −O. Normally, n is 2 or 3. Because of the high conductivity, large specific surface area, and tunable surface composition, it has attracted extensive attention in the field of electrocatalysis and is regarded to be an ideal substrate to support the active center (Liu et al., 2020b; Wang et al., 2021). For example, Mo@Mo2CO2 Mo2TiC2-based systems, Mo2C, and W2C have been reported as novel electrocatalysts for the NRR (Shao et al., 2018; Huang et al., 2019; Gao et al., 2020). Previous works mainly focused on three- and five-layered M2XO2 as electrocatalyst for the NRR, such as Mo@Ti2NO2 (Cheng et al., 2019), Mo@Mo2CO2 (Huang et al., 2019), Ru@Mo2CO2 (Peng et al., 2020), or B@W2CO2 (Zheng et al., 2019). In this work, we further consider MXenes composed of more layers that possess high thermodynamic and dynamic stability. The nonmetal B atom is doped into M3X2Ox or M4X3Ox which were selected out from the C2DB database (Rasmussen and Thygesen, 2015; Gjerding et al., 2021). Fourteen boron-doped MXenes (B@MXenes) have been studied as potential electrocatalysts. Three candidates are screened out, labeled as B@Nb4C3O2, B@Ti4N3O2, and B@Ti3N2O2. The nitrogen reduction processes are explored to evaluate the catalytic performance. In order to understand the promoted activity of the boron-containing systems, charge density differences and projected density of states (PDOS) are analyzed. Moreover, AIMD simulation is employed to verify the stability. This work may broaden the application of MXene-based materials and has great significance for the future development of electrocatalyst for the NRR. We hoped our work can inspire more researches on M3X2Ox or M4X3Ox that have the potential to be implemented experimentally.
Computational Methods
All density functional theory (DFT) computations were performed by using the Vienna ab initio simulation package (VASP) with the projector augmented wave (PAW) (Blöchl, 1994; Kresse and Furthmüller, 1996; Kresse and Joubert, 1999; Grimme, 2006). The van der Waals interaction was considered by the empirical correction of the Grimme’s scheme (DFT-D2). The generalized gradient approximation (GGA) in Perdew–Burke–Ernzerh (PBE) functional was adopted (Perdew and Wang, 1992; Perdew et al., 1997). Energy cutoff of 450 eV for the plane wave basis was applied. The Brillouin zone was sampled by using a 3 × 3 × 1 Monkhorst-Pack k-points mesh. Both energy cutoff and k-points mesh have been checked based on calculations of a typical system, as shown in Supplementary Figures S1, S2. To avoid interactions between periodic images, a vacuum distance of 15 Å was imposed between two periodic units. All structures were optimized within energy and force convergences of 10−5 eV/atom and 0.02 eV/Å, respectively. AIMD was performed under NVT ensemble by using Nosé–Hoover thermostat to control the temperature at 300 K. The time step is 2 fs, and each simulation run for 5,000 steps, namely, 10 ps. The energy barrier for B atom escaping from the vacancy is simulated by conducting the climbing-image–nudged elastic band (CI-NEB) methods. For each path, three intermediate images were inserted. The convergence criteria for energy and force were set to be 10−7 eV and 0.05 eV/Å, respectively.
The computational hydrogen electrode model (CHE) was employed to calculate the Gibbs free energy change (ΔG) of each elementary step (Norskov et al., 2004; Skúlason et al., 2012). According to the CHE model, the ΔG value can be determined by the following Eq. 1:
where ΔE is the internal energy change directly calculated from DFT, ΔEZPE is the change of zero-point energy, T is the room temperature (300 K), ΔS is the entropy change, and ΔGU is the free energy contribution related to electrode potential U. ΔGpH is the free energy correction of pH, which can be expressed as follows:
where kB is the Boltzmann constant and the value of pH is assumed to be zero. The activity was evaluated through the limiting potential (UL) vs. RHE that was defined as follows:
where ΔG is the free energy of the potential-determining step.
To evaluate the interaction between catalyst and nitrogen molecule, the adsorption energy (∆Ead) is calculated as follows:
where Etotal, EB@MXene, and EN2 are the energies of the B@MXene-N2 complex, B@MXene catalyst, and N2 molecule directly obtained from DFT calculation, respectively.
The binding energy of B atom (Eb) is calculated to evaluate the feasibility of the designed structures:
where EB, EMXene(Ov), and EB@MXene represent the energies of a single B atom, MXene with an oxygen vacancy, and B-doped MXene, respectively. A more positive value of Eb indicates a stronger binding between boron and the MXene substrate.
Results and Discussion
In experiment, the oxygen-covered MXenes are stable, while the coverage of surface groups is normally far from uniform, that is, there can be plenty of vacancy (Xie et al., 2014; Karlsson et al., 2015). It is possible to introduce boron dopant into oxygen vacancy, shown as a B-doped Mn + 1XnOx system, or labeled as B@Mn + 1XnTx in this work. Herein, fourteen thermodynamically stable MXenes were selected from structure database (Groom et al., 2016) to construct corresponding B@MXenes models. In Figure 1, B@Ti3C2O2 is taken as an example to show the typical atomic model.
