 Carla Luciana Padilla Franzotti1,2
Carla Luciana Padilla Franzotti1,2 Nicolas Palopoli1,2
Nicolas Palopoli1,2 Juliana Palma1,2*
Juliana Palma1,2* Gustavo Pierdominici-Sottile1,2*
Gustavo Pierdominici-Sottile1,2*- 1Departamento de Ciencia y Tecnología, Universidad Nacional de Quilmes, Buenos Aires, Argentina
- 2Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Ciudad Autónoma de Buenos Aires (CABA), Buenos Aires, Argentina
Introduction: The retinoblastoma protein (pRb) is a key regulator of the cell cycle that suppresses cell proliferation by binding to E2F transcription factors. Disruption of this pathway, commonly through mutations or interactions with viral oncoproteins, can lead to uncontrolled cell growth and cancer. The large T antigen of simian virus 40 (LTSV40) is known to bind pRb, thereby inhibiting its interaction with E2F transcription factors. However, the structural and dynamic mechanisms underlying this inhibition remain incompletely understood.
Methods: We employed molecular dynamics (MD) simulations, principal component analysis, and cluster analysis to investigate the conformational dynamics of pRb, LTSV40, and their complex. Our study focused on an intrinsically disordered region on the C-terminal side of the LFCSE motif of LTSV40, referred to as Linker 1.
Results: Our simulations reveal that Linker 1 undergoes a significant conformational shift upon binding to pRb. While this region adopts a predominantly bent structure in the unbound state, it transitions into an extended conformation in the complex. As a consequence of this change, the C-terminal segment of LTSV40 obstructs access to the AB-cleft of pRb, the binding site for E2F.
Discussion: Our findings suggest that the inactivation mechanism of pRb by LTSV40, as unveiled by MD simulations, could represent a broader strategy employed by other viral oncoproteins containing similar LXCXE motifs and adjacent disordered regions. This mechanism may even extend to endogenous pRb inactivation. As our conclusions are based on computational modeling, they require experimental validation. Such confirmation would pave the way for developing therapeutic strategies aimed at reactivating pRb function in pathologies where it is compromised.
1 Introduction
pRb belongs to the so-called Pocket Proteins family whose functions include preventing cell proliferation, regulating genomic stability, inhibiting apoptosis, modulating chromatin structure, and participating in the DNA damage response (Harrington et al., 1998; Hickman et al., 2002; Manning and Dyson, 2011). By binding E2F transcription factors (E2Fs), pRb acts as a negative regulator of the eukaryotic cell cycle1 (Morris and Dyson, 2001; Dick and Rubin, 2013). Particularly, the formation of the pRb-E2F complex inhibits the G1/S transition phase of the cell cycle. The interaction between these proteins depends on the phosphorylation state of pRb. Hypophosphorylated pRb binds E2Fs, inhibiting cell proliferation. When growth signals are present, pRb undergoes phosphorylation by cyclin-dependent kinases (CDKs), inactivating it. This prevents the formation of the complex by a still unclear mechanism. E2Fs in their free form promote activation of genes involved in cell cycle progression (Konagaya et al., 2024; Dyson, 1998; Weinberg, 1995; Konagaya et al., 2024). The main interactions between E2Fs and pRb occur within the pRb AB cleft, located at the interface of boxes A and B of the pocket domain of pRb (Xiao et al., 2003).
It is established that mutations in the RB genes can interfere with these pRb-E2F regulatory mechanisms leading to uncontrolled cell growth and cancer development (Goodrich, 2006; Odemis et al., 2023). The interaction of viral oncoproteins with pRb can also interrupt the formation of the pRb-E2F complex. Human papillomavirus E7 (HPV E7) and the large T antigen from simian virus 40 (LTSV40), for instance, bind pRb, disrupt the pRb-E2F pathway, and activate cell proliferation and viral replication (Ahuja et al., 2005; Gouw et al., 2017; Borchert et al., 2014; Fanning and Knippers, 1992; Lilyestrom et al., 2006). All these viral proteins contain a short linear motif, called the LXCXE motif, that mediates their interaction with pRb. The region of pRb involved in this interaction is known as the LXCXE binding cleft and represents a conserved region located within the B box (Palopoli et al., 2018). This binding site is approximately 30.0 Å apart from the pRb AB cleft (Xiao et al., 2003).
Given the crucial role of pRb in the development of a wide range of human malignancies, numerous studies have sought to understand its interactions with partner proteins (Burkhart and Sage, 2008; Weinberg, 1995; Morris and Dyson, 2001). However, many key aspects of its functioning remain unclear. Most experimental studies—likely due to the complexity of the system—focused on interactions involving sequences that bind to either the pRb AB domain or the LXCXE-binding cleft, while neglecting the N-terminal, C-terminal, or neighbouring regions (Chow and Dean, 1996; Lee and Cho, 2002; Lee et al., 1998; Liu and Marmorstein, 2007; Whyte et al., 1988). This omission may obscure important effects, as these regions represent a significant portion of pRb’s interactors. The elucidation of the crystal structure of the pRb-LTSV40 complex produced a significant advancement in our understanding of how viral oncoproteins hijack the cell cycle regulatory system (Kim et al., 2001). However, this structure does not include the region located at the C-terminal side of the LXCXE motif, restricting our ability to deduce how the rest of LTSV40 interacts with pRb.
In this article, we present the results of molecular dynamics (MD) simulations that investigate the structural and dynamical properties of the complete LTSV40 protein, the AB box of pRb, and the complex between them. Our analysis of the primary modes of motion in pRb revealed that fluctuations in its most flexible regions open access to the pRb AB cleft. On the other hand, the MD simulations of free LTSV40 provided insights into the relative positioning of its folded domains and the conformational changes necessary for binding. Finally, the characterization of the ensemble of conformations of the pRb-LTSV40 complex, helped us determine whether the attachment to the LXCXE binding cleft triggered allosteric effects on the AB cleft. This investigation also highlighted specific characteristics of the intrinsically disordered regions (IDRs) of LTSV40 that are crucial in preventing E2F binding. By assembling all these pieces of information, we propose a new mechanism for how LTSV40 deregulates cell-cycle repression. Additionally, based on the structural knowledge of alternative oncoproteins and pocket proteins, it seems likely that a similar mechanism could work for polyomaviruses and the inactivation of pRb through phosphorylation. Understanding the molecular details of how pRb is inactivated, whether by phosphorylation or through the binding of viral oncoproteins, not only illuminates the general mechanisms of protein-protein interactions in regulatory pathways but also paves the way for developing novel strategies to prevent and treat cancers related to viral infections or pRb deregulation.
2 Theoretical methods
In this section, we describe the methods we employed to prepare the computational models of pRb, LTSV40 and the complex between them. Also, we outline the protocols used to run the MD simulations and provide the numerical details required to reproduce them. Finally, we present the algorithms used to analyse the results.
2.1 Model construction
Human pRb (UniProtKB identifier - P06400) and LTSV40 (UniProtKB identifier - P03070) are multi domain monomeric proteins. pRb contains 928 amino acids organized into three domains. The central or “pocket” domain (Pro374-Ala772) is made up of two subdomains known as the A and B boxes. These boxes, primarily composed of
LTSV40, made up of 708 amino acids, is composed of four folded domains: the J-domain (Met1-Asn102), the origin-binding domain (OBD) (Pro135-Pro249), the Zn-binding domain (Lys271-Gln354) and the ATPase domain (Arg357-Thr708) (An et al., 2012). The J-domain is connected to the OBD by an intrinsically disordered region (IDR) which spans from Leu103 to Asp134. Hereafter, this region will be referred to as Linker 1. Another IDR, Linker 2, comprises the sequence Gly250-Trp270 that joins the OBD with the Zn-binding domain. This last domain and the ATPase domain are linked by a short segment of just three aminoacids. The first five residues of Linker 1, Leu103, Phe104, Cys105, Ser106, and Glu107 bind to the LXCXE binding cleft of pRb. In the following, this segment will be named as the LFCSE motif of LTSV40.
