 Fabian Morteo-Flores
Fabian Morteo-Flores Alberto Roldan
Alberto Roldan- Cardiff Catalysis Institute, School of Chemistry, Cardiff University, Cardiff, United Kingdom
Understanding the mechanisms of guaiacol’s catalytic hydrodeoxygenation (HDO) is essential to remove the oxygen excess in bio-oils. The present work systematically examines guaiacol’s HDO mechanisms to form benzene on six transition metal (TM) catalysts using density functional theory calculations. The results suggested a preferable Caryl−O bond scission on Ni (111) and Co (0001), whereas on Fe (110), the Caryl–OH bond scission is the most likely pathway. The C−O scission on Pd (111) and Pt (111) is not energetically feasible due to their high activation barriers and endothermic behaviour. Fe (110) also demonstrated its high oxophilic character by challenging the desorption of oxygenated products. A detailed analysis concludes that Co (0001) and Ni (111) are the most favourable in breaking phenolic compounds’ C−O type bonds. Brønsted-Evans-Polanyi (BEP) and transition state scaling (TSS) models were implemented on the catalytic results to derive trends and accelerate the catalyst design and innovation. TSS demonstrated a reliable trend in defining dissociation and association reaction energies. The phenyl ring-oxo-group and the metal-molecule distances complement the catalysts’ oxophilicity as selectivity descriptors in the HDO process.
1 Introduction
Currently, fossil fuels represent more than 70% of the world’s primary energy sources leading to significant pollution problems (Philippe et al., 2010). Biomass has emerged as a promising renewable resource within a sustainable circular economy to reduce society’s fossil fuel dependency and environmental concerns. Through fast pyrolysis technology, it is possible to transform biomass into liquid biofuels. However, one of the main disadvantages of fast pyrolysis is the high amount of oxygen compounds produced as aromatic alcohol and heterocyclic rings (around 20–50 wt%) (Mercader et al., 2010). These oxygenated compounds can polymerize under ambient conditions, causing an increase in the mixture’s viscosity and thermal instability during the combustion process. Therefore, oxygen content needs to be removed to yield biofuel with a high energy density (Furimsky, 2000). The catalytic hydrodeoxygenation (HDO) process is one of the most effective methods to remove oxygen from the pyrolytic mixture (Zakzeski et al., 2010). However, the process currently relies on high operational costs, and hence, new catalysts need to be developed with better stability, activity, and selectivity while increasing the HDO process efficiency (Jin et al., 2019).
Noble metals have been used as active phases in HDO catalysts for upgrading biofuels (Centeno et al., 1999; Gao et al., 2014; Yung et al., 2018). Although these metal catalysts show good performance, their implementation is limited because of their high cost. Transition metals (TMs) such as Ni and Co have emerged as alternative candidates due to their abundance and performance in hydrogenation reactions (Song et al., 2015; Fang et al., 2017). Indeed, several experiments have considered TMs as HDO catalysts, e.g., Fe (Olcese et al., 2012; Olcese et al., 2013), Co (Tran et al., 2016a; Tran et al., 2016b; Han et al., 2019), Ni (Zhao et al., 2011; Tran et al., 2016a), Cu (Deutsch and Shanks, 2012), Pd (Nimmanwudipong et al., 2011; Silva et al., 2021) and Pt (Nimmanwudipong et al., 2011; Silva et al., 2021), showing different selectivity as a function of the support, reaction temperature, and H2 partial pressure. These TMs have also been highlighted previously due to their oxygen and hydrogen affinity (Morteo-Flores et al., 2020).
Among the many species resulting from the pyrolytic treatment, guaiacol (1−hydroxy−2-methoxy benzene) has been used as a primary model compound for HDO investigations because it contains two types of O−functional groups, methoxy (−OCH3) and hydroxy (−OH), connected to an aromatic ring (Bui et al., 2011; Zhao et al., 2011; Bykova et al., 2012; Zhang et al., 2013; Morteo-Flores and Roldan, 2022). These features represent 39% of the components found in the phenolic fraction of biofuel after the fast-pyrolysis process (Lee et al., 2016). The most desired product upon guaiacol’s HDO is benzene because of the low hydrogen consumption; hence, reducing the production cost of unsaturated molecules (Garcia-Pintos et al., 2016).
The guaiacol HDO mechanism takes place through three different main pathways to form anisole, phenol and catechol compounds: dehydroxylation (Caryl−OH), demethylation (Calkyl–O) and demethoxylation (Caryl−OCH3) (Bui et al., 2011; Infantes-Molina et al., 2015; Teles et al., 2018; Phan et al., 2020). Different HDO experiments on TMs have also highlighted the presence of methane, water, and methanol in the products mixture (Tran et al., 2016b). Sun et al. studied the HDO of guaiacol on TM catalysts supported on carbon, e.g., Cu, Fe, Pd, Pt, and Ru. They found that catechol is the primary product, followed by phenol (Sun et al., 2013). According to DFT-based simulations on Pt (111) (Lu et al., 2015), the 3 C−O bonds have different dissociation energies, Caryl−OH (414 kJ/mol), Caryl−OCH3 (356 kJ/mol) and Calkyl–O (247 kJ/mol), being the demethylation route to catechol the most feasible pathway, also in agreement with previous publications (Bui et al., 2011; González-Borja and Resasco, 2011; Lan et al., 2018). Despite these results, the direct deoxygenation pathway is kinetically hindered (Lu et al., 2015; Lu and Heyden, 2015; Hensley et al., 2016; Zhou et al., 2019; Zhou and An, 2020). Co-adsorbed hydrogen on the catalysts is required to facilitate the C−O bond cleavage of oxy−compounds through the activation of the aromatic ring and the deoxygenation reaction (Zhu et al., 2016; Zhang et al., 2020).
A study based on understanding the mechanism characteristic of effective catalysts with a medium strength with oxy-compounds is necessary to engineer effective HDO catalysts performing at low temperatures. Previous studies on catalysts’ oxygen and hydrogen affinity drove us to select TMs catalysts favouring the deoxygenation and hydrogenation of products (Morteo-Flores et al., 2020; Morteo-Flores and Roldan, 2022). Therefore, in the present work, we employed density functional theory (DFT) to provide atomistic details on the guaiacol HDO mechanisms on six transition metal surfaces, Fe (110), Co (0001), Ni (111), Cu (111), Pd (111) and Pt (111). We proposed different reaction schemes based on the guaiacol C−O bond scission, i.e., i) Caryl−OH, ii) Caryl−OCH3, iii) Calkyl−O, as the first stage of the hydrogenation process. We selected and followed the most likely HDO pathway until producing benzene. The different C−O cleave energies are linked through the reaction energy profile, which allows rationalizing the catalyst’s performances and accelerating the design of new catalysts.
2 Computational Details
We have carried out spin-polarised density functional theory (DFT) calculations on transition metal slab models using the Vienna Ab initio Software Package (VASP) (Kresse and Furthmüller, 1996) to investigate the guaiacol HDO process on 6 TMs, i.e., Fe (110), Co (0001), Ni (111), Cu (111), Pd (111) and Pt (111). The exchange-correlation contributions were calculated using the generalized gradient approximation (GGA) with the revised functional of Perdew–Burke–Ernzerhof (RPBE) (Morteo-Flores et al., 2020). The core electrons were described using the Projected Augmented Wave (PAW) formalism (Blöchl, 1994), and a kinetic energy cut−off of 550 eV was chosen for the valence electron plane-wave basis set. The zero-damping Grimme’s empirical correction (D3) accounted for the long-range dispersion interactions (Grimme et al., 2011). Appropriate dipole correction was used perpendicular to the surfaces upon molecular adsorptions. The Brillouin zone was sampled with a Monkhorst−Pack 3 × 3 × 1 k-point grid. Isolated molecules were placed in a 20 × 20 × 20 Å box to avoid interactions with their periodic image. The convergence criteria were set to −0.03 eV Å−1 for the ionic and 10−5 eV for the electronic threshold. Slab models were generated with the atomic simulation environment (ASE) based on the optimized bulk lattice parameters (Hjorth Larsen et al., 2017). Each slab model is formed by a supercell size p(6 × 6) with five atomic layers; the top two layers were relaxed, and the bottom three were fixed to their bulk positions. A vacuum of 15 Å perpendicular to the surface was added to eliminate the interaction between periodic images.