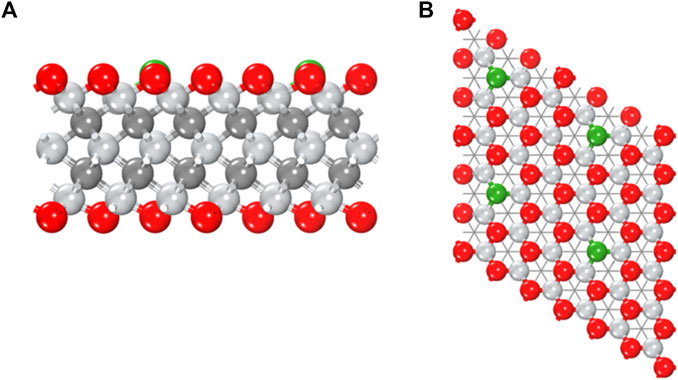
FIGURE 1. (A) Side and (B) top views of the B@Ti3C2O2 model. Color code: titanium in silver, carbon in gray, oxygen in red, and boron in green.
Intense adsorption of gas phase N2 on the surface of the catalyst is suggested to be the prerequisite for an efficient NRR process, which may activate the inert triple bond. Among the nonmetal elements, B atom possesses the combination of empty orbitals and occupied orbitals in specific direction; thus, the incorporated B site can serve as an active center for the capture of N2. Compared to previous results, the adsorption of N2 on B atom is preferable to that on O vacancy, indicating that the B center plays a key role in the electrocatalytic NRR process (Zheng et al., 2019). There are mainly four possible pathways for N2 electrochemical reduction to NH3: distal, alternative, enzymatic, and mixed paths. Two typical N2 adsorption configurations on B@MXenes systems are taken into account, including end-on and side-on configurations, which determine the following reduction path (Zhao and Chen, 2017). The simulated adsorption energies are shown in Figure 2. It is noticed that on all the fourteen MXene layers, the end-on pattern is considerably more stable than the side-on one and initializes all the following reduction paths. Furthermore, we also compare the hydrogen adsorption energy since the hydrogen evolution reaction (HER) is a competitive process. Both oxygen and boron sites are considered as the adsorption sites for some typical systems. As listed in Supplementary Table S1, the hydrogen adsorption on the O site is also weaker than that on B dopant. Although the desorption energy of hydrogen close to zero is normally related to high activity of the HER, the more negative adsorption energy of nitrogen indicates that the active center can be occupied by nitrogen, hindering the competitive hydrogen adsorption. The weaker hydrogen adsorption than nitrogen adsorption in the end-on configuration indicates a high selectivity of the NRR over the HER on all the considered B@MXenes.
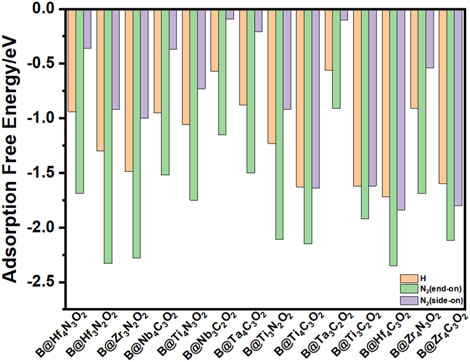
FIGURE 2. The adsorption energy of proton and N2 adsorption on fourteen B@MXenes. Both end-on and side-on configurations for N2 adsorption are simulated.
Normally, the potential-determining step (PDS) of the NRR occurs in the first hydrogenation step (*N2→*NNH) or the last one (*NH2→*NH3), which depends on the binding strength of the nitrogen-containing intermediate to the catalyst surface (Skúlason et al., 2012). If the bonding strength is too weak, the breaking of the nitrogen triple bond is probable to be the PDS of the whole process. On the contrary, the strong bonding strength leads to the difficulty of the *NH3 formation in the last hydrogenation step. We screened fourteen B@MXene systems according to the free energy changes of the first and the last hydrogenation steps as the criteria. As shown in Figure 3, Ti4N3O2, Ti3N2O2, and Nb4C3O2 in the lower left corner exhibit relatively low energy barriers in both the first and the last key steps. Subsequently, we further explore the whole NRR processes of these three systems.
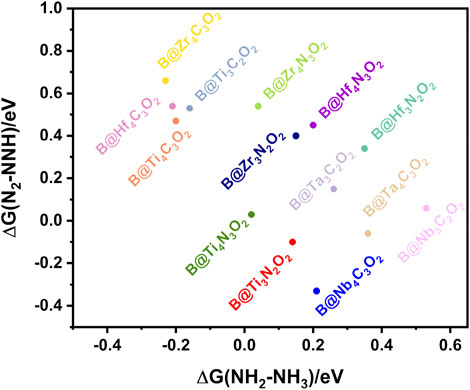
FIGURE 3. The free energy changes of the first and the last hydrogenation steps of the fourteen B@MXenes.
Due to the high energy barrier of N2 dissociation, the dissociative step was generally not considered in the hydrogenation process. As illustrated in Figure 4A, there are four possible associative NRR mechanisms, that is, distal, alternative, enzymatic, and mixed on the catalyst surface (Shi et al., 2019). The enzymatic path is excluded in our study since the side-on configuration is less stable than the corresponding end-on one that generally initializes the enzymatic mechanism. Starting from the end-on configuration, free energy diagrams including distal, alternative, and mixed paths for three candidates (B@Nb4C3O2, B@Ti4N3O2, and B@Ti3N2O2) are presented in Figures 4B–D.