The starting point to build the complex between pRb and LTSV40 was the structure reported in PDB entry 1GH6 (Kim et al., 2001). It contains the boxes A and B of the pRb pocket domain but lacks the flexible loop connecting them. This loop spans from residue Asp584 to Ser644 and is not part of any binding motif. Entry 1GH6 also contains a portion of LTSV40 ranging from Arg7 to Thr117. This segment accommodates the J-domain plus 15 of the 32 residues of Linker 1. The positions of residues in the loop between the boxes A and B of the pRb domain were predicted with Alphafold3 (Jumper et al., 2021; Varadi et al., 2024), using the known structure of pRb as a template. To complete the structure of LTSV40, we used information taken from PDB entries 1Q1S (Fontes et al., 2003) and 4GDF (Chang et al., 2013). Entry 1Q1S provides the conformation of the Asp119-Glu133 segment, almost completing the portion of Linker 1 that is absent in 1GH6. The only missing residue is Ala118. Accordingly, we used DS ViewerPro 5.0 to add Ala118 after Thr117 in structure 1GH6 and before Asp119 in structure 1Q1S, following the geometrical restrictions of the backbone atoms of Thr117 and Asp119, respectively. After that, we joined the two fragments by superimposing the backbone atoms of Ala118 of the two portions and then deleted one of the occurrences of this residue.
PDB entry 4GDF, on the other hand, contains the structure of the segment of LTSV40 that goes from Lys131 to Asp627. It encompasses part of the OBD, Linker 2, the Zn-binding domain and most of the ATPase domain. This sequence overlaps with the one in entry 1Q1S by the triad Lys131-Val132-Glu133. Therefore, to complete our model of LTSV40, we superimposed the overlapping sequences and then deleted one of them. In this way, our computational model of the pRb-LTSV40 complex comprised the pocket domain of pRb (Pro374-Ala772) and the four domains of LTSV40 (Arg7-Asp627) except for a small portion of the ATPase domain. Finally, to generate models of pRb and LTSV40 in their apo forms, we just removed pRb or LTSV40 from the PDB file of the pRb-LTSV40 complex. Figure 1 presents a graphical representation of the minimized conformations of pRb and LTSV40.
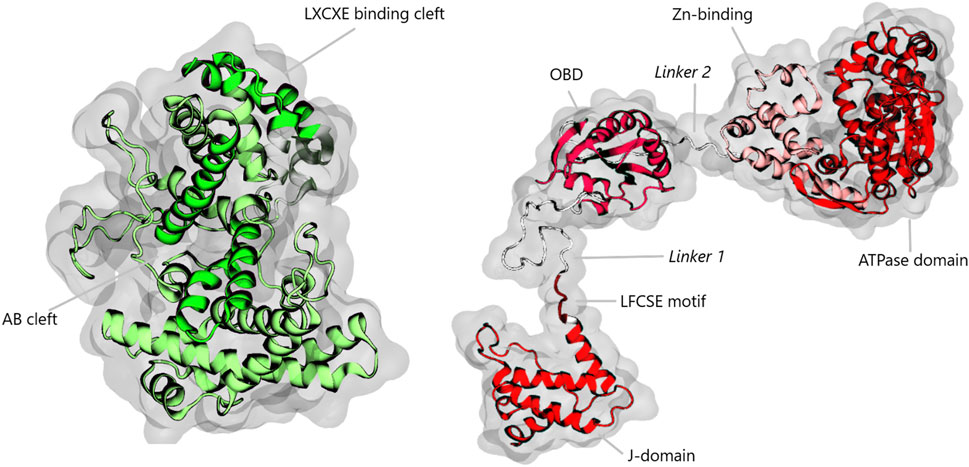
Figure 1. Diagram illustrating the pRb pocket domain and LTSV40. pRb is highlighted in green, emphasizing the AB and LXCXE binding clefts. LTSV40 is illustrated in various shades of red, representing the J-domain, the OBD, the Zn-binding domain, and the ATPase domain. Linker 1 and Linker 2 are shown in white. The LFCSE motif, located in the N-terminal region of Linker 1, is also marked for clarity.
2.2 Initial settings
The models were prepared using the LEAP module of AMBER18 (Case et al., 2018), where they were neutralized by adding
Simulations of models containing LTSV40 were performed using two different force field combinations to assess the robustness of their results (Rauscher et al., 2015). In half of the simulations, we employed the ff19SB force field for proteins (Tian et al., 2019) along with the OPC water model (Izadi et al., 2014), which has been shown to improve the accuracy of MD simulations of intrinsically disordered regions compared to the widely used TIP3P model (Shabane et al., 2019). In the remaining simulations, we used the
All simulations were conducted using the PMEMD module of AMBER18. Electrostatic interactions were computed with the Particle Mesh Ewald method, applying a 10.0 Å cutoff radius. Thus, direct-space calculations were performed for
2.3 Molecular dynamics simulations
Molecular dynamics (MD) simulations were performed on the computational models of pRb, LTSV40, and the pRb-LTSV40 complex. Because the LTSV40 model includes intrinsically disordered regions and was assembled using data from various experimental sources, we anticipated its initial conformation could deviate significantly from any representative structure of the molecule in solution. Additionally, we observed that applying the heating protocol repeatedly to the same minimized structure resulted in conformations whose differences could be detected by the naked eye. With these considerations in mind, and aiming to explore the conformational space of LTSV40 as extensively as our computational resources allow, we ran 50 separate heating, equilibration and production phases for models including this protein. Instead, for simulations of isolated pRb, we run one heating and one equilibration stage, followed by 50 alternative production stages. The numerical details of the complete protocols are provided below.
The heating stages took the minimized models from 0 to 303 K. They were carried out at constant volume and lasted for 100 ns. The temperature was controlled with a Langevin thermostat using a collision frequency of 1.0
2.4 Principal component analysis (PCA)
We used Principal Component Analysis (PCA) (Kitao, 2022; Palma and Pierdominici-Sottile, 2023) to identify the most significant deformations of pRb in its apo form and when complexed with LTSV40. Furthermore, we used PCA as a dimensionality reduction tool to allow the subsequent application of clustering algorithms (Sittel and Stock, 2018) on the conformations Linker 1 of LTSV40, both in its apo form and when complexed with pRb. In the first case, we performed PCA on the Cartesian coordinates of the
2.5 Cluster analysis
We performed a cluster analysis to identify the most representative conformations of Linker 1 of LTSV40. The metric we employed to that end was the Euclidean distance between the samples, in a reduced-dimensionality (RD) space determined with dPCA. Using this metrics is slightly more elaborated than the most widely employed RMSD, but has significant advantages. RMSD requires fitting all the structures to a reference structure to eliminate the effect of global rotation and translation. Unfortunately, this procedure cannot be unambiguously performed and problems become particularly serious for molecules presenting large deformations, such as those analysed here (Palma and Pierdominici-Sottile, 2023). On top of that, transforming from the high dimensional space of Cartesian coordinates to the 1D space of RMSD can introduce projection errors that tend to put structures differing in important features into the same cluster (Sittel and Stock, 2018). Because of these considerations, we decided to also implement a metric based on distances in the RD space of dPCA. Since this algorithm employs internal coordinates, it does not require any adjustment to a reference structure. Besides, in principle, the dimensions of the space to be used can be set as high as desired, aiming to include all directions with significant eigenvalues. In practice, however, the sample size,
To perform the cluster analysis, we utilized the DBSCAN algorithm to estimate the relevance of the regions in the configurational space that were explored during the simulations. Unlike the more commonly used k-means algorithm, DBSCAN is deterministic and does not require prior knowledge of the number of clusters in the dataset. Instead, it requires the setting of two parameters:
To determine both parameters, we calculated the distances from each point to its
2.6 Collinearity in an LTSV40 segment
Visual inspection of the LTSV40 configurations sampled from the MD simulations showed that an important parameter to characterize the state of this protein was the collinearity of the backbone of Linker 1. A quantitative assessment of this characteristic can be obtained by comparing two distances. One of them is the distance between the first and last
In Equation 1, the segment under analysis starts at residue
We applied Equation 2 to the residues of Linker 1 located on the C-terminal side of the LTSV40 LFCSE motif. This involved varying
2.7 Number of contacts
For the pRb-LTSV40 complex we measured the numbers of contacts between each residue of Linker 1, located on the C-terminal side of the LFCSE motif, and any residue of pRb pocket domain. To this end, we considered a contact existed when the intermolecular distance between any pair of atoms of the regions under consideration was shorter than 4.0 Å. This analysis was performed with module CPPTRAJ of AMBER18.