The adsorption energies (
The reaction energy of each reaction step (Er; Eq. 2) is given by the difference between the final (EFS) and the initial (EIS) state energies. We combined the climbing-image nudged elastic band (CI−NEB) (Henkelman et al., 2000; Henkelman and Jónsson, 2000) and the improved dimer method to find the saddle points of the transition states (TS) structures, linking the minima across the reaction profile (Henkelman and Jónsson, 1999). All transition states have been characterized using vibrational analysis to confirm one imaginary frequency along the reaction coordinate. Zero-point energy correction was not included in this work. We defined the activation barrier (Ea; Eq. 3) as the energy difference between the initial and transition state (ETS) energies.
3 Results and Discussion
3.1 Guaiacol Adsorption
Different guaiacol adsorption modes on the six transition metal surfaces selected were investigated, being the parallel configuration the most favourable in agreement with previous works (Lee et al., 2015; Lu et al., 2015; Hensley et al., 2016; Liu et al., 2018a; Liu et al., 2018b; Verma and Kishore, 2018; Konadu et al., 2021). The adsorption sites with the strongest affinity for oxo-groups, i.e., OH and −OCH3, are seen at the hollow sites, Figure 1; Supplementary Table S1. At the same time, the C−C bonds of the aromatic ring prefer the bridge position between metals atoms (Liu et al., 2013). In all cases, the molecule bonds the surface through ring π-electrons, tilting the H atoms and the oxo groups away from the surface. Figure 1 shows the most favourable guaiacol adsorptions on the transition metal surfaces; less favourable adsorbed structures are shown in Supplementary Table S2.
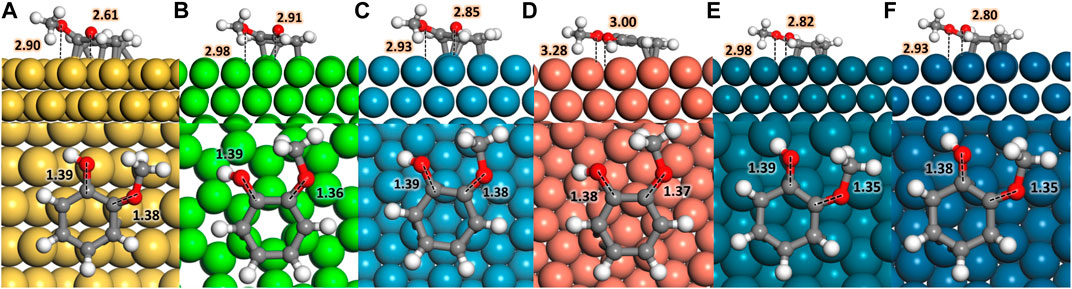
FIGURE 1. Top and side views of guaiacol adsorbed on (A) Fe (110), (B) Co (0001), (C) Ni (111), (D) Cu (111), (E) Pd (111), and (F) Pt (111); the metal atoms are represented in yellow, light green, blue, orange, dark green, and dark blue colour code, respectively. Red, white, and grey represent oxygen, hydrogen, and carbon. Inset distances are given in Å.
Table 1 shows the guaiacol adsorption energies on the most stable configurations in agreement with the literature’s results using comparable computational methods. The most favourable adsorption is on Pt (111), followed by Pd > Ni > Fe > Co > Cu. The guaiacol adsorption energy on Pt (111) is 0.37 eV stronger than previously reported (Lee et al., 2015) because of the different molecular coverage and different GGA pseudopotentials used. We took the most stable configurations as the initial guaiacol position to study the HDO reaction mechanism towards phenol, anisole, and catechol, which adsorptions follow the same metal preference as guaiacol. These results compare very well with different works, proving the validity of our methodology (Honkela et al., 2012; Lee et al., 2015; Hensley et al., 2016; Liu et al., 2018b; Porwal et al., 2019; Zhou et al., 2019; Zhou and An, 2020; Konadu et al., 2021). The interactions between the metallic surfaces and the phenolic molecules follow a similar trend, but not identical, as the metal oxophilicity (Morteo-Flores et al., 2020).
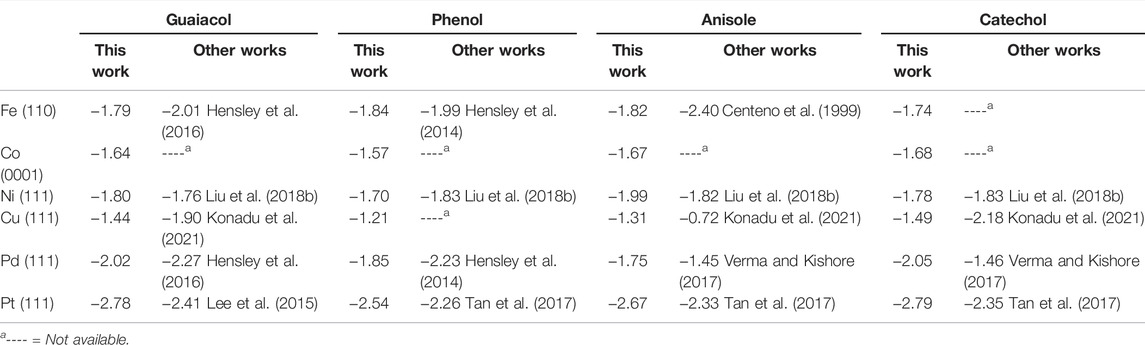
TABLE 1. Adsorption energy (in eV) for guaiacol, phenol, anisole, and catechol on the transition metal surfaces.
The interaction between the O atoms in guaiacol and the metal weakens the C−O bonds, indicating the bond activation (Gao et al., 2015; Liu et al., 2018a; Shi and Vohs, 2018). Many publications have considered the C−O bond elongation upon the guaiacol adsorption as a descriptor for the deoxygenation activity (Zhou et al., 2019; Liu et al., 2018b). The bond lengths within the molecule and the metal surface (M−O) were also examined, proving a clear relationship between the oxophilic nature of the metals with the M−O distances, Supplementary Table S3. Fe (110) has the shortest M−O distances, d (Fe−OH) = 2.61 Å and d (Fe−OCH3) = 2.90 Å, whereas Cu (111) present the largest one in the series with a d (Cu−OH) and d (Cu−OCH3) = 3.00 Å and 3.28 Å, respectively. Both metals, Fe and Cu, demonstrated their strong and weak interactions with the oxo-groups, making them poor candidates for the conversion of guaiacol according to Sabatier’s principle (Liu et al., 2018b).
We extended the bond activation concept across the oxo−groups in guaiacol, phenol, anisole, and catechol compounds, i.e., i) Caryl−OH, ii) Caryl−OCH3, and iii) Calkyl–O. The results are found in Supplementary Table S4. In all cases, Calkyl–O presented the most notorious elongation meaning that, in adsorbed guaiacol, it weakens more than the Caryl−OCH3, which showed little change. These trends agree with the reported Calkyl−O and Caryl−OCH3 dissociation bond energies of 2.56 and 3.69 eV, respectively (Bui et al., 2011; Jia et al., 2019). Namely, the demethylation route producing catechol seems to be the most favourable reaction in all the metals considered.
3.2 HDO Energy Profile
The guaiacol HDO reaction to form benzene may proceed through seven different pathways: the first three pathways (P1, P2, P3) lead to anisole, phenol, and catechol, respectively, pathways four and five convert catechol and anisole into phenol and pathways six and seven produce benzene as a final product, Scheme 1. The reaction mechanism starts with the guaiacol molecule adsorption and partial hydrogenation, activating the phenyl ring. The co-adsorbed H* (* denotes adsorbed species) promotes the C−O bond scission and saturates the ring’s dangling bonds.