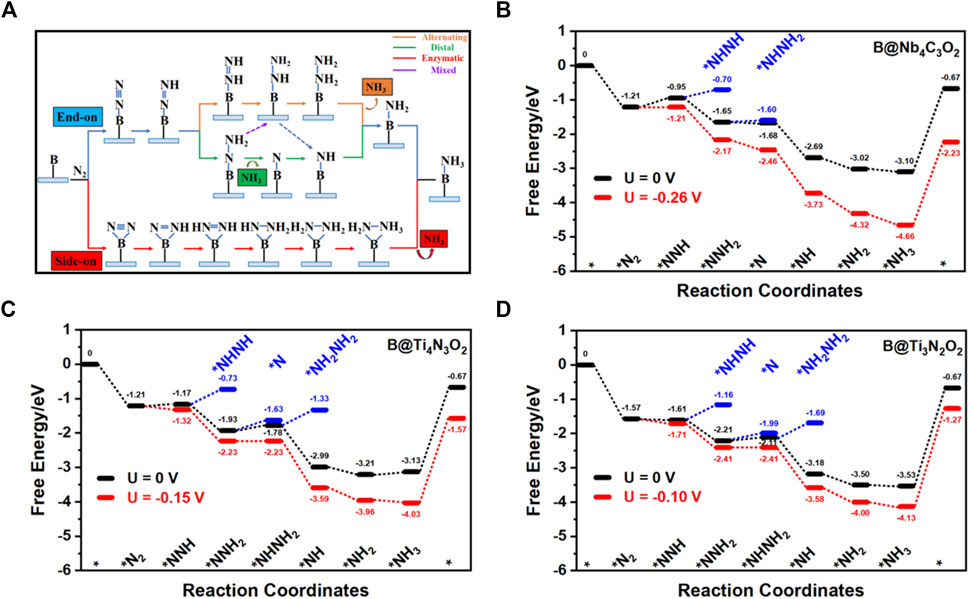
FIGURE 4. (A) Schematic illustration of the distal, alternating, and enzymatic pathways for the electrochemical NRR. Free energy diagrams for the nitrogen reduction to ammonia on (B) B@Nb4C3O2, (C) B@Ti4N3O2, and (D) B@Ti3N2O2.
On B@Nb4C3O2, the adsorbed N2 (*N2) can interact with the proton–electron (H+/e−) pair to form NNH*. The Gibbs free energy change of the first hydrogenation step (*N2→*NNH) is 0.26 eV. Then, the H+/e− pair can attack 1 N atom of the adsorbed NNH species to form *NHNH or *NNH2 intermediate, leading to the alternative or distal pathway, respectively. The calculated free energy change of the *NNH→*NNH2 step is −0.70 eV, rather lower than that of the *NNH→*NHNH step (0.25 eV). Therefore, B@Nb4C3O2 prefers to catalyze the NRR through the distal path. Once *NNH3 is formed, the first NH3 molecule can be readily desorbed from the surface, leaving a single nitrogen atom on the B site. In the subsequent reaction steps, three H+/e− pairs can continuously react with the remaining *N, forming *NH, *NH2, and *NH3 with energy changes of −1.01, −0.33, and −0.08 eV, respectively. Among all the elementary steps, the formation of *N2H (*N2→*NNH) is the PDS with the maximum free energy demand of 0.26 eV. In other words, when the external potential reaches to −0.26 V, the reaction can take place thermodynamically. The whole process proceeds the distal mechanism through the intermediates as *N2 → *NNH → *NNH2 → *N → *NH → *NH2 → *NH3. Even though NH3 desorption from the activation center requires a large amount of energy that may inhibit the regeneration of catalyst, we suppose that the strongly acidic electrolyte results in the formation of NH4+ and promotes desorption of the products (Ling et al., 2018; Zheng et al., 2019; Guo et al., 2020).
Similarly, the NRR pathways on B@Ti4N3O2 and B@Ti3N2O2 are examined from the end-on adsorption configuration. The Gibbs free energy changes of the first hydrogenation step (*N2→*NNH) on B@Ti4N3O2 and B@Ti3N2O2 are both 0.04 eV. Afterward, the *NNH is easily protonated to the energetically more favorable *NNH2, releasing energies of 0.76 and 0.60 eV, respectively. Then, the H+/e−pair can attack *NNH2 to form *NHNH2 or *NNH3.. On B@Ti4N3O2 and B@Ti3N2O2, the free energy changes of the *NNH2→*NHNH2 step are 0.15 and 0.10 eV, respectively, while the free energy changes of the *NNH2→*N step are 0.30 and 0.22 eV, respectively. Thus, mixed paths through *NNH2→*NHNH2 step are favorable on these two Ti-containing systems. In the following steps, three H+/e− pairs continuously attack *NHNH2, forming *NH, *NH2, and *NH3. Among all the elementary steps, the formation of NHNH2* (*NNH2→*NHNH2) is the PDS with maximum free energy changes of 0.15 or 0.10 eV on B@Ti4N3O2 or B@Ti3N2O2, respectively. Overall, the NRR processes proceed within the mixed mechanism through the following intermediates: *N2 → *NNH → *NNH2 → *NHNH2 → *NH → *NH2 → *NH3. All the three candidate systems exhibit quite low barriers of PDSs, requiring less negative limiting potentials compared with other reported investigations including metals (−0.45 V for Ru) and single-atom catalyst (−0.69 V for Ti on graphene) (Skúlason et al., 2012; Liu et al., 2021b).