2.8 Shortest path map
We used the Shortest Path Map (SPM) tool, available on the SPM server (Casadevall et al., 2024), to identify key residues influencing the structure and dynamics of Linker 1 in the pRb-LTSV40 complex. This analysis aimed to uncover critical residues involved in communication pathways between the molecular partners. To streamline computations, we focused on the
2.9 MM-GBSA calculations
To evaluate the stabilizing/destabilizing effects of pRb residues that bear the largest number of contacts with LTSV40 Linker 1 in the pRb-LTSV40 complex, we performed an alanine scanning calculation using the Molecular Mechanics-Generalized Born Surface Area (MM-GBSA) method (Genheden and Ryde, 2015). For each model of the pRb-LTSV40 complex, the analysis was conducted using snapshots taken at 1, 50, 100, and 150 ns of the production phase of each trajectory. Thus, with 50 trajectories per model, each MM-GBSA computation was based on 200 snapshots.
The protocol we followed computes the interaction energy and solvation free energy for both the complex and the isolated proteins, and the results are averaged to estimate the binding free energy. However, we did not include the entropic contribution in this estimation because the available procedure, which relies on normal mode analysis, is not suitable for proteins with disordered regions such as LTSV40. As a result, the computed
3 Results
We present, in the Supplementary Material section, information on the time evolution of key parameters used to assess the stability of the production phase of the simulations. This supplementary analysis also highlights similarities and differences in some general trends observed when using the ff19SB/OPC and ff99SB
Supplementary Figures S3–S5 depict the time evolution of the total number of hydrogen bonds and the radius of gyration for free pRb, free LTSV40, and LTSV40 in complex with pRb, respectively, using representative trajectories of each system. In all cases, both parameters remain stable showing the typical fluctuations. For free LTSV40, the total number of hydrogen bonds variates between 240 and 300 for both force field combinations. The radius of gyration exhibits a slightly narrower range in simulations with
Supplementary Figures S6, S7 show the secondary structure probabilities for residues in the Glu105–Glu138 sequence of LTSV40. They were computed from simulations of free LTSV40, carried out with the ff19SB/OPC and
Supplementary Figures S8, S9 present analogous results for LTSV40 in the pRb-LTSV40 complex. Again, the outcomes are similar between force fields. Compared to free LTSV40, the most notable difference is that
In the following sections, we first present the insights gained from the simulations regarding significant structural and dynamical characteristics of the pRb-LTSV40 complex. Then, we introduce the results corresponding to free LTSV40 and pRb.
3.1 The pRb-LTSV40 complex conformational ensemble
Our analysis of the pRb-LTSV40 complex mostly focused on the conformational ensemble of the viral protein, specifically examining the structures of Linker 1. As described in Section 2.4, we performed a dPCA analysis on this segment using the 30,000 structures obtained from the production phase of the MD simulations. The snapshots were then projected onto the first five dPCA eigenvectors, and the projections were subsequently used for DBSCAN cluster analysis. Employing the parameters outlined in Section 2.5, the algorithm identified 12 clusters that together account for 97.7% of the entire population of simulations done with the ff19SB/OPC force fields. The remaining 2.3% were classified as outliers. Using the same DBSCAN parameters, 9 clusters were identified in simulations employing the
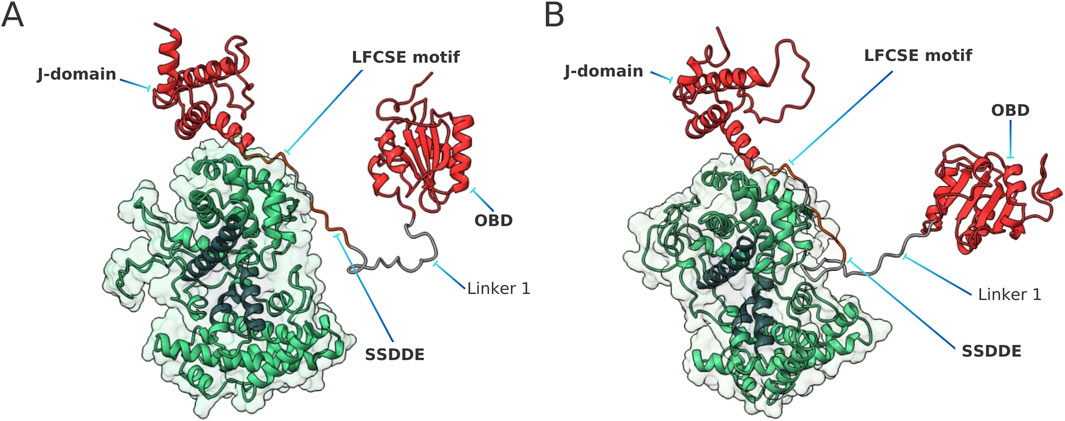
Figure 2. Representative structures of the pRb-LTSV40 complex, according to ff19SB/OPC simulations (A) and
A simple visual inspection reveals a noticeable characteristic in the most representative conformations of Linker 1 in the pRb-LTSV40 complex (see Figure 2). The conserved SSDDE pentapeptide and its neighbouring regions are situated close to a corner of the pRb AB cleft. This corner features a pair of loops: one of them connects
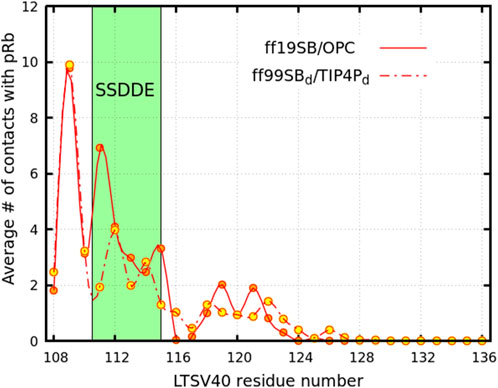
Figure 3. Average number of contacts between pRb and residues within Linker 1 of LTSV40 (excluding the LFCSE motif). The solid line connecting the orange circles represents results from trajectories performed using the ff19SB/OPC force field while the dashed line and yellow circles indicate results considering the
The pRb-Linker 1 interactions discussed in the previous paragraph influence the conformations the linker can adopt within the complex, as evidenced by the analysis in Figure 4. The curves in this figure represent the probability of each residue being part of a linear segment of at least five residues. In the pRb-LTSV40 complex (red curve), residues Glu108 to Asp113 exhibit a high likelihood of forming such segments, with probabilities exceeding 0.50 in simulations performed with both force field combinations. Notably, in ff19SB/OPC simulations, this probability surpasses 0.85 for the first four residues, while in
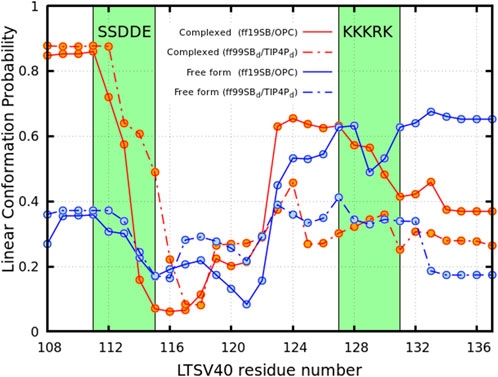