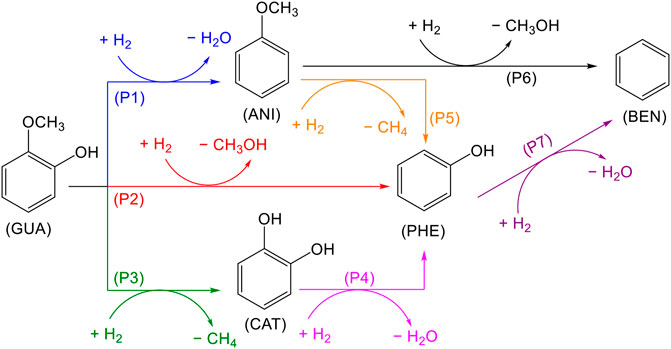
SCHEME 1. Proposed reaction network for the guaiacol HDO.
We examined the first three pathways (P1, P2, P3) on the six transition metal surfaces to identify the most favourable routes and continued the hydrogenation process to yield benzene. Upon C–O bond cleavage, there are two competing hydrogenation steps: (i) to saturate the dangling bonds in the ring and (ii) to reduce the cleavaged groups forming CH3OH, CH4 and H2O (e.g., −CH3 or −OH); Supplementary Tables S5–7 summarise the results on the different alternatives. Besides, each pathway in the HDO mechanism, described in Supplementary Table S8, has three TS. Reaction and activation energies (Er and Ea, respectively) are shown in Supplementary Tables S9, 10.
3.2.1 Guaiacol Dehydroxylation Pathway
The dehydroxylation (DHY) energy profile to produce anisole (P1) is shown in Supplementary Figure S2. The reaction is initiated with H* co-adsorption, which activates the ring through an electron transfer (from 0.37 e− to 0.09 e−). The extra electron in the molecular orbital disrupts the aromaticity and weakens the metal-phenolic interaction, provoking the Caryl−OH bond scission, Figure 2. The results show that breaking the Caryl−OH bond is exothermic and more likely on oxophilic catalysts. The activation barrier on Fe (110) is 1.13 eV, followed by Co > Ni > Pt > Cu > Pd. Indeed, guaiacol dehydroxylation is only exothermic on Fe and Co (Er = −0.83 eV and −0.18 eV, respectively). The Caryl−OH bond scission creates two co−adsorbed species: 2−methoxyphenyl and hydroxide. The formation of anisole is more feasible on Fe (110), which activation barrier is only 0.10 eV, followed by Pt, Ni, Pd, Cu, and Co. Finally, the last step is the formation of water as a subproduct. On Fe (110), it is endothermic (Er = +1.20 eV) and has a considerable activation barrier (Ea = 1.73 eV), thus, becoming the limiting reaction step.
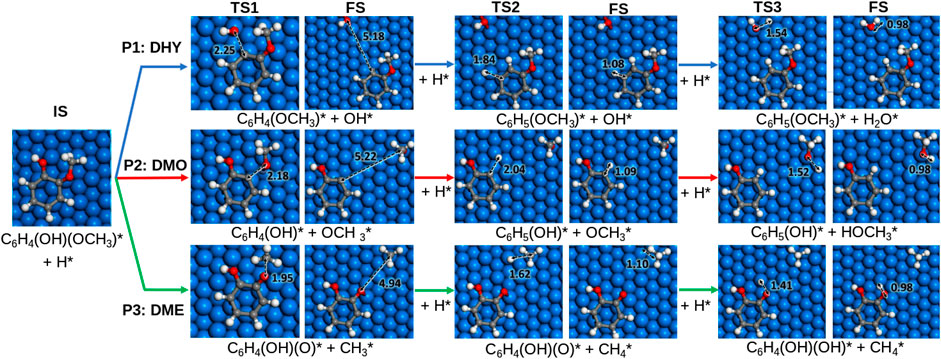
FIGURE 2. Proposed reaction pathways for the hydrodeoxygenation of guaiacol on Ni (111). Colour scheme: Oxygen, red; hydrogen, white; carbon, grey and nickel, blue. The asterisk (*) denotes adsorbed surface species. Functional groups are indicated in parenthesis.
3.2.2 Guaiacol Demethoxylation Pathway
Pathway 2 (P2) is the demethoxylation reaction (DMO) consisting of the guaiacol conversion into phenol and methanol. The energetic profile is shown in Supplementary Figure S3. The reaction pathway starts with the hydrogenation of guaiacol, provoking the Caryl−OCH3 scission, Figure 2. All the selected metals exhibit an endothermic behaviour except Fe (110), which shows an Er = −0.96 eV and the lowest activation energy (Ea = 1.28 eV) followed by Co > Ni > Pt > Pd, and Cu. The breaking of the bond creates two species: 2-hydroxyphenyl and methoxide. The formation of phenol is less energetically demanding for Ni with an activation barrier of 0.12 eV, followed by Co > Pt > Pd > Fe and Cu. The formation of methanol on Fe is also endothermic and kinetically unfavourable (Er = +1.15 eV and Ea = 1.78 eV). Accordingly, none of the metals explored promotes the demethoxylation pathway due to the high barrier to cleave the Caryl–OCH3 bond and the efficient CH3OH desorption.
3.2.3 Guaiacol Demethylation Pathway
The demethylation (DME) pathway (P3) converts guaiacol into catechol and methane. The pathway follows the scission of the Calkyl−O, as shown in Figure 2; Supplementary Figure S4 shows the DME energy profile. All the surfaces exhibit an exothermic behaviour except Cu (111) (Er = +0.04 eV). Indeed, Co (0001) and Ni (111) present the lowest activation energies (Ea = 1.23 and 1.21 eV) for the guaiacol demethylation. The results show the most accessible methane formation on Pt (111) (Ea = 0.36 eV) followed by Ni > Pd > Cu > Co and Fe; this is due to its good affinity with hydrogen, according to Supplementary Table S1. The catechol formation dominates the subsequent hydrogenation step, where Ni and Pd present the lowest activation barrier (Ea = 0.62 and 0.70 eV). Based on the DME results, metals with an average oxophilic character, like Ni, promote the cleavage of Calkyl−O.
The energy profiles (Supplementary Figures S2−4) and the results in Table 2 show that the guaiacol demethylation (DME) is the most likely pathway on Co, Ni, Cu, Pt, and Pd. It agrees with experimental data where catechol is the main product from guaiacol partial deoxygenation (Bykova et al., 2011; Lin et al., 2011; Sun et al., 2013). Still, it diverges from previous studies advocating for catalysts with high oxophilicity and hydrogen affinity (Chiu et al., 2014; Tan et al., 2017); one of the disadvantages of using a highly oxophilic metal is the difficulty formation and desorption of small molecular products, e.g., HO−CH3, CH4, and H2O, which hinders the overall reaction rate.
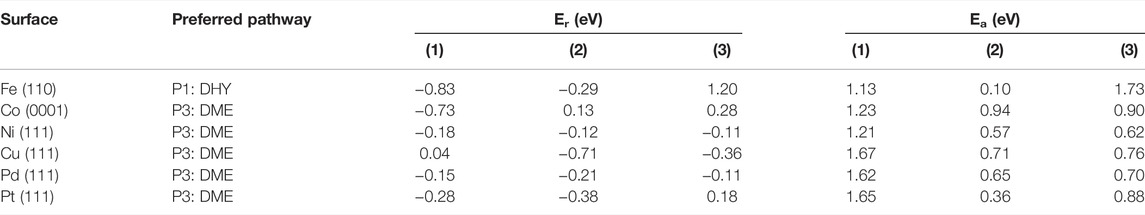
TABLE 2. Reaction (Er) and activation (Ea) energies (in eV) for the most feasible pathways on the six transition metal surfaces included in this work. DHY and DME stand for dehydroxylation and demethylation pathways, respectively.