To gain deep insight into the high activity of the three B-doped systems, we explored the electronic structures. Charge density differences of N2 adsorption configurations are analyzed as shown in Figure 5, indicating significant charge transfer from the B atom as the active site to the adsorbed N2 molecule. According to the so-called push-pull hypothesis (Geri et al., 2017), B@MXenes can “pull” the lone-pair electrons from the N2 and simultaneously “push” electrons back into the anti-bonding orbitals of N2. In other words, the N2 molecule donates its lone-pair electron to the empty 2pz orbital of boron. Meanwhile, the antibonding of N2 molecule can accept electrons from B@MXenes catalysts, which helps trigger the NRR. As shown in Figure 5, the increased electron density between nitrogen and the surface is related to the electrons that are pulled from nitrogen by the metal atom. On the other hand, electrons also accumulate on the two nitrogen atoms, representing the π* orbital that accepts “pushed” electrons from surface. In addition, we plotted the projected density of states (PDOS) of the adsorption configurations and free N2 molecules (Figure 6) (Guo et al., 2020). Compared to the molecular orbitals of free nitrogen, the interaction with the active B center apparently downshifts the band of nitrogen, stabilizing the adsorbed N2. The empty p-orbital of B atom receives electrons from nitrogen molecule, increasing the adsorption strength. Especially, in these two Ti-containing systems, the B dopants possess obvious occupation under and close to the Fermi level, which feedbacks electrons to the antibonding orbital of nitrogen and results in further activated nitrogen as well as lower barriers than that of the B@Nb4C3O2 system.
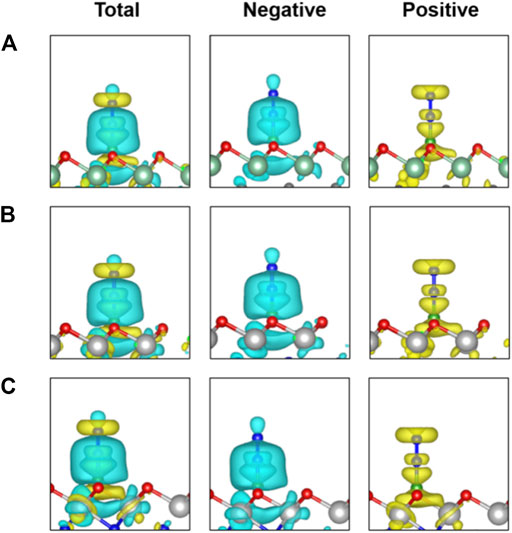
FIGURE 5. Charge density differences of N2 adsorbed on three B@MXenes surfaces. The charge depletion and accumulation are depicted in cyan and yellow, respectively. The isosurface value is set to 0.001 e/Å3.
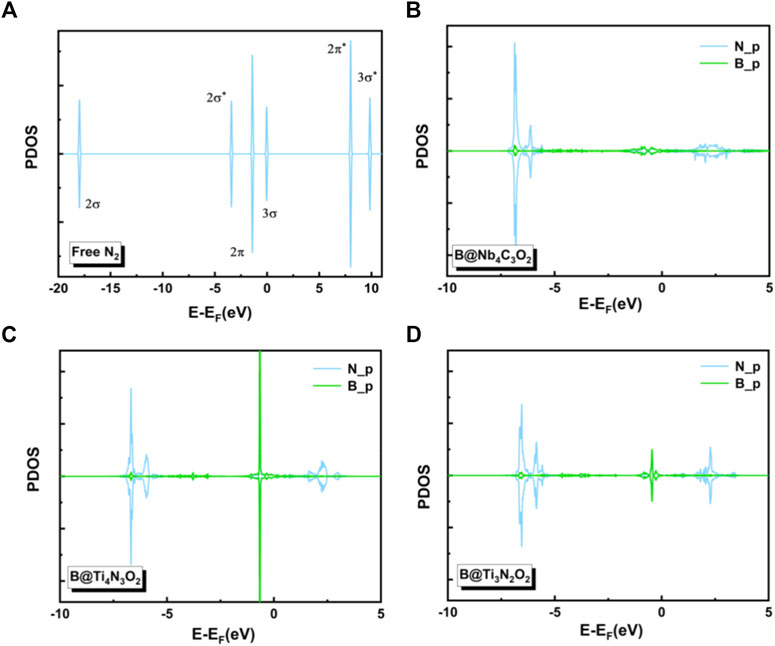
FIGURE 6. The projected densities of states (PDOSs) of (A) free N2 and N2 adsorbed on (B) B@Nb4C3O2, (C) B@Ti4N3O2, and (D) B@Ti3N2O2 surfaces.