Figure 4. Probability of the residues of linker 1 located at the C-terminal side of the LFCSE motif of being embedded in a linear conformation of more than five residues long. Blue circles describe the apo form of LTSV40 while red ones represent the viral protein complexed with pRb. Solid lines represents simulation performed using the ff19SB/OPC force field while the dashed lines show results corresponding to the
Figure 4 also shows that, for both force field combinations, the probability of residues between Glu115 and Asn121 being part of a linear segment is low in both, isolated LTSV40 and LTSV40 in complex with pRb. This probability then increases, likely due to the presence of two consecutive prolines at positions 125 and 126. Additionally, the stretch of five consecutive basic residues from positions 127 to 131, which constitute the protein’s nuclear localization signal, favours extended conformations over linear ones. To complete the analysis, we also identified the pRb residues that stablish the larger number of contacts with Linker 1, excluding those that interact with the LFCSE motif. For this analysis we considered side chain heavy atoms. The residues we spotted are presented in Table 1 along with their average number of contacts with Linker 1 and their stabilizing or destabilizing effect for the binding, as measured by the
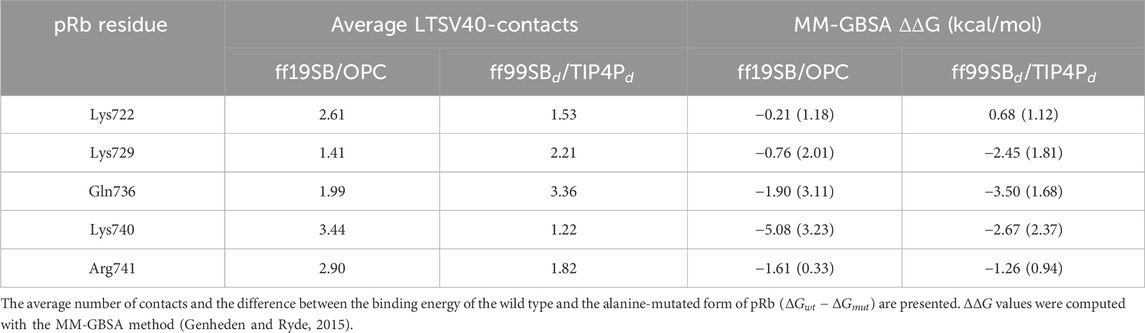
Table 1. pRb residues that exhibit the larger number of contacts with LTSV40 residues located on the C-terminal side of the LFCSE motif.
3.2 The conformational ensemble of isolated LTSV40
As outlined in Section 2.1, Linker 1 and Linker 2 of LTSV40 are long IDRs that connect the J-domain with the OBD, and the OBD with the Zn-binding domain, respectively. Examining the whole set of conformations visited by free LTSV40 in our MD simulations, we observed that the two linkers typically adopt relatively curved conformations, positioning the J-domain and the Zn-binding domain close to the OBD. This feature is illustrated in Figure 5, which displays the representative structures of the most populated clusters in the conformational ensemble of free LTSV40, according to each of the force field combinations used in this work. These structures should be compared with those of Figure 2.
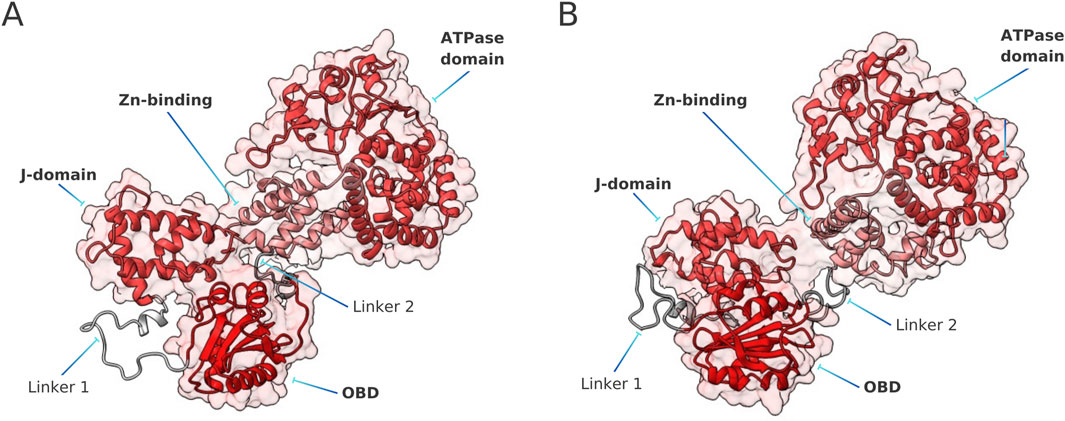
Figure 5. Representative structures of the apo form of LTSV40, according to ff19SB/OPC simulations (A) and
Another notable difference between the behaviour of free and complexed LTSV40 lies in the mobility of the LFCSE residues and those on its C-terminal side. This effect is illustrated in Supplementary Figure S12, which shows the difference in Root Mean Square Fluctuations (RMSF) between the free and complexed forms of the protein. As expected, fluctuations are significantly larger in the free form. The results obtained with both force field combinations are consistent.
3.3 pRb analysis
Unlike the intrinsically disordered regions of LTSV40, the residues in the AB box of pRb exhibit minimal conformational changes when transitioning from the free to the complexed form. This can be observed in Supplementary Figure S12, which presents the differences in RMSF between the two states. Additionally, Figure 6 highlights the most notable deformations in both the apo form (panel A) and the complexed form (panel B). Specifically, the figure depicts the movements generated by a linear combination of the first two principal component analysis (PCA) eigenvectors, each scaled by its corresponding eigenvalue. Animations of these vectors are available in the Supplementary Material.
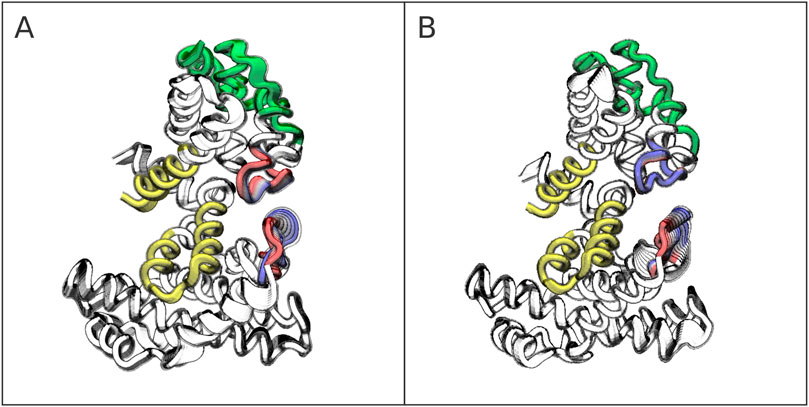
Figure 6. Most prominent pRb deformations in the apo form (A) and holo form (B). The AB cleft mostly composed by helices
The analysis reveals two key dynamical and structural characteristics of the pRb pocket domain. First, the AB cleft, highlighted in yellow in Figure 6, is a rigid structure that retains its shape and rigidity upon LTSV40 binding. Second, in both the free form of pRb and the complex with LTSV40, the most significant deformations occur in the loop connecting
4 Discussion
PRb functions as a negative regulator of the cell cycle, inhibiting cell proliferation by binding to E2F transcription factors through its AB cleft. Viral oncoproteins, such as adenovirus E1, Human Papillomavirus E7 (HPV E7) and LTSV40, can displace E2F from pRb, promoting cell division and thus boosting viral replication (Ahuja et al., 2005; Gouw et al., 2017; Borchert et al., 2014; Lilyestrom et al., 2006). LTSV40 and HPV E7, in particular, contain a LXCXE motif that mediates their interaction with pRb and other pocket proteins (Morris and Dyson, 2001; Gouw et al., 2017; Luna-Vargas et al., 2011; Kaustov et al., 2011; Vorobiev et al., 2009). The specific region of pRb involved in these interactions is referred to as the LXCXE binding cleft. Notably, the AB cleft, where E2F transcription factors bind, and the LXCXE binding cleft, where the viruses interact, are approximately 30.0 Å apart (Xiao et al., 2003). Therefore, it is unclear how these viruses can displace E2F from their complexes with pRb. Since the dissociation of these complexes is linked to cancer development, our limited understanding of this dissociation mechanism represents a significant obstacle to developing new drugs for cancer treatment (Nevins, 2001).