3.2.4 Catechol Direct Deoxygenation Pathway
The direct deoxygenation (DDO) pathway is the second step in the guaiacol reduction process, Figure 3. It consists of the catechol conversion into phenol (pathway 4). Co (0001) presented the most favourable process for the Caryl−OH scission (Er = +0.02 eV, Ea = 1.23 eV), Supplementary Tables S11–12. In contrast, the Caryl−OH bond requires more energy to break on Ni (111); still, it is kinetically more accessible than on Pd (111) and Pt (111) (Ea = 2.36 and 2.11 eV, respectively). The DDO results on the metals are very similar to those presented by Zhou et al. on NiFe (111) (Zhou and An, 2020). Upon phenol formation, the evolution of water is less favourable in terms of reaction and activation energies; on Co (0001), it presents reaction and activation energies of +0.55 and 1.36 eV, respectively. In contrast, the −OH hydrogenation on Ni (111) has a favourable behaviour (Er = 0.02 and Ea = 0.82 eV, respectively). Nevertheless, the reducing conditions during the HDO process should shift the reaction towards H2O formation.

FIGURE 3. Energy profile of the direct deoxygenation (DDO) converting catechol into phenol and water. The asterisk (*) represents adsorbed surface species; molecular functional groups are denoted in parenthesis. Inset distances are given in Å.
3.2.5 Anisole Pathway
According to the results in Table 2, on Fe (110), dehydroxylation is the preferable route to convert guaiacol into anisole. From this point, the following hydrogenation reactions may take two possible routes: i) anisole to phenol via demethylation (P5, DME) and ii) anisole to benzene (P6, DMO) via demethoxylation, Figure 4. Both pathways are tested, and their schematic results are shown in the reaction profile, Figure 5.
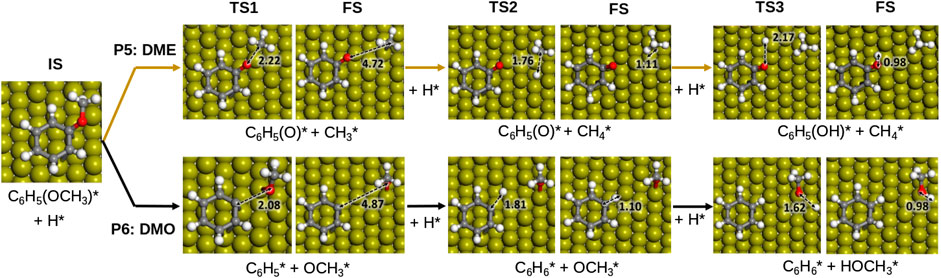
FIGURE 4. Proposed reaction scheme for the hydrodeoxygenation of anisole on Fe (110). Colour scheme: Oxygen, red; hydrogen, white; carbon, grey and Fe, dark yellow. Inset distances are given in Å.
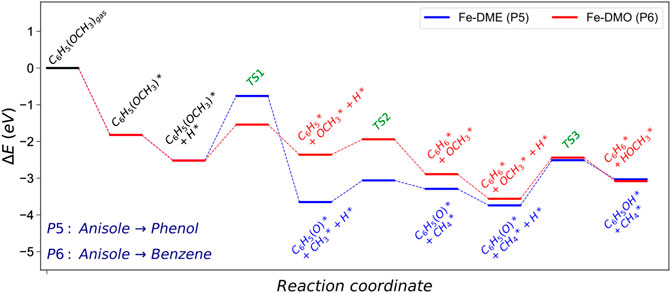
FIGURE 5. Energy profile of the demethylation (DME) and demethoxylation (DMO) pathways converting anisole into phenol and benzene on Fe (110). The asterisk (*) represents adsorbed surface species. Molecular functional groups are denoted in parenthesis.
The reaction mechanism starts with the co−adsorption of H* next to the phenyl ring, provoking the Calkyl−O bond activation. The process is exothermic (Er = −1.12 eV) with an activation energy of 1.76 eV, where the breaking of the bond creates two surface species: phenolate and methyl. The results show that the methane formation is an endothermic reaction (Er = 0.35 eV). The subsequent step is the phenol formation, which is also unfavourable (Er = 0.71 and Ea = 1.22 eV).
The demethoxylation reaction (P6) converts anisole into benzene and methanol (DMO). The H* co-adsorption reaction provokes the activation of the Caryl−OCH3, Figure 5 The scission step is endothermic with a relatively small activation energy (Er = 0.16 and Ea = 0.98 eV). The breaking of the bond creates two species: phenyl and methoxide. Benzene formation is an exothermic reaction (Er = −0.52 eV). However, the formation of methanol is an endothermic and kinetically hindered process (Er = 0.49 and Ea = 1.12 eV). The results demonstrate that Fe (110) preferentially breaks Caryl−OCH3 over Calkyl−O. This behaviour is also explained by the Fe high oxophilicity, which anchors O species such as, e.g., −OCH3, on its surface.
3.2.6 Phenol Hydrogenolysis Pathway
The hydrogenolysis (HGL) pathway is part of the second hydrogenation stage and forms benzene and water molecules from phenol. In this reaction, the cleavage of the phenol Caryl−OH is endothermic on all the metal catalysts studied, Figure 6. Co (0001) and Ni (111) are the metal surfaces that require less energy to break the Caryl−OH (Ea = 1.41 and 1.55 eV, respectively), although the energy barrier is still substantial, Supplementary Tables S13, 14. The noble metals, i.e., Pt and Pd, present the most hindered path, agreeing with previous experiments (Verma and Kishore, 2017). The Caryl−OH scission creates phenyl and hydroxide species, Supplementary Table S7. The phenyl hydrogenation is exothermic on all the surfaces investigated. Pt (111) shows the most feasible process (Er = −1.76 and Ea = 0.20 eV), although it is obstructed by the significant energy barrier to break Caryl−OH (Ea = 2.36 eV). Ni (111) is the most suitable catalyst for this pathway (P7) due to the relatively low energy required to break the Caryl−OH bond and hydrogenate the intermediates.
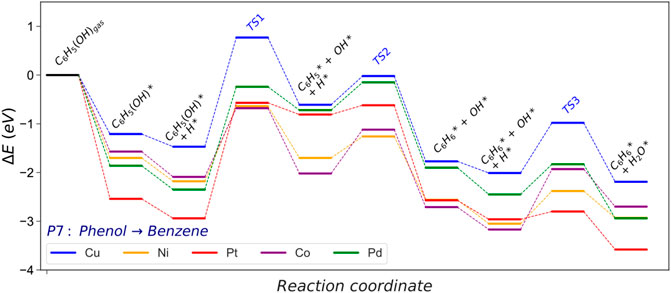
FIGURE 6. Energy profile of the hydrogenolysis (HGL) pathway converting phenol into benzene and water. The asterisk (*) represents adsorbed surface species; molecular functional groups are denoted in parenthesis.
3.3 BEP and TS Scaling Relations
Trends from the analysis performed on the different metal catalysts help develop eco-efficient catalysts by unravelling the scaling relation between reaction and activation energies. These trends describe crucial properties for engineering new catalysts of superior HDO activity towards the desired product. A sounded trend is represented by Brønsted-Evans-Polanyi (BEP), which shows a linear correlation between the reaction’s thermodynamics and kinetics (Logadottir et al., 2001; Alcala et al., 2003; Wang et al., 2011a; Wang et al., 2011b; Sutton and Vlachos, 2012). This relationship covers many essential reactions such as bond breaking and hydrogenations. An alternative method, which correlates the initial (EIS) or final state (EFS) energies with the transition state energy (ETS), is known as the transition state scaling (TSS). Supplementary Figure S5. Nevertheless, several publications have claimed these methods’ low accuracy trends (Wang et al., 2011a; Wang et al., 2014).