Besides, to evaluate the feasibility and stability of the designed systems, we calculate the dopant energies of the three potential systems. As shown in Supplementary Table S2, Eb values of the three candidates range from 4.0 to 4.5 eV, indicating an intense binding between the defective substrate and boron dopant. We assume that these systems could be prepared by using chemical vapor deposition technology. On the other hand, boron atoms can aggregate on the substrate. Considering the cohesive energies (EC) of boron as 6.24 eV, the (Eb−Ec) values of B@Nb4C3O2, B@Ti4N3O2, and B@Ti3N2O2 are −1.74, −2.00, and −2.21 eV, respectively. Herein, these B-doped MXenes may be thermodynamic instable if the density of dopant in the surface is too high to lead to B aggregation. Then we further studied the energy barrier for B atom escaping from the O vacancy, to evaluate the kinetic stability. The boron diffusion path is shown in Figure 7A. The kinetic barriers are 1.05, 1.59, and 1.80 eV for Nb4C3O2, Ti3N2O2, and Ti4N3O2, respectively. These relatively high barriers indicate that the boron localized in the defect hardly move out. Herein, these materials should be kinetically stable as catalysts. Moreover, ab initio molecular dynamic simulations are performed for these three B@MXenes. In 10 ps at 300 K, there is no obvious structural deformation, and the B atom stays in the vacancy, also confirming the stability of these systems (Figures 7B–D).
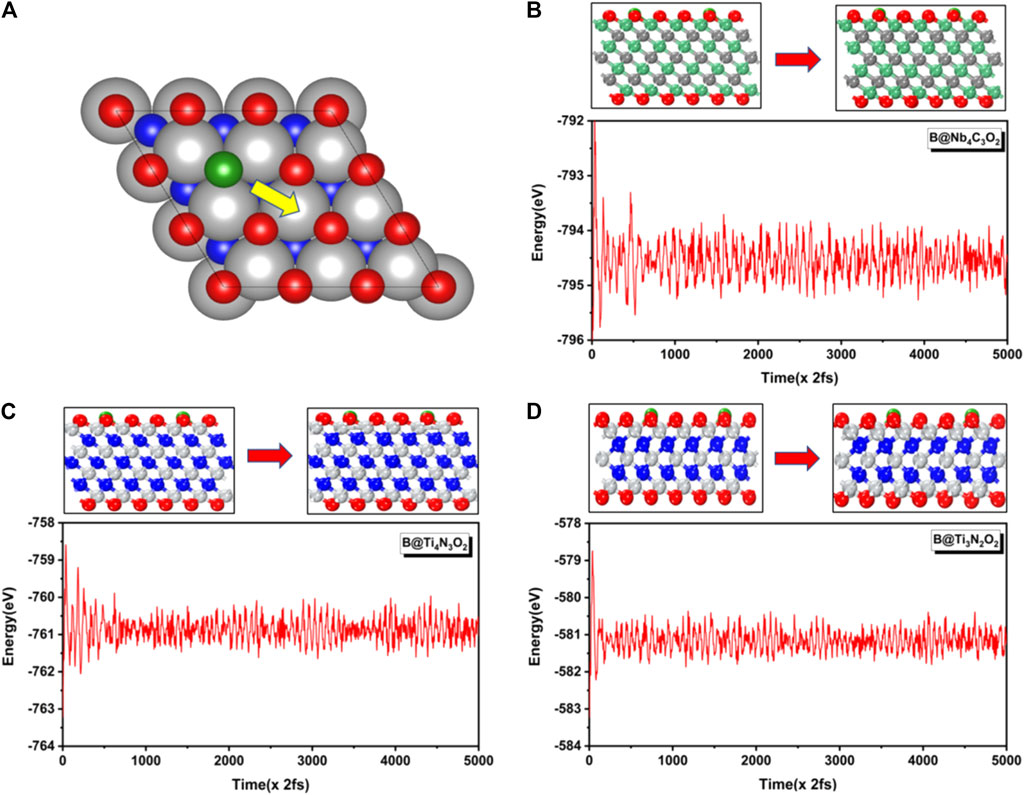
FIGURE 7. (A) The diffusion path of B atom from O vacancy to the adjacent adsorption site on MXenes. Variations of energy against the time for AIMD simulations of (B) B@Nb4C3O2, (C) B@Ti4N3O2, and (D) B@Ti3N2O2. The initial and final configurations are also depicted.
Conclusion
In summary, we conducted DFT calculations to evaluate the potential of B@MXenes systems in the electrochemical NRR. Based on energy changes of the first and last hydrogenation steps, B@Nb4C3O2, B@Ti4N3O2, and B@Ti3N2O2 are screened out as the most possible candidates with high catalytic activity. Subsequently, the reaction mechanisms are revealed to predict the nitrogen reduction performance of these three catalysts. On B@Nb4C3O2 catalyst, transfers of electron–proton pairs proceed following the distal mechanism, with the limiting potential of −0.26 V, whereas the NRR processes on the two Ti-containing systems go through the mixed pathway, with quite low limiting potentials of −0.15 and −0.10 V, respectively. In order to understand the good catalytic activity of the three structures on the NRR, their electronic structures are further analyzed. We found that N2 was captured by these three B@MXenes to trigger the NRR through the so-called push-pull hypothesis. The PDOSs show that the p-band of B atom close to the Fermi level in these Ti-containing systems contributes to the enhanced interaction with inert N2 and the low reaction barriers. Furthermore, the stabilities of the B-doped systems are verified by the kinetic barriers of B-dopant diffusion and AIMD simulation. Our findings not only prove that the three catalysts, namely, B@Nb4C3O2, B@Ti4N3O2, and B@Ti3N2O2, may exhibit outstanding performance on NRR catalysis but also provide further insight into the rational design of NRR catalysts. It is expected to stimulate deep research on exploration of novel catalysts in the field of nitrogen fixation and also expand the application range of MXenes-based materials.