To make things worse, this regulatory system’s structural information is scarce and incomplete, as the few structures that could be solved only contain fractions of the proteins involved. PDB structures 1N4M and 1O9K consist of pRb-E2F complexes that only contain 18 out of 437 residues of the C-terminal region of E2F. This is the part of the transcription factor that docks into the pRb AB cleft (Lee et al., 2002). On the other hand, PDB structures 1GH6 and 1GUX provide information on viral oncoproteins bound to pRb. 1GH6 illustrates the pRb-LTSV40 interaction through the LXCXE binding cleft of pRb (Kim et al., 2001). It includes residues Arg7 to Thr117 of LTSV40, corresponding to the J-domain and a fraction of Linker 1 containing the LXCXE motif (Leu103-Glu107). Similarly, 1GUX features the HPV E7 decapeptide Asp22-Asn31 interacting with the pRb LXCXE binding cleft (Lee et al., 1998).
The systems mentioned above share two common features. First, they lack structural information about the pRb interactors on one or both sides of the segment that attaches to pRb. Second, significant regions of the proteins not included in the available PDB files are identified as IDRs. Obtaining structural and dynamic insights into the behaviour of these missing parts, whether in isolation or as part of complexes with pRb, is essential for understanding the regulation of the pRb-E2F pathway.
4.1 Proposed mechanism of E2F liberation from pRb by LTSV40
Binding assays demonstrated that pRb can be complexed with a decapeptide of HPV E7 in its LXCXE cleft and a small portion of E2F in its AB cleft. This finding indicates that binding to the LXCXE and AB clefts can occur independently (Lee et al., 1998; Xiao et al., 2003). Additionally, experiments revealed that small peptides containing the LXCXE motif cannot inhibit E2F binding (Wu et al., 1993; Xiao et al., 2003). However, when full-length viral proteins or a significant portion of them are used in the assays, complexation with E2F does not occur (Wu et al., 1993; Xiao et al., 2003). This indicates that the inhibition of E2F binding is not directly due to the interaction of pRb with the LXCXE motif or its neighbouring segments but that more distant regions of the viral proteins are involved. Also, this raises the question of whether there is a mechanism common to proteins with a LXCXE motif and disordered regions towards their C-terminal side.
In this work, we present an analysis of the interaction between the pocket domain of pRb and the full viral protein LTSV40, to reveal information that could not be gained from experiments yet. To this end, we used MD to characterize the conformational ensembles of the isolated proteins as well as the complex between them. When bound to pRb, the LFCSE motif of LTSV40 adopts a nearly linear conformation to fit into the LXCXE binding cleft. Our simulations revealed that, in the complex, the adjacent residues Glu108–Asp113 also tend to adopt an extended conformation. This behaviour contrasts sharply with the free form of LTSV40, where the entire Leu103–Asp113 segment typically assumes a curved and more compact shape. We thus conclude that the linear conformations observed in the complex are caused by binding to pRb.
As a consequence of the linear shape, typical structures of the pRb-LTSV40 conformational ensemble have the C-terminal side of the LFCSE motif close to one corner of the AB cleft (see Figure 2). This corner should be occupied by E2F if the whole transcription factor was present Lee et al. (2002). In other words, due to the extended conformations adopted by Linker 1, the C-terminal side of the LFCSE motif blocks regions that would be occupied by E2F in the E2F-pRb complex. This findings would explain why small fragments of LTSV40 and E2F can simultaneously bind to the LXCXE and AB clefts of pRb, respectively, but simultaneous binding of longer constructs is not feasible. Supplementary Figure S13 of the SM section provides a graphical representation of the pRb-E2F-1 complex, as predicted by Alphafold3 using PDB structure 1O9K as a template. The template comprises the A and B boxes of pRB plus residues 409 to 426, which form the core of the binding region of the transcription factor. Although the conformation of the disordered region of E2F-1 in the figure is uncertain, according to Alphafold3 estimates, it is clear that the residues immediately adjacent to the fragment present in the PDB structure are placed in the same region as that occupied by part of LTSV40 Linker 1 in our simulations of the pRb-LTSV40 complex.
It is well established that the LXCXE motif is the minimum sequence required for binding the LXCXE cleft of pRb (Kim et al., 2001; Lee et al., 1998). However, viral oncoproteins have acidic residues on the C-terminal side of the LXCXE motif that also contribute to pRb binding (Lee et al., 1998). In LTSV40, this region contains the conserved pentapeptide SSDDE (Barbosa et al., 1990). According to our simulations, this motif mainly interacts with Lys722, Lys729, Gln736, Lys740 and Arg741 of the pRb B box. The two force field combinations employed in this study agree to indicate that Lys740 has a significant stabilizing effect in the complex, while the
Brown et. al. also refer to the possibility that the attachment of oncoproteins to the LXCXE binding cleft could lead to an allosteric conformational change in the AB cleft, potentially preventing E2F attachment. However, the authors suggested that this mechanism is unlikely due to the rigidity observed for these sites in the crystal structures. Supporting this suggestion, our examination of the primary deformations of pRb revealed that the two binding clefts remain relatively rigid and are not affected by the attachment of LTSV40. Instead, two flexible loops of pRb, one located between
We would like to conclude this section by clearly outlining the scope and limitations of our study. Molecular dynamics simulations have proven to be a highly valuable tool for studying the behaviour of proteins and other biomolecules, providing support and complementing experimental studies. However, they also come with significant limitations that must be taken into account when interpreting their results. Among these limitations are the shortcomings of additive force fields, which are used in most studies, including ours, and the challenges associated with adequately sampling the conformational space of most biomolecules. In this work, the issues arising from force field deficiencies were addressed by employing two alternative force field combinations, both of which have been reported to perform well for disordered proteins. To tackle the sampling problem, we conducted 50 alternating heating and equilibration stages for each model containing LTSV40. While these precautions increase the confidence in our results, they do not provide certainty, particularly because there are no alternative experimental data against which to validate the simulations predictions. Therefore, the results presented here should be regarded as a well-founded hypothesis rather than a definitive conclusion.
Having made this caveat, we must nevertheless point out that certain aspects of biomolecular behaviour are so robustly embedded in their sequence and structure that they consistently emerge in MD simulations, despite the limitations of the computational models employed. A notable example of this is the folded structure of Trp-cage, which was accurately predicted de novo over 20 years ago, even with the limitations of the force fields available at the time. The consistency of the ff19SB/OPC and
4.2 Can HPV E7 and other polyomaviruses use a mechanism similar to that of LTSV40?
HPV E7 is a viral oncoprotein consisting of 98 amino acids that exhibits significant similarities to LTSV40. Functionally, both proteins bind to pRb to disrupt the cell cycle regulation. Structurally, both have an LXCXE motif that interacts with the LXCXE binding cleft of pRb, as well as a conserved mostly acidic pentapeptide located on the C-terminal side of that motif (Chen and Paucha, 1990; Christensen and Imperiale, 1995). Additionally, in the two cases, the pentapeptide is located within an intrinsically disordered region (Pelka et al., 2008). These characteristics are also common to most polyomaviruses. Supplementary Figure S14, available at the SM section, presents an alignment of representative polyomavirus large antigens with the prediction of IDRs, highlighting the LXCXE motif position. These similarities suggest a common evolutionary background that has driven them to develop comparable strategies for manipulating the host cell machinery and promote tumorigenesis (Barbosa et al., 1990).