The heterogeneity of the processes, e.g., C−O scissions and hydrogenation, leads to a substantial mean absolute error (MAE) using BEP, TSS−IS or TSS−FS. Similarly, Wang et al. investigated the BEP and TSS models over different bond-breaking reactions occurring on the functional groups of furans on Pd (111). They found that the combination of all reactions decreases the accuracy (Wang et al., 2014). Our results combining all the steps are summarised in Supplementary Table S15; Supplementary Figure S6. Using these relationships to obtain activation barriers of C−O scissions and hydrogenations may lead to 1.70 eV errors. To improve the accuracy of these models, we divided the reaction data into two categories: (A) C−O bond cleavage and (B) hydrogenation reactions. A includes 1) Caryl−OH, 2) Caryl−OCH3 and 3) Calkyl−O. The B group consists of five hydrogenations: 4) Caryl−H, 5) CH3O−H, 6) H3C−H, 7) CarylO−H and 8) HO−H. We also used the root-mean-square deviation method (RMSE) and maximum absolute error (MiAE) to validate the accuracy of the methods. Supplementary Table S15 contains these trend series’s MAE, MiAE, RMSE and R2. The BEP relationship shows the lowest MAE for A cleavages, where the Caryl−OH and Caryl−OCH3 scissions reactions have the lowest mean error (0.16 and 0.21 eV, respectively). Although the MAE difference between TSS−IS and TSS−FS is considerable (≈0.09 eV), it is worth noting that Calkyl−O shows the lowest MAE for TSS-IS compared to the BEP relationship (0.16 eV). The BEP relationship again presents the lowest MAE (<0.20 eV) for B, whereas the MAE is up to 0.35 eV for TSS−IS and FS models, Figure 7.
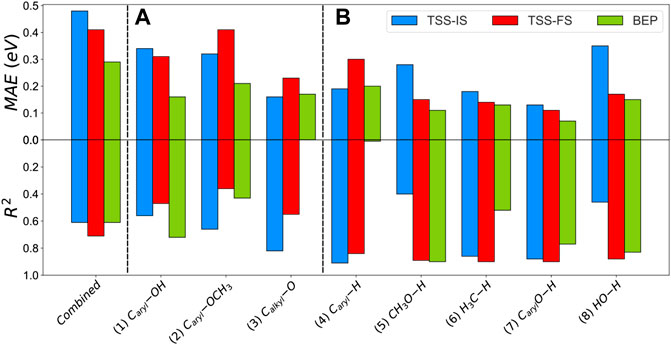
FIGURE 7. Mean absolute error (MAE) and R2 of the 1) combined, 2) scission of group (A) and 3) hydrogenation in group (B) for the TSS-IS, TSS-FS, and BEP relationships.
The BEP model does not correlate well with the bond scission data, provoking a low description of the activation energies for C−O cleavage. In general, TSS−IS and TSS−FS show low interdependence (R2 < 0.82) due to the variable nature of the transition state in group A. The TSS-FS model presents a good correlation factor for B, i.e., the hydrogenation series (R2 > 0.84).
We narrowed the A series down by considering only the fcc metals, i.e., excluding Fe (110) and Co. (0001), Supplementary Table S16. The BEP did not improve its predictability, contrarily to TSS−IS for Caryl−OCH3 and Calkyl−O (R2 = 0.95 and 0.89, respectively). The MAE also decreased considerably in the TSS models, making them more reliable to describe the C−O dissociation reactions. The same consideration in group B, i.e., only fcc metals, led to an accurate description between the initial and the transition state energies for the H3C−H and CarylO−H hydrogenation (R2 = 1.00 and 0.99, respectively). BEP shows acceptable accuracy only for forming small molecules such as CH3OH and H2O (R2 = 0.88 and 0.60, respectively).
Figure 8 shows the most accurate scaling models for each reaction (group A and B) for all the metals considered in this work. Figures 8A–C show the scaling models for the C−O bond scissions, where the TSS−IS model helps visualize the binding strength between lignin-derivate compounds and metals surfaces. For instance, Pt shows difficulties catalyzing the C−O bond scission due to its substantial activation energies and endothermic behaviour, Figures 8B,C. In contrast, Fe (110), Ni (111) and Co (0001) demonstrated exothermic C−O bond scissions with accessible energy barriers. Figures 8D–H) show the most robust scaling models for the hydrogenation reactions of small molecules and aromatic compounds where Fe (110) stands out due to its endothermic behaviour and a substantial energy barrier on the hydrogenation of Caryl−H bonds. Co (0001) displays good performance in the C−O bond scission but fails in the hydrogenation of small molecules such as CH3O−H. Whereas Ni (111) only shows acceptable energies for hydrogenation reactions. However, it is the most suitable for the C−O bond scission, both kinetically and thermodynamically, making it the most attractive catalysts candidate for the HDO of phenolic compounds.
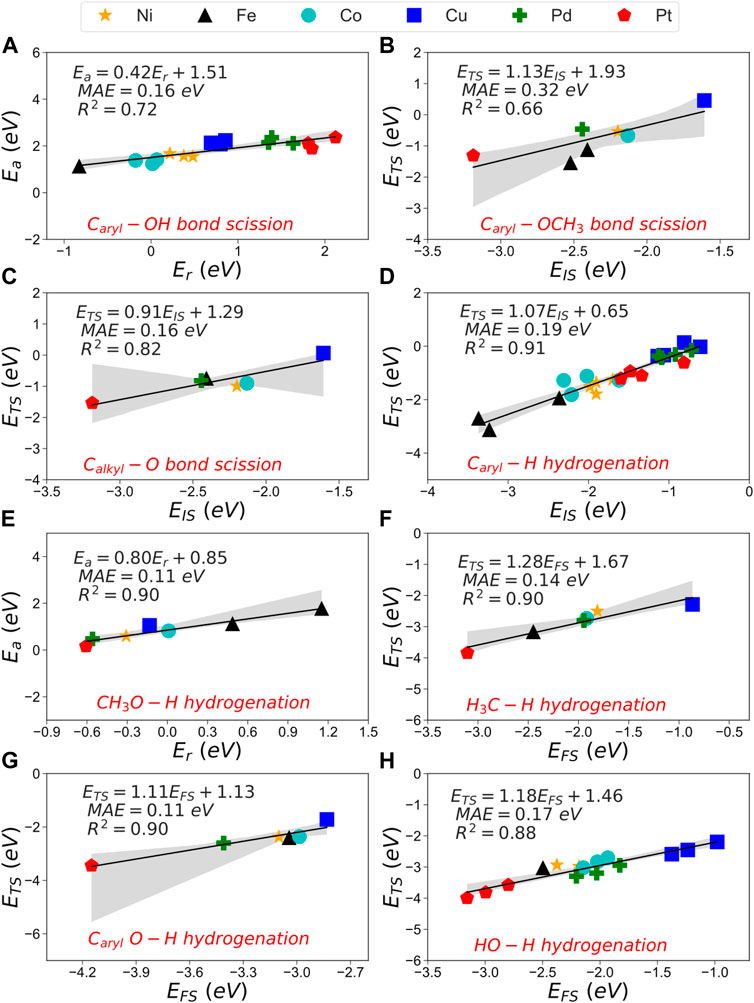
FIGURE 8. BEP (A,E), TSS-IS (B–D) and TSS-FS (F–H) of the C−O bond scission and hydrogenation reactions. Information on the linear equation, MAE and R2 are included in each graph.
4 Conclusion
Six transition metal surfaces, Fe (110), Co (0001), Ni (111), Cu (111), Pd (111) and Pt (111), were investigated as catalysts for the HDO of lignin derivates to benzene. As a model compound derived from lignin, guaiacol strongly interacts with Pt followed by Pd > Ni > Fe > Co > Cu. The adsorption energies are directly related to the metal-molecule distance and the metal oxophilicity for the functional groups such as −OH and −OCH3. The C−O bond elongation was evaluated as a descriptor to measure the C−O bond’s weakening (Caryl−OH, Caryl−OCH3 and Calkyl−O). These agreed with the guaiacol conversion into catechol as the most likely pathway. Three pathways were scrutinized in the first guaiacol reduction step (dihydroxylation, demethylation and demethoxylation). The results validated that the demethylation pathway (DME) to convert guaiacol into catechol is the most accessible mechanism on Co, Ni, Cu, Pd, and Pt follow the route guaiacol → catechol → phenol → benzene. In contrast, Fe (110) prefers producing anisole through the dehydroxylation (DHY) reaction pathway, following the route guaiacol → anisole → benzene.