Data Availability Statement
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
Author Contributions
YW, XQ, and GZ performed DFT calculation. ZT, YW, and QZ wrote the manuscript. All authors discussed the results on the manuscript.
Funding
This work was supported by the National Key Research and Development Program of China (Grant No. 2018YFB0704300), the National Science Foundation of China (Grant No. 21803074), the NingBo S&T Innovation 2025 Major Special Program (Grant No. 2018B10016), the Zhejiang Provincial Natural Science Foundation of China (Grant No. LY21B030006), and the Fujian Institute of Innovation, Chinese Academy of Sciences.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The author thanks the high-performance computing center of Nanjing University and the Supercomputer Center of Ningbo Institute of Materials Technology and Engineering, CAS, for providing computing resources.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fceng.2021.702129/full#supplementary-material
References
Blöchl, P. E. (1994). Projector Augmented-Wave Method. Phys. Rev. B 50 (24), 17953–17979. doi:10.1103/physrevb.50.17953
Cai, B. X., Liu, H. Z., and Han, W. F. (2020). Solution Combustion Synthesis of Fe2O3-Based Catalyst for Ammonia Synthesis. Catalysts 10 (9), 11. doi:10.3390/catal10091027
Cheng, Y., Dai, J., Song, Y., and Zhang, Y. (2019). Single Molybdenum Atom Anchored on 2D Ti2NO2 MXene as a Promising Electrocatalyst for N2 Fixation. Nanoscale 11 (39), 18132–18141. doi:10.1039/c9nr05402b
Cui, X. Y., Tang, C., and Zhang, Q. (2018). A Review of Electrocatalytic Reduction of Dinitrogen to Ammonia under Ambient Conditions. Adv. Energ. Mater. 8 (22), 25. doi:10.1002/aenm.201800369
Dai, C., Li, X., Zhang, M., Cui, Y., Zhao, B., and Ma, X. (2021). A Silica-Supported Ni-Based Catalyst Prepared Using TEPA for the Plasma Synthesis of Ammonia. Int. J. Hydrogen Energ. 46 (2), 2213–2224. doi:10.1016/j.ijhydene.2020.10.153
Gambarotta, S., and Scott, J. (2004). Multimetallic Cooperative Activation of N2. Angew. Chem. Int. Ed. 43 (40), 5298–5308. doi:10.1002/anie.200301669
Gao, Y., Cao, Y., Zhuo, H., Sun, X., Gu, Y., Zhuang, G., et al. (2020). Mo2TiC2 MXene: A Promising Catalyst for Electrocatalytic Ammonia Synthesis. Catal. Today 339, 120–126. doi:10.1016/j.cattod.2018.12.029
Geri, J. B., Shanahan, J. P., and Szymczak, N. K. (2017). Testing the Push-Pull Hypothesis: Lewis Acid Augmented N2 Activation at Iron. J. Am. Chem. Soc. 139 (16), 5952–5956. doi:10.1021/jacs.7b01982
Giddey, S., Badwal, S. P. S., and Kulkarni, A. (2013). Review of Electrochemical Ammonia Production Technologies and Materials. Int. J. Hydrogen Energ. 38 (34), 14576–14594. doi:10.1016/j.ijhydene.2013.09.054
Gjerding, M. N., Taghizadeh, A., Rasmussen, A., Ali, S., Bertoldo, F., Deilmann, T., et al. (2021). Recent Progress of the Computational 2D Materials Database. C2DB.
Grimme, S. (2006). Semiempirical GGA-type Density Functional Constructed with a Long-Range Dispersion Correction. J. Comput. Chem. 27 (15), 1787–1799. doi:10.1002/jcc.20495
Groom, C. R., Bruno, I. J., Lightfoot, M. P., and Ward, S. C. (2016). The Cambridge Structural Database. Acta Crystallogr. Sect B 72, 171–179. doi:10.1107/s2052520616003954
Guo, X., Gu, J., Lin, S., Zhang, S., Chen, Z., and Huang, S. (2020). Tackling the Activity and Selectivity Challenges of Electrocatalysts toward the Nitrogen Reduction Reaction via Atomically Dispersed Biatom Catalysts. J. Am. Chem. Soc. 142 (12), 5709–5721. doi:10.1021/jacs.9b13349
Hering-Junghans, C. (2018). Metal-Free Nitrogen Fixation at Boron. Angew. Chem. Int. Ed. 57 (23), 6738–6740. doi:10.1002/anie.201802675
Huang, B., Li, N., Ong, W.-J., and Zhou, N. (2019). Single Atom-Supported MXene: How Single-Atomic-Site Catalysts Tune the High Activity and Selectivity of Electrochemical Nitrogen Fixation. J. Mater. Chem. A. 7 (48), 27620–27631. doi:10.1039/c9ta09776g
Ji, S., Wang, Z., and Zhao, J. (2019). A boron-interstitial Doped C2N Layer as a Metal-free Electrocatalyst for N2 Fixation: a Computational Study. J. Mater. Chem. A. 7 (5), 2392–2399. doi:10.1039/c8ta10497b