In HPV E7, the LXCXE motif comprises the sequence Leu22-Glu26, while the segment of E2F that binds pRb is Asp410-Asp427 (Lee et al., 2002). Similar to the LTSV40-E2F case, short sequences of HPV E7 and E2F (HPV
Our findings regarding the position of the LTSV40 portion on the C-terminal side of the LXCXE motif, as well as the rigidity of the pRb clefts, align perfectly with the above-mentioned experiments that explore the mechanism by which HPV E7 blocks E2F binding to pRb. These similarities strongly suggest that both viral oncoproteins may employ the same blocking strategy. While there are no experimental studies akin to those conducted with HPV E7 for other polyomaviruses, sequence similarities and predictions of their intrinsically disordered regions suggest that they might utilize related strategies.
4.3 Is the mechanism common for pRb endogenous inactivation?
Hypophosphorylated pRb binds E2Fs, thereby inhibiting cell proliferation. However, in response to growth signals, pRb becomes phosphorylated and loses its ability to bind E2Fs. The phosphorylation sites of pRb are located on the C-terminal side of the pocket domain (Zhou et al., 2022; Zarkowska and Mittnacht, 1997; Ewens et al., 2017) and the region connecting that domain with the phosphorylation sites contains IDRs. Additionally, it is well established that the phosphorylated region interacts with basic residues in the B-box of the pocket domain, thereby inhibiting the formation of the E2F-pRb complex (Brown and Gallie, 2002; Kim et al., 2001; Lee et al., 1998). On the other hand, it has been shown that phosphorylated pRb cannot bind LTSV40 (Knudsen and Wang, 1996), and studies of the interaction between pRb and LXCXE-containing peptides suggest that pRb phosphorylation induces the release of these peptides via a competitive mechanism (Lee et al., 1998).
Although E2Fs and LXCXE-containing peptides/proteins bind to distinct regions of pRb, their release may be governed by a shared mechanism triggered by pRb phosphorylation. Previous experimental studies on the E2F/pRb complex suggest that phosphates attached to phosphorylation sites of pRb interact with the B-box, disrupting E2F binding and/or promoting its release (Brown and Gallie, 2002). We recall that the phosphorylation site and the B-box are connected by a lengthy IDR. On the other hand, our MD simulations revealed that in the LTSV40/pRb complex, the IDR in Linker 1 of LTSV40 sterically occludes the E2F-binding cleft of pRb. Integrating these findings suggests to extend Brown and Gallie’s hypothesis by proposing a model in which phosphorylation-dependent structural rearrangements of pRb, involving the IDR connecting the phosphorylation site to the B-box, block access to both the E2F and LXCXE binding sites, thereby preventing complex formation in any of them.
5 Conclusion
We have presented the results of MD simulations which provide significant insights into the structural and dynamical properties of the LTSV40 protein and its interaction with the pRb pocket domain. We have elucidated how fluctuations in pRb’s flexible regions facilitate access to the AB cleft, while the conformational dynamics of LTSV40 revealed essential binding characteristics. The characterization of the pRb-LTSV40 complex underscored the role of LTSV40 Linker 1 in inhibiting E2F binding, leading us to propose a novel mechanism for LTSV40-induced cell-cycle deregulation. According to the mechanism, in the pRb-LTSV40 complex, residues adjacent to the LXCXE binding motif adopt an extended conformation, causing the linker to block the entrance of the AB cleft. The SSDDE motif, located in the C-terminal side of the LXCXE motif, seems to play a significant role in achieving this conformation. This suggests that this motif could be a potential drug target aimed at mitigating the virus’s effects on E2F factor binding. Furthermore, our findings suggest that similar mechanisms may apply to other viral oncoproteins, highlighting a broader relevance in understanding pRb inactivation.
However, we must emphasize that the results presented in this article should be considered a reasonable hypothesis, which must be validated by experimental data, rather than a definitive conclusion. This is because the models used were assembled by combining parts of different experimentally determined structures, and because additive molecular dynamics force fields are not as reliable for describing intrinsically disordered proteins as they are for structured proteins. We have attempted to compensate for these limitations by using two alternative force field combinations and conducting a rather significant sampling. For this reason, we believe it is important to disclose the results, as they could inspire new experimental studies aimed at evaluating the proposed hypotheses. Summarizing, our work aims to contribute to our understanding of protein-protein interactions within regulatory pathways, hoping this can help open avenues for innovative therapeutic strategies against illnesses associated with viral infections and pRb dysfunction.
Data availability statement
Publicly available datasets were analyzed in this study. This data can be found here: https://gitlab.com/CLPF/prb-ltsv40.
Author contributions
CP: Data curation, Investigation, Visualization, Writing – original draft. NP: Conceptualization, Funding acquisition, Project administration, Resources, Writing – original draft. JP: Conceptualization, Investigation, Methodology, Software, Writing – original draft, Writing – review and editing. GP-S: Conceptualization, Investigation, Methodology, Supervision, Visualization, Writing – original draft, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Universidad Nacional de Quilmes (PP827-942/20), CONICET and ANPCyT (PICT 2020: 00192).
Acknowledgments
The authors thank to Dr. Hugo H. Juarez for the critical reading and comments on this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchbi.2025.1538350/full#supplementary-material
Footnotes
1A negative regulator is a protein that restrains or delays cell cycle progression at specific checkpoints, ensuring division occurs only under favorable conditions. An inhibitor, instead, is any substance that directly blocks cell cycle progression, regardless of conditions.
References
Ahuja, D., Sáenz-Robles, M. T., and Pipas, J. M. (2005). Sv40 large t antigen targets multiple cellular pathways to elicit cellular transformation. Oncogene 24, 7729–7745. doi:10.1038/sj.onc.1209046
Akdeniz Odemis, D., Kebudi, R., Bayramova, J., Kilic Erciyas, S., Kuru Turkcan, G., Tuncer, S. B., et al. (2023). Rb1 gene mutations and genetic spectrum in retinoblastoma cases. Medicine 102, e35068. doi:10.1097/md.0000000000035068
Altis, A., Otten, M., Nguyen, P. H., Hegger, R., and Stock, G. (2008). Construction of the free energy landscape of biomolecules via dihedral angle principal component analysis. J. Chem. Phys. 128, 245102. doi:10.1063/1.2945165
An, P., Sáenz Robles, M. T., and Pipas, J. M. (2012). Large t antigens of polyomaviruses: amazing molecular machines. Annu. Rev. Microbiol. 66, 213–236. doi:10.1146/annurev-micro-092611-150154
Barbosa, M., Edmonds, C., Fisher, C., Schiller, J., Lowy, D., and Vousden, K. (1990). The region of the hpv e7 oncoprotein homologous to adenovirus e1a and sv40 large t antigen contains separate domains for rb binding and casein kinase ii phosphorylation. EMBO J. 9, 153–160. doi:10.1002/j.1460-2075.1990.tb08091.x
Borchert, S., Czech-Sioli, M., Neumann, F., Schmidt, C., Wimmer, P., Dobner, T., et al. (2014). High-affinity rb binding, p53 inhibition, subcellular localization, and transformation by wild-type or tumor-derived shortened merkel cell polyomavirus large t antigens. J. virology 88, 3144–3160. doi:10.1128/jvi.02916-13
Brown, V. D., and Gallie, B. L. (2002). The b-domain lysine patch of prb is required for binding to large t antigen and release of e2f by phosphorylation. Mol. Cell. Biol. 22, 1390–1401. doi:10.1128/mcb.22.5.1390-1401.2002
Burkhart, D. L., and Sage, J. (2008). Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat. Rev. Cancer 8, 671–682. doi:10.1038/nrc2399
Casadevall, G., Casadevall, J., Duran, C., and Osuna, S. (2024). The shortest path method (spm) webserver for computational enzyme design. Protein Eng. Des. and Sel. PEDS 37, gzae005. doi:10.1093/protein/gzae005
Case, D., Ben-Shalom, I., Brozell, S., Cerutti, D., Cheatham, T., Cruzeiro, V., et al. (2018). Amber 18. San Francisco: University of California.