These results pointed at the distance between the oxygen and the metal surfaces to complement the C−O bond elongation as descriptors to identify the most suitable HDO routes. Moving forward to design superior catalysts for the HDO process, scaling models were generated to evaluate the overall catalytic activity, i.e., Bronsted-Evans-Polanyi and transition state scaling (TSS). These trends correlate positively with grouping fcc metals and reaction types, C−O scission, and hydrogenation. The scaling models exhibited the main trends in the studied catalysts where Ni (111) and Co (0001) displayed an accessible activation barrier for C−O scissions. The latter, however, fails to hydrogenate small oxygenated compounds, leaving Ni as the most feasible transition-metal catalyst for the HDO of phenolic species.
Data Availability Statement
The datasets presented in this study can be found in online repositories at the University of Cardiff Research Portal orca websites http://doi.org/10.17035/d.2021.0143885773.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We acknowledge the United Kingdom Interdisciplinary centre for Circular Chemical Economy (NIC3E), which is funded by EPSRC (EP/V011863/1). FM-F gratefully acknowledges CONACYT (National Council for Science and Technology) for the student scholarship with the reference number 440221. Via our membership of the UK’s HEC Materials Chemistry Consortium, funded by EPSRC (EP/R029431), this work used the United Kingdom Materials and Molecular Modelling Hub for computational resources, MMM Hub, which is partially funded by EPSRC (EP/T022213). We also acknowledge Supercomputing Wales for access to the Hawk HPC facility, part-funded by the European Regional Development Fund via the Welsh Government. Moreover, this work used the Isambard United Kingdom National Tier-2 HPC Service (http://gw4.ac.uk/isambard/) operated by GW4 and the United Kingdom Met Office and funded by EPSRC (EP/P020224/1).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fctls.2022.861364/full#supplementary-material
References
Alcala, R., Mavrikakis, M., and Dumesic, J. A. (2003). DFT Studies for Cleavage of Cõ C and Cõ O Bonds in Surface Species Derived from Ethanol on Pt (111). J. Catal. 218, 178–190. doi:10.1016/s0021-9517(03)00090-3
Bui, V. N., Laurenti, D., Afanasiev, P., and Geantet, C. (2011). Hydrodeoxygenation of Guaiacol with CoMo Catalysts. Part I: Promoting Effect of Cobalt on HDO Selectivity and Activity. Appl. Catal. B Environ. 101, 239–245. doi:10.1016/j.apcatb.2010.10.025
Bykova, M. V., Bulavchenko, O. A., Ermakov, D. Y., Lebedev, M. Y., Yakovlev, V. A., and Parmon, V. N. (2011). Guaiacol Hydrodeoxygenation in the Presence of Ni-Containing Catalysts. Catal. Ind. 3, 15–22. doi:10.1134/s2070050411010028
Bykova, M. V., Ermakov, D. Y., Kaichev, V. V., Bulavchenko, O. A., Saraev, A. A., Lebedev, M. Y., et al. (2012). Ni-based Sol-Gel Catalysts as Promising Systems for Crude Bio-Oil Upgrading: Guaiacol Hydrodeoxygenation Study. Appl. Catal. B Environ. 113-114, 296–307. doi:10.1016/j.apcatb.2011.11.051
Centeno, A., Maggi, R., and Delmon, B. (1999). “Use of Noble Metals in Hydrodeoxygenation Reactions,” in Studies in Surface Science and Catalysis. Editors B. Delmon, G. F. Froment, and P. Grange (Elsevier), 77–84.
Chiu, C.-c., Genest, A., Borgna, A., and Rösch, N. (2014). Hydrodeoxygenation of Guaiacol over Ru(0001): A DFT Study. ACS Catal. 4, 4178–4188. doi:10.1021/cs500911j
Deutsch, K. L., and Shanks, B. H. (2012). Hydrodeoxygenation of Lignin Model Compounds over a Copper Chromite Catalyst. Appl. Catal. Gen. 447-448, 144–150. doi:10.1016/j.apcata.2012.09.047
Fang, H., Zheng, J., Luo, X., Du, J., Roldan, A., Leoni, S., et al. (2017). Product Tunable Behavior of Carbon Nanotubes-Supported Ni-Fe Catalysts for Guaiacol Hydrodeoxygenation. Appl. Catal. Gen. 529, 20–31. doi:10.1016/j.apcata.2016.10.011
Furimsky, E. (2000). Catalytic Hydrodeoxygenation. Appl. Catal. Gen. 199, 147–190. doi:10.1016/s0926-860x(99)00555-4
Gao, D., Schweitzer, C., Hwang, H. T., and Varma, A. (2014). Conversion of Guaiacol on Noble Metal Catalysts: Reaction Performance and Deactivation Studies. Ind. Eng. Chem. Res. 53, 18658–18667. doi:10.1021/ie500495z
Gao, D., Xiao, Y., and Varma, A. (2015). Guaiacol Hydrodeoxygenation over Platinum Catalyst: Reaction Pathways and Kinetics. Ind. Eng. Chem. Res. 54, 10638–10644. doi:10.1021/acs.iecr.5b02940
Garcia-Pintos, D., Voss, J., Jensen, A. D., and Studt, F. (2016). Hydrodeoxygenation of Phenol to Benzene and Cyclohexane on Rh (111) and Rh (211) Surfaces: Insights from Density Functional Theory. J. Phys. Chem. C 120, 18529–18537. doi:10.1021/acs.jpcc.6b02970
González-Borja, M. Á., and Resasco, D. E. (2011). Anisole and Guaiacol Hydrodeoxygenation over Monolithic Pt–Sn Catalysts. Energy Fuels 25, 4155–4162. doi:10.1021/ef200728r
Grimme, S., Ehrlich, S., and Goerigk, L. (2011). Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 32, 1456–1465. doi:10.1002/jcc.21759
Han, G.-H., Lee, M. W., Park, S., Kim, H. J., Ahn, J.-P., Seo, M.-g., et al. (2019). Revealing the Factors Determining the Selectivity of Guaiacol HDO Reaction Pathways Using ZrP-Supported Co and Ni Catalysts. J. Catal. 377, 343–357. doi:10.1016/j.jcat.2019.07.034
Henkelman, G., and Jónsson, H. (1999). A Dimer Method for Finding Saddle Points on High Dimensional Potential Surfaces Using Only First Derivatives. J. Chem. Phys. 111, 7010–7022. doi:10.1063/1.480097