Karlsson, L. H., Birch, J., Halim, J., Barsoum, M. W., and Persson, P. O. Å. (2015). Atomically Resolved Structural and Chemical Investigation of Single MXene Sheets. Nano Lett. 15 (8), 4955–4960. doi:10.1021/acs.nanolett.5b00737
Kresse, G., and Furthmüller, J. (1996). Efficient Iterative Schemes Forab Initiototal-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 54 (16), 11169–11186. doi:10.1103/physrevb.54.11169
Kresse, G., and Joubert, D. (1999). From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B 59 (3), 1758–1775. doi:10.1103/physrevb.59.1758
Kugler, K., Luhn, M., Schramm, J. A., Rahimi, K., and Wessling, M. (2015). Galvanic Deposition of Rh and Ru on Randomly Structured Ti Felts for the Electrochemical NH3 Synthesis. Phys. Chem. Chem. Phys. 17 (5), 3768–3782. doi:10.1039/c4cp05501b
Légaré, M.-A., Bélanger-Chabot, G., Dewhurst, R. D., Welz, E., Krummenacher, I., Engels, B., et al. (2018). Nitrogen Fixation and Reduction at Boron. Science 359 (6378), 896–900. doi:10.1126/science.aaq1684
Ling, C., Niu, X., Li, Q., Du, A., and Wang, J. (2018). Metal-Free Single Atom Catalyst for N2 Fixation Driven by Visible Light. J. Am. Chem. Soc. 140 (43), 14161–14168. doi:10.1021/jacs.8b07472
Liu, A. M., Gao, M. F., Gao, Y. C., Ren, X. F., Yang, Y. N., Yang, Q. Y., et al. (2020). DFT Study of Ru/graphene as High-Performance Electrocatalyst for NRR. Inorg. Chem. Commun. 120, 6. doi:10.1016/j.inoche.2020.108169
Liu, A. M., Liang, X. Y., Ren, X. F., Guan, W. X., Gao, M. F., Yang, Y. N., et al. (2020). Recent Progress in MXene-Based Materials: Potential High-Performance Electrocatalysts. Adv. Funct. Mater. 30 (38), 22. doi:10.1002/adfm.202003437
Liu, C., Li, Q., Wu, C., Zhang, J., Jin, Y., MacFarlane, D. R., et al. (2019). Single-Boron Catalysts for Nitrogen Reduction Reaction. J. Am. Chem. Soc. 141 (7), 2884–2888. doi:10.1021/jacs.8b13165
Liu, D., Chen, M. P., Du, X. Y., Ai, H. Q., Lo, K. H., Wang, S. P., et al. (2021). Development of Electrocatalysts for Efficient Nitrogen Reduction Reaction under Ambient Condition. Adv. Funct. Mater. 31 (11), 36. doi:10.1002/adfm.202008983
Liu, T. Y., Dang, Q., Zhou, X. H., Li, J., Ge, Z., Che, H., et al. (2021). Synergistic Effect of Boron Nitride and Carbon Domains in Boron Carbide Nitride Nanotube Supported Single-Atom Catalysts for Efficient Nitrogen Fixation. Chem.-Eur. J. 27 (23), 6945–6953. doi:10.1002/chem.202005182
Manjunatha, R., and Schechter, A. (2018). Electrochemical Synthesis of Ammonia Using Ruthenium-Platinum Alloy at Ambient Pressure and Low Temperature. Electrochemistry Commun. 90, 96–100. doi:10.1016/j.elecom.2018.04.008
Norskov, J. K., Rossmeisl, J., Logadottir, A., Lindqvist, L., Kitchin, J. R., Bligaard, T., et al. (2004). Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B. 108 (46), 17886–17892. doi:10.1021/jp047349j
Peng, W., Luo, M., Xu, X. D., Jiang, K., Peng, M., Chen, D. C., et al. (2020). Spontaneous Atomic Ruthenium Doping in Mo2CTX MXene Defects Enhances Electrocatalytic Activity for the Nitrogen Reduction Reaction. Adv. Energ. Mater. 10 (25), 9. doi:10.1002/aenm.202070110
Perdew, J. P., Burke, K., and Ernzerhof, M. (1997). Generalized Gradient Approximation Made Simple [Phys. Rev. Lett. 77, 3865 (1996)]. Phys. Rev. Lett. 78 (7), 1396. doi:10.1103/physrevlett.78.1396
Perdew, J. P., and Wang, Y. (1992). Accurate and Simple Analytic Representation of the Electron-Gas Correlation Energy. Phys. Rev. B 45 (23), 13244–13249. doi:10.1103/physrevb.45.13244
Rao, C. N. R., and Rao, G. R. (1991). Nature of Nitrogen Adsorbed on Transition-Metal Surfaces as Revealed by Electron-Spectroscopy and Cognate Techniques. Surf. Sci. Rep. 13 (7), 221–263. doi:10.1016/0167-5729(91)90014-o
Rasmussen, F. A., and Thygesen, K. S. (2015). Computational 2D Materials Database: Electronic Structure of Transition-Metal Dichalcogenides and Oxides. J. Phys. Chem. C 119 (23), 13169–13183. doi:10.1021/acs.jpcc.5b02950