Chang, Y., Xu, M., Machado, A. C. D., Yu, X. J., Rohs, R., and Chen, X. S. (2013). Mechanism of origin dna recognition and assembly of an initiator-helicase complex by sv40 large tumor antigen. Cell Rep. 3, 1117–1127. doi:10.1016/j.celrep.2013.03.002
Chen, S., and Paucha, E. (1990). Identification of a region of simian virus 40 large t antigen required for cell transformation. J. virology 64, 3350–3357. doi:10.1128/jvi.64.7.3350-3357.1990
Chow, K., and Dean, D. C. (1996). Domains a and b in the rb pocket interact to form a transcriptional repressor motif. Mol. Cell. Biol. 16, 4862–4868. doi:10.1128/mcb.16.9.4862
Christensen, J. B., and Imperiale, M. J. (1995). Inactivation of the retinoblastoma susceptibility protein is not sufficient for the transforming function of the conserved region 2-like domain of simian virus 40 large t antigen. J. virology 69, 3945–3948. doi:10.1128/jvi.69.6.3945-3948.1995
Cossio-Pérez, R., Palma, J., and Pierdominici-Sottile, G. (2017). Consistent principal component modes from molecular dynamics simulations of proteins. J. Chem. Inf. Model. 57, 826–834. PMID: 28301154. doi:10.1021/acs.jcim.6b006461021/acs.jcim.6b00646
Darden, T., York, D., and Pedersen, L. (1993). Particle mesh ewald: an n.log(n) method for ewald sums in large systems. J. Chem. Phys. 98, 10089–10092. doi:10.1063/1.464397
Dick, F. A., and Rubin, S. M. (2013). Molecular mechanisms underlying rb protein function. Nat. Rev. Mol. cell Biol. 14, 297–306. doi:10.1038/nrm3567
Dyson, N. (1998). The regulation of e2f by prb-family proteins. Genes and Dev. 12, 2245–2262. doi:10.1101/gad.12.15.2245
Essmann, U., Perera, L., Berkowitz, M. L., Darden, T., Lee, H., and Pedersen, L. G. (1995). A smooth particle mesh ewald method. J. Chem. Phys. 103, 8577–8593. doi:10.1063/1.470117
Ewens, K. G., Bhatti, T. R., Moran, K. A., Richards-Yutz, J., Shields, C. L., Eagle, R. C., et al. (2017). Phosphorylation of prb: mechanism for rb pathway inactivation in mycn-amplified retinoblastoma. Cancer Med. 6, 619–630. doi:10.1002/cam4.1010
Fanning, E., and Knippers, R. (1992). Structure and function of simian virus 40 large tumor antigen. Annu. Rev. Biochem. 61, 55–85. doi:10.1146/annurev.bi.61.070192.000415
Fontes, M. R., Teh, T., Toth, G., John, A., Pavo, I., Jans, D. A., et al. (2003). Role of flanking sequences and phosphorylation in the recognition of the simian-virus-40 large t-antigen nuclear localization sequences by importin-α. Biochem. J. 375, 339–349. doi:10.1042/bj20030510
Genheden, S., and Ryde, U. (2015). The mm/pbsa and mm/gbsa methods to estimate ligand-binding affinities. Expert Opin. drug Discov. 10, 449–461. doi:10.1517/17460441.2015.1032936
Goodrich, D. W. (2006). The retinoblastoma tumor-suppressor gene, the exception that proves the rule. Oncogene 25, 5233–5243. doi:10.1038/sj.onc.1209616
Gouw, M., Michael, S., Sámano-Sánchez, H., Kumar, M., Zeke, A., Lang, B., et al. (2017). The eukaryotic linear motif resource – 2018 update. Nucleic Acids Res. 46, D428–D434. doi:10.1093/nar/gkx1077
Harrington, E. A., Bruce, J. L., Harlow, E., and Dyson, N. (1998). prb plays an essential role in cell cycle arrest induced by dna damage. Proc. Natl. Acad. Sci. 95, 11945–11950. doi:10.1073/pnas.95.20.11945
Hickman, E. S., Moroni, M. C., and Helin, K. (2002). The role of p53 and prb in apoptosis and cancer. Curr. Opin. Genet. and Dev. 12, 60–66. doi:10.1016/s0959-437x(01)00265-9
Huang, P. S., Patrick, D. R., Edwards, G., Goodhart, P. J., Huber, H. E., Miles, L., et al. (1993). Protein domains governing interactions between e2f, the retinoblastoma gene product, and human papillomavirus type 16 e7 protein. Mol. Cell. Biol. 13, 953–960. doi:10.1128/mcb.13.2.953
Izadi, S., Anandakrishnan, R., and Onufriev, A. V. (2014). Building water models: a different approach. J. Phys. Chem. Lett. 5, 3863–3871. doi:10.1021/jz501780a
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., et al. (2021). Highly accurate protein structure prediction with alphafold. nature 596, 583–589. doi:10.1038/s41586-021-03819-2
Kaustov, L., Ouyang, H., Amaya, M., Lemak, A., Nady, N., Duan, S., et al. (2011). Recognition and specificity determinants of the human cbx chromodomains. J. Biol. Chem. 286, 521–529. doi:10.1074/jbc.m110.191411
Kim, H.-Y., Ahn, B.-Y., and Cho, Y. (2001). Structural basis for the inactivation of retinoblastoma tumor suppressor by sv40 large t antigen. EMBO J. 20, 295–304. doi:10.1093/emboj/20.1.295
Kitao, A. (2022). Principal component analysis and related methods for investigating the dynamics of biological macromolecules. J 5, 298–317. doi:10.3390/j5020021
Knudsen, E. S., and Wang, J. Y. (1996). Differential regulation of retinoblastoma protein function by specific cdk phosphorylation sites. J. Biol. Chem. 271, 8313–8320. doi:10.1074/jbc.271.14.8313
Koder Hamid, M., Månsson, L. K., Meklesh, V., Persson, P., and Skepö, M. (2022). Molecular dynamics simulations of the adsorption of an intrinsically disordered protein: force field and water model evaluation in comparison with experiments. Front. Mol. Biosci. 9, 958175. doi:10.3389/fmolb.2022.958175
Konagaya, Y., Rosenthal, D., Ratnayeke, N., Fan, Y., and Meyer, T. (2024). An intermediate rb–e2f activity state safeguards proliferation commitment. Nature 631, 424–431. doi:10.1038/s41586-024-07554-2
Laskowski, R. A., Jabłońska, J., Pravda, L., Vařeková, R. S., and Thornton, J. M. (2018). Pdbsum: structural summaries of pdb entries. Protein Sci. 27, 129–134. doi:10.1002/pro.3289
Lee, C., Chang, J. H., Lee, H. S., and Cho, Y. (2002). Structural basis for the recognition of the e2f transactivation domain by the retinoblastoma tumor suppressor. Genes and Dev. 16, 3199–3212. doi:10.1101/gad.1046102
Lee, C., and Cho, Y. (2002). Interactions of sv40 large t antigen and other viral proteins with retinoblastoma tumour suppressor. Rev. Med. virology 12, 81–92. doi:10.1002/rmv.340
Lee, J.-O., Russo, A. A., and Pavletich, N. P. (1998). Structure of the retinoblastoma tumour-suppressor pocket domain bound to a peptide from hpv e7. Nature 391, 859–865. doi:10.1038/36038