Henkelman, G., and Jónsson, H. (2000). Improved Tangent Estimate in the Nudged Elastic Band Method for Finding Minimum Energy Paths and Saddle Points. J. Chem. Phys. 113, 9978–9985. doi:10.1063/1.1323224
Henkelman, G., Uberuaga, B. P., and Jónsson, H. (2000). A Climbing Image Nudged Elastic Band Method for Finding Saddle Points and Minimum Energy Paths. J. Chem. Phys. 113, 9901–9904. doi:10.1063/1.1329672
Hensley, A. J. R., Wang, Y., and McEwen, J.-S. (2016). Adsorption of Guaiacol on Fe (110) and Pd (111) from First Principles. Surf. Sci. 648, 227–235. doi:10.1016/j.susc.2015.10.030
Hensley, A. J. R., Wang, Y., and McEwen, J.-S. (2014). Adsorption of Phenol on Fe (110) and Pd (111) from First Principles. Surf. Sci. 630, 244–253. doi:10.1016/j.susc.2014.08.003
Hjorth Larsen, A., Jørgen Mortensen, J., Blomqvist, J., Castelli, I. E., Christensen, R., Dułak, M., et al. (2017). The Atomic Simulation Environment-A Python Library for Working with Atoms. J. Phys. Condens. Matter 29, 273002. doi:10.1088/1361-648x/aa680e
Honkela, M. L., Björk, J., and Persson, M. (2012). Computational Study of the Adsorption and Dissociation of Phenol on Pt and Rh Surfaces. Phys. Chem. Chem. Phys. 14, 5849–5854. doi:10.1039/c2cp24064e
Infantes-Molina, A., Pawelec, B., Fierro, J. L. G., Loricera, C. V., Jiménez-López, A., and Rodríguez-Castellón, E. (2015). Effect of Ir and Pt Addition on the HDO Performance of RuS2/SBA-15 Sulfide Catalysts. Top. Catal. 58, 247–257. doi:10.1007/s11244-015-0366-0
Jia, X., An, W., Wang, Z., and Zhou, J. (2019). Effect of Doped Metals on Hydrodeoxygenation of Phenol over Pt-Based Bimetallic Alloys: Caryl–OH Versus CaliphaticH–OH Bond Scission. J. Phys. Chem. C 123, 16873–16882. doi:10.1021/acs.jpcc.9b04457
Jin, W., Pastor-Pérez, L., Shen, D., Sepúlveda-Escribano, A., Gu, S., and Ramirez Reina, T. (2019). Catalytic Upgrading of Biomass Model Compounds: Novel Approaches and Lessons Learnt from Traditional Hydrodeoxygenation - a Review. ChemCatChem 11, 924–960. doi:10.1002/cctc.201801722
Konadu, D., Kwawu, C. R., Tia, R., Adei, E., and de Leeuw, N. H. (2021). Mechanism of Guaiacol Hydrodeoxygenation on Cu (111): Insights from Density Functional Theory Studies. Catalysts 11, 523. doi:10.3390/catal11040523
Kresse, G., and Furthmüller, J. (1996). Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mater. Sci. 6, 15–50. doi:10.1016/0927-0256(96)00008-0
Lan, X., Hensen, E. J. M., and Weber, T. (2018). Hydrodeoxygenation of Guaiacol over Ni2P/SiO2–Reaction Mechanism and Catalyst Deactivation. Appl. Catal. Gen. 550, 57–66. doi:10.1016/j.apcata.2017.10.018
Lee, H., Kim, H., Yu, M. J., Ko, C. H., Jeon, J. K., Jae, J., et al. (2016). Catalytic Hydrodeoxygenation of Bio-Oil Model Compounds over Pt/HY Catalyst. Sci. Rep. 6, 28765–28768. doi:10.1038/srep28765
Lee, K., Gu, G. H., Mullen, C. A., Boateng, A. A., and Vlachos, D. G. (2015). Guaiacol Hydrodeoxygenation Mechanism on Pt (111): Insights from Density Functional Theory and Linear Free Energy Relations. ChemSusChem 8, 315–322. doi:10.1002/cssc.201402940
Lin, Y.-C., Li, C.-L., Wan, H.-P., Lee, H.-T., and Liu, C.-F. (2011). Catalytic Hydrodeoxygenation of Guaiacol on Rh-Based and Sulfided CoMo and NiMo Catalysts. Energy fuels. 25, 890–896. doi:10.1021/ef101521z
Liu, W., Ruiz, V. G., Zhang, G.-X., Santra, B., Ren, X., Scheffler, M., et al. (2013). Structure and energetics of benzene adsorbed on transition-metal surfaces: density-functional theory with van der Waals interactions including collective substrate response. New J. Phys. 15, 053046. doi:10.1088/1367-2630/15/5/053046
Liu, X., An, W., Turner, C. H., and Resasco, D. E. (2018a). Hydrodeoxygenation of M-Cresol over Bimetallic NiFe Alloys: Kinetics and Thermodynamics Insight into Reaction Mechanism. J. Catal. 359, 272–286. doi:10.1016/j.jcat.2018.01.006
Liu, X., An, W., Wang, Y., Turner, C. H., and Resasco, D. E. (2018b). Hydrodeoxygenation of Guaiacol over Bimetallic Fe-Alloyed (Ni, Pt) Surfaces: Reaction Mechanism, Transition-State Scaling Relations and Descriptor for Predicting C–O Bond Scission Reactivity. Catal. Sci. Technol. 8, 2146–2158. doi:10.1039/c8cy00282g
Logadottir, A., Rod, T. H., Nørskov, J. K., Hammer, B., Dahl, S., and Jacobsen, C. J. H. (2001). The Brønsted-Evans-Polanyi Relation and the Volcano Plot for Ammonia Synthesis over Transition Metal Catalysts. J. Catal. 197, 229–231. doi:10.1006/jcat.2000.3087
Lu, J., Behtash, S., Mamun, O., and Heyden, A. (2015). Theoretical Investigation of the Reaction Mechanism of the Guaiacol Hydrogenation over a Pt (111) Catalyst. ACS Catal. 5, 2423–2435. doi:10.1021/cs5016244
Lu, J., and Heyden, A. (2015). Theoretical Investigation of the Reaction Mechanism of the Hydrodeoxygenation of Guaiacol over a Ru (0001) Model Surface. J. Catal. 321, 39–50. doi:10.1016/j.jcat.2014.11.003
Mercader, F. d. M., Groeneveld, M. J., Kersten, S. R. A., Venderbosch, R. H., and Hogendoorn, J. A. (2010). Pyrolysis Oil Upgrading by High Pressure Thermal Treatment. Fuel 89, 2829–2837. doi:10.1016/j.fuel.2010.01.026
Morteo-Flores, F., Engel, J., and Roldan, A. (2020). Biomass Hydrodeoxygenation Catalysts Innovation from Atomistic Activity Predictors. Phil. Trans. R. Soc. A 378, 20200056. doi:10.1098/rsta.2020.0056
Morteo-Flores, F., and Roldan, A. (2022). The Effect of Pristine and Hydroxylated Oxide Surfaces on the Guaiacol HDO Process: A DFT Study. ChemPhysChem 23, e202100583.