Shao, M., Shao, Y., Chen, W., Ao, K. L., Tong, R., Zhu, Q., et al. (2018). Efficient Nitrogen Fixation to Ammonia on MXenes. Phys. Chem. Chem. Phys. 20 (21), 14504–14512. doi:10.1039/c8cp01396a
Shi, L., Li, Q., Ling, C., Zhang, Y., Ouyang, Y., Bai, X., et al. (2019). Metal-free Electrocatalyst for Reducing Nitrogen to Ammonia Using a Lewis Acid Pair. J. Mater. Chem. A. 7 (9), 4865–4871. doi:10.1039/c8ta11025e
Singh, K., Gera, R., Sharma, R., Maithani, D., Chandra, D., Bhat, M. A., et al. (2021). Mechanism and Application of Sesbania Root-Nodulating Bacteria: An Alternative for Chemical Fertilizers and Sustainable Development. Arch. Microbiol. 203, 1259–1270. doi:10.1007/s00203-020-02137-x
Skúlason, E., Bligaard, T., Gudmundsdóttir, S., Studt, F., Rossmeisl, J., Abild-Pedersen, F., et al. (2012). A Theoretical Evaluation of Possible Transition Metal Electro-Catalysts for N2reduction. Phys. Chem. Chem. Phys. 14 (3), 1235–1245. doi:10.1039/c1cp22271f
Sun, J., Kong, W., Jin, Z., Han, Y., Ma, L., Ding, X., et al. (2020). Recent Advances of MXene as Promising Catalysts for Electrochemical Nitrogen Reduction Reaction. Chin. Chem. Lett. 31 (4), 953–960. doi:10.1016/j.cclet.2020.01.035
Wang, C., Zhao, Y.-N., Zhu, C.-Y., Zhang, M., Geng, Y., Li, Y.-G., et al. (2020). A Two-Dimensional Conductive Mo-Based Covalent Organic Framework as an Efficient Electrocatalyst for Nitrogen Fixation. J. Mater. Chem. A. 8 (44), 23599–23606. doi:10.1039/d0ta08676b
Wang, S., Li, B., Li, L., Tian, Z., Zhang, Q., Chen, L., et al. (2020). Highly Efficient N2 Fixation Catalysts: Transition-Metal Carbides M2C (MXenes). Nanoscale 12 (2), 538–547. doi:10.1039/c9nr09157b
Wang, Y., Nian, Y., Biswas, A. N., Li, W., Han, Y., and Chen, J. G. G. (2021). Challenges and Opportunities in Utilizing MXenes of Carbides and Nitrides as Electrocatalysts. Adv. Energ. Mater. 11 (3), 17. doi:10.1002/aenm.202002967
Xie, Y., Naguib, M., Mochalin, V. N., Barsoum, M. W., Gogotsi, Y., Yu, X., et al. (2014). Role of Surface Structure on Li-Ion Energy Storage Capacity of Two-Dimensional Transition-Metal Carbides. J. Am. Chem. Soc. 136 (17), 6385–6394. doi:10.1021/ja501520b
Yang, L., Ma, X., Xu, Y. Y., Xu, J. Y., and Song, Y. D. (2020). A DFT Study on Application of Dual-Atom Fe2/Phthalocyanine Catalyst for N2 Reduction Reaction. Int. J. Electrochem. Sci. 15 (10), 9698–9706. doi:10.20964/2020.10.46
Yu, X., Han, P., Wei, Z., Huang, L., Gu, Z., Peng, S., et al. (2018). Boron-doped Graphene for Electrocatalytic N2 Reduction. Joule 2 (8), 1610–1622. doi:10.1016/j.joule.2018.06.007
Zhan, S., and Zhang, F. (2021). Recent Progress on Electrocatalytic Synthesis of Ammonia under Amibent Conditions. Acta Chim. Sinica 79 (2), 146–157. doi:10.6023/a20090412
Zhao, J., and Chen, Z. (2017). Single Mo Atom Supported on Defective Boron Nitride Monolayer as an Efficient Electrocatalyst for Nitrogen Fixation: A Computational Study. J. Am. Chem. Soc. 139 (36), 12480–12487. doi:10.1021/jacs.7b05213
Zheng, G. K., Li, L., Hao, S. Y., Zhang, X. W., Tian, Z. Q., and Chen, L. (2020). Double Atom Catalysts: Heteronuclear Transition Metal Dimer Anchored on Nitrogen-Doped Graphene as Superior Electrocatalyst for Nitrogen Reduction Reaction. Adv. Theor. Simul. 3 (12), 8. doi:10.1002/adts.202000190
Keywords: electrocatalysis, nitrogen reduction, MXene, boron-containing compounds, DFT, single atom catalyst
Citation: Wang Y, Qian X, Zheng G, Tian Z and Zhang Q (2021) Boron-Doped MXenes as Electrocatalysts for Nitrogen Reduction Reaction: A Theoretical Study. Front. Chem. Eng. 3:702129. doi: 10.3389/fceng.2021.702129
Received: 29 April 2021; Accepted: 09 June 2021;
Published: 29 June 2021.
Edited by:
Cheng Lian, East China University of Science and Technology, ChinaReviewed by:
Haolan Tao, East China University of Science and Technology, ChinaShengwei Deng, Zhejiang University of Technology, China
Yun Tian, Zhengzhou University, China
Kai Huang, East China University of Science and Technology, China
Copyright © 2021 Wang, Qian, Zheng, Tian and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ziqi Tian, dGlhbnppcWlAbmltdGUuYWMuY24=; Qiuju Zhang, emhhbmdxakBuaW10ZS5hYy5jbg==