Lilyestrom, W., Klein, M. G., Zhang, R., Joachimiak, A., and Chen, X. S. (2006). Crystal structure of sv40 large t-antigen bound to p53: interplay between a viral oncoprotein and a cellular tumor suppressor. Genes and Dev. 20, 2373–2382. doi:10.1101/gad.1456306
Liu, X., and Marmorstein, R. (2007). Structure of the retinoblastoma protein bound to adenovirus e1a reveals the molecular basis for viral oncoprotein inactivation of a tumor suppressor. Genes and Dev. 21, 2711–2716. doi:10.1101/gad.1590607
Luna-Vargas, M. P. A., Faesen, A. C., Van Dijk, W. J., Rape, M., Fish, A., and Sixma, T. K. (2011). Ubiquitin-specific protease 4 is inhibited by its ubiquitin-like domain. EMBO Rep. 12, 365–372. doi:10.1038/embor.2011.33
Manning, A. L., and Dyson, N. J. (2011). prb, a tumor suppressor with a stabilizing presence. Trends cell Biol. 21, 433–441. doi:10.1016/j.tcb.2011.05.003
Morris, E., and Dyson, N. (2001). Retinoblastoma protein partners. Adv. Cancer Res. 82, 1–54. PMID: 11447760. doi:10.1016/s0065-230x(01)82001-7s0065-230x(01)82001-7
Mu, Y., Nguyen, P. H., and Stock, G. (2005). Energy landscape of a small peptide revealed by dihedral angle principal component analysis. Proteins Struct. Funct. Bioinforma. 58, 45–52. doi:10.1002/prot.20310
Nevins, J. R. (2001). The rb/e2f pathway and cancer. Hum. Mol. Genet. 10, 699–703. doi:10.1093/hmg/10.7.699
Palma, J., and Pierdominici-Sottile, G. (2023). On the uses of pca to characterise molecular dynamics simulations of biological macromolecules: basics and tips for an effective use. ChemPhysChem 24, e202200491. doi:10.1002/cphc.2022004911002/cphc.202200491
Palopoli, N., González Foutel, N. S., Gibson, T. J., and Chemes, L. B. (2018). Short linear motif core and flanking regions modulate retinoblastoma protein binding affinity and specificity. Protein Eng. Des. Sel. 31, 69–77. doi:10.1093/protein/gzx068
Pelka, P., Ablack, J. N., Fonseca, G. J., Yousef, A. F., and Mymryk, J. S. (2008). Intrinsic structural disorder in adenovirus e1a: a viral molecular hub linking multiple diverse processes. J. virology 82, 7252–7263. doi:10.1128/jvi.00104-08
Piana, S., Donchev, A. G., Robustelli, P., and Shaw, D. E. (2015). Water dispersion interactions strongly influence simulated structural properties of disordered protein states. J. Phys. Chem. B 119, 5113–5123. doi:10.1021/jp508971m
Rauscher, S., Gapsys, V., Gajda, M. J., Zweckstetter, M., de Groot, B. L., and Grubmüller, H. (2015). Structural ensembles of intrinsically disordered proteins depend strongly on force field: a comparison to experiment. J. Chem. Theory Comput. 11, 5513–5524. doi:10.1021/acs.jctc.5b00736
Robustelli, P., Piana, S., and Shaw, D. E. (2018). Developing a molecular dynamics force field for both folded and disordered protein states. Proc. Natl. Acad. Sci. U. S. A. 115, E4758-E4766–E4766. doi:10.1073/pnas.1800690115
Shabane, P. S., Izadi, S., and Onufriev, A. V. (2019). General purpose water model can improve atomistic simulations of intrinsically disordered proteins. J. Chem. Theory Comput. 15, 2620–2634. doi:10.1021/acs.jctc.8b01123
Sittel, F., Jain, A., and Stock, G. (2014). Principal component analysis of molecular dynamics: on the use of cartesian vs. internal coordinates. J. Chem. Phys. 141 (07B605_1), 014111. doi:10.1063/1.4885338
Sittel, F., and Stock, G. (2018). Perspective: identification of collective variables and metastable states of protein dynamics. J. Chem. Phys. 149, 150901. doi:10.1063/1.5049637
Tian, C., Kasavajhala, K., Belfon, K. A., Raguette, L., Huang, H., Migues, A. N., et al. (2019). ff19sb: amino-acid-specific protein backbone parameters trained against quantum mechanics energy surfaces in solution. J. Chem. theory Comput. 16, 528–552. doi:10.1021/acs.jctc.9b00591
Varadi, M., Bertoni, D., Magana, P., Paramval, U., Pidruchna, I., Radhakrishnan, M., et al. (2024). Alphafold protein structure database in 2024: providing structure coverage for over 214 million protein sequences. Nucleic acids Res. 52, D368–D375. doi:10.1093/nar/gkad1011
Vorobiev, S. M., Su, M., Seetharaman, J., Huang, Y. J., Chen, C. X., Maglaqui, M., et al. (2009). Crystal structure of human retinoblastoma binding protein 9. Proteins 74, 526–529. doi:10.1002/prot.22278
Weinberg, R. A. (1995). The retinoblastoma protein and cell cycle control. cell 81, 323–330. doi:10.1016/0092-8674(95)90385-2
Whyte, P., Buchkovich, K. J., Horowitz, J. M., Friend, S. H., Raybuck, M., Weinberg, R. A., et al. (1988). Association between an oncogene and an anti-oncogene: the adenovirus e1a proteins bind to the retinoblastoma gene product. Nature 334, 124–129. doi:10.1038/334124a0
Wu, E. W., Clemens, K., Heck, D., and Münger, K. (1993). The human papillomavirus e7 oncoprotein and the cellular transcription factor e2f bind to separate sites on the retinoblastoma tumor suppressor protein. J. virology 67, 2402–2407. doi:10.1128/jvi.67.4.2402-2407.1993
Xiao, B., Spencer, J., Clements, A., Ali-Khan, N., Mittnacht, S., Broceno, C., et al. (2003). Crystal structure of the retinoblastoma tumor suppressor protein bound to e2f and the molecular basis of its regulation. Proc. Natl. Acad. Sci. 100, 2363–2368. doi:10.1073/pnas.0436813100
Zarkowska, T., and Mittnacht, S. (1997). Differential phosphorylation of the retinoblastoma protein by g1/s cyclin-dependent kinases. J. Biol. Chem. 272, 12738–12746. doi:10.1074/jbc.272.19.12738
Keywords: pRb, E2F, LTSV40, retinoblastoma protein, viral oncoproteins, MD simulations
Citation: Padilla Franzotti CL, Palopoli N, Palma J and Pierdominici-Sottile G (2025) Structural and dynamical characterization of pRb, LTSV40 and the pRb-LTSV40 complex suggests a common mechanism for pRb inactivation. Front. Chem. Biol. 4:1538350. doi: 10.3389/fchbi.2025.1538350
Received: 02 December 2024; Accepted: 31 March 2025;
Published: 23 April 2025.
Edited by:
Andrea Ilari, National Research Council (CNR), ItalyCopyright © 2025 Padilla Franzotti, Palopoli, Palma and Pierdominici-Sottile. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gustavo Pierdominici-Sottile, Z3NvdHRpbGVAdW5xLmVkdS5hcg==; Juliana Palma, anVsaWFuYUB1bnEuZWR1LmFy