Nimmanwudipong, T., Runnebaum, R. C., Block, D. E., and Gates, B. C. (2011). Catalytic Conversion of Guaiacol Catalyzed by Platinum Supported on Alumina: Reaction Network Including Hydrodeoxygenation Reactions. Energy fuels. 25, 3417–3427. doi:10.1021/ef200803d
Olcese, R., Bettahar, M. M., Malaman, B., Ghanbaja, J., Tibavizco, L., Petitjean, D., et al. (2013). Gas-phase Hydrodeoxygenation of Guaiacol over Iron-Based Catalysts. Effect of Gases Composition, Iron Load and Supports (Silica and Activated Carbon). Appl. Catal. B Environ. 129, 528–538. doi:10.1016/j.apcatb.2012.09.043
Olcese, R. N., Bettahar, M., Petitjean, D., Malaman, B., Giovanella, F., and Dufour, A. (2012). Gas-phase Hydrodeoxygenation of Guaiacol over Fe/SiO2 Catalyst. Appl. Catal. B Environ. 115-116, 63–73. doi:10.1016/j.apcatb.2011.12.005
Phan, D.-P., Vo, T. K., Le, V. N., Kim, J., and Lee, E. Y. (2020). Spray Pyrolysis Synthesis of Bimetallic NiMo/Al2O3–TiO2 Catalyst for Hydrodeoxygenation of Guaiacol: Effects of Bimetallic Composition and Reduction Temperature. J. Industrial Eng. Chem. 83, 351–358. doi:10.1016/j.jiec.2019.12.008
Philippe, M., Richard, F., Hudebine, D., and Brunet, S. (2010). Inhibiting Effect of Oxygenated Model Compounds on the HDS of Dibenzothiophenes over CoMoP/Al2O3 Catalyst. Appl. Catal. Gen. 383, 14–23. doi:10.1016/j.apcata.2010.04.055
Porwal, G., Gupta, S., Sreedhala, S., Elizabeth, J., Khan, T. S., Haider, M. A., et al. (2019). Mechanistic Insights into the Pathways of Phenol Hydrogenation on Pd Nanostructures. ACS Sustain. Chem. Eng. 7, 17126–17136. doi:10.1021/acssuschemeng.9b03392
Shi, D., and Vohs, J. M. (2018). Mechanistic Study of the Hydrodeoxygenation of Lignin-Derived Oxygenates on a CoPt Bimetallic Catalyst: Reaction of Anisole on Co-Modified Pt (111). J. Phys. Energy 1, 015003. doi:10.1088/2515-7655/aadf55
Silva, N. K. G., Ferreira, R. A. R., Ribas, R. M., Monteiro, R. S., Barrozo, M. A. S., and Soares, R. R. (2021). Gas-phase Hydrodeoxygenation (HDO) of Guaiacol Over Pt/Al2O3 Catalyst Promoted by Nb2O5. Fuel 287, 119509. doi:10.1016/j.fuel.2020.119509
Song, W., Liu, Y., Baráth, E., Zhao, C., and Lercher, J. A. (2015). Synergistic Effects of Ni and Acid Sites for Hydrogenation and C-O Bond Cleavage of Substituted Phenols. Green Chem. 17, 1204–1218. doi:10.1039/c4gc01798f
Sun, J., Karim, A. M., Zhang, H., Kovarik, L., Li, X. S., Hensley, A. J., et al. (2013). Carbon-supported Bimetallic Pd-Fe Catalysts for Vapor-phase Hydrodeoxygenation of Guaiacol. J. Catal. 306, 47–57. doi:10.1016/j.jcat.2013.05.020
Sutton, J. E., and Vlachos, D. G. (2012). A Theoretical and Computational Analysis of Linear Free Energy Relations for the Estimation of Activation Energies. ACS Catal. 2, 1624–1634. doi:10.1021/cs3003269
Tan, Q., Wang, G., Long, A., Dinse, A., Buda, C., Shabaker, J., et al. (2017). Mechanistic Analysis of the Role of Metal Oxophilicity in the Hydrodeoxygenation of Anisole. J. Catal. 347, 102–115. doi:10.1016/j.jcat.2017.01.008
Teles, C. A., de Souza, P. M., Rabelo-Neto, R. C., Griffin, M. B., Mukarakate, C., Orton, K. A., et al. (2018). Catalytic Upgrading of Biomass Pyrolysis Vapors and Model Compounds Using Niobia Supported Pd Catalyst. Appl. Catal. B Environ. 238, 38–50. doi:10.1016/j.apcatb.2018.06.073
Tran, N. T. T., Uemura, Y., Chowdhury, S., and Ramli, A. (2016a). Vapor-phase Hydrodeoxygenation of Guaiacol on Al-MCM-41 Supported Ni and Co Catalysts. Appl. Catal. Gen. 512, 93–100. doi:10.1016/j.apcata.2015.12.021
Tran, N. T. T., Uemura, Y., and Ramli, A. (2016b). Hydrodeoxygenation of Guaiacol over Al-MCM-41 Supported Metal Catalysts: A Comparative Study of Co and Ni. Procedia Eng. 148, 1252–1258. doi:10.1016/j.proeng.2016.06.488
Verma, A. M., and Kishore, N. (2018). Molecular Modeling Approach to Elucidate Gas Phase Hydrodeoxygenation of Guaiacol over a Pd (111) Catalyst within DFT Framework. J. Mol. Model 24, 254–270. doi:10.1007/s00894-018-3803-8
Verma, A. M., and Kishore, N. (2017). Molecular Simulations of Palladium Catalysed Hydrodeoxygenation of 2-hydroxybenzaldehyde Using Density Functional Theory. Phys. Chem. Chem. Phys. 19, 25582–25597. doi:10.1039/c7cp05113a
Wang, S., Petzold, V., Tripkovic, V., Kleis, J., Howalt, J. G., Skúlason, E., et al. (2011a). Universal Transition State Scaling Relations for (de) Hydrogenation Over Transition Metals. Phys. Chem. Chem. Phys. 13, 20760–20765. doi:10.1039/c1cp20547a
Wang, S., Temel, B., Shen, J., Jones, G., Grabow, L. C., Studt, F., et al. (2011b). Universal Brønsted-Evans-Polanyi Relations for C-C, C-O, C-N, N-O, N-N, and O-O Dissociation Reactions. Catal. Lett. 141, 370–373. doi:10.1007/s10562-010-0477-y
Wang, S., Vorotnikov, V., Sutton, J. E., and Vlachos, D. G. (2014). Brønsted–Evans–Polanyi and Transition State Scaling Relations of Furan Derivatives on Pd (111) and Their Relation to Those of Small Molecules. ACS Catal. 4, 604–612. doi:10.1021/cs400942u
Yung, M. M., Foo, G. S., and Sievers, C. (2018). Role of Pt during Hydrodeoxygenation of Biomass Pyrolysis Vapors over Pt/HBEA. Catal. Today 302, 151–160. doi:10.1016/j.cattod.2017.03.014
Zakzeski, J., Bruijnincx, P. C. A., Jongerius, A. L., and Weckhuysen, B. M. (2010). The Catalytic Valorization of Lignin for the Production of Renewable Chemicals. Chem. Rev. 110, 3552–3599. doi:10.1021/cr900354u
Zhang, J., Sun, J., and Wang, Y. (2020). Recent Advances in the Selective Catalytic Hydrodeoxygenation of Lignin-Derived Oxygenates to Arenes. Green Chem. 22, 1072–1098. doi:10.1039/c9gc02762a
Zhang, X., Wang, T., Ma, L., Zhang, Q., Yu, Y., and Liu, Q. (2013). Characterization and Catalytic Properties of Ni and NiCu Catalysts Supported on ZrO2-SiO2 for Guaiacol Hydrodeoxygenation. Catal. Commun. 33, 15–19. doi:10.1016/j.catcom.2012.12.011
Zhao, H. Y., Li, D., Bui, P., and Oyama, S. T. (2011). Hydrodeoxygenation of Guaiacol as Model Compound for Pyrolysis Oil on Transition Metal Phosphide Hydroprocessing Catalysts. Appl. Catal. Gen. 391, 305–310. doi:10.1016/j.apcata.2010.07.039
Zhou, J., and An, W. (2020). Unravelling the Role of Oxophilic Metal in Promoting the Deoxygenation of Catechol on Ni-Based Alloy Catalysts. Catal. Sci. Technol. 10, 6849–6859. doi:10.1039/d0cy01361g
Zhou, J., An, W., Wang, Z., and Jia, X. (2019). Hydrodeoxygenation of Phenol over Ni-Based Bimetallic Single-Atom Surface Alloys: Mechanism, Kinetics and Descriptor. Catal. Sci. Technol. 9, 4314–4326. doi:10.1039/c9cy01082c
Keywords: hydrodeoxygenation (HDO), scaling relationships, transition states (TSs), heterogeneous catalysis, density functional theory, guaiacol, lignin
Citation: Morteo-Flores F and Roldan A (2022) Mechanisms and Trends of Guaiacol Hydrodeoxygenation on Transition Metal Catalysts. Front. Catal. 2:861364. doi: 10.3389/fctls.2022.861364
Received: 24 January 2022; Accepted: 14 April 2022;
Published: 23 May 2022.
Edited by:
Cong Liu, Argonne National Laboratory (DOE), United StatesReviewed by:
Mal-Soon Lee, Pacific Northwest National Laboratory (DOE), United StatesJose Luis Pinilla, Spanish National Research Council (CSIC), Spain
Copyright © 2022 Morteo-Flores and Roldan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alberto Roldan, cm9sZGFubWFydGluZXphQGNhcmRpZmYuYWMudWs=